Abstract
Incidence of cardiac arrhythmias increases significantly with age. In order to effectively stratify arrhythmic risk in the aging population it is crucial to elucidate the relevant underlying molecular mechanisms. The changes underlying age-related electrophysiological disruption appear to be closely associated with mitochondrial dysfunction. Thus, the present review examines the mechanisms by which age-related mitochondrial dysfunction promotes arrhythmic triggers and substrate. Namely, via alterations in plasmalemmal ionic currents (both sodium and potassium), gap junctions, cellular Ca2+ homeostasis, and cardiac fibrosis. Stratification of patients' mitochondrial function status permits application of appropriate anti-arrhythmic therapies. Here, we discuss novel potential anti-arrhythmic pharmacological interventions that specifically target upstream mitochondrial function and hence ameliorates the need for therapies targeting downstream changes which have constituted traditional antiarrhythmic therapy.
Introduction
Aging is the progressive decline in the fitness of an organism due to cumulative organ-specific physiological deterioration (1, 2). The advancement of modern medicine is thus reflected in increasing human life expectancy (3). However, an aging population offers novel medical challenges with increasing prevalence of a number of conditions including cardiovascular, oncological, and neurological diseases. The incidence of cardiovascular diseases increases exponentially in the elderly population (4, 5). In the aging population, cardiovascular diseases are the leading cause of morbidity and mortality (3, 5, 6). Thus, cardiovascular diseases have a prevalence of 82.6 million (36.2%) in the United States (4) carrying a greater financial burden than any other group of diseases including cancer and benign neoplasms (4). In 2007, 33.6% of all deaths (~814,000 people) in the United States had cardiovascular disease as the underlying cause of death (4). It is estimated that eliminating mortality from cardiovascular diseases would add between 5.5 and 7 years to mean life expectancy (4, 7). As the aging population continues to increase, with the number of elderly people predicted to double in the next 25 years in the United States age related cardiovascular diseases will continue to represent a major public health concern (5, 8). As such, it is increasingly important to be able to stratify risk of cardiovascular diseases by age and understand their underlying age-related molecular mechanisms in order to develop effective pharmacological therapies.
Within cardiovascular diseases, cardiac arrhythmias arise due to disruption in the orderly sequence of cardiomyocyte action potential activation and recovery through successive regions of the myocardium compromising cardiac function (9, 10). Of atrial arrhythmias, atrial fibrillation (AF) is the most common type. It is associated with major morbidity by increasing the risk of stroke and heart failure, as well as all-cause mortality (11–13). Ventricular arrhythmias such as ventricular tachycardia often degenerating into ventricular fibrillation (VF) are also a major public health concern. They constitute the primary cause of sudden cardiac death (SCD), which accounts for 4–5 million deaths/year worldwide (14) representing over 5% of overall mortality (15).
Incidence of cardiac rhythm abnormalities increases exponentially with age (6, 16, 17). Hence, incidence of AF in the general population increases 23-fold from the 20–24 to the 55–59 years age group (18, 19) and reaches a prevalence of over 13% in the >80 years age group (20). Similarly, incidence of VF in the general population increases 18-fold from the 20–24 to the 55–59 years age group (21).
Primary electrical abnormalities due to congenital channelopathies represent an important cause of arrhythmias and SCD (15, 22, 23). These include long QT syndrome 3 (LQT3) arising from a gain-of-function mutation in the cardiac sodium NaV1.5 channel gene SCN5A, Brugada Syndrome (BrS) arising from a loss-of-function mutation in the SCN5A gene, and catecholaminergic polymorphic ventricular tachycardia (CPVT) arising from a gain-of-function mutation in RyR2 gene or loss-of-function mutation in CASQ2 gene encoding cardiac calcium homeostasis proteins (23, 24). Proarrhythmic inherited channelopathies demonstrate how each component of the cardiac electrophysiological system contributes to arrhythmogenesis. Thus, studying those channelopathies has been crucial to elucidating the mechanisms underlying arrhythmogenesis in the general and aging population.
Interestingly, arrhythmic risk in individuals with many inherited channelopathies, such as BrS and LQT3, increases markedly with age, despite these individuals carrying the proarrhythmic mutation from birth (25). For example, LQT3 patients show significantly increased arrhythmic risk after the 40 years of age (26, 27). In CPVT however, patients are usually diagnosed in the first or second decade of life with the mean age of onset of symptoms, usually a syncopal episode, is between age seven and 12 years (28). Therefore, select channelopathies demonstrate an excellent paradigm to study the effects of age-related molecular changes on susceptible hearts with inherent proarrhythmic tendency. This will elucidate the molecular mechanisms underlying proarrhythmic changes with age and hence offer novel anti-arrhythmic pharmacological targets.
Aging and Energetic Dysfunction
It has long been established that central to the aging process of any organ is energetic dysfunction giving rise to free radical reactive oxygen species (ROS) that cause damage to cellular macromolecules, accumulation of this damage leads to the physiological compromise seen in aging (5, 29). Current evidence suggests that mitochondrial dysregulation is the cause and primary target of energetic dysfunction and free radical production (5, 30). Thus, transgenic mice overexpressing the cellular antioxidant catalase targeted to the mitochondria had a reduced ROS-induced damage of the mitochondria and significantly increased lifespan (31).
A clear link exists between aging and mitochondrial dysfunction, occurring through various mechanism which include mitochondrial DNA damage, clonal expansion of deleterious mutations in mitochondrial DNA and deficiencies in the enzymes of the mitochondrial respiratory chain, such as cytochrome-c-oxidase (32–36). This phenomenon of aging driving mitochondrial genetic instability has thus been observed not just in humans but several other mammalian species, including in mice, rats, and rhesus monkeys (37–39). The link between aging and mitochondrial dysfunction appears to be bidirectional. For example, increased levels of mitochondrial DNA mutations are associated with a premature aging syndrome in mice (34, 40). Therefore, it is apparent that understanding the biology of mitochondrial instability via mitochondrial DNA mutations and enzyme deficiencies is key to understanding cellular- and tissue-level changes that underlie aging-related pathology.
As such, damaged and dysfunctional mitochondria result in production of high levels of ROS, disrupted mitochondrial membrane potentials, reduced ATP production capacity, and altered cellular redox potential (5, 41–44). The consequent aberrant mitochondrial signaling predisposes the myocardium to arrhythmias (9, 43).
This is demonstrated clinically and experimentally. Mitochondria from human AF patients are abnormal in terms of morphology and function and show DNA damage (45–48). Abnormal mitochondria are also seen in animal models of AF and ventricular arrhythmia (49–51). Additionally, inherited errors of metabolism involving mitochondria such as Kearns-Sayre syndrome manifest symptomatically as fatal rhythm abnormalities (52). Detailed electrophysiological studies in peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc-1α) and Pgc-1β knockout models of mitochondrial dysfunction yield similar overt arrhythmic phenotypes whilst also yielding information on the ionic basis of these arrhythmias. For example, Pgc-1β-/- mice show decreased atrial and ventricular conduction velocity, which may be attributed to reduced voltage gated inward Na+ currents (53–60).
The present review separates the pro-arrhythmic molecular changes in aging into multiple pathways. However, this is largely to make the topic more accessible and easier to conceptualize. In reality these pathways are dependent upon and interact with each other through complex feedback loops. Physiological interactions which are important to the arrhythmic process are also highlighted.
Mice Models
Animal models have been pivotal in studying arrhythmias, permitting experimentation on the cellular and system level. Mice, often with electrophysiologically stable 129/Sv or C57BL/6 genetic backgrounds, have thus far represented the main transgenic system for modeling arrhythmic syndromes (61, 62) typically via well-defined mutations strategically positioned to reflect the genotypes associated with these syndromes and reliably reflecting their phenotype (9, 23, 63). From a practical aspect, mice are inexpensive, easily maintained, and reproduce rapidly thus allowing provision of aged mice over relatively short periods (25). Mice also reflect the human aging process such that they complete their growth before reproduction commences (1, 64). Furthermore, confounding risk factors which influence cardiovascular health (e.g., smoking and hypercholesterolaemia) are absent in murine models and as such their hearts reflect intrinsic cardiac aging (1). Together, these features of murine models make them valuable in the study of the mechanisms underlying cardiac aging.
Despite differences with regards to heart rate, heart size, as well as calcium- and potassium-mediated repolarization currents, which limits their ability to model conditions such as LQT1 and LQT2, murine and human hearts show significant structural and physiological resemblances (65). Structural similarities include similar conducting, sinoatrial and atrioventricular nodes, His-Purkinje systems and contracting atrial and ventricular chambers (25, 65, 66). Important electrophysiological similarities exist especially with respect to their action potential (AP) waveforms where they both share the same role of the inward sodium current in mediating phase 0 depolarization (25, 65) as well as similar transmural differences in AP duration and AP conduction velocities (65, 67, 68). These similarities are critical in permitting mice to effectively model LQT3 and BrS (9).
Mitochondrial Dysfunction and Disrupted Surface Membrane Ionic Currents
Mitochondrial Dysfunction and Sodium Currents
ROS promote both arrhythmic triggers and substrates and hence exert numerous proarrhythmic actions through modulation of intracellular and cell surface ion channels. Firstly, ROS modifies the expression and function of voltage gated Na+ carrying channel, NaV1.5, causing a decrease in the fast depolarizing component of the sodium current (INa) but an increase in the late sodium current (INa−L) (9, 69–72). Thus, in human embryonic kidney (HEK) cells and C57BL/6 murine cardiomyocytes, application of cytosolic NADH and mitochondrial ROS-generating molecules, such as the complex III inhibitor Anti-mycin A, reduced INa (69, 70). However, this effect was blocked by application of mitoTEMPO a specific scavenger of mitochondrial superoxide (69, 70). In murine hearts modeling mitochondrial dysfunction, increased age and Pgc-1β-/- genotype interacted to decrease atrial NaV1.5 channel expression (36). Furthermore, the A280V mutation in glycerol-3-phosphate dehydrogenase 1-like (GPD1-L) protein, which causes Brugada syndrome, reduces INa via increasing cytosolic NADH and mitochondrial ROS levels (73, 74). Additionally, through oxidation of the Ca2+-/calmodulin-dependent kinase II (CaMKII), ROS has also been shown to enhance INa−L (75–78). Together, these alterations in the cardiomyocyte sodium current promote arrhythmogenesis through increased triggered activity and arrhythmic substrate. These findings are summarized in Figure 1.
Figure 1
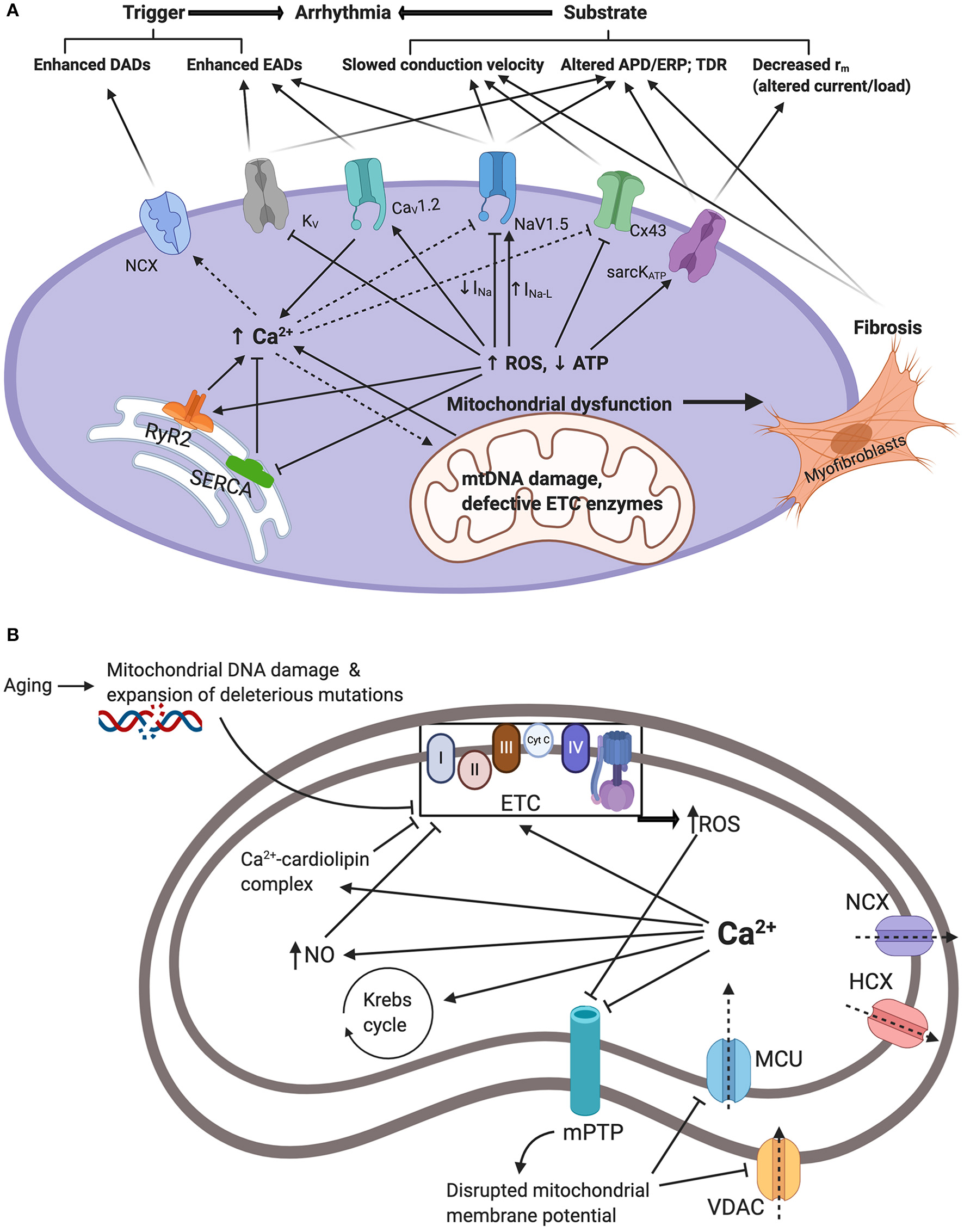
(A) Summarizes the interacting mechanisms by which mitochondrial dysfunction promotes arrhythmogenesis. Mitochondrial dysfunction driven by mitochondrial DNA damage and defective electron transport chain (ETC) enzymes results in reduced ATP and increased reactive oxygen species (ROS) production. In turn, this modifies function and/or expression of sarcolemmal ATP-sensitive K+ channels (sarcKATP), gap junction proteins (Cx43), cardiac sodium channels (NaV1.5), cardiac L-type voltage gated Ca2+ channels (CaV1.2), and cardiac voltage gated potassium channels (KV). Moreover, ROS modify endoplasmic reticulum Ca2+ homeostasis proteins ryanodine receptor (RyR) and sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) resulting in elevated cellular Ca2+ levels which drives the depolarizing Na+-Ca2+ exchanger (NCX). Together, these changes increase arrhythmic triggers and substrates. APD/ERP: action potential duration/effective refractory period ratios; DADs: delayed after-depolarizations; EADs: early after-depolarizations; TDR: transmural dispersion of repolarization. (B) Summarizes mitochondrial Ca2+ handling and its relation to mitochondrial dysfunction and elevated cytosolic Ca2+. Ca2+ enters the mitochondria via voltage dependent anion channels (VDAC) and mitochondrial calcium uniporter (MCU). This is driven by the negative mitochondrial membrane potential (~−180mV). Increased cytosolic Ca2+ through mechanisms demonstrated in (A) will result in increased uptake and concentration of mitochondrial Ca2+. Mitochondrial Ca2+ overload contributes to mitochondrial dysfunction. Firstly, Ca2+ stimulation of the Krebs cycle and the oxidative phosphorylation electron transport chain increasing electron leakage and ROS by-products. Secondly, Ca2+-cardiolipin complexation disrupting mitochondrial lipid and protein arrangement causing proteins such as cytochrome c dislocation and inhibition of the electron transport chain and ROS production. Thirdly, through activation of nitric oxide synthase generating NO radicals which inhibit components of the ETC and promote ROS production. Increased ROS and cytosolic Ca2+ also inhibit mitochondrial permeability transition pores (mPTP) impairing Ca2+ uptake and contributing to increased cytosolic Ca2+. Ca2+ is also extruded through the hydrogen-calcium exchanger (HCX) and the mitochondrial sodium-calcium exchanger (NCX).
Increased INa−L prolongs membrane repolarization and as such allows the development of early-after depolarizations (EADs) through reactivation of voltage-gated Ca2+ channels (VGCC), and in turn, EADs can trigger arrhythmic events (79, 80). In addition, repolarization defects caused by increased INa−L promote spatiotemporal heterogeneity and transmural dispersion of repolarization arrhythmic substrate (79, 80). The changes in INa have profound consequences on ordered action potential propagation through the myocardium. Cardiac conduction velocity is largely determined by the maximum rate of membrane depolarization (dV/dt)max, which in turn is determined by INa and conducted by the NaV1.5 channel. Reduced conduction velocity forms the arrhythmic substrate associated with re-entrant arrhythmias (81, 82). Interestingly, these findings may explain the change in phenotype with age in certain channelopathies. For example, an overlap syndrome in aging LQT3 patients describes the emergence of Brugada syndrome patterns on surface ECGs in addition to the prolonged QT interval indicative of LQT3 (83, 84). Similarly, electrophysiological studies on murine LQT3 models report decreased conduction velocity in aged, but not young, hearts (85, 86). Therefore, age-related mitochondrial dysfunction and ROS generation may account for the activation abnormalities that appear later in life in LQT3 patients and associated with increased arrhythmic risk. Another important consideration is the close link between intracellular Na+ and Ca2+ regulation. Hence, the increase in INa−L causing increased [Na+]i has been shown to increase [Ca2+]i largely through reversing the activity of the sodium-calcium exchanger (NCX) (87, 88). In turn, as discussed later, elevated [Ca2+]i promotes proarrhythmic electrophysiological changes including inhibition of INa (89, 90).
Mitochondrial Dysfunction and Gap Junctions
Cardiac conduction velocity is also influenced by the axial resistance (ra) to local current flow between cells as determined by intercellular gap junction channels formed by connexin (Cx) proteins (82, 91). ACE8/8 mice are produced by placing the angiotensin-converting enzyme (ACE) gene under the control of the α-myosin heavy chain promoter using targeted homologous recombination. This results in significantly increased cardiac ACE and angiotensin II levels. Studies on ACE8/8 mice demonstrated that increased ROS production through renin-angiotensin system (RAS) activation, increased expression and activation of the redox-sensitive tyrosine kinase cSrc in ventricular cardiomyocytes resulting in reduced Cx43 function and expression (74, 92, 93). This reduced conduction velocity and increased risk of ventricular arrhythmias (74, 94). Similarly, Pgc-1β-/- transgenic mice reflecting mitochondrial dysfunction showed reduced atrial Cx protein expression (36). The latter finding may represent a direct consequence of ROS induced pathophysiology, although it may also be linked to the increased cardiac fibrosis which is seen in these mice, and discussed later in this review.
Mitochondrial Dysfunction and Potassium Currents
Voltage gated potassium (KV) channels are regulated by cellular metabolism. KV channels give rise to the transient outward K+ current (Ito) underlying phase 1 repolarization, and the delayed rectifier K+ current (IK) underlying repolarization during phases 2 and 3 of the action potential (95). Electrophysiological studies on diabetic rats have demonstrated repolarization abnormalities resulting from downregulation KV currents (96, 97). Experimentally, increased ROS has been shown to reduce Ito and IK (including IKr, IKs and IKur) currents (74, 98, 99). This inhibition can be reversed through application of cellular antioxidant glutathione (74, 97, 100). ROS reduces KV currents through reducing channel mRNA and protein expression levels (74, 99, 101). Peroxisome proliferator-activated receptor α (PPARα) upregulation during metabolic dysfunction has specifically been associated with reduced transcription of KV channels (102). Additionally, ROS modulates KV channel function by altering their phosphorylation status particularly acting through PKC and PKA (74, 103). Reduced KV currents results in repolarization abnormalities resulting in prolonged action potential duration (APD) promoting EAD arrhythmic triggers (9, 95). Furthermore, altered APD/ effective refractory period (ERP) ratios result in spatiotemporal heterogeneity in activation and repolarization hence furnishing an arrhythmic substrate for re-entry arrhythmias (9, 81, 95).
Another group of K+ channels conduct an inwardly rectifying K+ current (Kir). These include sarcolemmal ATP-sensitive K+ channels. (sarcKATP) predominantly formed by Kir6.2 and SUR2A and are important in the electrophysiological response to stresses such as ischemia (104). These are activated by a reduced ATP/ADP ratio during metabolic stress (105). The high density of sarcKATP channels means that only 1% of those channels need to open to significantly shorten the APD and hence the ERP and action potential wavelength (9, 106). Furthermore, opening of a large number of channels drives the membrane potential toward EK causing the cardiomyocyte to become hyperpolarized and unexcitable (107). Thus, opening of sarcKATP channels generates a “current sink” which can slow or block action potential propagation (108). Together, these changes promote re-entrant arrhythmias (9, 106, 108, 109).
Mitochondrial Dysfunction and Disrupted Calcium Homeostasis
With a 10,000-fold transmembrane gradient, Ca2+ is the most tightly regulated intracellular ion being utilized virtually ubiquitously in cellular signaling pathways (110, 111). Cardiomyocyte Ca2+ homeostasis is heavily influenced by cellular metabolism with increased ROS levels increasing cytosolic Ca2+ concentration ([Ca2+]i) (74, 112). These findings are summarized in Figure 1. The addition of H2O2 generating ROS in guinea pig ventricular myocytes resulted in increased current through the L-type voltage gated Ca2+ channels (ICaL) and hence significantly increased [Ca2+]i (113). This, however, was reversed by application of the mitochondrial inhibitor myxothiazol or the L-type channel inhibitor nisoldipine (113). CAMKII activated by ROS has been shown to increase ICaL via phosphorylation of the CaV1.2 subunit (114) and similar accentuating effects on ICaL were induced by oxidized LDL in rat ventricular cardiomyocytes (115). Furthermore, L-type channel appear to undergo direct redox modification and glutathionylation at cysteine residues in the alpha interacting domain (116, 117). Interestingly, the effect of ROS accentuating ICaL has been challenged by other findings obtained under different experimental conditions which reported reduced ICaL following oxidative stress (118).
In addition to sarcolemmal Ca2+ entry, ROS modulates intracellular Ca2+ handling proteins. Both canine and rat cardiomyocytes show increased opening of RyR2 in response to elevated ROS which triggers RyR2 Ca2+ sparks and accentuated Ca2+ efflux from the sarcoplasmic reticulum (119–121). Similarly, old rabbit hearts had more depolarized mitochondria membrane potential and increased rate of ROS production associated with increased RyR activity and Ca2+ leak which was accentuated under conditions of β-adrenergic stimulation (122). Treatment with antioxidant dithiothreitol reduced RyR-mediated SR Ca2+ leak to levels of young hearts highlighting the role of thiol-oxidation of RyR in underlying pathological SR Ca2+ release (122). This response also appears to depend on calmodulin as a functional mediator of ROS-triggered Ca2+ release (119). In contrast to RyR2, the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) activity is reduced in response to ROS (123, 124). SERCA inhibition by oxidative stress appears to arise through multiple mechanisms including reduced ATP supply (125), direct oxidation of thiol groups by ROS (123), and CAMKII-dependent phosphorylation (74, 126). Interestingly, adult rat ventricular myocytes expressing redox-insensitive SERCA where C674 is replaced by serine (C674S) decreased basal SR calcium content, attenuated the rise in mitochondrial Ca2+, and prevented cytochrome c release and apoptosis (127). Furthermore, beyond ROS generation, dysfunctional mitochondria contribute to disrupted Ca2+ homeostasis through reduced Ca2+ storage capacity. Mitochondria function as an important cellular Ca2+ store with Ca2+ ions entering the inner mitochondrial membrane via the mitochondrial Ca2+ uniporter (MCU) (128). However, under conditions of metabolic stress, mitochondrial Ca2+ handling is disrupted (101). This results in increased size and frequency of cytosolic Ca2+ transients resulting in arrhythmogenic Ca2+ alternans (129). For example, in rat ventricular myocytes, stress induced by thoracic aortic banding enhanced mitochondrial Ca2+ accumulation and hence disrupted global Ca2+ handling, increased spontaneous Ca2+ waves, shortened RyR refractoriness and decreased SR Ca2+ content (130). These effects were inhibited by MCU inhibitor Ru360 which normalized RyR oxidation state, improved intracellular Ca2+ homeostasis and reduced triggered activity (130). However, other studies have produced contradicting evidence (130–133). In rabbit atrial myocytes MCU inhibitor Ru360 increased the severity of Ca2+ alternans whereas stimulation of Ca2+ uptake was protective (133). In fact, diabetic cardiomyopathy has been associated with abnormal mitochondrial Ca2+ handling with altered MCU expression and reduced mitochondrial Ca2+ levels (134, 135). As such, the mechanisms by which mitochondrial dysfunction contributes to abnormal Ca2+ transients remain controversial with further experiments required to clarify this relationship particularly in the context of the pro-arrhythmic aging heart.
Intriguingly, mitochondrial Ca2+ overload itself contributes to mitochondrial dysfunction and ROS generation (136, 137). This perpetuates a positive feedback cycle of ROS-induced Ca2+ overload, Ca2+ -induced ROS generation, and ROS-induced ROS release (74, 138, 139). This occurs via multiple mechanisms. Firstly, Ca2+ stimulation of the Krebs cycle and the electron transport chain increasing electron leakage and ROS by-products (140, 141). Secondly, Ca2+-cardiolipin complexation disrupting mitochondrial lipid and protein arrangement causing proteins such as cytochrome c dislocation and inhibition of the electron transport chain and ROS production (142, 143). Thirdly, through activation of nitric oxide synthase generating NO radicals which itself has been shown to disrupt Ca2+ handling proteins (144, 145) but also to inhibit components of the respiratory chain and promote ROS production (146, 147).
Therefore, age-related mitochondrial dysfunction results in disrupted cellular Ca2+ handling causing elevated [Ca2+]i. The pro-arrhythmic consequences of elevated [Ca2+]i are evident in CPVT hearts occurring due to mutations in cellular Ca2+ handling components, typically RyR2 or calsequestrin (148–150). This leads to potentially fatal ventricular arrhythmic episodes, often biventricular or polymorphic ventricular tachycardia and VF (151, 152). Interestingly, compared to aging mice where mitochondrial dysfunction disrupts multiple aspects of Ca2+ homeostasis, 129/Sv mice modeling CPVT demonstrated that altered function of only one of the Ca2+ handling proteins is sufficient to result in the proarrhythmic phenotype (150, 153).
Disrupted cardiomyocyte Ca2+ homeostasis develops a number of pro-arrhythmic pathways. Firstly, elevated [Ca2+]i promotes the activity of the electrogenic NCX resulting in the generation of delayed after-depolarizations (DAD) which act as arrhythmic triggers (154, 155). As such, ROS causing cytosolic Ca2+ overload has been shown to stimulate NCX activity in guinea pig ventricular myocytes (112, 156). Secondly, dysregulation of Ca2+ handling allows pathological Ca2+ cycling which has been associated with APD alternans and spatiotemporal heterogeneities in repolarization (9, 157, 158). Thirdly, cytosolic Ca2+ interacts with surface membrane NaV1.5 and Cx channels causing reduced conduction velocity. Thus, Ca2+ regulates NaV1.5 and reduces INa through (1) directly binding to the EF hand motif, (2) associating with calmodulin and binding to the IQ domain, and (3) CAMKII-mediated phosphorylation (89, 90, 159). Inhibition of Cx function occurs through activating calcineurin-dependent Cx phosphorylation (160). Finally, increased cytosolic Ca2+, through increased ROS production, promotes tissue fibrosis which is associated with slowed conduction velocity (161).
Mitochondrial Dysfunction and Cardiac Fibrosis
Aging is associated with increased cardiac fibrosis. Histological analysis of human hearts also demonstrates age-related progressive increase in collagen content and myocardial fibrosis (162, 163). Clinically, this is reflected in echocardiographic studies in both males and females which showed increased left ventricular wall thickness representing increased left ventricular hypertrophy (LVH) with age even in the absence of cardiovascular risk factors such as hypertension (5, 164). As such, age-related myocardial fibrosis has been shown to reduce ventricular elasticity, compromise left ventricular filling, and cause diastolic dysfunction (164, 165). Similarly, experimental mouse models also demonstrate increased collagen deposition in the aging myocardium (166). Transgenic premature aging (Polgm/m) mice show increased interstitial and subendocardial fibrosis along with greater amyloid deposition, vacuolization of cytoplasm and hyaline cytoplasmic change (5, 167).
Increased cardiac fibrosis with age reflects a disruption in the equilibrium of extracellular matrix (ECM) synthesis and degradation. ECM synthesis is stimulated by fibrogenic growth factors, such as transforming growth factor (TGF)-β which induce fibroblast production of matrix proteins and protease inhibitors such as tissue inhibitors of metalloproteinases (TIMPs) (168). However, ECM degradation is dependent on tumor necrosis factor (TNF)-α and interleukin (IL)-1β stimulating fibroblast production of matrix metalloproteinases (MMPs) (168). Hence, reduced MMP expression and inhibited ECM degradation appears to play a pivotal role in increased tissue fibrosis. As such, aging in murine models was associated with reduced MMP-1 and MMP-2 transcription and activity (169, 170).
With age, elevated ROS generation increases TGF-β and its downstream effector connective tissue growth factor (CTGF) (168). TGF-β in turn activates Smad2/3 signaling inducing fibroblast proliferation, differentiation into myofibroblasts, and the production of ECM components such as fibrillar collagen, fibronectin, and proteoglycans (168, 171). This is supported by studies in C57BL/6 mice which found increased cardiac fibrosis under conditions of immune dysregulation and tissue inflammation known to promote ROS production (172). Additionally, mice overexpressing catalase targeted to the mitochondria shoed reduced cardiomyocyte hypertrophy and significantly diminished cardiac fibrosis (167). Similarly, knock-out of SIRT3, which deacetylates the regulatory component of the mitochondrial permeability transition pore (mPTP), resulted in mitochondrial dysfunction, increased ROS production and accelerated signs of cardiac hypertrophy and fibrosis (173). Cardiac fibrosis also appears to be regulated by the renin-angiotensin system (RAS) signaling (174, 175). Angiotensin II (ANG II) activates the pro-fibrotic Smad2/3 signaling directly by acting on the ANG II type 1 receptor (AT1) and indirectly by promoting TGF-β production (168, 176). Furthermore, RAS has been shown to increase ROS levels through mechanisms including activation of NADPH oxidase (168, 177). ROS in turn have been found to promote the pro-fibrotic effects of ANG II which were suppressed through the application of antioxidants and AT1 antagonist losartan (177). Consistent with this, aged rat hearts demonstrate significantly increased angiotensin converting enzyme (ACE) and ANG II concentrations (167, 178, 179). Hence, mice carrying a gain-of-function mutation in the Ang II receptor type 1A developed early and progressive cardiac fibrosis (180).
Clinical studies strongly associate fibrosis with increased arrhythmic risk. For example, the origin of arrhythmia post-myocardial infarction is often mapped to the fibrotic border of the infarcted zone in patients undergoing ablation for recurrent VT (181). Similarly, most cases of AF are thought to originate from the atrial myocardial sleeve extending into the pulmonary veins. Histological analysis of pulmonary veins of patients with AF demonstrates increased myocardial content characterized by severe hypertrophy and fibrosis (182). Isolated Langendorff-perfused explanted human hearts with extensive infarction or dilated cardiomyopathy demonstrated increased vulnerability to triggering of VT due to cardiac fibrosis facilitating re-entry mechanisms (183, 184). Correspondingly, aged (24 months) Kunming mice had greater electrocardiographic abnormalities and inducibility of AF compared to young (2 months) mice which was associated to age-related increase in atrial fibrosis (185). Furthermore, transgenic Mkk4 knockout mice had dysregulated MMP function and upregulated TGF- β signaling causing increased susceptibility to atrial tachyarrhythmias (186). Importantly, these effects were more prominent in aged than young mice (186).
Interestingly, despite the primary biophysical defect of NaV1.5 haploinsufficiency being present from birth, BrS symptoms occur mainly in adulthood with mean age of SCD in BrS patients being 40 years (187, 188). The increased arrhythmogenicity later in age has thus been attributed to age-related structural changes primarily cardiac fibrosis (25, 188). Hence, old Scn5a+/− BrS mice demonstrated reduced conduction velocity and increased myocardial fibrosis compared to young mice (189, 190).
Age-related cardiac fibrosis increases arrhythmic tendency through a variety of mechanisms. Firstly, fibrosis causes slowed cardiac conduction velocity (82). Fibrosis creates strands of cardiomyocytes which are electrically isolated from each other by collagenous septa (191). Thus, this forces the action potential waves to follow a “zigzag” pattern, conducting circuitously from one strand to the other resulting in slowed conduction velocity (181, 192). Fibrosis also results in Cx-mediated cardiomyocyte-fibroblast coupling which increases cardiomyocyte membrane capacitance (Cm) slowing down action potential propagation (193, 194). Additionally, fibrosis reduces myocyte-myocyte coupling by decreasing Cx expression and promoting their redistribution away from the intercalated discz and hence increasing axial resistance resulting in slowed conduction velocity (195–198). Secondly, spatial heterogeneity in cardiac fibrosis and hence in compromised Cx function and altered ionic currents, including reduced Na+ current density, promotes APD alternans and dispersions of refractoriness causing unidirectional conduction block arrhythmic substrate (199–202). Additionally, patchy or interstitial fibrosis creates cardiomyocyte strands that are electrically coupled to nonfibrotic regions. Hence, creating a situation that reflects a 1 dimensional cable entering a 3 dimensional syncytium at which the interface acts as a “current sink” generating a “current-sink mismatch” due to the unequal transfer of depolarizing charge (191). Thus, if charge transfer to the syncytium is insufficient to depolarize the syncytium then action potential propagation fails (203). On the other hand, conduction from the syncytium to the 1-dimensional cable will succeed as the source-to-sink ratio is reversed. Therefore, this establishes a unidirectional conduction block facilitating arrhythmic re-entry circuits (191, 204).
Targeted Pharmacological Therapy
Elucidation of the mechanisms by which age-related mitochondrial dysfunction and ROS generation increases arrhythmic risk offers a number of potential anti-arrhythmic pharmacological targets. Some of these targeted therapies, differentiated from non-targeted antioxidant therapies, are highlighted in Table 1.
Table 1
| Mitochondria-targeted pharmacological therapy | Mechanism | Therapeutic effect |
|---|---|---|
| Antioxidants | ||
| TPP+ conjugated antioxidants | Highly lipophilic antioxidants conjugated to strongly positive cations accumulate in mitochondria | Reduce ROS production Reduce mitochondrial component oxidation Reduce ROS-induced apoptosis and necrosis |
| Szeto-Schiller peptides | Cationic tetrapeptides, accumulate in the inner mitochondrial membrane | Scavenge ROS Reduce lipid peroxidation Reduce Ca2+ induced mitochondrial swelling Reduce reperfusion injury |
| Modifiers of mitochondrial biogenesis | ||
| SIRT1 activators | Upregulation of SIRT1 transcription | Increasing PGC-1α expression Antioxidant properties (see above) Reduced NF-κB activation Electrophysiological modifications: Inhibition of INa−L, ICa−L Reduction of intracellular Ca2+ transients |
| Rapamycin | Inhibition of mTOR signaling | Reduced ROS production Reduced cardiac hypertrophy Normalization of age-related Ca2+ homeostasis disruption Increased SERCA expression Reduced RyR current amplitude Increased mitophagy |
Potential targeted therapeutics which may alleviate arrhythmogenic mitochondrial dysfunction.
PGC-1a, Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; ROS, reactive oxygen species; RyR, ryanodine receptor; SERCA, sarco-/endoplasmic reticulum Ca2+-ATPase; TPP, triphenylalkylphosphonium ion.
Antioxidant Therapy
Since mechanisms of ionic current dysregulation, disrupted Ca2+ homeostasis, and increased fibrosis all occur downstream of mitochondrial dysfunction, then it is likely that targeting upstream mitochondrial dysfunction and ROS generation will result in significant anti-arrhythmic effects.
Non-targeted Antioxidant Therapy
The first attempts to counteract oxidative damage in aging has been with the administration of non-targeted antioxidants such as vitamins E and C and β-carotene. While initial small studies indicated some protective effects of non-targeted antioxidants on cardiac function, meta-analysis of larger clinical randomized controlled trials collectively involving tens of thousands of patients found no significant positive effects of non-targeted antioxidants on cardiovascular health or overall mortality (205–207). This may be due to the types of antioxidants investigated by clinical studies. For example, vitamin E has been shown to have pro-oxidant effects (208). Endogenous non-targeted antioxidant enzymes such as superoxide dismutase and catalase which were used in experiments to support the use of antioxidant therapy are not feasible in a clinical setting due to their size, rapid degradation, and potential antigenicity (5).
The failure of non-targeted antioxidants in clinical studies coupled to experimental findings that the source of ROS in aging arises primarily from the mitochondria has motivated the development of mitochondria-targeted antioxidants.
Triphenylalkylphosphonium Ion (TPP+) Conjugated Antioxidants
The highly negative mitochondrial membrane potential (150-180 mV) has been utilized to target molecules to the mitochondria. Thus, coupling lipophilic antioxidants to strongly positive cations such as TPP+ increases accumulation in the mitochondria by 100- to 1000-fold compared to the cytosol (5). Such antioxidants include coenzyme Q (MitoQ), plastoquinone (SkQ1), and piperidine nitroxide in combination with triphenylphosphonium chloride (MitoTEMPO) (209, 210). Experimental studies found that they significantly reduce ROS generation, oxidation of mitochondrial components such as cardiolipin, ROS-induced apoptosis and necrosis, and prolonged lifespan of the fungus Podospora anserina, the crustacean Ceriodaphnia affinis, Drosophila, and mice models (210–212). Additionally, SkQ1 inhibited development of age-related conditions including retinopathy and osteoporosis in mammalian models of those conditions (210). In a rat model of H2O2- and ischemia/reperfusion-induced cardias arrhythmias, treatment with SkQ1 for 3 weeks abolished the steady heart arrhythmia (213). Furthermore, experiments in a guinea pig model of non-ischemic heart failure that recapitulates features of prolonged QT interval and high incidence of spontaneous arrhythmic SCD, MitoTEMPO normalized cellular ROS levels, avoided and reversed heart failure, and prevented SCD by decreasing dispersion of repolarization and ventricular arrhythmias (214). So far, clinical trials are yet to investigate the anti-arrhythmic effects of TPP+ conjugated antioxidants on human patients.
Limitations of TPP+ conjugated antioxidants include their reliance on the mitochondrial membrane potential gradient. This gradient is disrupted with mitochondrial dysfunction in aging, as well as a direct effect of the antioxidants at high concentrations hence limiting their uptake and effectiveness (5, 211, 212). Additionally, at higher micromolar concentrations, these molecules appear to show pro-oxidant rather than antioxidant effects (212). It is thus important to clarify the window between anti- and pro-oxidant concentrations before proceeding to clinical trials.
Szeto-Schiller (SS) Peptides
SS peptides are synthetic aromatic-cationic tetrapeptides that selectively target and concentrate in the inner mitochondrial membrane (215, 216). Hence, in vitro experiments have shown that SS peptides scavenge ROS including hydrogen peroxide, hydroxyl radical, and peroxynitrite (215, 217). As such, they prevent lipid peroxidation as well as Ca2+-mediated mitochondrial swelling or reperfusion injury by inhibiting mitochondrial permeability transition and cytochrome c release (215, 216, 218, 219). In mouse models of ANG II-induced cardiomyopathy and Gαq-overexpression induced heart failure, SS peptide administration prevented mitochondrial dysfunction and ROS generation, downregulated pro-oxidative pathways, and reduced cardiac hypertrophy and fibrosis (220). Similarly, in a rat model of ischemia-reperfusion injury, SS peptides significantly reduced myocardial lipid peroxidation and infarct size as well as reducing the frequency and severity of cardiac arrhythmias (221).
A significant advantage of SS peptides over MitoQ and SkQ1, is that SS peptides do not depend on the mitochondrial membrane potential gradient for accumulation in the mitochondria as they have been shown to concentrate in dysfunctional depolarized mitochondria (5, 215). Additionally, unlike most oligopeptides, SS peptides are water soluble and have good transcellular permeability (215, 222).
Targeting Mitochondrial Biogenesis
SIRT1 Activators (Caloric Restriction Mimetics)
Caloric restriction (CR) has been identified as one of the most potent interventions to improve health and slow down aging (223). Though the beneficial effects of CR are likely multifactorial, the sirtuin family of NAD+-dependent histone deacetylases, of which the predominant mammalian isoform is SIRT1, appear to be responsible for a large number of those beneficial effects (224, 225). SIRT1 acts through multiple pathways to regulate inflammatory responses, cellular senescence and its associated secretory phenotype, telomere attrition, and DNA damage responses (161, 226). As such, aging and its related mitochondrial dysfunction and ROS production are associated with decreased SIRT1 expression and activity (227). Thus, expression of SIRT1 was induced in rats undergoing CR and in human cells exposed to serum from CR rats, and in turn SIRT1 deacetylated the DNA repair factor Ku70 and sequestered the proapoptotic factor Bax away from mitochondria (224, 227). In mice, gain-of-function mutation of SIRT1 improved endothelial function through activating endothelial NO synthase (eNOS), preventing ROS production, inhibiting NF-κB signaling by deacetylating RelA/p65, and reducing the inflammatory response (228). Similarly, other experiments replicated the positive effects of SIRT1 activation including enhanced mitochondrial biogenesis by inducing eNOS expression (229). Therefore, it is expected that compounds capable of activating SIRT1 will recapitulate the protective anti-aging effects of caloric restriction and hence prolong life and improve cardiovascular health including reduced arrhythmic risk. Resveratrol is one such compound being investigated. Its presence in red wine is thought to account for the cardiovascular protective effects of red wine drinking particularly in southern France (230). Resveratrol induced similar transcription profiles as SIRT1 and CR and promoted the same protective effects in heart, skeletal muscle and brain tissue in mice where it also prolonged lifespan and prevented age-related cardiac dysfunction (231).
One of the main mechanisms through which SIRT1 acts is through stimulating PGC-1α expression which acts as an important regulator of mitochondria bioenergetics (232, 233). In rats, resveratrol demonstrated significant antioxidant properties in cultured aortic segments and endothelial cells through reducing ROS production and damage by reducing H2O2 levels and H2O2-mediated apoptosis, preventing UV-induced DNA damage, as well as increasing expression of antioxidant enzymes glutathione peroxidase, catalase, and heme oxygenase-1 (230). Similar antioxidant effects were reported in experiments using guinea pigs (234). Furthermore, it inhibited NF-κB activation and reduced vascular tissue inflammation (235). As such, it has been shown to block age-related cardiac hypertrophy and fibrosis in animal models (173, 236, 237). Interestingly, resveratrol has been suggested to normalize intracellular Ca2+ in a murine model of chronic diabetes through increasing SERCA2a expression (238, 239). Moreover, resveratrol exerts its antiarrhythmic effects on cardiac electrophysiology through regulating a number of ionic currents including inhibition of INa−L, inhibition of ICaL and reduction in the amplitude of intracellular Ca2+ transients, (232, 237, 240, 241). Intriguingly, resveratrol effects on repolarization currents appear more complex with studies finding contradictory changes, nonetheless, in all of those studies the change exerted antiarrhythmic effects (232, 233, 241–243). Additionally, resveratrol promotes the inotropic effect of sympathetic stimulation, without enhancing their proarrhythmic effects and hence evading sinoatrial tachycardia (244).
Therefore, in a rat model where ventricular arrhythmias are enhanced via ischemia-reperfusion, application of resveratrol significantly reduced the severity of ventricular arrhythmia and mortality rate (245, 246). Similarly, in a rabbit model of heart failure, inducibility of atrial fibrillation was markedly reduced by treatment with resveratrol (237). These antiarrhythmic properties have been demonstrated under a number of different experimental models (232, 233, 240, 243) confirming the potential of resveratrol to act as an effective cardioprotective antiarrhythmic agent. While significant clinical data regarding the protective effects of resveratrol, particularly its antiarrhythmic potential, are yet to be obtained, initial clinical trials focusing pharmacokinetics and metabolism of resveratrol have found it to be safe and reasonably well-tolerated at doses of up to 5 g/day (247).
Rapamycin and mTOR
In addition, mammalian target of rapamycin (mTOR) is an important component of nutrient signaling pathways implicated in the aging process (248). mTOR is a protein kinase that forms the core of two protein complexes, mTOR complex 1 and mTOR complex 2, which play an important role in aging through regulation of a variety of cellular pathways controlling cell growth and proliferation (249). Of those, complex 1 appears to be more important in cardiac aging accelerating ribosomal synthesis and cap-dependent translation through phosphorylation of p70S6K (S6K1) and 4E binding protein 1, respectively (5, 249). mTOR signaling is increased with age reflecting its role in the aging mechanism but is normalized with caloric restriction in mice (250).
Inhibition of mTOR signaling through rapamycin has been shown to prolong lifespan in numerous animal models including mice (251). In a murine model of load-induced cardiac hypertrophy via aortic constriction, rapamycin application suppressed S6K1 levels and prevented cardiac hypertrophy (252). Furthermore, application of rapamycin following established cardiac fibrosis improved ventricular function and reversed cardiac fibrosis (253, 254). Similar results were replicated clinically where patients who received rapamycin following cardiac transplant had reduced cardiac hypertrophy and improved cardiac function (255). Rapamycin has also been shown to normalize age-related disruption in ion homeostasis particularly of Ca2+. As such, rapamycin increased SERCA expression, and reduced RyR current amplitude, elevation in [Ca2+]i and activation of downstream Ca2+ pathways such as mitogen-activated protein (MAP) kinases (253, 256, 257). Mitochondrial ROS production and pro-arrhythmic disturbances in Ca2+ homeostasis are also caused by age-related decrease in mitochondrial autophagy (mitophagy) (258–260). Autophagy is negatively regulated by mTOR. Hence enhancing autophagy via Torin1 potent mTOR inhibitor in aged rabbit hearts reduced the rate of ROS production and restored both the depolarized mitochondrial membrane potential and defective Ca2+ handling (261). Therefore, rapamycin pharmacological inhibition of mTOR may offer feasible anti-aging and hence anti-arrhythmic therapy. However, the anti-arrhythmic effects are yet to be explored by laboratory and clinical studies.
Conclusion
Aging is a cardinal risk factor for arrhythmic incidence in the general population and in individuals with inherited channelopathies. Aging is closely related to mitochondrial dysfunction which promotes arrhythmogenesis whereby it increases arrhythmic triggers and substrates via modifying sodium (NaV1.5) and potassium (KV, sarcKATP) ion channels, gap junctions, Ca2+ homeostasis (CaV1.2, SERCA, RyR), and tissue fibrosis. Hence, stratification using “mitochondrial health” as a marker of arrhythmic risk such as through the utilization of metabolomics to analyze biopsy samples allows identification of vulnerable patients amenable to pharmacological therapy. As such, a number of exciting pharmacological therapies targeting mitochondrial dysfunction have been discussed including targeted antioxidants (TPP+-conjugated antioxidants and Szeto-Schiller peptides), SIRT1 activators (resveratrol), and mTOR inhibitors (rapamycin).
Statements
Author contributions
KS: conceptualization. KS and IF: writing—original draft preparation, writing—review, and editing. Both authors have read and agreed to the published version of the manuscript.
Acknowledgments
Biorender.com was used in the creation of Figure 1.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
Partridge L . The new biology of ageing. Philos Trans R Soc Lond B Biol Sci. (2010) 365:147–54. 10.1098/rstb.2009.0222
2.
Rose MR Flatt T Graves JL Greer LF Martinez DE Matos M et al . What is aging?Front Genet. (2012) 3:134. 10.3389/fgene.2012.00134
3.
Crimmins EM . Lifespan and healthspan: past, present, and promise. Gerontologist. (2015) 55:901–911. 10.1093/geront/gnv130
4.
Roger VL Go AS Lloyd-Jones DM Adams RJ Berry JD Brown TM et al . Heart disease and stroke statistics−2011 update: a report from the American Heart Association. Circulation. (2011) 123:e18–e209. 10.1161/CIR.0b013e3182009701
5.
Dai DF Chen T Johnson SC Szeto H Rabinovitch PS . Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal. (2012) 16:1492–526. 10.1089/ars.2011.4179
6.
Khane RS Surdi AD Bhatkar RS . Changes in ECG pattern with advancing age. J Basic Clin Physiol Pharmacol. (2011) 22:97–101. 10.1515/JBCPP.2011.017
7.
Arias E Heron M Tejada-Vera B . United States life tables eliminating certain causes of death, 1999-2001. Natl Vital Stat Rep. (2013) 61:1–28.
8.
Kanasi E Ayilavarapu S Jones J . The aging population: demographics and the biology of aging. Periodontol. (2016) 72:13–8. 10.1111/prd.12126
9.
Huang CL . Murine electrophysiological models of cardiac arrhythmogenesis. Physiol Rev. (2017) 97:283–409. 10.1152/physrev.00007.2016
10.
Huang CL Wu L Jeevaratnam K Lei M . Update on antiarrhythmic drug pharmacology. J Cardiovasc Electrophysiol. (2020) 31:579–92. 10.1111/jce.14347
11.
Wolf PA Dawber TR Thomas HE Kannel WB . Epidemiologic assessment of chronic atrial fibrillation and risk of stroke: the Framingham study. Neurology. (1978) 28:973–7. 10.1212/wnl.28.10.973
12.
Benjamin EJ Wolf PA D'Agostino RB Silbershatz H Kannel WB Levy D . Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. (1998) 98:946–52. 10.1161/01.cir.98.10.946
13.
Stewart S Hart CL Hole DJ McMurray JJ . A population-based study of the long-term risks associated with atrial fibrillation: 20-year follow-up of the Renfrew/Paisley study. Am J Med. (2002) 113:359–64. 10.1016/s0002-9343(02)01236-6
14.
Chugh SS Reinier K Teodorescu C Evanado A Kehr E Al Samara M et al . Epidemiology of sudden cardiac death: clinical and research implications. Prog Cardiovasc Dis. (2008) 51:213–28. 10.1016/j.pcad.2008.06.003
15.
Kuriachan VP Sumner GL Mitchell LB . Sudden cardiac death. Curr Probl Cardiol. (2015) 40:133–200. 10.1016/j.cpcardiol.2015.01.002
16.
Josephson ME . Sudden cardiac arrest. Indian Heart J. (2014) 66 Suppl. 1:S2–3. 10.1016/j.ihj.2014.01.001
17.
Khurshid S Choi SH Weng LC Wang EY Trinquart L Benjamin EJ et al . Frequency of cardiac rhythm abnormalities in a half million adults. Circ Arrhythm Electrophysiol. (2018) 11:e006273. 10.1161/CIRCEP.118.006273
18.
Wilke T Groth A Mueller S Pfannkuche M Verheyen F Linder R et al . Incidence and prevalence of atrial fibrillation: an analysis based on 8.3 million patients. Europace. (2013) 15:486–493. 10.1093/europace/eus333
19.
Andrade J Khairy P Dobrev D Nattel S . The clinical profile and pathophysiology of atrial fibrillation: relationships among clinical features, epidemiology, and mechanisms. Circ Res. (2014) 114:1453–68. 10.1161/CIRCRESAHA.114.303211
20.
Rahman F Kwan GF Benjamin EJ . Global epidemiology of atrial fibrillation. Nat Rev Cardiol. (2014) 11:639–54. 10.1038/nrcardio.2014.118
21.
Tseng WC Wu MH Chen HC Kao FY Huang SK . Ventricular fibrillation in a general population - a national database study. Circ J. (2016) 80:2310–6. 10.1253/circj.CJ-16-0602
22.
Tung RT Shen WK Hammill SC Gersh BJ . Idiopathic ventricular fibrillation in out-of-hospital cardiac arrest survivors. Pacing Clin Electrophysiol. (1994) 17:1405–12. 10.1111/j.1540-8159.1994.tb02460.x
23.
Martin CA Matthews GD Huang CL . Sudden cardiac death and inherited channelopathy: the basic electrophysiology of the myocyte and myocardium in ion channel disease. Heart. (2012) 98:536–43. 10.1136/heartjnl-2011-300953
24.
Behere SP Weindling SN . Inherited arrhythmias: the cardiac channelopathies. Ann Pediatr Cardiol. (2015) 8:210–20. 10.4103/0974-2069.164695
25.
Jeevaratnam K Chadda KR Salvage SC Valli H Ahmad S Grace AA et al . Ion channels, long QT syndrome and arrhythmogenesis in ageing. Clin Exp Pharmacol Physiol 44 Suppl. (2017) 1:38–45. 10.1111/1440-1681.12721
26.
Goldenberg I Moss AJ Bradley J Polonsky S Peterson DR McNitt S et al . Long-QT syndrome after age 40. Circulation. (2008) 117:2192–201. 10.1161/CIRCULATIONAHA.107.729368
27.
Moss AJ Goldenberg I . Importance of knowing the genotype and the specific mutation when managing patients with long QT syndrome. Circ Arrhythm Electrophysiol. (2008) 1:213–26; discussion 226. 10.1161/CIRCEP.108.796599
28.
Adam MP Ardinger HH Pagon RA Wallace SE Bean LJH Mirzaa G et al . GeneReviews. (1993).
29.
HARMAND . Aging: a theory based on free radical and radiation chemistry. J Gerontol. (1956) 11:298–300. 10.1093/geronj/11.3.298
30.
Harman D . The biologic clock: the mitochondria?J Am Geriatr Soc. (1972) 20:145–7. 10.1111/j.1532-5415.1972.tb00787.x
31.
Schriner SE Linford NJ Martin GM Treuting P Ogburn CE Emond M et al . Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. (2005) 308:1909–11. 10.1126/science.1106653
32.
Muller-Hocker J . Cytochrome-c-oxidase deficient cardiomyocytes in the human heart–an age-related phenomenon. A histochemical ultracytochemical study. Am J Pathol. (1989) 134:1167–73.
33.
Krishnan KJ Greaves LC Reeve AK Turnbull D . The ageing mitochondrial genome. Nucleic Acids Res. (2007) 35:7399–405. 10.1093/nar/gkm635
34.
Trifunovic A Larsson NG . Mitochondrial dysfunction as a cause of ageing. J Intern Med. (2008) 263:167–78. 10.1111/j.1365-2796.2007.01905.x
35.
Ademowo OS Dias HKI Burton DGA Griffiths HR . Lipid (per) oxidation in mitochondria: an emerging target in the ageing process?Biogerontology. (2017) 18:859–79. 10.1007/s10522-017-9710-z
36.
Edling CE Fazmin IT Saadeh K Chadda KR Ahmad S Valli H . Molecular basis of arrhythmic substrate in ageing murine peroxisome proliferator-activated receptor γ co-activator deficient hearts modelling mitochondrial dysfunction. Biosci Rep. (2019) 39: BSR20190403. 10.1042/BSR20190403
37.
Piko L Hougham AJ Bulpitt KJ . Studies of sequence heterogeneity of mitochondrial DNA from rat and mouse tissues: evidence for an increased frequency of deletions/additions with aging. Mech Ageing Dev. (1988) 43:279–93. 10.1016/0047-6374(88)90037-1
38.
Schwarze SR Lee CM Chung SS Roecker EB Weindruch R Aiken JM . High levels of mitochondrial DNA deletions in skeletal muscle of old rhesus monkeys. Mech Ageing Dev. (1995) 83:91–101. 10.1016/0047-6374(95)01611-3
39.
Wanagat J Cao Z Pathare P Aiken JM . Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. (2001) 15:322–32. 10.1096/fj.00-0320com
40.
Larsson NG . Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem. (2010) 79:683–706. 10.1146/annurev-biochem-060408-093701
41.
Muscari C Caldarera CM Guarnieri C . Age-dependent production of mitochondrial hydrogen peroxide, lipid peroxides and fluorescent pigments in the rat heart. Basic Res Cardiol. (1990) 85:172–8. 10.1007/BF01906970
42.
Nakada K Inoue K Ono T Isobe K Ogura A Goto YI et al . Inter-mitochondrial complementation: mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med. (2001) 7:934–40. 10.1038/90976
43.
Terman A Brunk UT . The aging myocardium: roles of mitochondrial damage and lysosomal degradation. Heart Lung Circ. (2005) 14:107–14. 10.1016/j.hlc.2004.12.023
44.
Terman A Kurz T Gustafsson B Brunk UT . The involvement of lysosomes in myocardial aging and disease. Curr Cardiol Rev. (2008) 4:107–15. 10.2174/157340308784245801
45.
Tsuboi M Hisatome I Morisaki T Tanaka M Tomikura Y Takeda S et al . Mitochondrial DNA deletion associated with the reduction of adenine nucleotides in human atrium and atrial fibrillation. Eur J Clin Invest. (2001) 31:489–96. 10.1046/j.1365-2362.2001.00844.x
46.
Lin PH Lee SH Su CP Wei YH . Oxidative damage to mitochondrial DNA in atrial muscle of patients with atrial fibrillation. Free Radic Biol Med. (2003) 35:1310–8. 10.1016/j.freeradbiomed.2003.07.002
47.
Ad N Schneider A Khaliulin I Borman JB Schwalb H . Impaired mitochondrial response to simulated ischemic injury as a predictor of the development of atrial fibrillation after cardiac surgery: in vitro study in human myocardium. J Thorac Cardiovasc Surg. (2005) 129:41–5. 10.1016/j.jtcvs.2004.03.058
48.
Bukowska A Schild L Keilhoff G Hirte D Neumann M Gardemann A et al . Mitochondrial dysfunction and redox signaling in atrial tachyarrhythmia. Exp Biol Med. (2008) 233:558–74. 10.3181/0706-RM-155
49.
Morillo CA Klein GJ Jones DL Guiraudon CM . Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation. (1995) 91:1588–95. 10.1161/01.cir.91.5.1588
50.
Ausma J Wijffels M Thone F Wouters L Allessie M Borgers M . Structural changes of atrial myocardium due to sustained atrial fibrillation in the goat. Circulation. (1997) 96:3157–63. 10.1161/01.cir.96.9.3157
51.
Brown DA Aon MA Frasier CR Sloan RC Maloney AH Anderson EJ et al . Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J Mol Cell Cardiol. (2010) 48:673–9. 10.1016/j.yjmcc.2009.11.011
52.
Kabunga P Lau AK Phan K Puranik R Liang C Davis RL et al . Systematic review of cardiac electrical disease in Kearns-Sayre syndrome and mitochondrial cytopathy. Int J Cardiol. (2015) 181:303–10. 10.1016/j.ijcard.2014.12.038
53.
Ahmad S Valli H Edling CE Grace AA Jeevaratnam K Huang CL . Effects of ageing on pro-arrhythmic ventricular phenotypes in incrementally paced murine Pgc-1β-/– hearts. Pflügers Archiv-Eur J Physiol. (2017) 469:1579–90. 10.1007/s00424-017-2054-3
54.
Valli H Ahmad S Chadda KR Al-Hadithi AB Grace AA Jeevaratnam K et al . Age-dependent atrial arrhythmic phenotype secondary to mitochondrial dysfunction in Pgc-1β deficient murine hearts. Mechan Ag Dev. (2017) 167:30–45. 10.1016/j.mad.2017.09.002
55.
Valli H Ahmad S Fraser JA Jeevaratnam K Huang CLH . Pro-arrhythmic atrial phenotypes in incrementally paced murine Pgc1β-/– hearts: effects of age. Exp Physiol. (2017) 102:1619–34. 10.1113/EP086589
56.
Ahmad S Valli H Chadda KR Cranley J Jeevaratnam K Huang LH et al . Ventricular pro-arrhythmic phenotype, arrhythmic substrate, ageing and mitochondrial dysfunction in peroxisome proliferator activated receptor-γ coactivator-1β deficient (Pgc-1β-/–) murine hearts. Mechan Ag Dev. (2018) 173:92–103. 10.1016/j.mad.2018.05.004
57.
Ahmad S Valli H Salvage SC Grace AA Jeevaratnam K Huang CLH . Age-dependent electrocardiographic changes in Pgc-1β deficient murine hearts. Clin Exp Pharm Physiol. (2018) 45:174–86. 10.1111/1440-1681.12863
58.
Valli H Ahmad S Jiang AY Smyth R Jeevaratnam K Matthews HR et al . Cardiomyocyte ionic currents in intact young and aged murine Pgc-1β-/– atrial preparations. Mech Ag Dev. (2018) 169:1–9. 10.1016/j.mad.2017.11.016
59.
Valli H Ahmad S Sriharan S Dean LD Grace AA Jeevaratnam K et al . Epac-induced ryanodine receptor type 2 activation inhibits sodium currents in atrial and ventricular murine cardiomyocytes. Clin Exp Pharmacol Physiol. (2018) 45:278–92. 10.1111/1440-1681.12870
60.
Ahmad S Valli H Smyth R Jiang AY Jeevaratnam K Matthews HR et al . Reduced cardiomyocyte Na+ current in the age-dependent murine Pgc-1β-/– model of ventricular arrhythmia. J Cell Physiol. (2019) 234:3921–32. 10.1002/jcp.27183
61.
Killeen MJ Thomas G Sabir IN Grace AA Huang CL . Mouse models of human arrhythmia syndromes. Acta Physiol. (2008) 192:455–69. 10.1111/j.1748-1716.2007.01822.x
62.
Dobrev D Wehrens XHT . Mouse models of cardiac arrhythmias. Circ Res. (2018) 123:332–4. 10.1161/CIRCRESAHA.118.313406
63.
Riley G Syeda F Kirchhof P Fabritz L . An introduction to murine models of atrial fibrillation. Front Physiol. (2012) 3:296. 10.3389/fphys.2012.00296
64.
Vaupel JW Baudisch A Dölling M Roach DA Gampe J . The case for negative senescence. Theor Popul Biol. (2004) 65:339–351. 10.1016/j.tpb.2003.12.003
65.
Sabir IN Killeen MJ Grace AA Huang CL . Ventricular arrhythmogenesis: insights from murine models. Prog Biophys Mol Biol. (2008) 98:208–18. 10.1016/j.pbiomolbio.2008.10.011
66.
Rentschler S Vaidya DM Tamaddon H Degenhardt K Sassoon D Morley GE et al . Visualization and functional characterization of the developing murine cardiac conduction system. Development. (2001) 128:1785–92.
67.
Knollmann BC Katchman AN Franz MR . Monophasic action potential recordings from intact mouse heart: validation, regional heterogeneity, and relation to refractoriness. J Cardiovasc Electrophysiol. (2001) 12:1286–94. 10.1046/j.1540-8167.2001.01286.x
68.
Liu G Iden JB Kovithavongs K Gulamhusein R Duff HJ Kavanagh KM . In vivo temporal and spatial distribution of depolarization and repolarization and the illusive murine T wave. J Physiol. (2004) 555:267–279. 10.1113/jphysiol.2003.054064
69.
Liu M Liu H Dudley SC . Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res. (2010) 107:967–74. 10.1161/CIRCRESAHA.110.220673
70.
Liu M Gu L Sulkin MS Liu H Jeong EM Greener I et al . Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy. J Mol Cell Cardiol. (2013) 54:25–34. 10.1016/j.yjmcc.2012.10.011
71.
Sag CM Wagner S Maier LS . Role of oxidants on calcium and sodium movement in healthy and diseased cardiac myocytes. Free Radic Biol Med. (2013) 63:338–49. 10.1016/j.freeradbiomed.2013.05.035
72.
Liu M Shi G Yang KC Gu L Kanthasamy AG Anantharam V et al . Role of protein kinase C in metabolic regulation of the cardiac Na. Heart Rhythm. (2017) 14:440–7. 10.1016/j.hrthm.2016.12.026
73.
London B Michalec M Mehdi H Zhu X Kerchner L Sanyal S et al . Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. (2007) 116:2260–8. 10.1161/CIRCULATIONAHA.107.703330
74.
Yang KC Bonini MG Dudley SC . Mitochondria and arrhythmias. Free Radic Biol Med. (2014) 71:351–61. 10.1016/j.freeradbiomed.2014.03.033
75.
Valdivia CR Chu WW Pu J Foell JD Haworth RA Wolff MR et al . Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. (2005) 38:475–83. 10.1016/j.yjmcc.2004.12.012
76.
Wagner S Ruff HM Weber SL Bellmann S Sowa T Schulte T et al . Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ Res. (2011) 108:555–65. 10.1161/CIRCRESAHA.110.221911
77.
Viatchenko-Karpinski S Kornyeyev D El-Bizri N Budas G Fan P Jiang Z et al . Intracellular Na+ overload causes oxidation of CaMKII and leads to Ca2+ mishandling in isolated ventricular myocytes. J Mol Cell Cardiol. (2014) 76:247–56. 10.1016/j.yjmcc.2014.09.009
78.
Dietl A Maack C . Targeting mitochondrial calcium handling and reactive oxygen species in heart failure. Curr Heart Fail Rep. (2017) 14:338–49. 10.1007/s11897-017-0347-7
79.
Thomas G Killeen MJ Grace AA Huang CL . Pharmacological separation of early afterdepolarizations from arrhythmogenic substrate in DeltaKPQ Scn5a murine hearts modelling human long QT 3 syndrome. Acta Physiol. (2008) 192:505–17. 10.1111/j.1748-1716.2007.01770.x
80.
Antzelevitch C Nesterenko V Shryock JC Rajamani S Song Y Belardinelli L . The role of late I Na in development of cardiac arrhythmias. Handb Exp Pharmacol. (2014) 221:137–68. 10.1007/978-3-642-41588-3_7
81.
Kalin A Usher-Smith J Jones VJ Huang CL Sabir IN . Cardiac arrhythmia: a simple conceptual framework. Trends Cardiovasc Med. (2010) 20:103–7. 10.1016/j.tcm.2010.09.003
82.
King JH Huang CL Fraser JA . Determinants of myocardial conduction velocity: implications for arrhythmogenesis. Front Physiol. (2013) 4:154. 10.3389/fphys.2013.00154
83.
Baroudi G Chahine M . Biophysical phenotypes of SCN5A mutations causing long QT and Brugada syndromes. FEBS Lett. (2000) 487:224–228. 10.1016/s0014-5793(00)02360-7
84.
Sandhu A Borne RT Mam C Bunch TJ Aleong RG . Double jeopardy: long QT3 and Brugada syndromes. Clin Case Rep. (2017) 5:1315–9. 10.1002/ccr3.1064
85.
Guzadhur L Pearcey SM Duehmke RM Jeevaratnam K Hohmann AF Zhang Y et al . Atrial arrhythmogenicity in aged Scn5a+/DeltaKPQ mice modeling long QT type 3 syndrome and its relationship to Na+ channel expression and cardiac conduction. Pflugers Arch. (2010) 460:593–601. 10.1007/s00424-010-0851-z
86.
Chadda KR Ahmad S Valli H den Uijl I Al-Hadithi AB Salvage SC et al . The effects of ageing and adrenergic challenge on electrocardiographic phenotypes in a murine model of long QT syndrome type 3. Sci Rep. (2017) 7:11070. 10.1038/s41598-017-11210-3
87.
Mochizuki S MacLeod KT . Effects of hypoxia and metabolic inhibition on increases in intracellular Ca2+ concentration induced by Na+/Ca2+ exchange in isolated guinea-pig cardiac myocytes. J Mol Cell Cardiol. (1997) 29:2979–2987. 10.1006/jmcc.1997.0542
88.
Armoundas AA Hobai IA Tomaselli GF Winslow RL O'Rourke B . Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res. (2003) 93:46–53. 10.1161/01.RES.0000080932.98903.D8
89.
Tan HL Kupershmidt S Zhang R Stepanovic S Roden DM Wilde AA et al . A calcium sensor in the sodium channel modulates cardiac excitability. Nature. (2002) 415:442–7. 10.1038/415442a
90.
Kim J Ghosh S Liu H Tateyama M Kass RS Pitt GS . Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem. (2004) 279:45004–12. 10.1074/jbc.M407286200
91.
Lo CW . Role of gap junctions in cardiac conduction and development: insights from the connexin knockout mice. Circ Res. (2000) 87:346–8. 10.1161/01.res.87.5.346
92.
Donoghue M Wakimoto H Maguire CT Acton S Hales P Stagliano N et al . Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins. J Mol Cell Cardiol. (2003) 35:1043–53. 10.1016/s0022-2828(03)00177-9
93.
Xiao HD Fuchs S Campbell DJ Lewis W Dudley SC Kasi VS et al . Mice with cardiac-restricted angiotensin-converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, sudden death. Am J Pathol. (2004) 165:1019–32. 10.1016/S0002-9440(10)63363-9
94.
Sovari AA Rutledge CA Jeong EM Dolmatova E Arasu D Liu H et al . Mitochondria oxidative stress, connexin43 remodeling, and sudden arrhythmic death. Circ Arrhythm Electrophysiol. (2013) 6:623–31. 10.1161/CIRCEP.112.976787
95.
Nerbonne JM Kass RS . Molecular physiology of cardiac repolarization. Physiol Rev. (2005) 85:1205–53. 10.1152/physrev.00002.2005
96.
Shimoni Y Firek L Severson D Giles W . Short-term diabetes alters K+ currents in rat ventricular myocytes. Circ Res. (1994) 74:620–8. 10.1161/01.res.74.4.620
97.
Xu Z Patel KP Lou MF Rozanski GJ . Up-regulation of K(+) channels in diabetic rat ventricular myocytes by insulin and glutathione. Cardiovasc Res. (2002) 53:80–8. 10.1016/s0008-6363(01)00446-1
98.
Cerbai E Ambrosio G Porciatti F Chiariello M Giotti A Mugelli A . Cellular electrophysiological basis for oxygen radical-induced arrhythmias. A patch-clamp study in guinea pig ventricular myocytes. Circulation. (1991) 84:1773–82. 10.1161/01.cir.84.4.1773
99.
Li X Tang K Xie B Li S Rozanski GJ . Regulation of Kv4 channel expression in failing rat heart by the thioredoxin system. Am J Physiol Heart Circ Physiol. (2008) 295:H416–24. 10.1152/ajpheart.91446.2007
100.
Li S Li X Li YL Shao CH Bidasee KR Rozanski GJ . Insulin regulation of glutathione and contractile phenotype in diabetic rat ventricular myocytes. Am J Physiol Heart Circ Physiol. (2007) 292:H1619–29. 10.1152/ajpheart.00140.2006
101.
Yang KC Kyle JW Makielski JC Dudley SC . Mechanisms of sudden cardiac death: oxidants and metabolism. Circ Res. (2015) 116:1937–55. 10.1161/CIRCRESAHA.116.304691
102.
Marionneau C Aimond F Brunet S Niwa N Finck B Kelly DP et al . PPARalpha-mediated remodeling of repolarizing voltage-gated K+ (Kv) channels in a mouse model of metabolic cardiomyopathy. J Mol Cell Cardiol. (2008) 44:1002–15. 10.1016/j.yjmcc.2008.03.023
103.
Li GR Feng J Wang Z Fermini B Nattel S . Adrenergic modulation of ultrarapid delayed rectifier K+ current in human atrial myocytes. Circ Res. (1996) 78:903–15. 10.1161/01.res.78.5.903
104.
Zhuo ML Huang Y Liu DP Liang CC . KATP channel: relation with cell metabolism and role in the cardiovascular system. Int J Biochem Cell Biol. (2005) 37:751–64. 10.1016/j.biocel.2004.10.008
105.
O'Rourke B . Myocardial K(ATP) channels in preconditioning. Circ Res. (2000) 87:845–55. 10.1161/01.res.87.10.845
106.
Faivre JF Findlay I . Action potential duration and activation of ATP-sensitive potassium current in isolated guinea-pig ventricular myocytes. Biochim Biophys Acta. (1990) 1029:167–72. 10.1016/0005-2736(90)90450-3
107.
Bhatnagar A . Contribution of ATP to oxidative stress-induced changes in action potential of isolated cardiac myocytes. Am J Physiol. (1997) 272:H1598–608. 10.1152/ajpheart.1997.272.4.H1598
108.
Akar FG O'Rourke B . Mitochondria are sources of metabolic sink and arrhythmias. Pharmacol Ther. (2011) 131:287–94. 10.1016/j.pharmthera.2011.04.005
109.
Fosset M De Weille JR Green RD Schmid-Antomarchi H Lazdunski M . Antidiabetic sulfonylureas control action potential properties in heart cells via high affinity receptors that are linked to ATP-dependent K+ channels. J Biol Chem. (1988) 263:7933–6.
110.
Bers DM . Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. (2008) 70:23–49. 10.1146/annurev.physiol.70.113006.100455
111.
Bagur R Hajnóczky G . Intracellular Ca. Mol Cell. (2017) 66:780–8. 10.1016/j.molcel.2017.05.028
112.
Goldhaber JI Ji S Lamp ST Weiss JN . Effects of exogenous free radicals on electromechanical function and metabolism in isolated rabbit and guinea pig ventricle. Implications for ischemia and reperfusion injury. J Clin Invest. (1989) 83:1800–9. 10.1172/JCI114085
113.
Viola HM Arthur PG Hool LC . Transient exposure to hydrogen peroxide causes an increase in mitochondria-derived superoxide as a result of sustained alteration in L-type Ca2+ channel function in the absence of apoptosis in ventricular myocytes. Circ Res. (2007) 100:1036–44. 10.1161/01.RES.0000263010.19273.48
114.
Wu Y MacMillan LB McNeill RB Colbran RJ Anderson ME . CaM kinase augments cardiac L-type Ca2+ current: a cellular mechanism for long Q-T arrhythmias. Am J Physiol. (1999) 276:H2168–78. 10.1152/ajpheart.1999.276.6.H2168
115.
Fearon IM . OxLDL enhances L-type Ca2+ currents via lysophosphatidylcholine-induced mitochondrial reactive oxygen species (ROS) production. Cardiovasc Res. (2006) 69:855–64. 10.1016/j.cardiores.2005.11.019
116.
Johnstone VP Hool LC . Glutathionylation of the L-type Ca2+ channel in oxidative stress-induced pathology of the heart. Int J Mol Sci. (2014) 15:19203–25. 10.3390/ijms151019203
117.
Muralidharan P Cserne Szappanos H Ingley E Hool LC . The cardiac L-type calcium channel alpha subunit is a target for direct redox modification during oxidative stress-the role of cysteine residues in the alpha interacting domain. Clin Exp Pharmacol Physiol. (2017) 44 Suppl. 1:46–54. 10.1111/1440-1681.12750
118.
Hammerschmidt S Wahn H . The effect of the oxidant hypochlorous acid on the L-type calcium current in isolated ventricular cardiomyocytes. J Mol Cell Cardiol. (1998) 30:1855–67. 10.1006/jmcc.1998.0749
119.
Kawakami M Okabe E . Superoxide anion radical-triggered Ca2+ release from cardiac sarcoplasmic reticulum through ryanodine receptor Ca2+ channel. Mol Pharmacol. (1998) 53:497–503. 10.1124/mol.53.3.497
120.
Terentyev D Györke I Belevych AE Terentyeva R Sridhar A Nishijima Y et al . Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. (2008) 103:1466–72. 10.1161/CIRCRESAHA.108.184457
121.
Yan Y Liu J Wei C Li K Xie W Wang Y et al . Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc Res. (2008) 77:432–41. 10.1093/cvr/cvm047
122.
Cooper LL Li W Lu Y Centracchio J Terentyeva R Koren G et al . Redox modification of ryanodine receptors by mitochondria-derived reactive oxygen species contributes to aberrant Ca2+ handling in ageing rabbit hearts. J Physiol. (2013) 591:5895–911. 10.1113/jphysiol.2013.260521
123.
Scherer NM Deamer DW . Oxidative stress impairs the function of sarcoplasmic reticulum by oxidation of sulfhydryl groups in the Ca2+-ATPase. Arch Biochem Biophys. (1986) 246:589–601. 10.1016/0003-9861(86)90314-0
124.
Xu KY Zweier JL Becker LC . Hydroxyl radical inhibits sarcoplasmic reticulum Ca(2+)-ATPase function by direct attack on the ATP binding site. Circ Res. (1997) 80:76–81. 10.1161/01.res.80.1.76
125.
Aggarwal NT Makielski JC . Redox control of cardiac excitability. Antioxid Redox Signal. (2013) 18:432–68. 10.1089/ars.2011.4234
126.
Rokita AG Anderson ME . New therapeutic targets in cardiology: arrhythmias and Ca2+/calmodulin-dependent kinase II (CaMKII). Circulation. (2012) 126:2125–39. 10.1161/CIRCULATIONAHA.112.124990
127.
Goodman JB Qin F Morgan RJ Chambers JM Croteau D Siwik DA et al . Redox-Resistant SERCA [Sarco(endo)plasmic reticulum calcium ATPase] attenuates oxidant-stimulated mitochondrial calcium and apoptosis in cardiac myocytes and pressure overload-induced myocardial failure in mice. Circulation. (2020) 142:2459–69. 10.1161/CIRCULATIONAHA.120.048183
128.
Xu Z Zhang D He X Huang Y Shao H . Transport of calcium ions into mitochondria. Curr Genomics. (2016) 17:215–9. 10.2174/1389202917666160202215748
129.
Florea SM Blatter LA . The role of mitochondria for the regulation of cardiac alternans. Front Physiol. (2010) 1:141. 10.3389/fphys.2010.00141
130.
Hamilton S Terentyeva R Kim TY Bronk P Clements RTO Uchi J et al . Pharmacological modulation of mitochondrial Ca. Front Physiol. (2018) 9:1831. 10.3389/fphys.2018.01831
131.
Maack C Cortassa S Aon MA Ganesan AN Liu T O'Rourke B . Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. (2006) 99:172–82. 10.1161/01.RES.0000232546.92777.05
132.
Hamilton S Terentyeva R Martin B Perger F Li J Stepanov A et al . Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS. Basic Res Cardiol. (2020) 115:38. 10.1007/s00395-020-0797-z
133.
Oropeza-Almazán Y Blatter LA . Mitochondrial calcium uniporter complex activation protects against calcium alternans in atrial myocytes. Am J Physiol Heart Circ Physiol. (2020) 319:H873–81. 10.1152/ajpheart.00375.2020
134.
Gollmer J Zirlik A Bugger H . Mitochondrial mechanisms in diabetic cardiomyopathy. Diabetes Metab J. (2020) 44:33–53. 10.4093/dmj.2019.0185
135.
Diaz-Juarez J Suarez JA Dillmann WH Suarez J . Mitochondrial calcium handling and heart disease in diabetes mellitus. Biochim Biophys Acta Mol Basis Dis. (2021) 1867:165984. 10.1016/j.bbadis.2020.165984
136.
Kowaltowski AJ Naia-da-Silva ES Castilho RF Vercesi AE . Ca2+-stimulated mitochondrial reactive oxygen species generation and permeability transition are inhibited by dibucaine or Mg2+. Arch Biochem Biophys. (1998) 359:77–81. 10.1006/abbi.1998.0870
137.
Przygodzki T Sokal A Bryszewska M . Calcium ionophore A23187 action on cardiac myocytes is accompanied by enhanced production of reactive oxygen species. Biochim Biophys Acta. (2005) 1740:481–8. 10.1016/j.bbadis.2005.03.009
138.
Brookes PS Yoon Y Robotham JL Anders MW Sheu SS . Calcium, ATP and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. (2004) 287:C817–33. 10.1152/ajpcell.00139.2004
139.
Feissner RF Skalska J Gaum WE Sheu SS . Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci. (2009) 14:1197–218. 10.2741/3303
140.
Sohal RS Allen RG . Relationship between metabolic rate, free radicals, differentiation and aging: a unified theory. Basic Life Sci. (1985) 35:75–104. 10.1007/978-1-4899-2218-2_4
141.
Perez-Campo R López-Torres M Cadenas S Rojas C Barja G . The rate of free radical production as a determinant of the rate of aging: evidence from the comparative approach. J Comp Physiol B. (1998) 168:149–58. 10.1007/s003600050131
142.
Grijalba MT Vercesi AE Schreier S . Ca2+-induced increased lipid packing and domain formation in submitochondrial particles. A possible early step in the mechanism of Ca2+-stimulated generation of reactive oxygen species by the respiratory chain. Biochemistry. (1999) 38:13279–87. 10.1021/bi9828674
143.
Ott M Robertson JD Gogvadze V Zhivotovsky B Orrenius S . Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA. (2002) 99:1259–63. 10.1073/pnas.241655498
144.
Gödecke A Heinicke T Kamkin A Kiseleva I Strasser RH Decking UK et al . Inotropic response to beta-adrenergic receptor stimulation and anti-adrenergic effect of ACh in endothelial NO synthase-deficient mouse hearts. J Physiol. (2001) 532(Pt 1):195–204. 10.1111/j.1469-7793.2001.0195g.x
145.
Sun J Picht E Ginsburg KS Bers DM Steenbergen C Murphy E . Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ Res. (2006) 98:403–11. 10.1161/01.RES.0000202707.79018.0a
146.
Cleeter MW Cooper JM Darley-Usmar VM Moncada S Schapira AH . Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. (1994) 345:50–4. 10.1016/0014-5793(94)00424-2
147.
Cassina A Radi R . Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys. (1996) 328:309–16. 10.1006/abbi.1996.0178
148.
Lahat H Pras E Olender T Avidan N Ben-Asher E Man O et al . A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. (2001) 69:1378–84. 10.1086/324565
149.
Laitinen PJ Brown KM Piippo K Swan H Devaney JM Brahmbhatt B et al . Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. (2001) 103:485–90. 10.1161/01.cir.103.4.485
150.
Saadeh K Achercouk Z Fazmin IT Nantha Kumar N Salvage SC Edling CE . Protein expression profiles in murine ventricles modeling catecholaminegic polymorphic ventricular tachycardia: effects of genotype and sex. Ann N Y Acad Sci. (2020) 1478:63–74. 10.1111/nyas.14426
151.
Abriel H Zaklyazminskaya EV . Cardiac channelopathies: genetic and molecular mechanisms. Gene. (2013) 517:1–11. 10.1016/j.gene.2012.12.061
152.
Webster G Berul CI . An update on channelopathies: from mechanisms to management. Circulation. (2013) 127:126–40. 10.1161/CIRCULATIONAHA.111.060343
153.
Zhang Y Wu J Jeevaratnam K King JH Guzadhur L Ren X et al . Conduction slowing contributes to spontaneous ventricular arrhythmias in intrinsically active murine RyR2-P2328S hearts. J Cardiovasc Electrophysiol. (2013) 24:210–8. 10.1111/jce.12015
154.
Kass RS Lederer WJ Tsien RW Weingart R . Role of calcium ions in transient inward currents and aftercontractions induced by strophanthidin in cardiac Purkinje fibres. J Physiol. (1978) 281:187–208. 10.1113/jphysiol.1978.sp012416
155.
January CT Fozzard HA . Delayed afterdepolarizations in heart muscle: mechanisms and relevance. Pharmacol Rev. (1988) 40:219–27.
156.
Eigel BN Gursahani H Hadley RW . ROS are required for rapid reactivation of Na+/Ca2+ exchanger in hypoxic reoxygenated guinea pig ventricular myocytes. Am J Physiol Heart Circ Physiol. (2004) 286:H955–63. 10.1152/ajpheart.00721.2003
157.
Edwards JN Blatter LA . Cardiac alternans and intracellular calcium cycling. Clin Exp Pharmacol Physiol. (2014) 41:524–32. 10.1111/1440-1681.12231
158.
Landstrom AP Dobrev D Wehrens XHT . Calcium Signaling and Cardiac Arrhythmias. Circ Res. (2017) 120:1969–93. 10.1161/CIRCRESAHA.117.310083
159.
Ashpole NM Herren AW Ginsburg KS Brogan JD Johnson DE Cummins TR et al . Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J Biol Chem. (2012) 287:19856–69. 10.1074/jbc.M111.322537
160.
Jabr RI Hatch FS Salvage SC Orlowski A Lampe PD Fry CH . Regulation of gap junction conductance by calcineurin through Cx43 phosphorylation: implications for action potential conduction. Pflugers Arch. (2016) 468:1945–55. 10.1007/s00424-016-1885-7
161.
Wu J Xia S Kalionis B Wan W Sun T . The role of oxidative stress and inflammation in cardiovascular aging. Biomed Res Int. (2014) 2014:615312. 10.1155/2014/615312
162.
Song Y Yao Q Zhu J Luo B Liang S . Age-related variation in the interstitial tissues of the cardiac conduction system; and autopsy study of 230 Han Chinese. Forensic Sci Int. (1999) 104:133–142. 10.1016/s0379-0738(99)00103-6
163.
Burkauskiene A Mackiewicz Z Virtanen I Konttinen YT . Age-related changes in myocardial nerve and collagen networks of the auricle of the right atrium. Acta Cardiol. (2006) 61:513–8. 10.2143/AC.61.5.2017765
164.
Lakatta EG Levy D . Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation. (2003) 107:346–54. 10.1161/01.cir.0000048893.62841.f7
165.
Burlew BS . Diastolic dysfunction in the elderly–the interstitial issue. Am J Geriatr Cardiol. (2004) 13:29–38. 10.1111/j.1076-7460.2004.00059.x
166.
Mukherjee D Sen S . Collagen phenotypes during development and regression of myocardial hypertrophy in spontaneously hypertensive rats. Circ Res. (1990) 67:1474–80. 10.1161/01.res.67.6.1474
167.
Dai DF Santana LF Vermulst M Tomazela DM Emond MJ MacCoss MJ et al . Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. (2009) 119:2789–97. 10.1161/CIRCULATIONAHA.108.822403
168.
Biernacka A Frangogiannis NG . Aging and cardiac fibrosis. Aging Dis. (2011) 2:158–73.
169.
Robert V Besse S Sabri A Silvestre JS Assayag P Nguyen VT et al . Differential regulation of matrix metalloproteinases associated with aging and hypertension in the rat heart. Lab Invest. (1997) 76:729–38.
170.
Gagliano N Arosio B Grizzi F Masson S Tagliabue J Dioguardi N et al . Reduced collagenolytic activity of matrix metalloproteinases and development of liver fibrosis in the aging rat. Mech Ageing Dev. (2002) 123:413–25. 10.1016/s0047-6374(01)00398-0
171.
Mauviel A . Transforming growth factor-beta: a key mediator of fibrosis. Methods Mol Med. (2005) 117:69–80. 10.1385/1-59259-940-0:069
172.
Cieslik KA Taffet GE Carlson S Hermosillo J Trial J Entman ML . Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. J Mol Cell Cardiol. (2011) 50:248–56. 10.1016/j.yjmcc.2010.10.019
173.
Hafner AV Dai J Gomes AP Xiao CY Palmeira CM Rosenzweig A et al . Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging. (2010) 2:914–23. 10.18632/aging.100252
174.
Sadoshima J Izumo S . Molecular characterization of angiotensin II–induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res. (1993) 73:413–23. 10.1161/01.res.73.3.413
175.
Rosenkranz S . TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. (2004) 63:423–32. 10.1016/j.cardiores.2004.04.030
176.
Weber KT Swamynathan SK Guntaka RV Sun Y . Angiotensin II and extracellular matrix homeostasis. Int J Biochem Cell Biol. (1999) 31:395–403. 10.1016/s1357-2725(98)00125-3
177.
Cheng TH Cheng PY Shih NL Chen IB Wang DL Chen JJ . Involvement of reactive oxygen species in angiotensin II-induced endothelin-1 gene expression in rat cardiac fibroblasts. J Am Coll Cardiol. (2003) 42:1845–54. 10.1016/j.jacc.2003.06.010
178.
Lakatta EG . Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part III: cellular and molecular clues to heart and arterial aging. Circulation. (2003) 107:490–7. 10.1161/01.cir.0000048894.99865.02
179.
Groban L Pailes NA Bennett CD Carter CS Chappell MC Kitzman DW et al . Growth hormone replacement attenuates diastolic dysfunction and cardiac angiotensin II expression in senescent rats. J Gerontol A Biol Sci Med Sci. (2006) 61:28–35. 10.1093/gerona/61.1.28
180.
Billet S Bardin S Verp S Baudrie V Michaud A Conchon S et al . Gain-of-function mutant of angiotensin II receptor, type 1A, causes hypertension and cardiovascular fibrosis in mice. J Clin Invest. (2007) 117:1914–25. 10.1172/JCI28764
181.
de Bakker JM van Capelle FJ Janse MJ Tasseron S Vermeulen JT de Jonge N et al . Slow conduction in the infarcted human heart. 'Zigzag' course of activation. Circulation. (1993) 88:915–26. 10.1161/01.cir.88.3.915
182.
Hassink RJ Aretz HT Ruskin J Keane D . Morphology of atrial myocardium in human pulmonary veins: a postmortem analysis in patients with and without atrial fibrillation. J Am Coll Cardiol. (2003) 42:1108–14. 10.1016/s0735-1097(03)00918-5
183.
de Bakker JM Coronel R Tasseron S Wilde AA Opthof T Janse MJ et al . Ventricular tachycardia in the infarcted, Langendorff-perfused human heart: role of the arrangement of surviving cardiac fibers. J Am Coll Cardiol. (1990) 15:1594–607. 10.1016/0735-1097(90)92832-m
184.
Wu TJ Ong JJ Hwang C Lee JJ Fishbein MC Czer L et al . Characteristics of wave fronts during ventricular fibrillation in human hearts with dilated cardiomyopathy: role of increased fibrosis in the generation of reentry. J Am Coll Cardiol. (1998) 32:187–96. 10.1016/s0735-1097(98)00184-3
185.
Luo T Chang CX Zhou X Gu SK Jiang TM Li YM . Characterization of atrial histopathological and electrophysiological changes in a mouse model of aging. Int J Mol Med. (2013) 31:138–46. 10.3892/ijmm.2012.1174
186.
Davies L Jin J Shen W Tsui H Shi Y Wang Y et al . Mkk4 is a negative regulator of the transforming growth factor beta 1 signaling associated with atrial remodeling and arrhythmogenesis with age. J Am Heart Assoc. (2014) 3:e000340. 10.1161/JAHA.113.000340
187.
Antzelevitch C Brugada P Brugada J Brugada R . Brugada syndrome: from cell to bedside. Curr Probl Cardiol. (2005) 30:9–54. 10.1016/j.cpcardiol.2004.04.005
188.
Jeevaratnam K Guzadhur L Goh YM Grace AA Huang CL . Sodium channel haploinsufficiency and structural change in ventricular arrhythmogenesis. Acta Physiol. (2016) 216:186–202. 10.1111/apha.12577
189.
Jeevaratnam K Poh Tee S Zhang Y Rewbury R Guzadhur L Duehmke R et al . Delayed conduction and its implications in murine Scn5a(+/-) hearts: independent and interacting effects of genotype, age, and sex. Pflugers Arch. (2011) 461:29–44. 10.1007/s00424-010-0906-1
190.
Jeevaratnam K Rewbury R Zhang Y Guzadhur L Grace AA Lei M et al . Frequency distribution analysis of activation times and regional fibrosis in murine Scn5a+/- hearts: the effects of ageing and sex. Mech Ageing Dev. (2012) 133:591–9. 10.1016/j.mad.2012.07.006
191.
Nguyen TP Qu Z Weiss JN . Cardiac fibrosis and arrhythmogenesis: the road to repair is paved with perils. J Mol Cell Cardiol. (2014) 70:83–91. 10.1016/j.yjmcc.2013.10.018
192.
Gardner PI Ursell PC Fenoglio JJ Wit AL . Electrophysiologic and anatomic basis for fractionated electrograms recorded from healed myocardial infarcts. Circulation. (1985) 72:596–611. 10.1161/01.cir.72.3.596
193.
Miragoli M Gaudesius G Rohr S . Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ Res. (2006) 98:801–10. 10.1161/01.RES.0000214537.44195.a3
194.
Maleckar MM Greenstein JL Giles WR Trayanova NA . Electrotonic coupling between human atrial myocytes and fibroblasts alters myocyte excitability and repolarization. Biophys J. (2009) 97:2179–90. 10.1016/j.bpj.2009.07.054
195.
van Veen TA Stein M Royer A Le Quang K Charpentier F Colledge WH et al . Impaired impulse propagation in Scn5a-knockout mice: combined contribution of excitability, connexin expression, and tissue architecture in relation to aging. Circulation. (2005) 112:1927–35. 10.1161/CIRCULATIONAHA.105.539072
196.
Spach MS . Mounting evidence that fibrosis generates a major mechanism for atrial fibrillation. Circ Res. (2007) 101:743–5. 10.1161/CIRCRESAHA.107.163956
197.
Burstein B Comtois P Michael G Nishida K Villeneuve L Yeh YH et al . Changes in connexin expression and the atrial fibrillation substrate in congestive heart failure. Circ Res. (2009) 105:1213–22. 10.1161/CIRCRESAHA.108.183400
198.
Duffy HS . The molecular mechanisms of gap junction remodeling. Heart Rhythm. (2012) 9:1331–4. 10.1016/j.hrthm.2011.11.048
199.
Spach MS Dolber PC Heidlage JF . Influence of the passive anisotropic properties on directional differences in propagation following modification of the sodium conductance in human atrial muscle. A model of reentry based on anisotropic discontinuous propagation. Circ Res. (1988) 62:811–32. 10.1161/01.res.62.4.811
200.
Pu J Boyden PA . Alterations of Na+ currents in myocytes from epicardial border zone of the infarcted heart. A possible ionic mechanism for reduced excitability and postrepolarization refractoriness. Circ Res. (1997) 81:110–9. 10.1161/01.res.81.1.110
201.
Qu Z Karagueuzian HS Garfinkel A Weiss JN . Effects of Na(+) channel and cell coupling abnormalities on vulnerability to reentry: a simulation study. Am J Physiol Heart Circ Physiol. (2004) 286:H1310–21. 10.1152/ajpheart.00561.2003
202.
Turner I Huang LHC Saumarez RC . Numerical simulation of paced electrogram fractionation: relating clinical observations to changes in fibrosis and action potential duration. J Cardiovasc Electrophysiol. (2005) 16:151–61. 10.1046/j.1540-8167.2005.30490.x
203.
Rohr S Kucera JP Fast VG Kléber AG . Paradoxical improvement of impulse conduction in cardiac tissue by partial cellular uncoupling. Science. (1997) 275:841–4. 10.1126/science.275.5301.841
204.
Ciaccio EJ Coromilas J Wit AL Peters NS Garan H . Source-sink mismatch causing functional conduction block in re-entrant ventricular tachycardia. JACC Clin Electrophysiol. (2018) 4:1–16. 10.1016/j.jacep.2017.08.019
205.
Vivekananthan DP Penn MS Sapp SK Hsu A Topol EJ . Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet. (2003) 361:2017–23. 10.1016/S0140-6736(03)13637-9
206.
Kris-Etherton PM Lichtenstein AH Howard BV Steinberg D Witztum JL Nutrition Committee of the American Heart Association Council on Nutrition PA . Metabolism antioxidant vitamin supplements and cardiovascular disease. Circulation. (2004) 110:637–41. 10.1161/01.CIR.0000137822.39831.F1
207.
Bjelakovic G Nikolova D Gluud LL Simonetti RG Gluud C . Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA. (2007) 297:842–57. 10.1001/jama.297.8.842
208.
Pearson P Lewis SA Britton J Young IS Fogarty A . The pro-oxidant activity of high-dose vitamin E supplements in vivo. BioDrugs. (2006) 20:271–3. 10.2165/00063030-200620050-00002
209.
Murphy MP Smith RA . Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. (2007) 47:629–56. 10.1146/annurev.pharmtox.47.120505.105110
210.
Skulachev VP Anisimov VN Antonenko YN Bakeeva LE Chernyak BV Erichev VP et al . An attempt to prevent senescence: a mitochondrial approach. Biochim Biophys Acta. (2009) 1787:437–61. 10.1016/j.bbabio.2008.12.008
211.
Kelso GF Porteous CM Coulter CV Hughes G Porteous WK Ledgerwood EC et al . Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem. (2001) 276:4588–96. 10.1074/jbc.M009093200
212.
Antonenko YN Avetisyan AV Bakeeva LE Chernyak BV Chertkov VA Domnina LV et al . Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: synthesis and in vitro studies. Biochemistry. (2008) 73:1273–87. 10.1134/s0006297908120018
213.
Bakeeva LE Barskov IV Egorov MV Isaev NK Kapelko VI Kazachenko AV et al . Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 2. Treatment of some ROS- and age-related diseases (heart arrhythmia, heart infarctions, kidney ischemia, and stroke). Biochemistry. (2008) 73:1288–99. 10.1134/s000629790812002x
214.
Dey S DeMazumder D Sidor A Foster DB O'Rourke B . Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ Res. (2018) 123:356–71. 10.1161/CIRCRESAHA.118.312708
215.
Zhao K Zhao GM Wu D Soong Y Birk AV Schiller PW et al . Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, reperfusion injury. J Biol Chem. (2004) 279:34682–90. 10.1074/jbc.M402999200
216.
Rocha M Hernandez-Mijares A Garcia-Malpartida K Bañuls C Bellod L Victor VM . Mitochondria-targeted antioxidant peptides. Curr Pharm Des. (2010) 16:3124–31. 10.2174/138161210793292519
217.
Dai DF Rabinovitch PS Ungvari Z . Mitochondria and cardiovascular aging. Circ Res. (2012) 110:1109–24. 10.1161/CIRCRESAHA.111.246140
218.
Zhao K Luo G Giannelli S Szeto HH . Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines. Biochem Pharmacol. (2005) 70:1796–806. 10.1016/j.bcp.2005.08.022
219.
Whiteman M Spencer JP Szeto HH Armstrong JS . Do mitochondriotropic antioxidants prevent chlorinative stress-induced mitochondrial and cellular injury?Antioxid Redox Signal. (2008) 10:641–50. 10.1089/ars.2007.1879
220.
Dai DF Chen T Szeto H Nieves-Cintrón M Kutyavin V Santana LF et al . Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. (2011) 58:73–82. 10.1016/j.jacc.2010.12.044
221.
Cho J Won K Wu D Soong Y Liu S Szeto HH et al . Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. (2007) 18:215–20. 10.1097/01.mca.0000236285.71683.b6
222.
Zhao K Luo G Zhao GM Schiller PW Szeto HH . Transcellular transport of a highly polar 3+ net charge opioid tetrapeptide. J Pharmacol Exp Ther. (2003) 304:425–32. 10.1124/jpet.102.040147
223.
Ungvari Z Parrado-Fernandez C Csiszar A de Cabo . R. Mechanisms underlying caloric restriction and lifespan regulation: implications for vascular aging. Circ Res. (2008) 102:519–28. 10.1161/CIRCRESAHA.107.168369
224.
Cohen HY Miller C Bitterman KJ Wall NR Hekking B Kessler B et al . Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. (2004) 305:390–2. 10.1126/science.1099196
225.
Guarente L Picard F . Calorie restriction–the SIR2 connection. Cell. (2005) 120:473–82. 10.1016/j.cell.2005.01.029
226.
Yao H Rahman I . Perspectives on translational and therapeutic aspects of SIRT1 in inflammaging and senescence. Biochem Pharmacol. (2012) 84:1332–9. 10.1016/j.bcp.2012.06.031
227.
Csiszar A Labinskyy N Jimenez R Pinto JT Ballabh P Losonczy G et al . Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: role of circulating factors and SIRT1. Mech Ageing Dev. (2009) 130:518–27. 10.1016/j.mad.2009.06.004
228.
Stein S Schäfer N Breitenstein A Besler C Winnik S Lohmann C et al . SIRT1 reduces endothelial activation without affecting vascular function in ApoE-/- mice. Aging. (2010) 2:353–60. 10.18632/aging.100162
229.
Nisoli E Tonello C Cardile A Cozzi V Bracale R Tedesco L et al . Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. (2005) 310:314–7. 10.1126/science.1117728
230.
Ungvari Z Orosz Z Rivera A Labinskyy N Xiangmin Z Olson S et al . Resveratrol increases vascular oxidative stress resistance. Am J Physiol Heart Circ Physiol. (2007) 292:H2417–24. 10.1152/ajpheart.01258.2006
231.
Barger JL Kayo T Vann JM Arias EB Wang J Hacker TA et al . A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS ONE. (2008) 3:e2264. 10.1371/journal.pone.0002264
232.
Sutanto H Dobrev D Heijman J . Resveratrol: an effective pharmacological agent to prevent inflammation-induced atrial fibrillation?Naunyn Schmiedebergs Arch Pharmacol. (2018) 391:1163–1167. 10.1007/s00210-018-1566-5
233.
Zhang Y Zhang S Liu Z Zhao X Yuan Y Sheng L et al . Resveratrol prevents atrial fibrillation by inhibiting atrial structural and metabolic remodeling in collagen-induced arthritis rats. Naunyn Schmiedebergs Arch Pharmacol. (2018) 391:1179–90. 10.1007/s00210-018-1554-9
234.
Floreani M Napoli E Quintieri L Palatini P . Oral administration of trans-resveratrol to guinea pigs increases cardiac DT-diaphorase and catalase activities, and protects isolated atria from menadione toxicity. Life Sci. (2003) 72:2741–50. 10.1016/s0024-3205(03)00179-6
235.
Csiszar A Smith K Labinskyy N Orosz Z Rivera A Ungvari Z . Resveratrol attenuates TNF-alpha-induced activation of coronary arterial endothelial cells: role of NF-kappaB inhibition. Am J Physiol Heart Circ Physiol. (2006) 291:H1694–9. 10.1152/ajpheart.00340.2006
236.
Sundaresan NR Gupta M Kim G Rajamohan SB Isbatan A Gupta MP . Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. (2009) 119:2758–71. 10.1172/JCI39162
237.
Chong E Chang SL Hsiao YW Singhal R Liu SH Leha T et al . Resveratrol, a red wine antioxidant, reduces atrial fibrillation susceptibility in the failing heart by PI3K/AKT/eNOS signaling pathway activation. Heart Rhythm. (2015) 12:1046–56. 10.1016/j.hrthm.2015.01.044
238.
Sulaiman M Matta MJ Sunderesan NR Gupta MP Periasamy M Gupta M . Resveratrol, an activator of SIRT1, upregulates sarcoplasmic calcium ATPase and improves cardiac function in diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. (2010) 298:H833–43. 10.1152/ajpheart.00418.2009
239.
Liu W Chen P Deng J Lv J Liu J . Resveratrol and polydatin as modulators of Ca. Ann N Y Acad Sci. (2017) 1403:82–91. 10.1111/nyas.13386
240.
Baczkó I Light PE . Resveratrol and derivatives for the treatment of atrial fibrillation. Ann N Y Acad Sci. (2015) 1348:68–74. 10.1111/nyas.12843
241.
Frommeyer G Wolfes J Ellermann C Kochhäuser S Dechering DG Eckardt L . Acute electrophysiologic effects of the polyphenols resveratrol and piceatannol in rabbit atria. Clin Exp Pharmacol Physiol. (2019) 46:94–8. 10.1111/1440-1681.13005
242.
Liew R Stagg MA MacLeod KT Collins P . The red wine polyphenol, resveratrol, exerts acute direct actions on guinea-pig ventricular myocytes. Eur J Pharmacol. (2005) 519:1–8. 10.1016/j.ejphar.2005.06.017
243.
Baczko I Liknes D Yang W Hamming KC Searle G Jaeger K et al . Characterization of a novel multifunctional resveratrol derivative for the treatment of atrial fibrillation. Br J Pharmacol. (2014) 171:92–106. 10.1111/bph.12409
244.
Hernandez-Cascales J . Resveratrol enhances the inotropic effect but inhibits the proarrhythmic effect of sympathomimetic agents in rat myocardium. PeerJ. (2017) 5:e3113. 10.7717/peerj.3113
245.
Hung LM Chen JK Huang SS Lee RS Su MJ . Cardioprotective effect of resveratrol, a natural antioxidant derived from grapes. Cardiovasc Res. (2000) 47:549–555. 10.1016/s0008-6363(00)00102-4
246.
Hung LM Su MJ Chen JK . Resveratrol protects myocardial ischemia-reperfusion injury through both NO-dependent and NO-independent mechanisms. Free Radic Biol Med. (2004) 36:774–81. 10.1016/j.freeradbiomed.2003.12.016
247.
Patel KR Brown VA Jones DJ Britton RG Hemingway D Miller AS et al . Clinical pharmacology of resveratrol and its metabolites in colorectal cancer patients. Cancer Res. (2010) 70:7392–9. 10.1158/0008-5472.CAN-10-2027
248.
Saxton RA Sabatini DM . mTOR signaling in growth, metabolism, and disease. Cell. (2017) 168:960–76. 10.1016/j.cell.2017.02.004
249.
Hay N Sonenberg N . Upstream and downstream of mTOR. Genes Dev. (2004) 18:1926–45. 10.1101/gad.1212704
250.
Donato AJ Walker AE Magerko KA Bramwell RC Black AD Henson GD et al . Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell. (2013) 12:772–83. 10.1111/acel.12103
251.
Flynn JM O'Leary MN Zambataro CA Academia EC Presley MP Garrett BJ et al . Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell. (2013) 12:851–62. 10.1111/acel.12109
252.
Shioi T McMullen JR Tarnavski O Converso K Sherwood MC Manning WJ et al . Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation. (2003) 107:1664–70. 10.1161/01.CIR.0000057979.36322.88
253.
McMullen JR Sherwood MC Tarnavski O Zhang L Dorfman AL Shioi T et al . Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. (2004) 109:3050–5. 10.1161/01.CIR.0000130641.08705.45
254.
Gao XM Wong G Wang B Kiriazis H Moore XL Su YD et al . Inhibition of mTOR reduces chronic pressure-overload cardiac hypertrophy and fibrosis. J Hypertens. (2006) 24:1663–70. 10.1097/01.hjh.0000239304.01496.83
255.
Raichlin E Chandrasekaran K Kremers WK Frantz RP Clavell AL Pereira NL et al . Sirolimus as primary immunosuppressant reduces left ventricular mass and improves diastolic function of the cardiac allograft. Transplantation. (2008) 86:1395–400. 10.1097/TP.0b013e318189049a
256.
Sadoshima J Qiu Z Morgan JP Izumo S . Angiotensin II and other hypertrophic stimuli mediated by G protein-coupled receptors activate tyrosine kinase, mitogen-activated protein kinase, and 90-kD S6 kinase in cardiac myocytes. The critical role of Ca(2+)-dependent signaling. Circ Res. (1995) 76:1–15. 10.1161/01.res.76.1.1
257.
Kaftan E Marks AR Ehrlich BE . Effects of rapamycin on ryanodine receptor/Ca(2+)-release channels from cardiac muscle. Circ Res. (1996) 78:990–7. 10.1161/01.res.78.6.990
258.
Taneike M Yamaguchi O Nakai A Hikoso S Takeda T Mizote I et al . Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. (2010) 6:600–6. 10.4161/auto.6.5.11947
259.
Terman A Kurz T Navratil M Arriaga EA Brunk UT . Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal. (2010) 12:503–35. 10.1089/ars.2009.2598
260.
Ashrafi G Schwarz TL . The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. (2013) 20:31–42. 10.1038/cdd.2012.81
261.
Murphy KR Baggett B Cooper LL Lu YO Uchi J Sedivy JM et al . Enhancing autophagy diminishes aberrant Ca. Front Physiol. (2019) 10:1277. 10.3389/fphys.2019.01277
Summary
Keywords
arrhythmias, mitochondrial dysfunction, ROS, aging, ion channels
Citation
Saadeh K and Fazmin IT (2021) Mitochondrial Dysfunction Increases Arrhythmic Triggers and Substrates; Potential Anti-arrhythmic Pharmacological Targets. Front. Cardiovasc. Med. 8:646932. doi: 10.3389/fcvm.2021.646932
Received
28 December 2020
Accepted
26 January 2021
Published
15 February 2021
Volume
8 - 2021
Edited by
Gary Tse, Second Hospital of Tianjin Medical University, China
Reviewed by
Dmitry Terentyev, Brown University, United States; Ursula Ravens, Technische Universität Dresden, Germany
Updates
Copyright
© 2021 Saadeh and Fazmin.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Khalil Saadeh ks802@cam.ac.uk
This article was submitted to Cardiac Rhythmology, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.