Abstract
IHD is a significant cause of mortality and morbidity worldwide. In the acute phase, it's demonstrated as myocardial infarction and ischemia-reperfusion injury, while in the chronic stage, the ischemic heart is mainly characterised by adverse myocardial remodelling. Although interventions such as thrombolysis and percutaneous coronary intervention could reduce the death risk of these patients, the underlying cellular and molecular mechanisms need more exploration. Mitochondria are crucial to maintain the physiological function of the heart. During IHD, mitochondrial dysfunction results in the pathogenesis of ischemic heart disease. Ischemia drives mitochondrial damage not only due to energy deprivation, but also to other aspects such as mitochondrial dynamics, mitochondria-related inflammation, etc. Given the critical roles of mitochondrial quality control in the pathological process of ischemic heart disease, in this review, we will summarise the efforts in targeting mitochondria (such as mitophagy, mtROS, and mitochondria-related inflammation) on IHD. In addition, we will briefly revisit the emerging therapeutic targets in this field.
Introduction
AMI is myocardial necrosis due to acute obstruction of a coronary artery or induced hypoperfusion of myocardial tissue, which is attributed to millions of deaths worldwide every year. The longer duration of ischemia usually leads to much more severe myocardial damage. Therefore, timely reestablishment of blood flow is the critical factor for rescuing the ischemic tissue. However, another form of strike, called IRI is also responsible for local damage (1, 2) (Figure 1). According to previous evidence, IRI describes tissue ischemia with inadequate oxygen supply after successful reperfusion of the culprit artery. Currently, there are no efficient strategies to prevent the damage caused by IRI (3). Thus, more explorations of the underlying molecular mechanisms are urgent to foster the identification of novel agents to improve outcomes following MI or IRI.
Figure 1
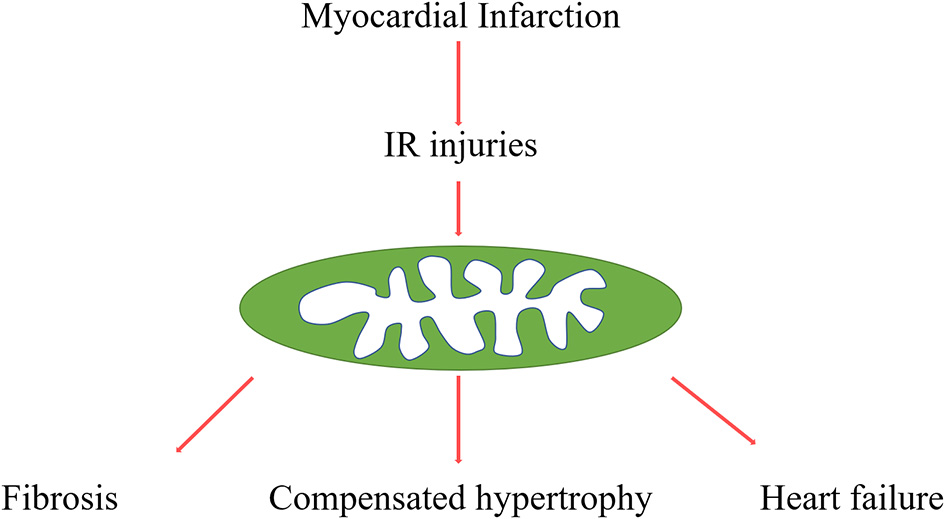
Procedures from myocardial infarction to post-infarction remodelling. Myocardial infarction and reperfusion attributed mitochondria damage, mitochondria function, and structure disorders are involved in various pathophysiological processes, such as cardiac fibrosis, compensated hypertrophy, and final heart failure.
Adverse myocardial remodelling is a significant feature of acute myocardial infarction, characterised by various gradual changes of left ventricular morphology, such as infarcted zone expansion and chamber dilatation. HF is the end stage of myocardial remodelling after AMI. It's diagnosed in ~13% of patients at 30 days and 20–30% at 1 year after discharge for MI (4, 5) (Figure 1). In addition, accumulated evidence indicated that myocardial inflammation and myocardial fibroblasts play critical roles in the process of cardiac repairment after AMI, but excessive inflammation and fibrosis also lead to cardiac remodelling (6, 7). Cardiac fibrosis has been an independent risk factor in HF, which attributed HF patients to sudden cardiac death and increased overall mortality independently of the ejection fraction (8). Although various studies focus on the underlying mechanisms of this pathological process, there are still many unsolved problems in this field, and there are no effective strategies to reverse this process.
Mitochondria is the energy house of cardiomyocytes, generating ATP to maintain normal heart contractile function (9). Mitochondria metabolic dysfunction is a key characteristic of ischemic heart disease. In addition, with more studies focusing on mitochondria, it's reported that mitochondria is not only an energy organelle, but also closely connected with apoptosis (10), ROS generation (11–13), lipid metabolism (14–16), and inflammation (17). All these mechanisms contribute to acute phase and post-infarction remodelling (18). In this review, we discuss the roles of mitochondria in the pathological of ischemic heart disease and the potential in translating mito-protective strategies into the clinical setting.
Mitochondrial Remodelling in Ischemic Heart Disease
Mitochondrial remodelling in ischemic heart disease includes structural and metabolic changes, both of which are identified to play key roles through each stage of the pathogenesis of ischemic heart disease. In cardiomyocytes, mitochondria are highly dynamic organelles, in response to environmental or metabolic changes, they underwent continuous fission, fusion and cristae remodelling (Figure 2). Fusion is an essential dynamic process to maintain the equilibration of matrix metabolites, intact mtDNA, and even membrane components (19–21). In reverse, mitochondrial fission exerts the function to segregate dysfunctional mitochondria to clean damaged proteins and mtDNA (19, 22). These mitochondrial activities are strictly regulated by a group of GTPases related to the dynamin family (23). Through the control of these proteins, mitochondria can maintain a dynamic fission-fusion balance to exert physiological functions. The structural disequilibrium is closely related to acute and chronic heart diseases, involving various molecular mechanisms (discussed in the following parts).
Figure 2
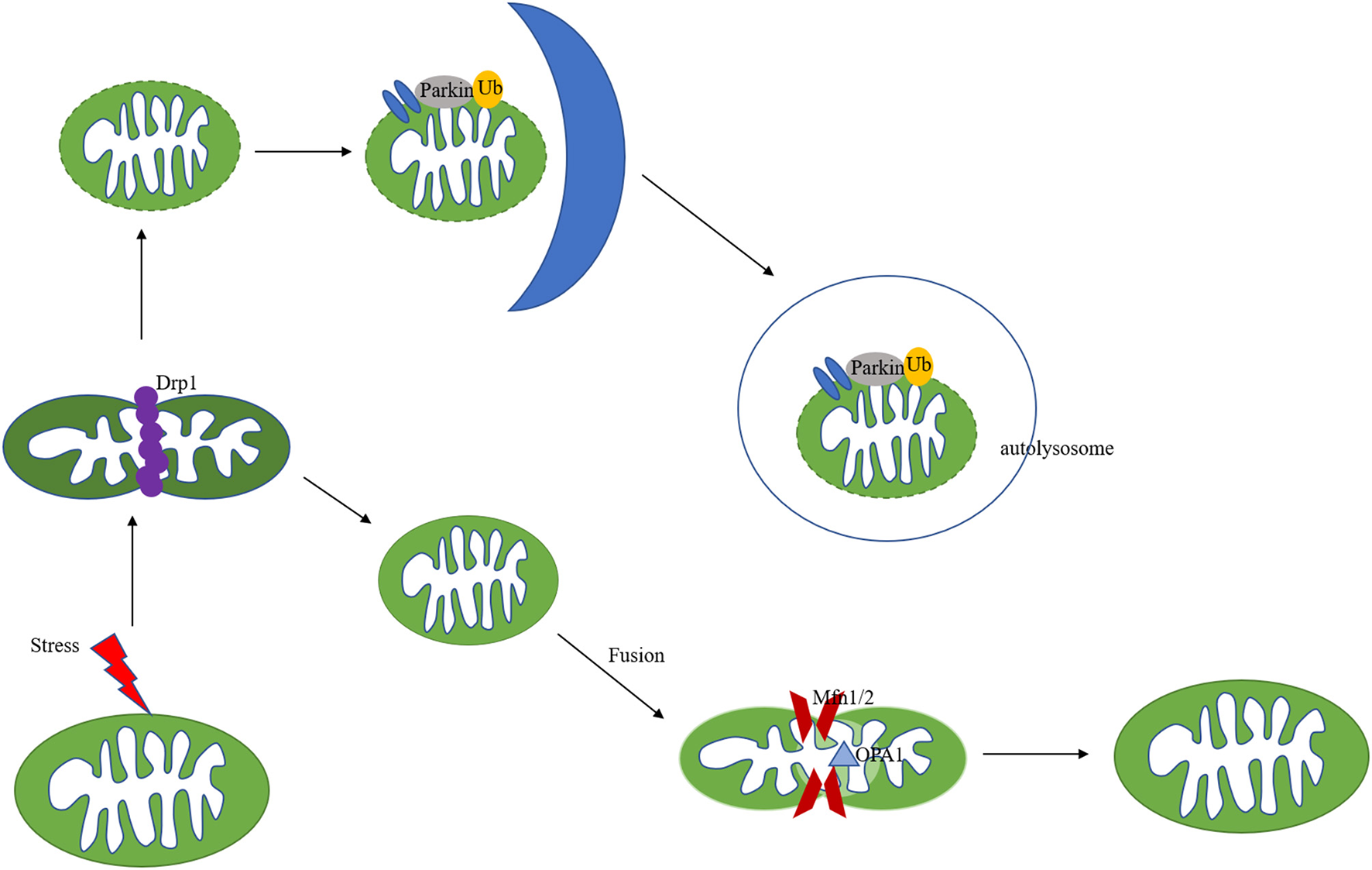
Mechanisms of mitochondria dynamics and mitophagy. Mitochondria are highly dynamic organelles undergoing coordinated cycles of fission and fusion. A series of GTPase-related proteins are involved in the dynamic process. Drp1 is the main regulator of mitochondrial fission, and Mfn1/2, combined with OPA1 regulate the fusion process. After mitochondrial fission, the mitochondrial fragments could be cleared out via PINK/Parkin-mediated mitophagy pathway.
The double-membrane mitochondria mediate OXPHOS, coupling the substrate oxidation to ATP generation, which is also known as the electron transport chain, ETC (24, 25). Mitochondrial energy remodelling is a key characteristic of myocardial ischemia. In the early ischemia stage, FAO, the main metabolic way of heart, increases slightly and provides 60–90% of cardiac ATP production. In addition, the rapid depletion of oxygen switches mitochondria metabolism to glycolysis (26), resulting in pyruvate and lactate accumulation, followed by intracellular acidification. After reperfusion, the restoration of oxygen may initiate the burst of ROS, resulting in severe intracellular damage. ROS, combined with calcium overload, will trigger the opening of the mPTP. Despite various researches on this topic, the exact molecular composition of mPTP is still controversial (Figure 3). Previous evidence indicated that mPTP includes ANT, VDAC, CyPD and PiC (27, 28), but genetic ablation of these proteins revealed that they are the regulators but not the pore of mPT (29, 30). Mitochondrial F1F0 ATP synthase is known to form dimers in the inner mitochondrial membrane (31, 32). Some studies indicated that the ablation of the main membrane-embedded component of ATP synthase, c-subunit, resulted in no change of the sensitivity of mPT (33, 34), however, other evidence found that c-subunit knockout lead to attenuate mPT (35, 36). Previous studies found that mitochondrial F1F0 ATP synthase dimers were essential to form the inner mitochondrial membrane channel, maintaining their physical function (37, 38). However, Nelli et al. found that ATP synthase monomer is sufficient, and dimer formation is not required, for mPTP activity (39). Anyway, despite the controversial components of mPTP, current evidence indicated that mPTP opening could lead to the depolarization of mitochondrial membrane potential followed by cell death. Upon heart failure, FAO and mitochondrial OXPHOS decrease, resulting in cardiac ATP behind the requirement. Although the slight increase of glucose uptake and glycolysis could exert a compensatory response, this upregulation is insufficient to restore ATP production (40–42). Accumulating evidence suggest that mitochondrial respiration disturbance is a potential contributory factor to ischemic heart disease due to its generation of ROS (43). Mitochondrial remodelling participates in many regulating processes in ischemic heart disease, which will be discussed in the following parts.
Figure 3
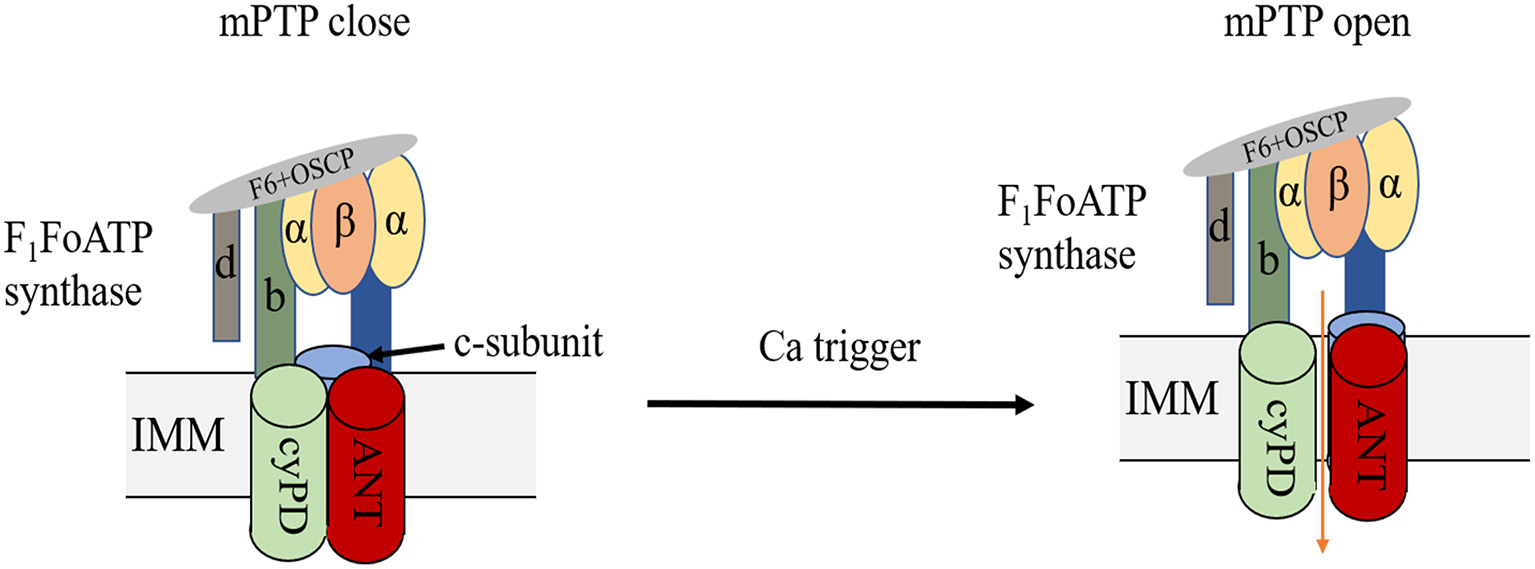
Structure of mPTP. mPTP is a non-specific and -selective channel composed of multiple proteins, which is voltage-dependent and spans cytoplasm, OMM, IMM, and mitochondrial matrix. F1F0 (F)-ATP synthase is the main component of the pore and that the regulatory molecule CypD is a protein modulator of the mPTP.
Mitochondria Mitophagy in Ischemic Heart Disease
Mitophagy is a specific subtype of autophagy, which is also an important mitochondria quality control system to maintain mitochondrial homeostasis (44, 45). The mechanisms of mitophagy induced by mitochondrial stress are complicated (Figure 2). Mitophagy impairment causes the accumulation of defective organelles, leading to cell and tissue damage. Previous studies focused on autophagosome-mediated mitophagy via LC3 adapters. Recently, many studies have investigated a PINK/Parkin pathway involved in mitophagy. PINK and Parkin were first reported as genetic factors of Parkinson's disease (46). PINK is a mitochondrial serine/threonine-protein kinase and Parkin is a cytosolic E3-ubiquitin ligase (47, 48). Under physical conditions, PINK is transported to the inner mitochondrial membrane and cleaved by MPP. The auto-phosphorylation of PINK recruited Parkin translocation to the mitochondrial surface (49, 50). Pathological stresses cause mitochondrial membrane depolarization and reduce the cleavage of PINK. Accumulated PINK could be self-phosphorylated and activated, recruiting Parkin to damaged mitochondria and triggering its E3 ligase activity (51, 52). PINK could also phosphorylate ubiquitin (Ub), forming poly-Ub chains on dysfunctional mitochondria. Parkin would be activated by PINK after binding with phospho-Ub, amplifying mitophagy signals (53). Parkin could polyubiquitinate its substrates, such as VDAC1 and Mfn1/2, leading to their degradation by the proteasomes (54, 55), followed by mitochondria fission and mitophagy (Figure 2). PINK-Parkin pathway also interferes with other mitochondrial quality control mechanisms, mitochondrial fusion, and fission play an essential role in mitophagy, mediating by dynamics-related proteins (the fission and fusion proteins) (56). Mitochondrial fission results in small fragmented mitochondria while fusion forms the elongated interconnected network. Mitochondria are divided into polarised and depolarised daughter mitochondria. Mitochondrial fission is usually considered as the prerequisite for the occurrence of mitophagy. Drp1 knockout disrupts mitochondria fission, promotes elongated mitochondria, and inhibits mitophagy, which aggravates cardiac dysfunction during IR injury (57). Fission inhibition resulted in the progression of cardiac injury due to impaired mitophagy, in addition, overexpression of Drp1 could promote mitophagy-mediated cell death (58, 59). Song et al. reported that Drp1 ablation interrupts mitochondrial fission and increases the activation of Parkin-mediated mitophagy, and Parkin deletion in Drp1-knockout mice rescues heart function and alleviates cardiac remodelling (60). NR4A1 could aggravate IR injury via increasing mitochondrial fission through Drp1 translocation and mitophagy suppression, NR4A1 ablation could protect against pathological fission and mitochondrial dysfunction. Novel therapeutic targeting the balance among NR4A1, fission, and mitophagy may improve cardiac function following IR injury (61).
In addition to increased mitochondrial fission during I/R injury, the decreased mitochondrial fusion promoted mitochondrial fragmentation, resulting in cardiac cell death and dysfunction. Mitochondrial fusion, mediated by mitofusin 1/2 and OPA1, could prevent damaged mitochondria from fusing with healthy ones. The dynamin-related protein OPA1, located on the inner mitochondrial membrane, protects against apoptosis by preventing the release of cytochrome c from the mitochondria (62). Chen et al. found that OPA1 decreased in samples from human hearts with ischemic cardiomyopathy (63). Increased ROS reduced the expression of OPA1 and aggravated cardiomyocytes apoptosis in response to I/R injury (64). OPA1 overexpression protected cardiomyocytes against hypoxia-induced damage and enhanced cell viability by inducing mitophagy (65), and melatonin could attenuate IRI via improving mitophagy and activating the AMPK-OPA1 signalling pathway (66). Additionally, Lichun et al. (67) found that increased expression of MCU induced calpain activation, down-regulating OPA1 and leading to myocardial IRI.
Current evidence indicated that impaired mitophagy participated in cardiac IRI. PTENα deficiency could disrupt mitophagy and lead to the accumulation of damaged mitochondria, followed by the higher risk of IR injury (68). WDR26 is a scaffolding protein that was found to increase after cardiac ischemia. Increasing the expression of WDR26 could increase mitochondria potential, thereby inhibit cardiomyocyte apoptosis via promoting Parkin-mediated mitophagy (69). Many agents such as antioxidants from grapes were reported to exert protection against IR injury by promoting the PINK/Parkin pathway (70, 71). Zinc ion also demonstrated cardiac protection from IR injury via promoting PINK-dependent mitophagy through the MAPK/ERK pathway, the activation of PINK/Parkin-dependent mitophagy could significantly decrease mitochondrial superoxide generation and oxidative stress (72). Cardiac Drp1 heterozygous knockout mice suffer disturbed mitophagy and are more susceptible to IR injury (57). FUNDC1 is a mitophagy receptor after hypoxia (73), exerting a protective property in cardiac IR injury. A decrease of FUNDC1 could increase ROS levels and promote apoptosis, leading to an increase in cardiac IR injury via MAPK/ERK-CREB pathway. Restoration of FUNDC1 levels could reduce myocardial infarct size (74). In addition, platelet activation and thrombosis formation is the key step in cardiac ischemia (75). Platelet-specific FUNDC1 ablation induces worse cardiac damage via mitophagy interruption and platelet activation (76, 77).
In addition to Parkin, there are several other ubiquitin E3 ligases, such as SMURF1, SIAH1, Gp78 also involve in mitophagy regulation (78, 79). All these factors could generate ubiquitin chains after being located on the mitochondrial surface, followed by the recruitment of autophagy adaptors such as optineurin, nuclear dot protein 52, and p62. These adaptors interact directly with LC3, anchoring Ub-tagged mitochondria into autophagosomes. PINK1 ubiquitin kinase mediates optineurin, nuclear dot protein 52 recruitment on damaged mitochondria, stimulating mitophagy. The serine/threonine-protein kinase TBK1 modulates the phosphorylation status of the adaptors, followed by their increasing binding affinity to Ub chains, and promoting mitochondrial removal (80–82). Choong et al. (83) reported that damaged mitochondria could release into extracellular space in free naked form or in membrane-surrounded vesicles. Mitochondrial stress may enhance this extracellular release process. Extracellular mitochondrial release acts as an alternative pathway to PRKN-dependent and independent mitophagy to help with the clearance of damaged mitochondria.
Macrophages are the most heterogeneous immune cell population, which could be activated by a variety of cytokines. A recent work examining macrophage transcriptome in the mice heart post- MI showed a robust reprogramming of mitochondrial genes, suggesting that mitochondrial function may lie at the heart of macrophage function and cardiac remodelling. Another primary function of macrophages is to eliminate unwanted material through phagocytosis (84). Nicolás-Ávila et al. (85) identified a non-canonical route of elimination of abnormal mitochondria from cardiomyocytes in vesiculated structures (exophers). The exophers is then taken up and processed by macrophages surround cardiomyocytes through the phagocytic receptor Mertk. In cardiac stress such as AMI and hypertrophy, failure to eliminate mitochondria-laden exophers results in activation of the inflammasome and autophagy arrest, ultimately compromising mitochondrial fitness.
Cardiac remodelling is also a canonical pathological process after myocardial infarction, characterised with a large number of cardiomyocytes undergoing cell death. To maintain normal cardiac output, surviving cardiomyocytes will increase in cellular size, mass, and volume. In addition to cardiomyocytes, cardiac fibroblasts are also activated to secret components of the ECM, which promotes the differentiation to myofibroblasts and exert increased migratory, proliferative and secretory properties (86). BNIP3 is an apoptosis-inducing protein, Diwan et al. (87) and Dorn et al. (88) reported that BNIP3 ablation in unstressed mice reveals no essential function, but BNIP3 specific knockout mice suffered reduced apoptosis and cardiac remodelling after myocardial infarction. It is possible that BNIP3 regulates mitochondrial quality through mitophagy under baseline conditions. However, during cardiac injury, BNIP3 may act as a death promoter. Pingjun and colleagues found that RIPK3 mediated cardiomyocyte necroptosis via AMPK/Parkin-mitophagy axis in post-MI heart failure (89). Moshi et al. (90) found that conditional ablation of Drp1 in mouse embryonic fibroblasts promoted mPTP-mediated mitophagy. In summary, mitophagy is indispensable for physiological mitochondrial function, interruption of which may reduce mitochondrial dysfunction both in ischemia and post-ischemic reperfusion.
Mitochondrial ROS in Ischemic Heart Disease
It's undoubtful that ROS is a toxic product of aerobic metabolism, involving in various physiological and pathological processes (91, 92). Mitochondria is the major cellular source of ROS. On the one hand, mitochondria consume more than 95% of the oxygen to generate the energy required to sustain life (93). During ischemia, oxygen transported to mitochondrial ETC reduced sharply, after blood restoration bring back oxygen, an electron back-up primer the soluble ubiquinone component of the ETC (especially complex I and complex III) to generate oxygen free radicals. On the other hand, NADPH oxidase was another source of ROS. NADPH oxidase could not deal with the superfluous ROS, causing damage to DNA, proteins, lipids or modulate cellular signalling pathways (Figure 4).
Figure 4
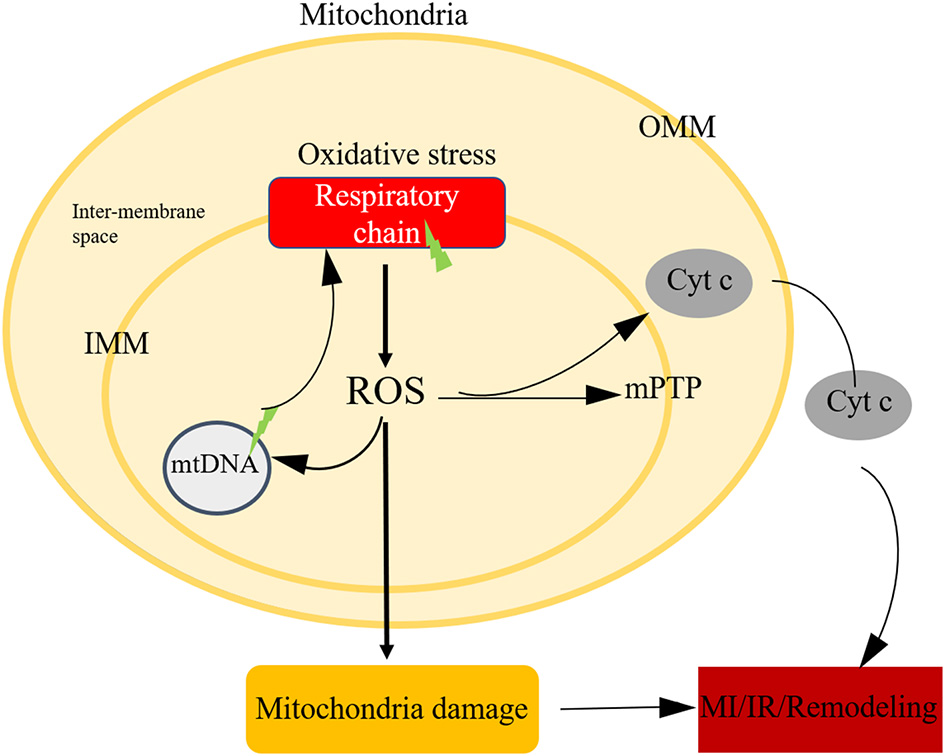
Mitochondria oxidative stress in ischemic heart disease. Mitochondria is the main source of ROS, after oxidative stress, the respiratory chain could generate ROS, the imbalance level of ROS could attribute to mtDNA damage, initiate the open of mPTP, leading to cell death.
Increased ROS levels could activate various second message pathways, such as the ERK, P38, protein kinase C, and PI3-kinase pathways (94–96). Zorov et al. reported that mtROS could dissipate the mitochondrial membrane potential and lead to mPTP opening (97). In the following study, Aon et al. (98) demonstrated that during ischemia-reperfusion injury, excessive ROS from ETC could activate the inner mitochondrial anion channel, causing the release of ROS into the cytoplasm and simultaneous dissipation of the membrane potential. As mentioned above, Nox is another important source of ROS. Braunersreuther et al. reported that in Nox1/Nox2 knockout mice, myocardial infarct size was significantly smaller than that in wild-type mice subjected to IR (30 min of ischemia and 24 h of reperfusion), the underlying pathways include Akt/ERK in Nox1-knockout mice and STAT3/ERK in Nox2-knockout mice (99). Matsushima et al. (100) aimed to figure out the role of Nox-4 in mediating IR injury and they found that Nox-4 knockout mice suffered reduced ROS production and attenuation of the infarct size after IR via the HIF-1α/PPARα pathway.
In addition to the harmful aspects, redox signalling also contributes to protective or adaptive responses during IR injury (101, 102). Many potential signal pathways have been reported in this process, HIF signalling is one of the most important ones. During ischemia, cardiomyocyte energy metabolism switches from FAO to glycolysis, under control of HIF, followed by the activated expression of several glycolytic genes (103–105). In addition, it's reported that oxidative stress following IR injury is neutralised by the CNC -bZIP transcription factor Nrf2, which could regulate intracellular redox homeostasis. With the accumulation of intracellular oxidants, the levels of Nrf2 increased in the nucleus, binding to ARE in the upstream regulatory regions of genes encoding detoxification and antioxidant enzymes, enhancing their transcription. This has been shown to protect the heart from IR injury (106, 107). Recently, some studies investigated that MAOs, including MAO-A and MAO-B, is another source of ROS. Located on the OMM, MAOs could generate O2 and H2O2 (108). During IR injury, the increased activity of the MAO-A isoform significantly deteriorated myocardial injury (109, 110) and promoted the cardiac remodelling (110).
Cardiac fibrosis is a significant feature of adverse cardiac remodelling after myocardial infarction, sustained fibrosis could result in myocardial stiffness, decrease of heart function, and increased risk of arrhythmias (111). After myocardial infarction, cardiac fibroblasts transform to a proinflammatory state, secreting cytokines and MMPs, later post-MI phase, fibroblasts transform to anti-inflammatory phenotype and generate ECM (112). It's reported that ROS is an important regulator of MMPs, the increased levels of ROS could increase the activity of MMPs, decrease tissue inhibitors of MMPs and increase collagen synthesis (113, 114). In vitro tests indicated that ROS could decrease collagen synthesis and increase transcriptional and posttranslational levels of MMPs (115, 116). In vivo, mice receiving ROS scavengers after MI could preserve left ventricular function via decreasing the activity of MMPs (117, 118). The evidence indicated that MMPs are key regulators in the process that ROS influences cardiac fibrosis. Gpx is an antioxidant enzyme, which could scavenge H2O2, meanwhile prevent the formation of other kinds of toxic radicals. Gpx transgenic mice presented improved heart function via attenuating apoptosis, fibrosis, and decreasing MMP-9 activation after MI (119). Some studies also identified the effect of mitochondrial oxidative stress on remote myocardium after MI. Overexpression of Prx3, a mitochondrial antioxidant enzyme, could inhibit cardiac remodelling and failure (120). Cardiolipin inhibition could prevent adverse cardiac remodelling in the non-infarcted MI border zone via the restoration of ETC and reduced ROS (121). In addition, another study demonstrated that increased lipid peroxidation products could be detected in post-MI heart failure (122). These studies indicated that mitochondrial oxidative stress is an important factor regulating ischemic heart disease.
Mitochondria-Related Inflammation in Ischemic Heart Disease
Current evidence demonstrated that inflammatory cell recruitment, together with the activation of innate and adaptive immune reactions, are the features of MI and IR injury (123, 124). Inflammation is an important component of tissue repair. However, recent studies suggested that excessive inflammation-related processes contributed to poor outcomes. In a steady-state, leukocytes and macrophages are the most prevalent subset in adult mouse hearts. After MI, B-, and T-cells were recruited to myocardium, leading to their increase of 5–10-folds. Circulating blood monocytes migrate into the infarcted heart and differentiate into macrophages. Many signalling pathways have been identified to be involved in mediating inflammation in acute and chronic myocardial injury. In this section, we mainly focused on the relationship between mitochondria and inflammation during cardiac injury (Figure 5).
Figure 5
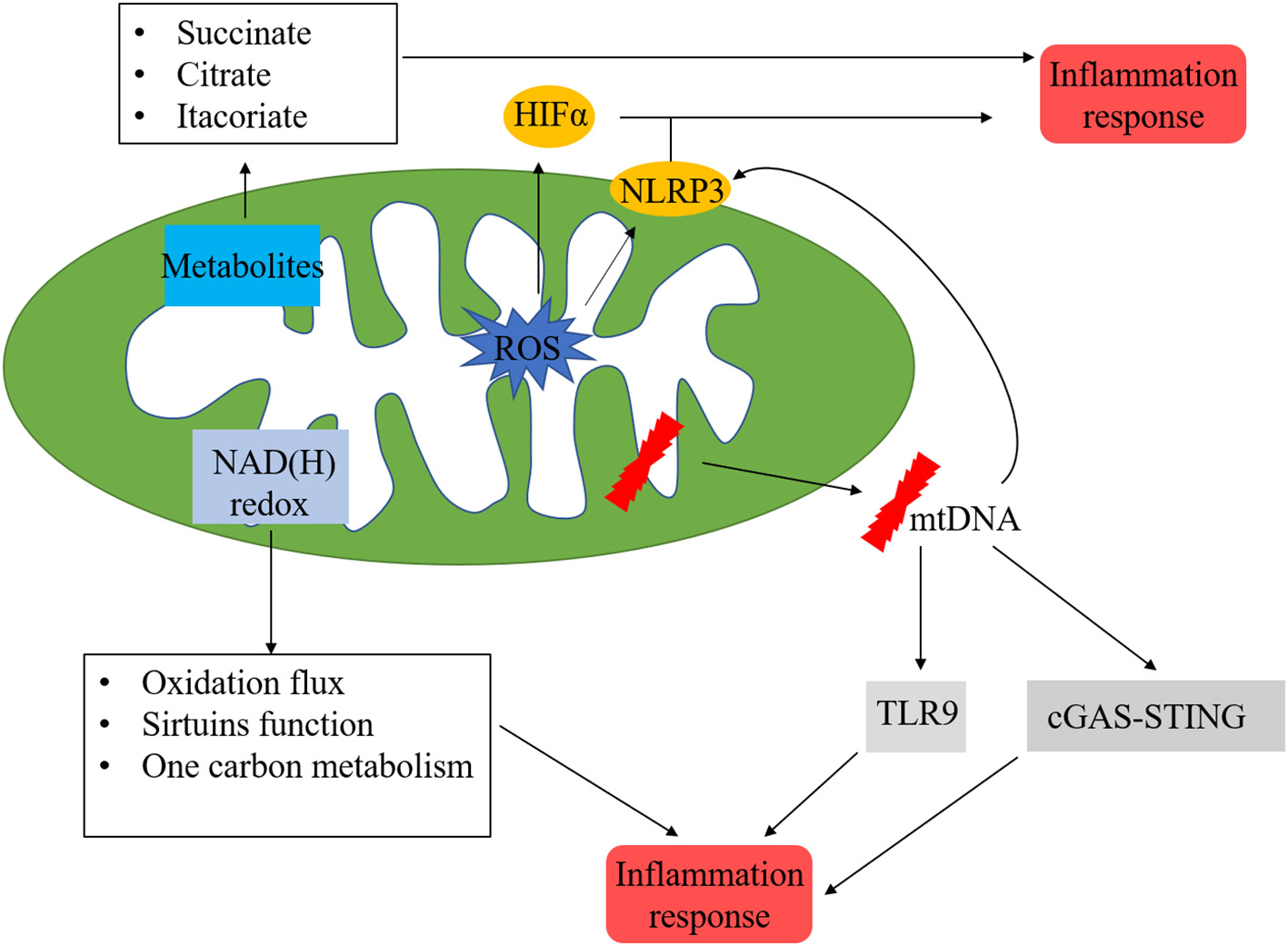
Mitochondria-related inflammation in ischemic heart disease. Many factors in mitochondria could initiate mitochondria-related inflammation, ROS from mitochondrial respiratory chain could directly activate HIFα and NLRP3, which are vital regulators in inflammation process. In addition, metabolites from mitochondria, such as succinate, citrate, and itacoriate also cause inflammation. mtROS could lead to mtDNA damage, which trigger the inflammation response via NLRP3, cGAS/STING, and TLR9 pathways. NAD(H) redox is another initiator of inflammation response via the regulation of oxidation flux, sirtuins function, and one carbon metabolism.
The inflammasome is a cytoplasmic multiprotein complex that contributes to the release of mature cytokines during the innate immune response. Inflammasome could recognise PAMPs or host-derived DAMPs, recruiting, and activating the pro-inflammatory protease caspase-1. NLRP3 inflammasome consists of NLRP3, ASC, and caspase-1 proteins, which play important roles in the pathophysiology of MI. Mitochondrial events and NLRP3 inflammasome activation are tightly bounded. The NLRP3 inflammasome could induce mitochondrial damage via mtROS (125), decreased production of mtROS could effectively inhibit the activation of NLRP3 inflammasome (126). In addition, the insufficiency of damaged mitochondria clearance due to disturbed mitophagy flow would strengthen the activation of the NLRP3 inflammasome. mtDNA is a potential pro-inflammatory trigger for immune cells and is widely accepted as a member of DAMPs (127). Kiichi et al. (128) indicated that mtDNA released into cytoplasm might activate NLRP3 inflammasome, mitophagy could clear damaged mitochondria, followed by the inhibition of NLRP3 inflammasome. Depleting the autophagic proteins LC3B and beclin 1 increased the activation of caspase-1along with secretion of IL-1 β and IL-18. Calcium homeostasis is a critical factor for maintaining mitochondrial function. NLRP3 stimulators (such as ATP) may result in calcium inflow and lead to mitochondrial damage, followed by an increase of mtROS and depletion of mitochondrial membrane potential (129). cGAS/STING is another reported cytosolic mtDNA-sensing pathway, when mitochondria damage leads to the release of fragmented mtDNA into the cytosol, cGAS activates STING, followed by the activation of TBK1, resulting in the translation of interferon genes (130). The inflammatory process initiated by mtDNA is a critical mechanism of ischemic heart disease.
Emerging evidence suggests that macrophage function is closely associated with its mitochondrial metabolism (131, 132). Changes in mitochondrial function have been observed in activated macrophages. In pro-inflammatory macrophages, impairment of TCA flux leads to the accumulation of metabolic intermediates such as succinate and malate (133), overload of succinate is linked to abnormal ROS production (134). The generation of αKG from glutaminolysis is important for alternative M2 activation of macrophages via JMJD3-dependent epigenetic reprogramming of M2 genes (135). Shuang et al. (136) reported a new mechanism of macrophage in the process of myocardial repair after MI, in which efferocytosis increased the level of cellular fatty acids, the increased fatty acids fueled mitochondrial respiration and activated an NAD+-dependent signal transduction cascade, and this process is positive for wound healing. In 2015, Xu et al. (137) reported that the NOTCH signalling pathway is involved in mitochondrial metabolism remodelling, resulting in mtROS generation and pro-inflammatory gene expressions, such as TNF α and IL-1β.
There are other participants reported to be related to the mitochondria-related inflammation process. Mst1 is a stress-activated, pro-apoptotic kinase, Jing et al. (138) reported that the SRV2 deletion inactivated the Mst1-mROS signalling pathway in cardiomyocytes, which could regulate the inflammation and oxidative stress. Another regulator of inflammatory process-S100a8/a9 caused cell death in the early stage of IR injury via mitochondrial respiratory dysfunction (139). Mechanistically, S100a8/a9 downregulated NDUF gene expression which will inhibit the activity of mitochondrial complex I via Toll-like receptor 4/Erk–mediated Pparg coactivator 1 alpha/nuclear respiratory factor 1 signalling suppression.
Mitochondrial Protein Post-Translational Modification in Ischemic Heart Disease
PTMs are alterations of proteins occurring after the translational process catalysed by numerous enzymes. Protein PTMs are important in various physiological and cellular processes, such as differentiation (140), protein degradation (141), gene expression (142, 143). PTMs of proteins have been identified to affect mitochondrial quality control, leading to the exacerbation, or alleviation of ischemic heart disease (144–146) (Figure 2).
Protein phosphorylation plays a critical regulatory role in cardiomyocytes via mediating protein activation or deactivation. Phosphorylation of mitochondrial proteins is vital to maintain mitochondrial function. Phosphorylation of mitochondrial complex IV subunit mediated its physical function in myocardial mitochondria. During IR injury, protein kinase A-dependent phosphorylation of complex IV increased and resulted in a decrease of its activity, followed by an increase in ROS production (147, 148). The STAT3 is a key regulator of mitochondrial metabolism via the interaction with mitochondrial proteins. In ischemic conditions, STAT3 phosphorylation improved mitochondrial function via preserving mitochondrial complex I, preventing mPTP opening with the result of infarction area reduction (149–151).
In eukaryotic cells, the UPS is a primary system of protein degradation. The ubiquitination occurred via ubiquitin-binding its COOH group with the target protein (152, 153). Ubiquitination has been widely accepted as one of the major ways of protein degradation to maintain mitochondrial quality. As we presented previously, PINK/Parkin-mediated mitophagy is a critical pathway for mitochondrial quality control. Parkin is an E3 ligase, which can ubiquitinate several mitochondrial outer membrane proteins via E3 ligase activity to recruit the p62 protein. CypD ubiquitinated by Parkin could inhibit mPTP opening, alleviating myocardial injury (154). In the process of heart remodelling after IR injury, exogenous ubiquitin supplement could reduce caspase-9 expression in the mitochondrial death pathway, increasing mitochondrial production, reducing infarct area, and finally restoring heart function (155). Wangxing et al. (156) demonstrated that leptin-overexpressing hMSCs into the infarcted heart could improve cardiac function. Further mechanical exploration indicated that leptin restored mitochondrial respiratory function via enhancing OPA1 expression by inhibiting the activity of OMA1, a mitochondrial protease. In addition, phosphorylation of GSK3 is a prerequisite for ubiquitination-depended degradation of OMA1 and attenuation of long-OPA1 cleavage.
SUMO is a member of the large family of ubiquitin-like proteins. SUMOylation is a classic ubiquitination-like PTMs, linking the SUMO protein to the lysine residue of the substrate protein (157). In heart IR injury, the binding of SUMO with Drp1 increased to form a complex, increasing the acidification of the complex could maintain mitochondrial quality and improve cardiac function (158). Another deSUMOylation-related enzyme, SENP3 could alleviate IR injury via the inhibition of Drp1 localisation in the mitochondria (159). Acetylation is one of the major PTMs in cell biology, SIRT3 and SIRT5 are sirtuins found in mitochondria. Angela et al. reported that SIRT3 could reduce the activity of CypD, inhibiting the opening of mPTP. Increasing SIRT3 expression in the failing heart could improve cardiac function (160). Studies showed that decreased expression of SIRT3 in heart increased susceptibility to IR injury (161, 162). In addition, SIRT1 was also found to be involved in cardiac IR injury, study found that increased expression of SIRT1 could restore left ventricular function during the construction of myocardial IR models (163). Zhao et al. invested the role of HDACs inhibition in myocardial IR injury and found that HDACs inhibition protected the heart against I/R injury (164). Moreover, SIRT5 (–/–) mouse hearts are more tendentious to suffer IRI due to the increase of lysine succinylation followed by the accumulation of mitochondrial ROS, and mtROS scavenged by SDH inhibition could reverse this process (165). Accordingly, the exploration of the interaction of PTMs in ischemic heart disease by modulating mitochondrial quality control has a bright future to investigate novel therapeutic targets.
MiRNAs are short non-coding RNA binding to the 3' UTR sequences and regulating targeted gene expression either by mRNA degradation or translational repression (166). Mitochondria contain miRNAs that are termed as mitomiRs in several species and cell types (167). MitomiRs target either mitochondrial or nuclear proteincoding mRNAs, thereby influencing mitochondrial metabolism and dynamics through regulation of the main mitochondrial pathways, such as OXPHOS, ETC components, TCA cycle (168). Overexpression of miRNA-34a in AMI patients' serum enhances cardiomyocyte apoptosis by down-regulating mitochondrial anti-apoptotic protein aldehyde dehydrogenase 2 (169). miRNA-1 could enter mitochondria and regulate mitochondrial ETC via targeting proteins in ETC networks, while increased miRNA-1 expression has been found in the remote myocardium of AMI patients (170). Mitochondrial miRNA-762 regulates cardiomyocyte apoptosis via impairing the core subunit of mitochondrial complex I (171). In addition, lncRNAs are also a heterogeneous class of transcripts involved in the epigenetic regulation of gene and genome activity (172, 173). Recent data indicated that lncRNAs localised to mitochondria, regulating mitochondrial function (174–176). Circulating levels of mitochondrial lncRNA LIPCAR were downregulated early after AMI and upregulated during later stages and were associated with adverse cardiac remodelling and death (175). In addition to lncRNAs, growing evidence also reports that circRNAs are involved in the regulation of the mitochondrial dynamics and cardiomyocyte apoptosis. Kun et al. (177) found that MFACR regulated mitochondrial fission and apoptosis in the heart by targeting and downregulation of miR-652-3p. To sum up, mitochondrial non-coding RNAs are involved in the pathogenesis of myocardial infarction via regulating various pathways.
Targeted Therapies
Early reperfusion of occluded coronary arteries is the most effective strategy of AMI over the past decades. However, there is no effective therapy for reperfusion injury and alleviation of cardiac remodellin. Therefore, with a growing understanding of the molecular mechanisms of ischemia, IR injury, and chronic remodelling, we may develop more novel therapeutic targets to protect the heart from IHD and improve clinical outcomes of these patients. Considering numerous studies in this field, we only discussed the agents involved in mitochondrial targets in animal studies or clinical trials.
Over decades, mtROS is one of the most popular targets of heart protection. A reduced generation or increased scavenge of mtROS have been reported to increase outcomes. MitoQ is the first mitochondria-targeted antioxidant, which is bioavailable orally without toxicity detected. Rat received MitoQ for 2 weeks suffered reduced oxidative stress and resisted heart ischemia-reperfusion injury (178). SS-31 is a kind of small artificial peptides with therapeutic potential due to its antioxidant properties (179–181), in rat tests, SS-31 could attenuate the strike of ischemia and reperfusion via reducing MI size (180, 182, 183). SODs are metal-containing antioxidant enzymes, protecting cells from damage by converting superoxide radicals to H2O2 and O2. Mito-specific SOD mimetics exert protection under oxidative stress (184). Clinical trials of SS-31 in patients with heart failure and acute myocardial infarction have been tested, although the myocardial infarct size did not show an improvement, it showed acceptable safety and tolerability (185, 186). Cerrato's group two analogues of SS-31 (mtCPP-1 and mtgCPP) and reported greater efficiency and antioxidant capacity than SS-31 (187, 188). In an animal model of post-infarction heart failure, activation of ALDH2 with Alda-1 improves the clinical outcomes via the decrease of reactive aldehydes (189).
mPTP is a mitochondria voltage- and Ca2+-dependent high-conductance channel. Piot et al. exerted a clinical trial that included 58 patients who suffered acute ST-elevation myocardial infarction to receive cyclosporine immediately before undergoing PCI, it came out that cyclosporine could decrease the infarct size to some extent (190). Currently, some larger clinical trials are ongoing to test the effect of mPTP inhibition on short- and long-term patients' outcomes. Trehalose is a small molecule from mushrooms, which could activate mitophagy. Studies indicated that administration of trehalose for 4 weeks could reverse cardiac remodelling and fibrosis in MI model mice (191, 192). Spermidine is another natural compound that activates autophagy. In the MI rat model, oral supplementation of spermidine is inversely associated with all-cause mortality and MI risk via enhancement of mitochondrial respiration (193). In addition, Jing et al. reported that spermidine supplement improved MI-induced cardiac dysfunction through AMPK/mTOR mediated autophagic flux (194). Mdivi-1 is an inhibitor of mitochondria-related fission protein-Drp1, which is proved to reduce infarct size, rescue cardiac function in the IRI mouse model (195). In addition, mdivi-1 treatment ameliorated IRI via the inhibition of connexin 43 loss and suppression of MMP3 (196). UPRmt is critical to maintain mitochondrial proteostasis under cellular stress, UPRmt induced by oligomycin or doxycycline has been identified to reduce MI size in mice models (197). The expression of AMPK increased in failing heart, metformin is considered as a pharmacological activator of AMPK, which could reduce cardiac infarct size and improve heart contractibility in a rat MI model. Artificial compounds targeting AMPK have been designed, Abbott lab generated an artificial agent, named A769662, which could reduce infarct size in rats via specific activating AMPK on β subunit (198, 199).
Although various experimental studies developed effective cardioprotective strategies, there are few successful clinical translations. And this has been attributed to different factors: firstly, there are no effective delivery systems for agents to carry out their functions. Secondly, animal models are different from patients with ischemic heart disease, which usually also suffered from other co-morbidities such as hypertension, diabetes. Recently, some clinical agents such as DPP-4 and SGLT2 have been identified to protect cardiomyocytes from IRI damage via mitochondrial function preservation. GLP-1 is an incretin hormone with cardioprotective capacities and was markedly increased in acute myocardial infarction. In physical conditions, GLP-1 could be degraded by DPP-4, Sebastian et al. (200) found that DPP-4 inhibitor could maintain the serum concentration of GLP-1, increasing AMPK activity and mitochondrial respiratory capacity of non-infarcted tissues. In addition, GLP-1 agonist, liraglutide could reduce cardiac infarct size, protected cardiomyocytes from injury and preserved contractile function via suppressing ROS generation, NADPH oxidase and proinflammatory signals (201). SGLT-2 inhibitors are a new generation of anti-diabetic agents, which have been recommended in cardio-protection (202). Various studies proved that SGLT-2 inhibitors could ameliorate cardiac remodelling and increase mitochondrial function (203, 204). Another type of anti-diabetic agent, metformin is also reported to exert cardio-protection by restoring mitochondria function and dynamics in cardiac I/R injury (205). Some natural agents or analogues such as taurine, fisetin, and humanin also exert cardio-protection against IRI by reducing mitochondrial dysfunction (206–208). In summary, despite there is much evidence supporting the targeting of mitochondria as a therapy strategy in IHD, more efforts are needed to promote the basic to clinical translation.
Conclusions
Mitochondrial homeostasis is critical for the maintain of the mitochondria network. In this review, we summarised the advances supporting the view that mitochondrial disorder is a major contributor to cardiac injury, IRI, as well as chronic remodelling. Mitophagy disorder, increased mtROS, mitochondria-related inflammation and post-translation of mitochondrial proteins are considered contributory factors to mitochondrial dysfunction in ischemic heart disease. Accordingly, many targeted modulations involved in mitochondrial quality control provide great chances for the design of novel therapies. Although there are no drugs with successful clinical transformation, which are directly targeted mitochondria-related mechanisms, many regulatory proteins or peptides and miRNAs possess significant potential.
Funding
This work was supported by Beijing Natural Science Foundation (No.7214219); Natural Science Foundation of Liao Ning Province (2019-MS-386).
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
YX, JinL, HG, and JuL contributed to the first and second version of the manuscript. XZ and HL contributed to the second version. HL supervised the project. JiaL made great contribution to our revision manuscript and provided helpful opinions. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Glossary
ALDH2, Aldehyde dehydrogenase 2; ANT, adenosine nucleotide translocase; AMPK, Adenosine 5′-monophosphate (AMP)-activated protein kinase; AMI, Acute Myocardial Infarction; ARE, Antioxidant response elements; ASC, Apoptosis-associated speck-like protein; α-KG, α-ketoglutarate; ATP, Adenosine triphosphate; BNIP3, BCL2 Interacting Protein 3; cGAS, GMP-AMP synthase; circRNA, covalently closed, single-stranded RNA; CNC, Cap “n” collar; CREB, cAMP-response element binding protein; CypD, Cyclophilin D; DAMP3, Danger signalling molecules 3; DPP4, Dipeptidyl peptidase-4; Drp1, Dynamin-related protein1; ECM, Extracellular matrix; ETC, electron transport chain; ERK, Extracellular signal-regulated kinas; FAO, Fatty acid β-oxidation; FUNDC1, FUN14 Domain Containing 1; GLP1, Glucagon-like peptide 1; Gpx, Glutathione peroxidase; GSK3, Glycogen synthase kinase-3; GTPase, Guanosine triphosphatases; HDAC, Histone deacetylases; HF, Heart failure; HIF, Hypoxia-inducible factor; hMSC, Human mesenchymal stem cells; IHD, Ischemic heart disease; IL-1β, Interleukin-1β; IRI, Ischemia reperfusion injury; LC3, Microtubule- associated protein1 light chain 3; lncRNA, Long noncoding RNAs; MAO, Monoamine oxidases; MAPK, Mitogen - activated protein kinase; MCU, Mitochondrial Calcium Uniporter; MFACR, Mitochondrial fission and apoptosis-related circRNA; Mfn1/2, Mitofusin 1/2; miRNA, MicroRNA; mitomiRs, Mitochondria miRNAs; MMP, Matrix processing peptidases; MPP, Matrix metalloproteinases; mPTP, Mitochondrial permeability transition pore; mRNA, Messenger RNA; Mst1, Mammalian STE20-like protein kinase 1; mTOR, Mammalian target of rapamycin; NAPDH, Nicotinamide adenine dinucleotide phosphate; NLRP3, Nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3; Nox, NADPH oxidases; NR4A1, Nuclear receptor subfamily 4 group A member 1; OMM, Outer mitochondrial membrane; OPA, Optica atrophy protein 1; OXPHOS, Oxidative phosphorylation; PAMP, Pathogen-associated molecular patterns; PCI, Percutaneous coronary intervention; PINK, PTEN-induced putative kinase 1; PTM, Post-translational modification; RIPK3, Receptor-interacting protein 3; ROS, Reactive oxygen species; SDH, Succinate dehydrogenase; SENP3, Sentrin-specific protease 3; SGLT2, Sodium-glucose cotransporter 2; SIRT, Sirtuin; SIAH1, Siah E3 ubiquitin protein ligase 1; SMURF1, Smad ubiquitination regulatory factor-1; SOD, Superoxide dismutase; SRV, Suppressor of ras val-2; STAT3, Signal transducer and activator of transcription 3; STING, Stimulator of interferon genes; SUMO, Small ubiquitin-like modifier; TBK1, TANK-binding kinase 1; TCA, Tricarboxylic acid cycle; TNF-α, Tumour necrosis factor α; UPRmt, Mitochondrial unfolded protein response; UPS, Ubiquitin-proteasome system; UTR, untranslated region; VDAC1, Voltage-dependent anion-selective channel protein 1; WDR26, WD repeat domain 26.
References
1.
Dorweiler B Pruefer D Andrasi TB Maksan SM Schmiedt W Neufang A et al . Ischemia-reperfusion injury: pathophysiology and clinical implications. Eur J Trauma Emerg Surg. (2007) 33:600–12. 10.1007/s00068-007-7152-z
2.
Murphy E Steenbergen C . Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. (2008) 88:581–609. 10.1152/physrev.00024.2007
3.
Heusch G Gersh BJ . The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge. Eur Heart J. (2017) 38:774–84. 10.1093/eurheartj/ehw224
4.
Hung J Teng TH Finn J Knuiman M Briffa T Stewart S et al . Trends from 1996 to 2007 in incidence and mortality outcomes of heart failure after acute myocardial infarction: a population-based study of 20,812 patients with first acute myocardial infarction in Western Australia. J Am Heart Assoc. (2013) 2:e000172. 10.1161/JAHA.113.000172
5.
Sulo G Igland J Vollset SE Nygård O Ebbing M Sulo E et al . Heart failure complicating acute myocardial infarction; burden and timing of occurrence: a nation-wide analysis including 86 771 patients from the cardiovascular disease in Norway (CVDNOR) Project. J Am Heart Assoc. (2016) 5:e002667. 10.1161/JAHA.115.002667
6.
Kaneko H Anzai T Takahashi T Kohno T Shimoda M Sasaki A et al . Role of vascular endothelial growth factor-A in development of abdominal aortic aneurysm. Cardiovasc Res. (2011) 91:358–67. 10.1093/cvr/cvr080
7.
Hara H Takeda N Komuro I . Pathophysiology and therapeutic potential of cardiac fibrosis. Inflamm Regen. (2017) 37:13. 10.1186/s41232-017-0046-5
8.
Gulati A Jabbour A Ismail TF Guha K Khwaja J Raza S et al . Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA. (2013) 309:896–908. 10.1001/jama.2013.1363
9.
Neubauer S . The failing heart–an engine out of fuel. N Engl J Med. (2007) 356:1140–51. 10.1056/NEJMra063052
10.
Jeong SY Seol DW . The role of mitochondria in apoptosis. BMB Rep. (2008) 41:11–22. 10.5483/BMBRep.2008.41.1.011
11.
Yang Y Karakhanova S Hartwig W D'Haese JG Philippov PP Werner J et al . Mitochondria and mitochondrial ROS in cancer: novel targets for anticancer therapy. J Cell Physiol. (2016) 231:2570–81. 10.1002/jcp.25349
12.
Zorov DB Juhaszova M Sollott SJ . Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. (2014) 94:909–50. 10.1152/physrev.00026.2013
13.
Cadenas S . Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim Biophys Acta Bioenerg. (2018) 1859:940–50. 10.1016/j.bbabio.2018.05.019
14.
Bartel K Pein H Popper B Schmitt S Janaki-Raman S Schulze A et al . Connecting lysosomes and mitochondria - a novel role for lipid metabolism in cancer cell death. Cell Commun Signal. (2019) 17:87. 10.1186/s12964-019-0399-2
15.
D'Souza K Nzirorera C Kienesberger PC . Lipid metabolism and signaling in cardiac lipotoxicity. Biochim Biophys Acta. (2016) 1861:1513–24. 10.1016/j.bbalip.2016.02.016
16.
Begriche K Massart J Robin MA Borgne-Sanchez A Fromenty B . Drug-induced toxicity on mitochondria and lipid metabolism: mechanistic diversity and deleterious consequences for the liver. J Hepatol. (2011) 54:773–94. 10.1016/j.jhep.2010.11.006
17.
Kolmychkova KI Zhelankin AV Karagodin VP Orekhov AN . Mitochondria and inflammation. Patol Fiziol Eksp Ter. (2016) 60:114–21.
18.
Bugger H Pfeil K . Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165768. 10.1016/j.bbadis.2020.165768
19.
Hoppins S Lackner L Nunnari J . The machines that divide and fuse mitochondria. Annu Rev Biochem. (2007) 76:751–80. 10.1146/annurev.biochem.76.071905.090048
20.
Busch KB Bereiter-Hahn J Wittig I Schagger H Jendrach M . Mitochondrial dynamics generate equal distribution but patchwork localization of respiratory Complex I. Mol Membr Biol. (2006) 23:509–20. 10.1080/09687860600877292
21.
Dimmer KS Scorrano L . (De)constructing mitochondria: what for?Physiology. (2006) 21:233–41. 10.1152/physiol.00010.2006
22.
Twig G Hyde B Shirihai OS . Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta. (2008) 1777:1092–7. 10.1016/j.bbabio.2008.05.001
23.
Kuzmicic J Del Campo A López-Crisosto C Morales PE Pennanen C Bravo-Sagua R et al . [Mitochondrial dynamics: a potential new therapeutic target for heart failure]. Rev Esp Cardiol. (2011) 64:916–23. 10.1016/j.rec.2011.05.022
24.
Balaban RS . Domestication of the cardiac mitochondrion for energy conversion. J Mol Cell Cardiol. (2009) 46:832–41. 10.1016/j.yjmcc.2009.02.018
25.
Solaini G Harris DA . Biochemical dysfunction in heart mitochondria exposed to ischaemia and reperfusion. Biochem J. (2005) 390:377–94. 10.1042/BJ20042006
26.
Lejay A Fang F John R Van JA Barr M Thaveau F et al . Ischemia reperfusion injury, ischemic conditioning and diabetes mellitus. J Mol Cell Cardiol. (2016) 91:11–22. 10.1016/j.yjmcc.2015.12.020
27.
Halestrap AP Brenner C . The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem. (2003) 10:1507–25. 10.2174/0929867033457278
28.
Kwong JQ Davis J Baines CP Sargent MA Karch J Wang X et al . Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. (2014) 21:1209–17. 10.1038/cdd.2014.36
29.
Baines CP Kaiser RA Sheiko T Craigen WJ Molkentin JD . Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. (2007) 9:550–5. 10.1038/ncb1575
30.
Halestrap AP . Mitochondrial permeability: dual role for the ADP/ATP translocator?Nature. (2004) 430:983. 10.1038/nature02816
31.
Davies KM Anselmi C Wittig I Faraldo-Gómez JD Kühlbrandt W . Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc Natl Acad Sci USA. (2012) 109:13602–7. 10.1073/pnas.1204593109
32.
Davies KM Daum B Gold VA Mühleip AW Brandt T Blum TB et al . Visualization of ATP synthase dimers in mitochondria by electron cryo-tomography. J Vis Exp. (2014) 14:51228. 10.3791/51228
33.
He J Carroll J Ding S Fearnley IM Walker JE . Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc Natl Acad Sci USA. (2017) 114:9086–91. 10.1073/pnas.1711201114
34.
He J Ford HC Carroll J Ding S Fearnley IM Walker JE . Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc Natl Acad Sci USA. (2017) 114:3409–14. 10.1073/pnas.1702357114
35.
Bonora M Bononi A De Marchi E Giorgi C Lebiedzinska M Marchi S et al . Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle. (2013) 12:674–83. 10.4161/cc.23599
36.
Azarashvili T Odinokova I Bakunts A Ternovsky V Krestinina O Tyynelä J et al . Potential role of subunit c of F0F1-ATPase and subunit c of storage body in the mitochondrial permeability transition. effect of the phosphorylation status of subunit c on pore opening. Cell Calcium. (2014) 55:69–77. 10.1016/j.ceca.2013.12.002
37.
Mannella CA Lederer WJ Jafri MS . The connection between inner membrane topology and mitochondrial function. J Mol Cell Cardiol. (2013) 62:51–7. 10.1016/j.yjmcc.2013.05.001
38.
Daum B Walter A Horst A Osiewacz HD Kühlbrandt W . Age-dependent dissociation of ATP synthase dimers and loss of inner-membrane cristae in mitochondria. Proc Natl Acad Sci USA. (2013) 110:15301–6. 10.1073/pnas.1305462110
39.
Mnatsakanyan N Llaguno MC Yang Y Yan Y Weber J Sigworth FJ et al . A mitochondrial megachannel resides in monomeric F(1)F(O) ATP synthase. Nat Commun. (2019) 10:5823. 10.1038/s41467-019-13766-2
40.
Luptak I Balschi JA Xing Y Leone TC Kelly DP Tian R . Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation. (2005) 112:2339–46. 10.1161/CIRCULATIONAHA.105.534594
41.
Neglia D De Caterina A Marraccini P Natali A Ciardetti M Vecoli C et al . Impaired myocardial metabolic reserve and substrate selection flexibility during stress in patients with idiopathic dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. (2007) 293:H3270–8. 10.1152/ajpheart.00887.2007
42.
Kolwicz SC Jr Tian R . Glucose metabolism and cardiac hypertrophy. Cardiovasc Res. (2011) 90:194–201. 10.1093/cvr/cvr071
43.
Nediani C Raimondi L Borchi E Cerbai E . Nitric oxide/reactive oxygen species generation and nitroso/redox imbalance in heart failure: from molecular mechanisms to therapeutic implications. Antioxid Redox Signal. (2011) 14:289–331. 10.1089/ars.2010.3198
44.
Yoo SM Jung YK . A molecular approach to mitophagy and mitochondrial dynamics. Mol Cells. (2018) 41:18–26. 10.14348/molcells.2018.2277
45.
Bravo-San Pedro JM Kroemer G Galluzzi L . Autophagy and mitophagy in cardiovascular disease. Circ Res. (2017) 120:1812–24. 10.1161/CIRCRESAHA.117.311082
46.
Pickrell AM Youle RJ . The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. (2015) 85:257–73. 10.1016/j.neuron.2014.12.007
47.
Matsuda N Sato S Shiba K Okatsu K Saisho K Gautier CA et al . PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. (2010) 189:211–21. 10.1083/jcb.200910140
48.
Narendra D Tanaka A Suen DF Youle RJ . Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. (2008) 183:795–803. 10.1083/jcb.200809125
49.
Harper JW Ordureau A Heo JM . Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol. (2018) 19:93–108. 10.1038/nrm.2017.129
50.
Sekine S Youle RJ . PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. (2018) 16:2. 10.1186/s12915-017-0470-7
51.
Pickles S Vigié P Youle RJ . Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. (2018) 28:R170–85. 10.1016/j.cub.2018.01.004
52.
Aguirre JD Dunkerley KM Mercier P Shaw GS . Structure of phosphorylated UBL domain and insights into PINK1-orchestrated parkin activation. Proc Natl Acad Sci USA. (2017) 114:298–303. 10.1073/pnas.1613040114
53.
Ordureau A Sarraf SA Duda DM Heo JM Jedrychowski MP Sviderskiy VO et al . Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. (2014) 56:360–75. 10.1016/j.molcel.2014.09.007
54.
Chan NC Salazar AM Pham AH Sweredoski MJ Kolawa NJ Graham RL et al . Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. (2011) 20:1726–37. 10.1093/hmg/ddr048
55.
Tanaka A Cleland MM Xu S Narendra DP Suen DF Karbowski M et al . Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. (2010) 191:1367–80. 10.1083/jcb.201007013
56.
Santulli G Lewis DR Marks AR . Physiology and pathophysiology of excitation-contraction coupling: the functional role of ryanodine receptor. J Muscle Res Cell Motil. (2017) 38:37–45. 10.1007/s10974-017-9470-z
57.
Ikeda Y Shirakabe A Maejima Y Zhai P Sciarretta S Toli J et al . Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. (2015) 116:264–78. 10.1161/CIRCRESAHA.116.303356
58.
Otera H Miyata N Kuge O Mihara K . Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J Cell Biol. (2016) 212:531–44. 10.1083/jcb.201508099
59.
Zhou H Zhang Y Hu S Shi C Zhu P Ma Q et al . Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res. (2017) 63:e12413. 10.1111/jpi.12413
60.
Song M Gong G Burelle Y Gustafsson ÅB Kitsis RN Matkovich SJ et al . Interdependence of parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ Res. (2015) 117:346–51. 10.1161/CIRCRESAHA.117.306859
61.
Zhou H Wang J Zhu P Zhu H Toan S Hu S et al . NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2α. Basic Res Cardiol. (2018) 113:23. 10.1007/s00395-018-0682-1
62.
Frezza C Cipolat S Martins de Brito O Micaroni M Beznoussenko GV Rudka T et al . OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. (2006) 126:177–89. 10.1016/j.cell.2006.06.025
63.
Chen L Gong Q Stice JP Knowlton AA . Mitochondrial OPA1, apoptosis, heart failure. Cardiovasc Res. (2009) 84:91–9. 10.1093/cvr/cvp181
64.
Shen T Zheng M Cao C Chen C Tang J Zhang W et al . Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chem. (2007) 282:23354–61. 10.1074/jbc.M702657200
65.
Xin T Lu C . Irisin activates Opa1-induced mitophagy to protect cardiomyocytes against apoptosis following myocardial infarction. Aging. (2020) 12:4474–88. 10.18632/aging.102899
66.
Zhang Y Wang Y Xu J Tian F Hu S Chen Y et al . Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J Pineal Res. (2019) 66:e12542. 10.1111/jpi.12542
67.
Guan L Che Z Meng X Yu Y Li M Yu Z et al . MCU Up-regulation contributes to myocardial ischemia-reperfusion Injury through calpain/OPA-1-mediated mitochondrial fusion/mitophagy Inhibition. J Cell Mol Med. (2019) 23:7830–43. 10.1111/jcmm.14662
68.
Li G Yang J Yang C Zhu M Jin Y McNutt MA et al . PTENα regulates mitophagy and maintains mitochondrial quality control. Autophagy. (2018) 14:1742–60. 10.1080/15548627.2018.1489477
69.
Feng Y Zhao J Hou H Zhang H Jiao Y Wang J et al . WDR26 promotes mitophagy of cardiomyocytes induced by hypoxia through Parkin translocation. Acta Biochim Biophys Sin. (2016) 48:1075–84. 10.1093/abbs/gmw104
70.
Das S Mitrovsky G Vasanthi HR Das DK . Antiaging properties of a grape-derived antioxidant are regulated by mitochondrial balance of fusion and fission leading to mitophagy triggered by a signaling network of Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid Med Cell Longev. (2014) 2014:345105. 10.1155/2014/345105
71.
Wang Y Lu X Zhao P Jiang J Yao L . [Overexpression of PPENK reduces myocardial ischemia reperfusion injury by promoting mitophagy in rats]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. (2017) 33:1335–40.
72.
Bian X Teng T Zhao H Qin J Qiao Z Sun Y et al . Zinc prevents mitochondrial superoxide generation by inducing mitophagy in the setting of hypoxia/reoxygenation in cardiac cells. Free Radic Res. (2018) 52:80–91. 10.1080/10715762.2017.1414949
73.
Yang M Linn BS Zhang Y Ren J . Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:2293–302. 10.1016/j.bbadis.2019.05.007
74.
Yu W Xu M Zhang T Zhang Q Zou C . Mst1 promotes cardiac ischemia-reperfusion injury by inhibiting the ERK-CREB pathway and repressing FUNDC1-mediated mitophagy. J Physiol Sci. (2019) 69:113–27. 10.1007/s12576-018-0627-3
75.
Zhang W Chen C Wang J Liu L He Y Chen Q . Mitophagy in cardiomyocytes and in platelets: a major mechanism of cardioprotection against ischemia/reperfusion injury. Physiology. (2018) 33:86–98. 10.1152/physiol.00030.2017
76.
Zhang W Siraj S Zhang R Chen Q . Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy. (2017) 13:1080–1. 10.1080/15548627.2017.1300224
77.
Zhang W Ren H Xu C Zhu C Wu H Liu D et al . Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. Elife. (2016) 5:e21407. 10.7554/eLife.21407
78.
Fu M St-Pierre P Shankar J Wang PT Joshi B Nabi IR . Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol Biol Cell. (2013) 24:1153–62. 10.1091/mbc.e12-08-0607
79.
Szargel R Shani V Abd Elghani F Mekies LN Liani E Rott R et al . The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum Mol Genet. (2016) 25:3476–90. 10.1093/hmg/ddw189
80.
Heo JM Ordureau A Paulo JA Rinehart J Harper JW . The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. (2015) 60:7–20. 10.1016/j.molcel.2015.08.016
81.
Moore AS Holzbaur EL . Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci USA. (2016) 113:E3349–58. 10.1073/pnas.1523810113
82.
Richter B Sliter DA Herhaus L Stolz A Wang C Beli P et al . Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA. (2016) 113:4039–44. 10.1073/pnas.1523926113
83.
Choong CJ Okuno T Ikenaka K Baba K Hayakawa H Koike M et al . Alternative mitochondrial quality control mediated by extracellular release. Autophagy. (2020) 17:2962–74. 10.1080/15548627.2020.1848130
84.
Gordon S . Phagocytosis: an immunobiologic process. Immunity. (2016) 44:463–75. 10.1016/j.immuni.2016.02.026
85.
Nicolás-Ávila JA Lechuga-Vieco AV Esteban-Martínez L Sánchez-Díaz M Díaz-García E Santiago DJ et al . A network of macrophages supports mitochondrial homeostasis in the heart. Cell. (2020) 183:94–109.e23. 10.1016/j.cell.2020.08.031
86.
Porter KE Turner NA . Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. (2009) 123:255–78. 10.1016/j.pharmthera.2009.05.002
87.
Diwan A Krenz M Syed FM Wansapura J Ren X Koesters AG et al . Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest. (2007) 117:2825–33. 10.1172/JCI32490
88.
Dorn GW II . Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res. (2010) 3:374–83. 10.1007/s12265-010-9174-x
89.
Zhu P Wan K Yin M Hu P Que Y Zhou X et al . RIPK3 induces cardiomyocyte necroptosis via inhibition of AMPK-parkin-mitophagy in cardiac remodelling after myocardial infarction. Oxid Med Cell Longev. (2021) 2021:6635955. 10.1155/2021/6635955
90.
Song M Mihara K Chen Y Scorrano L Dorn GW II . Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. (2015) 21:273–86. 10.1016/j.cmet.2014.12.011
91.
Finkel T . Signal transduction by reactive oxygen species. J Cell Biol. (2011) 194:7–15. 10.1083/jcb.201102095
92.
D'Autréaux B Toledano MB . ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. (2007) 8:813–24. 10.1038/nrm2256
93.
Duchen MR . Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol Aspects Med. (2004) 25:365–451. 10.1016/j.mam.2004.03.001
94.
Dougherty CJ Kubasiak LA Frazier DP Li H Xiong WC Bishopric NH et al . Mitochondrial signals initiate the activation of c-Jun N-terminal kinase (JNK) by hypoxia-reoxygenation. Faseb j. (2004) 18:1060–70. 10.1096/fj.04-1505com
95.
Halestrap AP Clarke SJ Khaliulin I . The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta. (2007) 1767:1007–31. 10.1016/j.bbabio.2007.05.008
96.
Frazier DP Wilson A Dougherty CJ Li H Bishopric NH Webster KA . PKC-alpha and TAK-1 are intermediates in the activation of c-Jun NH2-terminal kinase by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. (2007) 292:H1675–84. 10.1152/ajpheart.01132.2006
97.
Zorov DB Filburn CR Klotz LO Zweier JL Sollott SJ . Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. (2000) 192:1001–14. 10.1084/jem.192.7.1001
98.
Aon MA Cortassa S Marbán E O'Rourke B . Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem. (2003) 278:44735–44. 10.1074/jbc.M302673200
99.
Braunersreuther V Montecucco F Asrih M Pelli G Galan K Frias M et al . Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury. J Mol Cell Cardiol. (2013) 64:99–107. 10.1016/j.yjmcc.2013.09.007
100.
Matsushima S Kuroda J Ago T Zhai P Ikeda Y Oka S et al . Broad suppression of NADPH oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1α and upregulation of peroxisome proliferator-activated receptor-α. Circ Res. (2013) 112:1135–49. 10.1161/CIRCRESAHA.111.300171
101.
Pagliaro P Penna C . Redox signalling and cardioprotection: translatability and mechanism. Br J Pharmacol. (2015) 172:1974–95. 10.1111/bph.12975
102.
Heusch G . Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, remote conditioning. Circ Res. (2015) 116:674–99. 10.1161/CIRCRESAHA.116.305348
103.
Semenza GL . Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J. (2007) 405:1–9. 10.1042/BJ20070389
104.
Semenza GL . Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology. (2009) 24:97–106. 10.1152/physiol.00045.2008
105.
Kim JW Tchernyshyov I Semenza GL Dang CV . HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) 3:177–85. 10.1016/j.cmet.2006.02.002
106.
Xu B Zhang J Strom J Lee S Chen QM . Myocardial ischemic reperfusion induces de novo Nrf2 protein translation. Biochim Biophys Acta. (2014) 1842:1638–47. 10.1016/j.bbadis.2014.06.002
107.
Itoh K Chiba T Takahashi S Ishii T Igarashi K Katoh Y et al . An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. (1997) 236:313–22. 10.1006/bbrc.1997.6943
108.
Kaludercic N Mialet-Perez J Paolocci N Parini A Di Lisa F . Monoamine oxidases as sources of oxidants in the heart. J Mol Cell Cardiol. (2014) 73:34–42. 10.1016/j.yjmcc.2013.12.032
109.
Bianchi P Kunduzova O Masini E Cambon C Bani D Raimondi L et al . Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation. (2005) 112:3297–305. 10.1161/CIRCULATIONAHA.104.528133
110.
Carpi A Menabò R Kaludercic N Pelicci P Di Lisa F Giorgio M . The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim Biophys Acta. (2009) 1787:774–80. 10.1016/j.bbabio.2009.04.001
111.
Cohn JN Ferrari R Sharpe N . Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. behalf of an international forum on cardiac remodeling. J Am Coll Cardiol. (2000) 35:569–82. 10.1016/S0735-1097(99)00630-0
112.
Ma Y Iyer RP Jung M Czubryt MP Lindsey ML . Cardiac fibroblast activation post-myocardial infarction: current knowledge gaps. Trends Pharmacol Sci. (2017) 38:448–58. 10.1016/j.tips.2017.03.001
113.
Siwik DA Colucci WS . Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail Rev. (2004) 9:43–51. 10.1023/B:HREV.0000011393.40674.13
114.
Hori M Nishida K . Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res. (2009) 81:457–64. 10.1093/cvr/cvn335
115.
Siwik DA Pagano PJ Colucci WS . Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol. (2001) 280:C53–60. 10.1152/ajpcell.2001.280.1.C53
116.
Tyagi SC Ratajska A Weber KT . Myocardial matrix metalloproteinase(s): localization and activation. Mol Cell Biochem. (1993) 126:49–59. 10.1007/BF01772207
117.
Kinugawa S Tsutsui H Hayashidani S Ide T Suematsu N Satoh S et al . Treatment with dimethylthiourea prevents left ventricular remodeling and failure after experimental myocardial infarction in mice: role of oxidative stress. Circ Res. (2000) 87:392–8. 10.1161/01.RES.87.5.392
118.
Lacerda D Ortiz V Türck P Campos-Carraro C Zimmer A Teixeira R et al . Stilbenoid pterostilbene complexed with cyclodextrin preserves left ventricular function after myocardial infarction in rats: possible involvement of thiol proteins and modulation of phosphorylated GSK-3β. Free Radic Res. (2018) 52:988–99. 10.1080/10715762.2018.1506115
119.
Shiomi T Tsutsui H Matsusaka H Murakami K Hayashidani S Ikeuchi M et al . Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. (2004) 109:544–9. 10.1161/01.CIR.0000109701.77059.E9
120.
Matsushima S Ide T Yamato M Matsusaka H Hattori F Ikeuchi M et al . Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. (2006) 113:1779–86. 10.1161/CIRCULATIONAHA.105.582239
121.
Dai W Shi J Gupta RC Sabbah HN Hale SL Kloner RA . Bendavia, a mitochondria-targeting peptide, improves postinfarction cardiac function, prevents adverse left ventricular remodeling, and restores mitochondria-related gene expression in rats. J Cardiovasc Pharmacol. (2014) 64:543–53. 10.1097/FJC.0000000000000155
122.
Hill MF Singal PK . Right and left myocardial antioxidant responses during heart failure subsequent to myocardial infarction. Circulation. (1997) 96:2414–20. 10.1161/01.CIR.96.7.2414
123.
Nahrendorf M Pittet MJ Swirski FK . Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. (2010) 121:2437–45. 10.1161/CIRCULATIONAHA.109.916346
124.
Swirski FK Nahrendorf M . Leukocyte behavior in atherosclerosis, myocardial infarction, heart failure. Science. (2013) 339:161–6. 10.1126/science.1230719
125.
Zhou R Yazdi AS Menu P Tschopp J . A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469:221–5. 10.1038/nature09663
126.
Ip WK Medzhitov R . Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat Commun. (2015) 6:6931. 10.1038/ncomms7931
127.
Collins LV Hajizadeh S Holme E Jonsson IM Tarkowski A . Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol. (2004) 75:995–1000. 10.1189/jlb.0703328
128.
Nakahira K Haspel JA Rathinam VA Lee SJ Dolinay T Lam HC et al . Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222–30. 10.1038/ni.1980
129.
Lee GS Subramanian N Kim AI Aksentijevich I Goldbach-Mansky R Sacks DB et al . The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. (2012) 492:123–7. 10.1038/nature11588
130.
West AP Khoury-Hanold W Staron M Tal MC Pineda CM Lang SM et al . Mitochondrial DNA stress primes the antiviral innate immune response. Nature. (2015) 520:553–7. 10.1038/nature14156
131.
Palmieri EM Menga A Martín-Pérez R Quinto A Riera-Domingo C De Tullio G et al . Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an m1-like phenotype and inhibits tumor metastasis. Cell Rep. (2017) 20:1654–66. 10.1016/j.celrep.2017.07.054
132.
Yurdagul A Jr Subramanian M Wang X Crown SB Ilkayeva OR et al . Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. (2020) 31:518–33.e10. 10.1016/j.cmet.2020.01.001
133.
Ryan DG O'Neill LAJ . Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. (2017) 591:2992–3006. 10.1002/1873-3468.12744
134.
Mills EL Kelly B Logan A Costa ASH Varma M Bryant CE et al . Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. (2016) 167:457–70.e13. 10.1016/j.cell.2016.08.064
135.
Liu PS Wang H Li X Chao T Teav T Christen S et al . α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. (2017) 18:985–94. 10.1038/ni.3796
136.
Zhang S Weinberg S DeBerge M Gainullina A Schipma M Kinchen JM et al . Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. (2019) 29:443–56.e5. 10.1016/j.cmet.2018.12.004
137.
Xu J Chi F Guo T Punj V Lee WN French SW et al . NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J Clin Invest. (2015) 125:1579–90. 10.1172/JCI76468
138.
Zhang J Zhang F . Suppressor of ras val-2 promotes inflammation-mediated oxidative stress and cell apoptosis in cardiomyocytes through activating Mst1-mROS signaling pathway. J Recept Signal Transduct Res. (2020) 40:224–30. 10.1080/10799893.2020.1726953
139.
Li Y Chen B Yang X Zhang C Jiao Y Li P et al . S100a8/a9 signaling causes mitochondrial dysfunction and cardiomyocyte death in response to ischemic/reperfusion injury. Circulation. (2019) 140:751–64. 10.1161/CIRCULATIONAHA.118.039262
140.
Grotenbreg G Ploegh H . Chemical biology: dressed-up proteins. Nature. (2007) 446:993–5. 10.1038/446993a
141.
Geiss-Friedlander R Melchior F . Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. (2007) 8:947–56. 10.1038/nrm2293
142.
Fukuda H Sano N Muto S Horikoshi M . Simple histone acetylation plays a complex role in the regulation of gene expression. Brief Funct Genomic Proteomic. (2006) 5:190–208. 10.1093/bfgp/ell032
143.
Chu S Ferro TJ . Sp1: regulation of gene expression by phosphorylation. Gene. (2005) 348:1–11. 10.1016/j.gene.2005.01.013
144.
Fukushima A Lopaschuk GD . Acetylation control of cardiac fatty acid β-oxidation and energy metabolism in obesity, diabetes, heart failure. Biochim Biophys Acta. (2016) 1862:2211–20. 10.1016/j.bbadis.2016.07.020
145.
Parry TL Willis MS . Cardiac ubiquitin ligases: their role in cardiac metabolism, autophagy, cardioprotection and therapeutic potential. Biochim Biophys Acta. (2016) 1862:2259–69. 10.1016/j.bbadis.2016.07.002
146.
Nan J Zhu W Rahman MS Liu M Li D Su S et al . Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim Biophys Acta Mol Cell Res. (2017) 1864:1260–73. 10.1016/j.bbamcr.2017.03.006
147.
Fang JK Prabu SK Sepuri NB Raza H Anandatheerthavarada HK Galati D et al . Site specific phosphorylation of cytochrome c oxidase subunits I, IVi1 and Vb in rabbit hearts subjected to ischemia/reperfusion. FEBS Lett. (2007) 581:1302–10. 10.1016/j.febslet.2007.02.042
148.
Prabu SK Anandatheerthavarada HK Raza H Srinivasan S Spear JF Avadhani NG . Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J Biol Chem. (2006) 281:2061–70. 10.1074/jbc.M507741200
149.
Wegrzyn J Potla R Chwae YJ Sepuri NB Zhang Q Koeck T et al . Function of mitochondrial Stat3 in cellular respiration. Science. (2009) 323:793–7. 10.1126/science.1164551
150.
Heusch G Musiolik J Gedik N Skyschally A . Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res. (2011) 109:1302–8. 10.1161/CIRCRESAHA.111.255604
151.
Boengler K Hilfiker-Kleiner D Drexler H Heusch G Schulz R . The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther. (2008) 120:172–85. 10.1016/j.pharmthera.2008.08.002
152.
Nandi D Tahiliani P Kumar A Chandu D . The ubiquitin-proteasome system. J Biosci. (2006) 31:137–55. 10.1007/BF02705243
153.
Schmidt M Finley D . Regulation of proteasome activity in health and disease. Biochim Biophys Acta. (2014) 1843:13–25. 10.1016/j.bbamcr.2013.08.012
154.
Sun T Ding W Xu T Ao X Yu T Li M et al . Parkin regulates programmed necrosis and myocardial ischemia/reperfusion injury by targeting Cyclophilin-D. Antioxid Redox Signal. (2019) 31:1177–93. 10.1089/ars.2019.7734
155.
Dalal S Daniels CR Li Y Wright GL Singh M Singh K . Exogenous ubiquitin attenuates hypoxia/reoxygenation-induced cardiac myocyte apoptosis via the involvement of CXCR4 and modulation of mitochondrial homeostasis. Biochem Cell Biol. (2020) 98:492–501. 10.1139/bcb-2019-0339
156.
Yang F Wu R Jiang Z Chen J Nan J Su S et al . Leptin increases mitochondrial OPA1 via GSK3-mediated OMA1 ubiquitination to enhance therapeutic effects of mesenchymal stem cell transplantation. Cell Death Dis. (2018) 9:556. 10.1038/s41419-018-0579-9
157.
Yeh ET . SUMOylation and De-SUMOylation: wrestling with life's processes. J Biol Chem. (2009) 284:8223–7. 10.1074/jbc.R800050200
158.
Bian X Xu J Zhao H Zheng Q Xiao X Ma X et al . Zinc-induced SUMOylation of dynamin-related protein 1 protects the heart against ischemia-reperfusion injury. Oxid Med Cell Longev. (2019) 2019:1232146. 10.1155/2019/1232146
159.
Gao L Zhao Y He J Yan Y Xu L Lin N et al . The desumoylating enzyme sentrin-specific protease 3 contributes to myocardial ischemia reperfusion injury. J Genet Genomics. (2018) 45:125–35. 10.1016/j.jgg.2017.12.002
160.
Hafner AV Dai J Gomes AP Xiao CY Palmeira CM Rosenzweig A et al . Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging. (2010) 2:914–23. 10.18632/aging.100252
161.
Parodi-Rullán RM Chapa-Dubocq X Rullán PJ Jang S Javadov S . High sensitivity of SIRT3 deficient hearts to ischemia-reperfusion is associated with mitochondrial abnormalities. Front Pharmacol. (2017) 8:275. 10.3389/fphar.2017.00275
162.
Porter GA Urciuoli WR Brookes PS Nadtochiy SM . SIRT3 deficiency exacerbates ischemia-reperfusion injury: implication for aged hearts. Am J Physiol Heart Circ Physiol. (2014) 306:H1602–9. 10.1152/ajpheart.00027.2014
163.
Lu Y Feng Y Liu D Zhang Z Gao K Zhang W et al . Thymoquinone attenuates myocardial ischemia/reperfusion injury through activation of SIRT1 signaling. Cell Physiol Biochem. (2018) 47:1193–206. 10.1159/000490216
164.
Zhao TC Cheng G Zhang LX Tseng YT Padbury JF . Inhibition of histone deacetylases triggers pharmacologic preconditioning effects against myocardial ischemic injury. Cardiovasc Res. (2007) 76:473–81. 10.1016/j.cardiores.2007.08.010
165.
Boylston JA Sun J Chen Y Gucek M Sack MN Murphy E . Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J Mol Cell Cardiol. (2015) 88:73–81. 10.1016/j.yjmcc.2015.09.005
166.
Duarte FV Palmeira CM Rolo AP . The emerging role of MitomiRs in the pathophysiology of human disease. Adv Exp Med Biol. (2015) 888:123–54. 10.1007/978-3-319-22671-2_8
167.
Bandiera S Rüberg S Girard M Cagnard N Hanein S Chrétien D et al . Nuclear outsourcing of RNA interference components to human mitochondria. PLoS One. (2011) 6:e20746. 10.1371/journal.pone.0020746
168.
Baradan R Hollander JM Das S . Mitochondrial miRNAs in diabetes: just the tip of the iceberg. Can J Physiol Pharmacol. (2017) 95:1156–62. 10.1139/cjpp-2016-0580
169.
Fang G Qi J Huang L Zhao X . LncRNA MRAK048635_P1 is critical for vascular smooth muscle cell function and phenotypic switching in essential hypertension. Biosci Rep. (2019) 39:BSR20182229. 10.1042/BSR20182229
170.
Bostjancic E Zidar N Stajner D Glavac D . MicroRNA miR-1 is up-regulated in remote myocardium in patients with myocardial infarction. Folia Biol. (2010) 56:27–31.
171.
Yan K An T Zhai M Huang Y Wang Q Wang Y et al . Mitochondrial miR-762 regulates apoptosis and myocardial infarction by impairing ND2. Cell Death Dis. (2019) 10:500. 10.1038/s41419-019-1734-7
172.
Devaux Y Zangrando J Schroen B Creemers EE Pedrazzini T Chang CP et al . Long noncoding RNAs in cardiac development and ageing. Nat Rev Cardiol. (2015) 12:415–25. 10.1038/nrcardio.2015.55
173.
Gomes CPC Spencer H Ford KL Michel LYM Baker AH Emanueli C et al . The function and therapeutic potential of long non-coding RNAs in cardiovascular development and disease. Mol Ther Nucleic Acids. (2017) 8:494–507. 10.1016/j.omtn.2017.07.014
174.
Dong Y Yoshitomi T Hu JF Cui J . Long noncoding RNAs coordinate functions between mitochondria and the nucleus. Epigenetics Chromatin. (2017) 10:41. 10.1186/s13072-017-0149-x
175.
Kumarswamy R Bauters C Volkmann I Maury F Fetisch J Holzmann A et al . Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ Res. (2014) 114:1569–75. 10.1161/CIRCRESAHA.114.303915
176.
Li HJ Sun XM Li ZK Yin QW Pang H Pan JJ et al . LncRNA UCA1 promotes mitochondrial function of bladder cancer via the MiR-195/ARL2 signaling pathway. Cell Physiol Biochem. (2017) 43:2548–61. 10.1159/000484507
177.
Wang K Gan TY Li N Liu CY Zhou LY Gao JN et al . Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death Differ. (2017) 24:1111–20. 10.1038/cdd.2017.61
178.
Adlam VJ Harrison JC Porteous CM James AM Smith RA Murphy MP et al . Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. Faseb J. (2005) 19:1088–95. 10.1096/fj.05-3718com
179.
Zhao K Zhao GM Wu D Soong Y Birk AV Schiller PW et al . Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, reperfusion injury. J Biol Chem. (2004) 279:34682–90. 10.1074/jbc.M402999200
180.
Szeto HH . Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. (2008) 10:601–19. 10.1089/ars.2007.1892
181.
Cassarino DS Parks JK Parker WD Jr Bennett JP Jr . The parkinsonian neurotoxin MPP+ opens the mitochondrial permeability transition pore and releases cytochrome c in isolated mitochondria via an oxidative mechanism. Biochim Biophys Acta. (1999) 1453:49–62. 10.1016/S0925-4439(98)00083-0
182.
Cho J Won K Wu D Soong Y Liu S Szeto HH et al . Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. (2007) 18:215–20. 10.1097/01.mca.0000236285.71683.b6
183.
Sloan RC Moukdar F Frasier CR Patel HD Bostian PA Lust RM et al . Mitochondrial permeability transition in the diabetic heart: contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J Mol Cell Cardiol. (2012) 52:1009–18. 10.1016/j.yjmcc.2012.02.009
184.
Iranzo O . Manganese complexes displaying superoxide dismutase activity: a balance between different factors. Bioorg Chem. (2011) 39:73–87. 10.1016/j.bioorg.2011.02.001
185.
Daubert MA Yow E Dunn G Marchev S Barnhart H Douglas PS et al . Novel mitochondria-targeting peptide in heart failure treatment: a randomized, placebo-controlled trial of elamipretide. Circ Heart Fail. (2017) 10:e004389. 10.1161/CIRCHEARTFAILURE.117.004389
186.
Gibson CM Giugliano RP Kloner RA Bode C Tendera M Jánosi A et al . EMBRACE STEMI study: a Phase 2a trial to evaluate the safety, tolerability, and efficacy of intravenous MTP-131 on reperfusion injury in patients undergoing primary percutaneous coronary intervention. Eur Heart J. (2016) 37:1296–303. 10.1093/eurheartj/ehv597
187.
Cerrato CP Pirisinu M Vlachos EN Langel Ü . Novel cell-penetrating peptide targeting mitochondria. Faseb J. (2015) 29:4589–99. 10.1096/fj.14-269225
188.
Cerrato CP Langel Ü . Effect of a fusion peptide by covalent conjugation of a mitochondrial cell-penetrating peptide and a glutathione analog peptide. Mol Ther Methods Clin Dev. (2017) 5:221–31. 10.1016/j.omtm.2017.04.010
189.
Gomes KM Campos JC Bechara LR Queliconi B Lima VM Disatnik MH et al . Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodelling. Cardiovasc Res. (2014) 103:498–508. 10.1093/cvr/cvu125
190.
Piot C Croisille P Staat P Thibault H Rioufol G Mewton N et al . Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. (2008) 359:473–81. 10.1056/NEJMoa071142
191.
LaRocca TJ Henson GD Thorburn A Sindler AL Pierce GL Seals DR . Translational evidence that impaired autophagy contributes to arterial ageing. J Physiol. (2012) 590:3305–16. 10.1113/jphysiol.2012.229690
192.
Sciarretta S Yee D Nagarajan N Bianchi F Saito T Valenti V et al . Trehalose-induced activation of autophagy improves cardiac remodeling after myocardial infarction. J Am Coll Cardiol. (2018) 71:1999–2010. 10.1016/j.jacc.2018.02.066
193.
Eisenberg T Abdellatif M Schroeder S Primessnig U Stekovic S Pendl T et al . Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. (2016) 22:1428–1438. 10.1038/nm.4222
194.
Yan J Yan JY Wang YX Ling YN Song XD Wang SY et al . Spermidine-enhanced autophagic flux improves cardiac dysfunction following myocardial infarction by targeting the AMPK/mTOR signalling pathway. Br J Pharmacol. (2019) 176:3126–42. 10.1111/bph.14706
195.
Ding M Dong Q Liu Z Liu Z Qu Y Li X et al . Inhibition of dynamin-related protein 1 protects against myocardial ischemia-reperfusion injury in diabetic mice. Cardiovasc Diabetol. (2017) 16:19. 10.1186/s12933-017-0501-2
196.
Veeranki S Tyagi SC . Mdivi-1 induced acute changes in the angiogenic profile after ischemia-reperfusion injury in female mice. Physiol Rep. (2017) 5:e13298. 10.14814/phy2.13298
197.
Wang YT Lim Y McCall MN Huang KT Haynes CM Nehrke K et al . Cardioprotection by the mitochondrial unfolded protein response requires ATF5. Am J Physiol Heart Circ Physiol. (2019) 317:H472–78. 10.1152/ajpheart.00244.2019
198.
Sanders MJ Ali ZS Hegarty BD Heath R Snowden MA Carling D . Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J Biol Chem. (2007) 282:32539–48. 10.1074/jbc.M706543200
199.
Song T Lv LY Xu J Tian ZY Cui WY Wang QS et al . Diet-induced obesity suppresses sevoflurane preconditioning against myocardial ischemia-reperfusion injury: role of AMP-activated protein kinase pathway. Exp Biol Med. (2011) 236:1427–36. 10.1258/ebm.2011.011165
200.
Diebold S Moellmann J Kahles F Haj-Yehia E Liehn EA Nickel A et al . Myocardial infarction is sufficient to increase GLP-1 secretion, leading to improved left ventricular contractility and mitochondrial respiratory capacity. Diabetes Obes Metab. (2018) 20:2911–18. 10.1111/dom.13472
201.
Wang X Ding Z Yang F Dai Y Chen P Theus S et al . Modulation of myocardial injury and collagen deposition following ischaemia-reperfusion by linagliptin and liraglutide, both together. Clin Sci. (2016) 130:1353–62. 10.1042/CS20160061
202.
McDonagh TA Metra M Adamo M Gardner RS Baumbach A Böhm M et al . 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. (2021) 42:3599–726. 10.1093/eurheartj/ehab368
203.
Shao Q Meng L Lee S Tse G Gong M Zhang Z et al . Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc Diabetol. (2019) 18:165. 10.1186/s12933-019-0964-4
204.
Hu Z Ju F Du L Abbott GW . Empagliflozin protects the heart against ischemia/reperfusion-induced sudden cardiac death. Cardiovasc Diabetol. (2021) 20:199. 10.1186/s12933-021-01392-6
205.
Palee S Higgins L Leech T Chattipakorn SC Chattipakorn N . Acute metformin treatment provides cardioprotection via improved mitochondrial function in cardiac ischemia / reperfusion injury. Biomed Pharmacother. (2020) 130:110604. 10.1016/j.biopha.2020.110604
206.
Thummasorn S Shinlapawittayatorn K Chattipakorn SC Chattipakorn N . High-dose Humanin analogue applied during ischemia exerts cardioprotection against ischemia/reperfusion injury by reducing mitochondrial dysfunction. Cardiovasc Ther. (2017) 35. 10.1111/1755-5922.12289
207.
Shanmugam K Ravindran S Kurian GA Rajesh M . Fisetin confers cardioprotection against myocardial ischemia reperfusion injury by suppressing mitochondrial oxidative stress and mitochondrial dysfunction and inhibiting glycogen synthase kinase 3β activity. Oxid Med Cell Longev. (2018) 2018:9173436. 10.1155/2018/9173436
208.
Yang Y Zhang Y Liu X Zuo J Wang K Liu W et al . Exogenous taurine attenuates mitochondrial oxidative stress and endoplasmic reticulum stress in rat cardiomyocytes. Acta Biochim Biophys Sin. (2013) 45:359–67. 10.1093/abbs/gmt034
Summary
Keywords
mitochondria, myocardial infarction, metabolism, remodelling, inflammation
Citation
Xin Y, Zhang X, Li J, Gao H, Li J, Li J, Hu W and Li H (2021) New Insights Into the Role of Mitochondria Quality Control in Ischemic Heart Disease. Front. Cardiovasc. Med. 8:774619. doi: 10.3389/fcvm.2021.774619
Received
12 September 2021
Accepted
09 November 2021
Published
26 November 2021
Volume
8 - 2021
Edited by
Jian Wu, Fudan University, China
Reviewed by
Xu Chen, University of Mississippi Medical Center, United States; Yasuhiro Maejima, Tokyo Medical and Dental University, Japan
Updates
Copyright
© 2021 Xin, Zhang, Li, Gao, Li, Li, Hu and Li.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongwei Li lhw19656@sina.comWenyu Hu huwen0320@163.com
†These authors have contributed equally to this work
This article was submitted to Cardiovascular Metabolism, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.