Abstract
Peripheral artery disease (PAD) is caused by occluded or narrowed arteries that reduce blood flow to the lower limbs. The treatment focuses on lifestyle changes, management of modifiable risk factors and vascular surgery. In this review we focus on how Endothelial Cell (EC) dysfunction contributes to PAD pathophysiology and describe the largely untapped potential of correcting endothelial dysfunction. Moreover, we describe current treatments and clinical trials which improve EC dysfunction and offer insights into where future research efforts could be made. Endothelial dysfunction could represent a target for PAD therapy.
Introduction
Peripheral artery disease (PAD) is defined by the partial or total blockage of the arteries supplying the lower extremities. More than 200 million people world-wide are affected with >6.5 million people living with PAD in the United Stated of America (USA) alone (1). PAD has significant impact because of the frequent need for medical and surgical treatment. Using 2014 data from National Inpatient Sample, Kohn and colleagues recently identified that the cost burden of hospitalization for PAD patients in the USA was ~$6.3 billion per year (2). This medical and economic cost will rise; PAD prevalence is increasing due to the obesity and diabetes pandemic. People presenting with PAD have a higher risk of all-cause and cardiovascular mortality than those presenting with risk in coronary artery diseases (CAD) (3), but PAD has received limited attention in the development of treatments.
PAD may be classified into 3 clinical presentations—asymptomatic or atypical, intermittent claudication, and chronic limb-threatening ischemia (CLTI) (4). Most patients, ~20–50%, are asymptomatic or display atypical symptoms whereas 10–30% of patients display typical features of intermittent claudication i.e., exertional leg pain in ≥1 muscle group(s), relieved by rest (5). CLTI is the severe stage of PAD, including ischemic foot pain at rest, non-healing wounds/ulcerations and in severe cases, gangrene due to arterial insufficiency, necessitating revascularization surgery or amputation. The presentation of PAD may not reflect the severity of limb ischemia and because of the varying presentations, many patients are misdiagnosed or underdiagnosed.
PAD is generally viewed as a large vessel atherosclerotic disease but studies report that its pathophysiology differs from atherosclerosis in other vessel beds. Narula and colleagues recently summarized histological differences of plaques in patients with PAD and CAD (6). The authors suggested that the blockage of the coronary vessels in acute coronary syndromes was principally caused by luminal thrombus, with 65–75% suggested to be due to plaque rupture and 25–35% to plaque erosion (defined as the presence of luminal thrombosis in the absence of plaque rupture). In contrast, ~66% of peripheral arteries from CLTI were occluded by thrombosis in the absence of significant atherosclerosis, defined as normal intima or adaptive intimal thickening (6). The same authors also identified blood clots in the small distal arteries, proposing that local changes in these vessels could precipitate thromboembolic events, however it is unclear if these events were due to the process of amputation or if they occurred prior. The microvasculature also plays a significant role in PAD pathophysiology since microvascular dysfunction can increase amputation risk (7). These studies suggest that our knowledge in PAD pathophysiology is limited. Endothelial cell (EC) heterogeneity and plasticity has been confirmed in different organs and vascular beds in peripheral arteries (8–12), suggesting that EC dysfunction may represent a spectrum of EC phenotypes (13). This isn't surprising since the pattern and behavior of ECs are shaped by their environment and the tissues they reside in. Given ECs line the entire vascular tree, the study of EC function and dysfunction in peripheral arteries could be key to understanding some of these differences. Certainly, a healthy endothelium not only acts as a barrier between blood and surrounding tissues, but is considered an endocrine organ, regulating exchanges between the blood stream and tissues to control constriction and dilation and maintain vessel tone. The endothelium also inhibits thrombosis, reduces leukocyte adhesion and transmigration, limits atherogenesis, and is responsible for the formation of new blood vessels necessary for repair during damage.
In this review, we summarize EC function(s) that are altered in PAD. We highlight current therapeutics and treatments being investigated in clinical trials that impact EC function(s) as well as offer insight into where future research efforts could be made. Knowledge of how EC function and dysfunction contributes to PAD pathophysiology could have significant implications for therapeutic and diagnostic approaches for this disease.
EC dysfunction in pad
Inflammation
In homeostasis, leukocytes move in and out of the vascular system and tissues and are in constant surveillance of their microenvironment waiting for a signal. In response to stimuli, these cells are recruited to inflamed tissues, where they “clean up” the injury and contribute to repair. In atherosclerosis, damage to the endothelium (e.g., increased turbulence of blood flow, high blood pressure, high cholesterol, high glucose, oxidation etc.,) can upregulate multiple mediators governing leukocyte recruitment. Chemokines, cytokines, and other inflammatory mediators regulate the expression of adhesion molecules on both the endothelium, neutrophil, and monocyte surface to influence the three-step process of leukocyte recruitment: rolling, activation, and adhesion on the endothelium which involves E-, L- and P-selectins. While rolling is essential for leukocyte adherence, it does not necessarily lead to firm adhesion; firm adhesion requires activation of integrins (by selectin or chemokine engagement) and their interaction with ICAM-1 and VCAM-1 (intercellular adhesion molecule-1 and vascular cell adhesion molecule-1), which results in the complete arrest of the leukocyte. Leukocytes then transmigrate between the endothelium into the interstitial space toward a chemotactic stimulus, such as, CC-chemokine ligand-2 (CCL-2) (14).
Some of these cellular interactions have been implicated in PAD. For example, circulating levels of monocytes are significantly and independently associated with PAD (15), with high neutrophil-lymphocyte ratio a potential predictor of PAD severity (16, 17). Circulating chemokines and inflammatory markers expressed by leukocytes including high sensitivity C-reactive protein (hs-CRP), interleukins (IL), and matrix metalloproteinases (MMPs) are also upregulated in PAD patients, and in some cases associate with severity of disease (18–21). Indeed, a systematic review and meta-analysis of 47 studies involving >21,000 PAD patients, identified high levels of hs-CRP to predict the risk of major adverse cardiovascular events and mortality (22). Platelets release low abundance, highly active molecules including chemokines/chemokine ligands and angiogenic factors including chemokine ligand-5 (CCL-5) and platelet-derived growth factor (PDGF), which support leukocyte-platelet interaction and migration of neutrophils and monocytes to the developing atherosclerotic site (23). Indeed, Barrett et al., recently identified that platelets induced the migration of monocytes into atherosclerotic lesions of Ldlr−/− mice via upregulation of monocyte suppressor of cytokine signaling 3 (SOCS3) (24). Thus, platelets also contribute to inflammation, and with increased inflammatory burden, there is an increase in PAD prevalence (25). Another cytokine important for migration of leukocytes into the vessel wall is CCL-2. CCL-2 infusion of the femoral artery of rabbits following hindlimb ischemia increased monocyte accumulation in the vessel wall (26); a finding that was inhibited with ICAM-1 monoclonal antibody treatment (27), suggesting that monocyte adhesion to the endothelium in ischemia involves CCL-2 and ICAM-1. Importantly, circulating CCL-2 levels are increased in PAD (18, 28), associating with increased CCL-2 protein expression and macrophage content in limb tissues from patients (29).
Additional reports of the role of cell adhesion molecules come from the Edinburgh Artery Study, which described increased soluble levels of ICAM-1 associating with PAD diagnosis (30). In another study, soluble VCAM-1 levels were associated with worse PAD prognosis (31). From the selectins: E-selectin is EC specific whereas P-selectin is expressed by both ECs and platelets. Higher levels of soluble E-selectin have been reported in PAD patients, particularly in diabetes, and reflecting endothelial activation (32). Circulating P-selectin levels are also associated with PAD severity (33–35) and in the Multi-Ethnic Study of Atherosclerosis, a prospective large cohort study involving >6,800 participants, P-selectin levels were significantly associated with lower ankle-brachial index ratios as well as PAD prevalence (36). P-selectin's involvement in leukocyte adhesion was confirmed in vitro; human recombinant P-selectin increased neutrophil adhesion to platelets in the presence of plasma from healthy individuals (34), suggesting that neutrophil adhesion could also occur with activated endothelium expressing P-selectin in PAD patients. Moreover, a link between platelets and SOCS3-mediated activation in PAD was observed, with the SOCS1:SOCS3 ratio negatively correlating with IL-1β, but also with monocyte-platelet aggregates, P-selectin and CD40 (24). Indeed, increased circulating leukocyte-platelet aggregates are proposed as a biomarker of PAD severity (17). An in-depth summary of inflammatory biomarkers in PAD was recently reviewed (37). Figure 1 summarizes the endothelial, leukocyte and platelet contribution to inflammation in PAD.
Figure 1
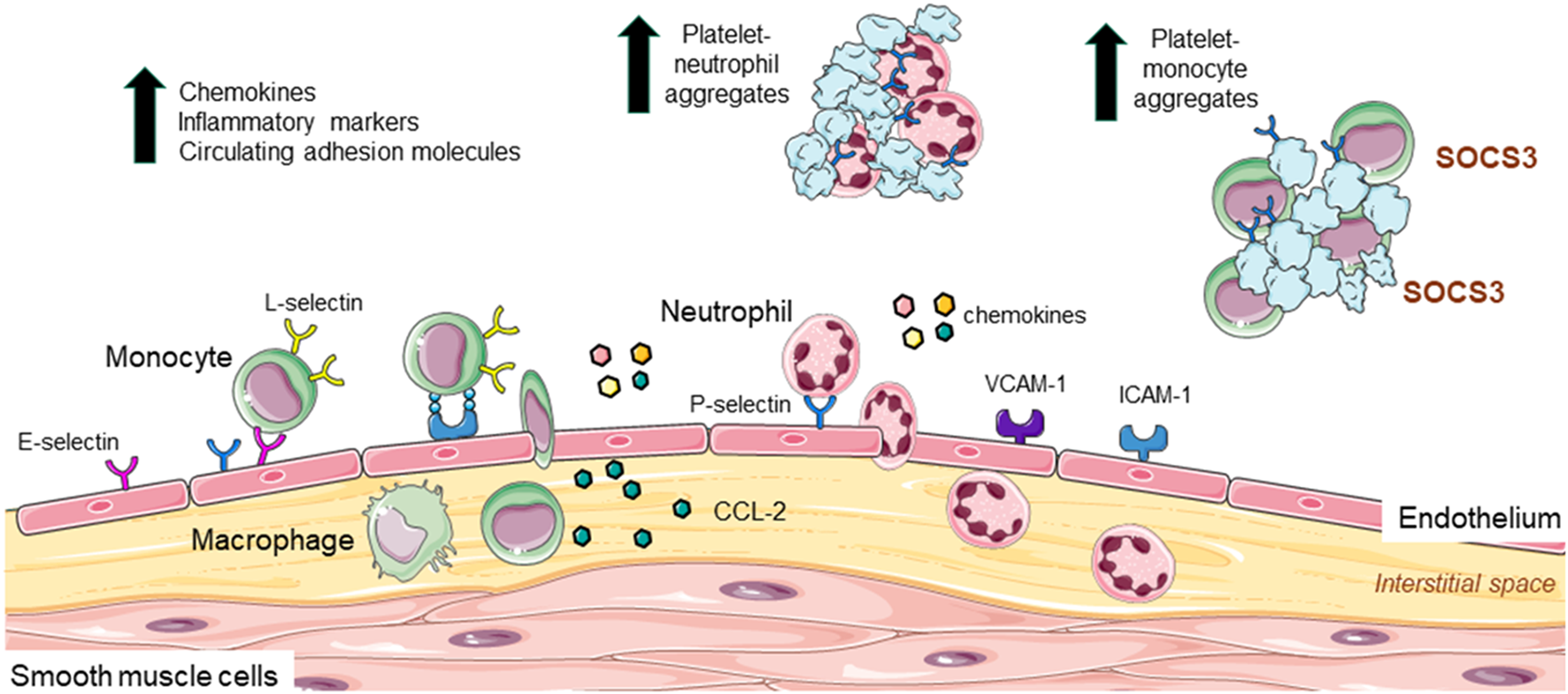
Leukocyte and platelet adhesion, recruitment to the endothelium, and contribution to inflammation in PAD. In atherosclerosis increased inflammation upregulates the expression of VCAM-1 and ICAM-1 as well as E- and P- selectin on endothelial cells. These adhesion markers are involved in the process of leukocyte recruitment (rolling, activation, and adhesion to the endothelium), with L-selectin also implicated in adhesive interactions between leukocytes and endothelial cells. Once adhered, leukocytes migrate into the interstitial space toward a chemotactic mediator such as CCL-2. Here, monocytes differentiate into macrophages. Monocytes are independently associated with PAD and increased neutrophil: lymphocyte ratio is linked to PAD severity. Circulating chemokines, inflammatory markers (e.g., CRP, interleukins, and matrix metalloproteinases), and adhesion molecules (ICAM and P-selectin) are up regulated in PAD and associated with disease severity. Platelets also secrete molecules that support platelet-neutrophil and platelet-monocyte interaction and adhesion. Circulating platelet-leukocyte interactions are considered a biomarker for PAD.
Platelet activation and thrombosis
Under normal circumstances, the endothelium exquisitely controls endothelial-platelet interactions and the balance between coagulation and anticoagulation in the vessel wall. ECs generate nitric oxide (NO) and prostacyclin, molecules which directly inhibit platelet activation. They express tissue factor pathway inhibitor, a potent anticoagulant, which limits tissue factor-inducible activation of factor VII and X (38). ECs also express co-factors for antithrombin III, or synthesize thrombomodulin (a thrombin receptor), which can directly reduce plasma thrombin levels upon thrombin binding, an effect that can increase the activity of the anticoagulant, protein C (39). Furthermore, ECs secrete tissue-type and urokinase-type plasminogen activator (t-PA and u-PA, respectively) to activate fibrinolysis and fibrin degradation. Because thrombosis is strongly implicated in PAD (6), the EC-platelet interaction may be more central to PAD pathogenesis. Indeed, PAD patients have elevated levels of circulating platelets (40) and increased platelet aggregability (41), with mean platelet volume increasing with PAD severity (42). Circulating tissue factor (43), t-PA (40) and factors IX and XI (44) are also increased, whereas tissue factor pathway inhibitor (43) and protein C levels are decreased (44); the latter associating with endothelial injury (44). Elevated von Willebrand factor and fibrinogen levels are also independently associated with the risk of development of PAD (45), with increased fibrinogen and D-dimer levels predictive of increased risk of mortality in PAD patients (22). Soluble thrombomodulin levels are increased in symptomatic PAD vs. asymptomatic age-matched control subjects (46), which is significant since elevated levels may reflect EC dysfunction in PAD (47, 48).
Interestingly, medial calcification and calcified nodules were identified in 70% of CLTI peripheral arteries examined (6, 49). Calcified nodules, accompanied by fibrin could pierce or disrupt the fibrous cap causing EC loss and plaque rupture (50). This is important since vascular calcification may predict poorer outcomes in PAD (51). However, reports suggest that calcification may also stabilize plaque (52). The role of calcification in PAD is not well established and requires further study.
Vessel tone
To meet physiological demand and manage blood flow, the endothelium dilates arteries by relaxing the underlying smooth muscle via a range of mechanisms. The most well studied is NO. Under normal physiological conditions, the formation of NO is dependent on calcium facilitated calmodulin binding to homodimeric endothelial nitric oxide synthase (eNOS). Bound calmodulin then facilitates, 6R-tetrahydrobiopterin (BH4)-dependent electron transfer across eNOS to catalyze the conversion of L-arginine to NO and L-citrulline. However, roles for cyclooxygenase (COX)-derived prostoglandins (53), membrane hyperpolarization (54), epoxyeicosatrienoic acids (55), myoendothelial gap junctions (56) and oxidants (57) can also control arterial tone in an endothelial-dependent manner. The specific mechanism of regulation differs depending on how the artery is stimulated, sex, and the location of the artery in the circulatory system. However, arguably the biggest variable that dictates which mechanism is employed is the health state of the artery.
In PAD patients many of the above mechanisms become dysfunctional. For instance, flow mediated dilation (FMD), a surrogate for endothelial function, and commonly associated with endothelial production of NO, is reduced (58). Consistent with this, PAD patient plasma and urine showed decreased BH4, cyclic guanosine monophosphate (cGMP) and decreased NOX's, pointing toward a decrease in endothelial NO bioavailability (59). Ismaeel et al. (60) has since shown a range of increased oxidative stress markers in PAD, proposing these as mechanisms by which NO bioavailability is lost, and is somewhat in agreement with other studies. For example, administration of L-arginine to patients to stimulate NO production, or oxypurinol to decrease oxidative stress (and thus increase NO bioavailability) increased FMD, restored blood flow and decreased patient symptoms (61, 62). However, despite the promise of these short-term studies, longer term clinical trials over 6 months in the NO-PAIN study showed that oral administration of L-arginine did not improve FMD or improve any NO biochemical parameters in patients with intermittent claudication (63). An additional strategy to increase NO bioavailability is to modulate the eNOS enzyme directly. In this regard the β-adrenergic receptors (βARs) may have therapeutic potential, particularly the β3AR isoform. β3AR agonists stimulate vasodilation via their ability to modulate eNOS activity and NO production (64, 65). Moreover, we showed that activation of β3AR restored NO and the redox balance, improving vasodilation and EC function in a mouse model of diabetic PAD (66).
The impact of PAD on other vasodilatory pathways is less investigated but should not be overlooked. Evidence already exists of other pathways such as the COX-dependent regulation of vascular tone that may be disrupted in PAD models (67), and that selective inhibition of COX-2 offers clinical improvement in intermittent claudication (68). Given that the contribution to vascular tone of NO decreases as vessel size decreases (69), future research should also investigate the effects of PAD on non-NO mechanisms of arterial dilation. A summary of these pathways is described in Figure 2.
Figure 2
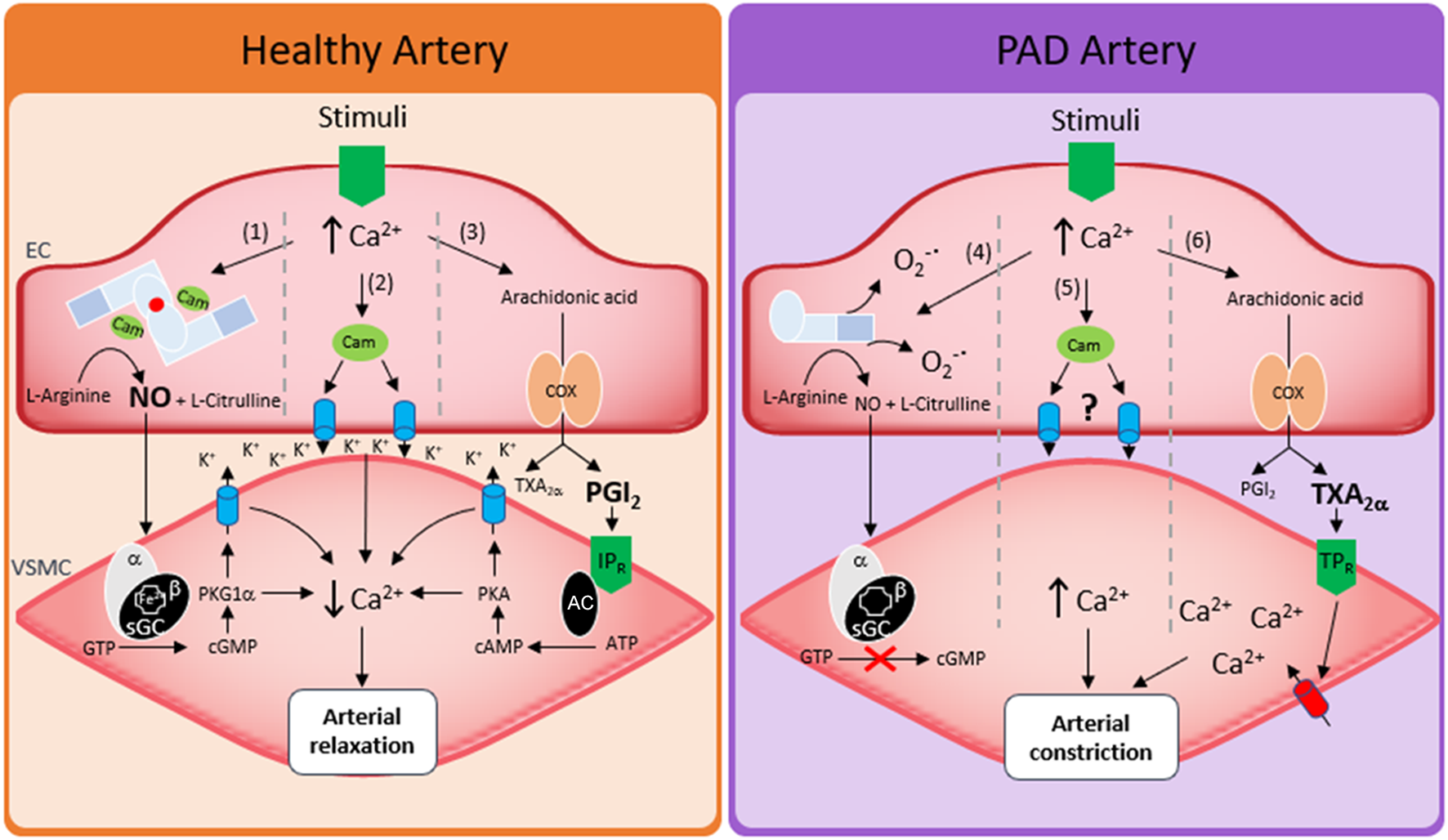
Regulation of vessel tone in healthy arteries and in PAD. Left hand side shows three mechanisms of arterial relaxation in healthy arteries. (1) Nitric oxide induced arterial relaxation, endothelial calcium binds calmodulin which in turn facilitates eNOS mediated oxidation of L-Arginine forming NO and citrulline. NO then diffuses from the endothelial cell (EC) to the vascular smooth muscle cell (VSMC) where it activates soluble guanylate cyclase (sGC). sGC then converts guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP). cGMP activates protein kinase G1a (PKG1α) which lowers smooth muscle calcium through potassium channel mediated hyperpolarization of smooth muscle cells. (2) Increased endothelial calcium binds calmodulin which in turn activates small and intermediate conductance calcium activated potassium channels. Potassium efflux through these channels hyperpolarizes vascular smooth muscle via direct spreading of membrane potential or via activation of smooth muscle ion channels. (3) Endothelial calcium increases and increases arachidonic acid formation from membrane phospholipids. Arachidonic acid is the metabolized via cyclooxygenase 1 or 2 (COX) to form prostaglandins. The dominant prostaglandin formed is prostacyclin (PGI2). PGI2 then activates the IP receptor (IPR) which converts adenosine triphosphate (ATP) into cyclic adenosine monophosphate (cAMP) in an adenylyl cyclase (AC)-dependent manner, cAMP then activates protein kinase A (PKA) which lowers smooth muscle calcium through potassium channel mediated hyperpolarization of smooth muscle cells. Right hand side shows how peripheral artery disease affects arterial relaxation. (4) Increased endothelial calcium increases production of superoxide (O2−•) via stimulation of uncoupled eNOS or stimulation of other oxidant sources such as NADPH oxidases (NOX's). Any NO that has been produced may be ineffective as its target sGC has been oxidized or become heme free. (5) Mechanisms of potassium channel induced hyperpolarization have not been investigated. (6) arachidonic acid breakdown results in increased thromboxane A2 (TXA2α) or increased activation of thromboxane receptor (TPR) activation.
Angiogenesis
In hemostasis, injury to blood vessels (e.g., ischemia) activate ECs, which then sprout, migrate, proliferate, and form EC tubules, driven by hypoxia-induced mediators, the most characterized being vascular endothelial growth factor (VEGF). Interestingly, in clinical practice and in animal models of cardiovascular disease, the endogenous angiogenic responses are impaired, particularly with aging (70, 71), in diabetes (72–74) or in dyslipidemia (71, 75, 76). Thus, stimulating angiogenesis in local ischemic tissues could be beneficial in PAD and other vascular diseases. However, to date, all large clinical trials delivering angiogenic factors, including VEGF to people suffering from ischemic diseases such as PAD have shown little benefit (77). Furthermore, the role of angiogenesis in atherosclerosis is conflicting. For example, in a rabbit carotid artery collar model of intimal hyperplasia, adenoviral delivery of VEGF-A, -B, -C and -D increased intimal thickening which was positively correlated with neovascularisation (78). VEGF and other angiogenic molecules including fibroblast growth factor (FGF) were shown to accelerate atherosclerosis in animal models (79, 80), whereas anti-angiogenic therapies reduced atherosclerosis development (81). In contrast, systemic inhibition of the VEGF receptor attenuated established atherosclerosis in high fat diet-fed Apoe−/− mice (82). Interestingly, plasma concentrations of VEGF-A, but not VEGF receptor-1, are significantly elevated in PAD patients vs. healthy controls (83). In support of this, a significant increase in plasma VEGF was observed in patients with intermittent claudication vs. CLTI, suggesting that VEGF may act as a biomarker or causal factor in disease (83). Indeed, Stehr et al. found increased VEGF levels associated with increased PAD severity (84). What is clear from these studies is that our knowledge of the complex angiogenic pathways and responses to ischemia in PAD is limited.
Other molecules of interest that can promote angiogenesis include TNF-related apoptosis-inducing ligand (TRAIL) and β3AR. TRAIL is a protein discovered for its ability to selectively kill tumor cells but leave normal cells resistant to its cytotoxic actions (85, 86). Interestingly, in ischemic cardiovascular diseases including PAD, TRAIL levels in the circulation are suppressed (87–90) and in the cardiovascular system TRAIL appears to have homeostatic rather than cytotoxic properties. For example, the presence of TRAIL attenuated atherosclerosis in mice (87, 91), in part by resolving inflammation and improving macrophage function (87), reducing oxidative-stress-induced EC dysfunction (92), and increasing eNOS activity to stimulate intracellular NO production in ECs (93). In the latter study, TRAIL stimulated NO production via a NOX-4-dependent mechanism (93). Furthermore, we and others showed that exogenous TRAIL treatment stimulated in vitro EC processes of angiogenesis (proliferation, migration and tubule formation) (93–95) and promoted stable collateral vessels in the ischemic limb of mice (93). Activation of the β3AR can also stimulate processes of angiogenesis. For example, the specific β3AR agonist BRL37344 increased retinal EC proliferation and migration in vitro, whereas stimulation of the β1 isoform, β1AR had no effect (96). Furthermore, nebivolol-induced angiogenesis in a mouse model of aortic sprouting was abrogated with β3AR deletion, demonstrating the importance of β3AR in neoangiogenesis (97). More recently, we showed that administration of CL316,243, a specific β3AR agonist, stimulated human umbilical vein EC migration and tubule formation in a NOS-dependent manner in vitro, and crucially, CL316,243 improved blood perfusion and angiogenesis in a mouse model of diabetic PAD (98). These findings support TRAIL and β3AR agonist modulators as promising new therapeutic agents for the treatment of PAD.
Current treatments and clinical trials which target and improve EC function
Current gold-standard treatment of PAD includes the management of symptoms such as exercise therapy, medical therapy (e.g., lipid control, blood pressure control, anti-platelet therapy and diabetes control), and revascularization to reduce the risk of myocardial infarction (MI) and stroke. Interventions for PAD are guided by disease stage; guidelines for management have been published by the European Society of Vascular Surgery and European Society of Cardiology in 2018 (99), the AHA and American College of Cardiology (ACC), 2016 (100), the Global Vascular Guidelines in 2019 (4) and the Asia-Pacific Consensus Statement on PAD Project Committee (APPADC) (101). These guidelines are summarized in Table 1. Below, we focus our attention on some of these therapies as well as describe emerging therapies known to impact EC functions.
Table 1
| Recommendation | Level of evidence | |||
|---|---|---|---|---|
|
AHA/ACC
2016 |
ESC/ESVC
2018 |
GVG 2019 |
APPACD
2021 |
|
| Lipid lowering drugs for all PAD patients | A | A | A | A |
| Antihypertensive therapy exclusively for hypertensive PAD patients to reduce chance of MI, stroke, heart failure or cardiovascular death | A | B | B | A |
| Diabetes therapies to maintain | C | C | B | B |
| Smoking cessation for PAD patients who smoke cigarettes or other forms of tobacco | A | B | A | A |
| Antiplatelet therapy to reduce risk of MI or stroke | A | C | A | A |
| Antithrombotic therapy to reduce risk of MI or stroke | A | A | A | A |
| Exercise therapy to improve claudication symptoms | A | C | N/A | A |
| Annual influenza shot | C | N/A | N/A | C |
| Healthy diet | N/A | C | N/A | N/A |
| Endovascular treatment for claudication | N/A | C | B | B |
Summary of Class I PAD treatment guidelines.
AHA/ACC, American Heart association and American College of Cardiology; ESC/ESVS, European Society of Vascular Surgery and European Society of Cardiology; GVG, Global Vascular Guidelines; APSAVD, Asia-Pacific Society of Atherosclerosis and Vascular Disease. A, data derived from multiple randomized control trials or meta data; B, data derived from a single randomized clinical trial or large non-randomized clinical trials; C, consensus of expert opinion and/or small studies, retrospective studies and registries; N/A, not applicable.
Exercise
Exercise training has positive effects on endothelial function. This is particularly evident in CAD where exercise training improved endothelium-dependent vasodilation that resulted in a 4-fold phosphorylation of eNOS1,177 from isolated arteries 4 weeks later (102). Increased endothelial function after exercise is also observed in patients with hypertension (103), and in type-1 and type-2 diabetes (104, 105). Furthermore, moderate-intensive exercise (>10 h/week) stimulated coronary collateral blood flow and improved diastolic heart function in CAD patients (106). In this prospective study, 60 patients were randomly assigned to high intensity exercise, moderate intensity exercise or control for 4 weeks. Angiography identified significantly increased coronary flow index in the exercise treated groups (39 and 41%, respectively) vs. control which was associated with increased VO2 peak (maximal oxygen uptake; measure of aerobic fitness) (106). The authors proposed two mechanisms: recruitment of pre-existing vessels or improved EC function of small intramyocardial vessels (106). Because supervised or home exercise programs are first-line therapy for PAD (101), these mechanisms may also be relevant in limbs. Indeed, exercise training improved brachial artery dilator function in older sedentary females, (107) but these findings are not as apparent in PAD. There is also potential for long-term exercise therapy to improve systemic inflammation since an inverse correlation between exercise, inflammation and plasma CRP levels exist (108). The role of exercise treatment on inflammation in PAD and its effect on EC function is unclear and requires further elucidation.
Cilostazol
Phosphodiesterases (PDE) play important role(s) in barrier function by inactivating the messenger cyclic nucleotides cyclic adenosine monophosphate (cAMP) and cGMP and ECs express 5 PDEs, namely, PDE1, PDE2, PDE3, PDE4, and PDE5. Cilostazol is a PDE3 inhibitor and antiplatelet medication used to relieve PAD patients from symptoms of intermittent claudication, which improves walking distance (109), in part by acting as a vasodilator and its ability to stimulate NO release. The most recent Cochrane review, which included 8 placebo-controlled randomized controlled trials involving 2,360 participants with PAD, reported that cilostazol significantly increased maximum walking distance (mean difference 39.6 m. 95% CI 21.8, 57.3; GRADE criteria very low certainty evidence) (110). However, cilostazol was associated with an increased odds of headache which is a common reason for discontinuation. It was suggested that the effects of cilostazol may vary depending on its ability to convert into its active metabolite via the cytochrome P450 system (111). Interestingly, cilostazol may have sex-dependent effects in ECs since female ECs express more Pde3b mRNA than male cells (112). However, no reports of differing responses in men and women have been identified. How cilostazol effects EC function(s) in PAD is not fully established.
Mirabegron
Mirabegron is a second generation β3AR agonist used to treat overactive bladder. The STAR-PAD trial is a Phase II, multicenter, double-blind, randomized, placebo-controlled trial of mirabegron vs. placebo on walking distance in patients with PAD that is currently recruiting by Figtree and colleagues (ACTRN12619000423112) (113). A total of 120 patients aged ≥40 years with stable PAD and intermittent claudication will be randomly assigned (1:1 ratio) to receive either mirabegron (50 mg orally once a day) or matched placebo for 12 weeks. The primary endpoint is change in peak walking distance assessed by a graded treadmill test. Secondary endpoints include: (i) initial claudication distance; (ii) average daily step count and total step count and (iii) functional status and quality of life assessment. Mechanistic sub-studies will examine potential effects of mirabegron on vascular function, including brachial artery FMD, arterial stiffness and angiogenesis. Given that mirabegron is well-tolerated and clinically available for alternative purposes, a positive study is positioned to immediately impact patient care.
Medications used for coronary microvascular dysfunction (CMD)
Large vessel blockages may not be the only mechanism contributing to PAD pathogenesis. Interestingly, microvascular dysfunction in the limb can increase amputation risk by ~20-fold, even in the absence of large vessel atherosclerosis (7). This is somewhat reminiscent of female patients with CMD who present with dysfunction of the small coronary vessels in the absence of atherosclerosis. These women have worse heart function and blood perfusion (114), with EC dysfunction the primary cause (115). Similar mechanisms may be at play with PAD, given majority of patients do not present with typical symptoms.
Central mechanisms thought to govern CMD include enhanced vasoreactivity at both epicardial and microvascular levels, impaired coronary vasodilator capacity, and increased microvascular resistance; effects of dysfunctional ECs (116). The mainstay of therapies for CMD are β-blockers, statins, calcium channel blockers and angiotensin converting enzyme (ACE)-inhibitors. These are also recommended as secondary prevention for PAD (Table 1). β-blockers and calcium channel blockers reduce severity of anginal symptoms and improve exercise stress test performance (117) and this may be due, in part, to the fact that they also block oxidative stress, improve EC survival (118), reduce EC activation and inflammation, and stimulate eNOS production (119, 120). Furthermore, β-blockers reduce FMD in people with cardiovascular diseases (121) whereas ACE-inhibitors have only showed modest improvement in FMD in patients with CAD (122), even though they modulate survival of ECs (123). Statins not only reduce cholesterol synthesis, but also dampen inflammation. They also do this via their direct effect on ECs. For example, low dose statins improved viability, reduced VCAM-1 and ICAM-1 expression, and atherosclerosis in pre-clinical models (124, 125). They also promoted NO release and repair mechanisms following EC injury (126). Although these medications are recommended for PAD treatment, adherence to these is variable amongst patients (127).
Anti-inflammatory treatment
As described earlier, inflammation contributes to atherosclerosis pathophysiology, in part, via endothelial activation, and recruitment of leukocytes to the vessel wall. The Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS), a randomized double-blind, placebo-controlled trial of canakinumab, a monoclonal antibody targeting IL-1β showed that 150 mg of canakinumab reduced recurrent cardiovascular events in patients with stable CAD when compared to placebo (128). Thus, anti-inflammatories could also hold promise for PAD therapy not only in their ability to reduce inflammation, but also since many anti-inflammatories impact EC function(s). For example, anakinra, an inhibitor of IL-1, reduced endothelial dysfunction in diabetic rats; a finding that was associated with decreased NOX and circulating inflammatory cytokines including IL-1β and TNFα (129). Anakinra treatment for 30-days also improved FMD in patients with rheumatoid arthritis (130). Similar findings were also observed with TNFα inhibitors (131). More recently, colchicine has shown considerable promise as a relatively safe, inexpensive dedicated agent which targets inflammation by attenuating NLRP3 activity and IL-1β expression; a recent meta-analysis demonstrating reduced MACE, MI, stroke, and the need for coronary revascularization in patients with coronary disease (132). Data on direct effects of colchicine on ECs is limited, however an older study showed that colchicine reduced the number of E-selectin molecules on the endothelium and subsequent adhesiveness of the ECs to IL-1 or TNFα (133). Although one recent study found no difference in FMD between low-dose colchicine and placebo in patients with CAD (134), another study showed improvement in FMD in patients with a white blood cell count ≥7,500 mm3 (135). Never-the-less, these studies suggest that anti-inflammatory agents could be used to reduce inflammation in PAD and have potential direct effects on EC dysfunction.
Platelet and thrombosis inhibitors to prevent PAD complications
As described earlier PAD is associated with dysregulated platelet activation and coagulation, which could precipitate major thrombotic events, observed not only in large vessel disease, but also in small vessels with microthrombi resulting in reduced tissue perfusion. Although current guidelines recommend the use of antiplatelet and antithrombotic medication to reduce the risk of MI or stroke (4, 99–101) there is a significant lack of guideline adherence (136), especially for newly-diagnosed PAD patients (137). Generally speaking, aspirin and clopidogrel are the two most studied antiplatelet medications. Aspirin inhibits COX and subsequent thromboxane A2, which is not only vasoconstrictive, but also activates platelets. Clopidogrel on the other hand prevents platelet activation by blocking the P2Y12 receptor on the surface of the platelet. Both have direct effects on ECs. For example, aspirin protected ECs against oxidized low-density lipoprotein, high glucose, angiotensin II, and H2O2-induced injury (138, 139). It also improved impaired acetylcholine-induced vasodilation in patients with atherosclerosis (140) and in pre-clinical models of aging (141). LPS-induced mRNA expression of inflammatory cytokines was attenuated with clopidogrel, associating with improved EC viability, migration, proliferation, and angiogenesis (142). Clopidogrel also prevented endothelial dysfunction in hypertensive rats (143).
Unlike anti-platelet medications, anti-coagulants inhibit the coagulation cascade and the formation of fibrin. An example is Rivaroxaban, a specific inhibitor of factor Xa. In the COMPASS trial which included >27,000 patients with stable CAD or PAD, patients were given low-dose rivaroxaban (5 mg, twice daily) and aspirin (100 mg, once daily), or aspirin alone. Patients assigned to low-dose rivaroxaban plus aspirin had better cardiovascular outcomes after ~2 years, including reduction in the combined risk of cardiovascular death, stroke, and MI (144). However, the risk of major bleeding events increased (144). In a sub-study, rivaroxaban plus aspirin reduced the incidence of major adverse limb events including amputations, when compared to aspirin alone (145). While the risk of major bleeding events was increased, the risk of fatal bleeding was not (145). In the more recent VOYAGER PAD trial (146), >6,500 PAD patients undergoing revascularization received either rivaroxaban (2.5 mg, twice daily) plus aspirin (100 mg, once daily), or aspirin alone for 3 years (147). A significant reduction in ischemic limb events including acute limb ischemia, amputation as well as cardiovascular outcomes (death, MI, stroke) were observed (147). Despite women having higher total cholesterol and greater prevalence in hypertension, diabetes and chronic kidney disease, the net clinical benefit of rivaroxaban plus aspirin was similar with sex, with comparable rates for cardiovascular outcomes and bleeding between men and women (148). The risk of major bleeding was still higher with rivaroxaban treatment.
Interestingly, rivaroxaban also has direct effects on ECs and the endothelium. Rivaroxaban administration improved vasodilation in diabetic wildtype mice, in part by increasing aortic eNOS activity (149). Forearm blood flow was also improved in diabetic patients administered rivaroxaban for 20 weeks, although treatment was associated with higher bleeding events (150). In vitro, rivaroxaban reduced ROS (reactive oxygen species), improved DNA repair (151) and reduced inflammatory gene expression in ECs exposed to hydroxycholesterol (152, 153). Rivaroxaban also stimulated blood flow and increased capillary density in a mouse model of diabetic PAD (154). The same group demonstrated improvement in endothelial progenitor cell migration and senescence, associating with increased eNOS activity in a hyperglycemic environment (154), suggesting that rivaroxaban has pleiotropic functions in ECs. These findings demonstrate that antiplatelet and anticoagulants may improve EC dysfunction in PAD, however additional studies are needed to fully characterize these effects.
Glucose lowering treatments
It is well established that diabetes mellitus increases the risk of PAD and accelerates atherogenesis. Although it is unclear if intensive glucose control reduces the risk of PAD, studies describe positive outcomes in lower-extremity events including a 31% reduction in risk of amputation with intensive glucose lowering (155). As such, glucose lowering is a recommended PAD guideline pharmacotherapy; its impact on PAD has been reviewed (156). Because hyperglycemia and insulin resistance can facilitate EC dysfunction (157), many glucose-lowering therapies can impact ECs directly. Insulin for example can directly regulate eNOS expression and NO release to cause vasodilation (158), and also via this pathway, inhibit platelet hyperactivity (159). Further, insulin has regenerative and healing capacity in ECs by stimulating angiogenesis (160). Similarly, metformin was shown to improve endothelial-dependent vasodilation in diabetic atherosclerotic mice (161), however, its role in angiogenesis is conflicting (162, 163). More recent studies demonstrate that diabetic patients treated with glucagon-like peptide agonists, sodium-glucose co-transporter-2 inhibitors or in combination, showed improved systolic blood pressure, endothelial glycocalyx thickness and cardiac function (164). This may in part be due to the direct effect of these therapies on EC functions. For example, glucagon-like peptide 1 and sodium-glucose co-transporter-2 inhibitors reduce EC ROS production, reduce adhesion molecule expression and inflammation, improve vasodilation, and stimulate angiogenesis (165, 166). Additional studies are needed to fully comprehend the effect of glucose-lowering agents on EC functions and their cardioprotective effects.
Emerging diagnostics and therapies
MicroRNAs (miRNAs) are small ~20–25 nucleotide long endogenous non-coding RNA sequences; their main function to regulate protein expression post-transcriptionally. miRNAs are emerging as a biomarker and potential PAD therapeutic. Using next generation genome-wide sequencing, a recent study led by Syed and colleagues identified miRNA-1827 to be significantly upregulated in the blood and plasma of patients with CLTI (167). miRNA-1827 was shown to inhibit cell proliferation and tumor angiogenesis in zebrafish (168) and may in part, contribute to impaired angiogenesis and EC function observed in PAD. miRNA-503 is also upregulated in amputated ischemic limb tissues from diabetic CLTI patients, in ischemic tissues of diabetic mice as well as in ECs exposed to diabetic conditions in vitro (169). Indeed, the authors found that miRNA-504 overexpression inhibited glucose-induced in vitro processes of angiogenesis, whereas inhibition of miRNA-503 improved blood perfusion and increased EC capillary density in diabetic mice following ischemic injury (169). These findings imply that miRNA-503 could be targeted for improving EC function in PAD. Other miRNAs are also altered in PAD patients including miRNA-130a,−27b and−210. Interestingly, miRNA-130a suppression was shown to increase angiogenesis and improve neurological function in ischemic stroke (170) and miRNA-27b inhibited human umbilical vein EC proliferation, migration and tubulogenesis by directly suppressing VEGF-C (171). In contrast, miRNA-210 stimulated pro-angiogenic processes in hypoxia in the brain and in ECs in vitro (172). The mRNA expression of all 3 miRNAs were increased in serum from atherosclerotic obliterans/PAD patients at I-III Fontaine stages (173), however, their role in EC functions in PAD is unclear. miRNAs could be the future in PAD diagnostics and gene therapy [reviewed in (174–176)].
In addition to miRNAs, dysfunction to the endothelium can stimulate the release of endothelial-derived microvesicles (EMVs). These are small vesicles (~0.5–2 μm) released by activated ECs during inflammation to regulate multiple cellular and vascular functions (177), playing a role in immunity, inflammation, and thrombosis (178). They can also carry miRNAs (178, 179). Because of these functions, EMVs are emerging biomarkers with therapeutic potential, particularly in atherosclerosis. For example, EMVs isolated from patients with CAD stimulated permeability and increased the mRNA expression of ICAM-1, VCAM-1, and CCl-2 in ECs in vitro (180). The role of EMVs in PAD is not elucidated, however, similar mechanisms may be at play. Further study is needed to understand their role in PAD.
Future perspectives and concluding remarks
The etiology of PAD is multifactorial, and the endothelium may hold clues into pathogenesis. From its anti-inflammatory, anti-thrombotic, anti-atherogenic and pro-repair and regeneration role, the endothelium is critical in mediating cardiovascular homeostasis. Currently, there are limited treatments for limb ischemia. Alternate or novel treatments that could restore EC function(s) could have significant therapeutic implications for PAD given that EC dysfunction is a common factor facilitating pathogenesis. New therapies reducing symptoms and the risk of amputation could be life changing for these patients.
Many questions remain. For example, is the pathogenesis of PAD and other atherosclerotic disease distinct? What triggers thrombosis in PAD patients? How does the spectrum of EC phenotypes affect EC function in PAD pathogenesis? What about the potential role of antioxidants in PAD therapy or EMVs and microRNAs? What about epigenetic changes which involve miRNAs, histone modification and DNA methylation (176)? Can biomarkers such as hs-CRP, fibrinogen and D-dimer (amongst others), recently identified to predict major adverse cardiovascular outcomes in PAD (22) be used for treatment selection? For example, could PAD patients with fibrinogen levels ≥446.35 mg/L, increasing risk of cardiovascular mortality (22) be given more intensive treatment with anti-coagulants? Indeed, biomarkers could potentially be used to identify PAD patients with greater risk of adverse outcomes and those patients who may show benefit from intensive treatment (22).
Sex-dependent differences are also emerging in PAD. PAD appears to be more prevalent in women >20 years of age (181), with women more likely to be asymptomatic and have worse outcomes to treatment (182). How EC dysfunction contributes to sex-dependent differences is completely unknown and it is tantalizing to speculate that mechanisms occurring in CMD may also play a role in women with PAD. More studies are needed to identify whether EC dysfunction and the spectrum of EC phenotypes reflect sex-dependent differences in pathogenesis. Since many patients are asymptomatic or with atypical symptoms, also raises the question of identifying a non-invasive technique to measure EC functions in the clinic, particularly those that may be high risk of CLTI. FMD is used as a current strategy, however, more improved assessments with greater sensitivity and specificity are needed to take into consideration macrovascular vs. microvascular effects of ECs in PAD. Indeed, a universal method for FMD measurements and newer technologies for assessing EC functions were recently proposed in a position statement by the European Society of Cardiology Working Groups (13).
There is also a gap in knowledge in our understanding of the interaction of the endothelium with the cellular and humoral immune system in PAD, which requires further investigation. Medical therapy and secondary risk prevention for PAD described earlier include statins, antiplatelets, antihypertensives, control of diabetes, and cessation of smoking, with many of these directly affecting EC functions(s), however there is a substantial lack of guideline adherence, with only ~11–67% reported to adhere to PAD recommended guideline therapy (136). Surprisingly, PAD patients are also less likely to receive these medications than patients with CAD (127), thus, the benefit and impact of these on PAD pathogenesis including effects on EC function(s) is not fully established and requires greater study.
Finally, a greater understanding of PAD pathogenesis and mechanisms of EC dysfunction are essential. Multi-omic approaches, combining genomics, proteomics, metabolomics with phenotypic data and network biology analysis, are underway to decipher these mechanisms in PAD. Pathogenic characterization at the molecular and cellular levels could identify strategic targets leading to improvements in diagnosis, management, and treatment of PAD.
Funding
MK and SC are supported by grants from the Australian National Health and Medical Research (NHMRC; 1188218) and the Heart Research Institute. JG is supported by grants from the NHMRC (1180736), the Medical Research Future Fund (2015979/2015817/MRAF000042) and Australian Heart Foundation (105529).
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
MK was responsible for conception and design. MK, CB, CS, FP, SP, JL, GF, JG, SA, and DR drafted the manuscript. CS and SC designed the figures. All authors provided intellectual content and approved the manuscript for publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
Virani SS Alonso A Aparicio HJ Benjamin EJ Bittencourt MS Callaway CW et al . Heart disease and stroke statistics-2021 update: a report from the American heart association. Circulation. (2021) 143:e254–743. 10.1161/CIR.0000000000000950
2.
Kohn CG Alberts MJ Peacock WF Bunz TJ Coleman C . I cost and inpatient burden of peripheral artery disease: findings from the national inpatient sample. Atherosclerosis. (2019) 286:142–6. 10.1016/j.atherosclerosis.2019.05.026
3.
Agnelli G Belch JJF Baumgartner I Giovas P Hoffmann U . Morbidity and mortality associated with atherosclerotic peripheral artery disease: a systematic review. Atherosclerosis. (2020) 293:94–100. 10.1016/j.atherosclerosis.2019.09.012
4.
Conte MS Bradbury AW Kolh P White JV Dick F Fitridge R et al . Global vascular guidelines on the management of chronic limb-threatening ischemia. Eur J Vasc Endovasc Surg. (2019) 58:S1–S109 e133. 10.1016/j.jvs.2019.06.102
5.
Davies JH Richards J Conway K Kenkre JE Lewis JE Mark Williams E et al . Peripheral arterial disease detection, awareness, and treatment in primary care. JAMA. (2001) 286:1317–24. 10.1001/jama.286.11.1317
6.
Narula N Olin JW Narula N . Pathologic disparities between peripheral artery disease and coronary artery disease. Arterioscler Thromb Vasc Biol. (2020) 40:1982–89. 10.1161/ATVBAHA.119.312864
7.
Behroozian A Beckman J . A microvascular disease increases amputation in patients with peripheral artery disease arterioscler. Arterioscler Thromb Vasc Biol. (2020) 40:534–40. 10.1161/ATVBAHA.119.312859
8.
Paik DT Tian L Williams IM Rhee S Zhang H Liu C et al . Single-cell RNA sequencing unveils unique transcriptomic signatures of organ-specific endothelial cells. Circulation. (2020) 142:1848–62. 10.1161/CIRCULATIONAHA.119.041433
9.
Chen PY Qin L Baeyens N Li G Afolabi T Budatha M et al . Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. (2015) 125:4514–28. 10.1172/JCI82719
10.
Dejana E Hirschi KK Simons M . The molecular basis of endothelial cell plasticity. Nat Commun. (2017) 8:14361. 10.1038/ncomms14361
11.
Evrard SM Lecce L Michelis KC Nomura-Kitabayashi A Pandey G Purushothaman KR et al . Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. (2016) 7:11853. 10.1038/ncomms11853
12.
Kovacic JC Dimmeler S Harvey RP Finkel T Aikawa E Krenning G et al . Endothelial to mesenchymal transition in cardiovascular disease: JACC state-of-the-art review. J Am Coll Cardiol. (2019) 73:190–209. 10.1016/j.jacc.2018.09.089
13.
Alexander Y Osto E Schmidt-Trucksäss A Shechter M Trifunovic D Duncker DJ et al . Endothelial function in cardiovascular medicine: a consensus paper of the European society of cardiology working groups on atherosclerosis and vascular biology, aorta and peripheral vascular diseases, coronary pathophysiology and microcirculation, and thrombosis. Cardiovasc Res. (2021) 117:29–42. 10.1093/cvr/cvaa085
14.
Sluiter TJ van Buul JD Huveneers S Quax PHA de Vries M . R endothelial barrier function and leukocyte transmigration in atherosclerosis. Biomedicines. (2021) 9:328. 10.3390/biomedicines9040328
15.
Nasir K Guallar E Navas-Acien A Criqui MH Lima JA . Relationship of monocyte count and peripheral arterial disease: results from the national health and nutrition examination survey 1999–2002. Arterioscler Thromb Vasc Biol. (2005) 25:1966–71. 10.1161/01.ATV.0000175296.02550.e4
16.
Aykan AÇ Hatem E Kalaycioglu E Karabay CY Zehir R Gökdeniz T et al . Neutrophil-to-lymphocyte ratio may be a marker of peripheral artery disease complexity. Anatol J Cardiol. (2016) 16:497–503. 10.5152/AnatolJCardiol.2015.6240
17.
Dopheide JF Rubrech J Trumpp A Geissler P Zeller GC Bock K et al . Leukocyte-platelet aggregates-a phenotypic characterization of different stages of peripheral arterial disease. Platelets. (2016) 27:658–67. 10.3109/09537104.2016.1153619
18.
Petrkova J Szotkowska J Hermanova Z Lukl J Petrek M . Monocyte chemoattractant protein-1 in patients with peripheral arterial disease. Mediators Inflamm. (2004) 13:39–43. 10.1080/09629350410001664752
19.
Signorelli SS Anzaldi M Fiore V . Inflammation in peripheral arterial disease (PAD). Current Pharmaceutical Des. (2012) 18:4350–57. 10.2174/138161212802481273
20.
Signorelli SS Fiore V Malaponte G . Inflammation and peripheral arterial disease: the value of circulating biomarkers (Review). Int J Mol Med. (2014) 33:777–83. 10.3892/ijmm.2014.1657
21.
Ridker PM Cushman M Stampfer MJ Tracy RP Hennekens CH . Plasma concentration of C-reactive protein and risk of developing peripheral vascular disease. Circulation. (1998) 97:425–8. 10.1161/01.CIR.97.5.425
22.
Kremers B Wübbeke L Mees B Cate HT Spronk H Cate-Hoek AT et al . Plasma biomarkers to predict cardiovascular outcome in patients with peripheral artery disease: a systematic review and meta-analysis. Arterioscler Thromb Vasc Biol. (2020) 40:2018–32. 10.1161/ATVBAHA.120.314774
23.
Nording HM Seizer P Langer HF . Platelets in inflammation and atherogenesis. Front Immunol. (2015) 6:98. 10.3389/fimmu.2015.00098
24.
Barrett TJ Schlegel M Zhou F Gorenchtein M Bolstorff J Moore KJ et al . Platelet regulation of myeloid suppressor of cytokine signaling 3 accelerates atherosclerosis. Sci Transl Med. (2019) 11:eaax0481. 10.1126/scitranslmed.aax0481
25.
Brevetti G Giugliano G Brevetti L Hiatt WR . Inflammation in peripheral artery disease. Circulation. (2010) 122:1862–75. 10.1161/CIRCULATIONAHA.109.918417
26.
Scholz D Ito W Fleming I Deindl E Sauer A Wiesnet M et al . Ultrastructure and molecular histology of rabbit hind-limb collateral artery growth (arteriogenesis). Virchows Arch. (2000) 436:257–70. 10.1007/s004280050039
27.
Hoefer IE Royen Nv Rectenwald JE Deindl E Hua J Jost M et al . Arteriogenesis proceeds via ICAM-1/Mac-1- mediated mechanisms. Circ Res. (2004) 94:1179–85. 10.1161/01.RES.0000126922.18222.F0
28.
Rull A Hernandez-Aguilera A Fibla M Sepulveda J Rodríguez-Gallego E Riera-Borrull M et al . Understanding the role of circulating chemokine (C-C motif) ligand 2 in patients with chronic ischemia threatening the lower extremities. Vasc Med. (2014) 19:442–51. 10.1177/1358863X14554034
29.
Hernández-Aguilera A Sepúlveda J Rodríguez-Gallego E Guirro M García-Heredia A Cabré N et al . Immunohistochemical analysis of paraoxonases and chemokines in arteries of patients with peripheral artery disease. Int J Mol Sci. (2015) 16:11323–38. 10.3390/ijms160511323
30.
Tzoulaki I Murray GD Lee AJ Rumley A Lowe GDO Fowkes FGR et al . Inflammatory, haemostatic, and rheological markers for incident peripheral arterial disease: edinburgh artery study. Eur Heart J. (2007) 28:354–62. 10.1093/eurheartj/ehl441
31.
Silvestro A Brevetti G Schiano V Scopacasa F Chiariello M . Adhesion molecules and cardiovascular risk in peripheral arterial disease Soluble vascular cell adhesion molecule-1 improves risk stratification. Thromb Haemost. (2005) 93:559–63. 10.1160/TH04-07-0440
32.
Boulbou MS Koukoulis GN Vasiou KG Petinaki EA Gourgoulianis KI Fezoulidis IB et al . Increased soluble E-selectin levels in type 2 diabetic patients with peripheral arterial disease. Int Angiol. (2004) 23:18–24.
33.
Rajagopalan S Mckay I Ford I Bachoo P Greaves M Brittenden J et al . Platelet activation increases with the severity of peripheral arterial disease: implications for clinical management. J Vasc Surg. (2007) 46:485–90. 10.1016/j.jvs.2007.05.039
34.
Woollard KJ Kling D Kulkarni S Dart AM Jackson S Chin-Dusting J et al . Raised plasma soluble P-selectin in peripheral arterial occlusive disease enhances leukocyte adhesion. Circ Res. (2006) 98:149–56. 10.1161/01.RES.0000199295.14073.69
35.
Cassar K Bachoo P Ford I Greaves M Brittenden J . Platelet activation is increased in peripheral arterial disease. J Vasc Surg. (2003) 38:99–103. 10.1016/S0741-5214(03)00129-0
36.
Wassel CL Berardi C Pankow JS Larson NB Decker PA Hanson NQ et al . Soluble P-selectin predicts lower extremity peripheral artery disease incidence and change in the ankle brachial index: the Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis. (2015) 239:405–11. 10.1016/j.atherosclerosis.2015.01.022
37.
Saenz-Pipaon G Martinez-Aguilar E Orbe J González Miqueo A Fernandez-Alonso L Paramo JA et al . The role of circulating biomarkers in peripheral arterial disease. Int J Mol Sci. (2021) 22:3601. 10.3390/ijms22073601
38.
Wood JP Ellery PE Maroney SA Mast AE . Biology of tissue factor pathway inhibitor. Blood. (2014) 123:2934–43. 10.1182/blood-2013-11-512764
39.
Martin FA Murphy RP Cummins PM . Thrombomodulin and the vascular endothelium: insights into functional, regulatory, and therapeutic aspects. Am J Physiol Heart Circ Physiol. (2013) 304:H1585–1597. 10.1152/ajpheart.00096.2013
40.
Wieczor R Wieczor AM Rosc D . Tissue-type plasminogen activator and plasminogen activator inhibitor type 1 in patients with symptomatic lower extremity artery disease. Minerva Cardiol Angiol. (2021) 69:161–71. 10.23736/S2724-5683.20.05114-2
41.
Polonsky TS McDermott MM . Increased platelet aggregation and activation in peripheral arterial disease. Eur J Vasc Endovasc Surg. (2003) 25:16–22. 10.1053/ejvs.2002.1794
42.
Berger JS Eraso LH Xie D Sha D Mohler ER . Mean platelet volume and prevalence of peripheral artery disease, the National Health and Nutrition Examination Survey, 1999–2004. Atherosclerosis. (2010) 213:586–91. 10.1016/j.atherosclerosis.2010.09.010
43.
Blann AD Amiral J McCollum CN Lip GY . Differences in free and total tissue factor pathway inhibitor, and tissue factor in peripheral artery disease compared to healthy controls. Atherosclerosis. (2000) 152:29–34. 10.1016/S0021-9150(99)00444-X
44.
Zamzam A Syed MH Rand ML Singh K Hussain MA Jain S et al . Altered coagulation profile in peripheral artery disease patients. Vascular. (2020) 28:368–77. 10.1177/1708538120915997
45.
Smith FB Lee AJ Hau CM Rumley A Lowe GD Fowkes FG et al . Plasma fibrinogen, haemostatic factors and prediction of peripheral arterial disease in the edinburgh artery study. Blood Coagul Fibrinolysis. (2000) 11:43–50. 10.1097/00001721-200011010-00005
46.
Blann AD Seigneur M Steiner M Boisseau MR McCollum CN . Circulating endothelial cell markers in peripheral vascular disease: relationship to the location and extent of atherosclerotic disease. Eur J Clin Invest. (1997) 27:916–21. 10.1046/j.1365-2362.1997.2180766.x
47.
Salomaa V Matei C Aleksic N Sansores-Garcia L Folsom AR Juneja H et al . Cross-sectional association of soluble thrombomodulin with mild peripheral artery disease; the ARIC study. Atherosclerosis risk in communities. Atherosclerosis. (2001) 157:309–14. 10.1016/S0021-9150(00)00729-2
48.
Drozdz D Latka M Drozdz T Sztefko K Kwinta P . Thrombomodulin as a new marker of endothelial dysfunction in chronic kidney disease in children. Oxid Med Cell Longev. (2018) 2018:1619293. 10.1155/2018/1619293
49.
Narula N Dannenberg AJ Olin JW Bhatt DL Johnson KW Nadkarni G et al . Pathology of peripheral artery disease in patients with critical limb ischemia. J Am Coll Cardiol. (2018) 72:2152–63. 10.1016/j.jacc.2018.08.002
50.
Yahagi K Davis HR Arbustini E Virmani R . Sex differences in coronary artery disease: pathological observations. Atherosclerosis. (2015) 239:260–7. 10.1016/j.atherosclerosis.2015.01.017
51.
Ho CY Shanahan CM . Medial arterial calcification: an overlooked player in peripheral arterial disease. Arterioscler Thromb Vasc Biol. (2016) 36:1475–82. 10.1161/ATVBAHA.116.306717
52.
Akers EJ Nicholls SJ Di Bartolo BA . Plaque calcification: do lipoproteins have a role?Arterioscler Thromb Vasc Biol. (2019) 39:1902–10. 10.1161/ATVBAHA.119.311574
53.
Mulder BJ . Not too old to be closed. Neth Heart J. (2010) 18:520–1. 10.1007/s12471-010-0826-x
54.
Edwards G Dora KA Gardener MJ Garland CJ Weston AH . K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. (1998) 396:269–72. 10.1038/24388
55.
Fleming I . Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. (1999) 401:493–7. 10.1038/46816
56.
Emerson GG Segal SS . Electrical activation of endothelium evokes vasodilation and hyperpolarization along hamster feed arteries. Am J Physiol Heart Circ Physiol. (2001) 280:H160–7. 10.1152/ajpheart.2001.280.1.H160
57.
Shimokawa H Morikawa K . Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. (2000) 106:1521–30. 10.1172/JCI10506
58.
Bellamkonda K Williams M Handa A Lee R . Flow mediated dilatation as a biomarker in vascular surgery research. J Atherosclerosis Thrombosis. (2017) 24:779–87. 10.5551/jat.40964
59.
Böger RH Bode-Böger SM Thiele W Junker W Alexander K Frölich JC et al . Biochemical evidence for impaired nitric oxide synthesis in patients with peripheral arterial occlusive disease. Circulation. (1997) 95:2068–74. 10.1161/01.CIR.95.8.2068
60.
Ismaeel A Papoutsi E Miserlis D Lavado R Haynatzki G Casale GP et al . The nitric oxide system in peripheral artery disease: connection with oxidative stress and biopterins. Antioxidants. (2020) 9:590. 10.3390/antiox9070590
61.
Baldus S Köster R Chumley P Heitzer T Rudolph V Ostad MA et al . Oxypurinol improves coronary and peripheral endothelial function in patients with coronary artery disease. Free Radic Biol Med. (2005) 39:1184–90. 10.1016/j.freeradbiomed.2005.06.004
62.
Bode-Böger SM Böger RH Alfke H Heinzel D Tsikas D Creutzig A et al . L-arginine induces nitric oxide-dependent vasodilation in patients with critical limb ischemia. A randomized, controlled study. Circulation. (1996) 93:85–90. 10.1161/01.CIR.93.1.85
63.
Wilson AM Harada R Nair N Balasubramanian N Cooke JP . L-arginine supplementation in peripheral arterial disease: no benefit and possible harm. Circulation. (2007) 116:188–95. 10.1161/CIRCULATIONAHA.106.683656
64.
Bundgaard H Liu C Garcia A Hamilton EJ Huang Y Chia KKM et al . Beta(3) adrenergic stimulation of the cardiac Na+-K+ pump by reversal of an inhibitory oxidative modification. Circulation. (2010) 122:2699–708. 10.1161/CIRCULATIONAHA.110.964619
65.
Galougahi KK Liu CC Bundgaard H Rasmussen HH . Beta-adrenergic regulation of the cardiac Na+-K+ ATPase mediated by oxidative signaling. Trends Cardiovasc Med. (2012) 22:83–7. 10.1016/j.tcm.2012.06.017
66.
Galougahi KK Liu C Garcia A Gentile C Fry NA Hamilton EJ et al . Beta3 adrenergic stimulation restores nitric oxide/redox balance and enhances endothelial function in hyperglycemia. J Am Heart Assoc. (2016) 5:e002824. 10.1161/JAHA.115.002824
67.
Bagi Z Ungvari Z Szollar L Koller A . Flow-induced constriction in arterioles of hyperhomocysteinemic rats is due to impaired nitric oxide and enhanced thromboxane A(2) mediation. Arterioscler Thromb Vasc Biol. (2001) 21:233–7. 10.1161/01.ATV.21.2.233
68.
Flórez A Haro Jd Martínez E Varela C Bleda S Acín F et al . Selective cyclooxygenase-2 inhibition reduces endothelial dysfunction and improves inflammatory status in patients with intermittent claudication. Rev Esp Cardiol. (2009) 62:851–7. 10.1016/S1885-5857(09)72649-0
69.
Bolton TB Lang RJ Takewaki T . Mechanisms of action of noradrenaline and carbachol on smooth muscle of guinea-pig anterior mesenteric artery. J Physiol. (1984) 351:549–72. 10.1113/jphysiol.1984.sp015262
70.
Hodges NA Suarez-Martinez AD Murfee WL . Understanding angiogenesis during aging: opportunities for discoveries and new models. J Appl Physiol. (2018) 125:1843–50. 10.1152/japplphysiol.00112.2018
71.
Nakae I Fujita M Miwa K Hasegawa K Kihara Y Nohara R et al . Age-dependent impairment of coronary collateral development in humans. Heart Vessels. (2000) 15:176–80. 10.1007/PL00007269
72.
Fadini GP Albiero M Bonora BM Avogaro A . Angiogenic abnormalities in diabetes mellitus: mechanistic and clinical aspects. J Clin Endocrinol Metab. (2019) 104:5431–44. 10.1210/jc.2019-00980
73.
Kolluru GK Bir SC Kevil CG . Endothelial dysfunction and diabetes: effects on angiogenesis, vascular remodeling, and wound healing. Int J Vasc Med. (2012) 2012:918267. 10.1155/2012/918267
74.
Okonkwo UA Chen L Ma D Haywood VA Barakat M Urao N et al . Compromised angiogenesis and vascular Integrity in impaired diabetic wound healing. PLoS ONE. (2020) 15:e0231962. 10.1371/journal.pone.0231962
75.
Jang JJ Ho HK Kwan HH Fajardo LF Cooke JP . Angiogenesis is impaired by hypercholesterolemia: role of asymmetric dimethylarginine. Circulation. (2000) 102:1414–9. 10.1161/01.CIR.102.12.1414
76.
Zechariah A ElAli A Hagemann N Jin F Doeppner TR Helfrich I et al . Hyperlipidemia attenuates vascular endothelial growth factor-induced angiogenesis, impairs cerebral blood flow, and disturbs stroke recovery via decreased pericyte coverage of brain endothelial cells. Arterioscler Thromb Vasc Biol. (2013) 33:1561–7. 10.1161/ATVBAHA.112.300749
77.
Emanueli C Madeddu P . Angiogenesis gene therapy to rescue ischaemic tissues: achievements and future directions. Br J Pharmacol. (2001) 133:951–8. 10.1038/sj.bjp.0704155
78.
Bhardwaj S Roy H Heikura T Yla-Herttuala S . VEGF-A, VEGF-D and VEGF-D(DeltaNDeltaC) induced intimal hyperplasia in carotid arteries. Eur J Clin Invest. (2005) 35:669–76. 10.1111/j.1365-2362.2005.01555.x
79.
Celletti FL Waugh JM Amabile PG Brendolan A Hilfiker PR Dake MD et al . Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat Med. (2001) 7:425–9. 10.1038/86490
80.
Che J Okigaki M Takahashi T Katsume A Adachi Y Yamaguchi S et al . Endothelial FGF receptor signaling accelerates atherosclerosis. Am J Physiol Heart Circ Physiol. (2011) 300:H154–161. 10.1152/ajpheart.00075.2010
81.
Folkman J . Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation. (1999) 99:1726–32. 10.1161/01.CIR.99.13.1726
82.
Winnik S Lohmann C Siciliani G Lukowicz Tv Kuschnerus K Kraenkel N et al . Systemic VEGF inhibition accelerates experimental atherosclerosis and disrupts endothelial homeostasis–implications for cardiovascular safety. Int J Cardiol. (2013) 168:2453–61. 10.1016/j.ijcard.2013.03.010
83.
Findley CM Mitchell RG Duscha BD Annex BH Kontos CD . Plasma levels of soluble Tie2 and vascular endothelial growth factor distinguish critical limb ischemia from intermittent claudication in patients with peripheral arterial disease. J Am Coll Cardiol. (2008) 52:387–93. 10.1016/j.jacc.2008.02.045
84.
Upadhya R Zingg W Shetty S Shetty AK . VEGF: a surrogate marker for peripheral vascular disease. Eur J Vasc Endovasc Surg. (2010) 39:330–2. 10.1016/j.ejvs.2009.09.025
85.
Pitti RM Marsters SA Ruppert S Donahue CJ Moore A Ashkenazi A et al . Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. (1996) 271:12687–90. 10.1074/jbc.271.22.12687
86.
Wiley SR Schooley K Smolak PJ Din WS Huang CP Nicholl JK et al . Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. (1995) 2:673–82. 10.1016/1074-7613(95)90057-8
87.
Cartland SP Genner SW Martínez GJ Robertson S Kockx M Lin RC et al . TRAIL-expressing monocyte/macrophages are critical for reducing inflammation and atherosclerosis. iScience. (2019) 12:41–52. 10.1016/j.isci.2018.12.037
88.
Shrimali D Shanmugam MK Kumar AP Zhang J Tan BK Ahn KS et al . The involvement of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) in atherosclerosis. J Am Coll Cardiol. (2005) 45:1018–24.
89.
Moon AR Park Y Chang JH Lee SS . Inverse regulation of serum osteoprotegerin and tumor necrosis factor-related apoptosis-inducing ligand levels in patients with leg lesional vascular calcification: an observational study. Medicine. (2019) 98:e14489. 10.1097/MD.0000000000014489
90.
Schoppet M Sattler AM Schaefer JR Hofbauer LC . Osteoprotegerin (OPG) and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) levels in atherosclerosis. Atherosclerosis. (2006) 184:446–7. 10.1016/j.atherosclerosis.2005.10.028
91.
Zauli G Pandolfi A Gonelli A Pietro RD Guarnieri S Ciabattoni G et al . Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) sequentially upregulates nitric oxide and prostanoid production in primary human endothelial cells. Circ Res. (2003) 92:732–40. 10.1161/01.RES.0000067928.83455.9C
92.
Cholan PM Cartland SP Dang L Rayner BS Patel S Thomas SR et al . TRAIL protects against endothelial dysfunction in vivo and inhibits angiotensin-II-induced oxidative stress in vascular endothelial cells in vitro. Free Radic Biol Med. (2018) 126:341–9. 10.1016/j.freeradbiomed.2018.08.031
93.
Bartolo BAD Cartland SP Prado-Lourenco L Griffith TS Gentile C Ravindran J et al . Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) promotes angiogenesis and ischemia-induced neovascularization via NADPH oxidase 4 (NOX4) and nitric oxide-dependent mechanisms. J Am Heart Assoc. (2015) 4:e002527. 10.1161/JAHA.115.002527
94.
Secchiero P Gonelli A Carnevale E Corallini F Rizzardi C Zacchigna S et al . Evidence for a proangiogenic activity of TNF-related apoptosis-inducing ligand. Neoplasia. (2004) 6:364–73. 10.1593/neo.03421
95.
Cartland SP Genner SW Zahoor A Kavurma MM . Comparative evaluation of TRAIL, FGF-2 and VEGF-A-induced angiogenesis in vitro and in vivo. Int J Mol Sci. (2016) 17:2025. 10.3390/ijms17122025
96.
Steinle JJ Booz GW Meininger CJ Day JN Granger HJ . Beta 3-adrenergic receptors regulate retinal endothelial cell migration and proliferation. J Biol Chem. (2003) 278:20681–6. 10.1074/jbc.M300368200
97.
Dessy C Saliez J Ghisdal P Daneau G Lobysheva II Frérart F et al . Endothelial beta3-adrenoreceptors mediate nitric oxide-dependent vasorelaxation of coronary microvessels in response to the third-generation beta-blocker nebivolol. Circulation. (2005) 112:1198–205. 10.1161/CIRCULATIONAHA.104.532960
98.
Bubb KJ Ravindran D Cartland SP Finemore M Clayton ZE Tsang M et al . Beta 3 adrenergic receptor stimulation promotes reperfusion in ischemic limbs in a murine diabetic model. Front Pharmacol. (2021) 12:666334. 10.3389/fphar.2021.666334
99.
Aboyans V Ricco JB Bartelink MEL Björck M Brodmann M Cohnert T et al . 2017 ESC guidelines on the diagnosis and treatment of peripheral arterial diseases, in collaboration with the European Society for Vascular Surgery (ESVS). Rev Esp Cardiol (Engl Ed). (2018) 71:111. 10.1016/j.rec.2017.12.014
100.
Gerhard-Herman MD Gornik HL Barrett C Barshes NR Corriere MA Drachman DE et al . 2016 AHA/ACC guideline on the management of patients with lower extremity peripheral artery disease: a report of the american college of cardiology/american heart association task force on clinical practice guidelines. Circulation. (2017) 135:e726–79. 10.1161/CIR.0000000000000502
101.
Abola MTB Golledge J Miyata T Rha S Yan BP Dy TC et al . Asia-Pacific consensus statement on the management of peripheral artery disease: a report from the Asian Pacific society of atherosclerosis and vascular disease Asia-Pacific peripheral artery disease consensus statement project committee. J Atheroscler Thromb. (2020) 27:809–907. 10.5551/jat.53660
102.
Hambrecht R Adams V Erbs S Linke A Kränkel N Shu Y et al . Regular physical activity improves endothelial function in patients with coronary artery disease by increasing phosphorylation of endothelial nitric oxide synthase. Circulation. (2003) 107:3152–8. 10.1161/01.CIR.0000074229.93804.5C
103.
Pedralli ML Marschner RA Kollet DP Neto SG Eibel B Tanaka H et al . Different exercise training modalities produce similar endothelial function improvements in individuals with prehypertension or hypertension: a randomized clinical trial Exercise, endothelium and blood pressure. Sci Rep. (2020) 10:7628. 10.1038/s41598-020-64365-x
104.
Fuchsjäger-Mayrl G Pleiner J Wiesinger GF Sieder AE Quittan M Nuhr MJ et al . Exercise training improves vascular endothelial function in patients with type 1 diabetes. Diabetes Care. (2002) 25:1795–801. 10.2337/diacare.25.10.1795
105.
Qiu S Cai X Yin H Sun Z Zügel M Steinacker JM et al . Exercise training and endothelial function in patients with type 2 diabetes: a meta-analysis. Cardiovasc Diabetol. (2018) 17:64. 10.1186/s12933-018-0711-2
106.
Möbius-Winkler S Uhlemann M Adams V Sandri M Erbs S Lenk K et al . Coronary collateral growth induced by physical exercise: results of the impact of intensive exercise training on coronary collateral circulation in patients with stable coronary artery disease (EXCITE) trial. Circulation. (2016) 133:1438–48. 10.1161/CIRCULATIONAHA.115.016442
107.
Black MA Cable NT Thijssen DH Green DJ . Impact of age, sex, and exercise on brachial artery flow-mediated dilatation. Am J Physiol Heart Circ Physiol. (2009) 297:H1109–1116. 10.1152/ajpheart.00226.2009
108.
Kasapis C Thompson PD . The effects of physical activity on serum C-reactive protein and inflammatory markers: a systematic review. J Am Coll Cardiol. (2005) 45:1563–9. 10.1016/j.jacc.2004.12.077
109.
Pande RL Hiatt WR Zhang P Hittel N Creager MA . A pooled analysis of the durability and predictors of treatment response of cilostazol in patients with intermittent claudication. Vasc Med. (2010) 15:181–8. 10.1177/1358863X10361545
110.
Brown T Forster RB Cleanthis M Mikhailidis DP Stansby G Stewart M et al . Cilostazol for intermittent claudication. Cochrane Database Syst Rev. (2021) 6:CD003748. 10.1002/14651858.CD003748.pub5
111.
Suri A Forbes WP Bramer SL . Pharmacokinetics of multiple-dose oral cilostazol in middle-age and elderly men and women. J Clin Pharmacol. (1998) 38:144–150. 10.1002/j.1552-4604.1998.tb04403.x
112.
Wang J Bingaman S Huxley VH . Intrinsic sex-specific differences in microvascular endothelial cell phosphodiesterases. Am J Physiol Heart Circ Physiol. (2010) 298:H1146–1154. 10.1152/ajpheart.00252.2009
113.
Bubb KJ Harmer JA Finemore M Aitken SJ Ali ZS Billot L et al . Protocol for the stimulating beta3-adrenergic receptors for peripheral artery disease (STAR-PAD) trial: a double-blinded, randomised, placebo-controlled study evaluating the effects of mirabegron on functional performance in patients with peripheral arterial disease. BMJ Open. (2021) 11:e049858. 10.1136/bmjopen-2021-049858
114.
Haas AV Rosner BA Kwong RY Rao AD Garg R Di Carli MF et al . Sex differences in coronary microvascular function in individuals with type 2 diabetes. Diabetes. (2019) 68:631–6. 10.2337/db18-0650
115.
Vancheri F Longo G Vancheri S Henein M . Coronary microvascular dysfunction. J Clin Med. (2020) 9:830–40. 10.3390/jcm9092880
116.
Godo S Takahashi J Yasuda S Shimokawa H . Role of inflammation in coronary epicardial and microvascular dysfunction. Eur Cardiol. (2021) 16:e13. 10.15420/ecr.2020.47
117.
Leonardo F Fragasso G Rossetti E Dabrowski P Pagnotta P Rosano GM et al . Comparison of trimetazidine with atenolol in patients with syndrome X: effects on diastolic function and exercise tolerance. Cardiologia. (1999) 44:1065–9.
118.
Haas MJ Kurban W Shah H Onstead-Haas L Mooradian AD . Beta blockers suppress dextrose-induced endoplasmic reticulum stress, oxidative stress, and apoptosis in human coronary artery endothelial cells. Am J Therapeutics. (2016) 23:e1524–31. 10.1097/MJT.0000000000000200
119.
Chen Q Guo F Liu S Xiao J Wang C Snowise S et al . Calcium channel blockers prevent endothelial cell activation in response to necrotic trophoblast debris: possible relevance to pre-eclampsia. Cardiovasc Res. (2012) 96:484–93. 10.1093/cvr/cvs279
120.
Ding Y Vaziri ND . Calcium channel blockade enhances nitric oxide synthase expression by cultured endothelial cells. Hypertension. (1998) 32:718–23. 10.1161/01.HYP.32.4.718
121.
Peller M Ozierański K Balsam P Grabowski M Filipiak KJ Opolski G et al . Influence of beta-blockers on endothelial function: a meta-analysis of randomized controlled trials. Cardiol J. (2015) 22:708–16. 10.5603/CJ.a2015.0042
122.
Bots ML Remme WJ Lüscher TF Fox KM Bertrand M Ferrari R et al . ACE inhibition and endothelial function: main findings of PERFECT, a sub-study of the EUROPA trial. Cardiovasc Drugs Ther. (2007) 21:269–79. 10.1007/s10557-007-6041-3
123.
Hamdi HK Castellon R . ACE inhibition actively promotes cell survival by altering gene expression. Biochem Biophys Res Commun. (2003) 310:1227–35. 10.1016/j.bbrc.2003.09.149
124.
He Z Du X Wu Y Hua L Wan L Yan N et al . Simvastatin promotes endothelial dysfunction by activating the Wnt/betacatenin pathway under oxidative stress. Int J Mol Med. (2019) 44:1289–98. 10.3892/ijmm.2019.4310
125.
Li X Xiao H Lin C Sun W Wu T Wang J et al . Synergistic effects of liposomes encapsulating atorvastatin calcium and curcumin and targeting dysfunctional endothelial cells in reducing atherosclerosis. Int J Nanomedicine. (2019) 14:649–65. 10.2147/IJN.S189819
126.
Ii M Losordo DW . Statins and the endothelium. Vascul Pharmacol. (2007) 46:1–9. 10.1016/j.vph.2006.06.012
127.
Krishnamurthy V Munir K Rectenwald JE Mansour A Hans S Eliason JL et al . Contemporary outcomes with percutaneous vascular interventions for peripheral critical limb ischemia in those with and without poly-vascular disease. Vasc Med. (2014) 19:491–9. 10.1177/1358863X14552013
128.
Ridker PM Everett BM Thuren T MacFadyen JG Chang WH Ballantyne C et al . Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. 10.1056/NEJMoa1707914
129.
Vallejo S Palacios E Romacho T Villalobos L Peiró C Sánchez-Ferrer CF et al . The interleukin-1 receptor antagonist anakinra improves endothelial dysfunction in streptozotocin-induced diabetic rats. Cardiovasc Diabetol. (2014) 13:158. 10.1186/s12933-014-0158-z
130.
Ikonomidis I Lekakis JP Nikolaou M Paraskevaidis I Andreadou I Kaplanoglou T et al . Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation. (2008) 117:2662–9. 10.1161/CIRCULATIONAHA.107.731877
131.
Brezinski EA Follansbee MR Armstrong EJ Armstrong AW . Endothelial dysfunction and the effects of TNF inhibitors on the endothelium in psoriasis and psoriatic arthritis: a systematic review. Current Pharmaceutical Des. (2014) 20:513–28. 10.2174/138161282004140213123852
132.
Fiolet ATL Opstal TSJ Mosterd A Eikelboom JW Jolly SS Keech AC et al . Efficacy and safety of low-dose colchicine in patients with coronary disease: a systematic review and meta-analysis of randomized trials. Eur Heart J. (2021) 42:2765–75. 10.1093/eurheartj/ehab115
133.
Cronstein BN Molad Y Reibman J Balakhane E Levin RI Weissmann G et al . Colchicine alters the quantitative and qualitative display of selectins on endothelial cells and neutrophils. J Clin Invest. (1995) 96:994–1002. 10.1172/JCI118147
134.
Hays AG Schär M Bonanno G Lai S Meyer J Afework Y et al . Randomized trial of anti-inflammatory medications and coronary endothelial dysfunction in patients with stable coronary disease. Front Cardiovasc Med. (2021) 8:728654. 10.3389/fcvm.2021.728654
135.
Kajikawa M Higashi Y Tomiyama H Maruhashi T Kurisu S Kihara Y et al . Effect of short-term colchicine treatment on endothelial function in patients with coronary artery disease. Int J Cardiol. (2019) 281:35–9. 10.1016/j.ijcard.2019.01.054
136.
Criqui MH Matsushita K Aboyans V Hess CN Hicks CW Kwan TW et al . Lower extremity peripheral artery disease: contemporary epidemiology, management gaps, and future directions: a scientific statement from the american heart association. Circulation. (2021) 144:e171–91. 10.1161/CIR.0000000000001005
137.
Brand AR Houben E Bezemer ID Visseren FLJ Bots ML Herings RM et al . Platelet aggregation inhibitor prescription for newly diagnosed peripheral arterial disease in the Netherlands: a cohort study. BMJ Open. (2021) 11:e041715. 10.1136/bmjopen-2020-041715
138.
Chen J Wang L Liu WH Shi J Zhong Y Liu SJ et al . Aspirin protects human coronary artery endothelial cells by inducing autophagy. Physiol Int. (2020) 107:294–305. 10.1556/2060.2020.00029
139.
Podhaisky HP Abate A Polte T Oberle S Schroder H . Aspirin protects endothelial cells from oxidative stress—possible synergism with vitamin E. FEBS Lett. (1997) 417:349–51. 10.1016/S0014-5793(97)01307-0
140.
Husain S Andrews NP Mulcahy D Panza JA Quyyumi AA . Aspirin improves endothelial dysfunction in atherosclerosis. Circulation. (1998) 97:716–20. 10.1161/01.CIR.97.8.716
141.
Bulckaen H Prévost G Boulanger E Robitaille G Roquet V Gaxatte C et al . Low-dose aspirin prevents age-related endothelial dysfunction in a mouse model of physiological aging. Am J Physiol Heart Circ Physiol. (2008) 294:H1562–1570. 10.1152/ajpheart.00241.2007
142.
Jia Z Huang Y Ji X Sun J Fu G . Ticagrelor and clopidogrel suppress NF-kappaB signaling pathway to alleviate LPS-induced dysfunction in vein endothelial cells. BMC Cardiovasc Disord. (2019) 19:318. 10.1186/s12872-019-01287-1
143.
Giachini FR Leite R Osmond DA Lima VV Inscho EW Webb RC et al . Anti-platelet therapy with clopidogrel prevents endothelial dysfunction and vascular remodeling in aortas from hypertensive rats. PLoS ONE. (2014) 9:e91890. 10.1371/journal.pone.0091890
144.
Eikelboom JW Connolly SJ Bosch J Dagenais GR Hart RG Shestakovska O et al . Rivaroxaban with or without aspirin in stable cardiovascular disease. N Engl J Med. (2017) 377:1319–30. 10.1056/NEJMoa1709118
145.
Anand SS Bosch J Eikelboom JW Connolly SJ Diaz R Widimsky P et al . Rivaroxaban with or without aspirin in patients with stable peripheral or carotid artery disease: an international, randomised, double-blind, placebo-controlled trial. Lancet. (2018) 391:219–29. 10.1016/S0140-6736(17)32409-1
146.
Capell WH Bonaca MP Nehler MR Chen E Kittelson JM Anand SS et al . Rationale and design for the vascular outcomes study of ASA along with rivaroxaban in endovascular or surgical limb revascularization for peripheral artery disease (VOYAGER PAD). Am Heart J. (2018) 199:83–91. 10.1016/j.ahj.2018.01.011
147.
Bonaca MP Bauersachs RM Anand SS Debus ES Nehler MR Patel MR et al . Rivaroxaban in peripheral artery disease after revascularization. N Engl J Med. (2020) 382:1994–2004. 10.1056/NEJMoa2000052
148.
Firnhaber JM Powell CS . Efficacy and safety of rivaroxaban plus aspirin in women and men with chronic coronary or peripheral artery disease. Cardiovasc Res. (2021) 117:942–9. 10.1093/cvr/cvaa100
149.
Pham PT Fukuda D Yagi S Kusunose K Yamada H Soeki T et al . Rivaroxaban, a specific FXa inhibitor, improved endothelium-dependent relaxation of aortic segments in diabetic mice. Sci Rep. (2019) 9:11206. 10.1038/s41598-019-47474-0
150.
Pistrosch F Matschke JB Schipp D Schipp B Henkel E Weigmann I et al . Rivaroxaban compared with low-dose aspirin in individuals with type 2 diabetes and high cardiovascular risk: a randomised trial to assess effects on endothelial function, platelet activation and vascular biomarkers. Diabetologia. (2021) 64:2701–12. 10.1007/s00125-021-05562-9
151.
Wozniak E Broncel M Bukowska B Gorzelak-Pabis P . The protective effect of dabigatran and rivaroxaban on DNA oxidative changes in a model of vascular endothelial damage with oxidized cholesterol. Int J Mol Sci. (2020) 21:1953. 10.3390/ijms21061953
152.
Gorzelak-Pabis P Pawlos A Broncel M Wojdan K Wozniak E . Expression of anti and pro-inflammatory genes in human endothelial cells activated by 25-hydroxycholesterol: A comparison of rivaroxaban and dabigatran. Clin Exp Pharmacol Physiol. (2022) 49:805–12. 10.1111/1440-1681.13668
153.
Gorzelak-Pabis P Broncel M Wojdan K Gajewski A Chalubinski M Gawrysiak M et al . Rivaroxaban protects from the oxysterol-induced damage and inflammatory activation of the vascular endothelium. Tissue Barriers. (2021) 9:1956284. 10.1080/21688370.2021.1956284
154.
Wu T Chan J Lee C Leu H Huang P Chen J et al . Rivaroxaban, a factor Xa inhibitor, improves neovascularization in the ischemic hindlimb of streptozotocin-induced diabetic mice. Cardiovasc Diabetol. (2015) 14:81. 10.1186/s12933-015-0243-y
155.
Singh N Armstrong DG Lipsky BA . Effect of intensive glycemic control on risk of lower extremity amputation. J Am Coll Surg. (2018) 227:596–604. 10.1016/j.jamcollsurg.2018.09.021
156.
Suades R Cosentino F Badimon L . Glucose-lowering treatment in cardiovascular and peripheral artery disease. Curr Opin Pharmacol. (2018) 39:86–98. 10.1016/j.coph.2018.03.001
157.
Calles-Escandon J Cipolla M . Diabetes and endothelial dysfunction: a clinical perspective. Endocr Rev. (2001) 22:36–52. 10.1210/edrv.22.1.0417
158.
Kearney MT Duncan ER Kahn M Wheatcroft SB . Insulin resistance and endothelial cell dysfunction: studies in mammalian models. Exp Physiol. (2008) 93:158–63. 10.1113/expphysiol.2007.039172
159.
Vinik AI Erbas T Park TS Nolan R Pittenger GL . Platelet dysfunction in type 2 diabetes. Diabetes Care. (2001) 24:1476–85. 10.2337/diacare.24.8.1476
160.
Escudero CA Herlitz K Troncoso F Guevara K Acurio J Aguayo C et al . Pro-angiogenic role of insulin: from physiology to pathology. Front Physiol. (2017) 8:204. 10.3389/fphys.2017.00204
161.
Wang Q Zhang M Torres G Wu S Ouyang C Xie Z et al . Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes. (2017) 66:193–205. 10.2337/db16-0915
162.
Bakhashab S Ahmed F Schulten H Ahmed FW Glanville M Al-Qahtani MH et al . Proangiogenic effect of metformin in endothelial cells is via upregulation of VEGFR1/2 and their signaling under hyperglycemia-hypoxia. Int J Mol Sci. (2018) 19:293. 10.3390/ijms19010293
163.
Dallaglio K Bruno A Cantelmo AR Esposito AI Ruggiero L Orecchioni S et al . Paradoxic effects of metformin on endothelial cells and angiogenesis. Carcinogenesis. (2014) 35:1055–66. 10.1093/carcin/bgu001
164.
Ikonomidis I Pavlidis G Thymis J Birba D Kalogeris A Kousathana F et al . Effects of glucagon-like peptide-1 receptor agonists, sodium-glucose cotransporter-2 inhibitors, and their combination on endothelial glycocalyx, arterial function, and myocardial work index in patients with type 2 diabetes mellitus after 12-month treatment. J Am Heart Assoc. (2020) 9:e015716. 10.1161/JAHA.119.015716
165.
Li X Preckel B Hermanides J Hollmann MW Zuurbier CJ Weber NC et al . Amelioration of endothelial dysfunction by sodium glucose co-transporter 2 inhibitors: pieces of the puzzle explaining their cardiovascular protection. Br J Pharmacol. (2022) 179:4047–62. 10.1111/bph.15850
166.
Ceriello A Novials A Ortega E Canivell S La Sala L Pujadas G et al . Glucagon-like peptide 1 reduces endothelial dysfunction, inflammation, and oxidative stress induced by both hyperglycemia and hypoglycemia in type 1 diabetes. Diabetes Care. (2013) 36:2346–50. 10.2337/dc12-2469
167.
Syed MH Zamzam A Valencia J Khan H Jain S Singh KK et al . MicroRNA profile of patients with chronic limb-threatening ischemia. Diagnostics. (2020) 10:230. 10.3390/diagnostics10040230
168.
Ho CS Noor SM Nagoor NH . MiR-378 and MiR-1827 regulate tumor invasion, migration and angiogenesis in human lung adenocarcinoma by targeting RBX1 and CRKL, respectively. J Cancer. (2018) 9:331–45. 10.7150/jca.18188
169.
Caporali A Meloni M Völlenkle C Bonci D Sala-Newby GB Addis R et al . Deregulation of microRNA-503 contributes to diabetes mellitus-induced impairment of endothelial function and reparative angiogenesis after limb ischemia. Circulation. (2011) 123:282–91. 10.1161/CIRCULATIONAHA.110.952325
170.
Deng W Fan C Zhao Y Mao Y Li J Zhang Y et al . MicroRNA-130a regulates neurological deficit and angiogenesis in rats with ischaemic stroke by targeting XIAP. J Cell Mol Med. (2020) 24:10987–1000. 10.1111/jcmm.15732
171.
Liu H Xing A Chen X Ma R Wang Y Shi D et al . MicroRNA-27b, microRNA-101 and microRNA-128 inhibit angiogenesis by down-regulating vascular endothelial growth factor C expression in gastric cancers. Oncotarget. (2015) 6:37458–70. 10.18632/oncotarget.6059
172.
Zeng L He X Wang Y Tang Y Zheng C Cai H et al . MicroRNA-210 overexpression induces angiogenesis and neurogenesis in the normal adult mouse brain. Gene Ther. (2014) 21:37–43. 10.1038/gt.2013.55
173.
Li T Cao H Zhuang J Wan J Guan M Yu B et al . Identification of miR-130a, miR-27b and miR-210 as serum biomarkers for atherosclerosis obliterans. Clin Chim Acta. (2011) 412:66–70. 10.1016/j.cca.2010.09.029
174.
Vogiatzi G Oikonomou E Deftereos S Siasos G Tousoulis D . Peripheral artery disease: a micro-RNA-related condition?Curr Opin Pharmacol. (2018) 39:105–12. 10.1016/j.coph.2018.04.001
175.
Ring A Ismaeel A Wechsler M Fletcher E Papoutsi E Miserlis D et al . MicroRNAs in peripheral artery disease: potential biomarkers and pathophysiological mechanisms. Ther Adv Cardiovasc Dis. (2022) 16:17539447221096940. 10.1177/17539447221096940
176.
Golledge J Biros E Bingley J Iyer V Krishna SM . Epigenetics and peripheral artery disease. Curr Atheroscler Rep. (2016) 18:15. 10.1007/s11883-016-0567-4
177.
Vitkova V Zivny J Janota J . Endothelial cell-derived microvesicles: potential mediators and biomarkers of pathologic processes. Biomark Med. (2018) 12:161–75. 10.2217/bmm-2017-0182
178.
Paone S Baxter AA Hulett MD Poon IKH . Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell Mol Life Sci. (2019) 76:1093–106. 10.1007/s00018-018-2983-9
179.
Shu Z Tan J Miao Y Zhang Q . The role of microvesicles containing microRNAs in vascular endothelial dysfunction. J Cell Mol Med. (2019) 23:7933–45. 10.1111/jcmm.14716
180.
Jia L Fan J Cui W Liu S Li N Lau WB et al . Endothelial cell-derived microparticles from patients with obstructive sleep apnea hypoxia syndrome and coronary artery disease increase aortic endothelial cell dysfunction. Cell Physiol Biochem. (2017) 43:2562–70. 10.1159/000484508
181.
Song P Rudan D Zhu Y Fowkes FJI Rahimi K Fowkes FGR et al . Global, regional, and national prevalence and risk factors for peripheral artery disease in 2015: an updated systematic review and analysis. Lancet Glob Health. (2019) 7:e1020–30. 10.1016/S2214-109X(19)30255-4
182.
Hirsch AT Allison MA Gomes AS Corriere MA Duval S Ershow AG et al . A call to action: women and peripheral artery disease: a scientific statement from the American heart association. Circulation. (2012) 125:1449–72. 10.1161/CIR.0b013e31824c39ba
Summary
Keywords
peripheral artery disease (PAD), inflammation, therapeutics, endothelial cell dysfunction, thrombosis, angiogenesis, vascular tone
Citation
Kavurma MM, Bursill C, Stanley CP, Passam F, Cartland SP, Patel S, Loa J, Figtree GA, Golledge J, Aitken S and Robinson DA (2022) Endothelial cell dysfunction: Implications for the pathogenesis of peripheral artery disease. Front. Cardiovasc. Med. 9:1054576. doi: 10.3389/fcvm.2022.1054576
Received
27 September 2022
Accepted
24 October 2022
Published
16 November 2022
Volume
9 - 2022
Edited by
Zhao-Jun Liu, University of Miami, United States
Reviewed by
Hugo Ten Cate, Maastricht University Medical Centre, Netherlands; Rosa Suades, Sant Pau Institute for Biomedical Research, Spain
Updates
Copyright
© 2022 Kavurma, Bursill, Stanley, Passam, Cartland, Patel, Loa, Figtree, Golledge, Aitken and Robinson.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mary M. Kavurma mary.kavurma@hri.org.au
This article was submitted to Atherosclerosis and Vascular Medicine, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.