Abstract
Background:
Left ventricular noncompaction (LVNC) cardiomyopathy is a disorder that can be complicated by heart failure, arrhythmias, thromboembolism, and sudden cardiac death. The aim of this study is to clarify the genetic landscape of LVNC in a large cohort of well-phenotyped Russian patients with LVNC, including 48 families (n=214).
Methods:
All index patients underwent clinical examination and genetic analysis, as well as family members who agreed to participate in the clinical study and/or in the genetic testing. The genetic testing included next generation sequencing and genetic classification according to ACMG guidelines.
Results:
A total of 55 alleles of 54 pathogenic and likely pathogenic variants in 24 genes were identified, with the largest number in the MYH7 and TTN genes. A significant proportion of variants −8 of 54 (14.8%) −have not been described earlier in other populations and may be specific to LVNC patients in Russia. In LVNC patients, the presence of each subsequent variant is associated with increased odds of having more severe LVNC subtypes than isolated LVNC with preserved ejection fraction. The corresponding odds ratio is 2.77 (1.37 −7.37; p <0.001) per variant after adjustment for sex, age, and family.
Conclusion:
Overall, the genetic analysis of LVNC patients, accompanied by cardiomyopathy-related family history analysis, resulted in a high diagnostic yield of 89.6%. These results suggest that genetic screening should be applied to the diagnosis and prognosis of LVNC patients.
1. Introduction
Left ventricular noncompaction (LVNC) cardiomyopathy is characterized by the presence of a two-layer structure in the myocardium. Its main layer is represented by a compact myocardium, and the other layer is a spongy structure with multiple trabeculae (1). Patients are diagnosed with LVNC after an echocardiography (ECHO) or cardiac magnetic resonance (CMR) examination (2, 3). LVNC, due to the development of diagnostic methods and increased awareness of the disease, has become more often diagnosed in both children and adults (4). Due to the variety of clinical subtypes of the disease, the tendency to develop life-threatening arrhythmias, sudden cardiac death (SCD), heart failure, and the underestimation of the disease by many clinicians, LVNC is a disease that requires better clinical identification, as well as an understanding of pathology for the timely initiation of therapy (1). Initially, for the diagnosis of LVNC, the identification and description of trabeculations were most crucial, but later other features became important in defining specific LVNC subtypes (5). There are several subtypes of LVNC: isolated, dilated, hypertrophic, and restrictive. The genetic nature of the disease is detected in only half of the cases; however, as sequencing data accumulates, their number will grow (6). About 180 genes associated with LVNC, with varying levels of evidence, were reported (7). For heterogeneous disorders such as LVNC, a better understanding of its genetic background could improve outcome prediction and patient management. The aim of this study was to clarify the genetic landscape of LVNC in a large cohort of well-phenotyped Russian patients listed in the multicenter LVNC register (8).
2. Materials and methods
2.1. Selection of participants and clinical data
Index patients with LVNC and burdened family history of LVNC and their relatives were included in this study. The inclusion criteria were: the presence of LVNC in an index patient and at least one relative of 1st, 2nd, or 3rd degree with LVNC or other cardiomyopathies. All index patients underwent clinical examination and genetic analysis, as well as family members who agreed to participate in the clinical study and/or in the genetic testing. Clinical examination included general examination, electrocardiography using 24-h Holter monitoring electrocardiogram, CMR, ECHO, and blood sample collection for biochemical and genetic analyses. For ECHO and CMR imaging, the criteria of LVNC suggested by Jenni et al. (2) and Petersen et al. (3) were used. The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Boards of the National Medical Research Center for Therapy and Preventive Medicine (Moscow, Russia). Every participant and/or their legal representative gave their written informed consent to be involved in this study.
The patients were classified into the 7 subtypes of LVNC: (1) isolated LVNC with preserved ejection fraction (EF) of the left ventricular (LV), if they had normal LV dimensions without LV hypertrophy; (2) isolated LVNC with reduced EF of the LV, if the patient had normal LV dimensions with EF < 50%; (3) dilated LVNC if the patient had LV dilatation; (4) hypertrophic LVNC, if the patient also had LV wall hypertrophy of ≥13 mm; (5) hypertrophic dilated LVNC, if the patient had LV dilation and LV wall hypertrophy; (6) LVNC with congenital heart disease (CHD); (7) restrictive LVNC is characterized by left atrial or biatrial dilation and diastolic dysfunction.
For pedigrees creation, the CeGaT Pedigree Chart Designer v.3.0 (CeGaT GmbH, Tübingen, Germany) was used (9).
2.2. Echocardiography and cardiac magnetic resonance
ECHO was performed using the ultrasound system Philips IE33 (Philips Medical Systems, Eindhoven, Netherlands). In the parasternal position on the short axis at the end of the systole were evaluated the presence or absence of a two-layer structure of the myocardium, the presence of trabeculae and blood flow between them, and the ratio of the thickness of the noncompact layer to the thickness of the compact layer were evaluated more than twice (2).
A CMR was performed using a 1.5 T scanner (Avanto, Siemens Medical Solutions, Erlangen, Germany) with retrospective ECG-gating. Standard protocols consisted of breath-hold cine-imaging (SSFP) and late gadolinium enhancement (LGE) were implemented. We analyzed the results using CVI 42 software. LV end-diastolic volume (EDV), LV end-systolic volume (ESV), myocardial mass, and non-compacted myocardial mass were calculated as well as indexed values. Patterns of contrast enhancement were analyzed.
2.3. Genetic analysis
2.3.1. DNA extraction
The blood samples and buccal swabs were stored at −30°C and +4°C, respectively, at the Biobank of the National Medical Research Center for Therapy and Preventive Medicine (Moscow, Russia) (10). DNA was extracted from whole blood and, in one case, buccal swab samples using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). The DNA concentration was measured with a Qubit 4 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA).
2.3.2. NGS
Next generation sequencing (NGS) was performed with three platforms (HiSeq 1500, NextSeq 550, and Ion S5) for 141 patients, including 48 index patients (see Supplementary Table S1). Sanger sequencing was performed for other relatives. The list of studied genes is presented in Supplementary Table S2. All sequencing stages were carried out in accordance with the manufacturers' protocols.
2.3.2.1. Exome sequencing
Exome sequencing was done on two platforms, HiSeq 1500 and NextSeq 550 (Illumina, San Diego, CA, USA).
For the NextSeq 550 platform, exome libraries were prepared with the TruSeq DNA Library Preparation Kit (Illumina, San Diego, CA, USA) and the xGen Exome Research Panel (IDT, Integrated DNA Technologies, Coralville, IA, USA) according to the IDT-Illumina TruSeq DNA Exome protocol (Illumina, San Diego, CA, USA). Sequencing was done using NextSeq 550 (Illumina, San Diego, CA, USA) with paired-end sequencing (150 bp) (11, 12).
For the HiSeq 1500 platform, exome libraries were prepared using the Kapa Library Amplification Kit (Roche, Basel, Switzerland) and NimbleGen SeqCap EZ Exome v3.0 (Roche, Basel, Switzerland). Sequencing was performed on HiSeq 1500 (Illumina, San Diego, CA, USA) with paired-end sequencing (250 bp).
2.3.2.2. Custom panel sequencing
We developed a list of genes for analysis on the basis of information about association with cardiomyopathies published in the HPO, ClinGen, OMIM, and ClinVar databases (13–16) and literature (17–19). The intersection of this list with the lists of genes included in the panels used in this study is presented in Supplementary Table S2. In the course of exome sequencing, all 297 genes were analyzed. For the two custom panels, 137 and 200 genes, respectively, were analyzed.
For the Ion S5 platform, a custom panel was used. It included 137 genes (CDS + 10 bp padding) associated with LVNC or other cardiomyopathies (Supplementary Table S2) and was designed in the Ion AmpliSeq Designer software (Thermo Fisher Scientific, Waltham, MA, USA). The preparation of AmpliSeq libraries was done on the Ion Chef System (Thermo Fisher Scientific, Waltham, MA, USA), the 200 bp sequencing was performed on the Ion S5 (Thermo Fisher Scientific, Waltham, MA, USA) (20).
For the NextSeq 550 platform, a custom panel was used that included exon sequences of 200 (CDS + 25 bp padding) genes, associated with LVNC or other cardiomyopathies (Supplementary Table S2). The libraries were prepared with the SeqCap EZ Prime Choice Library Kit (Roche, Basel, Switzerland). Sequencing was performed on NextSeq 550 (Illumina, San Diego, CA, USA) with paired-end sequencing (150 or 300 bp) (21).
2.3.2.3. Bioinformatic analysis
For the NextSeq 550 and HiSeq 1500 platforms, the first step of sequencing analysis generated fastq files. The GRCh37 reference genome was chosen for the alignment of paired-end reads. We used the custom-designed pipeline based on GATK 3.8 (22) for data processing and quality control evaluation. ENSEMBL Variant Effect Predictor (23), ClinVar (2021/01/10) (16), gnomAD (v2.1.1) (24), and dbSNP (25) databases were applied for the annotation of single-nucleotide variants and short indels. PLINK v1.90 (26) was used to get identity by state (IBS) values and identity by descent (IBD) proportion to estimate relatedness for all pairs of individuals.
For the Ion S5 platform, sequencing and bioinformatic analysis resulted in bam files. We used the Torrent Server (Thermo Fisher Scientific, Waltham, MA, USA) with default parameters to obtain the vcf files. For the annotation of the vcf files Ion Reporter (Thermo Fisher Scientific, Waltham, MA, USA) with the Annotate Variants analysis tool was applied.
2.3.3. Clinical interpretation
Variants in cardiomyopathy-associated genes (Supplementary Table S2) with allele frequencies <0.005 or missing in the gnomAD database were analyzed (24). The pathogenicity evaluation of the variants was done according to the recommendations of the American College of Medical Genetics and Genomics (27). In this study, the following types of variants are included: pathogenic (P), likely pathogenic (LP), and variant of unknown significance (VUS). For all variants, their presence in the ClinVar database (16) was studied.
2.3.4. Sanger sequencing
Sanger sequencing was performed according to the manufacturer's protocol for the purpose of verifying NGS results. PCR-amplified fragments were purified with ExoSAP-IT (Affymetrix, Santa Clara, CA, USA) and then sequenced on the Applied Biosystem 3500 Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA, USA) using the ABI PRISM BigDye Terminator reagent kit v. 3.1 (Thermo Fisher Scientific, Waltham, MA, USA).
2.4. Statistical analysis
Statistical analysis was performed using R software (version 3.5.1). For the representation of continuous variables, median (Me) and interquartile range (Q1; Q3) were used; for categorical variables, absolute numbers and percentages were used. For comparison of continuous variables, the Mann–Whitney U test was performed, and the two-sided Fisher's exact test was used for categorical variables. Numbers of pathogenic variants in the groups of patients are presented as mean ± standard deviation. A comparison of the number of variants between groups of patients with different LVNC subtypes was carried out using linear and logistic regressions with mixed effects implemented in the lme4 package (28). We used sex and age as fixed effects and the family variable as a random intercept effect. We considered the differences statistically significant if the p-value was <0.05. The diagram was created using the ggplot2 package (29) and the Viridis palette (30).
2.5. Limitations of the study
Not all study participants underwent NGS genetic testing. NGS genetic testing was carried out using three panels, each including a different number of genes (from 137 to 297). Moreover, it is possible that age-dependent penetrance may play a role in the clinical picture of LVNC, and it can occur that more relatives will be affected in the future. Functional studies to confirm the causal relationship of variants in the VCL, SLC22A5, and FHOD3 genes with LVNC development have not been performed.
3. Results
3.1. Clinical features of LVNC index patients and their relatives
A total of 48 families with LVNC (48 index patients and 166 relatives) were included in the study. Clinical characteristics of the participants are presented in Table 1. Overall, a total of 121 people had manifestations of LVNC (n = 111) or other cardiomyopathies (n = 10). Children (persons under 18 years old) and male participants accounted for 29.9% and 48.6% of all participants, respectively. The proportion of men and children did not differ significantly between the groups of patients and healthy people. The group of patients was significantly younger than the group of healthy participants, 32 years vs. 38 years. The LVNC subtypes of the studied patients are shown in the diagram (Figure 1). In individuals with LVNC (n = 111), the most common subtypes were dilated and isolated LVNC with preserved EF (76.6% in total). DCM or HCM were diagnosed in 6 and 4 relatives without LVNC, respectively.
Figure 1
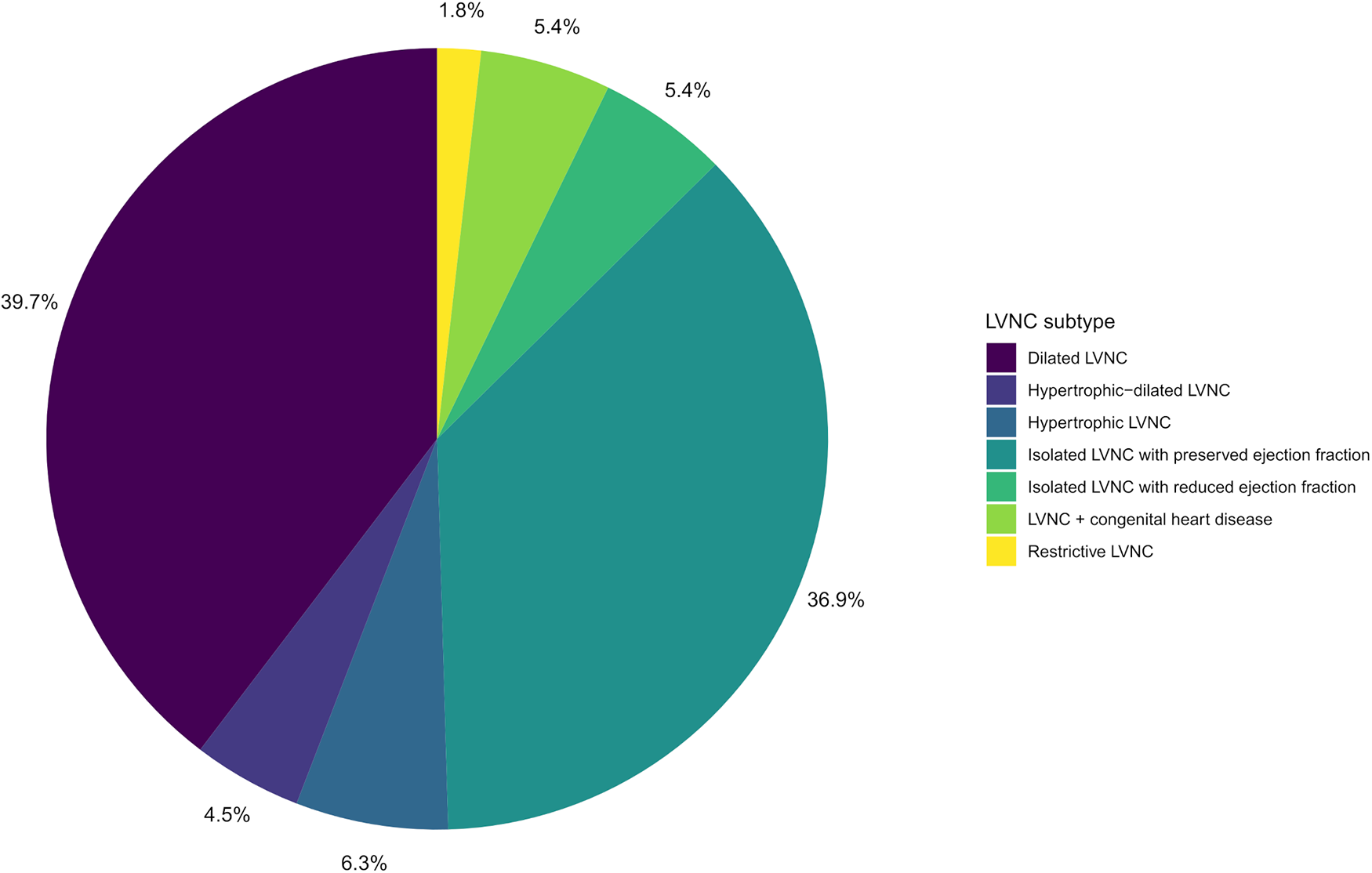
LVNC subtypes diagram of the studied patients.
Table 1
| Parameter | All participants (n = 214) | LVNC and cardiomyopathy patients (n = 121) | Healthy participants (n = 93) | p-value |
|---|---|---|---|---|
| Index patients, n | 48 | 48 | 0 | — |
| Relatives, n | 166 | 73 | 93 | — |
| Patients with LVNC subtypes, n (%) | 111 (51.9) | 111 (91.7) | 0 | — |
| Patients with other cardiomyopathies | 10 (4.7) | 10 (8.3) | 0 | — |
| Children, n (%) | 64 (29.9) | 38 (29.8) | 26 (28) | 0.65 |
| Men, n (%) | 104 (48.6) | 64 (52,9) | 40 (43) | 0.17 |
| Age, years, Me (25; 75) | 35.5 (14; 47.3) | 32 (13; 42) | 38 (17; 56.5) | 0.01 |
| BMI, kg/m2, >18 y.o., Me (25; 75) | 25.8 (22,2; 29.2) | 24.9 (21.6; 28.8) | 27.3 (23; 31.4) | 0.01 |
| EF LV, %, Me (25; 75) | 57 (45; 65) | 51 (38; 58.5) | 63 (60; 68.5) | <0.001 |
| EDV, ml, >18 y.o., Me (25;75) | 119.5 (99; 153.3) | 135.5 (112; 172.8) | 100.5 (87; 125.8) | <0.001 |
| EDD, ml, >18y.o., Me (25;75) | 51 (47; 55) | 53 (50; 58.8) | 48 (45; 51) | <0.001 |
| Nonsustained VT, n (%) | 30 (16) | 30 (26.1) | 0 | <0.001 |
| Sustained VT n (%) | 4 (2.3) | 4 (3.6) | 0 | 0.07 |
| Thromboembolic event, n (%) | 4 (2.1) | 4 (3.5) | 0 | 0.3 |
| Heart failure, n (%) | 80 (42.6) | 79 (68.7) | 1 (1.4) | <0.001 |
| Heart transplantation, n (%) | 6 (3.1) | 6 (5.2) | 0 | 0.08 |
| Neuromuscular diseases, n (%) | 9 (4.9) | 8 (7) | 1 (1.4) | 0.16 |
| Primary end point, n (%) | 14 (6.6) | 14 (11.7) | 0 | <0.001 |
| Death, n (%) | 8 (3.8) | 8 (6.7) | 0 | <0.001 |
| CVD death, n (%) | 6 (2.8) | 6 (5) | 0 | <0.001 |
| SCD, n (%) | 2 (0.9) | 2 (1.7) | 0 | <0.001 |
Clinical characteristics of the study participants.
BMI, body mass index; CVD, cardiovascular disease; EDD, end-diastolic diameter; EDV, end-diastolic volume; EF, ejection fraction; Me, median; SCD, sudden cardiac death; VT, ventricular tachycardia.
3.2. Genetic findings in LVNC index patients and their relatives
In 48 index patients, a total of 55 alleles of 54 P/LP variants have been identified in the genes associated with hereditary cardiomyopathies. Results are presented in Table 2, where they are grouped according to the classification given in a recent review (7). Corresponding separately published clinical cases are listed in the reference column. Only one variant was found in two unrelated families (TPM1, p.Ala242Val); each of the other variants was found in one family only. The largest number of variants was found in the MYH7 (27.3%, 15 variants, one of them de novo rs730880156) and TTN (14.5%, eight variants) genes. We have also identified pathogenic variants in the VCL and SLC22A5 genes that have a limited number of publications (31–33) confirming their association with LVNC but were not listed in the last review of the LVNC genes (6) (see Table 2, Supplementary Table S3). Moreover, we identified one pathogenic variant in the FHOD3 gene that was previously associated only with hypertrophic cardiomyopathy (HCM) (34–36). No P or LP variants were identified in the five families (10.4%). In 13 index patients, more than one variant was identified (27.1%). Pedigrees of index patients with more than one variant, clinical and genetic information are presented in Supplementary Table S3. In five index patients (10.4%), we additionally identified six rare variants classified as VUS. We suppose that they can modify the course of the disease (see Supplementary Table S4). A total of 14.8% (8 of 54) variants have not been described earlier in other populations and may be specific to LVNC patients in Russia: DSP:p.Gln948LysfsTer29, MYH7:p.Glu632Lys, MYH7:p.Glu497Lys, TTN:p.Gln23676HisfsTer16, ACTN2:p.Ile190Ser, ACTN2: p.Leu70del, ACTN2:p.Leu184Pro, SLC22A5:p.Asp388lfs*11.
Table 2
| Index patient | Gene | Pathogenicity | Variant (GRCh37) | dbSNP1 | Consequence1 | HGVSc1 | HGVSp1 | gnomADe AF (NFE) (24) | References2 |
|---|---|---|---|---|---|---|---|---|---|
| Definitive3 | |||||||||
| Fam017 | ACTC1 | LP | chr15:35084293A > G | rs397517071 | missense_variant, splice_region_variant | ENST00000290378.4:c.806T > C | ENSP00000290378.4:p.Ile269Thr | — | |
| Fam010 | DES | LP | chr2:220283520_220283528del | rs1553603239 | inframe_deletion | ENST00000373960.3:c.336_344del | ENSP00000363071.3:p.Gln113_Leu115del | — | (37, 38) |
| Fam023 | DES | LP | chr2:220285661G > C | rs59962885 | missense_variant | ENST00000373960.3:c.1009G > C | ENSP00000363071.3:p.Ala337Pro | — | (20) |
| Fam023 | DSP | LP | chr6:7580465del | — | frameshift_variant | ENST00000379802.3:c.4042del | ENSP00000369129.3:p.Leu1348Ter | — | (20) |
| Fam042 | DSP | LP | chr6:7580777A > T | — | stop_gained | ENST00000379802.3:c.4354A > T | ENSP00000369129.3:p.Arg1452Ter | — | (39) |
| Fam042 | DSP | LP | chr6:7581427dup | — | frameshift_variant | ENST00000379802.3:c.5004dup | ENSP00000369129.3:p.Leu1669ThrfsTer15 | — | (39) |
| Fam306 | DSP | P | chr6:7577240del* | — | frameshift_variant | ENST00000379802.3:c.2842del | ENSP00000369129.3:p.Gln948LysfsTer29 | — | |
| Fam031 | MIB1 | P | chr18:19378128C > A | rs748226232 | stop_gained | ENST00000261537.6:c.1176C > A | ENSP00000261537.6:p.Tyr392Ter | 0.000008803 | |
| Fam005 | MYBPC3 | P | chr11:47353740G > A | rs397516037 | stop_gained | ENST00000545968.1:c.3697C > T | ENSP00000442795.1:p.Gln1233Ter | 0.00001770 | (40) |
| Fam045 | MYBPC3 | P | chr11:47356592C > T | rs397515991 | splice_donor_variant | ENST00000545968.1:c.2905 + 1G > A | — | — | |
| Fam103 | MYBPC3 | LP | chr11:47363542C > T | rs727503195 | missense_variant, splice_region_variant | ENST00000545968.1:c.1790G > A | ENSP00000442795.1:p.Arg597Gln | 0 | |
| Fam001 | MYH7 | LP | chr14:23887575T > G | — | missense_variant | ENST00000355349.3:c.4013A > C | ENSP00000347507.3:p.His1338Pro | — | (41) |
| Fam002 | MYH7 | LP | chr14:23894202G > A | rs1064793206 | missense_variant | ENST00000355349.3:c.2455C > T | ENSP00000347507.3:p.Arg819Trp | — | |
| Fam003 | MYH7 | LP | chr14:23894584C > T | — | missense_variant | ENST00000355349.3:c.2330G > A | ENSP00000347507.3:p.Arg777Lys | — | (39) |
| Fam007 | MYH7 | LP | chr14:23899016C > T | rs397516089 | missense_variant | ENST00000355349.3:c.1106G > A | ENSP00000347507.3:p.Arg369Gln | — | |
| Fam018 | MYH7 | LP | chr14:23897054G > A | — | missense_variant | ENST00000355349.3:c.1628C > T | ENSP00000347507.3:p.Ala543Val | — | (39) |
| Fam022 | MYH7 | LP | chr14:23887513G > A | rs45451303 | missense_variant | ENST00000355349.3:c.4075C > T | ENSP00000347507.3:p.Arg1359Cys | 0.000008793 | |
| Fam025 | MYH7 | LP | chr14:23901007A > G | rs397516258 | missense_variant | ENST00000355349.3:c.602T > C | ENSP00000347507.3:p.Ile201Thr | — | |
| Fam027 | MYH7 | LP | chr14:23896511C > T* | — | missense_variant | ENST00000355349.3:c.1894G > A | ENSP00000347507.3:p.Glu632Lys | — | |
| Fam0314 | MYH7 | LP | chr14:23901077C > T | rs730880156 | missense_variant, splice_region_variant | ENST00000355349.3:c.532G > A | ENSP00000347507.3:p.Gly178Arg | 0 | |
| Fam034 | MYH7 | LP | chr14:23897798C > T* | — | missense_variant | ENST00000355349.3:c.1489G > A | ENSP00000347507.3:p.Glu497Lys | — | |
| Fam049 | MYH7 | LP | chr14:23888432G > A | — | missense_variant | ENST00000355349.3:c.3926C > T | ENSP00000347507.3:p.Thr1309Ile | — | (39) |
| Fam056 | MYH7 | LP | chr14:23900824C > G | — | missense_variant | ENST00000355349.3:c.702G > C | ENSP00000347507.3:p.Lys234Asn | — | |
| Fam062 | MYH7 | LP | chr14:23885272C > T | rs565663412 | missense_variant | ENST00000355349.3:c.4894G > A | ENSP00000347507.3:p.Ala1632Thr | 0.00002638 | (39) |
| Fam122 | MYH7 | LP | chr14:23900109C > T | rs111547156 | splice donor | NM_000257.4:c.895 + 1G > A | — | — | (42) |
| Fam135 | MYH7 | LP | chr14:23893175C > T | rs886039204 | missense_variant | ENST00000355349.3:c.2863G > A | ENSP00000347507.3:p.Asp955Asn | 0.000008790 | |
| Fam013, Fam242 | TPM1 | LP | chr15:63354797C > T | rs397516387 | missense_variant | ENST00000358278.3:c.725C > T | ENSP00000351022.3:p.Ala242Val | 0.000008792 | (43) |
| Fam009 | TTN | P | chr2:179406990C > G | rs727505319 | splice_donor_variant | ENST00000589042.1:c.97492 + 1G > C | — | — | |
| Fam017 | TTN | P | chr2:179463948G > A | rs745376275 | stop_gained | ENST00000589042.1:c.56572C > T | ENSP00000467141.1:p.Arg18858Ter | 0.00001110 | (39) |
| Fam019 | TTN | P | chr2:179477578G > A | rs1471414348 | stop_gained | ENST00000589042.1:c.49870C > T | ENSP00000467141.1:p.Arg16624Ter | — | (44) |
| Fam021 | TTN | P | chr2:179439830_179439830delinsG* | — | frameshift_variant | ENST00000589042.1:c.71028_71029delinsC | ENSP00000467141.1:p.Gln23676HisfsTer16 | — | |
| Fam022 | TTN | LP | chr2:179449453G > A | rs1432889079 | stop_gained | ENST00000589042.1:c.64915C > T | ENSP00000467141.1:p.Arg21639Ter | — | |
| Fam062 | TTN | P | chr2:179478953G > A | rs570046043 | stop_gained | ENST00000589042.1:c.49171C > T | ENSP00000467141.1:p.Arg16391Ter | — | |
| Fam070 | TTN | P | chr2:179604264G > A | rs775072385 | stop_gained | ENST00000589042.1:c.13696C > T | ENSP00000467141.1:p.Gln4566Ter | 0.000008909 | |
| Fam089 | TTN | LP | chr2:179397983del | rs760768093 | frameshift_variant | ENST00000589042.1:c.103360del | ENSP00000467141.1:p.Glu34454AsnfsTer3 | 0.00004446 | |
| Moderate3 | |||||||||
| Fam029 | ALPK3 | LP | chr15:85406781A > C | rs1963971731 | splice_acceptor_variant | ENST00000258888.6:c.4411-2A > C | — | — | |
| Fam004 | PRDM16 | P | chr1:3329197del | — | frameshift_variant | ENST00000270722.5:c.2436del | ENSP00000270722.5:p.Ala813ProfsTer58 | — | (45) |
| Fam006 | RBM20 | LP | chr10:112572068C > T | rs267607003 | missense_variant | ENST00000369519.3:c.1913C > T | ENSP00000358532.3:p.Pro638Leu | — | (46) |
| Fam033 | RBM20 | LP | chr10:112581114G > A | rs397516607 | missense_variant | ENST00000369519.3:c.2737G > A | ENSP00000358532.3:p.Glu913Lys | — | |
| Fam008 | TBX20 | LP | chr7:35271175_35271176dup | — | frameshift_variant | ENST00000408931.3:c.830_831dup | ENSP00000386170.3:p.Asp278Ter | — | (11, 39, 47) |
| Fam064 | TNNT2 | LP | chr1:201331109_201331111del | rs45578238 | inframe_deletion | ENST00000509001.1:c.629_631del | ENSP00000422031.1:p.Lys210del | — | |
| Limited3 | |||||||||
| Fam037 | ACTN2 | LP | chr1:236891010T > G* | — | missense_variant | ENST00000366578.4:c.569T > G | ENSP00000355537.4:p.Ile190Ser | — | |
| Fam131 | ACTN2 | LP | chr1:236881238_236881240del* | — | inframe_deletion | ENST00000366578.4:c.207_209del | ENSP00000355537.4:p.Leu70del | — | |
| Fam132 | ACTN2 | LP | chr1:236890992T > C* | — | missense_variant | ENST00000366578.4:c.551T > C | ENSP00000355537.4:p.Leu184Pro | — | |
| Fam008 | DSG2 | P | chr18:29111023C > A | rs751527714 | stop_gained | ENST00000261590.8:c.1088C > A | ENSP00000261590.8:p.Ser363Ter | 0 | (11, 39) |
| Fam024 | FBN2 | LP | chr5:127614327C > T | rs759198660 | missense_variant, splice_region_variant | ENST00000508053.1:c.7345G > A | ENSP00000424571.1:p.Asp2449Asn | 0.00003548 | |
| Fam011 | FLNC | LP | chr7:128481344A > C | rs1554398369 | missense_variant | ENST00000325888.8:c.1934A > C | ENSP00000327145.8:p.Asp645Ala | — | (48) |
| Fam031 | MTMR14 | LP | chr3:9695454G > A | rs1413765461 | splice_donor_variant | ENST00000296003.4:c.308 + 1G > A | — | 0 | |
| Fam003 | MYL2 | LP | chr12:111352058A > G | — | missense_variant | ENST00000228841.8:c.206T > C | ENSP00000228841.7:p.Met69Thr | — | (39) |
| Fam024 | PLEC | P | chr8:144994943_144994944del | rs782329610 | frameshift_variant | ENST00000322810.4:c.9458_9459del | ENSP00000323856.4:p.Val3153AlafsTer77 | 0 | |
| Fam245 | PRKAG2 | LP | chr7:151273498C > T | rs121908987 | missense_variant | ENST00000287878.4:c.905G > A | ENSP00000287878.3:p.Arg302Gln | — | |
| Genes, that may indicate an association with LVNC | |||||||||
| Fam026 | FHOD3 | P | chr18:34232893G > A | rs2036163874 | splice_donor_variant | ENST00000590592.6:c.1646 + 1G > A | — | — | (49) |
| Fam003 | VCL | P | chr10:75855578C > T | rs794729191 | stop_gained | ENST00000211998.4:c.1708C > T | ENSP00000211998.4:p.Arg570Ter | 0.000008796 | (39, 50) |
| Fam062 | SLC22A5 | P | chr5:131726419del* | — | frameshift_variant | ENST00000245407.3:c.1090del | ENSP00000245407.3:p.Asp364IlefsTer11 | — | |
List of the variants associated with LVNC found in the studied families.
dbSNP identifiers (database version 153), HGVS and consequences are assigned by ENSEMBL VEP.
References to the more detailed description of the clinical case.
According to the study (7).
de novo variant, the relationship between index patient and parents was confirmed.
The variants that have not been described earlier; AF, allele frequency; LP, likely pathogenic; NFE, non-finnish European; P, pathogenic.
Patients with isolated LVNC with preserved EF had 0.74 ± 0.59 causal variants vs. 1.16 ± 0.63 causal variants in patients with other LVNC subtypes. The difference in average numbers of causal variants is equal to 0.42 (0.17–0.66; p = 0.001) and decreases to 0.38 (0.18–0.57; p < 0.001) after adjustment for sex, age, and family. In patients with LVNC, the presence of each subsequent variant is associated with increased odds of having more severe LVNC subtypes than isolated LVNC with preserved EF. The corresponding odds ratio after adjustments is 3.59 (1.64–10.23; p = 0.001) per variant.
4. Discussion
The presented work is the largest study on LVNC genetics in Russia in terms of the number of included patients and is comparable with other studies in this field worldwide (6, 17, 51).
This study resulted in several remarkable findings. First of all, genetic analysis in 48 index patients with familial form of LVNC resulted in the diagnostic yield of nearly 90%: 10.4% of index patients had no LP/P variants, 62.5% had one LP/P variant, and 27.1% had more than one variant. This can be compared to other studies, where the percentage of index patients with the familial form of LVNC and no detected pathogenic variants ranged from 34% to 52% (6, 17, 52). In contrast, the absence of a family history of LVNC (sporadic cases) is usually associated with a low diagnostic yield – from 9% to 46% (17, 51). In our study, we used large gene panels containing 137, 200, and 297 genes, which was also the reason for the high diagnostic yield, apparently because it increases with the number of genes for which the analysis of variants is carried out (5, 53). Besides, 27.1% of the studied index patients had more than one variant, which is somewhat higher than in the other studies, e.g., 9.5% in Richard et al. (17) and 16.1% in Miszalski-Jamka et al. (31). This can also be explained by the number of genes analyzed.
Moreover, the high genetic heterogeneity of LVNC in Russia was shown: 54 variants were located in 24 genes, 8 (14.8%) of them were new, and only one variant was found in two unrelated families; each of the other variants was found in one family only. The heterogeneity of LVNC can be confirmed by other studies (5, 54). In our study, the most prevalent genes were MYH7 (27.3% of variants or 31.3% of index patients) and TTN (14.5% of variants or 16.7% of index patients), which is quite common for LVNC studies (17, 31, 52, 55). The next most prevalent genes were DSP, MYBPC3, ACTN2. Most of the 15 variants found in MYH7 were located in the myosin motor domain (53.3%, eight variants) and in the coiled coil region (33.3%, five variants) (56). Among the eight TTN variants, six (75%) were found in the A-band region, one (12.5%) in the I-band region, and one (12.5%) in the M-band region (57), which is similar to other studies (17). The fraction of 76.4% of all variants is located in genes from the “definitive” and “moderate” categories according to the Rojanasopondist et al. classification (7), which indicates the need to prioritize these genes for the genetic diagnostics. At the same time, given the great genetic heterogeneity of LVNC, it may be necessary to evaluate not only the genes from the “limited” category but also the genes associated with other monogenic cardiomyopathies (7).
Here we have described three pathogenic variants in the VCL, SLC22A5, and FHOD3 genes. These three cases are rather interesting because variants in other genes that have stronger evidence of association with LVNC did not show full family segregation. From our point of view, it is important to report and describe these cases, which can help clarify the association of these particular genes with LVNC in the future (see Table 2, Supplementary Table S3). The variants in VCL, SLC22A5, and FHOD3 have been shown earlier to be associated with HCM or dilated cardiomyopathies (DCM) (13–16, 34–36, 58–61). In the case of VCL and SLC22A5, there exist only three publications confirming their association with LVNC (31–33). These data support the overlap of genetic pathogenesis between the various cardiomyopathies.
The VCL gene codes for vinculin, a membrane-cytoskeletal actin-binding protein. In this study, one family with LVNC had a pathogenic VCL variant (p.Arg570Ter) in the index patient, her mother, and sister (ACMG criteria: PVS1, PM2, PP1), two LP variants—MYH7:p.Arg777Lys (PM1, PM2, PP2, PP3) and MYL2:p.Met69Thr (PM1, PM2, PP2, PP3)—were found in the index patient, her sister, and their healthy father (see Supplementary Table S3). We can only suggest that these LP variants can modify the course of the disease, because the mother of the index patient has a less severe LVNC subtype, but the variant that segregates in all family members with LVNC is in the VCL gene.
The SLC22A5 gene (also known as OCTN2) codes for the organic cation transporter novel 2 and is associated with the development of primary carnitine deficiency and cardiomyopathy (62, 63). Here we present a family with the index patient diagnosed with LVNC and three family members diagnosed with DCM (see Supplementary Table S3). Unfortunately, we did not have the opportunity to perform genetic testing for the index patient's husband and sister. Genetic analysis showed that the index patient with LVNC had the LP variant SLC22A5:p.D388lfs*11 (ACMG criteria: PVS1, PM2), and that the patient's daughter with DCM is homozygous for this variant. The index patient also had one LP variant MYH7:p.Ala1632Thr (PM1, PM2, PP2, PP3) and one P variant TTN:p.Arg16391Ter (PVS1, PM2, PP5).
The FHOD3 gene plays a role in the regulation of the actin cytoskeleton. The clinical description of the FHOD3 variant clinical case was published earlier (49). In this case, the CMR data allowed for an update of the diagnosis of the index patient and his sister from HCM to LVNC (49).
We believe that the expansion of our knowledge about new genes and variants associated with LVNC will help increase the effectiveness of genetic testing for this disease.
Another finding is related to the risk prediction of the LVNC subtype and associated risk of adverse events, which is important for counseling relatives of patients with LVNC. In this work, the most common LVNC subtypes were dilated LVNC (n = 44, 39.6%) and isolated LVNC with preserved EF (n = 41, 36.9%). These two subtypes seem to reflect the natural course of the disease and are stages in the same pathogenetic process. In the study by van Waning et al. (52), the share of isolated and dilated LVNC was even higher and amounted to 95%. In the work by Hirono et al., the proportion of these two subtypes was 67% (5).
Different subtypes of LVNC may occur in the same family, and the severity of the disease subtypes may be due to the presence of several variants in the patient (64, 65). Here, we have also shown that the presence of multiple pathogenic variants in one patient is accompanied by the presence of a more severe LVNC subtype. The presence of neuromuscular diseases can also influence the severity of LVNC. In this work, we observed several families with neuromuscular diseases (n = 6) who had severe LVNC with progressive heart failure, orthotopic heart transplantation, and fatal outcomes (in 2 cases). In the systematic review by Hirono and Ichida, it was noted that neuromuscular disorders were present in an average of 5% of LVNC patients, and apart from lower EF LV, concomitant neuromuscular disease and heart failure with LV dilation also led to poor prognosis and increased mortality (54). It is worth mentioning that in the group of patients with MYH7 variants, there were no deaths or orthotopic transplants, which is also consistent with the results of van Waning et al. (6). Besides, patients with CHD had LP variants in MYH7 or ACTN2, while in one of the previous studies only MYH7 variants were found in such patients (52). Another difference with the study by van Waning et al. is the presence of TTN variants among pediatric patients (see Table 2: Fam21, Fam70, and Fam89) (52).
In conclusion, we would like to summarize our findings. The genetic analysis in LVNC patients and the family history of LVNC have a high diagnostic yield. A total of 55 P or LP variants were identified in 44 index patients, with the largest number in the MYH7 and TTN genes. A total of 8 (14.8%) variants were new. Three pathogenic variants in the VCL, SLC22A5, and FHOD3 genes were identified, which may indicate an association with LVNC. Different LVNC subtypes may occur in the same family, and the severity of the phenotype may be due to the presence of several variants in the patient. These results support the idea that genetic screening should be applied to the diagnosis and prognosis of LVNC patients.
Statements
Data availability statement
The data presented in this study are available on request from the corresponding authors. Individual genotype information cannot be made available in order to protect participant privacy.
Ethics statement
The studies involving human participants were reviewed and approved by Ethics Committees in clinical cardiology of the National Research Center for Therapy and Preventive Medicine (a statement on ethics approval №06-21/17, 12 October 2017). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
Conceptualization: ANM, RPM, AVK, and OVK; methodology: ANM, RPM, AVK, and OVK; software and data analysis: AAZ, VER, MZ, YVV, and VAK; validation: AVK, EAS, MGD, and OPS; investigation: ANM, RPM, AVK, OVK, EAS, MMK, AAZ, SNK, EAM, VER, MZ, YVV, MSK, TGN, VES, MGD, VAK, ENB, VIB, NAS, OPS, IAE, and MSP; resources: ANM, RPM, AVK, OVK, VER, MSP, and OMD; data curation: ANM, RPM, AVK, OVK, AAZ, and VER; writing—original draft preparation: ANM, RPM, AVK, OVK, and EAS; writing—review and editing: ANM, RPM, AVK, OVK, VER, and VAK; visualization: AVK and EAS; supervision: ANM, RPM, and OMD; project administration: ANM and RPM; funding acquisition: ANM, RPM, VER, and OMD. All authors contributed to the article and approved the submitted version.
Acknowledgments
The authors are grateful to the patients and their families for their cooperation and support of our research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1205787/full#supplementary-material
References
1.
Stöllberger C Wegner C Finsterer J . Left ventricular hypertrabeculation/noncompaction, cardiac phenotype, and neuromuscular disorders. Herz. (2019) 44:659–65. 10.1007/s00059-018-4695-1
2.
Jenni R Oechslin E Schneider J Jost CA Kaufmann PA . Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart. (2001) 86:666–71. 10.1136/heart.86.6.666
3.
Petersen SE Selvanayagam JB Wiesmann F Robson MD Francis JM Anderson RH et al Left ventricular non-compaction: insights from cardiovascular magnetic resonance imaging. J Am Coll Cardiol. (2005) 46:101–5. 10.1016/j.jacc.2005.03.045
4.
Ichida F . Left ventricular noncompaction−risk stratification and genetic consideration. J. Cardiol. (2020) 75:1–9. 10.1016/j.jjcc.2019.09.011
5.
Hirono K Hata Y Miyao N Okabe M Takarada S Nakaoka H et al Increased burden of ion channel gene variants is related to distinct phenotypes in pediatric patients with left ventricular noncompaction. Circ Genom Precis Med. (2020) 13:e002940. 10.1161/CIRCGEN.119.002940
6.
van Waning JI Caliskan K Michels M Schinkel AF Hirsch A Dalinghaus M et al Cardiac phenotypes, genetics, and risks in familial noncompaction cardiomyopathy. J Am Coll Cardiol. (2019) 73:1601–11. 10.1016/j.jacc.2018.12.085
7.
Rojanasopondist P Nesheiwat L Piombo S Porter GA Jr Ren M Phoon CK . Genetic basis of left ventricular noncompaction. Circ Genom Precis Med. (2022) 15:e003517. 10.1161/CIRCGEN.121.003517
8.
Kulikova OV Myasnikov RP Mershina EA Pilus PS Koretskiy SN Meshkov AN et al Familial left ventricular noncompaction: phenotypes and clinical course. Results of the multicenter registry. Ter Arkh. (2021) 93:381–8. 10.26442/00403660.2021.04.200677
9.
CeGaT Pedigree Chart Designer Software. Available at:https://www.cegat.com/for-physicians/pedigree-chart-designer/(Accessed April 13, 2023).
10.
Anisimov SV Meshkov AN Glotov AS Borisova AL Balanovsky OP Belyaev VE et al National association of biobanks and biobanking specialists: new community for promoting biobanking ideas and projects in Russia. Biopreserv Biobank. (2021) 19:73–82. 10.1089/bio.2020.0049
11.
Myasnikov R Brodehl A Meshkov A Kulikova O Kiseleva A Pohl GM et al The double mutation DSG2-p. S363X and TBX20-p. D278X is associated with left ventricular non-compaction cardiomyopathy: case report. Int J Mol Sci. (2021) 22:6775. 10.3390/ijms22136775
12.
Brodehl A Meshkov A Myasnikov R Kiseleva A Kulikova O Klauke B et al Hemi-and homozygous loss-of-function mutations in dsg2 (desmoglein-2) cause recessive arrhythmogenic cardiomyopathy with an early onset. Int J Mol Sci. (2021) 22:3786. 10.3390/ijms22073786
13.
Köhler S Gargano M Matentzoglu N Carmody LC Lewis-Smith D Vasilevsky NA et al The human phenotype ontology in 2021. Nucleic Acids Res. (2021) 49:D1207–17. 10.1093/nar/gkaa1043
14.
Rehm HL Berg JS Brooks LD Bustamante CD Evans JP Landrum MJ et al Clingen—the clinical genome resource. N Engl J Med. (2015) 372:2235–42. 10.1056/NEJMsr1406261
15.
Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans institute of genetic medicine, Johns Hopkins university. Available at:https://omim.org/(Accessed April 13, 2023).
16.
Landrum MJ Lee JM Benson M Brown G Chao C Chitipiralla S et al Clinvar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. (2016) 44:D862–8. 10.1093/nar/gkv1222
17.
Richard P Ader F Roux M Donal E Eicher JC Aoutil N et al Targeted panel sequencing in adult patients with left ventricular non-compaction reveals a large genetic heterogeneity. Clin Genet. (2019) 95:356–67. 10.1111/cge.13484
18.
van Waning JI Moesker J Heijsman D Boersma E Majoor-Krakauer D . Systematic review of genotype-phenotype correlations in noncompaction cardiomyopathy. J Am Heart Assoc. (2019) 8:e012993. 10.1161/JAHA.119.012993
19.
Arbustini E Favalli V Narula N Serio A Grasso M . Left ventricular noncompaction: a distinct genetic cardiomyopathy?J Am Coll Cardiol. (2016) 68:949–66. 10.1016/j.jacc.2016.05.096
20.
Kulikova O Brodehl A Kiseleva A Myasnikov R Meshkov A Stanasiuk C et al The desmin (DES) mutation p. A337P is associated with left-ventricular non-compaction cardiomyopathy. Genes. (2021) 12:121. 10.3390/genes12010121
21.
Ramensky VE Ershova AI Zaicenoka M Kiseleva AV Zharikova AA Vyatkin YV et al Targeted sequencing of 242 clinically important genes in the Russian population from the ivanovo region. Front Genet. (2021) 12:709419. 10.3389/fgene.2021.709419
22.
Van der Auwera GA O’Connor BD . Genomics in the cloud: Using Docker, GATK, and WDL in Terra. Sebastopol, CA: O'Reilly Media (2020).
23.
McLaren W Gil L Hunt SE Riat HS Ritchie GR Thormann A et al The ensembl variant effect predictor. Genome Biol. (2016) 17:1–4. 10.1186/s13059-016-0974-4
24.
Karczewski KJ Francioli LC Tiao G Cummings BB Alföldi J Wang Q et al The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581:434–43. 10.1038/s41586-020-2308-7
25.
Sherry ST Ward MH Kholodov M Baker J Phan L Smigielski EM et al dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. (2001) 29:308–11. 10.1093/nar/29.1.308
26.
Chang CC Chow CC Tellier LC Vattikuti S Purcell SM Lee JJ . Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. (2015) 4:s13742-015. 10.1186/s13742-015-0047-8
27.
Richards S Aziz N Bale S Bick D Das S Gastier-Foster J et al Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–23. 10.1038/gim.2015.30
28.
Bates DM . Lme4: Mixed-Effects Modeling with R. New York: Springer (2010).
29.
Wickham H . GGPLOT2: elegant graphics for data analysis. New York: Springer-Verlag. R Package Version. (2016) 3(5).
30.
Garnier S Ross N Rudis R Camargo PA Sciaini M Scherer C . viridis—colorblind-friendly color maps for R. R Package Version 0.6. (2021). 10.5281/zenodo.4679424. Available online: https://zenodo.org/record/4679424#.ZGI1kHZBwuV (Accessed April 13, 2023)
31.
Miszalski-Jamka K Jefferies JL Mazur W Głowacki J Hu J Lazar M et al Novel genetic triggers and genotype–phenotype correlations in patients with left ventricular noncompaction. Circ Cardiovasc Genet. (2017) 10:e001763. 10.1161/CIRCGENETICS.117.001763
32.
Mokhtar A Mahrous N Elamin A Nawar M . Familial left ventricular noncompaction and conduction abnormalities. A case report with genetic mutation. J Clin Case Rep. (2017) 7:2. 10.4172/2165-7920.1000980
33.
Bhattacharya D Sasikumar D Kurup H Krishnamoorthy KM . Left ventricular noncompaction in primary systemic carnitine deficiency: a rare association. Ann Pediatr Cardiol. (2021) 14:521. 10.4103/apc.APC_152_20
34.
Ochoa JP Sabater-Molina M García-Pinilla JM Mogensen J Restrepo-Córdoba A Palomino-Doza J et al Formin homology 2 domain containing 3 (FHOD3) is a genetic basis for hypertrophic cardiomyopathy. J Am Coll Cardiol. (2018) 72:2457–67. 10.1016/j.jacc.2018.10.001
35.
Ochoa JP Lopes LR Perez-Barbeito M Cazón-Varela L de la Torre-Carpente MM Sonicheva-Paterson N et al Deletions of specific exons of FHOD3 detected by next-generation sequencing are associated with hypertrophic cardiomyopathy. Clin Genet. (2020) 98:86–90. 10.1111/cge.13759
36.
Wu G Ruan J Liu J Zhang C Kang L Wang J et al Variant spectrum of formin homology 2 domain-containing 3 gene in Chinese patients with hypertrophic cardiomyopathy. J Am Heart Assoc. (2021) 10:e018236. 10.1161/JAHA.120.018236
37.
Marakhonov AV Brodehl A Myasnikov RP Sparber PA Kiseleva AV Kulikova OV et al Noncompaction cardiomyopathy is caused by a novel in-frame desmin (DES) deletion mutation within the 1A coiled-coil rod segment leading to a severe filament assembly defect. Hum Mutat. (2019) 406:734–41. 10.1002/humu.23747
38.
Myasnikov RP Shcherbakova NV Kulikova ОV Meshkov АN Kharlap MS Kiseleva АV et al DES Gene mutation in a family of proband with myofibrillary myopathy and non-compaction cardiomyopathy resulted in cardiac transplantation. Russ J Cardiol. (2017) 10:9–16. 10.15829/1560-4071-2017-10-9-16
39.
Savostyanov KV Namazova-Baranova LS Basargina EN Vashakmadze ND Zhurkova NV Pushkov AA et al The new genome variants in Russian children with genetically determined cardiomyopathies revealed with massive parallel sequencing. Ann Russ Acad Med Sci. (2017) 72:242–53. 10.15690/vramn872
40.
Myasnikov RP Kulikova OV Meshkov AN Mershina EA Kiseleva AV Klimushina MV et al The combination of left ventricular non-compaction and hypertrophic cardiomyopathy in one family with a pathogenic variant in the MYBPC3 gene (rs397516037). Russ J Cardiol. (2020) 25(10):4115. 10.15829/1560-4071-2020-4115
41.
Myasnikov RP Kulikova OV Meshkov AN Kiseleva AV Shumarina AO Koretskiy SN et al New variant of MYH7 gene nucleotide sequence in familial non-compaction cardiomyopathy with benign course. Ration Pharmacother Cardiol. (2020) 16:383–91. 10.20996/1819-6446-2020-06-01
42.
Myasnikov RP Kulikova OV Meshkov AN Bukaeva AA Kiseleva AV Ershova AI et al A splice variant of the MYH7 gene is causative in a family with isolated left ventricular noncompaction cardiomyopathy. Genes. (2022) 13:1750. 10.3390/genes13101750
43.
Kudryavtseva MM Myasnikov RP Kiseleva AV Kulikova OV Meshkov AN Mershina EA et al Nucleotide sequence variant of the TPM1 gene in a family with different phenotypes of left ventricular noncompaction cardiomyopathy. Cardiovasc Ther Prev. (2023) 21:3471. 10.15829/1728-8800-2022-3471
44.
Kulikova OV Meshkov AN Myasnikov RP Kiseleva AV Koretsky SN Zharikova AA et al Pathogenic variant rs1471414348of the TTN gene in the patient with familial left venticular noncompaction cardiomyopathy. Ration Pharmacother Cardiol. (2019) 15:524–9. 10.20996/1819-6446-2019-15-4-524-529
45.
Myasnikov RP Bukaeva AA Kulikova OV Ershova AI Petukhova AV Zotova ED et al New variant of PRDM16 gene nucleotide sequence in a family with various phenotypic manifestations of the non-compacted myocardium. Russ J Cardiol. (2021) 26:4315. 10.15829/1560-4071-2021-4315
46.
Kulikova OV Myasnikov RP Meshkov AN Mershina EA Kiseleva AV Sotnikova EA et al Nucleotide sequence variant of the RBM20 gene in a family with a dilated phenotype of left ventricular noncompaction cardiomyopathy. Cardiovasc Ther Prev. (2023) 21:3470. 10.15829/1728-8800-2022-3470
47.
Savostyanov KV Basargina EN Ryabova EE Pushkov AA Zhanin IS Basargina EY et al Molecular genetic features of the development of restrictive cardiomyopathy in Russian children. Russ J Cardiol. (2021) 26:4590. 10.15829/1560-4071-2021-4590
48.
Kulikova OV Myasnikov RP Meshkov AN Kudryavtseva MM Mershina EA Kiseleva AV et al Variant of the FLNC gene nucleotide sequence in a family with different phenotypic manifestations of left ventricular non-compaction. Russ J Cardiol. (2021) 10:79–85. 10.15829/1560-4071-2021-4748
49.
Myasnikov R Bukaeva A Kulikova O Meshkov A Kiseleva A Ershova A et al A case of severe left-ventricular noncompaction associated with splicing altering variant in the FHOD3 gene. Genes. (2022) 13:309. 10.3390/genes13020309
50.
Meshkov AN Kulikova OV Myasnikov RP Shcherbakova NV Zharikova AA Koretsky SN et al Exome sequencing in Russian families with noncompaction cardiomyopathy. Proceedings of the Eur J Hum Genet; 2019 Jul 1; Nature publishing group (2019). p. 151–52).
51.
Ross SB Singer ES Driscoll E Nowak N Yeates L Puranik R et al Genetic architecture of left ventricular noncompaction in adults. Hum Genome Var. (2020) 7:33. 10.1038/s41439-020-00120-y
52.
van Waning JI Caliskan K Hoedemaekers YM van Spaendonck-Zwarts KY Baas AF Boekholdt SM et al Genetics, clinical features, and long-term outcome of noncompaction cardiomyopathy. J Am Coll Cardiol. (2018) 71:711–22. 10.1016/j.jacc.2017.12.019
53.
van Waning JI Caliskan K Chelu RG van der Velde N Pezzato A Michels M et al Diagnostic cardiovascular magnetic resonance imaging criteria in noncompaction cardiomyopathy and the yield of genetic testing. Can J Cardiol. (2021) 37:433–42. 10.1016/j.cjca.2020.05.021
54.
Hirono K Ichida F . Left ventricular noncompaction: a disorder with genotypic and phenotypic heterogeneity—a narrative review. Cardiovasc Diagn Ther. (2022) 12:495. 10.21037/cdt-22-198
55.
Sedaghat-Hamedani F Haas J Zhu F Geier C Kayvanpour E Liss M et al Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur Heart J. (2017) 38:3449–60. 10.1093/eurheartj/ehx545
56.
MYH7 - Myosin-7 - Homo sapiens (Human) | UniProtKB | UniProt. Available at: https://www.uniprot.org/uniprotkb/P12883/entry (Accessed April 13, 2023).
57.
Titin variation in dilated cardiomyopathy - cardiovascular genetics & genomics, imperial college London. Available at: https://www.cardiodb.org/titin/ (Accessed April 13, 2023).
58.
Kilic MU Özgül RK Coşkun TU Yücel D Karaca M Sivri HS et al Identification of mutations and evaluation of cardiomyopathy in Turkish patients with primary carnitine deficiency. JIMD Reports-Case and Research Reports, 2011/3. (2012) 3:17–23. 10.1007/8904_2011_36
59.
Lahrouchi N Lodder EM Mansouri M Tadros R Zniber L Adadi N et al Exome sequencing identifies primary carnitine deficiency in a family with cardiomyopathy and sudden death. Eur J Hum Genet. (2017) 25:783–7. 10.1038/ejhg.2017.22
60.
Papadopoulou-Legbelou K Gogou M Dokousli V Eboriadou M Evangeliou A . Dilated cardiomyopathy as the only clinical manifestation of carnitine transporter deficiency. Indian J Pediatr. (2017) 84:231–3. 10.1007/s12098-016-2250-8
61.
Yamak AA Bitar F Karam P Nemer G . Exclusive cardiac dysfunction in familial primary carnitine deficiency cases: a genotype–phenotype correlation. Clin Genet. (2007) 72:59–62. 10.1111/j.1399-0004.2007.00814.x
62.
Yang XF Liu GS Yi B . Primary carnitine deficiency in two sisters with intractable epilepsy and reversible metabolic cardiomyopathy: two case reports. Exp Ther Med. (2020) 20:1. 10.3892/etm.2020.9246
63.
Grünert SC Tucci S Schumann A Schwendt M Gramer G Hoffmann GF et al Primary carnitine deficiency–diagnosis after heart transplantation: better late than never!. Orphanet J Rare Dis. (2020) 15:1–6. 10.1186/s13023-020-01371-2
64.
Liu S Xie Y Zhang H Feng Z Huang J Huang J et al Multiple genetic variants in adolescent patients with left ventricular noncompaction cardiomyopathy. Int J Cardiol. (2020) 302:117–23. 10.1016/j.ijcard.2019.12.001
65.
Paluszkiewicz J Milting H Kałużna-Oleksy M Pyda M Janus M Körperich H et al Left ventricular non-compaction cardiomyopathy-still more questions than answers. J Clin Med. (2022) 11:4135. 10.3390/jcm11144135
Summary
Keywords
LVNC, left ventricular noncompaction cardiomyopathy, genetic screening, family form, MYH7 , TTN
Citation
Meshkov AN, Myasnikov RP, Kiseleva AV, Kulikova OV, Sotnikova EA, Kudryavtseva MM, Zharikova AA, Koretskiy SN, Mershina EA, Ramensky VE, Zaicenoka M, Vyatkin YV, Kharlap MS, Nikityuk TG, Sinitsyn VE, Divashuk MG, Kutsenko VA, Basargina EN, Barskiy VI, Sdvigova NA, Skirko OP, Efimova IA, Pokrovskaya MS and Drapkina OM (2023) Genetic landscape in Russian patients with familial left ventricular noncompaction. Front. Cardiovasc. Med. 10:1205787. doi: 10.3389/fcvm.2023.1205787
Received
14 April 2023
Accepted
09 May 2023
Published
24 May 2023
Volume
10 - 2023
Edited by
Keiichi Hirono, University of Toyama, Japan
Reviewed by
Sun Chen, Shanghai Jiao Tong University, China Luiza Guilherme, University of São Paulo, Brazil
Updates
Copyright
© 2023 Meshkov, Myasnikov, Kiseleva, Kulikova, Sotnikova, Kudryavtseva, Zharikova, Koretskiy, Mershina, Ramensky, Zaicenoka, Vyatkin, Kharlap, Nikityuk, Sinitsyn, Divashuk, Kutsenko, Basargina, Barskiy, Sdvigova, Skirko, Efimova, Pokrovskaya and Drapkina.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Alexey N. Meshkov meshkov@lipidclinic.ru Anna V. Kiseleva sanyutabe@gmail.com
†These authors have contributed equally to this work and share first authorship
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.