Abstract
Introduction:
It has been found that programmed cell death protein-1 (PD-1) or its ligand PD-L1 may play an important role in the onset and progression of coronary heart disease (CHD). Thus, we conducted this mendelian randomization analysis (MR) to estimate the causal relationship between PD-1/PD-L1 and 5 specific CHDs (chronic ischemic heart disease, acute myocardial infarction, angina pectoris, coronary atherosclerosis, and unstable angina pectoris), complemented by gene set enrichment analysis (GSEA) for further validation.
Methods:
Publicly available summary-level data were attained from the UK Biobank with genetic instruments obtained from the largest available, nonoverlapping genome-wide association studies (GWAS). Our analysis involved various approaches including inverse variance-weighted meta-analysis, alternative techniques like weighted median, MR-Egger, MR-multipotency residuals and outliers detection (PRESSO), along with multiple sensitivity assessments such as MR-Egger intercept test, Cochran's Q test, and leave-one-out sensitivity analysis to evaluate and exclude any anomalies.
Results:
Gene expression profile (GSE71226) was obtained from Gene Expression Omnibus (GEO) database for GSEA. IVW analysis showed a causal association between PD-1 and chronic ischemic heart disease (OR, 0.997; 95%CI, 0.995-0.999; P, 0.009), chronic ischemic heart disease and PD-1 (beta, −3.1; 95%CI, −6.017 to −0.183; P, 0.037), chronic ischemic heart disease and PD-L1 (beta, −3.269; 95%CI, −6.197 to −0.341; P, 0.029). No significant causal relationship was found between PD-1/PD-L1 and other 4 CHDs. The accuracy and robustness of these findings were confirmed by sensitivity tests. GSEA found that the KEGG pathway and related core genes of “PD-L1 expression and PD-1 checkpoint pathway in cancer” pathway were downregulated in CHD.
Discussion:
This study provided evidence of a bidirectional causal relationship between PD-1 and chronic ischemic heart disease and a protective association between chronic ischemic heart disease and PD-L1.
1 Introduction
Coronary heart disease (CHD) remains the leading cause of death worldwide (1). According to statistics released by the World Health Organization (WHO), CHD accounts for 16% of total deaths in the world, ranking as the foremost cause of death of the top 10 most deadly diseases globally. Since 2000, the number of deaths CHD deaths has increased significantly, with 2 million more deaths in 2019, totaling 8.9 million (2).
The basic pathological basis for the development of CHD is coronary atherosclerosis formation and plaque deposition, where inflammation and immune response have been shown to play an important role (3, 4). Previous studies have focused on cancer patients, claiming that PD-1/PD-L1 inhibitors accelerate atherosclerosis in cancer patients (5, 6). The role of PD-1/PD-L1 in the development of CHD in the general population needs to be further investigated. Some studies in mice have shown that low expression of PD-1/PD-L1 or anti-PD-1/PD-L1 treatment exacerbated atherosclerotic plaque formation and accelerated the immune process of CHD (7, 8). Clinical studies in patients with atherosclerosis have shown that abnormal PD-1/PD-L1 expression is associated with CHD (9, 10). Nevertheless, no trustworthy evidence exists to support the therapeutic effect of either upregulating or inhibiting the PD-1/PD-L1 pathway in CHD patients (11). Considering the limitations of different immune backgrounds, small sample sizes, single centers, and other confounding factors, previous studies can only suggest an association of PD-1/PD-L1 deficiency or low expression with CHD, rather than establishing a definitive causal relationship between PD-1/PD-L1 and CHD.
Mendelian randomization (MR) is an important method widely used in epidemiology for the assessment of potential causal relationships between exposure factors and clinical disease. The independent segregation of alleles at conception means that they are not affected by potential confounders, forming a natural experiment similar to a randomized trial, and thus Mendelian randomization analyses may provide more reliable insights into the potential causal relationship between PD-1/PD-L1 and CHD than traditional observational analyses (12–14). Gene set enrichment analysis (GSEA) is a powerful tool to associate a disease phenotype to a group of genes/proteins. Therefore, we conducted a mendelian randomized study to explore the causal relationship between PD-1/PD-L1 and five specific types of CHD (chronic ischemic heart disease, acute myocardial infarction, angina pectoris, coronary atherosclerosis, and unstable angina pectoris), complemented by GSEA for further validation.
2 Materials and methods
2.1 Study design
MR uses genetic instrumental variables (IVs) to evaluate the causal relationship between exposure and outcome. The basic principle of MR design is that genetic variations are fixed at conception and randomly assigned to individuals. Thus, it can overcome problems of unmeasured confounding and reverse causation typical of conventional observational epidemiology. In two sample MR, it combines data from multiple sources and uses two different research samples to estimate instrumental risk factors and instrumental outcome associations.
MR should be performed under three basic assumptions: (1) genetic variation is strongly associated with exposure; (2) genetic variation is independent of any potential confounding factors; and (3) genetic variation is independent of outcome except by means of exposure.
The design of our study had three key components: (1) the identification of genetic variants to serve as IVs; (2) the estimation of overall causal effect utilizing two sample, multivariable and bidirectional MR strategy; (3) the assessment of horizontal pleiotropy and validation of results conducting sensitivity analysis.
We further performed Gene Set Enrichment Analysis (GSEA) to further consolidate the causal relationship detected, checking whether CHD is associated with genes related to PD-1/PD-L1 pathway.
All relevant data we used are publicly downloadable on the website. All of these data are anonymous, freely downloadable, and can be used without restriction.
2.2 Data source
2.2.1 Data for exposure
The exposure of this study is defined as the plasma level of PD-1 and PD-L1. We used the summary-level data source from a genome-wide test of 10.6 million imputed autosomal variants against levels of 2,994 plasma proteins in 3,301 individuals of European descent, whose number of SNP was 10,534,735 (GWAS ID: prot-a-2214 and prot-a-431). This GWAS employed a complex method using special aptamers to measure the levels of 3,622 plasma proteins or protein complexes with 4,034 modified aptamers. The assay enhances the detection threshold for protein abundance beyond what traditional methods, such as immunoassays, typically allow. It is capable of measuring both extracellular and intracellular proteins, including the soluble domains of proteins associated with membranes (15). Detailed information is shown in Supplementary Table S1.
2.2.2 Data for outcome (Supplementary Table S1)
The outcome is defined as 5 self-reported doctor-diagnosed CHD: chronic ischemic heart disease (ICD10: I25), acute myocardial infarction (ICD10: I21), angina pectoris (ICD10: I20), coronary atherosclerosis, unstable angina pectoris. The GWAS summary statistics of chronic ischemic heart disease, acute myocardial infarction and angina pectoris were extracted from Neale lab analysis of UK Biobank phenotypes, round 1 (http://www.nealelab.is/blog/2017/7/19/rapid-gwas-of-thousands-of-phenotypes-for-337000-samples-in-the-uk-biobank). In particular, The GWAS summary statistics of chronic ischemic heart disease (GWAS ID: ukb-a-534) contained 8,755 cases and 328,444 controls; The GWAS summary statistics of acute myocardial infarction (GWAS ID: ukb-a-533) included 3,927 cases and 333,272 controls; The GWAS summary statistics of angina pectoris (GWAS ID: ukb-a-532) included 4,837 cases and 332,362 controls. Meanwhile, we extracted data on associations of relevant SNPs with coronary atherosclerosis and unstable angina pectoris from the Neale lab analysis of UK Biobank phenotypes, round 2 (http://www.nealelab.is/uk-biobank/). The GWAS summary statistics of coronary atherosclerosis (GWAS ID: ukb-d-I9_CORATHER) was based on 361,194 samples with 13,586,589 SNPs, including 14,334 cases and 346,860 controls. The GWAS summary statistics of unstable angina pectoris (GWAS ID: ukb-d-I9_UAP) were derived from 361,194 samples with 11,385,655 SNPs, including 3,439 cases and 357,755 controls.
2.2.3 Data for GSEA
Expression data set (GSE71226) was obtained from Gene Expression Omnibus (GEO) database, including 3 patients with coronary heart disease and 3 healthy people. Total RNA of each samples were extracted from peripheral blood to hybridize with Affymetrix microarrays (https://www.ncbi.nlm.nih.gov/geo/).
2.3 Selection of instrumental variables (IVs)
The effect allele for each SNP was defined as the allele associated with increased level of the relevant exposure. The genetic IVs were obtained based on the following three criteria: (1)For standard Mendelian randomization, single-nucleotide polymorphisms (SNPs) associated with the exposure, as well as the reverse study, were selected at the genome-wide significance level (p ≤ 5 × 10–6); (2)SNPs were clumped based on linkage disequilibrium (r2 < 0.001 and kB = 10,000); (3)Duplicate SNPs and palindromic SNPs were removed.
We also calculated the conditional F statistic to characterize instrument strengths. For a single variant, the F statistic is equal to the square of the genetic association with the exposure divided by the square of its standard deviation, calculated by the formula F = βi2/se(βi)2 and a value ≥ 10 was considered sufficient (16). All selected SNP were with F statistic ≥10.
2.4 Statistical analysis
2.4.1 Univariable MR
Inverse-variance weight (IVW) were the principal analyses for our study as it is the most efficient analysis method with valid instrumental variables, accounting for heterogeneity in the variant-specific casual estimates. This method provided a high-powered estimate and relied on the assumption that all SNPs were valid genetic instruments (17).
For sensitivity analysis, we performed multiple methods including Mendelian randomization-Egger (MR-Egger), Robust adjusted profile score (RAPS), the weighted median approach, the Mendelian Randomization Pleiotropy RESidual Sum and Outlier (MR-PRESSO), Cochran's Q statistic, leave-one-SNP-out analysis, and horizontal pleiotropy analysis.
The MR-Egger method offers a relatively stable estimate, independent of the instrumental variables’ validity, and adjusts for potential horizontal pleiotropy through regression slope and intercept adjustments (17). We calculated the intercept of MR-Egger to provide a measurement of horizontal pleiotropy, confirming if the variant has a direct effect on the target outcome (18).
Robust adjusted profile score (RAPS) is a recently recommended method which is robust to both systematic and idiosyncratic pleiotropy and can give a robust inference for MR analysis with many weak instruments (19).
The weighted median approach gives consistent estimates of the causal effect under the assumption that genetic variants representing over 50% of the weight in the analysis are valid instruments. The method is provided to ensure the median of all the instrumental variable estimates based on the individual genetic variants will be a consistent estimate (20).
MR-PRESSO method was used to test, and correct, if needed, for possible horizontal pleiotropic outliers in the analysis by removing SNPs that contribute to the heterogeneity disproportionately more than expected (18).
Cochran's Q statistic is a statistical test for heterogeneity which is derived from the IVW estimate, which follows ×2 distribution with degrees of freedom equal to the number of SNPs minus 1.
Leave-one-SNP-out analysis may provide us a dominant estimate of the casual effect when there is one genetic variant that is particularly strongly associated with the exposure.
2.4.2 Multivariable MR
The Multivariable Mendelian Randomization technique is suitable for employing numerous genetic instruments irrespective of their linkage to the exposure (21). While the instrumental variables might be linked to multiple risk factors, they must meet the equivalent instrumental-variable assumptions. Consequently, we utilized this approach, incorporating all instrumental variables pertaining to PD-1 and PD-L1, to ascertain their distinct impacts on CHD, employing IVW as our primary analytical method and MR-Egger as a sensitivity analysis.
2.4.3 Bidirectional MR
We additionally performed Bidirectional Mendelian randomization to assesses the effect of the outcome on the exposure (22), aiming to clarify whether the presence of CHD has a negative effect on PD-1/PD-L1, or whether the correlation between the two is due to latent confounding. The primary analyses for our bidirectional MR were IVW with multiple methods (MR-Egger, RAPS, the weighted median approach, MR-PRESSO, Cochran's Q statistic, leave-one-SNP-out analysis, horizontal pleiotropy analysis) performed as sensitivity analysis.
2.4.4 GSEA analysis
Initially, the Limma R package was employed to determine the log2(fold change) and P values for the expression data set. Subsequently, using clusterProfiler and org.Hs.eg.db R package, Gene Set Enrichment Analysis (GSEA) was performed to identify the core differentially expressed genes (DEGs), utilizing the KEGG gene set as the predefined set for enrichment analysis. Criteria for selecting significant enrichment pathways associated with coronary disease traits were established at a P-value of less than 0.05 and a false discovery rate (FDR) q value of less than 0.25.
3 Results
3.1 Univariable MR
The number of SNPs selected for PD-1 or PD-L1 ranged from 6 to 13. All SNPs selected for inclusion in univariable MR analysis are presented in Supplementary Table S2–S11. The IVW analysis found clear evidence of a protective causal effect of PD-1 on chronic ischemic heart disease (OR, 0.997; 95%CI, 0.995–0.999; P, 0.009), which was replicated by RAPS and WM (Table 1). MR-Egger analysis also showed a protective but not significant effect of PD-L1 on the risk of myocarditis (Table 1). In addition, there was no evidence in favour of an association between PD-1 and other four types of CHD, including acute myocardial infarction, angina pectoris, unstable angina pectoris, and coronary atherosclerosis (Table 1). We found no statistically significant effect of PD-L1 on the 5 types of CHD mentioned above (Table 2). No outlier between PD-1/PD-L1 and the risk of myocarditis was identified by the MR-PRESSO test (Tables 1 and 2). No directional pleiotropy was found in the MR-Egger regression (Tables 1 and 2). No heterogeneity, except for estimates of effect of PD-1 and PD-L1 on coronary atherosclerosis, was found by Cochran's Q statistic (Tables 1 and 2). The scatter plot and leave-one-out plot for PD-1/PD-L1's effects on the 5 CHD mentioned above are available in Figure 1, Supplementary Figures S1, S2, S7, S8.
Table 1
| Outcome | N SNP | Methods | OR (95% CI) | P-value | Q-statistics | Ph | Pp |
|---|---|---|---|---|---|---|---|
| Chronic ischemic heart disease | 12 | IVW | 0.997 (0.995–0.999) | 0.009* | 16.414 | 0.13 | |
| MR Egger | 0.997 (0.992–1.001) | 0.18 | 16.326 | 0.09 | 0.82 | ||
| RAPS | 0.998 (0.996–0.999) | 0.009* | |||||
| Weighted median | 0.998 (0.995–0.999) | 0.043* | |||||
| MR Presso | NA | NA | |||||
| Acute myocardial infarction | 12 | IVW | 1.000 (0.998–1.001) | 0.83 | 19.007 | 0.06 | |
| MR Egger | 1.001 (0.998–1.004) | 0.49 | 17.428 | 0.07 | 0.36 | ||
| RAPS | 1.000 (0.999–1.002) | 0.82 | |||||
| Weighted median | 1.001 (0.999–1.002) | 0.49 | |||||
| MR Presso | NA | NA | |||||
| Angina pectoris | 12 | IVW | 1.000 (0.999–1.001) | 0.73 | 6.504 | 0.84 | |
| MR Egger | 0.999 (0.997–1.002) | 0.67 | 6.024 | 0.81 | 0.50 | ||
| RAPS | 1.000 (0.999–1.002) | 0.74 | |||||
| Weighted median | 1.000 (0.998–1.002) | 0.94 | |||||
| MR Presso | NA | NA | |||||
| Unstable angina pectoris | 13 | IVW | 1.000 (0.999–1.001) | 0.52 | 10.854 | 0.54 | |
| MR Egger | 1.000 (0.998–1.002) | 0.87 | 10.574 | 0.48 | 0.61 | ||
| RAPS | 1.000 (0.999–1.001) | 0.46 | |||||
| Weighted median | 1.000 (0.999–1.002) | 0.69 | |||||
| MR Presso | NA | NA | |||||
| Coronary atherosclerosis | 13 | IVW | 0.998 (0.995–1.001) | 0.19 | 29.53 | 0.003 | |
| MR Egger | 0.999 (0.993–1.006) | 0.82 | 29.103 | 0.002 | 0.70 | ||
| RAPS | 0.998 (0.996–1.001) | 0.20 | |||||
| Weighted median | 0.999 (0.996–1.001) | 0.29 | |||||
| MR Presso | 0.999 (0.997–1.001) | 0.35 |
Single-variable MR results of PD-1 on risk of chronic ischemic heart disease, acute myocardial infarction, angina pectoris, unstable angina pectoris and coronary atherosclerosis.
IVW, inverse variance weighted; RAPS, robust adjusted profile score; MR Presso, Mendelian randomization pleiotropy RESidual sum and outlier; N SNP, number of genetic instruments; OR, odds ratio; CI, confidence interval; Ph, P-value for heterogeneity; Pp, P-value for Pleiotropy; NA, not applicable.
*P < 0.05.
Table 2
| Outcome | SNPS | Methods | OR (95% CI) | P-value | Q-statistics | Ph | Pp |
|---|---|---|---|---|---|---|---|
| Chronic ischemic heart disease | 6 | IVW | 1.000 (0.997–1.002) | 0.70 | 4.961 | 0.42 | |
| MR Egger | 1.003 (0.992–1.015) | 0.61 | 4.494 | 0.34 | 0.55 | ||
| RAPS | 1.000 (0.997–1.002) | 0.71 | |||||
| Weighted median | 1.001 (0.998–1.004) | 0.57 | |||||
| MR Presso | NA | NA | |||||
| Acute myocardial infarction | 6 | IVW | 0.999 (0.997–1.000) | 0.16 | 3.989 | 0.55 | |
| MR Egger | 1.001 (0.993–1.008) | 0.88 | 3.757 | 0.44 | 0.66 | ||
| RAPS | 0.999 (0.997–1.001) | 0.18 | |||||
| Weighted median | 0.998 (0.996–1.000) | 0.12 | |||||
| MR Presso | NA | NA | |||||
| Angina pectoris | 6 | IVW | 1.000 (0.998–1.002) | 0.87 | 2.925 | 0.71 | |
| MR Egger | 0.999 (0.991–1.008) | 0.88 | 2.909 | 0.57 | 0.90 | ||
| RAPS | 1.000 (0.998–1.002) | 0.87 | |||||
| Weighted median | 1.000 (0.998–1.002) | 0.84 | |||||
| MR Presso | NA | NA | |||||
| Unstable angina pectoris | 8 | IVW | 1.000 (0.999–1.001) | 0.77 | 4.535 | 0.72 | |
| MR Egger | 0.999 (0.993–1.004) | 0.64 | 4.196 | 0.65 | 0.58 | ||
| RAPS | 1.000 (0.999–1.001) | 0.82 | |||||
| Weighted median | 1.000 (0.999–1.002) | 0.70 | |||||
| MR Presso | NA | NA | |||||
| Coronary atherosclerosis | 8 | IVW | 0.998 (0.995–1.002) | 0.40 | 14.545 | 0.04 | |
| MR Egger | 1.011 (0.998–1.024) | 0.14 | 8.664 | 0.19 | 0.09 | ||
| RAPS | 0.999 (0.995–1.003) | 0.64 | |||||
| Weighted median | 1.000 (0.997–1.004) | 0.81 | |||||
| MR Presso | NA | NA |
Single-Variable MR results of PD-L1 on risk of chronic ischemic heart disease, acute myocardial infarction, angina pectoris, unstable angina pectoris and coronary atherosclerosis.
IVW, inverse variance weighted; RAPS, robust adjusted profile score; MR Presso, Mendelian randomization pleiotropy RESidual sum and outlier; N SNP, number of genetic instruments; OR, odds ratio; CI, confidence interval; Ph, P-value for heterogeneity; Pp, P-value for Pleiotropy; NA, not applicable.
Figure 1
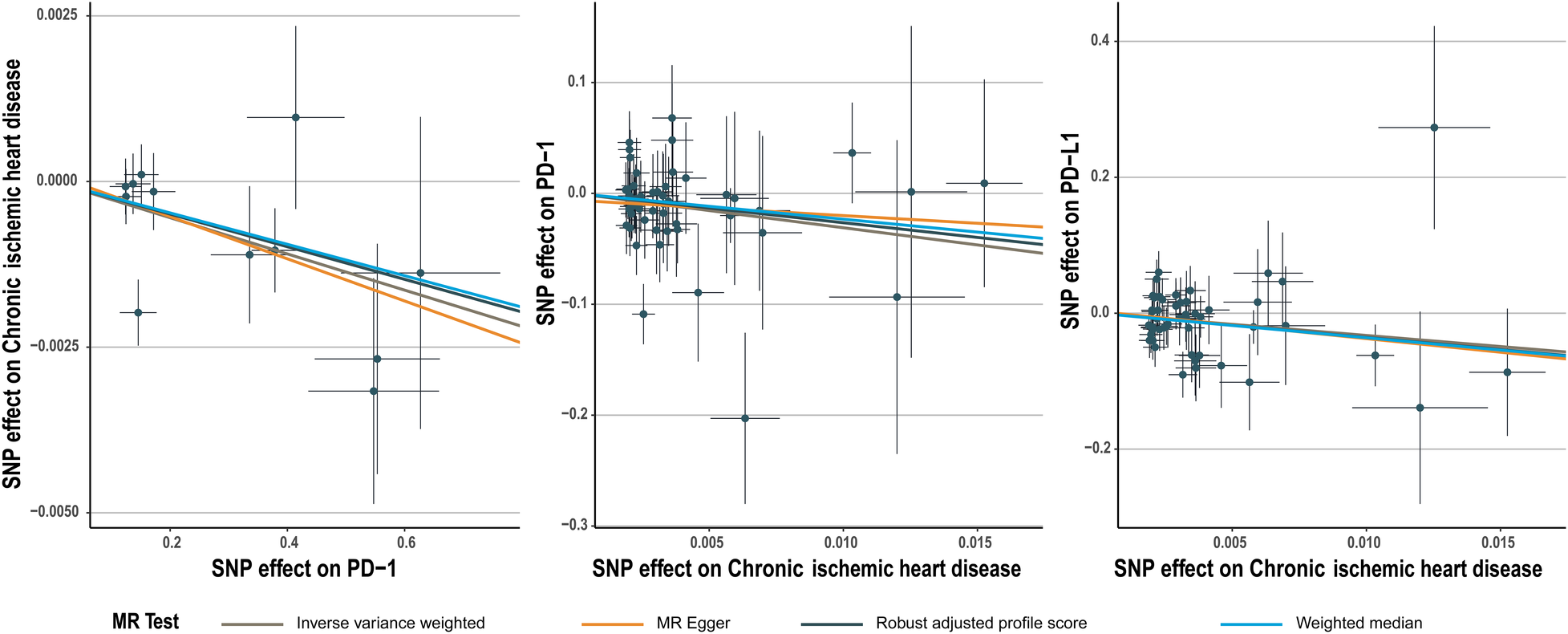
Scatter plot for PD-1 effect on chronic ischemic heart disease and chronic ischemic heart disease effect on PD-1/PD-L1.
3.2 Multivariable MR
In the multivariable MR that adjusted for the effect of PD-1 and PD-L1, the strong negative association between PD-1 and chronic ischemic heart disease remained and the association with other 4 types of CHD was still not significant. The multivariable-adjusted OR was 0.998 (95CI%, 0.996–0.999; P, 0.015) for association between PD-1 and chronic ischemic heart disease (Table 3). The causal relationship between PD-L1 and the 5 CHD remained insignificant (Table 3). The selected SNPs were listed in Supplementary Table S12–S16.
Table 3
| Exposure | Outcome | SNPS | Methods | OR (95% CI) | P-value |
|---|---|---|---|---|---|
| PD-1 | Chronic ischemic heart disease | 18 | IVW | 0.998 (0.996–0.999) | 0.02* |
| MR Egger | 0.997 (0.994–1.000) | 0.06 | |||
| PD-L1 | Chronic ischemic heart disease | 18 | IVW | 0.999 (0.996–1.002) | 0.50 |
| MR Egger | 0.999 (0.996–1.002) | 0.61 | |||
| PD-1 | Acute myocardial infarction | 18 | IVW | 1.000 (0.999–1.001) | 0.94 |
| MR Egger | 1.000 (0.998–1.002) | 0.90 | |||
| PD-L1 | Acute myocardial infarction | 18 | IVW | 0.998 (0.997–1.000) | 0.09 |
| MR Egger | 0.998 (0.997–1.000) | 0.12 | |||
| PD-1 | Angina pectoris | 18 | IVW | 1.000 (0.999–1.002) | 0.68 |
| MR Egger | 1.000 (0.998–1.002) | 0.72 | |||
| PD-L1 | Angina pectoris | 18 | IVW | 1.000 (0.998–1.002) | 0.88 |
| MR Egger | 1.000 (0.999–1.002) | 0.72 | |||
| PD-1 | Unstable angina pectoris | 21 | IVW | 1.000 (0.999–1.001) | 0.47 |
| MR Egger | 1.000 (0.999–1.002) | 0.71 | |||
| PD-L1 | Unstable angina pectoris | 21 | IVW | 1.000 (0.999–1.002) | 0.62 |
| MR Egger | 1.000 (0.999–1.002) | 0.62 | |||
| PD-1 | Coronary atherosclerosis | 21 | IVW | 0.999 (0.996–1.001) | 0.32 |
| MR Egger | 0.997 (0.993–1.002) | 0.22 | |||
| PD-L1 | Coronary atherosclerosis | 21 | IVW | 0.998 (0.994–1.001) | 0.23 |
| MR Egger | 0.998 (0.994–1.002) | 0.44 |
Multivariable MR results of PD-1/PD-L1 on risk of chronic ischemic heart disease, acute myocardial infarction, angina pectoris, unstable angina pectoris and coronary atherosclerosis.
IVW, inverse variance weighted; N SNP, number of genetic instruments; OR, odds ratio; CI, confidence interval.
*P < 0.05.
3.3 Bidirectional MR (effect of CHD on Pd-1/Pd-L1)
Chronic ischemic heart disease was shown by IVW to be significantly associated with PD-1 (beta, −3.1; 95%CI, −6.017 to −0.183, P, 0.037) and PD-L1(beta, −3.269; 95%CI, −6.197 to −0.341; P, 0.029) (Tables 4, 5). This result was consistent with other sensitivity analysis. The causal relationships between the other 4 CHDs and PD-1/PD-L1 were not statistically significant (Tables 4, 5). No outlier between CHD and PD-1/PD-L1 was identified by the MR-PRESSO test (Tables 4, 5). No directional pleiotropy was found in the MR-Egger regression (Tables 4, 5). No heterogeneity was found by Cochran's Q statistic (Tables 4, 5). The scatter plot and leave-one-out plot for the effects of 5 CHD on PD-1/PD-L1 are available in Figure 1, Supplementary Figures S3–S6, S9–S13. The selected SNPs were listed in Supplementary Table S17–S26.
Table 4
| Exposure | SNPS | Methods | Beta (95% CI) | P-value | Q-statistics | Ph | Pp |
|---|---|---|---|---|---|---|---|
| Chronic ischemic heart disease | 50 | IVW | −3.100 (−6.017 to −0.183) | 0.04 | 46.150 | 0.59 | |
| MR Egger | −1.384 (−7.645 to 4.876) | 0.67 | 45.782 | 0.56 | 0.55 | ||
| RAPS | −2.648 (−5.709 to 0.413) | 0.09 | |||||
| Weighted median | −2.328 (−6.635 to 1.978) | 0.29 | |||||
| MR Presso | NA | NA | |||||
| Acute myocardial infarction | 21 | IVW | −1.415 (−9.592 to 6.762) | 0.73 | 22.354 | 0.32 | |
| MR Egger | −1.032 (−18.413 to 16.349) | 0.91 | 22.352 | 0.27 | 0.96 | ||
| RAPS | −2.474 (−10.765 to 5.818) | 0.56 | |||||
| Weighted median | 1.706 (−9.363 to 12.775) | 0.76 | |||||
| MR Presso | NA | NA | |||||
| Angina pectoris | 23 | IVW | 3.203 (−3.823 to 10.23) | 0.37 | 16.525 | 0.79 | |
| MR Egger | 8.315 (−6.240 to 22.871) | 0.28 | 15.907 | 0.77 | 0.44 | ||
| RAPS | 3.197 (−4.267 to 10.661) | 0.40 | |||||
| Weighted median | 1.643 (−8.318 to 11.605) | 0.75 | |||||
| MR Presso | NA | NA | |||||
| Unstable angina pectoris | 20 | IVW | 4.999 (−4.446 to 14.445) | 0.30 | 16.612 | 0.62 | |
| MR Egger | 6.449 (−13.764 to 26.662) | 0.54 | 16.587 | 0.55 | 0.88 | ||
| RAPS | 5.710 (−5.177 to 16.597) | 0.30 | |||||
| Weighted median | 5.082 (−7.763 to 17.927) | 0.44 | |||||
| MR Presso | NA | NA | |||||
| Coronary atherosclerosis | 82 | IVW | −0.912 (−2.905 to 1.081) | 0.37 | 87.717 | 0.29 | |
| MR Egger | −3.169 (−7.594 to 1.256) | 0.16 | 86.366 | 0.29 | 0.27 | ||
| RAPS | −0.676 (−2.692 to 1.340) | 0.51 | |||||
| Weighted median | 0.230 (−2.658 to 3.118) | 0.88 | |||||
| MR Presso | NA | NA |
Single-Variable MR results of risk of chronic ischemic heart disease, acute myocardial infarction, angina pectoris, unstable angina pectoris and coronary atherosclerosis on PD-1.
IVW, inverse variance weighted; RAPS, robust adjusted profile score; MR Presso, Mendelian randomization pleiotropy RESidual sum and outlier; N SNP, number of genetic instruments; OR, odds ratio; CI, confidence interval; Ph, P-value for heterogeneity; Pp, P-value for Pleiotropy; NA, not applicable.
Table 5
| Exposure | SNPS | Methods | Beta (95% CI) | P-value | Q-statistics | Ph | Pp |
|---|---|---|---|---|---|---|---|
| Chronic ischemic heart disease | 50 | IVW | −3.269 (−6.197 to −0.341) | 0.03 | 49.362 | 0.46 | |
| MR Egger | −4.008 (−10.352 to 2.337) | 0.22 | 49.294 | 0.42 | 0.80 | ||
| RAPS | −3.563 (−6.685 to −0.441) | 0.03 | |||||
| Weighted median | −3.615 (−8.136 to 0.907) | 0.12 | |||||
| MR Presso | NA | NA | |||||
| Acute myocardial infarction | 21 | IVW | −1.775 (−9.509 to 5.959) | 0.65 | 16.186 | 0.71 | |
| MR Egger | −11.635 (−27.658 to 4.389) | 0.17 | 14.290 | 0.77 | 0.18 | ||
| RAPS | −0.771 (−8.951 to 7.409) | 0.85 | |||||
| Weighted median | −1.314 (−11.928 to 9.300) | 0.81 | |||||
| MR Presso | NA | NA | |||||
| Angina pectoris | 23 | IVW | −4.750 (−11.928 to 2.429) | 0.19 | 22.951 | 0.40 | |
| MR Egger | −0.972 (−16.079 to 14.135) | 0.90 | 22.614 | 0.36 | 0.58 | ||
| RAPS | −4.502 (−11.996 to 2.993) | 0.24 | |||||
| Weighted median | −2.394 (−11.921 to 7.133) | 0.62 | |||||
| MR Presso | NA | NA | |||||
| Unstable angina pectoris | 20 | IVW | 0.356 (−10.754 to 11.467) | 0.95 | 26.302 | 0.12 | |
| MR Egger | 2.445 (−21.958 to 26.848) | 0.85 | 26.249 | 0.09 | 0.85 | ||
| RAPS | −1.764 (−13.321 to 9.793) | 0.76 | |||||
| Weighted median | −5.880 (−19.714 to 7.954) | 0.40 | |||||
| MR Presso | NA | NA | |||||
| Coronary atherosclerosis | 82 | IVW | −0.694 (−2.703 to 1.315) | 0.50 | 89.168 | 0.25 | |
| MR Egger | −3.354 (−7.802 to 1.095) | 0.14 | 87.289 | 0.27 | 0.19 | ||
| RAPS | −1.084 (−3.146 to 0.979) | 0.30 | |||||
| Weighted median | −2.607 (−5.756 to 0.542) | 0.10 | |||||
| MR Presso | NA | NA |
Single-Variable MR results of risk of chronic ischemic heart disease, acute myocardial infarction, angina pectoris, unstable angina pectoris and coronary atherosclerosis on PD-L1.
IVW, inverse variance weighted; RAPS, robust adjusted profile score; MR Presso, Mendelian randomization pleiotropy RESidual sum and Outlier; N SNP, number of genetic instruments; OR, odds ratio; CI, confidence interval; Ph, P-value for heterogeneity; Pp, P-value for Pleiotropy; NA, not applicable.
3.4 GSEA analysis
Gene sets (GSE71226) from both the control and CHD groups were screened under the conditions of an q-value < 0.25, and P < 0.05, resulting in 77 differentially expressed gene sets (Supplementary eTable S27). Among these 77 gene sets, the gene set of “PD-L1 expression and PD-1 checkpoint pathway in cancer” pathway was included and downregulated in CHD group compared to control group with an enrichment score of −0.396 (P = 0.005; q = 0.019) (Figure 2).
Figure 2
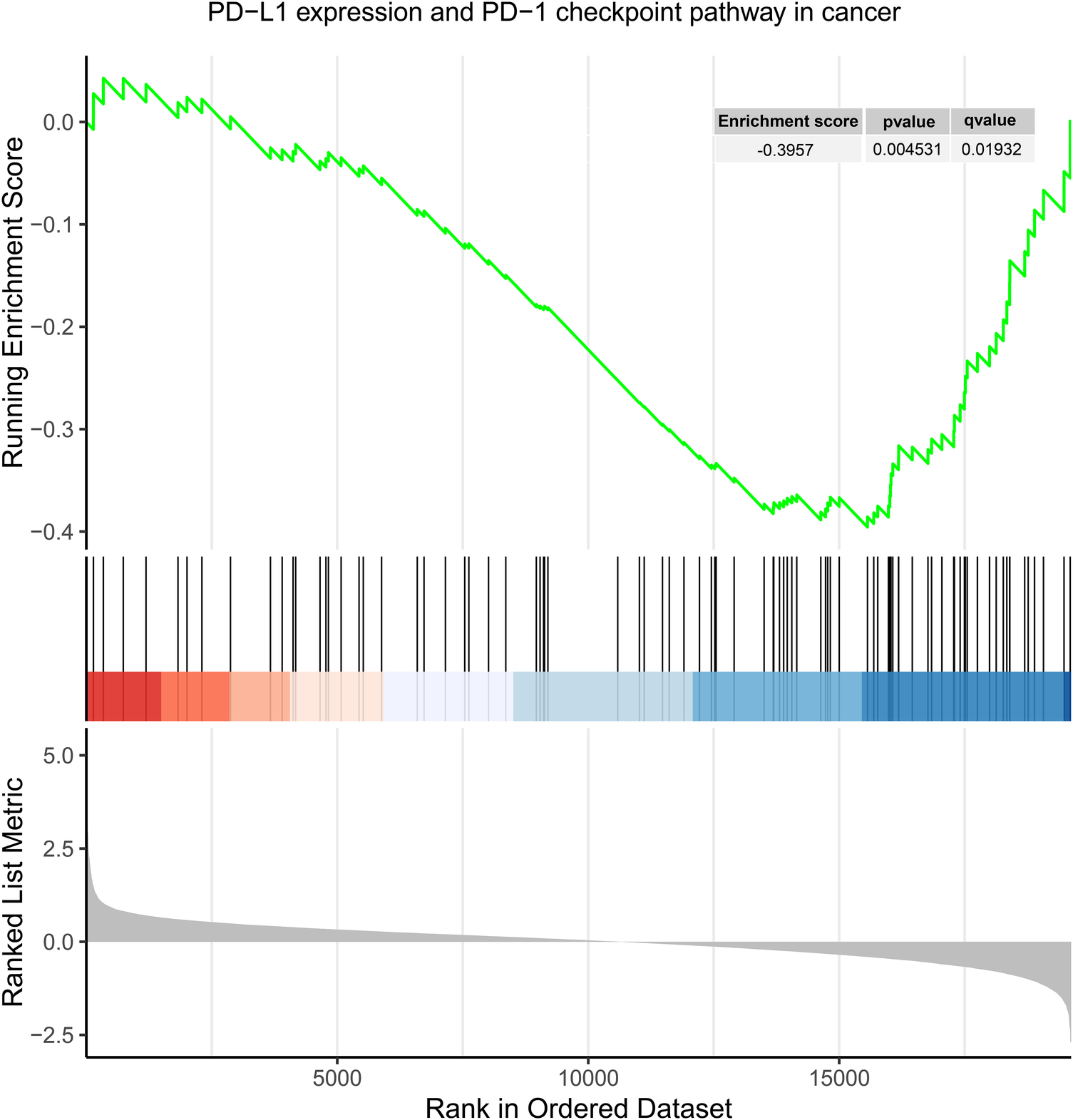
Enrichment plot for “PD-L1 expression and PD-1 checkpoint pathway in cancer” in GSE71226.
4 Discussion
This study revealed a protective causal relationship between PD-1 and chronic ischemic heart disease. In contrast, there was no evidence of a causal association between PD-1/PD-L1 and acute myocardial infarction, angina pectoris, unstable angina pectoris, or coronary atherosclerosis. After adjustment for PD-1 and PD-L1 through multivariable MR, the protective causal relationship between PD-1 and chronic ischemic heart disease remained. In reverse MR analysis, it was demonstrated that chronic ischemic heart disease was significantly associated with PD-1 and PD-L1 and no evidence was found in favor of association between other 4 CHD and PD-1 or PD-L1.
As well-characterized immune checkpoints, PD-1 and PD-L1 play a crucial role in the development of vascular inflammatory disorders by regulating T-cell activity (23, 24). Immune checkpoint inhibitors (ICIs) have become a well-established option for selected cancer therapies by conferring overall survival benefits to patients with advanced cancer, with an attendant focus on the cardiovascular adverse events that occur with ICI therapy. Since 2016, studies have reported cardiotoxicity due to ICI therapy (25, 26), PD-1/PD-L1 blockers provide a robust link to atherosclerosis, and lack of PD-1/PD-L1 exacerbates atherosclerotic patch formation by mechanisms that may be influenced through vascular inflammation, as well as T-cell activation and effector function (6, 11). PD-1 inhibitors can also induce cardiac injury via polarized Macrophages (27). This suggests a possible direct link between PD-1/PD-L1 itself for associated cardiovascular disease.
Previous studies demonstrated that patients with a history of myocardial infarction have significantly up-regulated PD-L1 expression (10). PD-L1 expression on peripheral blood T cells is significantly under-regulated in patients with coronary artery disease and might contribute to the pathogenesis and development of atherosclerosis (28). Fujisue et al. (29) proved that soluble PD-L1 (sPD-L1) levels were elevated in patients with coronary artery disease, and Miyazaki et al. (30) prospectively found elevated levels of sPD-L1 increased cardiovascular risk in these patients. Notably, a certain amount of circulating sPD-L1 acts to suppress the PD-1/PD-L1 pathway, thereby fostering chronic immune responses and inflammation, ultimately accelerating the pathological progression of CHD (31). The earlier studies, however, were mostly single-center and observed, failing to explain the causal relationship. Whereas to our knowledge there are no large randomized controlled trials assessing the causal link between PD-1/PD-L1 expression and CHD. Our study revealed a protective causal effect of PD-1 against chronic ischemic heart disease, a result that was held after multivariate analysis. Meanwhile, inverse MR also revealed that chronic ischemic heart disease was significantly associated with PD-1/PD-L1. The pathophysiological basis of chronic ischemic heart disease lies in coronary atherosclerosis, while the interaction of PD-1 and PD-L1 influences its onset and progression through various potential mechanisms. The PD-1/PD-L1 pathway restricts T cell activation, suppresses T cell responses, and promotes atherosclerosis. Simultaneously, PD-1 binding PD-L1 induces regulatory T cells, inhibiting the release of pro-inflammatory cytokines(e.g., IFN-γ and TNF-α), thereby aiding in reducing plaque volume. In addition, high PD-L1 expression diminishes T cell-mediated endothelial cytolytic damage, reducing vascular injury (11, 32, 33). Targeting the PD-1/PD-L1 pathway provides a novel perspective for prevention and treatment of chronic ischemic heart disease. Future research should delve into the intricate mechanisms of the PD-1/PD-L1 axis to optimize therapeutic or preventive strategies for chronic ischemic heart disease.
For other types of CHD, there is no clear causal relationship with PD-1/PD-L1, conceivably for the relevant genetic markers actually cause a broader susceptibility to coronary artery disease rather than being determinants, but the specific mechanisms need to be further explored. Only some subtypes of CHD are correlated with PD-1/PD-L1, possibly due to a combination of factors such as variations in the pathophysiological mechanisms of different disease subtypes and genetic influences. Firstly, each subtype of CHD exhibits distinct pathophysiological mechanisms (34, 35). Chronic ischemic heart disease may be more linked to chronic vascular inflammation, endothelial dysfunction, and atherosclerotic plaque formation, whereas acute myocardial infarction may be more associated with plaque rupture, thrombus formation, and acute coronary artery occlusion. The PD-1/PD-L1 signaling pathway plays a crucial role in immune regulation, potentially reducing inflammation by inhibiting T-cell activity (32). This effect might act as a protective mechanism in chronic ischemic heart disease, reducing ongoing vascular wall damage and plaque progression. However, in acute coronary events, this inhibitory effect may not be the primary pathological process, possibly overshadowed by more urgent physiological responses. Furthermore, the polymorphism of PD-1/PD-L1 genes may exert varying effects in different disease subtypes (36, 37). The interaction between genetic variations and environmental factors may also influence the role of PD-1/PD-L1 across different disease subtypes (38). Future research focusing on elucidating the specific mechanisms of the PD-1/PD-L1 signaling pathway in different CHD subtypes will contribute to a more accurate understanding of its causal relationships.
The current investigation offered several notable strengths. Initially, we delved into the reciprocal causal link between PD-1/PD-L1 and 5 types of CHD utilizing a bidirectional MR framework, thereby mitigating concerns regarding confounding factors, reverse causation, and exposure biases (39). Additionally, sensitivity assessments including MR-Egger, weighted median, and MR-PRESSO were conducted to bolster the consistency and resilience of our findings. Moreover, the integration of multivariable MR to adjust for confounding variables enhanced the reliability of inferring causal connections between PD-1/PD-L1 and CHD. Last, an additional validation of GSEA analysis was conducted to check whether CHD is associated with genes related to PD-1/PD-L1 pathway, increasing the rigorness of study.
Notwithstanding these strengths, it's essential to acknowledge certain limitations. Initially, a limited number of SNPs met the conventional bioinformatic threshold of p < 5 × 10−8. This scarcity of SNPs could pose challenges in matching instrumental variables (IVs) to the outcome, potentially weakening associations. Therefore, we opted for SNPs identified using a less stringent significance level of 5 × 10−6, a method recommended in prior research (40, 41). However, it's important to acknowledge that this approach may introduce a degree of weak instrumental variable bias. To assess this risk, we calculated F statistics, finding no substantial evidence supporting the presence of such bias (All IVs showed an F value greater than 10). Furthermore, our study focused on the European population, thus caution is warranted when generalizing our results to other demographic groups. Last, due to the aggregate nature of the GWAS data utilized in this study, we lacked access to data stratified by sex and age or individual-level data. Consequently, our investigation was unable to explore the potential causal associations between PD-1/PD-L1 and CHD across various age and sex subgroups.
5 Conclusion
This MR study supported a bidirectional causal relationship between PD-1 and chronic ischemic heart disease and protective association between chronic ischemic heart disease and PD-L1. Gene set related to PD-1/PD-L1 was revealed downregulated in CHD by GSEA, which consolidate the MR result. Though our results did not support a causal association between PD-1/PD-L1 and acute myocardial infarction, angina pectoris, coronary atherosclerosis, as well as unstable angina pectoris, further investigations are needed to clarify the mutual effect between PD-1/PD-L1 and CHD, as well as predictive models that detect intricate relationships and interactions among genetic variations, environmental influences, and the risk of CHD.
Statements
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
LZ: Writing – review & editing, Writing – original draft, Project administration, Formal Analysis, Conceptualization. YL: Writing – review & editing, Writing – original draft, Investigation. RZ: Writing – review & editing, Writing – original draft, Investigation, Funding acquisition, Conceptualization. WY: Writing – original draft, Visualization. KC: Writing – original draft, Investigation. BH: Writing – original draft, Methodology, Investigation. YQ: Writing – original draft, Investigation. LL: Writing – original draft, Methodology, Investigation. DZ: Writing – original draft, Methodology, Investigation. ZX: Writing – original draft, Investigation. HL: Writing – original draft, Software, Investigation. BZ: Writing – original draft, Methodology, Investigation. RL: Writing – original draft, Investigation. YL: Writing – review & editing, Supervision, Project administration. MY: Writing – review & editing, Resources. XL: Writing – review & editing, Resources, Funding acquisition.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by the College Students' Innovation Entrepreneurship Training Plan Program of China (Grant Nos. 202310570047), Guangzhou Medical University Student Innovation Ability Promoting Program (02-408-240603131023) and Medical Scientific Research Foundation of Guangdong Province of China (B2024115).
Acknowledgments
We thank the GWAS, UKB for providing these data.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2024.1424770/full#supplementary-material
References
1.
Shaya GE Leucker TM Jones SR Martin SS Toth PP . Coronary heart disease risk: low-density lipoprotein and beyond. Trends Cardiovasc Med. (2022) 32(4):181–94. 10.1016/j.tcm.2021.04.002
2.
WHO. The top 10 causes of death [updated 9 December 2020]. Available online at:https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death
3.
Hansson GK . Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. (2005) 352(16):1685–95. 10.1056/NEJMra043430
4.
Gotsman I Grabie N Dacosta R Sukhova G Sharpe A Lichtman AH . Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J Clin Invest. (2007) 117(10):2974–82. 10.1172/JCI31344
5.
Kythreotou A Siddique A Mauri FA Bower M Pinato DJ . PD-L1. J Clin Pathol. (2018) 71(3):189–94. 10.1136/jclinpath-2017-204853
6.
Dong M Yu T Tse G Lin Z Lin C Zhang N et al PD-1/PD-L1 blockade accelerates the progression of atherosclerosis in cancer patients. Curr Probl Cardiol. (2023) 48(3):101527. 10.1016/j.cpcardiol.2022.101527
7.
Bu DX Tarrio M Maganto-Garcia E Stavrakis G Tajima G Lederer J et al Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler Thromb Vasc Biol. (2011) 31(5):1100–7. 10.1161/ATVBAHA.111.224709
8.
Cochain C Chaudhari SM Koch M Wiendl H Eckstein HH Zernecke A . Programmed cell death-1 deficiency exacerbates T cell activation and atherogenesis despite expansion of regulatory T cells in atherosclerosis-prone mice. PLoS One. (2014) 9(4):e93280. 10.1371/journal.pone.0093280
9.
Lee J Zhuang Y Wei X Shang F Wang J Zhang Y et al Contributions of PD-1/PD-L1 pathway to interactions of myeloid DCs with T cells in atherosclerosis. J Mol Cell Cardiol. (2009) 46(2):169–76. 10.1016/j.yjmcc.2008.10.028
10.
Kushnareva E Kushnarev V Artemyeva A Mitrofanova L Moiseeva O . Myocardial PD-L1 expression in patients with ischemic and non-ischemic heart failure. Front Cardiovasc Med. (2021) 8:759972. 10.3389/fcvm.2021.759972
11.
Sun Y Li L Wu Y Yang K . PD-1/PD-L1 in cardiovascular disease. Clin Chim Acta. (2020) 505:26–30. 10.1016/j.cca.2020.02.019
12.
Wu F Huang Y Hu J Shao Z . Mendelian randomization study of inflammatory bowel disease and bone mineral density. BMC Med. (2020) 18(1):312. 10.1186/s12916-020-01778-5
13.
Davies NM Holmes MV Davey Smith G . Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. Br Med J. (2018) 362:k601. 10.1136/bmj.k601
14.
Larsson SC Butterworth AS Burgess S . Mendelian Randomization for cardiovascular diseases: principles and ap plications. Eur Heart J. (2023) 44(47):4913–24. 10.1093/eurheartj/ehad736
15.
Sun BB Maranville JC Peters JE Stacey D Staley JR Blackshaw J et al Genomic atlas of the human plasma proteome. Nature. (2018) 558(7708):73–9. 10.1038/s41586-018-0175-2
16.
Burgess S Thompson SG . Mendelian Randomization: Methods for Causal Inference Using Genetic Variants. Boca Raton, FL: CRC Press (2021).
17.
Burgess S Scott RA Timpson NJ Davey Smith G Thompson SG . Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. (2015) 30(7):543–52. 10.1007/s10654-015-0011-z
18.
Verbanck M Chen C-Y Neale B Do R . Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and disea ses. Nat Genet. (2018):50(5):693–8. 10.1038/s41588-018-0099-7
19.
Zhao Q Wang J Hemani G Bowden J Small DS . Statistical inference in two-sample summary-data Mendelian randomization using robust adjusted profile score. Ann Statist. (2020) 48(3):1742–69. 10.1214/19-AOS1866
20.
Bowden J Davey Smith G Haycock PC Burgess S . Consistent estimation in Mendelian randomization with some invalid ins truments using a weighted median estimator. Genet Epidemiol. (2016) 40(4):304–14. 10.1002/gepi.21965
21.
Sanderson E Davey Smith G Windmeijer F Bowden J . An examination of multivariable Mendelian randomization in the single- sample and two-sample summary data settings. Int J Epidemiol. (2019) 48(3):713–27. 10.1093/ije/dyy262
22.
Burgess S Daniel RM Butterworth AS Thompson SG . Network Mendelian randomization: using genetic variants as instrumental variables to investigate mediation in causal pathways. Int J Epidemiol. (2015) 44(2):484–95. 10.1093/ije/dyu176
23.
Nagaraju GP Malla RR Basha R Motofei IG . Contemporary clinical trials in pancreatic cancer immunotherapy targeting PD-1 and PD-L1. Semin Cancer Biol. (2022) 86(Pt 3):616–21. 10.1016/j.semcancer.2021.11.003
24.
Liang Y Li L Chen Y Xiao J Wei D . PD-1/PD-L1 immune checkpoints: tumor vs atherosclerotic progression. Clin Chim Acta. (2021) 519:70–5. 10.1016/j.cca.2021.04.010
25.
Johnson DB Balko JM Compton ML Chalkias S Gorham J Xu Y et al Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. (2016) 375(18):1749–55. 10.1056/NEJMoa1609214
26.
Semper H Muehlberg F Schulz-Menger J Allewelt M Grohé C . Drug-induced myocarditis after nivolumab treatment in a patient with PDL1- negative squamous cell carcinoma of the lung. Lung Cancer. (2016) 99:117–9. 10.1016/j.lungcan.2016.06.025
27.
Xia W Zou C Chen H Xie C Hou M . Immune checkpoint inhibitor induces cardiac injury through polarizing macrophages via modulating microRNA-34a/Kruppel-like factor 4 signaling. Cell Death Dis. (2020) 11(7):575. 10.1038/s41419-020-02778-2
28.
Zhang C Yang K Yang Y Zhao G . PD-L1 expression on peripheral T-cells and association with coronary heart disease patients: a protocol for systematic reviews and meta-analysis. Medicine (Baltimore). (2021) 100(12):e25157. 10.1097/MD.0000000000025157
29.
Fujisue K Yamamoto E Sueta D Takae M Nishihara T Komorita T et al Increased soluble programed cell death-ligand 1 is associated with acu te coronary syndrome. Int J Cardiol. (2022) 349:1–6. 10.1016/j.ijcard.2021.11.060
30.
Miyazaki S Fujisue K Yamanaga K Sueta D Usuku H Tabata N et al Prognostic significance of soluble PD-L1 on cardiovascular outcomes in patients with coronary artery disease. J Atheroscler Thromb. (2024) 31(4):355–67. 10.5551/jat.64183
31.
Li Y Xiao Y Su M Zhang R Ding J Hao X et al Role of soluble programmed death-1 (sPD-1) and sPD-ligand 1 in patients with cystic echinococcosis. Exp Ther Med. (2016) 11(1):251–6. 10.3892/etm.2015.2876
32.
Veluswamy P Wacker M Scherner M Wippermann J . Delicate role of PD-L1/PD-1 axis in blood vessel inflammatory diseases: current insight and future significance. Int J Mol Sci. (2020) 21(21):8159. 10.3390/ijms21218159
33.
Li SH Chen WJ Yan M Shu YW Liao YH . Expression of coinhibitory PD-L1 on CD4+CD25+FOXP3+ regulatory T cells is elevated in patients with acute coronary syndrome. Coron Artery Dis. (2015) 26(7):598–603. 10.1097/MCA.0000000000000282
34.
Bentzon JF Otsuka F Virmani R Falk E . Mechanisms of plaque formation and rupture. Circ Res. (2014) 114(12):1852–66. 10.1161/CIRCRESAHA.114.302721
35.
Wentzel JJ Chatzizisis YS Gijsen FJ Giannoglou GD Feldman CL Stone PH . Endothelial shear stress in the evolution of coronary atherosclerotic plaque and vascular remodelling: current understanding and remaining questions. Cardiovasc Res. (2012) 96(2):234–43. 10.1093/cvr/cvs217
36.
Qian C Guo H Chen X Shi A Li S Wang X et al Association of PD-1 and PD-L1 genetic polymorphyisms with type 1 diabetes susceptibility. J Diabetes Res. (2018) 2018:1614683. 10.1155/2018/1614683
37.
Yang M Liu Y Zheng S Geng P He T Lu L et al Associations of PD-1 and PD-L1 gene polymorphisms with cancer risk: a meta-analysis based on 50 studies. Aging (Albany NY). (2024) 16(7):6068–97. 10.18632/aging.205689
38.
Prasher D Greenway SC Singh RB . The impact of epigenetics on cardiovascular disease. Biochem Cell Biol. (2020) 98(1):12–22. 10.1139/bcb-2019-0045
39.
Davey Smith G Hemani G . Mendelian Randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. (2014) 23(R1):R89–98. 10.1093/hmg/ddu328
40.
Ference BA Majeed F Penumetcha R Flack JM Brook RD . Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2×2 factorial Mendelian randomization study. J Am Coll Cardiol. (2015) 65(15):1552–61. 10.1016/j.jacc.2015.02.020
41.
Zou X-L Wang S Wang L-Y Xiao L-X Yao T-X Zeng Y et al Childhood obesity and risk of stroke: a Mendelian randomisation analys is. Front Genet. (2021) 12:727475. 10.3389/fgene.2021.727475
Summary
Keywords
Mendelian randomization (MR), coronary heart disease (CHD), PD-1, PD-L1, GSEA
Citation
Zeng L, Liang Y, Zhou R, Yang W, Chen K, He B, Qiu Y, Liu L, Zhou D, Xiao Z, Liang H, Zhang B, Li R, Yu L, Yi M and Lin X (2024) PD-1/PD-L1 and coronary heart disease: a mendelian randomization study. Front. Cardiovasc. Med. 11:1424770. doi: 10.3389/fcvm.2024.1424770
Received
28 April 2024
Accepted
25 September 2024
Published
18 October 2024
Volume
11 - 2024
Edited by
Ioanna Koniari, Manchester University NHS Foundation Trust, United Kingdom
Reviewed by
Feng Jiang, Stanford University, United States
Tianhui Yuan, Guangzhou University of Chinese Medicine, China
Updates
Copyright
© 2024 Zeng, Liang, Zhou, Yang, Chen, He, Qiu, Liu, Zhou, Xiao, Liang, Zhang, Li, Yu, Yi and Lin.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Xiaozhen Lin linxz021@163.com Min Yi smu_min@126.com Lihong Yu ylh84@126.com
†These authors have contributed equally to this work and share last authorship
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.