Abstract
Despite the increasing number of anti-hypertensive drugs have been developed and used in the clinical setting, persistent deficiencies persist, including issues such as lifelong dosage, combination therapy. Notwithstanding receiving the treatment under enduring these deficiencies, approximately 4 in 5 patients still fail to achieve reliable blood pressure (BP) control. The application of neuromodulation in the context of hypertension presents a pioneering strategy for addressing this condition, con-currently implying a potential central nervous mechanism underlying hypertension onset. We hypothesize that neurological networks, an essential component of maintaining appropriate neurological function, are involved in hypertension. Drawing on both peer-reviewed research and our laboratory investigations, we endeavor to investigate the underlying neural mechanisms involved in hypertension by identifying a close relationship between its onset of hypertension and an excitation and inhibition (E/I) imbalance. In addition to the involvement of excitatory glutamatergic and GABAergic inhibitory system, the pathogenesis of hypertension is also associated with Voltage-gated sodium channels (VGSCs, Nav)-mediated E/I balance. The overloading of glutamate or enhancement of glutamate receptors may be attributed to the E/I imbalance, ultimately triggering hypertension. GABA loss and GABA receptor dysfunction have also proven to be involved. Furthermore, we have identified that abnormalities in sodium channel expression and function alter neural excitability, thereby disturbing E/I balance and potentially serving as a mechanism underlying hypertension. These insights are expected to furnish potential strategies for the advancement of innovative anti-hypertensive therapies and a meaningful reference for the exploration of central nervous system (CNS) targets of anti-hypertensives.
1 Introduction
Hypertension has caused a huge burden in mortality and healthcare worldwide due to its huge contribution to the pathogenesis of multiple cardiovascular diseases, such as coronary heart disease, stroke, aortic dissection, and renal events (1, 2). Billions of adults worldwide suffer from hypertension when using a threshold of systolic blood pressure (SBP) ≥140 mmHg and/or diastolic blood pressure (DBP) ≥90 mmHg (3). The magnitude of issue escalates significantly with the adoption of the new threshold of SBP ≥130 mmHg and/or DBP ≥80 mmHg (4). The staggering toll of up to 10.4 million deaths attributed to hypertension in 2017 (5), underscores the imperative for timely assessment and implementation of antihypertensive interventions globally. The majority of hypertension (≥90%) is essential hypertension, of which the pathogenesis is unclear. The central modulation of heart rate (HR) and BP offers a foundation for understanding neurogenic mechanisms underlying hypertension. Besides lifestyle improvements including exercise, reduced alcohol consumption, quitting smoking, and lowering sodium and saturated fats intake could be efficient in controlling hypertension (6), guidelines also recommend the use of diuretics, Angiotensin-Converting Enzyme Inhibitors (ACE/Is), angiotensin II receptor blockers, and calcium channel antagonists, etc. (7). Nonetheless, only about one-fifth of hypertensive patients live with controlled BP (8). Some hypertension that proved refractory escapes from various antihypertensive drugs, which can be effectively managed using deep brain stimulation according to N K Patel et al.'s report (9). This further confirms the potential involvement of a central nervous mechanism in hypertension pathogenesis. With deeper studies in neurology, scientists have found that neural function is not solely reliant on individual neurons but rather on complex neural networks composed of neuronal clusters (10). The dynamic balance of excitation and inhibition (E/I) is necessary for ensuring the optimal functionality of neural networks (11, 12).
Here we review the role of network E/I imbalance in the pathogenesis of hypertension and its potential mechanisms, to improve the understanding of neural mechanisms and narrow the gap in this knowledge. Based on the published research of peers and our lab, the possible neural mechanisms are explored for different etiopathogenesis of hypertension. We focus on hypertension that has received widespread attention over recent years including, but no limited to stress-induced, salt-induced, Angiotensin (Ang) II-induced, and neurogenic hypertension. First, we address the brain regions underlying the genesis of hypertension, and then explore the impact of alternations in these regions at the whole-brain level, such as neural network and neural connection. Accordingly, we have attempted to provide researchers with a new perspective on the pathogenesis of hypertension and with a view to providing some insights into curing hypertension.
2 The involvement of the network in hypertension
Previous studies have identified that prenatal malnutrition is tied to an increased risk of hypertension (13, 14). Offspring born to mothers exposed to Dutch famine during pregnancy exhibited heightened vulnerability to hypertension in middle age (15). It may be attributed to the fact that undernutrition during fetal life leads to lower body weight and brain changes, including neuronal loss, impaired neuronal differentiation even neuroplasticity deficits (16), all contributing to the functional aberrations observed in neural networks. Various studies establish that BP regulation by the CNS relies on the coordinated activity of a highly interconnected network of neurons (17, 18). Cayupe et al. reviewed the unfavorable Paraventricular-Coerulear Network arising in the genesis of hypertension in prenatally malnourished adult animals (19). Similarly, hypertension attributed to affective disorders involves neural networks. Hypertension is well-known to be influenced by genetic and environmental factors (20), and so is anxiety formation, especially generalized anxiety disorder (21). A significant anxiety-hypertension association in cross-sectional and prospective studies was pointed out from 59 studies by pooling data recently, even though the underlying pathway is still elusive (22). The pandemic of COVID-19 gave rise to half of the UK adults increased anxiety, depression, or stress due to fear of contracting (23). This heightened anxiety had been associated with a marked increase in cardiovascular events especially hypertension (24). A functional neuroimaging study of anxiety in humans hints at the frequent observation of amygdala and insular cortex (lc) hyperactivation in anxiety disorder (25), change of neural unit in both of which lead to neurogenic hypertension in animals (26, 27). Further animal experiments had made efforts to reveal the mechanism by which the alternation in the brain causes hypertension. They found occlusion of the right middle cerebral artery induces neurochemical disturbances in the amygdala and lc (28), thereby increasing sympathetic outflow and contributing to hypertension in mice (26). Sympathetic overflow increases myocardial contractility and thus stoke volume. Moreover, it also causes vasoconstriction leading increased vascular resistance via the effect of norepinephrine on postsynaptic α-adrenergic receptors of sympathetic nerves innervating the peripheral vasculature. Sympathetic blockade in hypertensive mice decreases cardiac output and peripheral vascular resistance, hence ameliorating hypertension (29). Another research in the context of menopausal women revealed lower intrinsic resting-state functional brain connectivity of the midbrain-brainstem-cerebellar network with bilateral anterior middle insula in hypertension of this population (30).
Sympathovagal imbalance in the central autonomic network may be involved in the pathophysiology of hypertension, given recent evidence confirming the autonomic regulation of the cardiovascular system, which encompasses the insular cortex and anterior cingulate (31, 32). The dense neural projections connect relevant brain regions such as the insula to the autonomic nucleus (33), known for their role in cardiac autonomic regulation (34, 35), offer insights into the underlying mechanism by which these neural connections cause cardiovascular events. A previous animal study confirmed that a high-fat diet leads to changes in neuropeptide gene expression within the CNS, possibly contributing to obesity-induced hypertension and autonomic dysregulation (36). While not directly establishing a causal relationship between autonomic network imbalance and hypertension, this evidence gives few hints. Shiina and his colleagues figured out that improving sympathovagal imbalance is effective in alleviating obstructive sleep apnea-associated hypertension and other adverse cardiovascular events (37). Naiara A Herrera et al. came to a similar conclusion that exercise ameliorates the autonomic balance of dexamethasone-induced hypertensive mice thereby improving the arterial pressure (38). Another acupuncture-based study found that stimulating bilateral TaiChong points in rats enhances the functional connection between the hypothalamus and brain regions related to BP regulation, such as the entorhinal cortex, hippocampal dentate gyrus, brain stem, etc., accompanied by anti-hypertensive effects (39). All these clinical studies corroborate that interventions targeting autonomic network balance alleviate hypertension, confirming to an extent the role of the autonomic network in hypertension pathogenesis. A recent animal experiment proved that autonomic network dysfunction causes hypertension which would be relieved when the autonomic network dysfunction is rescued (40).
3 Excitatory glutamatergic system in hypertension
Stress normally produces a temporary rise in arterial BP, with untreated chronic stress triggering persistent hypertension, especially in individuals with a genetic risk for hypertension (41). As the major excitatory neurotransmitter in the vertebrate nervous system, glutamate, along with the excitatory glutamatergic system it comprises, accounts for over 90% of the excitatory synaptic connections in the human brain (42). Glutamate makes its way to the synaptic cleft and acts on downstream receptors and ion channels, either synthesized de novo in glutamatergic neurons or recycled through the glutamate-glutamine cycle before being released into the synaptic cleft (43). Excess extracellular glutamate causes neuroexcitotoxicity resulting in an excitatory-inhibitory imbalance in the brain network, thereby precipitating pathological processes (44). The imbalance of the neural network induces excessive sympathetic outflow to trigger hypertension. Given the essential role of the in maintaining neural network homeostasis, its contribution to hypertension warrants investigation.
3.1 Stress-induced hypertension
Evidence suggests an important role for chronic stress in the pathogenesis of hypertension, cardiovascular disease, and stroke (45). Our recent publication confirms that chronic stress-induced activation of neurons in the dorsomedial prefrontal cortex (dmPFC) leads to increased glutamate concentrations in the hippocampal vCA1 projection areas of these neurons and elevated BP in mice (46). While not assessing alternations in neural networks excitability, our study observed a rise in glutamate, the main excitatory neurotransmitter, without significant changes in GABA. These results may also hint at the imbalance of E/I in neural networks (42). Basting et al. assessed the effects of acute stimulation of excitatory neurons in the paraventricular nucleus (PVN) selectively on resting BP in awake mice and found it was sufficient to drive an elevation of BP, akin to hypertension (47). PVN integrates the effects of endocrine and autonomic response to stress and ultimately causes hypertension by projecting to sympathetic-related areas in the brainstem and spinal cord leading to a sympathetic overflow (48, 49). A study published recently showed that chronic stress increases sympathetic outflow and ultimately induces hypertension through activation of glutamatergic receptors in PVN and increased binding to the corresponding ligands (41). Overactivation of glutamatergic receptors in PVN is a major source of elevated sympathetic outflow in hypertension (49). Ma et al. have further confirmed that excessive glutamatergic excitatory inputs are strongly linked to the pathogenesis of hypertension (50). Other studies delved into the mechanism of increased glutamatergic tone and found that it was maintained by presynaptic glutamate release and enhanced postsynaptic NMDAR activity (51, 52). Alternatively, research has suggested the upstream glutamatergic output that is projected to the PVN, leading to the activation of the vasopressin (VAP) neurons in it and raising BP (53).
Beyond the PVN, the glutamatergic tonic input of the rostral ventrolateral medulla (RVLM) also results in hypertension (54), as RVLM is a critical site central control of sympathetic outflow playing an essential role in maintaining basal BP (55). Neurons in the RVLM are proposed to send downward impulses to sympathetic preganglionic neurons in the intermediolateral column of the spinal cord (56). Additional glutamatergic input to the RVLM facilitates BP and sympathetic nervous activity (57). ROS-SAPK/JNK signaling pathway regulates the pressor effect of glutamatergic neurons in RVLM in stress-induced hypertensive mice (58). A study modeling stress-induced hypertension based on foot shock combined with noise exposure similarly confirmed the correlation between alterations in the glutamatergic system of RVLM and hypertension. The authors suggested that angiotensin (Ang) II-mediated glutamatergic effects as well as the upregulation of NMDA could activate glutamatergic neurons in the RVLM, releasing glutamate into the intermediolateral column (IML) and altering sympathetic outflow in stress-induced hypertension (59).
3.2 Salt-induced hypertension
Numerous epidemiologic, clinical, and experimental studies have confirmed that salt intake is associated with BP and that reducing dietary salt intake could lower BP (60). Normally, salt is often referred to as sodium salt, even though salt constituting only 40% of its composition. Salt-induced increases in BP are in part believed to be mediated by enhanced NaCl in cerebrospinal fluid (CSF) and sympathetic nerve activity (61, 62). Central hypernatremia initiates hypothalamic pathways increasing glutamatergic drive to RVLM neurons, thereby increasing arterial BP (63). An earlier study also found a significant elevation of BP when injecting glutamate into the RVLM of salt-sensitized rats, suggesting a relationship between excessive glutamatergic input and the development of hypertension (64). Changes in gut microbiota are believed to play an important role in salt-induced hypertension. Healthy animals exhibited elevated BP following transplantation of gut bacteria from hypertensive patients or animals. Zheng et al. found that salt overconsumption caused the upregulation of glutamate and its derivatives from comparing the differences between intestinal flora, metabolites, and metabolites pathways in salt-induced rats and controls (65). Subsequent KEGG analysis revealed that the salt-induced alternations were associated with the glutamatergic synaptic pathway, potentially culminating in disruption of E/I balance and consequent hypertension (52). Blocking the angiotensin type 1 receptors (AT1Rs) in the PVN fails to further decrease the peak BP response to the glutamate receptor blockers in salt-induced hypertension while conversely works, probably suggesting the activation of AT1 is primarily mediated by locally released glutamate (66). Also, some research concluded that prohypertensive signals, such as salt can be perceived by autonomic brain regions enhancing resident microglia mainly located in PVN to promote hypertension (67, 68). Instead, attenuation of microglial activation in PVN by administration of minocycline produces antihypertensive effects (69). Stabilized microglia prevent the overactivation of pre-sympathetic neurons in PVN, overactivated microglial constitutive releases platelet-derived growth factor B promoting neuronal potassium current conduction, which in turn triggers excess sympathetic outflow disrupting the excitatory balance of the autonomic system (70). In addition, some researchers stated that brain-derived neurotrophic factor from microglia increases the expression of NMDARs at postsynaptic terminals (71), which might underlie the association of microglial activation with the pathogenesis of hypertension.
3.3 Neurogenic hypertension
Sympathetic strain serves as a major pathway in the development of neurogenic hypertension. Basting et al. confirmed the critical role of glutamatergic neurons of PVN in the development of neurogenic hypertension in conscious mice using optogenetics (47). They found a frequency dependent increase in BP upon direct photoactivation of these PVN neurons. Whereas the model of deoxycorticosterone acetate-induced neurogenic hypertension exhibited near-normal BP when these PVN neurons were impaired. The use of calcineurin inhibitors in constructing neurogenic hypertension models has been employed in several studies (72, 73). Elevated synaptic NMDAR activity in pre-sympathetic neurons increased excitability inputs to the PVN, sustaining excessive sympathetic outflow, which may contribute to the genesis of neurogenic hypertension (72). This was supported by an earlier study that NMDAR activation normalize the higher frequency and amplitude of excitatory postsynaptic currents in PVN neurons, which elicits sympathetic tension, a crucial factor in the development of neurogenic hypertension (74). Besides NMDAR, also AMPA-type glutamate receptors (AMPARs) in the postsynaptic membrane, was shown involved in the occurrence of neurogenic hypertension (73). Calcineurin inhibitors initiated an altered phenotype of AMPARs in PVN pre-sympathetic neurons, resulting in an increased Ca2+-permeable AMPARs. These changes triggered an increased postsynaptic currents mediated by AMPAR, leading to excessive sympathetic outflow and the induction of neurogenic hypertension (73). AMPARs are homomeric or heteromeric tetramer consisting of a combination of four pore-forming subunits, GluA1, GluA2, GluA3, and GluA4. A study from Zhou et al. also found that increased synaptic Ca2+-permeable AMPARs in PVN presympathetic neuron maintain the sympathetic outflow in neurogenic hypertension by inhibiting the assembly of GluA1/GluA2 heteromers (75). In spite of being glutamate receptors, metabotropic glutamate receptors (mGluRs) do not play exactly the same role in hypertension as the above two. The mGluRs in PVN affect hypothalamic presympathetic neurons via opposing presynaptic and postsynaptic actions (76). Activation of presynaptic group III mGluRs depresses the excitability of PVN presympathetic neurons, attenuating sympathetic vasomotor activity. Conversely, postsynaptic group III mGluRs excite PVN presympathetic neurons, increasing sympathetic vasomotor activity (76).
3.4 Ang II-induced hypertension and other hypertension
As a critical site in the development of hypertension, PVN is also identified in other hypertensive models (52, 77, 78). Increased mineralocorticoid receptor leads to enhanced AT1 and glutamate receptor-dependent signaling in the PVN, thereby promoting a chronic increase in circulating Ang II to maintain BP in Ang II-dependent hypertension (77). Another study equally found that enhanced glutamate signaling of PVN, for instance, NMDA-mediated elevation of excitatory currents was present in Ang II-dependent hypertension (78). Central Ang II facilitated glutamate release, that enhanced glutamatergic neuron activation in the RVLM via AT1R binding to enhanced NMDAR and AMPAR, as well as increased sympathetic excitation, causing hypertension in mice (59). Glutamatergic connection activation between the nucleus tract solitarius and the ventral-lateral segment of the medulla oblongata may be one of the baroreflex pathways that produce hypertension (79). Qiao et al. demonstrated that presynaptic and postsynaptic NMDAR activity of RVLM-projecting PVN neurons in essential hypertensive rats whereas blocking this increase lowered BP (52). Nicotine possibly accelerates and deteriorates hypertension by enhancing glutamatergic transmission in spontaneously hypertensive rats (SHR) (80). The glutamatergic signaling network is as well involved in obesity-related hypertension, as the elevated BP induced by excessive central leptin was blocked by glutamatergic inhibition in mice fed a high-fat diet (81). Even blocking the glutamate receptor in postnatal period rescued some of the leptin-induced hypertension from regular feeding (82). All these findings above prompt the pivotal role of the glutamatergic system in the pathophysiology of hypertension and suggests its potential as a reliable strategy for the treatment of essential hypertension.
4 Inhibitory GABAergic system in hypertension
The GABAergic (gamma-aminobutyric acid) system serves as the major inhibitory system in the brain, working along with the excitatory system to maintain the delicate balance in the brain for adequate neurological function. Despite making up only 20% of the neurons, GABAergic neurons are vital in generating inhibitory inputs over others (83). As the main bearers of GABAergic signaling, GABAergic receptors (GABARs) were involved in numerous physiopathological processes such as pain, emotion, and cognition (84). As we reviewed above, E/I balance is thought to be implicated in the pathogenesis of hypertension. And what role does the GABAergic system, as an important component of E/I balance, play in achieving hypertension?
4.1 Stress-induced hypertension
Over the past decades, it has been well established that stress induces cardiovascular problems such as hypertension, and the regulation of various neurogenic pathways bridges the gap between stress and hypertension as well as cardiovascular disease (85). An MRI-based study showed that GABA levels in the hypothalamus of inherited SHR were significantly lower than in controls, accompanied by an elevated in excitability and energetic activity (86). Another study identified a precise brain region in SHR where E/I imbalance occurs, arguing that the decline in GABA and elevation of glutamate in RVLM may be closely related to the pathogenesis of hypertension (87). They examined neurotransmitter concentrations only in homogenates of tissue from RVLM. It would be more convincing to detect the levels of intercellular neurotransmitter which affect E/I balance in the brain network. Xia et al. examined the intercellular neurotransmitters in the RVLM of SHR treated with PVN injection of melatonin. They discovered that melatonin ameliorated hypertension in SHR while increasing intercellular GABA in the RVLM (88). It hints that altered intercellular GABA-mediated E/I balance in the PVN projective site RVLM closely correlates with the pathogenesis and modulation of hypertension.
To elucidate the mechanisms of neurotransmitter imbalance in the RVLM of SHR, numerous studies are underway. As one of the participants in hypertension, Ang II acts on various receptors, such as AT1R and AT2R. Du et al. found activation of the Ang II pathway plays an important role in SHR by reducing the GABA release (89). However, they did not explain how Ang II caused changed GABA release, which was given by Laura Légat and his team in their recent study. Ang II may cause AT2R activation at synapses in GABAergic neurons, thus altering GABA release (90). It has also been noted that GAD67 expression is decreased in hypertensive rats, ultimately leading to increased vesicle-dependent GABA release (91). Despite these intensive studies were conducted, none of them gave direct evidence to confirm their conclusions. This may await advances in experimental techniques or the launch of extensive research.
4.2 Salt-induced hypertension
Given that our daily salt intake far exceeds the recommended limit, health hazards especially cardiovascular risks such as hypertension, are increasingly being emphasized (92). Zheng et al. showed that high salt intake not only affected the excitatory glutamatergic system but also led to a downregulation of GABA. The KEGG enrichment analysis suggested that high salt intake may impact the relevant pathways of GABA metabolism (65). Distinctly, some researchers found that suppressing GABAergic excitation inhibits salt-induced hypertension via reducing the output of AVP neurons (93, 94). Given the inhibitory effect of GABA on neurons, it follows that activating the GABAergic system should inhibit the release of AVP from AVP neurons to attenuate hypertension. They believed that when GABAergic inhibition converts to excitation, the baroreflex loses its function in buffering BP and instead contributes to the development of hypertension via promoting AVP release (94). Kim and his colleague revealed that GABA does not consistently function as an inhibitory neurotransmitter in the brain network according to their study (95). These contradictory studies just indicate that it is unclear what the role of the GABAergic system is in the pathogenesis of hypertension, which is exactly the question that later researchers need to answer.
4.3 Neurogenic hypertension
Increased sympathetic outflow is considered to be a major contributor in the pathogenesis of hypertension, with both glutamatergic and GABAergic system being involved (72, 74, 96). PVN pre-sympathetic neurons receive tonic and inhibitory inputs, the balance of which determines the excitability of PVN pre-sympathetic neurons (49). The GABAergic current in PVN neurons of SHR was markedly lower compared with controls, a phenomenon that could be reversed by antagonizing GABARs concomitant with an improvement in hypertension (97). In another study using BPH/2J mice as subjects, it was found that maintenance of hypertension may be due to GABAAR structure and function alteration, reducing tonic neuronal inhibition of amygdalohypothalamic network and leading to subsequent sympathetic tension (98). This might account for the disrupted function of GABA receptors and GABAergic inputs to PVN that may lead to increased hypertensive sympathetic outflow in the neurogenic hypertension. It has also been noted a disruption of chloride homeostasis in PVN presympathetic neurons reduces GABAergic inhibition and plays a key role in the hyperexcitability of presympathetic neurons and elevated sympathetic vasomotor tone (96). Numerous studies have identified chronic intermittent hypoxia (CIH) contributes to neurogenic hypertension as well. Short term CIH induces a relatively rapid adaptive response that drives the tonic activity of PVN sympathetically regulated neurons and their acute excitability, a possible mechanism by which CIH evokes neurogenic hypertension (99). Farmer et al. further explored the reason behind this uncontrolled excitation, highlighting the possibility of CIH disrupting GABAA-mediated inhibition (100). While a lot of research pointed to a regulatory role for GABAergic system in neurogenic hypertension, its promise as therapeutic target has not been fully demonstrated. Even if some studies have confirmed the therapeutic potential of targeting the GABAergic system through specific injections of the drug in mice brains, an unbridgeable gap remains before an actually use of it for the cure of neurogenic hypertension.
4.4 Ang II-induced hypertension and other hypertension
The bed nucleus of the stria terminalis (BST) is a region rich in GABAergic neurons known to regulate cardiovascular parameters. Ang II upregulates the E3 ubiquitination ligase that targets it and promotes angiotensin-converting enzyme 2 (ACE2) ubiquitination and degradation. Onset of this ACE2 ubiquitination in BNST GABAergic neurons decreases PVN presympathetic inputs, promoting hypertension (101). In addition, brain Ang II increases mean arterial pressure and sympathetic outflow by working on AT1R in diverse parts of the brain, including the PVN, RVLM and nucleus of the solitary tract (NTS) (90). Ang II mediated stimulation of AT1R in the NTS leads to activation of eNOS or (and) nNOS, followed by local production of NO, enhancing GABA release from NTS interneurons (90). GABA release in the NTS inhibits glutamatergic neuronal projections to the caudal ventrolateral medulla (CVLM), reducing activation of inhibitory GABAergic nerves in CVLM and subsequently deregulating sympathetically-driven glutamatergic neurons in the RVLM (102). Control of GABAergic is also exhibited in other forms of hypertension. Haywood et al. confirmed that injection of GABA receptor inhibition in bilateral PVN resulted in a significant increase in BP and HR in renal-wrap hypertensive rats (103). They also concluded stronger GABAergic activity is the outcome of increased GABA release from the PVN (104). Maycon et al. instead uncovered that peripheral GABAergic control may also be part of the pathogenesis of renal hypertension, suggesting the equal importance of peripheral and central E/I balance in the pathogenesis of hypertension (105). Likewise, exercise could elevate glutamic acid decarboxylase (GAD) expression, which increases GABA levels to attenuate renal sympathoexcitability and hypertension by diminishing GABAergic deficits in the caudal hypothalamus (106, 107).
5 Nav isoforms and the potential effects of Nav-mediated E/I in hypertension
Chronic stress has garnered significant attention due to its capacity to trigger a cascade of pathophysiologic alterations, thereby contributing to kinds of cardiovascular disease via the (CNS) (85). For instance, sympathetic overactivity and neuronal excitation imbalance in the brain are involved in the regulation of hypertension (108). Except for chronic stress, sodium (Na+) intake is another environmental factor known to influence BP; increased Na+ heightens an individual's susceptibility to hypertension while the reduction of Na+ intake alleviates hypertensives (109). In human beings, there is a continuum of BP sensitivity to sodium, which is related to vascular endothelium and smooth muscle, renin-angiotensin-aldosterone system, immune responsive system. Additionally, the specific regions of the brain would influence the salt-sensing variability (110–112). It has been reported that epithelial sodium channels contribute to the salt sensitivity of BP in the kidney (113), the organ primarily responsible for sodium homeostasis.
Recent studies have found that function or expression changes of sodium, potassium, and calcium plasma channels in specific brain regions are related to the regulation of hypertension (114). Among these channels, Nav are normally responsible for the generation and propagation of action potentials in neurons, which directly affects the excitability of neurons and is involved in the regulation of Ca2+ transfer and the release of glutamate and/or GABA at the end of axons (115). The mammalian Nav family consists of 10 α-subunit genes encoding Nav1.1–Nav1.9. Nav1.1–1.9, each of which is expressed in different tissues and exert different functions. Mutations in Nav1.1 result in altered sodium channel activity occurring primarily on inhibitory interneurons with consequent slow recovery from inactivation, greater use-dependent inactivation and reduced action potential firing in interneurons. These changes ultimately lead to diminished inhibition and E/I imbalance within the brain (116). By contrast, Nav1.2 mutations caused a reduction in Na+ currents primarily on pyramidal neurons, with decreased excitatory synaptic inputs to pyramidal neurons in brain slices, whereas interneurons were unaffected, which biased the brain towards an inhibitory imbalance (117). Mice heterozygous of Nav1.6 had neurons in hippocampal CA1 in a state of hyperexcitability with a significant increase in the rate of rising rate of action potentials, spike amplitude and input resistance. Neuronal excitability in the cortex was also increased, which have led to the neural network hyperexcitability. Apart from this, these mice exhibited simultaneous tonic extension and bradycardia on ECG (118). Some other types of Nav introduce E/I imbalance response as well, which we will elaborate on in the following sections. Along with the direct alterations in Nav, the auxiliary protein fibroblast growth factor 13, which encodes Nav, would lead to decreased inhibitory synaptic inputs and increased excitatory synaptic inputs, leading to an E/I imbalance in the neural networks (119). In the CNS, Nav-mediated abnormal electrical activity of neurons can induce the neurological network E/I imbalance and lead to anxiety (120, 121). Then, would the Nav mutations in the brain affect the BP and the formation of hypertension? Our laboratory has long been involved in research related to ion channels, and we hypothesize the existence of additional central mechanisms involvement of ion channels in hypertension.
5.1 Stress-induced hypertension
Various Nav isoforms expressed in different tissues and exert different functions. Na1.1, Na1.2, Nav1.3, and Na1.6 are the primary isoforms expressed in the CNS, Nav1.4 is distributed in skeletal muscle, and Nav1.5 is only expressed in cardiac muscle. Especially, Nav1.6 is highly expressed in excitatory neurons, and is also shown in inhibitory neurons. We have reported that downregulating or inhibiting Nav1.6 suppresses intracellular Ca2+ accumulation, thereby attenuating the pathogenesis of Alzheimer's disease (122) and 6-OHDA-induced neurotoxicity and neuroinflammation (123). Recent findings by Lei Tong et al. declared that Nav1.6 is overexpressed in the RVLM neurons in SHR, accompanied by increased vesicular glutamate transporter 1 (VGluT1) expression and VGluT1-positive neurons, alongside decreased GAD67 expression and GAD67-labeled inhibitory neurons (124). In stress-induced hypertensive rats subjected to electrical foot and noise, the Nav1.6 expression in RVLM is markedly increased, whereas knockdown the Nav1.6 in RVLM significantly ameliorates stress-induced in SBP, HR and sympathetic nerve activity in rats (125). Calcium ion is the switch for glutamate release, and GAD catalyzes the conversion of glutamate to GABA. These indicate the E/I imbalance, resulting from the overactivated glutamate system and (or) inhibited GABAergic system, may be induced by increased Nav1.6 and Ca2+ accumulation, thereby mediating the stress-induced elevation of BP.
No reports are available to reveal a correlation between Nav1.7 and hypertension, although attempts to explore this area have concluded a lack of association (126, 127). Nav1.8 in the brain is mainly expressed in a few regions including lateral septal nucleus, BST, dorsal striatum, amygdala, hypothalamus, and the ventral periaqueductal gray (128), several of which may be involved in the pathogenesis of hypertension (26, 39, 101). The involvement of Nav1.8 in prolactin-regulated stress-induced behavior response was found in a study on preclinical migraine models done by Bianca and his colleagues (129). Another study also confirmed that stress-induced an upregulation of neuronal Nav1.8 expression, although this alteration was transient (130), which was corroborated by Helia et al. on their discovery that neurons transiently expressing Nav1.8 were able to sense noxious stimuli in the brain (128). Upregulated of Nav1.8 in prefrontal neurons undergoes excitatory deficits including reduced spiking frequency of action potential and depolarization of resting membrane potentials (131). Knockout out of Nav1.8 on dorsal root ganglion neurons prevents them from emitting all-or-none action potentials at the resting potential state (132). Above studies confirm the involvement of Nav1.8 on CNS and peripheral neurons trigger deficits in neuronal excitability, but lack evidence tying it to E/I imbalance and hypertension which deserves further exploration.
5.2 Salt-induced hypertension
Neuronal activity is affected by the concentration of Na+ in the CSF, which is extremely sensitive. An increase of 2 mmol/L in the Na+ concentration in the CSF triggers neuronal discharges (133), whereas chronic elevation of CSF [Na+] by 5 mmol/L results in sympathetic hyperactivity and hypertension (134). The sodium concentration in this fluid depends on the sodium transport between plasma and CSF by CP epithelial cells through the epithelial sodium channels (ENaCs) and Na + -K + -ATPase. High-salt diet increased the concentration of sodium ions in the CSF of rats, which led to the increase of sympathetic nervous system excitability, BP and HR, and these effects were more significant in Dahl-sensitive rats (135). The abnormal regulation of Na+ transport and Na + -K + -ATPase activity may be one of the reasons for the increase of [Na+] in CSF in Dahl salt-sensitive rats fed a high-salt diet, but not related to the abnormal regulation of ENaCs (136). They also found the reduction of [Na+] influx caused by blocking ENaCs by benzamil still resulted in elevated [Na+] in CSF even leaving the salt diet (136). It reflects the key role of ENaCs of transporting Na+ in salt-induced hypertension. Furthermore, the sympathetic and pressor responses to intracerebroventricular injection of sodium-rich artificial CSF were greater in Dahl S Rats than in salt-insensitive (R) or Wistar rats (135). This may be due to the acute and chronic increases in [Na+] that reduce NO release, leading to an imbalance of E/I and increasing Na + -induced hypertension via enhancing Ang-II release and AT1R activation in PVN (137).
5.3 Neurogenic hypertension and Ang II-induced hypertension
Some debate over the notion that excessive sodium intake leads to elevated [Na+] or CSF [Na+] which later causes hypertension. Nomura et al., in studying neurogenic hypertension, found that Nax (encoded by SCN7A) knock out mice failed to manifest mean blood pressure increases even after high-salt ingestion (138). Elevated extracellular [Na+] in the organum vasculosum lamina terminalis (OVLT) activates Nax, which stimulates the release of H+ from Nax-positive glial cells. The released H+ provokes OVLT (projecting to PVN) neurons to further activate PVN (projecting to RVLM) neurons, thereby increasing sympathetic activity and leading to hypertension. The OVLT's significant role in neurogenic hypertension likely stems from its lack of a typical blood-brain barrier and proximity to the third ventricle, allowing it to sense increased [Na+] in both the blood and CSF (138). This strained sympathetic activity is associated with elevated Nav1.6 expression in the RVLM, and reducing Nav1.6 expression results in decreased sympathetic activity (125). Overexpression of Nav1.6 leads to intracellular calcium accumulation via Na + -Ca2+ exchange in the brain (123), triggering excessive release of excitatory neurotransmitters thus initiating E/I imbalance (139). However, definitive evidence linking Nav1.6 to neurogenic hypertension through these mechanisms is still lacking, requiring further research. Gabor et al. demonstrate that glutamate receptor-dependent signaling in the PVN contributes to the BP maintenance in Ang II-hypertensive Wistar rats (77). Despite overexpression of Nav1.6 could bring excessive glutamate release based on previous studies (122, 123), it is difficult to justify the role of Nav1.6 in development of Ang II-dependent hypertension. More than just neurogenic hypertension, the role of Nav in the pathogenesis of Ang II-induced hypertension needs more exploration.
6 Conclusion
Hypertension is so far incurable that current treatment relying on medications to control the BP. Most hypertensive patients are required lifelong use of two or more antihypertensive drugs, leading to reduced compliance and disruption of normal routines. We reviewed the important role of neural network E/I imbalance in the pathogenesis of hypertension (Figure 1), suggesting its potential as a therapeutic target. This opens the possibility of exploring approaches such as transcutaneous or transcranial electrical stimulation targeting specific brain regions, nano-delivery systems, and targeted drugs to optimize disturbed brain network. These interventions could potentially lead to a curable outcome for hypertension, enabling patients to live a routine life free from lifelong medication.
Figure 1
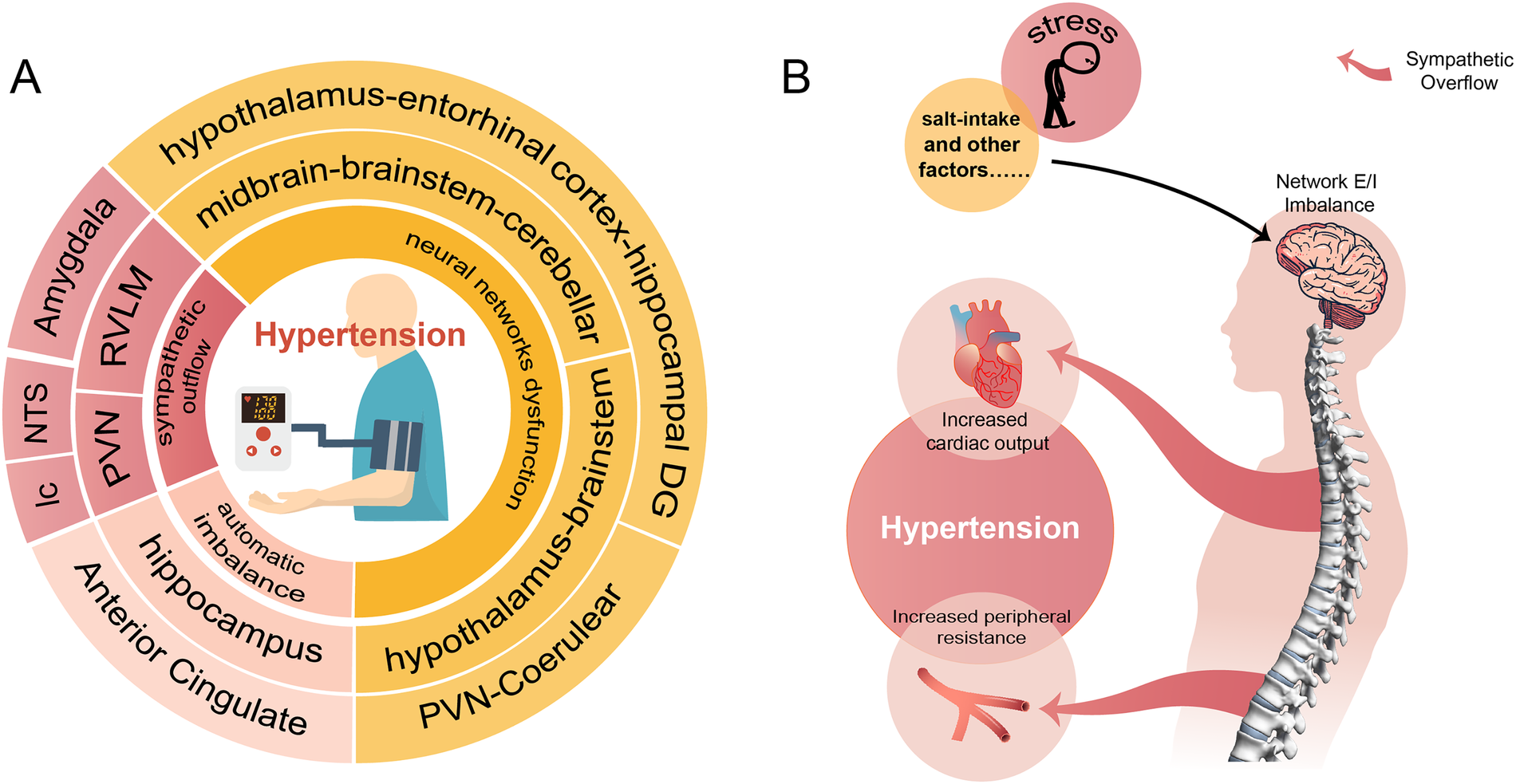
A conclusion of the neural network involved in the pathogenesis of hypertension. (A) Shows that hypertension involves neural mechanisms (innermost circle) and corresponding brain regions (two outer circles of the same color). (B) Demonstrates the process of external stimuli causing hypertension through disturbed neural networks and sympathetic overflow.
Statements
Author contributions
MX: Investigation, Writing – original draft, Writing – review & editing. TW: Data curation, Investigation, Writing – original draft. YW: Data curation, Writing – original draft. TH: Writing – original draft. DC: Supervision, Writing – review & editing. BW: Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the National Natural Sciences Foundation of China (82301700), the Natural Science Foundation of Liaoning Province of China (2024-MS-157), Scientific Research Projects from Wuhan Municipal Health Commission (WX23Z26).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Lewington S Clarke R Qizilbash N Peto R Collins R , Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. (2002) 360:1903–13. 10.1016/s0140-6736(02)11911-8
2.
Ettehad D Emdin CA Kiran A Anderson SG Callender T Emberson J et al Blood pressure lowering for prevention of cardiovascular disease and death: a systematic review and meta-analysis. Lancet. (2016) 387:957–67. 10.1016/S0140-6736(15)01225-8
3.
Forouzanfar MH Liu P Roth GA Ng M Biryukov S Marczak L et al Global burden of hypertension and systolic blood pressure of at least 110 to 115 mm hg, 1990–2015. JAMA. (2017) 317:165–82. 10.1001/jama.2016.19043
4.
Whelton PK Carey RM Aronow WS Casey DE Collins KJ Dennison Himmelfarb C et al 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation. (2018) 138:e426–83. 10.1161/CIR.0000000000000597
5.
GBD 2017 Risk Factor Collaborators. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet. (2018) 392:1923–94. 10.1016/S0140-6736(18)32225-6
6.
Williams B Mancia G Spiering W Agabiti Rosei E Azizi M Burnier M et al 2018 practice guidelines for the management of arterial hypertension of the European society of hypertension and the European Society of Cardiology: ESH/ESC task force for the management of arterial hypertension. J Hypertens. (2018) 36:2284–309. 10.1097/HJH.0000000000001961
7.
National Clinical Guideline Centre (UK). Hypertension: The Clinical Management of Primary Hypertension in Adults: Update of Clinical Guidelines 18 and 34. London: Royal College of Physicians (UK). (2011). Available online at:http://www.ncbi.nlm.nih.gov/books/NBK83274/(accessed October 7, 2023).
8.
Harris E . Majority of people live with uncontrolled hypertension worldwide. JAMA. (2023) 330(16):1515. 10.1001/jama.2023.19204
9.
Patel NK Javed S Khan S Papouchado M Malizia AL Pickering AE et al Deep brain stimulation relieves refractory hypertension. Neurology. (2011) 76:405–7. 10.1212/WNL.0b013e3182088108
10.
Yuste R . From the neuron doctrine to neural networks. Nat Rev Neurosci. (2015) 16:487–97. 10.1038/nrn3962
11.
Niciu MJ Kelmendi B Sanacora G . Overview of glutamatergic neurotransmission in the nervous system. Pharmacol Biochem Behav. (2012) 100:656–64. 10.1016/j.pbb.2011.08.008
12.
Oh WC Smith KR . Activity-dependent development of GABAergic synapses. Brain Res. (2019) 1707:18–26. 10.1016/j.brainres.2018.11.014
13.
Goland RS Jozak S Warren WB Conwell IM Stark RI Tropper PJ . Elevated levels of umbilical cord plasma corticotropin-releasing hormone in growth-retarded fetuses. J Clin Endocrinol Metab. (1993) 77:1174–9. 10.1210/jcem.77.5.8077309
14.
Bertram CE Hanson MA . Animal models and programming of the metabolic syndrome. Br Med Bull. (2001) 60:103–21. 10.1093/bmb/60.1.103
15.
Stein AD Zybert PA van der Pal-de Bruin K Lumey LH . Exposure to famine during gestation, size at birth, and blood pressure at age 59 y: evidence from the Dutch famine. Eur J Epidemiol. (2006) 21:759–65. 10.1007/s10654-006-9065-2
16.
Barra R Morgan C Sáez-Briones P Reyes-Parada M Burgos H Morales B et al Facts and hypotheses about the programming of neuroplastic deficits by prenatal malnutrition. Nutr Rev. (2019) 77:65–80. 10.1093/nutrit/nuy047
17.
Wang M Pan W Xu Y Zhang J Wan J Jiang H . Microglia-mediated neuroinflammation: a potential target for the treatment of cardiovascular diseases. J Inflamm Res. (2022) 15:3083–94. 10.2147/JIR.S350109
18.
Guyenet PG . The sympathetic control of blood pressure. Nat Rev Neurosci. (2006) 7:335–46. 10.1038/nrn1902
19.
Cayupe B Troncoso B Morgan C Sáez-Briones P Sotomayor-Zárate R Constandil L et al The role of the paraventricular-coerulear network on the programming of hypertension by prenatal undernutrition. Int J Mol Sci. (2022) 23:11965. 10.3390/ijms231911965
20.
Lauder L Mahfoud F Böhm M . Management of resistant hypertension. Annu Rev Med. (2024) 75:443–57. 10.1146/annurev-med-050922-052605
21.
Penninx BW Pine DS Holmes EA Reif A . Anxiety disorders. Lancet. (2021) 397:914–27. 10.1016/S0140-6736(21)00359-7
22.
Hung K-C Chu C-C Hsing C-H Chang Y-P Li Y-Y Liu W-C et al Association between perioperative intravenous lidocaine and subjective quality of recovery: a meta-analysis of randomized controlled trials. J Clin Anesth. (2021) 75:110521. 10.1016/j.jclinane.2021.110521
23.
Duffy B . Life Under Lockdown: Coronavirus in the UK. London: King’s College London: The Policy Institution (2020).
24.
Zhang S Zhong Y Wang L Yin X Li Y Liu Y et al Anxiety, home blood pressure monitoring, and cardiovascular events among older hypertension patients during the COVID-19 pandemic. Hypertens Res. (2022) 45:856–65. 10.1038/s41440-022-00852-0
25.
Etkin A Wager TD . Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. Am J Psychiatry. (2007) 164:1476–88. 10.1176/appi.ajp.2007.07030504
26.
Sheng Z-F Zhang H Zheng P Chen S Gu Z Zhou J-J et al Impaired Kv7 channel activity in the central amygdala contributes to elevated sympathetic outflow in hypertension. Cardiovasc Res. (2022) 118:585–96. 10.1093/cvr/cvab031
27.
Marins FR Iddings JA Fontes MAP Filosa JA . Evidence that remodeling of insular cortex neurovascular unit contributes to hypertension-related sympathoexcitation. Physiol Rep. (2017) 5:e13156. 10.14814/phy2.13156
28.
Cheung RT Cechetto DF . Neuropeptide changes following excitotoxic lesion of the insular cortex in rats. J Comp Neurol. (1995) 362:535–50. 10.1002/cne.903620408
29.
Biancardi VC Bergamaschi CT Lopes OU Campos RR . Sympathetic activation in rats with L-NAME-induced hypertension. Braz J Med Biol Res. (2007) 40:401–8. 10.1590/S0100-879X2006005000077
30.
McIntosh RC Lobo JD Yang A Schneiderman N . Brainstem network connectivity with mid-anterior insula predicts lower systolic blood pressure at rest in older adults with hypertension. J Hum Hypertens. (2021) 35:1098–108. 10.1038/s41371-020-00476-2
31.
Nagai M Hoshide S Ishikawa J Shimada K Kario K . Insular cortex atrophy as an independent determinant of disrupted diurnal rhythm of ambulatory blood pressure in elderly hypertension. Am J Hypertens. (2009) 22:723–9. 10.1038/ajh.2009.71
32.
Gianaros PJ Jennings JR Sheu LK Derbyshire SWG Matthews KA . Heightened functional neural activation to psychological stress covaries with exaggerated blood pressure reactivity. Hypertension. (2007) 49:134–40. 10.1161/01.HYP.0000250984.14992.64
33.
Gianaros PJ Onyewuenyi IC Sheu LK Christie IC Critchley HD . Brain systems for baroreflex suppression during stress in humans. Hum Brain Mapp. (2012) 33:1700–16. 10.1002/hbm.21315
34.
Hyam JA Kringelbach ML Silburn PA Aziz TZ Green AL . The autonomic effects of deep brain stimulation–a therapeutic opportunity. Nat Rev Neurol. (2012) 8:391–400. 10.1038/nrneurol.2012.100
35.
Shivkumar K Ardell JL . Cardiac autonomic control in health and disease. J Physiol. (2016) 594:3851–2. 10.1113/JP272580
36.
Chaar LJ Coelho A Silva NM Festuccia WL Antunes VR . High-fat diet-induced hypertension and autonomic imbalance are associated with an upregulation of CART in the dorsomedial hypothalamus of mice. Physiol Rep. (2016) 4:e12811. 10.14814/phy2.12811
37.
Shiina K Tomiyama H Takata Y Yoshida M Kato K Saruhara H et al Effects of CPAP therapy on the sympathovagal balance and arterial stiffness in obstructive sleep apnea. Respir Med. (2010) 104:911–6. 10.1016/j.rmed.2010.01.010
38.
Herrera NA Jesus I Shinohara AL Dionísio TJ Santos CF Amaral SL . Exercise training attenuates dexamethasone-induced hypertension by improving autonomic balance to the heart, sympathetic vascular modulation and skeletal muscle microcirculation. J Hypertens. (2016) 34:1967–76. 10.1097/HJH.0000000000001032
39.
Li Y-Y Liu J-P Shi S-F Yang K-Z Gong Y Sun J et al Acupuncture with twirling reinforcing and reducing manipulation shows a control of hypertension and regulation of blood pressure-related target brain regions in spontaneously hypertensive rat: a preliminary resting-state functional MRI study. Front Neurosci. (2023) 17:1161578. 10.3389/fnins.2023.1161578
40.
Guo Q Feng X Xue H Jin S Teng X Duan X et al Parental renovascular hypertension-induced autonomic dysfunction in male offspring is improved by prenatal or postnatal treatment with hydrogen sulfide. Front Physiol. (2019) 10:1184. 10.3389/fphys.2019.01184
41.
Zhou J-J Shao J-Y Chen S-R Li D-P Pan H-L . α2δ-1-dependent NMDA receptor activity in the hypothalamus is an effector of genetic-environment interactions that drive persistent hypertension. J Neurosci. (2021) 41:6551–63. 10.1523/JNEUROSCI.0346-21.2021
42.
Meldrum BS . Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. (2000) 130:1007S–15. 10.1093/jn/130.4.1007S
43.
McKenna MC . The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J Neurosci Res. (2007) 85:3347–58. 10.1002/jnr.21444
44.
Yadava N Nicholls DG . Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J Neurosci. (2007) 27:7310–7. 10.1523/JNEUROSCI.0212-07.2007
45.
Rozanski A Blumenthal JA Kaplan J . Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation. (1999) 99:2192–217. 10.1161/01.cir.99.16.2192
46.
Wang Y Xia M Lu J Wang T Zhang X Ntim M et al TIP60 mediates stress-induced hypertension via promoting glutamatedmPFC-to-vCA1 release. Clin Exp Hypertens. (2023) 45:2259130. 10.1080/10641963.2023.2259130
47.
Basting T Xu J Mukerjee S Epling J Fuchs R Sriramula S et al Glutamatergic neurons of the paraventricular nucleus are critical contributors to the development of neurogenic hypertension. J Physiol. (2018) 596:6235–48. 10.1113/JP276229
48.
Ulrich-Lai YM Herman JP . Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. (2009) 10:397–409. 10.1038/nrn2647
49.
Dampney RA Michelini LC Li D-P Pan H-L . Regulation of sympathetic vasomotor activity by the hypothalamic paraventricular nucleus in normotensive and hypertensive states. Am J Physiol Heart Circ Physiol. (2018) 315:H1200–14. 10.1152/ajpheart.00216.2018
50.
Ma H Chen S-R Chen H Pan H-L . Endogenous AT1 receptor-protein kinase C activity in the hypothalamus augments glutamatergic input and sympathetic outflow in hypertension. J Physiol. (2019) 597:4325–40. 10.1113/JP278427
51.
Ye Z-Y Li D-P Li L Pan H-L . Protein kinase CK2 increases glutamatergic input in the hypothalamus and sympathetic vasomotor tone in hypertension. J Neurosci. (2011) 31:8271–9. 10.1523/JNEUROSCI.1147-11.2011
52.
Qiao X Zhou J-J Li D-P Pan H-L . Src kinases regulate glutamatergic input to hypothalamic presympathetic neurons and sympathetic outflow in hypertension. Hypertension. (2017) 69:154–62. 10.1161/HYPERTENSIONAHA.116.07947
53.
Frazier CJ Harden SW Alleyne AR Mohammed M Sheng W Smith JA et al An angiotensin-responsive connection from the lamina terminalis to the paraventricular nucleus of the hypothalamus evokes vasopressin secretion to increase blood pressure in mice. J Neurosci. (2021) 41:1429–42. 10.1523/JNEUROSCI.1600-20.2020
54.
Fan J-F Wang W Tan X Ye P Li J-K Niu L-Y et al Contribution of cyclooxygenase-2 overexpression to enhancement in tonically active glutamatergic inputs to the rostral ventrolateral medulla in hypertension. J Hypertens. (2022) 40:2394–405. 10.1097/HJH.0000000000003268
55.
Guyenet PG Stornetta RL Holloway BB Souza GMPR Abbott SBG . Rostral ventrolateral medulla and hypertension. Hypertension. (2018) 72:559–66. 10.1161/HYPERTENSIONAHA.118.10921
56.
Xiang H-B Liu C Liu T-T Xiong J . Central circuits regulating the sympathetic outflow to lumbar muscles in spinally transected mice by retrograde transsynaptic transport. Int J Clin Exp Pathol. (2014) 7:2987–97.
57.
Zha Y-P Wang Y-K Deng Y Zhang R-W Tan X Yuan W-J et al Exercise training lowers the enhanced tonically active glutamatergic input to the rostral ventrolateral medulla in hypertensive rats. CNS Neurosci Ther. (2013) 19:244–51. 10.1111/cns.12065
58.
Jiang L Zhou X Yang H Guan R Xin Y Wang J et al Upregulation of AT1 receptor mediates a pressor effect through ROS-SAPK/JNK signaling in glutamatergic neurons of rostral ventrolateral medulla in rats with stress-induced hypertension. Front Physiol. (2018) 9:1860. 10.3389/fphys.2018.01860
59.
Zhou X Yang H Song X Wang J Shen L Wang J . Central blockade of the AT1 receptor attenuates pressor effects via reduction of glutamate release and downregulation of NMDA/AMPA receptors in the rostral ventrolateral medulla of rats with stress-induced hypertension. Hypertens Res. (2019) 42:1142–51. 10.1038/s41440-019-0242-6
60.
Frisoli TM Schmieder RE Grodzicki T Messerli FH . Salt and hypertension: is salt dietary reduction worth the effort?Am J Med. (2012) 125:433–9. 10.1016/j.amjmed.2011.10.023
61.
Blaustein MP Leenen FHH Chen L Golovina VA Hamlyn JM Pallone TL et al How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol. (2012) 302:H1031–49. 10.1152/ajpheart.00899.2011
62.
Stocker SD Monahan KD Browning KN . Neurogenic and sympathoexcitatory actions of NaCl in hypertension. Curr Hypertens Rep. (2013) 15:538–46. 10.1007/s11906-013-0385-9
63.
Stocker SD Lang SM Simmonds SS Wenner MM Farquhar WB . Cerebrospinal fluid hypernatremia elevates sympathetic nerve activity and blood pressure via the rostral ventrolateral medulla. Hypertension. (2015) 66:1184–90. 10.1161/HYPERTENSIONAHA.115.05936
64.
Ito S Hiratsuka M Komatsu K Tsukamoto K Kanmatsuse K Sved AF . Ventrolateral medulla AT1 receptors support arterial pressure in dahl salt-sensitive rats. Hypertension. (2003) 41:744–50. 10.1161/01.HYP.0000052944.54349.7B
65.
Zheng T Wu Y Peng M-J Xiao N-Q Tan Z-J Yang T . Hypertension of liver-yang hyperactivity syndrome induced by a high salt diet by altering components of the gut microbiota associated with the glutamate/GABA-glutamine cycle. Front Nutr. (2022) 9:964273. 10.3389/fnut.2022.964273
66.
Gabor A Leenen FHH . Cardiovascular effects of angiotensin II and glutamate in the PVN of dahl salt-sensitive rats. Brain Res. (2012) 1447:28–37. 10.1016/j.brainres.2012.01.060
67.
Yang T Richards EM Pepine CJ Raizada MK . The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat Rev Nephrol. (2018) 14:442–56. 10.1038/s41581-018-0018-2
68.
Santisteban MM Kim S Pepine CJ Raizada MK . Brain-gut-bone marrow axis: implications for hypertension and related therapeutics. Circ Res. (2016) 118:1327–36. 10.1161/CIRCRESAHA.116.307709
69.
Yang T Santisteban MM Rodriguez V Li E Ahmari N Carvajal JM et al Gut dysbiosis is linked to hypertension. Hypertension. (2015) 65:1331–40. 10.1161/HYPERTENSIONAHA.115.05315
70.
Bi Q Wang C Cheng G Chen N Wei B Liu X et al Microglia-derived PDGFB promotes neuronal potassium currents to suppress basal sympathetic tonicity and limit hypertension. Immunity. (2022) 55:1466–82.e9. 10.1016/j.immuni.2022.06.018
71.
Parkhurst CN Yang G Ninan I Savas JN Yates JR Lafaille JJ et al Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. (2013) 155:1596–609. 10.1016/j.cell.2013.11.030
72.
Zhou J-J Shao J-Y Chen S-R Pan H-L . Brain α2δ-1-bound NMDA receptors drive calcineurin inhibitor-induced hypertension. Circ Res. (2023) 133:611–27. 10.1161/CIRCRESAHA.123.322562
73.
Zhou J Shao J Chen S Chen H Pan H . Calcineurin regulates synaptic ca 2+ -permeable AMPA receptors in hypothalamic presympathetic neurons via α2δ-1-mediated GluA1/GluA2 assembly. J Physiol (Lond). (2024) 602:2179–97. 10.1113/JP286081
74.
Li D-P Zhou J-J Zhang J Pan H-L . CaMKII regulates synaptic NMDA receptor activity of hypothalamic presympathetic neurons and sympathetic outflow in hypertension. J Neurosci. (2017) 37:10690–9. 10.1523/JNEUROSCI.2141-17.2017
75.
Zhou J-J Shao J-Y Chen S-R Chen H Pan H-L . α2δ-1 protein promotes synaptic expression of Ca2+permeable-AMPA receptors by inhibiting GluA1/GluA2 heteromeric assembly in the hypothalamus in hypertension. J Neurochem. (2022) 161:40–52. 10.1111/jnc.15573
76.
Zhou J-J Pachuau J Li D-P Chen S-R Pan H-L . Group III metabotropic glutamate receptors regulate hypothalamic presympathetic neurons through opposing presynaptic and postsynaptic actions in hypertension. Neuropharmacology. (2020) 174:108159. 10.1016/j.neuropharm.2020.108159
77.
Gabor A Leenen FHH . Central mineralocorticoid receptors and the role of angiotensin II and glutamate in the paraventricular nucleus of rats with angiotensin II-induced hypertension. Hypertension. (2013) 61:1083–90. 10.1161/HYPERTENSIONAHA.111.00797
78.
Woods C Marques-Lopes J Contoreggi NH Milner TA Pickel VM Wang G et al Tumor necrosis factor α receptor type 1 activation in the hypothalamic paraventricular nucleus contributes to glutamate signaling and angiotensin II-dependent hypertension. J Neurosci. (2021) 41:1349–62. 10.1523/JNEUROSCI.2360-19.2020
79.
Weston M Wang H Stornetta RL Sevigny CP Guyenet PG . Fos expression by glutamatergic neurons of the solitary tract nucleus after phenylephrine-induced hypertension in rats. J Comp Neurol. (2003) 460:525–41. 10.1002/cne.10663
80.
Ferrari MFR Fior-Chadi DR . Chronic nicotine administration. Analysis of the development of hypertension and glutamatergic neurotransmission. Brain Res Bull. (2007) 72:215–24. 10.1016/j.brainresbull.2006.09.013
81.
Yu B Cai D . Neural programmatic role of leptin, TNFα, melanocortin, and glutamate in blood pressure regulation vs obesity-related hypertension in male C57BL/6 mice. Endocrinology. (2017) 158:1766–75. 10.1210/en.2016-1872
82.
Shi Z Li B Brooks VL . Role of the paraventricular nucleus of the hypothalamus in the sympathoexcitatory effects of leptin. Hypertension. (2015) 66:1034–41. 10.1161/HYPERTENSIONAHA.115.06017
83.
Sahara S Yanagawa Y O’Leary DDM Stevens CF . The fraction of cortical GABAergic neurons is constant from near the start of cortical neurogenesis to adulthood. J Neurosci. (2012) 32:4755–61. 10.1523/JNEUROSCI.6412-11.2012
84.
Knabl J Witschi R Hösl K Reinold H Zeilhofer UB Ahmadi S et al Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature. (2008) 451:330–4. 10.1038/nature06493
85.
Hering D Lachowska K Schlaich M . Role of the sympathetic nervous system in stress-mediated cardiovascular disease. Curr Hypertens Rep. (2015) 17:80. 10.1007/s11906-015-0594-5
86.
Seryapina AA Shevelev OB Moshkin MP Markel AL Akulov AE . Stress-sensitive arterial hypertension, haemodynamic changes and brain metabolites in hypertensive ISIAH rats: MRI investigation. Exp Physiol. (2017) 102:523–32. 10.1113/EP086064
87.
Du D Hu L Wu J Wu Q Cheng W Guo Y et al Neuroinflammation contributes to autophagy flux blockage in the neurons of rostral ventrolateral medulla in stress-induced hypertension rats. J Neuroinflammation. (2017) 14:169. 10.1186/s12974-017-0942-2
88.
Xia C-M Shao C-H Xin L Wang Y-R Ding C-N Wang J et al Effects of melatonin on blood pressure in stress-induced hypertension in rats. Clin Exp Pharmacol Physiol. (2008) 35:1258–64. 10.1111/j.1440-1681.2008.05000.x
89.
Du D Chen J Liu M Zhu M Jing H Fang J et al The effects of angiotensin II and angiotensin-(1–7) in the rostral ventrolateral medulla of rats on stress-induced hypertension. PLoS One. (2013) 8:e70976. 10.1371/journal.pone.0070976
90.
Légat L Smolders I Dupont AG . AT1 receptor mediated hypertensive response to Ang II in the nucleus tractus solitarii of normotensive rats involves NO dependent local GABA release. Front Pharmacol. (2019) 10:460. 10.3389/fphar.2019.00460
91.
Gao H-L Yu X-J Liu K-L Shi X-L Qi J Chen Y-M et al PVN blockade of p44/42 MAPK pathway attenuates salt-induced hypertension through modulating neurotransmitters and attenuating oxidative stress. Sci Rep. (2017) 7:43038. 10.1038/srep43038
92.
Yan X Jin J Su X Yin X Gao J Wang X et al Intestinal flora modulates blood pressure by regulating the synthesis of intestinal-derived corticosterone in high salt-induced hypertension. Circ Res. (2020) 126:839–53. 10.1161/CIRCRESAHA.119.316394
93.
Jin X Kim WB Kim M-N Jung WW Kang HK Hong E-H et al Oestrogen inhibits salt-dependent hypertension by suppressing GABAergic excitation in magnocellular AVP neurons. Cardiovasc Res. (2021) 117:2263–74. 10.1093/cvr/cvaa271
94.
Kim Y-B Kim YS Kim WB Shen F-Y Lee SW Chung HJ et al GABAergic excitation of vasopressin neurons: possible mechanism underlying sodium-dependent hypertension. Circ Res. (2013) 113:1296–307. 10.1161/CIRCRESAHA.113.301814
95.
Kim JS Kim WB Kim Y-B Lee Y Kim YS Shen F-Y et al Chronic hyperosmotic stress converts GABAergic inhibition into excitation in vasopressin and oxytocin neurons in the rat. J Neurosci. (2011) 31:13312–22. 10.1523/JNEUROSCI.1440-11.2011
96.
Ye Z-Y Li D-P Byun HS Li L Pan H-L . NKCC1 upregulation disrupts chloride homeostasis in the hypothalamus and increases neuronal activity-sympathetic drive in hypertension. J Neurosci. (2012) 32:8560–8. 10.1523/JNEUROSCI.1346-12.2012
97.
Li D-P Pan H-L . Role of gamma-aminobutyric acid (GABA)A and GABAB receptors in paraventricular nucleus in control of sympathetic vasomotor tone in hypertension. J Pharmacol Exp Ther. (2007) 320:615–26. 10.1124/jpet.106.109538
98.
Stevenson ER Johns EMC Marques FZ Jackson KL Davern PJ Evans RG et al Positive allosteric modulation of GABAA receptors attenuates high blood pressure in Schlager hypertensive mice. J Hypertens. (2017) 35:546–57. 10.1097/HJH.0000000000001210
99.
Sharpe AL Calderon AS Andrade MA Cunningham JT Mifflin SW Toney GM . Chronic intermittent hypoxia increases sympathetic control of blood pressure: role of neuronal activity in the hypothalamic paraventricular nucleus. Am J Physiol Heart Circ Physiol. (2013) 305:H1772–80. 10.1152/ajpheart.00592.2013
100.
Farmer GE Little JT Marciante AB Cunningham JT . AT1a-dependent GABAA inhibition in the MnPO following chronic intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol. (2021) 321:R469–81. 10.1152/ajpregu.00030.2021
101.
Mohammed M Ogunlade B Elgazzaz M Berdasco C Lakkappa N Ghita I et al Nedd4–2 up-regulation is associated with ACE2 ubiquitination in hypertension. Cardiovasc Res. (2023) 119:2130–41. 10.1093/cvr/cvad070
102.
Schreihofer AM Guyenet PG . Baro-activated neurons with pulse-modulated activity in the rat caudal ventrolateral medulla express GAD67 mRNA. J Neurophysiol. (2003) 89:1265–77. 10.1152/jn.00737.2002
103.
Martin DS Haywood JR . Reduced GABA inhibition of sympathetic function in renal-wrapped hypertensive rats. Am J Physiol. (1998) 275:R1523–9. 10.1152/ajpregu.1998.275.5.R1523
104.
Haywood JR Mifflin SW Craig T Calderon A Hensler JG Hinojosa-Laborde C . gamma-Aminobutyric acid (GABA)–A function and binding in the paraventricular nucleus of the hypothalamus in chronic renal-wrap hypertension. Hypertension. (2001) 37:614–8. 10.1161/01.hyp.37.2.614
105.
Milanez MIO Nishi EE Rocha AA Bergamaschi CT Campos RR . Interaction between angiotensin II and GABA in the spinal cord regulates sympathetic vasomotor activity in Goldblatt hypertension. Neurosci Lett. (2020) 728:134976. 10.1016/j.neulet.2020.134976
106.
Kramer JM Beatty JA Little HR Plowey ED Waldrop TG . Chronic exercise alters caudal hypothalamic regulation of the cardiovascular system in hypertensive rats. Am J Physiol Regul Integr Comp Physiol. (2001) 280:R389–97. 10.1152/ajpregu.2001.280.2.R389
107.
Ferreira-Junior NC Ruggeri A Silva SD Zampieri TT Ceroni A Michelini LC . Exercise training increases GAD65 expression, restores the depressed GABAA receptor function within the PVN and reduces sympathetic modulation in hypertension. Physiol Rep. (2019) 7:e14107. 10.14814/phy2.14107
108.
Pinto IS Mourão AA da Silva EF Camargo AS Marques SM Gomes KP et al Blockade of rostral ventrolateral medulla (RVLM) bombesin receptor type 1 decreases blood pressure and sympathetic activity in anesthetized spontaneously hypertensive rats. Front Physiol. (2016) 7:205. 10.3389/fphys.2016.00205
109.
Blumenthal JA Sherwood A Smith PJ Hinderliter A . The role of salt reduction in the management of hypertension. J Am Coll Cardiol. (2018) 71:1597–8. 10.1016/j.jacc.2018.01.071
110.
Kelly TN He J . Genomic epidemiology of blood pressure salt sensitivity. J Hypertens. (2012) 30:861–73. 10.1097/HJH.0b013e3283524949
111.
Pitzer AL Van Beusecum JP Kleyman TR Kirabo A . ENaC in salt-sensitive hypertension: kidney and beyond. Curr Hypertens Rep. (2020) 22:69. 10.1007/s11906-020-01067-9
112.
Mutchler SM Kleyman TR . New insights regarding epithelial na+ channel regulation and its role in the kidney, immune system and vasculature. Curr Opin Nephrol Hypertens. (2019) 28:113–9. 10.1097/MNH.0000000000000479
113.
Mutchler SM Kirabo A Kleyman TR . Epithelial sodium channel and salt-sensitive hypertension. Hypertension. (2021) 77:759–67. 10.1161/HYPERTENSIONAHA.120.14481
114.
Geraldes V Laranjo S Rocha I . Hypothalamic ion channels in hypertension. Curr Hypertens Rep. (2018) 20:14. 10.1007/s11906-018-0814-x
115.
Zhang J Chen X Eaton M Wu J Ma Z Lai S et al Severe deficiency of the voltage-gated sodium channel NaV1.2 elevates neuronal excitability in adult mice. Cell Rep. (2021) 36:109495. 10.1016/j.celrep.2021.109495
116.
Martin MS Dutt K Papale LA Dubé CM Dutton SB de Haan G et al Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J Biol Chem. (2010) 285:9823–34. 10.1074/jbc.M109.078568
117.
Wang H-G Bavley CC Li A Jones RM Hackett J Bayleyen Y et al Scn2a severe hypomorphic mutation decreases excitatory synaptic input and causes autism-associated behaviors. JCI Insight. (2021) 6:e150698. 10.1172/jci.insight.150698
118.
Bunton-Stasyshyn RKA Wagnon JL Wengert ER Barker BS Faulkner A Wagley PK et al Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain. (2019) 142:362–75. 10.1093/brain/awy324
119.
Puranam RS He XP Yao L Le T Jang W Rehder CW et al Disruption of Fgf13 causes synaptic excitatory-inhibitory imbalance and genetic epilepsy and febrile seizures plus. J Neurosci. (2015) 35:8866–81. 10.1523/JNEUROSCI.3470-14.2015
120.
Zhang J-Y Liu T-H He Y Pan H-Q Zhang W-H Yin X-P et al Chronic stress remodels synapses in an amygdala circuit-specific manner. Biol Psychiatry. (2019) 85:189–201. 10.1016/j.biopsych.2018.06.019
121.
Liu W-Z Zhang W-H Zheng Z-H Zou J-X Liu X-X Huang S-H et al Identification of a prefrontal cortex-to-amygdala pathway for chronic stress-induced anxiety. Nat Commun. (2020) 11:2221. 10.1038/s41467-020-15920-7
122.
Yuan D-J Yang G Wu W Li Q-F Xu D-E Ntim M et al Reducing Nav1.6 expression attenuates the pathogenesis of Alzheimer’s disease by suppressing BACE1 transcription. Aging Cell. (2022) 21:e13593. 10.1111/acel.13593
123.
Li X Wu X Li N Li D Sui A Khan K et al Scorpion venom heat-resistant synthesized peptide ameliorates 6-OHDA-induced neurotoxicity and neuroinflammation: likely role of Nav 1.6 inhibition in microglia. Br J Pharmacol. (2021) 178:3553–69. 10.1111/bph.15502
124.
Tong L Xing M Wu J Zhang S Chu D Zhang H et al Overexpression of NaV1.6 in the rostral ventrolateral medulla in rats mediates stress-induced hypertension via glutamate regulation. Clin Exp Hypertens. (2022) 44:134–45. 10.1080/10641963.2021.2007942
125.
Wu J-X Tong L Hu L Xia C-M Li M Chen Q-H et al Upregulation of Nav1.6 expression in the rostral ventrolateral medulla of stress-induced hypertensive rats. Hypertens Res. (2018) 41:1013–22. 10.1038/s41440-018-0105-6
126.
Fong R Ballow CH Naik H Steiner D Palmer J White WB . Effects of a state- and use-dependent Nav1.7 channel blocker on ambulatory blood pressure: a randomized, controlled crossover study. J Clin Pharmacol. (2019) 59:90–7. 10.1002/jcph.1298
127.
Morales PS Escobar RG Lizama M Aglony M Salomon J Drenth JPH et al Paediatric hypertension-associated erythromelalgia responds to corticosteroids and is not associated with SCN9A mutations. Rheumatology (Oxford). (2012) 51:2295–6. 10.1093/rheumatology/kes098
128.
Tenza-Ferrer H Collodetti M Nicolau ES de Birbrair A Magno LAV Romano-Silva MA . Transiently Nav1.8-expressing neurons are capable of sensing noxious stimuli in the brain. Front Cell Neurosci. (2022) 16:933874. 10.3389/fncel.2022.933874
129.
Mason BN Kallianpur R Price TJ Akopian AN Dussor GO . Prolactin signaling modulates stress-induced behavioral responses in a preclinical mouse model of migraine. Headache. (2022) 62:11–25. 10.1111/head.14248
130.
Zheng G Hong S Hayes JM Wiley JW . Chronic stress and peripheral pain: evidence for distinct, region-specific changes in visceral and somatosensory pain regulatory pathways. Exp Neurol. (2015) 273:301–11. 10.1016/j.expneurol.2015.09.013
131.
Rannals MD Hamersky GR Page SC Campbell MN Briley A Gallo RA et al Psychiatric risk gene transcription factor 4 regulates intrinsic excitability of prefrontal neurons via repression of SCN10a and KCNQ1. Neuron. (2016) 90:43–55. 10.1016/j.neuron.2016.02.021
132.
Nascimento de Lima AP Zhang H Chen L Effraim PR Gomis-Perez C Cheng X et al Nav1.8 in small dorsal root ganglion neurons contributes to vincristine-induced mechanical allodynia. Brain. (2024) 147(9):3157–70. 10.1093/brain/awae071
133.
Honda K Negoro H Dyball RE Higuchi T Takano S . The osmoreceptor complex in the rat: evidence for interactions between the supraoptic and other diencephalic nuclei. J Physiol. (1990) 431:225–41. 10.1113/jphysiol.1990.sp018328
134.
Huang BS Van Vliet BN Leenen FHH . Increases in CSF [Na+] precede the increases in blood pressure in Dahl S rats and SHR on a high-salt diet. Am J Physiol Heart Circ Physiol. (2004) 287:H1160–6. 10.1152/ajpheart.00126.2004
135.
Huang BS Wang H Leenen FH . Enhanced sympathoexcitatory and pressor responses to central na+ in Dahl salt-sensitive vs. -resistant rats. Am J Physiol Heart Circ Physiol. (2001) 281:H1881–9. 10.1152/ajpheart.2001.281.5.H1881
136.
Amin MS Reza E Wang H Leenen FHH . Sodium transport in the choroid plexus and salt-sensitive hypertension. Hypertension. (2009) 54:860–7. 10.1161/HYPERTENSIONAHA.108.125807
137.
Gabor A Leenen FHH . Mechanisms mediating sodium-induced pressor responses in the PVN of dahl rats. Am J Physiol Regul Integr Comp Physiol. (2011) 301:R1338–49. 10.1152/ajpregu.00246.2011
138.
Nomura K Hiyama TY Sakuta H Matsuda T Lin C-H Kobayashi K et al [Na+] increases in body fluids sensed by central nax induce sympathetically mediated blood pressure elevations via H+-dependent activation of ASIC1a. Neuron. (2019) 101:60–75.e6. 10.1016/j.neuron.2018.11.017
139.
Jaime Tobón LM Moser T . Ca2+regulation of glutamate release from inner hair cells of hearing mice. Proc Natl Acad Sci U S A. (2023) 120:e2311539120. 10.1073/pnas.2311539120
Summary
Keywords
hypertension, neural network, E/I imbalance, neural connection, sympathetic outflow
Citation
Xia M, Wang T, Wang Y, Hu T, Chen D and Wang B (2024) A neural perspective on the treatment of hypertension: the neurological network excitation and inhibition (E/I) imbalance in hypertension. Front. Cardiovasc. Med. 11:1436059. doi: 10.3389/fcvm.2024.1436059
Received
21 May 2024
Accepted
29 August 2024
Published
11 September 2024
Volume
11 - 2024
Edited by
Gino Seravalle, Italian Auxological Institute (IRCCS), Italy
Reviewed by
Xing Tan, Naval Medical Center, China
Robert A. Larson, Michigan Technological University, United States
Updates
Copyright
© 2024 Xia, Wang, Wang, Hu, Chen and Wang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Min Xia shiu911@163.com Defang Chen chdfc@126.com Bin Wang wb101900@126.com
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.