Abstract
The protease thrombin, which increases its levels with various pathologies, can signal through the G protein-coupled receptors protease-activated receptors 1 and 4 (PAR1/PAR4). PAR1 is a high-affinity receptor for thrombin, whereas PAR4 is a low-affinity receptor. Finding functions for PAR4 in endothelial cells (ECs) has been an elusive goal over the last two decades. Several studies have demonstrated a lack of functionality for PAR4 in ECs, with many claiming that PAR4 function is confined mostly to platelets. A recent study from our lab identified low expressing but functional PAR4 in hepatic ECs in vivo. We also found that PAR4 likely has a higher signaling potency than PAR1. Given this potency, ECs seem to limit PAR4 signaling except for extreme cases. As a result, we claim PAR4 is not an impotent receptor because it is low expressing, but rather PAR4 is low expressing because it is a very potent receptor. Since we have finally shown PAR4 to be present and functional on ECs in vivo, it is important to outline why such controversy arose over the last two decades and, more importantly, why the receptor was undervalued on ECs. This timely review aims to inspire investigators in the field of vascular biology to study the regulatory aspect of endothelial PAR4 and its relationship with the more highly expressed PAR1.
Introduction
The vascular endothelium, which is composed of the innermost lining of cells in blood vessels, covers a surface area of 270–720 square meters in humans (1, 2). Endothelial cells (ECs) play numerous roles in regulating tissue structure (3) and function (4) by impacting inflammation (5), permeability (6), and trafficking of proteins and nutrients (6). Given these roles, it is no surprise that dysfunctional ECs can contribute to pathological insults (7). One of these insults is a proteolytic storm; like a cytokine storm, it is a sudden and rapid rise of protease activity. In the vasculature, this can lead to disseminated intravascular coagulopathy (8), sepsis (9), and some cancers (10). Given that proteases and their inhibitors constitute over 2% of the genes in the human genome (11), understanding how these proteases signal, particularly in ECs, is critical to understanding disease progression.
Protease-activated receptors (PARs) act as cellular sensors for the proteolytic state of the extracellular environment. The PAR family of G protein-coupled receptors (GPCRs) is comprised of four variants in mammals (PAR1-4) (Figures 1A–D), which can convert an extracellular cleavage event from a variety of proteases (12) into a transmembrane signaling event. This is mechanistically accomplished by the receptor carrying its internal ligand [i.e., tethered ligand (TL)] on its N-terminus (13). The ligand is masked by an exodomain, which prevents the receptor from signaling, with a protease cleavage site bridging the two domains (Figures 1E–H). Upon proteolytic cleavage, the exodomain is released, and the receptor signals through the interaction of the TL with the body of the receptor, specifically the second extracellular loop (ECL2) domain of the receptor (Figure 1H) (14). Dysregulation of PARs has been linked to numerous pathological conditions, including cancer, inflammation, and thrombosis (15).
Figure 1
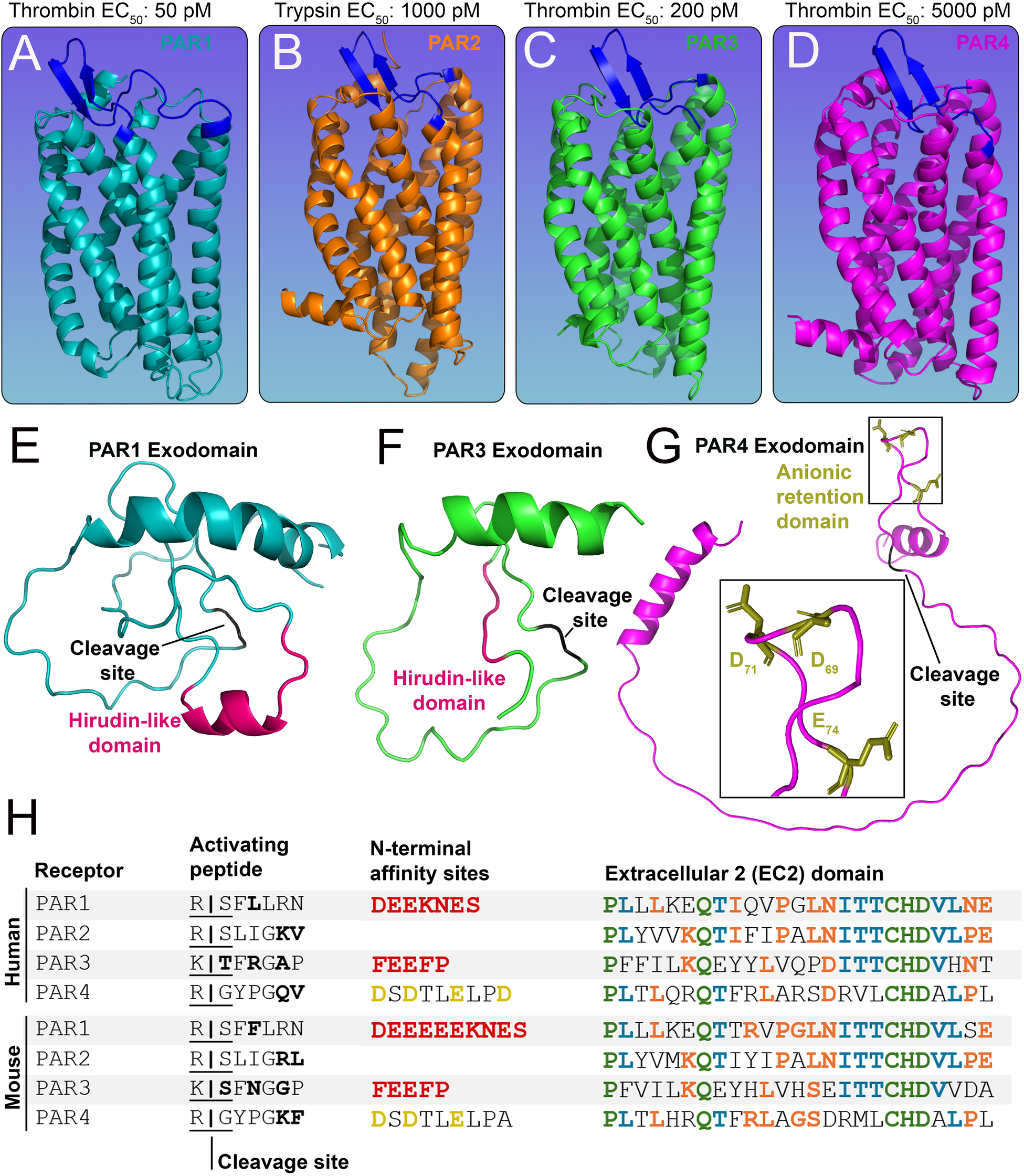
Structures of the PAR family of receptors. (A-D)In silico models of murine (A) PAR1 (cyan) (B) PAR2 (orange) (C) PAR3 (green) and (D) PAR4 (pink) with EC50 values for predominant activating proteases shown above. The extracellular 2 (ECL2) domain of each receptor is shown in blue. (E-G) The N-terminal exodomain of (E) PAR1 and (F) PAR3 with a red hirudin-like domain (HLD) and a black cleavage site. The N-terminal exodomain of (G) PAR4 shows the anionic retention domain in gold and the cleavage site in black. (H) Tables of human and mouse PAR1-4 protein sequences showing the activating peptide, cleavage sites (residues variable between mouse and human receptors are shown in bold), and N-terminal affinity sites showing HLDs (red) for PAR1, PAR3, and the anion retention domain (gold) for PAR4. Also shown are protein sequences of the ECL2 domain; residues shared by four (green), three (blue), and two (orange) PARs are highlighted accordingly.
PAR1 (Figure 1A), PAR3 (Figure 1C), and PAR4 (Figure 1D) can be activated by thrombin, whereas PAR2 (Figure 1B) is primarily activated by trypsin (15, 16). PAR3 is a co-receptor (17) and has a limited capacity to signal by itself (14), but it can enhance PAR1 and PAR4 signaling (17). PAR1 and PAR3 possess a hirudin-like-domain (HLD) on their N-termini (18, 19) (Figures 1E–F) and are the high-affinity receptors for thrombin (EC50: 50 pM and EC50: 200 pM, respectively) (16, 20). PAR4, on the other hand, is the low-affinity receptor for thrombin (EC50: 5,000 pM) (16) (Figure 1). The HLD mimics hirudin, a protein produced by leeches that acts as an anticoagulant (21); it binds exosite I of thrombin, which is distinct from the enzyme's active site that cleaves substrate peptides. Although PAR4 lacks an HLD, it does contain an anionic retention cluster (human: D57, D59, E62, D65, mice: D69, D71, E74) (Figure 1G), which slows the dissociation of cationic thrombin and prolongs the interaction time of thrombin with the receptor (22), allowing for bound thrombin to cleave the receptor more efficiently to initiate PAR4 signaling.
PAR1 is well characterized on ECs as the predominant mediator of thrombin signaling. PAR4, on the other hand, has minimal characterization in ECs, and there is significant controversy on whether it is even expressed in these cells (6, 23, 24). This review outlines what is known about PAR4 on ECs and provides a vision of how this controversial receptor signals in the endothelium based on recent breakthroughs in endothelial PAR4 research.
A brief history of thrombin signaling
The ancient Greek medical philosophers Galen, Hippocrates, Plato, and Aristotle all attempted to interpret the phenomena of blood clotting and tried to understand its functional consequences (25). One of the earliest references to the word thrombus is found in Hippocrates's Corpus Hippocraticum, which refers to the “lump” rising from the coagulation of bodily fluids (25). By 1872, the German physiologist Alexander Schmit hypothesized the existence of an enzyme that converts fibrinogen into fibrin (26). At the time Schmit referred to the enzyme as fibrin ferment (27). By 1894, inactive prothrombin was isolated from plasma by the Dutch physiologist Cornelis Pekelharing (28). By 1954, platelets were shown to be activated by thrombin, demonstrating that the enzyme could not only mediate proteolytic cleavage but also cellular effects (29). Following the successful culture of ECs in vitro in the early 1970s (30), thrombin was also shown to have cellular effects on ECs (31).
However, it was not until 1991 that the thrombin receptor (later renamed PAR1) was identified (20). Yet, almost immediately, a limitation of the receptor was noted: given that PAR1 is quickly and irreversibly activated at extremely low concentrations of thrombin (EC50: 50 pM), how can it modulate variable responses to different thrombin concentrations (32)? Theoretically, PAR1 should always signal with zero-order kinetics at physiologic concentrations of thrombin, which ranges from 1 nM (0.1 U/ml) to 500 nM (50 U/ml) during coagulation (27). Furthermore, given that the majority of PAR1 molecules are rapidly removed from the cell surface following the receptor's activation (33), it is unlikely that persistent thrombin concentrations would have variable signaling responses. In 1993, Ishii and colleagues postulated that “quantums” of second messenger were produced following PAR1 activation, and cells may be able to detect balances between different rates of receptor activation and second messenger clearance, thus allowing for variable thrombin responses. However, a simpler answer was determined in 1998, with the identification of the low-affinity thrombin receptor, PAR4 (34, 35). With the identification of PAR4, it was then understood how cells could mediate responsiveness to high and low concentrations of thrombin using a system of dual receptors (36, 37).
Controversy surrounding endothelial PAR4
By the early 2000s, PAR4 was shown to be present in murine pulmonary ECs, with PAR1 and PAR4 serving partially redundant roles in mediating thrombin responses in these cells (24). However, since 2003 there has been a paucity of studies identifying roles for endothelial PAR4. A PubMed search for “Endothelial”, “Protease”, and “PAR4” yields only 88 publications in the last 21 years; by comparison, the same period yields 615 publications related to endothelial PAR1. Of those 88 PAR4 publications, 16 claim they could not find a function for endothelial PAR4 in their respective models (38–53). This is likely due to a lack of suitable in vitro models since PAR4 shows limited expression and responses in human umbilical vein EC (HUVECs) (23), which are frequently used for in vitro EC studies. Additionally, endothelial PAR4 studies have been difficult to reproduce between labs (6, 54). For example, Vogel and colleagues found that a thrombin-induced increase in endothelial permeability was fully abrogated in Par1-deficient mice (54), unlike a similar study by Kataoka and colleagues that found partial redundancy between both PAR1 and PAR4 on ECs (24). The combination of low expression on ECs and mixed results regarding functionality has relegated PAR4 to be viewed as an irrelevant receptor on the endothelium—until now.
PAR4 is a potent and functional receptor in the hepatic endothelium
We have recently shown that murine liver ECs express functional PAR4, albeit at low levels, with the Par1:Par4 expression ratio in hepatic ECs being 153:1 (1). In a model of acetaminophen (APAP) overdose, the hepatic vasculature becomes compromised, which presents as increased permeability and erythrocyte congestion in the liver (1). Simultaneously, there is a rise in thrombin generation (1), which can lead to endothelial PAR activation. Using mice with conditional deletions for Par1 and/or Par4 in ECs, we showed that both receptors contribute independently to APAP-induced bleeding and permeability and that endothelial PAR1 and PAR4 act synergistically to drive APAP-induced permeability in the liver (1). Most importantly, we found that the loss of PAR4 in ECs was comparable to the loss of PAR1 in terms of phenocopying vascular protection against APAP-induced vascular dysfunction. Thus, even though Par4 constitutes <1% of Par transcripts in hepatic ECs, it mediates a response equivalent to the other 99%, suggesting that endothelial PAR4 is not only functional but is extremely potent compared to endothelial PAR1. This may be due to PAR4 potentially being able to generate massive second messenger levels due to its lack of receptor desensitization following activation (55–57). Altogether, our studies show PAR4 to be a low-expressing but potent receptor on hepatic ECs (1, 58).
Organotypic endothelial heterogeneity of PARs
Organotypic expression of endothelial PARs
ECs show remarkable heterogeneity in structure and function between different tissue beds (59). This also extends to organotypic gene expression. Given that PAR4 is functional at extremely low levels, an obvious question is whether these levels vary between ECs in an organotypic fashion. There were indications of this only a few years following the discovery of PAR4. In 2004, Fujiwara and colleagues reported the presence of PAR4 on aortic ECs but not on pulmonary artery ECs and HUVECs (60). However, there has never been a comprehensive study on this topic until our recent report (1). Using translating ribosome affinity purification (TRAP) datasets, we showed that Par4 is expressed at low levels in ECs of most murine tissue beds in vivo, with transcripts per million mapped reads (TPM) values being substantially lower than Par1 (1, 59). Furthermore, endothelial Par4 has organotypic differences in expression, with transcripts being limited to only a few of the organs that we analyzed in mice (Figure 2), unlike Par1, which is expressed ubiquitously among ECs of different organs (1, 59).
Figure 2
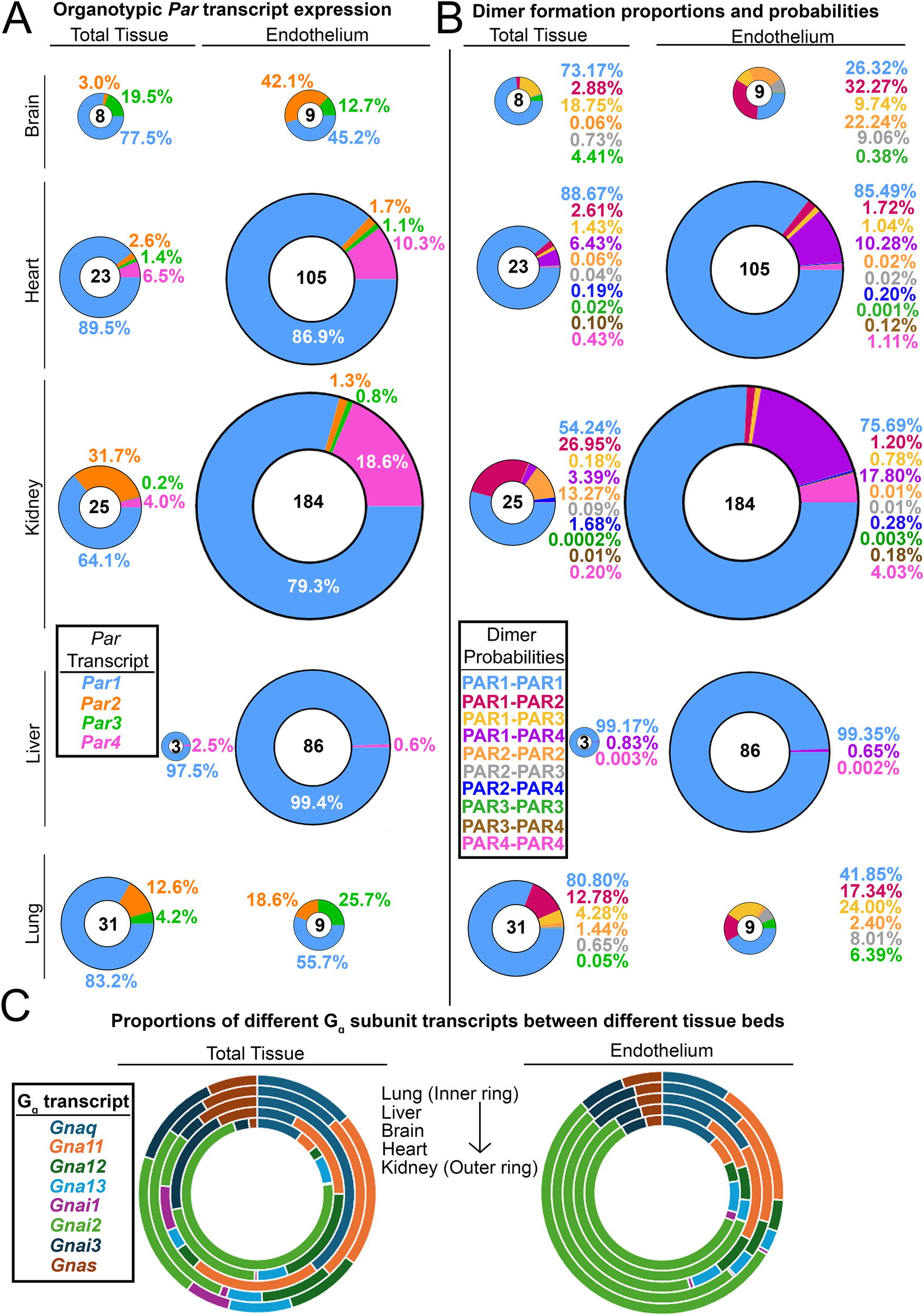
Organotypic differences in endothelial PARs and their G proteins. (A) Donut charts of transcript expression of all PARs in total tissues from different organs and the endothelium isolated from those organs. (B) Donut charts of PAR dimer formation probabilities in total tissues and endothelium from different organs. For A and B, the total transcript count for all PARs is shown in the center of each donut, and all donuts are sized to scale. (C) The proportion of G protein alpha subunit transcripts found in different organs and the endothelium isolated from those organs. Data for all organs in A–C (except for the liver) were generated by Cleuren et al. (59); liver data were generated by Rajala et al. (1).
What appears to be more interesting is the organs in which PAR4 expression is undetectable in vivo, such as the brain and the lung. These are two organs in which alterations in permeability lead to severe injury via stroke or pulmonary edema, respectively. Given that PAR4 is highly potent, lung and brain ECs may rely only on PAR1 for thrombin signaling, as signaling with impunity (as PAR4 does) could be incompatible with preserving barrier function in these organs.
Organotypic heterodimerization of endothelial PARs
All PARs have been shown to homo- and heterodimerize (61–63). This dimerization requires allosteric changes induced via thrombin-mediated receptor cleavage (61). However, the functional relevance of this dimerization is still an open question. Potential effects may include increased efficiency in activating protease recruitment (37), modified G-protein activity/second messenger production, altered dimerization-mediated coupling to different G-proteins (64), and differential internalization and trafficking of these receptors. Given the many possible variations in PAR dimer formations, careful combinatorial approaches to designing PAR experiments should take precedence over the assumption that specific dimers are responsible for cellular events.
As mentioned above, endothelial PAR expression ratios vary organotypically in vivo (Figure 2A). Assuming that homo- and heterodimerization kinetics are equivalent between all four receptors, one can easily determine the predicted formation probability of particular dimer products in organ-specific ECs based solely on expression ratios (Figure 2B). PAR1 homodimers are the most likely to form, with PAR1-PAR4 heterodimers being the second most likely in ECs of the kidney (∼18%) and heart (∼10%). In fact, the Par1:Par4 expression ratio on renal ECs is 4:1, which is higher than the 5:1 PAR1:PAR4 receptor ratio on human platelets [PAR1; 2,500 copies/platelet (65): PAR4; 500 copies/platelet (66)]. Since the likelihood of PAR1:PAR4 heterodimer formation is high in renal ECs, this tissue may serve as a model system to study how PAR1/4 heterodimers influence EC function and signaling. Altogether, these results highlight the fact that since dimer formation has different probabilities in different tissues, identifying the function of endothelial PAR dimers may need to be done in an organ-specific manner.
Organotypic expression of endothelial G proteins
One final category of organotypic variation that may affect endothelial PAR signaling is that of G protein alpha (Gα) subunits. It has been hypothesized that the complexity that arises from GPCR signaling is due in part to variations in Gα subunits (67). Theoretically, all receptors, when stimulated with the same ligand, should elicit similar responses. However, variations in how GPCRs couple to G proteins can result in diverse functional responses to the same ligand. We were curious if Gα subunits—like PARs—varied between ECs of different tissue beds; if so, this could contribute to organotypic PAR responses. Interestingly, we found that the expression of different Gα subunits does not vary in ECs of different organs (Figure 2C), although Gα subunit expression does vary when compared across total tissues of different organs (Figure 2C). This suggests that if ECs display diversity in PAR responses, it may be driven more by organotypic variations in PAR expression as opposed to variations in the expression of the Gα transducers to which PARs couple.
Regulation of PAR4 expression in endothelial cells
Given that PAR4 is expressed at low levels but is functional in hepatic ECs, an obvious question is how its expression is regulated in ECs? Answers to this question may reveal reasons why PAR4 functions the way it does.
Shear stress
We recently reported that PAR4 expression increases around 13-fold in HUVECs exposed to shear stress and that this upregulation is comparable to that seen with the known shear stress responsive endothelial genes (Krüppel-like factor 2) KLF2 and KLF4 (1). Likewise, PAR4 expression is increased in vivo in regions of high flow in the liver (1), as well as in vessels of high caliber (high flow) in the lung (68). If shear is a prerequisite for PAR4 expression, this may explain why static culture models have consistently failed to reveal the expression of PAR4 in ECs in vitro (23, 60).
This shear-induced upregulation of endothelial PAR4 should be considered in the context of thrombin availability, which is diluted by blood flow. In an injured blood vessel, thrombin is generated from the cleavage of the zymogen prothrombin by FV or FX (69). This activation occurs locally near the injury site and does not occur systemically in circulation, as that could lead to disseminated coagulation. Additionally, thrombin is only active for less than one minute following prothrombin conversion (70) due to rapid inhibition by antithrombin in circulation (27). If high flow is maintained around the site of thrombin activation, it further decreases the local residency time of thrombin. Given that the activating protease residence time is proportional to the probability of thrombin encountering lumenal endothelial PAR4, the constant dilution of locally generated thrombin would decrease the likelihood of PAR4 being activated. Thus, under high shear stress—when ECs are likely to increase expression of PAR4—the likelihood of PAR4 being activated is decreased unless local thrombin concentrations are high enough to overcome dilution by blood flow (Figure 3A). Otherwise, under slow flow conditions, the low expression of PAR4 would help limit its activation.
Figure 3
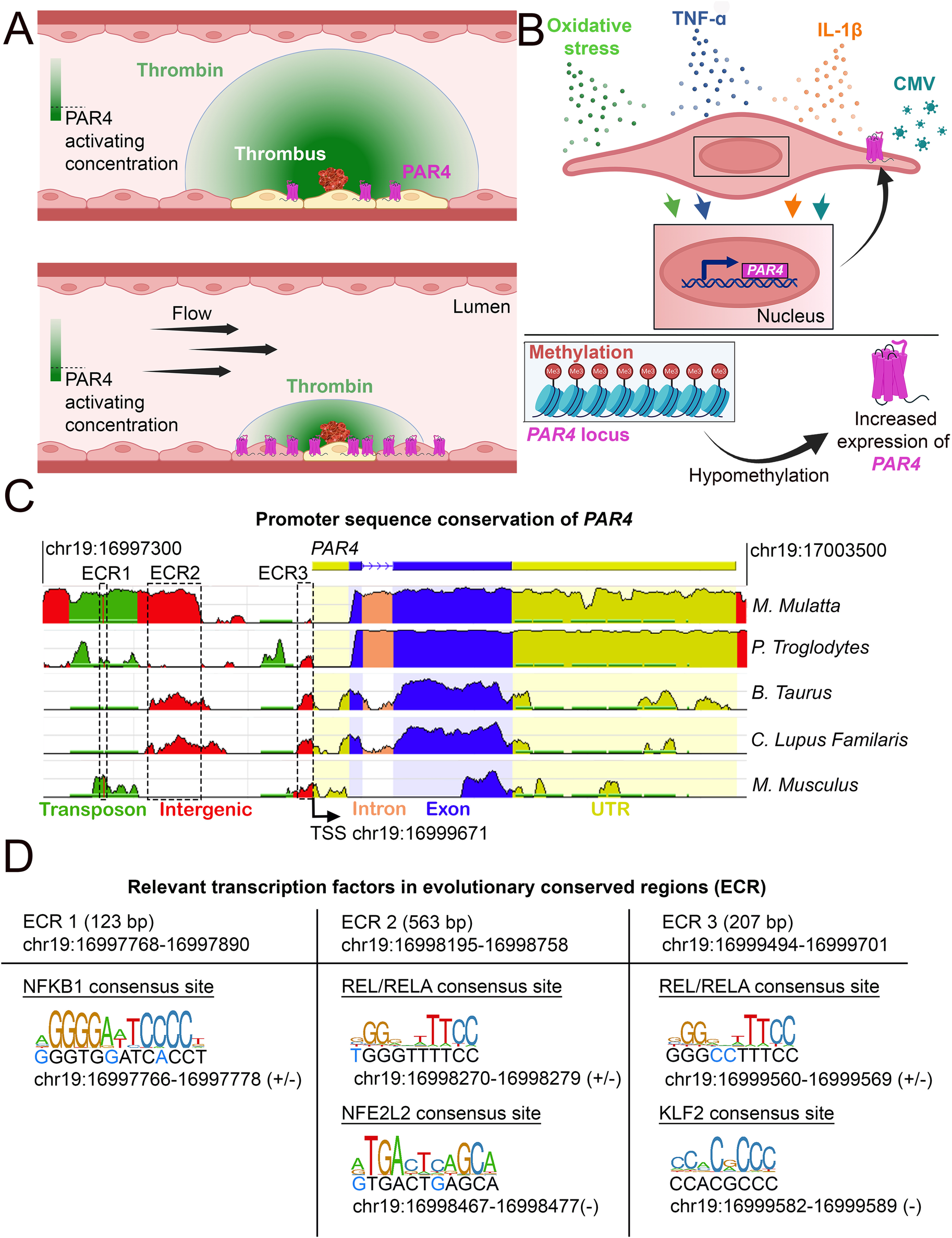
Conditions under which PAR4 expression is altered. (A) Schematic of how endothelial PAR4 (pink) increases with shear stress but how shear stress (flow) would limit the number of ECs activated by a point source of thrombin. The thrombin gradient is depicted in green. The use of differential color in ECs (yellow) depicts ECs engaging in PAR4 signaling. (B) Conditions under which PAR4 expression has been shown to increase in ECs. (C) Analysis of evolutionarily conserved regions (ECRs) in the promoter of PAR4 (human) between different species. Sequence homology was assessed using the NCBI DCODE website (http://www.dcode.org). Peak heights indicate the degree of sequence homology between species. Transposons (green), intergenic regions (red), intron (salmon), exons (blue), untranslated regions (yellow), and ECRs are annotated with dashed lines. (D) Relevant transcription factor binding sites (TFBS) in the three ECRs of PAR4. The sizes and locations of each ECR are given. Locations and strand(s) of each TFBS in the human genome are provided underneath each motif diagram. Nucleotides in the human genome that diverge from the motif sequence are identified with blue text.
Inflammation and oxidative stress
Another major driver of PAR4 expression in ECs is inflammation (Figure 3B). Cytomegalovirus-infected HUVECs demonstrate increased PAR4 expression (71). Studies have also shown that both human coronary arteries and HUVECs treated with the inflammatory cytokines IL-1β or TNF-α have increased PAR4 expression (72–75). In models of diabetic inflammation, increased vascular expression of PAR4 occurs (76–79). Similar effects can be seen in mice on a high-fat diet; elevated cardiac PAR4 expression correlates with increased IL-1β levels (80). Interestingly, PAR4 activation in cardiomyocytes results in increased TNF-α and IL-1β production, suggesting potential feedback between expression of PAR4 and inflammatory cytokines (81). However, this feedback mechanism has not yet been shown in ECs.
Cardiac microvascular ECs treated with homocysteine, which causes oxidative stress, show increased Par4 expression (82). ECs isolated from patients with cerebral cavernous malformations, which originate from an environment of both inflammation and oxidative stress, also show increased PAR4 expression (83). Thus, cytokine-mediated inflammation and/or oxidative stress are associated with increased PAR4 expression. One open question, however, is whether PAR4 signals the same under conditions of inflammation or shear stress. Future studies should focus on addressing this question.
Epigenetic regulation
Cigarette smoking leads to DNA hypomethylation at the PAR4 locus; this reduction in methylation is associated with increases in gene and protein expression for PAR4 (Figure 3B) (84). This hypomethylation has also been associated with an increased risk of death due to myocardial infarction (84). Furthermore, platelets from individuals with hypomethylation of the PAR4 locus show increased reactivity to a PAR4 agonist (84). Independently, it has been shown that PAR4 hypomethylation in DNA from blood cells is a strong predictor of all-cause mortality (85). Similar effects can be observed in cancer cells, in which PAR4 is expressed only when the promoter is hypomethylated (86–88).
It is also noteworthy that PAR4 is located on a different chromosome from the other PAR genes. PAR1, PAR2, and PAR3 all map to Ch.5q13.3 and Ch.13D2 in humans and mice, respectively, whereas PAR4 maps to Ch.19p12 in humans and Ch.8B3.3 in mice (89). Given the known methylation of the PAR4 genomic locus, it is possible that PAR4's spatial separation from the other PAR genes further facilitates distinct and tight epigenetic and transcription control over its expression. Altogether, we predict that methylation is another way of suppressing potent PAR4 expression on ECs, and it would be interesting to determine whether shear stress, inflammation, and oxidative stress can reverse this silencing epigenetic mark in pathological contexts.
PAR4 promoter conservation
Our cross-species analysis of the human PAR4 promoter region revealed three evolutionary conserved regions (ECRs) (Figure 3C). Within these regions, we identified transcription factor binding sites (TFBS) for transcription factors linked to inflammation (NFBK1, REL/RELA), flow (KLF2), and oxidative stress [Nuclear factor erythroid 2-related factor 2 (NFE2L2)] (Figure 3D). These findings are consistent with these stimulatory effects leading to increased PAR4 expression, as described above, and may indicate a role for these transcription factors in PAR4 regulation.
Heterologous downregulation
Using EC-TRAP, we have recently shown in murine hepatic ECs in vivo that PAR1 activation decreases Par4 mRNA levels and that PAR4 activation decreases Par1 mRNA (1). This can be interpreted as a form of heterologous downregulation (90) between both receptors. If endothelial PAR1 activation negatively regulates endothelial Par4 expression at concentrations of thrombin that are suboptimal for PAR4 activation, it could mean that PAR1 serves as an extracellular thrombin sensor on ECs to limit accidental PAR4 activation.
PAR4 trafficking habits and structural motifs reveal it to be a minimalistic receptor
GPCRs are regulated not only during transcription but also during the trafficking of the receptors to and from the cell surface, which controls their availability for signaling. Unlike other PARs, PAR4 appears to lack nuanced mechanisms for trafficking. However, we believe this is by design. Given the receptor's low expression and high potency, it would be unnecessary for PAR4 to evolve nuanced trafficking for various conditions. Rather, the receptor appears to only have one major function: transducing thrombin signaling at high thrombin concentrations. As such, we refer to PAR4 as a minimalistic receptor—one that is stripped of most of the trafficking levers found on other PARs.
PAR4 lacks many post-translational modification sites required for trafficking
PAR4 is smaller in terms of residues (Human: 385; Mouse: 386) than PAR1 (Human: 425; Mouse: 430) and PAR2 (Human: 397; Mouse: 398). This indicates that PAR4 is a receptor that lacks many motifs which are present in PAR1 and PAR2. This size reduction is particularly noteworthy when observing the carboxy tail terminus (CTT) of PAR4, which is particularly short when compared to the CTTs of PAR1 and PAR2 (Figure 4). The CTTs of GPCRs often possess residues that engage with intercellular trafficking machinery (91).
Figure 4
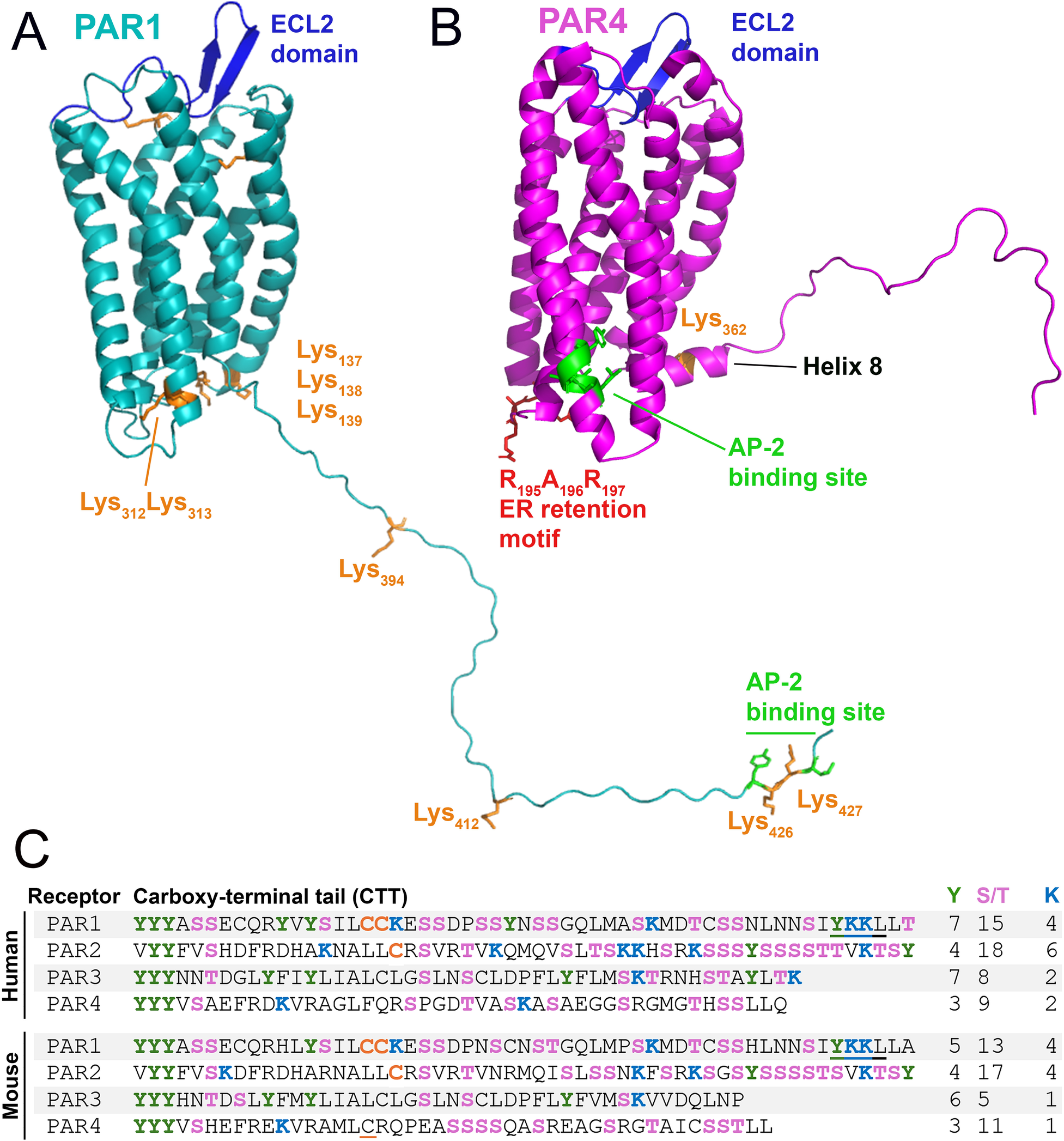
Structural elements of PAR1 and PAR4 that control trafficking. Major receptor trafficking elements of murine (A) PAR1 and (B) PAR4 are annotated. AP-2 binding site (green) and cytosolic lysines (orange), RXR retention motif (red). (C) Sequences of the C-terminal tail (CTT) of human and mouse PAR1-4: serine/threonine (pink); tyrosine (green); lysine (blue); palmitoylated cysteine (orange); putative palmitoylated cysteine is underlined in orange; AP-2 binding site (underlined). Numbers of tyrosine, serine/threonine, and lysine residues are listed at the right.
Phosphorylation
Phosphorylation of GPCR CTTs by G protein-coupled receptor kinases (GRKs) is required for β-arrestin (βarr) recruitment, which aids in receptor desensitization and internalization. Notably, PAR4 contains fewer Ser/Thr sites on its CTT (Human: 9; Mouse: 11) compared to PAR1 (Human: 15; Mouse: 13) and PAR2 (Human: 18; Mouse: 17). PAR2 has the highest quantity of Ser/Thr sites and unsurprisingly has a greater dependency placed on βarr and CTT phosphorylation for its internalization (92, 93). The lack of phosphorylation sites on the CTT of PAR4 suggests that phosphorylation is not a major modality of internalization the receptor employs.
Ubiquitination
PAR1 and PAR2 trafficking is regulated by ubiquitination (94, 95). Human PAR1 has 10 cytosolic lysines (murine PAR1 has 9 cytosolic lysines) (Figure 4: Orange), and PAR2 has 14 cytosolic lysines (96). Ubiquitination of these lysines can alter internalization and degradation; in the case of PAR1, ubiquitination prevents degradation, while it promotes degradation of PAR2 (96, 97). However, PAR4 has very few lysines present on the receptor's CTT. In silico models of murine PAR4 (1) show only a single lysine (K362) to be present on the Helix 8 domain (H8) on the CTT (Figure 4). There are only two lysines on the CTT of human PAR4: one at K350, which matches the single lysine present on the mouse receptor on H8, and a second lysine at K367 (Figure 4C). This paucity of lysines on PAR4 may suggest the receptor has internalization dynamics that are not robustly affected by ubiquitination.
Palmitoylation
Palmitoylation is the addition of a palmitic acid moiety predominantly to a cysteine (or occasionally serine/threonine) residue via a thioester linkage. Palmitoylation of the CTT affects receptor trafficking in PAR1 and PAR2 (98–100). The palmitoylation of PAR1 occurs on dual cysteines (99) (Human: Cys387Cys388; Mouse: Cys392Cys393) in the CTT (Figure 4C). PAR2 also undergoes palmitoylation of a cysteine (Human: Cys361; Mouse: Cys363) on its CTT (100) (Figure 4C). Palmitoylation of both receptors has been shown to stabilize their expression on the cell surface (99, 100). It should be noted that human PAR4 lacks any cysteine residues on the CTT and that mouse PAR4 possesses only two cysteine residues (Figure 4C). Given that palmitoylation increases receptor stability at the cell surface, and that cells like ECs may seek to limit the expression of PAR4 except in circumstances of high thrombin levels, limiting opportunities for PAR4 palmitoylation may reduce the likelihood of its unintentional activation.
AP-2 internalization
Following activation, PAR1 is rapidly internalized at a rate that is equivalent to or shorter than the time required for desensitization of its function (33). Although activated PAR1 is desensitized by βarr, it is not internalized in a βarr-dependent manner but in a distinct phosphorylation-, dynamin-, and clathrin-dependent pathway (101). This internalization is dependent on the clathrin adaptor protein complex-2 (AP-2) (102, 103). Following internalization, PAR1 rapidly passes through early and late endosomes before it is degraded in lysosomes (33, 104). The AP-2 binding domain of PAR1 (Human: Y420KKL423; Mouse: Y425KKL428) is located on the extreme C-terminus of the receptor (Figure 4) and binds the μ2 subunit of AP-2 (103).
Like PAR1, the internalization of PAR4 is not dependent on βarr but is dependent on AP-2 (105). However, unlike PAR1, in which the AP-2 binding site is found in the CTT, the AP-2 binding domain for PAR4 (Human: Y264GATL268; Mouse: Y276GATL280) is found in the intracellular loop 3 (ICL3) (105) (Figure 4). The fixed position of the ICL3 as opposed to the CTT, which has a free range of motion, would likely provide less AP-2 accessibility to its cognate binding site on PAR4 compared to PAR1. This limitation in AP-2 binding site accessibility may explain why PAR4 internalization and degradation have been observed to be slower than PAR1 (33, 105, 106). This in turn may explain why PAR4 has high potency on ECs and other cells, as the signaling half-life of the receptor on the cell surface is extended due to delayed internalization.
PAR4 lacks N-terminal proteolytic termination sequences
PAR1 and PAR2 each contain numerous proteolytic cleavage sites on the distal end of their N-terminus, downstream of the tethered ligand (TL) and proximal to the first transmembrane domain of the receptor (12). These sites—often cleaved by neutrophil elastase, proteinase 3, and cathepsin G—are deactivation sites for these receptors (12), and upon cleavage, the TL is released from the receptor. Although the respective EC50 for thrombin and trypsin is high for PAR1 and PAR2 (i.e., pM-nM range), the equivalent EC50 for their TLs is relatively low (i.e., µM range). However, since the TL is physically attached to the receptor it triggers maximal signaling despite this low binding affinity. Using previously published models (1), we calculated the effective concentration for the TL of PAR1 to range from ∼71.3 mM - 39.4 M. However, proteolytic cleavage of the TL dissociates it from the receptor and leads to rapid receptor deactivation (15). It is noteworthy that PAR4 lacks these deactivation sites on its distal N-terminus, unlike PAR1 and PAR2. This lack of PAR4 deactivation sites suggests that the receptor was not designed for rapid signal termination and further explains its potency.
Does PAR4 have an internal pool in ECs?
PAR1 is internalized both constitutively and upon activation. The constitutive internalization (i.e., tonic cycling) of PAR1 is critical for the generation of an internal pool of uncleaved receptors (107), which allows for the rapid replenishment of fresh receptors on the cell surface after proteolytic cleavage (33, 108). In ECs, this internal pool of PAR1 is ten times larger than the cell surface pool of the receptor (109). However, there is limited information on whether PAR4 has an internal pool. Given the receptor's high potency on ECs, we speculate that an internal pool of PAR4 may be unnecessary since there may be a lesser need for rapid replenishment of PAR4 on the cell surface following a PAR4 signaling event. Cells may choose to space out their PAR4 signaling events due to the receptor's potency. Furthermore, given the low expression of Par4 transcripts in ECs, even if an internal pool existed, its small size may preclude our ability to detect it. Future studies focusing on whether ECs lack an internal pool of PAR4 would be beneficial for determining how the receptor is trafficked and whether cell surface repopulation requires de novo synthesis of the receptor.
Is PAR4 a molecular hitchhiker?
PAR4 appears to take advantage of the movement of other receptors to traffic itself (110, 111). We term this behavior as receptor phoresy. Phoresy is defined as an interaction in which a phoretic animal (i.e., a phoront) latches itself onto a host for travel. The word is derived from the Greek phorein, meaning “to carry” (112). In the case of PARs, we argue that PAR4 is likely a phoront to other PARs. As a phoront, PAR4 would benefit from the regulation and trafficking of other receptors, without needing to carry specific trafficking motifs on itself. This behavior has been directly observed in the case of PAR2 and PAR4, with heterodimerization of these receptors resulting in more efficient PAR4 migration to the cell surface (110). In the absence of PAR2, PAR4 is retained in the ER via its arginine-based (RXR) ER retention motif in the second intracellular loop (ICL2) of the receptor (Figure 4). This suggests that PAR4 requires the assistance of a cofactor to migrate from the ER to the cell surface unless this domain is masked. However, in murine hepatic ECs—where PAR4 is known to be functional—we detect little to no expression of PAR2 (1) (Figure 2A). Thus, it may be an unlikely trafficking partner for PAR4 in these cells. Alternatively, PAR4 may be trafficked alongside PAR1 to the cell surface in hepatic ECs, particularly if stable heterodimers of PAR1 and PAR4 can form without thrombin-mediated cleavage, as seen in platelets (63).
Similarly, there is an open question as to whether heterodimerization of PARs alters their internalization. Overexpression studies have shown that PAR1 and PAR4 heterodimerize upon thrombin-induced cleavage (61). This allows the HLD of PAR1 to capture exosite I of thrombin and localize its active site to the exodomain of PAR4, effectively amplifying PAR4 signaling and decreasing its EC50 ∼2.9-fold (63). However, since proteolytic cleavage results in receptor internalization, it is not known whether the PAR1/4 heterodimer gets internalized as a complete unit or whether each monomer is internalized separately. In the former case, PAR4 may be internalized as a phoront alongside PAR1 with more rapid kinetics, thus limiting the signaling lifetime of PAR4. Future studies focusing on whether PAR4 is a phoretic receptor would be beneficial to determine how exactly this receptor is trafficked to and from the membrane.
How does PAR4 signal in endothelial cells—is it redundant to PAR1?
Different roads that lead to Rome
PAR1 and PAR4 are promiscuous receptors with the ability to couple with multiple G protein subunits [Gαi (113, 114), Gαq/11 (56, 114), Gα12/13 (115, 116)], as well as βarr (117, 118) and likely Gβγ (119–121) (Figure 5A). Traditionally, PAR4 was seen as a providing redundant function to PAR1 (122); however, studies have shown nuanced differences in the signaling of these receptors. Notably, these receptors can utilize different mechanistic paths to reach the same cellular destination (i.e., functional outcome). For example, PAR1 and PAR4 can also mediate the same function using different G proteins (Figure 5B). In bovine arterial ECs, PAR4 uses Gαi/adenylyl cyclase signaling to mediate nitric oxide production, whereas PAR1 utilizes Gαq/calcium signaling for this function (114). Similarly, in platelets, PAR4 activation mediates AKT phosphorylation independent of phosphoinositide 3-kinase (PI3K) (123), whereas PAR1 mediates AKT phosphorylation through PI3K (124, 125). In cultured human pulmonary ECs, PAR4 regulates actin cytoskeletal rearrangement, which can also potentially regulate permeability (126). The mechanism for this cytoskeletal rearrangement is likely via Gαq/phospholipase C-β (PLC-β)/calmodulin/myosin light chain kinase (MLCK) (127) or Gαq/RhoA-GTPase/p38/ERK/MAPK (128) or Gα12/13/Rho-GTPases signaling, in a similar manner to PAR1 (129). However, PAR4 has also been shown to regulate cytoskeleton rearrangement independently of G proteins in a βarr/RhoA-dependent pathway (118) or through a PI3K/Rho-dependent pathway (130). Overall, we hypothesize that even though PAR4 can be redundant to PAR1, it can utilize alternative signaling pathways to achieve the same functional outcome.
Figure 5
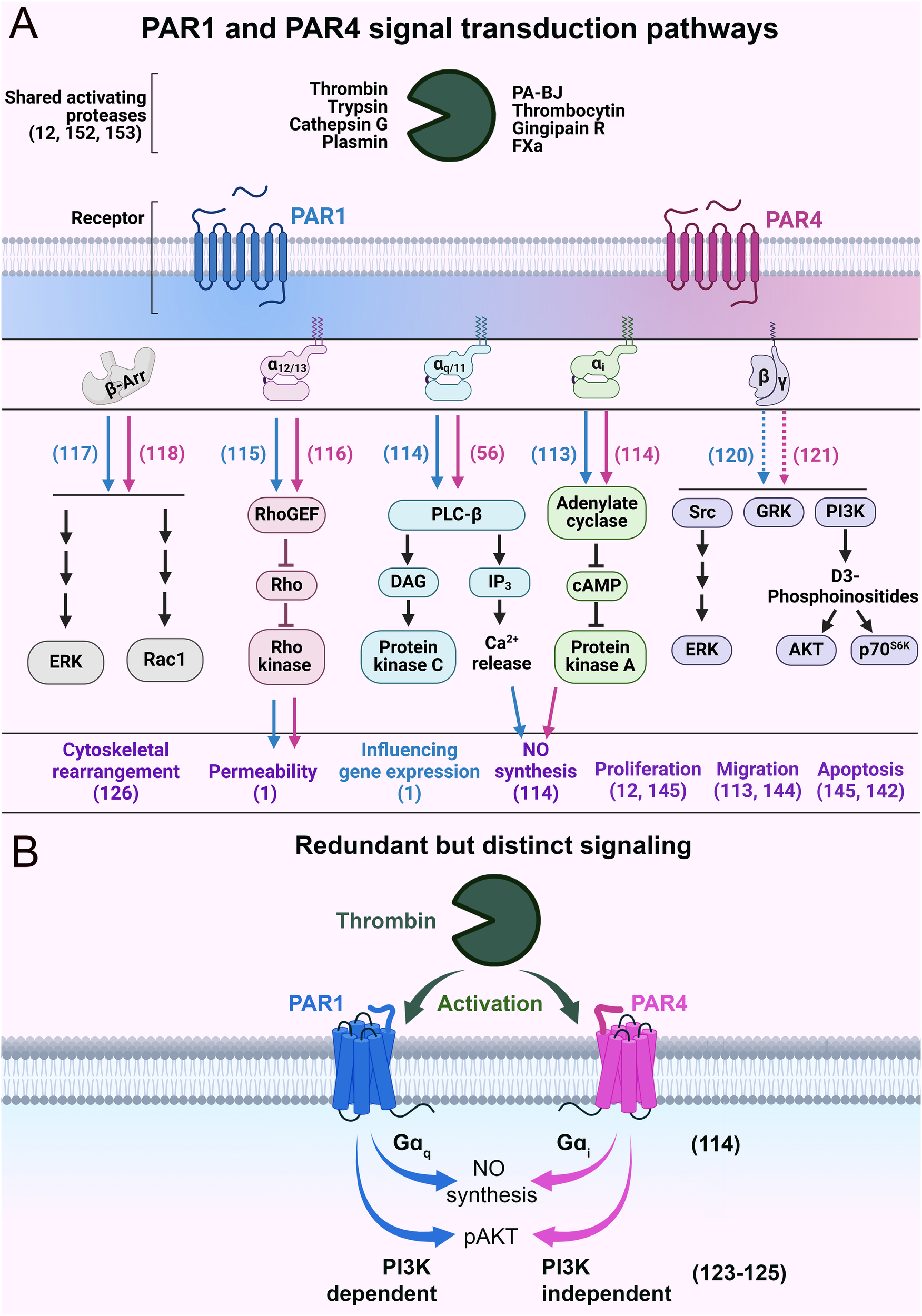
Schematics of PAR1 and PAR4 signal transduction pathways. (A) The major intracellular signaling events and biological effects following the activation of PAR1 and PAR4. Blue and pink arrows indicate PAR1- and PAR4-specific signaling, respectively. The purple text refers to the biological impacts mediated by PAR1 and PAR4. The blue text refers to biological effects mediated by PAR1 alone. Dashed lines indicate hypothesized behavior. (B) Schematic of functionally redundant but mechanistically distinct signaling of PAR1 and PAR4. Relevant citations are shown in parentheses.
These distinct mechanisms are reasonable given that PAR4 is a low-affinity receptor when compared to PAR1 (100-fold). Teleologically, a scenario can be imagined in which thrombin concentrations are high enough for PAR4 to be activated, but such concentrations would also result in nearly all the PAR1 on the cell surface getting cleaved and activated. In such a scenario, having PAR4 compete for second messenger resources with PAR1 would be counterproductive for signal transduction. Therefore, allowing for alternate pathways/G-proteins to transduce signal would allow PAR4 to signal efficiently even when PAR1 signaling is saturated. This is akin to a higher gear setting in a vehicle. As switching from a low gear to a high gear allows for more efficient transmission of power from the engine to the wheels, switching from PAR1 to PAR4 signaling allows for more efficient transduction of thrombin signaling to an EC.
PAR4 has limited roles in influencing endothelial gene expression
Recently we showed that in hepatic ECs, PAR1 can influence gene expression following activation, but PAR4 cannot. We found PAR1 activation resulted in upregulation in transcripts linked to permeability and cytoskeletal rearrangement, while PAR4 activation did not alter transcription in ECs (1). This is reasonable given that PAR4, unlike PAR1, is a potent receptor with limited desensitization (55). If PAR4 could influence gene expression, its persistent signaling could result in significant fluctuations in gene expression (56). Given that ECs can survive for years in vivo (131) and may encounter multiple thrombin signaling events during their lifetime, PAR4-mediated persistent transcriptional events in these cells could be deleterious to their long-term function and homeostasis.
Open questions in endothelial PAR4 biology
Is endothelial PAR4 mechanosensitive?
GPCRs are defined as heptahelical proteins, but some have an eighth alpha helix (H8) in their CTT. This H8 domain can have mechanosensing properties. Although there is a lack of sequence conservation or defined length for the mechanotranducing H8, the domain is always parallel to the inner leaflet of the cell surface (132) and is critical for sensing mechanical stretch forces in cells. For example, the mechanosensitive histamine H1 receptor (H1R) is an endothelial sensor of fluid shear stress through its H8 domain (133). In silico models of murine PAR2, PAR4, and to a lesser extent PAR3 also show the presence of this domain (Figures 1A-D). Interestingly, the PAR4 H8 domain (Figure 6A: green) has a similar homology (Mouse: EFREKVRAML | Human: EFRDKVRAGL) to the mechanosensitive H1R GPCR (ENFRKTFKRIL), with 30% fully conserved residues and 70% conservation of amino acid groups of similar properties. However, there are no studies that focus on whether/how flow affects PAR4 signaling. As mentioned previously, PAR4 expression is regulated by shear stress in HUVECs, but whether PAR4 signaling is altered during flow is still an open question. Given the fact that this highly potent receptor is expressed on ECs, which are constantly exposed to and respond robustly to variable flow rates, future studies focusing on how flow alters endothelial PAR4 signaling in different contexts would be beneficial.
Figure 6
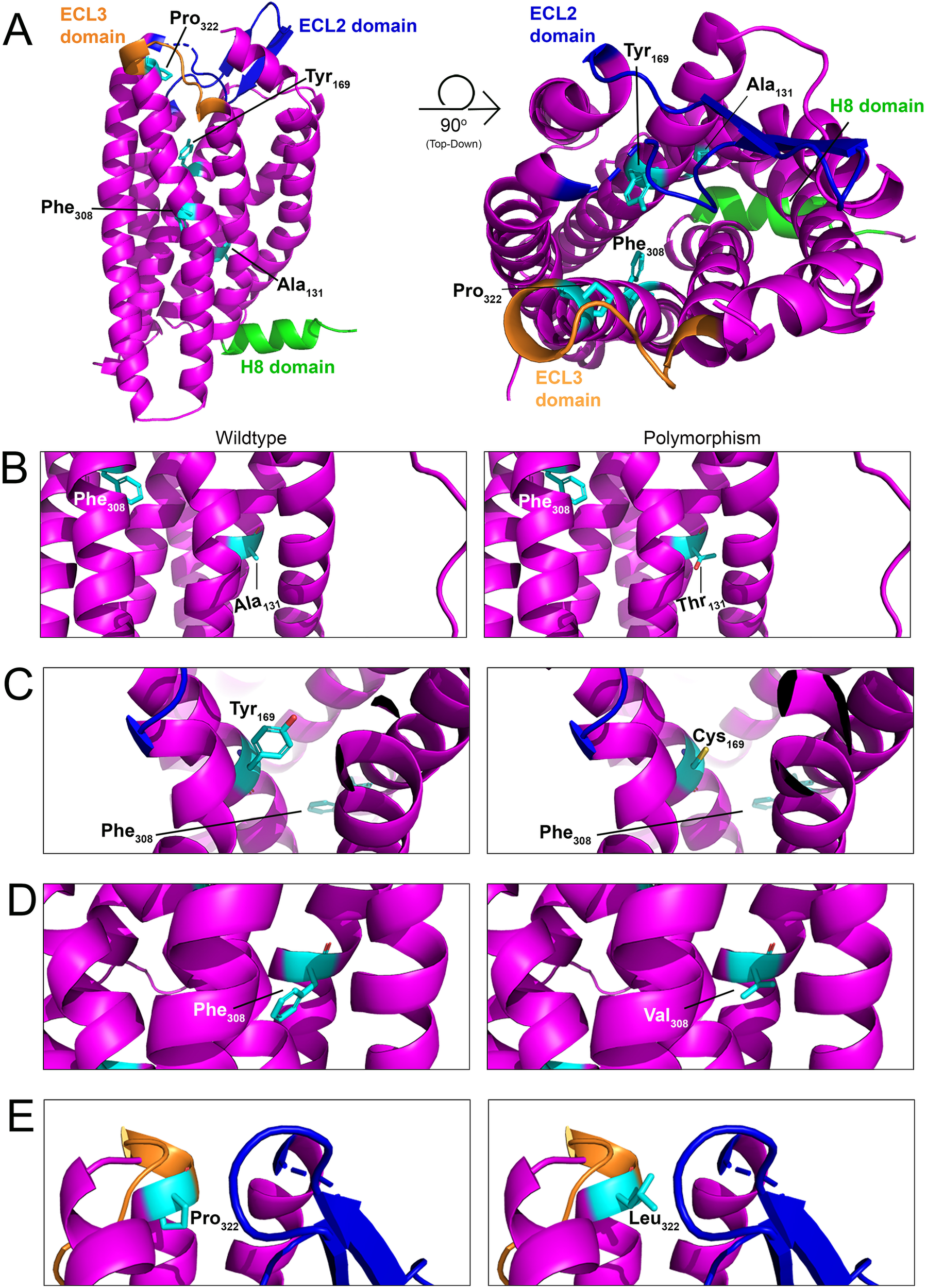
Structural model of PAR4 single nucleotide polymorphisms (SNPs). (A)In silico models with 90-degree rotation (top-down view; right) of murine PAR4 with common SNPs shown in cyan. The Helix 8 (H8) domain of the receptor is shown in green, the extracellular loop (ECL) 2 domain is shown in blue, and the ECL3 domain is shown in orange. Wildtype receptor (left) and polymorphism-carrying receptor (right) are depicted for (B) Ala131Thr, (C) Tyr169Cys, (D) Phe308Val, and (E) Pro322Leu.
Do PAR4 SNPs affect endothelial cell function?
Four single nucleotide polymorphisms (SNPs) have been identified within PAR4 that result in its altered expression and function in platelets (17, 134). However, nothing is known about how these SNPs affect PAR4 in ECs. Ala120Thr (rs773902): This mutation is found on the second transmembrane of PAR4 and leads to its increased functional responses in platelets (135). The equivalent residue in mice is Ala131 (Figure 6B). Tyr157Cys: This mutation, which is found on the third transmembrane domain of the receptor, leads to an attenuation of PAR4 functional responses in platelets (135). The equivalent residue in mice is Tyr169 (Figure 6C). Phe296Val (rs2227346): This mutation is less common and has only been observed in Black individuals (17, 136). The mutation is present on the sixth transmembrane domain and leads to decreased PAR4 second messenger production in response to receptor activation (135). The equivalent residue in mice is Phe308 (Figure 6D). Pro310Leu (rs2227376): This mutation is found on the extracellular loop 3 of the receptor and leads to an attenuation of PAR4 functional responses in platelets (137, 138). The equivalent residue in mice is Pro322 (Figure 6E).
Most of these SNPs produce inactivating mutations. Given that endothelial PAR4 appears to signal as a thrombin relief receptor (i.e., it signals only when PAR1 is saturated), there may be sufficient PAR1 on ECs to mitigate the loss of PAR4 function. The more concerning mutant is the Ala120Thr, which is a hyperactive mutation that increases PAR4 activity. Given the high basal potency of PAR4 on ECs, hyperactivation may lead to ECs that are more sensitive to thrombin-mediated endothelial dysfunction (79). Future studies focusing on whether the Thr120 variant of PAR4 results in EC hyperreactivity to thrombin would be beneficial.
Does PAR4 mediate apoptosis and regression in ECs?
Another interesting finding is that endothelial PAR4 expression increases in response to inflammatory cytokines, such as IL-1β and TNF-α. Also of note is that the combination of IL-1β, TNF-α, and thrombin treatment robustly drives the regression of capillary tubes grown in a 3D culture model (139). In the case of lymphatics, TNF-α, IFN-γ, and thrombin also promote capillary tube regression (140). Given that proinflammatory cytokines selectively increase PAR4 expression in ECs, it is possible that capillary regression may be driven less by thrombin activation of PAR1 than by thrombin activation of PAR4.
Similar effects are observed in cancer cells, in which PAR1 activation promotes tumor growth and metastasis, while PAR4 acts as a tumor suppressor that inhibits tumor growth and metastasis (141). This could be explained, in part, by the fact that PAR4 activation increases protein expression of apoptosis factors (e.g., caspase 9) (142). This apoptotic function of PAR4 is present in esophageal (142), gastric (87) and lung (143) cancers, although this effect is not universal for all cancers as colorectal cancers show increased proliferation with increased expression of PAR4 (144). Furthermore, other studies have shown that in certain cases PAR1 activation can also induce apoptosis, suggesting PARs can diversely influence cell death depending on varying conditions (145).
Do endothelial PARs affect thrombosis and hemostasis?
It is well-known that thrombin activates PAR1 and PAR4. However, what is less understood is the effect endothelial PARs have on thrombin generation and hypercoagulability in general. We have recently shown that in a model of APAP overdose (1)—which presents with increased thrombin levels and hypercoagulability, as measured by plasma thrombin-antithrombin (TAT) levels—that loss of both endothelial PAR1 and PAR4 reduces thrombin generation. However, mice lacking in endothelial PAR1 or PAR4 alone do not demonstrate reduced thrombin levels after APAP overdose, suggesting that both endothelial PAR1 and PAR4 work synergistically to promote thrombin generation. We speculate that endothelial PAR1/4-mediated thrombin generation results in cytoskeletal rearrangement of ECs (Figure 7A), resulting in exposure of subendothelial coagulation initiators such as collagen and tissue factor. This results in a feedback mechanism in which thrombin generation can be amplified by the actions of endothelial PAR1 and PAR4. In a separate study, we also found that PARs act synergistically between multiple cell types, including the endothelium, to promote hemostasis (146), further suggesting bona fide roles of endothelial PARs in mediating hemostatic responses.
Figure 7
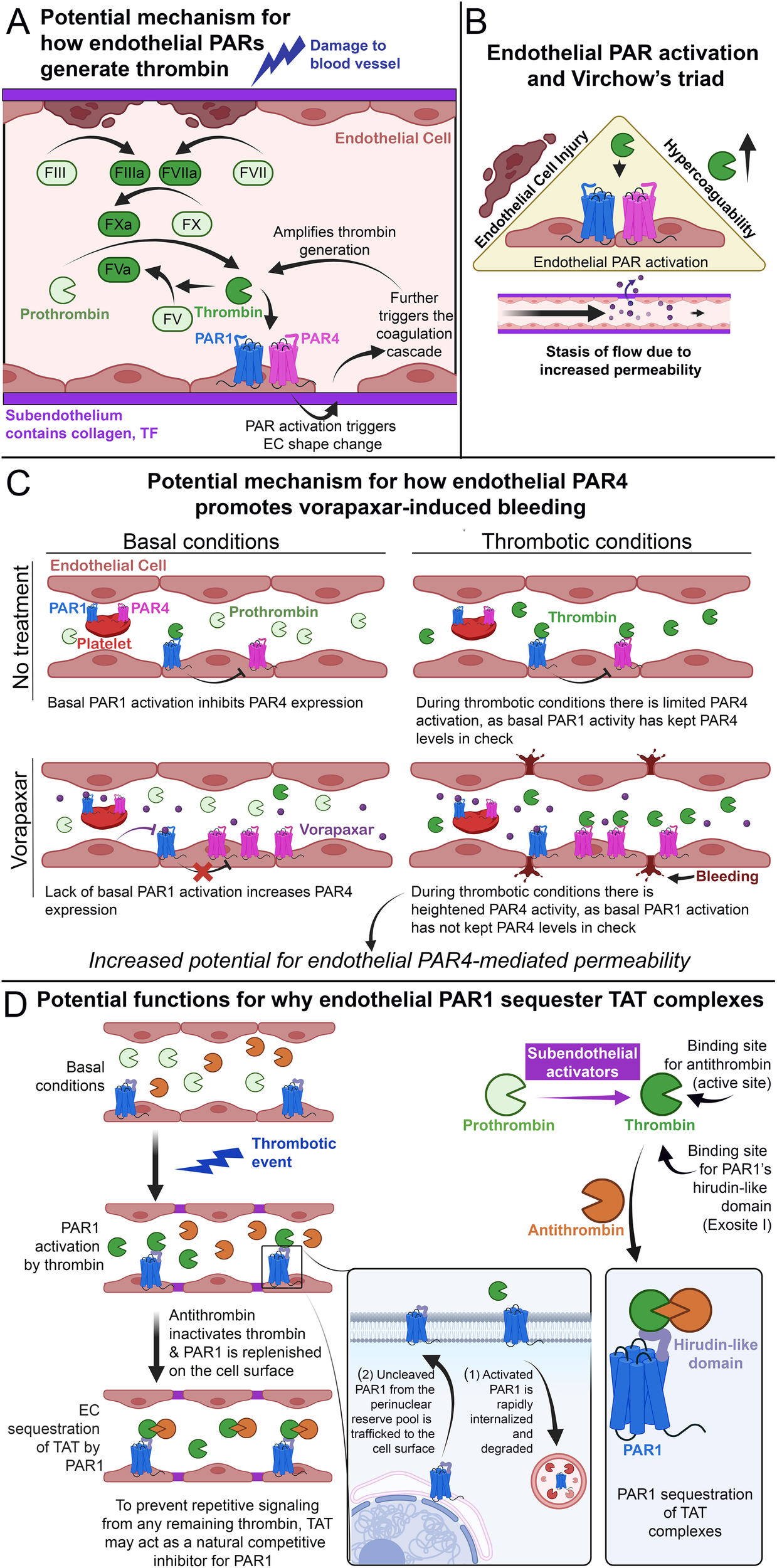
Endothelial PAR4 and thrombosis. (A) Schematic of how endothelial PARs potentially amplify thrombin generation after initial blood vessel damage. (B) Schematic of how endothelial PAR activation potentially increases thrombosis by promoting the factors of Virchow's triad. (C) Schematic of how endothelial PAR4 may potentially contribute to vorapaxar-induced bleeding in humans. (D) Schematic of how endothelial PAR1 may act as a molecular sink for TAT, subpanel adapted from Rajala et al. (1).
This form of regulation can also directly impact thrombosis. The modern understanding of thrombosis can be observed in the lens of Virchow's triad (147), which outlines the three broad contributing factors: hypercoagulability, endothelial injury, and stasis of flow (Figure 7B). All three of these factors are promoted by the activation of endothelial PARs. Hypercoagulability: Our data from APAP-overdosed mice indicate that endothelial PAR4 can contribute to hypercoagulability in vivo (1). EC injury: Overpotentiating PARs has been shown to mediate pro-inflammatory signaling (128) that can lead to EC injury (76). Stasis of flow: It has been shown that alterations in permeability can also alter flow rates in ECs (5, 148, 149). This is most notably observed in tumor vasculature, in which leaky vessels demonstrate increased angiogenesis alongside impaired flow (150). Furthermore, our recent work demonstrates that endothelial PAR1 and PAR4 promote vascular permeability after APAP overdose (1), and a result of this permeability may be impaired vascular flow. Thus, endothelial PARs fit the bill as factors that promote thrombosis and may represent novel therapeutic targets for reducing thrombotic activity.
Does endothelial PAR4 contribute to vorapaxar-induced bleeding?
Vorapaxar is an orthosteric PAR1 inhibitor that was designed as an antithrombotic drug because of PAR1 expression on human platelets (151). The principle of inhibition stemmed from PAR1 and PAR4 being dual thrombin receptors on human platelets (152, 153). By inhibiting high-affinity PAR1 with vorapaxar, accidental thrombosis triggered by low concentrations of thrombin can be prevented (122). Meanwhile, since low-affinity PAR4 is still functional, high concentrations of thrombin generated during injury can still allow for hemostasis via platelet PAR4.
However, the use of vorapaxar presents with risks of increased bleeding (154). Our recent article on APAP overdose demonstrated that ECs have low-expressing but functional PAR4 in vivo. These low levels are further reduced by PAR1 activation and heterologous desensitization (1). We argue this is a negative feedback loop between thrombin/PAR1 signaling and Par4 expression to limit PAR4 activation. Given that PAR4 is a potent receptor, ECs are invested in limiting its activation. Thus, in the case of low thrombin levels, subsequent endothelial PAR1 activation may act as a brake on PAR4 activation. Furthermore, studies in oligodendrocyte progenitor cells and astrocytes indicate that PAR1 has basal activity independent of thrombin stimulation (155). This suggests that PAR1 may be a leaky receptor, and perhaps PAR1 basal activity may be sufficient to act as a brake on PAR4 activity. Regardless of the mode of PAR1 activation, since vorapaxar effectively inhibits global PAR1 activation [including in ECs (156)], this may release the brake on endothelial PAR4. This potential for increased PAR4 activity on ECs may contribute to the increased bleeding seen in patients treated with vorapaxar, as it would result in unmitigated PAR4-mediated vascular dysfunction (Figure 7C). Overall, these findings raise questions about the interplay between PAR1 and PAR4. Understanding how these receptors interact with each other will be necessary to design effective and safe PAR therapeutics.
Can a TAT complex act as a PAR1 inhibitor?
In a recent study from our lab, we found evidence that endothelial PAR1 may act as a molecular “sink” to bind and sequester TAT complexes in vivo (1). TAT complexes are formed by the binding of antithrombin to thrombin's active site (157). In contrast, PAR1 binds to thrombin's exosite I via the receptor's HLD (19) (Figure 1E). Given that exosite I is still accessible in TAT complexes (157), we hypothesized that TAT could bind and be sequestered by endothelial PAR1 on the lumenal surfaces of blood vessels (1). Leger and colleagues have previously proposed functional sequestration of thrombin by PAR1 in a model of PAR1/4 dimerization (63). In this model, PAR1 cleavage and activation are followed by retention of thrombin via HLD-exosite I interactions. This retention allows for thrombin-mediated cleavage of neighboring PAR4 molecules and lowers the EC50 for PAR4 activation ∼2-fold (56, 63). Whether PAR1 similarly binds TAT as it does thrombin is unknown. However, we believe PAR1-TAT interactions could represent a novel mechanism for PAR1 inhibition (Figure 7D).
In the endothelium, following a thrombotic event, thrombin cleaves PAR1 and drives rapid internalization and degradation of the receptor (33). PAR1 is subsequently replaced on the cell surface with new and uncleaved PAR1 molecules from an intracellular pool (106). At the same time, circulating antithrombin binds and inactivates thrombin, forming TAT complexes. We propose that the newly formed TAT complexes might then bind the newly trafficked PAR1 on endothelial surfaces to inhibit further thrombin-mediated activation and prevent repeated endothelial PAR1 signaling in response to the same stimulus. Circulating TAT might also bind PAR1 on human platelets or PAR3—which also contains an HLD (Figure 1F)—on mouse platelets, potentially inhibiting thrombin-mediated platelet activation. This may explain why we observed that mice with elevated circulating TAT also displayed protection against thrombocytopenia following an acetaminophen-induced thrombotic challenge (1).
Currently, the only FDA-approved PAR1 inhibitor, vorapaxar, is no longer available in the U.S. due to significant adverse bleeding events (154). Issues with vorapaxar included its high binding affinity and long half-life in the body, which resulted in the drug acting as a functionally irreversible inhibitor (158) and likely contributed to its side effects. If TAT complexes act as natural and competitive inhibitors to PAR1, new compounds designed to mimic how TATs inhibit PAR1 may avoid some of the negative effects associated with vorapaxar. Such compounds—which we refer to as inactive thrombin mimetics (ITMs)—would be proteolytically inert and carry the exosite I motif of thrombin, thus allowing for reversible binding and inhibition of PAR1 while not interfering with endogenous thrombin activity.
Discussion: PAR4 is a PARsimonious receptor on the endothelium
Over the last two decades, endothelial PAR4 has been an enigma. Although some studies have shown the presence of PAR4 on ECs, its lack of expression in vitro and a lack of sensitive tools to assess receptor function in vivo have hindered research in this field. In this review, we show that the low expression of PAR4 may be purposeful. PAR4 is a very potent receptor, and ECs appear to employ numerous mechanisms to prevent its unintended activation except in cases of exceptional thrombin activity. As a result, ECs appear to take a parsimonious approach when employing PAR4 signaling by limiting the expression of the receptor. This is in stark contrast to platelets, in which PAR4 is robustly expressed (159) and was first detected (34). This lack of limitation on platelet PAR4 expression is reasonable when you consider that thrombin-mediated activation of platelets is a terminal event (160); platelets cease functioning and irreversibly aggregate following PAR4 activation. Meanwhile, thrombin activation of ECs is a temporary event; ECs are functional for years (131) and likely experience multiple PAR4 signaling events during their lifetime. Furthermore, unlike platelets, ECs need to survive to maintain blood vessel integrity, and an extensive second messenger response could be harmful to endothelial function and survival. The dichotomy between the terminal and the temporary could explain why tight regulation and low expression for PAR4 exist in ECs but not in platelets. Understanding this dichotomy is fundamental to understanding how PAR4 functions on ECs. Thus, for endothelial PAR4, we argue its low expression is not indicative of a failure to function, but rather, it appears to be a feature of the receptor to ensure that PAR4 signaling does not have deleterious consequences.
However, in cases when PAR4 is allowed to signal, it signals with impunity—using distinct signaling pathways from PAR1. This allows PAR4 to mediate continuous transduction of thrombin signaling, even in high thrombin environments, where PAR1 signaling is saturated (Figure 8). Therefore, the presence of PAR4 on ECs allows for access to higher powerbands of thrombin signaling. Nevertheless, this potency comes at a cost. PAR4 appears as a minimalistic receptor, in that the receptor does not appear to be designed to engage in nuanced post-translational regulation, unlike PAR1 and PAR2. There is a lack of cysteine, lysine, serine, and threonine residues on the CTT as well as a lack of lysines in the AP-2 binding site, suggesting a lesser role for palmitoylation, ubiquitination, and phosphorylation of the receptor. PAR4 may also be reliant on other PARs for trafficking. This may all reflect the likelihood that endothelial PAR4 is designed with a singular task in mind: transducing massive amounts of second messenger at high concentrations of thrombin. Thus, there is no need for PAR4 to demonstrate the nimbleness and elegance seen in PAR1 and PAR2 trafficking. In the case of PAR4, the cell needs only to place a low number of receptors on the cell surface and allow the receptor to signal with impunity. However, to compensate for the lack of posttranslational control placed on PAR4, ECs likely exercise significant transcriptional control on the receptor, which may explain why its levels are so low in ECs.
Figure 8
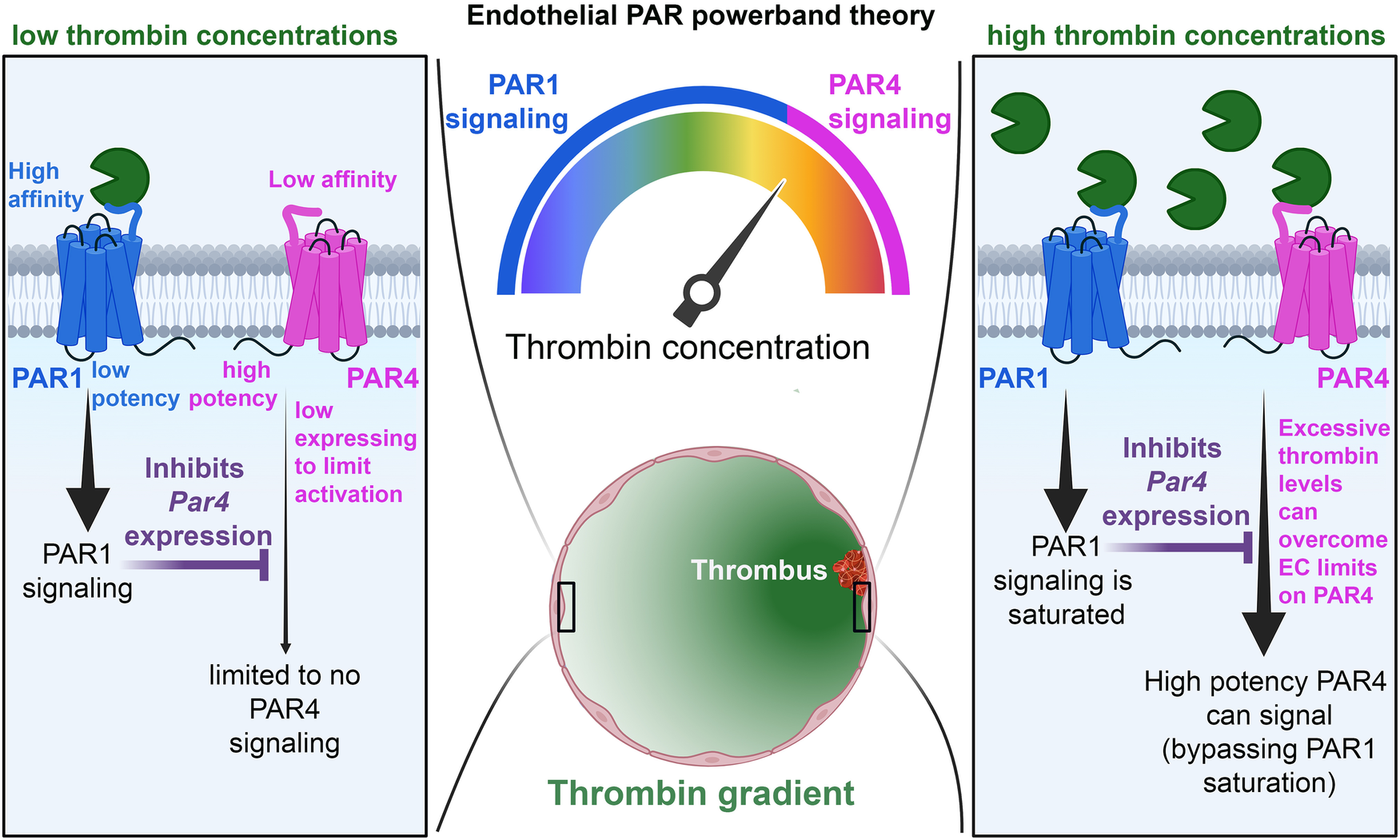
Schematic of the PAR powerband theory. In conditions of low thrombin activity (left), PAR1 is allowed to signal, and its activity inhibits PAR4 expression. This, coupled with low PAR4 expression and affinity for thrombin, prevents PAR4 from signaling on ECs. In conditions of high thrombin activity (right), PAR1 activation becomes saturated, and endothelial PAR4 is allowed to signal with high potency.
Taking all this into consideration, we argue that PAR4 is not an impotent receptor on the endothelium, but rather an important one whose regulation reflects its roles and functions.
Future directions
In vivo focus for endothelial PAR4 studies
Although mechanistic insights about PAR4 would theoretically be easier to explore in vitro, assigning roles for receptor function and expression based on cell culture models appears to result in spurious and contradictory results. Future studies should focus more on the receptor in an in vivo context with organotypic expression in mind, as this approach yields the most accuracy in assigning/identifying the cellular functions of the receptor.
How does PAR4 affect the endothelial phosphoproteome/kinome?
Protein phosphorylation is a significant regulator of protein function in all cells; kinases, phosphatases, and relevant regulatory subunits constitute 2.5% of all human genes (161). Given that we have previously shown that endothelial PAR4 has an extremely limited ability to influence transcription (1), PAR4 likely only mediates downstream signaling effects through post-translational modifications such as phosphorylation. Currently, there have been a few studies that focus on the impact of PAR1 activation on protein phosphorylation (156, 162, 163). Notably, Lin and colleagues looked at a bias in the phosphoproteome between activated protein C (aPC) and thrombin-mediated PAR1 activation (162). Identifying phosphoproteomic signatures following endothelial PAR4 activation and comparing them to signatures from PAR1 activation would allow us to gain a deeper understanding of receptor-specific actions. This was attempted in a recent study by Groten and colleagues using cultured ECs and PAR1/4-specific agonist peptides, and the authors found limited PAR4-mediated effects on protein phosphorylation (163). However, this may be due to the lack of flow (i.e., shear stress) used in these studies, which we detailed above as being important for PAR4 expression on ECs. Future studies may need to employ shear to determine PAR4-specific phosphorylation events more accurately. Additionally, given the advent of genetic mouse models to label endogenous proteins in vivo [e.g., biorthogonal noncanonical amino-acid tagging (BONCAT) (164)], coupled with new techniques that allow for kinome profiling (165, 166), future studies may also be able to identify endothelial PAR1/PAR4-specific kinases in vivo following receptor activation.
Limitations
This review focuses on the understudied area of PAR4 on the endothelium. We have sought to contextualize our new findings with past reports about PAR4 functions on ECs and other cell types. We acknowledge that some sections of this review related to possible functions for PAR4 on ECs are postulative; these sections are labeled with headings in the form of questions. We intend for these sections to highlight gaps in the field of endothelial PAR4 biology and hope they inspire future studies from other labs.
Statements
Author contributions
RR: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. CG: Funding acquisition, Supervision, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by a grant from the National Institutes of Health (R35HL144605) to CG and predoctoral fellowships from the American Heart Association (23PRE1014240) and OMRF to RR.
Acknowledgments
The authors regret that work from some colleagues could not be referenced or discussed due to space limitations. The authors would like to thank Drs. Audrey Cleuren and Charmain Fernando (OMRF: Cardiovascular Biology Research Program) for their helpful discussions during the preparation of this review, as well as Ms. Madelynn Dudgeon (University of Oklahoma: Dodge College of Arts and Sciences) for her help in acquiring copies of certain references. Data processing and analysis were supported by the OMRF Center for Biomedical Data Sciences. Images were made using BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AKT, Ak strain transforming; APAP, Acetaminophen; AP-2, adaptor protein complex-2; βarr, β-arrestin; CTT, Carboxy-terminal tail; EC, Endothelial cell; ECL2, Extracellular loop 2; ECR, Evolutionary conserved region; GPCR, G protein-coupled receptor; H1R, Histamine H1 receptor; H8, Helix 8 domain; HLD, Hirudin-like domain; HUVECs, Human umbilical vein endothelial cell; ICL3, Intracellular loop 3; IL-1β, Interleukin 1β; KLF2, Krüppel-like factor 2; KLF4, Krüppel-like factor 4; PAR1, Protease-activated receptor 1; PAR2, Protease-activated receptor 2; PAR3, Protease-activated receptor 3; PAR4, Protease-activated receptor 4; TAT, Thrombin-antithrombin TFBS, Transcription factor binding sites; TL, Tethered ligand; TNF-α, Tumor necrosis factor α; TPM, Transcripts per million mapped reads; TRAP, Translating ribosome affinity purification; (F2r and F2rl3 are the gene names for PAR1 and PAR4, respectively. For simplicity, we hereafter refer to these genes as Par1 and Par4).
References
1.
Rajala R Cleuren ACA Griffin CT . Acetaminophen overdose reveals Par4 as a low-expressing but potent receptor on the hepatic endothelium in mice. Arterioscler Thromb Vasc Biol. (2025) 45(1):53–71. 10.1161/ATVBAHA.124.321353
2.
Rajala R . How big is the endothelium? Comment on “spatial and temporal dynamics of the endothelium”. J Thromb Haemost. (2021) 19(10):2634–5. 10.1111/jth.15469
3.
Sabin FR . Studies on the Origin of Blood-Vessels and of Red Blood-Corpuscles as Seen in the Living Blastoderm of Chicks During the Second Day of Incubation. Washington, DC: Carnegie Institution of Washington (1920). p. 213–62, 6 plates.
4.
Trimm E Red-Horse K . Vascular endothelial cell development and diversity. Nat Rev Cardiol. (2022) 20:1–14. 10.1038/s41569-022-00770-1
5.
Rickman M Ghim M Pang K von Huelsen Rocha AC Drudi EM Sureda-Vives M et al Disturbed flow increases endothelial inflammation and permeability via a frizzled-4-Β-catenin-dependent pathway. J Cell Sci. (2023) 136(6):1–16. 10.1242/jcs.260449
6.
Mehta D Malik AB . Signaling mechanisms regulating endothelial permeability. Physiol Rev. (2006) 86(1):279–367. 10.1152/physrev.00012.2005
7.
Park KH Park WJ . Endothelial dysfunction: clinical implications in cardiovascular disease and therapeutic approaches. J Korean Med Sci. (2015) 30(9):1213–25. 10.3346/jkms.2015.30.9.1213
8.
Iba T Saitoh D . Efficacy of antithrombin in preclinical and clinical applications for sepsis-associated disseminated intravascular coagulation. J Intensive Care. (2014) 2(1):66. 10.1186/s40560-014-0051-6
9.
Schouten M Wiersinga WJ Levi M van der Poll T . Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol. (2008) 83(3):536–45. 10.1189/jlb.0607373
10.
Phillips-Mason PJ Craig SE Brady-Kalnay SM . A protease storm cleaves a cell-cell adhesion molecule in cancer: multiple proteases converge to regulate ptpmu in glioma cells. J Cell Biochem. (2014) 115(9):1609–23. 10.1002/jcb.24824
11.
Puente XS Gutiérrez-Fernández A Ordóñez GR Hillier LW López-Otín C . Comparative genomic analysis of human and chimpanzee proteases. Genomics. (2005) 86(6):638–47. 10.1016/j.ygeno.2005.07.009
12.
Heuberger DM Schuepbach RA . Protease-activated receptors (pars): mechanisms of action and potential therapeutic modulators in par-driven inflammatory diseases. Thromb J. (2019) 17:4. 10.1186/s12959-019-0194-8
13.
Coughlin SR . How the protease thrombin talks to cells. Proc Natl Acad Sci U S A. (1999) 96(20):11023–7. 10.1073/pnas.96.20.11023
14.
Macfarlane SR Seatter MJ Kanke T Hunter GD Plevin R . Proteinase-activated receptors. Pharmacol Rev. (2001) 53(2):245–82. 10.1016/S0031-6997(24)01493-5
15.
Peach CJ Edgington-Mitchell LE Bunnett NW Schmidt BL . Protease-activated receptors in health and disease. Physiol Rev. (2023) 103(1):717–85. 10.1152/physrev.00044.2021
16.
Steinberg SF . The cardiovascular actions of protease-activated receptors. Mol Pharmacol. (2005) 67(1):2–11. 10.1124/mol.104.003103
17.
Han X Nieman MT Kerlin BA . Protease-activated receptors: an illustrated review. Res Pract Thromb Haemost. (2021) 5(1):17–26. 10.1002/rth2.12454
18.
Bah A Chen Z Bush-Pelc LA Mathews FS Di Cera E . Crystal structures of murine thrombin in complex with the extracellular fragments of murine protease-activated receptors PAR3 and PAR4. Proc Natl Acad Sci U S A. (2007) 104(28):11603–8. 10.1073/pnas.0704409104
19.
Gandhi PS Chen Z Di Cera E . Crystal structure of thrombin bound to the uncleaved extracellular fragment of PAR1. J Biol Chem. (2010) 285(20):15393–8. 10.1074/jbc.M110.115337
20.
Vu TK Hung DT Wheaton VI Coughlin SR . Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. (1991) 64(6):1057–68. 10.1016/0092-8674(91)90261-V
21.
Haycraft JB . Iv. On the action of a secretion obtained from the medicinal leech on the coagulation of the blood. Proc R Soc Lond. (1883) 36(228–231):478–87. 10.1098/rspl.1883.0135
22.
Mumaw MM de la Fuente M Noble DN Nieman MT . Targeting the anionic region of human protease-activated receptor 4 inhibits platelet aggregation and thrombosis without interfering with hemostasis. J Thromb Haemost. (2014) 12(8):1331–41. 10.1111/jth.12619
23.
O'Brien PJ Prevost N Molino M Hollinger MK Woolkalis MJ Woulfe DS et al Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved Par1. J Biol Chem. (2000) 275(18):13502–9. 10.1074/jbc.275.18.13502
24.
Kataoka H Hamilton JR McKemy DD Camerer E Zheng YW Cheng A et al Protease-activated receptors 1 and 4 mediate thrombin signaling in endothelial cells. Blood. (2003) 102(9):3224–31. 10.1182/blood-2003-04-1130
25.
Tsoucalas G Karamanou M Papaioannou TG Sgantzos M . Theories about blood coagulation in the writings of ancient Greek medico-philosophers. Curr Pharm Des. (2017) 23(9):1275–8. 10.2174/1381612822666161205120848
26.
Schmidt AF . Neue untersuchungen Über die faserstoffgerinnung. Arch Gesamte Physiol Menschen Tiere. (1872) 6:413–538. 10.1007/BF01612263
27.
Larsen JB Hvas AM . Thrombin: a pivotal player in hemostasis and beyond. Semin Thromb Hemost. (2021) 47(7):759–74. 10.1055/s-0041-1727116
28.
Buckmaster GA . The coagulation of the blood. Sci Prog Twent Century. (1907) 2(5):51–75.
29.
Desforges JF Bigelow FS . An action of thrombin on platelets in accelerating clotting. Blood. (1954) 9(2):153–62. 10.1182/blood.V9.2.153.153
30.
Jaffe EA Nachman RL Becker CG Minick CR . Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. (1973) 52(11):2745–56. 10.1172/JCI107470
31.
D'Amore P Shepro D . Stimulation of growth and calcium influx in cultured, bovine, aortic endothelial cells by platelets and vasoactive substances. J Cell Physiol. (1977) 92(2):177–83. 10.1002/jcp.1040920206
32.
Ishii K Hein L Kobilka B Coughlin SR . Kinetics of thrombin receptor cleavage on intact cells. Relation to signaling. J Biol Chem. (1993) 268(13):9780–6. 10.1016/S0021-9258(18)98415-2
33.
Hoxie JA Ahuja M Belmonte E Pizarro S Parton R Brass LF . Internalization and recycling of activated thrombin receptors. J Biol Chem. (1993) 268(18):13756–63. 10.1016/S0021-9258(18)86921-6
34.
Kahn ML Zheng YW Huang W Bigornia V Zeng D Moff S et al A dual thrombin receptor system for platelet activation. Nature. (1998) 394(6694):690–4. 10.1038/29325
35.
Xu WF Andersen H Whitmore TE Presnell SR Yee DP Ching A et al Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A. (1998) 95(12):6642–6. 10.1073/pnas.95.12.6642
36.
Coughlin SR . Protease-activated receptors and platelet function. Thromb Haemost. (1999) 82(2):353–6. 10.1055/s-0037-1615853
37.
Sveshnikova AN Balatskiy AV Demianova AS Shepelyuk TO Shakhidzhanov SS Balatskaya MN et al Systems biology insights into the meaning of the platelet’s dual-receptor thrombin signaling. J Thromb Haemost. (2016) 14(10):2045–57. 10.1111/jth.13442
38.
Abdallah RT Keum JS El-Shewy HM Lee MH Wang B Gooz M et al Plasma kallikrein promotes epidermal growth factor receptor transactivation and signaling in vascular smooth muscle through direct activation of protease-activated receptors. J Biol Chem. (2010) 285(45):35206–15. 10.1074/jbc.M110.171769
39.
Aman M Hirano M Kanaide H Hirano K . Upregulation of proteinase-activated receptor-2 and increased response to trypsin in endothelial cells after exposure to oxidative stress in rat aortas. J Vasc Res. (2010) 47(6):494–506. 10.1159/000313877
40.
Billich A Urtz N Reuschel R Baumruker T . Sphingosine kinase 1 is essential for proteinase-activated receptor-1 signalling in epithelial and endothelial cells. Int J Biochem Cell Biol. (2009) 41(7):1547–55. 10.1016/j.biocel.2009.01.001
41.
Bochenek ML Gogiraju R Großmann S Krug J Orth J Reyda S et al Epcr-Par1 biased signaling regulates perfusion recovery and neovascularization in peripheral ischemia. JCI Insight. (2022) 7(14):1–20. 10.1172/jci.insight.157701
42.
Camerer E Cornelissen I Kataoka H Duong DN Zheng YW Coughlin SR . Roles of protease-activated receptors in a mouse model of endotoxemia. Blood. (2006) 107(10):3912–21. 10.1182/blood-2005-08-3130
43.
D'Hondt C Ponsaerts R Srinivas SP Vereecke J Himpens B . Thrombin inhibits intercellular calcium wave propagation in corneal endothelial cells by modulation of hemichannels and gap junctions. Invest Ophthalmol Vis Sci. (2007) 48(1):120–33. 10.1167/iovs.06-0770
44.
Kondo N Ogawa M Wada H Nishikawa S . Thrombin induces rapid disassembly of claudin-5 from the tight junction of endothelial cells. Exp Cell Res. (2009) 315(17):2879–87. 10.1016/j.yexcr.2009.07.031
45.
Lafleur MA Hollenberg MD Atkinson SJ Knäuper V Murphy G Edwards DR . Activation of pro-(matrix metalloproteinase-2) (pro-mmp-2) by thrombin is membrane-type-mmp-dependent in human umbilical vein endothelial cells and generates a distinct 63 kda active Species. Biochem J. (2001) 357(Pt 1):107–15. 10.1042/bj3570107
46.
Liu Y Mueller BM . Protease-activated receptor-2 regulates vascular endothelial growth factor expression in mda-mb-231 cells via mapk pathways. Biochem Biophys Res Commun. (2006) 344(4):1263–70. 10.1016/j.bbrc.2006.04.005
47.
Papadaki S Sidiropoulou S Moschonas IC Tselepis AD . Factor Xa and thrombin induce endothelial progenitor cell activation. The effect of direct oral anticoagulants. Platelets. (2020) 32(6):1–8. 10.1080/09537104.2020.1802413
48.
Rajput PS Lyden PD Chen B Lamb JA Pereira B Lamb A et al Protease activated receptor-1 mediates cytotoxicity during ischemia using in vivo and in vitro models. Neuroscience. (2014) 281:229–40. 10.1016/j.neuroscience.2014.09.038
49.
Rullier A Senant N Kisiel W Bioulac-Sage P Balabaud C Le Bail B et al Expression of protease-activated receptors and tissue factor in human liver. Virchows Arch. (2006) 448(1):46–51. 10.1007/s00428-005-0078-0
50.
Su X Camerer E Hamilton JR Coughlin SR Matthay MA . Protease-activated receptor-2 activation induces acute lung inflammation by neuropeptide-dependent mechanisms. J Immunol. (2005) 175(4):2598–605. 10.4049/jimmunol.175.4.2598
51.
Syeda F Grosjean J Houliston RA Keogh RJ Carter TD Paleolog E et al Cyclooxygenase-2 induction and prostacyclin release by protease-activated receptors in endothelial cells require cooperation between mitogen-activated protein kinase and nf-kappab pathways. J Biol Chem. (2006) 281(17):11792–804. 10.1074/jbc.M509292200
52.
Takeuchi T Harris JL Huang W Yan KW Coughlin SR Craik CS . Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J Biol Chem. (2000) 275(34):26333–42. 10.1074/jbc.M002941200
53.
Tognetto M D'Andrea MR Trevisani M Guerrini R Salvadori S Spisani L et al Proteinase-activated receptor-1 (par-1) activation contracts the isolated human renal artery in vitro. Br J Pharmacol. (2003) 139(1):21–7. 10.1038/sj.bjp.0705215
54.
Vogel SM Gao X Mehta D Ye RD John TA Andrade-Gordon P et al Abrogation of thrombin-induced increase in pulmonary microvascular permeability in par-1 knockout mice. Physiol Genomics. (2000) 4(2):137–45. 10.1152/physiolgenomics.2000.4.2.137
55.
Shapiro MJ Weiss EJ Faruqi TR Coughlin SR . Protease-activated receptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. J Biol Chem. (2000) 275(33):25216–21. 10.1074/jbc.M004589200
56.
Covic L Gresser AL Kuliopulos A . Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry. (2000) 39(18):5458–67. 10.1021/bi9927078
57.
Ando S Otani H Yagi Y Kawai K Araki H Nakamura T et al Protease-activated receptor 4-mediated Ca2+ signaling in mouse lung alveolar epithelial cells. Life Sci. (2007) 81(10):794–802. 10.1016/j.lfs.2007.06.026
58.
Kendrick NC Nieman MT . Hidden power: PAR4’S role in liver damage from Acetaminophen overdose in mice. Arterioscler Thromb Vasc Biol. (2024) 45(1):72–3. 10.1161/atvbaha.124.321881
59.
Cleuren ACA van der Ent MA Jiang H Hunker KL Yee A Siemieniak DR et al The in vivo endothelial cell translatome is highly heterogeneous across vascular beds. Proc Natl Acad Sci U S A. (2019) 116(47):23618–24. 10.1073/pnas.1912409116
60.
Fujiwara M Jin E Ghazizadeh M Kawanami O . Differential expression of protease-activated receptors 1, 2, and 4 on human endothelial cells from different vascular sites. Pathobiology. (2004) 71(1):52–8. 10.1159/000072962
61.
Arachiche A Mumaw MM de la Fuente M Nieman MT . Protease-activated receptor 1 (PAR1) and PAR4 heterodimers are required for PAR1-enhanced cleavage of PAR4 by Α-thrombin. J Biol Chem. (2013) 288(45):32553–62. 10.1074/jbc.M113.472373
62.
Lin H Liu AP Smith TH Trejo J . Cofactoring and dimerization of proteinase-activated receptors. Pharmacol Rev. (2013) 65(4):1198–213. 10.1124/pr.111.004747
63.
Leger AJ Jacques SL Badar J Kaneider NC Derian CK Andrade-Gordon P et al Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation. (2006) 113(9):1244–54. 10.1161/CIRCULATIONAHA.105.587758
64.
Zhao P Metcalf M Bunnett NW . Biased signaling of protease-activated receptors. Front Endocrinol (Lausanne). (2014) 5:67. 10.3389/fendo.2014.00067
65.
Clemetson KJ Clemetson JM . Chapter 9 - platelet receptors. In: MichelsonAD, editor. Platelets. 3rd ed. Amsterdam, Netherlands: Academic Press (2013). p. 169–94.
66.
Li S Tarlac V Christanto RBI French SL Hamilton JR . Determination of PAR4 numbers on the surface of human platelets: no effect of the single nucleotide polymorphism Rs773902. Platelets. (2020) 32(7):1–4. 10.1080/09537104.2020.1810654
67.
Offermanns S . G-Proteins as transducers in transmembrane signalling. Prog Biophys Mol Biol. (2003) 83(2):101–30. 10.1016/S0079-6107(03)00052-X
68.
Jesmin S Gando S Zaedi S Sakuraya F . Differential expression, time course and distribution of four pars in rats with endotoxin-induced acute lung injury. Inflammation. (2007) 30(1-2):14–27. 10.1007/s10753-006-9017-8
69.
Palta S Saroa R Palta A . Overview of the coagulation system. Indian J Anaesth. (2014) 58(5):515–23. 10.4103/0019-5049.144643
70.
Rühl H Müller J Harbrecht U Fimmers R Oldenburg J Mayer G et al Thrombin inhibition profiles in healthy individuals and thrombophilic patients. Thromb Haemost. (2012) 107(5):848–53. 10.1160/TH11-10-0719
71.
Popovic M Paskas S Zivkovic M Burysek L Laumonnier Y . Human cytomegalovirus increases huvec sensitivity to thrombin and modulates expression of thrombin receptors. J Thromb Thrombolysis. (2010) 30(2):164–71. 10.1007/s11239-010-0447-7
72.
Megyeri M Mako V Beinrohr L Doleschall Z Prohaszka Z Cervenak L et al Complement protease masp-1 activates human endothelial cells: par4 activation is a link between complement and endothelial function. J Immunol. (2009) 183(5):3409–16. 10.4049/jimmunol.0900879
73.
Ritchie E Saka M Mackenzie C Drummond R Wheeler-Jones C Kanke T et al Cytokine upregulation of proteinase-activated-receptors 2 and 4 expression mediated by P38 map kinase and inhibitory kappa B kinase Beta in human endothelial cells. Br J Pharmacol. (2007) 150(8):1044–54. 10.1038/sj.bjp.0707150
74.
Hamilton JR Frauman AG Cocks TM . Increased expression of protease-activated receptor-2 (PAR2) and PAR4 in human coronary artery by inflammatory stimuli unveils endothelium-dependent relaxations to Par2 and Par4 agonists. Circ Res. (2001) 89(1):92–8. 10.1161/hh1301.092661
75.
Wang Z Chen D Zhang R An S Zhang Z Ma Q . Interleukin-1β increased the expression of protease-activated receptor 4 mrna and protein in dorsal root ganglion neurons. Neurochem Res. (2013) 38(9):1895–903. 10.1007/s11064-013-1095-z
76.
Fender AC Rauch BH Geisler T Schror K . Protease-activated receptor par-4: an inducible switch between thrombosis and vascular inflammation?Thromb Haemost. (2017) 117(11):2013–25. 10.1160/TH17-03-0219
77.
Rahadian A Fukuda D Salim HM Yagi S Kusunose K Yamada H et al Thrombin inhibition by dabigatran attenuates endothelial dysfunction in diabetic mice. Vascul Pharmacol. (2020) 124:106632. 10.1016/j.vph.2019.106632
78.
Scridon A Marginean A Hutanu A Chinezu L Gheban D Perian M et al Vascular protease-activated receptor 4 upregulation, increased platelet aggregation, and coronary lipid deposits induced by long-term dabigatran administration - results from a diabetes animal model. J Thromb Haemost. (2019) 17(3):538–50. 10.1111/jth.14386
79.
Pavic G Grandoch M Dangwal S Jobi K Rauch BH Doller A et al Thrombin receptor protease-activated receptor 4 is a key regulator of exaggerated intimal thickening in diabetes mellitus. Circulation. (2014) 130(19):1700–11. 10.1161/CIRCULATIONAHA.113.007590
80.
Fender AC Kleeschulte S Stolte S Leineweber K Kamler M Bode J et al Thrombin receptor PAR4 drives canonical Nlrp3 inflammasome signaling in the heart. Basic Res Cardiol. (2020) 115(2):10. 10.1007/s00395-019-0771-9
81.
Mittendorff C Abu-Taha I Kassler L Hustedt T Wolf S Bode JG et al Thrombin receptor PAR4 cross-activates the tyrosine kinase C-met in atrial cardiomyocytes. Naunyn Schmiedebergs Arch Pharmacol. (2024):1–14. 10.1007/s00210-024-03436-6
82.
Tyagi N Sedoris KC Steed M Ovechkin AV Moshal KS Tyagi SC . Mechanisms of homocysteine-induced oxidative stress. Am J Physiol Heart Circ Physiol. (2005) 289(6):H2649–56. 10.1152/ajpheart.00548.2005
83.
Scimone C Alibrandi S Donato L De Gaetano GV Fusco C Nardella G et al Amplification of protease-activated receptors signaling in sporadic cerebral cavernous malformation endothelial cells. Biochim Biophys Acta Mol Cell Res. (2023) 1870(5):119474. 10.1016/j.bbamcr.2023.119474
84.
Corbin LJ White SJ Taylor A Williams CM Taylor K van den Bosch MT et al Epigenetic regulation of F2rl3 associates with myocardial infarction and platelet function. Circ Res. (2022) 130:384–400. 10.1161/circresaha.121.318836
85.
Zhang Y Yang R Burwinkel B Breitling LP Holleczek B Schottker B et al F2rl3 methylation in blood DNA is a strong predictor of mortality. Int J Epidemiol. (2014) 43(4):1215–25. 10.1093/ije/dyu006
86.
Yu G Jiang P Xiang Y Zhang Y Zhu Z Zhang C et al Increased expression of protease-activated receptor 4 and trefoil factor 2 in human colorectal cancer. PLoS One. (2015) 10(4):e0122678. 10.1371/journal.pone.0122678
87.
Zhang Y Yu G Jiang P Xiang Y Li W Lee W et al Decreased expression of protease-activated receptor 4 in human gastric cancer. Int J Biochem Cell Biol. (2011) 43(9):1277–83. 10.1016/j.biocel.2011.05.008
88.
Zhang Y Schottker B Ordonez-Mena J Holleczek B Yang R Burwinkel B et al F2rl3 methylation, lung cancer incidence and mortality. Int J Cancer. (2015) 137(7):1739–48. 10.1002/ijc.29537
89.
Kahn ML Hammes SR Botka C Coughlin SR . Gene and locus structure and chromosomal localization of the protease-activated receptor gene family. J Biol Chem. (1998) 273(36):23290–6. 10.1074/jbc.273.36.23290
90.
Nishida M Ogushi M Suda R Toyotaka M Saiki S Kitajima N et al Heterologous down-regulation of angiotensin type 1 receptors by purinergic P2y2 receptor stimulation through S-nitrosylation of nf-kappab. Proc Natl Acad Sci U S A. (2011) 108(16):6662–7. 10.1073/pnas.1017640108
91.
Kunselman JM Lott J Puthenveedu MA . Mechanisms of selective G protein-coupled receptor localization and trafficking. Curr Opin Cell Biol. (2021) 71:158–65. 10.1016/j.ceb.2021.03.002
92.
DeFea KA Zalevsky J Thoma MS Déry O Mullins RD Bunnett NW . Beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated Erk1/2. J Cell Biol. (2000) 148(6):1267–81. 10.1083/jcb.148.6.1267
93.
Déry O Thoma MS Wong H Grady EF Bunnett NW . Trafficking of proteinase-activated receptor-2 and Beta-arrestin-1 tagged with green fluorescent protein. Beta-arrestin-dependent endocytosis of a proteinase receptor. J Biol Chem. (1999) 274(26):18524–35. 10.1074/jbc.274.26.18524
94.
Dores MR Trejo J . Atypical regulation of G protein-coupled receptor intracellular trafficking by ubiquitination. Curr Opin Cell Biol. (2014) 27:44–50. 10.1016/j.ceb.2013.11.004
95.
Marchese A Trejo J . Ubiquitin-dependent regulation of G protein-coupled receptor trafficking and signaling. Cell Signal. (2013) 25(3):707–16. 10.1016/j.cellsig.2012.11.024
96.
Jacob C Cottrell GS Gehringer D Schmidlin F Grady EF Bunnett NW . C-Cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J Biol Chem. (2005) 280(16):16076–87. 10.1074/jbc.M500109200
97.
Wolfe BL Marchese A Trejo J . Ubiquitination differentially regulates clathrin-dependent internalization of protease-activated receptor-1. J Cell Biol. (2007) 177(5):905–16. 10.1083/jcb.200610154
98.
Adams MN Christensen ME He Y Waterhouse NJ Hooper JD . The role of palmitoylation in signalling, cellular trafficking and plasma membrane localization of protease-activated receptor-2. PLoS One. (2011) 6(11):e28018. 10.1371/journal.pone.0028018
99.
Botham A Guo X Xiao YP Morice AH Compton SJ Sadofsky LR . Palmitoylation of human proteinase-activated receptor-2 differentially regulates receptor-triggered Erk1/2 activation, calcium signalling and endocytosis. Biochem J. (2011) 438(2):359–67. 10.1042/BJ20101958
100.
Canto I Trejo J . Palmitoylation of protease-activated receptor-1 regulates adaptor protein complex-2 and -3 interaction with tyrosine-based motifs and endocytic sorting. J Biol Chem. (2013) 288(22):15900–12. 10.1074/jbc.M113.469866
101.
Paing MM Stutts AB Kohout TA Lefkowitz RJ Trejo J . Β-Arrestins regulate protease-activated receptor-1 desensitization but not internalization or down-regulation. J Biol Chem. (2002) 277(2):1292–300. 10.1074/jbc.M109160200
102.
Paing MM Temple BR Trejo J . A tyrosine-based sorting signal regulates intracellular trafficking of protease-activated receptor-1: multiple regulatory mechanisms for agonist-induced G protein-coupled receptor internalization. J Biol Chem. (2004) 279(21):21938–47. 10.1074/jbc.M401672200
103.
Paing MM Johnston CA Siderovski DP Trejo J . Clathrin adaptor AP2 regulates thrombin receptor constitutive internalization and endothelial cell resensitization. Mol Cell Biol. (2006) 26(8):3231–42. 10.1128/MCB.26.8.3231-3242.2006
104.
Wang Y Zhou Y Szabo K Haft CR Trejo J . Down-regulation of protease-activated receptor-1 is regulated by sorting nexin 1. Mol Biol Cell. (2002) 13(6):1965–76. 10.1091/mbc.e01-11-0131
105.
Smith TH Coronel LJ Li JG Dores MR Nieman MT Trejo J . Protease-activated receptor-4 signaling and trafficking is regulated by the clathrin adaptor protein Complex-2 independent of Beta-arrestins. J Biol Chem. (2016) 291(35):18453–64. 10.1074/jbc.M116.729285
106.
Soh UJ Dores MR Chen B Trejo J . Signal transduction by protease-activated receptors. Br J Pharmacol. (2010) 160(2):191–203. 10.1111/j.1476-5381.2010.00705.x
107.
Hein L Ishii K Coughlin SR Kobilka BK . Intracellular targeting and trafficking of thrombin receptors. A novel mechanism for resensitization of a G protein-coupled receptor. J Biol Chem. (1994) 269(44):27719–26. 10.1016/S0021-9258(18)47045-7
108.
Shapiro MJ Trejo J Zeng D Coughlin SR . Role of the thrombin receptor’s cytoplasmic tail in intracellular trafficking. Distinct determinants for agonist-triggered versus tonic internalization and intracellular localization. J Biol Chem. (1996) 271(51):32874–80. 10.1074/jbc.271.51.32874
109.
Horvat R Palade GE . The functional thrombin receptor is associated with the plasmalemma and a large endosomal network in cultured human umbilical vein endothelial cells. J Cell Sci. (1995) 108(Pt 3):1155–64. 10.1242/jcs.108.3.1155
110.
Cunningham MR McIntosh KA Pediani JD Robben J Cooke AE Nilsson M et al Novel role for proteinase-activated receptor 2 (PAR2) in membrane trafficking of proteinase-activated receptor 4 (PAR4). J Biol Chem. (2012) 287(20):16656–69. 10.1074/jbc.M111.315911
111.
Chandrabalan A Ramachandran R . Molecular mechanisms regulating proteinase activated receptors (pars). Febs J. (2021) 288(8):2697–726. 10.1111/febs.15829
112.
White PS Morran L de Roode J . Phoresy. Curr Biol. (2017) 27(12):R578–80. 10.1016/j.cub.2017.03.073
113.
McCoy KL Traynelis SF Hepler JR . PAR1 and PAR2 couple to overlapping and distinct sets of G proteins and linked signaling pathways to differentially regulate cell physiology. Mol Pharmacol. (2010) 77(6):1005–15. 10.1124/mol.109.062018
114.
Hirano K Nomoto N Hirano M Momota F Hanada A Kanaide H . Distinct Ca2+ requirement for no production between proteinase-activated receptor 1 and 4 (PAR1 and PAR4) in vascular endothelial cells. J Pharmacol Exp Ther. (2007) 322(2):668–77. 10.1124/jpet.107.121038
115.
Ruppel KM Willison D Kataoka H Wang A Zheng YW Cornelissen I et al Essential role for Galpha13 in endothelial cells during embryonic development. Proc Natl Acad Sci U S A. (2005) 102(23):8281–6. 10.1073/pnas.0503326102
116.
Woulfe DS . Platelet G protein-coupled receptors in hemostasis and thrombosis. J Thromb Haemost. (2005) 3(10):2193–200. 10.1111/j.1538-7836.2005.01338.x
117.
Molinar-Inglis O Birch CA Nicholas D Orduña-Castillo L Cisneros-Aguirre M Patwardhan A et al APC/PAR1 confers endothelial anti-apoptotic activity via a discrete, Β-arrestin-2-mediated Sphk1-S1pr1-akt signaling axis. Proc Natl Acad Sci U S A. (2021) 118(49):1–11. 10.1073/pnas.2106623118
118.
Vanderboor CM Thibeault PE Nixon KC Gros R Kramer JM Ramachandran R . Proteinase-activated receptor 4 (PAR4) activation triggers cell membrane blebbing through rhoa and Beta-arrestin. Mol Pharmacol. (2020) 97(6):365–76. 10.1124/mol.119.118232
119.
Touhara K Hawes BE van Biesen T Lefkowitz RJ . G protein Beta gamma subunits stimulate phosphorylation of shc adapter protein. Proc Natl Acad Sci U S A. (1995) 92(20):9284–7. 10.1073/pnas.92.20.9284
120.
Duan ZZ Zhang F Li FY Luan YF Guo P Li YH et al Protease activated receptor 1 (PAR1) enhances src-mediated tyrosine phosphorylation of nmda receptor in intracerebral hemorrhage (ich). Sci Rep. (2016) 6:29246. 10.1038/srep29246
121.
Sabri A Guo J Elouardighi H Darrow AL Andrade-Gordon P Steinberg SF . Mechanisms of protease-activated receptor-4 actions in cardiomyocytes. Role of SRC tyrosine kinase. J Biol Chem. (2003) 278(13):11714–20. 10.1074/jbc.M213091200
122.
Han X Nieman MT . Par4 (protease-activated receptor 4): particularly important 4 antiplatelet therapy. Arterioscler Thromb Vasc Biol. (2018) 38(2):287–9. 10.1161/ATVBAHA.117.310550
123.
Dangelmaier C Kunapuli SP . Protease-activated receptor 4 causes akt phosphorylation independently of Pi3 kinase pathways. Platelets. (2021) 32(6):832–7. 10.1080/09537104.2020.1802415
124.
Holinstat M Preininger AM Milne SB Hudson WJ Brown HA Hamm HE . Irreversible platelet activation requires protease-activated receptor 1-mediated signaling to phosphatidylinositol phosphates. Mol Pharmacol. (2009) 76(2):301–13. 10.1124/mol.109.056622
125.
Voss B McLaughlin JN Holinstat M Zent R Hamm HE . PAR1, but not PAR4, activates human platelets through a gi/O/phosphoinositide-3 kinase signaling axis. Mol Pharmacol. (2007) 71(5):1399–406. 10.1124/mol.106.033365
126.
Fujiwara M Jin E Ghazizadeh M Kawanami O . Activation of PAR4 induces a distinct actin fiber formation via P38 mapk in human lung endothelial cells. J Histochem Cytochem. (2005) 53(9):1121–9. 10.1369/jhc.4A6592.2005
127.
Andrianova I Kowalczyk M Denorme F . Protease activated receptor-4: ready to be part of the antithrombosis spectrum. Curr Opin Hematol. (2024) 31(5):238–44. 10.1097/moh.0000000000000828
128.
Rezaie AR . Protease-activated receptor signalling by coagulation proteases in endothelial cells. Thromb Haemost. (2014) 112(5):876–82. 10.1160/th14-02-0167
129.
Grimsey NJ Trejo J . Integration of endothelial protease-activated receptor-1 inflammatory signaling by ubiquitin. Curr Opin Hematol. (2016) 23(3):274–9. 10.1097/MOH.0000000000000232
130.
Heo Y Jeon H Namkung W . Par4-mediated Pi3k/akt and rhoa/rock signaling pathways are essential for thrombin-induced morphological changes in meg-01 cells. Int J Mol Sci. (2022) 23(2):1–15. 10.3390/ijms23020776
131.
Pearson JD . Plasticity of adult endothelium: how frequent and to what extent?Cardiovasc Res. (2015) 108(3):319–20. 10.1093/cvr/cvv240
132.
Hardman K Goldman A Pliotas C . Membrane force reception: mechanosensation in GPCRs and tools to address it. Curr Opin Physiol. (2023) 35:1–10. 10.1016/j.cophys.2023.100689
133.
Erdogmus S Storch U Danner L Becker J Winter M Ziegler N et al Helix 8 is the essential structural motif of mechanosensitive GPCRs. Nat Commun. (2019) 10(1):5784. 10.1038/s41467-019-13722-0
134.
Han X Nieman MT . The domino effect triggered by the tethered ligand of the protease activated receptors. Thromb Res. (2020) 196:87–98. 10.1016/j.thromres.2020.08.004
135.
Norman JE Cunningham MR Jones ML Walker ME Westbury SK Sessions RB et al Protease-activated receptor 4 variant P.Tyr157cys reduces platelet functional responses and alters receptor trafficking. Arterioscler Thromb Vasc Biol. (2016) 36(5):952–60. 10.1161/ATVBAHA.115.307102
136.
Edelstein LC Simon LM Lindsay CR Kong X Teruel-Montoya R Tourdot BE et al Common variants in the human platelet PAR4 thrombin receptor alter platelet function and differ by race. Blood. (2014) 124(23):3450–8. 10.1182/blood-2014-04-572479
137.
Han X Knauss EA Fuente M Li W Conlon RA LePage DF et al A mouse model of the protease activated receptor 4 (Par4) Pro310leu variant has reduced platelet reactivity. J Thromb Haemost. (2024) 22(6):1715–26. 10.1016/j.jtha.2024.03.004
138.
Han X Hofmann L de la Fuente M Alexander N Palczewski K Invent Consortium I et al PAR4 activation involves extracellular loop-3 and transmembrane residue Thr153. Blood. (2020) 136(19):2217–28. 10.1182/blood.2019004634
139.
Koller GM Schafer C Kemp SS Aguera KN Lin PK Forgy JC et al Proinflammatory mediators, il (interleukin)-1beta, tnf (tumor necrosis factor) alpha, and thrombin directly induce capillary tube regression. Arterioscler Thromb Vasc Biol. (2020) 40(2):365–77. 10.1161/ATVBAHA.119.313536
140.
Kemp SS Penn MR Koller GM Griffin CT Davis GE . Proinflammatory mediators, tnfα, ifnγ, and thrombin, directly induce lymphatic capillary tube regression. Front Cell Dev Biol. (2022) 10:937982. 10.3389/fcell.2022.937982
141.
Zhao J Jin G Liu X Wu K Yang Y He Z et al PAR1 and PAR4 exert opposite effects on tumor growth and metastasis of esophageal squamous cell carcinoma via STAT3 and NF-ΚB signaling pathways. Cancer Cell Int. (2021) 21(1):637. 10.1186/s12935-021-02354-4
142.
Wang M An S Wang D Ji H Geng M Guo X et al Quantitative proteomics identify the possible tumor suppressive role of protease-activated receptor-4 in esophageal squamous cell carcinoma cells. Pathol Oncol Res. (2019) 25(3):937–43. 10.1007/s12253-018-0395-7
143.
Jiang P Yu GY Zhang Y Xiang Y Hua HR Bian L et al Down-regulation of protease-activated receptor 4 in lung adenocarcinoma is associated with a more aggressive phenotype. Asian Pac J Cancer Prev. (2013) 14(6):3793–8. 10.7314/APJCP.2013.14.6.3793
144.
Zhang H Jiang P Zhang C Lee S Wang W Zou H . PAR4 overexpression promotes colorectal cancer cell proliferation and migration. Oncol Lett. (2018) 16(5):5745–52. 10.3892/ol.2018.9407
145.
Flynn AN Buret AG . Proteinase-activated receptor 1 (par-1) and cell apoptosis. Apoptosis. (2004) 9(6):729–37. 10.1023/B:APPT.0000045784.49886.96
146.
Rajala R Griffin CT . Intercellular synergy between protease-activated receptors in mouse development. J Thromb Haemostasis. (2024):1–3. 10.1016/j.jtha.2024.11.022
147.
Kumar DR Hanlin E Glurich I Mazza JJ Yale SH . Virchow’s contribution to the understanding of thrombosis and cellular biology. Clin Med Res. (2010) 8(3-4):168–72. 10.3121/cmr.2009.866
148.
Claesson-Welsh L Dejana E McDonald DM . Permeability of the endothelial barrier: identifying and reconciling controversies. Trends Mol Med. (2021) 27(4):314–31. 10.1016/j.molmed.2020.11.006
149.
McDonald DM Choyke PL . Imaging of angiogenesis: from microscope to clinic. Nat Med. (2003) 9(6):713–25. 10.1038/nm0603-713
150.
Folkman J . Tumor angiogenesis: therapeutic implications. N Engl J Med. (1971) 285(21):1182–6. 10.1056/NEJM197111182852108
151.
Jaffer IH Weitz JI . Chapter 149 - antithrombotic drugs. In: HoffmanRBenzEJSilbersteinLEHeslopHEWeitzJIAnastasiJet al, editors. Hematology. 7th ed. Philadelphia, PA: Elsevier (2018). p. 2168–88.
152.
Mao Y Jin J Daniel JL Kunapuli SP . Regulation of plasmin-induced protease-activated receptor 4 activation in platelets. Platelets. (2009) 20(3):191–8. 10.1080/09537100902803635
153.
Fu Q Cheng J Gao Y Zhang Y Chen X Xie J . Protease-activated receptor 4: a critical participator in inflammatory response. Inflammation. (2015) 38(2):886–95. 10.1007/s10753-014-9999-6
154.
Morrow DA Braunwald E Bonaca MP Ameriso SF Dalby AJ Fish MP et al Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. (2012) 366(15):1404–13. 10.1056/NEJMoa1200933
155.
Yoon H Choi CI Triplet EM Langley MR Kleppe LS Kim HN et al Blocking the thrombin receptor promotes repair of demyelinated lesions in the adult brain. J Neurosci. (2020) 40(7):1483–500. 10.1523/JNEUROSCI.2029-19.2019
156.
van den Eshof BL Hoogendijk AJ Simpson PJ van Alphen FPJ Zanivan S Mertens K et al Paradigm of biased PAR1 (protease-activated receptor-1) activation and inhibition in endothelial cells dissected by phosphoproteomics. Arterioscler Thromb Vasc Biol. (2017) 37(10):1891–902. 10.1161/ATVBAHA.117.309926
157.
Li W Johnson DJ Esmon CT Huntington JA . Structure of the antithrombin-thrombin-heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat Struct Mol Biol. (2004) 11(9):857–62. 10.1038/nsmb811
158.
Fala L . Zontivity (vorapaxar), first-in-class par-1 antagonist, receives FDA approval for risk reduction of heart attack, stroke, and cardiovascular death. Am Health Drug Benefits. (2015) 8(Spec Feature):148–51.
159.
Tabula Muris Consortium; Overall Coordination; Logistical Coordination; Organ Collection and Processing; Library Preparation and Sequencing; Computational Data Analysis; Cell Type Annotation; Writing Group; Supplemental Text Writing Group; Principal Investigators.Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. (2018) 562(7727):367–72. 10.1038/s41586-018-0590-4
160.
Aggarwal A Jennings CL Manning E Cameron SJ . Platelets at the vessel wall in non-thrombotic disease. Circ Res. (2023) 132(6):775–90. 10.1161/CIRCRESAHA.122.321566
161.
Hunter T . A journey from phosphotyrosine to phosphohistidine and beyond. Mol Cell. (2022) 82(12):2190–200. 10.1016/j.molcel.2022.05.007
162.
Lin Y Wozniak JM Grimsey NJ Girada S Patwardhan A Molinar-Inglis O et al Phosphoproteomic analysis of protease-activated receptor-1 biased signaling reveals unique modulators of endothelial barrier function. Proc Natl Acad Sci U S A. (2020) 117(9):5039–48. 10.1073/pnas.1917295117
163.
Groten SA van den Eshof BL van Alphen FPJ Meijer AB van den Biggelaar M Hoogendijk AJ . Integrative phosphoproteomic analyses reveal hemostatic-endothelial signaling interplay. J Thromb Haemost. (2024):1–14. 10.1016/j.jtha.2024.10.011
164.
Alvarez-Castelao B Schanzenbächer CT Hanus C Glock C Tom Dieck S Dörrbaum AR et al Cell-type-specific metabolic labeling of nascent proteomes in vivo. Nat Biotechnol. (2017) 35(12):1196–201. 10.1038/nbt.4016
165.
Johnson JL Yaron TM Huntsman EM Kerelsky A Song J Regev A et al An atlas of substrate specificities for the human serine/threonine kinome. Nature. (2023) 613:759–66. 10.1038/s41586-022-05575-3
166.
Yaron-Barir TM Joughin BA Huntsman EM Kerelsky A Cizin DM Cohen BM et al The intrinsic substrate specificity of the human tyrosine kinome. Nature. (2024) 629(8014):1174–81. 10.1038/s41586-024-07407-y
Summary
Keywords
PAR4, protease, vascular biology, G protein-coupled receptor (GPCR), endothelial cell, thrombin, signal transduction
Citation
Rajala R and Griffin CT (2025) Endothelial protease-activated receptor 4: impotent or important?. Front. Cardiovasc. Med. 12:1541879. doi: 10.3389/fcvm.2025.1541879
Received
11 December 2024
Accepted
09 January 2025
Published
28 January 2025
Volume
12 - 2025
Edited by
Philipp von Hundelshausen, Ludwig Maximilian University of Munich, Germany
Reviewed by
Antonia Follenzi, Università degli Studi del Piemonte Orientale, Italy
Marvin T. Nieman, Case Western Reserve University, United States
Updates
Copyright
© 2025 Rajala and Griffin.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Courtney T. Griffin courtney-griffin@omrf.org
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.