Abstract
Atherosclerosis has been defined as an inflammatory disease. As observed during acute infections, excess inflammatory activity is associated with disease severity and mortality. After myocardial infarction, several waves of inflammatory cells play a crucial role in infarct size and cardiac remodeling. In the short and long term, subtypes of inflammatory cells and cytokines released orchestrate the healing and stability of coronary disease. In recent years, some anti-inflammatory therapies have been shown to reduce the residual cardiovascular risk. Furthermore, some medications for treating risk factors and adoption of healthy lifestyle have decreased inflammatory markers and cardiovascular outcomes. In this complex network of possibilities, multiple interventions and not just on specific cell type or cytokine may provide better results. Finally, mild or moderate inflammatory activity appears necessary for better recovery and survival after acute myocardial infarction.
Introduction
Role of inflammation in main mechanisms of acute myocardial infarction
Despite continuous progress in therapeutic strategies, cardiovascular disease remains the leading cause of death worldwide (1). These deaths are mainly related to atherosclerosis, which can be defined as an inflammatory disease (2). Acute myocardial infarction with or without ST segment elevation are the most common thrombotic complications of coronary heart disease and are mainly related to plaque rupture or endothelial erosion, respectively (3). However, there are differences in the inflammatory mechanisms of these conditions in the set of acute coronary sindromes (3).
Vulnerable plaque is typically recognized in lesions characterized by a large lipid core within macrophages associated with apoptosis of these foam cells, forming debris in the intima. The imbalance between pro- and anti-inflammatory stimuli promoted by subtypes of lymphocytes and macrophages in the intima layer seems crucial to plaque rupture due to increased breakdown of matrix collagen (4). These proinflammatory stimuli are also related to thin fibrous cap. After rupture, the vulnerable plaque exposes highly thrombogenic constituents of plaque, leading to vessel occlusion (3, 4).
Plaque erosion has become increasingly common as a cause of acute coronary syndromes. Marked pathophysiological differences have been described between plaque erosion and plaque rupture. Eroded plaques usually occur in lipid poor plaques with increased matrix tissue (3). These plaques have increased content of proteoglycan and glycosaminoglycans (5, 6) and few inflammatory cells (7). Endothelial apoptosis may contribute to superficial erosion. Myeloperoxidase, a potent oxidant specie released by inflammatory cells, may promote endothelial death (8). More recently, the role of neutrophil extracellular traps was reported, showing endothelial cells activation and increased thrombogenicity through increased tissue factor expression (9).
Cytokines and inflammatory cells in acute myocardial infarction
Acute myocardial infarction triggers waves of circulating inflammatory cells, in part beneficial but harmful when in excess (10). The first wave is characterized by the presence of polymorphonuclear neutrophils in the damaged myocardium. The second wave is dominated by the recruitment of macrophages that seem important for removal of cell debris contributing to myocardial healing (10). In parallel, there is an increased participation of lymphocytes, which may raise the presence of macrophages of pro-inflammatory phenotype (11). Alongside macrophages, there is an important participation of lymphocytes for changes in the phenotype of M1 pro-inflammatory macrophages into M2 macrophages and release of protective cytokines such as interleukins (IL) – 2, IL-4, and IL-10, involved in the myocardial repair (10). Conversely, the release of IL-6 in the first day of myocardial infarction seems related to increased infarcted mass, and reduced left ventricular ejection fraction, quantified by cardiac magnetic resonance imaging (12).
In patients with plaque erosion, the presence of neutrophil extracellular traps is associated with endothelium activation, promoting macrophage recruitment and increased thrombogenicity associated with augmented expression of IL-1α and interferon type 1 (IFN-1) (13). In addition, neutrophil extracellular traps can activate the NOD-,LRR-, and pyrin domain-containing protein (NLRP)3 inflammasome (14). Further, this inflammatory platform activates caspase 1, with subsequent release of IL-1β, and pro-IL-18, triggering the inflammatory pathway related to cardiovascular disease. Circulating IL-1β amplifies inflammatory and pro-thrombotic pathways due to increased expression of IL-6 and also due to its own expression by many inflammatory cells (15). Interestingly, the effects of NLRP3 inflammasome seem attenuated in the acute phase of myocardial infarction, modulated by enzymes released by monocytes, avoiding excessive inflammatory stimuli (16).
Lymphocytes, monocytes, neutrophils, dendritic cells in acute myocardial infarction
The first stimulus for inflammatory cells recruitment is provided by necrotic myocytes with DNA fragments that act as danger-associated molecular patterns (DAMPs) (17). Next, the innate immune response is activated to clear cell debris from the region of myocardial infarction (18). The first mobilization of inflammatory cells is provided by neutrophils that are present at the myocardial infarct region in the first 24 h (18). In the same area, pro inflammatory monocytes and macrophages can be seen in the next 48–72 h, but are replaced by anti-inflammatory monocytes and macrophages for the days 4–7, which are important during the healing process of this phase (18) (Figure 1). Some studies conducted to evaluate early inhibition of neutrophils did not support protective effects in myocardial infarction size (19, 20). According to Nahrendorf (21), the initial infiltrate of monocytes are pro-inflammatory (M1) or C-C chemokine receptor type 2+(CCR2+), producing IL-1β and tumor necrosis factor alpha. Conversely, with the time, the monocyte phenotype that predominates is anti-inflammatory M2 or CCR2- (21, 22). Specimens obtained from patients who died in different post-infarction periods revealed a temporal accumulation of monocyte subsets. In the early inflammatory phase predominates classical monocytes CD14+CD16- in the infarct border region. In contrast, in the late proliferative phase after myocardial infarction, the monocytes subsets have comparable distribution. In the same study, a marked depletion of monocytes from spleen was described in the acute phase of myocardial infarction (23). These monocyte subsets were examined in patients with STEMI, stable coronary heart disease and healthy volunteers. Intermediate monocyte subset is considered the most inflammatory subtype and was associated with peak troponin and IL-6. Classical monocyte subset was also associated with IL-6. Some years after the publication of Nahrendorf (21), it was identified a third monocyte subtype and currently, the monocytes can be classified in three subsets, CD14++CD16- (classical monocytes), CD14++CD16+ (intermediate monocytes), and CD14+CD16++ (non-classical monocytes). Higher counts of intermediate monocytes appear to be related to more extensive myocardial infarction (24). Reduced CD16 expression in the first day was an independent predictor of higher left ventricular ejection fraction (25). On the other hand, late recruitment of CD16+ monocyte subset seems important for the myocardial repair (26). In elderly patients, an increase of CD14+CD16+ monocyte subset has been reported (27). This type of monocytes release pro-inflammatory cytokines, contributing for a chronic systemic inflammation in these patients (27). The persistence of pro-inflammatory subsets of monocytes was reported among STEMI patients examined at baseline, one month and six months, despite optimal medical therapy with statins, antiplatelet, betablockers, and renin angiotensin system blockers (28).
Figure 1
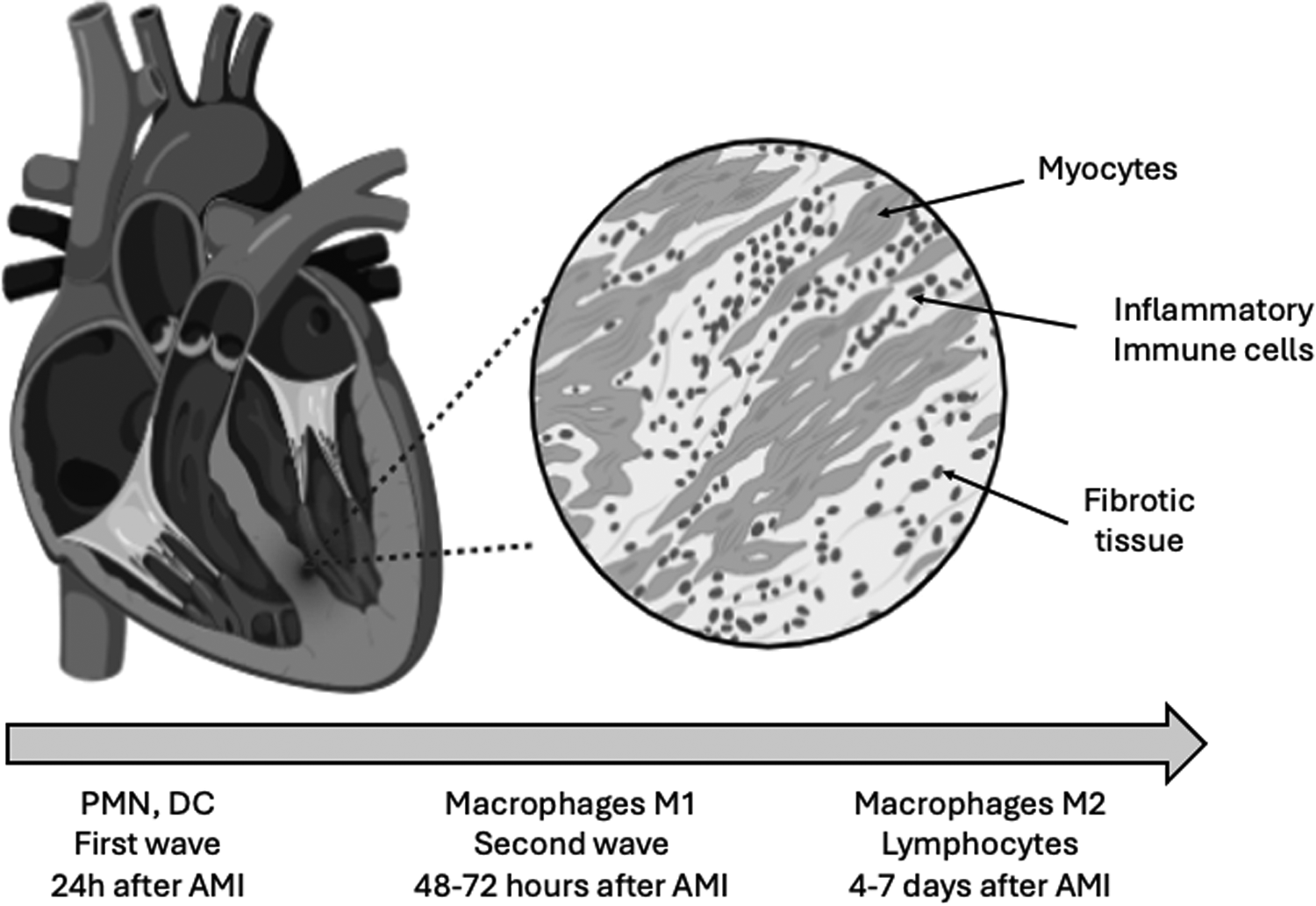
Waves of inflammatory cells after acute myocardial infarction. After coronary occlusion, the first wave of inflammatory cells are dominated by neutrophils (PMN) that can be seen in the first hours in the damaged myocardial. Dendritic cells (DC) are also early noted in the injured myocardial after reperfusion and these cells modulate the macrophage phenotype. The second wave is characterized by the presence of macrophages and, in parallel, lymphocytes. Macrophages are crucial for removal of cell debris. The lymphocytes are capable to modify the phenotype of macrophages into protective cells. Macrophages M2 are implicate in the release of anti-inflammatory cytokines involved in the myocardial healing (10–32).
Circulating lymphocytes decrease 90 min after reperfusion and is associated with worse prognostic (29). This drop in B and T lymphocytes seem related to the presence of these cells in the injured myocardial (18). B2 lymphocytes appear to be protective, and 30 days after myocardial infarction in humans, there was an association between these classical B2 cells (B2 memory plus B2 naïve) with better left ventricular ejection fraction examined by cardiac magnetic resonance imaging (12). T regulatory cells also appear to be beneficial for the healing phenotype of monocytes/macrophages, in part due to higher expression of transforming growth factor-beta 1 (11).
Secondary lymphoid organs are reservoirs for a variety of inflammatory cells, including B and T lymphocytes, and dendritic cells. Inflammatory cells present in the peritoneal cavity or even in the pericardium can influence tissue repair after myocardial infarction (30) as well as in atherosclerosis (31).
Excessive pro-inflammatory responses after acute myocardial infarction contributes to adverse ventricular remodeling. The effects of regulatory B cells in heart failure were examined in an experimental myocardial infarction model. The authors reported that regulatory B cells decreased the CCR2 in monocytes, reducing the mobilization of inflammatory monocytes to the heart, decreasing fibrosis, and promoting better ventricular function (32). In humans, reduced circulating regulatory B cells were found among AMI patients compared with stable coronary artery disease patients (33).
Dendritic cells are antigen-presenting cells, with crucial role in adaptive and innate immunity. These cells are present shortly after reperfusion myocardial injury, and contribute for a better cardiac remodeling. Dendritic cells modulate the inflammatory responses decreasing pro-inflammatory monocytes/macrophages and their release of pro-inflammatory cytokines (34, 35). Table 1 summarizes the role of inflammatory cells.
Table 1
| Cell types | Role in AMI |
|---|---|
| CD14++CD16- monocytes | Classical |
| CD14++CD16+ monocytes | Intermediate |
| CD14+CD16++ monocytes | Non-classical |
| B2 (naïve plus memory) lymphocytes | Beneficial |
| Regulatory B lymphocytes | Beneficial |
| Regulatory T lymphocytes | Beneficial |
| Dendritic cells | Beneficial |
Monocyte and lymphocyte subsets and role in acute myocardial infarction.
Higher amounts of intermediate monocytes are related to more extensive myocardial infarction (24); B2 cells related to better left ventricular ejection fraction (12); Regulatory B cells related to smaller fibrosis and better left ventricular ejection fraction (32); Regulatory T cells beneficial for the healing of injured myocardial (11); Dendritic cells change monocyte phenotypes to less inflammatory cells.
Microbiota and systemic inflammation in myocardial infarction
After myocardial infarction, an increase in intestinal permeability to bacteria products contributes to systemic inflammation and cardiac remodeling (36). Recently, bacteria translocation and lipopolysaccharides were associated with STEMI and poor prognosis (37, 38). Besides, dysbiosis and decrease in gut microcirculation after myocardial infarction seems related not only to systemic inflammation, but also to increased thrombus formation (39). Circulating lipopolysaccharides are increased in patients with myocardial infarction and are also present in coronary thrombi (37). The mechanism linking lipopolysaccharides to thrombus formation involves platelet activation by cathepsin G (37).
The heart gut microbiome immune axis was examined in humans and by an experimental model of ischemia/reperfusion (40). Compared to healthy controls, patients with myocardial infarction had augmented circulating levels of markers of increased gut permeability such as lipopolysaccharides (40). The authors found that lipopolysaccharides positively correlated with myocardial infarct size and negatively with left ventricular ejection fraction (40). In the experimental model, an increased intestinal mucosa injury was observed following myocardial ischemia/reperfusion (40). Taken together, both studies reinforce the relevance of the heart gut microbiome immune axis (Figure 2).
Figure 2
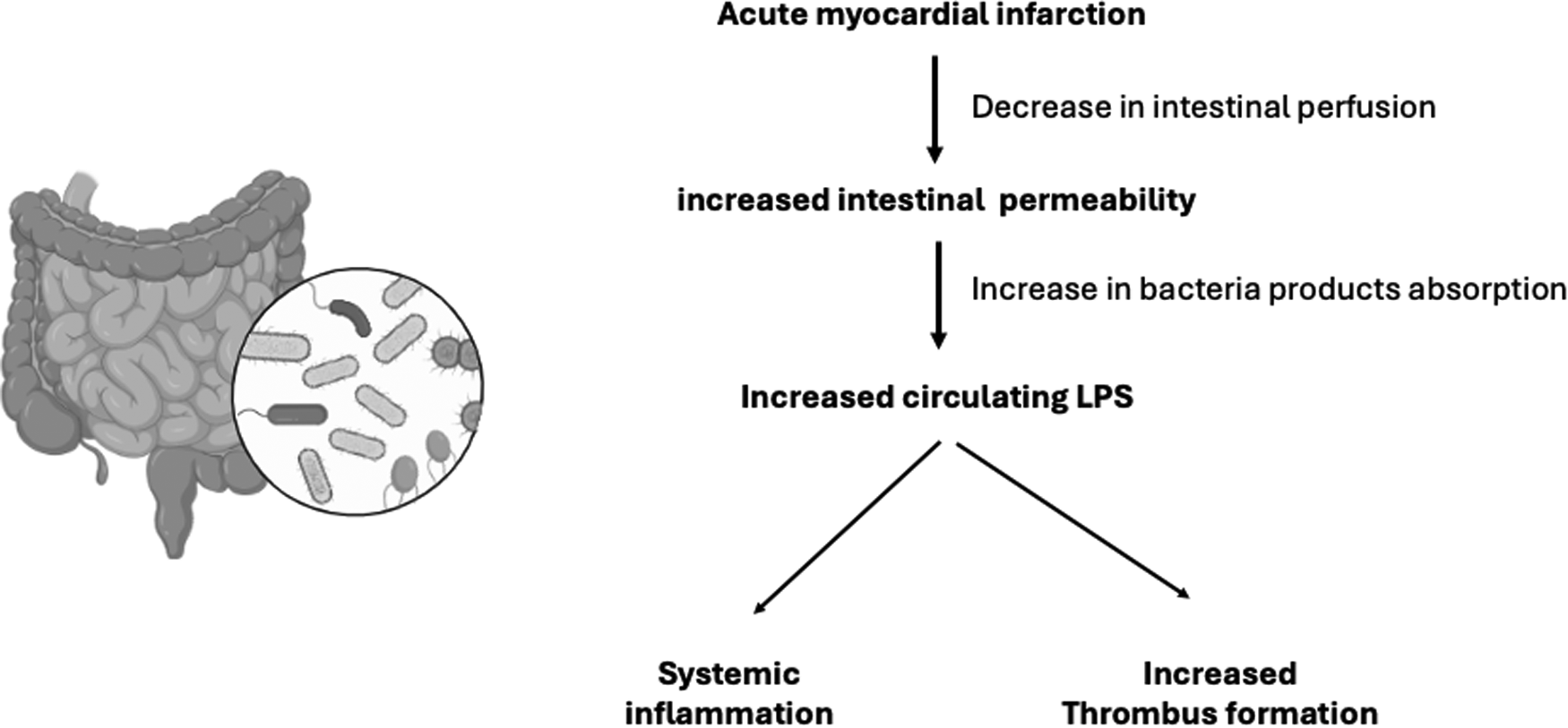
The heart-gut-microbiome-immune axis. After myocardial infarction, decrease in intestinal perfusion contributes to increase in intestinal permeability of bacteria products, including lipopolysaccharides (LPS). Increased LPS has been associated to poor ventricular function and increased infarction size. Circulating LPS is also related to increased thrombus formation (31–40).
Role of the immune system in myocardial infarction
In patients with myocardial infarction, determinants of ventricular remodeling are not only related to early reperfusion, but also to the degree of inflammation and immune responses (41). The innate immune system was developed to ward off infections through a rapid protective response provided by a variety of inflammatory cells. However, following myocardial infarction, even in the absence of pathogens, the release of DAMPs by injured myocytes can activate the immune system (41). In the healthy myocardial, there are few resident mast cells, but after ischemia/reperfusion, these cells can release pro-inflammatory mediators capable to activate endothelium, monocytes/macrophages and neutrophils (42, 43). In fact, smaller infarct size was observed after ischemia/reperfusion, in mast cell deficient mice (44).
The innate immune responses after myocardial infarction can be activated by toll-like receptors and nucleotide-binding oligomerization domain-like receptors after recognition of DAMPs and inflammatory markers due to ischemia/reperfusion (41).
Several interventions on toll-like receptors (TLR2, TLR3, TLR4) have been examined in their role for cardiac remodeling after myocardial infarction (41). Among these toll-like receptors, the TLR4 antagonist eritoran revealed promising results (45).
The innate immune system can also be activated by the inflammasome platform (NLRP3). Several stimuli, including ischemia/reperfusion, activates NOD-like receptors promoting the release of IL-1β and IL-18 (15, 46). Once in the circulation, IL-1β interacts with inflammatory cells increasing the expression of IL-6 (15, 46). In CANTOS trial (47), the monoclonal human antibody canakinumab decreased high-sensitivity C-reactive protein (hsCRP) and IL-6 levels and main cardiovascular events, in the long term after myocardial infarction (48, 49). Inhibition of NLRP3 is an interesting target and may be associated with smaller myocardial infarct size (50–53). Common cardiovascular risk factors have been associated with inflammasome activation, including traditional risk factors linked to atherosclerosis. In this scenario, cholesterol crystals, ischemia/reperfusion, neutrophil extracellular traps, atheroprone flow, and local hypoxia are capable to activate inflammasome triggering the inflammatory cascade mediated by IL-1β and IL-6 (54) (Figure 3).
Figure 3
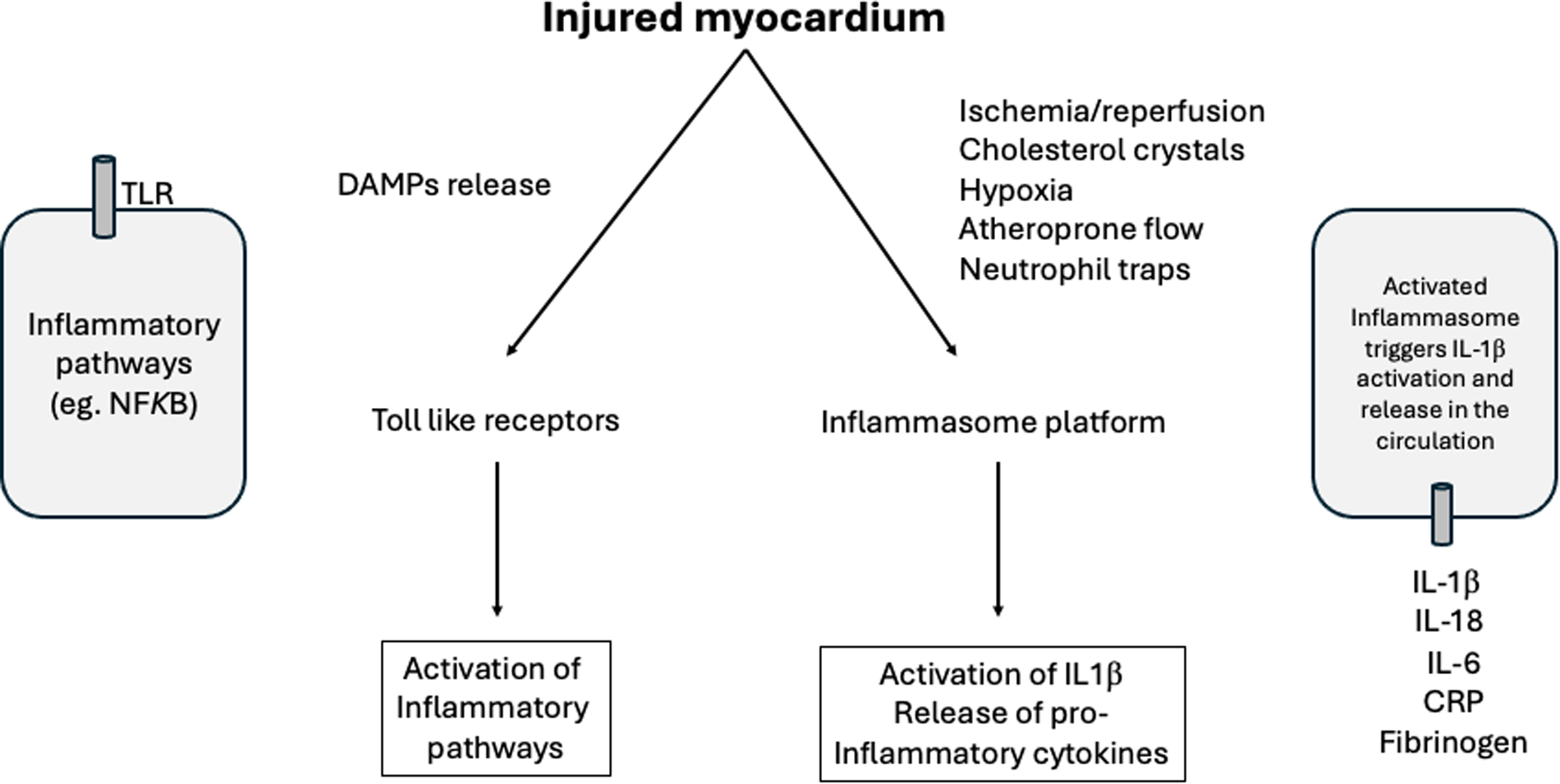
Role of the immune system in myocardial infarction. After ischemia/reperfusion there is a release from the injured myocardial of DAMPs that activate the immune system. The amount of resident mast cells are implicated in the degree of immune and inflammatory responses. After the release of DAMPs, the innate immune responses can be activated by toll-like receptors (TLR) and by the inflammasome platform. Several stimuli can trigger the activation of inflammasome, including ischemia/reperfusion, cholesterol crystals, neutrophil extracellular traps, atheroprone flow and hypoxia with subsequent release of pro-inflammatory cytokines. DAMPs, damage-associated molecular patterns; IL, interleukin; CRP, C-reactive protein; NFKB, Nuclear factor kappa beta (41–54).
How to estimate systemic inflammation?
Despite being a non-specific marker of inflammation, hsCRP is a very useful marker for cardiovascular risk stratification and for monitoring the treatment of cardiovascular disease (55). Plasma CRP is produced by the liver under transcriptional control by IL-6 (55). In patients with acute myocardial infarction, increase in hsCRP levels in the first 24 h was correlated with microvascular infarction estimated by cardiac magnetic resonance (56).
After myocardial infarction, hsCRP levels also predict adverse ventricular remodeling (12, 57–59). Recently, a large primary prevention population followed over a period of 20 years in the EPIC-NORFOLK cohort confirmed the independent association of hsCRP with major adverse cardiovascular events (60). In other large cohort involving US women, hsCRP was also independent predictor of major adverse cardiovascular events in a 30-year follow-up (61).
Interleukin 6 is also a strong marker for future myocardial infarction, supporting an important role of this cytokine in the complications of coronary atherosclerosis (62). In the CANTOS trial, decrease in IL-6 levels by canakinumab was associated with reduction in major cardiovascular events (49).
The effects of IL-6 receptor inhibition was also examined in the setting of acute myocardial infarction. Patients were randomized 1:1 to receive tocilizumab or matching placebo during percutaneous coronary intervention and the myocardial salvage index was quantified by cardiac magnetic resonance imaging 3–7 days after intervention (63). An increase in the myocardial salvage index (primary objective) was observed in the tocilizumab arm (63).
Inflammation detected by magnetic resonance imaging
In patients with acute myocardial infarction, impaired right ventricular ejection fraction and higher NT-proBNP values were related by T1 mapping by hepatic magnetic resonance, an useful biomarker of cardio-hepatic axis to be explored in the setting of inflammation (64). Myocardial edema in non-injured tissue after myocardial infarction may indicate inflammation and adverse outcomes. On this regard, cardiac magnetic resonance performed in patients with myocardial infarction showed that higher T2 mapping values in non-infarcted myocardial or surrounding tissue were related to larger infarct size, microvascular obstruction, left ventricular dysfunction, and adverse cardiovascular outcomes (65).
Role of adipokines in acute myocardial infarction
The role of adipokines in myocardial infarction has been reviewed. In the Copenhagen General Population Study (66), adiponectin was measured in 30.034 individuals. This observational study revealed that elevated plasma adiponectin was associated with heart failure, atrial fibrillation, aortic valve stenosis, and myocardial infarction. In the same study, genetic analysis did not show causality (66). There are pro-inflammatory adipokines beyond adiponectin, such as visfatin and resistin and anti-inflammatory adipokines as omentin and ghrelin, and some of uncertain effects such as leptin or apelin (67). Therefore, as in the case of cytokines, the imbalance of pro- and anti-inflammatory adipokines may affect the occurrence of myocardial infarction and its evolution.
New perspectives for inflammation control and cardiovascular outcomes
A comprehensive review of major findings from several anti-inflammatory clinical trials has already been reported (68). In our review we chose some of the most relevant to clinical practice or those that served as proof of concept.
In CANTOS trial, involving patients with previous myocardial infarction, baseline hsCRP levels were predictors of hospitalization due to heart failure (69). Treatment with canakinumab not only decreased atherothrombotic events, but also rates of hospitalization due to heart failure (69). However, neutropenia was more common among patients treated with canakinumab than those assigned to placebo, and more deaths were attributed to infections in patients treated with the canakinumab pooled groups (incidence rate, 0.31 vs. 0.18 per 100 person-years) (47).
Ziltivekimab is a fully human monoclonal antibody against IL-6 ligand. In the RESCUE-2 trial, involving high risk patients with chronic kidney disease (stages 3–5) also presenting hsCRP levels ≥2 mg/L, a substantial decrease in hsCRP levels (> 90%) was found (68). In a similar study, the RESCUE trial, a comparable decrease in hsCRP was reported (71). In both studies the treatment was well tolerated and additional benefits were described, such as decrease in fibrinogen, serum amyloid A, haptoglobin, phospholipase A2, and lipoprotein (a) (70, 71). Based on these findings, large outcome studies are currently ongoing. The ZEUS trial enrolling patients with established atherosclerotic disease, chronic kidney disease (stages 3–4) and elevated hsCRP levels, aims to evaluate the effects of ziltivekimab compared to placebo in major cardiovascular and renal outcomes (72). Ziltivekimab is also currently tested among patients with preserved or moderately decreased left ventricular ejection fraction in the HERMES trial (73). In addition to these studies, in patients with acute myocardial infarction, early therapy with ziltivekimab is being tested in the ARTEMIS trial (NCT06118281).
Colchicine is an inexpensive anti-inflammatory drug that has been tested in patients with chronic and acute coronary disease. In the setting of acute myocardial infarction, the use of colchicine (0.5 mg once daily) or placebo, started within the first 30 days was examined in 4,745 patients with a median follow-up of 22.6 months (74). Those assigned to colchicine had 23% relative risk reduction on major cardiovascular events. The drug was well tolerated, but a modest increase in pneumonia rate was reported (74). More recently, a new large trial with a 2-by-2 factorial design, in patients with myocardial infarction, tested the effects of colchicine or placebo and spironolactone or placebo (75). In the trial, treatment with these drugs started soon after myocardial infarction, but neither spironolactone nor colchicine reduced major cardiovascular events (76, 77). Thus, after the CLEAR SYNERGY (OASIS 9) trial results, the effects of colchicine in major cardiovascular events after AMI seem controversial (76, 77) (Table 2).
Table 2
| Trial | Therapy | Main results | References |
|---|---|---|---|
| CANTOS | Three doses of Canakinumab (monoclonal antibody against IL-1beta) vs. placebo, median follow-up of 3.7 years | Decrease CV death, non-fatal MI, non-fatal stroke (primary objective) and secondary end point including hospitalization for UA leading to urgent revascularization | (58, 66) |
| COLCOT | Colchicine 0.5 mg once daily vs. placebo, median follow-up of 22.6 months | Decrease in 23% CV death, resuscitated cardiac arrest, myocardial infarction, stroke, urgent hospitalization due to UA leading to coronary revascularization (primary objective) | (71) |
| CLEAR SYNERGY | 2 by 2 factorial design including spironolactone vs. placebo and either colchicine vs. placebo, median follow-up of 3 years | Negative results for the primary objective (CV death, recurrent MI, stroke, unplanned ischemia-driven revascularization) | (75–77) |
| RESCUE and RESCUE-2 | Ziltivekimab (monoclonal antibody against IL-6 ligand), three doses vs. placebo, every 4 weeks, follow-up 24 weeks | Decrease in C-reactive protein, fibrinogen, serum amyloid A, haptoglobin, phospholipase A2, lipoprotein(a) | (67, 68) |
Cardiovascular outcomes in clinical trials with anti-inflammatory therapy.
CV, cardiovascular; MI, myocardial infarction; UA, unstable angina.
Vaccines and immune therapies
Notably, the incidence of recurrent CV events is disproportionately higher within the first 30 days post-acute coronary syndrome compared to the long-term period, highlighting a critical window of vulnerability (78). Influenza and pneumococcal vaccines are associated with decrease in the risk of cardiovascular disease (79). Influenza infection has been identified as a potential catalyst for systemic inflammation and plaque destabilization, particularly during seasonal outbreaks. The virus may act as an external trigger that exacerbates the inflammatory milieu associated with unstable atherosclerotic lesions, thereby elevating the risk of both cardiovascular and cerebrovascular events during the influenza season (80). Particularly in the elderly, influenza vaccination is related to lower rates of acute coronary syndromes and stroke, or new cardiovascular events (80–82). The mechanism of cardiovascular protection after influenza vaccination seems related to decrease in plaque rupture and in the prothrombotic stimuli (83, 84). The hypothesis that preventing influenza infection during or shortly after an acute myocardial infarction (AMI) may reduce subsequent cardiovascular events was prospectively evaluated in the Influenza Vaccination After Myocardial Infarction (IAMI) trial, conducted across Scandinavian countries. This multicenter, double-blind, placebo-controlled trial investigated the in-hospital administration of a standard-dose (15 μg per strain) quadrivalent influenza vaccine vs. placebo in patients with acute myocardial infarction who were eligible for percutaneous coronary intervention (85). The trial was designed to test whether influenza vaccination, administered during the peak of immune activation—within 72 h of coronary angiography—could reduce major adverse cardiovascular events over 12 months. Findings from the IAMI trial were promising. Influenza vaccination, compared to placebo, was associated with a 28% relative reduction in the composite primary endpoint of all-cause mortality, recurrent myocardial infarction, or stent thrombosis [hazard ratio 0·72 (95% CI 0·52–0·99)]. Meta-analysis comprising six randomized controlled trials (RCTs) evaluated the impact of influenza vaccination compared with placebo in patients at high cardiovascular risk, encompassing a total of 6,735 participants (mean age 67 years; 51.3% women; 36.2% with established cardiac disease). The analysis demonstrated an association between influenza vaccination and a lower incidence of subsequent cardiovascular events, with a more pronounced effect observed in patients with recent acute coronary syndrome (86). In the context of patients with acute coronary syndromes, a large, multicenter, randomized study, evaluated the effects of double-dose influenza vaccine vs. standard dose, started during the first week after the coronary acute event. The patients were followed up to 12 months but no differences in cardiopulmonary outcomes were observed between groups (87). Therefore, the strategy of doubling the dose of influenza seems insufficient to enhance cardiopulmonary protection. These findings are in agreement with other large study showing neutral effects of the high-dose trivalent influenza vaccine compared with standard dose of quadrivalent influenza vaccine for mortality or cardiopulmonary hospitalization (88). A recent updated meta-analysis, incorporating the most recently published randomized trials, demonstrated that influenza vaccination is associated with a 34% reduction in the risk of major cardiovascular events compared to placebo or standard care. This protective effect was particularly pronounced in patients with recent acute coronary syndrome, among whom vaccination conferred a 45% lower risk of cardiovascular events within 12 months post-vaccination (89).
Obesity and hypertension are common cardiovascular risk factors that are also associated with interesting differences in the immune responses against oxidized LDL. Among hypertensive patients, body mass index (BMI) and abdominal circumference were inversely related to the antibodies (Abs) anti oxidized LDL (90). Systolic and diastolic blood pressure were also inversely related to the titers of oxidized LDL-Abs and increased titers of inflammatory cytokines (91). Furthermore, treatment of hypertension increased the titers of oxidized LDL-Abs (89). In fact, high titers of autoantibodies against oxidized LDL appear to be a health marker, as suggested by the findings of their elevated titers in stable clinical cardiovascular conditions and lower titers in unstable patients (92). In this scenario of immune strategies, vaccination based on epitopes of apoB has been investigated, and promising anti-atherosclerotic results have been reported experimentally, suggesting a protective role mediated by regulatory T cells (93, 94) (Figure 4).
Figure 4
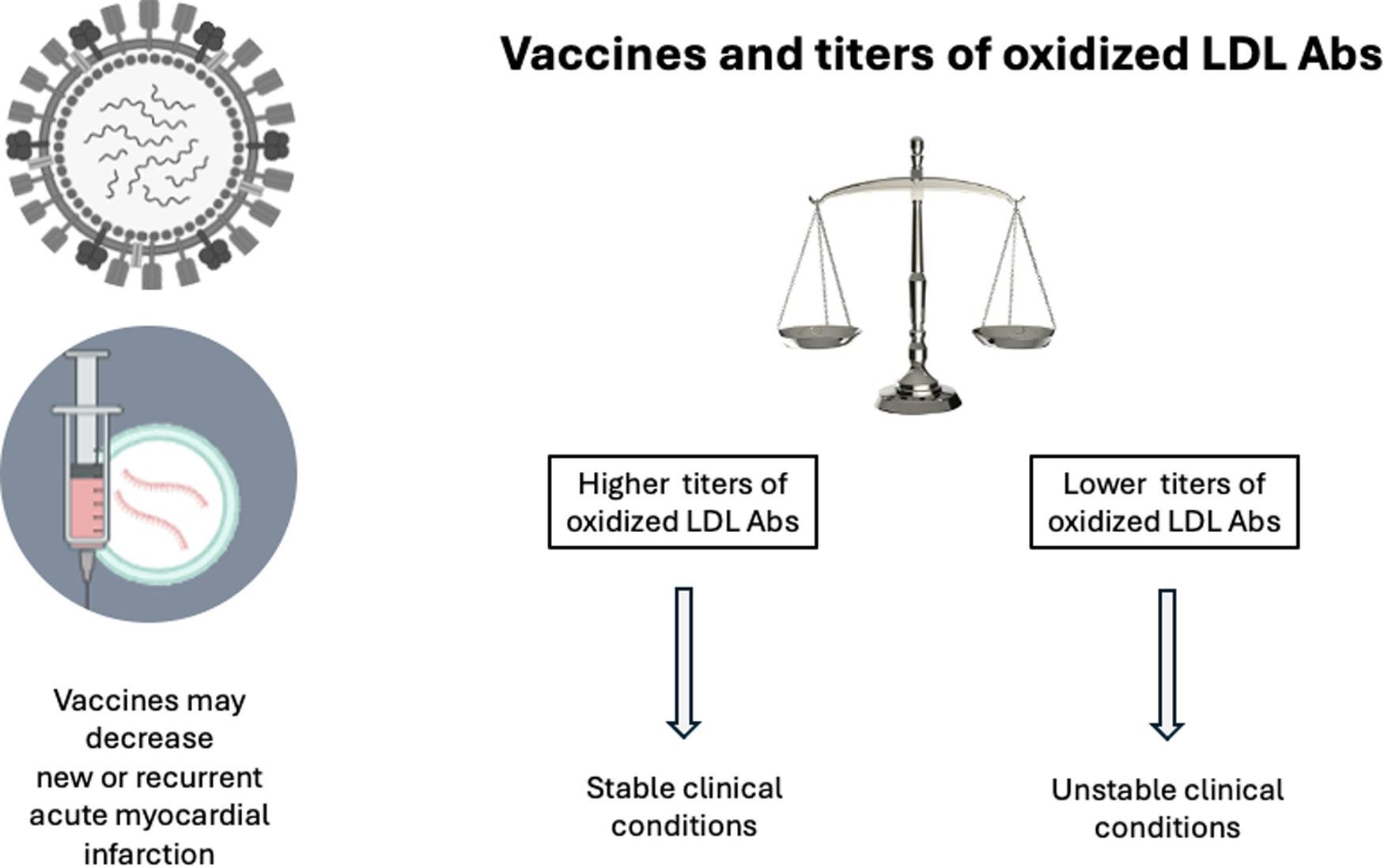
Vaccines and oxidized LDL Abs. Vaccines reduce the incidence of myocardial infarction due to decrease in inflammatory stimuli for plaque rupture and thrombosis. Uncontrolled cardiovascular risk factors seem related to decreased titers of oxidized LDL Abs. Conversely, stable clinical conditions are associated with higher titers of oxidized LDL Abs. Vaccination based on apo B epitopes have shown promising initial studies. Abs, antibodies (73–85).
Inflammation and perspectives with photobiomodulation
The possibility of modulating inflammatory responses after myocardial infarction has also been described in experimental model using photobiomodulation by laser. The authors reported transcriptional and post-transcriptional changes that can modify ventricular remodeling (95). As mentioned before, recovery of the heart after myocardial infarction is a complex process involving various inflammatory components and cardiomyocyte responses to ischemia (96). In this context, photobiomodulation, a non-invasive therapeutic modality that utilizes low-level light sources — typically low-power lasers or light-emitting diodes (LEDs) has proven to be a promising alternative (97). Oron's research team pioneered the demonstration of reduced mitochondrial damage and increased ATP content in the infarcted myocardial region of dogs treated with low-power lasers (96). The authors also demonstrated that low-power lasers reduced mortality and infarct size compared to untreated dogs. In addition to improving cellular energy potential, the mechanisms targeted by photobiomodulation to achieve a cardioprotective effect may include increased release of nitric oxide, vascular endothelial growth factor and new blood vessel formation (98, 99). Preclinical studies have also shown that photobiomodulation can modulate the enhanced inflammatory response after myocardial infarction. Our group has previously shown that application of low-power lasers to the myocardium immediately after coronary occlusion effectively reduces infarct size and the incidence of large infarcts and attenuates systolic dysfunction in rats at 3 days (100, 101). These findings were associated with reduced myocardial expression of IL-1β and IL-6 compared to non-irradiated rats. In two subsequent studies, our group carried out high-throughput gene expression analysis to identify differentially expressed genes in infarcted myocardium between 24 h and 3 days post-infarction, with low-power lasers therapy initiated approximately 60 s after coronary occlusion. Notably, low-power lasers induced a marked reduction in the mRNA expression of key mediators involved in post-MI inflammation and extracellular matrix remodeling, including IL-6, tumor necrosis factor receptor, transforming growth factor beta 1, and collagens type I and III (95, 102). Finally, additional studies utilizing prolonged low-power lasers therapy in infarcted rodent models have demonstrated improved outcomes in heart failure progression, including attenuation of myocardial hypertrophy and fibrosis, reduced pulmonary congestion, and enhanced left ventricular function (103, 104). These findings were associated with a potent antioxidant and anti-inflammatory effect of low-power lasers.
In summary, the immunomodulatory role of photobiomodulation, particularly low-power lasers therapy, may hold significant promise in attenuating the inflammatory response and promoting favorable post-infarction cardiac remodeling. This therapeutic strategy may be especially beneficial for individuals exhibiting an overactive and prolonged post-infarction inflammatory state, where improved inflammatory regulation could contribute to enhanced cardiac structure and function, reduced fibrosis, and decreased electrical instability via the suppression of pro-inflammatory cytokines (105–107).
Treatment of hypertension, diabetes, and chronic kidney disease as a key inflammatory and neglected concomitant diseases
Together, several inflammatory pathways lead to atherosclerosis and its complications, but in addition to specific therapies, many drugs in clinical practice have anti-inflammatory effects. In this context, meta-analysis of inhibitors of the renin-angiotensin system showed a significant decrease in markers of inflammation (108). However, decrease in hsCRP obtained with a renin-angiotensin system blocker may be abolished by concomitant use of hydrochlorothiazide (109).
Several antidiabetic drugs have anti-inflammatory properties, such as pioglitazone (110), glucagon like peptide-1 receptor agonists (111), and dipeptidyl peptidase-4 inhibitors (112). Conversely, meta-analysis with 38 randomized controlled studies evaluated the effects of inflammatory markers among sodium-glucose cotransporter-2 inhibitors and did not find anti-inflammatory effects, including effects on hsCRP levels (113).
Lipid-lowering agents such as statins, present anti-inflammatory properties, reducing hsCRP and cardiovascular events (114, 115). The combination of statin with ezetimibe promoted an additional reduction on concentrations of hsCRP and in cardiovascular events when compared to statin monotherapy (116). Despite the benefits on cardiovascular parameters across hsCRP strata, the inhibitor of proprotein convertase subtilisin/kexin type 9 (PCSK9) evolocumab did not change C-reactive protein levels in the Fourier trial (117, 118). Inclisiran, a novel small-interfering RNA against PCSK9 did not show effects on markers of inflammation or adverse events in immune parameters (119).
In addition to the anti-inflammatory properties of renin-angiotensin system inhibitors, finerenone, a nonsteroidal selective mineralocorticoid antagonist presents anti-inflammatory effects, with proven renal and cardiovascular benefits (120–122). In the ischemia/reperfusion model of kidney injury, finerenone showed favorable effect increasing the M2 protective macrophages in glomeruli (122).
A healthy lifestyle also reduces inflammation markers. In fact, lower levels of hsCRP were observed among professional runners, despite their high-intensity training, and they showed lower intima-media thickness, and higher percentage of circulating endothelial progenitor cells (123, 124). In a large cohort, changes in lifestyle with better risk factor control were strongly and independently associated with lower hsCRP levels (125).
Is there a link between inflammation and bleeding?
Acute coronary syndromes are related to increased bleeding risk after percutaneous coronary intervention compared to chronic coronary syndromes (126). In the JUPITER trial, among primary prevention patients with hsCRP levels ≥2 mg/L and relatively normal LDL-cholesterol levels, those treated with rosuvastatin had an impressive decrease in cardiovascular events (114). In the same trial a pre-specified secondary outcome was the effect of rosuvastatin in the rate of venous thromboembolism. Surprisingly, a marked decrease of venous thromboembolism was found among these patients, with elevated levels of hsCRP, receiving rosuvastatin (127). The link between C-reactive protein and thrombosis seems related to C-reactive protein destabilized isoforms that are not only pro-inflammatory but atherothrombotic (128).
The link between inflammation and bleeding was assessed in 1,864 consecutive patients with acute coronary syndromes. Patients were followed for one year, and baseline hsCRP levels were predictive of major cardiovascular outcomes, but not for bleeding risk (129). Therefore, inflammation per se does not seem related to bleeding, but possibly to increased thrombotic risk.
Conclusions
In brief, addressing the residual inflammatory cardiovascular risk requires a comprehensive understanding of the intricate network of inflammatory pathways, whose relevance may vary between the acute and chronic phases of coronary artery disease. While lifestyle modifications and control of traditional risk factors remain fundamental, particularly in primary prevention, targeted modulation of inflammation, whether through specific cytokine inhibition or broader immunomodulatory approaches, holds significant promise, especially in the acute setting of myocardial infarction. Although biomarkers like hsCRP are valuable for risk stratification, they lack causal specificity. In contrast, IL-6 has emerged as a particularly promising therapeutic target, given its more direct mechanistic involvement in atherosclerotic inflammation. Furthermore, growing evidence supports the potential of leveraging immune-modulatory strategies, including vaccines, to achieve long-term reduction in cardiovascular events. Future research should focus on refining these interventions to balance efficacy and safety, ultimately translating into more personalized and effective cardiovascular care.
Statements
Author contributions
FAHF: Conceptualization, Data curation, Formal analysis, Funding acquisition, Resources, Supervision, Writing – original draft, Writing – review & editing. CNF: Investigation, Writing – review & editing. HARF: Investigation, Writing – review & editing. AJS: Investigation, Writing – review & editing. MCI: Conceptualization, Investigation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
This review was supported in part by the Sao Paulo Research Foundation (FAPESP grant # 2012/51692-7 and by the National Council for Scientific and Technological Development (CNPq grant # 302462/2023-6).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
GBD 2017 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 359 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet. (2018) 392:1859–922. 10.1016/S0140-6736(18)32335-3
2.
Ross R . Atherosclerosis—an inflammatory disease. N Engl J Med. (1999) 340(2):115–26. 10.1056/NEJM199901143400207
3.
Libby P Pasterkamp G Crea F Jang IK . Reassessing the mechanisms of acute coronary syndromes. Circ Res. (2019) 124(1):150–60. 10.1161/CIRCRESAHA.118.311098
4.
Libby P . Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med. (2013) 368:2004–13. 10.1056/NEJMra1216063
5.
Kolodgie FD Burke AP Farb A Weber DK Kutys R Wight TN et al Differential accumulation of proteoglycans and hyaluronan in culprit lesions: insights into plaque erosion. Arterioscl Thromb Vasc Biol. (2002) 22(10):1642–8. 10.1161/01.ATV.0000034021.92658.4C
6.
Kolodgie FD Burke AP Wight TN Virmani R . The accumulation of specific types of proteoglycans in eroded plaques: a role in coronary thrombosis in the absence of rupture. Curr Opin Lipidol. (2004) 15:575–82. 10.1097/00041433-200410000-00012
7.
Virmani R Burke AP Farb A . Plaque rupture and plaque erosion. Thromb Haemost. (1999) 82(Suppl 1):1–3. 10.1055/s-0037-1615543
8.
Sugiyama S Kugiyama K Aikawa M Nakamura S Ogawa H Libby P . Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscl Thromb Vasc Biol. (2004) 24(7):1309–14. 10.1161/01.ATV.0000131784.50633.4f
9.
Folco EJ Mawson TL Vromman A Bernardes-Souza B Franck G Persson O et al Neutrophil extracellular traps induce endothelial cell activation and tissue factor production through interleukin-1α and cathepsin G. Arterioscl Thromb Vasc Biol. (2018) 38(8):1901–12. 10.1161/ATVBAHA.118.311150
10.
Matter MA Paneni F Libby P Frantz S Stahli BE Templin C et al Inflammation in acute myocardial infarction: the good, the bad and the ugly. Eur Heart J. (2024) 45(2):89–103. 10.1093/eurheartj/ehad486
11.
Weirather J Hofmann UDW Beyersdorf N Ramos GC Vogel B Frey A et al Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. (2014) 115:55–67. 10.1161/CIRCRESAHA.115.303895
12.
Casarotti ACA Teixeira D Longo-Maugeri IM Ishimura ME Coste MER Bianco HT et al Role of B lymphocytes in the infarcted mass in patients with acute myocardial infarction. Biosci Rep. (2021) 41(2):BSR20203413. 10.1042/BSR20203413
13.
Moschonas IC Tselepis AD . The pathway of neutrophil extracellular traps towards atherosclerosis and thrombosis. Atherosclerosis. (2019) 288:9–16. 10.1016/j.atherosclerosis.2019.06.919
14.
Olsen MB Gregersen I Sandanger O Yang K Sokolova M Halvorsen BE et al Targeting the inflammasome in cardiovascular disease. JACC Basic Transl Sci. (2022) 7:84–98. 10.1016/j.jacbts.2021.08.006
15.
Libby P . Interleukin-1 Beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J Am Coll Cardiol. (2017) 70:2278–89. 10.1016/j.jacc.2017.09.028
16.
Giral H Franke V Moobed M Müller MF Lübking L James DM et al Rapid inflammasome activation is attenuated in post-myocardial infarction monocytes. Frontiers Immunol. (2022) 13:857455. 10.3389/fimmu.2022.857455
17.
Yang XM Cui L White J Kuck J Ruchko MV Wilson GL et al Mitochondrially targeted endonuclease III has a powerful anti-infarct effect in an in vivo rat model of myocardial ischemia/reperfusion. Basic Res Cardiol. (2015) 110(2):3. 10.1007/s00395-014-0459-0
18.
Andreadou I Cabrera-Fuentes HA Devaux Y Frangogiannis NG Frantz S Guzik T et al Immune cells as targets for cardioprotection: new players and novel therapeutic opportunities. Cardiovasc Res. (2019) 115(7):1117–30. 10.1093/cvr/cvz050
19.
Baran KW Nguyen M McKendall GR Lambrew CT Dykstra G Palmeri ST et al , Limitation of Myocardial Infarction Following Thrombolysis in Acute Myocardial Infarction (LIMIT AMI) Study Group. Double-blind, randomized trial of an anti-CD18 antibody in conjunction with recombinant tissue plasminogen activator for acute myocardial infarction: limitation of myocardial infarction following thrombolysis in acute myocardial infarction (LIMIT AMI) study. Circulation. (2001) 104(23):2778–83. 10.1161/hc4801.100236
20.
Faxon DP Gibbons RJ Chronos NA Gurbel PA Sheehan F , HALT-MI Investigators. The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT-MI study. J Am Coll Cardiol. (2002) 40(7):1199–204. 10.1016/S0735-1097(02)02136-8
21.
Nahrendorf M Swirski FK Aikawa E Stangenberg L Wurdinger T Figueiredo J-L et al The healing myocardium sequentially mobilizes two monocyte subsets with divergente and complementary functions. J Exp Med. (2007) 204:3037–47. 10.1084/jem.20070885
22.
Nahrendorf M Swirski FK . Monocyte and macrophage heterogeneity in the heart. Circ Res. (2013) 112(12):1624–33. 10.1161/CIRCRESAHA.113.300890
23.
van der Laan AM Ter Horst EN Delewi R Begieneman MP Krijnen PA Hirsch A et al Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur Heart J. (2014) 35(6):376–85. 10.1093/eurheartj/eht331
24.
Askari N Lipps C Voss S Staubach N Grün D Klingenberg R et al Circulating monocyte subsets are associated with extent of myocardial injury but not with type of myocardial infarction. Front Cardiovasc Med. (2021) 8:741890. 10.3389/fcvm.2021.741890
25.
Tapp LD Shantsila E Wrigley BJ Pamukcu B Lip GYH . The CD14++CD16+ monocyte subset and monocyte-platelet interactions in patients with ST-elevation myocardial infarction. J Thromb Haemost. (2012) 10(7):1231–41. 10.1111/j.1538-7836.2011.04603.x
26.
Idzkowska E Eljaszewicz A Miklasz P Musial WJ Tycinska AM Moniuszko M . The role of different monocyte subsets in the pathogenesis of atherosclerosis and acute coronary syndromes. Scand J Immunol. (2015) 82(3):163–73. 10.1111/sji.12314
27.
Merino A Buendia P Martin-Malo A Aljama P Ramirez R Carracedo J . Senescent CD14 + CD16+ monocytes exhibit proinflammatory and proatherosclerotic activity. J Immunol. (2011) 186(3):1809–15. 10.4049/jimmunol.1001866
28.
Carvalho DC Fonseca FAH Izar MC Assunção ALP Tuleta ID Amaral JB et al Monocytes presenting a pro-inflammatory profile persist in patients submitted to a long-term pharmacological treatment after acute myocardial infarction. Frontiers Physiol. (2023) 13:1056466. 10.3389/fphys.2022.1056466
29.
Boag SE Das R Shmeleva EV Bagnall A Egred M Howard N et al T lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients. J Clin Invest. (2015) 125(8):3063–76. 10.1172/JCI80055
30.
Horckmans M Bianchini M Santovito D Megens RTA Springael JY Negri I et al Pericardial adipose tissue regulates granulopoiesis, fibrosis, and cardiac function after myocardial infarction. Circulation. (2018) 137(9):948–60. 10.1161/CIRCULATIONAHA.117.028833
31.
Relvas WG Izar MC Segreto HR Giordani AJ Ihara SS Mariano M et al Resident peritoneal inflammatory cells are pivotal in the development of experimental atherosclerosis. J Atheroscl Thromb. (2010) 17(4):378–85. 10.5551/jat.3418
32.
Jiao J He S Wang Y Lu Y Gu M Li D et al Regulatory B cells improve ventricular remodeling after myocardial infarction by modulating monocyte migration. Basic Res Cardiol. (2021) 116(1):46. 10.1007/s00395-021-00886-4
33.
Volodarsky I Shimoni S Haberman D Mirkin V Frabrikant Y Mashriki TY et al Circulating regulatory B-lymphocytes in patients with acute myocardial infarction: a pilot study. J Cardiovasc Dev Dis. (2022) 10(1):2. 10.3390/jcdd10010002
34.
Anzai A Anzai T Nagai S Maekawa Y Naito K Kaneko H et al Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation. (2012) 125(10):1234–45. 10.1161/CIRCULATIONAHA.111.052126
35.
Nagai T Honda S Sugano Y Matsuyama TA Ohta-Ogo K Asaumi Y et al Decreased myocardial dendritic cells is associated with impaired reparative fibrosis and development of cardiac rupture after myocardial infarction in humans. J Am Heart Assoc. (2014) 3(3):e000839. 10.1161/JAHA.114.000839
36.
Zhou X Li J Guo J Geng B Ji W Zhao Q et al Gut-dependent microbial translocation induces inflammation and cardiovascular events after ST-elevation myocardial infarction. Microbiome. (2018) 6(1):66. 10.1186/s40168-018-0441-4
37.
Carnevale R Sciarretta S Valenti V di Nonno F Calvieri C Nocella C et al Low-grade endotoxaemia enhances artery thrombus growth via toll-like receptor 4: implication for myocardial infarction. Eur Heart J. (2020) 41:3156–65. 10.1093/eurheartj/ehz893
38.
Amar J Lelouvier B Servant F Lluch J Burcelin R Bongard V et al Blood Microbiota modification after myocardial infarction Depends upon low-density lipoprotein cholesterol levels. J Am Heart Assoc. (2019) 8:e011797. 10.1161/JAHA.118.011797
39.
Violi F Nocella C Carnevale R . Gut microbiota and myocardial infarction. Eur Heart J. (2020) 41(23):2221–2. 10.1093/eurheartj/ehaa222
40.
Zhao J Zhang Q Cheng W Dai Q Wei Z Guo M et al Heart-gut microbiota communication determines the severity of cardiac injury after myocardial ischaemia/reperfusion. Cardiovasc Res. (2023) 119(6):1390–402. 10.1093/cvr/cvad023
41.
Zuurbier CJ Abbate A Cabrera-Fuentes HA Cohen MV Collino M De Kleijn DPV et al Innate immunity as a target for acute cardioprotection. Cardiovasc Res. (2019) 115(7):1131–42. 10.1093/cvr/cvy304
42.
Janicki JS Brower GL Levick SP . The emerging prominence of the cardiac mast cell as a potent mediator of adverse myocardial remodeling. Methods Mol Biol. (2015) 1220:121–39. 10.1007/978-1-4939-1568-2_8
43.
Frangogiannis NG Lindsey ML Michael LH Youker KA Bressler RB Mendoza LH et al Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. (1998) 98(7):699–710. 10.1161/01.CIR.98.7.699
44.
Bhattacharya K Farwell K Huang M Kempuraj D Donelan J Papaliodis D et al Mast cell deficient W/wv mice have lower serum IL-6 and less cardiac tissue necrosis than their normal littermates following myocardial ischemia-reperfusion. Int J Immunopathol Pharmacol. (2007) 20(1):69–74. 10.1177/039463200702000108
45.
Shimamoto A Chong AJ Yada M Shomura S Takayama H Fleisig AJ et al Inhibition of toll-like receptor 4 with eritoran attenuates myocardial ischemia-reperfusion injury. Circulation. (2006) 114(1Suppl):I270–4. 10.1161/CIRCULATIONAHA.105.000901
46.
Fonseca FA Izar MC . Role of inflammation in cardiac remodeling after myocardial infarction. Front Physiol. (2022) 13:927163. 10.3389/fphys.2022.927163
47.
Ridker PM Everett BM Thuren T MacFadyen JG Chang WH Ballantyne C et al Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377(12):1119–31. 10.1056/NEJMoa1707914
48.
Ridker PM MacFadyen JG Everett BM Libby P Thuren T Glynn RJ , CANTOS Trial Group. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomized controlled trial. Lancet. (2018) 391(10118):319–28. 10.1016/S0140-6736(17)32814-3
49.
Ridker PM Libby P MacFadyen JG Thuren T Ballantyne C Fonseca F et al Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: analyses from the canakinumab anti-inflammatory thrombosis outcomes study (CANTOS). Eur Heart J. (2018) 39(38):3499–507. 10.1093/eurheartj/ehy310
50.
Mezzaroma E Toldo S Farkas D Seropian IM Van Tassell BW Salloum FN et al The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A. (2011) 108(49):19725–30. 10.1073/pnas.1108586108
51.
Kawaguchi M Takahashi M Hata T Kashima Y Usui F Morimoto H et al Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. (2011) 123(6):594–604. 10.1161/CIRCULATIONAHA.110.982777
52.
Sandanger O Ranheim T Vinge LE Bliksoen M Alfsnes K Finsen AV et al The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. (2013) 99(1):164–74. 10.1093/cvr/cvt091
53.
Liu Y Lian K Zhang L Wang R Yi F Gao C et al TXNIP Mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res Cardiol. (2014) 109(5):415. 10.1007/s00395-014-0415-z
54.
Ridker PM . From C-reactive protein to interleukin-6 to interleukin-1: moving upstream to identify novel targets for atheroprotection. Circ Res. (2016) 118(1):145–56. 10.1161/CIRCRESAHA.115.306656
55.
Pepys MB Hirschfield GM . C-reactive protein: a critical update. J Clin Invest. (2003) 111(12):1805–12. 10.1172/JCI200318921
56.
Holzknecht M Tiller C Reindl M Lechner I Troger F Hosp M et al C-reactive protein velocity predicts microvascular pathology after acute ST-elevation myocardial infarction. Int J Cardiol. (2021) 338:30–6. 10.1016/j.ijcard.2021.06.023
57.
Seferović PM Ašanin M Ristić AD . Acute stress disorder and C-reactive protein in patients with acute myocardial infarction. Eur J Prev Cardiol. (2018) 25(7):702–5. 10.1177/2047487318761091
58.
Reindl M Reinstadler SJ Feistritzer HJ Klug G Tiller C Mair J et al Relation of inflammatory markers with myocardial and microvascular injury in patients with reperfused ST-elevation myocardial infarction. Eur Heart J Acute Cardiovasc Care. (2017) 6(7):640–9. 10.1177/2048872616661691
59.
Tiller C Reindl M Holzknecht M Lechner I Simma F Schwaiger J et al High sensitivity C-reactive protein is associated with worse infarct healing after revascularized ST-elevation myocardial infarction. Int J Cardiol. (2021) 328:191–6. 10.1016/j.ijcard.2020.12.006
60.
Kraaijenhof JM Nurmohamed NS Nordestgaard AT Reeskamp LF Stroes ESG Hovingh GK et al Low-density lipoprotein cholesterol, C-reactive protein, and lipoprotein(a) universal one-time screening in primary prevention: the EPIC-norfolk study. Eur Heart J. (2025):1–10. 10.1093/eurheartj/ehaf209
61.
Ridker PM Moorthy MV Cook NR Rifai N Lee IM Buring JE . Inflammation, cholesterol, lipoprotein(a), and 30-year cardiovascular outcomes in women. N Engl J Med. (2024) 391(22):2087–97. 10.1056/NEJMoa2405182
62.
Ridker PM Rifai N Stampfer MJ Hennekens CH . Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. (2000) 101(15):1767–72. 10.1161/01.CIR.101.15.1767
63.
Broch K Anstensrud AK Woxholt S Sharma K Tøllefsen IM Bendz B et al Randomized trial of interleukin-6 receptor inhibition in patients with acute ST-segment elevation myocardial infarction. J Am Coll Cardiol. (2021) 77(15):1845–55. 10.1016/j.jacc.2021.02.049
64.
Bergamaschi L Arangalage D Maurizi N Pizzi C Valgimigli M Iglesias JF et al Hepatic T1 mapping as a novel cardio-hepatic axis imaging biomarker early after ST-elevation myocardial infarction. Eur Heart J Cardiovasc Imaging. (2025) 26(2):229–38. 10.1093/ehjci/jeae256
65.
Bergamaschi L Landi A Maurizi N Pizzi C Leo LA Arangalage D et al Acute response of the noninfarcted myocardium and surrounding tissue assessed by T2 mapping after STEMI. JACC Cardiovasc Imaging. (2024) 17(6):610–21. 10.1016/j.jcmg.2023.11.014
66.
Nielsen MB Çolak Y Benn M Mason A Burgess S Nordestgaard BG . Plasma adiponectin levels and risk of heart failure, atrial fibrillation, aortic valve stenosis, and myocardial infarction: large-scale observational and Mendelian randomization evidence. Cardiovasc Res. (2024) 120(1):95–107. 10.1093/cvr/cvad162
67.
Mitsis A Kadoglou NPE Lambadian V Alexiou S Theodoropoulos KC Avraamides P et al Prognostic role of inflammatory cytokines and novel adipokines in acute myocardial infarction: an updated and comprehensive review. Cytokine. (2022) 153:155848. 10.1016/j.cyto.2022.155848
68.
Mitsis A Kyriakou M Sokratous S Karmioti G Drakomathioulakis M Myrianthefs M et al Exploring the landscape of anti-inflammatory trials: a comprehensive review of strategies for targeting inflammation in acute myocardial infarction. Biomedicines. (2024) 12(3):701. 10.3390/biomedicines12030701
69.
Burger PM Koudstaal S Mosterd A Fiolet ATL Teraa M van der Meer MG et al C-Reactive protein and risk of incident heart failure in patients with cardiovascular disease. J Am Coll Cardiol. (2023) 82(5):414–26. 10.1016/j.jacc.2023.05.035
70.
Wada Y Jensen C Meyer ASP Zonoozi AAM Honda H . Efficacy and safety of interleukin-6 inhibition with ziltivekimab in patients at high risk of atherosclerotic events in Japan (RESCUE-2): a randomized, double-blind, placebo-controlled, phase 2 trial. J Cardiol. (2023) 82(4):279–85. 10.1016/j.jjcc.2023.05.006
71.
Ridker PM Devalaraja M Baeres FMM Engelmann MDM Hovingh GK Ivkovic M et al IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomized, placebo-controlled, phase 2 trial. Lancet. (2021) 397(10289):2060–9. 10.1016/S0140-6736(21)00520-1
72.
Ridker PM . From RESCUE to ZEUS: will interleukin-6 inhibition with ziltivekimab prove effective for cardiovascular event reduction?Cardiovasc Res. (2021) 117(11):e138. 10.1093/cvr/cvab231
73.
Petrie M Borlaug B Buchholtz K Ducharme A Hvelplund A Ping CLS et al HERMES: effects of ziltivekimab versus placebo on morbidity and mortality in patients with heart failure with mildly reduced or preserved ejection fraction and systemic inflammation. J Card Fail. (2024) 30(1):126. 10.1016/j.cardfail.2023.10.024
74.
Tardif JC Kouz S Waters DD Bertrand OF Diaz R Maggioni AP et al Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. (2019) 381(26):2497–505. 10.1056/NEJMoa1912388
75.
d’Entremont MA Lee SF Mian R Kedev S Montalescot G Cornel JH et al Design and rationale of the CLEAR SYNERGY (OASIS 9) trial: a 2 ( 2 factorial randomized controlled trial of colchicine versus placebo and spironolactone vs placebo in patients with myocardial infarction. Am Heart J. (2024) 275:173–82. 10.1016/j.ahj.2024.06.007
76.
Jolly SS d’Entremont MA Pitt B Lee SF Mian R Tyrwhitt J et al Routine spironolactone in acute myocardial infarction. N Engl J Med. (2025) 392(7):643–52. 10.1056/NEJMoa2405923
77.
Jolly SS d’Entremont MA Lee SF Mian R Tyrwhitt J Kedev S et al Colchicine in acute myocardial infarction. N Engl J Med. (2025) 392(7):633–42. 10.1056/NEJMoa2405922
78.
Pilgrim T Vranckx P Valgimigli M Stefanini GG Piccolo R Rat J et al Risk and timing of recurrent ischemic events among patients with stable ischemic heart disease, non-ST-segment elevation acute coronary syndrome, and ST-segment elevation myocardial infarction. Am Heart J. (2016) 175:56–65. 10.1016/j.ahj.2016.01.021
79.
Addario A Célarier T Bongue B Barth N Gavazzi G Botelho-Nevers E . Impact of influenza, herpes zoster, and pneumococcal vaccinations on the incidence of cardiovascular events in subjects aged over 65 years: a systematic review. Geroscience. (2023) 45(6):3419–47. 10.1007/s11357-023-00807-4
80.
Kwong JC Schwartz KL Campitelli MA Chung H Crowcroft NS Karnauchow T et al Acute myocardial infarction after laboratory-confirmed influenza infection. N Engl J Med. (2018) 378:345–53. 10.1056/NEJMoa1702090
81.
Chiang MH Wu HH Shih CJ Chen YT Kuo SC Chen TL . Association between influenza vaccination and reduced risks of major adverse cardiovascular events in elderly patients. Am Heart J. (2017) 193:1–7. 10.1016/j.ahj.2017.07.020
82.
Phrommintikul A Kuanprasert S Wongcharoen W Kanjanavanit R Chaiwarith R Sukonthasarn A . Influenza vaccination reduces cardiovascular events in patients with acute coronary syndrome. Eur Heart J. (2011) 32(14):1730–5. 10.1093/eurheartj/ehr004
83.
Madjid M Awan I Ali M Frazier L Casscells W . Influenza and atherosclerosis: vaccination for cardiovascular disease prevention. Expert Opin Biol Ther. (2005) 5(1):91–6. 10.1517/14712598.5.1.91
84.
Harskamp RE van Ginkel MW . Acute respiratory tract infections: a potential trigger for the acute coronary syndrome. Ann Med. (2008) 40(2):121–8. 10.1080/07853890701753672
85.
Fröbert O Götberg M Erlinge D Akhtar Z Christiansen EH MacIntyre CR et al Influenza vaccination after myocardial infarction: a randomized, double-blind, placebo-controlled, multicenter trial. Circulation. (2021) 144(18):1476–84. 10.1161/CIRCULATIONAHA.121.057042
86.
Udell JA Zawi R Bhatt DL Keshtkar-Jahromi M Gaughran F Phrommintikul A et al Association between influenza vaccination and cardiovascular outcomes in high-risk patients: a meta-analysis. JAMA. (2013) 310(16):1711–20. 10.1001/jama.2013.279206
87.
Fonseca HAR Furtado RHM Zimerman A Lemos PA Franken M Monfardini F et al Influenza vaccination strategy in acute coronary syndromes: the VIP-ACS trial. Eur Heart J. (2022) 43(41):4378–88. 10.1093/eurheartj/ehac472
88.
Vardeny O Kim K Udell JA Joseph J Desai AS Farkouh ME et al Effect of high-dose trivalent vs standard-dose quadrivalent influenza vaccine on mortality or cardiopulmonary hospitalization in patients with high-risk cardiovascular disease: a randomized clinical trial. JAMA. (2021) 325(1):39–49. 10.1001/jama.2020.23649
89.
Behrouzi B Bhatt DL Cannon CP Vardeny O Lee DS Solomon SD et al Association of influenza vaccination with cardiovascular risk: a meta-analysis. JAMA Netw Open. (2022) 5(4):e228873. 10.1001/jamanetworkopen.2022.8873
90.
Fonseca HA Fonseca FA Monteiro AM Bianco HT Boschcov P Brandão SA et al Obesity modulates the immune response to oxidized LDL in hypertensive patients. Cell Biochem Biophys. (2013) 67(3):1451–60. 10.1007/s12013-013-9585-9
91.
Brandão SA Izar MC Fischer SM Santos AO Monteiro CM Póvoa RM et al Early increase in autoantibodies against human oxidized low-density lipoprotein in hypertensive patients after blood pressure control. Am J Hypertens. (2010) 23(2):208–14. 10.1038/ajh.2009.214
92.
Santos AO Fonseca FA Fischer SM Monteiro CM Brandão SA Póvoa RM et al High circulating autoantibodies against human oxidized low-density lipoprotein are related to stable and lower titers to unstable clinical situation. Clin Chim Acta. (2009) 406(1-2):113–8. 10.1016/j.cca.2009.06.005
93.
Gisterå A Hermansson A Strodthoff D Klement ML Hedin U Fredrikson GN et al Vaccination against T-cell epitopes of native ApoB100 reduces vascular inflammation and disease in a humanized mouse model of atherosclerosis. J Intern Med. (2017) 281(4):383–97. 10.1111/joim.12589
94.
Nilsson J Hansson GK . Vaccination strategies and immune modulation of atherosclerosis. Circ Res. (2020) 126(9):1281–96. 10.1161/CIRCRESAHA.120.315942
95.
Feliciano RDS Atum ALB Ruiz ÉGDS Serra AJ Antônio EL Manchini MT et al Photobiomodulation therapy on myocardial infarction in rats: transcriptional and posttranscriptional implications to cardiac remodeling. Lasers Surg Med. (2021) 53(9):1247–57. 10.1002/lsm.23407
96.
Oron U Yaakobi T Oron A Mordechovitz D Shofti R Hayam G et al Low-energy laser irradiation reduces formation of scar tissue after myocardial infarction in rats and dogs. Circulation. (2001) 103(2):296–301. 10.1161/01.CIR.103.2.296
97.
Ganipineni VDP Gutlapalli SD Ajay Sai Krishna Kumar I Monica P Vagdevi M Samuel Sowrab T . Exploring the potential of energy-based therapeutics (photobiomodulation/low-level Laser light therapy) in cardiovascular disorders: a review and perspective. Cureus. (2023) 15(4):e37880. 10.7759/cureus.37880
98.
Mirsky N Krispel Y Shoshany Y Maltz L Oron U . Promotion of angiogenesis by low energy laser irradiation. Antioxid Redox Signal. (2002) 4(5):785–90. 10.1089/152308602760598936
99.
Tuby H Maltz L Oron U . Modulations of VEGF and iNOS in the rat heart by low level laser therapy are associated with cardioprotection and enhanced angiogenesis. Lasers Surg Med. (2006) 38(7):682–8. 10.1002/lsm.20377
100.
Manchini MT Antonio EL Silva Junior JA de Carvalho PT Albertini R Pereira FC et al Low-Level Laser application in the early myocardial infarction stage has No beneficial role in heart failure. Front Physiol. (2017) 8:23. 10.3389/fphys.2017.00023
101.
Manchini MT Serra AJ Feliciano Rdos S Santana ET Antonio EL de Tarso Camillo de Carvalho P et al Amelioration of cardiac function and activation of anti-inflammatory vasoactive peptides expression in the rat myocardium by low level laser therapy. PLoS One. (2014) 9(7):e101270. 10.1371/journal.pone.0101270
102.
Feliciano RDS Manchini MT Atum ALB da Silva GA Antonio EL Serra AJ et al Photobiomodulation therapy’s effects on cardiac fibrosis activation after experimental myocardial infarction. Lasers Surg Med. (2022) 54(6):883–94. 10.1002/lsm.23544
103.
Grandinetti V Carlos FP Antonio EL de Oliveira HA Dos Santos LFN Yoshizaki A et al Photobiomodulation therapy combined with carvedilol attenuates post-infarction heart failure by suppressing excessive inflammation and oxidative stress in rats. Sci Rep. (2019) 9(1):9425. 10.1038/s41598-019-46021-1
104.
Luiz Antonio E de Oliveira HA Albuquerque-Pontes GM Teixeira ILA Yoshizaki AP Dos Santos LFN et al Examining the impact of varying low-level laser dose on cardiac failure. Photochem Photobiol. (2025) 101(2):483–93. 10.1111/php.14012
105.
Frangogiannis NG . The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. (2014) 11(5):255–65. 10.1038/nrcardio.2014.28
106.
Toldo S Abbate A . The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. (2018) 15(4):203–14. 10.1038/nrcardio.2017.161
107.
Westman PC Lipinski MJ Luger D Waksman R Bonow RO Wu E et al Inflammation as a driver of adverse left ventricular remodeling after acute myocardial infarction. J Am Coll Cardiol. (2016) 67(17):2050–60. 10.1016/j.jacc.2016.01.073
108.
Awad K Zaki MM Mohammed M Lewek J Lavie CJ Banach M , Lipid and Blood Pressure Meta-analysis Collaboration Group. Effect of the renin-angiotensin system inhibitors on inflammatory markers: a systematic review and meta-analysis of randomized controlled trials. Mayo Clin Proc. (2022) 97(10):1808–23. 10.1016/j.mayocp.2022.06.036
109.
Ridker PM Danielson E Rifai N Glynn RJ . Val-MARC investigators. Valsartan, blood pressure reduction, and C-reactive protein: primary report of the val-MARC trial. Hypertension. (2006) 48(1):73–9. 10.1161/01.HYP.0000226046.58883.32
110.
Pfützner A Schöndorf T Hanefeld M Forst T . High-sensitivity C-reactive protein predicts cardiovascular risk in diabetic and nondiabetic patients: effects of insulin-sensitizing treatment with pioglitazone. J Diab Sci Technol. (2010) 4(3):706–16. 10.1177/193229681000400326
111.
Bendotti G Montefusco L Lunati ME Usuelli V Pastore I Lazzaroni E et al The anti-inflammatory and immunological properties of GLP-1 receptor agonists. Pharmacol Res. (2022) 182:106320. 10.1016/j.phrs.2022.106320
112.
Theofilis P Sagris M Oikonomou E Antonopoulos AS Siasos G Tsioufis K et al The anti-inflammatory effect of novel antidiabetic agents. Life (Basel). (2022) 12(11):1829. 10.3390/life12111829
113.
Buttice L Ghani M Suthakar J Gnanalingham S Carande E Kennedy BWC et al The effect of sodium-glucose cotransporter-2 inhibitors on inflammatory biomarkers: a meta-analysis of randomized controlled trials. Diabetes Obes Metab. (2024) 26(7):2706–21. 10.1111/dom.15586
114.
Ridker PM Danielson E Fonseca FA Genest J Gotto AM Jr Kastelein JJ et al Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. (2008) 359(21):2195–207. 10.1056/NEJMoa0807646
115.
Ridker PM Danielson E Fonseca FA Genest J Gotto AM Jr Kastelein JJ et al Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. (2009) 373(9670):1175–82. 10.1016/S0140-6736(09)60447-5
116.
Bohula EA Giugliano RP Cannon CP Zhou J Murphy SA White JA et al Achievement of dual low-density lipoprotein cholesterol and high-sensitivity C-reactive protein targets more frequent with the addition of ezetimibe to simvastatin and associated with better outcomes in IMPROVE-IT. Circulation. (2015) 132(13):1224–33. 10.1161/CIRCULATIONAHA.115.018381
117.
Bohula EA Giugliano RP Leiter LA Verma S Park JG Sever PS et al Inflammatory and cholesterol risk in the FOURIER trial. Circulation. (2018) 138(2):131–40. 10.1161/CIRCULATIONAHA.118.034032
118.
Ruscica M Tokgözoğlu L Corsini A Sirtori CR . PCSK9 Inhibition and inflammation: a narrative review. Atherosclerosis. (2019) 288:146–55. 10.1016/j.atherosclerosis.2019.07.015
119.
Landmesser U Haghikia A Leiter LA Wright RS Kallend D Wijngaard P et al Effect of inclisiran, the small-interfering RNA against proprotein convertase subtilisin/kexin type 9, on platelets, immune cells, and immunological biomarkers: a pre-specified analysis from ORION-1. Cardiovasc Res. (2021) 117(1):284–91. 10.1093/cvr/cvaa077
120.
González-Juanatey JR Górriz JL Ortiz A Valle A Soler MJ Facila L . Cardiorenal benefits of finerenone: protecting kidney and heart. Ann Med. (2023) 55(1):502–13. 10.1080/07853890.2023.2171110
121.
Rayego-Mateos S Rodrigues-Diez RR Fernandez-Fernandez B Mora-Fernández C Marchant V Donate-Correa J et al Targeting inflammation to treat diabetic kidney disease: the road to 2030. Kidney Int. (2023) 103(2):282–96. 10.1016/j.kint.2022.10.030
122.
Barrera-Chimal J Estrela GR Lechner SM Giraud S El Moghrabi S Kaaki S et al The myeloid mineralocorticoid receptor controls inflammatory and fibrotic responses after renal injury via macrophage interleukin-4 receptor signaling. Kidney Int. (2018) 93(6):1344–55. 10.1016/j.kint.2017.12.016
123.
Bittencourt CRO Izar MCO França CN Schwerz VL Póvoa RMDS Fonseca FAH . Effects of chronic exercise on endothelial progenitor cells and microparticles in professional runners. Arq Bras Cardiol. (2017) 108(3):212–6. 10.5935/abc.20170022
124.
Bittencourt CR Izar MC Schwerz VL Póvoa RM Fonseca HA Fonseca MI et al Effects of high-intensity training of professional runners on myocardial hypertrophy and subclinical atherosclerosis. PLoS One. (2016) 11(11):e0166009. 10.1371/journal.pone.0166009
125.
Blaum C Brunner FJ Kröger F Braetz J Lorenz T Goßling A et al Modifiable lifestyle risk factors and C-reactive protein in patients with coronary artery disease: implications for an anti-inflammatory treatment target population. Eur J Prev Cardiol. (2021) 28(2):152–8. 10.1177/2047487319885458
126.
Gragnano F Spirito A Corpataux N Vaisnora L Galea R Gargiulo G et al Impact of clinical presentation on bleeding risk after percutaneous coronary intervention and implications for the ARC-HBR definition. Eurointervention. (2021) 17(11):e898–909. 10.4244/EIJ-D-21-00181
127.
Glynn RJ Danielson E Fonseca FAH Genest J Gotto AM Jr Kastelein JJP et al A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med. (2009) 360(18):1851–61. 10.1056/NEJMoa0900241
128.
Dix C Zeller J Stevens H Eisenhardt SU Shing KSCT Nero TL et al C-reactive protein, immunothrombosis and venous thromboembolism. Front Immunol. (2022) 13:1002652. 10.3389/fimmu.2022.1002652
129.
Nanchen D Klingenberg R Gencer B Räber L Carballo D von Eckardstein A et al Inflammation during acute coronary syndromes—risk of cardiovascular events and bleeding. Int J Cardiol. (2019) 287:13–8. 10.1016/j.ijcard.2019.03.049
Summary
Keywords
B cells, T cells, macrophages/monocytes, neutrophils, dendritic cells, cytokines, C-reactive protein, microbiota
Citation
Fonseca FAH, França CN, Fonseca HAR, Serra AJ and Izar MC (2025) Key inflammatory players for infarcted mass and cardiac remodeling after acute myocardial infarction. Front. Cardiovasc. Med. 12:1609705. doi: 10.3389/fcvm.2025.1609705
Received
10 April 2025
Accepted
27 June 2025
Published
18 July 2025
Volume
12 - 2025
Edited by
Tommaso Gori, Johannes Gutenberg University Mainz, Germany
Reviewed by
Luca Bergamaschi, University of Bologna, Italy
Andreas Mitsis, Nicosia General Hospital, Cyprus
Updates
Copyright
© 2025 Fonseca, França, Fonseca, Serra and Izar.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Francisco A. H. Fonseca fah.fonseca@unifesp.br
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.