 Jinqiao Wu
Jinqiao Wu Min Chen
Min Chen Ze Peng
Ze Peng Yao Sun
Yao Sun Juan Jin
Juan Jin- 1First Clinical Medical College, Heilongjiang University of Chinese Medicine, Harbin, Heilongjiang, China
- 2Department of Cardiovascular Diseases 1, The First Affiliated Hospital of Heilongjiang University of Chinese Medicine, Harbin, Heilongjiang, China
Myocardial fibrosis (MF) is a hallmark pathological outcome of many cardiovascular diseases and a key component of cardiac remodeling. The p38 MAPK, JNK, and ERK signaling pathways are central to this process. This review summarizes the roles and interactions of these factors in MF and identifies relevant drug and non-pharmacological therapies targeting these pathways.
1 Introduction
Myocardial fibrosis (MF), a significant pathological basis for heart failure (HF), is characterized by excessive collagen deposition in the myocardial interstitium driven by abnormal fibroblast activation (1). These pathological changes impair systolic and diastolic functions. Three pathological subtypes are recognized: replacement fibrosis, interstitial fibrosis, and perivascular fibrosis (2). Early MF may be clinically silent and detectable only through imaging. With progression, it can lead to HF, arrhythmias, and diastolic dysfunction. Early intervention may slow disease progression; however, established MF is usually irreversible. The extent of fibrosis strongly correlates with cardiovascular mortality and hospitalization for HF. Therefore, preventive and therapeutic strategies are essential.
The mitogen-activated protein kinase (MAPK) signaling pathway is a central intracellular signal transduction network that regulates cell proliferation, differentiation, apoptosis, and stress responses (3). The MAPK pathway uses a three-kinase cascade to transmit signals. In MF, diverse pathological stimuli activate this cascade, modulate transcription factors, and alter the expression of connected genes. The principal kinases in this pathway are p38 MAPK, c-Jun N-terminal kinase (JNK), and extracellular signal-regulated kinase (ERK) (4). Furthermore, atrial fibrosis is the core pathological feature of atrial cardiomyopathy (5). Studies have demonstrated that p38 protein expression in the atrial muscle is associated with an increase in the number of myofibroblasts (6). This review synthesizes the core roles and recent progress of MAPK signaling in MF and discusses the related findings with broad relevance for understanding atrial fibrosis.
The TGF-β/Smad3 signaling pathway is also important in MF (7). TGF-β ligand binding at the cell surface triggers the phosphorylation of TGF-β receptor II, which then activates receptor I. Receptor I phosphorylates Smad2/3. Phosphorylated Smad2/3 and Smad4 can form a complex and translocates to the nucleus, affecting the expression of various profibrotic genes, including collagens (COL1A1, COL3A1, COL5A2, COL6A1, COL6A3, COL7A1) (8), PAI-1 (9), proteoglycans (10), integrins (11), connective tissue growth factor (CTGF) (12) and matrix metallopeptidase (MMP) (13). This is the classical Smad pathway. TGF-β also signals through non-Smad routes by activating MAPKs, namely p38 MAPK, JNK, and ERK, which promote MF formation.
2 Mechanisms of MAPK in MF, therapeutic strategies, and research progress
2.1 Roles of MAPK subtypes in MF
2.1.1 Mechanisms of p38 MAPK promoting MF
p38 MAPK is the most extensively studied subtype. The p38 MAPK family consists of four subtypes (α, β, γ, and δ), among which p38α is the most abundantly expressed in the heart and has the closest relationship with MF. In MF, p38 MAPK is mainly activated by cytokines, growth factors, and oxidative stress. Subsequently, it promotes the development of MF by regulating processes, including inflammation, oxidative stress, apoptosis, and fibroblast activation. Lastly, it phosphorylates downstream transcription factors, enhances the production of various pro-fibrotic factors, and forms a positive feedback loop.
2.1.1.1 Activation of myofibroblast differentiation
p38 MAPK is a major signaling effector pathway downstream of the TGF-β non-Smad pathway. p38 MAPK can also activate Smad3, leading to MF (14). α-smooth muscle actin(α-SMA) is the main marker protein of myofibroblasts. When cardiac fibroblasts (CFs) are activated by injury or pathological factors, such as TGF-β and angiotensin(Ang) Ⅱ, they transform into myofibroblasts expressing α-SMA, thereby enhancing collagen secretion and excessive extracellular matrix (ECM) deposition, leading to MF. Through two distinct mechanisms, p38 MAPK increases ACTA2 expression α-SMA, thereby driving cardiac fibroblast-to-myofibroblast differentiation (15).
2.1.1.2 Mediation of inflammation
NF-κB and AP-1 can serve as binding sites for transcription factors within the promoter region of inflammatory cytokine genes, both of which can be activated downstream of p38 MAPK. p38 MAPK can also regulate the transcription levels of pro-inflammatory factors secreted by CFs. It has been found that p38 MAPK can raise the mRNA expression of inflammatory factors in cultured human CFs (16–18). Fisetin can improve left atrial inflammation and fibrosis after myocardial infarction (MI) via the p38 MAPK signaling pathway (19).
2.1.1.3 Regulation of oxidative stress
In the heart, reactive oxygen species (ROS) can activate the p38 MAPK pathway (20). Activated p38 MAPK can induce the expression of α-SMA (21). Hypoxia can also stimulate mitochondria to produce ROS, which, in turn, activate p38 MAPK. Furthermore, p38 MAPK can inhibit the function and expression of superoxide dismutase (SOD). In chronic intermittent hypoxia, p38 MAPK inhibitors can elevate SOD activity and reduce oxidative damage indicators (22). Mendelian randomization analysis further supports a negative correlation between MAPK14 (the gene encoding p38 MAPK) and SOD levels, indicating a bidirectional inhibitory relationship (23).
2.1.1.4 Induction of apoptosis and myocardial remodeling
p38 MAPK can activate the expression of pro-apoptotic proteins, such as Bax and caspase-3, whereas the inhibition of p38 MAPK may result in elevated levels of anti-apoptotic proteins, such as Bcl (24, 25). These findings indicate that p38 MAPK induces cardiomyocyte apoptosis. Moreover, p38 MAPK can increase the stability of the mRNAs for MMP-1, MMP-3, and MMP-9 (26, 27). It can also induce the transcription of MMP1 (28) and MMP9 (29), leading to ECM deposition and exacerbating MF.
2.1.2 Mechanisms of JNK promoting MF
JNK plays an important role in MF, and the main subtypes in the heart are JNK1/2.
2.1.2.1 Activation of myofibroblast differentiation
c-JUN plays an important role in the pathological processes of MF. It can directly bind to the promoter region of collagen genes, promote ECM deposition, and amplify the inflammatory response by mediating the signal transduction of inflammatory factors, indirectly promoting MF. JNK enhances the phosphorylation of c-JUN and transcriptional activity of AP-1, elevates the levels of fibrosis-related genes, and promotes the differentiation of myofibroblasts (30). JNK inhibitors can significantly reduce the expression of fibrosis-related genes (31).
2.1.2.2 Mediation of inflammation and oxidative stress
JNK stimulates NF-κB and AP-1, facilitates the production of inflammatory mediators, intensifies the process of inflammation within the cardiac milieu, and promotes MF. JNK phosphorylates the N-terminus of c-JUN, enhancing its transcriptional activity and regulating collagen deposition. In hypertensive HF mice, Ang Ⅱ induction increased the phosphorylation of JNK and c-JUN nuclear translocation. These effects were eliminated by 20(S)-ginsenoside Rh2 in a dose-dependent manner (32). Moreover, Rg5 can mitigate inflammation in the hearts of mice and cultured myocardial cells by blocking the JNK/AP-1 pathway activated by Ang Ⅱ (33). Theophylline can also reduce c-JUN levels and inhibit Ang Ⅱ-induced MF (34).
2.1.2.3 Regulation of extracellular matrix metabolism
JNK is a driving factor in the activation of collagen synthesis in MF. JNK can stimulate CF proliferation, leading to an increase in COL Ⅰ/Ⅲ (35). Inflammatory factors [IL-22 (36) and tryptase (37)]can stimulate JNK, resulting in enhanced collagen synthesis and fibrosis. Moreover, tryptase can also stimulate the upregulation of Fn, MMP-1, and TIMP-1 through the JNK pathway, leading to excessive ECM deposition. Blocking JNK signal transduction significantly alleviates these effects (38).
2.1.2.4 Interaction with other pathways
In vitro studies have indicated that resistin can stimulate the production of pro-fibrotic genes via JAK2/STAT3 and JNK/c-Jun pathways. In vivo studies have demonstrated that overexpression of resistin significantly increases the phosphorylation of the above pathways, indicating that resistin regulates fibrosis through network pathways (31). Epigallocatechin gallate can improve MF via the TGF-β1/JNK pathway (39).
2.1.3 Mechanisms of ERK promoting MF
ERK1/2 participates in cardiac remodeling and can be activated by growth factors and G protein-coupled receptor ligands (40, 41). The main types of receptors for ERK1/2 on the surface of myocardial cells are RTK (42) and GPCR (43). The binding of a growth factor to the extracellular domain of an RTK promotes tail-to-tail contact and kinase activation, which propagates signals to ERK1/2 (44).
2.1.3.1 Activation of the proliferation and differentiation of cardiac myofibroblasts
Studies have demonstrated that endothelin-1 leads to the activation of fibroblasts and differentiation of myofibroblasts through the ERK1/2 signaling pathway (45). TGF-β/Smad can activate the ERK1/2 pathway, upregulate CTGF/CCN2, and promote MF (46). ERK is also a key signaling effector pathway downstream of the TGF-β non-Smad pathway. Ang Ⅱ can promote the differentiation of fibroblasts into myofibroblasts via the TGF-β1/ERK1/2 pathway, manifested as fibroblast proliferation and formation of stress fibers (47).
2.1.3.2 Regulation of extracellular matrix metabolism
Hyperglycemia can activate ERK1/2 in CFs, resulting in elevated COL Ⅰ/Ⅲ mRNA and protein levels. Inhibiting ERK1/2 activation significantly diminishes collagen production (48). LPS can increase the production of MMP-2 and MMP-9 via the ERK1/2 pathway in CFs (49). TIMPs can directly inhibit MMPs and form complexes with them to regulate their activation and stability. In HF, factors such as Wnt5a and VEGF-D activate ERK signaling in CFs and myofibroblasts, resulting in the elevated production of TIMP-1 and TIMP-2 (50).
Angiotensin II type 1 receptor (AT1R) is a member of the GPCR family. When excessive Ang Ⅱ accumulates in the body and is not metabolized, it links to amino acids on the fibroblast membrane, facilitating the interaction between AT1R and different proteins, thereby activating ERK1/2 signal transduction (51). Persistent stimulation by excess growth factors and Ang II activates receptor tyrosine kinases (RTKs) and the AT1R. This drives positive feedback phosphorylation of upstream kinases and downstream effectors in the ERK1/2 pathway and promotes MF (35). An in vitro experiment demonstrated that stimulation with TGF-β1 can induce fibroblast proliferation, α-SMA expression, and collagen synthesis, and pretreatment with PD98059 (an ERK1/2 inhibitor) can significantly inhibit these effects (52).
2.2 Therapeutic strategies and research progress in regulating the MAPK signaling pathway
The following mechanistic studies provide a solid foundation for the treatment of MF by inhibiting the p38 MAPK/JNK/ERK signaling pathway with targeted drugs, conventional drugs, Chinese medicine monomers, Chinese patent medicines and decoctions, and non-pharmacological therapies (Figure 1). These studies provide proof-of-concept evidence for this hypothesis (Table 1).

Figure 1. Schematic diagram of MAPK signaling pathway.
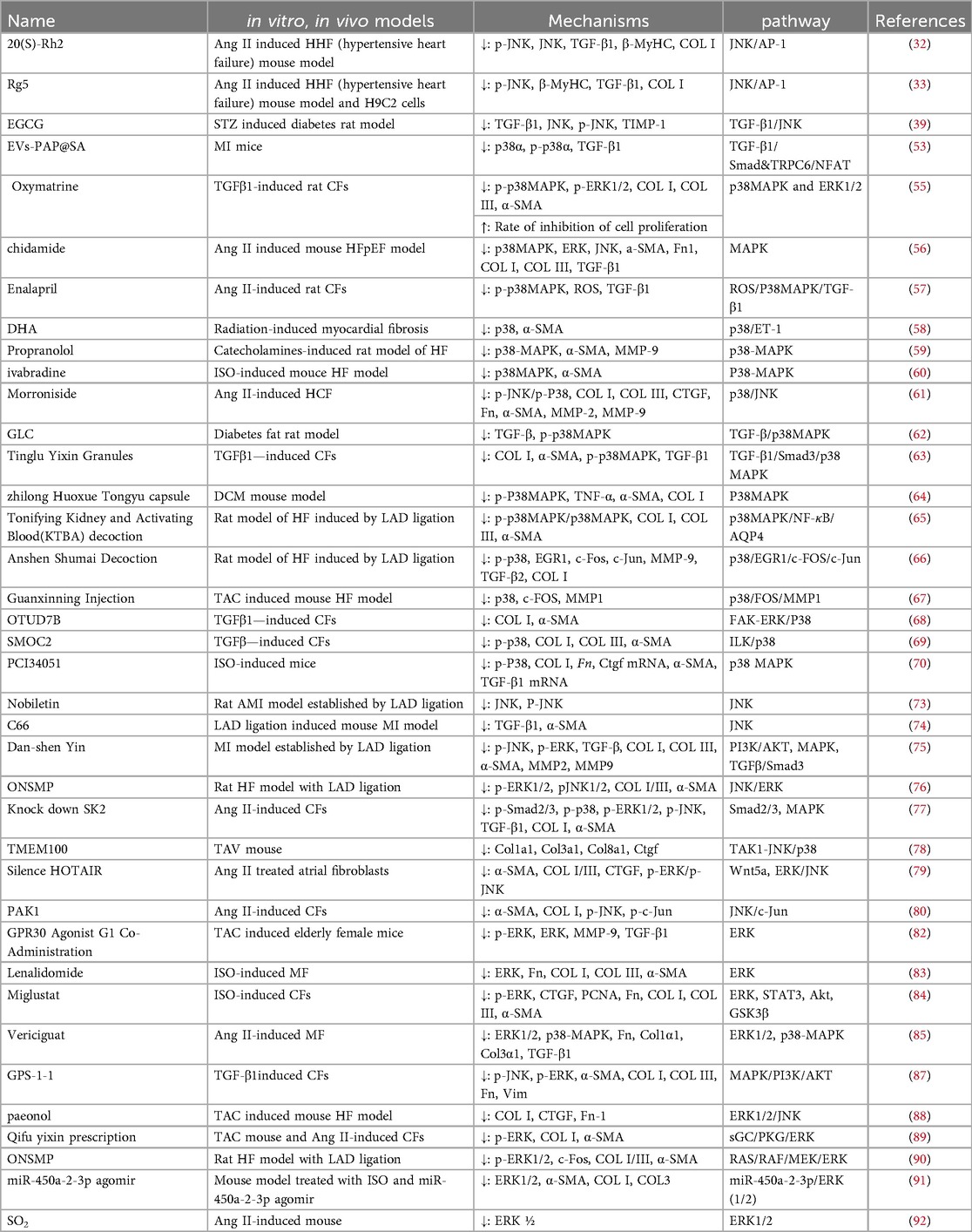
Table 1. Drugs or methods to improve myocardial fibrosis by inhibiting the MAPK signaling pathway.
2.2.1 Agents and approaches acting on the p38 MAPK pathway
2.2.1.1 P38 MAPK-targeted drugs
SB203580 is a selective p38 MAPK inhibitor. Studies have found that SB203580 can reduce the expression of α-SMA, COL Ⅰ, TGF-β1, and TNF-α, and inhibit the progression of MF. Novel delivery systems, such as hydrogels loaded with p38α antagonistic peptides or nanoparticles targeting activated fibroblasts, enhance the local efficacy of anti-fibrotic drugs (53, 54). Oxymatrine simultaneously inhibits the TGF-β1 and p38 MAPK pathways. Compared with SB203580 alone, this combination produced a significantly stronger anti-fibrotic effect, indicating synergistic inhibition (55).
2.2.1.2 Conventional drugs targeting the p38 MAPK
In the Ang Ⅱ-induced HFpEF mouse model, chidamide improved myocardial hypertrophy and fibroblast proliferation and differentiation by inhibiting p38 MAPK. Subsequent studies have demonstrated that chidamide administration markedly diminishes the expression of cardiac fibrosis markers (PCNA, COL Ⅰ, COL Ⅲ, TGF-β1, and α-SMA) (56). Enalapril can inhibit the proliferation of Ang Ⅱ-induced rat fibroblasts through the ROS/p38 MAPK/TGF-β1 pathway (57). Liu found that DHA can reduce collagen deposition and α-SMA expression via blocking the p38/ET-1 pathway in cardiomyocytes, and alleviate radiation-induced MF (58). Propranolol can alleviate MF caused by excessive catecholamines by modulating p38 MAPK (59). Sun found that ivabradine exhibits a protective effect against isoproterenol-induced cardiac injury, which is related to its blocking of the p38 MAPK signaling pathway, reduction of MF, decrease in cardiomyocyte apoptosis, and increase in autophagy (60).
2.2.1.3 Chinese medicine monomers targeting the p38 MAPK
Oxymatrine can inhibit TGF-β1-induced cardiac fibroblast proliferation and fibroblast-myofibroblast transformation by modulating p38 MAPK and ERK1/2 pathways (55). Zheng found that Morroniside, the active ingredient of Fructus Corni, can block the p38/JNK pathway by downregulating KLF5, which can improve the proliferation, migration, and extracellular matrix deposition of CFs, thereby exerting a protective effect on MF (61). GLC can ameliorate MF in murine models of diabetic cardiomyopathy via the TGF-β/p38 MAPK pathway (62).
2.2.1.4 Chinese patent medicines and decoctions targeting the p38 MAPK
Tinglu Yixin Granules can inhibit the TGF-β1/Smad3/p38 MAPK signal transduction, reduce the transformation of fibroblasts, and inhibit the expression of collagen and α-SMA. In vivo experiments have also confirmed that Tinglu Yixin Granules can ameliorate MF in diabetic mouse models (63). Zhilong Huoxue Tongyu Capsules can improve MF animals suffering from diabetic cardiomyopathy. The results of enrichment analysis point to MAPK. In vivo experiments have confirmed that Zhilong Huoxue Tongyu Capsules downregulate the expression of fibrosis-related proteins (64). In a study of the HF rat model with left anterior descending ligation, Xu found that Tonifying Kidney and Activating Blood Decoction can regulate the p38 MAPK/NF-κB/AQP4 axis to delay MF (65). Another study found that Anshen Shumai Decoction mainly exerts its effect by down-regulating the gene expression of FOS and EGR1 and the p38 MAPK pathway, which significantly inhibits myocardial cell apoptosis and MF in infarcted rats (66). Furthermore, Guanxinning Injection inhibits myocardial hypertrophy and fibrosis in HF mice by modulating the p38/FOS/MMP1 pathway (67).
2.2.1.5 Non-pharmacological therapies targeting the p38 MAPK
OTUD7B is a drug target that can mitigate MF by decreasing the phosphorylation of ERK/p38. Research indicates that silencing OTUD7B with siRNA increases the levels of α-SMA and COL Ⅰ in CFs, while overexpressing OTUD7B with adenovirus reduces their expression, thereby producing an anti-fibrotic action (68). Therapeutic SMOC2 silencing inhibits the production of COL Ⅰ, COL Ⅲ, and α-SMA through the ILK/p38 pathway in vitro (69). Moreover, the delivery of mBMSCs-EVs and PAP using SA hydrogel mitigated MF in mice with MI (53). Treatment with PCI34051 suppresses the expression of murine fibrosis markers by diminishing isoproterenol-induced activation of p38 MAPK (70).
2.2.2 Agents and approaches acting on the JNK pathway
2.2.2.1 JNK's targeted drugs and conventional drugs
Studies have demonstrated that SP600125 (a JNK inhibitor) can alleviate MF by inhibiting JNK phosphorylation, reducing apoptosis, and decreasing oxidative stress in animals and cells (71). In a murine model of MI, SP600125 can modulate the JNK pathway and counteract the pro-fibrotic effect of mCRP (72). MicroRNAs targeting the JNK pathway can alleviate hypoxia-induced MF by reducing collagen expression, further supporting the therapeutic potential of the JNK pathway (38). Guì et al. found that epigallocatechin gallate can improve MF in streptozotocin-diabetic rats with intraperitoneal injection by modulating the TGF-β1/JNK signaling pathway (39).
2.2.2.2 Chinese herbal monomers targeting the JNK
Nobiletin improves MF in a rat model of acute myocardial infarction (AMI) by inhibiting the JNK pathway. It was also found that intraperitoneal injection of nobiletin at a medium dose exhibited the best improvement effect (73). C66, a curcumin analog, can improve MF in rats following AMI by inhibiting the JNK pathway (74). 20(S)-Ginsenoside Rh2 [20(S)-Rh2] is a chemical present in Radix Ginseng and studies have found that it can ameliorate MF in hypertensive rats and those with HF induced by Ang II, as well as in ventricular myocytes. Studies have demonstrated that 20(S)-Rh2 can inhibit the levels of TGF-β1, β-MyHC, and COL Ⅰ in model rats and ventricular myocytes in a dose-dependent manner at both the transcriptome and proteomic levels by inhibiting JNK/AP-1 (32). Furthermore, Ginsenoside Rg5 can mitigate Ang Ⅱ-induced MF by suppressing the JNK/AP-1 pathway (33).
2.2.2.3 Chinese patent medicines and decoctions targeting the JNK
Danshen Yin can diminish the expression of COL Ⅰ, COL Ⅲ, α-SMA, MMP 2, MMP9, and TGF-β by inhibiting JNK and ERK pathways, thereby alleviating MF following MI in rats (75). Optimized new Shengmai powder (ONSMP) ameliorated MF in rats with HF. The study found that medium and high doses of ONSMP reduced the expression levels of serum p-ERK1/2 and p-JNK1/2 in the myocardial tissue of rats with HF (76).
2.2.2.4 Non-pharmacological therapies targeting the JNK
Research has found that SK2 channel knockdown can inhibit the differentiation of fibroblasts and the secretion of collagen induced by Ang Ⅱ. Moreover, knocking down SK2 significantly inhibits the activity of signaling molecules associated with the TGF-β pathway, resulting in a substantial reduction in the phosphorylation levels of Smad2/3, p38, ERK1/2, and JNK (77). Zhang's research found that the overexpression of transmembrane protein 100 can alleviate MF in mice with transverse aortic constriction via the TAK1-JNK/p38 pathway (78). HOTAIR silencing exhibits anti-migratory and anti-proliferative effects on primary atrial fibroblasts by inhibiting the Wnt5a/ERK/JNK pathway (79). Moreover, Zhou found that decreased PAK1 expression can inhibit Ang Ⅱ-induced proliferation, migration, and differentiation of HCFs through the JNK/c-Jun pathway (80).
2.2.3 Agents and approaches acting on the ERK pathway
2.2.3.1 ERK-targeted inhibitors
In the cardiac transplantation paradigm, ERK inhibition using U0126 attenuates graft fibrosis (81). PD98059 can reduce the levels of phosphorylated ERK1/2, MMP-9, and TGF-β1 in CFs, demonstrating its ability to resist MF (82).
2.2.3.2 Conventional drugs modulating the ERK pathway
Lenalidomide inhibits β-adrenergic receptor-induced MF by diminishing the gene and protein expression of Fn, COL Ⅰ, COL Ⅲ, and α-SMA via the PI3K/AKT and JNK pathways (83). Moreover, Miglustat can improve β-adrenergic receptor-induced MF by partially inhibiting the ERK pathway (84). Studies have determined that high-dose vericiguat can significantly improve Ang Ⅱ-induced left ventricular MF in murine models. This effect is accomplished by regulating the ERK1/2 or p38 MAPK pathway to attenuate the expression of Col1a1, Col3a1, and Tgfb1 (85). Tetrandrine improves aortic constriction-induced MF in mice via the MAPK/NF-κB pathway, where MAPK mainly refers to JNK and ERK pathways (86).
2.2.3.3 Chinese medicine monomers targeting the ERK
In vitro experiments have confirmed that GSP-1-1 inhibits the activation of the MAPK/PI3K/AKT signaling pathway by downregulating the expression of PDGFB and reducing the protein expression of Vim, Fn, α-SMA, COL Ⅰ, and COL Ⅲ, thereby inhibiting the fibrosis of fibroblasts (87). In vivo experiments have indicated that paeonol can inhibit fibrosis in TAC-induced HF mice via the ERK1/2/JNK pathway (88).
2.2.3.4 Chinese patent medicines and decoctions modulating the ERK pathway
The Qifu Yixin prescription worsens MF by activating sGC to suppress ERK phosphorylation, thereby slowing the progression of HF in mice caused by transverse aortic constriction (TAC) (89). ONSMP can diminish MF in HF mice (90).
2.2.3.5 Non-pharmacological interventions targeting the ERK pathway
miR-450a-2-3p overexpression can reduce the elevation of ISO-induced α-SMA, COL Ⅰ and COL Ⅲ by lowering ERK1/2, indicating that it can inhibit collagen formation in cardiac tissue via the ERK pathway (91). Sulfur dioxide can inhibit cardiac fibroblast proliferation by sulfenylating ERK1/2 and phosphorylating ERK1/2 (92). Wang et al. found that overexpression of GPR30 and combined administration of its agonist G1 can reduce MF induced by TAC in aged female mice (82).
3 Conclusion and future perspectives
The MAPK-p38 MAPK/JNK/ERK pathway is significant in MF. This process is triggered by stimuli, such as growth factors, initiating MF in a cascading manner. This process involves mechanisms such as oxidative stress, signal transduction, gene expression, and extracellular matrix deposition. Fibroblasts can proliferate and differentiate into myofibroblasts via the MAPK pathway, and the intervention of drugs on this pathway is significant. These findings indicate that MAPK serves as a key target driving MF. Our study implicates p38 MAPK in atrial fibrosis, suggesting that MAPK signaling may be a shared mechanism linking myocardial and atrial fibrosis. As atrial fibrosis sustains atrial fibrillation (AF), targeting this pathway could delay or reverse fibrosis and offer a disease-modifying approach to AF. If MAPK proves to be a shared pathway, the targeted drugs developed could also delay or reverse atrial fibrosis, offering a potential avenue for fundamental treatment of AF.
This review summarizes the therapeutic modalities that regulate MAPK signaling. These include targeted agents, conventional drugs, Chinese patent medicines, and non-pharmacological interventions. Across studies, these approaches reduce myocardial hypertrophy, limit fibroblast proliferation and differentiation, decrease collagen deposition, and suppress MF. In our study, morroniside and oxymatrine acted on multiple pathways and provided synergistic benefits against MF. Compound herbal formulations can also slow down MF progression by simultaneously regulating MAPK, inflammation, and apoptosis. Combination therapy is appropriate for multi-target regulation using herbal compounds. Pairing MAPK inhibitors with other anti-fibrotic agents can also allow dose reductions and may help prevent drug resistance.
Furthermore, p38 MAPK, JNK, and ERK have various subtypes, and the expression scenarios of these subtypes are also different. For example, the primary subtype of p38 MAPK expressed in the myocardium is p38α. The development of subtype-selective inhibitors should be a key goal for future studies. Disease heterogeneity limits the generalizability of treatment effects, and different etiologies activate distinct MAPK pathways. For instance, MF in diabetic cardiomyopathy is marked by elevated p38 activity, whereas JNK and ERK are more active in HF models. These differences support individualized or stratified anti-fibrotic strategies across diseases. Most current studies have emphasized oral agents. Although oral dosing is convenient, it often fails to achieve adequate drug concentrations in target tissues. Cardiac or fibroblast-targeted nanocarriers and ligand-guided delivery systems can increase early cardiac drug levels while reducing systemic exposure and toxicity.
In mice with MI, a fibronectin gel-loaded Gouqi-derived nanovessel can target the p38-MAPK signaling pathway to attenuate myocardial cell apoptosis and limit the progression of MF (93). In preclinical studies, CRISPR/Cas9 gene-editing technology has been proven to effectively correct genetic mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy, reduce MF, and improve heart function, indicating the potential of this technology in treating hereditary cardiomyopathies (94). Furthermore, in in vitro experiments, MicroRNA29a can inhibit the proliferation of fibroblasts by targeting ERK1/2 (95), and miR-43 (14), miR-32-5p (96), miR-338-3p (97), and miR-155 (98) can affect the activation of the MAPK pathway and regulate the differentiation of fibroblasts. These findings illustrate the potential of selectively targeting CFs.
With the advancement of technology and an in-depth understanding of the mechanism, the MAPK pathway remains an attractive target for MF research. This review examines the fundamental role of the MAPK pathway in MF and introduces relevant drugs. There is a solid scientific basis for targeting the MAPK pathway in MF.
Although preclinical studies of MAPK-targeted therapy for MF are encouraging, the current evidence has clear limitations. Most reports are short-term animal or cellular experiments. These designs cannot predict drug effects across the entire disease course, and in vitro systems do not replicate the physiological complexity of humans. Research also focuses on a few key nodes within MAPK; accordingly, its role in the broader disease network remains incomplete. To close these gaps, future studies should prioritize well-designed phase I/II clinical trials, include long-term follow-up endpoints, and comprehensively assess durability and long-term risks. These findings would provide clinicians with more precise and diverse treatment options.
Author contributions
JW: Writing – original draft. MC: Writing – review & editing. ZP: Writing – review & editing. YS: Writing – review & editing. JJ: Writing – review & editing, Conceptualization.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Poddi S, Lefter CL, Linardi D, Ardigò A, Luciani GB, Rungatscher A. Myocardial fibrosis: assessment, quantification, prognostic signification, and anti-fibrosis targets: a state-of-the-art review. J Cardiovasc Dev Dis. (2025) 12(5):192. doi: 10.3390/jcdd12050192
2. Lunde IG, Rypdal KB, Van Linthout S, Diez J, González A. Myocardial fibrosis from the perspective of the extracellular matrix: mechanisms to clinical impact. Matrix Biol. (2024) 134:1–22. doi: 10.1016/j.matbio.2024.08.008
3. Yue J, López JM. Understanding MAPK signaling pathways in apoptosis. Int J Mol Sci. (2020) 21(7):2346. doi: 10.3390/ijms21072346
4. Mai L, Zhu X, Huang F, He H, Fan W. P38 mitogen-activated protein kinase and pain. Life Sci. (2020) 256:117885. doi: 10.1016/j.lfs.2020.117885
5. Pierucci N, Mariani MV, Iannetti G, Maffei L, Coluccio A, Laviola D, et al. Atrial cardiomyopathy: new pathophysiological and clinical aspects. Minerva Cardiol Angiol. (2025). doi: 10.23736/S2724-5683.25.06725-0
6. Kira S, Abe I, Ishii Y, Miyoshi M, Oniki T, Arakane M, et al. Role of angiopoietin-like protein 2 in atrial fibrosis induced by human epicardial adipose tissue: analysis using an organo-culture system. Heart Rhythm. (2020) 17(9):1591–601. doi: 10.1016/j.hrthm.2020.04.027
7. Zeng Z, Wang Q, Yang X, Ren Y, Jiao S, Zhu Q, et al. Qishen granule attenuates cardiac fibrosis by regulating TGF-β/Smad3 and GSK-3β pathway. Phytomedicine. (2019) 62:152949. doi: 10.1016/j.phymed.2019.152949
8. Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF-beta/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem. (2001) 276(20):17058–62. doi: 10.1074/jbc.M100754200
9. Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. (1998) 17(11):3091–100. doi: 10.1093/emboj/17.11.3091
10. Dadlani H, Ballinger ML, Osman N, Getachew R, Little PJ. Smad and p38 MAP kinase-mediated signaling of proteoglycan synthesis in vascular smooth muscle. J Biol Chem. (2008) 283(12):7844–52. doi: 10.1074/jbc.M703125200
11. Margadant C, Sonnenberg A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. (2010) 11(2):97–105. doi: 10.1038/embor.2009.276
12. Chen Y, Blom IE, Sa S, Goldschmeding R, Abraham DJ, Leask A. CTGF expression in mesangial cells: involvement of SMADs, MAP kinase, and PKC. Kidney Int. (2002) 62(4):1149–59. doi: 10.1111/j.1523-1755.2002.kid567.x
13. Yuan W, Varga J. Transforming growth factor-beta repression of matrix metalloproteinase-1 in dermal fibroblasts involves Smad3. J Biol Chem. (2001) 276(42):38502–10. doi: 10.1074/jbc.M107081200
14. Tao L, Bei Y, Chen P, Lei Z, Fu S, Zhang H, et al. Crucial role of miR-433 in regulating cardiac fibrosis. Theranostics. (2016) 6(12):2068–83. doi: 10.7150/thno.15007
15. Turner NA, Blythe NM. Cardiac fibroblast p38 MAPK: a critical regulator of myocardial remodeling. J Cardiovasc Dev Dis. (2019) 6(3):27. doi: 10.3390/jcdd6030027
16. Soni S, Anand P, Padwad YS. MAPKAPK2: the master regulator of RNA-binding proteins modulates transcript stability and tumor progression. J Exp Clin Cancer Res. (2019) 38(1):121. doi: 10.1186/s13046-019-1115-1
17. Turner NA, Das A, Warburton P, O'Regan DJ, Ball SG, Porter KE. Interleukin-1alpha stimulates proinflammatory cytokine expression in human cardiac myofibroblasts. Am J Physiol Heart Circ Physiol. (2009) 297(3):H1117–27. doi: 10.1152/ajpheart.00372.2009
18. Turner NA, Mughal RS, Warburton P, O'Regan DJ, Ball SG, Porter KE. Mechanism of TNFalpha-induced IL-1alpha, IL-1beta and IL-6 expression in human cardiac fibroblasts: effects of statins and thiazolidinediones. Cardiovasc Res. (2007) 76(1):81–90. doi: 10.1016/j.cardiores.2007.06.003
19. Liu L, Gan S, Li B, Ge X, Yu H, Zhou H. Fisetin alleviates atrial inflammation, remodeling, and vulnerability to atrial fibrillation after myocardial infarction. Int Heart J. (2019) 60(6):1398–406. doi: 10.1536/ihj.19-131
20. Zhang Q, Deng Y, Lai W, Guan X, Sun X, Han Q, et al. Maternal inflammation activated ROS-p38 MAPK predisposes offspring to heart damages caused by isoproterenol via augmenting ROS generation. Sci Rep. (2016) 6:30146. doi: 10.1038/srep30146
21. Kulisz A, Chen N, Chandel NS, Shao Z, Schumacker PT. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am J Physiol Lung Cell Mol Physiol. (2002) 282(6):L1324–9. doi: 10.1152/ajplung.00326.2001
22. Lu HD, Liu ZC, Zhou LY, Zhou J, Feng XR, Wang B. Influence of the TLR4-mediated p38MAPK signaling pathway on chronic intermittent hypoxic-induced rat’s oxidative stress and inflammatory cytokines in rats. Eur Rev Med Pharmacol Sci. (2019) 23(1):352–60. doi: 10.26355/eurrev_201901_16783
23. Qiu M, Huang Y, Zhou X, Yu J, Li J, Wang W, et al. Hyperlipidemia exacerbates acute pancreatitis via interactions between P38MAPK and oxidative stress. Cell Signal. (2025) 125:111504. doi: 10.1016/j.cellsig.2024.111504
24. Xiong W, Chen H, Lu J, Ren J, Nie C, Liang R, et al. IL-39 increases ROS production and promotes the phosphorylation of p38 MAPK in the apoptotic cardiomyocytes. Folia Histochem Cytobiol. (2021) 59(3):195–202. doi: 10.5603/FHC.a2021.0019
25. Kaiser RA, Bueno OF, Lips DJ, Doevendans PA, Jones F, Kimball TF, et al. Targeted inhibition of p38 mitogen-activated protein kinase antagonizes cardiac injury and cell death following ischemia-reperfusion in vivo. J Biol Chem. (2004) 279(15):15524–30. doi: 10.1074/jbc.M313717200
26. Reunanen N, Li SP, Ahonen M, Foschi M, Han J, Kähäri VM. Activation of p38 alpha MAPK enhances collagenase-1 [matrix metalloproteinase (MMP)-1] and stromelysin-1 (MMP-3) expression by mRNA stabilization. J Biol Chem. (2002) 277(35):32360–8. doi: 10.1074/jbc.M204296200
27. Kumar B, Koul S, Petersen J, Khandrika L, Hwa JS, Meacham RB. P38 mitogen-activated protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer Res. (2010) 70(2):832–41. doi: 10.1158/0008-5472.CAN-09-2918
28. Cortez DM, Feldman MD, Mummidi S, Valente AJ, Steffensen B, Vincenti M, et al. IL-17 stimulates MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK- and ERK1/2-dependent C/EBP-beta, NF-kappaB, and AP-1 activation. Am J Physiol Heart Circ Physiol. (2007) 293(6):H3356–65. doi: 10.1152/ajpheart.00928.2007
29. Simon C, Simon M, Vucelic G, Hicks MJ, Plinkert PK, Koitschev A, et al. The p38 SAPK pathway regulates the expression of the MMP-9 collagenase via AP-1-dependent promoter activation. Exp Cell Res. (2001) 271(2):344–55. doi: 10.1006/excr.2001.5374
30. Zhou M, Chen J-y, Chao M-L, Zhang C, Shi Z-g, Zhou X-c, et al. S-nitrosylation of c-Jun N-terminal kinase mediates pressure overload-induced cardiac dysfunction and fibrosis. Acta Pharmacol Sin. (2022) 43(3):602–12. doi: 10.1038/s41401-021-00674-9
31. Singh R, Kaundal RK, Zhao B, Bouchareb R, Lebeche D. Resistin induces cardiac fibroblast-myofibroblast differentiation through JAK/STAT3 and JNK/c-Jun signaling. Pharmacol Res. (2021) 167:105414. doi: 10.1016/j.phrs.2020.105414
32. Yu T, Xu J, Wang Q, Han X, Tu Y, Wang Y, et al. 20(S)-ginsenoside Rh2 inhibits angiotensin-2 mediated cardiac remodeling and inflammation associated with suppression of the JNK/AP-1 pathway. Biomed Pharmacother. (2023) 169:115880. doi: 10.1016/j.biopha.2023.115880
33. Yu T, Xu X, Wei J, Xu J, Luo W, Li A, et al. Ginsenoside Rg5 alleviates Ang II-induced cardiac inflammation and remodeling by inhibiting the JNK/AP-1 pathway. Int Immunopharmacol. (2023) 120:110408. doi: 10.1016/j.intimp.2023.110408
34. Zhang SQ, Bao YN, Lv LY, Du XH, Wang YC. Conophylline suppresses angiotensin II-induced myocardial fibrosis in vitro via the BMP4/JNK pathway. Bull Exp Biol Med. (2021) 171(3):305–11. doi: 10.1007/s10517-021-05217-0
35. Zhang Z, Yang Z, Wang S, Wang X, Mao J. Targeting MAPK-ERK/JNK pathway: a potential intervention mechanism of myocardial fibrosis in heart failure. Biomed Pharmacother. (2024) 173:116413. doi: 10.1016/j.biopha.2024.116413
36. Wu Y, Tan L, Shi L, Yang Z, Xue Y, Zeng T, et al. Interleukin-22 is elevated in the atrium and plasma of patients with atrial fibrillation and increases collagen synthesis in transforming growth factor-β1-treated cardiac fibroblasts via the JNK pathway. Exp Ther Med. (2020) 20(2):1012–20. doi: 10.3892/etm.2020.8778
37. Tan H, Chen Z, Chen F, Xu W, Liu X. CKAP4 participates in tryptase-induced phenotypic conversion in atrial fibroblasts through PAR2/p38/JNK pathway. Am J Transl Res. (2021) 13(4):2270–82.34017388
38. Lo C-H, Li L-C, Yang S-F, Tsai C-F, Chuang Y-T, Chu H-J, et al. MicroRNA let-7a, -7e and -133a attenuate hypoxia-induced atrial fibrosis via targeting collagen expression and the JNK pathway in HL1 cardiomyocytes. Int J Mol Sci. (2022) 23(17):9636. doi: 10.3390/ijms23179636
39. Gui L, Wang F, Hu X, Liu X, Yang H, Cai Z, et al. Epigallocatechin gallate protects diabetes mellitus rats complicated with cardiomyopathy through TGF-β1/JNK signaling pathway. Curr Pharm Des. (2022) 28(33):2758–70. doi: 10.2174/1381612828666220902115437
40. Lei Y, Chen X, Mo JL, Lv LL, Kou ZW, Sun FY. Vascular endothelial growth factor promotes transdifferentiation of astrocytes into neurons via activation of the MAPK/Erk-Pax6 signal pathway. Glia. (2023) 71(7):1648–66. doi: 10.1002/glia.24361
41. Liu Q, Liu Y, Li X, Wang D, Zhang A, Pang J, et al. Perfluoroalkyl substances promote breast cancer progression via ERα and GPER mediated PI3K/Akt and MAPK/Erk signaling pathways. Ecotoxicol Environ Saf. (2023) 258:114980. doi: 10.1016/j.ecoenv.2023.114980
42. Zhou R, Han B, Xia C, Zhuang X. Membrane-associated periodic skeleton is a signaling platform for RTK transactivation in neurons. Science. (2019) 365(6456):929–34. doi: 10.1126/science.aaw5937
43. Jain R, Watson U, Vasudevan L, Saini DK. ERK activation pathways downstream of GPCRs. Int Rev Cell Mol Biol. (2018) 338:79–109. doi: 10.1016/bs.ircmb.2018.02.003
44. Shin WS, Oh SW, Park HN, Kim JH, Lee ST. Knockdown of PTK7 reduces the oncogenic potential of breast cancer cells by impeding receptor tyrosine kinase signaling. Int J Mol Sci. (2023) 24(15):12173. doi: 10.3390/ijms241512173
45. Duangrat R, Parichatikanond W, Likitnukul S, Mangmool S. Endothelin-1 induces cell proliferation and myofibroblast differentiation through the ETAR/gαq/ERK signaling pathway in human cardiac fibroblasts. Int J Mol Sci. (2023) 24(5):4475. doi: 10.3390/ijms24054475
46. Chatzifrangkeskou M, Le Dour C, Wu W, Morrow JP, Joseph LC, Beuvin M, et al. ERK1/2 Directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum Mol Genet. (2016) 25(11):2220–33. doi: 10.1093/hmg/ddw090
47. Parichatikanond W, Duangrat R, Mangmool S. Gαq protein-biased ligand of angiotensin II type 1 receptor mediates myofibroblast differentiation through TGF-β1/ERK axis in human cardiac fibroblasts. Eur J Pharmacol. (2023) 951:175780. doi: 10.1016/j.ejphar.2023.175780
48. Tang M, Zhang W, Lin H, Jiang H, Dai H, Zhang Y. High glucose promotes the production of collagen types I and III by cardiac fibroblasts through a pathway dependent on extracellular-signal-regulated kinase 1/2. Mol Cell Biochem. (2007) 301(1-2):109–14. doi: 10.1007/s11010-006-9401-6
49. Han C-K, Tien Y-C, Jine-Yuan Hsieh D, Ho T-J, Lai C-H, Yeh Y-L, et al. Attenuation of the LPS-induced, ERK-mediated upregulation of fibrosis-related factors FGF-2, uPA, MMP-2, and MMP-9 by Carthamus tinctorius L in cardiomyoblasts. Environ Toxicol. (2017) 32(3):754–63. doi: 10.1002/tox.22275
50. Abraityte A, Vinge LE, Askevold ET, Lekva T, Michelsen AE, Ranheim T, et al. Wnt5a is elevated in heart failure and affects cardiac fibroblast function. J Mol Med (Berl). (2017) 95(7):767–77. doi: 10.1007/s00109-017-1529-1
51. Zhao T, Zhao W, Meng W, Liu C, Chen Y, Bhattacharya SK, et al. Vascular endothelial growth factor-D mediates fibrogenic response in myofibroblasts. Mol Cell Biochem. (2016) 413(1-2):127–35. doi: 10.1007/s11010-015-2646-1
52. Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, et al. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev. (2018) 98(3):1627–738. doi: 10.1152/physrev.00038.2017
53. Chen S, Zeng X, Wu M, Zhu J, Wu Y. Sodium alginate hydrogel infusion of bone marrow mesenchymal stem cell-derived extracellular vesicles and p38α antagonistic peptides in myocardial infarction fibrosis mitigation. J Am Heart Assoc. (2025) 14(8):e036887. doi: 10.1161/JAHA.124.036887
54. Ji X, Meng Y, Wang Q, Tong T, Liu Z, Lin J, et al. Cysteine-based redox-responsive nanoparticles for fibroblast-targeted drug delivery in the treatment of myocardial infarction. ACS Nano. (2023) 17(6):5421–34. doi: 10.1021/acsnano.2c10042
55. Xu Y, Xiao H, Luo H, Chen Y, Zhang Y, Tao L, et al. Inhibitory effects of oxymatrine on TGF-β1-induced proliferation and abnormal differentiation in rat cardiac fibroblasts via the p38MAPK and ERK1/2 signaling pathways. Mol Med Rep. (2017) 16(4):5354–62. doi: 10.3892/mmr.2017.7277
56. Tian J, Li W, Zeng L, Li Y, Du J, Li Y, et al. HBI-8000 improves heart failure with preserved ejection fraction via the TGF-β1/MAPK signalling pathway. J Cell Mol Med. (2024) 28(7):e18238. doi: 10.1111/jcmm.18238
57. Yu M, Zheng Y, Sun HX, Yu DJ. Inhibitory effects of enalaprilat on rat cardiac fibroblast proliferation via ROS/P38MAPK/TGF-β1 signaling pathway. Molecules. (2012) 17(3):2738–51. doi: 10.3390/molecules17032738
58. Liu Y, Chen P, Liu T, Cheng B, Sun C, Xin H, et al. Docosahexaenoic acid attenuates radiation-induced myocardial fibrosis by inhibiting the p38/ET-1 pathway in cardiomyocytes. Int J Radiat Oncol Biol Phys. (2023) 115(5):1229–43. doi: 10.1016/j.ijrobp.2022.11.007
59. Liu TH, Hsieh RJ, Chen HH, Kuo TJ, Lee JC, Lu WH. Propranolol alleviates cardiac injury after acute catecholamine infusion through p38-MAPK pathways. J Cardiovasc Pharmacol. (2024) 84(1):110–7. doi: 10.1097/FJC.0000000000001571
60. Sun M, Yin F, Wu X, Sun S, An Y, Zhu M, et al. Effects of ivabradine on myocardial autophagia and apoptosis in isoprenaline-induced heart failure in mice. Iran J Basic Med Sci. (2024) 27(1):107–13. doi: 10.22038/IJBMS.2023.70060.15236
61. Zheng H, Yang L, Huang H, Lin Y, Chen L. Morroniside improves AngII-induced cardiac fibroblast proliferation, migration, and extracellular matrix deposition by blocking p38/JNK signaling pathway through the downregulation of KLF5. Naunyn Schmiedebergs Arch Pharmacol. (2024) 397(9):6611–21. doi: 10.1007/s00210-024-03039-1
62. Thakur V, Alcoreza N, Delgado M, Joddar B, Chattopadhyay M. Cardioprotective effect of glycyrrhizin on myocardial remodeling in diabetic rats. Biomolecules. (2021) 11(4):569. doi: 10.3390/biom11040569
63. Zhang M, Sun X, Zhao F, Chen Z, Liu M, Wang P, et al. Tinglu Yixin granule inhibited fibroblast-myofibroblast transdifferentiation to ameliorate myocardial fibrosis in diabetic mice. J Ethnopharmacol. (2025) 337(Pt 3):118980. doi: 10.1016/j.jep.2024.118980
64. Yang F, Luo G, Liu MN, Liu P, Wu D, Chen HL, et al. Network pharmacology and experimental validation to investigate the mechanism of action of Zhilong Huoxue Tongyu capsule in the prevention and treatment of diabetic cardiomyopathy. PLoS One. (2025) 20(5):e0323745. doi: 10.1371/journal.pone.0323745
65. Xu R, Bi Y, Ju Y, Yin W, Zhao S, Zhang Y, et al. Uncovering the molecular mechanisms of tonifying kidney and activating blood decoction against myocardial fibrosis using network pharmacology and experimental validation. Sci Rep. (2025) 15(1):18912. doi: 10.1038/s41598-025-01276-9
66. Wang J, Ye X, Wang Y. Anshen Shumai decoction inhibits post-infarction inflammation and myocardial remodeling through suppression of the p38 MAPK/c-FOS/EGR1 pathway. J Mol Histol. (2024) 55(4):437–54. doi: 10.1007/s10735-024-10214-4
67. Fan S, Xiao G, Ni J, Zhao Y, Du H, Liang Y, et al. Guanxinning injection ameliorates cardiac remodeling in HF mouse and 3D heart spheroid models via p38/FOS/MMP1-mediated inhibition of myocardial hypertrophy and fibrosis. Biomed Pharmacother. (2023) 162:114642. doi: 10.1016/j.biopha.2023.114642
68. Zhang J, Zha Y, Jiao Y, Li Y, Wang J, Zhang S. OTUD7B (Cezanne) ameliorates fibrosis after myocardial infarction via FAK-ERK/P38 MAPK signaling pathway. Arch Biochem Biophys. (2022) 724:109266. doi: 10.1016/j.abb.2022.109266
69. Rui H, Zhao F, Yuhua L, Hong J. Suppression of SMOC2 alleviates myocardial fibrosis via the ILK/p38 pathway. Front Cardiovasc Med. (2023) 9:951704. doi: 10.3389/fcvm.2022.951704
70. Zhao T, Kee HJ, Bai L, Kim MK, Kee SJ, Jeong MH. Selective HDAC8 inhibition attenuates isoproterenol-induced cardiac hypertrophy and fibrosis via p38 MAPK pathway. Front Pharmacol. (2021) 12:677757. doi: 10.3389/fphar.2021.677757
71. Chang HY, Hsu HC, Fang YH, Liu PY, Liu YW. Empagliflozin attenuates doxorubicin-induced cardiotoxicity by inhibiting the JNK signaling pathway. Biomed Pharmacother. (2024) 176:116759. doi: 10.1016/j.biopha.2024.116759
72. Zha Z, Cheng Y, Cao L, Qian Y, Liu X, Guo Y, et al. Monomeric CRP aggravates myocardial injury after myocardial infarction by polarizing the macrophage to pro-inflammatory phenotype through JNK signaling pathway. J Inflamm Res. (2021) 14:7053–64. doi: 10.2147/JIR.S316816
73. Liu Z, Gao Z, Zeng L, Liang Z, Zheng D, Wu X. Nobiletin ameliorates cardiac impairment and alleviates cardiac remodeling after acute myocardial infarction in rats via JNK regulation. Pharmacol Res Perspect. (2021) 9(2):e00728. doi: 10.1002/prp2.728
74. Hao H, Yuan T, Li Z, Zhang C, Liu J, Liang G, et al. Curcumin analogue C66 ameliorates mouse cardiac dysfunction and structural disorders after acute myocardial infarction via suppressing JNK activation. Eur J Pharmacol. (2023) 946:175629. doi: 10.1016/j.ejphar.2023.175629
75. Gao X, Ni C, Song Y, Xie X, Zhang S, Chen Y, et al. Dan-shen Yin attenuates myocardial fibrosis after myocardial infarction in rats: molecular mechanism insights by integrated transcriptomics and network pharmacology analysis and experimental validation. J Ethnopharmacol. (2025) 338(Pt 2):119070. doi: 10.1016/j.jep.2024.119070
76. Zhang Z, Song Y, Zhang X, Wang S, Jia Z, Wang L, et al. Optimized new Shengmai powder ameliorates myocardial fibrosis in rats with heart failure by inhibition of the MAPK signaling pathway. J Ethnopharmacol. (2024) 319(Pt 1):117210. doi: 10.1016/j.jep.2023.117210
77. Chen Y, Bao L, Dong F, Xv M, Li W, Luo T, et al. Effect of fibroblasts small- conductance Ca2+ -activated potassium channel subtype 2 (SK2) on myocardial fibrosis in pressure overload mouse. Cell Signal. (2024) 124:111401. doi: 10.1016/j.cellsig.2024.111401
78. Zhang B-B, Zhao Y-L, Lu Y-Y, Shen J-H, Li H-Y, Zhang H-X, et al. TMEM100 acts as a TAK1 receptor that prevents pathological cardiac hypertrophy progression. Cell Commun Signal. (2024) 22(1):438. doi: 10.1186/s12964-024-01816-2
79. Tan W, Wang K, Yang X, Wang K, Wang N, Jiang TB. LncRNA HOTAIR promotes myocardial fibrosis in atrial fibrillation through binding with PTBP1 to increase the stability of Wnt5a. Int J Cardiol. (2022) 369:21–8. doi: 10.1016/j.ijcard.2022.06.073
80. Zhou Y, Xie Y, Li T, Zhang P, Chen T, Fan Z, et al. P21-activated kinase 1 mediates angiotensin II-induced differentiation of human atrial fibroblasts via the JNK/c-Jun pathway. Mol Med Rep. (2021) 23(3):207. doi: 10.3892/mmr.2021.11846
81. Zheng H, Su Y, Zhu C, Quan D, Skaro AI, McAlister V, et al. An addition of U0126 protecting heart grafts from prolonged cold ischemia-reperfusion injury in heart transplantation: a new preservation strategy. Transplantation. (2021) 105(2):308–17. doi: 10.1097/TP.0000000000003402
82. Wang X, Ma J, Zhang S, Li Z, Hong Z, Jiang L, et al. G protein-coupled estrogen receptor 30 reduces transverse aortic constriction-induced myocardial fibrosis in aged female mice by inhibiting the ERK1/2 -MMP-9 signaling pathway. Front Pharmacol. (2021) 12:731609. doi: 10.3389/fphar.2021.731609
83. Jiao R, Li W, Gu X, Liu J, Liu Z, Hu Y, et al. Lenalidomide attenuates cardiac fibrosis and inflammation induced by β-adrenergic receptor activation. Int Immunopharmacol. (2025) 158:114848. doi: 10.1016/j.intimp.2025.114848
84. Liu J, Li W, Jiao R, Liu Z, Zhang T, Chai D, et al. Miglustat ameliorates isoproterenol-induced cardiac fibrosis via targeting UGCG. Mol Med. (2025) 31(1):55. doi: 10.1186/s10020-025-01093-w
85. Harada T, Kondo H, Nakamura K, He Y, Goto S, Takahashi M, et al. Soluble guanylate cyclase stimulator vericiguat attenuates angiotensin II-induced oxidative stress and cardiac remodeling. Circ J. (2025) 89(7):982–91. doi: 10.1253/circj.CJ-24-0659
86. Wang Y, Zhang R, Li J, Guo S, Yuan Y, Zheng R, et al. Tetrandrine improves ventricular remodeling and inflammation via inhibition of the MAPK/NF-κB pathway. Int Heart J. (2025) 66(3):463–74. doi: 10.1536/ihj.24-697
87. Niu P, Zhang X, Zhang G, Jing R, Qiao Y, Zhou X, et al. A polysaccharide from Glycyrrhiza uralensis attenuates myocardial fibrosis via modulating the MAPK/PI3K/AKT signaling pathway. Int J Biol Macromol. (2025) 286:138207. doi: 10.1016/j.ijbiomac.2024.138207
88. Chen X, Zhang Z, Zhang X, Jia Z, Liu J, Chen X, et al. Paeonol attenuates heart failure induced by transverse aortic constriction via ERK1/2 signalling. Pharm Biol. (2022) 60(1):562–9. doi: 10.1080/13880209.2022.2040543
89. Xu Z, Yang J, Hu Y, Wan Q, Wang X, Lu C, et al. Qifu yixin prescription ameliorates cardiac fibrosis by activating soluble guanylate cyclase (sGC) in heart failure. J Ethnopharmacol. (2025) 340:119229. doi: 10.1016/j.jep.2024.119229
90. Zeyu Z, Zhuangzhuang J, Yuwei S, Xuan Z, Ci W, Shuai W, et al. Optimized new Shengmai powder inhibits myocardial fibrosis in heart failure by regulating the rat sarcoma/rapidly accelerated fibrosarcoma/mitogen-activated protein kinase kinase/extracellular regulated protein kinases signaling pathway. J Tradit Chin Med. (2024) 44(3):448–57. doi: 10.19852/j.cnki.jtcm.20240402.004
91. Liu L, Luo F. miR-450a-2-3p targets ERK(1/2) to ameliorate ISO-induced cardiac fibrosis in mice. Genes Nutr. (2024) 19(1):16. doi: 10.1186/s12263-024-00753-6
92. Ge M, Zhang L, Du J, Jin H, Lv B, Huang Y. Sulfenylation of ERK1/2: a novel mechanism for SO2-mediated inhibition of cardiac fibroblast proliferation. Heliyon. (2024) 10(14):e34260. doi: 10.1016/j.heliyon.2024.e34260
93. Zhou H-H, Zhou X, Pei J, Xu S, Jin B, Chen J, et al. A fibrin gel-loaded gouqi-derived nanovesicle (GqDNV) repairs the heart after myocardial infarction by inhibiting p38 MAPK/NF-κB p65 pathway. J Nanobiotechnology. (2025) 23(1):535. doi: 10.1186/s12951-025-03615-4
94. Moradi A, Khoshniyat S, Nzeako T, Khazeei Tabari MA, Olanisa OO, et al. The future of clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 gene therapy in cardiomyopathies: a review of its therapeutic potential and emerging applications. Cureus. (2025) 17(2):e79372. doi: 10.7759/cureus.79372
95. Tao H, Chen ZW, Yang JJ, Shi KH. MicroRNA-29a suppresses cardiac fibroblasts proliferation via targeting VEGF-A/MAPK signal pathway. Int J Biol Macromol. (2016) 88:414–23. doi: 10.1016/j.ijbiomac.2016.04.010
96. Shen J, Xing W, Liu R, Zhang Y, Xie C, Gong F. MiR-32-5p influences high glucose-induced cardiac fibroblast proliferation and phenotypic alteration by inhibiting DUSP1. BMC Mol Biol. (2019) 20(1):21. doi: 10.1186/s12867-019-0135-x
97. Huang C, Wang R, Lu J, He Y, Wu Y, Ma W, et al. MicroRNA-338-3p as a therapeutic target in cardiac fibrosis through FGFR2 suppression. J Clin Lab Anal. (2022) 36(8):e24584. doi: 10.1002/jcla.24584
Keywords: MAPK, myocardial fibrosis, research progress, mechanism, drugs
Citation: Wu J, Chen M, Peng Z, Sun Y and Jin J (2025) Mechanism and research progress of MAPK signaling pathway in myocardial fibrosis. Front. Cardiovasc. Med. 12:1667568. doi: 10.3389/fcvm.2025.1667568
Received: 17 July 2025; Accepted: 10 November 2025;
Published: 21 November 2025.
Edited by:
Dongze Qin, Albert Einstein College of Medicine, United StatesReviewed by:
Nicola Pierucci, Sapienza University of Rome, ItalyQingyuan Yang, Shanghai Jiao Tong University, China
Copyright: © 2025 Wu, Chen, Peng, Sun and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Jin, amluanVhbl83MjNAMTYzLmNvbQ==