Abstract
Significant improvements in cancer survival rates have been achieved through advancements in treatment and early diagnosis. However, non-cancer-related mortality among cancer survivors continues to rise each year. Cardiovascular diseases (CVDs) related to cancer therapy now rank as the second leading cause of death in survivors, sometimes surpassing the cancer itself. Among these, cardiovascular aging represents one of the most severe side effects, often leading to detrimental structural changes such as cardiac atrophy or fibrosis, which ultimately impair cardiac function and reduce survival. Preventing or treating cardiovascular aging has emerged as a promising strategy to mitigate Cancer Therapy-Related Cardiovascular Toxicity (CTR-CVT). This review offers a comprehensive analysis of the characteristics and mechanisms underlying cancer therapy-induced accelerated cardiovascular cellular senescence, outlines current monitoring and intervention strategies, and explores future research opportunities and challenges. Enhancing the understanding of Cancer Therapy-Related Cardiovascular Cellular Senescence and Cancer Therapy-Related Cardiovascular Aging (CTR-CVA) is crucial for optimizing cancer treatment, advancing medical research, and improving clinical practice, all of which are vital for preserving cardiac health and improving the quality of life of patients with cancers.
1 Introduction
Advancements in treatment modalities and early precision diagnosis have significantly improved the survival rates of patients with cancers. According to the Cancer Research Institute, the number of cancer survivors in the United States grew from 18 million in 2022 to nearly 26 million by 2024 (1). Despite these gains, long-term survivors face the challenge of cancer treatment-related diseases, particularly cardiovascular diseases (CVDs). Cancer Therapy-Related Cardiovascular Toxicity (CTR-CVT), referring to a series of acute and chronic cardiovascular system injuries occurring after cancer treatment, has now emerged as the second leading cause of long-term morbidity and mortality among cancer survivors (2). Childhood cancer survivors (CCS) underwent chest radiotherapy are at a 10.6-fold increased risk of myocardial infarction and congestive heart failure later in life compared to their siblings (3). Consequently, exploration of the long-term underlying pathophysiological mechanisms, early prediction, diagnosis, and treatment of CTR-CVT remain key research priorities in the field of CTR-CVT (4). The establishment Cardio-Oncology marked the inception of this specialized discipline (5). Recent research has highlighted cellular senescence as a critical driver of Cancer Therapy-Related Cardiovascular Diseases (CTR-CVD). Senescent cardiovascular cells contribute to the remodeling of the extracellular matrix (ECM), disrupting normal cellular function and development, thereby increasing disease susceptibility (6, 7). This, in turn, elevates the risk of major adverse cardiovascular events (MACE) in the patients with cancers (7).
Aging is a process in which the overall functions of an organism gradually decline over time. As a key intrinsic driving mechanism underlying macroscopic aging, cellular senescence leads to the functional decline of tissues and organs and promotes the continuous progression of the aging process by inducing cell cycle arrest and senescence-associated metabolic changes. Cardiovascular aging is a physiological or pathological process characterized by structural degradation and functional decline of the cardiovascular system, with cellular senescence as its fundamental cytological basis, occurring under the influence of aging or pathological factors. With the continuous expansion and in-depth development of relevant research, the academic view that the arrest of the cellular senescence cycle is irreversible is gradually being questioned and revised (8–10), opening up a breakthrough for the intervention in cell senescence-related diseases. In recent years, the American Institute for Cancer Research has begun to pay attention to the potential value of eliminating senescent cells, including those in normal tissues (11). Previous articles have put forward the concept of the Aging-Cancer Cycle, suggesting that there is a two-way relationship between cancer treatment and aging (12). In 2024, articles in the JACC series proposed the existence of cardiovascular aging under tumor treatment (13). This type of cardiovascular aging, caused by the continuous accumulation of senescent cells in the cardiovascular system induced by cancer therapy, can be defined as Cancer Therapy-Related Cardiovascular Aging (CTR-CVA). It represents the long-term progression of acute or subacute injuries in CTR-CVT, as well as a branching manifestation of chronic injuries in CTR-CVT (Figure 1).
Figure 1
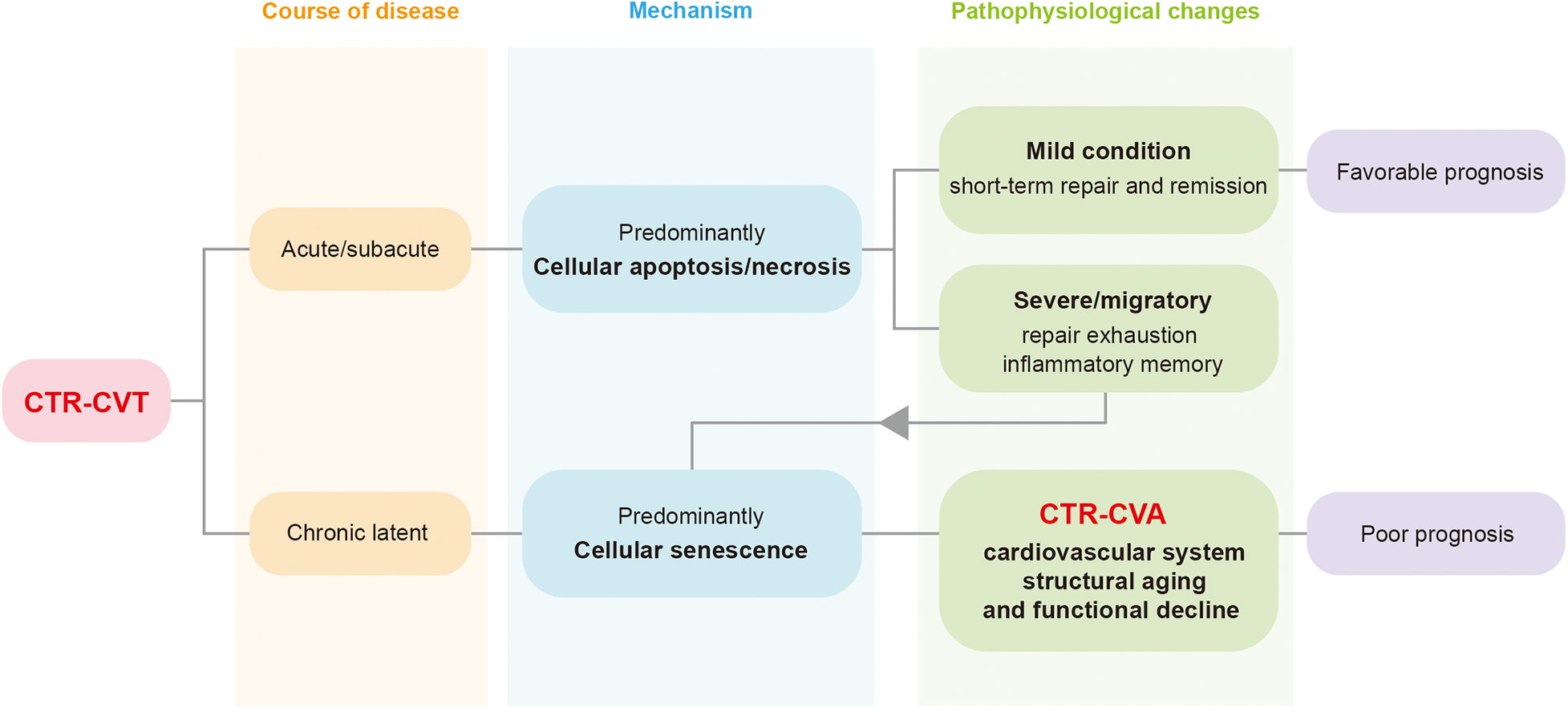
The relationship between Cancer Therapy-Related Cardiovascular Toxicity (CTR-CVT) and Cancer Therapy-Related Cardiovascular Aging (CTR-CVA). CTR-CVT is a collection of a series of acute and chronic injuries after cancer treatment. CTR-CVA essentially represents the long-term progression of acute/subacute injuries and the chronic phenotype of latent injuries in CTR-CVT. CTR-CVA is characterized by the accumulation of senescent cells, manifested as structural degeneration and functional decline of the cardiovascular system. CTR-CVT, Cancer Therapy-Related Cardiovascular Toxicity; CTR-CVA, Cancer Therapy-Related Cardiovascular Aging.
This review focuses on exploring the mechanisms of CTR-CVA, advancements in detecting and monitoring cardiovascular aging, and potential directions for future research. This article updates the existing reviews in this field, with a focus on the advanced imaging techniques currently used for identifying senescent cells and the popular therapeutic approaches. Strengthening interdisciplinary collaboration across Cardio-Oncology, materials science, molecular imaging, and artificial intelligence is crucial to support early diagnosis and intervention for CTR-CVT. In particular, effective treatments for cellular senescence could significantly improve the quality of life for cancer survivors (Figure 2).
Figure 2
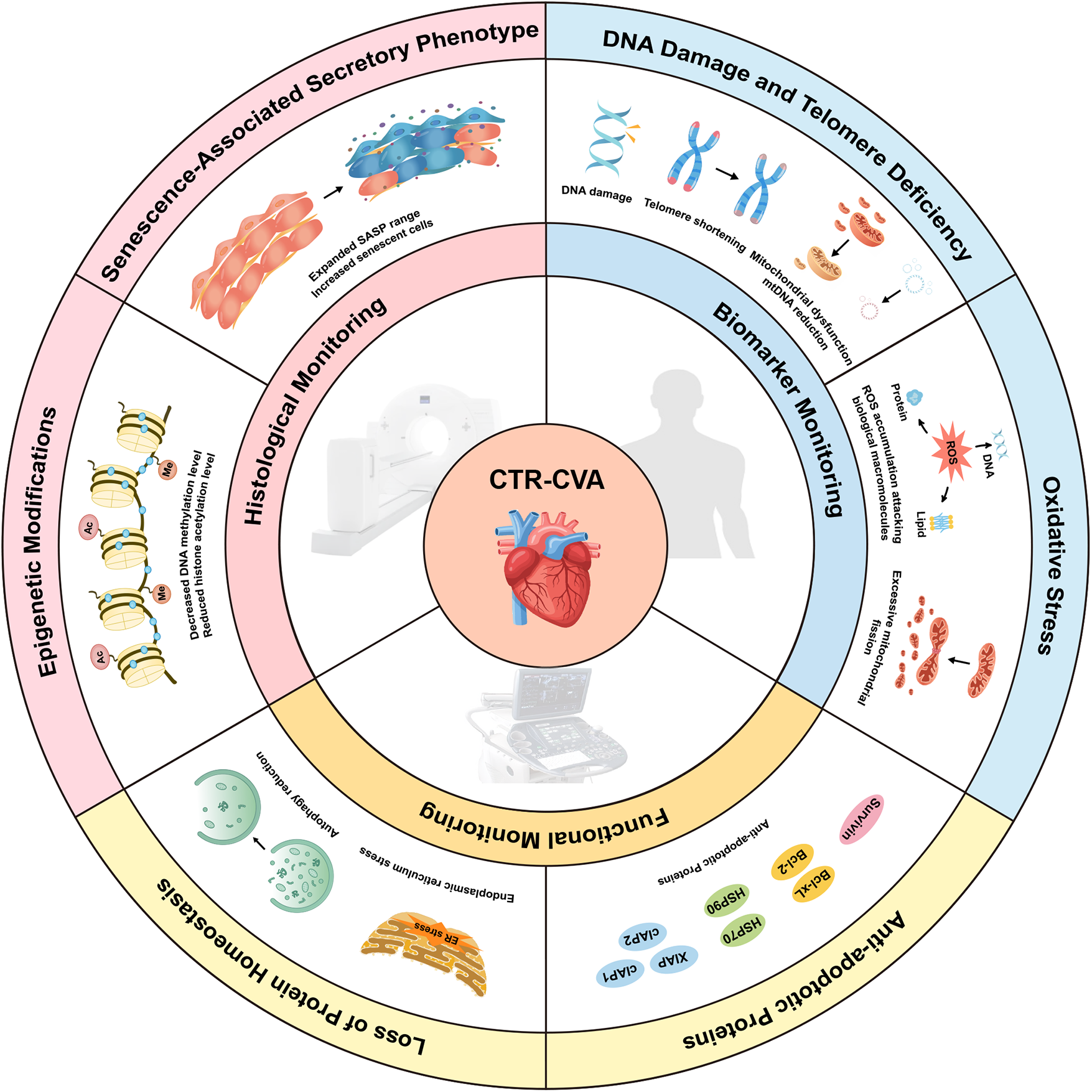
Mechanisms and Monitoring of Cancer Therapy-Related Cardiovascular Aging (CTR-CVA). CTR-CVA is based on cellular senescence and involves six mechanisms: DNA damage and telomere deficiency, oxidative stress, upregulation of anti-apoptotic proteins, disruption of protein homeostasis, epigenetic modifications, and the senescence-associated secretory phenotype. In this review, the monitoring of CTR-CVA is categorized into biomarker-based monitoring, functional monitoring, and histological monitoring. CTR-CVA, Cancer Therapy-Related Cardiovascular Aging; ER, endoplasmic reticulum; mtDNA, mitochondrial DNA; SASP, senescence-associated secretory phenotype.
2 Cancer therapy-related cardiovascular disease and cardiovascular aging
With the evolution of Cardio-Oncology, there is a growing understanding of the complexities surrounding CTR-CVD, leading to the development of comprehensive strategies for their diagnosis, prevention, therapy, and management (14). CTR-CVD encompasses a broad spectrum of diseases, including heart failure, arrhythmia, coronary artery disease (CAD), pericardial diseases, and valvular disorders (15, 16). Several factors influence the incidence of CTR-CVD: (i) Treatment modalities: patients treated with anthracyclines often experience severe cardiac adverse events, while liposomal doxorubicin (DOX) treatment tends to have fewer cardiac complications (17–19). (ii) Therapy-related drug accumulation and dosage cycles (20, 21): High-dose DOX is primarily associated with acute declines in cardiac function or an increased incidence of arrhythmias. In contrast, low-dose DOX is linked to long-term, atypical cardiac symptoms or cardiovascular dysfunction (22). The risk of heart failure increases with cumulative DOX dosage, with incidences of 5%, 26%, and 48% when the cumulative dose reaches 400 mg/m2, 550 mg/m2, and 700 mg/m2, respectively, with most cases manifesting within the first year of treatment (23). This study supplied the evidence that the cumulative DOX dose should not exceed 500–550 mg/m2 in clinical trials (20, 21). (iii) Early and delayed side effects: The onset and pattern of CTR-CVD symptoms varied according to the cancer treatment. Radiation-induced pericarditis typically occurs shortly after exposure, while other radiation-related heart diseases often emerge 10–15 years later (24). (iv) Individual differences: Even under the same therapeutic regimen, individuals may experience distinct manifestations. Elderly patients, particularly those undergoing combination therapies, are more prone to aging-related phenomena such as palpitations, fatigue, and elevated serum biomarkers indicative of injury (7). Epidemiological studies have shown that childhood cancer survivors experience a significantly higher incidence of severe cardiovascular adverse events compared to their siblings (25). Imaging and pathological assessments of these survivors reveal alterations in cardiovascular structure and function, including ventricular remodeling, myocardial fibrosis, and valvular calcification (26).
Cellular senescence is a key factor in the onset, progression, and outcomes of various diseases, influencing the entire pathophysiological process (27). Various cancer treatments can induce celluar senescence in healthy, non-cancerous cells. The accumulation of senescent cells plays a central role in driving premature tissue aging. In CTR-CVD, the importance of cardiovascular aging is increasingly recognized by researchers. Cardiovascular aging and cellular senescence can be detected in the peripheral blood (28), cardiac electrophysiology (29), or tissue specimens (30) of patients with cancers long after treatment (Table 1). Similar to natural aging, CTR-CVA can trigger tissue remodeling phenomena such as vascular wall thickening and cardiac fibrosis, accompanied by functional deterioration manifestations such as increased arterial stiffness and decreased cardiac diastolic function. Although some pathological features and changes in clinical indicators are similar to those of natural cardiovascular aging, CTR-CVA is often more severe. The essential difference between the two is that natural aging is a slow and progressive process that follows biological laws, while cancer treatments such as radiotherapy and chemotherapy can activate cellular senescence and related signaling pathways earlier through exogenous stimulation, thereby accelerating the process of cardiovascular aging. Therefore, Cardiovascular cellular senescence has become a key link in the pathogenesis of CTR-CVD (31), a comprehensive understanding of its mechanisms is essential for the prevention and treatment of CTR-CVT.
Table 1
| Sample types | Grouping of research subjects | Evidence of aging | Specific indicators | Literature results in detail | Year |
|---|---|---|---|---|---|
| Patient Cardiac Specimens | Treatment group: Autopsy cardiac specimens of 6 tumor patients who had received DOX treatment and died of congestive heart failure; Control group: Autopsy cardiac specimens of 6 patients with non-cardiovascular causes of death | Cell cycle arrest and cardiac fibrosis | p16INK4a, γ-H2AX, Masson trichrome staining area | In the cardiac progenitor cells (CPCs) of the cardiomyopathy cardiac specimens exposed to DOX treatment, the expressions of p16INK4a and γ-H2AX increased | 2013 (30) |
| Patient Blood Specimens | 10 blood samples at different time points from patients receiving combination chemotherapy of DOX and cyclophosphamide | Decrease in circulating endothelial progenitor cells (CEPs) | CEPs positive for CD-133 and Vascular Endothelial Growth Factor Receptor-2 (VEGFR-2) | In patients exposed to DOX and cyclophosphamide, the number of serum CD-133 and VEGFR-2 positive CEPs showed a linear downward trend from baseline to the 12th week | 2013 (32) |
| Cross-sectional survivor cohort: 176 female survivors aged 50 and above who had undergone surgical resection for stage I-III breast cancer, with 69 receiving adjuvant chemotherapy and the rest not; Prospective adjuvant treatment cohort: 33 female patients diagnosed with stage I-III breast cancer and planned to receive adjuvant chemotherapy | Cell cycle arrest and increased SASP expression | p16INK4a and Alternative Reading Frame (ARF), Interleukin-6 (IL-6), Interleukin-7 (IL-7), Interleukin-8 (IL-8), Vascular Endothelial Growth Factor A (VEGFA), and Monocyte Chemoattractant Protein-1 (MCP1) | In the peripheral blood T lymphocytes and serum samples of breast cancer survivors who had received adjuvant chemotherapy, the expressions of p16INK4a and ARF increased, and the expressions of multiple SASP factors in the serum were significantly higher than those in patients who had not received adjuvant chemotherapy | 2,014 (33) | |
| Treatment group: 87 asymptomatic childhood acute lymphoblastic leukemia (ALL) survivors, of which 48.3% had received combined treatment of chemotherapy and radiotherapy, and the rest had only received chemotherapy; Control group: 87 healthy volunteers | Telomere shortening and increased SASP expression | Telomere length, Interleukin-2 (IL-2), Interleukin-10 (IL-10), Interleukin-17a (IL-17a), High-sensitivity C-reactive protein (hsCRP) | The peripheral blood leukocyte telomere length of ALL survivors who had received chemotherapy or combined treatment of chemotherapy and radiotherapy was significantly shorter than that of the control group, and the levels of multiple inflammatory factors in the plasma increased. Among them, the telomeres of patients who had received radiotherapy were shorter than those of survivors who had only received chemotherapy | 2017 (34) | |
| 15 post-menopausal female breast cancer survivors, Non-lateralized cerebral oxygenation group: The difference in cerebral oxygenation between the left and right hemispheres was less than 3%; Lateralized cerebral oxygenation group: The difference in cerebral oxygenation between the left and right hemispheres was ≥ 3% | Decline in cerebral cognitive function and increased SASP expression | IL-6, Tumor Necrosis Factor α (TNF-α), CRP, and Insulinlike Growth Factor-1 (IGF-1) | The increase in the levels of SASP and other factors and the decrease in the level of IGF-1 in the peripheral blood samples of breast cancer patients who had received chemotherapy indicated accelerated cerebrovascular aging, which led to the decline in cerebral cognition | 2018 (35) | |
| Treatment group: 72 newly-diagnosed stage 0-IIIA breast cancer patients after treatment, of which 35 had received combined treatment of chemotherapy and radiotherapy, and the remaining 37 had only received radiotherapy; Control group: The above 72 newly-diagnosed stage 0-IIIA breast cancer patients before treatment | DNA methylation and epigenetic changes | Extrinsic Epigenetic Age Acceleration (EEAA), Phenotypic Epigenetic Age Acceleration (PEAA), and Grim Epigenetic Age Acceleration (GEAA) | In the DNA methylation detection results of the peripheral whole blood samples of breast cancer patients after radiotherapy and chemotherapy, it was observed that indicators such as EEAA, PEAA, and GEAA all increased | 2020 (28) | |
| 55 blood samples from patients with metastatic testicular cancer before and after treatment with cisplatin | Telomere shortening | Telomere length | The peripheral blood leukocyte telomeres of 7/55 patients with metastatic testicular cancer shortened one year after cisplatin treatment | 2024 (36) | |
| Patient Electrocardiogram Analysis | Group1: 15 and 22 childhood survivors who had only received anthracycline-based treatment of ≥300 mg/m2 and <300 mg/m2 respectively; Group2: 15 and 22 childhood survivors who had received thoracic radiotherapy and anthracycline-based treatment of ≥300 mg/m2 and <300 mg/m2 respectively | Prolongation of QT interval | Corrected QT interval (QTc) | The incidence of QTc prolongation (≥ 0.43) in children cancer patients who received higher doses of anthracyclines was higher than that in the low-dose group, and chest radiotherapy may have a synergistic effect. | 1993 (29) |
Clinical evidence associated with cancer therapy-related cardiovascular aging.
3 Mechanisms of cancer therapy-related cardiovascular aging
Cellular senescence is the key underlying mechanism of tissue aging. Cellular senescence is characterized by a permanent proliferation arrest in response to both endogenous and exogenous stress or damage, triggering a range of intracellular phenotypic changes (37–39). The hallmarks of cellular senescence include cell cycle arrest, activation of the DNA damage response (DDR), nuclear alterations, upregulation of anti-apoptotic factors, the senescence-associated secretory phenotype (SASP), metabolic adaptation, morphological changes, increased lysosomal content, and the expression of specific cell surface markers (31, 39). Recent studies have also identified the accumulation of immunoglobulins as an important marker of senescence (40). Under prolonged stress or disease conditions, the continuous accumulation of senescent cells will lead to a decline in essential repair functions and immune clearance capabilities, alterations in intercellular communication, changes in the extracellular matrix (41), chronic inflammation, and so on. This will continuously accelerate the accumulation of cellular senescence and form a negative feedback loop, further exacerbating tissue aging. Baker et al. developed an “Inducible and Regulatable Targeted Apoptosis of Senescent Cells (INK-ATTAC)” system using transgenic technology, which targets cells expressing p16INK4a (42). This technique allowed for the anatomical localization of senescent cells in the kidneys of aged mice and demonstrated significant efficacy in their targeted clearance. By eliminating senescent cardiac cells with AP20187, cardiomyocyte hypertrophy in aged mice was alleviated without compromising heart function (42). These findings provided valuable insight into the link between cellular senescence and cardiac aging. Similar studies lay the theoretical groundwork for the targeted clearance of senescent cells as a strategy to mitigate CTR-CVT (43). Therefore, the mechanisms underlying CTR-CVA, particularly those induced by conventional chemotherapy and radiotherapy, will be discussed in relation to markers of cellular senescence in the following sections (Figure 3).
Figure 3
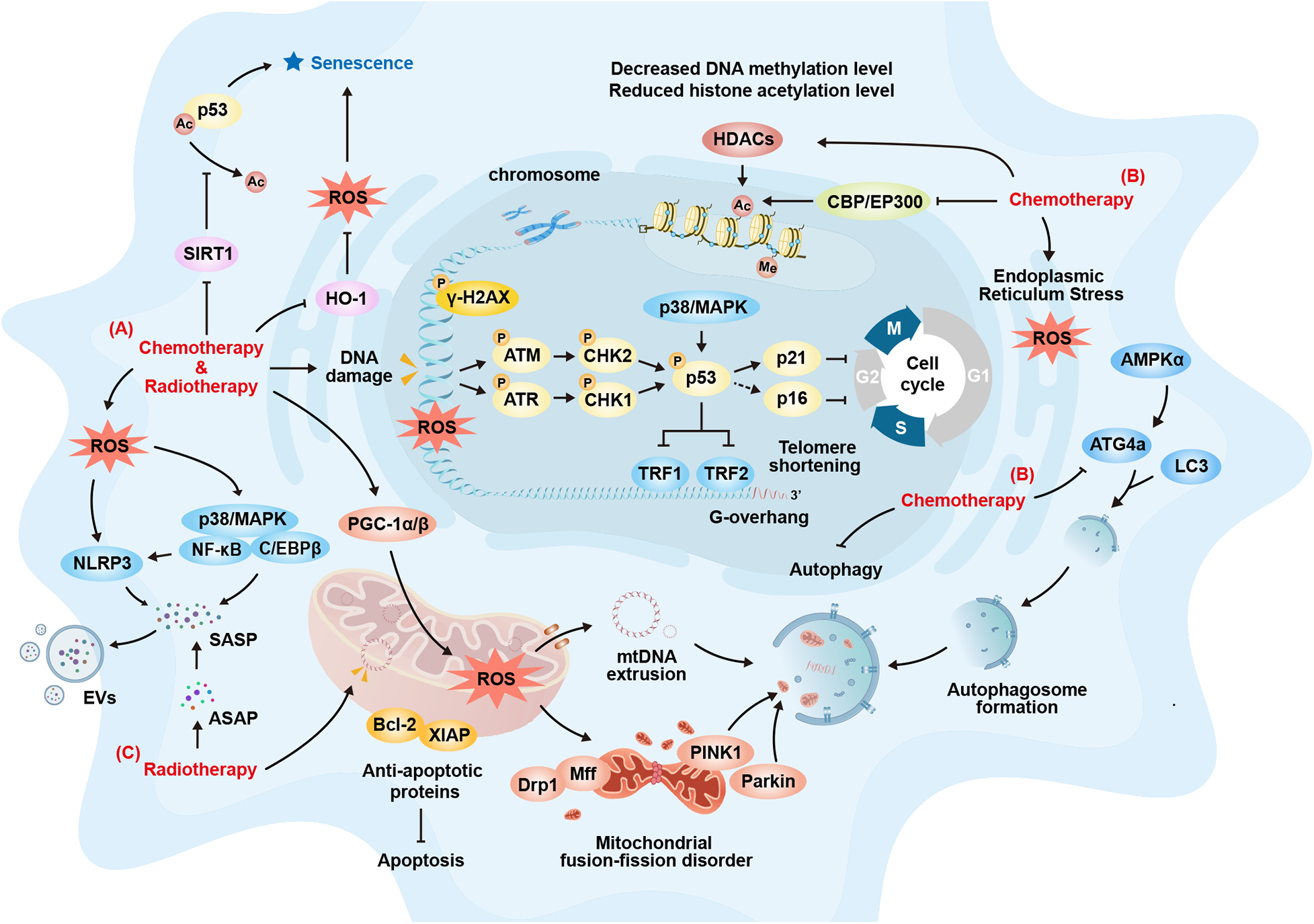
Cardiovascular Cellular Senescence Signaling during Chemotherapy and Radiotherapy. (A) Common Mechanisms of Cellular Senescence Induced by Chemotherapy and Radiotherapy: Both can induce DNA damage and abnormal repair, activating classical cellular senescence pathways such as the p53-p21 and p16-Rb pathways, which promotes cell-cycle arrest, leading cells to enter a state of cellular senescence; Both can induce oxidative stress related cellular senescence by activating the P38/MAPK-NFκB pathway or the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome; Both can result in the secretion of various cytokines through pro-inflammatory reactions, causing the accumulation and spread of the senescence-associated secretory phenotype (SASP), thus inducing cellular senescence in adjacent cells; Both can increase the expression of anti-apoptotic proteins such as Bcl-2 and X-linked inhibitor of apoptosis protein (XIAP) to counteract damage. Cells then enter a state of cellular senescence to avoid excessive apoptosis; Both can affect the mitochondrial oxidative respiratory chain, causing the accumulation of ROS within mitochondria, which exacerbates cellular senescence. (B) Other Mechanisms of Cellular Senescence Induced by Chemotherapy: Chemotherapy can promote telomere shortening and cellular senescence by downregulating the expression of telomeric repeat-binding factor 1 (TRF1) and telomeric repeat-binding factor 2 (TRF2); Chemotherapy can induce cellular senescence by promoting endoplasmic reticulum dysfunction, leading to protein homeostasis imbalance and exacerbating endoplasmic reticulum stress; Chemotherapy can promote cellular senescence by reducing DNA methylation; Chemotherapy can promote cellular senescence by upregulating histone deacetylases (HDACs), downregulating histone-acetylation-regulating genes such as CBP and Ep300, and thus decreasing the level of histone acetylation. It can also induce senescence by inhibiting SIRT1 to activate p53. (C) Other Mechanisms of Cellular Senescence Induced by Radiotherapy: Radiotherapy can impact mitochondrial DNA (mtDNA), affecting normal biosynthesis and promoting cellular senescence; Radiotherapy can trigger the release and maintenance of short-term acute-stress-associated phenotypes (ASAP). This prolongs the duration of inflammatory stimulation and gradually transforms it into SASP, promoting cellular senescence and its spread. ATG4a, autophagy-related gene 4a; ATM, ataxia-telangiectasia mutated; ASAP, acute stress-associated phenotype; DRP1, dynamin-related protein 1; EVs, extracellular vesicles; HDACs, histone deacetylases; HO-1, heme oxygenase-1; MFF, mitochondrial fission factor; mtDNA, mitochondrial DNA; NLRP3, NOD-like receptor family pyrin domain containing 3; PGC-1α/β, peroxisome proliferator-activated receptor gamma co-activator 1-Alpha/Beta; PINK1, PTEN-induced putative kinase 1; ROS, reactive oxygen species; SASP, senescence-associated secretory phenotype; SIRT1, sirtuin 1; TRF1/2, telomeric repeat-binding factor 1/2; XIAP, X-linked inhibitor of apoptosis protein.
3.1 Mechanisms of anthracycline-induced cardiovascular aging
Anthracyclines are first-line chemotherapy agents used to treat various malignancies, including breast cancer, lymphoma, and acute leukemia (44). DOX, epirubicin, and daunorubicin belong to the anthracycline. Among these, DOX is the earliest discovered and most widely used. Unfortunately, the cardiovascular adverse effects of DOX, a first-generation anthracycline, have been recognized in a dose-dependent manner since 1976 (45, 46). The molecular mechanisms underlying DOX-induced cardiac senescence are complex, involving DNA damage and telomere deficiency, oxidative stress, upregulation of anti-apoptotic proteins, loss of protein homeostasis, epigenetic modifications, and SASP (Table 2). It is important to note that in current cellular and animal experiments designed to establish stable cellular senescence models, the drug dosages employed in most studies—after relevant conversion—exceed the clinical threshold (<500–550 mg/m2). Consequently, the experimental results may not accurately recapitulate the authentic pathological conditions and underlying mechanisms of cardiovascular cellular senescence induced by cancer therapy in clinical practice.
Table 2
| Treatment modalities | Experimental subjects | Treatment protocols | Detection of senescence | Mechanisms | Year |
|---|---|---|---|---|---|
| Anthracycline Drugs | In vitro: Isolated neonatal rat cardiomyocytes | In vitro: Treatment with 10−7 mol/L DOX for 7 days | Increased SA-β-gal activity; Decreased in telomerase activity; Shortened telomere length; Increased protein levels of P27kip1 and p21cip1/waf1; Increased mRNA levels of p16INK4a | The activation of the PML-acetylated p53 complex | 2008 (47) |
| In vivo: 4-week-old male Wistar rats | In vivo: Intraperitoneal administration of DOX, divided into six equal injections (2.5 mg/kg each) over 2 weeks, with a total cumulative dose of 15 mg/kg body weight, with sample collection for monitoring at 11 months of age | ||||
| In vitro: Isolated Ventricular Myocytes from 2-Day-Old Sprague-Dawley Rats (Lonza) and H9c2 Rat Cardiomyoblasts | In vitro: Treatment with 0.1 μM DOX for 3 h, followed by assessment at 24 and 48 h | Increased SA-β-gal activity; Cellular senescence morphology; Increased micronuclei; Chromosomal abnormalities; Increased protein levels of p53 | The downregulation of TRF1 and TRF2 via MAPK and p53-mediated pathways | 2009 (48) | |
| In vitro: Isolated c-kit positive human cardiac progenitor cells | In vitro: Treatment with 0.1, 0.5, and 1.0 μM DOX for 24 and 48 h, followed by 7 days of culture | Increased SA-β-gal activity; Increased tissue immunofluorescence labeling of p16INK4a and γ-H2AX; Decreased expression of BrdU and Ki67; Activation of DNA damage pathways | The activation of the p16-Rb pathway-triggered cell cycle arrest | 2013 (30) | |
| In vivo: Human cardiac autopsy specimens | In vivo: Standard clinical doses for cancer patients | ||||
| In vitro: Human primary umbilical artery vascular smooth muscle cells (VSMCs) and mouse aortic VSMCs. | In vitro: Treatment with 0.25, 0.5, and 1 µM DOX for 3 h, followed by replacement with normal culture medium and monitoring after 3 days | Increased SA-β-Gal activity; Decreased cell viability; Increased protein levels of p53, p21Cip1/Waf1, and p16INK4a | The upregulation of uPAR-triggered TRF2 ubiquitination and proteasomal degradation | 2013 (49) | |
| In vitro: Human vascular smooth muscle cells | In vitro: Treatment of passages 5–8 cells with 100 nM DOX to induce premature senescence, followed by analysis at 1, 3, and 7 days | Increased SA-β-gal activity; Cellular senescence morphology; Cell cycle arrest; Increased DNA damage-associated 53BP1 foci; Increased micronuclei formation; Increased γ-H2AX levels; Increased protein levels of Increased p21cip1/waf1 protein levels; Increased expression of SASP; Increased superoxide production; DNA methylation inhibition | The Activation of DNA Damage Repair-related ATM Pathway-coupled with Superoxide Level Accumulation | 2014 (50) | |
| In vitro: H9c2 cells and isolated primary cardiomyocytes from mice. | In vitro: Pre-treatment with 0.1 μmol/L DOX for 3 h, followed by sample collection after 21 h, 45 h, 7 days, 14 days, and 21 days in normal culture medium | Increased SA-β-gal activity; Increased protein levels of p53, and p16INK4a | The downregulation of TRF2-induced reduction in telomere protection | 2016 (51) | |
| In vivo: 4-week-old male Wistar rats | In vivo: Subcutaneous injection of DOX at 2 mg/kg once weekly for 7 weeks, total cumulative dose of 14 mg/kg, with sample collection in week 9 | Reduced mitochondrial DNA (mtDNA); Decreased DNA methylation levels | The depletion of mtDNA-coupled with DNA methylation reduction | 2017 (52) | |
| In vitro: HL-1 mouse cardiomyocytes | In vitro: Treatment with 5 µM DOX for 0, 24, 48, and 72 h | Decreased cell viability; Shortened telomere length; Decreased in telomerase activity; Elevated intracellular ROS; Elevated mitochondrial superoxide; Increased mRNA levels of p53, p27Kip1, and p16INK4a | The increase in lincRNA-p21 expression-coupled with Wnt/β-catenin Signaling Pathway Activation and ROS-related Indicator Increase | 2018 (53) | |
| In vitro: H9c2 cells | In vitro: Treatment with 0.5 µM DOX for 72 h | Decreased cell viability; Increased mRNA levels of p53 and p16INK4a; Shortened telomere length; Decreased in telomerase activity | The Elevation of TGF-β1 Expression | 2018 (54) | |
| In vitro: H9c2 rat embryonic cardiomyocytes | In vitro: Treatment with 0.1 µM DOX for 24 h | Increased SA-β-gal activity | The miR-34a/PNUTS axis | 2019 (55) | |
| In vitro: H9c2 cells | In vitro: Treatment with 0.1 µM DOX for 48 h | Increased SA-β-gal activity; Decreased cell viability; Increased protein levels of p21cip1/waf1 and p16INK4a; Increased expression of SASP; Elevated intracellular ROS | The upregulation of TXNIP-triggering intracellular redox imbalance and NLRP3 inflammasome activation | 2020 (56) | |
|
In vitro: Isolated neonatal rat cardiomyocytes and H9c2 cells; In vivo: 3–4 week old Wistar rats |
In vitro: Treatment with 50 nM DOX for 3 h, followed by monitoring after 7 days; In vivo: Treatment with DOX through intraperitoneal injections 9 times every other day at 5 mg/kg, with a total cumulative dose of 45 mg/kg, collect samples after 8 months |
Increased SA-β-gal activity; Reduced mtDNA; Increased numbers of p16INK4a positive cells and myosin positive cells under flow cytometry. | The damage of mtDNA | 2020 (57) | |
| In vitro: Cardiomyocytes differentiated from human induced pluripotent stem cells (iPSC) | In vitro: Treatment with 0.5 μM DOX for 24 h | Increased SA-β-gal activity; Cell cycle arrest; Increased mRNA levels of p53 and p21cip1/waf1 | The upregulation of miR-92a-3p and inhibition of ATG4a expression-induced attenuation of mitochondrial metabolism | 2020 (58) | |
|
In vitro: Isolated mouse ventricular myocytes; In vivo: eight-week-old male C57BL/6 mice |
In vitro: Treatment with 1 μM DOX for 72 h; In vivo: Intraperitoneal injection of DOX was carried out on Mondays, Wednesdays and Fridays within one week, at a dose of 4 mg/kg each time, with a total cumulative dose of 12 mg/kg. Sample collection for monitoring was initiated on the 14th day |
Increased SA-β-gal activity; Increased mRNA levels of p27kip1, p16INK4a, and p21cip1/waf1 | The activation of the miR-221–3p/Sirt2 pathway | 2020 (59) | |
| In vitro: Isolated neonatal mouse cardiomyocytes | In vitro: Treatment with 1 μM DOX for 24 h and 7 days | Increased SA-β-gal activity; Increased γ-H2AX foci; Decreased cell viability; Increased protein levels of p53 and p21cip1/waf1; Elevated mitochondrial ROS; Elevated intracellular ROS; Increased expression of SASP | The downregulation of mitochondrial autophagy via TBK1 K63-linked polyubiquitination facilitation | 2021 (60) | |
| In vitro: H9c2 cells | In vitro: Treatment with 0.5 µM DOX for 24 h, followed by replacement with normal culture medium and monitoring after 10 days | Increased SA-β-gal activity; Decreased cell viability; Shortened telomere length; Increased intracellular ROS and mitochondrial superoxide; Increased mRNA levels of p53 and p21cip1/waf1 | The depletion of Sirt6-caused mitochondrial damage, telomere dysfunction, increased H3K9 acetylation and upregulated NF-κB-related oxidative stress | 2021 (61) | |
|
In vitro: H9c2 cells; In vivo: C57BL/6 mice |
In vitro: Pre-treatment with 0.1 µM DOX for 3 h, followed by replacement with normal culture medium and monitoring after 0, 24, 48 and 72 h; In vivo: Intraperitoneal injections at 2.5 mg/kg, three times a week for two consecutive weeks, with a cumulative dose of 15 mg/kg, collect samples for monitoring 4 months after completion of injections |
Increased SA-β-gal activity; Decreased cell viability; Increased protein levels of p53, p21Cip1/Waf1, and p16INK4a; Increased expression of SASP | The p38 MAPK-Redd1-NF-κB pathway | 2021 (62) | |
| In vitro: H9c2 rat embryonic cardiomyocytes and AC16 human cardiomyocyte-like cells | In vitro: Treatment with 0.1 μmol/l DOX for 24 h | Increased SA-β-gal activity; Shortened telomere length; Decreased in telomerase activity; Increased protein levels of p53, p21cip1/waf1, p16INK4a, and IGFBP3 | The upregulation of C5a and C5aR-induced elevation of TNF-α and IFN-γ expression and ROS level | 2021 (63) | |
|
In vivo: Human ventricular cardiac fibroblasts (HCF), human umbilical vein endothelial cells (HUVECs); In vitro: 6-week-old wild-type BALB/c female mice |
In vivo: Treatment with 100 nM DOX for 7 days, followed by replacement with serum-free medium and monitoring after 2 days; In vitro: Inject dox dissolved in 0.9% saline subcutaneously every other day starting on the 7th day after tumor formation, with a cumulative dose of 22 mg/kg, collect samples after 3 weeks |
Increased protein levels of p21cip1/waf1 and p16INK4a | / | 2022 (64) | |
| In vitro: H9c2 cells | In vitro: Pre-treatment with 0.3 μM DOX for 24 h | Increased SA-β-gal activity; Decreased cell viability; Shortened telomere length; Increased intracellular ROS and mitochondrial superoxide; Increased mRNA levels of p53 and p21cip1/waf1 | The Klotho/SIRT1 signaling pathway. | 2022 (65) | |
| In vitro: Immortalized human umbilical vein endothelial cell line EA.hy926 and primary HUVECs | In vitro: Treatment with 0.5µM DOX for 24 h, followed by replacement with normal culture medium and monitoring after 72 h and 5 days | Increased SA-β-gal activity; Cell cycle arrest; Increased protein levels of p53 and p21cip1/waf1; Increased expression of SASP; | The promotion of anti-apoptotic protein expression | 2022 (66) | |
|
In vitro: Cardiomyocytes differentiated from iPSC (iCM) and isolated mouse cardiomyocytes; In vivo: Cardiac tissue transcriptomics data |
In vitro: Treatment with sub-lethal concentration of 0.2 μM DOX for 3 h, followed by replacement with normal culture medium and monitoring after 4 days | Increased SA-β-gal activity; Cell cycle arrest; Increased γ-H2AX positive nuclear foci; Increased mRNA and protein levels of p21cip1/waf1 and p16INK4a; Increased expression of SASP; Increased cell size; Elevated intracellular ROS | The loss of mitochondrial membrane potential-coupled with ROS increase | 2022 (67) | |
| In vitro: HL-1 cardiomyocytes |
In vitro: Treatment with 100 nM DOX for 72 h; In vivo: Administer a total cumulative dose of 20 mg/kg intraperitoneally in 8 injections over 4 weeks at 2.5 mg/kg per injection, and collect samples on the 44th day |
Increased SA-β-gal activity; Cell cycle arrest; Decreased Ki67 proliferation; Increased mRNA and protein levels of p53 and p21cip1/waf1 | The promotion of anti-apoptotic protein expression | 2022 (68) | |
| In vivo: 10-week-old male and female C57BL/6 J mice | |||||
| In vitro: HUVECs | In vitro: Treatment with 100 nM DOX for 24 h. | Increased SA-β-gal activity; Decreased cell proliferation; Increased protein levels of p21cip1/waf1 and p16INK4a | The ALDH1A2/AKT/ERK1/2-p21 pathway | 2022 (69) | |
| In vitro: HUVECs and EA.hy926 human endothelial-derived cell line | In vitro: Treatment with 0.5 μM DOX for 24 h, followed by replacement with normal culture medium and monitoring after 72 and 120 h | Increased SA-β-gal activity; Increased mRNA levels of p53 and p21cip1/waf1 | The activation of MAPK and JNK pathways-coupled with NF-κB-related oxidative stress and SASP | 2023 (70) | |
|
In vitro: H9c2 cells; In vivo: 8-week-old C57BL/6 female mice |
In vitro: Treatment with 0.1 μM DOX for 6 days; In vivo: Intraperitoneal injections at 3 mg/kg, once daily for 7 days, with a cumulative dose of 21 mg/kg, collect samples for monitoring on the 9th day |
Increased SA-β-gal activity; Decreased cell viability; Increased mRNA levels of p53 and p16INK4a; Increased expression of SASP; Elevated intracellular ROS | The promotion of mTOR protein phosphorylation | 2023 (71) | |
|
In vitro: Isolated neonatal mouse cardiomyocytes (NMCM); In vivo: 6–8-week-old ICR mice |
In vitro: Treatment with 1 μM DOX for 72 h; In vivo: Treat ICR mice with intraperitoneal injections of DOX at 3 mg/kg, 6 times within two weeks, with a total cumulative dose of 18 mg/kg, collect samples on the 35th day |
Increased SA-β-gal activity; Increased mRNA and protein levels of p21cip1/waf1 and p16INK4a; Increased mitochondrial fragmentation | The activation of the VPO1/ERK pathway-triggering enhanced mitochondrial fission | 2024 (72) | |
|
In vitro: AC16 cells; In vivo: C57BL/6 mice |
In vitro: Treatment with 1.25 μM, 2.5 μM, and 5 μM DOX for 24 h; In vivo: Intraperitoneal injections at 2.5 mg/kg, three times a week for two consecutive weeks, with a cumulative dose of 15 mg/kg, collect samples for monitoring 4 months after completion of injections |
Increased SA-β-gal activity; Decreased cell viability; Increased mRNA and protein levels of p27kip1, p16INK4a, and p21cip1/waf1; Increased expression of SASP | The suppression of CRIF1 expression and promotion of PXDN expression-facilitating mitochondrial fission and oxidative stress | 2024 (73) | |
|
In vitro: H9c2 cells; In vivo: 8-week-old male C57/Bl6 mice |
In vitro: Pre-treatment with 1 μM DOX for 24 h; In vivo: Single intraperitoneal injection of 20 mg/kg DOX, collect samples for monitoring after 2 weeks |
Increased SA-β-gal activity; Increase γ-H2AX foci; Increased mRNA levels of IGFBP3, p21cip1/waf1 and p16INK4a; Increased expression of SASP; Elevated intracellular ROS | The upregulation of PARP-2 expression, suppression of SIRT1 expression and activity, and activation of the FOXO1/p53 signaling pathway | 2024 (74) | |
|
In vitro: H9c2 cells; In vivo: C57BL/6 mice |
In vitro: Treatment with 1.0 μM DOX for 0, 12, 24, and 48 h; In vivo: single intraperitoneal injection of 20 mg/kg, collect samples for monitoring after two weeks |
Increased SA-β-gal activity; Increase γ-H2AX foci; Decreased cell viability; Increased mRNA levels of p53, p21cip1/waf1 and p16INK4a; Increased expression of SASP; Increased superoxide dismutase (SOD) activity; Elevated intracellular ROS | The depletion of SIRT6 and downregulation of PPARα | 2024 (75) | |
| In vivo: C57BL/6 mice | In vivo: Administer a total cumulative dose of 24 mg/kg intraperitoneally in 6 injections over 6 weeks at 4 mg/kg per injection, and collect samples after 4 days | Increased mRNA levels of p53, p21cip1/waf1, p16INK4a and p19Arf; Increased expression of SASP; | The activation of the p38/MAPK signaling pathway | 2024 (76) | |
| In vitro: Human cardiac organoids (hCOs or hCardioids) | In vitro: Treatment with 0.5 μM DOX for 3 and 32 days; | Decreased expression of Ki67; Increased mRNA levels of p15, p21cip1/waf1, p16INK4a and p19Arf; Elevated intracellular ROS | The activation of the oxidative stress pathway | 2025 (77) | |
|
In vitro: Cardiomyocytes differentiated from human induced pluripotent stem cells (iPSC); In vivo: C57BL/6 mice |
In vitro: Treatment with 1.0 μM DOX for 24 h; In vivo: Administer a total cumulative dose of 20 mg/kg intraperitoneally in 4 injections over 4 weeks at 5 mg/kg per injection, and collect samples at the end of the 6th week |
Increased SA-β-gal activity; Increased mRNA and protein levels of p53, p16INK4a, and p21cip1/waf1; Elevated mitochondrial ROS | The leakage of mtRNA and the activation of the cGAS-STING pathway | 2025 (78) | |
| Radiotherapy | In vitro: Bovine Aortic Endothelial Cells (BAECs) and HUVECs | In vitro: Sample collection 3–5 days after 8 Gy ionizing radiation | Increased SA-β-gal activity; Cellular senescence morphology; Cell cycle arrest; Increased γ-H2AX levels; Increased expression of SASP | The damage of DNA | 2007 (79) |
| In vitro: HL-1 mouse cardiomyocytes and H9c2 rat cardiomyocytes | In vitro: Sample collection at 72 h and 96 h post-exposure to 0, 2, and 8 Gy ionizing radiation | Increased SA-β-gal activity; Decreased cell proliferation; Cellular senescence morphology; Elevated intracellular ROS | The accumulation of ROS | 2015 (80) | |
| In vitro: Human aortic endothelial cells | In vitro: Sample collection at 4 and 6 days post-irradiation with unknown Gy dose | Increased SA-β-gal activity; Decreased cell proliferation; Cellular senescence morphology; Increased γ-H2AX levels; Increased protein levels of p53, p21Cip1/Waf1, and p16INK4a | The increase of GDF15 expression, ROS accumulation and activation of ERK signaling pathway along with p16/Rb pathway | 2016 (81) | |
| In vitro: Human cardiomyocytes (HCMs) | In vitro: Sample collection at 24, 48, and 72 h post-exposure to 5 Gy ionizing radiation | Increased SA-β-gal activity; Decreased cell proliferation; Increased mRNA levels of p21Cip1/Waf1 and p16INK4a; Elevated intracellular ROS | The inhibition of SIRT1, upregulation of miR-34a expression and induction of oxidative stress | 2018 (82) | |
| In vivo: Carotid arteries of apolipoprotein E knockout (ApoE−/−) mice | In vivo: After 2 weeks of inducing atherosclerotic lesions by ligating the left carotid artery, perform 6Gy whole-body irradiation on mice, collect samples after 4 weeks | Increase DNA damage-related 53BP1 foci; Increased γ-H2AX levels; Increased protein levels of p16INK4a; Increased mRNA levels of p21Cip1/Waf1 and p16INK4a | The DNA damage-coupled with SASP and other inflammatory factors | 2021 (83) |
Mechanisms associated with cancer therapy-related cardiovascular aging.
3.1.1 DNA damage and telomere deficiency
DOX exerts its cardiotoxicity effects by intercalating base pairs in DNA or forming a ternary complex with topoisomerase II and DNA, which leads to DNA double-strand breaks and replication arrest—key mechanisms contributing to cardiotoxicity, cell cycle arrest, and cellular senescence (48, 84–86). Abnormalities in the DDR also play a role in DOX-induced senescence (87). Low-dose DOX activates the DDR, recruits the ataxia-telangiectasia mutated (ATM) protein, and triggers the p53-p21 pathway, ultimately inducing cellular senescence.
As telomeres shorten with each cell division, they eventually reach a critical length that triggers entry into cellular senescence (14). DOX exposure accelerated this process by aggravating telomere attrition. Telomere attrition, a progressive loss of protective caps at chromosome ends, is another key factor in DOX-induced cardiomyocyte senescence. A low dose of 0.1 µM DOX downregulates the expression of telomeric repeat-binding factor 1 (TRF1) and telomeric repeat-binding factor 2 (TRF2) through MAPK and p53-mediated pathways, impairing telomere protection and exacerbating telomere damage and cellular senescence (48, 51). Moreover, telomere attrition activates the DDR, culminating in senescence in neonatal rat cardiomyocytes. In contrast, high doses of DOX promote early cardiomyocyte apoptosis by reducing TRF2 levels and increasing TRF1 expression (49). Similar results have been observed in senescent smooth muscle cells.
3.1.2 Oxidative stress
Redox reactions are central to fundamental biological processes in humans. According to the “Free Radical Theory of Aging,” aging is primarily driven by the overactivation of free radical reactions or excessive accumulation of ROS (88). The overaccumulation of ROS can damage biological macromolecules, such as lipids and proteins, leading to oxidative stress, cell senescence, and ultimately organ failure. In a DOX-induced mouse model of cardiomyopathy, cardiomyocyte senescence was linked to the upregulation of thioredoxin-interacting protein (TXNIP), which inhibited thioredoxin activity and disrupted intracellular antioxidant balance (56). DOX-treated HL-1 cardiomyocytes exhibited features of cellular senescence, including reduced mitochondrial membrane potential, increased ROS, and elevated malondialdehyde (MDA) levels, which were associated with higher expression of lincRNA-p21 and activation of the Wnt/β-catenin signaling pathway (53).
3.1.3 Anti-apoptotic proteins
Apoptosis and cellular senescence represent two distinct cellular responses to damage. Senescent cells are highly resistant to apoptosis due to the upregulation of various anti-apoptotic proteins, such as members of the Bcl-2 family, Survivin, and XIAP (X-linked inhibitor of apoptosis protein) (89). In DOX-treated cardiomyocytes, the upregulation of these anti-apoptotic proteins contributes to a senescent, pro-inflammatory phenotype (90). Furthermore, activation of anti-apoptotic signaling pathways, including the PI3K/Akt pathway (90, 91), the Nrf2 pathway, and the NF-κB-IAP pathway, has also been observed in the senescence cardiomyocytes (92, 93). Clearing senescent cells by downregulating these anti-apoptotic proteins can alleviate chemotherapy-induced cardiovascular aging (66, 68). The novel senolytic agent ABT-263 (Navitoclax), which targeted the Bcl-2 family, effectively induced apoptosis in DOX-induced senescent cardiomyocytes and primary human umbilical vein endothelial cells (HUVECs) (68). However, ABT-263 failed to induce apoptosis in senescent somatic hybrid cells, likely due to differences in the levels of anti-apoptotic proteins (66).
3.1.4 Protein homeostasis
Protein homeostasis is essential for maintaining cellular structure and function, preventing the accumulation of metabolic waste, and playing a pivotal role in cardiovascular aging and cardiac myocyte renewal. Autophagy, the lysosomal degradation of misfolded and aggregated proteins as well as damaged organelles, is a key process in protein homeostasis. Therefore, autophagy regulates cardiovascular aging and the renewal of cardiac myocytes by maintaining protein homeostasis. In DOX-treated cardiomyocytes, a loss of autophagy-related gene 4a (ATG4a), a mediator of autophagosome formation and maturation, triggered cardiomyocyte senescence (58). The senescence induced by DOX treatment can be alleviated by exosomes derived from mesenchymal stem cells (MSCs) that express LncRNA-MALAT1+. This effect is mediated by inhibiting miR-92a-3p and promoting ATG4a expression and autophagosome formation. AMPK, a key kinase involved in energy homeostasis and a known positive regulator of autophagy, plays a key role in regulating cardiomyocyte senescence. Activation of AMPK can delay or prevent cancer therapy-related cardiovascular senescence and diseases. Yusei Fujioka et al. found that DOX-induced senescent fibroblasts increased the secretion of extracellular vesicles (EVs) with characteristics of autophagy activation, possibly through AMPKα activation (94). However, whether autophagy induced by EVs is protective or exacerbates cardiac pathology requires further investigation.
3.1.5 Epigenetic modifications
Epigenetic alterations, such as modifications in the genome, heterochromatin disruption, DNA methylation patterns, and histone modifications (e.g., histone acetylation), are critical for gene expression regulation. Loss of epigenetic information has been demonstrated in progeroid mice (8, 95). Compared to newborns, aged populations exhibit lower DNA methylation levels, with a 7% reduction in the methylation status of neighboring cytosine—phosphate—guanine (CpG) sites (96, 97). DOX has been shown to alter the metabolism of S-adenosylmethionine (SAM), a key methylation donor, and decrease global DNA m5C levels in cardiac tissue (52). Given the role of DNA methylation in senescence, it is plausible to speculate that DNA methylation plays a role in DOX-induced cardiovascular senescence.
In addition to DNA methylation, histone acetylation has recently been implicated in the development of cardiovascular aging (98). A decline in histone acetylation alters chromatin structure, which may disrupt the binding of transcription factors and repair enzymes to DNA, interfering with the normal cell cycle and promoting cellular senescence (99). In DOX-treated rats, an upregulation of histone deacetylases (HDACs) and downregulation of histone acetylation regulatory genes, such as CBP and Ep300, were observed (52, 100). Together, these alterations in DNA methylation and histone acetylation play a pivotal role in cardiovascular aging. Targeting these specific epigenetic modifications may offer a novel strategy for mitigating cytotoxic drug-induced senescence.
3.1.6 Senescence-associated secretory phenotype (SASP)
Senescent cells typically exhibit a distinctive secretion profile, including various proinflammatory cytokines, chemokines, growth factors, and proteases, collectively referred to as SASP. The SASP both results from and drives cellular senescence. DOX-induced senescent cardiomyocytes secrete SASP factors that contribute to chronic low-grade inflammation, disrupting cardiac structure and function (70). Furthermore, inflammation-induced SASP can exacerbate DNA and telomere damage, while directly influencing cell cycle arrest. Key inflammatory regulators, including NF-κB, p38/MAPK, and C/EBPβ, are activated and promote SASP secretion in DOX-treated mice and cells (101–103). Additionally, SASP factors secreted by DOX-induced senescent cardiomyocytes can propagate senescence in neighboring cells through exosomal delivery (103). The oxidative stress associated with SASP factors may also be driven by the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome (104). The SASP is diverse, with both pro-inflammatory and anti-inflammatory factors potentially arising from the same stressor. Analyzing, evaluating, and strategically targeting the elimination of SASP represents a critical approach to alleviate the burden of cardiac aging.
In addition to the mechanisms mentioned earlier, alterations in mitochondrial structure, quantity, and function are also critical contributors to cardiac aging. Mitochondria, the primary energy-producing organelles, play an essential role in DOX-induced cardiovascular senescence (85, 105). DOX treatment disrupts mitochondrial quality control (MQC) and suppresses mitochondrial biogenesis through the peroxisome proliferator-activated receptor gamma co-activator 1-Alpha/Beta (PGC-1α/β) pathway (106), leading to a reduction in mitochondrial DNA and insufficient ATP production in the heart (52), thereby promoting the senescence phenotype (107, 108). Notably, DOX induces mitochondrial membrane destabilization via the activation of BAX/BAK-mediated mitochondrial permeability transition pores (mPTPs), where VDAC serves as a critical component. This cascade leads to the extrusion of mtDNA into the cytosol, thereby triggering the cGAS-STING inflammatory axis and driving cardiomyocyte senescence (78). The processes of mitochondrial fusion and fission are also vital in maintaining mitochondrial ROS homeostasis (109). A decrease in fusion coupled with an increase in fission promotes excessive ROS accumulation in cells, accelerating cellular senescence (73). Prolonged low-dose DOX treatment recruits dynamin-related protein 1 (DRP1) via overactivated ERK, which interacts with fission proteins such as Fis1 and mitochondrial fission factor (MFF) on the outer mitochondrial membrane, leading to exacerbated mitochondrial fragmentation in cardiomyocytes. This fragmentation causes electron leakage and excessive ROS accumulation, which further induces cardiomyocyte senescence (72). Mitochondrial autophagy, a key aspect of MQC, plays a critical role in removing damaged mitochondria. This process involves the PTEN-induced putative kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin. Parkin has been shown to mitigate DOX-induced cardiac aging by promoting K63-linked polyubiquitination of TBK1, activating the TBK1/P62 signaling pathway, and facilitating type II mitophagy (60, 110).
3.2 Mechanisms of other chemotherapeutic agents-induced cardiovascular aging
Various chemotherapeutic agents, including platinum-based drugs, taxanes, antimetabolites and alkylating agents, contribute to treatment-induced senescence. Cisplatin can inhibit the CaMKKβ/AMPK signaling pathway and suppresses Sirtuin 3 (SIRT3) expression. As a key mitochondrial deacetylase, SIRT3 inhibition can lead to ROS accumulation and induces senescence in renal tubular cells (111). Paclitaxel, a microtubule-stabilizing agent, is known to induce cell cycle arrest and cause premature senescence in MSCs (112). Yet, Macías et al. reported that paclitaxel ameliorated abnormal heart rate variability and sinoatrial block in mice with Hutchinson-Gilford Progeria Syndrome (HGPS) (113). This indicates that cell type, disease background and other variables can be used in the study of paclitaxel in the field of aging, and its effect and mechanism of aging in the cardiovascular system warrant further investigation.
5-fluorouracil (5-FU) inhibits the expression of nitric oxide synthase (eNOS) and Sirtuin 1 (SIRT1) by activating the p38/JNK pathway, inducing endothelial senescence and dysfunction. Conversely, glucagon-like peptide-1 (GLP-1) alleviates 5-FU-induced endothelial senescence by moderately activating ERK1/2, PI3K, and PKA pathways (114). Alkylating agents, such as cyclophosphamide, ifosfamide, and busulfan, may induce premature ovarian cellular senescence by inhibiting the TrkB/Akt/ERK axis (115). The activation of ERK1/2 in the aging heart is typically impaired; Research have suggested that transduction of constitutively active MEK (CaMEK) can activate ERK1/2 signaling, inhibit mitochondrial fragmentation, and thereby alleviate ischemic damage in the aging heart (116). All the above studies indicate that balancing the MEK/ERK pathway may play an important role in protecting cellular functions, maintaining mitochondrial homeostasis, and delaying CTR-CVA (117, 118). Chen et al. demonstrated in a chronic rhinosinusitis (CRS) model that MEK1/2 and WNT2B signaling can synergistically promote mucosal repair and epithelial remodeling (119). The core logic of “MEK/WNT synergistic regulation of tissue repair” may provide a crucial regulatory node for improving tumor therapy-induced vascular endothelial dysfunction and CTR-CVA. Additionally, the impact of numerous other drugs on cardiovascular aging, including tyrosine kinase inhibitors (TKIs) and immune checkpoint inhibitors, remains insufficiently explored.
3.3 Mechanisms of radiotherapy-induced cardiovascular aging
Approximately 50% of patients with cancers undergo radiation therapy (120), including those with advanced nasopharyngeal carcinoma, lung cancer, liver cancer, esophageal cancer, and breast cancer (121, 122). Similar to DOX, high-energy ionizing radiation directly damages DNA and telomeres, leading to genomic instability, oxidative stress, and cellular senescence. Consequently, senescence plays a detrimental role in the onset and progression of radiation-induced cardiotoxicity (RIC). Endothelial cells, key components of the cardiovascular system, are particularly prone to senescence or permanent cell-cycle arrest after exposure to moderate or high radiation doses (123). These cells are critical initiators of RIC and are highly susceptible to radiation-induced senescence (124–126). Park et al. demonstrated that overexpression of GDF15 in human aortic endothelial cells (HAECs) promoted ROS production, overactivated the ERK signaling pathway, and induced the cellular senescence via the p16INK4a-Rb pathway following ionizing radiation exposure (81).
In addition to direct radiation-induced damage, the radiation-induced by-stander effect (RIBE) also plays a significant role in cellular senescence (127, 128). The ROS and SASP factors released by irradiated cells can cause oxidative damage and chronic inflammation in neighboring cells. Zhang et al. observed a shift from an acute stress-associated phenotype (ASAP) to SASP in stromal cells exposed to radiation (129). Various cytokines and chemokines, including interleukins, TNF-α, and TGF-β, are secreted by irradiated cells under radiation stress and can affect adjacent non-irradiated cells via gap junctions, paracrine signaling, or exosome-mediated communication (128, 130). Consequently, the bystander effect has significant implications for radiation-induced cardiovascular aging.
4 Monitoring of cancer treatment-related cellular senescence and cardiovascular aging
Cardiovascular aging and cellular senescence are primarily monitored through biomarkers, histological analysis, and functional evaluations. Non-invasive tracking of cardiovascular aging and cellular senescence is essential for diagnosis and predicting disease progression. Currently, most non-invasive tracking senescence indicators are indirect evidence, such as using cine-MRI to assess arterial strain as a surrogate for vascular aging. Common imaging indicators for monitoring cardiovascular aging include myocardial mass, tissue composition, ventricular volumes, systolic and diastolic function, left ventricular (LV) strain, and left atrial volumes and function (131). The following sections will highlight the latest research in three key areas of cardiovascular aging monitoring: cytological, functional, and histological assessments.
4.1 Biomarker-based cancer treatment-related cardiovascular aging monitoring
Imaging and monitoring of senescent cells have gained significant attention in recent years, with the development of simplified tools for effective detection. The combination of cellular senescence biomarkers with optical or imaging techniques has enabled the creation of specific tracers for real-time monitoring of senescent cell distribution and dynamic changes in living organisms, providing more intuitive guidance for clinical applications. Beyond ex vivo analyses, such as blood or omics studies, the primary in vivo monitoring tools for cellular senescence are largely based on biomarkers like SA-β-gal (132), ROS, and p16INK4a, often using PET or MRI imaging. However, while these tools excel at detecting senescent cancer cells, their application for monitoring cardiovascular senescence remains limited.
4.1.1 Monitoring tools based on β-galactosidase (SA-β-gal)
β-Galactosidase is a classic biomarker for cellular senescence monitoring. β-Gal-based probes typically consist of two key components: a recognition group (β-galactoside) and a fluorescent reporting group. Upon hydrolysis by the elevated β-galactosidase in senescent cells, the probe is activated, emitting fluorescence.
Among various materials, aggregation-induced emission (AIE) materials are distinguished by their exceptional photostability and sensitivity to aggregation environments. Cen et al. developed the QM-β-gal probe, incorporating an AIE luminogen (AIEgen), which effectively detects DOX-induced senescent cancer cells (132). Notably, the QM-β-gal probe maintains stable in vivo imaging for up to 14 days post-injection, offering significant potential for monitoring cancer cell recurrence and metastasis. However, the probe was unable to detect cellular senescence in the heart, likely due to the lower cumulative dose of DOX compared to conventional doses used in cardiovascular aging models, coupled with a relatively short tissue sampling time (132). Recently, the β-Gal-targeting PET imaging probe, 68Ga-BGal, was designed to directly monitor cellular senescence in live animals, highlighting its considerable clinical translation potential (133).
Notably, not all β-galactosidase-positive cells are necessarily senescent, indicating that false-positive results must be considered in clinical practice.
4.1.2 Monitoring tools based on reactive oxygen species
Excessive accumulation of hypochlorous acid (HClO) is a major contributor to elevated intracellular ROS levels, which serve as a hallmark of senescent cells. Consequently, HClO represents a promising target for monitoring cellular senescence. Li et al. developed a novel probe to detect HClO levels both in vitro and in vivo, which can effectively differentiate senescent cells from non-senescent ones in cardiac tissues. The near-infrared fluorescent probe SA-HCy-1 responds to three key indicators—SA-β-gal, high ROS levels, and elevated lysosomal pH—enabling the visualization of short-term senescence in live cells within a skin photoaging mouse model (134). However, its clinical applicability requires further investigation, particularly regarding its correlation with classical markers, such as p21cip1 and p16INK4a.
4.1.3 Other monitoring tools
Radioactive tracers targeting p16INK4a allow for in vivo imaging of cellular senescence via positron emission tomography (PET) (135). Monitoring telomerase activity and the expression of human telomerase reverse transcriptase (hTERT) in CD34(+) circulating progenitor cells has emerged as a simple and effective method for assessing cardiac aging (136). Moreover, SASP expression in plasma has been more strongly linked to cardiovascular aging, suggesting SASPs as potential biomarkers for monitoring this process (137).
4.2 Function-based cancer treatment-related cardiovascular aging monitoring
Cardiovascular aging and cellular senescence are typically associated with functional changes, including reduced cardiac contractility, impaired diastolic function, decreased vascular elasticity, and abnormal cardiac conduction. Functional monitoring indicators for cardiovascular aging include left ventricular ejection fraction (LVEF), the E/A ratio, global longitudinal strain (GLS), and pulse-wave velocity (PWV) (16). Selecting appropriate and accessible functional monitoring metrics will aid in the indirect diagnosis and early prediction of cancer treatment-related cardiovascular aging.
4.2.1 Monitoring of cardiac contraction
Impaired cardiac contractility during cancer treatment is primarily attributed to factors such as cardiomyocyte loss, myocardial microvascular remodeling, reduced coronary perfusion, and collagen deposition in the myocardium. The conventional quantitative parameter for assessing cardiac contractile function is LVEF, which commonly declines in the early stages of cancer treatment-related cardiovascular aging (138). Thus, traditional CTR-CVT LVEF data can serve as a useful benchmark. Three-dimensional transthoracic echocardiography (3D TTE) is the preferred method for LVEF evaluation. In cases where 3D echocardiography is not feasible, the modified two-dimensional (2D) Simpson's biplane method can be used as an alternative (139, 140). Notably, cancer treatment-related cardiovascular aging often occurs with preserved ejection fraction. Therefore, a normal LVEF value cannot rule out cardiac contractility dysfunction.
4.2.2 Monitoring of diastole
Cancer treatment induces cardiac fibroblast transformation into myofibroblasts, which secrete excessive collagen, leading to cardiac fibrosis. This fibrosis alters the mechanical properties of the heart chambers, increasing stiffness and reducing elasticity (141). Additionally, cancer therapies can impair calcium transport proteins and disrupt calcium homeostasis in cardiomyocytes, contributing to diastolic dysfunction. Diastolic function indicators are therefore valuable for indirectly assessing cancer treatment-related cardiovascular aging. Early-stage DOX-induced cardiomyopathy is primarily characterized by diastolic dysfunction (142, 143). Radiotherapy can also cause diastolic dysfunction, often linked to cardiomyocyte damage, microvascular occlusion, and ECM remodeling. Additionally, DOX treatment induces collagen deposition in the atrial walls, leading to increased stiffness, which can be indexed by the diastolic capacity of the left atrium (LA). This suggests that LA diastolic capacity may also serve as a indicator for cancer treatment-related cardiovascular aging (144, 145). Magnetic resonance radiomics, based on major and minor axis lengths and 2D diameter imaging, has revealed a reduction in LA chamber size that is strongly associated with cardiovascular aging, highlighting the potential value of magnetic resonance radiomics in monitoring cancer treatment-related cardiovascular aging (146).
4.2.3 Monitoring of strain
Myocardial strain capacity is influenced by muscle length and angles, encompassing longitudinal, radial, and circumferential dimensions. A decline in myocardial strain capacity is a hallmark of aging cardiovascular systems, suggesting that strain capacity also serves as a functional indicator of cancer treatment-related cardiovascular aging. Key quantitative imaging indicators for myocardial strain capacity include global longitudinal strain (GLS), global circumferential strain (GCS), global radial strain (GRS), left ventricular twist (LV Twist), and Torsion (146). GLS, in particular, is recommended for evaluating left ventricular strain in CTR-CVD and can be used to indirectly monitor cancer treatment-related cardiovascular aging (147).
4.2.4 Monitoring of conduction
Dysfunction of the cardiac conduction system is associated with a range of physiological and pathological cardiovascular changes (148, 149). Cancer treatment-related cardiovascular aging exhibits similar characteristics to general organ aging (29), including a reduction in heart rate and QT interval prolongation (150, 151). Baseline electrocardiogram (ECG) monitoring prior to treatment, incorporating the Fridericia correction method (QTcF) (152), along with dynamic ECG (Holter monitoring), are common tools for tracking transient arrhythmias and conduction abnormalities.
The integration of AI, wearable devices, electronic stethoscopes, and ECG technology (153) has enabled the application of deep learning models to more precisely assess conduction system dysfunction, predict cancer treatment-related cardiovascular aging, and guide drug administration (154, 155). Significant alterations in the QT interval, exceeding established thresholds, signal the need to promptly discontinue non-essential medications during cancer treatment-related cardiovascular aging (150, 156, 157). However, it is important to recognize that current machine learning models are limited by data constraints, and the performance of external applications still requires optimization.
4.3 Histology-based cancer treatment-related cardiovascular aging monitoring
Histological monitoring offers more direct structural insights into CTR-CVA, facilitating dynamic evaluations. Given the high risks associated with pathological biopsy, current histological indicators primarily rely on imaging parameters to indirectly assess myocardial mass, cardiac fibrosis, myocardial perfusion, coronary artery calcification (CAC), and valvular calcification.
High levels of oxidative stress and chronic inflammation are potential mechanisms driving vascular aging associated with cancer therapies (158). CMR first-pass perfusion imaging, which assesses myocardial perfusion index (PI), may reveal alterations in cardiac microcirculation in patients undergoing prolonged chemotherapy, serving as a valuable reference for CTR-CVA evaluation (159). Radionuclide myocardial perfusion imaging, reflecting myocardial perfusion via radiolabeled compound uptake by myocardial cells, could also become a useful tool for CTR-CVA assessment. Additionally, automated coronary calcium scoring from CT scans offers significant advantages in evaluating the extent of coronary aging in patients with cancers. PET/CT has been shown to effectively monitor CAC (160, 161), though clinical studies specifically addressing CAC resulting from both aging and cancer treatment remain scarce. Certain chemotherapeutic agents, such as alkylating drugs, may induce ovarian insufficiency, leading to decreased estrogen levels (162). Therefore, changes in estrogen levels must be considered when monitoring arterial stiffness in patients undergoing cancer treatments. Addressing these challenges requires personalized assessment, monitoring, and follow-up to optimize patient care.
Given these indicators, refining conventional monitoring methods for CTR-CVA is essential. Establishing a comprehensive baseline for cardiovascular aging is critical. More high-quality randomized controlled trials (RCTs) should be prioritized, and long-term follow-up assessment protocols should be systematically adopted to achieve a more comprehensive, objective, and accurate comprehensive evaluation of CTR-CVA. Developing a non-invasive imaging system capable of monitoring aging from cellular senescence to histological and functional changes could significantly enhance clinicians' ability to detect early cardiovascular aging in patients and foster more precise interventions, ultimately improving survival outcomes.
5 Treatment of cancer treatment-induced cardiovascular aging
Cellular senescence is an important influencing factor of CTR-CVT. Therefore, anti-aging interventions may mitigate the progression of CTR-CVT and CTR-CVA. Current strategies for preventing and treating cardiovascular aging can be divided into pharmacological and non-pharmacological approaches. Currently, the prevention and management of CTR-CVA remain challenging.
5.1 Pharmacotherapy
Anti-aging drugs, or senotherapeutics, are designed to target both intracellular senescence-associated pathways and extracellular membrane proteins specifically upregulated in senescent cells. These agents are categorized into senolytics and senomorphics. Senolytics selectively eliminate senescent cells, whereas senomorphics modulate the SASP without inducing cell death. Immunotherapy, an emerging approach, aims to specifically eliminate senescent cells by targeting unique surface markers (Table 3).
Table 3
| Classification | Name | Experimental model | Treatment protocols | Literature results in detail | Year |
|---|---|---|---|---|---|
| Senolytics | Dasatinib + Quercetin (D + Q) |
In vivo: 1)6-week-old C57BL/6 mouse diabetes model; 2)Cardiac tissue and cardiac stem cells (CSCs) from clinical non-elderly type 2 diabetes mellitus (T2DM) patients; In vitro: High-glucose model of CSCs |
In vivo: After T2DM model establishment, intraperitoneal injection of 5 mg/kg D and 50 mg/kg Q for 3 consecutive days, repeated for 4 weeks, then sampling. | D + Q eliminated cardiac senescent cells (including CSCs), restored CSC function, promoted cardiomyocyte regeneration, reduced myocardial fibrosis, and improved diabetes-related cardiac diastolic function. | 2022 (163) |
| In vitro: After high glucose-induced senescence, treated with 0.25 μmol/L D and 10 μmol/L Q, repeated every 2 days for 4 days, sampling 2 days after the last treatment | |||||
| In vivo: 22–24-month-old female C57BL/6 mouse myocardial infarction (MI) model | In vivo: Intraperitoneal injection of 5 mg/kg D and 50 mg/kg Q for 3 consecutive days weekly starting after MI, sampling after 4 weeks | D + Q eliminated senescent cells, improved the inflammatory microenvironment, and activated cardiac stem cell function, which significantly enhanced cardiac function after myocardial infarction in aged female mice. | 2022 (164) | ||
| In vitro: a Human Model of Cardiac Fibrosis-on-a-Chip | In vitro: After 1 week of microarray construction, treated with 0.1 μM D and 2 μM Q for 2 weeks under continuous TGF-β stimulation, sampling after treatment | D + Q selectively induced apoptosis of senescent myofibroblasts, significantly improved systolic function, and reduced passive tension, with effects superior to traditional antifibrotic drugs (such as pirfenidone and losartan). | 2023 (165) | ||
| In vivo: 2-month-old rat MI model | In vivo: Gavage of 5 mg/kg D and 50 mg/kg Q starting 4 h after MI, once daily for 3 consecutive days, repeated after a 2-week interval, then sampling | D + Q significantly reduced the proportion of p16+αSMA+ myofibroblasts and p16+CD31+ endothelial cells in the atria of MI rats, reversed atrial remodeling, and decreased the atrial fibrillation induction rate (from 89% to 0%). | 2024 (166) | ||
| In vivo: 8-week-old female C57BL/6NTac mouse obesity model | In vivo: After obesity model establishment (week 16), gavage of 5 mg/kg D and 50 mg/kg Q for 3 consecutive days weekly until sampling at week 24 | D + Q reduced SA-β-Gal+ cells in cardiac tissue, decreased the protein expression of p16/p21/p53, and improved obesity-related cardiac dysfunction. | 2024 (167) | ||
| In vivo: 1) 22–24-month-old C57BL/6 mouse aging model; 2) Various disease models: MI, heart failure (HF), hypertension, atherosclerosis, abdominal aortic aneurysm (AAA). | In vivo: Intraperitoneal injection of 5 mg/kg D and 50 mg/kg Q 3 days weekly for 2–4 weeks after model establishment, sampling 24–48 h after last administration or 4–8 weeks after model induction | D + Q eliminated senescent cells in various models, improved function, and improved the microenvironment. | 2024 (168) | ||
| In vitro: DOX-induced senescence model of human induced pluripotent stem cell (hiPSC)-derived cardiac organoids (hCardioids) | In vitro: After establishing DOX-induced senescent cardiac organoid model, treated with 0.25 μM D and 2 μM Q for 2 weeks under continuous TGF-β stimulation, sampling after treatment | D + Q eliminated senescent cells, promoted recovery of progenitor cell function, inhibited fibrosis, and effectively reversed DOX-induced senescent cardiomyopathy. | 2025 (77) | ||
| Navitoclax (ABT263) | In vivo: 3–4-month-old male C57BL/6J mouse myocardial ischemia-reperfusion injury (IRI) model | In vivo: Gavage of 50 mg/kg Navitoclax on days 4–10 after model establishment, sampling after five weeks | Navitoclax and its targeted formulations eliminated DOX-induced senescent cardiomyocytes, alleviated cardiotoxicity, and restored cardiac function. | 2020 (169) | |
|
In vivo: 6–8-week-old male C57BL/6J mouse AngII-induced senescence model; In vitro: Primary cardiomyocytes and fibroblasts AngII-induced senescence models |
In vivo: Gavage of 50 mg/kg Navitoclax for 2 cycles (7 days per cycle, 2-week interval), sampling 1 week after last administration; In vitro: Treatment with 1 µmol/L Navitoclax for 72 h after model establishment, then sampling |
Navitoclax eliminated AngII-induced senescent cardiomyocytes and fibroblasts, inhibited SASP, inflammation, and fibrosis, and improved cardiac function and electrophysiological properties. | 2020 (170) | ||
|
In vivo: 10-week-old C57BL/6J mouse DOX-induced senescence model (chronic low-dose multiple administration); In vitro: HL-1 cardiomyocyte line DOX-induced senescence model |
In vivo: Intraperitoneal injection or gavage of three Navitoclax formulations after model establishment, sampling 30 days post-last administration In vitro: Treatment with 0.37 µM Navitoclax for 48 h after model establishment, then sampling | Navitoclax and its targeted formulations eliminated DOX-induced senescent cardiomyocytes, alleviated cardiotoxicity, and restored cardiac function. | 2022 (68) | ||
| In vitro: DOX-induced senescence models in EA.hy926 cells and HUVECs | In vitro: Treatment with 0.1–10 uM ABT-263 for 24–48 h after model establishment, then sampling | ABT-263 selectively eliminated senescent HUVECs by inhibiting anti-apoptotic proteins such as BCL-xL. | 2022 (66) | ||
| In vivo: 6-month-old Atg7cKO mouse autophagic deficiency model; | In vivo: Gavage of 50 mg/kg Navitoclax for 2 cycles (5 days per cycle, 1-week interval) after model establishment, sampling 1 week post-last administration | Navitoclax improved the function of autophagy-deficient hearts by eliminating senescent cells. | 2024 (171) | ||
|
In vivo: Male Sprague-Dawley (SD) rat abdominal aortic constriction (AAC)-induced HF model; In vitro: Neonatal rat cardiomyocytes (NRCMs) and mouse H9c2 cardiomyocyte line AngII-induced senescence models |
In vivo: Intravenous injection of 25 mg/kg ABT263 and equivalent dose of AP@ABT263 nanoassemblies at weeks 9, 12, 15, 18 after model establishment, sampling 1 week post-last administration; In vitro: Treatment with 5 μM ABT263 and equivalent dose of AP@ABT263 for 48–72 h after model establishment, then sampling |
AP@ABT263 achieved precise elimination of senescent cardiomyocytes, significantly improved cardiac function in the HF model, and avoided the systemic toxicity of free drugs. | 2024 (172) | ||
| Fisetin |
In vivo: 1) Ercc1−/Δ progeria mice; 2) 22–24-month-old C57BL/6 mice; In vitro: Oxidative stress-induced senescence model in mouse embryonic fibroblasts (MEFs); Etoposide-induced senescence model in human fibroblasts (IMR90) |
In vivo: 1) Ercc1−/Δ progeria mice intermittently fed with 500 ppm fisetin (≈60 mg/kg) diet at 6–8 and 12–14 weeks of age, sampling 1–2 weeks post-last intervention; 2) 22–24-month-old C57BL/6 mice gavaged with 100 mg/kg fisetin for 5 consecutive days, sampling 3 days after drug withdrawal; 3) C57BL/6 mice fed with 500 ppm fisetin diet starting at 85 weeks of age until natural death or 24 months, then sampling; In vitro: Treatment with 1–15 μM fisetin for 24–48 h after model establishment, then sampling |
The natural flavonoid fisetin exhibited highly efficient senolytic activity, improved cardiac ejection fraction in aged mice, increased vascular reactivity, and prolonged lifespan. | 2018 (173) | |
|
In vivo: 66-week-old aged mice; In vitro: DOX-induced senescence model in rat thoracic aortic smooth muscle cells (VSMCs) |
In vivo: Gavage of 50 mg/kg fisetin for 4 consecutive weeks after model establishment, sampling on day 28; In vitro: Treatment with 10/50 μM fisetin for 6 h after model establishment, immediate sampling |
Fisetin reversed VSMCs senescence via the PTEN/mTORC2-Akt (Ser473) signaling pathway. | 2021 (174) | ||
|
In vivo: DOX-induced senescence model in 7-week-old male C57BL/6 mice; In vitro: H₂O₂-induced senescence model in rat primary VSMCs |
In vivo: Gavage of 50 mg/kg fisetin once daily for 4 weeks after model establishment, then sampling; In vitro: Treatment with 10–100 μM fisetin for 12 h after model establishment, immediate sampling |
Fisetin alleviated oxidative stress-induced VSMCs senescence by activating PPARγ and regulating the mTORC2-FoxO3a-autophagy cascade. | 2023 (175) | ||
| In vitro: H₂O₂-induced senescence model in rat primary VSMCs | In vitro: Treatment with 10/20/50 μM fisetin for 12 h after model establishment, immediate sampling | Fisetin alleviated VSMCs senescence via the PTEN-mediated PKCδ-NOX1 pathway. | 2023 (176) | ||
|
In vivo: 1) 27-month-old wild-type C57BL/6N mice; 2) p16–3MR transgenic mice; In vitro: Replicative senescence model in HUVECs and human aortic endothelial cells (HAECs) |
In vivo: Gavage of 100 mg/kg fisetin for 1 week, followed by 2-week drug withdrawal and another 1-week administration after model establishment, sampling 1 week post-last administration | Intermittent supplementation with fisetin reversed arterial dysfunction in aged mice by eliminating vascular senescent cells, reducing oxidative stress and inflammation, and improving arterial structure. | 2024 (177) | ||
| In vitro: Treatment with 0–1 μM fisetin for 48 h after model establishment, immediate sampling | |||||
| In vivo: Rat arteriovenous fistula (AVF) model | In vivo: Intraperitoneal injection of 30 mg/kg fisetin for 3 weeks after model establishment, then sampling | Fisetin promoted outward remodeling of AVF and improved AVF blood flow by blocking heme-related pro-senescence effects. | 2024 (178) | ||
|
In vivo: 1) C57BL/6 mouse T2DM model; 2) 16-month-old C57BL/6 mice; In vitro: High glucose-induced senescence model in HUVECs and VSMCs |
In vivo: Gavage of 50/100/200 mg/kg fisetin or 100 mg/kg fisetin and 100 mg/kg Metformin (Metformin daily gavage for 12 consecutive weeks, fisetin treated for 3 days every 2 weeks for 12 weeks) after model establishment, sampling 3 days post-last administration; In vitro: Treatment with 10–50 μM fisetin for 48–72 h after model establishment, immediate sampling |
Fisetin induced apoptosis of senescent cells by targeting the Pi3k-Akt-Bcl-2/Bcl-xl pathway, reduced vascular remodeling, and alleviated aortic aging in T2DM mice. | 2025 (179) | ||
| Senomorphics | Metformin | In vitro: High glucose-induced senescence model in HUVECs | In vitro: Treatment with 5 μM resveratrol and 50 μM metformin after model establishment, including whole-course treatment (3 days of high glucose and 3 days of normal glucose) or stage-specific treatment (3 days of high glucose only or 3 days of normal glucose only), sampling on day 6 | Metformin and Resveratrol reversed the metabolic memory of endothelial cell senescence by activating SIRT1, regulating p53 acetylation, and resisting oxidative stress. | 2015 (180) |
|
In vivo: ApoE−/− mouse atherosclerosis model; In vitro: Replicative senescence model in HAECs |
In vivo: 50 mg/kg metformin added to drinking water after model establishment, sampling at 14 months; In vitro: Continuous passage with 2 mM metformin until P15, then sampling |
Metformin regulated mitochondrial biogenesis and endothelial cell senescence via AMPK-mediated H3K79 methylation. | 2018 (181) | ||
|
In vivo: DOX-induced senescence model in male C57BL/6 mice; In vitro: DOX-induced senescence model in rat thoracic aortic VSMCs |
In vivo: Gavage of 200 mg/kg metformin daily for 4 weeks after model establishment, sampling 5 days post-last administration; In vitro: Treatment with 2 mM metformin for 1 h after model establishment, then sampling |
The Metformin alleviated VSMCs senescence by regulating the AMPKα (Ser485)-SIRT3 pathway. | 2022 (182) | ||
|
In vivo: 1) AngII-induced senescence model in 10-week-old C57BL/6 mice; 2) 20-month-old aged C57BL/6 mice; In vitro: AngII-induced senescence model in mouse VSMCs; primary aortic VSMCs from elderly humans |
In vivo: 1) Gavage of 150 mg/kg metformin daily after model establishment, sampling on day 28; 2) 20-month-old mice treated with 3 g/L metformin in drinking water, sampling at 10 months; In vitro: Treatment with 200 μM metformin (daily medium change) after model establishment, sampling on day 12 |
Metformin delayed vascular aging by enhancing autophagic flux and inhibiting SASP and VSMCs senescence. | 2022 (183) | ||
|
In vivo: 1) 12-month-old ApoE⁻/⁻ mouse model; 2) 20-month-old C57BL/6 mice; In vitro: Replicative senescence model in HAECs |
In vivo: Gavage of low-dose (2.5 mg/kg) or physiological-dose (50 mg/kg) metformin daily for 10 weeks after model establishment, then sampling; In vitro: Continuous passage with 2 mM metformin until passage 15, then sampling |
Mito-Esc and metformin delayed vascular aging and improved atherosclerosis by activating mitochondrial function and inhibiting oxidative stress and inflammation via the AMPK-SIRT1/SIRT6 axis. | 2024 (184) | ||
| In vivo: Male Wistar rat diabetic cardiomyopathy model | In vivo: Gavage of 200 mg/kg metformin daily for 10 weeks after model establishment, then sampling | Metformin alleviated diabetic myocardial aging through multiple pathways, including regulating macrophage phenotype, enhancing Klotho and GDF-15 expression, improving immunometabolism, and inhibiting cellular senescence. | 2025 (185) | ||
| Rapamycin | In vivo: 22-month-old aged mice | In vivo: Intraperitoneal injection of 2 mg/kg rapamycin once daily for 8 weeks, then sampling | Rapamycin alleviated MIF deficiency-related cardiac aging by activating autophagy. | 2016 (186) | |
|
In vivo: 8–9-week-old C57BL/6 wild-type mouse atherosclerosis model; In vitro: Shear stress-induced senescence model in HUVECs |
In vivo: Intraperitoneal injection of 4 mg/kg rapamycin twice (24-hour interval), sampling 4 h post-last injection; In vitro: Addition of 0.5 μmol/L rapamycin under low shear stress conditions, sampling after 24 h |
Rapamycin mitigated the progression of atherosclerosis under high shear stress conditions by activating autophagy, restoring endothelial cell alignment, and inhibiting inflammation and senescence. | 2017 (187) | ||
| In vitro: DOX-induced senescence model in SD rat thoracic aortic VSMCs | In vitro: VSMCs pre-treated with 2 μg/mL rapamycin for 4 h, then treated with 500 nM DOX for 4 h before sampling | The mTOR inhibitor rapamycin alleviated DOX-induced senescence by activating autophagy and downregulating p53/p21/p16. | 2018 (188) | ||
| In vitro: Replicative senescence model in SD rat thoracic aortic VSMCs | In vitro: Treatment with 20 nM rapamycin for 12 h, then sampling | Rapamycin reversed cell cycle arrest and senescence phenotype in senescent VSMCs by downregulating miR-30a, relieving its inhibition on Beclin1, and activating the autophagy pathway. | 2019 (189) | ||
| In vitro: VSMCs isolated from 6-month-old male PPARGC1A-knockout (ppargc1a−/−) and SQSTM1-knockout (sqstm1−/−) mice | In vivo: Intraperitoneal injection of 2 mg/kg rapamycin twice weekly for 2–4 weeks, sampling 24–48 h after last injection | Rapamycin effectively reversed the senescence phenotype of vascular smooth muscle cells induced by PPARGC1A or SQSTM1 deficiency by activating autophagy and inhibiting the mTOR pathway. | 2020 (190) | ||
| In vitro: Human cardiac progenitor cells (hCPCs) | In vitro: Culturing with 1 nM, 10 nM, or 100 nM rapamycin (medium changed every 2 days), continuous passage beyond P17, sampling at each passage | Rapamycin significantly delayed replicative senescence of hCPCs and restored their proliferation, migration, and multilineage differentiation capacities by inhibiting the mTOR signaling pathway and activating the STAT3-PIM1 axis. | 2020 (191) | ||
| In vivo: 18-month-old SD rats | In vivo: Daily feeding with 14 mg/kg enteric-coated microencapsulated feed containing rapamycin (equivalent to 2.24 mg/kg) for 6 months, then sampling | Rapamycin significantly reduced myocardial lipofuscin accumulation, decreased the proportion of senescent cells, and delayed cardiac aging in aged rats by activating autophagy. | 2021 (192) | ||
| In vitro: Homocysteine-induced senescence model in rat VSMCs | In vitro: Pre-treatment with 10 ng/mL rapamycin for 30 min, followed by treatment with 500 μmol/L Hcy for 24 h before sampling | The autophagy activator rapamycin reversed homocysteine-induced VSMCs senescence. | 2021 (193) | ||
| In vitro: H₂O₂-induced senescence model in HUVECs | In vitro: Treatment with 182.83 ng/mL rapamycin-functionalized carbon dots (Rapa-CDs) and 200 nM free rapamycin (Rapa) for 6 h, then sampling | Rapa-CDs significantly improved the water solubility of rapamycin, enhanced ROS scavenging capacity, inhibited the mTOR pathway, and effectively suppressed HUVECs senescence. | 2024 (194) | ||
| In vivo: Mouse T2DM model | In vivo: Intraperitoneal injection of 4 mg/kg rapamycin every 2 days for 8 weeks, then sampling | Rapamycin inhibited NLRP3 signaling, alleviated immune senescence, and slowed diabetic vascular aging. | 2025 (195) | ||
| In vitro: Primary senescent activated valvular interstitial cells (aVICs) isolated from canine myxoid mitral valve degeneration (MMVD) | In vitro: Culturing with 25–300 nM rapamycin for 48–72 h, then sampling | mTOR inhibitors (such as rapamycin) alleviated MMVD by activating autophagy to degrade p21/p16. | 2025 (196) | ||
| Curcumin | In vivo: Diabetes model in C57BL/6 mice | In vivo: Gavage with 1000 mg/kg curcumin after model establishment, sampling after 14 days | Curcumin significantly improved blood flow recovery and capillary density in the hindlimb ischemia model of diabetic mice by regulating the function of endothelial progenitor cells (EPCs). | 2017 (197) | |
|
In vivo: 18-month-old male C57BL/6 mice; In vitro: Replicative senescence models of human atrial fibroblasts (HATs) and mouse atrial fibroblasts (MAFs) |
In vivo: After model establishment, daily feeding with 50/100 mg/kg curcumin dissolved in feed for 6 months, then sampling; In vitro: After model establishment, treatment with 6–12 µM curcumin for 48 h, then sampling |
Curcumin improved age-related atrial fibrillation (AF) by inhibiting p300. | 2023 (198) | ||
|
In vivo: 12–18-month-old C57BL/6 mice; In vitro: Replicative senescence model of human atrial fibroblasts |
In vivo: Gavage with 50 mg/kg curcumin after model establishment, sampling after 6 months; In vitro: Treatment with 6, 9, or 12 μM curcumin for 24–48 h after model establishment, then sampling |
The p300 inhibitor curcumin reversed age-related atrial fibrosis and reduced the risk of AF. | 2023 (199) | ||
|
In vivo: 18-month-old male C57BL/6 mice; In vitro: TBHP-induced senescence model in MAFs |
In vivo: After model establishment, daily feeding with 100 mg/kg curcumin dissolved in feed for 6 months, then sampling; In vitro: Treatment with unknown concentration of curcumin for 48 h after model establishment, then sampling |
Curcumin reduced the atrial fibrillation induction rate in senescent mice, shortened the sinus node recovery time (SNRT), and improved electrophysiological abnormalities by alleviating atrial senescence and fibrosis. | 2024 (200) | ||
| Resveratrol | In vitro: High glucose-induced senescence model in HUVECs | In vitro: Treatment with 50 μM metformin and 5 μM resveratrol after model establishment, including whole-course treatment (3 days of high glucose and 3 days of normal glucose) or stage-specific treatment (3 days of high glucose only or 3 days of normal glucose only), sampling on day 6 | Metformin and Resveratrol reversed the metabolic memory of endothelial cell senescence by activating SIRT1, regulating p53 acetylation, and resisting oxidative stress. | 2015 (180) | |
| In vivo: DOX-induced cardiac injury model in male senescence-accelerated mice (SAMP8) | In vivo: Single intraperitoneal injection of 18 mg/kg DOX on day 1, intraperitoneal injection of 20 mg/kg resveratrol on days 2–4, sampling on day 5 | Resveratrol alleviated cardiotoxicity in doxorubicin treatment of aged mice by regulating the SIRT1-USP7 axis. | 2015 (201) | ||
| In vivo: DOX-induced cardiomyopathy model in 3-month-old female Fischer 344 rats | In vivo: 1) One week after model establishment, intramyocardial injection of hCPCs pre-treated with DOX and resveratrol, sampling after 3 weeks of observation; 2) Gavage of resveratrol daily at the time of model establishment, sampling after 6 weeks of continuous administration | Resveratrol reversed DOX-induced senescence and apoptosis in hCPCs and partially restored their regenerative functions by activating SIRT1, promoting p53 deacetylation, repairing the IGF-1/Akt pathway, and enhancing antioxidant capacity. | 2015 (202) | ||
| In vivo: MI and IRI models in 9–11-week-old male C57BL/6J mice | In vivo: 1) MI model: Daily oral gavage of 320 mg/kg resveratrol for 1 week before MI model establishment, medication ceased after surgery, sampling on postoperative days 3, 7, and 14; 2) IRI model: Daily oral gavage of 320 mg/kg resveratrol for 1 week before IRI model establishment, sampling after 45 min of ischemia and 24 h of reperfusion during surgery | Resveratrol inhibited cardiomyocyte senescence by activating the Sirt1/p53 pathway and alleviated inflammatory responses by inhibiting the NLRP3 inflammasome, thereby improving ischemia-hypoxia-induced myocardial injury. | 2020 (203) | ||
| In vivo: Model of diabetes combined with MI in male SD rats | In vivo: Intramuscular injection of high-glucose rat bone marrow mesenchymal stem cells (BMSCs) pre-treated with 2 μM resveratrol for 96 h at 4 sites in the myocardial infarction border zone, sampling at 1 week and 3 weeks post-operation | Resveratrol reversed BMSCs senescence and enhanced their transplantation efficacy in diabetic myocardial infarction rats by downregulating miR-34a and activating SIRT1, as manifested by improved cardiac function, reduced myocardial fibrosis, and increased angiogenesis. | 2021 (204) | ||
| In vitro: 1) DOX-induced senescence model in human pluripotent stem cell-derived cardiomyocytes (hPSC-CMs); 2) DOX-induced senescence model in 3D dynamic engineered heart tissues (dyn-EHTs) constructed with hPSC-CMs and cardiac fibroblasts | In vitro: 1) Co-treatment with 10 μmol/L resveratrol and 0.1 μmol/L DOX for 48 h each, followed by 48 h recovery, repeated for 2 cycles, sampling on day 7; 2) Co-treatment with 10 μmol/L resveratrol and 0.1 μmol/L DOX, each cycle including a 48-hour administration period and a 5-day recovery period, repeated for 4 cycles, sampling on day 28 | Resveratrol inhibited the expression of senescence markers and improved mitochondrial function. | 2023 (205) | ||
| Rutin | In vivo: ApoE−/− mouse model of atherosclerosis |
In vivo: Daily gavage with 40 mg/kg Rutin after model establishment, sampling after 6 weeks; In vitro: Treatment with 50 μM Rutin for 72 h after model establishment, then sampling |
Rutin inhibited premature senescence of VSMCs in diabetic mice and reduced senescent cell accumulation in plaques by anti-oxidative stress and telomere protection, thereby increasing the number of VSMCs and stabilizing atherosclerotic plaques. | 2018 (206) | |
| In vitro: H₂O₂-induced senescence model in primary thoracic aortic VSMCs | |||||
| In vivo: MI model in aged rats | In vivo: Mesenchymal stem cells pre-treated with 10–200 μM Rutin for 1 h, then induced senescence by 400 μM H₂O₂ treatment for 2–4 h, followed by intramyocardial injection and sampling at 2 and 4 weeks | Rutin and quercetin significantly reduced ROS levels in young and senescent MSCs, decreased myocardial fibrosis, and promoted angiogenesis and cardiomyocyte regeneration. | 2023 (207) | ||
| Canagliflozin | In vitro: High fat-induced senescence model in HUVECs | In vitro: Co-treatment with 0.3 mM palmitic acid (PA) and 2.5/5/10 μg/mL Canagliflozin for 24 h, then sampling | Canagliflozin delayed vascular endothelial cell senescence by binding to p38/JNK and inhibiting their phosphorylation, reducing SASP secretion and cell cycle arrest, and scavenging ROS. | 2023 (208) | |
| In vitro: High fat-induced senescence model in HUVECs | In vitro: Co-treatment with 0.3 mM PA and 0.1/0.5 μM Canagliflozin for 24 h, then sampling | Canagliflozin delayed the senescence of vascular endothelial cells by scavenging ROS, inhibiting the ERK signaling pathway, and synergistically suppressing the ferroptosis pathway. | 2024 (209) | ||
| Immune-activating Therapies | Serum chemokine CCL17 | In vivo: AngII-induced vascular injury model in 4-month-old wild-type mice | In vivo: Daily intraperitoneal injection of 100 μg anti-CCL17 for 4 consecutive weeks, followed by sampling | CCL17 neutralizing antibody treatment improved vascular aging or AngII-induced vascular dysfunction, remodeling, and immune dysregulation. | 2023 (210) |
| Canagliflozin | In vivo: High-fat diet model in C57BL/6 mice | In vivo: Addition of 0.03% w/w Canagliflozin to diet from day 3 to week 20, sampling at day 7 or week 4 | The SGLT2 inhibitor canagliflozin improved age-related phenotypic changes by negatively regulating PD-L1 expression, enhancing endogenous immune surveillance, and clearing senescent cells. | 2024 (211) |
Applications of various anti-aging therapies in the cardiovascular field (2015–2025).
5.1.1 Senolytics
Senolytics primarily induce the death of senescent cells, effectively preventing their accumulation in tissues. Regimens for senolytic therapy include combinations such as dasatinib and quercetin (D + Q), navitoclax (ABT-263), fisetin, proxofim (FOXO4-DRI), and HSP90 inhibitors.
Among these, D + Q has emerged as the most promising senolytic combination for promoting the clearance of senescent cells (212, 213). D + Q facilitates senescent cell elimination by inhibiting anti-apoptotic pathways, including the Ephrin B (EFNB) serine protease inhibitors (Serpins) and the PI3K/AKT pathway (214). Wagner et al. demonstrated that D + Q effectively reverses cardiac dysfunction induced by cellular senescence in animal models (215), while Roos et al. confirmed its efficacy in improving age-related vascular dysfunction (216). Notably, different senescent cells show certain selectivity for anti-aging drugs. For instance, the advantage of dasatinib in dealing with senescent adipocytes cannot be transferred to senescent endothelial cells, which are obviously more sensitive to quercetin treatment (214).
Navitoclax (68), a BCL-2 family inhibitor, is a classic agent used to mitigate chemotherapy-induced cardiovascular cellular senescence. Despite a long research history, its clinical translation is currently unfeasible due to safety issues such as induction of acute platelet toxicity. Fisetin (173), a natural flavonoid, exhibits senolytic activity through its metabolic byproducts. FOXO4 (217) neutralizes drug-induced toxicity and eliminates senescent cells in a DOX-induced kidney aging model by modulating p53. HSP90 inhibitors, such as 17-DMAG and alvespimycin (218), affect the PI3K/AKT/mTOR senescence-related pathway, triggering senescent cell apoptosis (219).
5.1.2 Senomorphics
In addition to senolytic strategies, senomorphics offer another promising approach to mitigate age-related side effects. Most senomorphics exert their effects by modulating the signaling or transcriptional regulators of the SASP, including the NF-κB, JAK-STAT, and mTOR pathways (220). Metformin, a well-established senomorphics agent, reduces SASP expression and inhibits DOX-induced endothelial cell senescence by suppressing the JNK and NF-κB pathways (70). Long-term rapamycin treatment in senescent cardiac progenitor cells (CPCs) suppresses mTOR signaling, activates the JAK-STAT3-PIM1 pathway, decreases senescence marker expression, and restores clonogenic, migratory, and differentiative potential (221). Additionally, natural compounds such as curcumin and resveratrol (a SIRT1 activator) have been shown to regulate the mTOR pathway, mitigating low-dose anthracycline-induced cardiovascular aging (222). Rutin, an antioxidant plant compound, inhibits SASP expression across various cell lineages, including IL-6, IL-8, IL-1α, IL-1β, CXCL3, MMP3, and GM-CSF (223). In summary, while senomorphics exhibit milder anti-aging effects compared to senolytics, they are associated with lower cytotoxicity and fewer side effects.
5.1.3 Immune-activating therapies
Immunotherapy is an emerging strategy aimed at specifically eliminating senescent cells by targeting distinct surface markers. Under normal physiological conditions, immune cells can effectively clear senescent cells. However, in pathological or aging states, senescent cells may evade immune surveillance, hindering their clearance (224). Additionally, senescent cells actively create an immunosuppressive environment that facilitates their survival. Thus, immunotherapies targeting cellular senescence present a promising approach for treating CTR-CVA. Katsuumi G. et al. demonstrated that canagliflozin, an SGLT2 inhibitor, downregulates PD-L1, restoring immune surveillance and promoting the clearance of senescent cells (211, 225). Anti-aging vaccines have also been developed to induce specific immune memory and actively remove senescent cells. For example, the ATRQβ-001 vaccine has been shown to slow the decline in cardiac function associated with cardiovascular aging (226).
Selective neutralizing or blocking antibodies, often categorized as senomorphic agents, have emerged as another therapeutic avenue (227). Lister et al. demonstrated that an Ephrin-B2 neutralizing antibody (B11) could suppress the SASP in senescent fibroblasts, thereby mitigating pulmonary fibrosis in mice. Furthermore, serum chemokine CCL17 has been identified as a potential biomarker for diagnosing and monitoring vascular stiffness and aging. Knockout of CCL17 in mice has been shown to slow vascular remodeling and inhibit vascular aging (210). The integrin family also may provide novel targets for the prediction and intervention of CTR-CVA. Sun et al. demonstrated that integrin β4 (ITGB4) can induce celluar senescence of vascular endothelial cells by activating the H-ras/caveolin-1/AP-1 axis, thereby exacerbating endothelial dysfunction (228). In cancer therapy, the signaling of the integrin-β family (ITGB4/ITGA3/5/6) may be overactivated (117), which can lead to endothelial celluar senescence, vascular sclerosis, and myocardial dysfunction through signal transduction (229, 230). Therefore, identifying key cytokines in the context of cancer treatment and targeting them to improve the inflammatory microenvironment is critical, as this may enhance therapies aimed at addressing cardiovascular aging.
However, the low specificity of immunotherapy may lead to immune overactivation, particularly given the heterogeneity of senescent cells. Consequently, careful selection of antigens and rigorous safety testing of vaccines are essential for clinical applications. Recently, urokinase-type plasminogen activator receptor (uPAR)-specific CAR-T cells have shown promise in efficiently eliminating senescent cells in a pulmonary fibrosis mouse model, representing a potential strategy for aging-related diseases (231).
5.2 Non-pharmacological therapy
In addition to pharmacological interventions, supportive therapies—such as dietary modifications, physical exercise, and mental health support—are also recommended to mitigate the progression of cardiovascular aging (232).
Dietary interventions play a key role in reducing ROS production, limiting the accumulation of nuclear and mitochondrial DNA damage, and maintaining mitochondrial homeostasis. Long-term caloric restriction has been consistently linked to a reduced incidence of age-related diseases (233, 234). Intermittent fasting, caloric restriction, and glucose restriction may elevate NAD+ levels, potentially offering protective effects against stress-induced cardiovascular aging (235). Nevertheless, as current evidence primarily comes from preclinical studies, clinical translation should proceed with caution to avoid synergistic toxicity. Moreover, methionine restriction and ketogenic diets contribute to improved cellular metabolism and aging mitigation. Aerobic exercise training (AET) has been shown to promote myocardial ischemia recovery by preventing the reduction of circulating endothelial progenitor cells in mice treated with a combination of doxorubicin and cyclophosphamide (32). Given the similarities in the mechanisms between cancer treatment-induced cardiovascular aging and cellular senescence, the supportive therapies outlined above may offer promise in addressing cancer treatment-induced cardiovascular aging.
Additionally, insufficient sleep is a significant risk factor for accelerating cardiovascular aging (236). A quiet, safe sleep environment and adequate rest are essential to prevent cellular damage and senescence. Psychological distress, mental stress, and cognitive biases associated with cancer and its treatments also require attention and support from both society and family.
6 Discussion
Advancements in cancer therapy have significantly improved the survival rates of patients with cancers. However, this progress has been accompanied by an escalating risk of cardiovascular toxicities in these patients. Striking an optimal balance between minimizing cancer treatment-related cardiac events and maximizing antitumor efficacy remains a challenge. Numerous studies have highlighted cellular senescence as a critical initiator and mediator of therapy-induced late side effects. This review focuses on exploring the relationships among cellular senescence, CTR-CVA, and CTR-CVT, aiming to build a bridge between the field of Aging and the field of Cardio-Oncology.
Cellular senescence is considered as a key initiator and mediating factor of CTR-CVT. In the field of Cardio-Oncology, CTR-CVT is a broad term that encompasses various cardiac toxicities under different treatment modalities, including all the adverse effects of cancer treatments on the cardiovascular system. As one of the core subsets of CTR-CVT, CTR-CVA possesses non-negligible independent research value, which is mainly manifested in the following aspects. (i) Differences in intrinsic mechanisms. Traditional CTR-CVT mostly emphasizes the damage caused by tumor treatments to cardiovascular cells. The intrinsic mechanisms are mostly apoptosis or necrosis of cells. In contrast, CTR-CVA emphasizes the long-term maintenance of a non-lethal pathway. The intrinsic mechanism is the activation of cellular senescence and related pathway, which ultimately leads to the acceleration of tissue aging. It has a more obvious time dependence, so the latency period is longer. (ii) Differences in the focuses of monitoring and intervention. The monitoring of CTR-CVA, on the basis of that of CTR-CVT, introduces the focus on the pathology of cellular senescence and tissue aging. The key points of intervention are to reduce senescent cells, improve the senescent phenotype, and rejuvenate the cardiovascular environment. However, the precise molecular mechanisms of CTR-CVA remain unclear, and the role of senescence in some cancer drugs prone to causing cardiotoxicity (such as immunotherapeutic agents) has not been validated by basic experiments, which hinders the development of targeted therapies.
Additionally, the lack of accurate monitoring methodologies for CTR-CVA presents another major challenge. To mitigate the side effects of CTR-CVA, future research should focus on the following areas: (i) Large-cohort, multi-center studies to identify precise humoral and imaging biomarkers of cardiac aging, which would enable early prediction of CTR-CVA onset, diagnosis of its severity, and assessment of the efficacy of related treatments; (ii) The use of advanced models such as organoids and engineered mouse models to investigate the molecular mechanisms underlying the onset and progression of CTR-CVA, thereby identifying potential therapeutic targets (iii) The development of high-throughput screening technologies to identify targeted drugs for CTR-CVA treatment, enhancing treatment specificity while minimizing toxicity to normal tissues and organs. Emerging technologies like single-cell sequencing, spatial transcriptomics, and artificial intelligence are highly relevant to the rapidly evolving field of cardio-oncology, offering significant promise for both research and clinical care. Through interdisciplinary collaboration among oncologists, cardiologists, materials scientists, and engineers, we aim to integrate cancer treatment with cardiovascular aging management across clinical pathways. By combining multi-omics technologies with AI-assisted detection, this approach will enable precise diagnosis, effective prevention, and systematic management of CTR-CVA, ultimately improving long-term outcomes for patients with cancers and survivors.
Statements
Author contributions
XL: Investigation, Writing – original draft, Conceptualization, Funding acquisition, Writing – review & editing. ZW: Writing – original draft, Investigation. YL: Writing – review & editing, Supervision. XL: Supervision, Writing – review & editing, Funding acquisition.
Funding
The author(s) declared that financial support was received for this work and/or its publication. We discloses support for the research of this work from the National Natural Science Foundation of China (No. 82071984, China), the Special Fund for Science and Technology Program of Jiangsu Province (No. BE2023759, China), the Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. SJCX23_2106, China).
Acknowledgments
We thank all members of the team and acknowledge the support from various funds.
Conflict of interest
The author(s) declared that this work was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declared that generative AI was not used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Miller KD Nogueira L Devasia T Mariotto AB Yabroff KR Jemal A et al Cancer treatment and survivorship statistics, 2022. CA Cancer J Clin. (2022) 72(5):409–36. 10.3322/caac.21731
2.
Herrmann J Lenihan D Armenian S Barac A Blaes A Cardinale D et al Defining cardiovascular toxicities of cancer therapies: an international cardio-oncology society (IC-OS) consensus statement. Eur Heart J. (2022) 43(4):280–99. 10.1093/eurheartj/ehab674
3.
Oeffinger KC Mertens AC Sklar CA Kawashima T Hudson MM Meadows AT et al Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. (2006) 355(15):1572–82. 10.1056/NEJMsa060185
4.
Grunewald M Kumar S Sharife H Volinsky E Gileles-Hillel A Licht T et al Counteracting age-related vegf signaling insufficiency promotes healthy aging and extends life span. Science. (2021) 373(6554):eabc8479. 10.1126/science.abc8479
5.
Liu Y Zhang YL Liu JW Fang FQ Li JM Xia YL . Emergence, development, and future of cardio-oncology in China. Chin Med J. (2018) 131(21):2640–4. 10.4103/0366-6999.244101
6.
Demaria M O'Leary MN Chang J Shao L Liu S Alimirah F et al Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. (2017) 7(2):165–76. 10.1158/2159-8290.CD-16-0241
7.
Richter D Guasti L Walker D Lambrinou E Lionis C Abreu A et al Frailty in cardiology: definition, assessment and clinical implications for general cardiology. A consensus document of the council for cardiology practice (CCP), association for acute cardio vascular care (ACVC), association of cardiovascular nursing and allied professions (ACNAP), European association of preventive cardiology (EAPC), European heart rhythm association (ehra), council on valvular heart diseases (VHD), council on hypertension (CHT), council of cardio-oncology (CCO), working group (WG) aorta and peripheral vascular diseases, WG E-cardiology, WG thrombosis, of the European Society of Cardiology, European primary care cardiology society (EPCCS). Eur J Prev Cardiol. (2022) 29(1):216–27. 10.1093/eurjpc/zwaa167
8.
Sarkar TJ Quarta M Mukherjee S Colville A Paine P Doan L et al Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat Commun. (2020) 11(1):1545. 10.1038/s41467-020-15174-3
9.
Bi Y Qiao X Cai Z Zhao H Ye R Liu Q et al Exosomal mir-302b rejuvenates aging mice by reversing the proliferative arrest of senescent cells. Cell Metab. (2025) 37(2):527–41 e6. 10.1016/j.cmet.2024.11.013
10.
Beyret E Martinez Redondo P Platero Luengo A Izpisua Belmonte JC . Elixir of life: thwarting aging with regenerative reprogramming. Circ Res. (2018) 122(1):128–41. 10.1161/CIRCRESAHA.117.311866
11.
Abdellatif M Rainer PP Sedej S Kroemer G . Hallmarks of cardiovascular ageing. Nat Rev Cardiol. (2023) 20(11):754–77. 10.1038/s41569-023-00881-3
12.
Sedrak MS Cohen HJ . The aging-cancer cycle: mechanisms and opportunities for intervention. J Gerontol A Biol Sci Med Sci. (2023) 78(7):1234–8. 10.1093/gerona/glac247
13.
Ioffe D Bhatia-Patel SC Gandhi S Hamad EA Dotan E . Cardiovascular concerns, cancer treatment, and biological and chronological aging in cancer: jacc family series. JACC CardioOncol. (2024) 6(2):143–58. 10.1016/j.jaccao.2024.02.001
14.
Abe J Martin JF Yeh ET . The future of onco-cardiology: we are not just “Side effect hunters”. Circ Res. (2016) 119(8):896–9. 10.1161/CIRCRESAHA.116.309573
15.
GBD 2021 US Burden of Disease and Forecasting Collaborators, MokdadAHBisignanoCHsuJMBryazkaDCaoSBhattacharjeeNVet alBurden of disease scenarios by state in the USA, 2022–50: a forecasting analysis for the global burden of disease study 2021. Lancet. (2024) 404(10469):2341–70. 10.1016/S0140-6736(24)02246-3
16.
Lyon AR Lopez-Fernandez T Couch LS Asteggiano R Aznar MC Bergler-Klein J et al 2022 ESC guidelines on cardio-oncology developed in collaboration with the European hematology association (EHA), the European society for therapeutic radiology and oncology (ESTRO) and the international cardio-oncology society (IC-OS). Eur Heart J Cardiovasc Imaging. (2022) 23(10):e333–465. 10.1093/ehjci/jeac106
17.
Franco YL Vaidya TR Ait-Oudhia S . Anticancer and cardio-protective effects of liposomal doxorubicin in the treatment of breast cancer. Breast Cancer. (2018) 10:131–41. 10.2147/BCTT.S170239
18.
Gyongyosi M Lukovic D Zlabinger K Spannbauer A Gugerell A Pavo N et al Liposomal doxorubicin attenuates cardiotoxicity via induction of interferon-related DNA damage resistance. Cardiovasc Res. (2020) 116(5):970–82. 10.1093/cvr/cvz192
19.
Moles E Howard CB Huda P Karsa M McCalmont H Kimpton K et al Delivery of pegylated liposomal doxorubicin by bispecific antibodies improves treatment in models of high-risk childhood leukemia. Sci Transl Med. (2023) 15(696):eabm1262. 10.1126/scitranslmed.abm1262
20.
Lefrak EA Pitha J Rosenheim S Gottlieb JA . A clinicopathologic analysis of Adriamycin cardiotoxicity. Cancer. (1973) 32(2):302–14. 10.1002/1097-0142(197308)32:2/lt;302::aid-cncr2820320205%3E3.0.co;2-2
21.
Von Hoff DD Layard MW Basa P Davis HL Jr. Von Hoff AL Rozencweig M et al Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. (1979) 91(5):710–7. 10.7326/0003-4819-91-5-710
22.
Eom YW Kim MA Park SS Goo MJ Kwon HJ Sohn S et al Two distinct modes of cell death induced by doxorubicin: apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene. (2005) 24(30):4765–77. 10.1038/sj.onc.1208627
23.
Osataphan N Phrommintikul A Chattipakorn SC Chattipakorn N . Effects of doxorubicin-induced cardiotoxicity on cardiac mitochondrial dynamics and mitochondrial function: insights for future interventions. J Cell Mol Med. (2020) 24(12):6534–57. 10.1111/jcmm.15305
24.
Aleman BM Moser EC Nuver J Suter TM Maraldo MV Specht L et al Cardiovascular disease after cancer therapy. EJC Suppl. (2014) 12(1):18–28. 10.1016/j.ejcsup.2014.03.002
25.
Armstrong GT Oeffinger KC Chen Y Kawashima T Yasui Y Leisenring W et al Modifiable risk factors and Major cardiac events among adult survivors of childhood cancer. J Clin Oncol. (2013) 31(29):3673–80. 10.1200/JCO.2013.49.3205
26.
Liu CY Liu YC Wu C Armstrong A Volpe GJ van der Geest RJ et al Evaluation of age-related interstitial myocardial fibrosis with cardiac magnetic resonance contrast-enhanced T1 mapping: mesa (multi-ethnic study of atherosclerosis). J Am Coll Cardiol. (2013) 62(14):1280–7. 10.1016/j.jacc.2013.05.078
27.
Childs BG Gluscevic M Baker DJ Laberge RM Marquess D Dananberg J et al Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. (2017) 16(10):718–35. 10.1038/nrd.2017.116
28.
Sehl ME Carroll JE Horvath S Bower JE . The acute effects of adjuvant radiation and chemotherapy on peripheral blood epigenetic age in early stage breast cancer patients. NPJ Breast Cancer. (2020) 6:23. 10.1038/s41523-020-0161-3
29.
Schwartz CL Hobbie WL Truesdell S Constine LC Clark EB . Corrected QT interval prolongation in anthracycline-treated survivors of childhood cancer. J Clin Oncol. (1993) 11(10):1906–10. 10.1200/JCO.1993.11.10.1906
30.
Piegari E De Angelis A Cappetta D Russo R Esposito G Costantino S et al Doxorubicin induces senescence and impairs function of human cardiac progenitor cells. Basic Res Cardiol. (2013) 108(2):334. 10.1007/s00395-013-0334-4
31.
Suryadevara V Hudgins AD Rajesh A Pappalardo A Karpova A Dey AK et al Sennet recommendations for detecting senescent cells in different tissues. Nat Rev Mol Cell Biol. (2024) 25(12):1001–23. 10.1038/s41580-024-00738-8
32.
Jones LW Fels DR West M Allen JD Broadwater G Barry WT et al Modulation of circulating angiogenic factors and tumor biology by aerobic training in breast cancer patients receiving neoadjuvant chemotherapy. Cancer Prev Res. (2013) 6(9):925–37. 10.1158/1940-6207.CAPR-12-0416
33.
Sanoff HK Deal AM Krishnamurthy J Torrice C Dillon P Sorrentino J et al Effect of cytotoxic chemotherapy on markers of molecular age in patients with breast cancer. J Natl Cancer Inst. (2014) 106(4):dju057. 10.1093/jnci/dju057
34.
Ariffin H Azanan MS Abd Ghafar SS Oh L Lau KH Thirunavakarasu T et al Young adult survivors of childhood acute lymphoblastic leukemia show evidence of chronic inflammation and cellular aging. Cancer. (2017) 123(21):4207–14. 10.1002/cncr.30857
35.
Carlson BW Craft MA Carlson JR Razaq W Deardeuff KK Benbrook DM . Accelerated vascular aging and persistent cognitive impairment in older female breast cancer survivors. Geroscience. (2018) 40(3):325–36. 10.1007/s11357-018-0025-z
36.
Volders ELD Meijer C Steeneken LS Lubberts S Zwart N van Roon AM et al Change in telomere length and cardiovascular risk factors in testicular cancer survivors. Urol Oncol. (2024) 42(1):24.e1–8. 10.1016/j.urolonc.2023.10.010
37.
Zhang L Pitcher LE Yousefzadeh MJ Niedernhofer LJ Robbins PD Zhu Y . Cellular senescence: a key therapeutic target in aging and diseases. J Clin Invest. (2022) 132(15):e158450. 10.1172/JCI158450
38.
Huang W Hickson LJ Eirin A Kirkland JL Lerman LO . Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol. (2022) 18(10):611–27. 10.1038/s41581-022-00601-z
39.
Lopez-Otin C Blasco MA Partridge L Serrano M Kroemer G . Hallmarks of aging: an expanding universe. Cell. (2023) 186(2):243–78. 10.1016/j.cell.2022.11.001
40.
Ma S Ji Z Zhang B Geng L Cai Y Nie C et al Spatial transcriptomic landscape unveils immunoglobin-associated senescence as a hallmark of aging. Cell. (2024) 187(24):7025–44.e34. 10.1016/j.cell.2024.10.019
41.
Kroemer G Maier AB Cuervo AM Gladyshev VN Ferrucci L Gorbunova V et al From geroscience to precision geromedicine: understanding and managing aging. Cell. (2025) 188(8):2043–62. 10.1016/j.cell.2025.03.011
42.
Baker DJ Childs BG Durik M Wijers ME Sieben CJ Zhong J et al Naturally occurring P16(Ink4a)-positive cells shorten healthy lifespan. Nature. (2016) 530(7589):184–9. 10.1038/nature16932
43.
Pu S Wang Q Liu Q Zhao H Zhou Z Wu Q . Nr1d1 mediated cell senescence in mouse heart-derived Sca-1(+)Cd31(-) cells. Int J Mol Sci. (2022) 23(20):12455. 10.3390/ijms232012455
44.
Gradishar WJ Moran MS Abraham J Abramson V Aft R Agnese D et al Breast cancer, version 3.2024, nccn clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2024) 22(5):331–57. 10.6004/jnccn.2024.0035
45.
Shan K Lincoff AM Young JB . Anthracycline-Induced cardiotoxicity. Ann Intern Med. (1996) 125(1):47–58. 10.7326/0003-4819-125-1-199607010-00008
46.
Tan C Tasaka H Yu KP Murphy ML Karnofsky DA . Daunomycin, an antitumor antibiotic, in the treatment of neoplastic disease. Clinical evaluation with special reference to childhood leukemia. Cancer. (1967) 20(3):333–53. 10.1002/1097-0142(1967)20:3/lt;333::aid-cncr2820200302%3E3.0.co;2-k
47.
Maejima Y Adachi S Ito H Hirao K Isobe M . Induction of premature senescence in cardiomyocytes by doxorubicin as a novel mechanism of myocardial damage. Aging Cell. (2008) 7(2):125–36. 10.1111/j.1474-9726.2007.00358.x
48.
Spallarossa P Altieri P Aloi C Garibaldi S Barisione C Ghigliotti G et al Doxorubicin induces senescence or apoptosis in rat neonatal cardiomyocytes by regulating the expression levels of the telomere binding factors 1 and 2. Am J Physiol Heart Circ Physiol. (2009) 297(6):H2169–81. 10.1152/ajpheart.00068.2009
49.
Hodjat M Haller H Dumler I Kiyan Y . Urokinase receptor mediates doxorubicin-induced vascular smooth muscle cell senescence via proteasomal degradation of Trf2. J Vasc Res. (2013) 50(2):109–23. 10.1159/000343000
50.
Bielak-Zmijewska A Wnuk M Przybylska D Grabowska W Lewinska A Alster O et al A comparison of replicative senescence and doxorubicin-induced premature senescence of vascular smooth muscle cells isolated from human aorta. Biogerontology. (2014) 15(1):47–64. 10.1007/s10522-013-9477-9
51.
Altieri P Barisione C Lazzarini E Garuti A Bezante GP Canepa M et al Testosterone antagonizes doxorubicin-induced senescence of cardiomyocytes. J Am Heart Assoc. (2016) 5(1):e002383. 10.1161/JAHA.115.002383
52.
Ferreira A Cunha-Oliveira T Simões RF Carvalho FS Burgeiro A Nordgren K et al Altered mitochondrial epigenetics associated with subchronic doxorubicin cardiotoxicity. Toxicology. (2017) 390:63–73. 10.1016/j.tox.2017.08.011
53.
Xie Z Xia W Hou M . Long intergenic non-coding Rna-P21 mediates cardiac senescence via the wnt/Beta-catenin signaling pathway in doxorubicin-induced cardiotoxicity. Mol Med Rep. (2018) 17(2):2695–704. 10.3892/mmr.2017.8169
54.
Chen L Xia W Hou M . Mesenchymal stem cells attenuate doxorubicin-induced cellular senescence through the Vegf/Notch/Tgf-Β signaling pathway in H9c2 cardiomyocytes. Int J Mol Med. (2018) 42(1):674–84. 10.3892/ijmm.2018.3635
55.
Liu Y Liu Z Xie Y Zhao C Xu J . Serum extracellular vesicles retard H9c2 cell senescence by suppressing Mir-34a expression. J Cardiovasc Transl Res. (2019) 12(1):45–50. 10.1007/s12265-018-9847-4
56.
Huang PP Fu J Liu LH Wu KF Liu HX Qi BM et al Honokiol antagonizes doxorubicin-induced cardiomyocyte senescence by inhibiting Txnip-mediated Nlrp3 inflammasome activation. Int J Mol Med. (2020) 45(1):186–94. 10.3892/ijmm.2019.4393
57.
Mitry MA Laurent D Keith BL Sira E Eisenberg CA Eisenberg LM et al Accelerated cardiomyocyte senescence contributes to late-onset doxorubicin-induced cardiotoxicity. Am J Physiol Cell Physiol. (2020) 318(2):C380–91. 10.1152/ajpcell.00073.2019
58.
Xia W Chen H Xie C Hou M . Long-Noncoding rna Malat1 sponges microrna-92a-3p to inhibit doxorubicin-induced cardiac senescence by targeting Atg4a. Aging. (2020) 12(9):8241–60. 10.18632/aging.103136
59.
Zhuang L Xia W Chen D Ye Y Hu T Li S et al Exosomal Lncrna-Neat1 derived from Mif-treated mesenchymal stem cells protected against doxorubicin-induced cardiac senescence through sponging Mir-221-3p. J Nanobiotechnology. (2020) 18(1):157. 10.1186/s12951-020-00716-0
60.
Gao B Yu W Lv P Liang X Sun S Zhang Y . Parkin overexpression alleviates cardiac aging through facilitating K63-polyubiquitination of Tbk1 to facilitate mitophagy. Biochim Biophys Acta Mol Basis Dis. (2021) 1867(1):165997. 10.1016/j.bbadis.2020.165997
61.
Pillai VB Samant S Hund S Gupta M Gupta MP . The nuclear sirtuin Sirt6 protects the heart from developing aging-associated myocyte senescence and cardiac hypertrophy. Aging. (2021) 13(9):12334–58. 10.18632/aging.203027
62.
Huang P Bai L Liu L Fu J Wu K Liu H et al Redd1 knockdown prevents doxorubicin-induced cardiac senescence. Aging. (2021) 13(10):13788–806. 10.18632/aging.202972
63.
Wen Y Shen F Wu H . Role of C5a and C5ar in doxorubicin-induced cardiomyocyte senescence. Exp Ther Med. (2021) 22(4):1114. 10.3892/etm.2021.10548
64.
Hou J Yun Y Cui C Kim S . Ginsenoside Rh2 mitigates doxorubicin-induced cardiotoxicity by inhibiting apoptotic and inflammatory damage and weakening pathological remodelling in breast cancer-bearing mice. Cell Prolif. (2022) 55(6):e13246. 10.1111/cpr.13246
65.
Li C Jiang S Wang H Wang Y Han Y Jiang J . Berberine exerts protective effects on cardiac senescence by regulating the klotho/Sirt1 signaling pathway. Biomed Pharmacother. (2022) 151:113097. 10.1016/j.biopha.2022.113097
66.
Abdelgawad IY Agostinucci K Ismail SG Grant MKO Zordoky BN . EA.Hy926 cells and huvecs share similar senescence phenotypes but respond differently to the senolytic drug Abt-263. Cells. (2022) 11(13):1992. 10.3390/cells11131992
67.
Lazzarini E Lodrini AM Arici M Bolis S Vagni S Panella S et al Stress-Induced premature senescence is associated with a prolonged qt interval and recapitulates features of cardiac aging. Theranostics. (2022) 12(11):5237–57. 10.7150/thno.70884
68.
Lerida-Viso A Estepa-Fernandez A Morella-Aucejo A Lozano-Torres B Alfonso M Blandez JF et al Pharmacological senolysis reduces doxorubicin-induced cardiotoxicity and improves cardiac function in mice. Pharmacol Res. (2022) 183:106356. 10.1016/j.phrs.2022.106356
69.
Li C Liu Z Chen M Zhang L Shi R Zhong H . Critical role of cathepsin L/V in regulating endothelial cell senescence. Biology. (2022) 12(1):42. 10.3390/biology12010042
70.
Abdelgawad IY Agostinucci K Sadaf B Grant MKO Zordoky BN . Metformin mitigates sasp secretion and lps-triggered hyper-inflammation in doxorubicin-induced senescent endothelial cells. Front Aging. (2023) 4:1170434. 10.3389/fragi.2023.1170434
71.
Hua H Zhao Q Xia J Dai QL Bai SR Wang XB et al Peficitinib ameliorates doxorubicin-induced cardiotoxicity by suppressing cellular senescence and enhances its antitumor activity. Int Immunopharmacol. (2023) 122:110630. 10.1016/j.intimp.2023.110630
72.
Zheng H Liang X Liu B Huang X Shen Y Lin F et al Exosomal mir-9-5p derived from ipsc-mscs ameliorates doxorubicin-induced cardiomyopathy by inhibiting cardiomyocyte senescence. J Nanobiotechnology. (2024) 22(1):195. 10.1186/s12951-024-02421-8
73.
Zhou L Zhai G Tian G . Crif1 attenuates doxorubicin-mediated mitochondrial dysfunction and myocardial senescence via regulating pxdn. Aging. (2024) 16(6):5567–80. 10.18632/aging.205664
74.
Huang C Zhang X Wang S Shen A Xu T Hou Y et al Parp-2 mediates cardiomyocyte aging and damage induced by doxorubicin through Sirt1 inhibition. Apoptosis. (2024) 29(5-6):816–34. 10.1007/s10495-023-01929-y
75.
Wang S Zhang X Hou Y Zhang Y Chen J Gao S et al Sirt6 activates pparα to improve doxorubicin-induced myocardial cell aging and damage. Chem-Biol Interact. (2024) 392:110920. 10.1016/j.cbi.2024.110920
76.
Dabour MS Abdelgawad IY Sadaf B Daniel MR Grant MKO Seelig D et al Losmapimod ameliorates doxorubicin-induced cardiotoxicity through attenuating senescence and inflammatory pathways. Biomed Pharmacother. (2024) 179:117288. 10.1016/j.biopha.2024.117288
77.
Scalise M Cianflone E Quercia C Pagano L Chiefalo A Stincelli A et al Senolytics rejuvenate aging cardiomyopathy in human cardiac organoids. Mech Ageing Dev. (2025) 223:112007. 10.1016/j.mad.2024.112007
78.
Xiong W Li B Pan J Li D Yuan H Wan X et al Mitochondrial amount determines doxorubicin-induced cardiotoxicity in cardiomyocytes. Adv Sci. (2025) 12(12):e2412017. 10.1002/advs.202412017
79.
Igarashi K Sakimoto I Kataoka K Ohta K Miura M . Radiation-induced senescence-like phenotype in proliferating and plateau-phase vascular endothelial cells. Exp Cell Res. (2007) 313(15):3326–36. 10.1016/j.yexcr.2007.06.001
80.
Kim EJ Lee J Jung YR Park JJ Park MJ Lee JS et al Involvement of corin downregulation in ionizing radiation-induced senescence of myocardial cells. Int J Mol Med. (2015) 35(3):731–8. 10.3892/ijmm.2014.2048
81.
Park H Kim CH Jeong JH Park M Kim KS . Gdf15 contributes to radiation-induced senescence through the ros-mediated P16 pathway in human endothelial cells. Oncotarget. (2016) 7(9):9634–44. 10.18632/oncotarget.7457
82.
Hu Y Xia W Hou M . Macrophage migration inhibitory factor serves a pivotal role in the regulation of radiation-induced cardiac senescencethrough rebalancing the Microrna-34a/Sirtuin 1 signaling pathway. Int J Mol Med. (2018) 42(5):2849–58. 10.3892/ijmm.2018.3838
83.
Yamamoto Y Minami M Yoshida K Nagata M Miyata T Yang T et al Irradiation accelerates plaque formation and cellular senescence in flow-altered carotid arteries of apolipoprotein E knock-out mice. J Am Heart Assoc. (2021) 10(14):e020712. 10.1161/JAHA.120.020712
84.
Rawat PS Jaiswal A Khurana A Bhatti JS Navik U . Doxorubicin-Induced cardiotoxicity: an update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed Pharmacother. (2021) 139:111708. 10.1016/j.biopha.2021.111708
85.
Wu L Wang L Du Y Zhang Y Ren J . Mitochondrial quality control mechanisms as therapeutic targets in doxorubicin-induced cardiotoxicity. Trends Pharmacol Sci. (2023) 44(1):34–49. 10.1016/j.tips.2022.10.003
86.
Abdelgawad IY Sadak KT Lone DW Dabour MS Niedernhofer LJ Zordoky BN . Molecular mechanisms and cardiovascular implications of cancer therapy-induced senescence. Pharmacol Ther. (2021) 221:107751. 10.1016/j.pharmthera.2020.107751
87.
Zhao Y Simon M Seluanov A Gorbunova V . DNA damage and repair in age-related inflammation. Nat Rev Immunol. (2023) 23(2):75–89. 10.1038/s41577-022-00751-y
88.
Harman D . Aging: a theory based on free radical and radiation chemistry. J Gerontol. (1956) 11(3):298–300. 10.1093/geronj/11.3.298
89.
Paez-Ribes M Gonzalez-Gualda E Doherty GJ Munoz-Espin D . Targeting senescent cells in translational medicine. EMBO Mol Med. (2019) 11(12):e10234. 10.15252/emmm.201810234
90.
Zhang Z Shen C Wu N Wang J Du W Chen X . Gab1 overexpression alleviates doxorubicin-induced cardiac oxidative stress, inflammation, and apoptosis through Pi3k/Akt signaling pathway. J Cardiovasc Pharmacol. (2022) 80(6):804–12. 10.1097/fjc.0000000000001333
91.
Li X Luo W Tang Y Wu J Zhang J Chen S et al Semaglutide attenuates doxorubicin-induced cardiotoxicity by ameliorating Bnip3-mediated mitochondrial dysfunction. Redox Biol. (2024) 72:103129. 10.1016/j.redox.2024.103129
92.
Fuchs Y Steller H . Programmed cell death in animal development and disease. Cell. (2011) 147(4):742–58. 10.1016/j.cell.2011.10.033
93.
Manning BD Cantley LC . Akt/pkb signaling: navigating downstream. Cell. (2007) 129(7):1261–74. 10.1016/j.cell.2007.06.009
94.
Fujioka Y Otani K Kodama T Okada M Yamawaki H . Autocrine role of senescent cardiac fibroblasts-derived extracellular vesicles. J Vet Med Sci. (2023) 85(11):1157–64. 10.1292/jvms.23-0279
95.
Yang JH Hayano M Griffin PT Amorim JA Bonkowski MS Apostolides JK et al Loss of epigenetic information as a cause of mammalian aging. Cell. (2023) 186(2):305–26.e27. 10.1016/j.cell.2022.12.027
96.
Hannum G Guinney J Zhao L Zhang L Hughes G Sadda S et al Genome-Wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. (2013) 49(2):359–67. 10.1016/j.molcel.2012.10.016
97.
Bollati V Schwartz J Wright R Litonjua A Tarantini L Suh H et al Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev. (2009) 130(4):234–9. 10.1016/j.mad.2008.12.003
98.
Ghosh AK . Acetyltransferase P300 is a putative epidrug target for amelioration of cellular aging-related cardiovascular disease. Cells. (2021) 10(11):2839. 10.3390/cells10112839
99.
Fedintsev A Moskalev A . Stochastic non-enzymatic modification of long-lived macromolecules—a missing hallmark of aging. Ageing Res Rev. (2020) 62:101097. 10.1016/j.arr.2020.101097
100.
Wang W Pan K Chen Y Huang C Zhang X . The acetylation of transcription factor Hbp1 by P300/cbp enhances P16ink4a expression. Nucleic Acids Res. (2012) 40(3):981–95. 10.1093/nar/gkr818
101.
Childs BG Durik M Baker DJ van Deursen JM . Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. (2015) 21(12):1424–35. 10.1038/nm.4000
102.
Nelson G Wordsworth J Wang C Jurk D Lawless C Martin-Ruiz C et al A senescent cell bystander effect: senescence-induced senescence. Aging Cell. (2012) 11(2):345–9. 10.1111/j.1474-9726.2012.00795.x
103.
Xia W Chen H Yang H Zhu L Xie C Hou M . Depletion of sasp senescent cardiomyocytes with senolytic drugs confers therapeutic effects in doxorubicin-related cardiotoxicity. FEBS J. (2024) 291(18):4029–42. 10.1111/febs.17164
104.
Misuth S Uhrinova M Klimas J Vavrincova-Yaghi D Vavrinec P . Vildagliptin improves vascular smooth muscle relaxation and decreases cellular senescence in the aorta of doxorubicin-treated rats. Vascul Pharmacol. (2021) 138:106855. 10.1016/j.vph.2021.106855
105.
Picca A Mankowski RT Burman JL Donisi L Kim JS Marzetti E et al Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat Rev Cardiol. (2018) 15(9):543–54. 10.1038/s41569-018-0059-z
106.
Sahin E Colla S Liesa M Moslehi J Muller FL Guo M et al Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. (2011) 470(7334):359–65. 10.1038/nature09787
107.
Safdar A Bourgeois JM Ogborn DI Little JP Hettinga BP Akhtar M et al Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtdna mutator mice. Proc Natl Acad Sci U S A. (2011) 108(10):4135–40. 10.1073/pnas.1019581108
108.
Chaudhary KR El-Sikhry H Seubert JM . Mitochondria and the aging heart. J Geriatr Cardiol. (2011) 8(3):159–67. 10.3724/SP.J.1263.2011.00159
109.
Wang Y Hekimi S . Mitochondrial dysfunction and longevity in animals: untangling the knot. Science. (2015) 350(6265):1204–7. 10.1126/science.aac4357
110.
Sarraf SA Raman M Guarani-Pereira V Sowa ME Huttlin EL Gygi SP et al Landscape of the parkin-dependent ubiquitylome in response to mitochondrial depolarization. Nature. (2013) 496(7445):372–6. 10.1038/nature12043
111.
Yang L Wang B Guo F Huang R Liang Y Li L et al Ffar4 improves the senescence of tubular epithelial cells by ampk/Sirt3 signaling in acute kidney injury. Signal Transduct Target Ther. (2022) 7(1):384. 10.1038/s41392-022-01254-x
112.
Münz F Lopez Perez R Trinh T Sisombath S Weber KJ Wuchter P et al Human mesenchymal stem cells lose their functional properties after paclitaxel treatment. Sci Rep. (2018) 8(1):312. 10.1038/s41598-017-18862-1
113.
Macias A Diaz-Larrosa JJ Blanco Y Fanjul V Gonzalez-Gomez C Gonzalo P et al Paclitaxel mitigates structural alterations and cardiac conduction system defects in a mouse model of Hutchinson-Gilford progeria syndrome. Cardiovasc Res. (2022) 118(2):503–16. 10.1093/cvr/cvab055
114.
Altieri P Murialdo R Barisione C Lazzarini E Garibaldi S Fabbi P et al 5-Fluorouracil Causes endothelial cell senescence: potential protective role of glucagon-like peptide 1. Br J Pharmacol. (2017) 174(21):3713–26. 10.1111/bph.13725
115.
Qin X Zhao Y Zhang T Yin C Qiao J Guo W et al Trkb agonist antibody ameliorates fertility deficits in aged and cyclophosphamide-induced premature ovarian failure model mice. Nat Commun. (2022) 13(1):914. 10.1038/s41467-022-28611-2
116.
Zhao Q Liu F Zhao Q Zhang J Luo J Li X et al Constitutive activation of Erk1/2 signaling protects against myocardial ischemia via inhibition of mitochondrial fragmentation in the aging heart. Ann Transl Med. (2021) 9(6):479. 10.21037/atm-21-503
117.
Feng C Mao W Yuan C Dong P Liu Y . Nicotine-Induced Chrna5 activation modulates Ces1 expression, impacting head and neck squamous cell carcinoma recurrence and metastasis via mek/erk pathway. Cell Death Dis. (2024) 15(10):785. 10.1038/s41419-024-07178-4
118.
Gkioni L Nespital T Baghdadi M Monzo C Bali J Nassr T et al The geroprotectors trametinib and rapamycin combine additively to extend mouse healthspan and lifespan. Nat Aging. (2025) 5(7):1249–65. 10.1038/s43587-025-00876-4
119.
Feng C Zhao Y Yuan C Lu D Wang S Sun Z et al S Aureus upregulation of Muc13 modulates mucosal remodeling in chronic rhinosinusitis via Mek1/2 and Wnt2b. J Allergy Clin Immunol. (2025) 156(3):679–89. 10.1016/j.jaci.2025.06.023
120.
Baskar R Lee KA Yeo R Yeoh KW . Cancer and radiation therapy: current advances and future directions. Int J Med Sci. (2012) 9(3):193–9. 10.7150/ijms.3635
121.
Taylor C Correa C Duane FK Aznar MC Anderson SJ Bergh J et al Estimating the risks of breast cancer radiotherapy: evidence from modern radiation doses to the lungs and heart and from previous randomized trials. J Clin Oncol. (2017) 35(15):1641–9. 10.1200/jco.2016.72.0722
122.
Herrmann J . Adverse cardiac effects of cancer therapies: cardiotoxicity and arrhythmia. Nat Rev Cardiol. (2020) 17(8):474–502. 10.1038/s41569-020-0348-1
123.
Ungvari Z Orosz Z Labinskyy N Rivera A Xiangmin Z Smith K et al Increased mitochondrial H2o2 production promotes endothelial nf-kappab activation in aged rat arteries. Am J Physiol Heart Circ Physiol. (2007) 293(1):H37–47. 10.1152/ajpheart.01346.2006
124.
Azimzadeh O Sievert W Sarioglu H Merl-Pham J Yentrapalli R Bakshi MV et al Integrative proteomics and targeted transcriptomics analyses in cardiac endothelial cells unravel mechanisms of long-term radiation-induced vascular dysfunction. J Proteome Res. (2015) 14(2):1203–19. 10.1021/pr501141b
125.
Abe JI Allen BG Beyer AM Lewandowski D Mapuskar KA Subramanian V et al Radiation-Induced macrovessel/microvessel disease. Arterioscler Thromb Vasc Biol. (2024) 44(12):2407–15. 10.1161/atvbaha.124.319866
126.
Zhang Y Wang X Li XK Lv SJ Wang HP Liu Y et al Sirtuin 2 deficiency aggravates ageing-induced vascular remodelling in humans and mice. Eur Heart J. (2023) 44(29):2746–59. 10.1093/eurheartj/ehad381
127.
Burdak-Rothkamm S Rothkamm K . Radiation-Induced bystander and systemic effects serve as a unifying model system for genotoxic stress responses. Mutat Res Rev Mutat Res. (2018) 778:13–22. 10.1016/j.mrrev.2018.08.001
128.
Jokar S Marques IA Khazaei S Martins-Marques T Girao H Laranjo M et al The footprint of exosomes in the radiation-induced bystander effects. Bioengineering. (2022) 9(6):243. 10.3390/bioengineering9060243
129.
Zhang B Fu D Xu Q Cong X Wu C Zhong X et al The senescence-associated secretory phenotype is potentiated by feedforward regulatory mechanisms involving Zscan4 and Tak1. Nat Commun. (2018) 9(1):1723. 10.1038/s41467-018-04010-4
130.
Yang Z Zhong W Yang L Wen P Luo Y Wu C . The emerging role of exosomes in radiotherapy. Cell Commun Signal. (2022) 20(1):171. 10.1186/s12964-022-00986-1
131.
Raisi-Estabragh Z Szabo L Schuermans A Salih AM Chin CWL Vago H et al Noninvasive techniques for tracking biological aging of the cardiovascular system: jacc family series. JACC Cardiovasc Imaging. (2024) 17(5):533–51. 10.1016/j.jcmg.2024.03.001
132.
Cen P Cui C Huang J Chen H Wu F Niu J et al Cellular senescence imaging and senolysis monitoring in cancer therapy based on a Beta-galactosidase-activated aggregation-induced emission luminogen. Acta Biomater. (2024) 179:340–53. 10.1016/j.actbio.2024.03.027
133.
Xiang X Dong C Zhou L Liu J Rabinowitz ZM Zhang Y et al Novel pet imaging probe for quantitative detection of senescence in vivo. J Med Chem. (2024) 67(7):5924–34. 10.1021/acs.jmedchem.4c00179
134.
Liu H Lv R Song F Yang Y Zhang F Xin L et al A near-ir ratiometric fluorescent probe for the precise tracking of senescence: a multidimensional sensing assay of biomarkers in cell senescence pathways. Chem Sci. (2024) 15(15):5681–93. 10.1039/d4sc00595c
135.
Li J Bi Z Wang L Xia Y Xie Y Liu Y . Recent advances in strategies for imaging detection and intervention of cellular senescence. Chembiochem. (2023) 24(1):e202200364. 10.1002/cbic.202200364
136.
Topel ML Hayek SS Ko YA Sandesara PB Samman Tahhan A Hesaroieh I et al Sex differences in circulating progenitor cells. J Am Heart Assoc. (2017) 6(10):e006245. 10.1161/JAHA.117.006245
137.
Meenakshi-Siddharthan DV Livia C Peterson TE Stalboerger P Attia ZI Clavell AL et al Artificial intelligence-derived electrocardiogram assessment of cardiac age and molecular markers of senescence in heart failure. Mayo Clin Proc. (2023) 98(3):372–85. 10.1016/j.mayocp.2022.10.026
138.
Cheng S Fernandes VR Bluemke DA McClelland RL Kronmal RA Lima JA . Age-Related left ventricular remodeling and associated risk for cardiovascular outcomes: the multi-ethnic study of atherosclerosis. Circ Cardiovasc Imaging. (2009) 2(3):191–8. 10.1161/CIRCIMAGING.108.819938
139.
Santoro C Arpino G Esposito R Lembo M Paciolla I Cardalesi C et al 2d And 3d strain for detection of subclinical anthracycline cardiotoxicity in breast cancer patients: a balance with feasibility. Eur Heart J Cardiovasc Imaging. (2017) 18(8):930–6. 10.1093/ehjci/jex033
140.
Zhang KW Finkelman BS Gulati G Narayan HK Upshaw J Narayan V et al Abnormalities in 3-dimensional left ventricular mechanics with anthracycline chemotherapy are associated with systolic and diastolic dysfunction. JACC Cardiovasc Imaging. (2018) 11(8):1059–68. 10.1016/j.jcmg.2018.01.015
141.
Obas V Vasan RS . The aging heart. Clin Sci. (2018) 132(13):1367–82. 10.1042/cs20171156
142.
Octavia Y Tocchetti CG Gabrielson KL Janssens S Crijns HJ Moens AL . Doxorubicin-Induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. (2012) 52(6):1213–25. 10.1016/j.yjmcc.2012.03.006
143.
Raj S Franco VI Lipshultz SE . Anthracycline-Induced cardiotoxicity: a review of pathophysiology, diagnosis, and treatment. Curr Treat Options Cardiovasc Med. (2014) 16(6):315. 10.1007/s11936-014-0315-4
144.
Nagiub M Filippone S Durrant D Das A Kukreja RC . Long-Acting Pde5 inhibitor tadalafil prevents early doxorubicin-induced left ventricle diastolic dysfunction in juvenile mice: potential role of cytoskeletal proteins. Can J Physiol Pharmacol. (2017) 95(3):295–304. 10.1139/cjpp-2016-0551
145.
Evin M Redheuil A Soulat G Perdrix L Ashrafpoor G Giron A et al Left atrial aging: a cardiac magnetic resonance feature-tracking study. Am J Physiol Heart Circ Physiol. (2016) 310(5):H542–9. 10.1152/ajpheart.00504.2015
146.
Salih AM Pujadas ER Campello VM McCracken C Harvey NC Neubauer S et al Image-Based biological heart age estimation reveals differential aging patterns across cardiac chambers. J Magn Reson Imaging. (2023) 58(6):1797–812. 10.1002/jmri.28675
147.
Kleijn SA Pandian NG Thomas JD Perez de Isla L Kamp O Zuber M et al Normal reference values of left ventricular strain using three-dimensional speckle tracking echocardiography: results from a multicentre study. Eur Heart J Cardiovasc Imaging. (2015) 16(4):410–6. 10.1093/ehjci/jeu213
148.
Grune J Yamazoe M Nahrendorf M . Electroimmunology and cardiac arrhythmia. Nat Rev Cardiol. (2021) 18(8):547–64. 10.1038/s41569-021-00520-9
149.
Chadda KR Ajijola OA Vaseghi M Shivkumar K Huang CL Jeevaratnam K . Ageing, the autonomic nervous system and arrhythmia: from brain to heart. Ageing Res Rev. (2018) 48:40–50. 10.1016/j.arr.2018.09.005
150.
Kim PY Irizarry-Caro JA Ramesh T Iliescu C Lopez-Mattei JC . How to diagnose and manage qt prolongation in cancer patients. JACC CardioOncol. (2021) 3(1):145–9. 10.1016/j.jaccao.2021.01.002
151.
Reardon M Malik M . Qt interval change with age in an overtly healthy older population. Clin Cardiol. (1996) 19(12):949–52. 10.1002/clc.4960191209
152.
Porta-Sanchez A Gilbert C Spears D Amir E Chan J Nanthakumar K et al Incidence, diagnosis, and management of qt prolongation induced by cancer therapies: a systematic review. J Am Heart Assoc. (2017) 6(12):e007724. 10.1161/JAHA.117.007724
153.
Sengupta PP Kluin J Lee SP Oh JK Smits A . The future of valvular heart disease assessment and therapy. Lancet. (2024) 403(10436):1590–602. 10.1016/S0140-6736(23)02754-X
154.
Shah M de A Ináico MH Lu C Schiratti PR Zheng SL Clement A et al Environmental and genetic predictors of human cardiovascular ageing. Nat Commun. (2023) 14(1):4941. 10.1038/s41467-023-40566-6
155.
Lindow T Palencia-Lamela I Schlegel TT Ugander M . Heart age estimated using explainable advanced electrocardiography. Sci Rep. (2022) 12(1):9840. 10.1038/s41598-022-13912-9
156.
Attia ZI Friedman PA Noseworthy PA Lopez-Jimenez F Ladewig DJ Satam G et al Age and sex estimation using artificial intelligence from standard 12-lead ecgs. Circ Arrhythm Electrophysiol. (2019) 12(9):e007284. 10.1161/CIRCEP.119.007284
157.
Brant LCC Ribeiro AH Pinto-Filho MM Kornej J Preis SR Fetterman JL et al Association between electrocardiographic age and cardiovascular events in community settings: the framingham heart study. Circ Cardiovasc Qual Outcomes. (2023) 16(7):e009821. 10.1161/circoutcomes.122.009821
158.
Pietri P Vyssoulis G Vlachopoulos C Zervoudaki A Gialernios T Aznaouridis K et al Relationship between low-grade inflammation and arterial stiffness in patients with essential hypertension. J Hypertens. (2006) 24(11):2231–8. 10.1097/01.hjh.0000249701.49854.21
159.
Yang MX Li QL Wang DQ Ye L Li KM Lin XJ et al Myocardial microvascular function assessed by cmr first-pass perfusion in patients treated with chemotherapy for gynecologic malignancies. Eur Radiol. (2022) 32(10):6850–8. 10.1007/s00330-022-08823-2
160.
Shaw LJ Blankstein R . Next step for hybrid pet-ct imaging: automation of cac scores. JACC Cardiovasc Imaging. (2023) 16(5):688–90. 10.1016/j.jcmg.2023.02.021
161.
Rozanski A Han D Miller RJH Gransar H Slomka P Hayes SW et al Comparison of coronary artery calcium scores among patients referred for cardiac imaging tests. Prog Cardiovasc Dis. (2023) 81:24–32. 10.1016/j.pcad.2023.10.005
162.
Molina JR Barton DL Loprinzi CL . Chemotherapy-Induced ovarian failure: manifestations and management. Drug Saf. (2005) 28(5):401–16. 10.2165/00002018-200528050-00004
163.
Marino F Scalise M Salerno N Salerno L Molinaro C Cappetta D et al Diabetes-Induced cellular senescence and senescence-associated secretory phenotype impair cardiac regeneration and function independently of age. Diabetes. (2022) 71(5):1081–98. 10.2337/db21-0536
164.
Salerno N Marino F Scalise M Salerno L Molinaro C Filardo A et al Pharmacological clearance of senescent cells improves cardiac remodeling and function after myocardial infarction in female aged mice. Mech Ageing Dev. (2022) 208:111740. 10.1016/j.mad.2022.111740
165.
Mourad O Mastikhina O Khan S Sun X Hatkar R Williams K et al Antisenescence therapy improves function in a human model of cardiac fibrosis-on-a-chip. ACS Mater Au. (2023) 3(4):360–70. 10.1021/acsmaterialsau.3c00009
166.
Mehdizadeh M Naud P Abu-Taha IH Hiram R Xiong F Xiao J et al The role of cellular senescence in profibrillatory atrial remodelling associated with cardiac pathology. Cardiovasc Res. (2024) 120(5):506–18. 10.1093/cvr/cvae003
167.
de Oliveira Silva T Lunardon G Lino CA de Almeida Silva A Zhang S Irigoyen MCC et al Senescent cell depletion alleviates obesity-related metabolic and cardiac disorders. Mol Metab. (2025) 91:102065. 10.1016/j.molmet.2024.102065
168.
Suda M Katsuumi G Tchkonia T Kirkland JL Minamino T . Potential clinical implications of senotherapies for cardiovascular disease. Circ J. (2024) 88(3):277–84. 10.1253/circj.CJ-23-0657
169.
Dookun E Walaszczyk A Redgrave R Palmowski P Tual-Chalot S Suwana A et al Clearance of senescent cells during cardiac ischemia-reperfusion injury improves recovery. Aging Cell. (2020) 19(10):e13249. 10.1111/acel.13249
170.
Jia K Dai Y Liu A Li X Wu L Lu L et al Senolytic agent navitoclax inhibits angiotensin ii-induced heart failure in mice. J Cardiovasc Pharmacol. (2020) 76(4):452–60. 10.1097/FJC.0000000000000878
171.
Zhai P Sung EA Shiheido-Watanabe Y Takayama K Tian Y Sadoshima J . Suppression of autophagy induces senescence in the heart. J Mol Cell Cardiol. (2024) 195:83–96. 10.1016/j.yjmcc.2024.08.001
172.
Wu Z Zeng Y Chen H Xiao Z Guo J Abubakar MN et al Peptide-Amphiphilic nanoassemblies as a responsive senolytic navigator for targeted removal of senescent cardiomyocytes to ameliorate heart failure. ACS Appl Mater Interfaces. (2024) 16(38):50282–94. 10.1021/acsami.4c09734
173.
Yousefzadeh MJ Zhu Y McGowan SJ Angelini L Fuhrmann-Stroissnigg H Xu M et al Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. (2018) 36:18–28. 10.1016/j.ebiom.2018.09.015
174.
Kim SG Sung JY Kim JR Choi HC . Fisetin-Induced pten expression reverses cellular senescence by inhibiting the Mtorc2-akt Ser473 phosphorylation pathway in vascular smooth muscle cells. Exp Gerontol. (2021) 156:111598. 10.1016/j.exger.2021.111598
175.
Kim SG Sung JY Kang YJ Choi HC . PPARγ activation by fisetin mitigates vascular smooth muscle cell senescence via the Mtorc2-Foxo3a-autophagy signaling pathway. Biochem Pharmacol. (2023) 218:115892. 10.1016/j.bcp.2023.115892
176.
Kim SG Sung JY Kang YJ Choi HC . Fisetin alleviates cellular senescence through pten mediated inhibition of Pkcδ-Nox1 pathway in vascular smooth muscle cells. Arch Gerontol Geriatr. (2023) 108:104927. 10.1016/j.archger.2023.104927
177.
Mahoney SA Venkatasubramanian R Darrah MA Ludwig KR VanDongen NS Greenberg NT et al Intermittent supplementation with fisetin improves arterial function in old mice by decreasing cellular senescence. Aging Cell. (2024) 23(3):e14060. 10.1111/acel.14060
178.
Nath KA Juncos LA Singh RD Grande JP Croatt AJ Ackerman AW et al The occurrence of senescence in the arteriovenous Fistula in the rat. Kidney360. (2025) 6(1):27–37. 10.34067/KID.0000000605
179.
Ji XM Dong XX Li JP Tai GJ Qiu S Wei W et al Fisetin clears senescent cells through the Pi3k-Akt-Bcl-2/Bcl-Xl pathway to alleviate diabetic aortic aging. Phytother Res. (2025) 39:2757–75. 10.1002/ptr.8507
180.
Zhang E Guo Q Gao H Xu R Teng S Wu Y . Metformin and resveratrol inhibited high glucose-induced metabolic memory of endothelial senescence through Sirt1/P300/P53/P21 pathway. PLoS One. (2015) 10(12):e0143814. 10.1371/journal.pone.0143814
181.
Karnewar S Neeli PK Panuganti D Kotagiri S Mallappa S Jain N et al Metformin regulates mitochondrial biogenesis and senescence through ampk mediated H3k79 methylation: relevance in age-associated vascular dysfunction. Biochim Biophys Acta Mol Basis Dis. (2018) 1115(4 Pt A):1864–28. 10.1016/j.bbadis.2018.01.018
182.
Sung JY Kim SG Kang YJ Choi HC . Metformin mitigates stress-induced premature senescence by upregulating ampkα at Ser485 phosphorylation induced Sirt3 expression and inactivating mitochondrial oxidants. Mech Ageing Dev. (2022) 206:111708. 10.1016/j.mad.2022.111708
183.
Tai S Sun J Zhou Y Zhu Z He Y Chen M et al Metformin suppresses vascular smooth muscle cell senescence by promoting autophagic flux. J Adv Res. (2022) 41:205–18. 10.1016/j.jare.2021.12.009
184.
Pulipaka S Singuru G Sahoo S Shaikh A Thennati R Kotamraju S . Therapeutic efficacies of mitochondria-targeted esculetin and metformin in the improvement of age-associated atherosclerosis via regulating ampk activation. Geroscience. (2024) 46(2):2391–408. 10.1007/s11357-023-01015-w
185.
Almohaimeed GM Alonazi AS Alshammari TK Bin Dayel AF Alghibiwi HK Alamin MA et al Metformin-Mediated protection against immunosenescence in diabetic cardiomyopathy: the potential roles of gdf-15 and klotho proteins. Int Immunopharmacol. (2025) 153:114530. 10.1016/j.intimp.2025.114530
186.
Xu X Pang J Chen Y Bucala R Zhang Y Ren J . Macrophage migration inhibitory factor (Mif) deficiency exacerbates aging-induced cardiac remodeling and dysfunction despite improved inflammation: role of autophagy regulation. Sci Rep. (2016) 6:22488. 10.1038/srep22488
187.
Vion AC Kheloufi M Hammoutene A Poisson J Lasselin J Devue C et al Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc Natl Acad Sci U S A. (2017) 114(41):E8675–e84. 10.1073/pnas.1702223114
188.
Sung JY Lee KY Kim JR Choi HC . Interaction between mtor pathway inhibition and autophagy induction attenuates Adriamycin-induced vascular smooth muscle cell senescence through decreased expressions of P53/P21/P16. Exp Gerontol. (2018) 109:51–8. 10.1016/j.exger.2017.08.001
189.
Tan P Wang H Zhan J Ma X Cui X Wang Y et al Rapamycin-induced mir-30a downregulation inhibits senescence of vsmcs by targeting Beclin1. Int J Mol Med. (2019) 43(3):1311–20. 10.3892/ijmm.2019.4074
190.
Salazar G Cullen A Huang J Zhao Y Serino A Hilenski L et al Sqstm1/P62 and Ppargc1a/pgc-1alpha at the interface of autophagy and vascular senescence. Autophagy. (2020) 16(6):1092–110. 10.1080/15548627.2019.1659612
191.
Park JH Lee NK Lim HJ Ji ST Kim YJ Jang WB et al Pharmacological inhibition of mtor attenuates replicative cell senescence and improves cellular function via regulating the Stat3-Pim1 axis in human cardiac progenitor cells. Exp Mol Med. (2020) 52(4):615–28. 10.1038/s12276-020-0374-4
192.
Li WW Wang HJ Tan YZ Wang YL Yu SN Li ZH . Reducing lipofuscin accumulation and cardiomyocytic senescence of aging heart by enhancing autophagy. Exp Cell Res. (2021) 403(1):112585. 10.1016/j.yexcr.2021.112585
193.
Yan W Cao Y Zhen P Ji D Chai J Xue K et al Decreased autophagy of vascular smooth muscle cells was involved in hyperhomocysteinemia-induced vascular ageing. Clin Exp Pharmacol Physiol. (2021) 48(4):524–33. 10.1111/1440-1681.13442
194.
Dong J Wang Q Gu T Liu G Petrov YV Baulin VE et al Rapamycin functionalized carbon dots: target-oriented synthesis and suppression of vascular cell senescence. J Colloid Interface Sci. (2024) 660:534–44. 10.1016/j.jcis.2024.01.032
195.
Tai GJ Ma YJ Feng JL Li JP Qiu S Yu QQ et al Nlrp3 inflammasome-mediated premature immunosenescence drives diabetic vascular aging dependent on the induction of perivascular adipose tissue dysfunction. Cardiovasc Res. (2025) 121(1):77–96. 10.1093/cvr/cvae079
196.
Tang Q Tang K Markby GR Parys M Phadwal K MacRae VE et al Autophagy regulates cellular senescence by mediating the degradation of Cdkn1a/P21 and Cdkn2a/P16 through Sqstm1/P62-mediated selective autophagy in myxomatous mitral valve degeneration. Autophagy. (2025) 21:1–23. 10.1080/15548627.2025.2469315
197.
You J Sun J Ma T Yang Z Wang X Zhang Z et al Curcumin induces therapeutic angiogenesis in a diabetic mouse hindlimb ischemia model via modulating the function of endothelial progenitor cells. Stem Cell Res Ther. (2017) 8(1):182. 10.1186/s13287-017-0636-9
198.
Lai Y He J Gao X Peng D Zhou H Xu Y et al Involvement of plasminogen activator inhibitor-1 in P300/P53-mediated age-related atrial fibrosis. PeerJ. (2023) 11:e16545. 10.7717/peerj.16545
199.
Gao XY Lai YY Luo XS Peng DW Li QQ Zhou HS et al Acetyltransferase P300 regulates atrial fibroblast senescence and age-related atrial fibrosis through P53/Smad3 axis. Aging Cell. (2023) 22(1):e13743. 10.1111/acel.13743
200.
Luo X Liu P Ye X He J Lai Y Lv Y et al Curcumin improves atrial fibrillation susceptibility by regulating tsrna expression in aging mouse atrium. PeerJ. (2024) 12:e17495. 10.7717/peerj.17495
201.
Sin TK Tam BT Yung BY Yip SP Chan LW Wong CS et al Resveratrol protects against doxorubicin-induced cardiotoxicity in aged hearts through the Sirt1-Usp7 axis. J Physiol (Lond). (2015) 593(8):1887–99. 10.1113/jphysiol.2014.270101
202.
De Angelis A Piegari E Cappetta D Russo R Esposito G Ciuffreda LP et al Sirt1 activation rescues doxorubicin-induced loss of functional competence of human cardiac progenitor cells. Int J Cardiol. (2015) 189:30–44. 10.1016/j.ijcard.2015.03.438
203.
Feng H Mou SQ Li WJ Zhang N Zhou ZY Ding W et al Resveratrol inhibits ischemia-induced myocardial senescence signals and Nlrp3 inflammasome activation. Oxid Med Cell Longev. (2020) 2020:2647807. 10.1155/2020/2647807
204.
Zhang F Wang K Gao F Xuan Y Liu X Zhang Z . Resveratrol pretreatment improved heart recovery ability of hyperglycemic bone marrow stem cells transplantation in diabetic myocardial infarction by down-regulating microrna-34a. Front Pharmacol. (2021) 12:632375. 10.3389/fphar.2021.632375
205.
Linders AN Dias IB Ovchinnikova ES Vermeer M Hoes MF Markousis Mavrogenis G et al Evaluation of senescence and its prevention in doxorubicin-induced cardiotoxicity using dynamic engineered heart tissues. JACC CardioOncol. (2023) 5(3):298–315. 10.1016/j.jaccao.2023.03.012
206.
Li Y Qin R Yan H Wang F Huang S Zhang Y et al Inhibition of vascular smooth muscle cells premature senescence with rutin attenuates and stabilizes diabetic atherosclerosis. J Nutr Biochem. (2018) 51:91–8. 10.1016/j.jnutbio.2017.09.012
207.
Mustafa T Khan I Iqbal H Usman S Naeem N Faizi S et al Rutin and quercetagetin enhance the regeneration potential of young and aging bone marrow-derived mesenchymal stem cells in the rat infarcted myocardium. Mol Cell Biochem. (2023) 478(8):1759–70. 10.1007/s11010-022-04628-5
208.
Hao W Shan W Wan F Luo J Niu Y Zhou J et al Canagliflozin delays aging of huvecs induced by palmitic acid via the ros/P38/jnk pathway. Antioxidants. (2023) 12(4):838. 10.3390/antiox12040838
209.
Wan F He X Xie W . Canagliflozin inhibits palmitic acid-induced vascular cell aging in vitro through ros/erk and ferroptosis pathways. Antioxidants. (2024) 13(7):831. 10.3390/antiox13070831
210.
Zhang Y Tang X Wang Z Wang L Chen Z Qian JY et al The chemokine Ccl17 is a novel therapeutic target for cardiovascular aging. Signal Transduct Target Ther. (2023) 8(1):157. 10.1038/s41392-023-01363-1
211.
Katsuumi G Shimizu I Suda M Yoshida Y Furihata T Joki Y et al Sglt2 inhibition eliminates senescent cells and alleviates pathological aging. Nat Aging. (2024) 4(7):926–38. 10.1038/s43587-024-00642-y
212.
Schafer MJ White TA Iijima K Haak AJ Ligresti G Atkinson EJ et al Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. (2017) 8:14532. 10.1038/ncomms14532
213.
Ogrodnik M Miwa S Tchkonia T Tiniakos D Wilson CL Lahat A et al Cellular senescence drives age-dependent hepatic steatosis. Nat Commun. (2017) 8:15691. 10.1038/ncomms15691
214.
Zhu Y Tchkonia T Pirtskhalava T Gower AC Ding H Giorgadze N et al The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. (2015) 14(4):644–58. 10.1111/acel.12344
215.
Wagner JUG Tombor LS Malacarne PF Kettenhausen LM Panthel J Kujundzic H et al Aging impairs the neurovascular interface in the heart. Science. (2023) 381(6660):897–906. 10.1126/science.ade4961
216.
Roos CM Zhang B Palmer AK Ogrodnik MB Pirtskhalava T Thalji NM et al Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. (2016) 15(5):973–7. 10.1111/acel.12458
217.
Baar MP Brandt RMC Putavet DA Klein JDD Derks KWJ Bourgeois BRM et al Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. (2017) 169(1):132–147.e16. 10.1016/j.cell.2017.02.031
218.
Janssens GE Lin XX Millan-Ariño L Kavšek A Sen I Seinstra RI et al Transcriptomics-Based screening identifies pharmacological inhibition of Hsp90 as a means to defer aging. Cell Rep. (2019) 27(2):467–480.e6. 10.1016/j.celrep.2019.03.044
219.
Fuhrmann-Stroissnigg H Ling YY Zhao J McGowan SJ Zhu Y Brooks RW et al Identification of Hsp90 inhibitors as a novel class of senolytics. Nat Commun. (2017) 8(1):422. 10.1038/s41467-017-00314-z
220.
Spilman P Podlutskaya N Hart MJ Debnath J Gorostiza O Bredesen D et al Inhibition of mtor by rapamycin abolishes cognitive deficits and reduces amyloid-Beta levels in a mouse model of Alzheimer’s disease. PLoS One. (2010) 5(4):e9979. 10.1371/journal.pone.0009979
221.
Park JH Lee NK Lim HJ Ji ST Kim YJ Jang WB et al Author correction: pharmacological inhibition of mtor attenuates replicative cell senescence and improves cellular function via regulating the Stat3-Pim1 axis in human cardiac progenitor cells. Exp Mol Med. (2023) 55(11):2473. 10.1038/s12276-023-01127-5
222.
Zhang J Wang M Ding W Zhao M Ye J Xu Y et al Resolvin E1 protects against doxorubicin-induced cardiotoxicity by inhibiting oxidative stress, autophagy and apoptosis by targeting akt/mtor signaling. Biochem Pharmacol. (2020) 180:114188. 10.1016/j.bcp.2020.114188
223.
Liu H Xu Q Wufuer H Li Z Sun R Jiang Z et al Rutin is a potent senomorphic agent to target senescent cells and can improve chemotherapeutic efficacy. Aging Cell. (2024) 23(1):e13921. 10.1111/acel.13921
224.
Liu Z Liang Q Ren Y Guo C Ge X Wang L et al Immunosenescence: molecular mechanisms and diseases. Signal Transduct Target Ther. (2023) 8(1):200. 10.1038/s41392-023-01451-2
225.
Wang TW Johmura Y Suzuki N Omori S Migita T Yamaguchi K et al Blocking Pd-L1-Pd-1 improves senescence surveillance and ageing phenotypes. Nature. (2022) 611(7935):358–64. 10.1038/s41586-022-05388-4
226.
Pan Y Zhou Z Zhang H Zhou Y Li Y Li C et al The Atrqβ-001 vaccine improves cardiac function and prevents postinfarction cardiac remodeling in mice. Hypertens Res. (2019) 42(3):329–40. 10.1038/s41440-018-0185-3
227.
Widjaja AA Lim WW Viswanathan S Chothani S Corden B Dasan CM et al Inhibition of il-11 signalling extends mammalian healthspan and lifespan. Nature. (2024) 632(8023):157–65. 10.1038/s41586-024-07701-9
228.
Sun C Liu E X Qi L Xu J Zhao J Zhang Y et al Modulation of vascular endothelial cell senescence by integrin Beta4. J Cell Physiol. (2010) 225(3):673–81. 10.1002/jcp.22262
229.
Li S Jiang X Huang W Meng Q Pu L Sun B et al Oscillatory shear stress activates integrin Beta3, blocking autophagic flux in endothelial cells and promoting endothelial cells senescence. Biochim Biophys Acta Mol Cell Res. (2025) 1872(7):119991. 10.1016/j.bbamcr.2025.119991
230.
Lawkowska K Bonowicz K Jerka D Bai Y Gagat M . Integrins in cardiovascular health and disease: molecular mechanisms and therapeutic opportunities. Biomolecules. (2025) 15(2):233. 10.3390/biom15020233
231.
Amor C Feucht J Leibold J Ho YJ Zhu C Alonso-Curbelo D et al Senolytic car T cells reverse senescence-associated pathologies. Nature. (2020) 583(7814):127–32. 10.1038/s41586-020-2403-9
232.
Cagigas ML Twigg SM Fontana L . Ten tips for promoting cardiometabolic health and slowing cardiovascular aging. Eur Heart J. (2024) 45(13):1094–7. 10.1093/eurheartj/ehad853
233.
Colman RJ Anderson RM Johnson SC Kastman EK Kosmatka KJ Beasley TM et al Caloric restriction delays disease onset and mortality in rhesus monkeys. Science (2009) 325(5937):201–4. 10.1126/science.1173635
234.
Roth GS Lane MA Ingram DK Mattison JA Elahi D Tobin JD et al Biomarkers of caloric restriction may predict longevity in humans. Science. (2002) 297(5582):811. 10.1126/science.1071851
235.
Abdellatif M Sedej S Kroemer G . Nad(+) metabolism in cardiac health, aging, and disease. Circulation. (2021) 144(22):1795–817. 10.1161/CIRCULATIONAHA.121.056589
236.
Van Craenenbroeck EM . Sleep deprivation and increased cardiovascular risk: a wake-up call!. Eur J Prev Cardiol. (2021) 28(2):187–8. 10.1177/2047487319890765
Glossary
AAA abdominal aortic aneurysm AAC abdominal aortic constriction ABT-263 Navitoclax AET Aerobic exercise training AF atrial fibrillation AIE aggregation-induced emission AIEgen AIE luminogen ALL acute lymphoblastic leukemia ApoE−/− apolipoprotein E knockout ARF alternative reading frame ASAP acute stress-associated phenotype ATG4a autophagy-related gene 4a ATM ataxia-telangiectasia mutated AVF arteriovenous fistula aVICs activated valvular interstitial cells BAECs bovine aortic endothelial cells CAC coronary artery calcification CAD coronary artery disease CCS Childhood cancer survivor CEPs circulating endothelial progenitor cells CPCs cardiac progenitor cells CpG cytosine—phosphate—guanine CSCs cardiac stem cells CTR-CVA Cancer Therapy-Related Cardiovascular Aging CTR-CVD Cancer Therapy-Related Cardiovascular Diseases CTR-CVT Cancer Therapy-Related Cardiovascular Toxicity CVDs Cardiovascular diseases DDR DNA damage response DOX doxorubicin DRP1 dynamin-related protein 1 D + Q dasatinib and quercetin ECG electrocardiogram ECM extracellular matrix EEAA extrinsic epigenetic age acceleration EFNB Ephrin B EPCs endothelial progenitor cells EVs extracellular vesicles GCS global circumferential strain GEAA grim epigenetic ege ecceleration GLS global longitudinal strain GRS global radial strain HAECs human aortic endothelial cells HATs human atrial fibroblasts HCF human cardiac fibroblasts HClO hypochlorous acid HCMs Human cardiomyocytes HDACs histone deacetylases HF heart failure HGPS Hutchinson-Gilford Progeria Syndrome HO-1 heme oxygenase-1 HUVECs human umbilical vein endothelial cells hsCRP high-sensitivity C-reactive protein hTERT human telomerase reverse transcriptase iCM cardiomyocytes differentiated from iPSC IGF-1 insulinlike growth factor-1 IL-2 interleukin-2 IL-6 interleukin-6 IL-7 interleukin-7 IL-8 interleukin-8 IL-10 interleukin-10 IL-17a interleukin-17a INK-ATTAC Inducible and Regulatable Targeted Apoptosis of Senescent Cells iPSC human induced pluripotent stem cells IRI ischemia-reperfusion injury LA left atrium LV left ventricular LVEF left ventricular ejection fraction LV Twist left ventricular twist mPTPs mitochondrial permeability transition pores mtDNA mitochondrial DNA MACE major adverse cardiovascular event MAFs mouse atrial fibroblasts MCP1 monocyte chemoattractant protein-1 MDA malondialdehyde MEFs mouse embryonic fibroblasts MFF mitochondrial fission factor MI myocardial infarction MMVD myxoid mitral valve degeneration MQC mitochondrial quality control MSCs mesenchymal stem cells NLRP3 NOD-like receptor family pyrin domain containing 3 NMCM neonatal mouse cardiomyocytes NRCMs Neonatal rat cardiomyocytes PEAA phenotypic epigenetic age acceleration PET positron emission tomography PGC-1α/β peroxisome proliferator-activated receptor gamma co-activator 1-Alpha/Beta PI perfusion index PINK1 PTEN-induced putative kinase 1 PWV pulse-wave velocity QTc Corrected QT interval RIBE radiation-induced by-stander effect RIC radiation-induced cardiotoxicity ROS reactive oxygen species SASP senescence-associated secretory phenotype SD Sprague-Dawley Serpins serine protease inhibitors SIRT1 sirtuin1 SNRT sinus node recovery time SOD superoxide dismutase TKIs tyrosine kinase inhibitors TNF-α tumor necrosis factor-α TRF1 telomeric repeat-binding factor 1 TRF2 telomeric repeat-binding factor 2 TXNIP thioredoxin-interacting protein T2DM type 2 diabetes mellitus uPAR urokinase-type plasminogen activator receptor VEGFA vascular endothelial growth factor A VEGFR-2 vascular endothelial growth factor receptor-2 VSMCs vascular smooth muscle cells XIAP X-linked inhibitor of apoptosis protein 2D two-dimensional 3D TTE three-dimensional transthoracic echocardiography 5-FU 5-fluorouracil.
Summary
Keywords
cancer therapy, cardiovascular aging, cellular senescence, mechanism, monitoring
Citation
Lin X, Wu Z, Liu Y and Liao X (2026) Cancer therapy-related cardiovascular aging: mechanisms, monitoring, and intervention strategies. Front. Cardiovasc. Med. 12:1668623. doi: 10.3389/fcvm.2025.1668623
Received
18 July 2025
Revised
04 December 2025
Accepted
08 December 2025
Published
08 January 2026
Volume
12 - 2025
Edited by
Ariane Vieira Scarlatelli Macedo, Santa Casa of Sao Paulo, Brazil
Reviewed by
Chen Feng, Shandong University, China
Shimaa Hussein Kotb, Sphinx University, Egypt
Joao Pedro Passos Dutra, CEPON, Brazil
Updates
Copyright
© 2026 Lin, Wu, Liu and Liao.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Xiang Liao liaoxiang025@126.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.