 Zhen Zhang
Zhen Zhang Shasha Liu
Shasha Liu Bin Zhang
Bin Zhang Liang Qiao3
Liang Qiao3 Yi Zhang
Yi Zhang Yi Zhang
Yi Zhang- 1Biotherapy Center, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2Department of Hematology/Oncology, School of Medicine, Northwestern University, Chicago, IL, United States
- 3Department of Microbiology and Immunology, Stritch School of Medicine, Health Sciences Division, Loyola University Chicago, Maywood, IL, United States
- 4Fels Institute for Cancer Research and Molecular Biology, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, United States
- 5Cancer Center, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 6Henan Key Laboratory for Tumor Immunology and Biotherapy, Zhengzhou, China
- 7School of Life Sciences, Zhengzhou University, Zhengzhou, China
Tumor immunotherapy is a promising therapeutic strategy for patients with advanced cancers. T cells are key mediators of antitumor function that specifically recognize and react to tumor-expressing antigens and have proven critical for cancer immunotherapy. However, T cells are not as effective against cancer as expected. This is partly because T cells enter a dysfunctional or exhausted state, which is characterized by sustained expression of inhibitory receptors and a transcriptional state distinct from that of functional effector or memory T cells. T cell dysfunction induces the out of control of tumors. Recently, T cell dysfunction has been investigated in many experimental and clinical settings. The molecular definition of T cell dysfunction and the underlying causes of the T cell dysfunction has been advanced regardless of the fact that the pathways involved are not well elucidated, which proposing promising therapeutic opportunities in clinic. In this review, we will discuss the recent advances in the molecular mechanisms that affect TME and induce T cell dysfunction, and the development of promising immunotherapies to counteract the mechanisms of tumor-induced T cell dysfunction. Better understanding these underlying mechanisms may lead to new strategies to improve the clinical outcome of patients with cancer.
Introduction
Cancer immunotherapy is a transformative strategy that utilizes the immune system of the body to treat cancer. T cells destruct tumor cells by recognizing and reacting to tumor-associated antigens through their T cell receptors (TCRs) (Kishton et al., 2017). Considerable progress has been made in the development of immunotherapy techniques that enhance T cell anti-tumor immunity, including adoptive transfer of tumor infiltrating lymphocytes (TILs), endogenous peripheral blood-derived T cells (ETC), chimeric antigen receptor-engineered T cells (CAR-T), and TCR-engineered T cells (TCR-T). In addition, neoantigen vaccines and checkpoint blockade therapies using anti-programed cell death 1 (PD-1) and anti-PD-ligand 1 (PD-L1) have shown potent therapeutic effects in patients with advanced cancer (Chapuis et al., 2016b; Yee and Lizee, 2017; Boyiadzis et al., 2018; Yee, 2018; Riley et al., 2019). These encouraging results demonstrate that T cell-based cancer therapies offer great promise in inducing complete responses in patients against several types of cancer (Maude et al., 2014; Yee et al., 2015). However, many patients who have responded to T cell-based therapies do not achieve durable clinical responses. Mechanisms underlying resistance or short-term response to these therapies remain largely unknown. Some studies suggest that the efficacy of immunotherapy is limited by the generation of dysfunctional T cells in the tumor microenvironment (TME) (Thommen and Schumacher, 2018; Vodnala et al., 2019). Indeed, negative regulators that mediate T cell dysfunction have been identified in mice and humans. For example, treatment with anti-PD-1/PD-L1 and anti-CTLA-4 immune checkpoint inhibitors reinvigorate dysfunctional TILs and augment their anti-tumor effects (Wherry and Kurachi, 2015; Zarour, 2016; Miller et al., 2019; Wang et al., 2019a). Thus, a better understanding of the mechanisms underlying T cell dysfunction in the TME may lead to novel therapeutic interventions for patients with cancer.
Recent studies have shown that the exhaustion and functional impairment of T cells in the TME is a defining feature of many cancers (Jiang et al., 2015). The TME consists of cancer cells as well as immunosuppressive cells and their associated cytokines, i.e., interleukin-10 (IL-10) and transforming growth factor-β (TGF-β) that facilitate tumor progression and mediate T cell dysfunction. Many studies have shown that dysfunctional CD8+ T cells in cancer are characterized by high expression levels of inhibitory receptors, including PD-1, TIM-3, LAG-3 and immunoreceptor tyrosine-based inhibitory motif domain (TIGIT), which are positively associated with T cell exhaustion. In addition, these dysfunctional CD8+ T cells show impaired production of effector cytokines, such as IL-2, IFN-γ and tumor necrosis factor-α (TNF-α) (Chauvin et al., 2015; Wang et al., 2017). Moreover, T cell dysfunction results in impaired proliferation and decreased production of effector molecules in response to tumor antigens. In this review, we discuss the characteristics of T cell dysfunction in cancer with an aim to elucidate the molecular mechanisms through which TME-derived factors mediate T cell dysfunction factors, and this may promote the exploration of novel strategies for restoring intratumoral T cell function, which can further enhance immunotherapy efficacy.
Hallmarks of T Cell Dysfunction in Cancer
Within the heterogeneous TME, T cells are a major part of the immune infiltrate. The intratumoral T cell population comprises naive, memory, effector and regulatory T cells (Treg) (Hashimoto et al., 2018). Upon stimulation by an antigen, TCRs activate a cell-intrinsic program that guides T cell differentiation into cytotoxic effector cells capable of clearing the antigen. Following the peak of effector cell expansion and the clearance of the specific antigen, most effector T cells die, with the exception of a small number of memory T cells that survive and provide long-term protection against the antigen (Chang et al., 2014). However, when antigen-experienced T cells are chronically exposed to the same antigen, substantial alterations in T cell activation and differentiation may occur, leading to T cell “dysfunction” or “exhaustion” (Wherry, 2011; Schietinger and Greenberg, 2014). A previous study has shown that the tumor-specific T cell dysfunctional exhaustion state is initiated early after tumor initiation and antigen encounter in a murine model (Schietinger et al., 2016). Dysfunctional CD8+ T cells are characterized by a loss of effector functions, such as cytotoxicity and proliferation. In addition, the upregulation of immune checkpoints and changes in transcriptional and metabolic molecules have been described as hallmarks of T cell dysfunction (Table 1). For example, the single cell RNA sequencing of tumoral T cells from melanoma and lung cancer indicated that T cells expressed genes such as PDCD1 and LAG3 that are associated with T cell dysfunction (Guo et al., 2018; Li H. et al., 2019). Nevertheless, T cell function can be successfully reinvigorated by blocking PD-1 or PD-L1, highlighting the critical role of PD-1/PD-L1 axis in T cell dysfunction. However, activated and functional CD8+ T cells can also overexpress PD-1 in cancer patients (Fourcade et al., 2010), and not all PD-1+ cells might respond equally to anti-PD-1 therapy (Thommen et al., 2018). It has reported that PD-1+CD38+CD8+ T cells are a population of dysfunctional cells that fail to respond to anti-PD-1 therapy (Verma et al., 2019). Meanwhile, the TME contains a variety of cell types and cytokines (Table 1) that take part in tumor progression, which could contribute to T cell dysfunction (Xia et al., 2019). Therefore, there is growing interest in the identification of the molecular signatures and characteristics that are associated with dysfunctional T cells in cancer (Figure 1).
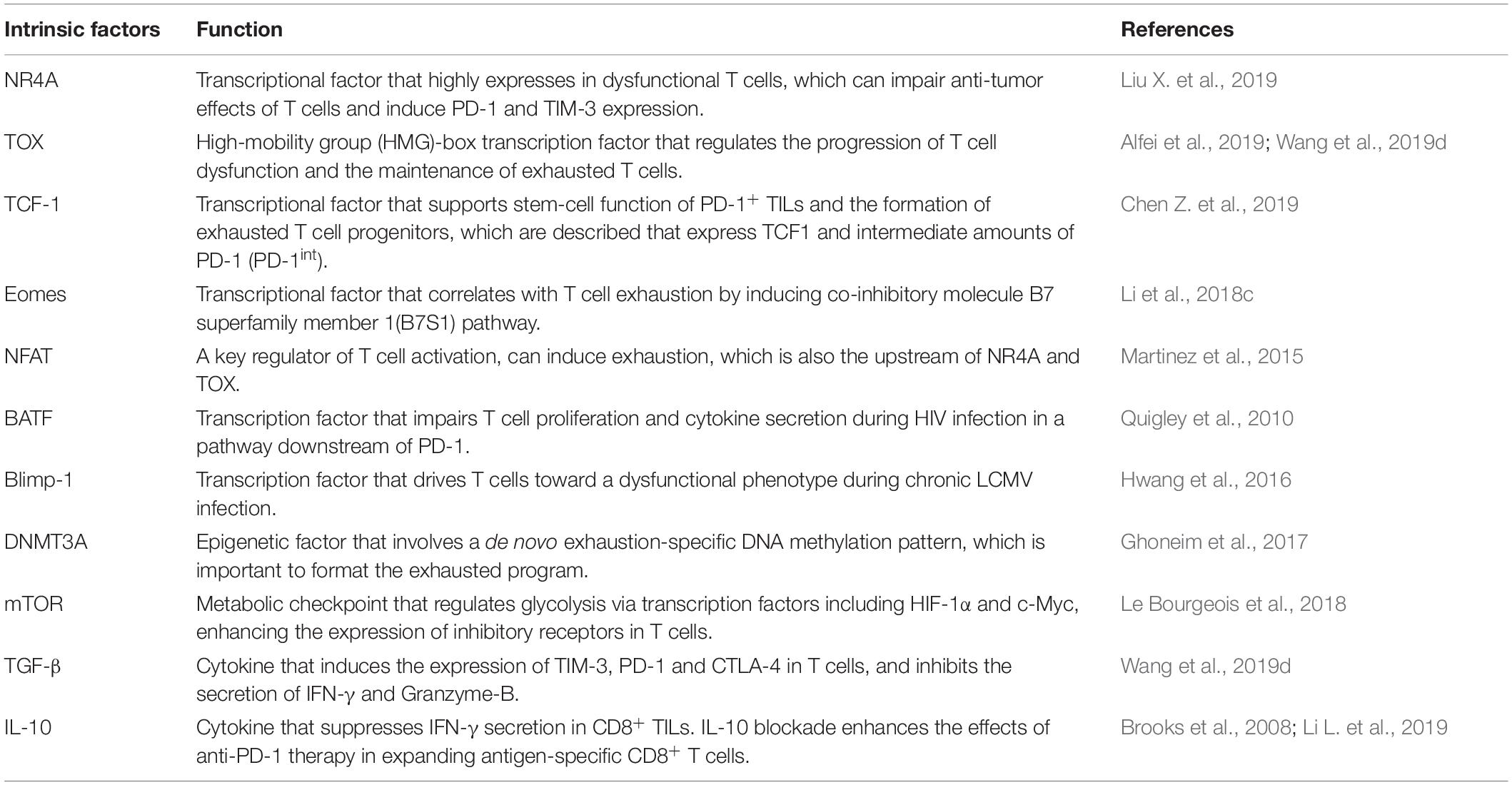
Table 1. Core molecular regulation of T cell dysfunction or exhaustion.
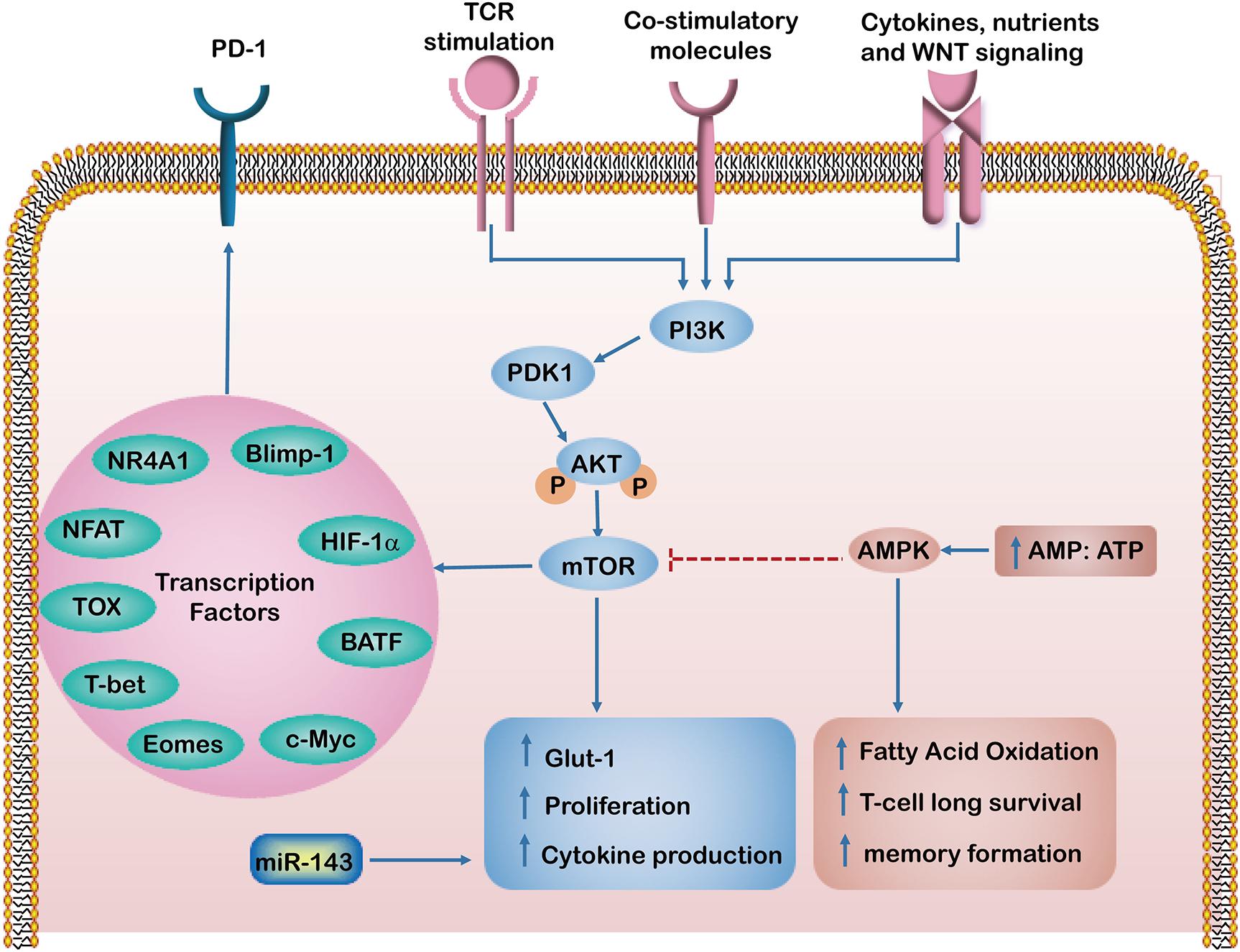
Figure 1. The intrinsic factors regulating T cell dysfunction. In response to T cell receptors (TCRs), co-stimulatory and growth factor cytokines activate PI3K/Akt/mTOR signaling pathways, which induce glucose transporter-1 (Glut-1) expression and enhance T cell proliferation and cytokine production. Activation of mTOR leads to the expression of downstream transcriptional regulators such as HIF-1α and c-Myc. However, an increased AMP to ATP ratio activates AMP-activated protein kinase (AMPK), which in turn inhibits mTOR activity and enhances fatty acid oxidation, which maintains long term T-cell survival and formation of memory T cells. The Transcription factors such as HIF-1α, NR4A1, TOX, Eomes, T-bet, Blimp-1, NFAT and BATF regulate PD-1 expression and have been implicated in T cell exhaustion and dysfunction.
Intrinsic Factors That Induced T Cell Dysfunction
Transcription Factors
It has become increasingly clear that several transcriptional factors, including NR4A1, TOX, Eomes, T-bet, Prdm1 (Blimp-1), NFAT and BATF, regulate the PD-1 expression and are implicated in T cell exhaustion and dysfunction (Wang et al., 2017; Liu X. et al., 2019). For example, NR4A1 was found highly expressed in tolerant or dysfunctional T cells in a mouse model. Overexpression of NR4A1 inhibits effector T cell differentiation, whereas deletion of NR4A1 overcomes T cell tolerance and increases T cell proliferation, enhancing anti-tumor effects. Moreover, expression levels of PD-1 and TIM-3 in T cells were found significantly decreased in NR4A1–/– mice. A mechanistic analysis suggested that NR4A1 is preferentially recruited to binding sites of the transcription factor activator protein 1 (AP-1), where it inhibits effector gene expression by reducing AP-1 function. These findings indicate that NR4A1 is important for inducing T cell dysfunction and represents a promising target for augmenting cancer immunotherapy (Liu X. et al., 2019).
Recently, the high-mobility group (HMG)-box transcription factor TOX was reported as a critical regulator in the progression of T cell dysfunction and the maintenance of exhausted T cells during chronic infection (Alfei et al., 2019). Several studies also showed that TOX may have a role in mediating transcriptional and epigenetic reprograming that are critical for the exhausted CD8+ T cells responses in cancer (Khan et al., 2019). Although the formation of effector and memory T cells is not dependent on TOX, the formation of exhausted T cells was failure without TOX. Robust expression of TOX can translate continuous stimulation that induces T cell exhaustion (Khan et al., 2019). Moreover, TOX and TOX2 as well as NR4A family members are highly induced PD-1 and TIM-3 expression in CAR+ TILs. TOX and TOX2 deficient CAR+ TILs can prevent tumor growth and prolong survival of tumor-bearing mice (Seo et al., 2019). In a mouse model of hepatocellular carcinoma (HCC), TOX was upregulated in exhausted CD8+ T cells, impairing their anti-tumor function. The underlying mechanism involves a TOX-induced decrease in PD-1 degradation and promotion of PD-1 endocytic recycling to the cell surface. Knocking down TOX in tumor-specific CD8+ T cells promoted the anti-tumor effects of these T cells, exhibiting the synergetic role of anti-PD-1therapy (Wang et al., 2019d).
TCF-1 has been implicated in the formation of memory precursor T cells mediated by Wnt signaling pathway (Jeannet et al., 2010). Similarly, TCF-1 is also required for the stem-like functions of TCF1+PD-1+ TILs, which were detected in the blood of patients with melanoma and who had responded to checkpoint blockade (Siddiqui et al., 2019). Most notably, TCF-1 enhances Bcl2 expression via c-Mycb and supports the establishment of exhausted T cell progenitors (Chen Z. et al., 2019). These progenitor cells were described that express TCF1 and intermediate amounts of PD-1 (PD-1int). These TCF1+PD-1int cells give rise to dysfunctional TCF1–PD-1hi TIM-3+ cells, which show resistance to PD-1 blocking therapy. Moreover, TCF1+PD-1int cell survival can be boost by upregulating TOX expression (Mann and Kaech, 2019). Scott found that TOX-deficient tumor-specific T cells failed to persist in cancers, and hypothesized that TOX-induced exhaustion serves as a negative feedback mechanism that prevents activation-induced T cell death and overstimulation of antigen-specific T cells (Scott et al., 2019). These findings suggest that TOX may play a two-blade function in T cell dysfunction or exhaustion.
T-bet and Eomes were found to operate in contrasting ways to facilitate the effector versus memory CD8+ T cell fates (Chang et al., 2014). Enhanced T-bet expression fosters effector differentiation of antigen-specific CD8+ T cells toward the terminally differentiated fate. In contrast, Eomes is highly expressed in memory T cells and is considered important for the maintenance of memory T cells (Knudson et al., 2017). Notably, recently studies identified high expression levels of Eomes in exhausted CD8+ T cells during chronic lymphocytic choriomeningitis virus (LCMV) infection. Interestingly, the CD8+ T cells producing high levels of Eomes also expressed high levels of PD-1. These EomeshiPD-1hi CD8+ T cells co-expressed other inhibitory receptors and displayed limited proliferative capacity (Li et al., 2018b). In addition, Eomes is directly involved in exhaustion of CD8+ TILs via the co-inhibitory molecule B7 superfamily member 1(B7S1) pathway (Li et al., 2018c). However, how increased expression of Eomes promotes CD8+ T cell exhaustion remains elusive.
Studies have shown that NFAT, a key regulator of T cell activation, can induce hyporesponsiveness (anergy and exhaustion) in both CD4+ and CD8+ T cells, if it does not bind AP-1 transcription factors (Martinez et al., 2015). Moreover, TOX and NR4A are important for the transcriptional program of CD8+ T cell exhaustion downstream of NFAT (Seo et al., 2019). Intriguingly, BATF, a transcription factor of the AP-1 family, was found to impair T cell proliferation and cytokine secretion during HIV infection in a pathway downstream of PD-1 (Quigley et al., 2010). Similarly, Blimp-1 was found to drive T cells toward a dysfunctional phenotype during chronic LCMV infection (Hwang et al., 2016). Further identifying how these transcription factors are integrated together to mediate CD4+ and CD8+ T cell exhaustion or dysfunction will provide molecular insights into T cell responses and immunity.
Epigenetic Factors
Emerging evidence indicates that epigenetic states and chromatin landscapes are closely associated with the functional state of dysfunctional or exhausted CD8+ T cells, which are abnormally expressed PD-1 (Pauken et al., 2016; Sen et al., 2016; Kartikasari et al., 2018). Epigenetic components including DNA methylation and histone modifications could control PD-1 expression and T cell exhaustion. For example, DNA methylation enzymes such as DNMT1 and DNMT3B are significantly upregulated in exhausted T cells (Schietinger et al., 2016). Meanwhile, DNA methyltransferase 3A (DNMT3A) has been demonstrated to functionally establish a de novo exhaustion-specific DNA methylation pattern. Inhibition of DNMT3A in these T cells can promote their differentiation toward memory cells. Critically, observations from studies of chronic viral infections indicated a critical role for the demethylation at the promoter region of PD-1 locus in mediating T cell dysfunction (Ghoneim et al., 2017). Inhibition of DNA methylation leads to a revitalized effect on the function of exhausted T cells.
The most widely studied histone lysine methylation sites in T cells are histone 3 lysine 4 (H3K4), and H3K27. H3K4 methylation is associated with transcriptional activation, and H3K27 trimethylation is associated with the repression of genes important for T-cell differentiation and survival (Liu H. et al., 2019). When TCR stimulation and IL-6 or IL-12 treatment were combined, both H3K4me1 and H3K27 acetylation were contributed to increased PD-1 expression (Bally et al., 2016). Moreover, overexpression of miR-155 significantly enhances polycomb repressor complex 2 (PRC2), which restrains T cell exhaustion and sustains CD8+ T cell antitumor responses (Ji et al., 2019). Additionally, EZH2 is a catalytic subunit of PRC2 that can alter gene expression by trimethylating H3K27 (Zhao et al., 2016; He et al., 2017). In a recent study, EZH2 was found to control the polyfunctionality and differentiation of effector T cells (He et al., 2017). Interestingly, inhibition of EZH2 in ovarian and colorectal cancer patients resulted in increased of CXCL9 and CXCL10 production and augmented the infiltration of T cells that eliminate tumors (Nagarsheth et al., 2016; Jones et al., 2018). EZH2 represses the expression of tumor suppressor genes in various cancer cells, thereby promoting cell invasion and driving tumor progression (Bohrer et al., 2010; Hayashi et al., 2011). Thus, EZH2 may be a promising target for cancer immunotherapy (Wang et al., 2019c). Meanwhile, treatment with JQ1-α specific inhibitor of the histone acetylation reader bromodomain-containing protein 4 (BRD4)-results in decreased PD-L1 expression on tumor cells and macrophages, which is correlated with an increase in antitumor T cell activity (Zhu et al., 2016). These findings indicate that pharmacological manipulation of epigenetic mechanisms can alter T cell exhaustion or dysfunction in a clinically relevant manner. Thus, therapies employing hypomethylating agents and PD-1 blockade are a promising strategy in cancer patients.
Metabolic Factors
The metabolic program is a set of biochemical reactions that allows T cells to acquire and utilize nutrients necessary for their survival, proliferation, and functions (Chang et al., 2015). It has been shown that effector T cells use glycolysis for anabolic metabolism for their growth and proliferation. In contrast, memory T cells switch to a non-proliferative form of metabolism, using FAO as a predominant metabolic program and obtaining ATP mainly via OXPHOS (Pearce et al., 2013). Interestingly, the link between antigenic stimulation and metabolic pathway activation appears to be altered in dysfunctional T cells (Le Bourgeois et al., 2018; Sugiura and Rathmell, 2018). Recently, metabolic checkpoints (e.g., AMPK, Myc, HIF-1α and mTOR) that control T cell differentiation have been highlighted as a novel therapeutic targets for immune modulation (Chang and Pearce, 2016). AMPK is a heterotrimeric serine/threonine kinase complex that senses the intracellular AMP/ATP ratio. Activated AMPK can enhance FAO and simultaneously inhibit glucose and mTOR activity. This results in maintaining long-term T cell survival and memory formation (Ma et al., 2017). A previous study demonstrated that the abrogation of phosphoenolpyruvate carboxykinase (Pck1)–glycogen–pentose phosphate pathway (PPP) decreases GSH/GSSG ratios and increases levels of ROS levels, leading to impairment of memory CD8+ T cell formation (Ma et al., 2018). Our previous work demonstrated that miR-143 enhances the anti-tumor effects of CD8+ T cells by promoting memory T cell differentiation and metabolism reprograming by inhibiting of glycolysis targeting glucose transporter-1 (Glut-1) (Zhang et al., 2018). TCR signaling, together with costimulatory molecules and growth factor cytokines, activates phosphatidylinositide 3 kinase (PI3K)/Akt/mTOR signaling pathways, which induce Glut-1 and enhance T cell proliferation and cytokine production (Salmond, 2018). Furthermore, activation of the mTOR pathway and engagement of glycolysis lead to the expression of downstream transcriptional regulators such as HIF-1α and c-Myc, enhancing the expression of inhibitory receptors on T cells (Le Bourgeois et al., 2018). Meanwhile, suppression of Akt and mTOR is required for augmenting activity of the transcription factor FoxO1. Importantly, FoxO1 sustains PD-1 expression, which promotes the differentiation of terminally exhausted T cells (Staron et al., 2014). Therefore, these intrinsic metabolic factors regulate T cell metabolism and activate pathways involved in effector function and exhaustion.
Extrinsic Factors: Tumor Microenvironment
Apart from T cell self-regulation, the interaction between other cells or cytokines in the TME is another important factor that induces T cell dysfunction. Various types of cancers and cytokines compose the TME, including tumor cells, immunosuppressive cells, stromal cells, IL-6 and IL-10, to name a few, which collectively form a complex of immunosuppressive network. These TME components exert potent effects on limiting T cell differentiation and driving T cell dysfunction. In addition, tumor cells and immunosuppressive cells within TME produced highly reactive soluble oxygen and toxic metabolites, which inhibit T cell responses (Maimela et al., 2019). Thus, it is challenging to precisely define the relative contribution of these potential extrinsic factors to T cell function and differentiation in the TME (Figure 2).
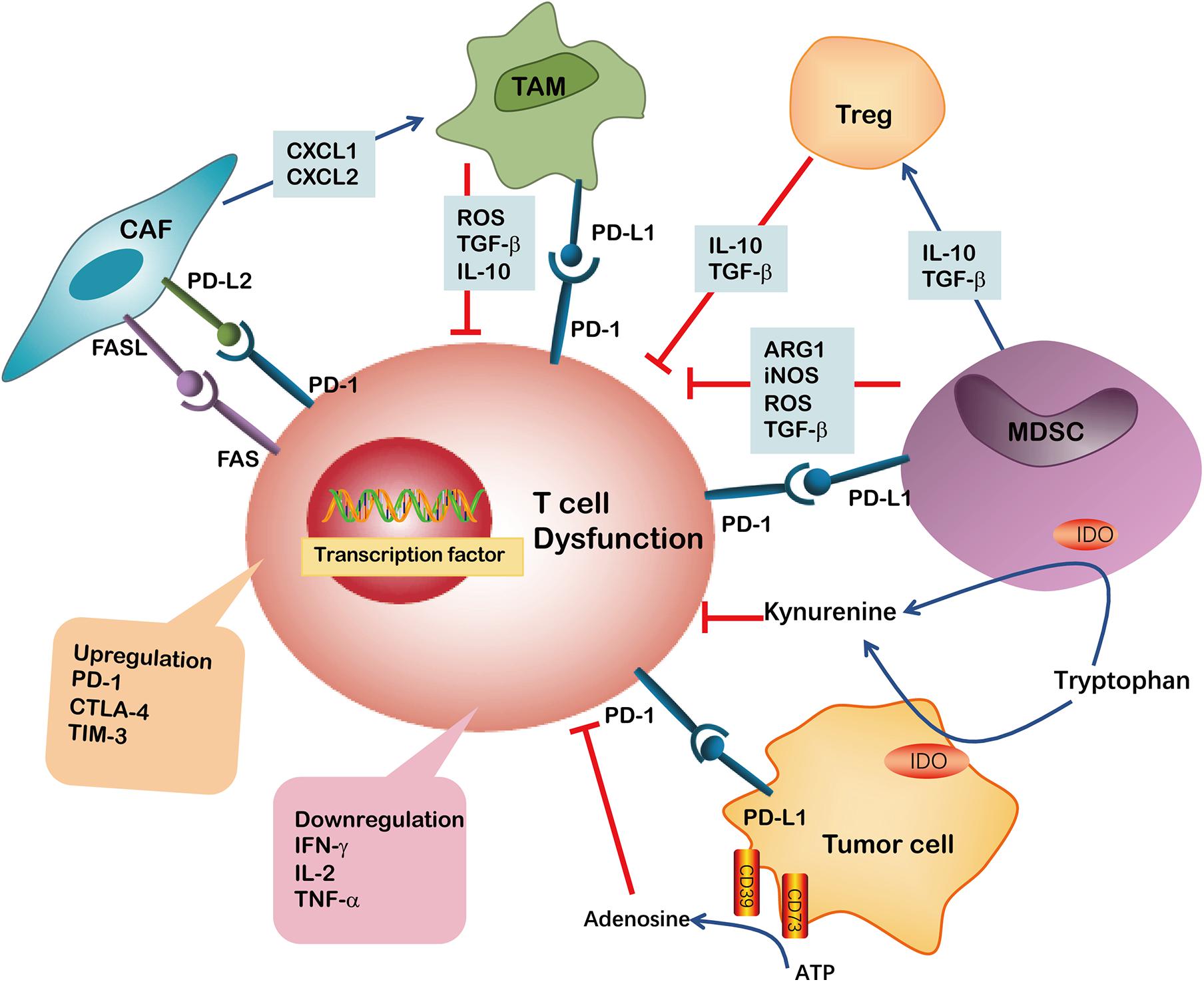
Figure 2. Immunosuppressive cells or factors have been implicated in CD8+ T cell dysfunction in TME. The ARG1, iNOS, TGF-β and ROS are secreted by MDSCs or TAMs and induce CD8+ T cell dysfunction. Both MDSCs and tumor cells may suppress CD8+ T cell proliferation through IDO hydrolyzation of tryptophan in the presence of IFN-γ. Kynurenine inhibits CD8+ T cell activation. MDSCs may additionally produce immunosuppressive cytokines like IL-10, TGF-β and induce Tregs. The upregulation of PD-L1 on MDSCs, TAMs and tumor cells induced CD8+ T cell exhaustion by binding to PD-1 on T cells. Tumor cells also express CD39 and CD73 on their surface, facilitating the metabolism of extracellular ATP into AMP and finally into adenosine, which induce CD8+ T cell dysfunction. CAFs are involved in impairing anti-tumor T cell responses by secreting chemokines such as CXCL1 and CXCL2 to tumors and polarizing them toward the M2 phenotype. Furthermore, the expression of PD-L2 or FASL on CAFs bind to corresponding PD-1 and FAS receptors, respectively, causing CD8+ T cell dysfunction.
Myeloid Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSCs) play pivotal roles in promoting tumor progression and contribute to immunosuppressive function (Ostrand-Rosenberg, 2010; Li et al., 2018e). MDSCs are a heterogeneous group of pathologically activated immature myeloid cells in the TME. The various mechanisms implicated in MDSC-mediated immune suppression include the release of high levels of arginase (ARG)-1, inducible nitric oxide synthase (iNOS), reactive oxygen species (ROS) and cyclooxygenase-2 (COX2) (Chen J. et al., 2019). For example, ARG-1 and iNOS, either separately or in combination, are used by MDSCs to impede CD8+ T cells response to antigens. Indeed, expression of ARG-1 has been reported to decrease CD3ζ-chain biosynthesis, thus impairing T cell function (Rodriguez et al., 2002). In addition, NO alone can suppress CD8+ T cells by inhibiting the phosphorylation and activation of JAK3 and STAT5 transcription factors, as well as inducing T-cell apoptosis (Wu et al., 2015). Elevated levels of ROS in the TME was involved in limiting T cell growth, differentiation, and ultimately promoting the exhaustion of T cells. ROS produced by MDSCs and other cells may interact with T cells and cause oxidative stress which may induce CD8+ T cell hypo-responsiveness in cancer (Chen et al., 2016).
Interestingly, MDSCs produce high levels of IDO, which catabolizes tryptophan and generates kynurenine. Depletion of tryptophan and induction of kynurenine lead to blockade of clonal expansion of activated T cells. Experimental studies indicate that IDO hydrolyzation of tryptophan represents an important mechanism by which MDSCs suppress proliferation and survival of tumor infiltrating CD8+ T cells and CD8+ T cells homed to the lymph nodes. Other reports demonstrated that STAT3-dependent IDO expression mediates immunosuppressive effects of MDSCs in breast cancer, in which MDSCs dramatically inhibit the proliferation of CD8+ T cells and their production of IFN-γ (Yu et al., 2013). Data from our previous studies indicate that CD11b+CD33+ MDSCs in tumor tissues from NSCLC patients express surface ectonucleotidases CD39 and CD73. Moreover, TGF-β stimulates CD39 and CD73 expression, thereby inhibiting autologous CD8+ T cell proliferation and function (Li et al., 2017). Thus, MDSCs play an important role in repressing CD8+ T cell proliferation thus inducing CD8+ T-cell exhaustion in TME.
Tumor-Associated Macrophages
Macrophages play a critical role in innate immunity and are responsible for defending the host against foreign pathogens. They can be further classified into pro-inflammatory M1 or anti-inflammatory M2 macrophages. M1 cells are characterized by the high expression of various pro-inflammatory cytokines and contribute to promoting the Th1 response. They also have strong microbicidal and tumoricidal activity (Sica and Mantovani, 2012; Ambade et al., 2016). M2 cells, also known as alternatively activated macrophages, are activated by Th2 cytokines (e.g., IL-4, IL-10, and IL-13) and secret high levels of anti-inflammatory cytokines such as IL-10, and TGF-β. M2 macrophages primarily contribute to immune-suppression and favor tumor promotion (Liu S. et al., 2019).
Tumor-associated macrophages (TAMs) are generally characterized as an M2-like macrophage phenotype in TME. They can be identified by the expression of CD163, CD204 or CD206 in human derived TAMs, and F4/80, CD163, CD206, ARG1 or Ym1 in murine-derived TAMs (Cassetta et al., 2016; Chen Y. et al., 2017; Benner et al., 2019). TAMs often accelerate the progression of untreated cancer and negatively influence the efficacy of anticancer drugs, including checkpoint blockade immunotherapies. Therefore, TAMs are shown to be closely correlated with a poor prognosis of patients with cancer. Li and colleagues demonstrated that TAMs-secreted IL-10 promotes cancer stem cell-like properties and tumor growth in NSCLC; High levels of IL-10 are associated with a poor prognosis of NSCLC patients (Yang and Zhang, 2017). TAMs can induce immunosuppression mainly through several ways: (1) TAMs may induce the expression of PD-L1 in monocytes, which binds to PD-1 on the surface of CD8+ T cells, inducing T cell exhaustion; (2) TAMs secret numerous immunosuppressive cytokines and factors, including IL-10, TGF-β and ROS, which induce CD8+ TIL exhaustion and dysfunction; and (3) TAMs can directly inhibit CD8+ T cells cytotoxicity through the depletion of the amino acids, such as L-arginine and tryptophan. In addition, these functions indicate that TAMs produce high levels of IDO to inhibit CD8+ T cells cytotoxicity. Collectively, TAMs are a highly active subset of immunosuppressive cells promoting tumor survival and immune evasion (Jiang et al., 2015; Yang and Zhang, 2017).
Efforts are underway to either deplete M2 cells or convert the M2 phenotype into M1 cells (inflammatory) in most tumors. In a mouse model of ovarian cancer, it was shown that tumor rejection by CAR-T cells required the presence of M1 macrophages, suggesting that tumor-reactive T cells were not sufficient to completely eliminate the tumor on their own (Yeku et al., 2017). We have recently reported that M1 macrophages converted from M2 macrophages by Pseudomonas aeruginosa-mannose-sensitive hemagglutinin (PA-MSHA) can enhance the anti-tumor immune response. This effect primarily relies on activation of toll like receptor 4 (TLR4) (Yang et al., 2015).
Cancer Associated Fibroblasts
Cancer-associated fibroblasts (CAFs), the most abundant stromal population, secrete immunomodulatory factors in TME and are emerging as suppressive mediators of T cell immunity. CAFs can recruit myeloid cells to tumors via the secretion of chemokines such as CXCL1 and CXCL2 to tumors. They can also polarize these recruited myeloid cells toward the M2 phenotype (Zhen et al., 2017). Similarly, CAFs can further bolster the immunosuppressive TME by recruiting MDSCs and Tregs. Thus, the crosstalk between CAFs and other cells contributes to the immunosuppressive TME. Other studies revealed a new biological function for CAFs, showing that these cells directly suppress anti-tumor T-cell responses by a mechanism dependent on immune checkpoint activation. One of the underlying mechanisms was the upregulation of FAS/FASL and PD-1/PD-L2 on CD8+ T cells and CAFs, respectively, which drives the dysfunction of tumor-specific CD8+ T cells (Lakins et al., 2018). Targeting CAFs or CAFs-related pathways can be considered as a powerful strategy for attenuating stromal barriers and promoting cancer immunotherapy.
Soluble Mediators
Immunosuppressive cytokines in TME can be produced by tumor cells, MDSCs or CAFs and are crucial factors that mediate T-cell exhaustion and dysfunction (Chen J. et al., 2017). For example, the TGF-β signaling pathway plays an important role in tumor suppression and paradoxically, plays a role in tumor promotion. In general, TGF-β mediates tumor suppression via the inhibition of cancer cell proliferation and induction of cancer stem cell senescence by diminishing their self-renewing capability during the early stages of tumor development. Additionally, TGF-β promotes tumor progression and metastasis through the modulation of immune responses in later stages (Colak and Ten Dijke, 2017). TGF-β derived TAMs exert its function by inducing TIM-3, PD-1 and CTLA-4 expression in T cells and inhibiting IFN-γ and Granzyme-B secretion in a dose-dependent manner. Treatment with anti-TGF-β antibody restored the impaired T cell cytotoxic function in MPE. Furthermore, inhibiting TGF-β signaling in CD8+ T cells using dominant negative receptors can improve the function of exhausted cells (Wang et al., 2019b).
IL-10 in TME is primarily secreted by cancer cells, TAMs, natural killer cells (NK) and CD4+ Tregs (Landskron et al., 2014). A study of patients with ovarian carcinoma demonstrated that the tumor-infiltrating follicular regulatory T (Tfr) cells exhibit significantly upregulated IL-10 expression, which is negatively associated with IFN-γ secretion in CD8+ TILs (Li L. et al., 2019). In chronic viral infections, PD-1 blockade augments IL-10R expression by antigen-specific CD8+ T cells, thereby increasing their sensitivity to the immunosuppressive effects of endogenous IL-10. Conversely, IL-10 blockade strengthens the effects of PD-1 blockade in expanding antigen-specific CD8+ T cells and reinforcing their function. Thus, IL-10 and PD-1 pathways act synergistically through distinct pathways to suppress T cell survival and function (Brooks et al., 2008).
Cancer Immunotherapy and T Cell Dysfunction in the TME
As the pivotal player in the adaptive immune system, T cells can recognize and eliminate tumor cells. However, tumor cells evade from the immune attack once the T cells enter dysfunctional state. The emergence of engineered T cells as a form of cancer therapy marks the beginning of a new era in medicine, providing a transformative way to combat tumors (Mikkilineni and Kochenderfer, 2017). To date, CD19-targeted CAR T-cell therapy has been largely successful in hematological malignancies, showing up to 90% complete response in relapsed or treatment-refractory acute lymphoblastic leukemia (ALL) patients (Maude et al., 2014). However, despite extensive research, the efficacy of CAR-T cell therapy on controlling solid tumors is limited effects due to the influence of TME (Yu et al., 2017; Li et al., 2018d; Yan et al., 2019). Studies suggest that solid tumors may induce hyporesponsiveness of CAR-T cells (Irving et al., 2017; Martinez and Moon, 2019). Three CAR-T cell trials that targeted IL13Rα2, Her2/CMV and EGFRvIII showed poor T cell persistence and an inability to prolong overall survival of patients with glioma (Brown et al., 2016; Ahmed et al., 2017; O’Rourke et al., 2017; Migliorini et al., 2018). Compared with pre-infusion tumor tissues, post-CAR-T cell infusion tumor specimens show markedly upregulated expression of many immunosuppressive molecules, particularly IDO1 and Foxp3, and in some cases, IL-10, PD-L1, and/or TGF-β (O’Rourke et al., 2017). Lim and June (2017) suggested that the TME of solid tumors is hostile for CAR-T cells, reporting that even though CAR-T cells successfully penetrate into the tumor, they are exposed to numerous suppressive factors and tumor-associated stromal cells, which contribute to limiting their function. Additionally, these infused CAR-T cells express high levels of PD-1, T-cell immunoglobulin domain and mucin domain protein 3 (TIM3) and CTLA-4, indicating that they become exhausted or dysfunctional cells (Lim and June, 2017).
Recent studies have identified the crucial role that NR4As play in mediating exhaustion of CAR-T cells. In the NR4A-knocked out mice, CAR-T cells demonstrated low expression levels of inhibitory receptors and increased anti-tumor activity in vivo (Chen J. et al., 2019). Other studies have demonstrated the critical roles of that metabolic barriers play in the TME. Low glucose levels in the TME produce particular challenges for memory CAR-T cells. Thus, novel strategies are needed to assist CAR-T cells in overcoming the immunosuppressive microenvironment presented of many solid tumors.
Cancer neoantigens are derived from random somatic mutations in tumor tissues and are attractive targets for cancer immunotherapies (Chu et al., 2018). Neoantigen-based personalized cancer vaccines have recently shown marked therapeutic potential in both preclinical and early phase clinical studies. Although the number of patients with advanced melanoma treated by neoantigen vaccines is small, results from several phase I clinical trials are quite encouraging. Reportedly, neoantigen-pulsed dendritic cells may induce neoantigen-specific T-cell responses in these patients. A phase I/Ib glioblastoma trial also verified that the circulating polyfunctional neoantigen-specific CD4+ and CD8+ T cell responses were generated in these patients (Ott et al., 2017). However, cancer types with a low mutation burden may not be eligible for this vaccine therapy. Meanwhile, the complicated TME possesses numerous immunosuppressive mechanisms that result in immune escape.
Importantly, tumor cell clones can generate tumors that recapitulated T cell-inflamed and non-T-cell-inflamed TMEs upon implantation in immunocompetent mice, with characteristic patterns of infiltration by immune cell subsets. CXCL1 was identified as a determinant of the non-T-cell-inflamed microenvironment, and ablation of CXCL1 promoted T cell infiltration and sensitivity to immunotherapy (Li et al., 2018a). Furthermore, lower expression levels of co-stimulatory molecules and higher expression levels of co-inhibitory receptors, such as PD-L1, have been shown to be correlated with T-cell dysfunction. Therefore, many efforts are currently focused on addressing challenges in the development of neoantigen-based cancer vaccines for wide clinical applications. Notably, two melanoma patients that experienced disease relapse after successful neoantigen vaccine treatments, and later, achieved complete response after subsequent anti-PD-1 antibody treatment (Ott et al., 2017).
One of the most popular and successful strategies to combat T cell exhaustion is the use of checkpoint inhibitors. The ICB such as anti-PD-1, anti-PD-L1 and anti-CTLA-4 are currently approved by the U.S. FDA for various of caner types. However, the response rate of ICB therapy is less than 30% in solid tumors. First, PD-1 expression levels have a distinct role in contributing to T cell dysfunction or resisting to PD-1 blockade. While intratumoral PD-1high CD8+ subsets share the properties of co-expression of inhibitory receptors and loss of effector function, these populations secreted high levels of CXCL13, which can recruit immune cells to TME. Moreover, the presence of PD-1high T cells was strongly predictive for clinical outcome in a small number of NSCLC patients treated with anti-PD-1 (Thommen et al., 2018). Second, cancers that are non-responsive to checkpoint blockade therapies usually already have decreased numbers of T cells infiltrating their tumors. Infiltrating T cells often co-express multiple inhibitory markers, and expression of the corresponding ligands is evident in tumor cells. In addition, factors beyond tumor genomics influence cancer development and therapeutic responses, including host factors such as the gastrointestinal (gut) microbiome and obesity (Gopalakrishnan et al., 2018; Popovic et al., 2018; Wang et al., 2019e). For example, the frequency of CD8+ TILs expressing PD-1and TIM3 is higher in the diet-induced obese mice (DIO) than that in control mice. Anti-PD-1 monotherapy had minimal to no effect on control mice but significantly reduced tumor burden and significantly improved the survival of DIO mice. It remains to be clinically described whether the environment in the obese state results in greater T cell function once checkpoint blockade is applied (Wang et al., 2019e). More importantly, understanding the relationship between heterogeneous dysfunctional T cells and the TME may significantly impact on the success of therapies like checkpoint blockade and could lead to the production of more functional CAR-T cells.
Reversing T Cell Dysfunction by Combination Cancer Immunotherapy
Great efforts have been made to characterize the intrinsic properties of dysfunctional T cells. Additionally, transcriptional regulators as well as metabolic and epigenetic factors have been investigated as possible targets to improve the anti-tumor efficacy of immunotherapies (Figure 3). New treatment strategies employing epigenetic drugs and immune checkpoint blockade therapies have been investigated in an effort to reverse T cell dysfunction. For instance, in a mouse model of epithelial ovarian cancer, the DNA methyltransferase and histone deacetylase inhibitors (DNMTi and HDACi, respectively) can reduce the immunosuppressive microenvironment through type I IFN signaling and improve response to anti-PD-1 therapy. Addition of HDACi and DNMTi 5-azacytidine (AZA) enhances the modulation of the immune microenvironment, specifically increasing T cell activation and reducing the percentage of macrophage in vivo (Stone et al., 2017).
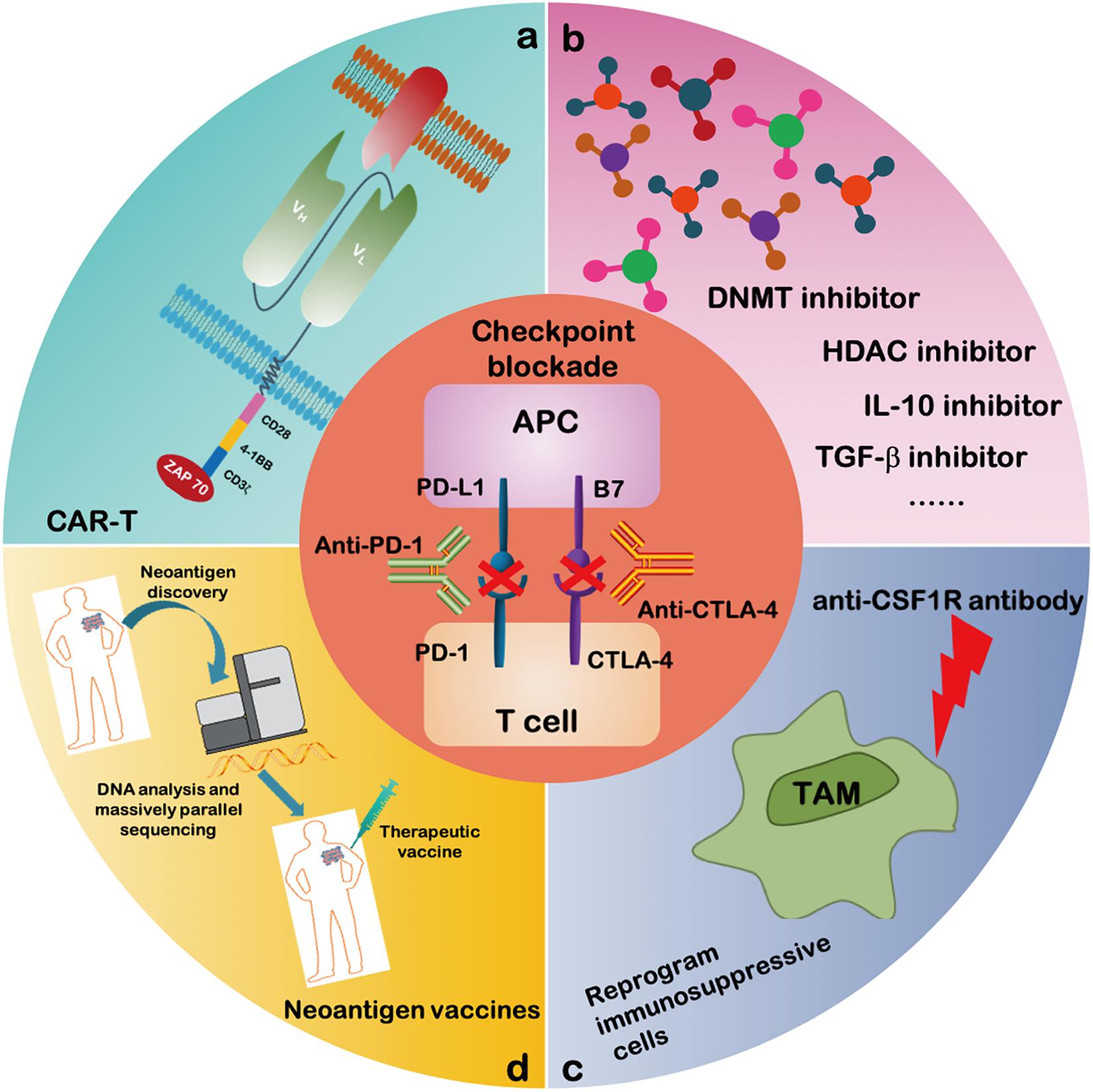
Figure 3. Combinatorial therapeutic strategies to reverse the T cell dysfunction. A growing number of studies propose to evaluate the efficacy of immune checkpoint blockade antibodies together with: (a) CAR-T cell therapy; (b) inhibitors of soluble mediators targeting IL-10, TGF-β, DNA methyltransferase and histone deacetylase; (c) CSF1R antibody targeting activating receptors on TAMs to inhibit the polarization of TAM; and (d) neoantigen vaccines.
Recent studies have also provided evidence that classical immune checkpoints can interact with metabolic checkpoints. In a mouse sarcoma model, glucose consumption by tumors metabolically restricts T cells, leading to their dampened mTOR activity and facilitating cancer progression. It was also discovered that PD-L1 blockade can act directly on tumor cells to inhibit mTOR activity, increasing extracellular glucose availability (Chang et al., 2015). These findings suggest that immune checkpoint blockades counteract T cell dysfunction not only by preventing intrinsic T-cell inhibitory signals but also by increasing T cell metabolic fitness. Meanwhile, the AMPK activator metformin, a first-line treatment drug for type 2 diabetes, was reported to have anti-cancer activity. In a mouse model, metformin was found to increase the number of CD8+ TILs and protected them from apoptosis and exhaustion characterized. Thus, a direct effect of metformin on CD8+ T cells is critical for protecting against the T cell exhaustion in TME. Thus, the combined use of metformin and cancer vaccines can improve TILs multifunctionality (Eikawa et al., 2015).
However, whether the combination of ICB and metformin can restore the dysfunctional T cells remains unclear. Transcriptional profiling of dysfunctional T cells revealed a set of transcription factors that are altered in expression compared with effector or memory T cells. Indeed, a growing list of transcription factors that can regulate the expression of inhibitory receptors has been identified, highlighting potential targets for immunotherapy. Two reports have identified NR4A transcription factors as key mediators of T cell function and demonstrated that NR4A deficiency leads to the downregulation of PD-1, which is functionally similar to the effects of PD-1 blockade (Chen J. et al., 2019; Liu X. et al., 2019). Thus, inhibiting the function of NR4A in TILs or CAR-T cells could be a promising strategy in cancer immunotherapy, similar to combination therapies with ICB against CTLA-4 or GITR (glucocorticoid-induced tumor necrosis factor receptor–related protein) antibodies. Additionally, TOX has been defined as an important transcription factor in regulating T cell exhaustion. Possibly, reducing TOX expression in combination with anti-PD-1 therapy can potentially provide a more effective strategy of abrogating the TOX-dependent pathway of CD8+ T cell exhaustion. Emerging data from a clinical study reported limited clinical activity for anti-GITR monotherapy but potentially promising data for the combination therapy. Combination treatments aimed at PD-1 inhibition and activation of GITR, decrease CD8+ T cell dysfunction and induce a highly proliferative precursor effector memory T cell phenotype (Wang et al., 2018). Monotherapy with CTLA-4 leads to disease control in 20–28% of patients with metastatic melanoma. However, the maintenance of T cell responses triggered by anti-CTLA-4 alone is in most cases insufficient to successfully eradicate tumors, and durable long-term complete remissions (CRs) are seen in a minority of patients. Similarly, adoptive transfer of peripheral blood-derived antigen-specific cytotoxic T cells (CTLs) alone is generally insufficient to eliminate tumors, whereas IL-21-primed CTLs with characteristics of a long-lived memory phenotype may enhance T cell survival after infusion to patients. Thus, the anti-CTLA-4 combined with IL-21-primed CTLs results in long term T cell persistence and durable anti-tumor function (Chapuis et al., 2016a, b).
Importantly, cancer immunotherapies aim to reinvigorate T cell function as well as target immunosuppressive and tumor-promoting pathways mediated by TME (Figure 3). Several specific strategies that target TME are being investigated in combination with ICB therapies in order to improve T cell mediated immunotherapy. Recently, there has been a significant new interest in using macrophage modulators to optimize TAMs. An anti-CSF1R antibody was shown to reprogram TAM polarization and improve the responses to ICB therapy in pancreatic cancer (Cassetta and Kitamura, 2018). Other strategies are focused on inhibiting MDSC function and depleting and/or reprograming MDSCs to enhance the efficacy of checkpoint agents. A clinical trial showed MDSC frequencies as potential biomarkers and reported on their correlation with clinical outcomes of melanoma patients treated with ipilimumab (Meyer et al., 2014). Interestingly, a host of cytokines released by immune and tumor cells have been found to negatively contribute to immunosuppression and have therefore been targeted toward reprograming the immunosuppressive TME. Thus, a combined treatment of IL-10 or TGF-β inhibitor(s) with ICB represents a promising strategy for immunotherapy strategy (Zarour, 2016).
Conclusion
CAR-T cells, neoantigen vaccines and immune checkpoint-modulating agents have increasingly been proven successful in driving antitumor immune responses. Despite these rapid advances in cancer immunotherapy, enormous challenges remain for the future development of cancer therapy for wide clinical applications. Most clinical and preclinical studies using immunotherapy have been focused on T cell exhaustion and dysfunction in TME. In this review, we discussed the unique transcriptional programs and the metabolic and epigenetic factors underlying tumor-induced T cell dysfunction, with the hope that a clearer understanding of TME may enable the development of novel targeted therapeutics, improving the efficacy of immunotherapies. Moreover, the following aspects should be given more attention, (1) identification of mechanisms that convert immunologically cold tumors to T cell rich hot tumors; (2) agents or strategies that reverse T cell exhaustion, and/or reprogram an otherwise immunosuppressive TME must be employed together with immune checkpoint modulators to achieve a robust and durable clinical response; and (3) utilizing RNA sequencing or NanoString tumor expression profiles, to identify gene signatures of T cell dysfunction and predict the outcome of patients treated with checkpoint modulators. This will aid in identifying new targets and advance our fundamental understanding of new targets or the optimal combination therapies for cancer patients.
Author Contributions
ZZ, SL, and YZ (sixth author) conceptualized this review, decided on the content, and wrote the manuscript. ZZ and SL prepared the figures. BZ, LQ, and YZ (fifth author) revised this review. All authors approved the final version of the manuscript and agreed to be accountable for all aspects of the work.
Funding
This study was supported by the National Key Research and Development Program of China (No. 2018YFC1313400) and the National Natural Science Foundation of China (Grant Nos. U1804281, 81771781, 81773046, 81702810, and 81773060).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Cassian Yee (Departments of Melanoma Medical Oncology and Immunology, University of Texas MD Anderson Cancer Center) for his valuable comments.
Abbreviations
ARG-1, arginase-1; CAFs, cancer-associated fibroblasts; CAR, chimeric antigen receptor; CAR-T, chimeric antigen receptor-engineered T cells; ETC, endogenous peripheral blood-derived T cells; EZH2, enhancer of zeste homolog 2; FAO, fatty acid oxidation; Glut-1, glycosis transporter-1; ICB, immune checkpoint blockers; IDO, indoleamine 2,3-dioxygenase-1; IFN- γ, interferon- γ; IL-2, interleukin-2; iNOS, inducible nitric oxide synthase; LAG-3, lymphocyte activation gene 3; MDSCs, myeloid derived suppressor cells; MPE, malignant pleural effusion; NSCLC, non-small cell lung cancer; OXPHOS, oxidative phosphorylation; PA-MSHA, Pseudomonas aeruginosa-mannose- sensitive hemagglutinin; PD-1, programed cell death 1; PD-L1, programed cell death ligand 1; ROS, reactive oxygen species; TAMs, tumor-associated macrophages; TCR, T cell receptor; TCR-T, TCR-engineered T cells; TGF- β, transforming growth factor- β; TIGIT, immunoreceptor tyrosine-based inhibitory motif domain; TIL, tumor infiltrated lymphocytes; TIM-3, T-cell immunoglobulin domain and mucin domain protein 3; TLR4, toll like receptor 4; TME, tumor microenvironment; TNF- α, tumor necrosis factor- α; Tregs, T regulatory cells.
References
Ahmed, N., Brawley, V., Hegde, M., Bielamowicz, K., Kalra, M., Landi, D., et al. (2017). HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 3, 1094–1101. doi: 10.1001/jamaoncol.2017.0184
Alfei, F., Kanev, K., Hofmann, M., Wu, M., Ghoneim, H. E., Roelli, P., et al. (2019). TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269. doi: 10.1038/s41586-019-1326-9
Ambade, A., Satishchandran, A., Saha, B., Gyongyosi, B., Lowe, P., Kodys, K., et al. (2016). Hepatocellular carcinoma is accelerated by NASH involving M2 macrophage polarization mediated by hif-1alphainduced IL-10. Oncoimmunology 5:e1221557. doi: 10.1080/2162402x.2016.1221557
Bally, A. P., Austin, J. W., and Boss, J. M. (2016). Genetic and epigenetic regulation of PD-1 expression. J. Immunol. 196, 2431–2437. doi: 10.4049/jimmunol.1502643
Benner, B., Scarberry, L., Suarez-Kelly, L. P., Duggan, M. C., Campbell, A. R., Smith, E., et al. (2019). Generation of monocyte-derived tumor-associated macrophages using tumor-conditioned media provides a novel method to study tumor-associated macrophages in vitro. J. Immunother. Cancer 7:140. doi: 10.1186/s40425-019-0622-0
Bohrer, L. R., Chen, S., Hallstrom, T. C., and Huang, H. (2010). Androgens suppress EZH2 expression via retinoblastoma (RB) and p130-dependent pathways: a potential mechanism of androgen-refractory progression of prostate cancer. Endocrinology 151, 5136–5145. doi: 10.1210/en.2010-0436
Boyiadzis, M. M., Dhodapkar, M. V., Brentjens, R. J., Kochenderfer, J. N., Neelapu, S. S., Maus, M. V., et al. (2018). Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: clinical perspective and significance. J. Immunother. Cancer 6:137. doi: 10.1186/s40425-018-0460-5
Brooks, D. G., Ha, S. J., Elsaesser, H., Sharpe, A. H., Freeman, G. J., and Oldstone, M. B. (2008). IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc. Natl. Acad. Sci. U.S.A. 105, 20428–20433. doi: 10.1073/pnas.0811139106
Brown, C. E., Alizadeh, D., Starr, R., Weng, L., Wagner, J. R., Naranjo, A., et al. (2016). Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 375, 2561–2569. doi: 10.1056/NEJMoa1610497
Cassetta, L., and Kitamura, T. (2018). Targeting tumor-associated macrophages as a potential strategy to enhance the response to immune checkpoint inhibitors. Front. Cell. Dev. Biol. 6:38. doi: 10.3389/fcell.2018.00038
Cassetta, L., Noy, R., Swierczak, A., Sugano, G., Smith, H., Wiechmann, L., et al. (2016). Isolation of mouse and human tumor-associated macrophages. Adv. Exp. Med. Biol. 899, 211–229. doi: 10.1007/978-3-319-26666-4_12
Chang, C. H., and Pearce, E. L. (2016). Emerging concepts of T cell metabolism as a target of immunotherapy. Nat. Immunol. 17, 364–368. doi: 10.1038/ni.3415
Chang, C. H., Qiu, J., O’Sullivan, D., Buck, M. D., Noguchi, T., Curtis, J. D., et al. (2015). Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241. doi: 10.1016/j.cell.2015.08.016
Chang, J. T., Wherry, E. J., and Goldrath, A. W. (2014). Molecular regulation of effector and memory T cell differentiation. Nat. Immunol. 15, 1104–1115. doi: 10.1038/ni.3031
Chapuis, A. G., Lee, S. M., Thompson, J. A., Roberts, I. M., Margolin, K. A., Bhatia, S., et al. (2016a). Combined IL-21-primed polyclonal CTL plus CTLA4 blockade controls refractory metastatic melanoma in a patient. J. Exp. Med. 213, 1133–1139. doi: 10.1084/jem.20152021
Chapuis, A. G., Roberts, I. M., Thompson, J. A., Margolin, K. A., Bhatia, S., Lee, S. M., et al. (2016b). T-cell therapy using interleukin-21-primed cytotoxic T-cell lymphocytes combined with cytotoxic T-cell lymphocyte antigen-4 blockade results in long-term cell persistence and durable tumor regression. J. Clin. Oncol. 34, 3787–3795. doi: 10.1200/jco.2015.65.5142
Chauvin, J. M., Pagliano, O., Fourcade, J., Sun, Z., Wang, H., Sander, C., et al. (2015). TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J. Clin. Invest. 125, 2046–2058. doi: 10.1172/jci80445
Chen, J., Lopez-Moyado, I. F., Seo, H., Lio, C. J., Hempleman, L. J., Sekiya, T., et al. (2019). NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534. doi: 10.1038/s41586-019-0985-x
Chen, J., Ye, Y., Liu, P., Yu, W., Wei, F., Li, H., et al. (2017). Suppression of T cells by myeloid-derived suppressor cells in cancer. Hum. Immunol. 78, 113–119. doi: 10.1016/j.humimm.2016.12.001
Chen, X., Song, M., Zhang, B., and Zhang, Y. (2016). Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxid. Med. Cell. Longev. 2016:1580967. doi: 10.1155/2016/1580967
Chen, Y., Zhang, S., Wang, Q., and Zhang, X. (2017). Tumor-recruited M2 macrophages promote gastric and breast cancer metastasis via M2 macrophage-secreted CHI3L1 protein. J. Hematol. Oncol. 10:36. doi: 10.1186/s13045-017-0408-0
Chen, Z., Ji, Z., Ngiow, S. F., Manne, S., Cai, Z., Huang, A. C., et al. (2019). TCF-1-centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity 51, 840–855.e5. doi: 10.1016/j.immuni.2019.09.013
Chu, Y., Liu, Q., Wei, J., and Liu, B. (2018). Personalized cancer neoantigen vaccines come of age. Theranostics 8, 4238–4246. doi: 10.7150/thno.24387
Colak, S., and Ten Dijke, P. (2017). Targeting TGF-β signaling in cancer. Trends Cancer 3, 56–71. doi: 10.1016/j.trecan.2016.11.008
Eikawa, S., Nishida, M., Mizukami, S., Yamazaki, C., Nakayama, E., and Udono, H. (2015). Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. U.S.A. 112, 1809–1814. doi: 10.1073/pnas.1417636112
Fourcade, J., Sun, Z., Benallaoua, M., Guillaume, P., Luescher, I. F., Sander, C., et al. (2010). Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 207, 2175–2186. doi: 10.1084/jem.20100637
Ghoneim, H. E., Fan, Y., Moustaki, A., Abdelsamed, H. A., Dash, P., Dogra, P., et al. (2017). De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell 170, 142–157.e19. doi: 10.1016/j.cell.2017.06.007
Gopalakrishnan, V., Spencer, C. N., Nezi, L., Reuben, A., Andrews, M. C., Karpinets, T. V., et al. (2018). Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103. doi: 10.1126/science.aan4236
Guo, X., Zhang, Y., Zheng, L., Zheng, C., Song, J., Zhang, Q., et al. (2018). Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 24, 978–985. doi: 10.1038/s41591-018-0045-3
Hashimoto, M., Kamphorst, A. O., Im, S. J., Kissick, H. T., Pillai, R. N., Ramalingam, S. S., et al. (2018). CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu. Rev. Med. 69, 301–318. doi: 10.1146/annurev-med-012017-043208
Hayashi, S., Kumai, T., Matsuda, Y., Aoki, N., Sato, K., Kimura, S., et al. (2011). Six-transmembrane epithelial antigen of the prostate and enhancer of zeste homolog 2 as immunotherapeutic targets for lung cancer. J. Transl. Med. 9:191. doi: 10.1186/1479-5876-9-191
He, S., Liu, Y., Meng, L., Sun, H., Wang, Y., Ji, Y., et al. (2017). Ezh2 phosphorylation state determines its capacity to maintain CD8(+) T memory precursors for antitumor immunity. Nat. Commun. 8:2125. doi: 10.1038/s41467-017-02187-8
Hwang, S., Cobb, D. A., Bhadra, R., Youngblood, B., and Khan, I. A. (2016). Blimp-1-mediated CD4 T cell exhaustion causes CD8 T cell dysfunction during chronic toxoplasmosis. J. Exp. Med. 213, 1799–1818. doi: 10.1084/jem.20151995
Irving, M., Vuillefroy de Silly, R., Scholten, K., Dilek, N., and Coukos, G. (2017). Engineering chimeric antigen receptor T-cells for racing in solid tumors: don’t forget the fuel. Front. Immunol. 8:267. doi: 10.3389/fimmu.2017.00267
Jeannet, G., Boudousquie, C., Gardiol, N., Kang, J., Huelsken, J., and Held, W. (2010). Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc. Natl. Acad. Sci. U.S.A. 107, 9777–9782. doi: 10.1073/pnas.0914127107
Ji, Y., Fioravanti, J., Zhu, W., Wang, H., Wu, T., Hu, J., et al. (2019). miR-155 harnesses Phf19 to potentiate cancer immunotherapy through epigenetic reprogramming of CD8(+) T cell fate. Nat. Commun. 10:2157. doi: 10.1038/s41467-019-09882-8
Jiang, Y., Li, Y., and Zhu, B. (2015). T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 6:e1792. doi: 10.1038/cddis.2015.162
Jones, B. A., Varambally, S., and Arend, R. C. (2018). Histone methyltransferase EZH2: a therapeutic target for ovarian cancer. Mol. Cancer Ther. 17, 591–602. doi: 10.1158/1535-7163.mct-17-0437
Kartikasari, A. E. R., Prakash, M. D., Cox, M., Wilson, K., Boer, J. C., Cauchi, J. A., et al. (2018). Therapeutic cancer vaccines-T cell responses and epigenetic modulation. Front. Immunol. 9:3109. doi: 10.3389/fimmu.2018.03109
Khan, O., Giles, J. R., McDonald, S., Manne, S., Ngiow, S. F., Patel, K. P., et al. (2019). TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 571, 211–218. doi: 10.1038/s41586-019-1325-x
Kishton, R. J., Sukumar, M., and Restifo, N. P. (2017). Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab. 26, 94–109. doi: 10.1016/j.cmet.2017.06.016
Knudson, K. M., Pritzl, C. J., Saxena, V., Altman, A., Daniels, M. A., and Teixeiro, E. (2017). NFkappaB-Pim-1-eomesodermin axis is critical for maintaining CD8 T-cell memory quality. Proc. Natl. Acad. Sci. U.S.A. 114, E1659–E1667. doi: 10.1073/pnas.1608448114
Lakins, M. A., Ghorani, E., Munir, H., Martins, C. P., and Shields, J. D. (2018). Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T cells to protect tumour cells. Nat. Commun. 9:948. doi: 10.1038/s41467-018-03347-0
Landskron, G., De la Fuente, M., Thuwajit, P., Thuwajit, C., and Hermoso, M. A. (2014). Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014:149185. doi: 10.1155/2014/149185
Le Bourgeois, T., Strauss, L., Aksoylar, H. I., Daneshmandi, S., Seth, P., Patsoukis, N., et al. (2018). Targeting T cell metabolism for improvement of cancer immunotherapy. Front. Oncol. 8:237. doi: 10.3389/fonc.2018.00237
Li, H., van der Leun, A. M., Yofe, I., Lubling, Y., Gelbard-Solodkin, D., van Akkooi, A. C. J., et al. (2019). Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 176, 775–789.e18. doi: 10.1016/j.cell.2018.11.043
Li, J., Byrne, K. T., Yan, F., Yamazoe, T., Chen, Z., Baslan, T., et al. (2018a). Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity 49, 178–193.e7. doi: 10.1016/j.immuni.2018.06.006
Li, J., He, Y., Hao, J., Ni, L., and Dong, C. (2018b). High levels of eomes promote exhaustion of anti-tumor CD8(+) T cells. Front. Immunol. 9:2981. doi: 10.3389/fimmu.2018.02981
Li, J., Lee, Y., Li, Y., Jiang, Y., Lu, H., Zang, W., et al. (2018c). Co-inhibitory molecule B7 superfamily member 1 expressed by tumor-infiltrating myeloid cells induces dysfunction of anti-tumor CD8(+) T cells. Immunity 48, 773–786.e5. doi: 10.1016/j.immuni.2018.03.018
Li, J., Li, W., Huang, K., Zhang, Y., Kupfer, G., and Zhao, Q. (2018d). Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: lessons learned and strategies for moving forward. J. Hematol. Oncol. 11:22. doi: 10.1186/s13045-018-0568-6
Li, L., Ma, Y., and Xu, Y. (2019). Follicular regulatory T cells infiltrated the ovarian carcinoma and resulted in CD8 T cell dysfunction dependent on IL-10 pathway. Int. Immunopharmacol. 68, 81–87. doi: 10.1016/j.intimp.2018.12.051
Li, L., Wang, L., Li, J., Fan, Z., Yang, L., Zhang, Z., et al. (2018e). Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. 78, 1779–1791. doi: 10.1158/0008-5472.can-17-2460
Li, J., Wang, L., Chen, X., Li, L., Li, Y., Ping, Y., et al. (2017). CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-βeta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 6:e1320011. doi: 10.1080/2162402x.2017.1320011
Lim, W. A., and June, C. H. (2017). The principles of engineering immune cells to treat cancer. Cell 168, 724–740. doi: 10.1016/j.cell.2017.01.016
Liu, H., Li, P., Wei, Z., Zhang, C., Xia, M., Du, Q., et al. (2019). Regulation of T cell differentiation and function by epigenetic modification enzymes. Semin. Immunopathol. 41, 315–326. doi: 10.1007/s00281-019-00731-w
Liu, S., Zhang, C., Maimela, N. R., Yang, L., Zhang, Z., Ping, Y., et al. (2019). Molecular and clinical characterization of CD163 expression via large-scale analysis in glioma. Oncoimmunology 8:1601478. doi: 10.1080/2162402x.2019.1601478
Liu, X., Wang, Y., Lu, H., Li, J., Yan, X., Xiao, M., et al. (2019). Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 567, 525–529. doi: 10.1038/s41586-019-0979-8
Ma, E. H., Poffenberger, M. C., Wong, A. H., and Jones, R. G. (2017). The role of AMPK in T cell metabolism and function. Curr. Opin. Immunol. 46, 45–52. doi: 10.1016/j.coi.2017.04.004
Ma, R., Ji, T., Zhang, H., Dong, W., Chen, X., Xu, P., et al. (2018). A Pck1-directed glycogen metabolic program regulates formation and maintenance of memory CD8(+) T cells. Nat. Cell Biol. 20, 21–27. doi: 10.1038/s41556-017-0002-2
Maimela, N. R., Liu, S., and Zhang, Y. (2019). Fates of CD8+ T cells in tumor microenvironment. Comput. Struct. Biotechnol. J. 17, 1–13. doi: 10.1016/j.csbj.2018.11.004
Mann, T. H., and Kaech, S. M. (2019). Tick-TOX, it’s time for T cell exhaustion. Nat. Immunol. 20, 1092–1094. doi: 10.1038/s41590-019-0478-y
Martinez, G. J., Pereira, R. M., Aijo, T., Kim, E. Y., Marangoni, F., Pipkin, M. E., et al. (2015). The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity 42, 265–278. doi: 10.1016/j.immuni.2015.01.006
Martinez, M., and Moon, E. K. (2019). CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 10:128. doi: 10.3389/fimmu.2019.00128
Maude, S. L., Frey, N., Shaw, P. A., Aplenc, R., Barrett, D. M., Bunin, N. J., et al. (2014). Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517. doi: 10.1056/NEJMoa1407222
Meyer, C., Cagnon, L., Costa-Nunes, C. M., Baumgaertner, P., Montandon, N., Leyvraz, L., et al. (2014). Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 63, 247–257. doi: 10.1007/s00262-013-1508-5
Migliorini, D., Dietrich, P. Y., Stupp, R., Linette, G. P., Posey, A. D. Jr., and June, C. H. (2018). CAR T-cell therapies in glioblastoma: a first look. Clin. Cancer Res. 24, 535–540. doi: 10.1158/1078-0432.ccr-17-2871
Mikkilineni, L., and Kochenderfer, J. N. (2017). Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood 130, 2594–2602. doi: 10.1182/blood-2017-06-793869
Miller, B. C., Sen, D. R., Al Abosy, R., Bi, K., Virkud, Y. V., LaFleur, M. W., et al. (2019). Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336. doi: 10.1038/s41590-019-0312-6
Nagarsheth, N., Peng, D., Kryczek, I., Wu, K., Li, W., Zhao, E., et al. (2016). PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res. 76, 275–282. doi: 10.1158/0008-5472.can-15-1938
O’Rourke, D. M., Nasrallah, M. P., Desai, A., Melenhorst, J. J., Mansfield, K., Morrissette, J. J. D., et al. (2017). A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
Ostrand-Rosenberg, S. (2010). Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol. Immunother. 59, 1593–1600. doi: 10.1007/s00262-010-0855-8
Ott, P. A., Hu, Z., Keskin, D. B., Shukla, S. A., Sun, J., Bozym, D. J., et al. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221. doi: 10.1038/nature22991
Pauken, K. E., Sammons, M. A., Odorizzi, P. M., Manne, S., Godec, J., Khan, O., et al. (2016). Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165. doi: 10.1126/science.aaf2807
Pearce, E. L., Poffenberger, M. C., Chang, C. H., and Jones, R. G. (2013). Fueling immunity: insights into metabolism and lymphocyte function. Science 342:1242454. doi: 10.1126/science.1242454
Popovic, A., Jaffee, E. M., and Zaidi, N. (2018). Emerging strategies for combination checkpoint modulators in cancer immunotherapy. J. Clin. Invest. 128, 3209–3218. doi: 10.1172/jci120775
Quigley, M., Pereyra, F., Nilsson, B., Porichis, F., Fonseca, C., Eichbaum, Q., et al. (2010). Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med. 16, 1147–1151. doi: 10.1038/nm.2232
Riley, R. S., June, C. H., Langer, R., and Mitchell, M. J. (2019). Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 18, 175–196. doi: 10.1038/s41573-018-0006-z
Rodriguez, P. C., Zea, A. H., Culotta, K. S., Zabaleta, J., Ochoa, J. B., and Ochoa, A. C. (2002). Regulation of T cell receptor CD3zeta chain expression by L-arginine. J. Biol. Chem. 277, 21123–21129. doi: 10.1074/jbc.M110675200
Salmond, R. J. (2018). mTOR regulation of glycolytic metabolism in T cells. Front. Cell. Dev. Biol. 6:122. doi: 10.3389/fcell.2018.00122
Schietinger, A., and Greenberg, P. D. (2014). Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 35, 51–60. doi: 10.1016/j.it.2013.10.001
Schietinger, A., Philip, M., Krisnawan, V. E., Chiu, E. Y., Delrow, J. J., Basom, R. S., et al. (2016). Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 45, 389–401. doi: 10.1016/j.immuni.2016.07.011
Scott, A. C., Dundar, F., Zumbo, P., Chandran, S. S., Klebanoff, C. A., Shakiba, M., et al. (2019). TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274. doi: 10.1038/s41586-019-1324-y
Sen, D. R., Kaminski, J., Barnitz, R. A., Kurachi, M., Gerdemann, U., Yates, K. B., et al. (2016). The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169. doi: 10.1126/science.aae0491
Seo, H., Chen, J., Gonzalez-Avalos, E., Samaniego-Castruita, D., Das, A., Wang, Y. H., et al. (2019). TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc. Natl. Acad. Sci. U.S.A. 116, 12410–12415. doi: 10.1073/pnas.1905675116
Sica, A., and Mantovani, A. (2012). Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795. doi: 10.1172/jci59643
Siddiqui, I., Schaeuble, K., Chennupati, V., Fuertes Marraco, S. A., Calderon-Copete, S., Pais Ferreira, D., et al. (2019). Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211.e10. doi: 10.1016/j.immuni.2018.12.021
Staron, M. M., Gray, S. M., Marshall, H. D., Parish, I. A., Chen, J. H., Perry, C. J., et al. (2014). The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity 41, 802–814. doi: 10.1016/j.immuni.2014.10.013
Stone, M. L., Chiappinelli, K. B., Li, H., Murphy, L. M., Travers, M. E., Topper, M. J., et al. (2017). Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc. Natl. Acad. Sci. U.S.A. 114, E10981–E10990. doi: 10.1073/pnas.1712514114
Sugiura, A., and Rathmell, J. C. (2018). Metabolic barriers to T cell function in tumors. J. Immunol. 200, 400–407. doi: 10.4049/jimmunol.1701041
Thommen, D. S., Koelzer, V. H., Herzig, P., Roller, A., Trefny, M., Dimeloe, S., et al. (2018). A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 24, 994–1004. doi: 10.1038/s41591-018-0057-z
Thommen, D. S., and Schumacher, T. N. (2018). T cell dysfunction in cancer. Cancer Cell 33, 547–562. doi: 10.1016/j.ccell.2018.03.012
Verma, V., Shrimali, R. K., Ahmad, S., Dai, W., Wang, H., Lu, S., et al. (2019). PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat. Immunol. 20, 1231–1243. doi: 10.1038/s41590-019-0441-y
Vodnala, S. K., Eil, R., Kishton, R. J., Sukumar, M., Yamamoto, T. N., Ha, N. H., et al. (2019). T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 363:eaau0135. doi: 10.1126/science.aau0135
Wang, B., Zhang, W., Jankovic, V., Golubov, J., Poon, P., Oswald, E. M., et al. (2018). Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8(+) T cell dysfunction and maintain memory phenotype. Sci. Immunol. 3:eaat7061. doi: 10.1126/sciimmunol.aat7061
Wang, C., Singer, M., and Anderson, A. C. (2017). Molecular dissection of CD8(+) T-cell dysfunction. Trends Immunol. 38, 567–576. doi: 10.1016/j.it.2017.05.008
Wang, D., Lin, J., Yang, X., Long, J., Bai, Y., Yang, X., et al. (2019a). Combination regimens with PD-1/PD-L1 immune checkpoint inhibitors for gastrointestinal malignancies. J. Hematol. Oncol. 12:42. doi: 10.1186/s13045-019-0730-9
Wang, D., Yang, L., Yue, D., Cao, L., Li, L., Wang, D., et al. (2019b). Macrophage-derived CCL22 promotes an immunosuppressive tumor microenvironment via IL-8 in malignant pleural effusion. Cancer Lett. 452, 244–253. doi: 10.1016/j.canlet.2019.03.040
Wang, X., Brea, L. T., and Yu, J. (2019c). Immune modulatory functions of EZH2 in the tumor microenvironment: implications in cancer immunotherapy. Am. J. Clin. Exp. Urol. 7, 85–91.
Wang, X., He, Q., Shen, H., Xia, A., Tian, W., Yu, W., et al. (2019d). TOX promotes the exhaustion of antitumor CD8(+) T cells by preventing PD1 degradation in hepatocellular carcinoma. J. Hepatol. 71, 731–741. doi: 10.1016/j.jhep.2019.05.015
Wang, Z., Aguilar, E. G., Luna, J. I., Dunai, C., Khuat, L. T., Le, C. T., et al. (2019e). Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 25, 141–151. doi: 10.1038/s41591-018-0221-5
Wherry, E. J., and Kurachi, M. (2015). Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499. doi: 10.1038/nri3862
Wu, A. A., Drake, V., Huang, H. S., Chiu, S., and Zheng, L. (2015). Reprogramming the tumor microenvironment: tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology 4:e1016700. doi: 10.1080/2162402x.2015.1016700
Xia, A., Zhang, Y., Xu, J., Yin, T., and Lu, X. J. (2019). T cell dysfunction in cancer immunity and immunotherapy. Front. Immunol. 10:1719. doi: 10.3389/fimmu.2019.01719
Yan, Z., Li, L., Wang, W., OuYang, B., Cheng, S., Wang, L., et al. (2019). Clinical efficacy and tumor microenvironment influence in a dose-escalation study of anti-CD19 chimeric antigen receptor T cells in refractory B-cell non-Hodgkin’s lymphoma. Clin. Cancer Res. 25, 6995–7003. doi: 10.1158/1078-0432.Ccr-19-0101
Yang, L., Wang, F., Wang, L., Huang, L., Wang, J., Zhang, B., et al. (2015). CD163+ tumor-associated macrophage is a prognostic biomarker and is associated with therapeutic effect on malignant pleural effusion of lung cancer patients. Oncotarget 6, 10592–10603. doi: 10.18632/oncotarget.3547
Yang, L., and Zhang, Y. (2017). Tumor-associated macrophages: from basic research to clinical application. J. Hematol. Oncol. 10:58. doi: 10.1186/s13045-017-0430-2
Yee, C. (2018). Adoptive T cell therapy: points to consider. Curr. Opin. Immunol. 51, 197–203. doi: 10.1016/j.coi.2018.04.007
Yee, C., Lizee, G., and Schueneman, A. J. (2015). Endogenous T-cell therapy: clinical experience. Cancer J. 21, 492–500. doi: 10.1097/ppo.0000000000000158
Yee, C., and Lizee, G. A. (2017). Personalized therapy: tumor antigen discovery for adoptive cellular therapy. Cancer J. 23, 144–148. doi: 10.1097/ppo.0000000000000255
Yeku, O. O., Purdon, T. J., Koneru, M., Spriggs, D., and Brentjens, R. J. (2017). Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 7:10541. doi: 10.1038/s41598-017-10940-8
Yu, J., Du, W., Yan, F., Wang, Y., Li, H., Cao, S., et al. (2013). Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 190, 3783–3797. doi: 10.4049/jimmunol.1201449
Yu, S., Li, A., Liu, Q., Li, T., Yuan, X., Han, X., et al. (2017). Chimeric antigen receptor T cells: a novel therapy for solid tumors. J. Hematol. Oncol. 10:78. doi: 10.1186/s13045-017-0444-9
Zarour, H. M. (2016). Reversing T-cell dysfunction and exhaustion in cancer. Clin. Cancer Res. 22, 1856–1864. doi: 10.1158/1078-0432.ccr-15-1849
Zhang, T., Zhang, Z., Li, F., Ping, Y., Qin, G., Zhang, C., et al. (2018). miR-143 regulates memory T cell differentiation by reprogramming T cell metabolism. J. Immunol. 201, 2165–2175. doi: 10.4049/jimmunol.1800230
Zhao, E., Maj, T., Kryczek, I., Li, W., Wu, K., Zhao, L., et al. (2016). Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat. Immunol. 17, 95–103. doi: 10.1038/ni.3313
Zhen, Z., Tang, W., Wang, M., Zhou, S., Wang, H., Wu, Z., et al. (2017). Protein nanocage mediated fibroblast-activation protein targeted photoimmunotherapy to enhance cytotoxic T cell infiltration and tumor control. Nano Lett. 17, 862–869. doi: 10.1021/acs.nanolett.6b04150
Keywords: T cells dysfunction, intrinsic factors, extrinsic factors, tumor microenvironment, cancer immunotherapy
Citation: Zhang Z, Liu S, Zhang B, Qiao L, Zhang Y and Zhang Y (2020) T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 8:17. doi: 10.3389/fcell.2020.00017
Received: 05 October 2019; Accepted: 10 January 2020;
Published: 11 February 2020.
Edited by:
Bin Li, Shanghai Jiao Tong University School of Medicine, ChinaReviewed by:
Shengtao Zhou, West China Second University Hospital of Sichuan University, ChinaLianjun Zhang, Center of Systems Medicine, Chinese Academy of Medical Sciences, Suzhou Institute of Systems Medicine (ISM), China
Copyright © 2020 Zhang, Liu, Zhang, Qiao, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Zhang, eWl6aGFuZ0B6enUuZWR1LmNu
†These authors have contributed equally to this work