 Shuwen Deng
Shuwen Deng Jia Liu
Jia Liu Wei Lu
Wei Lu- Department of Neurology, The Second Xiangya Hospital, Central South University, Changsha, China
Autophagy has dual effects in human diseases: appropriate autophagy may protect cells from stress, while excessive autophagy may cause cell death. Additionally, close interactions exist between autophagy and the Golgi. This review outlines recent advances regarding the role of the Golgi apparatus in autophagy. The signaling processes of autophagy are dependent on the normal function of the Golgi. Specifically, (i) autophagy-related protein 9 is mainly located in the Golgi and forms new autophagosomes in response to stressors; (ii) Golgi fragmentation is induced by Golgi-related proteins and accompanied with autophagy induction; and (iii) the endoplasmic reticulum-Golgi intermediate compartment and the reticular trans-Golgi network play essential roles in autophagosome formation to provide a template for lipidation of microtubule-associated protein 1A/1B-light chain 3 and induce further ubiquitination. Golgi-related proteins regulate formation of autophagosomes, and disrupted formation of autophagy can influence Golgi function. Notably, aberrant autophagy has been demonstrated to be implicated in neurological diseases. Thus, targeted therapies aimed at protecting the Golgi or regulating Golgi proteins might prevent or ameliorate autophagy-related neurological diseases. Further studies are needed to investigate the potential application of Golgi therapy in autophagy-based neurological diseases.
Functions of the Golgi Apparatus
The Golgi apparatus (Golgi hereafter) is a processing and dispatching station, whereby newly synthesized soluble and transmembrane proteins, as well as lipids, are sorted for subsequent transport to the cell surface, secretory granules, or the endosomal system (Rohn et al., 2000; Viotti, 2016). Morphologically, the Golgi comprises associated vesicles and numerous flattened, stacked sacs that are known as cisternae (Rambourg and Clermont, 1990; Koga and Ushiki, 2006). Golgi stacks are collected near the minus ends of microtubules and laterally linked by dynamic fusion events to form a ribbon-like network (Rambourg and Clermont, 1990; Jarvela and Linstedt, 2014). The functional structure of the Golgi includes the cis-Golgi, medial Golgi, and trans-Golgi networks. In the secretory pathway, endoplasmic reticulum (ER)-Golgi intermediate compartment (ERGIC) is a tubulo-vesicular compartment closely associated with the ER-exit sites (ERES) (Liu et al., 2020) and functions as post-ER sorting stations (Appenzeller-Herzog and Hauri, 2006). Cargos derived from the ER enter a Golgi stack at its cis-side and then sequentially pass through medial and trans-cisternae (Tie et al., 2017). The trans-Golgi network sorts a variety of transport carriers and secretory cargos for delivery to their final destinations at the exit face of the Golgi (Glick and Nakano, 2009; Suda and Nakano, 2012). Golgi proteins involved in vesicle transportation including golgins, conserved oligomeric Golgi (COG) complex and Golgi-associated retrograde protein (GARP) complex etc. (Figure 1).
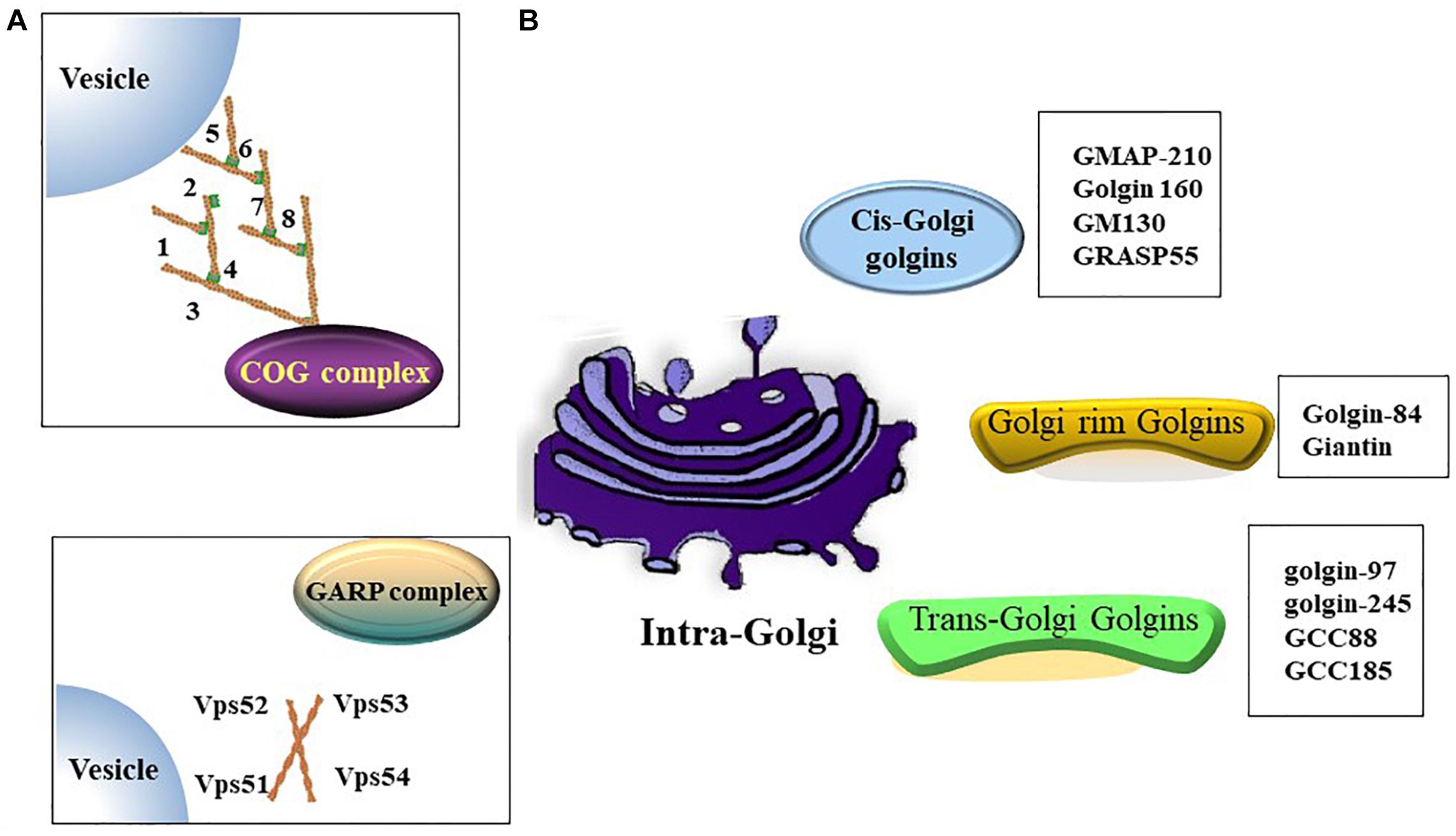
Figure 1. Golgi proteins involved in vesicles transportation. (A) Both of oligomeric Golgi (COG) complex (comprised of Cog1-8) and Golgi-associated retrograde protein (GARP) complex (comprised of VPS51, VPS52, VPS53 and VPS54) are tethering complex that functions as cargo trafficking complex. (B) A summary of golgins in different zones.
In addition to the membrane transport and glycosylation functions, the Golgi apparatus is also involved in various biological chemical processes, including mitosis, DNA repair, stress responses, autophagy, apoptosis, and inflammation (Kulkarni-Gosavi et al., 2019). Meanwhile, the biological function of the Golgi is derived from its central position in the secretory pathway and regulatory role in large amounts of signaling molecules (Cancino et al., 2013).
Formation of Autophagy
Autophagy is a degradation process of the damaged subcellular organelles and improperly folded proteins in the cytoplasm digested by lysosomal lytic enzymes (Arakawa et al., 2017). There are at least three types of autophagic processes in mammalian cells: (i) macro-autophagy coordinates the assembly of autophagy-related (ATG) proteins to generate autophagosomes that are subsequently fused with the lysosomal membrane (Nakatogawa et al., 2009), (ii) chaperone-mediated autophagy delivers KFERQ-like motif-bearing proteins to the lysosomes for degradation via chaperone heat shock cognate protein 70 and cochaperones (Kaushik and Cuervo, 2018), and (iii) micro-autophagy is involved in the bulk degradation of proteins and organelles through invaginations at the lysosomal membrane (Muller et al., 2000; Scrivo et al., 2018). In addition, macro-autophagy includes ATG5/7-dependent canonical and ATG5/7-independent non-canonical autophagy (Nishida et al., 2009). “Macro-autophagy” is referred to as “autophagy” hereafter in this review.
Nonselective autophagy may be divided into initiation, elongation/expansion of the isolation membrane, and completion and subsequent fusion of the autophagosome with the lysosome (Zahoor and Farhan, 2018). There are some differences in autophagy-related genes and functions between mammals and yeasts (Li and Zhang, 2019). For instance, ATG29 and ATG31 are yeast specific in the ATG1-complex during the induction phase of autophagy (Li and Zhang, 2019). Functionally, mammalian ATGs can be subdivided into six functional clusters (Wesselborg and Stork, 2015; Table 1). During the initial phase of autophagy, the mammalian autophagy complex- unc-51-like kinase 1 (ULK1) complex is activated and then binds to and phosphorylates ATG9 on the vesicles (Li and Zhang, 2019). Upon the induction of autophagy, ATG9 may first accumulate in tubular vesicular compartments known as ATG9 compartments/reservoir (De Tito et al., 2020). After induction of autophagy, Beclin-1 and its multiple modifiers (Kihara et al., 2001) associated with the ATG system fulfill the elongation of isolation membranes/phagophores (Kihara et al., 2001; Mizushima et al., 2011). Finally, ATG8 (including microtubule-associated protein 1A/1B-light chain [LC]3b and GABARAPS) recognizes ubiquitinated targets. P62 binds to ubiquitin and ATG8 proteins via its C-terminal UBA domain (Zaffagnini et al., 2018) and delivers ubiquitinated cargos for autophagic degradation (Johansen and Lamark, 2011; Liu et al., 2016; Figure 2).
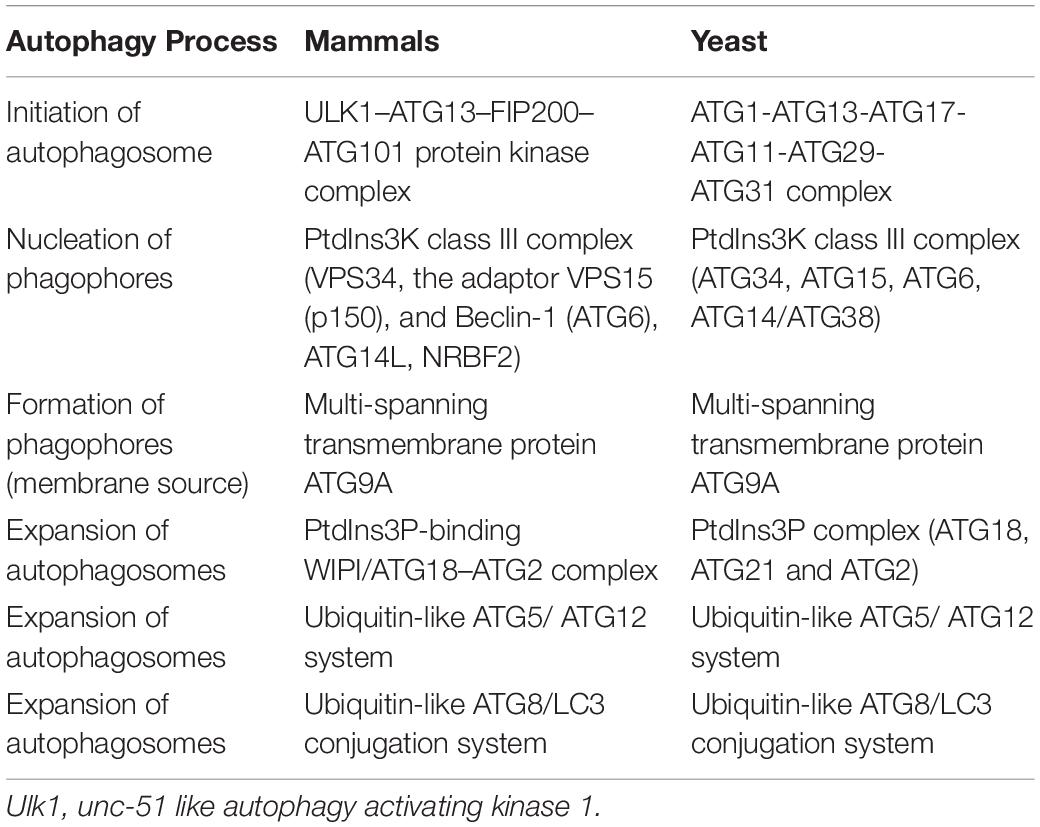
Table 1. Function of ATGs in different steps during autophagosome biogenesis.
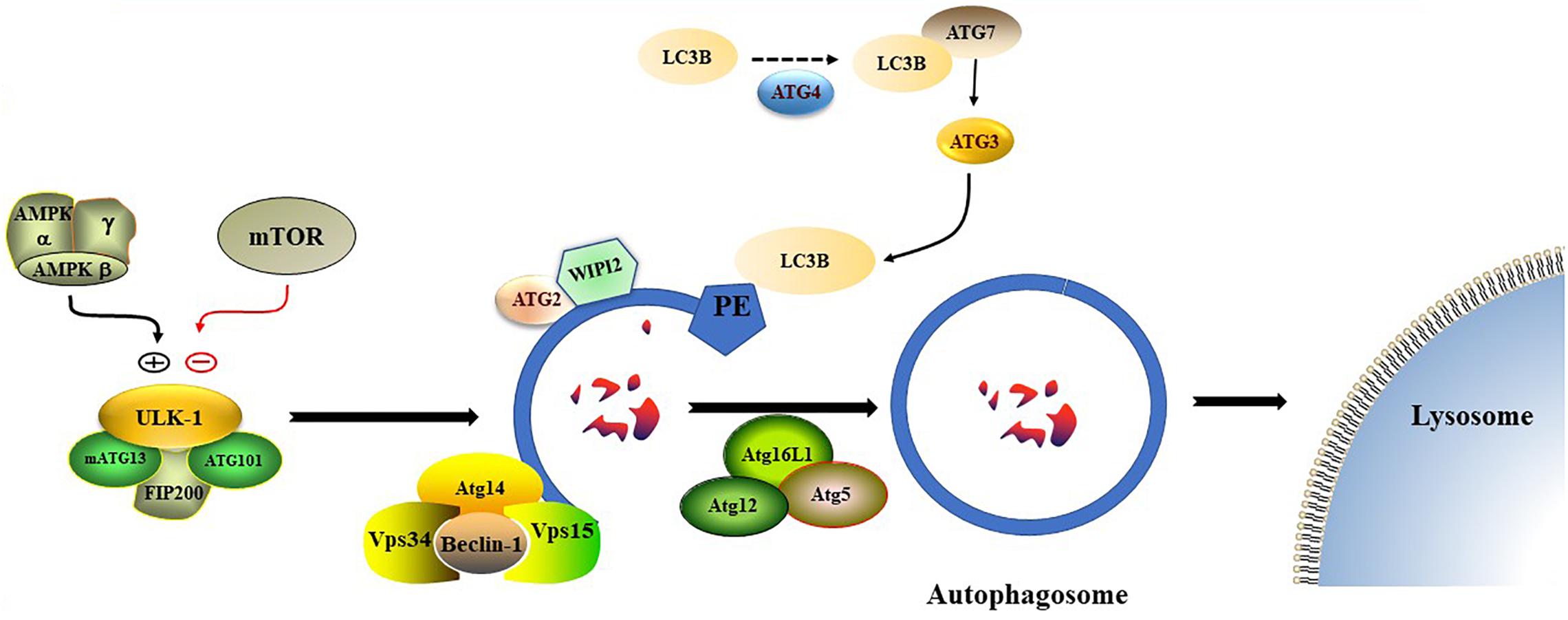
Figure 2. The formation process of autophagosome. Macro-autophagy coordinates the assembly of autophagy-related (ATG) proteins to generate autophagosomes that are subsequently fused with the lysosomal membrane.
Autophagy has a variety of signal regulatory pathways. Starvation-induced autophagy is regulated by inactivation of the mammalian target of rapamycin (mTOR) and activation of AMP kinase (AMPK) (Rubinsztein et al., 2012). In addition, ULK1 is an effector of the two kinases (Egan et al., 2011; Kim et al., 2011). Another regulatory pathway of autophagy is the cAMP–Epac–PLCε–IP3-calcium pathway (Metcalf et al., 2012).
Interaction Between the Golgi Apparatus and Autophagy
In mammalian cells, the Golgi is a cell sensor because various signaling factors are located in or transduced to the apparatus (Cancino et al., 2013). Autophagy could be influenced by dysfunction of the Golgi. As a signaling platform, the Golgi provides not only a membrane for autophagosome formation but also a location for induction and elongation of autophagosomes. Moreover, Golgi-related proteins participate in autophagosome formation directly. Conversely, moderate autophagy can help maintain the normal function of the Golgi apparatus.
Golgi Influences the Signaling Pathway of Autophagy
The Golgi Is Involved in Autophagosome Formation by Regulating Transport of ATG9
ATG9 is the sole multi-spanning transmembrane protein among core ATG proteins and is essential for autophagy (Kakuta et al., 2012; Imai et al., 2016). The mechanisms regulating ATG9 trafficking may differ between the species (Webber and Tooze, 2010). In mammalian cells, ATG9-containing vesicles cycle between the Trans-Golgi Network (TGN), post-Golgi, and endosomal compartments (Mari et al., 2010; Imai et al., 2016) and form new autophagosomes in response to stresses (Zhou et al., 2017). Under nutrient-rich conditions, ATG9 is mainly co-localizes with TGN markers of the Golgi and recycling endosomes (Orsi et al., 2012). During starvation-induced autophagy, membrane ATG9 (mATG9) localizes to both a juxtanuclear region, which corresponds to the TGN, and a peripheral population, which partially co-localizes with late endosomes (Zhou et al., 2017). Initiation and formation of the autophagosome in mammalian cells are through the formation of the omegasome (Mizushima et al., 2011; Yu et al., 2018). Retrieval of ATG9 to the Golgi from RAB11-positive recycling endosomes to RAB1-positive Golgi compartments is controlled by a complex including TBC1D14 and mammalian transport protein particle (TRAPP) III (Lamb and Tooze, 2016; Lamb et al., 2016). TRAPPC8 forms part of a mammalian TRAPPIII-like complex required for RAB1 activity and directed to the omegasome (Lamb and Tooze, 2016). In addition, TRAPPC8 is needed for maintenance of Golgi integrity (Lamb et al., 2016). Thus, abnormal function or morphology of the Golgi may influence the transportation of ATG9 from the Golgi to the destined organelle and ultimately disrupt autophagosome formation (Figure 3).
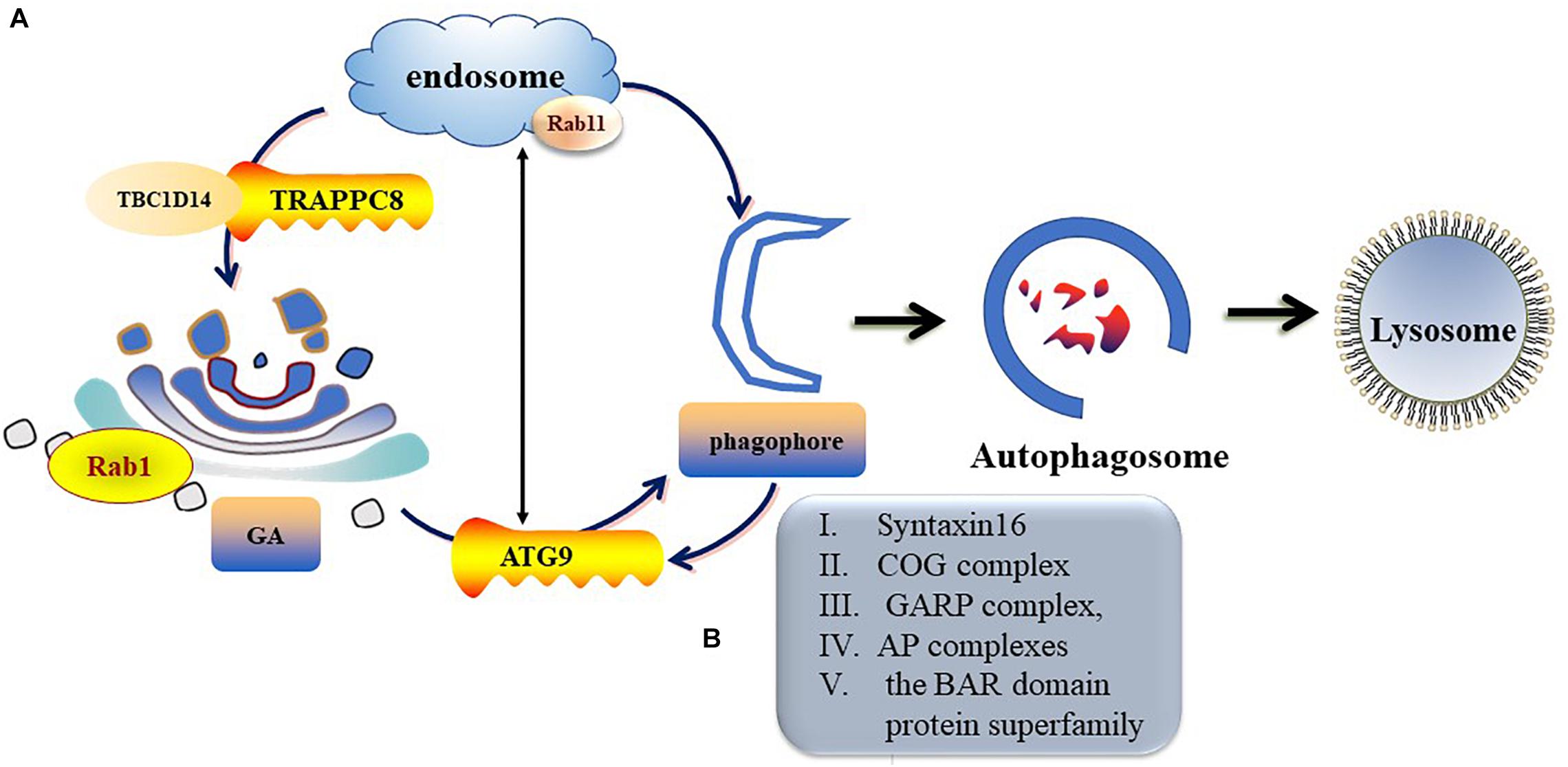
Figure 3. Trafficking of mATG9 in different conditions. (A) Under nutrient-rich conditions, ATG9 is mainly colocalizes with TGN and endosomes. During starvation-induced autophagy, mAtg9 redistributes from TGN to juxtanuclear region. ATG9 relocate to the Golgi from RAB11-positive recycling endosomes to RAB1-positive Golgi compartments is controlled by a complex including TBC1D14 and mammalian TRAPPIII. (B) Syntaxin16, oligomeric Golgi (COG) complex, Golgi-associated retrograde protein (GARP) complex, adaptor protein (AP) complexes and the BAR domain protein superfamily take part in the regulation of ATG9 trafficking.
Golgi Fragmentation Is Accompanied by Autophagy Induction
α Soluble N-ethylmaleimide sensitive factor attachment protein alpha (αSNAP) is a well-known component of the vesicle trafficking machinery (Naydenov et al., 2012a, 2014) and mediates vesicle transport from the ER to the cis-Golgi (Peter et al., 1998). Loss of αSNAP impairs Golgi-dependent glycosylation (Naydenov et al., 2014), triggers Golgi fragmentation, and induces autophagy (Naydenov et al., 2012b). By controlling the integrity of the Golgi, αSNAP inhibits mTOR-related signaling, limits the membrane supply for autophagosomal biogenesis, and negatively regulates autophagy (Naydenov et al., 2012b). Additionally, depletion of another multi-subunit tether factor, Golgi-associated retrograde protein (GARP), also causes autophagy defects (Perez-Victoria et al., 2010). The GARP complex is a multi-subunit complex made of four distinct proteins (VPS51, VPS52, VPS53, and VPS54) localized in the TGN and involved in endosome-to-TGN retrograde transport (Hirata et al., 2015; Uwineza et al., 2019). Dysfunction of the GARP complex may influence transport of the retrograde vesicle and sorting of proteins in the Golgi apparatus (Schmitt-John, 2015). GARP complex is the effector that connects Arl1 and Ypt6 with autophagy (Yang and Rosenwald, 2016), and it may cause autophagy defects in mATG9 that is not transported or is unable to obtain sufficient membrane support because of disrupted integrity of the Golgi (Eapen and Haber, 2013). Moreover, conserved oligomeric Golgi (COG) complex mutants exhibit defective autophagy (Wang I. H. et al., 2017). The COG complex, which consists of eight subunits (COG 1-8), is a multi-subunit vesicle tethering complex that functions in retrograde trafficking at the Golgi (D’Souza et al., 2019). Similar to the depletion effect of GARP mentioned above, defective autophagy that is caused by COG complex malfunction is also associated with blocked transportation of ATG9 and insufficient membrane support derived from abnormal function of the Golgi (Ohashi and Munro, 2010). Moreover, the COG complex localizes to the pre-autophagosomal structure and interacts with ATG12, ATG17, ATG20, and ATG24 (Yen et al., 2010). Thus, autophagy is dependent on normal function and integrity of the Golgi because the loss of Golgi-related proteins, including αSNAP, GARP, or COG, can result in aberrant autophagy.
ERGIC and TGN Provide a Location for Formation of Autophagosomes
The ERGIC functions as the membrane provider that triggers LC3 lipidation and is required for autophagosomal biogenesis (Ge et al., 2013). LC3 is linked to phosphatidylethanolamine on autophagosomal precursor membranes for lipidation which is mediated by the E3-like Atg12-Atg5⋅Atg16 complex (Ge et al., 2015). The above process is modulated by the phosphatidylinositol 3-kinase (PtdIns3K) complex, which consists of ATG14, Beclin-1, PIK3C3/VPS34, and PIK3R4/VPS15/p150. Beclin-1-PtdIns3K complexes are concentrated in the TGN (Shoji-Kawata et al., 2013) and play a role in the sorting of autophagosomal components and lysosomal proteins (Kihara et al., 2001). The ERGIC may play a role in an early stage of phagophore formation by providing a platform to recruit the class III PI3K complex and provide precursor membranes for phagophore initiation (Ge and Schekman, 2014; Ge et al., 2014). Additionally, ATG14 directs Beclin-1/Atg6 from the TGN to the autophagosomes (Sun et al., 2008). Therefore, ERGIC and TGN provide a location for the induction and elongation of autophagosomes.
Golgi-Related Proteins Are Directly Involved in Formation or Regulation of Autophagy
Different Golgi proteins play different roles in formation and degradation of autophagosomes. For example, Golgi coiled-coil protein (GCC)88 and Golgin subfamily A member 2 (GM130) play negative regulatory roles in formation and degradation of autophagosomes, while p230, WW domain containing adaptor with coiled-coil (WAC), Rabs, Golgi reassembly-stacking protein of 55 kDa (GRASP55), CLE6A, progestin and AdipoQ Receptor 3 (PAQR3), and Golgi phosphoprotein 3 (GOLPH3) play positive regulatory roles in formation and degradation of autophagosomes. The detailed underlying mechanisms, including transportation of mATG9; regulation of mTOR, ULK, homotypic fusion and protein sorting complex, or PtdIns3K UV radiation resistance-associated gene protein (UVRAG) complex; and late steps of macroautophagic and endocytic degradation, are described below (Figure 4).
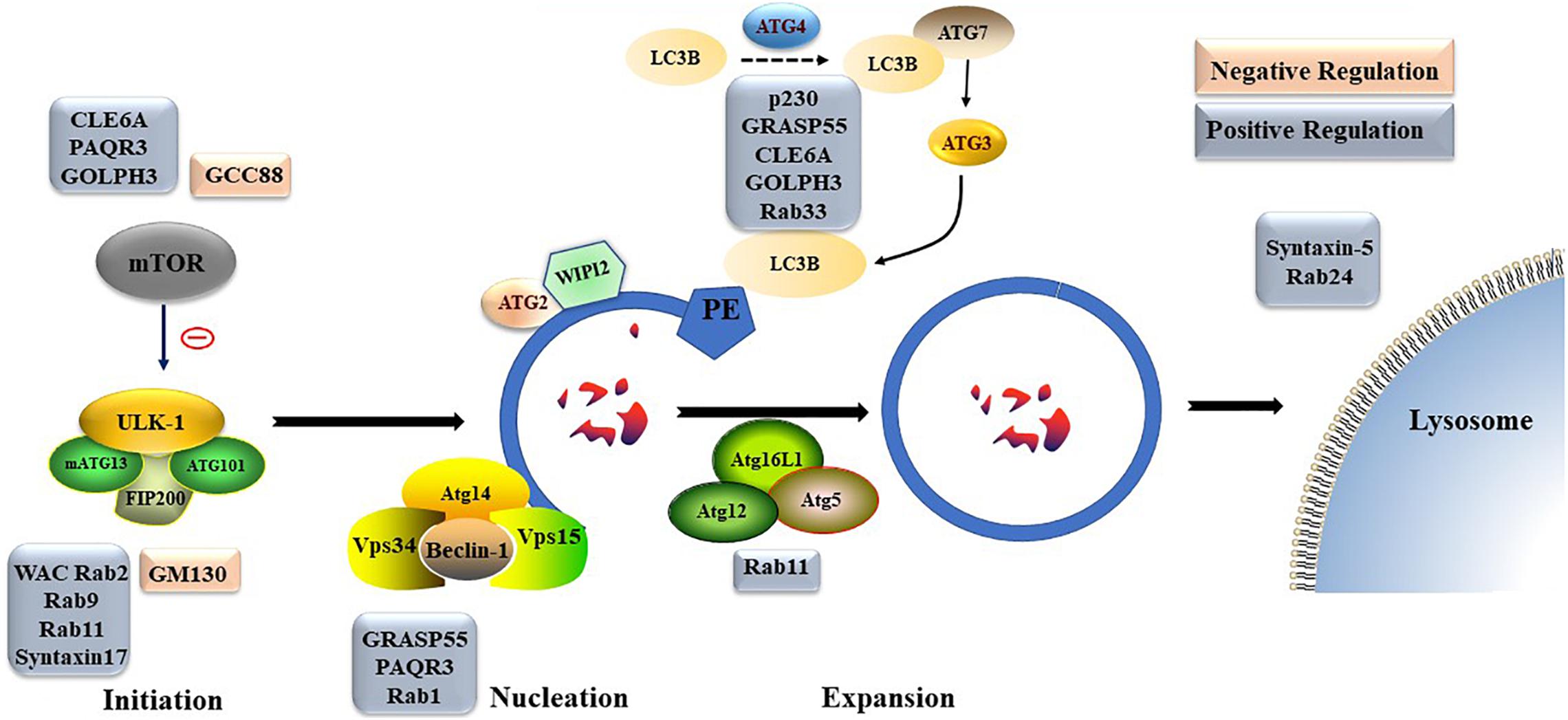
Figure 4. The detailed regulation mechanisms of Golgi proteins involved in autophagosome formation.
Golgin-245 (p230)
Golgin-245 or t-golgin-1 is a member of peripheral membrane proteins that associate with the TGN via a C-terminal GRIP domain (Yoshino et al., 2005), and dimerization of the GRIP domain is essential for its Golgi localization (Lu et al., 2006). P230 participates in cargo trafficking and regulates the position of the Golgi indirectly probably by regulating the retrograde movement of cargos or activation of dynein-dynactin complexes in newly formed Golgi elements (Yoshino et al., 2005). p230 poses a positive regulation in autophagy. During amino acid starvation, p230 is essential to the formation of LC3-positive structures in autophagosome (Sohda et al., 2015). The potential mechanism derived that p230 provides a molecular link of specific vesicles with mAtg9 from the TGN to the target structures (Sohda et al., 2015).
GCC88
GCC88 negatively regulates the Golgi ribbon, which involves the alteration in actin cytoskeleton. Studies have reported that knock-down of GCC88 results in a longer Golgi ribbon (Gosavi et al., 2018). Scaffold intersectin-1 (ITSN-1) is a scaffolding protein involved in endocytosis and has guanine nucleotide exchange activity for Cdc42 (Zamanian and Kelly, 2003), and it is a novel TGN component and a binding partner of GCC88 that links the Golgi complex to the actin cytoskeleton to regulate the Golgi structure (Makhoul et al., 2019). The GCC88-ITSN-1 pathway is relevant to several different processes that lead to abnormal morphology of the Golgi (Makhoul et al., 2019). Alteration in Golgi architecture does not affect membrane transport, while organization of the Golgi ribbon has a regulation effect on autophagy via mTOR signaling (Gosavi et al., 2018). mTOR is one of the main regulators of autophagy by controlling activity of the ULK1 complex (Al-Bari and Xu, 2020). Loss of the Golgi ribbon results in dramatic reduction in mTOR activity and induction of autophagy, presumably due to the inability to recruit mTOR to the Golgi pool (Gosavi et al., 2018).
GM130 and WAC
GM130 is located in the cis-Golgi, and knockout of GM130 causes Golgi fragmentation and impaired secretory trafficking in mice (Liu et al., 2017). WAC localizes in the nucleus and the Golgi, where it regulates epigenetics and post-mitotic Golgi reassembly, respectively (Joachim and Tooze, 2016). Two Golgi proteins, WAC and GM130, are interacted with the ATG8 homolog (GABARAP) and influence the autophagy signaling pathway by regulating the subcellular localization of GABARAP (Joachim et al., 2015). WAC promotes autophagy, while GM130 inhibits autophagy (Joachim et al., 2015). The GABARAP subfamily is critical to starvation-induced activation of autophagy and specifically promotes ULK kinase activation dependent on the ULK1 LIR motif (Slobodkin and Elazar, 2013). GABARAP located in the Golgi can inhibit autophagy, while the pericentriolar matrix (PCM) pool of GABARAP facilitates formation of autophagosome (Joachim et al., 2015; Joachim and Tooze, 2016). In steady-state conditions, GABARAP is bound to GM130 on the Golgi complex (Joachim et al., 2015). Amino acid starvation promotes activation of WAC by promoting its association with GM130 and dissociation of GABARAP from GM130 (Slobodkin and Elazar, 2013; Joachim and Tooze, 2018). Upon release from the Golgi complex, GABARAP accumulates in the PCM, and the reservoir of GABARAP in PCM activates the ULK complex and induces autophagy (Joachim and Tooze, 2018).
Rabs (Small GTP Binding Proteins)
Rab protein is the largest subfamily of the small molecule GTP-binding protein family, consisting of the conserved G domain and highly variable N-terminal and C-terminal. Rab proteins not only participate in maintaining integrity of the Golgi but also play a regulatory role in signal pathways or cargo trafficking in the Golgi. In addition, Rab GTPases that are critical to the function of the Golgi have been demonstrated to take part in induction, elongation, and degradation of autophagosomes, as shown in Table 2. Rab proteins, many with ATG proteins as interacting partners, modulate ULK1 kinase activity, recruit the ATG5–12/16L complex, and tether ATG9 vesicles or regulate endosomal degradation, which composes the necessary steps in the early or late stage of autophagy (Morgan et al., 2019).
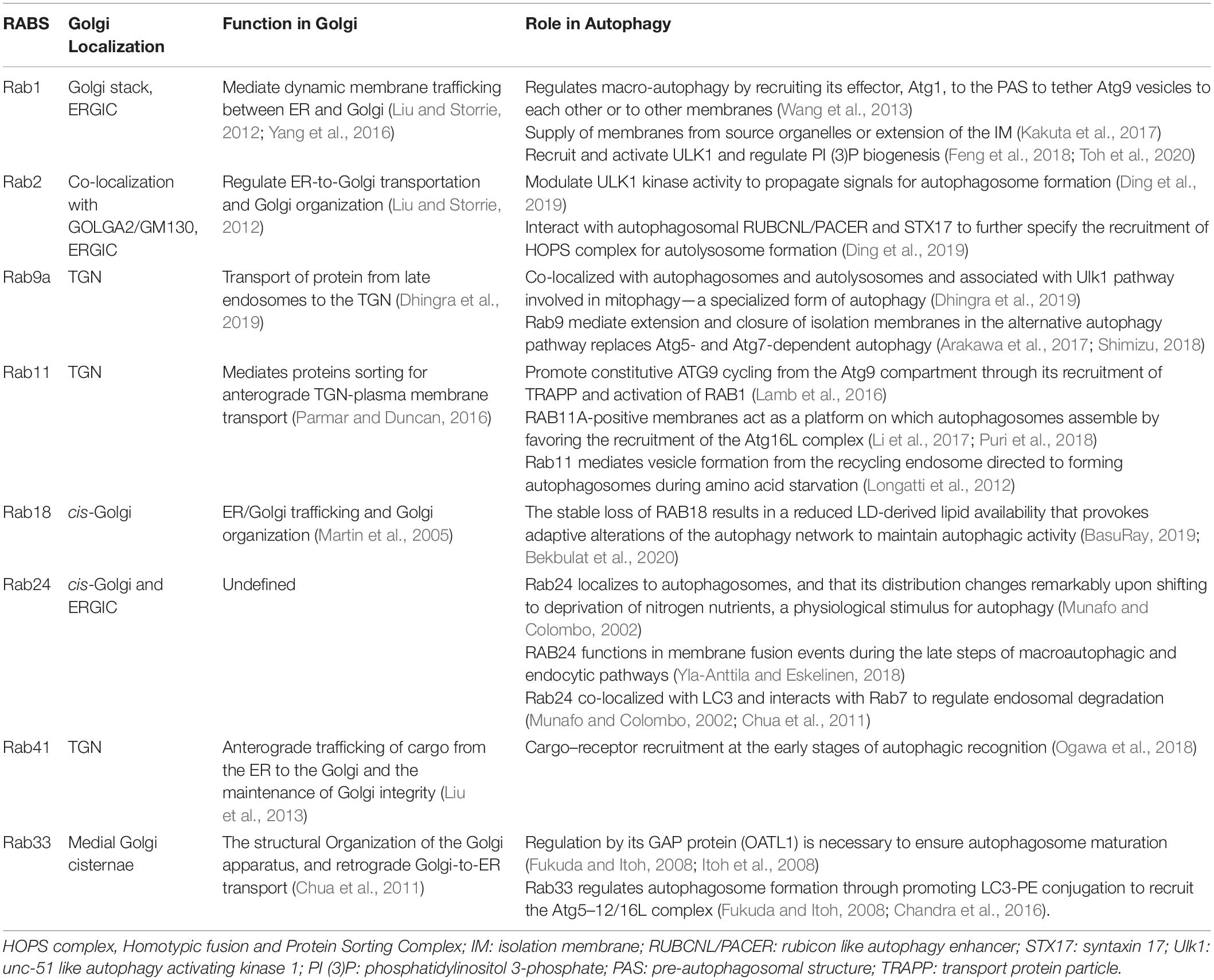
Table 2. The function of Rabs in autophagy.
GORASP2/GRASP55
GRASP55 is primarily localized on the medial-trans cisternae, plays a role in medial-trans Golgi stack formation to maintain the number of cisternae per stack, and links the Golgi ribbon (Xiang and Wang, 2010). Depletion of GRASP55 impairs accurate protein glycosylation and sorting (Zhang and Wang, 2015). Moreover, GRASP55 also functions as an energy sensor and membrane linker in autophagy (Zhang et al., 2018). Upon glucose starvation, GORASP2 is de-O-GlcNAcylated and acts as a membrane tether to facilitate autophagosome-lysosome fusion (Zhang et al., 2018). Additionally, GORASP55 interacts with BECN1 to facilitate the assembly and membrane association of the PtdIns3K UVRAG complex (Zhang et al., 2019). Furthermore, GRASP55 displays positive regulatory effects on autophagy induction and influences the lipidation of LC3b (Dupont et al., 2011).
CLEC16A
CLEC16A is important for the morphology maintenance of the Golgi in Purkinje cells, which may be derived from its function in maintaining or assisting other Golgi apparatus proteins in organelle structures (Redmann et al., 2016). Golgi-associated CLEC16A co-localizes with active mTOR complex and may function as a negative regulator in starvation-induced autophagy via enhancing the mTORC1 signaling pathway (Tam et al., 2017). Moreover, CLEC16A deficiency results in a failure of autolysosome function or clearance in the step-in autophagy downstream of autophagosome and lysosome fusion (Redmann et al., 2016).
PAQR3
PAQR3 is a 7-transmembrane protein specifically localized in the Golgi apparatus (Li et al., 2018). PAQR3 can augment autophagy via different mechanisms. PAQR3 can not only enhance the capacity of pro-autophagy class III PI3K but also integrate AMPK signal to activation of ATG14L-linked VPS34 complex upon glucose starvation (Xu et al., 2016). Besides the initiation role in the early stage of autophagy induced by glucose starvation, PAQR3 also negatively regulates amino acid-induced activation of mTORC1 (Wang L. et al., 2017). In addition, Cao et al. demonstrated that PAQR3 modulates tyrosine kinase inhibitor-induced autophagy in lung cancer cells by blocking epidermal growth factor receptor interaction with BECN1 and inhibiting BECN1 phosphorylation (Cao et al., 2020).
GOLPH3
GOLPH3, which is also known as MIDAS, GOPP1, or GPP34, is a peripheral membrane protein localized in the Golgi (Yu et al., 2016; Kuna and Field, 2019). GOLPH3 localized to the trans-Golgi provides a link from the trans-Golgi membrane to the actin cytoskeleton that plays a critical role in anterograde trafficking to the plasma membrane (Buschman et al., 2015). GOLPH3 affects autophagy through different mechanisms. For instance, Li et al. showed that during the initial oxygen-glucose deprivation injury, suppression of GOLPH3 led to a significant reduction in reactive oxygen species (ROS) generation and directly affected stress-related autophagy by suppressing lipidation of LC3b (Li et al., 2016). Moreover, GOLPH3 overexpression activates autophagy inhibitor mTOR-signaling and confers autophagy resistance (Scott et al., 2009; Salem et al., 2012).
Protein-Mediated Trafficking at the Golgi
The Golgi provides a locale for the formation of a variety of cargo-containing vesicles targeted to their destinations by binding with different protein coats (De Tito et al., 2020). Among various coats involved in TGN exit, adaptor protein (AP) complexes and the BAR domain protein superfamily, including bax-interacting factor 1 (Bif-1)/SH3GLB1, Arfaptins, and PI4KIII, are implicated in the regulation of ATG9A trafficking and are firmly connected with autophagy (De Tito et al., 2020). AP-1, 2, and 4, as sorting motifs, bind with ATG9 during trafficking from recycling endosomes to the Golgi (Imai et al., 2016). Bif-1-mediated fragmentation of the Golgi complex during nutrient starvation plays a crucial role in ATG9 trafficking and autophagosome biogenesis through the Bif-1-dynamin 2 interaction (Takahashi et al., 2011). Arfaptin2 regulates the starvation-dependent distribution of ATG9A vesicles, and PI4KIIIβ interacts with ATG9A and ATG13 to control PI4P production in the autophagic response (Imai et al., 2016).
Soluble N-Ethylmaleimide-Sensitive-Factor-Attachment Protein Receptors (SNAREs)
Accumulating evidence demonstrates that Syntaxin (Stx)5, Stx17, and Stx16 are involved in the autophagy pathway (Tang, 2019). Stx17 is a SNARE protein mainly localized in the ER and partly in the ERGIC (Muppirala et al., 2011) and is essential for maintaining the architecture of ERGIC and the Golgi (Muppirala et al., 2011), and it contributes to the autophagy pathway at several stages from initiation to maturation (Gu et al., 2019). Kumar et al. revealed that TBK1-phosphorylated Stx17 is important for assembly of the ULK1 complex and is critical for autophagy initiation (Kumar et al., 2019). In addition, Stx17 interacts with mAtg8s via the LIR motif and inserts into the autophagosomal membrane (Kumar et al., 2018; Shen et al., 2020). Stx16 is localized in the late Golgi compartments, receives retrograde transport from the endosomes (Xu et al., 2002), and is involved in autophagy by facilitating the transport of ATG9a-containing vesicles to growing autophagosomes (Tang, 2019). Moreover, stx16 is recruited by Atg8/LC3/GABARAP family proteins to autophagosomes and endolysosomes (Tang, 2019). Stx5 is involved in ER-Golgi trafficking of specialized cargo molecules, and its proper expression maintains the normal morphology and function of the Golgi in mammalian cells (Linders et al., 2019). The STX5 partner SNARE GS27 binds to both COG6 and COG8, and the COG complex plays a regulatory role in the cis-Golgi-localized STX5 SNARE complex (Willett et al., 2013). Depletion of syntaxin-5 complex components results in accumulation of autophagosomes due to lysosomal dysfunction, thereby leading to decreased degradation of autophagic substrates (Renna et al., 2011).
Effects of Disrupted Formation of Autophagy on Golgi Function
Autophagy May Induce the Golgi Stress Response by Activating TFE3
The Golgi stress response, a mechanism to meet cellular demands, maintains the homeostasis of the Golgi under the conditions of the overloading state (Taniguchi et al., 2015). Sensors of Golgi stress detect the accumulation of proteins that are not properly modified (Taniguchi and Yoshida, 2017). Then, sensors activate the following three downstream signaling pathways related to the Golgi response to stress: TFE3 (inducing Golgi expansion by regulating the transcriptional induction of Golgi-related genes), HSP47 (preventing Golgi stress-induced apoptosis), and CREB3 (inducing apoptosis through the transcriptional induction of ARF4) (Taniguchi and Yoshida, 2017). One previous study found that autophagy initiation allowed for the parallel activation of the TFE3 pathway (Mytych et al., 2020). Additionally, it has been proposed that core proteins accumulate in the Golgi, cause Golgi dysfunction, and induce autophagy of the Golgi apparatus. Subsequently, signal-inducing autophagy indirectly activates TFE3 (Taniguchi et al., 2015; Mytych et al., 2020). After activating TFE3, the Golgi expands in accordance with greater protein synthesis demands.
Non-Canonical Golgi Membrane-Associated Degradation (GOMED) Promotes Alternative Clearance in Autophagy-Deficient Cells
The molecular mechanisms of macro-autophagy involve more than 30 autophagy-related proteins (ATGs) (Mizushima et al., 2011), and ATG5 and ATG7 are key components in the formation of autophagosomes and are essential for macro-autophagy (Su et al., 2017). Nishida et al. (2009) identified two distinct autophagic pathways: the conventional autophagy pathway and the alternative autophagy pathway, the latter one was demonstrated as an ATG5/ATG7-independent type of autophagy and named “alternative autophagy.” Different from the conventional autophagy, the autophagic membrane source of “alternative autophagy” is exclusively derived from Golgi apparatus (Arakawa et al., 2017). A novel degradation pathway, named Golgi membrane-associated degradation (GOMED) pathway, is activated when Golgi-to-plasma membrane trafficking is disrupted due to lack of Atg5/Atg7-dependent autophagy (Yamaguchi et al., 2016). When this pathway is activated, the Golgi membranes become stacked, elongated, and curved and generate double-membrane compartments that enclose the cytoplasm and various organelles (Yamaguchi et al., 2016); this process is also classified as “alternative autophagy.” Furthermore, autophagy inhibition can trigger a compensatory clearance by GOMED when autophagic lysosomal degradation is impaired (Barthet and Ryan, 2018).
Aberrant Autophagy is Involved in Neurological Diseases
Autophagy is involved in tumor suppression, resistance to pathogens, and extension of lifespan (Dupont et al., 2011). It is worth noting that moderate levels of autophagy are important for the proper functioning of the nervous system. Autophagy plays physiological roles in neurodevelopment and maintenance of neuronal homeostasis, and it is involved in pathological processes that affect the brain, including neurodegenerative disorders and stroke (Corti et al., 2020).
Stroke
Ischemic stroke produces ROS and induces oxidative stress. In response to stress, autophagy exerts a two-way effect in ischemic stroke, and cerebral ischemic injury can be reduced by regulating autophagy (Mo et al., 2020). By enhancing autophagosome formation and promoting fusion of autophagosomes or attenuating autophagic-lysosomal defects, the autophagy-lysosome pathway exerts a protective effect on cerebral ischemia; however, promoting lysosomal dysfunction and autophagic defect exerts a detrimental effect (Chen et al., 2020). The major regulatory pathways of autophagy in cerebral ischemic injury involve the PI3K/Akt/mTOR pathway (inhibit), PI3K pathway (type I PI3K, inhibit; type III PI3K, enhance), the PPAR-γ/Beclin-1 pathway (enhance), the MAPK pathway (enhance), the NF-κB-dependent p53 signaling pathway (enhance) (Wolf et al., 2019), the HIF-1α/AMPK/mTOR signaling pathway (enhance), and the HIF-1α/BNIP3 signaling pathway (enhance) (Sun et al., 2018).
Neurodegenerative Disorders
Alzheimer’s Disease (AD)
Alzheimer’s disease is characterized by progressive dementia and cognitive impairment (Djajadikerta et al., 2020). Its pathogenesis is presented as intracellular accumulation of hyperphosphorylated tau protein and extracellular aggregates of amyloid β (Aβ) (Djajadikerta et al., 2020; Tiwari et al., 2019). Autophagy plays a dual role in Aβ degradation and secretion. In the early stages of AD, Aβ formation can activate autophagy, and Aβ can be degraded via transportation from autophagosomes to lysosomes. In the later stages of AD, the persistent accumulation of Aβ induces aberrant autophagy and excessive phagocytosis of normal proteins (Kuang et al., 2020). Further, Aβ-induced autophagy impairment that is mediated by the loss of Beclin-1 leads to microglial activation and increased production of proinflammatory cytokines that contribute to neuroinflammation (Thangaraj et al., 2020). Moreover, Aβ accumulation leads to decreased Bif-1 expression in neurons, which in turn elevates Aβ accumulation and plaque formation, possibly through impairment of autophagy (Wang et al., 2015).
Parkinson’s Disease (PD)
Parkinson’s disease is a progressive neurological disease characterized by Lewy bodies, which are composed of aggregates of α-synuclein (Dickson, 2018). α-synuclein is considered the typical pathologic correlate of PD (Dickson, 2018) and is composed of an N-terminal of amphipathic α-helical domain combining with the membrane (Deng et al., 2018). It is encoded by the synuclein alpha (SNCA) gene, and most SNCA duplication carriers are associated with parkinsonism and nonmotor symptoms (Chartier-Harlin et al., 2004; Deng et al., 2018). Mutations in α-synuclein, such as the mutant SNCA A53T, result in significantly decreased lipidation of LC3 and increased activity of mTOR signaling pathway, which inhibit the autophagic flux and reduce removal of abnormal proteins (Jiang et al., 2013). Besides mutations in SNCA, other genetic forms of PD involve mutations in LRRK2, VPS35, PRKN, PINK1, and PARK7 (Hou et al., 2020). Different PD genes/mutations affect autophagic function and contribution of autophagic impairment to PD pathogenesis (Hou et al., 2020).
Huntington’s Disease (HD)
Huntington’s disease is characterized by a pathologic mutation consisting of an expanded CAG repeat in the huntingtin gene (HTT) on chromosome 4, encoding the huntingtin (htt) protein (Dayalu and Albin, 2015). Htt functions as a scaffold protein that is required for selective autophagy (Ochaba et al., 2014; Rui et al., 2015). Htt positively modulates selective autophagy by facilitating p62-mediated cargo recognition or promoting ULK1 activation (Rui et al., 2015). Mutant HTT (mHTT) could affect mitophagy by interfering with ULK1 activation and shift from the mTORC1 to HTT scaffolding complexes and impairing the interaction of BECN1-PIK3R4/VPS15 (Franco-Iborra et al., 2020). Impaired autophagy leads to impaired clearance of abnormal proteins.
Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD)
Amyotrophic lateral sclerosis and FTD are mainly caused by genetic mutations that lead to abnormal protein aggregates, such as RNA-binding protein and TAR DNA-binding protein-43 kDa, in the brain (Hedl et al., 2019). More than 25 genes that are implicated in ALS, FTD, and ALS/FTD have been identified (Ling et al., 2013). There are a multitude of ALS genes with functions related to the autophagic system, including p62, optineurin, VCP, ubiquilin 2, and TBK1 (Nguyen et al., 2019). Among the most frequently identified mutations in ALS, mutations in superoxide dismutase 1 (SOD1) can influence autophagy (Casterton et al., 2020). Mutant SOD1 fails to interact with Beclin-1 and destabilizes Beclin-1-Bcl-xL complex or inhibits mTOR signaling, leading to a defective autophagy flux (Lee et al., 2015; Nguyen et al., 2019). Additionally, mutations in microtubule-associated protein tau or granulin (GRN) are associated with pure FTD. GRN is a gene that encodes progranulin (PGRN) (Holler et al., 2017). PGRN can regulate lysosome function, and PGRN deficiency in neurons increases autophagy and causes abnormally enlarged lysosomes (Elia et al., 2019).
Traumatic Brain Injury (TBI)
Traumatic brain injury is defined as a traumatically induced structural injury or physiological disruption of brain function as a result of external forces (Zeng et al., 2020). Neuron dysfunction induced by TBI is derived from primary and secondary brain injury mechanisms, and secondary injury results from delayed neurochemical, metabolic, and cellular changes (Feng et al., 2017). Further, disruption of autophagy flux is one of the mechanical attributes to secondary injury (Feng et al., 2017; Wu and Lipinski, 2019). Autophagy has a double regulating effect in the pathological process in TBI. Results from a previous study revealed that LC3-II, Beclin-1, p62, and autophagosomes were increased in human TBI autopsy samples (Sakai et al., 2014), suggesting enhancement of autophagy initiation. Conversely, TBI activates phospholipase A2 and attenuates lysosomal membrane permeability, as well as subsequent impairment of autophagy and neuronal loss (Sarkar et al., 2020).
Subarachnoid Hemorrhage (SAH)
Subarachnoid hemorrhage is a subtype of hemorrhagic stroke with high mortality and morbidity that are mainly caused by early brain injury (EBI) (Liu et al., 2014). Elevated autophagy levels and activated autophagy-related pathway proteins, which exert a protective effect, have been demonstrated in experimental SAH models (Wang et al., 2012). Li Y. et al. (2019) found that rapamycin inhibited mTOR activity and induced autophagy, which subsequently attenuated neuronal injury in EBI with improved neurological deficits, BBB permeability, and brain edema. Li and Han (2018) demonstrated that resveratrol (RSV) attenuated EBI after SAH, and the effects of RSV on SAH-induced EBI were mediated via the AMPK/SIRT1/autophagy pathway.
Regulation of Golgi Function May Be a Therapeutic Target for Aberrant Autophagy-Based Neurological Diseases
Proper regulation of autophagy can reduce neuronal damage and diminish the disease progression. As we mentioned before, the Golgi plays an important role in regulating autophagy. Therefore, therapies that affect Golgi morphology or function could be used as a therapeutic tool in aberrant autophagy-related diseases. Studies have reported that Golgi therapy alleviates the damage in cancers (Chang et al., 2012; Cao et al., 2019) and autoimmune diseases (Schuster et al., 2015) by influencing the formation of autophagosomes. Since the same autophagy mechanisms are observed in cancers, autoimmune diseases, and neurological diseases, regulation of the function of the Golgi apparatus may be a potential therapeutic target for neurology diseases, and neuron damage in neurological diseases could be alleviated by repairing the Golgi function or regulating Golgi proteins to keep moderate autophagy. In fact, a normal and appropriate level of autophagy plays a role in the maintenance of Golgi homeostasis. For example, (Li D. et al., 2019) found that the upregulation of miR-21-5p inhibited formation of autophagosomes in neurons by targeting Rab11a and subsequently attenuated autophagy-induced nerve injury in vitro, and (Li et al., 2016) demonstrated that suppression of GOLPH3 alleviated oxidative injury and directly affected oxidative stress-related autophagy by suppressing lipidation of LC3B in oxygen-glucose deprivation/reoxygenation injury. These findings support the hypothesis that the Golgi may be an ideal target for regulating autophagy disruption in neurodegenerative diseases or stroke. Thus, targeted therapies that are aimed at protecting the Golgi or regulating Golgi proteins may be effective therapeutic approaches that prevent autophagy-related neurological diseases in the future.
Conclusion
The Golgi plays an important role in autophagy from the early stages of formation to the final degradation. Autophagy-related proteins, including Beclin-1 and ATG9, are localized in the Golgi, and some regulators in autophagy signaling pathways, such as PtdIns3K class III complex and Rabs, are trafficking in ERGIC and TGN. In addition, Golgi-localized proteins, including GRASP55, Golgin230, GM130, and GOLPH3, maintain the function and integrity of the Golgi and regulate the transport and formation of autophagosomes. Moreover, aberrant autophagy is involved in neurological diseases. Therefore, Golgi-targeted therapies that overexpress or silence Golgi proteins involved in autophagy or that interfere with the transport or trafficking functions of the Golgi may be used to treat neurological diseases. Future studies should investigate the potential application of Golgi therapy in autophagy-based neurological diseases.
Author Contributions
SD carried out the literature review and drafted the manuscript. JL and XW helped draft the manuscript. WL contributed to and finalized the draft. All authors read and approved the final manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (#81571181).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Al-Bari, M. A. A., and Xu, P. (2020). Molecular regulation of autophagy machinery by mTOR-dependent and -independent pathways. Ann. N. Y. Acad. Sci. 1467, 3–20. doi: 10.1111/nyas.14305
Appenzeller-Herzog, C., and Hauri, H. P. (2006). The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J. Cell Sci. 119(Pt 11), 2173–2183. doi: 10.1242/jcs.03019
Arakawa, S., Honda, S., Yamaguchi, H., and Shimizu, S. (2017). Molecular mechanisms and physiological roles of Atg5/Atg7-independent alternative autophagy. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 93, 378–385. doi: 10.2183/pjab.93.023
Barthet, V. J. A., and Ryan, K. M. (2018). Autophagy in Neurodegeneration: Can’t Digest It. Spit It Out! Trends Cell Biol. 28, 171–173. doi: 10.1016/j.tcb.2018.01.001
BasuRay, S. (2019). RAB18 modulates autophagy in human stellate cells. J. Clin. Lipidol. 13, 832–838. doi: 10.1016/j.jacl.2019.07.006
Bekbulat, F., Schmitt, D., Feldmann, A., Huesmann, H., Eimer, S., Juretschke, T., et al. (2020). RAB18 loss interferes with lipid droplet catabolism and provokes autophagy network adaptations. J. Mol. Biol. 432, 1216–1234. doi: 10.1016/j.jmb.2019.12.031
Buschman, M. D., Xing, M., and Field, S. J. (2015). The GOLPH3 pathway regulates Golgi shape and function and is activated by DNA damage. Front. Neurosci. 9:362. doi: 10.3389/fnins.2015.00362
Cancino, J., Jung, J. E., and Luini, A. (2013). Regulation of Golgi signaling and trafficking by the KDEL receptor. Histochem. Cell Biol. 140, 395–405. doi: 10.1007/s00418-013-1130-9
Cao, Q., You, X., Xu, L., Wang, L., and Chen, Y. (2019). PAQR3 suppresses the growth of non-small cell lung cancer cells via modulation of EGFR-mediated autophagy. Autophagy 167, 1236–1247. doi: 10.1080/15548627.2019.1659654
Cao, Q., You, X., Xu, L., Wang, L., and Chen, Y. (2020). PAQR3 suppresses the growth of non-small cell lung cancer cells via modulation of EGFR-mediated autophagy. Autophagy 16, 1236–1247. doi: 10.1080/15548627.2019.1659654
Casterton, R. L., Hunt, R. J., and Fanto, M. (2020). Pathomechanism heterogeneity in the amyotrophic lateral sclerosis and frontotemporal dementia disease spectrum: providing focus through the lens of autophagy. J. Mol. Biol. 432, 2692–2713. doi: 10.1016/j.jmb.2020.02.018
Chandra, M., Saran, R., and Datta, S. (2016). Deciphering the role of Atg5 in nucleotide dependent interaction of Rab33B with the dimeric complex, Atg5-Atg16L1. Biochem. Biophys. Res. Commun. 473, 8–16. doi: 10.1016/j.bbrc.2016.03.043
Chang, S. H., Hong, S. H., Jiang, H. L., Minai-Tehrani, A., Yu, K. N., Lee, J. H., et al. (2012). GOLGA2/GM130, cis-Golgi matrix protein, is a novel target of anticancer gene therapy. Mol. Ther. 20, 2052–2063. doi: 10.1038/mt.2012.125
Chartier-Harlin, M. C., Kachergus, J., Roumier, C., Mouroux, V., Douay, X., Lincoln, S., et al. (2004). Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169. doi: 10.1016/s0140-6736(04)17103-1
Chen, C., Qin, H., Tan, J., Hu, Z., and Zeng, L. (2020). The role of ubiquitin-proteasome pathway and autophagy-lysosome pathway in cerebral ischemia. Oxid. Med. Cell. Longev. 2020:5457049. doi: 10.1155/2020/5457049
Chua, C. E., Gan, B. Q., and Tang, B. L. (2011). Involvement of members of the Rab family and related small GTPases in autophagosome formation and maturation. Cell. Mol. Life Sci. 68, 3349–3358. doi: 10.1007/s00018-011-0748-9
Corti, O., Blomgren, K., Poletti, A., and Beart, P. M. (2020). Autophagy in neurodegeneration: new insights underpinning therapy for neurological diseases. J. Neurochem. 154:e15002. doi: 10.1111/jnc.15002
Dayalu, P., and Albin, R. L. (2015). Huntington disease: pathogenesis and treatment. Neurol. Clin. 33, 101–114. doi: 10.1016/j.ncl.2014.09.003
De Tito, S., Hervás, J. H., van Vliet, A. R., and Tooze, S. A. (2020). The golgi as an assembly line to the autophagosome. Trends Biochem. Sci. 45, 484–496. doi: 10.1016/j.tibs.2020.03.010
Deng, H., Wang, P., and Jankovic, J. (2018). The genetics of Parkinson disease. Ageing Res. Rev. 42, 72–85. doi: 10.1016/j.arr.2017.12.007
Dhingra, R., Rabinovich-Nikitin, I., and Kirshenbaum, L. A. (2019). Ulk1/Rab9-mediated alternative mitophagy confers cardioprotection during energy stress. J. Clin. Invest. 129, 509–512. doi: 10.1172/JCI125980
Dickson, D. W. (2018). Neuropathology of Parkinson disease. Parkinsonism Relat. Disord. 46(Suppl. 1), S30–S33. doi: 10.1016/j.parkreldis.2017.07.033
Ding, X., Jiang, X., Tian, R., Zhao, P., Li, L., Wang, X., et al. (2019). RAB2 regulates the formation of autophagosome and autolysosome in mammalian cells. Autophagy 15, 1774–1786. doi: 10.1080/15548627.2019.1596478
Djajadikerta, A., Keshri, S., Pavel, M., Prestil, R., Ryan, L., and Rubinsztein, D. C. (2020). Autophagy induction as a therapeutic strategy for neurodegenerative diseases. J. Mol. Biol. 432, 2799–2821. doi: 10.1016/j.jmb.2019.12.035
D’Souza, Z., Blackburn, J. B., Kudlyk, T., Pokrovskaya, I. D., and Lupashin, V. V. (2019). Defects in COG-mediated golgi trafficking alter endo-lysosomal system in human cells. Front. Cell Dev. Biol. 7:118. doi: 10.3389/fcell.2019.00118
Dupont, N., Jiang, S., Pilli, M., Ornatowski, W., Bhattacharya, D., and Deretic, V. (2011). Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 30, 4701–4711. doi: 10.1038/emboj.2011.398
Eapen, V. V., and Haber, J. E. (2013). DNA damage signaling triggers the cytoplasm-to-vacuole pathway of autophagy to regulate cell cycle progression. Autophagy 9, 440–441. doi: 10.4161/auto.23280
Egan, D. F., Shackelford, D. B., Mihaylova, M. M., Gelino, S., Kohnz, R. A., Mair, W., et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. doi: 10.1126/science.1196371
Elia, L. P., Mason, A. R., Alijagic, A., and Finkbeiner, S. (2019). Genetic regulation of neuronal progranulin reveals a critical role for the autophagy-lysosome pathway. J. Neurosci. 39, 3332–3344. doi: 10.1523/jneurosci.3498-17.2019
Feng, Y., Gao, J., Cui, Y., Li, M., Li, R., Cui, C., et al. (2017). Neuroprotective effects of resatorvid against traumatic brain injury in rat: involvement of neuronal autophagy and tlr4 signaling pathway. Cell. Mol. Neurobiol. 37, 155–168. doi: 10.1007/s10571-016-0356-1
Feng, Z. Z., Jiang, A. J., Mao, A. W., Feng, Y., Wang, W., Li, J., et al. (2018). The Salmonella effectors SseF and SseG inhibit Rab1A-mediated autophagy to facilitate intracellular bacterial survival and replication. J. Biol. Chem. 293, 9662–9673. doi: 10.1074/jbc.M117.811737
Franco-Iborra, S., Plaza-Zabala, A., Montpeyo, M., Sebastian, D., Vila, M., and Martinez-Vicente, M. (2020). Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy. doi: 10.1080/15548627.2020.1728096 [Epub ahead of print].
Fukuda, M., and Itoh, T. (2008). Direct link between Atg protein and small GTPase Rab: Atg16L functions as a potential Rab33 effector in mammals. Autophagy 4, 824–826. doi: 10.4161/auto.6542
Ge, L., Melville, D., Zhang, M., and Schekman, R. (2013). The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife 2:e00947. doi: 10.7554/eLife.00947
Ge, L., and Schekman, R. (2014). The ER-Golgi intermediate compartment feeds the phagophore membrane. Autophagy 10, 170–172. doi: 10.4161/auto.26787
Ge, L., Wilz, L., and Schekman, R. (2015). Biogenesis of autophagosomal precursors for LC3 lipidation from the ER-Golgi intermediate compartment. Autophagy 11, 2372–2374. doi: 10.1080/15548627.2015.1105422
Ge, L., Zhang, M., and Schekman, R. (2014). Phosphatidylinositol 3-kinase and COPII generate LC3 lipidation vesicles from the ER-Golgi intermediate compartment. eLife 3:e04135. doi: 10.7554/eLife.04135
Glick, B. S., and Nakano, A. (2009). Membrane traffic within the Golgi apparatus. Annu. Rev. Cell Dev. Biol. 25, 113–132. doi: 10.1146/annurev.cellbio.24.110707.175421
Gosavi, P., Houghton, F. J., McMillan, P. J., Hanssen, E., and Gleeson, P. A. (2018). The Golgi ribbon in mammalian cells negatively regulates autophagy by modulating mTOR activity. J. Cell Sci. 131:jcs211987. doi: 10.1242/jcs.211987
Gu, Y., Princely Abudu, Y., Kumar, S., Bissa, B., Choi, S. W., Jia, J., et al. (2019). Mammalian Atg8 proteins regulate lysosome and autolysosome biogenesis through SNAREs. EMBO J. 38:e101994. doi: 10.15252/embj.2019101994
Hedl, T. J., San Gil, R., Cheng, F., Rayner, S. L., Davidson, J. M., De Luca, A., et al. (2019). Proteomics Approaches for Biomarker and Drug Target Discovery in ALS and FTD. Front. Neurosci. 13:548. doi: 10.3389/fnins.2019.00548
Hirata, T., Fujita, M., Nakamura, S., Gotoh, K., Motooka, D., Murakami, Y., et al. (2015). Post-Golgi anterograde transport requires GARP-dependent endosome-to-TGN retrograde transport. Mol. Biol. Cell 26, 3071–3084. doi: 10.1091/mbc.e14-11-1568
Holler, C. J., Taylor, G., Deng, Q., and Kukar, T. (2017). Intracellular Proteolysis of Progranulin Generates Stable, Lysosomal Granulins that Are Haploinsufficient in Patients with Frontotemporal Dementia Caused by GRN Mutations. eNeuro 4:ENEURO.0100-17.2017. doi: 10.1523/eneuro.0100-17.2017
Hou, X., Watzlawik, J. O., Fiesel, F. C., and Springer, W. (2020). Autophagy in Parkinson’s Disease. J. Mol. Biol. 432, 2651–2672. doi: 10.1016/j.jmb.2020.01.037
Imai, K., Hao, F., Fujita, N., Tsuji, Y., Oe, Y., Araki, Y., et al. (2016). Atg9A trafficking through the recycling endosomes is required for autophagosome formation. J. Cell Sci. 129, 3781–3791. doi: 10.1242/jcs.196196
Itoh, T., Fujita, N., Kanno, E., Yamamoto, A., Yoshimori, T., and Fukuda, M. (2008). Golgi-resident small GTPase Rab33B interacts with Atg16L and modulates autophagosome formation. Mol. Biol. Cell 19, 2916–2925. doi: 10.1091/mbc.E07-12-1231
Jarvela, T., and Linstedt, A. D. (2014). Isoform-specific tethering links the Golgi ribbon to maintain compartmentalization. Mol. Biol. Cell 25, 133–144. doi: 10.1091/mbc.E13-07-0395
Jiang, T. F., Zhang, Y. J., Zhou, H. Y., Wang, H. M., Tian, L. P., Liu, J., et al. (2013). Curcumin ameliorates the neurodegenerative pathology in A53T alpha-synuclein cell model of Parkinson’s disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. J. Neuroimmune Pharmacol. 8, 356–369. doi: 10.1007/s11481-012-9431-7
Joachim, J., Jefferies, H. B., Razi, M., Frith, D., Snijders, A. P., Chakravarty, P., et al. (2015). Activation of ULK Kinase and Autophagy by GABARAP Trafficking from the Centrosome Is Regulated by WAC and GM130. Mol. Cell 60, 899–913. doi: 10.1016/j.molcel.2015.11.018
Joachim, J., and Tooze, S. A. (2016). GABARAP activates ULK1 and traffics from the centrosome dependent on Golgi partners WAC and GOLGA2/GM130. Autophagy 12, 892–893. doi: 10.1080/15548627.2016.1159368
Joachim, J., and Tooze, S. A. (2018). Control of GABARAP-mediated autophagy by the Golgi complex, centrosome and centriolar satellites. Biol. Cell 110, 1–5. doi: 10.1111/boc.201700046
Johansen, T., and Lamark, T. (2011). Selective autophagy mediated by autophagic adapter proteins. Autophagy 7, 279–296. doi: 10.4161/auto.7.3.14487
Kakuta, S., Yamaguchi, J., Suzuki, C., Sasaki, M., Kazuno, S., and Uchiyama, Y. (2017). Small GTPase Rab1B is associated with ATG9A vesicles and regulates autophagosome formation. FASEB J. 31, 3757–3773. doi: 10.1096/fj.201601052R
Kakuta, S., Yamamoto, H., Negishi, L., Kondo-Kakuta, C., Hayashi, N., and Ohsumi, Y. (2012). Atg9 vesicles recruit vesicle-tethering proteins Trs85 and Ypt1 to the autophagosome formation site. J. Biol. Chem. 287, 44261–44269. doi: 10.1074/jbc.M112.411454
Kaushik, S., and Cuervo, A. M. (2018). The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 19, 365–381. doi: 10.1038/s41580-018-0001-6
Kihara, A., Kabeya, Y., Ohsumi, Y., and Yoshimori, T. (2001). Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2, 330–335. doi: 10.1093/embo-reports/kve061
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Koga, D., and Ushiki, T. (2006). Three-dimensional ultrastructure of the Golgi apparatus in different cells: high-resolution scanning electron microscopy of osmium-macerated tissues. Arch. Histol. Cytol. 69, 357–374. doi: 10.1679/aohc.69.357
Kuang, H., Tan, C. Y., Tian, H. Z., Liu, L. H., Yang, M. W., Hong, F. F., et al. (2020). Exploring the bi-directional relationship between autophagy and Alzheimer’s disease. CNS Neurosci. Ther. 26, 155–166. doi: 10.1111/cns.13216
Kulkarni-Gosavi, P., Makhoul, C., and Gleeson, P. A. (2019). Form and function of the Golgi apparatus: scaffolds, cytoskeleton and signalling. FEBS Lett. 593, 2289–2305. doi: 10.1002/1873-3468.13567
Kumar, S., Gu, Y., Abudu, Y. P., Bruun, J. A., Jain, A., Farzam, F., et al. (2019). Phosphorylation of Syntaxin 17 by TBK1 Controls Autophagy Initiation. Dev. Cell 49, 130–144.e6. doi: 10.1016/j.devcel.2019.01.027
Kumar, S., Jain, A., Farzam, F., Jia, J., Gu, Y., Choi, S. W., et al. (2018). Mechanism of Stx17 recruitment to autophagosomes via IRGM and mammalian Atg8 proteins. J. Cell Biol. 217, 997–1013. doi: 10.1083/jcb.201708039
Kuna, R. S., and Field, S. J. (2019). GOLPH3: a Golgi phosphatidylinositol(4)phosphate effector that directs vesicle trafficking and drives cancer. J. Lipid Res. 60, 269–275. doi: 10.1194/jlr.R088328
Lamb, C. A., Nuhlen, S., Judith, D., Frith, D., Snijders, A. P., Behrends, C., et al. (2016). TBC1D14 regulates autophagy via the TRAPP complex and ATG9 traffic. EMBO J. 35, 281–301. doi: 10.15252/embj.201592695
Lamb, C. A., and Tooze, S. A. (2016). TBC1D14 sets the TRAPP for ATG9. Autophagy 12, 1212–1213. doi: 10.1080/15548627.2016.1177696
Lee, J. K., Shin, J. H., Lee, J. E., and Choi, E. J. (2015). Role of autophagy in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1852, 2517–2524. doi: 10.1016/j.bbadis.2015.08.005
Li, D., Huang, S., Zhu, J., Hu, T., Han, Z., Zhang, S., et al. (2019). Exosomes from MiR-21-5p-increased neurons play a role in neuroprotection by suppressing Rab11a-mediated neuronal autophagy in vitro after traumatic brain injury. Med. Sci. Monit. 25, 1871–1885. doi: 10.12659/MSM.915727
Li, H., Wang, Y., Chen, B., and Shi, J. (2018). Silencing of PAQR3 suppresses extracellular matrix accumulation in high glucose-stimulated human glomerular mesangial cells via PI3K/AKT signaling pathway. Eur. J. Pharmacol. 832, 50–55. doi: 10.1016/j.ejphar.2018.05.032
Li, J., Chen, Z., Stang, M. T., and Gao, W. (2017). Transiently expressed ATG16L1 inhibits autophagosome biogenesis and aberrantly targets RAB11-positive recycling endosomes. Autophagy 13, 345–358. doi: 10.1080/15548627.2016.1256521
Li, T., You, H., Mo, X., He, W., Tang, X., Jiang, Z., et al. (2016). GOLPH3 Mediated Golgi Stress Response in Modulating N2A Cell Death upon Oxygen-Glucose Deprivation and Reoxygenation Injury. Mol. Neurobiol. 53, 1377–1385. doi: 10.1007/s12035-014-9083-0
Li, W., and Zhang, L. (2019). Regulation of ATG and Autophagy Initiation. Adv. Exp. Med. Biol. 1206, 41–65. doi: 10.1007/978-981-15-0602-4_2
Li, Y., Wu, P., Dai, J., Zhang, T., Bihl, J., Wang, C., et al. (2019). Inhibition of mTOR alleviates early brain injury after subarachnoid hemorrhage via relieving excessive mitochondrial fission. Cell. Mol. Neurobiol. 40, 629–642. doi: 10.1007/s10571-019-00760-x
Li, Z., and Han, X. (2018). Resveratrol alleviates early brain injury following subarachnoid hemorrhage: possible involvement of the AMPK/SIRT1/autophagy signaling pathway. Biol. Chem. 399, 1339–1350. doi: 10.1515/hsz-2018-0269
Linders, P. T., Horst, C. V., Beest, M. T., and van den Bogaart, G. (2019). Stx5-mediated ER-Golgi transport in mammals and yeast. Cells 8:780. doi: 10.3390/cells8080780
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Liu, C., Mei, M., Li, Q., Roboti, P., Pang, Q., Ying, Z., et al. (2017). Loss of the golgin GM130 causes Golgi disruption, Purkinje neuron loss, and ataxia in mice. Proc. Natl. Acad. Sci. U.S.A. 114, 346–351. doi: 10.1073/pnas.1608576114
Liu, L., Zhang, M., and Ge, L. (2020). Protein translocation into the ERGIC: an upstream event of secretory autophagy. Autophagy 16, 1358–1360. doi: 10.1080/15548627.2020.1768668
Liu, S., Hunt, L., and Storrie, B. (2013). Rab41 is a novel regulator of Golgi apparatus organization that is needed for ER-to-Golgi trafficking and cell growth. PLoS One 8:e71886. doi: 10.1371/journal.pone.0071886
Liu, S., and Storrie, B. (2012). Are Rab proteins the link between Golgi organization and membrane trafficking? Cell. Mol. Life Sci. 69, 4093–4106. doi: 10.1007/s00018-012-1021-6
Liu, W. J., Ye, L., Huang, W. F., Guo, L. J., Xu, Z. G., Wu, H. L., et al. (2016). p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 21:29. doi: 10.1186/s11658-016-0031-z
Liu, Y., Li, J., Wang, Z., Yu, Z., and Chen, G. (2014). Attenuation of early brain injury and learning deficits following experimental subarachnoid hemorrhage secondary to Cystatin C: possible involvement of the autophagy pathway. Mol. Neurobiol. 49, 1043–1054. doi: 10.1007/s12035-013-8579-3
Longatti, A., Lamb, C. A., Razi, M., Yoshimura, S., Barr, F. A., and Tooze, S. A. (2012). TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J. Cell Biol. 197, 659–675. doi: 10.1083/jcb.201111079
Lu, L., Tai, G., Wu, M., Song, H., and Hong, W. (2006). Multilayer interactions determine the Golgi localization of GRIP golgins. Traffic 7, 1399–1407. doi: 10.1111/j.1600-0854.2006.00473.x
Makhoul, C., Gosavi, P., Duffield, R., Delbridge, B., Williamson, N. A., and Gleeson, P. A. (2019). Intersectin-1 interacts with the golgin GCC88 to couple the actin network and Golgi architecture. Mol. Biol. Cell 30, 370–386. doi: 10.1091/mbc.E18-05-0313
Mari, M., Griffith, J., Rieter, E., Krishnappa, L., Klionsky, D. J., and Reggiori, F. (2010). An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 190, 1005–1022. doi: 10.1083/jcb.200912089
Martin, S., Driessen, K., Nixon, S. J., Zerial, M., and Parton, R. G. (2005). Regulated localization of Rab18 to lipid droplets: effects of lipolytic stimulation and inhibition of lipid droplet catabolism. J. Biol. Chem. 280, 42325–42335. doi: 10.1074/jbc.M506651200
Metcalf, D. J., García-Arencibia, M., Hochfeld, W. E., and Rubinsztein, D. C. (2012). Autophagy and misfolded proteins in neurodegeneration. Exp. Neurol. 238, 22–28. doi: 10.1016/j.expneurol.2010.11.003
Mizushima, N., Yoshimori, T., and Ohsumi, Y. (2011). The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132. doi: 10.1146/annurev-cellbio-092910-154005
Mo, Y., Sun, Y. Y., and Liu, K. Y. (2020). Autophagy and inflammation in ischemic stroke. Neural Regen. Res. 15, 1388–1396. doi: 10.4103/1673-5374.274331
Morgan, N. E., Cutrona, M. B., and Simpson, J. C. (2019). Multitasking Rab proteins in autophagy and membrane trafficking: a focus on Rab33b. Int. J. Mol. Sci. 20:3916. doi: 10.3390/ijms20163916
Muller, O., Sattler, T., Flotenmeyer, M., Schwarz, H., Plattner, H., and Mayer, A. (2000). Autophagic tubes: vacuolar invaginations involved in lateral membrane sorting and inverse vesicle budding. J. Cell Biol. 151, 519–528. doi: 10.1083/jcb.151.3.519
Munafo, D. B., and Colombo, M. I. (2002). Induction of autophagy causes dramatic changes in the subcellular distribution of GFP-Rab24. Traffic 3, 472–482. doi: 10.1034/j.1600-0854.2002.30704.x
Muppirala, M., Gupta, V., and Swarup, G. (2011). Syntaxin 17 cycles between the ER and ERGIC and is required to maintain the architecture of ERGIC and Golgi. Biol. Cell 103, 333–350. doi: 10.1042/bc20110006
Mytych, J., Solek, P., Bedzinska, A., Rusinek, K., Warzybok, A., Tabecka-Lonczynska, A., et al. (2020). Klotho-mediated changes in the expression of Atg13 alter formation of ULK1 complex and thus initiation of ER- and Golgi-stress response mediated autophagy. Apoptosis 25, 57–72. doi: 10.1007/s10495-019-01579-z
Nakatogawa, H., Suzuki, K., Kamada, Y., and Ohsumi, Y. (2009). Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell Biol. 10, 458–467. doi: 10.1038/nrm2708
Naydenov, N. G., Feygin, A., Wang, L., and Ivanov, A. I. (2014). N-ethylmaleimide-sensitive factor attachment protein alpha (alphaSNAP) regulates matrix adhesion and integrin processing in human epithelial cells. J. Biol. Chem. 289, 2424–2439. doi: 10.1074/jbc.M113.498691
Naydenov, N. G., Harris, G., Brown, B., Schaefer, K. L., Das, S. K., Fisher, P. B., et al. (2012a). Loss of soluble N-ethylmaleimide-sensitive factor attachment protein α (αSNAP) induces epithelial cell apoptosis via down-regulation of Bcl-2 expression and disruption of the Golgi. J. Biol. Chem. 287, 5928–5941. doi: 10.1074/jbc.M111.278358
Naydenov, N. G., Harris, G., Morales, V., and Ivanov, A. I. (2012b). Loss of a membrane trafficking protein alphaSNAP induces non-canonical autophagy in human epithelia. Cell Cycle 11, 4613–4625. doi: 10.4161/cc.22885
Nguyen, D. K. H., Thombre, R., and Wang, J. (2019). Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci. Lett. 697, 34–48. doi: 10.1016/j.neulet.2018.04.006
Nishida, Y., Arakawa, S., Fujitani, K., Yamaguchi, H., Mizuta, T., Kanaseki, T., et al. (2009). Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461, 654–658. doi: 10.1038/nature08455
Ochaba, J., Lukacsovich, T., Csikos, G., Zheng, S., Margulis, J., Salazar, L., et al. (2014). Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. U.S.A. 111, 16889–16894. doi: 10.1073/pnas.1420103111
Ogawa, M., Matsuda, R., Takada, N., Tomokiyo, M., Yamamoto, S., Shizukusihi, S., et al. (2018). Molecular mechanisms of Streptococcus pneumoniae-targeted autophagy via pneumolysin, Golgi-resident Rab41, and Nedd4-1-mediated K63-linked ubiquitination. Cell. Microbiol. 20, e12846. doi: 10.1111/cmi.12846
Ohashi, Y., and Munro, S. (2010). Membrane delivery to the yeast autophagosome from the Golgi-endosomal system. Mol. Biol. Cell 21, 3998–4008. doi: 10.1091/mbc.e10-05-0457
Orsi, A., Razi, M., Dooley, H. C., Robinson, D., Weston, A. E., Collinson, L. M., et al. (2012). Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 23, 1860–1873. doi: 10.1091/mbc.E11-09-0746
Parmar, H. B., and Duncan, R. (2016). A novel tribasic Golgi export signal directs cargo protein interaction with activated Rab11 and AP-1-dependent Golgi-plasma membrane trafficking. Mol. Biol. Cell 27, 1320–1331. doi: 10.1091/mbc.E15-12-0845
Perez-Victoria, F. J., Schindler, C., Magadan, J. G., Mardones, G. A., Delevoye, C., Romao, M., et al. (2010). Ang2/fat-free is a conserved subunit of the Golgi-associated retrograde protein complex. Mol. Biol. Cell 21, 3386–3395. doi: 10.1091/mbc.E10-05-0392
Peter, F., Wong, S. H., Subramaniam, V. N., Tang, B. L., and Hong, W. (1998). Alpha-SNAP but not gamma-SNAP is required for ER-Golgi transport after vesicle budding and the Rab1-requiring step but before the EGTA-sensitive step. J. Cell Sci. 111(Pt 17), 2625–2633.
Puri, C., Vicinanza, M., Ashkenazi, A., Gratian, M. J., Zhang, Q., Bento, C. F., et al. (2018). The RAB11A-Positive Compartment Is a Primary Platform for Autophagosome Assembly Mediated by WIPI2 Recognition of PI3P-RAB11A. Dev. Cell 45, 114–131.e8. doi: 10.1016/j.devcel.2018.03.008
Rambourg, A., and Clermont, Y. (1990). Three-dimensional electron microscopy: structure of the Golgi apparatus. Eur. J. Cell Biol. 51, 189–200.
Redmann, V., Lamb, C. A., Hwang, S., Orchard, R. C., Kim, S., Razi, M., et al. (2016). Clec16a is critical for autolysosome function and purkinje cell survival. Sci. Rep. 6:23326. doi: 10.1038/srep23326
Renna, M., Schaffner, C., Winslow, A. R., Menzies, F. M., Peden, A. A., Floto, R. A., et al. (2011). Autophagic substrate clearance requires activity of the syntaxin-5 SNARE complex. J. Cell Sci. 124(Pt 3), 469–482. doi: 10.1242/jcs.076489
Rohn, W. M., Rouillé, Y., Waguri, S., and Hoflack, B. (2000). Bi-directional trafficking between the trans-Golgi network and the endosomal/lysosomal system. J. Cell Sci. 113(Pt 12), 2093–2101.
Rubinsztein, D. C., Shpilka, T., and Elazar, Z. (2012). Mechanisms of autophagosome biogenesis. Curr. Biol. 22, R29–R34. doi: 10.1016/j.cub.2011.11.034
Rui, Y. N., Xu, Z., Patel, B., Chen, Z., Chen, D., Tito, A., et al. (2015). Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 17, 262–275. doi: 10.1038/ncb3101
Sakai, K., Fukuda, T., and Iwadate, K. (2014). Immunohistochemical analysis of the ubiquitin proteasome system and autophagy lysosome system induced after traumatic intracranial injury: association with time between the injury and death. Am. J. Forensic Med. Pathol. 35, 38–44. doi: 10.1097/paf.0000000000000067
Salem, A. F., Whitaker-Menezes, D., Lin, Z., Martinez-Outschoorn, U. E., Tanowitz, H. B., Al-Zoubi, M. S., et al. (2012). Two-compartment tumor metabolism: autophagy in the tumor microenvironment and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle 11, 2545–2556. doi: 10.4161/cc.20920
Sarkar, C., Jones, J. W., Hegdekar, N., Thayer, J. A., Kumar, A., Faden, A. I., et al. (2020). PLA2G4A/cPLA2-mediated lysosomal membrane damage leads to inhibition of autophagy and neurodegeneration after brain trauma. Autophagy 16, 466–485. doi: 10.1080/15548627.2019.1628538
Schmitt-John, T. (2015). VPS54 and the wobbler mouse. Front. Neurosci. 9:381. doi: 10.3389/fnins.2015.00381
Schuster, C., Gerold, K. D., Schober, K., Probst, L., Boerner, K., Kim, M. J., et al. (2015). The autoimmunity-associated gene CLEC16A modulates thymic epithelial cell autophagy and alters T Cell Selection. Immunity 42, 942–952. doi: 10.1016/j.immuni.2015.04.011
Scott, K. L., Kabbarah, O., Liang, M. C., Ivanova, E., Anagnostou, V., Wu, J., et al. (2009). GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature 459, 1085–1090. doi: 10.1038/nature08109
Scrivo, A., Bourdenx, M., Pampliega, O., and Cuervo, A. M. (2018). Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 17, 802–815. doi: 10.1016/s1474-4422(18)30238-2
Shen, Q., Shi, Y., Liu, J., Su, H., Huang, J., Zhang, Y., et al. (2020). Acetylation of STX17 (syntaxin 17) controls autophagosome maturation. Autophagy. doi: 10.1080/15548627.2020.1752471 [Epub ahead of print].
Shimizu, S. (2018). Biological roles of alternative autophagy. Mol. Cells 41, 50–54. doi: 10.14348/molcells.2018.2215
Shoji-Kawata, S., Sumpter, R., Leveno, M., Campbell, G. R., Zou, Z., Kinch, L., et al. (2013). Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494, 201–206. doi: 10.1038/nature11866
Slobodkin, M. R., and Elazar, Z. (2013). The Atg8 family: multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem. 55, 51–64. doi: 10.1042/bse0550051
Sohda, M., Misumi, Y., Ogata, S., Sakisaka, S., Hirose, S., Ikehara, Y., et al. (2015). Trans-Golgi protein p230/golgin-245 is involved in phagophore formation. Biochem. Biophys. Res. Commun. 456, 275–281. doi: 10.1016/j.bbrc.2014.11.071
Su, L. Y., Luo, R., Liu, Q., Su, J. R., Yang, L. X., Ding, Y. Q., et al. (2017). Atg5- and Atg7-dependent autophagy in dopaminergic neurons regulates cellular and behavioral responses to morphine. Autophagy 13, 1496–1511. doi: 10.1080/15548627.2017.1332549
Suda, Y., and Nakano, A. (2012). The yeast Golgi apparatus. Traffic 13, 505–510. doi: 10.1111/j.1600-0854.2011.01316.x
Sun, Q., Fan, W., Chen, K., Ding, X., Chen, S., and Zhong, Q. (2008). Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. U.S.A. 105, 19211–19216. doi: 10.1073/pnas.0810452105
Sun, Y., Zhu, Y., Zhong, X., Chen, X., Wang, J., and Ying, G. (2018). Crosstalk between autophagy and cerebral ischemia. Front. Neurosci. 12:1022. doi: 10.3389/fnins.2018.01022
Takahashi, Y., Meyerkord, C. L., Hori, T., Runkle, K., Fox, T. E., Kester, M., et al. (2011). Bif-1 regulates Atg9 trafficking by mediating the fission of Golgi membranes during autophagy. Autophagy 7, 61–73. doi: 10.4161/auto.7.1.14015
Tam, R. C., Li, M. W., Gao, Y. P., Pang, Y. T., Yan, S., Ge, W., et al. (2017). Human CLEC16A regulates autophagy through modulating mTOR activity. Exp. Cell Res. 352, 304–312. doi: 10.1016/j.yexcr.2017.02.017
Tang, B. L. (2019). Syntaxin 16’s Newly Deciphered Roles in Autophagy. Cells 8:1655. doi: 10.3390/cells8121655
Taniguchi, M., Nadanaka, S., Tanakura, S., Sawaguchi, S., Midori, S., Kawai, Y., et al. (2015). TFE3 is a bHLH-ZIP-type transcription factor that regulates the mammalian Golgi stress response. Cell Struct. Funct. 40, 13–30. doi: 10.1247/csf.14015
Taniguchi, M., and Yoshida, H. (2017). TFE3, HSP47, and CREB3 Pathways of the Mammalian Golgi Stress Response. Cell Struct. Funct. 42, 27–36. doi: 10.1247/csf.16023
Thangaraj, A., Sil, S., Tripathi, A., Chivero, E. T., Periyasamy, P., and Buch, S. (2020). Targeting endoplasmic reticulum stress and autophagy as therapeutic approaches for neurological diseases. Int. Rev. Cell Mol. Biol. 350, 285–325. doi: 10.1016/bs.ircmb.2019.11.001
Tie, H. C., Chen, B., Sun, X., Cheng, L., and Lu, L. (2017). Quantitative Localization of a Golgi Protein by Imaging Its Center of Fluorescence Mass. J. Vis. Exp. 126:55996. doi: 10.3791/55996
Tiwari, S., Atluri, V., Kaushik, A., Yndart, A., and Nair, M. (2019). Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 14, 5541–5554. doi: 10.2147/ijn.s200490
Toh, H., Nozawa, T., Minowa-Nozawa, A., Hikichi, M., Nakajima, S., Aikawa, C., et al. (2020). Group A Streptococcus modulates RAB1- and PIK3C3 complex-dependent autophagy. Autophagy 16, 334–346. doi: 10.1080/15548627.2019.1628539
Uwineza, A., Caberg, J. H., Hitayezu, J., Wenric, S., Mutesa, L., Vial, Y., et al. (2019). VPS51 biallelic variants cause microcephaly with brain malformations: a confirmatory report. Eur. J. Med. Genet. 62:103704. doi: 10.1016/j.ejmg.2019.103704
Viotti, C. (2016). ER to Golgi-dependent protein secretion: the conventional pathway. Methods Mol. Biol. 1459, 3–29. doi: 10.1007/978-1-4939-3804-9_1
Wang, D. B., Kinoshita, Y., Kinoshita, C., Uo, T., Sopher, B. L., Cudaback, E., et al. (2015). Loss of endophilin-B1 exacerbates Alzheimer’s disease pathology. Brain 138(Pt 7), 2005–2019. doi: 10.1093/brain/awv128
Wang, I. H., Chen, Y. J., Hsu, J. W., and Lee, F. J. (2017). The Arl3 and Arl1 GTPases co-operate with Cog8 to regulate selective autophagy via Atg9 trafficking. Traffic 18, 580–589. doi: 10.1111/tra.12498
Wang, J., Menon, S., Yamasaki, A., Chou, H. T., Walz, T., Jiang, Y., et al. (2013). Ypt1 recruits the Atg1 kinase to the preautophagosomal structure. Proc. Natl. Acad. Sci. U.S.A. 110, 9800–9805. doi: 10.1073/pnas.1302337110
Wang, L., Pan, Y., Huang, M., You, X., Guo, F., and Chen, Y. (2017). PAQR3 augments amino acid deprivation-induced autophagy by inhibiting mTORC1 signaling. Cell Signal. 33, 98–106. doi: 10.1016/j.cellsig.2017.02.017
Wang, Z., Shi, X. Y., Yin, J., Zuo, G., Zhang, J., and Chen, G. (2012). Role of autophagy in early brain injury after experimental subarachnoid hemorrhage. J. Mol. Neurosci. 46, 192–202. doi: 10.1007/s12031-011-9575-6
Webber, J. L., and Tooze, S. A. (2010). New insights into the function of Atg9. FEBS Lett. 584, 1319–1326. doi: 10.1016/j.febslet.2010.01.020
Wesselborg, S., and Stork, B. (2015). Autophagy signal transduction by ATG proteins: from hierarchies to networks. Cell. Mol. Life Sci. 72, 4721–4757. doi: 10.1007/s00018-015-2034-8
Willett, R., Kudlyk, T., Pokrovskaya, I., Schönherr, R., Ungar, D., Duden, R., et al. (2013). COG complexes form spatial landmarks for distinct SNARE complexes. Nat. Commun. 4:1553. doi: 10.1038/ncomms2535
Wolf, M. S., Bayir, H., Kochanek, P. M., and Clark, R. S. B. (2019). The role of autophagy in acute brain injury: A state of flux? Neurobiol. Dis. 122, 9–15. doi: 10.1016/j.nbd.2018.04.018
Wu, J., and Lipinski, M. M. (2019). Autophagy in neurotrauma: good, bad, or dysregulated. Cells 8:693. doi: 10.3390/cells8070693
Xiang, Y., and Wang, Y. (2010). GRASP55 and GRASP65 play complementary and essential roles in Golgi cisternal stacking. J. Cell Biol. 188, 237–251. doi: 10.1083/jcb.200907132
Xu, D. Q., Wang, Z., Wang, C. Y., Zhang, D. Y., Wan, H. D., Zhao, Z. L., et al. (2016). PAQR3 controls autophagy by integrating AMPK signaling to enhance ATG14L-associated PI3K activity. EMBO J. 35, 496–514. doi: 10.15252/embj.201592864
Xu, H., Boulianne, G. L., and Trimble, W. S. (2002). Drosophila syntaxin 16 is a Q-SNARE implicated in Golgi dynamics. J. Cell Sci. 115(Pt 23), 4447–4455. doi: 10.1242/jcs.00139
Yamaguchi, H., Arakawa, S., Kanaseki, T., Miyatsuka, T., Fujitani, Y., Watada, H., et al. (2016). Golgi membrane-associated degradation pathway in yeast and mammals. EMBO J. 35, 1991–2007. doi: 10.15252/embj.201593191
Yang, S., and Rosenwald, A. G. (2016). Autophagy in Saccharomyces cerevisiae requires the monomeric GTP-binding proteins. Arl1 and Ypt6. Autophagy 12, 1721–1737. doi: 10.1080/15548627.2016.1196316
Yang, X. Z., Li, X. X., Zhang, Y. J., Rodriguez-Rodriguez, L., Xiang, M. Q., Wang, H. Y., et al. (2016). Rab1 in cell signaling, cancer and other diseases. Oncogene 35, 5699–5704. doi: 10.1038/onc.2016.81
Yen, W. L., Shintani, T., Nair, U., Cao, Y., Richardson, B. C., Li, Z., et al. (2010). The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J. Cell Biol. 188, 101–114. doi: 10.1083/jcb.200904075
Yla-Anttila, P., and Eskelinen, E. L. (2018). Roles for RAB24 in autophagy and disease. Small GTPases 9, 57–65. doi: 10.1080/21541248.2017.1317699
Yoshino, A., Setty, S. R., Poynton, C., Whiteman, E. L., Saint-Pol, A., Burd, C. G., et al. (2005). tGolgin-1 (p230, golgin-245) modulates Shiga-toxin transport to the Golgi and Golgi motility towards the microtubule-organizing centre. J. Cell Sci. 118(Pt 10), 2279–2293. doi: 10.1242/jcs.02358
Yu, K. N., Kim, H. J., Kim, S., Dawaadamdin, O., Lee, A. Y., Hong, S. H., et al. (2016). Cigarette Smoking Condensate Disrupts Endoplasmic Reticulum-Golgi Network Homeostasis Through GOLPH3 Expression in Normal Lung Epithelial Cells. Nicotine Tob. Res. 18, 1877–1885. doi: 10.1093/ntr/ntw079
Yu, L., Chen, Y., and Tooze, S. A. (2018). Autophagy pathway: cellular and molecular mechanisms. Autophagy 14, 207–215. doi: 10.1080/15548627.2017.1378838
Zaffagnini, G., Savova, A., Danieli, A., Romanov, J., Tremel, S., Ebner, M., et al. (2018). p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J 37:e98308. doi: 10.15252/embj.201798308
Zahoor, M., and Farhan, H. (2018). Crosstalk of autophagy and the secretory pathway and its role in diseases. Int. Rev. Cell Mol. Biol. 337, 153–184. doi: 10.1016/bs.ircmb.2017.12.004
Zamanian, J. L., and Kelly, R. B. (2003). Intersectin 1L guanine nucleotide exchange activity is regulated by adjacent src homology 3 domains that are also involved in endocytosis. Mol. Biol. Cell 14, 1624–1637. doi: 10.1091/mbc.e02-08-0494
Zeng, Z., Zhang, Y., Jiang, W., He, L., and Qu, H. (2020). Modulation of autophagy in traumatic brain injury. J. Cell. Physiol. 235, 1973–1985. doi: 10.1002/jcp.29173
Zhang, X., Wang, L., Ireland, S. C., Ahat, E., Li, J., Bekier, M. E., et al. (2019). GORASP2/GRASP55 collaborates with the PtdIns3K UVRAG complex to facilitate autophagosome-lysosome fusion. Autophagy 15, 1787–1800. doi: 10.1080/15548627.2019.1596480
Zhang, X., Wang, L., Lak, B., Li, J., Jokitalo, E., and Wang, Y. (2018). GRASP55 senses glucose deprivation through O-GlcNAcylation to promote autophagosome-lysosome fusion. Dev. Cell 45, 245–261.e6. doi: 10.1016/j.devcel.2018.03.023
Zhang, X., and Wang, Y. (2015). GRASPs in Golgi structure and function. Front. Cell Dev. Biol. 3:84. doi: 10.3389/fcell.2015.00084
Keywords: autophagy, cellular processes, golgi, neurological diseases, therapy
Citation: Deng S, Liu J, Wu X and Lu W (2020) Golgi Apparatus: A Potential Therapeutic Target for Autophagy-Associated Neurological Diseases. Front. Cell Dev. Biol. 8:564975. doi: 10.3389/fcell.2020.564975
Received: 23 May 2020; Accepted: 17 August 2020;
Published: 09 September 2020.
Edited by:
Brian C. Schaefer, Uniformed Services University of the Health Sciences, United StatesReviewed by:
Yoshinori Takahashi, Penn State Health Milton S. Hershey Medical Center, United StatesMaurizio Renna, University of Cambridge, United Kingdom
Copyright © 2020 Deng, Liu, Wu and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Lu, bHV3ZWkwMzM4QGNzdS5lZHUuY24=