 Xiaona Chen1,2
Xiaona Chen1,2 Wenjun Fu
Wenjun Fu Amirhossein Sahebkar
Amirhossein Sahebkar Suowen Xu
Suowen Xu Hong Li
Hong Li- 1Department of Medical Biotechnology, School of Basic Medical Sciences, Guangzhou University of Chinese Medicine, Guangzhou, China
- 2The Research Center of Basic Integrative Medicine, Guangzhou University of Chinese Medicine, Guangzhou, China
- 3Biotechnology Research Center, Pharmaceutical Technology Institute, Mashhad University of Medical Sciences, Mashhad, Iran
- 4Neurogenic Inflammation Research Center, Mashhad University of Medical Sciences, Mashhad, Iran
- 5Polish Mother’s Memorial Hospital Research Institute, Łódź, Poland
- 6Department of Endocrinology, First Affiliated Hospital, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, China
Atherosclerosis (AS), the most common underlying pathology for coronary artery disease, is a chronic inflammatory, proliferative disease in large- and medium-sized arteries. The vascular endothelium is important for maintaining vascular health. Endothelial dysfunction is a critical early event leading to AS, which is a major risk factor for stroke and myocardial infarction. Accumulating evidence has suggested the critical roles of histone deacetylases (HDACs) in regulating vascular cell homeostasis and AS. The purpose of this review is to present an updated view on the roles of HDACs (Class I, Class II, Class IV) and HDAC inhibitors in vascular dysfunction and AS. We also elaborate on the novel therapeutic targets and agents in atherosclerotic cardiovascular diseases.
Introduction
Atherosclerosis (AS) is the critical underlying pathology of CVD, which ranks the first on the morbidity and mortality of diseases (Fanelli et al., 2017; Libby et al., 2019; Niu et al., 2019; Wang et al., 2019). It lessens the elasticity of the arteries and may lead to myocardial infraction, ischemic stroke, cerebrovascular incidents, and peripheral vascular disease (Ziegler et al., 2019). The most prominent characteristic of AS is plaque formation in the arteries. Although the cause of the spontaneous AS and its initially characteristic focal plaque morphology has not been well understood, the histology and the progression of the advanced plaque have been identified (Ross, 1993; Poston, 2019). The progression of AS includes low-density lipoprotein (LDL) oxidation, endothelial activation, monocytes recruitment, macrophage-derived foam cell formation, VSMC proliferation, and thrombus formation (Poston, 2019).
Among the multiple mechanisms that exist in the development of AS, endothelial dysfunction has been recognized as one of the major cardiovascular risk factors (Arcaro et al., 1995). The healthy endothelium is important to maintain vascular homeostasis. It possesses the function of generating bioactive NO, regulating vascular tone, protecting the endothelial cell (EC) integrity, repairing the injury and inducing angiogenesis (Huynh and Heo, 2019). However, impaired NO bioavailability, oxidative stress, inflammation cytokines, and vascular tone potentially disrupt the endothelium homeostasis with consequence of endothelial dysfunction. Once the endothelium function is altered, followed by increased permeability to lipoprotein that attracts more leukocytes, induced secretion of inflammation cytokines and ROS, but less NO production (Poston, 2019); it exacerbates the pathology lesion of the arteries. Ensuing events of endothelial dysfunction include the proliferation and migration of VSMCs and formation of foam cell (Tian K. et al., 2019).
The epigenetic modification on genes is a crucial mechanism for many diseases, including cancer and CVDs (Xu et al., 2018, 2019; Khan et al., 2020). As one of histone modifications, histone acetylation plays an important role in altering the condensation of chromatin (which is mainly composed of DNA and histones in the nucleus of cells) without changing DNA sequences and has been regarded as the potential therapeutic targets. Acetylation of histones and nonhistone proteins is achieved by histone acetylases but removed by HDACs, which can regulate the transcriptional activities of the specific genes via interaction with the histones and transcription factors (Shirodkar and Marsden, 2011). In fact, the role of HDACs in cancer has been extensively studied in vivo and in vitro. Particularly, there are several HDAC inhibitors (HDACi) that have been approved by the Food and Drug Administration (FDA) for clinical application in cancer. During last two decades, much attention has been focused on the critical involvement of HDACs in CVD. The purpose of this article is to provide a systematic review on the role of HDACs and their inhibitors in vascular function and the progression of AS and highlight the potential application of HDACi in treating AS.
Histone Deacetylases
Histone deacetylases remove the acetylated residues at lysine, redense the chromatin structures, and inhibit the transcription of target genes (Bae et al., 2009). They are divided into two families, HDAC family and sirtuin family, including 18 members. These members are characterized into four groups: Classes I, II, III, and IV (Parra and Verdin, 2010) (Figure 1). Class III consists of the sirtuin family (SIRT1-7), which has been identified since 21st century. They differ from other groups because of their specific conserved catalytic core domain that requires the binding of NAD+/NADH, whereas the others require zinc molecule as an activator (Mihaylova and Shaw, 2013). Correspondingly, HDACs in Classes I, II, and IV, other than Class III, are referred to as the classical HDACs. Class I (HDAC1/2/3/8) HDACs are similar to the yeast Rpd3 and mostly locate in the nucleus, except that HDAC3 can export to the cytoplasm. In addition, Class II HDACs were subclassified into Classes IIa and IIb based on their primary structures. Class IIa (HDAC4/5/7/9) subfamily contains only an N-terminal regulatory domain, whereas Class IIb (HDAC6/10) subfamily has two catalytic domains (Gray and Ekström, 2001). All Class IIa members shuttle between the nucleus and the cytoplasm, which interact with the kinase families such as the calcium-independent protein kinase and the MAPK, acting as a signal transducer, but Class IIb (HDAC6/10) is mainly located in the cytoplasm (Fischle et al., 2002). As for Class III, SIRT1/2 are located in both the nucleus and cytoplasm, SIRT6/7 are in the nucleus, and SIRT 3/4/5 are in the mitochondria (Luo et al., 2014) (Figure 1). Of note, the location of HDACs might vary from different types of cells. HDAC11 is the only member of Class IV, and it is a negative regulator of interleukin (IL) 10 and the activity of T cells, indicating the potential role of HDAC11 in treating AS, which occurs as an inflammatory process (Yanginlar and Logie, 2018).
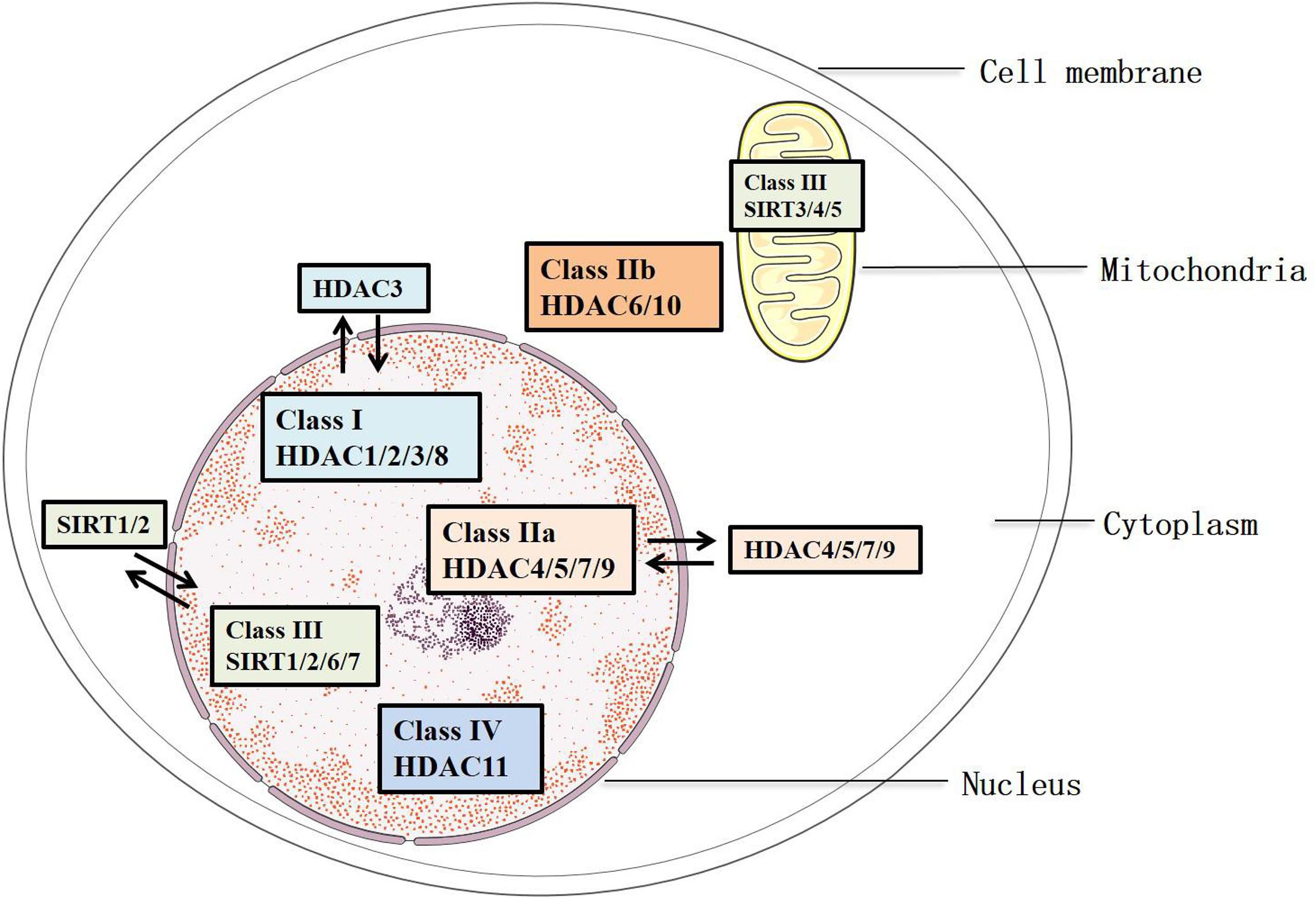
Figure 1. Classification and sublocation of HDACs.
Although HDACs were primarily identified as enzymes that deacetylate histones, further studies have identified many other nonhistone protein substrates, especially transcription factors such as the p65 subunit of NF-κB, E2F1, SP1, KLF 2/4, and signal transducers and activators of transcription 1 (STAT1) (Lyu et al., 2019). Because of multifunctional properties of HDACs, they have been involved in many cellular activities, tissue development, and various diseases, including embryonic development, tissue function, viral infections, CVD, cancer, kidney diseases, and autoimmune diseases (Gatla et al., 2019). Of note, the sirtuin family is well known for their effects in regulating vascular health, which have been deeply analyzed in recent reviews (Kane and Sinclair, 2018; Zhang et al., 2020). Hence, in this review, we mainly focus the role of classical HDACs in vascular function and AS, as well as the pharmacological effects of HDACi on AS.
HDAC Inhibitors
Because of the fact that HDACs are implicated in triggering the development of some diseases, especially cancer, HDACi have been designed or investigated. These inhibitors mainly bind with the catalytic sites of HDACs, resulting in genes re-expression (Zhang et al., 2018). Although HDACi are specific for HDACs, most of them are not specific to the HADC subclass. Based on their chemical structures, HDACi can be classified into four groups: short-chain fatty acids, benzamides, hydroxamic acids, and cyclic peptides (Marks, 2010). To date, five HDACi (SAHA, LBH589, PXD101, VPA, romidepsin) have been approved by the FDA for clinical treatment of cancer (Gatla et al., 2019). In addition, some HDACi are also in clinical trials for other diseases such as human immunodeficiency virus infection (Elliott et al., 2014), sickle cell disease (Okam et al., 2015), Duchenne muscular dystrophy (Bettica et al., 2016), polycythemia vera (Finazzi et al., 2013), and myeloproliferative diseases (Rambaldi et al., 2010). Although currently there is still no clinical usage of HDACi in CVD, plenty of evidence has shown the great potential of some HDACi in inhibiting endothelial dysfunction and AS (Zheng et al., 2015; Xu et al., 2017; Lee and Chiu, 2019). Notably, as some HDACs protect vascular cells against injury triggered by proatherogenic stimuli, it is necessary to clarify the specific role of each HDAC in different cellular context and activities in the vasculature, so that HDACi could be better repurposed for cardiovascular therapeutics.
Role of HDACs in Atherosclerosis
HDACs Are Involved in Endothelial Function and Dysfunction
HDACs and NO Production
Nitric oxide is a catalytic product of endothelial NO synthase (eNOS) in the endothelium, with the L-arginine as the substrate and BH4 as the cofactor (Förstermann et al., 2017). NO production can be inhibited by arginase 2 or AngII from VSMCs (Krause et al., 2016; Ryu et al., 2019). Once NO diffuses across the EC membrane, it can activate the sGC rapidly in VSMCs. The activated sGC catalyzes GTP to generate cGMP, a second messenger mediating the PKC signaling pathway, which results in a decline of the intracellular concentration of calcium and triggers the vasodilation of the VSMCs (Feil et al., 2003). The endogenous NO not only decreases the vascular tone but also inhibits the progression of the inflammation and the angiogenesis. Interestingly, HDACs and their inhibitors have been found to modulate the production of NO.
On the one hand, HDACs regulate the expression of eNOS. For example, HDAC1 was recruited at eNOS promoters and impaired eNOS expression in ECs subject to ischemic/reperfusion insult, leading to decrease in NO formation, which was ameliorated by the HDACi TSA treatment (Yang et al., 2012). In ECs from DJ-1/park7–/– mice, HDAC1 also inhibited eNOS transcription by inhibiting histone acetylation at the eNOS promoter (Won et al., 2014). HDAC3 (Zhang et al., 2008) and HDAC6 (Chen et al., 2019) had similar effects in inhibiting eNOS expression, and the effects could be reversed by tubacin, a selective inhibitor of HDAC6. By contrast, fluid shear stress induced phosphorylation of HDAC5, which was subsequently exported from nucleus and was involved in increased eNOS expression (Wang W. et al., 2010; Kwon et al., 2014). However, both of the HDACi-BuA and entinostat (MS-275) caused a decrease in eNOS protein (Rossig et al., 2002). In addition, the authors found that although TSA increased the eNOS promoter transcriptional activity, it reduced the production of NO through posttranscriptional suppression of eNOS protein levels. On the other hand, eNOS activity was regulated by HDACs via posttranslational modifications. HDAC1 (Hyndman et al., 2014) and HDAC3 (Jung et al., 2010) could reduce the lysine acetylation of eNOS and blocked NO expression. On the contrary, VPA could improve NO production by influencing eNOS phosphorylation (Cho et al., 2014). Of note, overexpression of HDAC2 could suppress the expression of arginase 2, a protein that counteracts eNOS activity, and the effect was reversed by TSA through increasing levels of H3K9 and H4K12 acetylation at arginase 2 proximal and core promoter (Pandey et al., 2014; Krause et al., 2016), indicating a protective role of HDAC2 in endothelial dysfunction and AS.
HDACs and Endothelial Oxidative Stress
BH4, the coactivator of the eNOS, can be easily oxidized to dihydrobiopterin, resulting in uncoupling of eNOS, leading to superoxide rather than NO production (Stocker and Keaney, 2004). Superoxide interacts with NO to produce ONOO–, which reduces the bioavailability of NO. In general, superoxide is mainly derived from the process when the membrane oxidase Nox transfers the electrons from NADPH to oxygen (Craige et al., 2015). Among the members of Nox family, Nox1/2/4/5 are expressed in the cardiovascular system with abundant expression of Nox4.
It was reported that the pan-HDACi varinostat (SAHA) reduced expression of Nox1/2/4 in the aorta of ApoE–/– mice, which contributed to its anti-AS effect (Manea et al., 2020). Recruitment of HDAC abolishes the interaction of RNA polymerase II and p300 to the promoter sites of Nox2/4/5, respectively, which inhibited the activation at those promoter regions and resulted in a decline of ROS (Chen et al., 2016; Hakami et al., 2016). In addition, HDAC3 could ameliorate the oxidative stress induced by AngII or disturbed flow (Martin et al., 2014). In another aspect, HDAC1 and HDACi could regulate the antioxidant enzymes including SOD and CAT, which scavenge the superoxide in vivo or in vitro. HDAC1 dissociation from SOD3 promoter was critically involved in SOD3 expression elicited by caffeic acid phenethyl ester (Ohashi et al., 2017). It is reported that TSA could induce the expression of SOD3 and reduce NOX expression robustly in human pulmonary artery ECs exposed to scriptaid (Zelko and Folz, 2015). However, MS-275 failed to influence the expression of Nox1, Nox2, and p47phox (Ryu et al., 2019).
Reactive oxygen species are the stimuli that reduce the bioavailability of NO and induce inflammation of ECs. It was found that IGF-1 enhanced the phosphorylation of HDAC5 that is associated with AS and led to nuclear export of HDAC5, which was mediated by Nox4-dependent ROS production, as well as the phosphatidylinositol 3-kinase (PI3K)/AKT pathways (Pietruczuk et al., 2019). HDAC6 expression and activity were upregulated in ox-LDL-treated ECs, which led to decreased expression of cystathionine γ-lyase and contributed to endothelial dysfunction (Leucker et al., 2017), and knockdown of HDAC6 or pharmacological inhibition with the dietary HDACi sodium butyrate (Hou et al., 2018; Wu et al., 2018), the inhibitor of Class I HDAC, could inhibit endothelial dysfunction. Moreover, loss of HDAC2 or specific inhibition of HDAC6 was found to activate HO-1/SIRT1 pathway and inhibit oxidative stress induced by high glucose (Gao et al., 2018; Abouhish et al., 2020).
HDACs and Endothelial Inflammation
Oxidative stress, eNOS uncoupling, and inflammation are potential contributors to endothelial dysfunction. Inflammation in vasculature causes alteration of vascular wall, which can trigger the CVD especially AS (Ali et al., 2018). In the development of AS, ECs and macrophages produce the proinflammatory cytokines, such as TNF-α, IL-β, and IL-6 (Barbour and Turner, 2014). These proinflammatory molecules cause the secretion of adherent molecules, including ICAM-1, VCAM-1, and E-selectin, to recruit the leukocytes and monocytes to endothelium (Xu et al., 2017). TNF-α mediated an important signaling pathway in increasing the expression of inflammatory cytokines and superoxide, thereby attracting more monocytes into the subendothelium space (Kleinbongard et al., 2010). Accumulating studies have demonstrated that NF-κB plays a crucial role in promoting the inflammation cytokines release. Activation of NF-κB leads to the upregulation of TNF-α, IL-β, IL-6, and adherent molecules in ECs (Stein et al., 2010). In human ECs treated with ox-LDL, HDAC1 and HDAC2 were downregulated, and the effect was reversed by simvastatin, which also inhibited the NF-κB pathway (Dje N’Guessan et al., 2009). HDAC2 is also a mediator of neutrophil migration and regulated multiple MMPs and CD16 gene expression in acute ischemic stroke patients (Li et al., 2020). Similarly, phosphorylation of HDAC5 led to HDAC5 nuclear export and upregulation of KLF2 and mediated the anti-inflammatory effects of metformin in ECs (Tian R. et al., 2019). On the contrary, several HDACs were also implicated in the proinflammatory response in ECs. Bedenbender et al. (2019) reported that in TNF-α-treated human umbilical vein ECs (HUVECs), HDAC2 was recruited to the RNase I promoter and reduced the histone acetylation, leading to downregulation of RNase I, a protective molecule in vascular homeostasis. HDAC3 mediated the inflammatory response in ECs by regulating galectin-9 expression (Alam et al., 2011). In human pulmonary ECs subjected to Staphylococcus aureus infection, HDAC6 was upregulated followed by elevated ROS, and knockout or pharmacological inhibition of HDAC6 in mice could inhibit vascular inflammation and protect the EC integrity (Karki et al., 2019). It was suggested that the detrimental effect of HDAC6 might be mediated by microtube destabilization. Moreover, HDAC7 could induce leucocyte adhesion to ECs (Ismail et al., 2012), and HDAC8 was involved in increased secretion of ICAM-1 and VCAM-1 in the aortas of mice infused with AngII (Kee et al., 2019).
Because of the proinflammatory effects of most classical HDACs, several HDACi have been employed to determine their anti-inflammatory effects. In human lung ECs, LPS upregulated IKBα mRNA, which might be mediated by deacetylation of H3K9 and could be blocked by TSA (Thangjam et al., 2014). By contrast, TSA could suppress COX-2 expression induced by LPS through inhibiting the phosphorylation of JNK and p38 MAPK (Hsu et al., 2011). Moreover, TSA suppressed VCAM-1 (but not ICAM-1) expression in TNF-α-induced HUVECs and sickle transgenic mice, whereas MS-275 inhibited VCAM-1 and MCP-1 expression in AngII-induced hypertensive mice (Inoue et al., 2006; Hebbel et al., 2010; Ryu et al., 2019). He et al. (2011) showed that TSA inhibited the expression of TLR4 and HDAC2 induced by LPS in cultured EA.hy926 cells, which protected the EC from injury. Li et al. (2018a) found that TSA inhibited IL-8 production, and VCAM-1 expression induced by TNF-α in HUVECs, and the adhesion of peripheral blood mononuclear cell to HUVECs was also blocked. Besides, butyrate (Ogawa et al., 2003), other short-chain fatty acids (Miller et al., 2005; Vinolo et al., 2011), and HDAC8 selective inhibitor PCI34051 (Kee et al., 2019) also showed anti-inflammatory effect in ECs. It is notable that although HDACi exhibited an anti-inflammatory effect in various ECs or animal models, the inflammatory markers detected in each study varied, and the regulatory mechanism was also not well illustrated. Further study is needed to demonstrate the specific HDAC isoforms involved in and to evaluate whether the anti-inflammatory effect of HDACi is dependent on increased acetylation of histones or specific transcription/repressive factors.
HDACs Regulate EC Proliferation
Endothelium works as a selective barrier between the blood and tissue, controlling exchange of the ions and cytokines. Under healthy conditions, ECs tend to be tight and are not proliferating, but they start to migrate and proliferate when subjected to hypoxia, injury, or stress (Zeng et al., 2009). Although EC proliferation is required to response to the hypoxia, excessive EC proliferation contributes to EC turnover, which is critically associated with the endothelium permeability (Caplan and Schwartz, 1973).
β-Catenin is a signal transducer that accelerates cell proliferation and growth via activating the Id2, T cell factor/lymphoid enhancer factor, and follistatin transcription factors. Margariti et al. (2010) found that HDAC7 was important in modulating the expression of genes related to EC proliferation via regulating the transcription activity of β-catenin. Overexpression of HDAC7 suppressed HUVEC proliferation through inhibition of nuclear translocation of β-catenin and downregulation of Id2 and cyclin D1 expression, causing G1 phase elongation. The effect of HDAC7 overexpression could be abolished by the VEGF, which degraded HDAC7 via PLC-PI3K signal pathway and disrupted the complex of HDAC7 and β-catenin, leading to β-catenin released into the nucleus. It was shown that VEGF induced phosphorylation and cytoplasm translocation of HDAC7, resulting in activation of VEGF-responsive genes, and EC proliferation was enhanced (Wang et al., 2008). Similarly, HDAC4 and HDAC5 were also critical in mediating EC proliferation during cardiovascular development (Kang et al., 2013). However, in most cases, the classical HDACs were deemed to promote abnormal EC proliferation. Lee et al. (2012; Chiu and Chien, 2011) found that HDAC1/2/3-specific siRNAs reversed the increased level of cyclin A and reduction of p21 induced by OSS, which is considered to be a contributor to endothelial dysfunction of arterial branches and curvatures. Meanwhile, the HDACi VPA could suppress OSS-induced EC proliferation in BrdU-infused rats (Lee et al., 2012). HDAC4 and HDAC5 in nuclear could inhibit myocyte-enhancer factor 2 (MEF2) and KLF2/4 activity (Yang et al., 2019), whereas HDAC5 in cytoplasm mediated KLF2 expression (Wang W. et al., 2010; Kwon et al., 2014). Normal shear stress induced HDAC6 activity, which reduced tubulin acetylation and promoted ECs migration (Wang Y.H. et al., 2010). The pro-proliferation effect of VEGF could be inhibited by expressing a signal-resistant HDAC7 mutant protein in ECs (Wang et al., 2008). In parallel, the HDACi such as SAHA (Cheng and Hung, 2013), tubacin (Li et al., 2016), TSA, and apicidin (Yang et al., 2015) inhibited EC proliferation in different conditions.
HDACs and Endothelium Integrity
The endothelial barrier possesses tight junction, endothelial glycocalyx, and efflux transporters that are all essential for protecting the endothelial integrity and cell permeability and control the entry of the chemicals exchange between the blood and tissues. The endothelial glycocalyx is on the endothelial surface (Weinbaum et al., 2007). The MMPs are produced by ECs, which could degrade the ECM and connective tissue proteins, including the ones that form the glycocalyx (Haas, 2005). HDACs play a crucial role in protecting the endothelial barrier function by regulating cell permeability, chemical transporter, tight junction protein, and MMPs (Gao et al., 2008; Joshi et al., 2015; Shi et al., 2016; Castro et al., 2018). Among the classical HDACs, it seems that only HDAC7 and HDAC3 exhibit a protective effect in endothelial integrity. It was demonstrated that HDAC7 could interact with MEF2, inhibiting its transcription activity, and thus suppressed the expression of target genes such as MMP-10 (Chang et al., 2006; Gao et al., 2008; Su et al., 2013). By contrast, RGFP-966, an HDAC3-selective inhibitor, was found to significantly attenuate the oxygen–glucose deprivation/reperfusion-induced transendothelial cell permeability and downregulate the tight junction protein claudin-5 via activating PPARγ signaling pathway (Zhao et al., 2019). HDAC4 and HDAC5 regulated expression of connexin 37 and 40 in PAECs (Kim et al., 2015), and increased HDAC5 activity was essential for MMP induction, glycocalyx remodeling, and reduced expression of TIMP (Ali et al., 2019). As for HDAC6, it was a mediator leading to S. aureus-induced (Karki et al., 2019) or TNF-α-induced (Yu et al., 2016) endothelial permeability and could attenuate tubulin acetylation, leading to reduced cell stability (Fernandes et al., 2015). Besides, inhibition of HDAC6 could attenuate CSE-induced EC permeability and acute lung injury (Borgas et al., 2016). As the only member of class IV HDACs, HDAC11 is least studied in endothelial function. It was found that HDAC11 was upregulated in PAR2 agonist-treated ECs and mediated the impaired barrier function via modulating VE-cadherin expression (Zhang and Ge, 2017). Moreover, inhibition of HDACs via TSA or SAHA increased the protein levels of TIMP-1 and TIMP-3 (Ali et al., 2019).
The multidrug resistance protein 1 (also known as MDR1) is a crucial efflux transporter located on the top surface of capillary ECs and prevents xenobiotics accumulating in the brain (Serlin et al., 2015). You et al. (2019) showed that VPA, apicidin, and SAHA could increase MDR1 expression in human brain ECs. Glucose transporters are responsible for the energetic supply at the blood–brain barrier (Castro et al., 2016). β-Hydroxybutyrate, an HDACi, could upregulate the glucose transporter gene Slc2a1 expression in brain microvascular EC s and NB2a neuronal cells by increasing acetylation of H3K9 at the promoter site of the Slc2a1 gene (Rafehi et al., 2017). Besides, TSA, VPA, short-chain fatty acid, and the specific Class IIa HDACs inhibitor TMP269 could reverse endothelial barrier dysfunction induced by hemorrhage, lethal scald injury, and acute lung injury, respectively (Miyoshi et al., 2008; Bruhn et al., 2018; Tang et al., 2018; Kovacs-Kasa et al., 2020).
HDACs and Endothelial Apoptosis
Endothelial cell apoptosis is not active under healthy conditions, but it is activated in the development of AS and contributes to the hyperpermeability of endothelium, as well as thrombus formation on eroded plaques (Tedgui and Mallat, 2003). Li et al. (2018c) found that HDAC1 was reduced in AS lesions and ox-LDL-treated human aortic ECs (HAECs), which was regulated by miR-34a and might mediate EC apoptosis. In the process of senescence induced by radiation, HDAC1 was also reduced (Okamoto et al., 2006). Similarly, HDAC3 is critical for endothelial survival, and knockdown of HDAC3 led to apoptosis in ECs (Zampetaki et al., 2010). However, HDAC6 was implicated in apoptotic response of lung ECs induced by TNF-α (Yu et al., 2016). VPA induced the Bcl-2 phosphorylation and release of the cytochrome c via activating ERK1/2 and consequently inhibited the serum starvation-induced HUVECs apoptosis (Michaelis et al., 2006). Comparatively, VPA was found to induce apoptosis of tumor cells (Yamanegi et al., 2015).
HDACs and Angiogenesis
Angiogenesis mainly refers to the formation of functional capillaries especially in the progression of tumor, and it is a complex biological process including alterations of gene expression. Numerous studies were focused on the role of HDAC/HDACi and angiogenesis in cancer, which have been reviewed elsewhere (Mastoraki et al., 2020). Actually, the angiogenic ability of EC is also very important for maintaining the cell or tissue function during development, ischemia, hypoxia, or injury. VEGF is considered as a predominant growth factor during the formation of capillaries, which has been a therapeutic target for tumor angiogenesis (Pepper et al., 1996). Notably, VEGF could be induced by HIF-1α activation in response to hypoxia (Ikeda, 2005). Inhibition of HDAC1/4/6/7 abolished the expression of VEGF via impairing the induction or activity of HIF-1α in hypoxia-exposed ECV304 cells, HaCaT cells, or patients with chronic obstructive pulmonary disease (Granger et al., 2008; Reynoso-Roldán et al., 2012; To et al., 2012; Kowshik et al., 2014).
Blood flow is a critical factor inducing angiogenic sprouting, in the process of which HDAC1 was phosphorylated and translocated from the nucleus to cytosol. Similarly, phosphorylation and nuclear export of HDAC5 also mediated VEGF-induced angiogenesis (Ha et al., 2008). In addition to the effect of inducing angiogenesis during myocardial infraction (Yang et al., 2019), HDAC6 also improved repair stimulated by injury (Wu et al., 2016). By targeting the antiangiogenic microRNA-17-92 cluster, HDAC9 accelerated angiogenesis in ECs (Kaluza et al., 2013). In addition, HDAC5 played an important role in inhibiting expression of the genes participating in angiogenesis including CYR61, PVRL2, FSTL1, and Slit2 in patients with systemic sclerosis, which could be reversed by silencing of HDAC5 (Urbich et al., 2009; Tsou et al., 2016). Endothelial migration is a key step of angiogenesis, and both HDAC6 (Li et al., 2011; Birdsey et al., 2012) and HDAC7 (Mottet et al., 2007; Yu et al., 2014) could promote angiogenesis by regulating cell migration. On the contrary, it was shown that HDAC3 was a negative regulator of angiogenesis (Park et al., 2014). Treatment with TSA or SAHA suppressed HIF-1α, VEGF, VEGF receptors (VEGFR1 and VEGFR2), and the formation of capillary-like structures, but increased semaphorin, a VEGF competitor, in rat lungs and cultured human pulmonary microvascular ECs (Deroanne et al., 2002; Mizuno et al., 2011). SAHA could upregulate the WNT-inducible secreted protein 1, a matricellular molecule that accelerates angiogenesis in TNF-α-stimulated HCAECs (Wright et al., 2018). Importantly, the HDACi such as VPA (Jin et al., 2011; Wang et al., 2012) and SAHA (Jin et al., 2011; Wright et al., 2018) mainly exerted proangiogenic effect, whereas TSA (Williams, 2001) could also inhibit angiogenesis.
HDACs and Cell Differentiation Into ECs
When the endothelium is damaged or nearly denuded, the circulating or local resident stem or progenitor cells would differentiate into ECs to rescue the denuded ECs (Rinkevich et al., 2011). Rossig et al. (2005) found that inhibition of HDACs abolished the endothelial differentiation from adult progenitor cells by inhibiting the expression of homeobox transcription factors HoxA9, which is a regulator of eNOS, VEGF-R2, VE-cadherin, and EC maturation induced by shear stress. TSA improved H3K9 acetylation and downregulated HDAC1 expression in bone marrow progenitor cells, which were further treated to generate cardiac progenitor cells and were able to differentiate into myocytes and ECs in the infarcted mouse heart (Rajasingh et al., 2011). Furthermore, TSA induced EC marker VE-cadherin, von Willebrand factor (vWF), and Flk in VEGF-treated multipotent adult progenitor cells and induced the differentiation into ECs (Mahapatra et al., 2010). However, in the progression of stem cell-derived ECs differentiating into ECs, TSA showed an opposite effect. While VEGF induced the differentiation accompanied by increased activity of HDAC, TSA or silence of HDAC3 reduced the EC lineage marker, indicating that the HDAC3 positively regulated the differentiation (Xiao et al., 2006; Zeng et al., 2009). Moreover, HDAC6 was important for maintaining mechanical sensing in human induced pluripotent stem cell-derived ECs (Smith et al., 2018).
HDACs and Vascular Tone
Endothelium is crucial in regulating vascular tone to control the local cardiovascular function, especially by NO, which determines the endothelium-dependent relaxation and thus, to some extent, prevents hypertension and PAH (Pan et al., 2018). PAH results in the unnatural proliferation of PAECs, triggering enhanced pulmonary vascular resistance, as well as right ventricular failure. It is recognized that PAECs, pulmonary arterial smooth muscle cells, fibroblasts, and pericytes are involved in the pathogenesis of PAH (Rabinovitch, 2012). Furthermore, the factors secreted from PAECs such as fibroblast growth factor 2, IL-6, and ET-1 induce proliferation, migration, and vascular remodeling by influencing other cells in the development of PAH (Ricard et al., 2014). Kim et al. (2015) identified that MEF2 acted as the key cis-acting factor that regulated the expression of target genes contributing to pulmonary vascular homeostasis, such as microRNAs-424 and -503, connexin 37, connexin 40, KLF2, and KLF4, which were evidently decreased in PAECs isolated from PAH patients. This action could be abolished by nuclear accumulation of HDAC4 and HDAC5, causing inhibition of MEF2 transcription activity. Importantly, several HDAC isoforms were upregulated in isolated pulmonary arteries in a monocrotaline-induced PAH in rat models, and HDACi inhibited Nox expression and PAH markers in isolated pulmonary arteries (Chen et al., 2016). In addition, HDAC1 was involved in increased systolic blood pressure in mice by regulating eNOS expression and NO production, which could be inhibited by VPA (Won et al., 2014). The increased NO is the major vasodilator that reduces the vascular tone, which plays a critical role in maintaining arterial patency (Nishida et al., 1992). Several studies have shown that HDAC6 was crucial in impairing vascular tone. On the one hand, HDAC6 regulated chromatin remodeling and promoted ET-1 expression (Li et al., 2016). On the other hand, it was upregulated in AngII-treated aortas and HAECs, which induced cystathionine γ-lyase ubiquitination and degradation, and was implicated in AngII-induced hypertension (Chi et al., 2019, 2020). Moreover, PAH was significantly ameliorated in HDAC6–/– mice. Similarly, HDAC8 was implicated in AngII-induced hypertension, and its selective inhibitor PCI34015 could reverse the effect and reduced vascular hypertrophy and inflammation (Kee et al., 2019). IGF-1 is a growth factor contributing to PAH in neonatal mice, through activating the AKT signaling pathway. Apicidin, an inhibitor of HDAC, suppressed pulmonary IGF-1/pAKT signaling pathway, which ameliorated right ventricular hypertrophy and vascular remodeling in lungs (Yang et al., 2015). And TSA could inhibit hypertension induced by abdominal aortic constriction in rats (Kang et al., 2015).
HDACs Regulate VSMC Proliferation and Migration in Atherosclerosis
During the development of AS, VSMCs migrate to the subendothelium space and switch to a collagen-secreting synthetic phenotype (Doran et al., 2008) to form the fibrous cap, with mass of the foamy macrophages and cholesterol on the surface. Moreover, the proliferating VSMCs can also remodel the ECM by degrading the matrix components through increasing expression and activity of MMPs, which disrupts the fibrous cap of the plaque, causing plaque vulnerability (Shah, 1996).
Increasing evidence has shown the important role of HDACs in regulating VSMC proliferation and migration. Laminar flow enhanced HDAC1 activity and the interaction between HDAC1 and p53, which resulted in p21WAFl activation in VSMCs (Zeng et al., 2003). Sun et al. (2020) found that HDAC1 was critical for the migration and phenotypic switch of aortic VSMCs. KLF4 and KLF5 acetylation in VSMCs was regulated by phosphorylated HDAC2, which was implicated in VSMC proliferation induced by retinoic acid receptor agonist (Meng et al., 2009; Zheng et al., 2011). HDAC4 also promoted VSMC proliferation and migration (Usui et al., 2014; Li et al., 2018b; Zhang et al., 2019), whereas interfering HDAC4 could inhibit the effect (Zheng et al., 2019). HDAC5 in VSMCs could be activated by AngII, a potent stimuli for VSMC proliferation (Pang et al., 2008). Moreover, Zhou et al. (2011a) showed that by regulating β-catenin translocation, splicing of HDAC7 induced SMC proliferation. In addition, HDAC2 and HDAC5 hypoacetylated histone H4 at the promoter site of α-smooth muscle actin and decreased the expression of marker genes of SMC differentiation induced by POVPC (Yoshida et al., 2008).
It is recognized that cyclic strain regulates phenotype switch, migration, and proliferation of VSMCs in the pathogenesis of AS (Haga et al., 2007). Cyclic strain induced the secretion of transforming growth factor β1 in VSMCs and the expression of contractile phenotype markers, such as smooth muscle protein 22α, α-smooth muscle actin, and calponin (Yao et al., 2014). It was shown that the cyclic strain induced the migration of VSMCs by up-regulating HDAC7 and down-regulating the levels of HDAC3/4. Tributyrin, a pan-inhibitor of HDAC, could suppress VSMC migration accompanied by reduced expression of HDAC7 (Yan et al., 2009). TSA could inhibit VSMC proliferation by inducing expression of p21WAF1, rather than p16INK4, p27KIP1, or p53, followed by cell cycle arrest through reducing Rb phosphorylation at the G1–S phase (Okamoto et al., 2006). It was reported that butyrate, a dietary HDACi, had the effect on arresting the proliferation of VSMCs. Furthermore, butyrate downregulated the G1-specfic CDKs including CDK4, CDK6, and CDK2 and induced the expression of CDK inhibitors, p15INK4b and p21Cip1, leading to cell cycle arrest of VSMCs (Mathew et al., 2010). The authors also found that the effect of the butyrate was mediated by acetylating H3K and phosphorylating H3-serine10, in addition to dimethylation of H3K9 and H3K4 (Mathew et al., 2010). Besides, HDAC4 and HDAC5 was involved in vascular calcification (Abend et al., 2017; Choe et al., 2020) and inflammatory response in VSMCs (Lee et al., 2008), whereas HDAC1 (Liu and Khachigian, 2009) and HDAC5 (Pietruczuk et al., 2019) acted as a proinflammatory molecule in VSMCs. TSA could inhibit VSMC calcification (Azechi et al., 2013). All these studies revealed that the classical HDACs mediate the proliferation and migration of VSMCs, which could be blocked by HDACi.
HDACs Are Involved in Macrophage-Derived Foam Cell Formation
Macrophage-derived foam cell formation is one of the major contributors to AS development. In the early stage of plaque formation, macrophages are mainly derived from the monocytes infiltrating into subintima (Ley et al., 2007). The macrophages stimulated by ox-LDL and its lipid components produce more inflammatory factors, such as IL-α, IL-β, IL-6, IL-18, and TNF-β (Poston, 2019), which trigger adhesion molecule secretion from ECs to attract more monocytes to differentiate into macrophages (Ley et al., 2007). By uptaking ox-LDL through binding to scavenger receptors such as CD36, macrophages became lipid-laden foam cells (Orekhov, 2018). MCP-1 expressed by VSMCs guides the foam cells to the atherosclerotic plaques (Tesfamariam and DeFelice, 2007). Unfortunately, when the foam cells underwent cell death (apoptosis or necrosis), the inner cholesterol components are released to the plaque. The released cholesterol, together with the collagen-secreted VSMCs and foam cells, contributes to the early plaque (Poston, 2019).
Treatment with TSA markedly upregulated the expression of CD36 in macrophages, which increased the uptake of oxLDL and accelerated AS (Choi et al., 2005). The lipid efflux, removing redundant cholesterol from macrophage, is a vital step in suppressing the development of AS (Li et al., 2010). ATP-binding transporters (ABCA1 and ABCG1) transfer the cholesterol from macrophage to lipid-poor apolipoprotein and high-density lipoprotein (HDL). Interestingly, inhibition of HDAC with TSA, ITF2357 (pan-HDACi), or RNA interference to silence genes of HDAC 1/2/3/6/8, respectively, increased histone acetylation and ABCA1/ABCG1 expression, which suppressed the accumulation of cholesterol in macrophage, and HDAC3 silence appeared the most effective (Van den Bossche et al., 2014). The precise role of HDAC isoforms and the pharmacological effects of HDACi in foam cell formation warrant further studies. Additionally, PPARγ is a transcriptional factor critically involved in lipid metabolism and possesses anti-inflammation properties (Vallée et al., 2019). Recent study showed that, by activating PPARγ through elevating acetylation of C/EBPα (CCAAT enhancer binding protein α), TSA increased ABCA1/ABCG1 expression and reduced TNF-α/IL-1β, which contributed to the inhibition of foam cell formation and atherogenesis (Gao et al., 2020).
HDACs and Thrombus Formation
In the development of AS, both prothrombotic molecules and procoagulant molecules contribute to thrombosis formation. Tissue factor (TF) produced by plaque macrophages activates platelets through the extrinsic pathway to initiate thrombin signaling (Collot-Teixeira et al., 2007). In addition, ox-LDL is considered to be another extrinsic factor that activates the platelets through its scavenger receptors (Ivanciu and Stalker, 2015). Thrombin activates ECs to release vWF, a factor that stimulates platelets (Spronk et al., 2013). The activated platelets potentially stimulate the platelets in a self-perpetuation manner, which produces more thrombin (Spronk et al., 2013). Wang et al. (2007) found that the HDACi (TSA, MS-275, sodium butyrate, and VPA) suppressed the expression of TF and its bioactivity. TF can enhance the release of vWF that causes platelet adhesion to ECs. The transcription factor nuclear factor Y (NE-Y) interacts with both HDAC1 and histone acetyltransferase PCAF in vWF gene promoter, causing dissociation of HDAC1 from the complex and leading to more PCAF recruited to vWF promoter, thereby promoting vWF expression (Peng et al., 2007). t-PA is generated by ECs, acting as an antithrombotic factor that is involved in clearance of intravascular fibrin deposits in ECs. The expression of t-PA could be inhibited by HDAC through deacetylating histone H3 and H4 in the t-PA gene promoter (Lappas, 2012). Furthermore, treatment with TSA, MS-275 or VPA increased the t-PA expression (Larsson et al., 2013), respectively, whereas the effect of VPA could be suppressed by knockdown of HDAC3/5/7 (Larsson et al., 2012).
HDACs Regulate Atherosclerosis in Human and Animals
Among the classical HDACs, HDAC9 is best known for its role in the development of AS. The rs2107595 HDAC9 gene polymorphism leads to increased expression of HDAC9 in the internal carotid artery (Grbić et al., 2020) and plasma (Wang et al., 2016) and modulated gene expression in the blood of patients suffering from large vessel atherosclerotic stroke (Shroff et al., 2019). The polymorphism variant is significantly associated with AS (Prestel et al., 2019; Grbić et al., 2020) and may contribute to coronary AS and coronary artery disease risk (Wang et al., 2016). Consistently, HDAC9 deficiency in ApoE–/– mice resulted in evidently reduced lesion size in aortas and less advanced lesions (Azghandi et al., 2015). Further studies showed that HDAC9 could activate inhibitory κB kinase and regulate atherosclerotic plaque vulnerability (Asare et al., 2020). Moreover, HDAC9 repressed cholesterol efflux by downregulating ABCA1, ABCG1, and PPARγ and alternatively promoted macrophage activation in AS (Cao et al., 2014). On the contrary, it was demonstrated that reduced expression of HDAC9 induced by miR-182 was implicated in increased levels of cholesterol, lipoprotein lipase, and proinflammatory cytokines in oxLDL-treated human THP-1 macrophages, which the author suggested might mediate the lipid accumulation in atherosclerotic lesions and thus promoted atherogenesis in ApoE–/– mice administered with miR-182 agomir (Cheng et al., 2017). Moreover, it was found that HDAC1/2/3/4/6/11 were all upregulated in atherosclerotic carotid arteries and aortas from human and ApoE–/– mice, respectively (Manea et al., 2020). By using aortic isografted model, Zampetaki et al. (2010) found that endothelial-specific knockdown of HDAC3 in aortas from ApoE–/– mice robustly promoted atherosclerotic lesion formation. However, Hoeksema et al. (2014) found that HDAC3 was the only isoform upregulated in human ruptured plaques, and myeloid deletion of HDAC3 in LDL receptor knockout mice led to more stabilized atherosclerotic lesions, which might be mediated by phenotype shift of macrophages to be anti-inflammatory and less lipid accumulation. The distinct effects of HDAC3 in these two studies suggest the cell-specific function of HDAC3 during the development of AS. In ox-LDL-treated HAECs and aortas from ApoE–/– mice, HDAC6 was upregulated, and its selective inhibitor tubacin prevented endothelial dysfunction and the development of AS (Leucker et al., 2017). Several HDACi such as TSA (Gao et al., 2020), metacept-1 (Vinh et al., 2008), BuA (He and Moreau, 2019), and SAHA (Ye et al., 2018; Manea et al., 2020) have been demonstrated to inhibit the development of AS.
Potential Combination of BRD4 Inhibitors With HDAC Inhibitors
Recent years have witnessed the important roles of bromodomain and extra terminal (BET) proteins, which are readers of acetylated histones. The BET family comprises four members, including BRD2, BRD3, BRD4, and the testis-restricted BRDT, among which BRD4 is the most characterized and critically implicated in transcriptional regulation and atherogenesis (Lin and Du, 2020; Wang et al., 2020). Intriguingly, the BRD4 inhibitor RVX-208 (now called apabetalone) showed potent effect in increasing apolipoprotein A-I and HDL levels/particles and reducing AS (McLure et al., 2013; Jahagirdar et al., 2014; Gilham et al., 2016; Ghosh et al., 2017), and has been tested in clinical trials (Nicholls et al., 2016, 2018; Shishikura et al., 2019). Pooling completed phase 2 trial data that suggested its clinical benefits on reducing major adverse cardiovascular events in treated patients (Borck et al., 2020). However, a phase III trial (BETonMACE) completed recently showed that addition of RVX 208 to contemporary standard of care for acute coronary syndrome (ACS) did not significantly reduce major adverse cardiovascular events in patients with a recent (7–90 days) ACS, type 2 diabetes, and low HDL cholesterol (Ray et al., 2020). To date, little is known about the direct interaction of BETs and HDACs or the combinational effects of BETi and HDACi in vascular cells and AS. Nevertheless, because HDACs and BETs share many common targets and affect similar cellular activities, it will be very interesting to study their molecular interplay during AS development. Those studies will be very helpful to evaluate whether combinational strategy using specific HDACi with BETi or developing dual BET/HDACi would be promising for AS treatment, as the strategy is rational and has been suggested in cancer studies (He et al., 2020; Liu et al., 2020).
Conclusion and Perspective
As a family of enzymes critically involved in chromatin remodeling and gene transcription, HDACs have multiple functions in ECs and other vascular cells that are implicated in AS (summarized in Figure 2 and Table 1) (Zhou et al., 2011b). Among the 11 classical HDACs, HDAC9 is the only subtype that has been well studied for its association with AS risk in both human and animals (Grbić et al., 2020). By contrast, little is known about the role of HDAC10 and HDAC11. With respect to other members, HDAC6 and HDAC8 mainly exhibit the role of mediating endothelial dysfunction and AS, whereas HDAC7 is mainly recognized as a protective isoform.
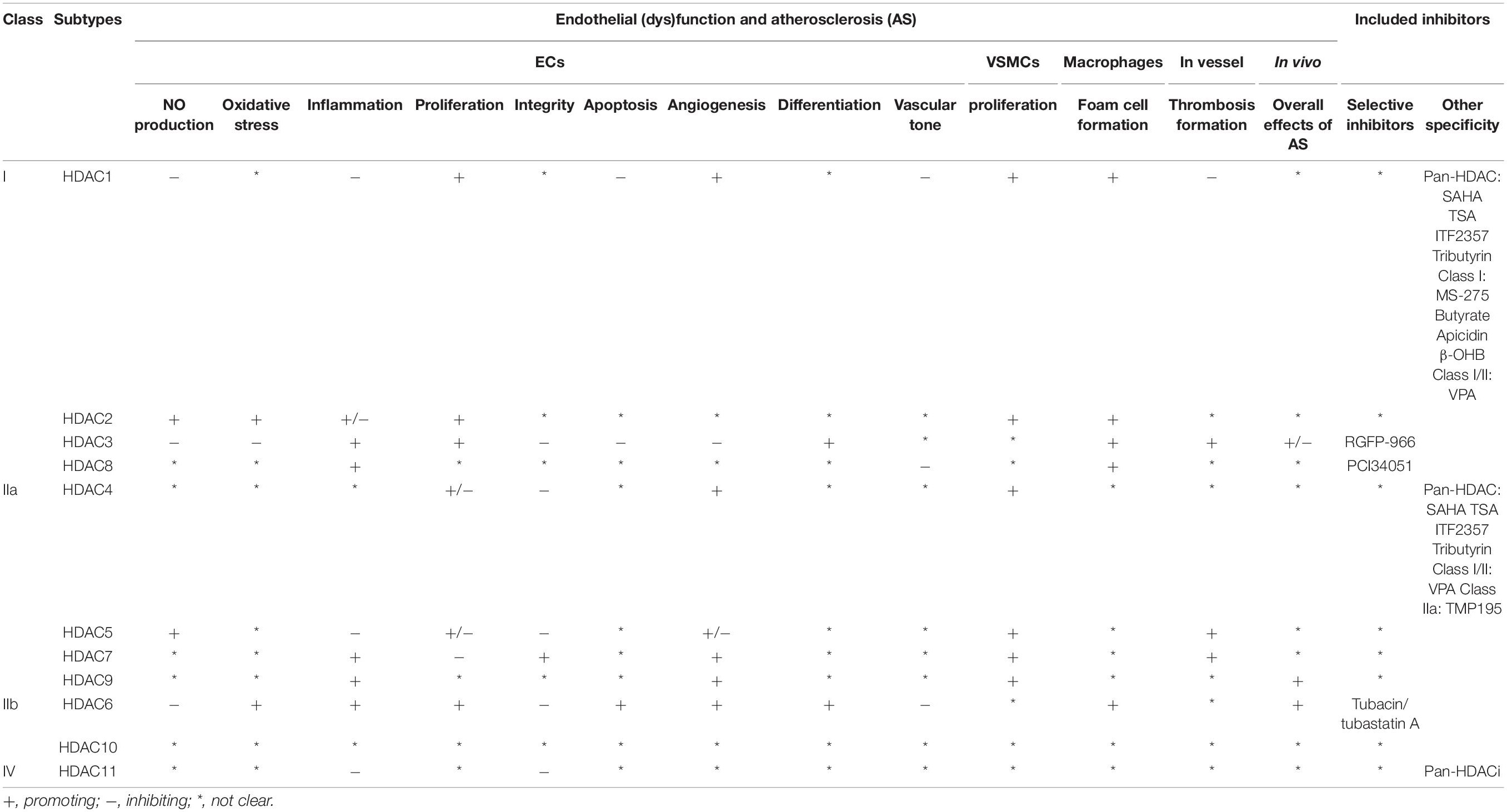
Table 1. Effect of HDAC members in endothelial (dys)function and atherosclerosis.
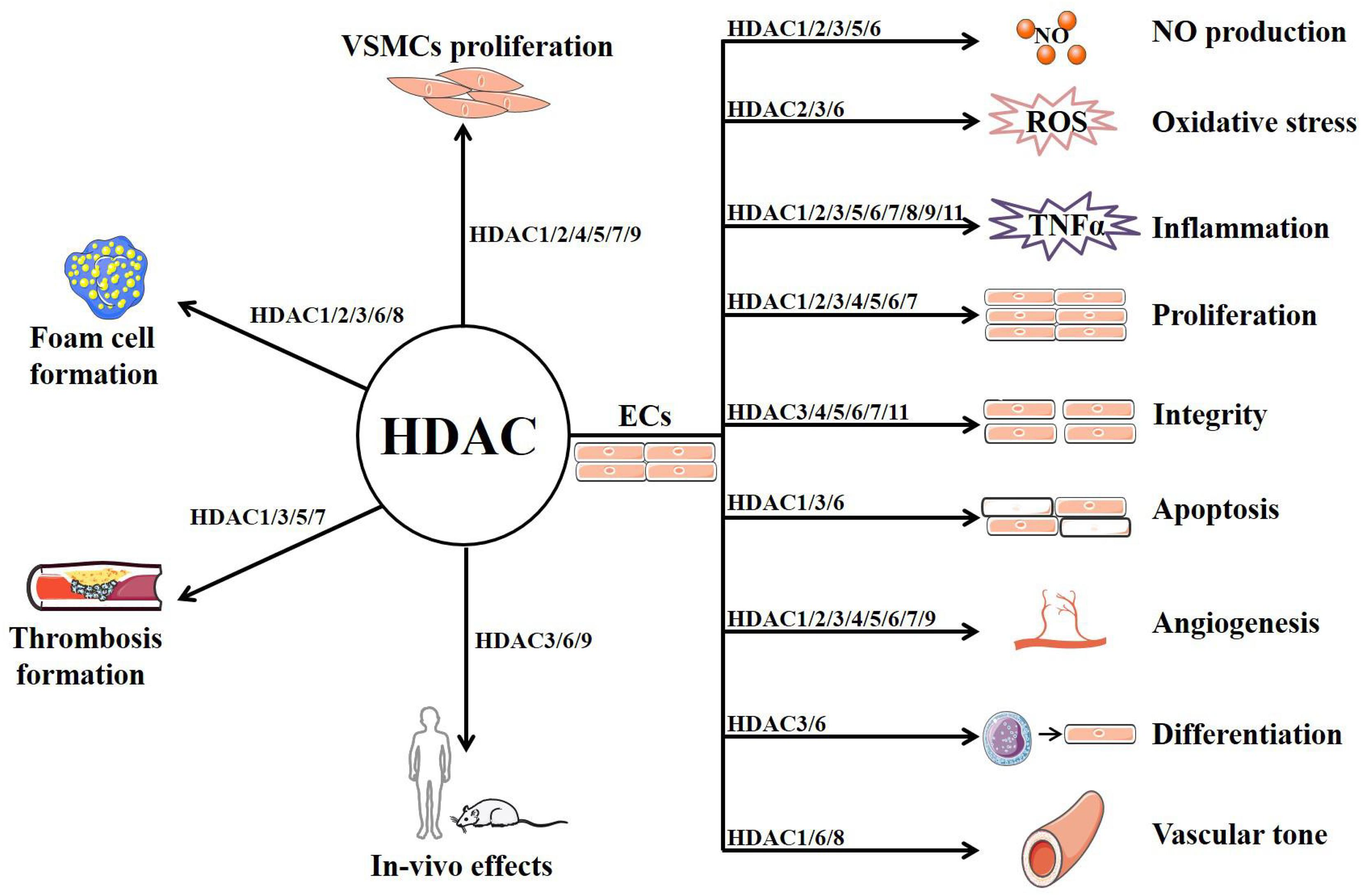
Figure 2. The potential mechanism of HDACs in atherosclerosis.
As exemplified in this review, many studies have been performed to investigate the effect of HDACi in vascular dysfunction and AS. Although in most cases, HDACi could prevent the pathological process, some HDACi might exacerbate it. The reason might be attributed to the unspecific property of HDACi in either HDAC subtypes or tissues/cells, which might also limit their applications in clinic. Moreover, the dosage used in the experiments and the off-target effect would also affect the results. Interestingly, inhibitors specific to HDAC3 (Zhao et al., 2019), HDAC6 (Chen et al., 2019), and HDAC8 (Kee et al., 2019) have been found to be effective in regulating vascular homeostasis, but further studies are still required to evaluate the pharmacological and pharmacokinetic profiles. Moreover, as some HDACs exhibit protective effects, it is suggested that HDAC analogs might be designed and investigated, such as the HDAC7-derived peptide (Pan et al., 2018). Nevertheless, extensive studies are urgently needed to elucidate and validate the mechanisms of HDACs in vascular function and AS to develop the more specific and targeted HDACi with less toxicity or side effects. Furthermore, chronic toxicity studies and randomized controlled trials with optimized dosage are also required.
Author Contributions
XC, YH, WF, YT, and HL wrote the manuscript. HL generated the illustration. AS revised the manuscript. SX conceptualized the manuscript, drafted the outline, and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (Grant No. 82070464 to SX and 81903606 to HL) and Natural Science Foundation of Guangdong Province Grant No. 2017A030310542 to HL.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ABCA1, ATP-binding cassette subfamily A1; ABCG1, ATP-binding cassette subfamily G1; AKT, protein kinase B; AMPK, adenosine 5 ′ -monophosphate (AMP)-activated protein kinase; AngII, angiotensin II; ApoE–/–, apolipoprotein E knockout; AS, atherosclerosis; Bcl-2, B-cell lymphoma-2; BH4, tetrahydrobiopterin; BuA, butyric acid; CAT, catalase; CD16, cluster of differentiation 16; cdk2, cyclin-dependent kinase2; cdk4, cyclin-dependent kinase4; cdk6, cyclin-dependent kinase6; COX-2, cyclooxygenase 2; CSE, cigarette smoke extract; CVD, cardiovascular disease; CYR61, cysteine rich angiogenic inducer 61; E2F1, E2F transcription factor 1; ECM, extracellular matrix; ECs, endothelial cells; ECV304 cells, human umbilical vein endothelial 304; eNOS, nitric oxide synthase; ERK 1/2, extracellular regulated protein kinases1/2; ET-1, endothelin 1; FSTL1, follistatin like 1; GLUT-1/4, glucose transport protein 1/4; H3K9, histone H3 lysine 9; HAECs, human aortic endothelial cells; HDAC6–/–, HDAC6 knockout; HDACi, histone deacetylases inhibitors; HDACs, histone deacetylases; HIF-1α, hypoxia inducible factor-1; HoxA9, homeobox A9; HPAECs, human pulmonary artery endothelial cells; HUVECs, human umbilical vein endothelial cells; ICAM-1, intercellular cell adhesion molecule 1; Id2, inhibitor of DNA binding 2; IGF-1, insulin-like growth factor 1; IKBα, NF-κB inhibitor; IL-10, interleukin 10; JNK, c-JunN-terminal kinase; KLF2/4, Krüppel-like factor 2/4; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MCP1, monocyte chemoattractant protein-1; MDR1, multidrug resistance protein 1; MEF2, myocyte enhancer factor 2; MMPs, matrix metalloproteinase; MS-275, entinostat; NAD+, nicotinamide adenine dinucleotide; NE-Y, nuclear factor-Y; NF-κB, nuclear factor κB; NO, nitric oxide; Nox, NADPH oxidase; ONOO–, peroxynitrite; OSS, oscillatory shear stress; ox-LDL, oxidized low density lipoprotein; PAECs, pulmonary artery vascular cells; PAH, pulmonary arterial hypertension; PAI-1, plasminogen activator inhibitor type 1; PCAF, p300/CBP-associated factor; PPARγ, peroxisome proliferator-activated receptors; PVRL2, poliovirus receptor related protein 2; ROS, reactive oxygen species; SAHA, varinostat; sGC, soluble guanylyl cyclase; SIRT, sirtuin; Slc2a1, solute carrier family 2 member 1; Slit2, Slit homolog 2 protein; SMC, smooth muscle cell; SOD, superoxide dismutase; SP1, Specificity Protein 1; STAT1, signal transducers and activators of transcription 1; TF, tissue factor; TIMP, the tissue inhibitor of metalloproteinase; TLR4, toll like receptor 4; TNF-α, tumor necrosis factor α; t-PA, tissue-type plasminogen activator; TSA, trichostatin; VCAM-1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor; VPA, valproic acid; VSMC, vascular smooth muscle cells; vWF, von-Willebrand factor.
References
Abend, A., Shkedi, O., Fertouk, M., Caspi, L. H., and Kehat, I. (2017). Salt-inducible kinase induces cytoplasmic histone deacetylase 4 to promote vascular calcification. EMBO Rep. 18, 1166–1185. doi: 10.15252/embr.201643686
Abouhish, H., Thounaojam, M. C., Jadeja, R. N., Gutsaeva, D. R., Powell, F. L., Khriza, M., et al. (2020). Inhibition of HDAC6 attenuates diabetes-induced retinal redox imbalance and microangiopathy. Antioxidants 9:599. doi: 10.3390/antiox9070599
Alam, S., Li, H., Margariti, A., Martin, D., Zampetaki, A., Habi, O., et al. (2011). Galectin-9 protein expression in endothelial cells is positively regulated by histone deacetylase 3. J. Biol. Chem. 286, 44211–44217. doi: 10.1074/jbc.M111.242289
Ali, L., Schnitzler, J. G., and Kroon, J. (2018). Metabolism: the road to inflammation and atherosclerosis. Curr. Opin. Lipidol. 29, 474–480. doi: 10.1097/mol.0000000000000550
Ali, M. M., Mahmoud, A. M., Le Master, E., Levitan, I., and Phillips, S. A. (2019). Role of matrix metalloproteinases and histone deacetylase in oxidative stress-induced degradation of the endothelial glycocalyx. Am. J. Physiol. Heart Circ. Physiol. 316, H647–H663. doi: 10.1152/ajpheart.00090.2018
Arcaro, G., Zenere, B. M., Travia, D., Zenti, M. G., Covi, G., Lechi, A., et al. (1995). Non-invasive detection of early endothelial dysfunction in hypercholesterolaemic subjects. Atherosclerosis 114, 247–254. doi: 10.1016/0021-9150(94)05489-6
Asare, Y., Campbell-James, T. A., Bokov, Y., Yu, L. L., Prestel, M., El Bounkari, O., et al. (2020). Histone Deacetylase 9 activates IKK to regulate atherosclerotic plaque vulnerability. Circ. Res. 127, 811–823. doi: 10.1161/circresaha.120.316743
Azechi, T., Kanehira, D., Kobayashi, T., Sudo, R., Nishimura, A., Sato, F., et al. (2013). Trichostatin, A., an HDAC class I/II inhibitor, promotes Pi-induced vascular calcification via up-regulation of the expression of alkaline phosphatase. J. Atheroscler Thromb. 20, 538–547. doi: 10.5551/jat.15826
Azghandi, S., Prell, C., van der Laan, S. W., Schneider, M., Malik, R., Berer, K., et al. (2015). Deficiency of the stroke relevant HDAC9 gene attenuates atherosclerosis in accord with allele-specific effects at 7p21.1. Stroke 46, 197–202. doi: 10.1161/strokeaha.114.007213
Bae, Y. S., Lee, J. H., Choi, S. H., Kim, S., Almazan, F., Witztum, J. L., et al. (2009). Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase 2. Circ. Res. 104, 210–218. 221p following 218. doi: 10.1161/circresaha.108.181040
Barbour, J. A., and Turner, N. (2014). Mitochondrial stress signaling promotes cellular adaptations. Int. J. Cell Biol. 2014:156020. doi: 10.1155/2014/156020
Bedenbender, K., Scheller, N., Fischer, S., Leiting, S., Preissner, K. T., Schmeck, B. T., et al. (2019). Inflammation-mediated deacetylation of the ribonuclease 1 promoter via histone deacetylase 2 in endothelial cells. Faseb J. 33, 9017–9029. doi: 10.1096/fj.201900451R
Bettica, P., Petrini, S., D’Oria, V., D’Amico, A., Catteruccia, M., Pane, M., et al. (2016). Histological effects of givinostat in boys with Duchenne muscular dystrophy. Neuromuscul. Disord. 26, 643–649. doi: 10.1016/j.nmd.2016.07.002
Birdsey, G. M., Dryden, N. H., Shah, A. V., Hannah, R., Hall, M. D., Haskard, D. O., et al. (2012). The transcription factor Erg regulates expression of histone deacetylase 6 and multiple pathways involved in endothelial cell migration and angiogenesis. Blood 119, 894–903. doi: 10.1182/blood-2011-04-350025
Borck, P. C., Guo, L. W., and Plutzky, J. (2020). BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ. Res. 126, 1190–1208. doi: 10.1161/circresaha.120.315929
Borgas, D., Chambers, E., Newton, J., Ko, J., Rivera, S., Rounds, S., et al. (2016). Cigarette smoke disrupted lung endothelial barrier integrity and increased susceptibility to acute lung injury via histone deacetylase 6. Am. J. Respir. Cell Mol. Biol. 54, 683–696. doi: 10.1165/rcmb.2015-0149OC
Bruhn, P. J., Nikolian, V. C., Halaweish, I., Chang, Z., Sillesen, M., Liu, B., et al. (2018). Tubastatin A prevents hemorrhage-induced endothelial barrier dysfunction. J. Trauma Acute Care Surg. 84, 386–392. doi: 10.1097/ta.0000000000001753
Cao, Q., Rong, S., Repa, J. J., St Clair, R., Parks, J. S., and Mishra, N. (2014). Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler Thromb. Vasc. Biol. 34, 1871–1879. doi: 10.1161/atvbaha.114.303393
Caplan, B. A., and Schwartz, C. J. (1973). Increased endothelial cell turnover in areas of in vivo Evans Blue uptake in the pig aorta. Atherosclerosis 17, 401–417. doi: 10.1016/0021-9150(73)90031-2
Castro, V., Bertrand, L., Luethen, M., Dabrowski, S., Lombardi, J., Morgan, L., et al. (2016). Occludin controls HIV transcription in brain pericytes via regulation of SIRT-1 activation. Faseb J. 30, 1234–1246. doi: 10.1096/fj.15-277673
Castro, V., Skowronska, M., Lombardi, J., He, J., Seth, N., Velichkovska, M., et al. (2018). Occludin regulates glucose uptake and ATP production in pericytes by influencing AMP-activated protein kinase activity. J. Cereb. Blood Flow Metab. 38, 317–332. doi: 10.1177/0271678x17720816
Chang, S., Young, B. D., Li, S., Qi, X., Richardson, J. A., and Olson, E. N. (2006). Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 126, 321–334. doi: 10.1016/j.cell.2006.05.040
Chen, F., Li, X., Aquadro, E., Haigh, S., Zhou, J., Stepp, D. W., et al. (2016). Inhibition of histone deacetylase reduces transcription of NADPH oxidases and ROS production and ameliorates pulmonary arterial hypertension. Free Radic. Biol. Med. 99, 167–178. doi: 10.1016/j.freeradbiomed.2016.08.003
Chen, J., Zhang, J., Shaik, N. F., Yi, B., Wei, X., Yang, X. F., et al. (2019). The histone deacetylase inhibitor tubacin mitigates endothelial dysfunction by up-regulating the expression of endothelial nitric oxide synthase. J. Biol. Chem. 294, 19565–19576. doi: 10.1074/jbc.RA119.011317
Cheng, H. P., Gong, D., Zhao, Z. W., He, P. P., Yu, X. H., Ye, Q., et al. (2017). MicroRNA-182 promotes lipoprotein lipase expression and atherogenesisby targeting histone deacetylase 9 in apolipoprotein E-Knockout Mice. Circ. J. 82, 28–38. doi: 10.1253/circj.CJ-16-1165
Cheng, H. T., and Hung, W. C. (2013). Inhibition of proliferation, sprouting, tube formation and Tie2 signaling of lymphatic endothelial cells by the histone deacetylase inhibitor SAHA. Oncol. Rep. 30, 961–967. doi: 10.3892/or.2013.2523
Chi, Z., Byeon, H. E., Seo, E., Nguyen, Q. T., Lee, W., Jeong, Y., et al. (2019). Histone deacetylase 6 inhibitor tubastatin A attenuates angiotensin II-induced hypertension by preventing cystathionine γ-lyase protein degradation. Pharmacol. Res. 146:104281. doi: 10.1016/j.phrs.2019.104281
Chi, Z., Le, T. P. H., Lee, S. K., Guo, E., Kim, D., Lee, S., et al. (2020). Honokiol ameliorates angiotensin II-induced hypertension and endothelial dysfunction by inhibiting HDAC6-mediated cystathionine γ-lyase degradation. J. Cell Mol. Med. 24, 10663–10676. doi: 10.1111/jcmm.15686
Chiu, J. J., and Chien, S. (2011). Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol. Rev. 91, 327–387. doi: 10.1152/physrev.00047.2009
Cho, D. H., Park, J. H., Joo Lee, E., Jong Won, K., Lee, S. H., Kim, Y. H., et al. (2014). Valproic acid increases NO production via the SH-PTP1-CDK5-eNOS-Ser(116) signaling cascade in endothelial cells and mice. Free Radic. Biol. Med. 76, 96–106. doi: 10.1016/j.freeradbiomed.2014.07.043
Choe, N., Shin, S., Joung, H., Ryu, J., Kim, Y. K., Ahn, Y., et al. (2020). The microRNA miR-134-5p induces calcium deposition by inhibiting histone deacetylase 5 in vascular smooth muscle cells. J. Cell Mol. Med. 24, 10542–10550. doi: 10.1111/jcmm.15670
Choi, J. H., Nam, K. H., Kim, J., Baek, M. W., Park, J. E., Park, H. Y., et al. (2005). Trichostatin A exacerbates atherosclerosis in low density lipoprotein receptor-deficient mice. Arterioscler Thromb. Vasc. Biol. 25, 2404–2409. doi: 10.1161/01.Atv.0000184758.07257.88
Collot-Teixeira, S., De Lorenzo, F., and McGregor, J. L. (2007). Scavenger receptor A and CD36 are implicated in mediating platelet activation induced by oxidized low- density lipoproteins. Arterioscler Thromb. Vasc. Biol. 27, 2491–2492. doi: 10.1161/atvbaha.107.154864
Craige, S. M., Kant, S., and Keaney, J. F. Jr. (2015). Reactive oxygen species in endothelial function - from disease to adaptation. Circ. J. 79, 1145–1155. doi: 10.1253/circj.CJ-15-0464
Deroanne, C. F., Bonjean, K., Servotte, S., Devy, L., Colige, A., Clausse, N., et al. (2002). Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 21, 427–436. doi: 10.1038/sj.onc.1205108
Dje N’Guessan, P., Riediger, F., Vardarova, K., Scharf, S., Eitel, J., Opitz, B., et al. (2009). Statins control oxidized LDL-mediated histone modifications and gene expression in cultured human endothelial cells. Arterioscler Thromb. Vasc. Biol. 29, 380–386. doi: 10.1161/atvbaha.108.178319
Doran, A. C., Meller, N., and McNamara, C. A. (2008). Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb. Vasc. Biol. 28, 812–819. doi: 10.1161/atvbaha.107.159327
Elliott, J. H., Wightman, F., Solomon, A., Ghneim, K., Ahlers, J., Cameron, M. J., et al. (2014). Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 10:e1004473. doi: 10.1371/journal.ppat.1004473
Fanelli, A., Ghisi, D., Aprile, P. L., and Lapi, F. (2017). Cardiovascular and cerebrovascular risk with nonsteroidal anti-inflammatory drugs and cyclooxygenase 2 inhibitors: latest evidence and clinical implications. Ther. Adv. Drug Saf. 8, 173–182. doi: 10.1177/2042098617690485
Feil, R., Lohmann, S. M., de Jonge, H., Walter, U., and Hofmann, F. (2003). Cyclic GMP-dependent protein kinases and the cardiovascular system: insights from genetically modified mice. Circ. Res. 93, 907–916. doi: 10.1161/01.Res.0000100390.68771.Cc
Fernandes, S., Salta, S., and Summavielle, T. (2015). Methamphetamine promotes α-tubulin deacetylation in endothelial cells: the protective role of acetyl-l-carnitine. Toxicol. Lett. 234, 131–138. doi: 10.1016/j.toxlet.2015.02.011
Finazzi, G., Vannucchi, A. M., Martinelli, V., Ruggeri, M., Nobile, F., Specchia, G., et al. (2013). A phase II study of Givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br. J. Haematol. 161, 688–694. doi: 10.1111/bjh.12332
Fischle, W., Dequiedt, F., Hendzel, M. J., Guenther, M. G., Lazar, M. A., Voelter, W., et al. (2002). Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 9, 45–57. doi: 10.1016/s1097-2765(01)00429-4
Förstermann, U., Xia, N., and Li, H. (2017). Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 120, 713–735. doi: 10.1161/circresaha.116.309326
Gao, C., Cheng, X., Lam, M., Liu, Y., Liu, Q., Chang, K. S., et al. (2008). Signal-dependent regulation of transcription by histone deacetylase 7 involves recruitment to promyelocytic leukemia protein nuclear bodies. Mol. Biol. Cell 19, 3020–3027. doi: 10.1091/mbc.e07-11-1203
Gao, J., Wang, Y., Li, W., Zhang, J., Che, Y., Cui, X., et al. (2018). Loss of histone deacetylase 2 inhibits oxidative stress induced by high glucose via the HO-1/SIRT1 pathway in endothelial progenitor cells. Gene 678, 1–7. doi: 10.1016/j.gene.2018.07.072
Gao, Q., Wei, A., Chen, F., Chen, X., Ding, W., Ding, Z., et al. (2020). Enhancing PPARγ by HDAC inhibition reduces foam cell formation and atherosclerosis in ApoE deficient mice. Pharmacol. Res. 160:105059. doi: 10.1016/j.phrs.2020.105059
Gatla, H. R., Muniraj, N., Thevkar, P., Yavvari, S., Sukhavasi, S., and Makena, M. R. (2019). Regulation of chemokines and cytokines by histone deacetylases and an update on histone decetylase inhibitors in human diseases. Int. J. Mol. Sci. 20:1110. doi: 10.3390/ijms20051110
Ghosh, G. C., Bhadra, R., Ghosh, R. K., Banerjee, K., and Gupta, A. (2017). RVX 208: a novel BET protein inhibitor, role as an inducer of apo A-I/HDL and beyond. Cardiovasc. Ther. 35:e12265. doi: 10.1111/1755-5922.12265
Gilham, D., Wasiak, S., Tsujikawa, L. M., Halliday, C., Norek, K., Patel, R. G., et al. (2016). RVX-208, a BET-inhibitor for treating atherosclerotic cardiovascular disease, raises ApoA-I/HDL and represses pathways that contribute to cardiovascular disease. Atherosclerosis 247, 48–57. doi: 10.1016/j.atherosclerosis.2016.01.036
Granger, A., Abdullah, I., Huebner, F., Stout, A., Wang, T., Huebner, T., et al. (2008). Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. Faseb J. 22, 3549–3560. doi: 10.1096/fj.08-108548
Gray, S. G., and Ekström, T. J. (2001). The human histone deacetylase family. Exp. Cell Res. 262, 75–83. doi: 10.1006/excr.2000.5080
Grbić, E., Gorkič, N., Pleskovič, A., Zorc, M., Ljuca, F., Gasparini, M., et al. (2020). Association between rs2107595 HDAC9 gene polymorphism and advanced carotid atherosclerosis in the Slovenian cohort. Lipids Health Dis. 19:71. doi: 10.1186/s12944-020-01255-1
Ha, C. H., Wang, W., Jhun, B. S., Wong, C., Hausser, A., Pfizenmaier, K., et al. (2008). Protein kinase D-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. J. Biol. Chem. 283, 14590–14599. doi: 10.1074/jbc.M800264200
Haas, T. L. (2005). Endothelial cell regulation of matrix metalloproteinases. Can. J. Physiol. Pharmacol. 83, 1–7. doi: 10.1139/y04-120
Haga, J. H., Li, Y. S., and Chien, S. (2007). Molecular basis of the effects of mechanical stretch on vascular smooth muscle cells. J. Biomech. 40, 947–960. doi: 10.1016/j.jbiomech.2006.04.011
Hakami, N. Y., Dusting, G. J., and Peshavariya, H. M. (2016). Trichostatin, A., a histone deacetylase inhibitor suppresses NADPH oxidase 4-Derived redox signalling and angiogenesis. J. Cell Mol. Med. 20, 1932–1944. doi: 10.1111/jcmm.12885
He, B., and Moreau, R. (2019). Lipid-regulating properties of butyric acid and 4-phenylbutyric acid: molecular mechanisms and therapeutic applications. Pharmacol. Res. 144, 116–131. doi: 10.1016/j.phrs.2019.04.002
He, H. M., Li, A., Zhang, S. W., and Duan, M. L. (2011). [The effects of a histone deacetylase (HDAC) inhibitor on endotoxin-induced endothelial cell injury]. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue 23, 602–604.
He, S., Dong, G., Li, Y., Wu, S., Wang, W., and Sheng, C. (2020). Potent dual BET/HDAC inhibitors for efficient treatment of pancreatic cancer. Angew. Chem. Int. Ed. Engl. 59, 3028–3032. doi: 10.1002/anie.201915896
Hebbel, R. P., Vercellotti, G. M., Pace, B. S., Solovey, A. N., Kollander, R., Abanonu, C. F., et al. (2010). The HDAC inhibitors trichostatin A and suberoylanilide hydroxamic acid exhibit multiple modalities of benefit for the vascular pathobiology of sickle transgenic mice. Blood 115, 2483–2490. doi: 10.1182/blood-2009-02-204990
Hoeksema, M. A., Gijbels, M. J., Van den Bossche, J., van der Velden, S., Sijm, A., Neele, A. E., et al. (2014). Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol. Med. 6, 1124–1132. doi: 10.15252/emmm.201404170
Hou, Q., Hu, K., Liu, X., Quan, J., and Liu, Z. (2018). HADC regulates the diabetic vascular endothelial dysfunction by targetting MnSOD. Biosci. Rep. 38:BSR20181042. doi: 10.1042/bsr20181042
Hsu, Y. F., Sheu, J. R., Lin, C. H., Chen, W. C., Hsiao, G., Ou, G., et al. (2011). MAPK phosphatase-1 contributes to trichostatin A inhibition of cyclooxygenase-2 expression in human umbilical vascular endothelial cells exposed to lipopolysaccharide. Biochim. Biophys. Acta 1810, 1160–1169. doi: 10.1016/j.bbagen.2011.08.015
Huynh, D. T. N., and Heo, K. S. (2019). Therapeutic targets for endothelial dysfunction in vascular diseases. Arch. Pharm. Res. 42, 848–861. doi: 10.1007/s12272-019-01180-7
Hyndman, K. A., Ho, D. H., Sega, M. F., and Pollock, J. S. (2014). Histone deacetylase 1 reduces NO production in endothelial cells via lysine deacetylation of NO synthase 3. Am. J. Physiol. Heart Circ. Physiol. 307, H803–H809. doi: 10.1152/ajpheart.00243.2014
Ikeda, E. (2005). Cellular response to tissue hypoxia and its involvement in disease progression. Pathol. Int. 55, 603–610. doi: 10.1111/j.1440-1827.2005.01877.x
Inoue, K., Kobayashi, M., Yano, K., Miura, M., Izumi, A., Mataki, C., et al. (2006). Histone deacetylase inhibitor reduces monocyte adhesion to endothelium through the suppression of vascular cell adhesion molecule-1 expression. Arterioscler Thromb. Vasc. Biol. 26, 2652–2659. doi: 10.1161/01.ATV.0000247247.89787.e7
Ismail, H., Mofarrahi, M., Echavarria, R., Harel, S., Verdin, E., Lim, H. W., et al. (2012). Angiopoietin-1 and vascular endothelial growth factor regulation of leukocyte adhesion to endothelial cells: role of nuclear receptor-77. Arterioscler Thromb. Vasc. Biol. 32, 1707–1716. doi: 10.1161/atvbaha.112.251546
Ivanciu, L., and Stalker, T. J. (2015). Spatiotemporal regulation of coagulation and platelet activation during the hemostatic response in vivo. J. Thromb. Haemost. 13, 1949–1959. doi: 10.1111/jth.13145
Jahagirdar, R., Zhang, H., Azhar, S., Tobin, J., Attwell, S., Yu, R., et al. (2014). A novel BET bromodomain inhibitor, RVX-208, shows reduction of atherosclerosis in hyperlipidemic ApoE deficient mice. Atherosclerosis 236, 91–100. doi: 10.1016/j.atherosclerosis.2014.06.008
Jin, G., Bausch, D., Knightly, T., Liu, Z., Li, Y., Liu, B., et al. (2011). Histone deacetylase inhibitors enhance endothelial cell sprouting angiogenesis in vitro. Surgery 150, 429–435. doi: 10.1016/j.surg.2011.07.001
Joshi, A. D., Barabutis, N., Birmpas, C., Dimitropoulou, C., Thangjam, G., Cherian-Shaw, M., et al. (2015). Histone deacetylase inhibitors prevent pulmonary endothelial hyperpermeability and acute lung injury by regulating heat shock protein 90 function. Am. J. Physiol. Lung. Cell Mol. Physiol. 309, L1410–L1419. doi: 10.1152/ajplung.00180.2015
Jung, S. B., Kim, C. S., Naqvi, A., Yamamori, T., Mattagajasingh, I., Hoffman, T. A., et al. (2010). Histone deacetylase 3 antagonizes aspirin-stimulated endothelial nitric oxide production by reversing aspirin-induced lysine acetylation of endothelial nitric oxide synthase. Circ. Res. 107, 877–887. doi: 10.1161/circresaha.110.222968
Kaluza, D., Kroll, J., Gesierich, S., Manavski, Y., Boeckel, J. N., Doebele, C., et al. (2013). Histone deacetylase 9 promotes angiogenesis by targeting the antiangiogenic microRNA-17-92 cluster in endothelial cells. Arterioscler Thromb. Vasc. Biol. 33, 533–543. doi: 10.1161/atvbaha.112.300415
Kane, A. E., and Sinclair, D. A. (2018). Sirtuins and NAD(+) in the development and treatment of metabolic and cardiovascular diseases. Circ. Res. 123, 868–885. doi: 10.1161/CIRCRESAHA.118.312498
Kang, G., Lee, Y. R., Joo, H. K., Park, M. S., Kim, C. S., Choi, S., et al. (2015). Trichostatin A modulates angiotensin II-induced vasoconstriction and blood pressure via inhibition of p66shc activation. Korean J. Physiol. Pharmacol. 19, 467–472. doi: 10.4196/kjpp.2015.19.5.467
Kang, Y., Kim, J., Anderson, J. P., Wu, J., Gleim, S. R., Kundu, R. K., et al. (2013). Apelin-APJ signaling is a critical regulator of endothelial MEF2 activation in cardiovascular development. Circ. Res. 113, 22–31. doi: 10.1161/circresaha.113.301324
Karki, P., Ke, Y., Tian, Y., Ohmura, T., Sitikov, A., Sarich, N., et al. (2019). Staphylococcus aureus-induced endothelial permeability and inflammation are mediated by microtubule destabilization. J. Biol. Chem. 294, 3369–3384. doi: 10.1074/jbc.RA118.004030
Kee, H. J., Ryu, Y., Seok, Y. M., Choi, S. Y., Sun, S., Kim, G. R., et al. (2019). Selective inhibition of histone deacetylase 8 improves vascular hypertrophy, relaxation, and inflammation in angiotensin II hypertensive mice. Clin. Hypertens 25:13. doi: 10.1186/s40885-019-0118-8
Khan, H., Belwal, T., Efferth, T., Farooqi, A. A., Sanches-Silva, A., Vacca, R. A., et al. (2020). Targeting epigenetics in cancer: therapeutic potential of flavonoids. Crit. Rev. Food Sci. Nutr. 1–24. doi: 10.1080/10408398.2020.1763910
Kim, J., Hwangbo, C., Hu, X., Kang, Y., Papangeli, I., Mehrotra, D., et al. (2015). Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation 131, 190–199. doi: 10.1161/CIRCULATIONAHA.114.013339
Kleinbongard, P., Heusch, G., and Schulz, R. (2010). TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol. Ther. 127, 295–314. doi: 10.1016/j.pharmthera.2010.05.002
Kovacs-Kasa, A., Kovacs, L., Cherian-Shaw, M., Patel, V., Meadows, M. L., Fulton, D. J., et al. (2020). Inhibition of Class IIa HDACs improves endothelial barrier function in endotoxin-induced acute lung injury. J. Cell Physiol. doi: 10.1002/jcp.30053 [Epub ahead of print].
Kowshik, J., Giri, H., Kishore, T. K., Kesavan, R., Vankudavath, R. N., Reddy, G. B., et al. (2014). Ellagic acid inhibits VEGF/VEGFR2, PI3K/Akt and MAPK signaling cascades in the hamster cheek pouch carcinogenesis model. Anticancer Agents. Med. Chem. 14, 1249–1260. doi: 10.2174/1871520614666140723114217
Krause, B. J., Hernandez, C., Caniuguir, A., Vasquez-Devaud, P., Carrasco-Wong, I., Uauy, R., et al. (2016). Arginase-2 is cooperatively up-regulated by nitric oxide and histone deacetylase inhibition in human umbilical artery endothelial cells. Biochem. Pharmacol. 99, 53–59. doi: 10.1016/j.bcp.2015.10.018
Kwon, I. S., Wang, W., Xu, S., and Jin, Z. G. (2014). Histone deacetylase 5 interacts with Krüppel-like factor 2 and inhibits its transcriptional activity in endothelium. Cardiovasc. Res. 104, 127–137. doi: 10.1093/cvr/cvu183
Lappas, M. (2012). Anti-inflammatory properties of sirtuin 6 in human umbilical vein endothelial cells. Mediators Inflamm 2012:597514. doi: 10.1155/2012/597514
Larsson, P., Bergh, N., Lu, E., Ulfhammer, E., Magnusson, M., Wahlander, K., et al. (2013). Histone deacetylase inhibitors stimulate tissue-type plasminogen activator production in vascular endothelial cells. J. Thromb. Thrombolysis. 35, 185–192. doi: 10.1007/s11239-012-0831-6
Larsson, P., Ulfhammer, E., Magnusson, M., Bergh, N., Lunke, S., El-Osta, A., et al. (2012). Role of histone acetylation in the stimulatory effect of valproic acid on vascular endothelial tissue-type plasminogen activator expression. PLoS One 7:e31573. doi: 10.1371/journal.pone.0031573
Lee, C. W., Lin, C. C., Luo, S. F., Lee, H. C., Lee, I. T., Aird, W. C., et al. (2008). Tumor necrosis factor-alpha enhances neutrophil adhesiveness: induction of vascular cell adhesion molecule-1 via activation of Akt and CaM kinase II and modifications of histone acetyltransferase and histone deacetylase 4 in human tracheal smooth muscle cells. Mol. Pharmacol. 73, 1454–1464. doi: 10.1124/mol.107.038091
Lee, D. Y., and Chiu, J. J. (2019). Atherosclerosis and flow: roles of epigenetic modulation in vascular endothelium. J. Biomed. Sci. 26:56. doi: 10.1186/s12929-019-0551-8
Lee, D. Y., Lee, C. I., Lin, T. E., Lim, S. H., Zhou, J., Tseng, Y. C., et al. (2012). Role of histone deacetylases in transcription factor regulation and cell cycle modulation in endothelial cells in response to disturbed flow. Proc. Natl. Acad. Sci. U.S.A. 109, 1967–1972. doi: 10.1073/pnas.1121214109
Leucker, T. M., Nomura, Y., Kim, J. H., Bhatta, A., Wang, V., Wecker, A., et al. (2017). Cystathionine γ-lyase protects vascular endothelium: a role for inhibition of histone deacetylase 6. Am. J. Physiol. Heart Circ. Physiol. 312, H711–H720. doi: 10.1152/ajpheart.00724.2016
Ley, K., Laudanna, C., Cybulsky, M. I., and Nourshargh, S. (2007). Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689. doi: 10.1038/nri2156
Li, C., Zhou, Y., Loberg, A., Tahara, S. M., Malik, P., and Kalra, V. K. (2016). Activated transcription factor 3 in association with histone deacetylase 6 negatively regulates MicroRNA 199a2 transcription by chromatin remodeling and reduces endothelin-1 expression. Mol. Cell Biol. 36, 2838–2854. doi: 10.1128/mcb.00345-16
Li, D., Wang, D., Wang, Y., Ling, W., Feng, X., and Xia, M. (2010). Adenosine monophosphate-activated protein kinase induces cholesterol efflux from macrophage-derived foam cells and alleviates atherosclerosis in apolipoprotein E-deficient mice. J. Biol. Chem. 285, 33499–33509. doi: 10.1074/jbc.M110.159772
Li, D., Xie, S., Ren, Y., Huo, L., Gao, J., Cui, D., et al. (2011). Microtubule-associated deacetylase HDAC6 promotes angiogenesis by regulating cell migration in an EB1-dependent manner. Protein Cell 2, 150–160. doi: 10.1007/s13238-011-1015-4
Li, F., Zhao, H., Li, G., Zhang, S., Wang, R., Tao, Z., et al. (2020). Intravenous antagomiR-494 lessens brain-infiltrating neutrophils by increasing HDAC2-mediated repression of multiple MMPs in experimental stroke. Faseb J. 34, 6934–6949. doi: 10.1096/fj.201903127R
Li, M., van Esch, B., Henricks, P. A. J., Folkerts, G., and Garssen, J. (2018a). The anti-inflammatory effects of short chain fatty acids on lipopolysaccharide- or tumor necrosis factor α-stimulated endothelial cells via activation of GPR41/43 and inhibition of HDACs. Front. Pharmacol. 9:533. doi: 10.3389/fphar.2018.00533
Li, Y., Li, L., Qian, Z., Lin, B., Chen, J., Luo, Y., et al. (2018b). Phosphatidylinositol 3-Kinase-DNA Methyltransferase 1-miR-1281-Histone Deacetylase 4 regulatory axis mediates platelet-derived growth factor-induced proliferation and migration of pulmonary artery smooth muscle cells. J. Am. Heart Assoc. 7:e007572. doi: 10.1161/jaha.117.007572
Li, Y., Zhang, K., and Mao, W. (2018c). Inhibition of miR-34a prevents endothelial cell apoptosis by directly targeting HDAC1 in the setting of atherosclerosis. Mol. Med. Rep. 17, 4645–4650. doi: 10.3892/mmr.2018.8411
Libby, P., Buring, J. E., Badimon, L., Hansson, G. K., Deanfield, J., Bittencourt, M. S., et al. (2019). Atherosclerosis. Nat. Rev. Dis. Primers 5:56. doi: 10.1038/s41572-019-0106-z
Lin, S., and Du, L. (2020). The therapeutic potential of BRD4 in cardiovascular disease. Hypertens Res. 86, 1006–1014. doi: 10.1038/s41440-020-0459-4
Liu, M. Y., and Khachigian, L. M. (2009). Histone deacetylase-1 is enriched at the platelet-derived growth factor-D promoter in response to interleukin-1beta and forms a cytokine-inducible gene-silencing complex with NF-kappab p65 and interferon regulatory factor-1. J. Biol. Chem. 284, 35101–35112. doi: 10.1074/jbc.M109.061903
Liu, T., Wan, Y., Xiao, Y., Xia, C., and Duan, G. (2020). Dual-target inhibitors BASED on HDACs: novel antitumor agents for cancer therapy. J. Med. Chem. 63, 8977–9002. doi: 10.1021/acs.jmedchem.0c00491
Luo, X. Y., Qu, S. L., Tang, Z. H., Zhang, Y., Liu, M. H., Peng, J., et al. (2014). SIRT1 in cardiovascular aging. Clin. Chim. Acta 437, 106–114. doi: 10.1016/j.cca.2014.07.019
Lyu, X., Hu, M., Peng, J., Zhang, X., and Sanders, Y. Y. (2019). HDAC inhibitors as antifibrotic drugs in cardiac and pulmonary fibrosis. Ther. Adv. Chronic Dis. 10:2040622319862697. doi: 10.1177/2040622319862697
Mahapatra, S., Firpo, M. T., and Bacanamwo, M. (2010). Inhibition of DNA methyltransferases and histone deacetylases induces bone marrow-derived multipotent adult progenitor cells to differentiate into endothelial cells. Ethn. Dis. 20, S1–S60.
Manea, S. A., Vlad, M. L., Fenyo, I. M., Lazar, A. G., Raicu, M., Muresian, H., et al. (2020). Pharmacological inhibition of histone deacetylase reduces NADPH oxidase expression, oxidative stress and the progression of atherosclerotic lesions in hypercholesterolemic apolipoprotein E-deficient mice; potential implications for human atherosclerosis. Redox Biol. 28:101338. doi: 10.1016/j.redox.2019.101338
Margariti, A., Zampetaki, A., Xiao, Q., Zhou, B., Karamariti, E., Martin, D., et al. (2010). Histone deacetylase 7 controls endothelial cell growth through modulation of beta-catenin. Circ. Res. 106, 1202–1211. doi: 10.1161/CIRCRESAHA.109.213165
Marks, P. A. (2010). The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin. Investig Drugs 19, 1049–1066. doi: 10.1517/13543784.2010.510514
Martin, D., Li, Y., Yang, J., Wang, G., Margariti, A., Jiang, Z., et al. (2014). Unspliced X-box-binding protein 1 (XBP1) protects endothelial cells from oxidative stress through interaction with histone deacetylase 3. J. Biol. Chem. 289, 30625–30634. doi: 10.1074/jbc.M114.571984
Mastoraki, A., Schizas, D., Charalampakis, N., Naar, L., Ioannidi, M., Tsilimigras, D., et al. (2020). Contribution of histone deacetylases in prognosis and therapeutic management of cholangiocarcinoma. Mol. Diagn. Ther. 24, 175–184. doi: 10.1007/s40291-020-00454-x
Mathew, O. P., Ranganna, K., and Yatsu, F. M. (2010). Butyrate, an HDAC inhibitor, stimulates interplay between different posttranslational modifications of histone H3 and differently alters G1-specific cell cycle proteins in vascular smooth muscle cells. Biomed. Pharmacother. 64, 733–740. doi: 10.1016/j.biopha.2010.09.017
McLure, K. G., Gesner, E. M., Tsujikawa, L., Kharenko, O. A., Attwell, S., Campeau, E., et al. (2013). RVX-208, an inducer of ApoA-I in humans, is a BET bromodomain antagonist. PLoS One 8:e83190. doi: 10.1371/journal.pone.0083190
Meng, F., Han, M., Zheng, B., Wang, C., Zhang, R., Zhang, X. H., et al. (2009). All-trans retinoic acid increases KLF4 acetylation by inducing HDAC2 phosphorylation and its dissociation from KLF4 in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 387, 13–18. doi: 10.1016/j.bbrc.2009.05.112
Michaelis, M., Suhan, T., Michaelis, U. R., Beek, K., Rothweiler, F., Tausch, L., et al. (2006). Valproic acid induces extracellular signal-regulated kinase 1/2 activation and inhibits apoptosis in endothelial cells. Cell Death Differ. 13, 446–453. doi: 10.1038/sj.cdd.4401759
Mihaylova, M. M., and Shaw, R. J. (2013). Metabolic reprogramming by class I and II histone deacetylases. Trends Endocrinol. Metab. 24, 48–57. doi: 10.1016/j.tem.2012.09.003
Miller, S. J., Zaloga, G. P., Hoggatt, A. M., Labarrere, C., and Faulk, W. P. (2005). Short-chain fatty acids modulate gene expression for vascular endothelial cell adhesion molecules. Nutrition 21, 740–748. doi: 10.1016/j.nut.2004.11.011
Miyoshi, M., Usami, M., and Ohata, A. (2008). Short-chain fatty acids and trichostatin A alter tight junction permeability in human umbilical vein endothelial cells. Nutrition 24, 1189–1198. doi: 10.1016/j.nut.2008.06.012
Mizuno, S., Yasuo, M., Bogaard, H. J., Kraskauskas, D., Natarajan, R., and Voelkel, N. F. (2011). Inhibition of histone deacetylase causes emphysema. Am. J. Physiol. Lung. Cell Mol. Physiol. 300, L402–L413. doi: 10.1152/ajplung.00207.2010
Mottet, D., Bellahcène, A., Pirotte, S., Waltregny, D., Deroanne, C., Lamour, V., et al. (2007). Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ. Res. 101, 1237–1246. doi: 10.1161/circresaha.107.149377
Nicholls, S. J., Puri, R., Wolski, K., Ballantyne, C. M., Barter, P. J., Brewer, H. B., et al. (2016). Effect of the BET protein inhibitor, RVX-208, on progression of coronary atherosclerosis: results of the phase 2b, randomized, double-blind, multicenter, ASSURE Trial. Am. J. Cardiovasc. Drugs 16, 55–65. doi: 10.1007/s40256-015-0146-z
Nicholls, S. J., Ray, K. K., Johansson, J. O., Gordon, A., Sweeney, M., Halliday, C., et al. (2018). Selective BET protein inhibition with apabetalone and cardiovascular events: a pooled analysis of trials in patients with coronary artery disease. Am. J. Cardiovasc. Drugs 18, 109–115. doi: 10.1007/s40256-017-0250-3
Nishida, K., Harrison, D. G., Navas, J. P., Fisher, A. A., Dockery, S. P., Uematsu, M., et al. (1992). Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J. Clin. Invest. 90, 2092–2096. doi: 10.1172/jci116092
Niu, N., Xu, S., Xu, Y., Little, P. J., and Jin, Z. G. (2019). Targeting mechanosensitive transcription factors in atherosclerosis. Trends Pharmacol. Sci. 40, 253–266. doi: 10.1016/j.tips.2019.02.004
Ogawa, H., Rafiee, P., Fisher, P. J., Johnson, N. A., Otterson, M. F., and Binion, D. G. (2003). Butyrate modulates gene and protein expression in human intestinal endothelial cells. Biochem. Biophys. Res. Commun. 309, 512–519. doi: 10.1016/j.bbrc.2003.08.026
Ohashi, A., Yasuda, H., Kamiya, T., Hara, H., and Adachi, T. (2017). CAPE increases the expression of SOD3 through epigenetics in human retinal endothelial cells. J. Clin. Biochem. Nutr. 61, 6–13. doi: 10.3164/jcbn.16-109
Okam, M. M., Esrick, E. B., Mandell, E., Campigotto, F., Neuberg, D. S., and Ebert, B. L. (2015). Phase 1/2 trial of vorinostat in patients with sickle cell disease who have not benefitted from hydroxyurea. Blood 125, 3668–3669. doi: 10.1182/blood-2015-03-635391
Okamoto, H., Fujioka, Y., Takahashi, A., Takahashi, T., Taniguchi, T., Ishikawa, Y., et al. (2006). Trichostatin, A., an inhibitor of histone deacetylase, inhibits smooth muscle cell proliferation via induction of p21(WAF1). J. Atheroscler Thromb. 13, 183–191. doi: 10.5551/jat.13.183
Orekhov, A. N. (2018). LDL and foam cell formation as the basis of atherogenesis. Curr. Opin. Lipidol. 29, 279–284. doi: 10.1097/mol.0000000000000525
Pan, Y., Yang, J., Wei, Y., Wang, H., Jiao, R., Moraga, A., et al. (2018). Histone deacetylase 7-derived peptides play a vital role in vascular repair and regeneration. Adv. Sci. 5:1800006. doi: 10.1002/advs.201800006
Pandey, D., Sikka, G., Bergman, Y., Kim, J. H., Ryoo, S., Romer, L., et al. (2014). Transcriptional regulation of endothelial arginase 2 by histone deacetylase 2. Arterioscler Thromb. Vasc. Biol. 34, 1556–1566. doi: 10.1161/ATVBAHA.114.303685
Pang, J., Yan, C., Natarajan, K., Cavet, M. E., Massett, M. P., Yin, G., et al. (2008). GIT1 mediates HDAC5 activation by angiotensin II in vascular smooth muscle cells. Arterioscler Thromb. Vasc. Biol. 28, 892–898. doi: 10.1161/atvbaha.107.161349
Park, D., Park, H., Kim, Y., Kim, H., and Jeoung, D. (2014). HDAC3 acts as a negative regulator of angiogenesis. BMB Rep. 47, 227–232. doi: 10.5483/bmbrep.2014.47.4.128
Parra, M., and Verdin, E. (2010). Regulatory signal transduction pathways for class IIa histone deacetylases. Curr. Opin. Pharmacol. 10, 454–460. doi: 10.1016/j.coph.2010.04.004
Peng, Y., Stewart, D., Li, W., Hawkins, M., Kulak, S., Ballermann, B., et al. (2007). Irradiation modulates association of NF-Y with histone-modifying cofactors PCAF and HDAC. Oncogene 26, 7576–7583. doi: 10.1038/sj.onc.1210565
Pepper, M. S., Montesano, R., Mandriota, S. J., Orci, L., and Vassalli, J. D. (1996). Angiogenesis: a paradigm for balanced extracellular proteolysis during cell migration and morphogenesis. Enzyme Protein 49, 138–162. doi: 10.1159/000468622
Pietruczuk, P., Jain, A., Simo-Cheyou, E. R., Anand-Srivastava, M. B., and Srivastava, A. K. (2019). Protein kinase B/AKT mediates insulin-like growth factor 1-induced phosphorylation and nuclear export of histone deacetylase 5 via NADPH oxidase 4 activation in vascular smooth muscle cells. J. Cell Physiol. 234, 17337–17350. doi: 10.1002/jcp.28353
Poston, R. N. (2019). Atherosclerosis: integration of its pathogenesis as a self-perpetuating propagating inflammation: a review. Cardiovasc. Endocrinol. Metab. 8, 51–61. doi: 10.1097/xce.0000000000000172
Prestel, M., Prell-Schicker, C., Webb, T., Malik, R., Lindner, B., Ziesch, N., et al. (2019). The atherosclerosis risk variant rs2107595 mediates allele-specific transcriptional regulation of HDAC9 via E2F3 and Rb1. Stroke 50, 2651–2660. doi: 10.1161/strokeaha.119.026112
Rabinovitch, M. (2012). Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Invest. 122, 4306–4313. doi: 10.1172/jci60658
Rafehi, H., Karagiannis, T. C., and El-Osta, A. (2017). Pharmacological histone deacetylation distinguishes transcriptional regulators. Curr. Top. Med. Chem. 17, 1611–1622. doi: 10.2174/1568026617666161104104341
Rajasingh, J., Thangavel, J., Siddiqui, M. R., Gomes, I., Gao, X. P., Kishore, R., et al. (2011). Improvement of cardiac function in mouse myocardial infarction after transplantation of epigenetically-modified bone marrow progenitor cells. PLoS One 6:e22550. doi: 10.1371/journal.pone.0022550
Rambaldi, A., Dellacasa, C. M., Finazzi, G., Carobbio, A., Ferrari, M. L., Guglielmelli, P., et al. (2010). A pilot study of the histone-deacetylase inhibitor givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br. J. Haematol. 150, 446–455. doi: 10.1111/j.1365-2141.2010.08266.x
Ray, K. K., Nicholls, S. J., Buhr, K. A., Ginsberg, H. N., Johansson, J. O., Kalantar-Zadeh, K., et al. (2020). Effect of apabetalone added to standard therapy on major adverse cardiovascular events in patients with recent acute coronary syndrome and type 2 diabetes: a randomized clinical trial. Jama 323, 1565–1573. doi: 10.1001/jama.2020.3308
Reynoso-Roldán, A., Roldán, M. L., Cancino-Diaz, J. C., Rodríguez-Martínez, S., and Cancino-Diaz, M. E. (2012). Vascular endothelial growth factor production is induced by histone deacetylase 1 and suppressed by von Hippel-Lindau protein in HaCaT cells. Clin. Invest. Med. 35, E340–E350. doi: 10.25011/cim.v35i6.19205
Ricard, N., Tu, L., Le Hiress, M., Huertas, A., Phan, C., Thuillet, R., et al. (2014). Increased pericyte coverage mediated by endothelial-derived fibroblast growth factor-2 and interleukin-6 is a source of smooth muscle-like cells in pulmonary hypertension. Circulation 129, 1586–1597. doi: 10.1161/circulationaha.113.007469
Rinkevich, Y., Lindau, P., Ueno, H., Longaker, M. T., and Weissman, I. L. (2011). Germ-layer and lineage-restricted stem/progenitors regenerate the mouse digit tip. Nature 476, 409–413. doi: 10.1038/nature10346
Ross, R. (1993). The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 362, 801–809. doi: 10.1038/362801a0
Rossig, L., Li, H., Fisslthaler, B., Urbich, C., Fleming, I., Forstermann, U., et al. (2002). Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ. Res. 91, 837–844. doi: 10.1161/01.res.0000037983.07158.b1
Rossig, L., Urbich, C., Bruhl, T., Dernbach, E., Heeschen, C., Chavakis, E., et al. (2005). Histone deacetylase activity is essential for the expression of HoxA9 and for endothelial commitment of progenitor cells. J. Exp. Med. 201, 1825–1835. doi: 10.1084/jem.20042097
Ryu, Y., Kee, H. J., Sun, S., Seok, Y. M., Choi, S. Y., Kim, G. R., et al. (2019). Class I histone deacetylase inhibitor MS-275 attenuates vasoconstriction and inflammation in angiotensin II-induced hypertension. PLoS One 14:e0213186. doi: 10.1371/journal.pone.0213186
Serlin, Y., Shelef, I., Knyazer, B., and Friedman, A. (2015). Anatomy and physiology of the blood-brain barrier. Semin. Cell Dev. Biol. 38, 2–6. doi: 10.1016/j.semcdb.2015.01.002
Shah, P. K. (1996). Pathophysiology of plaque rupture and the concept of plaque stabilization. Cardiol. Clin. 14, 17–29. doi: 10.1016/s0733-8651(05)70258-7
Shi, W., Wei, X., Wang, Z., Han, H., Fu, Y., Liu, J., et al. (2016). HDAC9 exacerbates endothelial injury in cerebral ischaemia/reperfusion injury. J. Cell Mol. Med. 20, 1139–1149. doi: 10.1111/jcmm.12803
Shirodkar, A. V., and Marsden, P. A. (2011). Epigenetics in cardiovascular disease. Curr. Opin. Cardiol. 26, 209–215. doi: 10.1097/HCO.0b013e328345986e
Shishikura, D., Kataoka, Y., Honda, S., Takata, K., Kim, S. W., Andrews, J., et al. (2019). The effect of bromodomain and extra-terminal inhibitor apabetalone on attenuated coronary atherosclerotic plaque: insights from the ASSURE Trial. Am. J. Cardiovasc. Drugs 19, 49–57. doi: 10.1007/s40256-018-0298-8
Shroff, N., Ander, B. P., Zhan, X., Stamova, B., Liu, D., Hull, H., et al. (2019). HDAC9 polymorphism alters blood gene expression in patients with large vessel atherosclerotic stroke. Transl. Stroke Res. 10, 19–25. doi: 10.1007/s12975-018-0619-x
Smith, Q., Macklin, B., Chan, X. Y., Jones, H., Trempel, M., Yoder, M. C., et al. (2018). Differential HDAC6 activity modulates ciliogenesis and subsequent mechanosensing of endothelial cells derived from pluripotent stem cells. Cell Rep. 24, 895.e6–908.e6. doi: 10.1016/j.celrep.2018.06.083
Spronk, H. M., Borissoff, J. I., and ten Cate, H. (2013). New insights into modulation of thrombin formation. Curr. Atheroscler Rep. 15:363. doi: 10.1007/s11883-013-0363-3
Stein, S., Schäfer, N., Breitenstein, A., Besler, C., Winnik, S., Lohmann, C., et al. (2010). SIRT1 reduces endothelial activation without affecting vascular function in ApoE-/- mice. Aging 2, 353–360. doi: 10.18632/aging.100162
Stocker, R., and Keaney, J. F. Jr. (2004). Role of oxidative modifications in atherosclerosis. Physiol. Rev. 84, 1381–1478. doi: 10.1152/physrev.00047.2003
Su, Y. T., Gao, C., Liu, Y., Guo, S., Wang, A., Wang, B., et al. (2013). Monoubiquitination of filamin B regulates vascular endothelial growth factor-mediated trafficking of histone deacetylase 7. Mol. Cell Biol. 33, 1546–1560. doi: 10.1128/mcb.01146-12
Sun, L., Wang, C., Yuan, Y., Guo, Z., He, Y., Ma, W., et al. (2020). Downregulation of HDAC1 suppresses media degeneration by inhibiting the migration and phenotypic switch of aortic vascular smooth muscle cells in aortic dissection. J. Cell Physiol. 235, 8747–8756. doi: 10.1002/jcp.29718
Tang, F. B., Dai, Y. L., Zhou, G. Y., Zhang, W. H., Wang, H. B., Li, Y. G., et al. (2018). Valproic acid treatment inhibits vasopermeability and improves survival in rats with lethal scald injury. J. Burn. Care Res. 39, 209–217. doi: 10.1097/bcr.0000000000000568
Tedgui, A., and Mallat, Z. (2003). Apoptosis, a major determinant of atherothrombosis. Arch. Mal. Coeur Vaiss 96, 671–675.
Tesfamariam, B., and DeFelice, A. F. (2007). Endothelial injury in the initiation and progression of vascular disorders. Vascul. Pharmacol. 46, 229–237. doi: 10.1016/j.vph.2006.11.005
Thangjam, G. S., Dimitropoulou, C., Joshi, A. D., Barabutis, N., Shaw, M. C., Kovalenkov, Y., et al. (2014). Novel mechanism of attenuation of LPS-induced NF-kappaB activation by the heat shock protein 90 inhibitor, 17-N-allylamino-17-demethoxygeldanamycin, in human lung microvascular endothelial cells. Am. J. Respir. Cell Mol. Biol. 50, 942–952. doi: 10.1165/rcmb.2013-0214OC
Tian, K., Ogura, S., Little, P. J., Xu, S. W., and Sawamura, T. (2019). Targeting LOX-1 in atherosclerosis and vasculopathy: current knowledge and future perspectives. Ann. N. Y. Acad. Sci. 1443, 34–53. doi: 10.1111/nyas.13984
Tian, R., Li, R., Liu, Y., Liu, J., Pan, T., Zhang, R., et al. (2019). Metformin ameliorates endotoxemia-induced endothelial pro-inflammatory responses via AMPK-dependent mediation of HDAC5 and KLF2. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 1701–1712. doi: 10.1016/j.bbadis.2019.04.009
To, M., Yamamura, S., Akashi, K., Charron, C. E., Haruki, K., Barnes, P. J., et al. (2012). Defect of adaptation to hypoxia in patients with COPD due to reduction of histone deacetylase 7. Chest 141, 1233–1242. doi: 10.1378/chest.11-1536
Tsou, P. S., Wren, J. D., Amin, M. A., Schiopu, E., Fox, D. A., Khanna, D., et al. (2016). Histone Deacetylase 5 Is overexpressed in scleroderma endothelial cells and impairs angiogenesis via repression of proangiogenic factors. Arthritis Rheumatol. 68, 2975–2985. doi: 10.1002/art.39828
Urbich, C., Rossig, L., Kaluza, D., Potente, M., Boeckel, J. N., Knau, A., et al. (2009). HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood 113, 5669–5679. doi: 10.1182/blood-2009-01-196485
Usui, T., Morita, T., Okada, M., and Yamawaki, H. (2014). Histone deacetylase 4 controls neointimal hyperplasia via stimulating proliferation and migration of vascular smooth muscle cells. Hypertension 63, 397–403. doi: 10.1161/hypertensionaha.113.01843
Vallée, A., Vallée, J. N., and Lecarpentier, Y. (2019). Metabolic reprogramming in atherosclerosis: opposed interplay between the canonical WNT/β-catenin pathway and PPARγ. J. Mol. Cell Cardiol. 133, 36–46. doi: 10.1016/j.yjmcc.2019.05.024
Van den Bossche, J., Neele, A. E., Hoeksema, M. A., de Heij, F., Boshuizen, M. C., van der Velden, S., et al. (2014). Inhibiting epigenetic enzymes to improve atherogenic macrophage functions. Biochem. Biophys. Res. Commun. 455, 396–402. doi: 10.1016/j.bbrc.2014.11.029
Vinh, A., Gaspari, T. A., Liu, H. B., Dousha, L. F., Widdop, R. E., and Dear, A. E. (2008). A novel histone deacetylase inhibitor reduces abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. J. Vasc. Res. 45, 143–152. doi: 10.1159/000110041
Vinolo, M. A., Rodrigues, H. G., Nachbar, R. T., and Curi, R. (2011). Regulation of inflammation by short chain fatty acids. Nutrients 3, 858–876. doi: 10.3390/nu3100858
Wang, D., Yang, Y., Lei, Y., Tzvetkov, N. T., Liu, X., Yeung, A. W. K., et al. (2019). Targeting foam cell formation in atherosclerosis: therapeutic potential of natural products. Pharmacol. Rev. 71, 596–670. doi: 10.1124/pr.118.017178
Wang, H., Fu, H., Zhu, R., Wu, X., Ji, X., Li, X., et al. (2020). BRD4 contributes to LPS-induced macrophage senescence and promotes progression of atherosclerosis-associated lipid uptake. Aging 12, 9240–9259. doi: 10.18632/aging.103200
Wang, J., Mahmud, S. A., Bitterman, P. B., Huo, Y., and Slungaard, A. (2007). Histone deacetylase inhibitors suppress TF-kappaB-dependent agonist-driven tissue factor expression in endothelial cells and monocytes. J. Biol. Chem. 282, 28408–28418. doi: 10.1074/jbc.M703586200
Wang, S., Li, X., Parra, M., Verdin, E., Bassel-Duby, R., and Olson, E. N. (2008). Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proc. Natl. Acad. Sci. U.S.A. 105, 7738–7743. doi: 10.1073/pnas.0802857105
Wang, W., Ha, C. H., Jhun, B. S., Wong, C., Jain, M. K., and Jin, Z. G. (2010). Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood 115, 2971–2979. doi: 10.1182/blood-2009-05-224824
Wang, Y. H., Yan, Z. Q., Qi, Y. X., Cheng, B. B., Wang, X. D., Zhao, D., et al. (2010). Normal shear stress and vascular smooth muscle cells modulate migration of endothelial cells through histone deacetylase 6 activation and tubulin acetylation. Ann. Biomed. Eng. 38, 729–737. doi: 10.1007/s10439-009-9896-6
Wang, X. B., Han, Y. D., Sabina, S., Cui, N. H., Zhang, S., Liu, Z. J., et al. (2016). HDAC9 Variant Rs2107595 modifies susceptibility to coronary artery disease and the severity of coronary atherosclerosis in a chinese han population. PLoS One 11:e0160449. doi: 10.1371/journal.pone.0160449
Wang, Z., Tsai, L. K., Munasinghe, J., Leng, Y., Fessler, E. B., Chibane, F., et al. (2012). Chronic valproate treatment enhances postischemic angiogenesis and promotes functional recovery in a rat model of ischemic stroke. Stroke 43, 2430–2436. doi: 10.1161/strokeaha.112.652545
Weinbaum, S., Tarbell, J. M., and Damiano, E. R. (2007). The structure and function of the endothelial glycocalyx layer. Annu. Rev. Biomed. Eng. 9, 121–167. doi: 10.1146/annurev.bioeng.9.060906.151959
Williams, R. J. (2001). Trichostatin, A., an inhibitor of histone deacetylase, inhibits hypoxia-induced angiogenesis. Expert Opin. Investig Drugs 10, 1571–1573. doi: 10.1517/13543784.10.8.1571
Won, K. J., Jung, S. H., Jung, S. H., Lee, K. P., Lee, H. M., Lee, D. Y., et al. (2014). DJ-1/park7 modulates vasorelaxation and blood pressure via epigenetic modification of endothelial nitric oxide synthase. Cardiovasc. Res. 101, 473–481. doi: 10.1093/cvr/cvt274
Wright, L. H., Herr, D. J., Brown, S. S., Kasiganesan, H., and Menick, D. R. (2018). Angiokine Wisp-1 is increased in myocardial infarction and regulates cardiac endothelial signaling. JCI Insight 3:e95824. doi: 10.1172/jci.insight.95824
Wu, H., Cheng, X. W., Hu, L., Takeshita, K., Hu, C., Du, Q., et al. (2016). Cathepsin S activity controls injury-related vascular repair in mice via the TLR2-mediated p38MAPK and PI3K-Akt/p-HDAC6 signaling pathway. Arterioscler Thromb. Vasc. Biol. 36, 1549–1557. doi: 10.1161/atvbaha.115.307110
Wu, J., Jiang, Z., Zhang, H., Liang, W., Huang, W., Zhang, H., et al. (2018). Sodium butyrate attenuates diabetes-induced aortic endothelial dysfunction via P300-mediated transcriptional activation of Nrf2. Free Radic. Biol. Med. 124, 454–465. doi: 10.1016/j.freeradbiomed.2018.06.034
Xiao, Q., Zeng, L., Zhang, Z., Margariti, A., Ali, Z. A., Channon, K. M., et al. (2006). Sca-1+ progenitors derived from embryonic stem cells differentiate into endothelial cells capable of vascular repair after arterial injury. Arterioscler Thromb. Vasc. Biol. 26, 2244–2251. doi: 10.1161/01.ATV.0000240251.50215.50
Xu, S., Kamato, D., Little, P. J., Nakagawa, S., Pelisek, J., and Jin, Z. G. (2019). Targeting epigenetics and non-coding RNAs in atherosclerosis: from mechanisms to therapeutics. Pharmacol. Ther. 196, 15–43. doi: 10.1016/j.pharmthera.2018.11.003
Xu, S., Pelisek, J., and Jin, Z. G. (2018). Atherosclerosis Is an Epigenetic Disease. Trends Endocrinol. Metab. 29, 739–742. doi: 10.1016/j.tem.2018.04.007
Xu, Y., Xu, S., Liu, P., Koroleva, M., Zhang, S., Si, S., et al. (2017). Suberanilohydroxamic acid as a pharmacological kruppel-like factor 2 activator that represses vascular inflammation and atherosclerosis. J. Am. Heart Assoc. 6:e007134. doi: 10.1161/jaha.117.007134
Yamanegi, K., Kawabe, M., Futani, H., Nishiura, H., Yamada, N., Kato-Kogoe, N., et al. (2015). Sodium valproate, a histone deacetylase inhibitor, modulates the vascular endothelial growth inhibitor-mediated cell death in human osteosarcoma and vascular endothelial cells. Int. J. Oncol. 46, 1994–2002. doi: 10.3892/ijo.2015.2924
Yan, Z. Q., Yao, Q. P., Zhang, M. L., Qi, Y. X., Guo, Z. Y., Shen, B. R., et al. (2009). Histone deacetylases modulate vascular smooth muscle cell migration induced by cyclic mechanical strain. J. Biomech. 42, 945–948. doi: 10.1016/j.jbiomech.2009.01.012
Yang, D., Xie, P., and Liu, Z. (2012). Ischemia/reperfusion-induced MKP-3 impairs endothelial NO formation via inactivation of ERK1/2 pathway. PLoS One 7:e42076. doi: 10.1371/journal.pone.0042076
Yang, L., Liu, N., Zhao, W., Li, X., Han, L., Zhang, Z., et al. (2019). Angiogenic function of astragaloside IV in rats with myocardial infarction occurs via the PKD1-HDAC5-VEGF pathway. Exp. Ther. Med. 17, 2511–2518. doi: 10.3892/etm.2019.7273
Yang, Q., Sun, M., Ramchandran, R., and Raj, J. U. (2015). IGF-1 signaling in neonatal hypoxia-induced pulmonary hypertension: role of epigenetic regulation. Vascul. Pharmacol. 73, 20–31. doi: 10.1016/j.vph.2015.04.005
Yanginlar, C., and Logie, C. (2018). HDAC11 is a regulator of diverse immune functions. Biochim. Biophys. Acta Gene Regul. Mech. 1861, 54–59. doi: 10.1016/j.bbagrm.2017.12.002
Yao, Q. P., Zhang, P., Qi, Y. X., Chen, S. G., Shen, B. R., Han, Y., et al. (2014). The role of SIRT6 in the differentiation of vascular smooth muscle cells in response to cyclic strain. Int. J. Biochem. Cell Biol. 49, 98–104. doi: 10.1016/j.biocel.2014.01.016
Ye, Y., Zhao, X., Lu, Y., Long, B., and Zhang, S. (2018). Varinostat alters gene expression profiles in aortic tissues from ApoE(-/-) Mice. Hum. Gene Ther. Clin. Dev. 29, 214–225. doi: 10.1089/humc.2018.141
Yoshida, T., Gan, Q., and Owens, G. K. (2008). Kruppel-like factor 4, Elk-1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids. Am. J. Physiol. Cell Physiol. 295, C1175–C1182. doi: 10.1152/ajpcell.00288.2008
You, D., Wen, X., Gorczyca, L., Morris, A., Richardson, J. R., and Aleksunes, L. M. (2019). Increased MDR1 transporter expression in human brain endothelial cells through enhanced histone acetylation and activation of aryl hydrocarbon receptor signaling. Mol. Neurobiol. 56, 6986–7002. doi: 10.1007/s12035-019-1565-7
Yu, D., Chen, W., Ren, J., Zhang, T., Yang, K., Wu, G., et al. (2014). VEGF-PKD1-HDAC7 signaling promotes endothelial progenitor cell migration and tube formation. Microvasc. Res. 91, 66–72. doi: 10.1016/j.mvr.2013.10.006
Yu, J., Ma, M., Ma, Z., and Fu, J. (2016). HDAC6 inhibition prevents TNF-α-induced caspase 3 activation in lung endothelial cell and maintains cell-cell junctions. Oncotarget 7, 54714–54722. doi: 10.18632/oncotarget.10591
Zampetaki, A., Zeng, L., Margariti, A., Xiao, Q., Li, H., Zhang, Z., et al. (2010). Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation 121, 132–142. doi: 10.1161/circulationaha.109.890491
Zelko, I. N., and Folz, R. J. (2015). Regulation of oxidative stress in pulmonary artery endothelium. modulation of extracellular superoxide dismutase and NOX4 expression using histone deacetylase class i inhibitors. Am. J. Respir. Cell Mol. Biol. 53, 513–524. doi: 10.1165/rcmb.2014-0260OC
Zeng, L., Zampetaki, A., Margariti, A., Pepe, A. E., Alam, S., Martin, D., et al. (2009). Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc. Natl. Acad. Sci. U.S.A. 106, 8326–8331. doi: 10.1073/pnas.0903197106
Zeng, L., Zhang, Y., Chien, S., Liu, X., and Shyy, J. Y. (2003). The role of p53 deacetylation in p21Waf1 regulation by laminar flow. J. Biol. Chem. 278, 24594–24599. doi: 10.1074/jbc.M301955200
Zhang, B., Dong, Y., Liu, M., Yang, L., and Zhao, Z. (2019). miR-149-5p inhibits vascular smooth muscle cells proliferation, invasion, and migration by targeting histone deacetylase 4 (HDAC4). Med. Sci. Monit. 25, 7581–7590. doi: 10.12659/msm.916522
Zhang, H. N., Dai, Y., Zhang, C. H., Omondi, A. M., Ghosh, A., Khanra, I., et al. (2020). Sirtuins family as a target in endothelial cell dysfunction: implications for vascular ageing. Biogerontology 21, 495–516. doi: 10.1007/s10522-020-09873-z
Zhang, L., Zhang, J., Jiang, Q., Zhang, L., and Song, W. (2018). Zinc binding groups for histone deacetylase inhibitors. J. Enzyme Inhib. Med. Chem. 33, 714–721. doi: 10.1080/14756366.2017.1417274
Zhang, M. X., Zhang, C., Shen, Y. H., Wang, J., Li, X. N., Chen, L., et al. (2008). Effect of 27nt small RNA on endothelial nitric-oxide synthase expression. Mol. Biol. Cell 19, 3997–4005. doi: 10.1091/mbc.e07-11-1186
Zhang, R., and Ge, J. (2017). Proteinase-activated receptor-2 modulates ve-cadherin expression to affect human vascular endothelial barrier function. J. Cell Biochem. 118, 4587–4593. doi: 10.1002/jcb.26123
Zhao, Q., Yu, Z., Zhang, F., Huang, L., Xing, C., Liu, N., et al. (2019). HDAC3 inhibition prevents oxygen glucose deprivation/reoxygenation-induced transendothelial permeability by elevating PPARgamma activity in vitro. J. Neurochem. 149, 298–310. doi: 10.1111/jnc.14619
Zheng, B., Han, M., Shu, Y. N., Li, Y. J., Miao, S. B., Zhang, X. H., et al. (2011). HDAC2 phosphorylation-dependent Klf5 deacetylation and RARα acetylation induced by RAR agonist switch the transcription regulatory programs of p21 in VSMCs. Cell Res. 21, 1487–1508. doi: 10.1038/cr.2011.34
Zheng, X., Wu, Z., Xu, K., Qiu, Y., Su, X., Zhang, Z., et al. (2019). Interfering histone deacetylase 4 inhibits the proliferation of vascular smooth muscle cells via regulating MEG3/miR-125a-5p/IRF1. Cell Adh. Migr. 13, 41–49. doi: 10.1080/19336918.2018.1506653
Zheng, X. X., Zhou, T., Wang, X. A., Tong, X. H., and Ding, J. W. (2015). Histone deacetylases and atherosclerosis. Atherosclerosis 240, 355–366. doi: 10.1016/j.atherosclerosis.2014.12.048
Zhou, B., Margariti, A., Zeng, L., Habi, O., Xiao, Q., Martin, D., et al. (2011a). Splicing of histone deacetylase 7 modulates smooth muscle cell proliferation and neointima formation through nuclear β-catenin translocation. Arterioscler Thromb. Vasc. Biol. 31, 2676–2684. doi: 10.1161/atvbaha.111.230888
Zhou, B., Margariti, A., Zeng, L., and Xu, Q. (2011b). Role of histone deacetylases in vascular cell homeostasis and arteriosclerosis. Cardiovasc. Res. 90, 413–420. doi: 10.1093/cvr/cvr003
Keywords: atherosclerosis, endothelial dysfunction, smooth muscle cells, macrophage, epigenetic, histone deacetylation, HDAC inhibitors
Citation: Chen X, He Y, Fu W, Sahebkar A, Tan Y, Xu S and Li H (2020). Histone Deacetylases (HDACs) and Atherosclerosis: A Mechanistic and Pharmacological Review. Front. Cell Dev. Biol. 8:581015. doi: 10.3389/fcell.2020.581015
Received: 07 July 2020; Accepted: 14 October 2020;
Published: 12 November 2020.
Edited by:
Nejat Dalay, Istanbul University, TurkeyReviewed by:
Silvio Zaina, University of Guanajuato, MexicoAmy Tsurumi, Massachusetts General Hospital and Harvard Medical School, United States
Copyright © 2020 Chen, He, Fu, Sahebkar, Tan, Xu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suowen Xu, c3h1MTk4NEB1c3RjLmVkdS5jbg==; c3Vvd2VuLnh1QGdtYWlsLmNvbQ==; Hong Li, bGloZ3p1Y21AZm94bWFpbC5jb20=