 Aitor Rodriguez-Casanova1,2,3†
Aitor Rodriguez-Casanova1,2,3† Nicolas Costa-Fraga1,3†
Nicolas Costa-Fraga1,3† Clara Castro-Carballeira4
Clara Castro-Carballeira4 Miriam González-Conde2,5Carmen Abuin2
Miriam González-Conde2,5Carmen Abuin2 Aida Bao-Caamano1,3
Aida Bao-Caamano1,3 Tomás García-Caballero6Elena Brozos-Vazquez7Carmela Rodriguez-López7Victor Cebey7Patricia Palacios7Juan F. Cueva5,7Rafael López-López2,5,7*
Tomás García-Caballero6Elena Brozos-Vazquez7Carmela Rodriguez-López7Victor Cebey7Patricia Palacios7Juan F. Cueva5,7Rafael López-López2,5,7* Clotilde Costa2,5‡
Clotilde Costa2,5‡ Angel Díaz-Lagares1,5*‡
Angel Díaz-Lagares1,5*‡- 1Epigenomics Unit, Cancer Epigenomics, Translational Medical Oncology Group (ONCOMET), Health Research Institute of Santiago de Compostela (IDIS), University Clinical Hospital of Santiago (CHUS/SERGAS), Santiago de Compostela, Spain
- 2Roche-Chus Joint Unit, Translational Medical Oncology Group (ONCOMET), Health Research Institute of Santiago (IDIS), Santiago de Compostela, Spain
- 3Universidade de Santiago de Compostela (USC), Santiago de Compostela, Spain
- 4Department of Oncology, Marqués de Valdecilla University Hospital, Santander, Spain
- 5Centro de Investigación Biomédica en Red Cáncer (CIBERONC), ISCIII, Madrid, Spain
- 6Department of Morphological Sciences, University of Santiago de Compostela and Xerencia de Xestión Integrada de Santiago (XXIS/SERGAS), Santiago de Compostela, Spain
- 7Translational Medical Oncology Group (ONCOMET), Health Research Institute of Santiago de Compostela (IDIS), University Clinical Hospital of Santiago (CHUS/SERGAS), Santiago de Compostela, Spain
Breast cancers of the luminal B subtype are frequent tumors with high proliferation and poor prognosis. Epigenetic alterations have been found in breast tumors and in biological fluids. We aimed to profile the cell-free DNA (cfDNA) methylome of metastatic luminal B breast cancer (LBBC) patients using an epigenomic approach to discover potential noninvasive biomarkers. Plasma cfDNA was analyzed using the Infinium MethylationEpic array in a cohort of 14 women, including metastatic LBBC patients and nontumor controls. The methylation levels of cfDNA and tissue samples were validated with droplet digital PCR. The methylation and gene expression data of 582 primary luminal breast tumors and 79 nontumor tissues were obtained from The Cancer Genome Atlas (TCGA). We found an episignature of 1,467 differentially methylated CpGs that clearly identified patients with LBBC. Among the genes identified, the promoter hypermethylation of WNT1 was validated in cfDNA, showing an area under the ROC curve (AUC) of 0.86 for the noninvasive detection of metastatic LBBC. Both paired cfDNA and primary/metastatic breast tumor samples showed hypermethylation of WNT1. TCGA analysis revealed significant WNT1 hypermethylation in the primary tumors of luminal breast cancer patients, with a negative association between WNT1 methylation and gene expression. In this proof-of-principle study, we discovered an episignature associated with metastatic LBBC using a genome-wide cfDNA methylation approach. We also identified the promoter hypermethylation of WNT1 in cfDNA as a potential noninvasive biomarker for luminal breast cancer. Our results support the use of EPIC arrays to identify new epigenetic noninvasive biomarkers in breast cancer.
Introduction
Breast cancer (BC) is the most frequently diagnosed cancer in women worldwide, with 2.3 million new cases (11.7% of all cancer cases) in 2020, representing the leading cause of cancer death in women (Sung et al., 2021). BC is a heterogeneous disease with several distinct clinical characteristics that, according to a gene expression profile, can be divided into four molecular subtypes: luminal, HER2-enriched, basal-like, and normal breast-like (Perou et al., 2000). In addition, luminal tumors can be divided into the luminal A and B subtypes according to the expression profile of the estrogen receptor (ER), progesterone (PR), HER2, and proliferation tumor status (Cheang et al., 2009). The luminal B subtype is a common BC subtype characterized by high proliferation, resistance to standard therapies, risk of early relapse, and poor prognosis (Tran and Bedard, 2011; Cornen et al., 2014; Li et al., 2016). In addition, this tumor subtype is more likely to exhibit local recurrence and single bone metastases than nonluminal BC. However, recent studies have not investigated this tumor subtype as thoroughly as other subtypes (Li et al., 2016). Notably, the incidence of luminal B tumors has increased in recent years in many racial/ethnic and age groups (Acheampong et al., 2020).
Cancer metastasis is characterized by highly variable clinical manifestations and is responsible for over 90% of cancer-related deaths (Gupta and Massague, 2006; Chaffer and Weinberg, 2011). However, despite recent advances, the clinical need to identify biomarkers in metastatic BC disease remains unmet (Gupta et al., 2020). In recent years, liquid biopsy has emerged as a good opportunity to address this clinical need. This noninvasive approach allows for the characterization of the molecular landscape of circulating tumor elements in body fluids, such as epigenetic modifications of cell-free DNA (cfDNA), to obtain biomarkers for the management of cancer patients (Siravegna et al., 2017).
The most well-known epigenetic modification is DNA methylation, which is an important regulator of gene expression originating from the addition of a methyl group (CH3) to the 5’ carbon of cytosines in cytosine–phosphate–guanine (CpG) dinucleotides (Bao-Caamano et al., 2020). The deregulation of this epigenetic mechanism in breast tumor cells has major implications for cancer development, progression, and therapy response (Gomez-Miragaya et al., 2019; Martin-Pardillos et al., 2019; Palomeras et al., 2019; Pineda et al., 2019). Notably, the analysis of DNA methylation in liquid biopsy has shown utility as a potential clinical biomarker for BC patients (Palanca-Ballester et al., 2021).
Recently published studies of other tumor types have shown that epigenomic approaches based on the Infinium MethylationEPIC array (EPIC array) technology, which covers over 850,000 CpG sites along the human genome, could be useful to profile the methylation of cfDNA in biological fluids (Gallardo-Gomez et al., 2018; Herrgott et al., 2022). Therefore, this proof-of-principle study aimed to profile the cfDNA methylome of luminal B breast cancer (LBBC) patients using an EPIC array approach to discover new noninvasive biomarkers. In this study, we identified an epigenetic signature (episignature) based on the methylation of cfDNA associated with metastatic LBBC. Among the genes of this episignature, we confirmed the hypermethylation of WNT1 in cfDNA and tumor tissues (primary and metastatic) as a potential new biomarker for LBBC patients. The results of our work support the application of the EPIC array technology as a noninvasive tool to identify new biomarkers in breast cancer.
Methods
Study participants
In this retrospective study, 9 luminal B metastatic breast cancer patients and 5 healthy controls (nontumor controls) were recruited between 2016 and 2018 at the Medical Oncology Department at the University Clinical Hospital of Santiago de Compostela (Spain). Most of the metastatic patients of this study (7 out of 9) had been diagnosed in the past at M0 stage. Two patients of our cohort had metastases at the time of primary tumor diagnosis. The study was approved by the Galician Ethical Committee (reference number 2015/772) and conducted in accordance with the guidelines for Good Clinical Practice and the Declaration of Helsinki. All participants included in the study signed the informed consent to participate.
Blood and tissue samples
Blood sample was obtained from all the patients at the time of diagnosis of metastasis and before starting the treatment. Blood samples were collected by phlebotomy into collection tubes containing K2EDTA as an anticoagulant. Plasma was isolated within 2 h of collection by initial centrifugation at 1,700 × g for 10 min at room temperature (RT), followed by a second centrifugation at 15,000 × g for 10 min at RT. Isolated plasma was stored at −80°C until analysis. All tumor tissues used were obtained according to standard-of-care (SOC) procedures. We used formalin-fixed and paraffin-embedded (FFPE) primary and/or metastatic tumor and matched nontumor tissue samples available from 4 patients included in the study. Whole slide FFPE tissue sections of 10 μm were obtained.
Isolation of DNA from plasma and tissue samples
We used the QIAamp® Circulating Nucleic Acid Kit (Qiagen) and the vacuum system QIAvac 24 Plus (Qiagen) following the manufacturer´s recommendations to isolate cfDNA from 2 ml of plasma. DNA was also isolated from 10-μm FFPE tissue sections using the AllPrep DNA/RNA FFPE Kit (Qiagen) following the manufacturer’s protocol. The quality and quantity of DNA from FFPE tissue sections were evaluated with a NanoDrop (Thermo Fisher), and cfDNA was quantified using the Qubit 1× dsDNA High-Sensitivity Assay Kit and a Qubit 4.0 Fluorometer (Thermo Fisher Scientific). The DNA from FFPE tissue sections and cfDNA from plasma were stored at −80°C until analysis.
Genome-wide cell-free DNA methylation analysis
Fifteen nanograms of each individual sample of plasma cfDNA was bisulfite-converted using the EZ DNA Methylation Lightning Kit (Zymo Research) following the manufacturer’s recommendations. Subsequently, the bisulfite-modified cfDNA was then subjected to whole genome amplification (WGA) using the EpiTect Whole Bisulfitome Kit (Qiagen) according to the manufacturer’s protocol. Briefly, the bisulfite-modified cfDNA of each individual sample was amplified with a reaction buffer containing REPLI-g Midi DNA Polymerase (Qiagen) at 28°C for 8 h, which was subsequently inactivated at 95 °C for 5 min. After the WGA of cfDNA, the Illumina Infinium HD methylation protocol was followed using MethylationEPIC BeadChips that were analyzed in a HiScan (Illumina). Samples with a mean detection p-value < 0.01 were considered valid for the analysis. The methylation data were processed in the R statistical environment using RnBeads 2.0 (Muller et al., 2019). Raw intensity data files (IDATs) were imported into RnBeads 2.0 for quality control and preprocessing. First, a greedycut algorithm was used to filter out low-quality probes. Probes overlapping with SNPs and probes whose sequences mapped to multiple genomic locations (cross-reactive) were removed. IDATs obtained in the array were normalized using the beta-mixture quantile (BMIQ) method. Hierarchical linear models were used to obtain the methylation differences between groups. p-values were corrected for multiple testing using the Benjamini‒Hochberg method, and a false discovery rate (FDR) < 10% was selected for significance. The DNA methylation level was represented as the average β-value, which was calculated as the ratio of the fluorescent signal intensity of the methylated probe to those of total (methylated and unmethylated) probes. Average β-values were used to calculate the mean methylation difference between groups as the Δβ-value (β-value Luminal B – β-value Control). An unsupervised hierarchical clustering heatmap of β-values was generated using the ComplexHeatmap package. Gene ontology (GO) enrichment analysis of biological pathways from the PANTHER database was performed using GENECODIS (Tabas-Madrid et al., 2012).
Methylation and expression analysis from The Cancer Genome Atlas
The DNA methylation (β-values) and expression data of WNT1 in luminal primary breast tumors and nontumor controls were obtained from the public datasets of The Cancer Genome Atlas (TCGA) Cancer Genome Atlas, N. (2012). The breast cancer subtype of patients was obtained from the clinical information available at TCGA and the classification of these TCGA patients based on PAM50 assay performed by Netanely et al. (2016).
Methylation analysis of the WNT1 promoter in cell-free DNA by droplet digital PCR
The methylation of the WNT1 promoter was analyzed by droplet digital PCR (ddPCR) in a QX200 system (Bio-Rad). Twenty nanograms of plasma cfDNA and 30 ng of DNA from FFPE tissue samples were bisulfite converted using the EZ DNA Methylation Lightning Kit (Zymo Research) following the manufacturer’s recommendations. A custom Bio-Rad assay to detect the methylation status of WNT1 (cg27196808) was designed: WNT1-M for methylation and WNT1-U for unmethylation (Supplementary Table S1). A multiplex preamplification reaction was performed with ∼2 ng of bisulfite-converted DNA using SsoAdvanced™ PreAmp Supermix (Bio-Rad), WNT1-M, and WNT1-U. The PCR conditions were as follows: 3 min at 95°C, 10 cycles of 95°C for 15 s and 50.6°C for 4 min, and a final hold step at 4°C. Next, a multiplex reaction mix was prepared with 2 µL of the preamplification product using ddPCR Supermix for Probes (No dUTP) (Bio-Rad), WNT1-M, and WNT1-U. The QX200™ Droplet Generator (Bio-Rad) was used to generate droplets. The thermocycling conditions were as follows: 10 min at 95°C, 40 cycles of 95°C for 15 s and 50.6°C for 30 s, 98°C for 10 min, and a final hold step at 4°C. The temperature ramp increment was 2.5°C/s for all steps. Droplets were counted and analyzed using the QX200™ Droplet Reader system (Bio-Rad), and the QuantaSoft analysis software (Bio-Rad) was used to acquire data. Water was included as a no-template control, and the Human Methylated and Non-Methylated DNA set (Zymo Research) was used as a positive control for methylation and unmethylation. Reactions were performed in triplicate. DNA methylation was expressed according to the following formula: Methylation (%) = [M/(U + M)] x 100, where M represents the copies/μl of methylated cfDNA, and U the copies/μl of unmethylated cfDNA.
Statistical analysis
The Kolmogorov‒Smirnov test was used to evaluate the normality of the distribution of the data. The nonparametric Mann‒Whitney U test was used to compare methylation data. To assess the diagnostic accuracy, a receiver operating characteristic (ROC) curve was generated. The greatest combination of sensitivity and specificity was obtained using the Youden index (J): J = sensitivity + specificity - 1. The association between DNA methylation and gene expression was evaluated with a Spearman correlation. The GraphPad Prism 6.0 software (GraphPad Software) and the R statistical environment (version 4.2.0) were used for statistical analysis and graphical representation. All expressed p-values were calculated with two-tailed tests and were considered significant when the p-value < 0.05.
Results
Clinical characteristics of patients
A retrospective cohort of 14 women was included in this study: 9 patients with LBBC at the time of metastatic disease diagnosis and 5 nontumor controls. The mean age of the patients was 66 ± 16 years, whereas the control group had a mean age of 53 ± 10 years. The main clinical characteristics of the analyzed cohort are described in Supplementary Table S2. All patients had distant metastases and invasive ductal carcinoma with a high Ki-67 proliferative index (≥20%), and they were positive for estrogen receptors (ER+). Eight out of nine patients were positive for progesterone receptors (PR+), and two patients had HER2 overexpression (Supplementary Table S2). Six out of the 9 patients (66%) included in the study had lung metastasis, 4 patients (44%) showed bone lesions, and 3 patients (33%) had liver affectation. In addition, 5 of the patients (55%) had multiple metastatic locations.
Genome-wide cell-free DNA methylation analysis of metastatic patients with luminal B breast cancer
The analysis of DNA methylome with the EPIC array methodology usually needs a high amount of DNA, which is difficult to obtain in the clinic from individual plasma samples. As a novelty in our study, to overcome this limitation, we have used small amounts of cfDNA from individual plasma samples, which were genome-wide amplified after bisulfite modification and then analyzed using EPIC arrays. Thus, using this approach we performed a genome-wide cell-free DNA methylation analysis in our cohort of 9 LBBC patients and 5 nontumor controls (Figure 1A). After hybridizing the samples in the EPIC array, 2 LBBC samples showed a mean detection p-value > 0.01 and were not considered valid for the analysis. Therefore, we ultimately compared the methylation status of cfDNA in 7 LBBC patients and 5 nontumor controls, leading to 28,799 differentially methylated CpGs (DMCpGs) (p < 0.05; FDR < 10%) between LBBC and nontumor controls. These DMCpGs showed a wide distribution throughout all chromosomes of the genome (Figure 1B). Of these DMCpGs, 92% (26,486) were hypomethylated and 8% (2,313) were hypermethylated in LBBC patients with respect to nontumor controls (Figure 1C). Most of the hypomethylated CpGs were distributed in regions with low CpG density (open sea) (Figure 1D) and outside promoter regions (Figure 1E), whereas hypermethylated CpGs were mainly located in CpG islands (CpGIs) (Figure 1F) and promoters (Figure 1G).
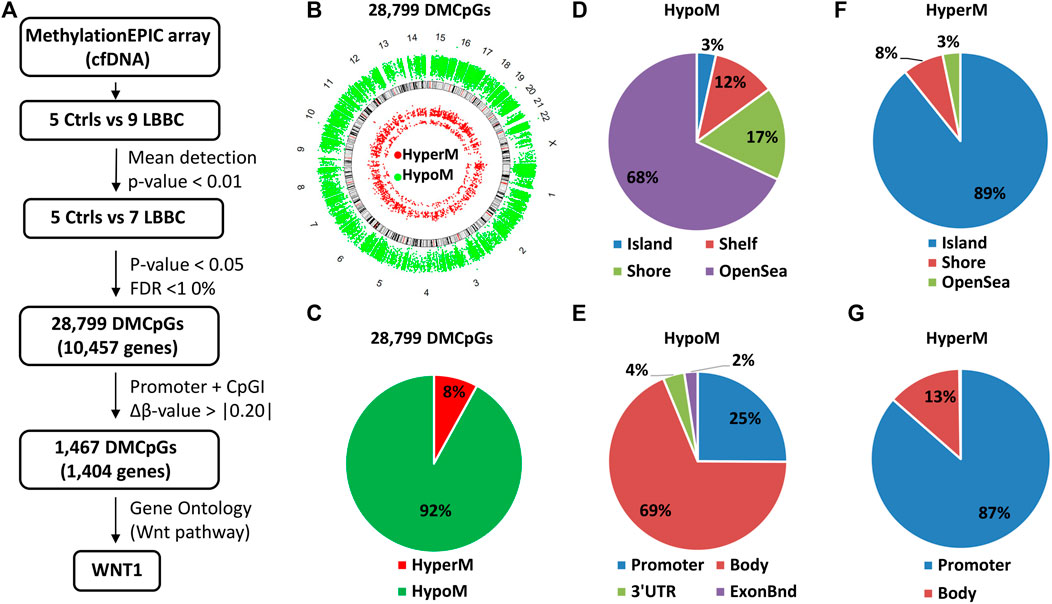
FIGURE 1. Methylation landscape of cell-free DNA in metastatic patients with luminal B breast cancer. (A) Flowchart of the cfDNA methylation analysis. Individual cfDNA samples from controls and luminal B breast cancer patients were analyzed with EPIC array. (B–G) Description of the 28,799 differentially methylated CpGs (DMCpGs) found in cfDNA of luminal B breast cancer patients according to (B) chromosome location, (C) methylation status, (D,F) CpG context, and (E,G) gene location. cfDNA, cell-free DNA; Ctrls, controls; LBBC, luminal B breast cancer; FDR, false discovery rate; HypoM, hypomethylated; HyperM, hypermethylated.
Identification of a cell-free DNA episignature in metastatic patients with luminal B breast cancer
The aberrant hypermethylation of CpGI promoters is a very relevant feature that usually occurs in tumor cells (Baylin and Chen, 2005). Therefore, we focused our study on analyzing the methylation profile of cfDNA at the CpGIs of promoters. In these regions of cfDNA, we identified 1,467 DMCpGs (p < 0.05; FDR < 10%) with a difference in methylation (Δβ-value) higher than 0.20 (Δβ-value > |0.20|) (Figure 1A). Notably, this epigenetic signature (episignature) of 1,467 DMCpGs was able to clearly differentiate LBBC patients from nontumor controls (Figure 2A). Next, to obtain information related to the functional pathways involved in the identified episignature, we performed a gene ontology (GO) enrichment analysis based on the PANTHER database. This analysis revealed that methylation differences in the cfDNA of LBBC patients and nontumor controls were mainly associated with genes related to the Wnt signaling pathway (Figure 2B). Table 1 shows the 34 DMCpGs (corresponding to 24 genes) of the episignature of cfDNA that are associated with the Wnt signaling pathway. Relevantly, the genes of these 34 DMCpGs that are associated with Wnt signaling belonged to a network significantly enriched in protein interactions (p < 0.001) according to a STRING analysis (Supplementary Figure S1).
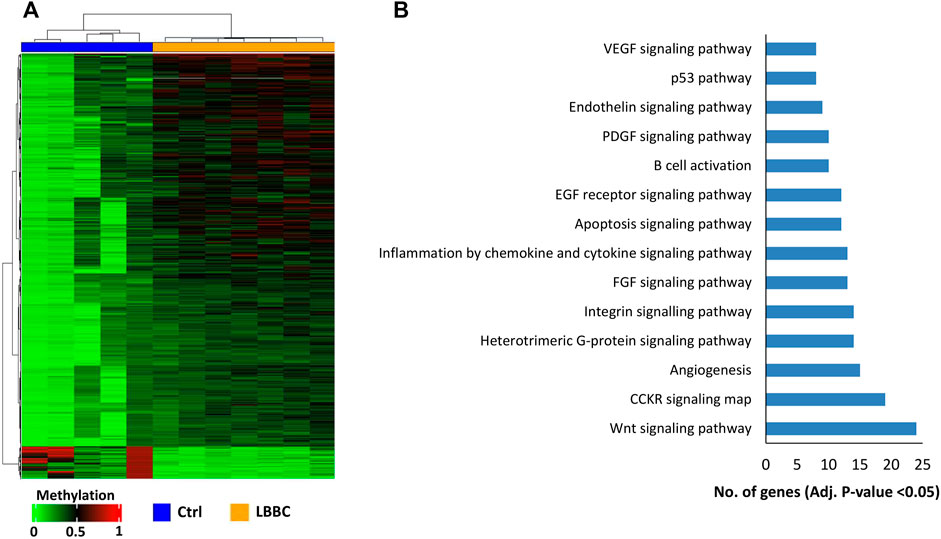
FIGURE 2. Episignature of cell-free DNA in metastatic patients with luminal B breast cancer. (A) Unsupervised hierarchical clustering heatmap of the episignature (1,467 DMCpGs) obtained in cfDNA that differentiates LBBC patients (n = 7) from nontumor controls (n = 5). (B) Gene Ontology enrichment analysis by the PANTHER database, showing the most representative pathways associated with the episignature of cfDNA in luminal B breast cancer patients. Ctrls, controls; LBBC, luminal B breast cancer; DMCpGs, differentially methylated CpGs; cfDNA, cell-free DNA.
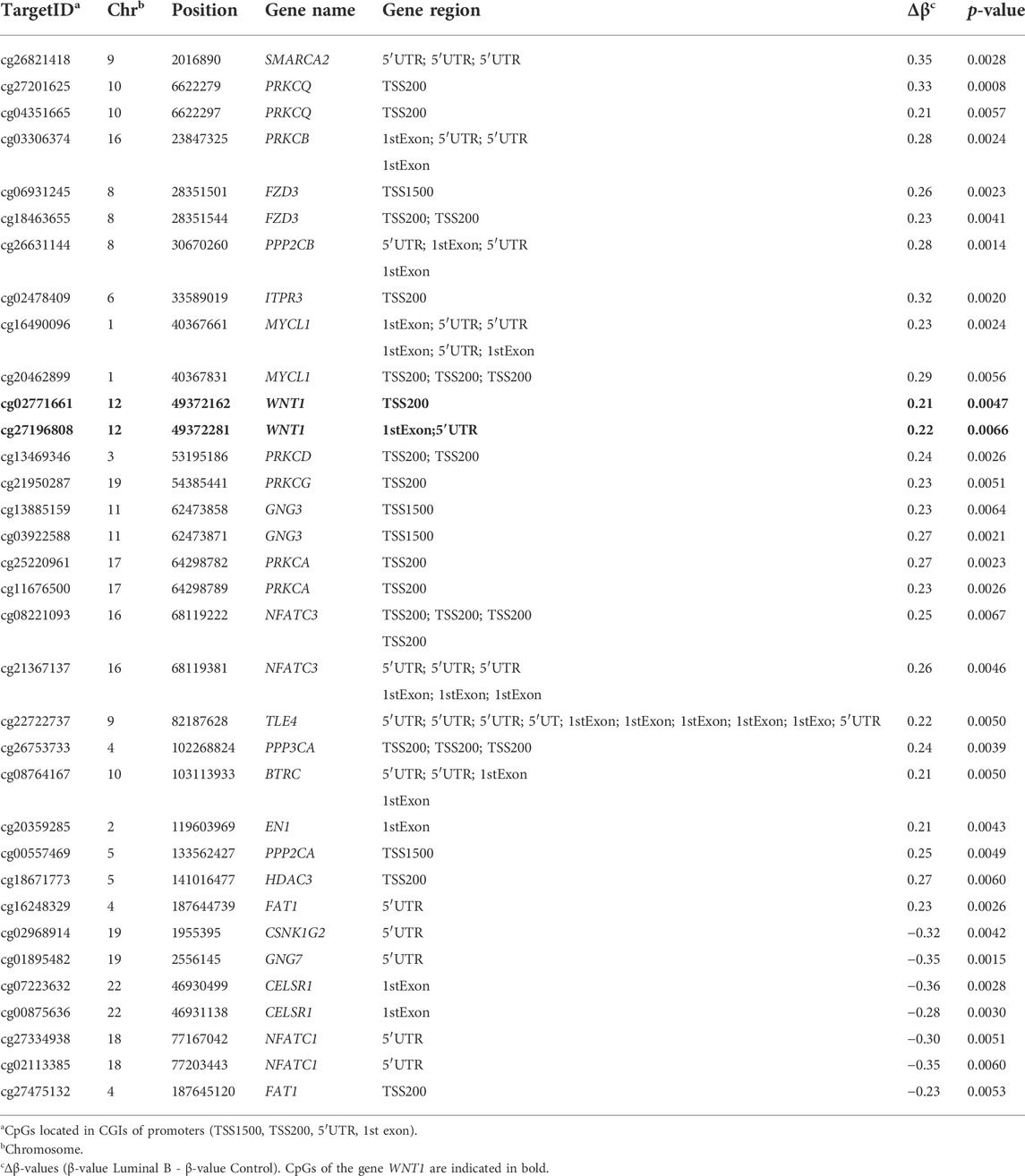
TABLE 1. The 34 CpGs of cfDNA episignature found in metastatic patients with luminal B breast cancer associated with the Wnt signaling pathway.
Hypermethylation of the WNT1 promoter in the cell-free DNA and tumor samples of patients with luminal B breast cancer
Among the DMCpGs in the episignature obtained from the cfDNA of LBBC patients that were associated with the Wnt signaling pathway (Table 1), we found 2 CpGs (cg27196808 and cg02771661) located in WNT1 that were hypermethylated in the cfDNA of LBBC patients with respect to nontumor controls. To confirm this aberrant methylation, we selected the most DMCpG of WNT1 (cg27196808), and we analyzed its methylation status in the cfDNA of our cohort using ddPCR. As expected, the methylation of WNT1 was significantly higher in LBBC patients than in nontumor controls (Figure 3A). In addition, using a ROC curve analysis, the methylation status of the WNT1 promoter analyzed by ddPCR accurately differentiated LBBC patients from nontumor controls, with an area under the ROC curve (AUC) of 0.86 (95% CI: 0.65–1.00, p = 0.045) (Figure 3B), a sensitivity of 78% (CI 95%: 40%–98%), and a specificity of 100% (CI 95%: 40%–100%).
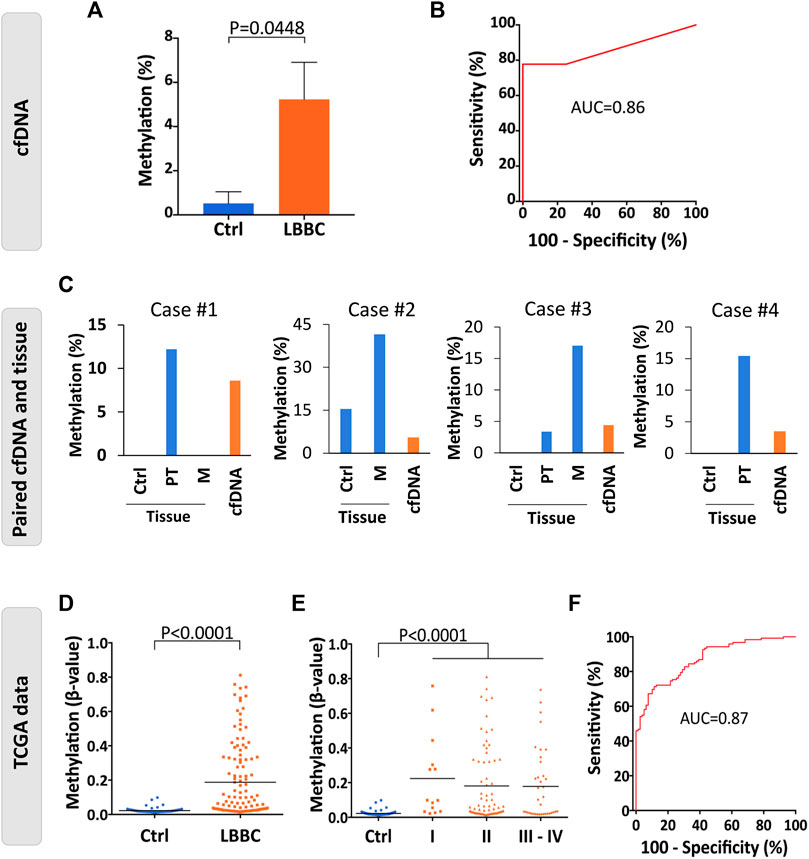
FIGURE 3. Methylation status of the WNT1 promoter in the cfDNA and tumor samples of patients with luminal B breast cancer. (A) Validation of the methylation levels of the WNT1 promoter (cg27196808) in cell-free DNA (cfDNA) of luminal B breast cancer patients (n = 9) and nontumor controls (n = 4) analyzed by droplet digital PCR (ddPCR). Methylation levels are represented as the mean ± SEM. (B) Diagnostic accuracy of the methylation of the WNT1 promoter using droplet digital PCR (ddPCR) in cfDNA for the detection of metastatic luminal B breast cancer patients (n = 9) with respect to nontumor controls (n = 4). (C) Methylation levels of WNT1 in cfDNA and paired breast primary and/or metastatic tumor samples of 4 luminal B breast cancer patients analyzed by droplet digital PCR (ddPCR). Nontumor tissues from the same patients were used as controls. (D–E) Methylation status of WNT1 in primary tumors of luminal B breast cancer patients (n = 122) and nontumor controls (n = 79) analyzed by 450K methylation array and obtained from TCGA considering (D) all TNM stages together (stages I–IV, n = 122) or (E) separated according to TNM stage (I, n = 14; II, n = 70; III-IV, n = 37). The horizontal line represents the mean methylation values. (F) ROC curve evaluating the methylation of WNT1 to detect primary tumors of luminal B breast cancer patients (stages I-IV, n = 122) with respect to nontumor controls (n = 79) from TCGA. Ctrl, control; P, p-value; AUC, area under the ROC curve; ROC curve, receiver operating characteristic curve. PT, primary tumor; M, metastasis.
To verify that the hypermethylation of WNT1 found in cfDNA (cg27196808) was also present in the tumor tissues of patients with LBBC, we assessed its methylation status by ddPCR in the available matched primary and/or metastatic tumor tissue samples (n = 4) of our cohort. This assay revealed that WNT1 hypermethylation was present not only in the cfDNA but also in the paired primary and/or metastatic tumor samples of the LBBC patients analyzed (Figure 3C). Next, we took advantage of public DNA methylation array data from The Cancer Genome Atlas (TCGA) to evaluate whether the hypermethylation of the WNT1 promoter was a frequent event in LBBC. This analysis showed that the methylation of the WNT1 promoter (cg27196808) was significantly higher in luminal B primary tumors (stages I–IV) than in nontumor controls (Figure 3D), and this observation was consistent across all TNM tumor stages (Figure 3E). However, the methylation status of WNT1 did not differ among the tumor stages of LBBC analyzed (Figure 3E). An ROC curve analysis showed that WNT1 methylation differentiated luminal B primary tumors (stages I-IV) from nontumor controls with high diagnostic accuracy, with an AUC of 0.87 (95% CI: 0.82–0.92, p < 0.0001) (Figure 3F). In addition, we also analyzed the WNT1 expression data (RNAseq) available from LBBC patients and nontumor controls included in TCGA, revealing that WNT1 was significantly downregulated in this BC subtype (Supplementary Figure S2).
Finally, we also evaluated in breast primary tumors from TCGA whether the hypermethylation of WNT1 was a specific event of LBBC patients. The methylation levels of WNT1 were significantly higher in LBBC than in the other breast tumor subtypes (Supplementary Figure S3). Interestingly, we observed that WNT1 was also significantly hypermethylated in other breast cancer subtypes (Luminal A, triple negative and HER2+) in comparison with nontumor controls.
Discussion
Alterations in epigenetic mechanisms, such as aberrant DNA methylation, are implicated in the development, progression, and therapy response of BC (Palomeras et al., 2019; Pineda et al., 2019; Glodzik et al., 2020). In recent years, the methylation analysis of liquid biopsy samples in BC patients has shown clinical utility as a biomarker for the detection, prognosis, and monitoring of the disease (Hoque et al., 2006; Mastoraki et al., 2018; Palanca-Ballester et al., 2021). However, a clinical need to find new noninvasive biomarkers associated with metastatic BC subtypes persists (Gupta et al., 2020). Herein, we focused our study on patients with advanced LBBC, since it represents a frequent, aggressive and poor prognosis BC subtype (Creighton, 2012). The characterization of liquid biopsy samples using epigenomic tools for genome-wide methylation analyses has been recently proposed as a good approach to discover new noninvasive biomarkers (Shen et al., 2018). Thus, we used a genome-wide DNA methylation approach based on the EPIC array methodology to profile the methylome of cfDNA and discover novel noninvasive biomarkers in metastatic LBBC patients.
Our work revealed that the cfDNA of patients with metastatic LBBC is characterized by the hypomethylation of regions with a low density of CpGs and the site-specific hypermethylation of CpGIs in promoter regions. Importantly, this pattern is similar to the deregulation of DNA methylation that has been previously described in cancer cells (Nishiyama and Nakanishi, 2021), suggesting that the methylation profile in the cfDNA of the patients in our cohort mirrors the epigenetic alterations of BC cells.
Specific genes, such as RASSF1A and BRCA1, have been previously described to exhibit aberrant hypermethylation of their promoter CpGIs in BC (Rice et al., 1998; Dammann et al., 2001). Accordingly, we focused our study on promoter CpGIs and were able to identify a novel noninvasive episignature in cfDNA based on 1,467 CpGs that was associated with LBBC patients. We found that the genes of this episignature were related to relevant biological pathways, mainly Wnt signalling pathway. Among these genes, we focused on WNT1, which is involved in the canonical Wnt signaling pathway (also known as Wnt/β-Catenin) in cancer cells (Ayyanan et al., 2006; Mehta et al., 2021). Thus, we confirmed that the promoter CpGI of WNT1 was hypermethylated in the cfDNA of patients with metastatic LBBC and that this aberrant methylation showed a high diagnostic accuracy to detect this BC subtype, suggesting that the hypermethylation of WNT1 could be a suitable biomarker for cancer detection and monitoring of metastatic patients. In line with this, it has been recently shown that methylation biomarkers of cfDNA with high diagnostic accuracy are useful not only for diagnosis but also for monitoring tumor burden dynamics under different therapeutic regimens in advanced disease (Barault et al., 2018). Importantly, evaluating prognosis and monitoring tumor response in real time during treatment continues to be an unmet clinical need in advanced BC (Gupta et al., 2020). Wnt signaling is a very relevant pathway in BC whose molecular alterations have clinical implications to establish the prognosis of the disease (Mukherjee et al., 2012) and has been associated to breast cancer therapy response (Abreu de Oliveira et al., 2022). Therefore, it would be interesting to evaluate in future studies whether the hypermethylation of WNT1 could be useful for the selection of patients susceptible to systemic therapies (CDK inhibitors for example) in the BC metastatic setting.
Of note, we also found that the promoter hypermethylation of WNT1 was present not only in cfDNA of LBBC patients but also in their primary and/or metastatic tumors. This finding is in accordance with our previous work and that of other authors showing that the molecular landscape present in liquid biopsy may also be detected in the corresponding tumor tissue of patients (Rahvar et al., 2020; Rodriguez-Casanova et al., 2021). In addition, when we extended our study to the analysis of breast primary tumors using the public TCGA database, we were able to confirm that the hypermethylation of WNT1 is a frequent event in early and advanced LBBC, suggesting that the epigenetic deregulation of WNT1 is not a specific biomarker of metastatic disease but rather a biomarker of breast cancer cells. In agreement with our results, the aberrant methylation of other genes involved in the Wnt signaling pathway (e.g., WNT5A and WNT7A) has previously been described in tumor cells from the BC luminal subtype (Shan et al., 2019) and in other tumor types, such as gastric cancer or chronic lymphocytic leukemia (Liu et al., 2019; Poppova et al., 2022).
Several studies have shown that aberrant promoter hypermethylation is a relevant mechanism that is able to repress the expression of key genes in breast tumor cells (Alvarez et al., 2013; Palomeras et al., 2019). Accordingly, the analysis of luminal breast tumors from the TCGA database also revealed that WNT1 promoter hypermethylation was associated with a downregulation of its gene expression in primary tumors, suggesting that WNT1 is epigenetically regulated in luminal BC. The downregulation of WNT1 observed in particular BC subtypes corroborates the work by Koval and Katanaev (2018), who reported low expression of this gene in primary tumors of nontriple-negative BC patients (ER+/PR + and HER+). Indeed, it has been reported that the deregulation of some Wnt signaling components depends on the BC subtype, with many being downregulated in the luminal B subtype (Smid et al., 2008).
One limitation of our study is that the epigenomic profiling of cfDNA performed is based on a retrospective cohort of patients with a small sample size. Although the results obtained in this work should be taken with caution, they provide the basis for further large, prospective and independent studies that validate the clinical utility of the potential epigenetic biomarkers identified herein. In addition, it would be interesting to evaluate in future works the implications of the epigenetic deregulation of WNT1 in the development of metastasis.
In summary, in this proof-of-principle study, we discovered an episignature associated with patients with advanced LBBC using a genome-wide cfDNA methylation approach. We also identified the promoter hypermethylation of WNT1 in cfDNA as a potential noninvasive biomarker for luminal BC. Our results support the use of EPIC array technology to identify new noninvasive biomarkers in BC.
Data availability statement
The methylation data of cfDNA analyzed in this study with EPIC Array are available at the NCBI GEO repository with accession number GSE214344.
Ethics statement
The studies involving human participants were reviewed and approved by Galician Ethical Committee (reference number 2015/772). The patients/participants provided their written informed consent to participate in this study.
Author contributions
Conceptualization, CC, RL-L, and AD-L; resources, TG-C, EB-V, CR-L, VC, PP, JFC, and RL-L; Data curation, AR-C, NC-F, and CA; methodology, AR-C, NC-F, CC-C, MG-C, CA, AB-C, and TG-C; formal analysis, AR-C, NC-F, CC, and AD-L; writing—original draft preparation, AR-C, NC-F, CC, and AD-L; writing—review and editing, AR-C, NC-F, JFC, RL-L, CC, and AD-L; supervision, CC, RL-L, and AD-L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by “Beca FSEOM/FECMA para Proyectos de Investigación en Cáncer de Mama 2017”, the Roche-Chus Joint Unit (IN853B 2018/03), “Axencia Galega de Innovación (GAIN), Vicepresidencia Segunda e Consellería de Economía, Empresa e Innovación”. AR-C is supported by the Roche-Chus Joint Unit (IN853B 2018/03) funded by GAIN, “Axencia Galega de Innovación (GAIN), Vicepresidencia Segunda e Consellería de Economía, Empresa e Innovación”. NC-F is funded by a predoctoral contract from “Xunta de Galicia” (IN606A-2020/004). AB-C is funded by a predoctoral contract PFIS (FI19/00240) from “Instituto de Salud Carlos III” (ISCIII) co-funded by “Fondo Social Europeo” (FSE). MG-C is supported by “Instituto de Salud Carlos III” (ISCIII), with the grant FI19/00140, co-financed by the European Regional Development Fund (FEDER). CC is supported by AECC (INVES211437COST). AD-L is funded by a contract “Juan Rodés” (JR17/00016) from “Instituto de Salud Carlos III” (ISCIII).
Acknowledgments
We would like to thank all the patients and healthy volunteers included in this study for their kind collaboration, and all personnel who provided technical support with sample collection. We also thank María Amalia Jácome, from MODES Group (CITIC, Faculty of Science, Universidade da Coruña), for the support with DNA methylome analysis.
Conflict of interest
RL-L has received honoraria for participation in Advisory Boards from Roche, AstraZeneca, Merck, Merck Sharp and; Dohme, Bayer, Bristol-Myers Squibb, Novartis, Janssen, Lilly, Pfizer, and Leo; travel, accommodations, and expenses from PharmaMar, Roche, Bristol-Myers Squibb, and Pierre Fabre; research funding from Roche and Merck; and is co-founder and shareholder in Nasasbiotech, S.L., Mtrap Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.1016955/full#supplementary-material
References
Abreu de Oliveira, W. A., El Laithy, Y., Bruna, A., Annibali, D., and Lluis, F. (2022). Wnt signaling in the breast: From development to disease. Front. Cell Dev. Biol. 10, 884467. doi:10.3389/fcell.2022.884467
Acheampong, T., Kehm, R. D., Terry, M. B., Argov, E. L., and Tehranifar, P. (2020). Incidence trends of breast cancer molecular subtypes by age and race/ethnicity in the US from 2010 to 2016. JAMA Netw. Open 3 (8), e2013226. doi:10.1001/jamanetworkopen.2020.13226
Alvarez, C., Tapia, T., Cornejo, V., Fernandez, W., Munoz, A., Camus, M., et al. (2013). Silencing of tumor suppressor genes RASSF1A, SLIT2, and WIF1 by promoter hypermethylation in hereditary breast cancer. Mol. Carcinog. 52 (6), 475–487. doi:10.1002/mc.21881
Ayyanan, A., Civenni, G., Ciarloni, L., Morel, C., Mueller, N., Lefort, K., et al. (2006). Increased Wnt signaling triggers oncogenic conversion of human breast epithelial cells by a Notch-dependent mechanism. Proc. Natl. Acad. Sci. U. S. A. 103 (10), 3799–3804. doi:10.1073/pnas.0600065103
Bao-Caamano, A., Rodriguez-Casanova, A., and Diaz-Lagares, A. (2020). Epigenetics of circulating tumor cells in breast cancer. Adv. Exp. Med. Biol. 1220, 117–134. doi:10.1007/978-3-030-35805-1_8
Barault, L., Amatu, A., Siravegna, G., Ponzetti, A., Moran, S., Cassingena, A., et al. (2018). Discovery of methylated circulating DNA biomarkers for comprehensive non-invasive monitoring of treatment response in metastatic colorectal cancer. Gut 67 (11), 1995–2005. doi:10.1136/gutjnl-2016-313372
Baylin, S. B., and Chen, W. Y. (2005). Aberrant gene silencing in tumor progression: Implications for control of cancer. Cold Spring Harb. Symp. Quant. Biol. 70, 427–433. doi:10.1101/sqb.2005.70.010
Cancer Genome Atlas, N. (2012). Comprehensive molecular portraits of human breast tumours. Nature 490 (7418), 61–70. doi:10.1038/nature11412
Chaffer, C. L., and Weinberg, R. A. (2011). A perspective on cancer cell metastasis. Science 331 (6024), 1559–1564. doi:10.1126/science.1203543
Cheang, M. C., Chia, S. K., Voduc, D., Gao, D., Leung, S., Snider, J., et al. (2009). Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J. Natl. Cancer Inst. 101 (10), 736–750. doi:10.1093/jnci/djp082
Cornen, S., Guille, A., Adelaide, J., Addou-Klouche, L., Finetti, P., Saade, M. R., et al. (2014). Candidate luminal B breast cancer genes identified by genome, gene expression and DNA methylation profiling. PLoS One 9 (1), e81843. doi:10.1371/journal.pone.0081843
Creighton, C. J. (2012). The molecular profile of luminal B breast cancer. Biologics 6, 289–297. doi:10.2147/BTT.S29923
Dammann, R., Yang, G., and Pfeifer, G. P. (2001). Hypermethylation of the cpG island of Ras association domain family 1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3 locus, occurs in a large percentage of human breast cancers. Cancer Res. 61 (7), 3105–3109.
Gallardo-Gomez, M., Moran, S., Paez de la Cadena, M., Martinez-Zorzano, V. S., Rodriguez-Berrocal, F. J., Rodriguez-Girondo, M., et al. (2018). A new approach to epigenome-wide discovery of non-invasive methylation biomarkers for colorectal cancer screening in circulating cell-free DNA using pooled samples. Clin. Epigenetics 10, 53. doi:10.1186/s13148-018-0487-y
Glodzik, D., Bosch, A., Hartman, J., Aine, M., Vallon-Christersson, J., Reutersward, C., et al. (2020). Comprehensive molecular comparison of BRCA1 hypermethylated and BRCA1 mutated triple negative breast cancers. Nat. Commun. 11 (1), 3747. doi:10.1038/s41467-020-17537-2
Gomez-Miragaya, J., Moran, S., Calleja-Cervantes, M. E., Collado-Sole, A., Pare, L., Gomez, A., et al. (2019). The altered transcriptome and DNA methylation profiles of docetaxel resistance in breast cancer PDX models. Mol. Cancer Res. 17 (10), 2063–2076. doi:10.1158/1541-7786.MCR-19-0040
Gupta, G., Lee, C. D., Guye, M. L., Van Sciver, R. E., Lee, M. P., Lafever, A. C., et al. (2020). Unmet clinical need: Developing prognostic biomarkers and precision medicine to forecast early tumor relapse, detect chemo-resistance and improve overall survival in high-risk breast cancer. Ann. Breast Cancer Ther. 4 (1), 48–57. doi:10.36959/739/525
Gupta, G. P., and Massague, J. (2006). Cancer metastasis: Building a framework. Cell 127 (4), 679–695. doi:10.1016/j.cell.2006.11.001
Herrgott, G. A., Asmaro, K. P., Wells, M., Sabedot, T. S., Malta, T. M., Mosella, M. S., et al. (2022). Detection of tumor-specific DNA methylation markers in the blood of patients with pituitary neuroendocrine tumors. Neuro. Oncol. 24 (7), 1126–1139. doi:10.1093/neuonc/noac050
Hoque, M. O., Feng, Q., Toure, P., Dem, A., Critchlow, C. W., Hawes, S. E., et al. (2006). Detection of aberrant methylation of four genes in plasma DNA for the detection of breast cancer. J. Clin. Oncol. 24 (26), 4262–4269. doi:10.1200/JCO.2005.01.3516
Koval, A., and Katanaev, V. L. (2018). Dramatic dysbalancing of the Wnt pathway in breast cancers. Sci. Rep. 8 (1), 7329. doi:10.1038/s41598-018-25672-6
Li, Z. H., Hu, P. H., Tu, J. H., and Yu, N. S. (2016). Luminal B breast cancer: Patterns of recurrence and clinical outcome. Oncotarget 7 (40), 65024–65033. doi:10.18632/oncotarget.11344
Liu, Y., Qiao, Y., Zhang, H., Li, W., and Zheng, J. (2019). Wnt7a, frequently silenced by CpG methylation, inhibits tumor growth and metastasis via suppressing epithelial-mesenchymal transition in gastric cancer. J. Cell. Biochem. 120 (10), 18142–18151. doi:10.1002/jcb.29118
Martin-Pardillos, A., Valls Chiva, A., Bande Vargas, G., Hurtado Blanco, P., Pineiro Cid, R., Guijarro, P. J., et al. (2019). The role of clonal communication and heterogeneity in breast cancer. BMC Cancer 19 (1), 666. doi:10.1186/s12885-019-5883-y
Mastoraki, S., Strati, A., Tzanikou, E., Chimonidou, M., Politaki, E., Voutsina, A., et al. (2018). ESR1 methylation: A liquid biopsy-based epigenetic assay for the follow-up of patients with metastatic breast cancer receiving endocrine treatment. Clin. Cancer Res. 24 (6), 1500–1510. doi:10.1158/1078-0432.CCR-17-1181
Mehta, S., Hingole, S., and Chaudhary, V. (2021). The emerging mechanisms of Wnt secretion and signaling in development. Front. Cell Dev. Biol. 9, 714746. doi:10.3389/fcell.2021.714746
Mukherjee, N., Bhattacharya, N., Alam, N., Roy, A., Roychoudhury, S., and Panda, C. K. (2012). Subtype-specific alterations of the Wnt signaling pathway in breast cancer: Clinical and prognostic significance. Cancer Sci. 103 (2), 210–220. doi:10.1111/j.1349-7006.2011.02131.x
Muller, F., Scherer, M., Assenov, Y., Lutsik, P., Walter, J., Lengauer, T., et al. (2019). RnBeads 2.0: Comprehensive analysis of DNA methylation data. Genome Biol. 20 (1), 55. doi:10.1186/s13059-019-1664-9
Netanely, D., Avraham, A., Ben-Baruch, A., Evron, E., and Shamir, R. (2016). Expression and methylation patterns partition luminal-A breast tumors into distinct prognostic subgroups. Breast Cancer Res. 18 (1), 74. doi:10.1186/s13058-016-0724-2
Nishiyama, A., and Nakanishi, M. (2021). Navigating the DNA methylation landscape of cancer. Trends Genet. 37 (11), 1012–1027. doi:10.1016/j.tig.2021.05.002
Palanca-Ballester, C., Rodriguez-Casanova, A., Torres, S., Calabuig-Farinas, S., Exposito, F., Serrano, D., et al. (2021). Cancer epigenetic biomarkers in liquid biopsy for high incidence malignancies. Cancers (Basel) 13 (12), 3016. doi:10.3390/cancers13123016
Palomeras, S., Diaz-Lagares, A., Vinas, G., Setien, F., Ferreira, H. J., Oliveras, G., et al. (2019). Epigenetic silencing of TGFBI confers resistance to trastuzumab in human breast cancer. Breast Cancer Res. 21 (1), 79. doi:10.1186/s13058-019-1160-x
Perou, C. M., Sorlie, T., Eisen, M. B., van de Rijn, M., Jeffrey, S. S., Rees, C. A., et al. (2000). Molecular portraits of human breast tumours. Nature 406 (6797), 747–752. doi:10.1038/35021093
Pineda, B., Diaz-Lagares, A., Perez-Fidalgo, J. A., Burgues, O., Gonzalez-Barrallo, I., Crujeiras, A. B., et al. (2019). A two-gene epigenetic signature for the prediction of response to neoadjuvant chemotherapy in triple-negative breast cancer patients. Clin. Epigenetics 11 (1), 33. doi:10.1186/s13148-019-0626-0
Poppova, L., Pavlova, S., Gonzalez, B., Kotaskova, J., Plevova, K., Dumbovic, G., et al. (2022). Memory B-cell like chronic lymphocytic leukaemia is associated with specific methylation profile of WNT5A promoter and undetectable expression of WNT5A gene. Epigenetics, 1–8. doi:10.1080/15592294.2022.2050004
Rahvar, F., Salimi, M., and Mozdarani, H. (2020). Plasma GBP2 promoter methylation is associated with advanced stages in breast cancer. Genet. Mol. Biol. 43 (4), e20190230. doi:10.1590/1678-4685-GMB-2019-0230
Rice, J. C., Massey-Brown, K. S., and Futscher, B. W. (1998). Aberrant methylation of the BRCA1 CpG island promoter is associated with decreased BRCA1 mRNA in sporadic breast cancer cells. Oncogene 17 (14), 1807–1812. doi:10.1038/sj.onc.1202086
Rodriguez-Casanova, A., Bao-Caamano, A., Lago-Leston, R. M., Brozos-Vazquez, E., Costa-Fraga, N., Ferreiros-Vidal, I., et al. (2021). Evaluation of a targeted next-generation sequencing panel for the non-invasive detection of variants in circulating DNA of colorectal cancer. J. Clin. Med. 10 (19), 4487. doi:10.3390/jcm10194487
Shan, M., Zhang, L., Liu, Y., Gao, C., Kang, W., Yang, W., et al. (2019). DNA methylation profiles and their diagnostic utility in BC. Dis. Markers 2019, 6328503. doi:10.1155/2019/6328503
Shen, S. Y., Singhania, R., Fehringer, G., Chakravarthy, A., Roehrl, M. H. A., Chadwick, D., et al. (2018). Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 563 (7732), 579–583. doi:10.1038/s41586-018-0703-0
Siravegna, G., Marsoni, S., Siena, S., and Bardelli, A. (2017). Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 14 (9), 531–548. doi:10.1038/nrclinonc.2017.14
Smid, M., Wang, Y., Zhang, Y., Sieuwerts, A. M., Yu, J., Klijn, J. G., et al. (2008). Subtypes of breast cancer show preferential site of relapse. Cancer Res. 68 (9), 3108–3114. doi:10.1158/0008-5472.CAN-07-5644
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca. Cancer J. Clin. 71 (3), 209–249. doi:10.3322/caac.21660
Tabas-Madrid, D., Nogales-Cadenas, R., and Pascual-Montano, A. (2012). GeneCodis3: A non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 40, W478–W483. doi:10.1093/nar/gks402
Keywords: DNA methylation, EPIC Array, cell-free DNA, liquid biopsy, metastasis, luminal B, breast cancer
Citation: Rodriguez-Casanova A, Costa-Fraga N, Castro-Carballeira C, González-Conde M, Abuin C, Bao-Caamano A, García-Caballero T, Brozos-Vazquez E, Rodriguez-López C, Cebey V, Palacios P, Cueva JF, López-López R, Costa C and Díaz-Lagares A (2022) A genome-wide cell-free DNA methylation analysis identifies an episignature associated with metastatic luminal B breast cancer. Front. Cell Dev. Biol. 10:1016955. doi: 10.3389/fcell.2022.1016955
Received: 11 August 2022; Accepted: 12 October 2022;
Published: 25 October 2022.
Edited by:
Xiao Wang, Konge Larsen ApS, DenmarkReviewed by:
Nagarajan Kannan, Mayo Clinic, United StatesMiodrag Guzvic, University Medical Center Regensburg, Germany
Copyright © 2022 Rodriguez-Casanova, Costa-Fraga, Castro-Carballeira, González-Conde, Abuin, Bao-Caamano, García-Caballero, Brozos-Vazquez, Rodriguez-López, Cebey, Palacios, Cueva, López-López, Costa and Díaz-Lagares. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafael López-López, cmFmYWVsLmxvcGV6LmxvcGV6QHNlcmdhcy5lcw==; Angel Díaz-Lagares, YW5nZWwuZGlhei5sYWdhcmVzQHNlcmdhcy5lcw==
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share senior authorship