 Antje Kampmeier1*
Antje Kampmeier1* Elsa Leitão1
Elsa Leitão1 Ilaria Parenti1Jasmin Beygo1
Ilaria Parenti1Jasmin Beygo1 Christel Depienne1Nuria C Bramswig1
Christel Depienne1Nuria C Bramswig1 Tzung-Chien Hsieh2Alexandra Afenjar3Stefanie Beck-Wödl4
Tzung-Chien Hsieh2Alexandra Afenjar3Stefanie Beck-Wödl4 Ute Grasshoff4Tobias B Haack4Emilia K Bijlsma5
Ute Grasshoff4Tobias B Haack4Emilia K Bijlsma5 Claudia Ruivenkamp5
Claudia Ruivenkamp5 Eva Lausberg6
Eva Lausberg6 Miriam Elbracht6
Miriam Elbracht6 Maria K Haanpää7,8
Maria K Haanpää7,8 Hannele Koillinen7,9Uwe Heinrich10Imma Rost10Rami Abou Jamra11
Hannele Koillinen7,9Uwe Heinrich10Imma Rost10Rami Abou Jamra11 Denny Popp11Margarete Koch-Hogrebe12
Denny Popp11Margarete Koch-Hogrebe12 Kevin Rostasy12Vanesa López-González13,14
Kevin Rostasy12Vanesa López-González13,14 María José Sanchez-Soler13Catarina Macedo15
María José Sanchez-Soler13Catarina Macedo15 Ariane Schmetz16
Ariane Schmetz16 Carmen Steinborn17Sabine Weidensee18Hellen Lesmann19
Carmen Steinborn17Sabine Weidensee18Hellen Lesmann19 Felix Marbach20
Felix Marbach20 Pilar Caro20Christian P. Schaaf20
Pilar Caro20Christian P. Schaaf20 Peter Krawitz2Dagmar Wieczorek16,21Frank J Kaiser1,22Alma Kuechler1,22*
Peter Krawitz2Dagmar Wieczorek16,21Frank J Kaiser1,22Alma Kuechler1,22*- 1Institut für Humangenetik, Universitätsmedizin Essen, Universität Duisburg-Essen, Essen, Germany
- 2Institut für Genomische Statistik und Bioinformatik, Universitätsklinikum Bonn, Rheinische Friedrich-Wilhelms-Universität Bonn, Bonn, Germany
- 3Département de génétique et embryologie médicale, Centre de Référence Malformations et maladies congénitales du cervelet et déficiences intellectuelles de causes rares, Hôpital Trousseau, APHP Sorbonne Université, Paris, France
- 4Institute of Medical Genetics and Applied Genomics, University of Tübingen, Tübingen, Germany
- 5Department of Clinical Genetics, Leiden University Medical Center, Leiden, Netherlands
- 6Institut für Humangenetik und Genommedizin, Uniklinik RWTH Aachen, Aachen, Germany
- 7Clinical Genetics Unit, Turku University Hospital, Turku, Finland
- 8Department of Genomics, Turku University Hospital, Turku, Finland
- 9Institute of Biomedicine, University of Turku, Turku, Finland
- 10Zentrum für Humangenetik und Laboratoriumsdiagnostik Dr. Klein Dr. Rost und Kollegen, Martinsried, Germany
- 11Institute of Human Genetics, University of Leipzig Medical Center, Leipzig, Germany
- 12Vestische Kinder- und Jugendklinik Datteln, Abteilung für Neuropädiatrie, Datteln, Germany
- 13Sección Genética Médica, Servicio de Pediatría, Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, Spain
- 14Sección de Genética Médica, Servicio de Pediatría, Hospital Clínico Universitario Virgen de la Arrixaca, IMIB-Arrixaca, CIBERER, Murcia, Spain
- 15Serviço de Genética, Departamento de Pediatria, Hospital de Santa Maria, Centro Hospitalar e Universitário Lisboa Norte, Centro Académico de Medicina de Lisboa, Lisboa, Portugal
- 16Institute of Human Genetics, Medical Faculty, University Hospital Düsseldorf, Heinrich Heine University Düsseldorf, Düsseldorf, Germany
- 17MVZ Mitteldeutscher Praxisverbund Humangenetik, Dresden, Germany
- 18MVZ Mitteldeutscher Praxisverbund Humangenetik, Erfurt, Germany
- 19Institut für Humangenetik, Universitätsklinikum Bonn, Universität Bonn, Bonn, Germany
- 20Institut für Humangenetik, Universitätsklinikum Heidelberg, Universität Heidelberg, Heidelberg, Germany
- 21Center for Rare Diseases, Medical Faculty, University Hospital Düsseldorf, Heinrich Heine University Düsseldorf, Düsseldorf, Germany
- 22Essener Zentrum für Seltene Erkrankungen (EZSE), Universitätsmedizin Essen, Essen, Germany
In 2016 and 2018, Chung, Jansen and others described a new syndrome caused by haploinsufficiency of PHIP (pleckstrin homology domain interacting protein, OMIM *612,870) and mainly characterized by developmental delay (DD), learning difficulties/intellectual disability (ID), behavioral abnormalities, facial dysmorphism and obesity (CHUJANS, OMIM #617991). So far, PHIP alterations appear to be a rare cause of DD/ID. “Omics” technologies such as exome sequencing or array analyses have led to the identification of distinct types of alterations of PHIP, including, truncating variants, missense substitutions, splice variants and large deletions encompassing portions of the gene or the entire gene as well as adjacent genomic regions. We collected clinical and genetic data of 23 individuals with PHIP-associated Chung-Jansen syndrome (CHUJANS) from all over Europe. Follow-up investigations (e.g. Sanger sequencing, qPCR or Fluorescence-in-situ-Hybridization) and segregation analysis showed either de novo occurrence or inheritance from an also (mildly) affected parent. In accordance with previously described patients, almost all individuals reported here show developmental delay (22/23), learning disability or ID (22/23), behavioral abnormalities (20/23), weight problems (13/23) and characteristic craniofacial features (i.e. large ears/earlobes, prominent eyebrows, anteverted nares and long philtrum (23/23)). To further investigate the facial gestalt of individuals with CHUJANS, we performed facial analysis using the GestaltMatcher approach. By this, we could establish that PHIP patients are indistinguishable based on the type of PHIP alteration (e.g. missense, loss-of-function, splice site) but show a significant difference to the average face of healthy individuals as well as to individuals with Prader-Willi syndrome (PWS, OMIM #176270) or with a CUL4B-alteration (Intellectual developmental disorder, X-linked, syndromic, Cabezas type, OMIM #300354). Our findings expand the mutational and clinical spectrum of CHUJANS. We discuss the molecular and clinical features in comparison to the published individuals. The fact that some variants were inherited from a mildly affected parent further illustrates the variability of the associated phenotype and outlines the importance of a thorough clinical evaluation combined with genetic analyses for accurate diagnosis and counselling.
Introduction
PHIP (pleckstrin homology domain interacting protein; OMIM *612870) was originally identified as a candidate gene for intellectual disability (ID) in one individual from a cohort of 100 ID-cases (de Ligt et al., 2012). Furthermore, microdeletions in the region 6q14.1, including PHIP, have been described in association with ID, developmental delay (DD) and dysmorphic features (Lespinasse et al., 2009; Van Esch et al., 2010; Becker et al., 2012). Webster et al. described in 2016 two individuals showing DD, ID, obesity and dysmorphisms, each having a de novo heterozygous predicted deleterious variant in PHIP (Webster et al., 2016). In 2018, Jansen et al. described 23 patients with different heterozygous mutations in PHIP and associated them with a syndrome mainly characterized by DD, learning difficulties/ID, behavioral abnormalities, facial dysmorphism and obesity (Jansen et al., 2018), later termed Chung-Jansen syndrome (CHUJANS, OMIM #617991) or DIDOD (Developmental delay, Intellectual Disability, Obesity, and Dysmorphism). Afterwards, Craddock et al. further characterized 10 individuals with predicted deleterious variants in PHIP and could show that the mutation spectrum is diverse and without any clustering or mutational hotspots (Craddock et al., 2019). Aoi et al. investigated a cohort of patients with suspected Cornelia-de-Lange syndrome (CdLS, OMIM #122470) and identified one individual with a missense substitution in PHIP (Aoi et al., 2019), while Kaur et al. extended the phenotypic spectrum with a case of CHUJANS also showing hypothyroidism and small kidneys (Kaur and Panigrahi, 2021). The latest publication reviewed 35 already reported individuals with PHIP variants together with one newly identified individual and sheds light on the impact of the variants on the structure of the protein by means of protein modeling (Dietrich et al., 2022).
PHIP encodes two protein isoforms, PHIP/DCAF14 (Pleckstrin Homology Domain Interacting Protein/DDB1- and CUL4-associated factor 14) and NDRP (Neuronal Differentiation-Related Protein). Both isoforms play a role in neurodevelopmental processes, such as E3 ubiquitination and neuronal differentiation (Jansen et al., 2018). Genotype/phenotype analyses suggest that PHIP haploinsufficiency is the cause of the described phenotypes of CHUJANS (Webster et al., 2016; Jansen et al., 2018; Craddock et al., 2019). Additionally, deficiency of CUL4B, a PHIP-interacting protein, has been previously associated with a very similar phenotype showing ID, central obesity, muscle wasting and dysmorphisms, due to disruption of the ubiquitin ligase pathway (syndromic X-linked intellectual developmental disorder; OMIM #300354) (Tarpey et al., 2007; Webster et al., 2016).
Altogether, 39 patients with variants in PHIP and five patients with deletions including PHIP have been published so far, and it still appears that PHIP variants are a rare cause of DD/ID.
In this study, we report 23 additional individuals with newly identified pathogenic or likely pathogenic PHIP variants or (partial) deletions of PHIP. Five of the described cases were proven to be inherited, either maternally or paternally, from a mildly affected parent. We performed facial phenotype analyses by the next-generation phenotyping approach GestaltMatcher. The phenotypic analysis was similarly extended to additional syndromes with molecular or clinical overlap with CHUJANS, namely CUL4B-related disorder and Prader-Willi syndrome (OMIM #176270).
Our findings further expand the mutational and phenotypic spectrum of PHIP. We discuss the molecular and clinical features in comparison to the already published individuals.
Materials and methods
Our cohort
Our cohort comprises 23 individuals of which thirteen were males, ten were females. They all presented in genetic departments due to DD/ID and/or behavioral changes. The data were collected thanks to a large international cooperation including Germany, Spain, Netherlands, Finland, France and Portugal. All individuals/legal guardians gave signed consent for anonymous publication of genetic and clinical data and 19 out of 23 individuals additionally consented to publication of photographs. Comprehensive clinical and genetic data were provided by the referring geneticists. Data were anonymized before sharing and ethics approvals were locally obtained.
Genetic testing
Individuals had been analyzed by means of microarrays, gene panels or exome sequencing according to standard protocols at their respective institutions. Variants are mapped on the NM_017934.7 transcript.
PHIP variants present in gnomAD
Known PHIP variants (ENST00000275034.4, corresponding to NM_017934) were retrieved from gnomAD v2.1.1 (Karczewski et al., 2020), limiting to loss-of-function, missense and synonymous single nucleotide variants (SNVs) or indels. The combined annotation-dependent depletion (CADD) score (Rentzsch et al., 2019) (https://cadd.gs.washington.edu/score) was calculated for each variant using GRCh37-v1.6 genomic coordinates. Amino acid positions of each protein region were retrieved from Uniprot (UniProt, 2021).
GestaltMatcher analysis
GestaltMatcher (Hsieh et al., 2022) is a deep learning framework that quantifies the facial dysmorphic similarities among patients. GestaltMatcher was first trained on 3,438 frontal images with 139 different disorders from GestaltMatcher Database (https://db.gestaltmatcher.org/) to learn the facial dysmorphic features. It later encoded the photo into a 320-dimensional facial phenotype descriptor (FPD and spanned a clinical face phenotype space (CFPS)). The similarity between two patients can be quantified by the cosine distance in the CFPS.
We performed GestaltMatcher analyses on the following three cohorts: individuals with variants in PHIP (36 images), in CUL4B (27 images), and with Prader-Willi syndrome (PWS, 11 images). The PHIP cohort consisted of 18 frontal images from the unpublished cohort recruited in this study and 18 frontal images from previously published works - 14 images from Jansen et al. (2018), one image from Webster et al. (2016), one image from Aoi et al. (2019), and two images from Craddock et al. (2019). We collected CUL4B patients from four different publications, 13 images from Vulto-van Silfhout et al. (2015), seven images from Tarpey et al. (2007), four images from Cabezas et al. (2000), and three images from Badura-Stronka et al. (2010). We recruited 11 images of patients with PWS from the patient support group.
We first encoded each of the 74 images into FPDs and performed t-distributed stochastic neighbor embedding tSNE to visualize the distribution of patients in two-dimensional space.
Average face analysis
We performed the average face analysis to visualize the different facial phenotypes in the three different cohorts (CHUJANS, CUL4B-related ID and PWS) and the healthy individuals. To simulate the cohort for the healthy individuals, we randomly selected ten healthy individuals that match the sex and age in UTKFace (Zhang et al., 2017) for each PHIP patient. Therefore, the healthy cohort contained 360 images. We then averaged the faces in the following four cohorts: PHIP (36 images), CUL4B (27 images), Prader-Willi syndrome (11 images), and healthy cohort (360 images) by first detecting the 68 facial landmarks and averaging over all the images in each cohort.
Results
PHIP variants
The point variants described in our cohort are shown in Figure 1A. Fourteen variants in PHIP identified in our cohort had not been described before. One variant had previously been described in the literature (g.79735299G>T (hg19), c.860C>A, p.(Ser287Tyr)), (Craddock et al., 2019). Ten of the sequence variants were loss-of-function variants, four were missense variants and one affected a splice site. Three variants were found in two individuals each: one variant shared by two siblings, another found in a father and his daughter and another in two individuals that were not related to each other. The variants were classified according to ACMG criteria (Richards et al., 2015). The criteria used for classification as well as the variants can also be found in Table 1 and the Supplementary Table S1.
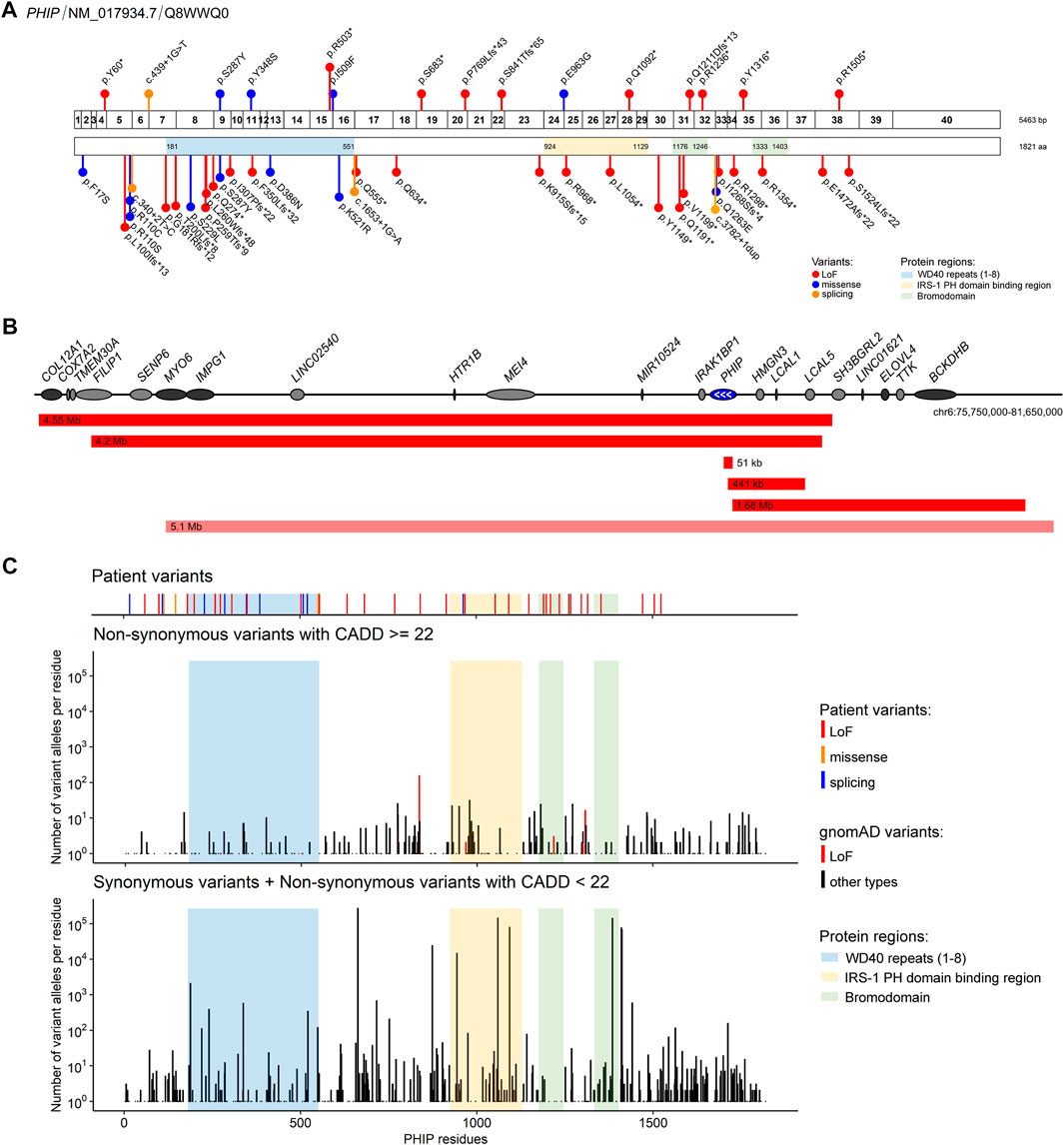
FIGURE 1. PHIP variants associated with Chung-Jansen syndrome identified in the present cohort and/or described in the literature (A) Schematic representation of PHIP exons (top; 1–40) and its encoded protein (bottom) showing the identified variants relative to the protein domains/regions. Variants identified in the present cohort are shown above, and the described in the literature are shown below. Variants are labelled with the nomenclature based on changes at protein levels except for splicing variants, for which coding DNA sequence nomenclature was used (B) Schematic representation of the deletions encompassing the PHIP gene (blue) identified in the present cohort (red) and in the literature (light red). Deletion sizes are shown. White arrows identify the direction of PHIP transcription. Genes in dark grey are, as PHIP, OMIM morbid genes (C) Comparison of the distribution of the PHIP variants identified in the present cohort and/or in the literature with the variants reported in gnomAD. Number of individuals with gnomAD variants were plotted for each combined codon. gnomAD variants were stratified by damage potential (based on variant type and CADD score).
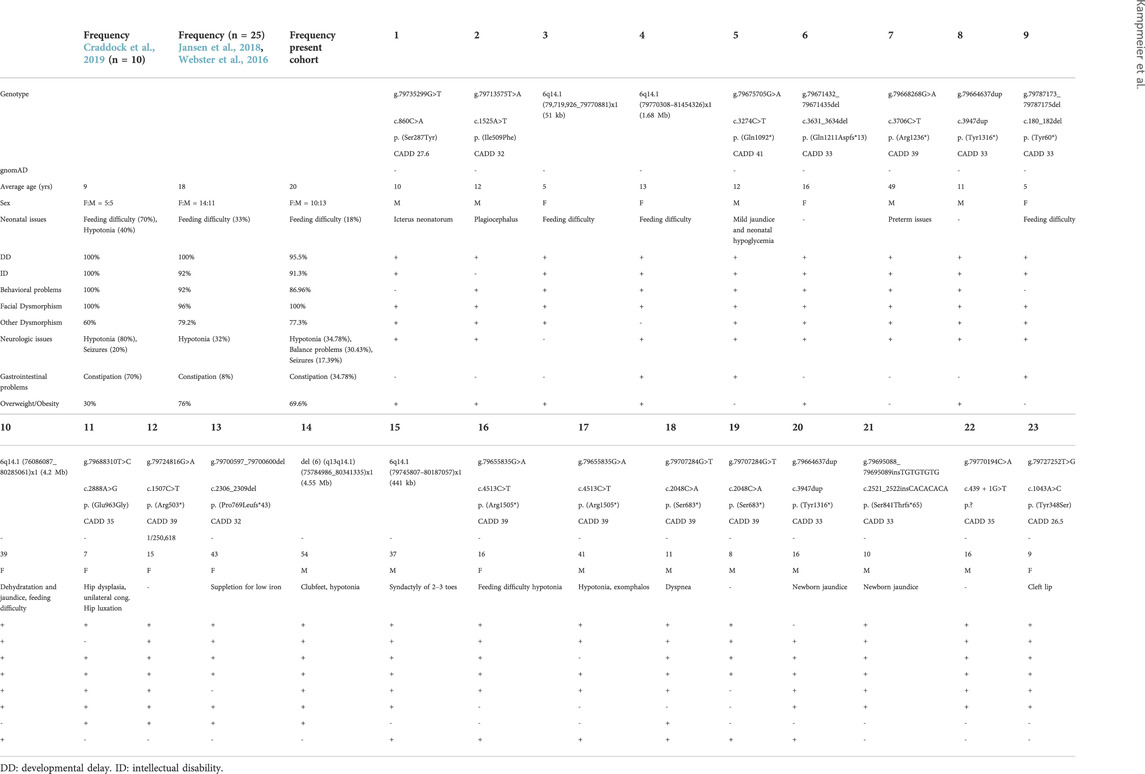
TABLE 1. Molecular and clinical features of PHIP individuals.
Five individuals showed PHIP haploinsufficiency due to a partial or complete deletion of PHIP as shown in Figure 1B. The size of the deletions varied from 51 kb to 4.55 Mb. Three of the deletions only partially affected PHIP: one intragenic deletion, another partially included PHIP in addition to three other genes (HMGN3, LCAL1, and LCAL5), and another partially included PHIP together with eight additional genes. The two remaining deletions encompassed the entire PHIP gene: one encompassed 16 genes, including PHIP, and the other included PHIP and 12 additional genes (one of which only partially). Although the deletion size and the number of genes involved vary among patients, the phenotypes are consistent and overlapping, hence suggesting that PHIP haploinsufficiency might be the major contributor to the disease of those patients.
Inheritance of the variants could be assessed in 15 individuals. Variants were found to be de novo in ten cases, whereas three variants were paternally inherited and two were maternally inherited. Due to unavailability of at least one of the parents, the origin of the variant could not be assessed in eight patients. In patients carrying a deletion affecting PHIP, one was proven to be de novo, one maternally inherited, one paternally inherited and in the two remaining cases inheritance was unknown.
In one individual (individual 1), whole exome analysis detected a second de novo variant in CUX1 (NM_181552.4: c.3334del; p.(Gln1112Serfs*19)) which was classified as class 3 (unknown significance). The impact of the respective variant on the phenotype cannot be defined more precisely at present.
All variants of our cohort are either absent from gnomAD or present at a very low allele frequency. PHIP variants available in gnomAD were subsequently retrieved to verify whether specific domains of the PHIP protein might be depleted from putatively damaging variants. As shown in Figure 1C, less residues are affected by putatively damaging variants in gnomAD (CADD score ≥22) in comparison to putatively benign variants (synonymous variants and variants with CADD score <22).
Most frequent clinical features
A summary of the main clinical features associated with PHIP alterations can be found in Table 1. Detailed clinical description of each patient of our cohort is available in Supplementary Table S1.
In our cohort of 23 individuals with PHIP variants (SNVs, partial deletion, or deletion) the most common clinical features are DD (96%), followed by ID (91%) and behavioral problems (87%), as shown in Figure 2A and Table 1 as well as in the Supplementary Table S1. Overweight/obesity is the fourth most common feature (70%) in our cohort. Vision problems occurred in 48% of the individuals while tapering fingers were present in 43%. Constipation and muscular hypotonia were seen in 35%, followed by balance problems in 30%.
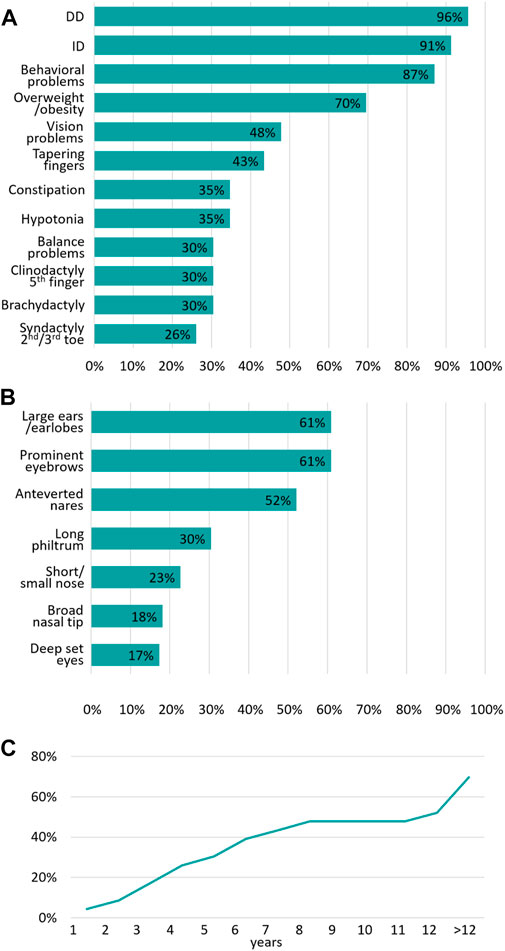
FIGURE 2. Summary of the main clinical features associated with PHIP variants (A) Summary of the most frequent clinical features of our cohort expressed as a percentage (n = 23) (B) Summary of the most frequent craniofacial dysmorphism of our cohort expressed as a percentage (n = 23) (C) Age of onset of overweight/obesity and the cumulative frequency expressed as a percentage.
Interestingly, neonatal muscular hypotonia was only reported in 13% of the individuals, while it was more often reported later in life when the individuals were presented to a clinical geneticist (35%), which is important for the comparison with PWS patients.
Craniofacial dysmorphisms
All 23 individuals presented with characteristic craniofacial dysmorphic features (Figure 3). As shown in Figure 2B, the most common features are large ears/earlobes (61%) and prominent eyebrows (61%), followed by anteverted nares (52%). Less frequent dysmorphism comprise a long philtrum (30%), a short nose (23%), a broad nasal tip (17%) and deep-set eyes.

FIGURE 3. Facial appearance of the individuals of our cohort (A) Individual 1 (10 years) (B,C) individual 2 (1 year, 12 years) (D,E) individual 4 (3 years, 6 years) (F–H) individual 5 (3 years, 12 years, profile 12 years) (I,J) individual 6 (16 years) (K) individual 7 (49 years) (L) individual 8 (11 years) (M,N) individual 10 (39 years) (O) individual 11 (7 years) (P,Q) individual 13 (38 years) (R,S) individual 15 (37 years) (T) individual 16 (16 years) (U) individual 17 (41 years) (V) individual 18 (age unknown) (W) individual 19 (age unknown) (X,Y) individual 20 (16 years) (Z,AA) individual 21 (10 years) (BB) individual 22 (16 years) (CC) individual 23 (9 years).
Limb dysmorphisms
Nearly half of the cohort presented tapering fingers (43%). 30% of the individuals showed brachydactyly, while clinodactyly of the fifth finger could be found in 30% of the individuals (Figure 4). 26% of the individuals showed syndactyly of the second/third toes.
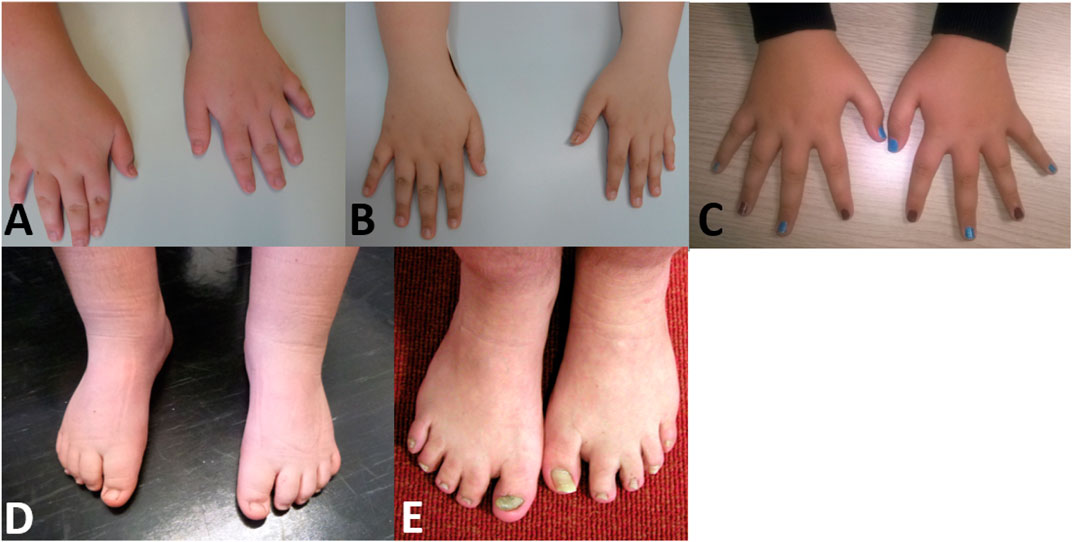
FIGURE 4. Pictures of individuals showing hand and/or feet anomalies (A) Hands of individual 1 (10 years) (B) hands of individual 2 (12 years) (C) hands of individual 6 (16 years) (D) feet of individual 1 (10 years) (E) feet of individual 17 (41 years).
Age of onset of overweight/obesity
The age of onset of overweight/obesity appears to be highly variable. The incidence of this clinical feature increases as patients grow older (Figure 2C). In our cohort, obesity was present in 70% of the individuals. However, some of the individuals were examined at an age below 12 years, an age where obesity onset is not always observed. Based on our data, the incidence of overweight/obesity rises sharply during puberty.
30% of the individuals already show overweight/obesity at the age of 5 years. Almost 50% showed overweight/obesity at the age of 8 years, followed by a plateau phase with a second, sharp rise of incidence from the age of 12 years and above. The highest BMI in our cohort was 36.26 kg/m2 at the age of 15 years in a female individual.
Facial gestalt analyzed by GestaltMatcher
We subsequently took advantage of the GestaltMatcher approach to verify whether facial dysmorphisms are associated with a specific variant type and whether patients with different variants (large deletions, loss-of-function point variants, and missense substitutions) can be distinguished based on their facial features. As shown in Figure 5A, PHIP patients do not cluster based on variant type. Importantly, subtle differences might not be evident due to the small sample size.
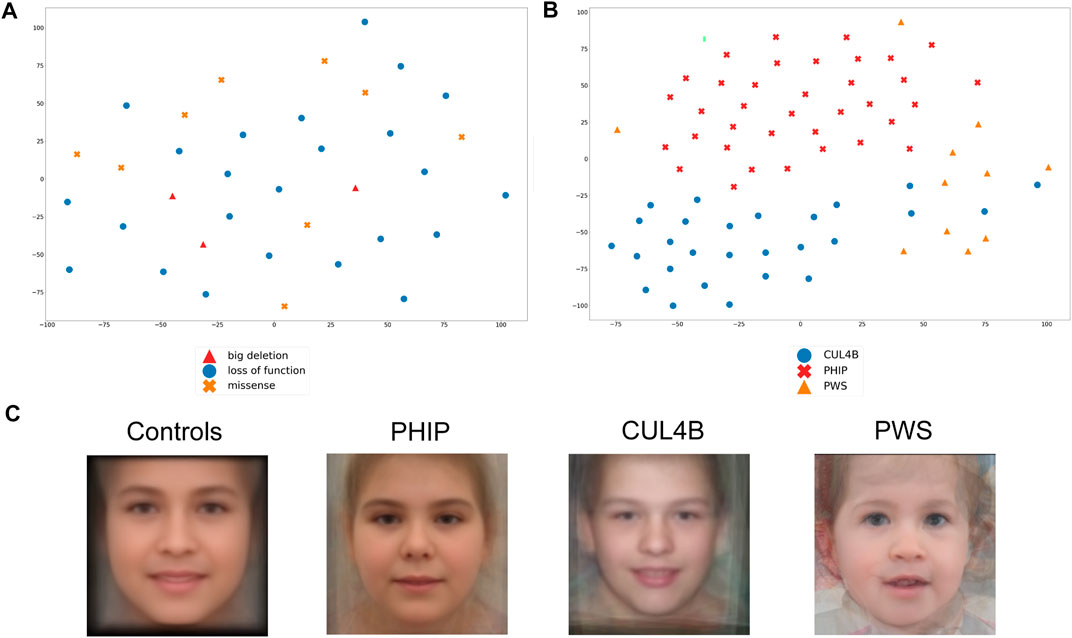
FIGURE 5. Analysis of the facial Gestalt with Gestaltmatcher. (A) tSNE analysis to validate whether PHIP individuals cluster based on the type of mutations (missense, loss-of-function, splice site). (B) tSNE analysis of patients with PHIP alteration, CUL4B alteration or PWS, which shows a clear phenotypic separation of the three conditions. (C) Average faces for healthy controls and each of the differential clinical diagnoses for CHUJANS, namely CUL4B-related disorders and PWS.
We then extended the analysis and compared the facial gestalt of our patients with those of individuals with the clinical diagnosis of PWS as well as individuals with CUL4B variants, as both syndromes are characterized by ID and overweight and represent important differential clinical diagnoses (Figure 5B). PWS is one of the main clinical differential diagnoses that was suspected in almost all patients at a certain point. Furthermore, CUL4B and PHIP functionally interact on protein level as binding partners within the same protein complex. We could show that individuals with PHIP variants as well as individuals with CUL4B variants or PWS patients form distinct clusters, although some of CUL4B patients localize within the PWS cluster. The low image quality of some of the patients might explain why some patients behave as outliers.
We further generated average faces for individuals with PHIP SNVs or (partial) deletions, CUL4B variants and PWS, as well as a healthy control face. By this, we could see a difference in the facial gestalt from the PHIP cohort in comparison to the CUL4B and PWS cohort and from all the three cohorts to the average healthy control face, as depicted in Figure 5C.
Discussion
Here we report on 23 new cases of PHIP associated CHUJANS. The shared clinical features include DD, ID, behavioral problems, overweight/obesity and some dysmorphic facial signs. Interestingly, the most common dysmorphic facial features are large ears/earlobes, prominent eyebrows and anteverted nares. This is in line with the previously reported cohorts of patients (Wentzel et al., 2010; Becker et al., 2012; Webster et al., 2016; Jansen et al., 2018; Craddock et al., 2019).
Developmental delay and ID
Overall, 96% of the individuals of our cohort showed some kind of DD. In some cases, either motor or speech development was affected. One case presented with normal development until the age of 3 years, followed by a delay of psychomotor development after this age. This reveals that developmental milestones up to the first years of life are not always disrupted, contrary to previous findings (Craddock et al., 2019). ID was observed in 91% of the individuals in our cohort, but only three of our patients presented with an IQ below 50. This is in agreement with the findings of Craddock and others, who reported that intellectual disability is generally mild and characterized by a better verbal performance than IQ (Craddock et al., 2019). Accordingly, five parents of our cohort were diagnosed as mildly affected only after the identification of the genetic cause of the disease of their respective children. In the previously published cohorts, only two inherited cases were described (Jansen et al., 2018).
Overweight/obesity
In our study, we found a different time of onset of overweight or obesity. Whereas some patients started gaining weight during childhood, others started being overweight only during puberty. Overall, nearly 70% of the individuals of our cohort showed overweight or obesity. Since obesity was defined as one of the main criteria of the also called “DIDOD-Syndrome” (developmental delay, intellectual disability, OBESITY and dysmorphism), one should be careful not to miss the diagnosis of CHUJANS as patients might not show obesity or overweight at a younger age. This should be considered for phenotype-based filtering processes of exome or genome data.
Marenne et al. (2020) were able to demonstrate that mutations in PHIP are causing overweight through PHIP interaction with Proopiomelanocortin (POMC). They could establish that loss-of-function variants in PHIP are more frequently associated with obesity or overweight than missense substitutions. Our study cannot confirm their finding that missense variants in PHIP do not lead to obesity, as two out of four individuals with a missense variant in our cohort showed obesity already during childhood.
In addition, Marenne and others also identified patients with PHIP variants and obesity but without developmental delay (Marenne et al., 2020). They concluded that PHIP variants might affect transcriptional regulation and different types of mutations result in variable clinical outcomes. They suggested that PHIP should be included in genetic testing recommended in clinical guidelines as part of the assessment for severe childhood-onset obesity (Marenne et al., 2020).
Our findings confirm the importance of PHIP variants in the context of obesity onset. However, as the age range of onset of obesity in all our obese patients was rather wide, from early childhood to puberty (>12 years), PHIP haploinsufficiency should be taken into account also in the absence of overweight/obesity. We also suggest following up on the overweight/obesity as longitudinal data are missing for this relatively newly defined syndrome (Chung-Jansen syndrome or DIDOD syndrome, OMIM #617991) (de Ligt et al., 2012; Webster et al., 2016; Jansen et al., 2018; Craddock et al., 2019). It would be interesting to assess if all individuals with a reduced dosage of functional PHIP gene product show overweight/obesity in later adult years.
As PWS is also a DD/ID/obesity syndrome, it is a common clinical differential diagnosis to CHUJANS. When PWS testing is negative, CHUJANS should be considered. Thus, PHIP should also be included in obesity panels.
Behavioral problems
Behavioral problems have been described as one of the most common features of patients with PHIP variants. Our data highlight this relevance since 87% showed behavioral problems which were not necessarily present at younger ages. The types of behavioral changes are very broad and include loss of self-control and impulsiveness, aggressions, anxiety, autism spectrum disorder and motor hyperactivity. A large variety of behavioral problems have also been described in other patients (Jansen et al., 2018; Craddock et al., 2019). Interestingly, longitudinal follow-up on one patient revealed that behavioral problems worsened with increasing age. By the late teenage years, the patient had already been in the psychiatric ward several times. The worsening of his behavioral disturbances made it impossible for this patient to live at home with his family. We suggest a regular follow-up on the behavioral problems of patients diagnosed with PHIP in childhood, adolescence and adulthood to learn more about this main feature of the syndrome.
Facial gestalt
The main craniofacial dysmorphisms are large ears/earlobes and prominent eyebrows, followed by anteverted nares and, much less often, by a long philtrum, a short nose, a broad nasal tip and deep-set eyes. This is in line with previous reports (Webster et al., 2016; Jansen et al., 2018; Craddock et al., 2019).
In our cohort, we could not show any clustering of the facial phenotype depending on the type of genetic variant or deletion affecting PHIP. This may also be due to the small cohort size.
Van der Donk and others had previously shown that patients with a pathogenic variant in PHIP have a characteristic facial gestalt (van der Donk et al., 2019). Because of possible overlapping facial features, we compared the facial gestalt of individuals with variants affecting PHIP with the facial gestalt of published individuals with CUL4B alterations. Since PHIP/DCAF14 is one of the multiple substrate receptors of the proteolytic CUL4-DDB1 (Jansen et al., 2018; Tirado-Class et al., 2022), we postulated a phenotypic overlap between patients with PHIP or CUL4B variants. Furthermore, both proteins are involved in fork stability and genome integrity (Tirado-Class et al., 2022). GestaltMatcher did not confirm this hypothesis and rather showed that each disorder shows its distinct characteristic facial phenotype. We also compared the facial phenotype to that of the clinically relevant differential diagnosis PWS (which was suspected in almost all patients of our cohort at a certain time point). GestaltMatcher confirmed the hypothesis that the facial gestalt of both conditions differs. The low image quality of some of the patients might explain why some patients behave as outliers.
In summary, we conclude that the facial gestalt of individuals with PHIP alterations leading to CHUJANS is quite specific and recognizable and can be identified by using next-generation phenotyping approaches like GestaltMatcher.
Comparison of phenotypes: CHUJANS vs. Cabezas syndrome and PWS
Since the two proteins PHIP and CUL4B belong to the same complex and are involved in the same processes (DNA repair, fork stability and genome integrity), as mentioned before, we chose individuals with CUL4B associated Cabezas syndrome to investigate whether their facial gestalt overlaps with that of CHUJANS or can be clearly distinguished by next-generation phenotyping approaches like GestaltMatcher (reported in 4.4). To compare the clinical phenotype of CHUJANS with that of Cabezas syndrome and that of PWS beyond facial features, we contrasted the clinical features of our cohort with those of published cases (2000–2019) of Cabezas syndrome, summarized by López et al., 2020, and the clinical features typical of PWS according to GeneReviews® (see Supplementary Table S2). This revealed that ID is the most common clinical feature of these syndromes while DD and speech delay are also found in nearly all individuals affected by Cabezas syndrome and PWS but only in about half of the individuals having CHUJANS. Other frequent findings in Cabezas syndrome seem to be short stature, sandal gap and short feet (Lopez et al., 2020) which are very rarely found in our cohort of CHUJANS. Development of short stature and obesity, key features in PWS, is favorably influenced by the growth hormone therapy that is part of PWS clinical management. Obesity, however, was described in about half of the affected individuals in Cabezas syndrome while nearly 70% of individuals with CHUJANS became overweight/obese. Tremor is common in Cabezas syndrome, which has not been reported in our cohort and in PWS. Nevertheless, balance problems have occurred in 7/23 individuals of our CHUJANS cohort. Behavioral changes are a little less frequent in Cabezas syndrome than in our CHUJANS cohort. Tarpey et al. reported that 12 out of 15 individuals showing Cabezas syndrome have aggressive outbursts (Tarpey et al., 2007). This feature has also been reported in our cohort but only in 6/23 individuals. Overall, 86.4% of our cohort show a kind of behavioral change, but aggressions seem not to be the most common one. An age dependent, characteristic behavior profile is observed in almost all individuals with PWS. In summary, not only facial gestalt but also clinical characteristics of CHUJANS differ from those of Cabezas syndrome and of PWS.
Conclusion
The phenotype of PHIP-associated disorders is variable, although some common features can be recognized. Even though it is still a rare finding, we suggest including PHIP in diagnostic gene panels specific for developmental delay, ID, behavioral abnormalities and/or obesity. This could help find the genetic cause for unsolved cases of DD/ID and/or behavioral problems as well as obesity in childhood and adulthood. In turn, the discovery of new patients with PHIP variants would elucidate further the variability of the associated clinical phenotype. In addition, we suggest taking advantage of facial gestalt software such as GestaltMatcher to assist with the clinical classification of patients.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by local commitees at the respective institutions. For facial gestalt analysis using GestaltMatcher, the ethics committee approval has been obtained by the institutional ethic committee of the medical faculty, University of Bonn (386/17). Written informed consent to participate in this study was provided by the participants; legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s); legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
Supervision: AK; Conceptualization: AK, AKM, IP, EL; Writing- original draft: AKM, AK, Investigation, formal analysis, writing-review and editing: all authors.
Funding
This work was partly done within the Zentrum für Seltene Erkrankungen of the University Hospital Düsseldorf (ZSED) and the Essener Zentrum für Seltene Erkrankungen (EZSE). Authors of this publication (DW, FK, AK) are members of the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability ERN-ITHACA [EU Framework Partnership Agreement ID: 3HP-HP-FPA ERN-01–2016/739516, 20191016-J3B7EI7J].
Acknowledgments
We acknowledge support by the Open Access Publication Fund of the University of Duisburg-Essen. We would like to thank all patients and their families for participation in this study and consent to publication. This work is generated within the European Reference Network ITHACA.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.1020609/full#supplementary-material
References
Aoi, H., Mizuguchi, T., Ceroni, J. R., Kim, V. E. H., Furquim, I., Honjo, R. S., et al. (2019). Comprehensive genetic analysis of 57 families with clinically suspected Cornelia de Lange syndrome. J. Hum. Genet. 64, 967–978. doi:10.1038/s10038-019-0643-z
Badura-Stronka, M., Jamsheer, A., Materna-Kiryluk, A., Sowinska, A., Kiryluk, K., Budny, B., et al. (2010). A novel nonsense mutation in CUL4B gene in three brothers with X-linked mental retardation syndrome. Clin. Genet. 77, 141–144. doi:10.1111/j.1399-0004.2009.01331.x
Becker, K., Di Donato, N., Holder-Espinasse, M., Andrieux, J., Cuisset, J. M., Vallée, L., et al. (2012). De novo microdeletions of chromosome 6q14.1-q14.3 and 6q12.1-q14.1 in two patients with intellectual disability - further delineation of the 6q14 microdeletion syndrome and review of the literature. Eur. J. Med. Genet. 55, 490–497. doi:10.1016/j.ejmg.2012.03.003
Cabezas, D. A., Slaugh, R., Abidi, F., Arena, J. F., Stevenson, R. E., Schwartz, C. E., et al. (2000). A new X linked mental retardation (XLMR) syndrome with short stature, small testes, muscle wasting, and tremor localises to Xq24-q25. J. Med. Genet. 37, 663–668. doi:10.1136/jmg.37.9.663
Craddock, K. E., Okur, V., Wilson, A., Gerkes, E. H., Ramsey, K., Heeley, J. M., et al. (2019). Clinical and genetic characterization of individuals with predicted deleterious PHIP variants. Cold Spring Harb. Mol. Case Stud. 5, a004200. doi:10.1101/mcs.a004200
De Ligt, J., Willemsen, M. H., Van Bon, B. W., Kleefstra, T., Yntema, H. G., Kroes, T., et al. (2012). Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929. doi:10.1056/NEJMoa1206524
Dietrich, J., Lovell, S., Veatch, O. J., and Butler, M. G. (2022). PHIP gene variants with protein modeling, interactions, and clinical phenotypes. Am. J. Med. Genet. A 188, 579–589. doi:10.1002/ajmg.a.62557
Hsieh, T. C., Bar-Haim, A., Moosa, S., Ehmke, N., Gripp, K. W., Pantel, J. T., et al. (2022). GestaltMatcher facilitates rare disease matching using facial phenotype descriptors. Nat. Genet. 54, 349–357. doi:10.1038/s41588-021-01010-x
Jansen, S., Hoischen, A., Coe, B. P., Carvill, G. L., Van Esch, H., Bosch, D. G. M., et al. (2018). A genotype-first approach identifies an intellectual disability-overweight syndrome caused by PHIP haploinsufficiency. Eur. J. Hum. Genet. 26, 54–63. doi:10.1038/s41431-017-0039-5
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alfoldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature 581, 434–443. doi:10.1038/s41586-020-2308-7
Kaur, H., and Panigrahi, I. (2021). Chung-jansen syndrome with obesity. Obes. Res. Clin. Pract. 15, 303–305. doi:10.1016/j.orcp.2021.03.015
Lespinasse, J., Gimelli, S., Bena, F., Antonarakis, S. E., Ansermet, F., and Paoloni-Giacobino, A. (2009). Characterization of an interstitial deletion 6q13-q14.1 in a female with mild mental retardation, language delay and minor dysmorphisms. Eur. J. Med. Genet. 52, 49–52. doi:10.1016/j.ejmg.2008.10.001
Lopez, M., Perez-Grijalba, V., Garcia-Cobaleda, I., and Dominguez-Garrido, E. (2020). A 22.5 kb deletion in CUL4B causing Cabezas syndrome identified using CNV approach from WES data. Clin. Case Rep. 8, 3184–3188. doi:10.1002/ccr3.3381
Marenne, G., Hendricks, A. E., Perdikari, A., Bounds, R., Payne, F., Keogh, J. M., et al. (2020). Exome sequencing identifies genes and gene sets contributing to severe childhood obesity, linking PHIP variants to repressed POMC transcription. Cell. Metab. 31, 1107–1119. doi:10.1016/j.cmet.2020.05.007
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). Cadd: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894. doi:10.1093/nar/gky1016
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Tarpey, P. S., Raymond, F. L., O'Meara, S., Edkins, S., Teague, J., Butler, A., et al. (2007). Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am. J. Hum. Genet. 80, 345–352. doi:10.1086/511134
Tirado-Class, N., Hathaway, C., Chung, W. K., and Dungrawala, H. (2022). PHIP variants associated with Chung-Jansen syndrome disrupt replication fork stability and genome integrity. Cold. Spring. Harb. Mol. Case Stud. 8, mcs.a006212. doi:10.1101/mcs.a006212
Uniprot, C. (2021). UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. doi:10.1093/nar/gkaa1100
Van Der Donk, R., Jansen, S., Schuurs-Hoeijmakers, J. H. M., Koolen, D. A., Goltstein, L., Hoischen, A., et al. (2019). Next-generation phenotyping using computer vision algorithms in rare genomic neurodevelopmental disorders. Genet. Med. 21, 1719–1725. doi:10.1038/s41436-018-0404-y
Van Esch, H., Rosser, E. M., Janssens, S., Van Ingelghem, I., Loeys, B., and Menten, B. (2010). Developmental delay and connective tissue disorder in four patients sharing a common microdeletion at 6q13-14. J. Med. Genet. 47, 717–720. doi:10.1136/jmg.2010.077586
Vulto-Van Silfhout, A. T., Nakagawa, T., Bahi-Buisson, N., Haas, S. A., Hu, H., Bienek, M., et al. (2015). Variants in CUL4B are associated with cerebral malformations. Hum. Mutat. 36, 106–117. doi:10.1002/humu.22718
Webster, E., Cho, M. T., Alexander, N., Desai, S., Naidu, S., Bekheirnia, M. R., et al. (2016). De novo PHIP-predicted deleterious variants are associated with developmental delay, intellectual disability, obesity, and dysmorphic features. Cold Spring Harb. Mol. Case Stud. 2, a001172. doi:10.1101/mcs.a001172
Wentzel, C., Lynch, S. A., Stattin, E. L., Sharkey, F. H., Annerén, G., and Thuresson, A. C. (2010). Interstitial deletions at 6q14.1-q15 associated with obesity, developmental delay and a distinct clinical phenotype. Mol. Syndromol. 1, 75–81. doi:10.1159/000314025
Keywords: Chung-Jansen syndrome, CHUJANS, PHIP, DIDOD syndrome, ID, DD, obesity, CUL4B
Citation: Kampmeier A, Leitão E, Parenti I, Beygo J, Depienne C, Bramswig NC, Hsieh T-C, Afenjar A, Beck-Wödl S, Grasshoff U, Haack TB, Bijlsma EK, Ruivenkamp C, Lausberg E, Elbracht M, Haanpää MK, Koillinen H, Heinrich U, Rost I, Jamra RA, Popp D, Koch-Hogrebe M, Rostasy K, López-González V, Sanchez-Soler MJ, Macedo C, Schmetz A, Steinborn C, Weidensee S, Lesmann H, Marbach F, Caro P, Schaaf CP, Krawitz P, Wieczorek D, Kaiser FJ and Kuechler A (2023) PHIP-associated Chung-Jansen syndrome: Report of 23 new individuals. Front. Cell Dev. Biol. 10:1020609. doi: 10.3389/fcell.2022.1020609
Received: 16 August 2022; Accepted: 16 November 2022;
Published: 16 January 2023.
Edited by:
Lidia Larizza, Italian Auxological Institute (IRCCS), ItalyReviewed by:
Manuela Priolo, Great Metropolitan Hospital of Reggio Calabria, ItalyMaria Giuseppina Miano, National Research Council (CNR), Italy
Copyright © 2023 Kampmeier, Leitão, Parenti, Beygo, Depienne, Bramswig, Hsieh, Afenjar, Beck-Wödl, Grasshoff, Haack, Bijlsma, Ruivenkamp, Lausberg, Elbracht, Haanpää, Koillinen, Heinrich, Rost, Jamra, Popp, Koch-Hogrebe, Rostasy, López-González, Sanchez-Soler, Macedo, Schmetz, Steinborn, Weidensee, Lesmann, Marbach, Caro, Schaaf, Krawitz, Wieczorek, Kaiser and Kuechler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antje Kampmeier, YW50amUua2FtcG1laWVyQHVuaS1kdWUuZGU=; Alma Kuechler, YWxtYS5rdWVjaGxlckB1bmktZHVlLmRl