Abstract
Cathepsin K (Ctsk) is a cysteine protease of the papain superfamily initially identified in differentiated osteoclasts; it plays a critical role in degrading the bone matrix. However, subsequent in vivo and in vitro studies based on animal models elucidate novel subpopulations of Ctsk-expressing cells, which display markers and properties of mesenchymal stem/progenitor cells. This review introduces the function, identity, and role of Ctsk+ cells and their therapeutic implications in related preclinical osseous disorder models. It also summarizes the available in vivo models for studying Ctsk+ cells and their progeny. Further investigations of detailed properties and mechanisms of Ctsk+ cells in transgenic models are required to guide potential therapeutic targets in multiple diseases in the future.
Introduction
As one of the most efficacious lysosomal cysteine proteases of the papain superfamily initially identified in differentiated osteoclasts (Drake et al., 1996), cathepsin K (Ctsk) is now known to be expressed in various tissues and stem/progenitor cells, such as periosteal stem cells (PSCs) (Debnath et al., 2018; Han et al., 2019), Ctsk+ chondroid progenitors (CCPs) (Yang et al., 2013), tendon-derived progenitor cells (TDPCs) (Feng et al., 2020), breast cancer cells, prostate cancer cells (Littlewood-Evans et al., 1997; Brubaker et al., 2003), neurons (Bernstein et al., 2007), failing heart, atherosclerotic lesions (Sukhova et al., 1998; Shi et al., 1999; Lutgens et al., 2006), bronchial and alveolar epithelial cells, alveolar macrophages (Bühling et al., 1999; Haeckel et al., 1999; Bühling et al., 2001), and white adipose tissue (Soukas et al., 2000; Chiellini et al., 2003). In addition, Ctsk plays a critical role in degrading the bone matrix and contributes to osteoclast-mediated bone resorption. Osteoporosis occurs when the Ctsk-induced bone resorption exceeds bone formation. Furthermore, Ctsk is involved in the pathogenesis of pulmonary fibrosis (Bühling et al., 2004), atherosclerotic plaque progression (Guo et al., 2009), and some tumor metastasis, such as epithelial ovarian cancer, melanoma, prostate cancer, and breast cancer (Brubaker et al., 2003; Munari et al., 2017; Petricevic et al., 2017; Fan et al., 2018; Gu et al., 2019). In this review, we mainly discuss the available animal models for studying Ctsk+ cells and their progeny, identity, role of Ctsk-labeled nonosteoclast cells in the skeletal system, and translational applications of Ctsk inhibitors in bone metabolic diseases.
Cathepsin K: Collagenolytic Cysteine Protease
Ctsk is a cysteine protease of the papain superfamily, and it is mainly known for its significant resorptive function in bones (Inaoka et al., 1995; Drake et al., 1996). Osteoclasts cling to the bone surface via a specialized cell-matrix adhesion structure, the sealing zone (Takito et al., 2018). Ctsk is predominantly expressed in mature osteoclasts and is secreted along with protons into the resorption lacunae. The low pH environment of the resorption lacunae acidifies the externally mineralized component of the bone so that Ctsk exerts its proteolytic function on the exposed organic matrix. The bone extracellular organic matrix is composed of collagen Ⅰ (90%), which is formed by a triple helix with a telopeptide end (Gistelinck et al., 2016). In addition, Ctsk has a stronger collagenolytic ability than other proteases, such as matrix metalloproteinases and neutrophil serine elastase. It cleaves the triple helix across all three chains and attacks the telopeptides, while other proteases can only degrade telopeptides (Garnero et al., 1998; Kafienah et al., 1998). Once collagen Ⅰ is degraded, the products of telopeptide crosslinks from the N- and C-terminal ends are released into the circulatory system. They are detected in the blood and urine samples as markers of bone resorptive activity in osteoporosis patients. Impressively, a cross-sectional study with 1752 postmenopausal Chinese women indicated no evident connection between serum Ctsk and bone mineral density (BMD) (Gao et al., 2020). In addition, some studies have shown that Ctsk has a productive capacity for dissolving collagen Ⅱ, the primary protein marker of cartilage (Kafienah et al., 1998; Hou et al., 2001; Mort et al., 2016). The function of Ctsk in osteoclasts has been studied extensively, but its exact role in the newly discovered stem/progenitor cells remains unclear.
In Vivo Models to Study Ctsk+ Cells and Their Progeny
The generation of transgenic models has contributed to visualizing and isolating Ctsk+ cells, and we can follow the action of Ctsk+ cells in vivo through reporter genes. The Cre recombinase technology can be used to genetically control the expression of additional genes under the Ctsk promoter. The interest focused initially on osteoclasts in bone remodeling, and it recently extended to other subtypes of stem/progenitor cells whose properties are consistent with the mesenchymal lineage. Although in vivo models remarkably target particular Ctsk+ cell subsets, they do overlap to a certain extent. Hence, a combination of models is preferable to study the Ctsk+ cells and their progeny.
Cathepsin K Knockout Mice
Genomic structure and intron sizes of Ctsk in mice indicate that the initiation codon ATG is located at exon 2, and the termination codon TGA is at exon 8, which is highly similar to the human homolog. The first cathepsin K knockout (Ctsk−/−) mouse model was established to explore the role of Ctsk in bone resorption. They inserted the neomycin phosphotransferase gene (Neo) cassette into a HindIII restriction site at exon 7, which mimicked a premature translational termination codon, to disrupt Ctsk transcription in embryonic stem cells (Gelb et al., 1996; Saftig et al., 1998). However, some abnormalities in pycnodysostosis, such as short stature and craniofacial defects, are not seen in Ctsk−/− mice. cathepsin K deficient mice proved to be a survivor with intact fertility. In addition, Ctsk−/− mice displayed no noticeable phenotypic abnormalities until they were 10 months old (Li et al., 2006). Another Ctsk−/− mouse model was set up by targeting the coding region of cathepsin exon 3 via homologous recombination (Gowen et al., 1999); the histological and radiographic analyses revealed osteopetrosis of the long bones and vertebrae. The Ctsk deletion in mice increased the RANKL/OPG ratio and mediated osteoclastogenesis (Kiviranta et al., 2005). Splenomegaly emerged in both pycnodysostosis and Ctsk−/− mouse models, thereby compensating for the reduced bone marrow space. Gowen et al. (Gowen et al., 1999) did not find any apparent abnormalities in other organs/tissues. Taken together, these mice may represent a valuable animal model for pycnodysostosis. In addition to osteoporosis, the Ctsk−/− mice are utilized to study the role of Ctsk in various skeletal disease models. In Ctsk−/− mice, a closed stabilized femoral fracture “Einhorn” model was developed to evaluate the effects of Ctsk inhibition on fracture healing (Gentile et al., 2014). Surprisingly, the Ctsk−/− mice had increased callus mineralization and improved callus strength compared to the wild-type mice (same sex and age). Kozawa et al. (Kozawa et al., 2012) reported that the progression of joint instability-induced osteoarthritis in Ctsk−/− mice was slower than that in the wild-type control group. In rheumatoid arthritis models, the C57BL/6J background Ctsk−/− mice hybridizing with human TNF-transgenic mice highlighted the crucial role of Ctsk in reducing inflammation and bone erosion (Hao et al., 2015). The bone volume of Ctsk−/−-prednisolone mice indicated that Ctsk deletion in part helps rescue glucocorticoid-induced osteoporosis (GIO) (Yang et al., 2018), thereby making the Ctsk inhibitors effective in GIO prevention. Moreover, no animal models of Ctsk overexpression have been established to date. Ctsk overexpression may enhance bone resorption of osteoclasts and cause an osteoporosis phenotype, thereby further confirming the therapeutic effects of Ctsk deletion or Ctsk inhibitor on various diseases.
Ctsk-GFP/Ctsk-YFP
To et al. (To et al., 2012) established an osteoclast reporter line in medaka wherein the Ctsk promoter controlled the expression of green fluorescent protein (GFP). A 3.18-kb upstream regulatory region of the medaka Ctsk gene, including 80 nucleotides of exon 1, was cloned in front of mEGFP in an I-SceI meganuclease vector. Chatani et al. used a 3-kb upstream sequence of the Ctsk gene, a slightly shorter promoter, to generate a Ctsk-GFP line comparable to the former (Chatani et al., 2011). Interestingly, Ctsk is endogenously expressed in various tissues, such as the heart, a single neuron in the brain, and the pineal gland, but it is not detected in transgenics. It shows selectivity in cell subsets, and the used promoter lacks regulatory elements that can drive the expression in these nonskeletal tissues. However, for the most part, in situ ribonucleic acid (RNA) hybridization revealed that Ctsk:mEGFP expression was comparable to endogenous Ctsk expression of the protein. The Ctsk:mEGFP expression in tartrate-resistant acid phosphatase-positive (TRAP+) cells started 12 days postfertilization (To et al., 2012). The author also used Ctsk:mEGFP and mCherry under the control of the Osterix promoter to observe the interaction between osteoclasts and osteoblasts in vivo. However, a medaka osteoclast reporter line that expressed mEGFP under the control of the Ctsk promoter reported revealed destructive osteoclast activities and vertebral deformations in adult medaka (To et al., 2015). After the termination of the Ctsk expression of the protein, fluorescence could still be detected due to the turnover of GFP. Reporter genes with an upstream floxed STOP cassette in mice help trace the lineage generations with Ctsk+ progenitor populations, thereby unavoidably making it difficult to distinguish the targeted Ctsk+ cells from the lineage-traced ones.
Ctsk-Cre/Ctsk-CreERT2
The Cre-loxP-mediated recombination system (the “Cre-loxP system”) is a powerful and broadly used genetic experimental tool for selectively deleting, exchanging, or inserting genes of interest in specific discrete cells. The “floxed” loci (flanking DNA segments) are readily formed in mice by targeting embryonic stem cells, although blastocysts are also workable in mice (Sauer, 1998; McLellan et al., 2017). Several Ctsk-Cre lines are generated to direct recombination to osteoclasts, chondrocytes, PSCs, CPCs, and TDPCs. Specifically, once phenotypic differences occur after conditionally deleting the targeted gene in Ctsk-Cre cells in the bone, identifying the exact Ctsk+ cell type that made the difference is controversial. Although the Ctsk gene promoter helps narrow the range of cells that express Cre, the potential off-target Cre expression is detected in other tissues, including the brain, colon, heart, kidneys, liver, lungs, spleen, stomach, and ovary (Chiu et al., 2004). There is no perfect solution at the moment, but the off-target effects need to be considered and rigorous tests need to be performed after a mouse model is constructed.
The Ctsk-CreERT2 mouse model is a sophisticated genetic model based on the Ctsk-Cre model that controls Ctsk lineage temporally. The Ctsk promoter drives the expressions of Cre and estrogen receptor ligand-binding domain fusion proteins (Cre-ER-LBD). In the absence of tamoxifen, the Cre-ER-LBD fusion proteins are sequestered in the cytoplasm using heat shock protein 90 (HSP90). Tamoxifen, after injection, replaces HSP90 and binds with cre-ER fusion proteins to initiate recombination. In this process, the cre-ER is not responsive to estrogen and is not influenced by estrogen fluctuation (McLellan et al., 2017). Cre-ERT2 is the most common tamoxifen-inducible Cre transgene, which showed ten-fold greater sensitivity to tamoxifen than cre-ERT. Polymerase chain reaction, histological analyses, and E17.5 embryos showed that Ctsk-cre-ERT2 was expressed predominantly in bones (Sanchez-Fernandez et al., 2012). Furthermore, this improved genetic tool avoided some embryonic and postnatal gene lethality and allowed the compilation of genes at any time during development or adulthood. However, tamoxifen served as an active, selective estrogen receptor modulator that affects bone homeostasis. It was required in the control group with the same amount to nullify the tamoxifen effect to obtain comparable consequences.
Identity of Ctsk+ Cells in the Skeletal System
The abovementioned transgenic models have helped identify different subsets of Ctsk+ cells and their progeny. This section discusses the novel nonosteoclast Ctsk+ cell subsets characterized to date in the skeletal system.
Ctsk+ Chondrocytes and Fibroblast-Like Synoviocytes
Random sequencing of expressed sequence tags from cDNA libraries and Ctsk immunostaining indicated that Ctsk is predominantly stained in mature osteoclasts. Moreover, Ctsk is granularly located at a single pole of the osteoclasts (Drake et al., 1996; Hussein et al., 2017; Debnath et al., 2018). Using the same methodology, Ctsk is moderately expressed in osteoblasts and in some osteocytes underlying the matrix of metaphyseal trabecular femoral heads. However, the Ctsk expression of the protein was reduced during bone matrix formation (Mandelin et al., 2006; Hussein et al., 2017; Lotinun et al., 2019). Immunohistochemical analysis conducted in the degradation area, weight-bearing areas of knee cartilage, and menisci in osteoarthritis patients showed that chondrocytes and fibroblast-like synoviocytes expressed Ctsk. Quantitative morphometric analysis showed enhanced expression compared with healthy subjects (Morko et al., 2004; Kozawa et al., 2016). However, the lack of genetic markers to identify stem/progenitor cells, such as PSCs, and the absence of distinction among various progenitor cell types restrict the further exploration of the properties of these cells (Figure 1).
FIGURE 1
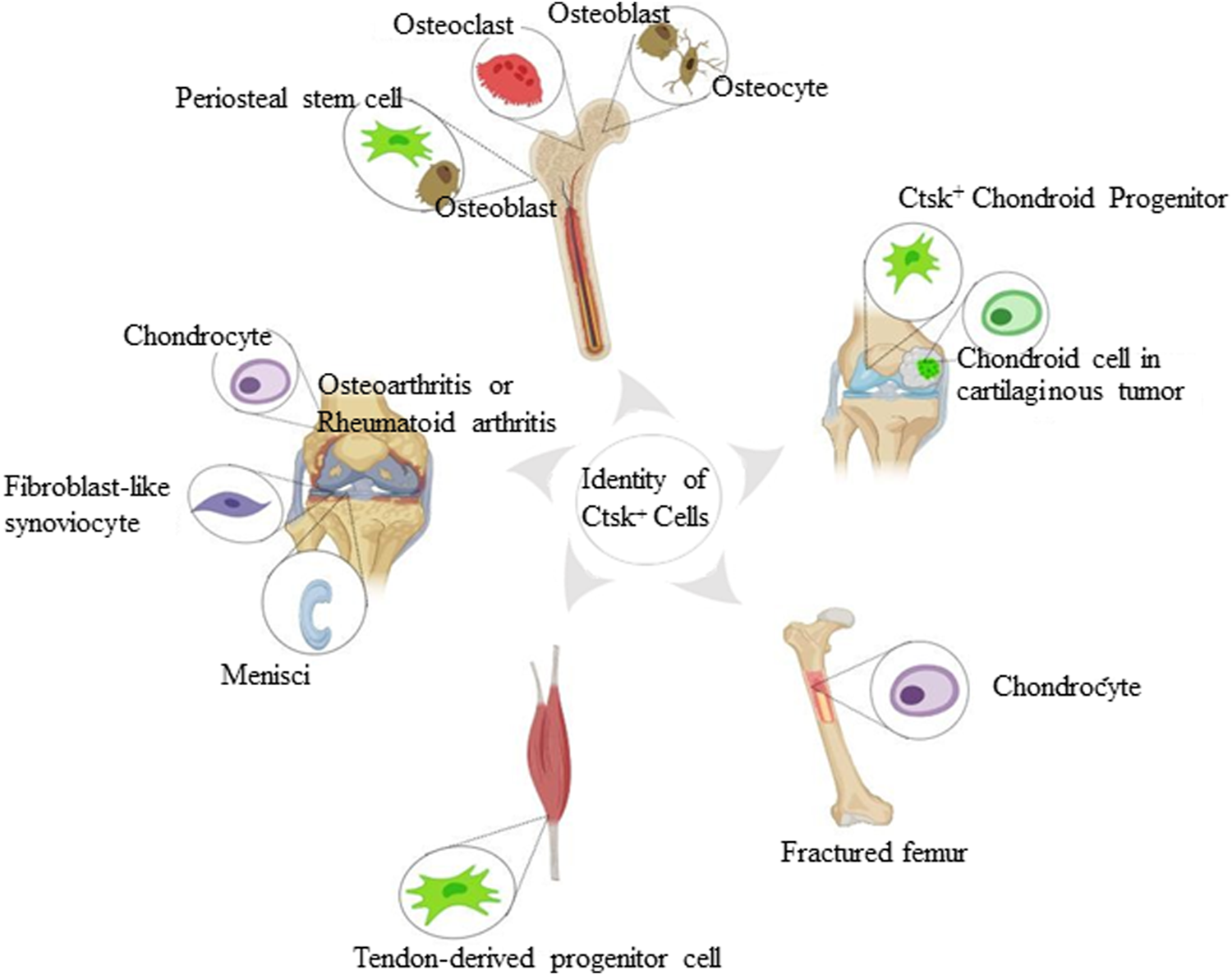
Identity of Ctsk+ cells found in the skeletal system. The schematic depiction shows subsets of Ctsk+ cells, including mature osteoclasts, osteoblasts, osteocytes, chondrocytes, and some mesenchymal stem/progenitor cells. Mature osteoclasts, osteoblasts, and osteocytes are displayed according to Ctsk immunostaining. Ctsk+YFP+ stem/progenitor cells are represented in green.
Ctsk+ Chondroid Progenitors
Yang et al., for the first time, found Ctsk-positive cells in extramedullary bones and confirmed the nonosteoclast nature of these cells using Ctsk-Cre transgenic mice (Yang et al., 2013). To trace and characterize this subpopulation of Ctsk+ cells, they used Rosa26LSL-lacZ (R26LSL-lacZ) or Rosa26LSL-YFP (R26LSL-YFP) Cre reporter mice. Ctsk-positive cells were a subpopulation of perichondrial cells in the “groove of Ranvier.” The perichondrial groove of Ranvier has been reported to serve as a potential reservoir of cartilaginous precursor stem cells. These cells could migrate to the growth plate and knee joint to maintain cartilage homeostasis (Fenichel et al., 2006; Karlsson et al., 2009). Furthermore, the Ctsk-yellow fluorescent protein (YFP+) was detected in the articular cartilage and growth plate (Yang et al., 2013). Usami et al. revealed that ZsG+ cells occupied the majority of cells in the perichondrial groove of Ranvier at the postnatal age of 7 days (P7) of the Ctsk-cre;Rosa26ZsGreen mouse system and showed up in the epiphyseal cartilage around P14. Unlike what Yang et al. had observed, these ZsG+ cells did not migrate to the growth plate at P28 or P70 (Usami et al., 2019). Few red fluorescent protein-positive (RFP+) cells in Col2-CreERT;RFP mice were detected in the perichondrial groove of Ranvier 3 or 7 days after injection, further indicating that chondrocytes in the growth plate did not originate from the perichondrial groove of Ranvier from the neonatal to the growth stage. These inconsistent arguments may be attributed to different reporter mice, and cells in the perichondrial groove of Ranvier are not the significant sources that produce cells in the growth plate during postnatal growth under physiological conditions. However, they may increase the odds of differentiating into chondrocytes and provide osteoblasts under pathological circumstances. Interestingly, CCPs have the capacity of in vitro osteogenesis, adipogenesis, and chondrogenesis. They display CD44, CD90, CD166 (mesenchymal progenitor markers), Stro1, and Jagged1 (presumptive chondroprogenitor markers) at a more intense level when CCPs are under pathological conditions. Nevertheless, CCPs are a small population, and their properties and detailed biochemistry need further study.
Ctsk+ Periosteal Mesenchymal Stem/Progenitor Cells
Bones contain discrete multipotent stem cell populations that can generate various lineages (Chan et al., 2015; Worthley et al., 2015; Debnath et al., 2018; Gao et al., 2019). Hence, the lack of genetic markers distinguishing periosteal mesenchyme from endosteal mesenchyme has been a critical limitation in identifying periosteal stem/progenitor cells. Intriguingly, Debnath et al. observed CTSK-mGFP cells in periosteal mesenchyme of Ctsk-cre;mTmG reporter mice on embryonic day 14.5 (E14.5), and these cells were detected in the endosteal marrow compartment as well on postnatal day 10. The CTSK-mGFP cells in the endosteal marrow compartment were confirmed to be osteoclasts by flow cytometry and co-staining for TRAP (Debnath et al., 2018; Han et al., 2019). The CTSK-mGFP cells in the periosteum manifested CD45−, TER119−, and CD31− (hereafter Lin−). Fractionation of CTSK-mGFP mesenchymal cells using multicolor flow cytometry showed three distinct populations, including PSCs (CD200+ and CD105−), periosteal progenitor 1 (PP1) (CD200− and CD105−), and periosteal progenitor 2 (PP2) (CD105+, CD200variable), where PSCs exhibited self-renewal ability and multipotency of mesenchymal stem cells, and ranked the most dedifferentiation (Table 1). Moreover, a subpopulation of CTSK-mGFP cells also expressed Gremlin1 and Nestin, but the CTSK-mGFP cells did not display markers of hematopoietic mesenchymal cells, such as LEPR, CD146, and CD140α (Sacchetti et al., 2007; Morikawa et al., 2009; Pinho et al., 2013; Zhou et al., 2014; Debnath et al., 2018). Compared with non-Ctsk MSCs (Lin−, 6C3−, THY−, CD200+, CD105−, and GFP−), PSCs mediated the intramembranous bone formation. In contrast, non-Ctsk MSCs processed endochondral ossification with hematopoietic recruitment and cartilage formation, although they expressed the same genetic markers of mesenchymal stem/progenitor cells, including Runx2 and Sox9 (Chan et al., 2009). PSCs were found in the sutures of the calvarium, while CTSK-mGFP PP1, PP2, THY+, CD146+, and SCA1+ cells appeared outside the sutures of the calvarium. Craniofacial bones are formed mainly through intramembranous ossification (Maruyama, 2019), thereby indicating that PSCs generate bones in the same way. In parallel, Han et al. further demonstrated that Ctsk could label periosteal mesenchyme (Han et al., 2019). Rosa26-Ai9 reporter mice showed that Ctsk-Ai9+ cells expressed the common stem cell markers, including Sca1, CD24, CD44, CD49f, and CD146. Moreover, alkaline phosphatase (ALP), Alcian blue, and oil red O staining indicated the multidirectional differentiation ability of the periosteal Ctsk+ cells, which was consistent with the observations of Debnath et al.
TABLE 1
| Ctsk-Labeled cells | Genetic markers | Related diseases | Potential therapeutic targets | References |
|---|---|---|---|---|
| Osteoclasts | TRAP+ | Osteoporosis | Cathepsin K inhibitor |
Drake et al. (1996) Brömme et al. (2016) McClung et al. (2019) |
| Chondrocytes and fibroblast-like synoviocytes | Not mentioned | Osteoarthritis (OA) and rheumatoid arthritis (RA) | Cathepsin K inhibitor |
Morko et al. (2004) Kozawa et al. (2016) |
| Ctsk + chondroid progenitors (CCPs) | CD44+, CD90+, CD166+, Stro1+, and Jagged1+ | Metachondromatosis (MC) | Smoothened inhibitor |
Karlsson et al. (2009) Yang et al. (2013) Usami et al. (2019) |
| Periosteal mesenchymal stem cells (PSCs) | CD200+ and CD105− | Osteosarcoma and bone regeneration | mTORC1 inhibition and cathepsin K inhibitor |
Gentile et al. (2014) Debnath et al. (2018) Han et al. (2019) |
| Periosteal progenitor 1 (PP1) | CD200− and CD105− | |||
| Periosteal progenitor 2 (PP2) | CD105+ and CD200variable | |||
| Tendon-derived progenitor cells (TDPCs) | Scx+, CD44+, CD105+, Nestin+, and Sca1+ | Heterotopic ossification (HO) | JQ1 |
Bi et al. (2007) Feng et al. (2020) |
Genetic markers of Ctsk-labeled cells and related diseases in the skeletal system.
Ctsk+ TDPCs
In vitro TDPCs are widely studied, thereby showing excellent self-renewal capacity and multipotency (Bi et al., 2007) of TDPCs. However, their in vivo studies are rare. Notably, Ai9 reporter mice showed that Ctsk-Cre could label a subpopulation of TDPCs (Feng et al., 2020). These Ctsk-cre–expressing cells were detected in most cells within the Achilles tendon, quadriceps tendon, and tendinous insertions of the patella in vivo. They also expressed tendon-related marker Scx after crossbreeding with ScxGFP mice. Ctsk+Scx+ cells classified using fluorescence-activated cell sorting were expressed as CD44, CD105, Nestin, Sca1, and other genetic markers of stem/progenitor cells, including CD24 and CD200. In addition, Ctsk+Scx+ cells exhibited higher differentiation potential into osteoblasts, chondrocytes, and adipocytes than Ctsk+Scx– tendon-derived cells, while neither Ctsk–Scx– nor Ctsk–Scx+ tendon-derived cells lacked the capacity.
Role of Ctsk+ Cells in Pathogenesis and Therapeutic Strategies
Many studies have worked on the role of Ctsk-expressing cells in bone remodeling and cellular regulation (especially progenitor/stem cells) (Figure 2). These studies suggest that Ctsk-labeled cells may be further investigated to understand the pathogenesis of various skeletal disorders and could provide an attractive therapeutic target in clinical practice.
FIGURE 2
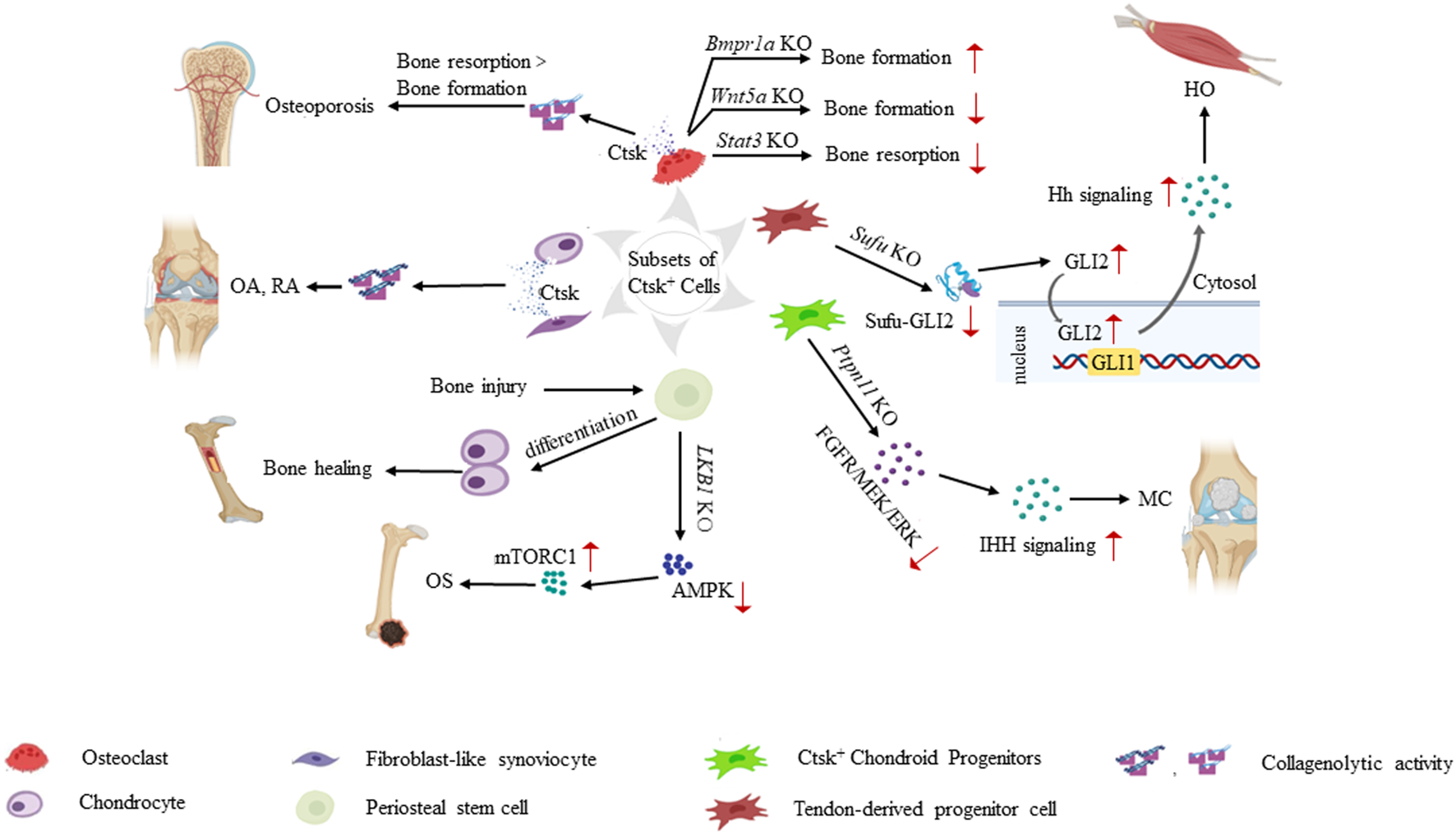
Role of Ctsk+ cells in the pathogenesis of various skeletal system diseases. Ctsk+ cells help determine the origin cells of some musculoskeletal system-related diseases. Increased secretion of Ctsk from mature osteoclasts and chondrocytes leads to enhanced collagenolytic activity and subsequently contributes to osteoporosis, OA, and RA. Knockouts of some key genes, such as Bmpr1a, Wnt5a, and Stat3 in Ctsk+ osteoclasts break the balance of bone formation and resorption. In Ctsk+ mesenchymal stem/progenitor cells, Ctsk serves as a genetic marker and helps us deeply understand the property and function of mesenchymal stem/progenitor cells. PSCs play a significant role in intramembranous bone formation under physiological conditions. They differentiate into chondrocytes after bone injury, thereby participating in endochondral bone formation to promote bone healing. PSCs also serve as pathological precursors in osteogenic tumors through the LKB1/AMPK/mTORC1 signaling pathways. Ptpn11 depletion in CCPs leads to MC via the ERK and Ihh signaling pathways. Ctsk+ TDPCs with targeted Sufu deletion result in heterotopic HO via Hh pathway activation. Translocation of GLI2 from the cytoplasm to the nucleus transactivates the GLI1 promoter. Increased Gli1/Gli2 in Ctsk+ TDPCs enhances the Hh signaling activity and promotes HO progression.
Ctsk+ Osteoclasts and Osteoporosis
Ctsk-Cre mice have been widely used to study the functional properties of osteoclasts in preclinical models of osteoporosis. BMP signal in osteoblasts is elucidated to promote bone formation via the canonical Wnt signaling pathway (Kamiya et al., 2008a; Kamiya et al., 2008b; Okamoto et al., 2011). Tamoxifen-induced osteoblast-specific ablation of Bmpr1a decreases the osteoclast number and bone resorption activity in mice. Conversely, conditional knockout of Bmpr1a in differentiated osteoclasts in Ctsk-Cre mouse models enhances osteogenesis. Hence, Ctsk+ osteoclasts can interfere with the activities of osteoblasts in a BMPR1A signaling-dependent manner. A previous study found that Wnt5a secreted by osteoblasts activated Rank expression to stimulate osteoclastogenesis (Maeda et al., 2012). Recently, Wnt5a loss-of-function in Ctsk+ osteoclast was tested by intercrossing Wnt5afl/f and Ctsk-Cre mouse lines. Ctsk-Cre;Wnt5afl/f mice exhibited reduced trabecular and cortical bone without osteoclast elevation (Roberts et al., 2020). Stat3 participated in maintaining bone homeostasis in osteoblasts (Zhou et al., 2011). Furthermore, Stat3 deletion in bone marrow macrophages was reported to reduce NFATc1 expression and decrease osteoclast differentiation, and Stat3 deletion in osteoclasts led to a decreased osteoclast activity (Yang et al., 2019; Yang et al., 2020). These molecules exerted exactly opposing effects on bone remodeling as they were derived from different types of cells. Mechanistically, the exact role of Ctsk in osteoclasts was elucidated by Lotinun et al. (Lotinun et al., 2013). They used a specific conditional knockout of CD11b-Cre to exclusively target the monocyte-macrophage lineage to generate CD11b-Cre;Ctskfl/fl. Ctsk deleted in osteoclasts revealed an increased bone formation rate, while cathepsin K knockout in Osx-Cre mice specifically targeted to osteoblasts did not exhibit any skeletal phenotype.
Ctsk+ Chondrocytes and Synovial Cells and Osteoarthritis and Rheumatoid Arthritis
Progressive cartilage damage is the key process that contributes to osteoarthritis (OA) and rheumatoid arthritis (RA) (Pap and Korb-Pap, 2015). Ctsk oligomerized with chondroitin sulfate, particularly chondroitin-4-sulfate molecules, was likely to degrade collagen Ⅱ (Li et al., 2000; Li et al., 2002). Moreover, cathepsin K mRNA expression increases in the articular chondrocytes near the matrix destruction of the transgenic osteoarthritic model at the onset of cartilage degeneration. In contrast, only a tiny minority of chondrocytes were observed expressing Ctsk in the control mice. With age, Ctsk also increased in the control group with developed OA (Morko et al., 2004). Accordingly, Ctsk−/− mice were used to investigate the role of Ctsk in OA progression (Li et al., 2006). The study showed that Ctsk deficiency slowed down OA development, particularly in the early to intermediate stages compared with Ctsk+/+ mice. Ctsk deficiency could cause delayed remodeling of subchondral and calcified cartilage, which protects the articular cartilage (Soki et al., 2018). There existed a positive correlation between the severity of OA and the expression of Ctsk in chondrocytes (Konttinen et al., 2002). Consistently, Ctsk was expressed in chondrocytes in humans, and its collagenolytic activity was augmented in OA patients (Kozawa et al., 2016). Interestingly, a recent study demonstrated that mechanical stress loading enhanced Ctsk expression of protein in human chondrocytic HCS-2/8 cells, which was suppressed by hyaluronan (Suzuki et al., 2020). Using the pharmacological approach, current treatments of OA act mainly on symptom improvement without slowing down the OA development.
Furthermore, RA-associated joint destruction is caused by synovial fibroblast-mediated collagen degradation. Cartilage degradation started at the most superficial layer in the monkey collagen-induced arthritis (CIA) model (Tanaka et al., 2016). Rheumatoid arthritis and periodontitis share many pathological features; Ctsk deletion in the RA and periodontitis mice models ameliorates inflammation and bone erosion in both of them owing to its shared osteoimmune role (Hao et al., 2015). Ctsk is activated by immune cells through the TLR4, 5, and 9 signaling pathways, and the inhibition of Ctsk reduces TLR9 expression and downregulates the autophagy signaling pathway (Wei et al., 2020). In addition, in the CIA mouse model, the inhibition of Ctsks alleviates bone erosion, cartilage degradation, and inflammation at joints and enhances bone strength (Svelander et al., 2009; Yamashita et al., 2018).
Ctsk+ Chondroid Progenitor Cells and Cartilaginous Tumor
Metachondromatosis (MC), a rare hereditary disease of the skeletal system, is characterized by multiple osteochondroma and enchondroma. Hereditary multiple osteochondroma is a benign tumor that grows outward from the metaphyses of long bones, and enchondroma is a benign cartilaginous tumor that grows endophytic to the bone (Pannier and Legeai-Mallet, 2008). Proteintyrosine phosphatase nonreceptor type 11 (ptpn11) encodes the nonreceptor proteintyrosine phosphatase SHP2. Targeted deleting of Ptpn11 in chondrocytes contributed to the MC phenotype (Kim et al., 2014). In addition, Ptpn11 depletion in Ctsk-expressing cells caused bone phenotypes consistent with MC using Ctsk-cre;Ptpn11 fl/fl (Ctsk-KO) transgenic mice (the Ctsk promoter is active only in mature osteoclasts). In contrast, mild osteopetrosis presented in lysozyme M-cre;Ptpn11 fl/fl mice (the LysM promoter is active in monocytes, macrophages, and osteoclast precursors) (Faust et al., 2000; Yang et al., 2013). Lineage tracing indicated that Ctsk+ perichondral cells within “Groove of Ranvier” are labeled pathological cells. Notably, YFP+ cells had entered and differentiated into ectopic cartilaginous tissues by postnatal week 2 in Ctsk-KO/YFP reporter mice. The CCPs differentiate into chondrocytes at different stages of development in lesions using cell morphology and Col2α1 and Col10α1 immunostaining, including proliferating, prehypertrophic, and hypertrophic chondrocytes. Ptpn11 is required for fibroblast growth factor/mitogen-activated protein kinase/extracellular signal-regulated kinase (FGFR/MEK/ERK)-dependent pathways in various growths and development-related signaling pathways (Grossmann et al., 2010). The Ctsk-cre;Ptpn11 fl/fl mice showed significantly decreased ERK expressions. Smoothened inhibitor PF-04449913 treatment is believed to suppress the Ihh target gene expression and reduce the number of exostoses. It slows down the MC progression in Ctsk-cre;Ptpn11 fl/fl mice. Almost all chondroid tumor cells were YFP+ in Ctsk-KO/YFP reporter mice, thereby indicating that specific genetic blockade of CCPs could serve as pathological cells that contribute to the pathogenesis of MC, although CCPs are rare. Liver kinase b1 (Lkb1) is a master serine/threonine kinase that plays a crucial role in energy homeostasis and cell growth (Shaw, 2009). Lkb1 deficiency in periosteum-derived Ctsk+ cells may cause an osteogenic tumor-like phenotype (Han et al., 2019). However, the loss of Lkb1 in chondrocytes contributes to enchondroma-like tumors (Lai et al., 2013). Accordingly, disruptions of Lkb1 in specific types of cells lead to different kinds of tumor-like phenotypes via respective signaling pathways. Further experimental studies on the detailed properties of CCPs are needed to understand how these crucial regulators mediate molecular actions in CCPs and seek more novel attractive targets for therapeutic strategies for cartilaginous diseases.
Ctsk+ Periosteal Mesenchymal Progenitors and Osteogenic Tumor
Osteosarcoma (OS) is a malignant neoplasm with an unfavorable prognosis that primarily occurs in children and adolescents. Previous studies have reported that mesenchymal progenitors (Prx1-cre), osteoblast precursors (Osx-cre), and osteoblast-committed cells (Col1a1-Cre and OCN-Cre), which are of mesenchymal origin and osteogenic lineage, can function as the pathological precursors contributing to OS (Walkley et al., 2008; Calo et al., 2010; Abarrategi et al., 2016; Del Mare et al., 2016). The periosteum-derived Ctsk+ cells congruously acted with mesenchymal stem cells; therefore, the deletion of LKB1 in Ctsk-expressing periosteal mesenchymal cells caused osteogenic tumor, as expected (Han et al., 2019). This finding advanced knowledge of the pathogenesis of osteogenic tumors in a specific type of cell. It stimulated targeted therapeutic approaches for new strategies, although the exact role of Ctsk in these stem cell-like periosteal cells remained unknown. Liver kinase B1 is a tumor suppressor, and a decreased Lkb1 protein expression was detected in 41% of patients with osteosarcoma (Presneau et al., 2017). Lkb1 deficiency stimulates the proliferation and osteogenesis of Ctsk+ periosteal mesenchymal cells. In addition, mTORC1 activation observed in OS patients is the downstream of LKB1-dependent adenosine monophosphate (AMP) kinases (AMPKs). Simultaneously, knockout of Lkb1 in Prx1+ mesenchymal progenitors resulted in osteoid tumors transgressing the cortex and BM cavity. However, both the femur and tibia of Prx1-cre;Lkb1fl/fl mice displayed an enchondroma-like phenotype, which was different from the observation in Ctsk-cre;Lkb1fl/fl mice. This may be due to the Prx1+ and Ctsk+ mesenchymal progenitor distribution; Prx1+ mesenchymal progenitors reside in both the periosteum and bone marrow, and Ctsk+ mesenchymal progenitors are seen only in the periosteum (Wang et al., 2016; Han et al., 2019). Conditional knockout of Osterix (Osx) in Ctsk-Cre mice produces a marginal effect on endosteal bone mass and significantly decreases the periosteal bone formation (Debnath et al., 2018). These observations defined periosteum-derived Ctsk+ cells as a novel subpopulation of mesenchymal progenitors. Inspiringly, mTORC1 inhibition used in Ctsk-cre;Lkb1fl/fl mice may slow down tumorigenesis progression. In the context of unsatisfactory traditional chemotherapy treatment, LKB1/AMPK/mTORC1 signaling provides a novel targeted therapeutic approach for treating osteosarcoma.
Ctsk+ Periosteal Cells and Bone Regeneration
The periosteum is a reservoir of quiescent mesenchymal progenitor cells that facilitate endochondral bone formation to promote bone regeneration after bone injury (Colnot, 2009). The available evidence from preclinical studies has demonstrated that a subset of Ctsk-positive (PSCs) participate in intramembranous bone formation to maintain bone homeostasis under physiological conditions. However, when the bone is damaged, PSCs differentiate into chondrocytes and participate in endochondral bone formation to promote fracture healing. Ctsk+ PSCs displayed more robust proliferation and osteogenic differentiation ability than non-Ctsk+ PSCs after fracture (Debnath et al., 2018). Injuries stimulate the proliferation and osteogenic differentiation of PSCs. Periostin, expressed explicitly in the periosteum, creates an osteogenic microenvironment (Horiuchi et al., 1999). Bonnet et al. described periostin as the substrate for degradation by Ctsk and Ctsk inhibitors or deletion stimulated periostin and β-catenin expression (Bonnet et al., 2017). Deleting Ctsk also increased TRAP+ mononuclear cells and platelet-derived growth factor (PDGF)-BB secretion (Chen et al., 2007; Xie et al., 2014). PDGF-BB is necessary for Nestin+ and LepR+ periosteum-derived cells to migrate to the periosteal surface for bone formation and periosteum homeostasis (Gao et al., 2019). Periosteal osteoclast precursors (OCPs), instead of bone marrow osteoclast precursors, were significantly upregulated in the Ctsk−/− periosteum upon fracture (Walia et al., 2018). Its secretion of PDGF-BB and S1P in the periosteum stimulated osteogenic differentiation, thereby intensifying bone mineralization and strength. Meanwhile, PDGF-BB binding to the PDGF receptor may trigger the PI3K/Akt/FAK signaling pathway and promote angiogenesis coupling with osteogenesis (Gentile et al., 2014; Xie et al., 2014). However, a study indicated that OCPs from Ctsk−/− mice were disabled to migrate and engraft in the bone callus in fractured mice (Jacome-Galarza et al., 2014).
Ctsk+ TDPCs and Heterotopic Ossification
Heterotopic ossification (HO) is an aberrant phenomenon where the bone cells surround soft tissues, such as muscles, tendons, ligaments, and joints (Xu et al., 2018). Scx+ progenitor lineages are responsible for spontaneous ligament, tendon, joint, and trauma-induced HO (Dey et al., 2016; Agarwal et al., 2017). Recently, Ctsk+ TDPCs have been identified as a novel subtype of progenitors contributing to HO (Feng et al., 2020). The Ihh signaling activation in Ctsk-Cre cells produces MC (Yang et al., 2013). However, a disruption in the suppressor of fused (Sufu) in Ctsk-expressing cells, a suppressor regulating the Hh pathway, did not generate the presumptive phenotype. Instead, pathological bone formation is present in ligaments, tendons, joints, and other soft tissues. Zinc finger transcription factors GLI1 and GLI2 are critical in Hh signaling, while Sufu inhibits the translocation of GLI2 from the cytoplasm to the nucleus to transactivate the GLI1 promoter (Varjosalo and Taipale, 2008). It is encouraging to learn that Gli1/Gli2 deficiency in Ctsk+ TDPCs disrupted the Hh signaling stability and ameliorated HO progression, thereby providing the rationale for HO clinical treatment as an attractive therapeutic target. The expressed Ctsk+ cells increased the chondrogenic differentiation marker COLII and the osteogenic differentiation markers Opn, Ocn, and Alp. However, they decreased the tendon-related genes Scx, Mkx, and Tnmd, thereby suggesting that Ctsk+ TDPCs could differentiate into chondrocytes and osteoblasts; the Hh signaling activation changed the fate of Ctsk+ TDPCs. Gant61 was used to treat Hh-overactive tumors in mice (Lauth et al., 2007; Fu et al., 2013), but it failed to ameliorate HO caused by Sufu deficiency in Ctsk+ TDPCs. Feng et al. found a small-molecule inhibitor, JQ1, as the pharmacological inhibitor of Hh signaling. JQ1 downregulated Hh signaling in Ctsk+ TDPCs from Ctsk-cre;Sufufl/fl mice. Disappointedly, no CreERT2 mouse line was designed to observe the specific time when the cells were marked and to determine the exact role of Ctsk+ lineage cells in trauma-induced HO. Cumulatively, the molecular mechanisms and mouse models remain to be explored further.
Cathepsin K Inhibitor in Translational Applications
Ctsk inhibitors can treat skeletal diseases, such as osteoporosis, osteoarthritis, RA, and bone fracture, with odanacatib being the most well-known inhibitor. Previous studies have shown that odanacatib has therapeutic effects on osteoporosis and bone fracture in postmenopausal women (Bone et al., 2010; Eisman et al., 2011; Langdahl et al., 2012; Brixen et al., 2013; Cheung et al., 2014; Rizzoli et al., 2016). However, odanacatib increased the risk of stroke in phase III clinical trials; therefore, its use was discontinued (McClung et al., 2019). There may be overlapping disease pathways in the bone and cardiovascular biology, which should be further researched to guide therapies for these two diseases. The other two Ctsk inhibitors that progressed to clinical studies include ONO-5334 (Phase 1 and 2 studies) and MIV-711 (Phase 1 and 2 studies). ONO-5334 effectively elevated the BMD in a phase II trial for postmenopausal osteoporosis (Tanaka et al., 2017). It also prevented joint destruction in the monkey CIA model (Yamada et al., 2019). There are no obvious safety implications in the phase II study for osteoporosis; thus, ONO-5334 can be a potential drug for preventing joint destruction in RA patients. Furthermore, MIV-711, as a novel selective inhibitor, alleviated cartilage degradation in a phase-IIa trial, with a marginal effect on pain relief (Conaghan et al., 2020). In general, a Ctsk inhibitor is still a researchable hotspot for osteoporosis.
Conclusion
Abundant evidence demonstrates that Ctsk is a pivotal lysosomal cysteine protease that participates in the physiological process of bone resorption. Furthermore, Ctsk, a biomarker of some subsets of stem/progenitor cells, assists in determining the origin cells of skeletal diseases. To date, the pathogenesis and promising therapeutic values of these diseases in Ctsk+ stem/progenitor cells are scarce. The precise function of Ctsk secreted by these stem/progenitor cells remains unclear. One study showed that as a metastasis-related protein upregulated by the imbalance of intestinal microbiota (Li et al., 2019), Ctsk enhances the migration and motility of colorectal cancer cells. We may speculate that Ctsk promotes the migration of Ctsk+ stem/progenitor cells in bone homeostasis and fracture healing. Hence, the true nature of Ctsk+ cells in the in vivo models is the target of extensive studies. Data recorded from these genetic models are precious but require scrutiny and comprehensive analysis of their pitfalls. The role of Ctsk in some osseous diseases is well proven, but its role in other types of bone tumors, such as osteomalacia and rachitis, is unknown. Future studies with improved in vivo models would help define the nature of Ctsk+ cells and seek more salutary targets for the clinical treatment of these diseases.
Statements
Author contributions
CL contributed to concepts, article review and revision, and final approval of the article; NZ drafted the review article; and RL prepared the figures and table. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Key R&D Program of China (Grant No. 2019YFA0111900) and the National Natural Science Foundation of China (Grant Nos. 81922017, 81873669, and 81802209).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
References
1
Abarrategi A. Tornin J. Martinez-Cruzado L. Hamilton A. Martinez-Campos E. Rodrigo J. P. et al (2016). Osteosarcoma: Cells-Of-Origin, Cancer Stem Cells, and Targeted Therapies. Stem Cells Int.2016, 3631764. 10.1155/2016/3631764
2
Agarwal S. Loder S. J. Cholok D. Peterson J. Li J. Breuler C. et al (2017). Scleraxis-Lineage Cells Contribute to Ectopic Bone Formation in Muscle and Tendon. Stem Cells35 (3), 705–710. 10.1002/stem.2515
3
Bernstein H.-G. Bukowska A. Dobrowolny H. Bogerts B. Lendeckel U. (2007). Cathepsin K and Schizophrenia. Synapse61 (4), 252–253. 10.1002/syn.20358
4
Bi Y. Ehirchiou D. Kilts T. M. Inkson C. A. Embree M. C. Sonoyama W. et al (2007). Identification of Tendon Stem/progenitor Cells and the Role of the Extracellular Matrix in Their Niche. Nat. Med.13 (10), 1219–1227. 10.1038/nm1630
5
Bone H. G. McClung M. R. Roux C. Recker R. R. Eisman J. A. Verbruggen N. et al (2010). Odanacatib, a Cathepsin-K Inhibitor for Osteoporosis: a Two-Year Study in Postmenopausal Women with Low Bone Density. J. Bone Min. Res.25 (5), 937–947. 10.1359/jbmr.091035
6
Bonnet N. Brun J. Rousseau J.-C. Duong L. T. Ferrari S. L. (2017). Cathepsin K Controls Cortical Bone Formation by Degrading Periostin. J. Bone Min. Res.32 (7), 1432–1441. 10.1002/jbmr.3136
7
Brixen K. Chapurlat R. Cheung A. M. Keaveny T. M. Fuerst T. Engelke K. et al (2013). Bone Density, Turnover, and Estimated Strength in Postmenopausal Women Treated with Odanacatib: a Randomized Trial. J. Clin. Endocrinol. Metab.98 (2), 571–580. 10.1210/jc.2012-2972
8
Brömme D. Panwar P. Turan S. (2016). Cathepsin K Osteoporosis Trials, Pycnodysostosis and Mouse Deficiency Models: Commonalities and Differences. Expert Opin. Drug Discov.11 (5), 457–472. 10.1517/17460441.2016.1160884
9
Brubaker K. Vessella R. True L. Thomas R. Corey E. (2003). Cathepsin K mRNA and Protein Expression in Prostate Cancer Progression. J. Bone Min. Res.18 (2), 222–230. 10.1359/jbmr.2003.18.2.222
10
Bühling F. Gerber A. Häckel C. Krüger S. Köhnlein T. Brömme D. et al (1999). Expression of Cathepsin K in Lung Epithelial Cells. Am. J. Respir. Cell Mol. Biol.20 (4), 612–619. 10.1165/ajrcmb.20.4.3405
11
Bühling F. Reisenauer A. Gerber A. Krüger S. Weber E. Brömme D. et al (2001). Cathepsin K - a Marker of Macrophage Differentiation?J. Pathol.195 (3), 375–382. 10.1002/path.959
12
Bühling F. Röcken C. Brasch F. Hartig R. Yasuda Y. Saftig P. et al (2004). Pivotal Role of Cathepsin K in Lung Fibrosis. Am. J. Pathology164 (6), 2203–2216. 10.1016/s0002-9440(10)63777-7
13
Calo E. Quintero-Estades J. A. Danielian P. S. Nedelcu S. Berman S. D. Lees J. A. (2010). Rb Regulates Fate Choice and Lineage Commitment In Vivo. Nature466 (7310), 1110–1114. 10.1038/nature09264
14
Chan C. K. Seo E. Y. Chen J. Y. Lo D. McArdle A. Sinha R. et al (2015). Identification and Specification of the Mouse Skeletal Stem Cell. Cell160 (1-2), 285–298. 10.1016/j.cell.2014.12.002
15
Chan C. K. F. Chen C.-C. Luppen C. A. Kim J.-B. DeBoer A. T. Wei K. et al (2009). Endochondral Ossification Is Required for Haematopoietic Stem-Cell Niche Formation. Nature457 (7228), 490–494. 10.1038/nature07547
16
Chatani M. Takano Y. Kudo A. (2011). Osteoclasts in Bone Modeling, as Revealed by In Vivo Imaging, Are Essential for Organogenesis in Fish. Dev. Biol.360 (1), 96–109. 10.1016/j.ydbio.2011.09.013
17
Chen W. Yang S. Abe Y. Li M. Wang Y. Shao J. et al (2007). Novel Pycnodysostosis Mouse Model Uncovers Cathepsin K Function as a Potential Regulator of Osteoclast Apoptosis and Senescence. Hum. Mol. Genet.16 (4), 410–423. 10.1093/hmg/ddl474
18
Cheung A. M. Majumdar S. Brixen K. Chapurlat R. Fuerst T. Engelke K. et al (2014). Effects of Odanacatib on the Radius and Tibia of Postmenopausal Women: Improvements in Bone Geometry, Microarchitecture, and Estimated Bone Strength. J. Bone Min. Res.29 (8), 1786–1794. 10.1002/jbmr.2194
19
Chiellini C. Costa M. Novelli S. E. Amri E.-Z. Benzi L. Bertacca A. et al (2003). Identification of Cathepsin K as a Novel Marker of Adiposity in White Adipose Tissue. J. Cell. Physiol.195 (2), 309–321. 10.1002/jcp.10253
20
Chiu W. S. M. McManus J. F. Notini A. J. Cassady A. I. Zajac J. D. Davey R. A. (2004). Transgenic Mice that Express Cre Recombinase in Osteoclasts. Genesis39 (3), 178–185. 10.1002/gene.20041
21
Colnot C. (2009). Skeletal Cell Fate Decisions within Periosteum and Bone Marrow during Bone Regeneration. J. Bone Mineral Res.24 (2), 274–282. 10.1359/jbmr.081003
22
Conaghan P. G. Bowes M. A. Kingsbury S. R. Brett A. Guillard G. Rizoska B. et al (2020). Disease-Modifying Effects of a Novel Cathepsin K Inhibitor in Osteoarthritis. Ann. Intern Med.172 (2), 86–95. 10.7326/m19-0675
23
Debnath S. Yallowitz A. R. McCormick J. Lalani S. Zhang T. Xu R. et al (2018). Discovery of a Periosteal Stem Cell Mediating Intramembranous Bone Formation. Nature562 (7725), 133–139. 10.1038/s41586-018-0554-8
24
Del Mare S. Husanie H. Iancu O. Abu-Odeh M. Evangelou K. Lovat F. et al (2016). WWOX and P53 Dysregulation Synergize to Drive the Development of Osteosarcoma. Cancer Res.76 (20), 6107–6117. 10.1158/0008-5472.can-16-0621
25
Dey D. Bagarova J. Hatsell S. J. Armstrong K. A. Huang L. Ermann J. et al (2016). Two Tissue-Resident Progenitor Lineages Drive Distinct Phenotypes of Heterotopic Ossification. Sci. Transl. Med.8 (366), 366ra163. 10.1126/scitranslmed.aaf1090
26
Drake F. H. Dodds R. A. James I. E. Connor J. R. Debouck C. Richardson S. et al (1996). Cathepsin K, but Not Cathepsins B, L, or S, Is Abundantly Expressed in Human Osteoclasts. J. Biol. Chem.271 (21), 12511–12516. 10.1074/jbc.271.21.12511
27
Eisman J. A. Bone H. G. Hosking D. J. McClung M. R. Reid I. R. Rizzoli R. et al (2011). Odanacatib in the Treatment of Postmenopausal Women with Low Bone Mineral Density: Three-Year Continued Therapy and Resolution of Effect. J. Bone Min. Res.26 (2), 242–251. 10.1002/jbmr.212
28
Fan X. Wang C. Song X. Liu H. Li X. Zhang Y. (2018). Elevated Cathepsin K Potentiates Metastasis of Epithelial Ovarian Cancer. Histol. Histopathol.33 (7), 673–680. 10.14670/HH-11-960
29
Faust N. Varas F. Kelly L. M. Heck S. Graf T. (2000). Insertion of Enhanced Green Fluorescent Protein into the Lysozyme Gene Creates Mice with Green Fluorescent Granulocytes and Macrophages. Blood96 (2), 719–726. 10.1182/blood.v96.2.719
30
Feng H. Xing W. Han Y. Sun J. Kong M. Gao B. et al (2020). Tendon-derived Cathepsin K-Expressing Progenitor Cells Activate Hedgehog Signaling to Drive Heterotopic Ossification. J. Clin. Invest.130 (12), 6354–6365. 10.1172/jci132518
31
Fenichel I. Evron Z. Nevo Z. (2006). The Perichondrial Ring as a Reservoir for Precartilaginous Cells. In Vivo Model in Young Chicks' Epiphysis. Int. Orthop. (SICOT)30 (5), 353–356. 10.1007/s00264-006-0082-2
32
Fu J. Rodova M. Roy S. K. Sharma J. Singh K. P. Srivastava R. K. et al (2013). GANT-61 Inhibits Pancreatic Cancer Stem Cell Growth In Vitro and in NOD/SCID/IL2R Gamma Null Mice Xenograft. Cancer Lett.330 (1), 22–32. 10.1016/j.canlet.2012.11.018
33
Gao B. Deng R. Chai Y. Chen H. Hu B. Wang X. et al (2019). Macrophage-lineage TRAP+ Cells Recruit Periosteum-Derived Cells for Periosteal Osteogenesis and Regeneration. J. Clin. Invest.129 (6), 2578–2594. 10.1172/jci98857
34
Gao L.-h. Li S.-s. Yue H. Zhang Z.-l. (2020). Associations of Serum Cathepsin K and Polymorphisms in CTSK Gene with Bone Mineral Density and Bone Metabolism Markers in Postmenopausal Chinese Women. Front. Endocrinol.11, 48. 10.3389/fendo.2020.00048
35
Garnero P. Borel O. Byrjalsen I. Ferreras M. Drake F. H. McQueney M. S. et al (1998). The Collagenolytic Activity of Cathepsin K Is Unique Among Mammalian Proteinases. J. Biol. Chem.273 (48), 32347–32352. 10.1074/jbc.273.48.32347
36
Gelb B. D. Moissoglu K. Zhang J. Martignetti J. A. Brömme D. Desnick R. J. (1996). Cathepsin K: Isolation and Characterization of the Murine cDNA and Genomic Sequence, the Homologue of the Human Pycnodysostosis Gene. Biochem. Mol. Med.59 (2), 200–206. 10.1006/bmme.1996.0088
37
Gentile M. A. Soung D. Y. Horrell C. Samadfam R. Drissi H. Duong L. T. (2014). Increased Fracture Callus Mineralization and Strength in Cathepsin K Knockout Mice. Bone66, 72–81. 10.1016/j.bone.2014.04.032
38
Gistelinck C. Gioia R. Gagliardi A. Tonelli F. Marchese L. Bianchi L. et al (2016). Zebrafish Collagen Type I: Molecular and Biochemical Characterization of the Major Structural Protein in Bone and Skin. Sci. Rep.6, 21540. 10.1038/srep21540
39
Gowen M. Lazner F. Dodds R. Kapadia R. Feild J. Tavaria M. et al (1999). Cathepsin K Knockout Mice Develop Osteopetrosis Due to a Deficit in Matrix Degradation but Not Demineralization. J. Bone Min. Res.14 (10), 1654–1663. 10.1359/jbmr.1999.14.10.1654
40
Grossmann K. S. Rosário M. Birchmeier C. Birchmeier W. (2010). The Tyrosine Phosphatase Shp2 in Development and Cancer. Adv. Cancer Res.106, 53–89. 10.1016/s0065-230x(10)06002-1
41
Gu X. Peng Y. Zhao Y. Liang X. Tang Y. Liu J. (2019). A Novel Derivative of Artemisinin Inhibits Cell Proliferation and Metastasis via Down-Regulation of Cathepsin K in Breast Cancer. Eur. J. Pharmacol.858, 172382. 10.1016/j.ejphar.2019.05.011
42
Guo J. Bot I. de Nooijer R. Hoffman S. J. Stroup G. B. Biessen E. A. et al (2009). Leucocyte Cathepsin K Affects Atherosclerotic Lesion Composition and Bone Mineral Density in Low-Density Lipoprotein Receptor Deficient Mice. Cardiovasc Res.81 (2), 278–285. 10.1093/cvr/cvn311
43
Haeckel C. Krueger S. Buehling F. Broemme D. Franke K. Schuetze A. et al (1999). Expression of Cathepsin K in the Human Embryo and Fetus. Dev. Dyn.216 (2), 89–95. 10.1002/(sici)1097-0177(199910)216:2<89::aid-dvdy1>3.0.co;2-9
44
Han Y. Feng H. Sun J. Liang X. Wang Z. Xing W. et al (2019). Lkb1 Deletion in Periosteal Mesenchymal Progenitors Induces Osteogenic Tumors through mTORC1 Activation. J. Clin. Invest.129 (5), 1895–1909. 10.1172/jci124590
45
Hao L. Zhu G. Lu Y. Wang M. Jules J. Zhou X. et al (2015). Deficiency of Cathepsin K Prevents Inflammation and Bone Erosion in Rheumatoid Arthritis and Periodontitis and Reveals its Shared Osteoimmune Role. FEBS Lett.589 (12), 1331–1339. 10.1016/j.febslet.2015.04.008
46
Horiuchi K. Amizuka N. Takeshita S. Takamatsu H. Katsuura M. Ozawa H. et al (1999). Identification and Characterization of a Novel Protein, Periostin, with Restricted Expression to Periosteum and Periodontal Ligament and Increased Expression by Transforming Growth Factor β. J. Bone Min. Res.14 (7), 1239–1249. 10.1359/jbmr.1999.14.7.1239
47
Hou W.-S. Li Z. Gordon R. E. Chan K. Klein M. J. Levy R. et al (2001). Cathepsin K Is a Critical Protease in Synovial Fibroblast-Mediated Collagen Degradation. Am. J. Pathology159 (6), 2167–2177. 10.1016/s0002-9440(10)63068-4
48
Hussein H. Boyaka P. Dulin J. Russell D. Smanik L. Azab M. et al (2017). Cathepsin K Localizes to Equine Bone In Vivo and Inhibits Bone Marrow Stem and Progenitor Cells Differentiation In Vitro. J. Stem Cells Regen. Med.13 (2), 45–53. 10.46582/jsrm.1302008
49
Inaoka T. Bilbe G. Ishibashi O. Tezuka K. Kumegawa M. Kokubo T. (1995). Molecular Cloning of Human cDNA for Cathepsin K: Novel Cysteine Proteinase Predominantly Expressed in Bone. Biochem. Biophysical Res. Commun.206 (1), 89–96. 10.1006/bbrc.1995.1013
50
Jacome-Galarza C. Soung D. Y. Adapala N. S. Pickarski M. Sanjay A. Duong L. T. et al (2014). Altered Hematopoietic Stem Cell and Osteoclast Precursor Frequency in Cathepsin K Null Mice. J. Cell. Biochem.115 (8), 1449–1457. 10.1002/jcb.24801
51
Kafienah W. Brömme D. Buttle D. J. Croucher L. J. Hollander A. P. (1998). Human Cathepsin K Cleaves Native Type I and II Collagens at the N-Terminal End of the Triple Helix. Biochem. J.331 (Pt 3), 727–732. 10.1042/bj3310727
52
Kamiya N. Ye L. Kobayashi T. Lucas D. J. Mochida Y. Yamauchi M. et al (2008). Disruption of BMP Signaling in Osteoblasts through Type IA Receptor (BMPRIA) Increases Bone Mass*. J. Bone Mineral Res.23 (12), 2007–2017. 10.1359/jbmr.080809
53
Kamiya N. Ye L. Kobayashi T. Mochida Y. Yamauchi M. Kronenberg H. M. et al (2008). BMP Signaling Negatively Regulates Bone Mass through Sclerostin by Inhibiting the Canonical Wnt Pathway. Development135 (22), 3801–3811. 10.1242/dev.025825
54
Karlsson C. Thornemo M. Henriksson H. B. Lindahl A. (2009). Identification of a Stem Cell Niche in the Zone of Ranvier within the Knee Joint. J. Anat.215 (3), 355–363. 10.1111/j.1469-7580.2009.01115.x
55
Kim H. K. Feng G.-S. Chen D. King P. D. Kamiya N. (2014). Targeted Disruption ofShp2in Chondrocytes Leads to Metachondromatosis with Multiple Cartilaginous Protrusions. J. Bone Min. Res.29 (3), 761–769. 10.1002/jbmr.2062
56
Kiviranta R. Morko J. Alatalo S. L. NicAmhlaoibh R. Risteli J. Laitala-Leinonen T. et al (2005). Impaired Bone Resorption in Cathepsin K-Deficient Mice Is Partially Compensated for by Enhanced Osteoclastogenesis and Increased Expression of Other Proteases via an Increased RANKL/OPG Ratio. Bone36 (1), 159–172. 10.1016/j.bone.2004.09.020
57
Konttinen Y. T. Mandelin J. Li T.-F. Salo J. Lassus J. Liljeström M. et al (2002). Acidic Cysteine Endoproteinase Cathepsin K in the Degeneration of the Superficial Articular Hyaline Cartilage in Osteoarthritis. Arthritis & Rheumatism46 (4), 953–960. 10.1002/art.10185
58
Kozawa E. Cheng X. W. Urakawa H. Arai E. Yamada Y. Kitamura S. et al (2016). Increased Expression and Activation of Cathepsin K in Human Osteoarthritic Cartilage and Synovial Tissues. J. Orthop. Res.34 (1), 127–134. 10.1002/jor.23005
59
Kozawa E. Nishida Y. Cheng X. W. Urakawa H. Arai E. Futamura N. et al (2012). Osteoarthritic Change Is Delayed in a Ctsk-Knockout Mouse Model of Osteoarthritis. Arthritis & Rheumatism64 (2), 454–464. 10.1002/art.33398
60
Lai L. P. Lilley B. N. Sanes J. R. McMahon A. P. (2013). Lkb1/Stk11 Regulation of mTOR Signaling Controls the Transition of Chondrocyte Fates and Suppresses Skeletal Tumor Formation. Proc. Natl. Acad. Sci. U.S.A.110 (48), 19450–19455. 10.1073/pnas.1309001110
61
Langdahl B. Binkley N. Bone H. Gilchrist N. Resch H. Rodriguez Portales J. et al (2012). Odanacatib in the Treatment of Postmenopausal Women with Low Bone Mineral Density: Five Years of Continued Therapy in a Phase 2 Study. J. Bone Min. Res.27 (11), 2251–2258. 10.1002/jbmr.1695
62
Lauth M. Bergström Å. Shimokawa T. Toftgård R. (2007). Inhibition of GLI-Mediated Transcription and Tumor Cell Growth by Small-Molecule Antagonists. Proc. Natl. Acad. Sci. U.S.A.104 (20), 8455–8460. 10.1073/pnas.0609699104
63
Li C. Y. Jepsen K. J. Majeska R. J. Zhang J. Ni R. Gelb B. D. et al (2006). Mice Lacking Cathepsin K Maintain Bone Remodeling but Develop Bone Fragility Despite High Bone Mass. J. Bone Min. Res.21 (6), 865–875. 10.1359/jbmr.060313
64
Li R. Zhou R. Wang H. Li W. Pan M. Yao X. et al (2019). Gut Microbiota-Stimulated Cathepsin K Secretion Mediates TLR4-dependent M2 Macrophage Polarization and Promotes Tumor Metastasis in Colorectal Cancer. Cell Death Differ.26 (11), 2447–2463. 10.1038/s41418-019-0312-y
65
Li Z. Hou W.-S. Brömme D. (2000). Collagenolytic Activity of Cathepsin K Is Specifically Modulated by Cartilage-Resident Chondroitin Sulfates. Biochemistry39 (3), 529–536. 10.1021/bi992251u
66
Li Z. Hou W.-S. Escalante-Torres C. R. Gelb B. D. Brömme D. (2002). Collagenase Activity of Cathepsin K Depends on Complex Formation with Chondroitin Sulfate. J. Biol. Chem.277 (32), 28669–28676. 10.1074/jbc.m204004200
67
Littlewood-Evans A. J. Bilbe G. Bowler W. B. Farley D. Wlodarski B. Kokubo T. et al (1997). The Osteoclast-Associated Protease Cathepsin K Is Expressed in Human Breast Carcinoma. Cancer Res.57 (23), 5386–5390.
68
Lotinun S. Kiviranta R. Matsubara T. Alzate J. A. Neff L. Lüth A. et al (2013). Osteoclast-specific Cathepsin K Deletion Stimulates S1P-dependent Bone Formation. J. Clin. Invest.123 (2), 666–681. 10.1172/JCI64840
69
Lotinun S. Ishihara Y. Nagano K. Kiviranta R. Carpentier V. T. Neff L. et al (2019). Cathepsin K-Deficient Osteocytes Prevent Lactation-Induced Bone Loss and Parathyroid Hormone Suppression. J. Clin. Invest.129 (8), 3058–3071. 10.1172/jci122936
70
Lutgens E. Lutgens S. P. M. Faber B. C. G. Heeneman S. Gijbels M. M. J. de Winther M. P. J. et al (2006). Disruption of the Cathepsin K Gene Reduces Atherosclerosis Progression and Induces Plaque Fibrosis but Accelerates Macrophage Foam Cell Formation. Circulation113 (1), 98–107. 10.1161/circulationaha.105.561449
71
Maeda K. Kobayashi Y. Udagawa N. Uehara S. Ishihara A. Mizoguchi T. et al (2012). Wnt5a-Ror2 Signaling between Osteoblast-Lineage Cells and Osteoclast Precursors Enhances Osteoclastogenesis. Nat. Med.18 (3), 405–412. 10.1038/nm.2653
72
Mandelin J. Hukkanen M. Li T.-F. Korhonen M. Liljeström M. Sillat T. et al (2006). Human Osteoblasts Produce Cathepsin K. Bone38 (6), 769–777. 10.1016/j.bone.2005.10.017
73
Maruyama T. (2019). Stem Cells of the Suture Mesenchyme in Craniofacial Bone Development, Repair and Regeneration. Keio J. Med.68 (2), 42. 10.2302/kjm.68-003-abst
74
McClung M. R. O'Donoghue M. L. Papapoulos S. E. Bone H. Langdahl B. Saag K. G. et al (2019). Odanacatib for the Treatment of Postmenopausal Osteoporosis: Results of the LOFT Multicentre, Randomised, Double-Blind, Placebo-Controlled Trial and LOFT Extension Study. Lancet Diabetes Endocrinol.7 (12), 899–911. 10.1016/S2213-8587(19)30346-8
75
McLellan M. A. Rosenthal N. A. Pinto A. R. (2017). Cre-loxP-Mediated Recombination: General Principles and Experimental Considerations. Curr. Protoc. Mouse Biol.7 (1), 1–12. 10.1002/cpmo.22
76
Morikawa S. Mabuchi Y. Kubota Y. Nagai Y. Niibe K. Hiratsu E. et al (2009). Prospective Identification, Isolation, and Systemic Transplantation of Multipotent Mesenchymal Stem Cells in Murine Bone Marrow. J. Exp. Med.206 (11), 2483–2496. 10.1084/jem.20091046
77
Morko J. P. Soderstrom M. Saamanen A. M. Salminen H. J. Vuorio E. I. (2004). Up Regulation of Cathepsin K Expression in Articular Chondrocytes in a Transgenic Mouse Model for Osteoarthritis. Ann. Rheumatic Dis.63 (6), 649–655. 10.1136/ard.2002.004671
78
Mort J. S. Beaudry F. Théroux K. Emmott A. A. Richard H. Fisher W. D. et al (2016). Early Cathepsin K Degradation of Type II Collagen In Vitro and In Vivo in Articular Cartilage. Osteoarthr. Cartil.24 (8), 1461–1469. 10.1016/j.joca.2016.03.016
79
Munari E. Cima L. Massari F. Bertoldo F. Porcaro A. B. Caliò A. et al (2017). Cathepsin K Expression in Castration-Resistant Prostate Carcinoma: a Therapeutical Target for Patients at Risk for Bone Metastases. Int. J. Biol. Markers32 (2), e243–e7. 10.5301/jbm.5000246
80
Okamoto M. Murai J. Imai Y. Ikegami D. Kamiya N. Kato S. et al (2011). Conditional Deletion of Bmpr1a in Differentiated Osteoclasts Increases Osteoblastic Bone Formation, Increasing Volume of Remodeling Bone in Mice. J. Bone Min. Res.26 (10), 2511–2522. 10.1002/jbmr.477
81
Pannier S. Legeai-Mallet L. (2008). Hereditary Multiple Exostoses and Enchondromatosis. Best Pract. Res. Clin. Rheumatology22 (1), 45–54. 10.1016/j.berh.2007.12.004
82
Pap T. Korb-Pap A. (2015). Cartilage Damage in Osteoarthritis and Rheumatoid Arthritis-Two Unequal Siblings. Nat. Rev. Rheumatol.11 (10), 606–615. 10.1038/nrrheum.2015.95
83
Petricevic S. J. Pavlovic A. Capkun V. Becic K. Durdov M. G. (2017). Cathepsin K Expression in Melanoma Is Associated with Metastases. Histol. Histopathol.32 (7), 711–716. 10.14670/HH-11-833
84
Pinho S. Lacombe J. Hanoun M. Mizoguchi T. Bruns I. Kunisaki Y. et al (2013). PDGFRα and CD51 Mark Human Nestin+ Sphere-Forming Mesenchymal Stem Cells Capable of Hematopoietic Progenitor Cell Expansion. J. Exp. Med.210 (7), 1351–1367. 10.1084/jem.20122252
85
Presneau N. Duhamel L. A. Ye H. Tirabosco R. Flanagan A. M. Eskandarpour M. (2017). Post-translational Regulation Contributes to the Loss of LKB1 Expression through SIRT1 Deacetylase in Osteosarcomas. Br. J. Cancer117 (3), 398–408. 10.1038/bjc.2017.174
86
Rizzoli R. Benhamou C.-L. Halse J. Miller P. D. Reid I. R. Rodríguez Portales J. A. et al (2016). Continuous Treatment with Odanacatib for up to 8 Years in Postmenopausal Women with Low Bone Mineral Density: a Phase 2 Study. Osteoporos. Int.27 (6), 2099–2107. 10.1007/s00198-016-3503-0
87
Roberts J. L. Liu G. Paglia D. N. Kinter C. W. Fernandes L. M. Lorenzo J. et al (2020). Deletion of Wnt5a in Osteoclasts Results in Bone Loss through Decreased Bone Formation. Ann. N.Y. Acad. Sci.1463 (1), 45–59. 10.1111/nyas.14293
88
Sacchetti B. Funari A. Michienzi S. Di Cesare S. Piersanti S. Saggio I. et al (2007). Self-renewing Osteoprogenitors in Bone Marrow Sinusoids Can Organize a Hematopoietic Microenvironment. Cell131 (2), 324–336. 10.1016/j.cell.2007.08.025
89
Saftig P. Hunziker E. Wehmeyer O. Jones S. Boyde A. Rommerskirch W. et al (1998). Impaired Osteoclastic Bone Resorption Leads to Osteopetrosis in Cathepsin-K-Deficient Mice. Proc. Natl. Acad. Sci. U.S.A.95 (23), 13453–13458. 10.1073/pnas.95.23.13453
90
Sanchez-Fernandez M. A. Sbacchi S. Correa-Tapia M. Naumann R. Klemm J. Chambon P. et al (2012). Transgenic Mice for a Tamoxifen-Induced, Conditional Expression of the Cre Recombinase in Osteoclasts. PLoS One7 (5), e37592. 10.1371/journal.pone.0037592
91
Sauer B. (1998). Inducible Gene Targeting in Mice Using the Cre/loxSystem. Methods14 (4), 381–392. 10.1006/meth.1998.0593
92
Shaw R. J. (2009). LKB1 and AMP-Activated Protein Kinase Control of mTOR Signalling and Growth. Acta Physiol. (Oxf).196 (1), 65–80. 10.1111/j.1748-1716.2009.01972.x
93
Shi G.-P. Sukhova G. K. Grubb A. Ducharme A. Rhode L. H. Lee R. T. et al (1999). Cystatin C Deficiency in Human Atherosclerosis and Aortic Aneurysms. J. Clin. Invest.104 (9), 1191–1197. 10.1172/jci7709
94
Soki F. N. Yoshida R. Paglia D. N. Duong L. T. Hansen M. F. Drissi H. (2018). Articular Cartilage Protection in Ctsk ‐/‐ Mice Is Associated with Cellular and Molecular Changes in Subchondral Bone and Cartilage Matrix. J. Cell Physiol.233 (11), 8666–8676. 10.1002/jcp.26745
95
Soukas A. Cohen P. Socci N. D. Friedman J. M. (2000). Leptin-specific Patterns of Gene Expression in White Adipose Tissue. Genes Dev.14 (8), 963–980. 10.1101/gad.14.8.963
96
Sukhova G. K. Shi G. P. Simon D. I. Chapman H. A. Libby P. (1998). Expression of the Elastolytic Cathepsins S and K in Human Atheroma and Regulation of Their Production in Smooth Muscle Cells. J. Clin. Invest.102 (3), 576–583. 10.1172/jci181
97
Suzuki M. Takahashi N. Sobue Y. Ohashi Y. Kishimoto K. Hattori K. et al (2020). Hyaluronan Suppresses Enhanced Cathepsin K Expression via Activation of NF-Κb with Mechanical Stress Loading in a Human Chondrocytic HCS-2/8 Cells. Sci. Rep.10 (1), 216. 10.1038/s41598-019-57073-8
98
Svelander L. Erlandsson-Harris H. Astner L. Grabowska U. Klareskog L. Lindstrom E. et al (2009). Inhibition of Cathepsin K Reduces Bone Erosion, Cartilage Degradation and Inflammation Evoked by Collagen-Induced Arthritis in Mice. Eur. J. Pharmacol.613 (1-3), 155–162. 10.1016/j.ejphar.2009.03.074
99
Takito J. Inoue S. Nakamura M. (2018). The Sealing Zone in Osteoclasts: A Self-Organized Structure on the Bone. Int. J. Mol. Sci.19 (4), 984. 10.3390/ijms19040984
100
Tanaka M. Yamada H. Nishikawa S. Mori H. Ochi Y. Horai N. et al (2016). Joint Degradation in a Monkey Model of Collagen-Induced Arthritis: Role of Cathepsin K Based on Biochemical Markers and Histological Evaluation. Int. J. Rheumatol.2016, 8938916. 10.1155/2016/8938916
101
Tanaka M. Hashimoto Y. Hasegawa C. Deacon S. Eastell R. (2017). Antiresorptive Effect of a Cathepsin K Inhibitor ONO-5334 and its Relationship to BMD Increase in a Phase II Trial for Postmenopausal Osteoporosis. BMC Musculoskelet. Disord.18 (1), 267. 10.1186/s12891-017-1625-y
102
To T. T. Witten P. E. Huysseune A. Winkler C. (2015). An Adult Osteopetrosis Model in Medaka Reveals the Importance of Osteoclast Function for Bone Remodeling in Teleost Fish. Comp. Biochem. Physiology Part C Toxicol. Pharmacol.178, 68–75. 10.1016/j.cbpc.2015.08.007
103
To T. T. Witten P. E. Renn J. Bhattacharya D. Huysseune A. Winkler C. (2012). Rankl-induced Osteoclastogenesis Leads to Loss of Mineralization in a Medaka Osteoporosis Model. Development139 (1), 141–150. 10.1242/dev.071035
104
Usami Y. Gunawardena A. T. Francois N. B. Otsuru S. Takano H. Hirose K. et al (2019). Possible Contribution of Wnt‐Responsive Chondroprogenitors to the Postnatal Murine Growth Plate. J. Bone Mineral Res.34 (5), 964–974. 10.1002/jbmr.3658
105
Varjosalo M. Taipale J. (2008). Hedgehog: Functions and Mechanisms. Genes Dev.22 (18), 2454–2472. 10.1101/gad.1693608
106
Walia B. Lingenheld E. Duong L. Sanjay A. Drissi H. (2018). A Novel Role for Cathepsin K in Periosteal Osteoclast Precursors during Fracture Repair. Ann. N.Y. Acad. Sci.1415 (1), 57–68. 10.1111/nyas.13629
107
Walkley C. R. Qudsi R. Sankaran V. G. Perry J. A. Gostissa M. Roth S. I. et al (2008). Conditional Mouse Osteosarcoma, Dependent on P53 Loss and Potentiated by Loss of Rb, Mimics the Human Disease. Genes Dev.22 (12), 1662–1676. 10.1101/gad.1656808
108
Wang C. Inzana J. A. Mirando A. J. Ren Y. Liu Z. Shen J. et al (2016). NOTCH Signaling in Skeletal Progenitors Is Critical for Fracture Repair. J. Clin. Invest.126 (4), 1471–1481. 10.1172/jci80672
109
Wei W. Ren J. Yin W. Ding H. Lu Q. Tan L. et al (2020). Inhibition of Ctsk Modulates Periodontitis with Arthritis via Downregulation of TLR9 and Autophagy. Cell Prolif.53 (1), e12722. 10.1111/cpr.12722
110
Worthley D. L. Churchill M. Compton J. T. Tailor Y. Rao M. Si Y. et al (2015). Gremlin 1 Identifies a Skeletal Stem Cell with Bone, Cartilage, and Reticular Stromal Potential. Cell160 (1-2), 269–284. 10.1016/j.cell.2014.11.042
111
Xie H. Cui Z. Wang L. Xia Z. Hu Y. Xian L. et al (2014). PDGF-BB Secreted by Preosteoclasts Induces Angiogenesis during Coupling with Osteogenesis. Nat. Med.20 (11), 1270–1278. 10.1038/nm.3668
112
Xu R. Hu J. Zhou X. Yang Y. (2018). Heterotopic Ossification: Mechanistic Insights and Clinical Challenges. Bone109, 134–142. 10.1016/j.bone.2017.08.025
113
Yamada H. Mori H. Nakanishi Y. Nishikawa S. Hashimoto Y. Ochi Y. et al (2019). Effects of the Cathepsin K Inhibitor ONO-5334 and Concomitant Use of ONO-5334 with Methotrexate on Collagen-Induced Arthritis in Cynomolgus Monkeys. Int. J. Rheumatol.2019, 5710340. 10.1155/2019/5710340
114
Yamashita T. Hagino H. Hayashi I. Hayashibara M. Tanida A. Nagira K. et al (2018). Effect of a Cathepsin K Inhibitor on Arthritis and Bone Mineral Density in Ovariectomized Rats with Collagen-Induced Arthritis. Bone Rep.9, 1–10. 10.1016/j.bonr.2018.05.006
115
Yang P. Lv S. Wang Y. Peng Y. Ye Z. Xia Z. et al (2018). Preservation of Type H Vessels and Osteoblasts by Enhanced Preosteoclast Platelet-Derived Growth Factor Type BB Attenuates Glucocorticoid-Induced Osteoporosis in Growing Mice. Bone114, 1–13. 10.1016/j.bone.2018.05.025
116
Yang W. Wang J. Moore D. C. Liang H. Dooner M. Wu Q. et al (2013). Ptpn11 Deletion in a Novel Progenitor Causes Metachondromatosis by Inducing Hedgehog Signalling. Nature499 (7459), 491–495. 10.1038/nature12396
117
Yang Y. Chen Q. Zhou S. Gong X. Xu H. Hong Y. et al (2020). Skeletal Phenotype Analysis of a Conditional Stat3 Deletion Mouse Model. J. Vis. Exp.2020 (161), e61390. 10.3791/61390
118
Yang Y. Chung M. R. Zhou S. Gong X. Xu H. Hong Y. et al (2019). STAT3 Controls Osteoclast Differentiation and Bone Homeostasis by Regulating NFATc1 Transcription. J. Biol. Chem.294 (42), 15395–15407. 10.1074/jbc.ra119.010139
119
Zhou B. O. Yue R. Murphy M. M. Peyer J. G. Morrison S. J. (2014). Leptin-receptor-expressing Mesenchymal Stromal Cells Represent the Main Source of Bone Formed by Adult Bone Marrow. Cell Stem Cell15 (2), 154–168. 10.1016/j.stem.2014.06.008
120
Zhou H. Newnum A. B. Martin J. R. Li P. Nelson M. T. Moh A. et al (2011). Osteoblast/osteocyte-specific Inactivation of Stat3 Decreases Load-Driven Bone Formation and Accumulates Reactive Oxygen Species. Bone49 (3), 404–411. 10.1016/j.bone.2011.04.020
Summary
Keywords
cathepsin K, Ctsk+ cells, animal model, genetic marker, bone metabolic disease
Citation
Zou N, Liu R and Li C (2022) Cathepsin K+ Non-Osteoclast Cells in the Skeletal System: Function, Models, Identity, and Therapeutic Implications. Front. Cell Dev. Biol. 10:818462. doi: 10.3389/fcell.2022.818462
Received
19 November 2021
Accepted
31 May 2022
Published
13 July 2022
Volume
10 - 2022
Edited by
Atsushi Asakura, University of Minnesota Twin Cities, United States
Reviewed by
Marco Ponzetti, University of L'Aquila, Italy
Manu Platt, Georgia Institute of Technology, United States
Updates
Copyright
© 2022 Zou, Liu and Li.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changjun Li, lichangjun@csu.edu.cn
This article was submitted to Stem Cell Research, a section of the journal Frontiers in Cell and Developmental Biology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.