 Yoshiyasu Ogura
Yoshiyasu Ogura Imari Mimura
Imari Mimura- Division of Nephrology and Endocrinology, The University of Tokyo Graduate School of Medicine, Tokyo, Japan
Accumulation of somatic hematopoietic stem cell mutations with aging has been revealed by the recent genome-wide analysis. Clonal expansion, known as clonal hematopoiesis of indeterminate potential (CHIP), is a premalignant condition of hematological cancers. It is defined as the absence of definitive morphological evidence of a hematological neoplasm and occurrence of ≥2% of mutant allele fraction in the peripheral blood. In CHIP, the most frequently mutated genes are epigenetic regulators such as DNMT3A, TET2, and ASXL1. CHIP induces inflammation. CHIP is shown to be associated with not only hematological malignancy but also non-malignant disorders such as atherosclerosis, cardiovascular diseases and chronic liver disease. In addition, recent several large clinical trials have shown that CHIP is also the risk factor for developing chronic kidney disease (CKD). In this review article, we proposed novel findings about CHIP and CHIP related kidney disease based on the recent basic and clinical research. The possible mechanism of the kidney injury in CHIP is supposed to be due to the clonal expansion in both myeloid and lymphoid cell lines. In myeloid cell lines, the mutated macrophages increase the inflammatory cytokine level and induce chronic inflammation. It leads to epigenetic downregulation of kidney and macrophage klotho level. In lymphoid cell lines, CHIP might be related to monoclonal gammopathy of renal significance (MGRS). It describes any B cell or plasma cell clonal disorder that does not fulfill the criteria for cancer yet produces a nephrotoxic monoclonal immunoglobulin that leads to kidney injury or disease. MGRS causes M-protein related nephropathy frequently observed among aged CKD patients. It is important to consider the CHIP-related complications such as hematological malignancy, cardiovascular diseases and metabolic disorders in managing the elderly CKD patients. There are no established therapies for CHIP and CHIP-related CKD yet. However, recent studies have supported the development of effective CHIP therapies, such as blocking the expansion of aberrant HSCs and inhibiting chronic inflammation. In addition, drugs targeting the epigenetic regulation of Klotho in the kidney and macrophages might be therapeutic targets of CHIP in the kidney.
1 Introduction
Clonal hematopoiesis of indeterminate potential (CHIP) was originally proposed in the field of hematology. Recent genome-wide analyses have revealed that accumulation of somatic hematopoietic stem cell mutations and clonal expansion affect healthy populations. Clonal expansion is considered a pre-malignant condition in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) (Genovese et al., 2014; Steensma et al., 2015; Miles et al., 2020).
Somatic mutations are the most common single-nucleotide variants (SNVs), small insertions or deletions, and copy number changes in large chromosomal regions (Jaiswal, 2020). Hematopoietic stem cells (HSCs) are estimated to acquire approximately 20 somatic mutations per year in their whole genome, most of which are SNVs (Osorio et al., 2018). Within the bone marrow, only long-lived HSCs can self-renew during an organism’s lifetime (Reya et al., 2001). Therefore, only mutations in the long-lived HSCs persist throughout the lifetime of an individual. The size of HSCs increases steadily in early life, reaching a stable plateau by adolescence. The estimated number of HSCs is 50,000–200,000 per person (Lee-Six et al., 2018). Humans are expected to harbor between 350,000 and 1,400,000 coding mutations within the HSC pool by 70 years of age (Jaiswal, 2020). If only one of these mutations is capable of providing a selective advantage to the HSC in which it arises, clonal expansion in the blood becomes prevalent during aging (Jaiswal and Ebert, 2019).
CHIP is different from hematological cancers and is defined by the absence of definitive morphological evidence of a hematological neoplasm and occurrence of ≥2% of mutant allele fraction in the peripheral blood (Steensma et al., 2015). The most commonly mutated genes in clonal hematopoiesis (CH) are DNMT3A, TET2, ASXL1, PPM1D, JAK2, TP53, and SF3B1, which are also commonly mutated in AML, MDS, and myeloproliferative neoplasms (Jaiswal, 2020). Loss-of-function mutations in two DNA methylation enzymes, DNMT3A and TET2, accounted for nearly two-thirds of CHIP cases. The third most commonly mutated gene is ASXL1, a chromatin regulator. The epigenetic regulators, DNMT3A, TET2 and ASXL1, are essential for maintaining proper gene expression of HSC regulatory genes. Mutations of these genes cause the aberrant proliferation of HSCs (Challen et al., 2011; Solary et al., 2014; Fujino and Kitamura, 2020) TP53 and PPM1D are DNA damage responsive genes. JAK2 is involved in cellular growth signaling and SF3B1 is a splicing factor (Jaiswal and Ebert, 2019). Mutations in CHIP can also be detected in circulating immune cells, such as granulocytes, monocytes, and lymphocytes (Jaiswal, 2020). This finding suggests that CHIP may lead to altered immune responses and non-malignant diseases associated with aging.
CHIP is an age-related disorder more commonly observed in older adults. Mutated HSCs and macrophages elevate inflammatory cytokine levels, leading to chronic inflammation (Jaiswal et al., 2014). A study showed that CHIP is associated with atherosclerotic cardiovascular disease (Jaiswal et al., 2017). Moreover, recent studies have shown that CHIP is a risk factor for non-malignant diseases, such as venous thrombosis (Hinds et al., 2016), type 2 diabetes (T2DM) (Fuster et al., 2020), age-related neurodegenerative diseases, such as Alzheimer’s disease (Bouzid et al., 2023), chronic obstructive pulmonary disease (COPD) (Buscarlet et al., 2017), and chronic liver disease (Jaiswal, 2020; Wong et al., 2023). Furthermore, CHIP has garnered attention as a few recent large clinical trials have reported that CHIP is a risk factor for chronic kidney disease (CKD) (Vlasschaert et al., 2022; Kestenbaum et al., 2023) (Figure 1).
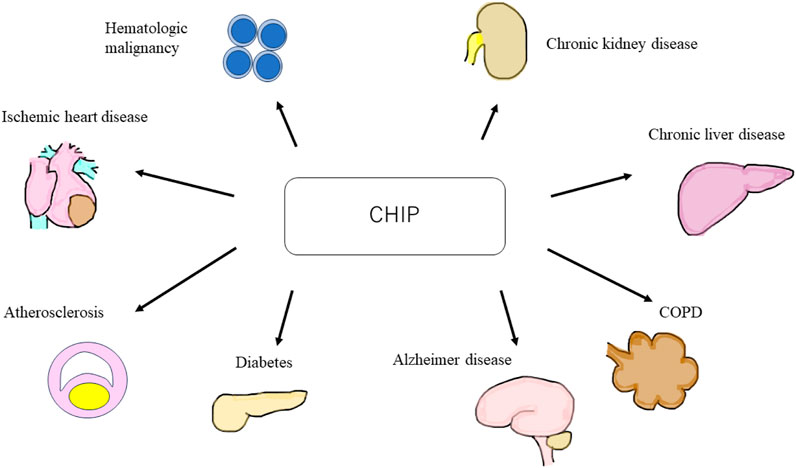
FIGURE 1. Schematic of clonal hematopoiesis of indeterminate potential (CHIP) related diseases. CHIP is a cause of hematologic malignant and non-malignant disorders, such as ischemic heart disease, atherosclerosis, diabetes, Alzheimer’s disease, chronic obstructive pulmonary disease, chronic liver disease, and chronic kidney disease. CHIP, clonal hematopoiesis of indeterminate potential; COPD, chronic obstructive pulmonary disease.
2 Risk factors for inducing CHIP
Aging is the strongest risk factor of CHIP. Detectable somatic mutations are rare in persons <40 years but rise in frequency with age (Jaiswal et al., 2014). It has been reported that 10% of persons >65 years and the majority (62%) of individuals ≥80 years had CHIP (Genovese et al., 2014; van Zeventer et al., 2021). The most common mutation observed in CHIP is a C-to-T single-nucleotide substitution in the coding region, which occurs due to an age-dependent increase in the rate of spontaneous deamination of 5-methyl-cytosines at CpG loci (Alexandrov et al., 2013). Both DNMT3A and TET2 mutations tend to be age-dependent (Buscarlet et al., 2017). DNMT3A and TET2 mutations confer an advantage by enhancing self-renewal of stem and progenitor cells and inhibiting their differentiation (Challen and Goodell, 2020). The second most important risk factor for CHIP is history of chemotherapy and radiation. Therapy-related CH is frequently observed in patients with solid tumors (Coombs et al., 2017). Cancer therapy with radiation is associated with mutations in DNA damage response (DDR) genes (TP53, PPM1D, and CHEK2) (Bolton et al., 2020). It has been reported that DDR mutations are selected for due to their enhanced resistance to cytotoxic exposures (Challen and Goodell, 2020). Additionally, CHIP has been observed in patients who underwent autologous stem cell transplantation (ASCT) for multiple myeloma (MM) and lymphoma. Especially, PPM1D mutations were associated with inferior overall survival after ASCT for lymphoma (Gibson et al., 2017). Furthermore, smoking is reported to be strongly associated with CH, especially ASXL1 mutation, in the United Kingdom Biobank cohort. The inflammatory environment induced by smoking might promote the outgrowth of ASXL1-mutant clones (Dawoud et al., 2020). Moreover, genetic factors have been reported to be risk factors for CHIP. Simultaneous analyses of germline and somatic whole-genome sequences revealed that mutations in the TET2 distal enhancer in the germline lead to increased self-renewal of HSCs and CHIP (Bick et al., 2020). A whole-genome shotgun study showed that a germline variant of the telomerase reverse transcriptase (TERT) gene was associated with CHIP (Zink et al., 2017). Furthermore, other genome-wide analyses of United Kingdom Biobank participants revealed that germline mutations in DNA damage repair (PARP1, ATM, and CHEK2), HSC migration/homing (CD164), and myeloid oncogenesis (SETBP1) genes were associated with CH (Kar et al., 2022) (Table.1)

TABLE 1. The risk factors of CHIP and related references. We summarize the association between risk factors and related CH mutations in Table 1.
3 Positive feedback loop of inflammation and CHIP
HSCs harboring driver mutations in CHIP accelerate chronic inflammation. And, CHIP-associated mutations provide HSCs with competitive advantages under inflammatory conditions. Therefore, a positive feedback loop exists between chronic inflammation and CHIP, which leads to the worsening of inflammation and atherosclerosis. Recent studies have described inflammatory responses in macrophages or HSCs from murine models of TET2, DNMT3A, ASXL1 and JAK2 mutations which are the most common clonal hematopoiesis mutations (Balasubramani et al., 2015; Sano et al., 2018a; Fidler et al., 2021).
Hematopoietic TET2 or DNMT3A disruption promotes both cardiac and renal fibrosis in hypertensive cardiac remodeling mice model via elevated expression of inflammatory cytokines such as IL-6 and C-C motif chemokine 5 (CCL5). TET2 or DNMT3A-deficiency in myeloid cell lines promoted the inflammatory renponses (Sano et al., 2018a). Furthermore, in murine models of heart failure with TET2 deficiency in hematopoietic cells, cardiac function worsened due to the elevation of IL-1β/NLRP3 inflammasome, and the models responded better to IL-1β/NLRP3 inflammasome inhibition (Sano et al., 2018b). Hypercholesterolemia-prone mice engrafted with bone marrow obtained from TET2 knock-out mice had larger atherosclerotic lesions in the aorta than mice that received control bone marrow. In addition, macrophages from TET2 knock-out mice secreted elevated levels of proinflammatory cytokines, such as IL1β and IL-6 (Jaiswal et al., 2017). Similarly, in dietary models of non-alcoholic steatohepatitis, mice transplanted with TET2-deficient hematopoietic cells demonstrated more severe liver inflammation and fibrosis due to elevated NLRP3 inflammasome and downstream inflammatory cytokine expression in TET2-deficient macrophages (Wong et al., 2023). It has been shown that DNMT3A caused DNA hyper-methylation in the IL-6 promoter regions in synovial fibroblasts from osteoarthritis patients and suppressed the IL-6 expression (Yang et al., 2017). TET2 has been reported to actively repress interleukin-6 (IL-6) transcription during inflammation resolution by recruiting histone deacetylase (HDAC) 2 in macrophages (Zhang et al., 2015). These papers suggested that DNMT3A or TET2 deficiency promotes the inflammation.
CRISPR-Cas9-mediated sequential editing of human induced pluripotent stem cell-derived hematopoietic stem progenitor cells (HSPCs) resulting in ASXL1, SRSF2, and NRAS mutations activates innate immunity signaling pathways (Wang et al., 2021). ASXL1-MT knock-in mice show age-related expansion of phenotypic HSCs via overactivation of Akt/mTOR signaling through deubiquitination of Akt1 by theASXL1-MT/Bap1 complex (Fujino et al., 2021). DNMT3A R878H knock-in mice, in which epigenetic regulation is disrupted, also develop hematologic malignancy through the overactivation of the mTOR pathway (Dai et al., 2017). The Akt/mTOR pathway is crucial for cell growth and survival (Porta et al., 2014), and its overactivation leads to mitochondrial activation, reactive oxygen species overproduction, and HSPC dysfunction (Fujino et al., 2021). This indicates that the mTOR pathway is a common pathology in CHIP. JAK2 mutation, one of the most commonly observed mutations in CHIP, are associated with inflammation and atherosclerosis. JAK2V617F(JAK2VF) mutations increase the risk of developing premature coronary artery disease (Jaiswal et al., 2017). Increased proliferation and glycolytic metabolism of JAK2VF macrophages leads to DNA replication stress and AIM2 inflammasome activation, thereby aggravating atherosclerosis (Fidler et al., 2021).
Clinical trials that used whole-genome sequencing data from several large cohorts showed that CHIP was strongly associated with epigenetic age acceleration (EAA), which is defined as the residual after regressing epigenetic clock age on chronological age, in several clocks, ranging from 1.31 years to 3.08 years (Nachun et al., 2021). Mutations in most CHIP genes, except DNA damage response genes, are associated with increases in several measures of age acceleration, and CHIP carriers are at a high risk of all-cause mortality and coronary heart disease (Nachun et al., 2021). In addition, EAA in adolescence is associated with obesity, inflammation (high sensitivity C-reactive protein and interferon-γ-inducible protein of 10 kDa (IP-10)), and future risk of cardiovascular diseases (Huang et al., 2019). IP-10 is a monocyte-derived proinflammatory chemokine that promotes the recruitment of lymphocytes and monocytes to site of inflammation. IP-10 is expressed in human atherosclerotic plaques (Profumo et al., 2010). Therefore, CHIP leads to EAA, systemic inflammation, and atherosclerosis. Aging and obesity are also reported to induce impaired hematopoietic regeneration by promoting expansion of the adipogenic lineage which produces an excess of dipeptidyl peptidase 4 (DPP4) (Ambrosi et al., 2017).
In contrast, chronic inflammation drives CH. For instance, IFNγ signaling induced during chronic infection drives DNMT3A-loss-of-function CH (Hormaechea-Agulla et al., 2021). Similarly, TET2 deficient mature myeloid cells and HSCs expand in response to inflammatory stress, which results in enhanced activation of IL-6, the Shp2-Stat3 signaling axis, and anti-apoptotic long non-coding RNA, Morrbid (Cai et al., 2018). In addition, chronic inflammation induced by the activation of noncanonical NF-κB pathway contributes to a competitive advantage in MDS HSCs (Muto et al., 2020) (Figure 2).
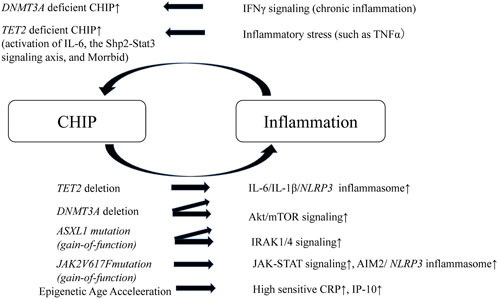
FIGURE 2. Schematic of positive feedback loop of inflammation and CHIP. A positive feedback loop exists between chronic inflammation and CHIP, which leads to the worsening of inflammation and atherosclerosis. TET2 or DNMT3A deleted HSCs elevate IL-6/IL-1β/NLRP3 inflammasome. DNMT3A deleted HSCs or ASXL1 mutant HSCs (gain-of-function) overactivates Akt/mTOR signaling. ASXL1 mutant HSCs also activates IRAK1/4 signaling. JAK2 mutant HSCs and macrophages activate JAK-STAT signaling and enhance AIM2/NLRP3 inflammasome. In addition, CHIP leads to pigenetic age acceleration, which results in elevation of high sensitive CRP and IP-10. In contrast, chronic inflammation drives CHIP. IFNγ under chronic inflammation drives DNMT3A deficient CHIP. TET2-deficient HSCs demonstrate a striking advantage in an inflammatory environment that contains TNFα and, TET2 deficient mature myeloid cells and HSCs expand in response to inflammatory stress, which results in enhanced activation of IL-6, the Shp2-Stat3 signaling axis, and anti-apoptotic long non-coding RNA, Morrbid. CHIP, clonal hematopoietic indeterminate potential; HSCs, hematopoietic stem cells; IRAK1/4, IL-1 receptor-associated kinase 1 and 4; IP-10, interferon-γ-inducible protein of 10 kDa.
4 CKD in CHIP and the possible mechanism of the kidney injury in CHIP
4.1 CHIP is associated with CKD
Several clinical trials have demonstrated the correlation between CHIP and CKD progression. In two independent cohorts of 87 and 85 adults, respectively, with an estimated glomerular filtration rate (eGFR) < 60 mL/min per 1.73 m2 (Kingston and CanPREDDICT cohorts), 25% of the individuals with advanced CKD had CHIP. Those with CHIP had a 2.2-fold greater risk of kidney failure over 5 years of follow-up and were more likely to have CKD complications, including anemia, than those without CHIP (Vlasschaert et al., 2022). In another study, CH was identified in 5449 (2.9%) eligible United Kingdom Biobank participants (n = 190,487), and it was found that CH was associated with CKD, which was defined by eGFR cystatin. Moreover, CH promotes adverse outcomes such as death, myocardial infarction, and stroke in CKD (Dawoud et al., 2022). In three community-based cohorts of the TOPMed Consortium (n = 12,004), the median baseline eGFR was 84 mL/min/1.73 m2, and the prevalence of CHIP was 6.6%, 9.0%, and 12.2% in persons aged 50–60, 60–70, and >70 years, respectively. A meta-analysis showed that CHIP was associated with a greater risk of 30% eGFR decline (17% [95%CI, 1%–36%] higher, p = 0.04). Additionally, the study revealed an association between CHIP and eGFR decline in three general population cohorts without known kidney disease (Kestenbaum et al., 2023). In contrast, a nested case-control study of the PROVALID study, which was a prospective cohort study including 4000 patients with T2DM in five European countries, showed that common risk factors (albuminuria, hemoglobin A1c, heart failure, and smoking) and microinflammation, but not CHIP, were associated with kidney function decline in T2DM (hazard ratio [HR] 1.06 [95% CI 0.57–1.96]) (Denicolo et al., 2022).
In experimental animal models, the recapitulation of CHIP has been shown to promote interstitial fibrosis and accumulation of macrophages in the kidneys (Sano et al., 2018a; Sano et al., 2018b). However, since these studies used a heart disease model (hypertensive cardiac remodeling or heart failure model), renal evaluation was a secondary endpoint. Therefore, the mechanisms underlying CHIP-related renal failure should be explored in animal models of kidney damage. Considering clinical and basic research, accumulation of mutated macrophages in the kidney might be the cause of renal fibrosis, glomerulosclerosis, and CKD, but the underlying mechanism remains unclear. Macrophages are the key regulators of tissue repair, regeneration, and fibrosis. Disturbances in macrophage function can lead to aberrant repair; deficient generation of anti-inflammatory macrophages; or failed crosstalk between macrophages and epithelial cells, endothelial cells, fibroblasts, and stem or tissue progenitor cells (Wynn and Vannella, 2016). Animal models have demonstrated that macrophages are major contributors to inflammatory responses in kidney injury and renal fibrosis (Huen and Cantley, 2017). Mutated macrophages generated from clonal expansion of myeloid cell line secrete cytokines such as IL-1β, IL-6, and TNFα and they might cause the injury of tubular epithelial cells or the activation of myofibroblast. In CHIP and kidney disease models, the pathology of impact of macrophages with CHIP on renal parenchymal cells should be verified.
4.2 A potential relationship between CHIP and monoclonal gammopathy of renal significance (MGRS)
A recent study showed that CHIP is associated with not only myeloid lineage malignancies but also lymphoid lineage ones, such as multiple myeloma (MM) and lymphoma (Husby et al., 2020; Wudhikarn et al., 2021). Recently, the kidney diseases caused by abnormal monoclonal antibodies generated by lymphoid lineage malignancies, which is called “monoclonal gammopathy of renal significance” (MGRS) has garnered attention among nephrologists and hematologists. MGRS is the disease concept that has recently been proposed (Leung et al., 2021). In this section, we propose the potential relationship between CHIP and MGRS.
Monoclonal gammopathy accompanied by lymphoid lineage malignancy is known as an important cause of kidney injury (Leung et al., 2021) Monoclonal gammopathy is defined as the presence of a monoclonal immunoglobulin in the plasma, urine, or both, which is often produced by clonal plasma cells, such as multiple myeloma (MM), and less commonly by B lymphocytes. Renal failure is a common complication of monoclonal gammopathy (Sy-Go et al., 2022). Until now, treatment for renal failure has not been recommended unless patients met the tumor burden criterion and had end-organ damage. Those patients had received careful monitoring as monoclonal gammopathy of undetermined significance (MGUS) because MGUS has risk of converting to malignancies such as MM, Waldenström macroglobulinaemia (WN), or chronic lymphocytic leukaemia (CLL) (Dimopoulos et al., 2009; Rajkumar et al., 2014; Strati and Shanafelt, 2015). However, it has been reported that there is B cell or plasma cell clonal disorder that does not fulfill the criteria for cancer yet produces a nephrotoxic monoclonal immunoglobulin that leads to kidney injury or disease, which was termed as MGRS (Leung et al., 2019).
Monoclonal immunoglobulins can cause kidney damage through various mechanisms that can be separated by the presence of a high or low tumor burden (Leung et al., 2021). Kidney injury from a high tumor burden is represented by light-chain cast nephropathy, which is characterized by monoclonal light chains that bind to Tamm-Horsfall protein through their variable domain to form obstructive casts (Sanders and Booker, 1992). However, in light chain cast nephropathy, high levels of serum free light chains are required, and almost all cases are accompanied by apparent hematological cancer such as multiple myeloma. Light-chain cast nephropathy is not considered an MGRS-related kidney lesion. MGRS is most commonly associated with low levels of monoclonal gammopathy. MGRS-associated renal lesions are initially separated by the presence or absence of monoclonal immunoglobulin deposits in kidney biopsy samples (Leung et al., 2019). Most MGRS-associated lesions are caused by the deposition of entire or parts of the monoclonal immunoglobulins or of various products of aggregation (Leung et al., 2019). The most common MGRS lesion is amyloid light chain (AL) amyloidosis. A small B-cell clone, most commonly a plasma cell clone, produces monoclonal light chains that exert organ toxicity and deposit in tissue in the form of amyloid fibrils (Palladini et al., 2020). Misfolding of a fragment of monoclonal immunoglobulin light chain occurs, resulting in the formation of toxic amyloid multimers and amyloid fibrils (Merlini and Bellotti, 2003). Renal involvement is very frequent in AL amyloidosis and can lead to the development of nephrotic syndrome followed by renal failure in some cases (Rysava, 2019). Other than AL amyloidosis, monoclonal immunoglobulin deposits related MGRS lesions include many diseases such as light-chain proximal tubulopathy (LCPT), immunotactoid glomerulonephritis, monoclonal fibrillary glomerulonephritis, cryoglobulinaemic glomerulonephritis, monoclonal immunoglobulin deposition disease (MIDD), and proliferative glomerulonephritis and monoclonal immunoglobulin deposits (PGNMID), which are relatively infrequent even among MGRS lesions (Leung et al., 2019). MGRS-associated lesions without deposits are C3 glomerulopathy with monoclonal gammopathy and thrombotic microangiopathy. Circulating monoclonal immunoglobulins are suspected to overactivate the alternative pathway, resulting in glomerular deposition of C3 or endothelial cell injury (Leung et al., 2021).
The diagnosis of MGRS can be established only by performing a kidney biopsy that either demonstrates the presence of monoclonal immunoglobulin deposits or infers their involvement with a circulating monoclonal immunoglobulin. Moreover, hematological evaluation such as peripheral blood flow cytometry and bone marrow biopsy is needed in order to detect a monoclonal immunoglobulin and predict treatment responses (Leung et al., 2019). Early diagnosis of MGRS is crucial, as it strongly impacts renal prognosis (Bridoux et al., 2015). Patients with MGRS require early effective treatment, such as clone-directed therapy, based on agents that were previously restricted to patients with overt hematologic cancers (Leung et al., 2021).
At the moment, the relationship between CHIP and MGRS has not verified. However, here is one clinical study which may indicate the relationship between CHIP and MGRS. CHIP mutations were observed in 21% among AL amyloidosis patients (Testa et al., 2022). In this study, CHIP was defined as the presence of DNMT3A, TET2, or ASXL1 mutations in the peripheral blood or bone marrow (DTA-CH). Among CHIP mutations, TET2 and DNMT3A were the most frequently mutated genes. And, DTA-CH did not predict worse overall survival (OS) or progression-free survival (PFS) in AL amyloidosis (Testa et al., 2022). However, this study included a relatively small sample size. And it did not include the data of proteinuria and renal function. Therefore, the clinical study focusing the relationship between CHIP and kidney function of MGRS patients is needed.
It has been reported that the type of CH mutations differs between myeloid and lymphoid lineage malignancies, and the type of mutation is highly predictive of patients being at risk of myeloid or lymphoid malignancies (Niroula et al., 2021). For example, it has been reported that the timing of CHIP related mutation occurrence differs in each gene. DNMT3A mutations occur in a multipotent HSC and affect both myeloid and lymphoid lineages. In contrast, TET2 mutations occur in a more myeloid lineage committed HSCs and have myeloid bias (Buscarlet et al., 2017; Buscarlet et al., 2018).
The type of CH mutations might influence the type of kidney injuries. In the first place, MGRS is difficult to diagnose and unfamiliar even among nephrologists. Many MGRS patients are thought to be undiagnosed among CKD patients. Further research on the association between CHIP and MGRS is warranted (Figure 3).
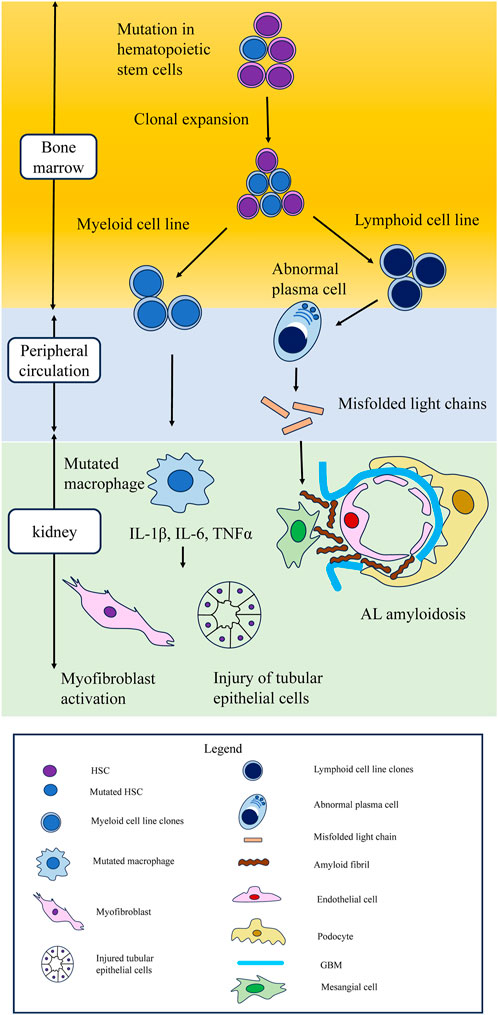
FIGURE 3. Schematic of hematopoietic stem cell mutations leading to clonal expansion in myeloid and lymphoid cell lines, leading to kidney damage. Possible mechanism of kidney injury in CHIP. Mutations in hematopoietic stem cells lead to clonal expansion in both myeloid and lymphoid cell lines. In myeloid cell line, cytokines such as IL-1β, IL-6, and TNFα secreted from mutated macrophages cause injury to tubular epithelial cells and myofibroblast activation. CHIP is also associated with lymphoid lineage malignancies, such as multiple myeloma and lymphoma. In lymphoid cell lines, aberrant monoclonal antibodies are related to monoclonal gammopathy of renal significance. In AL amyloidosis, which is the most typical MGRS lesion, abnormal plasma cells secrete misfolded light chains, resulting in the formation of toxic amyloid multimers and amyloid fibrils that deposit in the mesangium and the subepithelial space within glomeruli. CHIP, clonal hematopoietic indeterminate potential; MGRS, monoclonal gammopathy of renal significance; AL amyloidosis, amyloid light chain amyloidosis.
5 Epigenetic downregulation of Klotho in kidney and macrophage via inflammation
Kidney function steadily declines with age. Klotho, a renoprotective anti-aging protein secreted by the kidney, is involved in kidney aging process (Buchanan et al., 2020). Klotho has been identified as a central player in the aging-inflammation interface (Izquierdo et al., 2012). Enhanced inflammation is a common feature of CKD (Kadatane et al., 2023). Systemic or local renal inflammation decreases Klotho expression in the kidney (Izquierdo et al., 2012), and most kidney disorders are associated with a decline in circulating levels of Klotho mRNA and Klotho expression in the kidney (Wolf, 2014). As described previously, CHIP accelerates aging and induces systemic chronic inflammation, which leads to non-malignant diseases, including CKD. Thus, CHIP is regarded as one of the causes of decreased Klotho levels among the elderly.
In 1997, the Klotho gene was discovered when researchers observed that Klotho gene-disrupted mice exhibit a syndrome resembling human premature aging (Kuro-o et al., 1997). Klotho is mainly expressed in the kidneys, brain, parathyroid gland, and skeletal muscles. The distal convoluted tubule is the main site of Klotho expression in the kidneys (Kuro-o et al., 1997). Klotho exists in two general forms: as a transmembrane protein and a secretory form produced via alternative mRNA splicing of Klotho (Xu and Sun, 2015). Transmembrane Klotho is a co-receptor for fibroblast growth factor 23 (FGF23), which plays a critical role in the maintenance of phosphate and vitamin D homeostasis (Erben, 2016). In contrast, secreted Klotho protein regulates multiple growth factor signaling pathways, including insulin like growth-1, Wnt, and TGFβ1, and the activity of multiple ion channels and transporters (Izquierdo et al., 2012). Kidney Klotho expression responds to local and systemic inflammation, and epigenetic mechanisms, such as DNA methylation and histone modifications, and micro-RNA expression contribute to its downregulation (Ruiz-Andres et al., 2016).
DNA methylation suppresses gene expression and the subsequent protein translation. Inflammation-induced DNA hypermethylation resulted in Klotho promoter methylation, suppression of renal Klotho levels, and increased levels of the inflammatory marker CCL5 in animal models of renal interstitial fibrosis and human proximal tubular epithelial (HK-2) cells treated with CCL5 (Liu et al., 2022). Hypoxia and/or reoxygenation induced downregulation of Klotho expression has been observed in HK2 cells and renal tissues with ischemia-reperfusion injury (Zhao et al., 2022). In both the aforementioned studies, the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (Aza) effectively reversed Klotho expression. Aza reduced Klotho promoter DNA methylation and exerted anti-apoptotic and anti-inflammatory effects by increasing Klotho expression in renal injury (Liu et al., 2022; Zhao et al., 2022). Methylation of the Klotho promoter has been observed in injured kidneys and peripheral blood leukocytes (PBLs). CCL5 upregulation concomitant with Klotho downregulation in the serum and DNA hypermethylation in PBLs has been observed in CKD patients (Liu et al., 2022). In another study, higher levels of Klotho promoter methylation were observed in the renal tissue and peripheral blood mononuclear cells (PBMC) of patients with CKD than in controls. The degree of Klotho promoter methylation in PBMCs was associated with the clinical and histological severity of CKD. PBMC Klotho promoter methylation level has been reported as a potential biomarker of renal Klotho promoter hypermethylation (Chen et al., 2013). The most frequently observed mutations in CH are loss of function mutations of DNMT3A and TET2, which are involved in DNA methylation and demethylation, respectively (Jaiswal, 2020). The relationship between DNMT3A or TET2 mutations in CH and increase in Klotho promoter methylation levels in PBLs in CKD patients is unclear; therefore, further research is warranted.
Different combinations of histone modifications, such as methylation, acetylation, ubiquitination, and phosphorylation, regulate the chromatin structure and transcriptional status (Strahl and Allis, 2000). Histone acetylation relaxes the chromatin and facilitates transcription factor recruitment and transcription. Inflammatory cytokines, such as TNFα or TNF-like weak inducer of apoptosis (TWEAK), have been reported to promote the binding of NF-κB RelA (p65) to the Klotho promoter and thereby enhancing histone H3/H4 deacetylation of the murine Klotho promoter in cultured tubular cells, resulting in downregulation of Klotho expression. The histone deacetylase inhibitor trichostatin A prevents inflammatory cytokine induced Klotho downregulation (Moreno et al., 2011). The transcriptional activity of Klotho is also regulated by H3K9 modifications (Irifuku et al., 2016). In fibrotic kidney, TGFβ1, produced mainly by macrophages (Chevalier et al., 2009), increases the H3K9 methyltransferase G9a (Irifuku et al., 2016). Downregulation of Klotho levels, elevation of G9a, and monomethylation of H3K9 were observed in mouse models of unilateral ureteral obstruction and human kidney samples from patients with IgA nephropathy (Irifuku et al., 2016).
miRNAs are endogenous short non-coding RNAs of 22–25 base pairs that regulate gene expression through post-transcriptional repression of target mRNAs. miRNA binds to the 3′-untranslational region (UTR) of the target mRNA through base-pairing mechanism to suppress target gene expression by either mRNA degradation or inhibiting protein translation (Lu and Rothenberg, 2018). miRNAs regulate Klotho expression in renal diseases. miR-34a was reported to aggravate kidney fibrosis by downregulating Klotho expression in tubular epithelial cells (Liu et al., 2019). Oxidative stress-induced miR-200c binding to the Klotho mRNA 3′-UTR results in reduced Klotho expression in renal epithelial cells (Morii et al., 2019). miRNAs affect Klotho expression in renal cells and macrophages. Tissue macrophages are differentiated into M1 and M2 phenotypes on stimulation (Martinez et al., 2008). M1 macrophages (also called classically activated) are proinflammatory, which are activated by interferon gamma/lipopolysaccharide (IFN-γ/LPS). On the other hand, M2 macrophages (also called alternatively activated) are anti-inflammatory and promote wound healing and resolution of inflammation, which are activated by interleukin-4/interleukin-13 (IL-4/IL-13) (Huen and Cantley, 2017). miR-199a-5p from albumin-stimulated HK2 cell-derived extracellular vesicles (EVs) induces M1 polarization by suppressing Klotho in macrophages. In addition, Klotho polarizes M1 macrophages toward the M2 phenotype through Toll-like receptor 4 and ameliorates renal fibrosis in DM mouse models (Jia et al., 2019). In CKD with CHIP, mutated macrophages secrete inflammatory cytokines and damage renal cells. It has been speculated that miRNAs from damaged tubular epithelial cells affect mutated macrophages. miRNAs suppress Klotho expression in macrophages and exacerbate polarization of the M1 macrophage phenotype. A bidirectional worsening mechanism may exist between macrophages and tubular epithelial cells via Klotho and miRNAs (Figure 4).
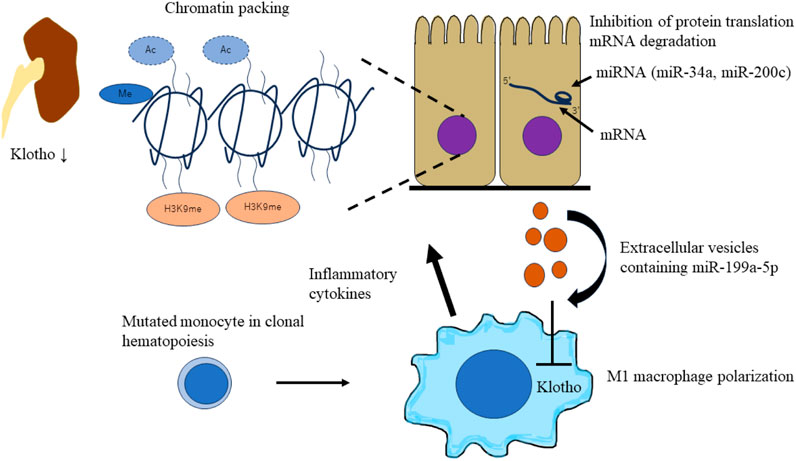
FIGURE 4. Schematic of epigenetic downregulation of kidney and macrophage Klotho levels by CHIP induced inflammation. CHIP-induced inflammation leads to epigenetic downregulation of kidney and macrophage Klotho levels. Inflammatory cytokines (TNFα or TWEAK) and TGFβ1 secreted by mutated macrophages induce chromatin packaging (Klotho promoter methylation, histone H3/H4 deacetylation, and H3K9 modification) in tubular epithelial cells. This leads to the decrease of Klotho levels in an injured kidney. miRNAs affect Klotho expression in renal cells and macrophages. miRNAs (miR-34a and miR-200c) aggravate kidney fibrosis by downregulating Klotho expression in tubular epithelial cells. Extracellular vesicles containing miRNAs (miR-100a-5p) from damaged tubular epithelial cells affect mutated macrophages. miRNAs suppress Klotho expression in macrophages and exacerbate the polarization of M1 macrophage phenotype. CHIP, clonal hematopoietic indeterminate potential; TWEAK, TNF-like weak inducer of apoptosis.
6 The role of CHIP related genes in the kidney
The most frequently observed mutations in CHIP (DNMT3A, TET2, and ASXL-1) are of epigenetic regulators (Jaiswal et al., 2014). Epigenetic alterations are related to acute kidney injury-to-chronic kidney disease transition (Nangaku et al., 2017; Tanemoto et al., 2022; Tanemoto and Mimura, 2022), and the roles of DNMT3A, TET2, and ASXL-1 in kidneys are reported as shown below.
DNA methylation involves DNA methyltransferase enzymes (DNMTs) such as DNMT1, DNMT3A, and DNMT3B. Maintenance of DNMT1 and de novo methyltransferases, DNMT3A and DNMT3B, are essential for mammalian development (Jin and Robertson, 2013). DNA methylation represses gene transcription and is associated with kidney injury (Tang and Zhuang, 2019). In studies on adenine-fed CKD mice, aberrant DNMT1/DNMT3A elevation, Klotho promoter hypermethylation, and Klotho suppression have been observed (Xia and Cao, 2021). DNMT3A and DNMT3B are responsible for the methylation of gene regulatory regions that act as enhancers during kidney development, but are decommissioned in adult mice. Although DNMT3A and DNMT3B knock-out mice displayed no obvious kidney abnormalities, they showed resistance to acute kidney injury (AKI). Human kidney disease risk loci were enriched in fetal regulatory regions (enhancers) that were decommissioned by DNMT3A/3B and were no longer active in adult kidneys. Genetic and epigenetic (DNMT3A/3B) factors may converge in the same genetic region, resulting in the development of kidney disease (Guan et al., 2020). Additionally, in renal fibrosis, the promoter region of PTEN is methylated and PTEN expression is downregulated. DNMT3A negatively regulates PTEN to activate the PI3K/AKT signaling pathway, induces epithelial-mesenchymal transition (EMT) in renal tubular epithelial cells, and aggravates renal fibrosis (Hu et al., 2022).
Ten eleven translocation (TET) methylcytosine dioxygenase family members catalyze the conversion of 5-methylcytosine to 5-hydroxymethylcytosine. The TET family of proteins, TET1, TET2, and TET3 are involved in DNA demethylation. In addition, TET proteins play a role in regulating immunity and inflammation. In a study on TET2 knock-out AKI mice model, it was observed that TET2 exerted a renal-protective effect during AKI by regulating metabolic and inflammatory responses through the PPAR signaling pathway (Bao et al., 2021). Additionally, in mesangial cells, TET2 activates TGFβ1 expression through demethylation of CpG islands in the TGFβ1 regulatory region under high-glucose conditions. Moreover, TET2 plays an important role in the pathogenesis of diabetic nephropathy (Yang et al., 2018).
The additional sex comb-like (ASXL) family of proteins comprises chromatin factors that function in transcriptional activation and repression. It was originally identified in Drosophila as an enhancer of the trithorax and polycomb group proteins. Three Asx homologs have been identified in mammals: ASXL1, ASXL2, and ASXL3. Among them, ASXL1 mutations are often found in a wide range of myeloid malignancies (Carbuccia et al., 2009) and Bohring-Opitz syndrome (Hoischen et al., 2011). ASXL1 is essential for normal hematopoiesis and wild-type ASXL1 epigenetically controls gene expression through its pivotal role in regulating the levels of H2AK119ub, H3K27me3, and H3K4me3 (Asada et al., 2019). In addition, wild-type ASXL1 interacts with AKT1, and ASXL1 deficiency in embryonic fibroblasts leads to p27-dependent cell cycle arrest, resulting in cellular senescence (Youn et al., 2017). Defects in kidney size and glomerular podocyte are observed in ASXL1-null mice; therefore, ASXL1 is essential for normal podocyte development. ASXL1 maintains podocyte structure via its association with Wilms’ tumor 1-interacting protein (WTIP) and the regulation of WT1 signaling during early kidney development (Moon et al., 2015). However, the role of ASXL1 in kidney injury remains unclear.
7 Potential therapeutic approaches for CHIP and CHIP-related CKD
There are neither established therapies for CHIP nor definite criteria to determine eligibility for CHIP therapy yet. However, recent studies have supported the development of effective CHIP therapies, such as blocking the expansion of aberrant HSCs and inhibiting chronic inflammation. For example, in TET2-deficient mice, high-dose vitamin C reversed the aberrant self-renewal of HSPCs via TET2 restoration (Cimmino et al., 2017). Additionally, since the mTOR pathway is a candidate pathway involved in CHIP pathology, Rapamycin, an mTOR inhibitor that effectively blocks the expansion of phenotypic HSCs in aged ASXL1-mutant knock-in mice (Fujino et al., 2021) can be a potential CHIP treatment.
Furthermore, the inhibition of inflammatory/innate signaling, such as the IRAK1/4 cascade, has been reported to prevent CHIP progression (Wang et al., 2021). In addition, anti-inflammatory drugs are effective against CHIP-related heart disease. In mouse models of heart failure with hematopoietic or myeloid TET2 deficiency, IL-1β/NLRP3 inflammasome inhibitor ameliorated heart failure (Sano et al., 2018b). CHIP-related CKD may be induced by mutated macrophages accumulating in the kidney, and anti-inflammatory drugs may be a treatment option for CKD. Renin-angiotensin system inhibitors (RASi) and sodium-glucose cotransporter-2 inhibitors (SGLT2i) are other potential candidates for CHIP treatment. RASi, angiotensin-converting enzyme inhibitors, and angiotensin receptor blockers are widely used to treat proteinuric CKD (Jafar et al., 2001). SGLT2i have been shown to delay CKD progression, regardless of the presence or absence of diabetes (Heerspink et al., 2020). Both RASi and SGLT2i have been reported to have anti-inflammatory effects (Cantero-Navarro et al., 2021; Winiarska et al., 2021), making them potential candidates for the treatment of CKD with CHIP. In addition, drugs targeting the epigenetic regulation of Klotho are a treatment option. EVs containing miRNAs (miR-199a-5p) from injured tubular epithelial cells have been shown to decrease Klotho expression and induce inflammation in macrophages (Jia et al., 2019). Therefore, inhibiting miRNAs in injured tubular epithelial cells may suppress the activation of inflammatory macrophages in patients with CKD with CHIP. Additionally, there is a potential relationship between CHIP and MGRS; therefore, if MGRS is diagnosed, clone-directed therapy may be required (Leung et al., 2021) (Figure 5).
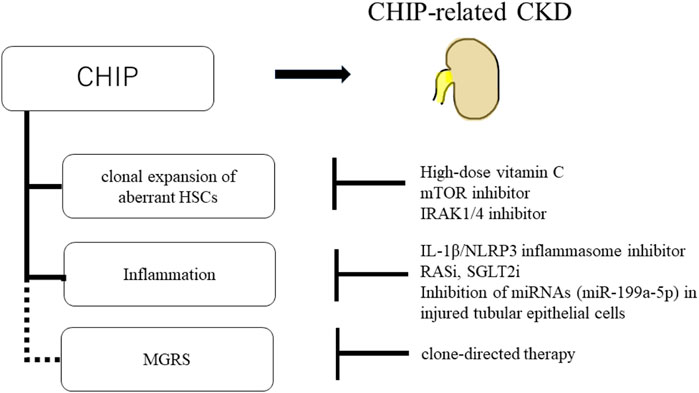
FIGURE 5. Schematic of potential therapeutic approaches for CHIP and CHIP-related CKD. High-dose vitamin C, mTOR inhibitor, and IRAK1/4 inhibitor prevent the clonal expansion of aberrant HSCs. And, IL-1β/NLRP3 inflammasome inhibitor, RASi, SGLT2i, and inhibition of miRNAs (miR-199a-5p) in injured tubular epithelial cells suppress the inflammation in CHIP. There is potential relationship between CHIP and MGRS. If MGRS is diagnosed, clone-directed therapy is the treatment option.
8 Conclusion
Recent studies have revealed that CHIP is a risk factor of non-malignant diseases including CKD. There is a positive feedback loop between CHIP and inflammation. The possible mechanism of the kidney injury in CHIP is supposed to be due to the clonal expansion in both myeloid and lymphoid cell lines. Mutated macrophages in CHIP secrete cytokines which damage the organs including kidney. Inflammation leads to epigenetic downregulation of kidney and macrophage klotho which is anti-aging factor. And there is potential relationship between CHIP and MGRS which has emerged as an important factor of CKD among elderly patients. It is important to consider the CHIP related complications such as hematological malignancy, cardiovascular diseases and metabolic disorders in managing the elderly CKD patients.
Author contributions
YO: Writing–original draft. IM: Conceptualization, Funding acquisition, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This review was in part supported by the Takeda Science Foundation (IM), The Naito Foundation (IM). This review was also supported by World-leading Innovative Graduate Study Program for Life Science and Technology (WINGS-LST), by the Ministry of Education, Culture, Sports, Science and Technology (to IM).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A., Behjati, S., Biankin, A. V., et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. doi:10.1038/nature12477
Ambrosi, T. H., Scialdone, A., Graja, A., Gohlke, S., Jank, A. M., Bocian, C., et al. (2017). Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell. Stem Cell. 20, 771–784. doi:10.1016/j.stem.2017.02.009
Asada, S., Fujino, T., Goyama, S., and Kitamura, T. (2019). The role of ASXL1 in hematopoiesis and myeloid malignancies. Cell. Mol. Life Sci. 76, 2511–2523. doi:10.1007/s00018-019-03084-7
Balasubramani, A., Larjo, A., Bassein, J. A., Chang, X., Hastie, R. B., Togher, S. M., et al. (2015). Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat. Commun. 6, 7307. doi:10.1038/ncomms8307
Bao, Y., Bai, M., Zhu, H., Yuan, Y., Wang, Y., Zhang, Y., et al. (2021). DNA demethylase Tet2 suppresses cisplatin-induced acute kidney injury. Cell. Death Discov. 7, 167. doi:10.1038/s41420-021-00528-7
Bick, A. G., Weinstock, J. S., Nandakumar, S. K., Fulco, C. P., Bao, E. L., Zekavat, S. M., et al. (2020). Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 586, 763–768. doi:10.1038/s41586-020-2819-2
Bolton, K. L., Ptashkin, R. N., Gao, T., Braunstein, L., Devlin, S. M., Kelly, D., et al. (2020). Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 52, 1219–1226. doi:10.1038/s41588-020-00710-0
Bouzid, H., Belk, J. A., Jan, M., Qi, Y., Sarnowski, C., Wirth, S., et al. (2023). Clonal hematopoiesis is associated with protection from Alzheimer's disease. Nat. Med. 29, 1662–1670. doi:10.1038/s41591-023-02397-2
Bridoux, F., Leung, N., Hutchison, C. A., Touchard, G., Sethi, S., Fermand, J. P., et al. (2015). Diagnosis of monoclonal gammopathy of renal significance. Kidney Int. 87, 698–711. doi:10.1038/ki.2014.408
Buchanan, S., Combet, E., Stenvinkel, P., and Shiels, P. G. (2020). Klotho, aging, and the failing kidney. Front. Endocrinol. (Lausanne) 11, 560. doi:10.3389/fendo.2020.00560
Buscarlet, M., Provost, S., Zada, Y. F., Barhdadi, A., Bourgoin, V., Lepine, G., et al. (2017). DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 130, 753–762. doi:10.1182/blood-2017-04-777029
Buscarlet, M., Provost, S., Zada, Y. F., Bourgoin, V., Mollica, L., Dube, M. P., et al. (2018). Lineage restriction analyses in CHIP indicate myeloid bias for TET2 and multipotent stem cell origin for DNMT3A. Blood 132, 277–280. doi:10.1182/blood-2018-01-829937
Cai, Z., Kotzin, J. J., Ramdas, B., Chen, S., Nelanuthala, S., Palam, L. R., et al. (2018). Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell. Stem Cell. 23, 833–849. doi:10.1016/j.stem.2018.10.013
Cantero-Navarro, E., Fernandez-Fernandez, B., Ramos, A. M., Rayego-Mateos, S., Rodrigues-Diez, R. R., Sanchez-Nino, M. D., et al. (2021). Renin-angiotensin system and inflammation update. Mol. Cell. Endocrinol. 529, 111254. doi:10.1016/j.mce.2021.111254
Carbuccia, N., Murati, A., Trouplin, V., Brecqueville, M., Adelaide, J., Rey, J., et al. (2009). Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 23, 2183–2186. doi:10.1038/leu.2009.141
Challen, G. A., and Goodell, M. A. (2020). Clonal hematopoiesis: mechanisms driving dominance of stem cell clones. Blood 136, 1590–1598. doi:10.1182/blood.2020006510
Challen, G. A., Sun, D., Jeong, M., Luo, M., Jelinek, J., Berg, J. S., et al. (2011). Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 44, 23–31. doi:10.1038/ng.1009
Chen, J., Zhang, X., Zhang, H., Lin, J., Zhang, C., Wu, Q., et al. (2013). Elevated Klotho promoter methylation is associated with severity of chronic kidney disease. PLoS One 8, e79856. doi:10.1371/journal.pone.0079856
Chevalier, R. L., Forbes, M. S., and Thornhill, B. A. (2009). Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 75, 1145–1152. doi:10.1038/ki.2009.86
Cimmino, L., Dolgalev, I., Wang, Y., Yoshimi, A., Martin, G. H., Wang, J., et al. (2017). Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell. 170, 1079–1095. doi:10.1016/j.cell.2017.07.032
Coombs, C. C., Zehir, A., Devlin, S. M., Kishtagari, A., Syed, A., Jonsson, P., et al. (2017). Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell. Stem Cell. 21, 374–382. doi:10.1016/j.stem.2017.07.010
Dai, Y. J., Wang, Y. Y., Huang, J. Y., Xia, L., Shi, X. D., Xu, J., et al. (2017). Conditional knockin of Dnmt3a R878H initiates acute myeloid leukemia with mTOR pathway involvement. Proc. Natl. Acad. Sci. U. S. A. 114, 5237–5242. doi:10.1073/pnas.1703476114
Dawoud, A. A. Z., Gilbert, R. D., Tapper, W. J., and Cross, N. C. P. (2022). Clonal myelopoiesis promotes adverse outcomes in chronic kidney disease. Leukemia 36, 507–515. doi:10.1038/s41375-021-01382-3
Dawoud, A. a.Z., Tapper, W. J., and Cross, N. C. P. (2020). Clonal myelopoiesis in the UK Biobank cohort: ASXL1 mutations are strongly associated with smoking. Leukemia 34, 2660–2672. doi:10.1038/s41375-020-0896-8
Denicolo, S., Vogi, V., Keller, F., Thoni, S., Eder, S., Heerspink, H. J. L., et al. (2022). Clonal hematopoiesis of indeterminate potential and diabetic kidney disease: a nested case-control study. Kidney Int. Rep. 7, 876–888. doi:10.1016/j.ekir.2022.01.1064
Dimopoulos, M. A., Gertz, M. A., Kastritis, E., Garcia-Sanz, R., Kimby, E. K., Leblond, V., et al. (2009). Update on treatment recommendations from the fourth international workshop on waldenstrom's macroglobulinemia. J. Clin. Oncol. 27, 120–126. doi:10.1200/JCO.2008.17.7865
Erben, R. G. (2016). Update on FGF23 and klotho signaling. Mol. Cell. Endocrinol. 432, 56–65. doi:10.1016/j.mce.2016.05.008
Fidler, T. P., Xue, C., Yalcinkaya, M., Hardaway, B., Abramowicz, S., Xiao, T., et al. (2021). The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 592, 296–301. doi:10.1038/s41586-021-03341-5
Fujino, T., Goyama, S., Sugiura, Y., Inoue, D., Asada, S., Yamasaki, S., et al. (2021). Mutant ASXL1 induces age-related expansion of phenotypic hematopoietic stem cells through activation of Akt/mTOR pathway. Nat. Commun. 12, 1826. doi:10.1038/s41467-021-22053-y
Fujino, T., and Kitamura, T. (2020). ASXL1 mutation in clonal hematopoiesis. Exp. Hematol. 83, 74–84. doi:10.1016/j.exphem.2020.01.002
Fuster, J. J., Zuriaga, M. A., Zorita, V., Maclauchlan, S., Polackal, M. N., Viana-Huete, V., et al. (2020). TET2-Loss-of-Function-Driven clonal hematopoiesis exacerbates experimental insulin resistance in aging and obesity. Cell. Rep. 33, 108326. doi:10.1016/j.celrep.2020.108326
Genovese, G., Kahler, A. K., Handsaker, R. E., Lindberg, J., Rose, S. A., Bakhoum, S. F., et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487. doi:10.1056/NEJMoa1409405
Gibson, C. J., Lindsley, R. C., Tchekmedyian, V., Mar, B. G., Shi, J., Jaiswal, S., et al. (2017). Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J. Clin. Oncol. 35, 1598–1605. doi:10.1200/JCO.2016.71.6712
Guan, Y., Liu, H., Ma, Z., Li, S. Y., Park, J., Sheng, X., et al. (2020). Dnmt3a and dnmt3b-decommissioned fetal enhancers are linked to kidney disease. J. Am. Soc. Nephrol. 31, 765–782. doi:10.1681/ASN.2019080797
Heerspink, H. J. L., Stefansson, B. V., Correa-Rotter, R., Chertow, G. M., Greene, T., Hou, F. F., et al. (2020). Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 383, 1436–1446. doi:10.1056/NEJMoa2024816
Hinds, D. A., Barnholt, K. E., Mesa, R. A., Kiefer, A. K., Do, C. B., Eriksson, N., et al. (2016). Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 128, 1121–1128. doi:10.1182/blood-2015-06-652941
Hoischen, A., Van Bon, B. W., Rodriguez-Santiago, B., Gilissen, C., Vissers, L. E., De Vries, P., et al. (2011). De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat. Genet. 43, 729–731. doi:10.1038/ng.868
Hormaechea-Agulla, D., Matatall, K. A., Le, D. T., Kain, B., Long, X., Kus, P., et al. (2021). Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNγ signaling. Cell. Stem Cell. 28, 1428–1442.e6. doi:10.1016/j.stem.2021.03.002
Hu, T., Chen, F., Chen, D., and Liang, H. (2022). DNMT3a negatively regulates PTEN to activate the PI3K/AKT pathway to aggravate renal fibrosis. Cell. Signal 96, 110352. doi:10.1016/j.cellsig.2022.110352
Huang, R. C., Lillycrop, K. A., Beilin, L. J., Godfrey, K. M., Anderson, D., Mori, T. A., et al. (2019). Epigenetic age acceleration in adolescence associates with BMI, inflammation, and risk score for middle age cardiovascular disease. J. Clin. Endocrinol. Metab. 104, 3012–3024. doi:10.1210/jc.2018-02076
Huen, S. C., and Cantley, L. G. (2017). Macrophages in renal injury and repair. Annu. Rev. Physiol. 79, 449–469. doi:10.1146/annurev-physiol-022516-034219
Husby, S., Favero, F., Nielsen, C., Sorensen, B. S., Baech, J., Grell, K., et al. (2020). Clinical impact of clonal hematopoiesis in patients with lymphoma undergoing ASCT: a national population-based cohort study. Leukemia 34, 3256–3268. doi:10.1038/s41375-020-0795-z
Irifuku, T., Doi, S., Sasaki, K., Doi, T., Nakashima, A., Ueno, T., et al. (2016). Inhibition of H3K9 histone methyltransferase G9a attenuates renal fibrosis and retains klotho expression. Kidney Int. 89, 147–157. doi:10.1038/ki.2015.291
Izquierdo, M. C., Perez-Gomez, M. V., Sanchez-Nino, M. D., Sanz, A. B., Ruiz-Andres, O., Poveda, J., et al. (2012). Klotho, phosphate and inflammation/ageing in chronic kidney disease. Nephrol. Dial. Transpl. 27 (4), iv6–10. doi:10.1093/ndt/gfs426
Jafar, T. H., Schmid, C. H., Landa, M., Giatras, I., Toto, R., Remuzzi, G., et al. (2001). Angiotensin-converting enzyme inhibitors and progression of nondiabetic renal disease. A meta-analysis of patient-level data. Ann. Intern Med. 135, 73–87. doi:10.7326/0003-4819-135-2-200107170-00007
Jaiswal, S. (2020). Clonal hematopoiesis and nonhematologic disorders. Blood 136, 1606–1614. doi:10.1182/blood.2019000989
Jaiswal, S., and Ebert, B. L. (2019). Clonal hematopoiesis in human aging and disease. Science 366, eaan4673. doi:10.1126/science.aan4673
Jaiswal, S., Fontanillas, P., Flannick, J., Manning, A., Grauman, P. V., Mar, B. G., et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498. doi:10.1056/NEJMoa1408617
Jaiswal, S., Natarajan, P., Silver, A. J., Gibson, C. J., Bick, A. G., Shvartz, E., et al. (2017). Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377, 111–121. doi:10.1056/NEJMoa1701719
Jia, Y., Zheng, Z., Xue, M., Zhang, S., Hu, F., Li, Y., et al. (2019). Extracellular vesicles from albumin-induced tubular epithelial cells promote the M1 macrophage phenotype by targeting klotho. Mol. Ther. 27, 1452–1466. doi:10.1016/j.ymthe.2019.05.019
Jin, B., and Robertson, K. D. (2013). DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 754, 3–29. doi:10.1007/978-1-4419-9967-2_1
Kadatane, S. P., Satariano, M., Massey, M., Mongan, K., and Raina, R. (2023). The role of inflammation in CKD. Cells 12, 1581. doi:10.3390/cells12121581
Kar, S. P., Quiros, P. M., Gu, M., Jiang, T., Mitchell, J., Langdon, R., et al. (2022). Genome-wide analyses of 200,453 individuals yield new insights into the causes and consequences of clonal hematopoiesis. Nat. Genet. 54, 1155–1166. doi:10.1038/s41588-022-01121-z
Kestenbaum, B., Bick, A. G., Vlasschaert, C., Rauh, M. J., Lanktree, M. B., Franceschini, N., et al. (2023). Clonal hematopoiesis of indeterminate potential and kidney function decline in the general population. Am. J. Kidney Dis. 81, 329–335. doi:10.1053/j.ajkd.2022.08.014
Kuro-O, M., Matsumura, Y., Aizawa, H., Kawaguchi, H., Suga, T., Utsugi, T., et al. (1997). Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51. doi:10.1038/36285
Lee-Six, H., Obro, N. F., Shepherd, M. S., Grossmann, S., Dawson, K., Belmonte, M., et al. (2018). Population dynamics of normal human blood inferred from somatic mutations. Nature 561, 473–478. doi:10.1038/s41586-018-0497-0
Leung, N., Bridoux, F., Batuman, V., Chaidos, A., Cockwell, P., D'agati, V. D., et al. (2019). The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat. Rev. Nephrol. 15, 45–59. doi:10.1038/s41581-018-0077-4
Leung, N., Bridoux, F., and Nasr, S. H. (2021). Monoclonal gammopathy of renal significance. N. Engl. J. Med. 384, 1931–1941. doi:10.1056/NEJMra1810907
Liu, Q., Li, S., Yu, L., Yin, X., Liu, X., Ye, J., et al. (2022). CCL5 suppresses klotho expression via p-STAT3/DNA methyltransferase1-mediated promoter hypermethylation. Front. Physiol. 13, 856088. doi:10.3389/fphys.2022.856088
Liu, Y., Bi, X., Xiong, J., Han, W., Xiao, T., Xu, X., et al. (2019). MicroRNA-34a promotes renal fibrosis by downregulation of klotho in tubular epithelial cells. Mol. Ther. 27, 1051–1065. doi:10.1016/j.ymthe.2019.02.009
Lu, T. X., and Rothenberg, M. E. (2018). MicroRNA. J. Allergy Clin. Immunol. 141, 1202–1207. doi:10.1016/j.jaci.2017.08.034
Martinez, F. O., Sica, A., Mantovani, A., and Locati, M. (2008). Macrophage activation and polarization. Front. Biosci. 13, 453–461. doi:10.2741/2692
Merlini, G., and Bellotti, V. (2003). Molecular mechanisms of amyloidosis. N. Engl. J. Med. 349, 583–596. doi:10.1056/NEJMra023144
Miles, L. A., Bowman, R. L., Merlinsky, T. R., Csete, I. S., Ooi, A. T., Durruthy-Durruthy, R., et al. (2020). Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 587, 477–482. doi:10.1038/s41586-020-2864-x
Moon, S., Um, S. J., and Kim, E. J. (2015). Role of Asxl1 in kidney podocyte development via its interaction with Wtip. Biochem. Biophys. Res. Commun. 466, 560–566. doi:10.1016/j.bbrc.2015.09.077
Moreno, J. A., Izquierdo, M. C., Sanchez-Nino, M. D., Suarez-Alvarez, B., Lopez-Larrea, C., Jakubowski, A., et al. (2011). The inflammatory cytokines TWEAK and TNFα reduce renal klotho expression through NFκB. J. Am. Soc. Nephrol. 22, 1315–1325. doi:10.1681/ASN.2010101073
Morii, K., Yamasaki, S., Doi, S., Irifuku, T., Sasaki, K., Doi, T., et al. (2019). microRNA-200c regulates KLOTHO expression in human kidney cells under oxidative stress. PLoS One 14, e0218468. doi:10.1371/journal.pone.0218468
Muto, T., Walker, C. S., Choi, K., Hueneman, K., Smith, M. A., Gul, Z., et al. (2020). Adaptive response to inflammation contributes to sustained myelopoiesis and confers a competitive advantage in myelodysplastic syndrome HSCs. Nat. Immunol. 21, 535–545. doi:10.1038/s41590-020-0663-z
Nachun, D., Lu, A. T., Bick, A. G., Natarajan, P., Weinstock, J., Szeto, M. D., et al. (2021). Clonal hematopoiesis associated with epigenetic aging and clinical outcomes. Aging Cell. 20, e13366. doi:10.1111/acel.13366
Nangaku, M., Hirakawa, Y., Mimura, I., Inagi, R., and Tanaka, T. (2017). Epigenetic changes in the acute kidney injury-to-chronic kidney disease transition. Nephron 137, 256–259. doi:10.1159/000476078
Niroula, A., Sekar, A., Murakami, M. A., Trinder, M., Agrawal, M., Wong, W. J., et al. (2021). Distinction of lymphoid and myeloid clonal hematopoiesis. Nat. Med. 27, 1921–1927. doi:10.1038/s41591-021-01521-4
Osorio, F. G., Rosendahl Huber, A., Oka, R., Verheul, M., Patel, S. H., Hasaart, K., et al. (2018). Somatic mutations reveal lineage relationships and age-related mutagenesis in human hematopoiesis. Cell. Rep. 25, 2308–2316. doi:10.1016/j.celrep.2018.11.014
Palladini, G., Milani, P., and Merlini, G. (2020). Management of AL amyloidosis in 2020. Blood 136, 2620–2627. doi:10.1182/blood.2020006913
Porta, C., Paglino, C., and Mosca, A. (2014). Targeting PI3K/Akt/mTOR signaling in cancer. Front. Oncol. 4, 64. doi:10.3389/fonc.2014.00064
Profumo, E., Buttari, B., Tosti, M. E., Alessandri, C., Valesini, G., Marcuccio, L., et al. (2010). Identification of IP-10 and IL-5 as proteins differentially expressed in human complicated and uncomplicated carotid atherosclerotic plaques. Int. J. Immunopathol. Pharmacol. 23, 775–782. doi:10.1177/039463201002300311
Rajkumar, S. V., Dimopoulos, M. A., Palumbo, A., Blade, J., Merlini, G., Mateos, M. V., et al. (2014). International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 15, e538–e548. doi:10.1016/S1470-2045(14)70442-5
Reya, T., Morrison, S. J., Clarke, M. F., and Weissman, I. L. (2001). Stem cells, cancer, and cancer stem cells. Nature 414, 105–111. doi:10.1038/35102167
Ruiz-Andres, O., Sanchez-Nino, M. D., Moreno, J. A., Ruiz-Ortega, M., Ramos, A. M., Sanz, A. B., et al. (2016). Downregulation of kidney protective factors by inflammation: role of transcription factors and epigenetic mechanisms. Am. J. Physiol. Ren. Physiol. 311, F1329–F1340. doi:10.1152/ajprenal.00487.2016
Rysava, R. (2019). AL amyloidosis: advances in diagnostics and treatment. Nephrol. Dial. Transpl. 34, 1460–1466. doi:10.1093/ndt/gfy291
Sanders, P. W., and Booker, B. B. (1992). Pathobiology of cast nephropathy from human Bence Jones proteins. J. Clin. Invest. 89, 630–639. doi:10.1172/JCI115629
Sano, S., Oshima, K., Wang, Y., Katanasaka, Y., Sano, M., and Walsh, K. (2018a). CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ. Res. 123, 335–341. doi:10.1161/CIRCRESAHA.118.313225
Sano, S., Oshima, K., Wang, Y., Maclauchlan, S., Katanasaka, Y., Sano, M., et al. (2018b). Tet2-Mediated clonal hematopoiesis accelerates heart failure through a Mechanism involving the IL-1β/NLRP3 Inflammasome. J. Am. Coll. Cardiol. 71, 875–886. doi:10.1016/j.jacc.2017.12.037
Solary, E., Bernard, O. A., Tefferi, A., Fuks, F., and Vainchenker, W. (2014). The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia 28, 485–496. doi:10.1038/leu.2013.337
Steensma, D. P., Bejar, R., Jaiswal, S., Lindsley, R. C., Sekeres, M. A., Hasserjian, R. P., et al. (2015). Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126, 9–16. doi:10.1182/blood-2015-03-631747
Strahl, B. D., and Allis, C. D. (2000). The language of covalent histone modifications. Nature 403, 41–45. doi:10.1038/47412
Strati, P., and Shanafelt, T. D. (2015). Monoclonal B-cell lymphocytosis and early-stage chronic lymphocytic leukemia: diagnosis, natural history, and risk stratification. Blood 126, 454–462. doi:10.1182/blood-2015-02-585059
Sy-Go, J. P. T., Herrmann, S. M., and Seshan, S. V. (2022). Monoclonal gammopathy-related kidney diseases. Adv. Chronic Kidney Dis. 29, 86–102.e1. doi:10.1053/j.ackd.2022.01.004
Tanemoto, F., and Mimura, I. (2022). Therapies targeting epigenetic alterations in acute kidney injury-to-chronic kidney disease transition. Pharm. (Basel) 15, 123. doi:10.3390/ph15020123
Tanemoto, F., Nangaku, M., and Mimura, I. (2022). Epigenetic memory contributing to the pathogenesis of AKI-to-CKD transition. Front. Mol. Biosci. 9, 1003227. doi:10.3389/fmolb.2022.1003227
Tang, J., and Zhuang, S. (2019). Histone acetylation and DNA methylation in ischemia/reperfusion injury. Clin. Sci. (Lond) 133, 597–609. doi:10.1042/CS20180465
Testa, S., Kumar, J., Goodell, A. J., Zehnder, J. L., Alexander, K. M., Sidana, S., et al. (2022). Prevalence, mutational spectrum and clinical implications of clonal hematopoiesis of indeterminate potential in plasma cell dyscrasias. Semin. Oncol. 49, 465–475. doi:10.1053/j.seminoncol.2022.11.001
Van Zeventer, I. A., Salzbrunn, J. B., De Graaf, A. O., Van Der Reijden, B. A., Boezen, H. M., Vonk, J. M., et al. (2021). Prevalence, predictors, and outcomes of clonal hematopoiesis in individuals aged ≥80 years. Blood Adv. 5, 2115–2122. doi:10.1182/bloodadvances.2020004062
Vlasschaert, C., Mcnaughton, A. J. M., Chong, M., Cook, E. K., Hopman, W., Kestenbaum, B., et al. (2022). Association of clonal hematopoiesis of indeterminate potential with worse kidney function and anemia in two cohorts of patients with advanced chronic kidney disease. J. Am. Soc. Nephrol. 33, 985–995. doi:10.1681/ASN.2021060774
Wang, T., Pine, A. R., Kotini, A. G., Yuan, H., Zamparo, L., Starczynowski, D. T., et al. (2021). Sequential CRISPR gene editing in human iPSCs charts the clonal evolution of myeloid leukemia and identifies early disease targets. Cell. Stem Cell. 28, 1074–1089.e7. doi:10.1016/j.stem.2021.01.011
Winiarska, A., Knysak, M., Nabrdalik, K., Gumprecht, J., and Stompor, T. (2021). Inflammation and oxidative stress in diabetic kidney disease: the targets for SGLT2 inhibitors and GLP-1 receptor agonists. Int. J. Mol. Sci. 22, 10822. doi:10.3390/ijms221910822
Wolf, M. (2014). In search of the fountain of youth. J. Am. Soc. Nephrol. 25, 2143–2145. doi:10.1681/ASN.2014040392
Wong, W. J., Emdin, C., Bick, A. G., Zekavat, S. M., Niroula, A., Pirruccello, J. P., et al. (2023). Clonal haematopoiesis and risk of chronic liver disease. Nature 616, 747–754. doi:10.1038/s41586-023-05857-4
Wudhikarn, K., Padrnos, L., Lasho, T., Laplant, B., Kumar, S., Dispenzieri, A., et al. (2021). Clinical correlates and prognostic impact of clonal hematopoiesis in multiple myeloma patients receiving post-autologous stem cell transplantation lenalidomide maintenance therapy. Am. J. Hematol. 96, E157–E162. doi:10.1002/ajh.26125
Wynn, T. A., and Vannella, K. M. (2016). Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462. doi:10.1016/j.immuni.2016.02.015
Xia, J., and Cao, W. (2021). Epigenetic modifications of Klotho expression in kidney diseases. J. Mol. Med. Berl. 99, 581–592. doi:10.1007/s00109-021-02044-8
Xu, Y., and Sun, Z. (2015). Molecular basis of Klotho: from gene to function in aging. Endocr. Rev. 36, 174–193. doi:10.1210/er.2013-1079
Yang, F., Zhou, S., Wang, C., Huang, Y., Li, H., Wang, Y., et al. (2017). Epigenetic modifications of interleukin-6 in synovial fibroblasts from osteoarthritis patients. Sci. Rep. 7, 43592. doi:10.1038/srep43592
Yang, L., Zhang, Q., Wu, Q., Wei, Y., Yu, J., Mu, J., et al. (2018). Effect of TET2 on the pathogenesis of diabetic nephropathy through activation of transforming growth factor β1 expression via DNA demethylation. Life Sci. 207, 127–137. doi:10.1016/j.lfs.2018.04.044
Youn, H. S., Kim, T. Y., Park, U. H., Moon, S. T., An, S. J., Lee, Y. K., et al. (2017). Asxl1 deficiency in embryonic fibroblasts leads to cellular senescence via impairment of the AKT-E2F pathway and Ezh2 inactivation. Sci. Rep. 7, 5198. doi:10.1038/s41598-017-05564-x
Zhang, Q., Zhao, K., Shen, Q., Han, Y., Gu, Y., Li, X., et al. (2015). Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393. doi:10.1038/nature15252
Zhao, Y., Zeng, X., Xu, X., Wang, W., Xu, L., Wu, Y., et al. (2022). Low-dose 5-aza-2'-deoxycytidine protects against early renal injury by increasing klotho expression. Epigenomics 14, 1411–1425. doi:10.2217/epi-2022-0430
Keywords: CHIP, aging, CKD, epigenetics, Klotho
Citation: Ogura Y and Mimura I (2023) Epigenetic roles in clonal hematopoiesis and aging kidney-related chronic kidney disease. Front. Cell Dev. Biol. 11:1281850. doi: 10.3389/fcell.2023.1281850
Received: 23 August 2023; Accepted: 11 October 2023;
Published: 20 October 2023.
Edited by:
Hongbing Liu, Tulane University, United StatesReviewed by:
Christian Kosan, Friedrich Schiller University Jena, GermanyNina Graffmann, Heinrich Heine University of Düsseldorf, Germany
Copyright © 2023 Ogura and Mimura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Imari Mimura, aW1pbXVyYUBtLnUtdG9reW8uYWMuanA=