Abstract
Myeloproliferative neoplasms (MPNs) are subdivided into Philadelphia (Ph) chromosome-positive chronic myeloid leukemia (CML) and Ph-negative MPNs. BCR::ABL1 translocation is essential for the development and diagnosis of CML; on the other hand, the majority of Ph-negative MPNs are characterized by generally mutually exclusive mutations of Janus kinase 2 (JAK2), calreticulin (CALR), or thrombopoietin receptor/myeloproliferative leukemia (MPL). CALR mutations have been described essentially in JAK2 and MPL wild-type essential thrombocythemia and primary myelofibrosis. Rarely coexisting CALR and MPL mutations have been found in Ph-negative MPNs. BCR::ABL1 translocation and JAK2 mutations were initially considered mutually exclusive genomic events, but a discrete number of cases with the combination of these genetic alterations have been reported. The presence of BCR::ABL1 translocation with a coexisting CALR mutation is even more uncommon. Herein, starting from a routinely diagnosed case of CALR-mutated primary myelofibrosis subsequently acquiring BCR::ABL1 translocation, we performed a comprehensive review of the literature, discussing the clinicopathologic and molecular features, as well as the outcome and treatment of cases with BCR::ABL1 and CALR co-occurrence.
1 Introduction
Myeloproliferative neoplasms (MPNs) are clonal disorders with proliferation of at least one hematopoietic lineage and are subdivided into Philadelphia (Ph)-positive and Ph-negative forms (WHO, 2017; Arber et al., 2022; Khoury et al., 2022). Ph-positive chronic myeloid leukemia (CML) is typically characterized by the presence of the pathognomonic Ph-chromosome with translocation t (9; 22), resulting in the BCR::ABL1 oncogene. In 2005, it was discovered that the activating mutation in Janus kinase 2 (JAK2), mostly at codon 617 (JAK2 V617F), is involved in the development of Ph-negative MPNs, including polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) (Cazzola and Kralovics, 2014; WHO, 2017; Arber et al., 2022; Khoury et al., 2022). Subsequently, novel insights into Ph-negative MPN development were provided by the identification of the activating mutations in the myeloproliferative leukemia (MPL) gene in 2006 and CALR gene in 2013 (Klampfl et al., 2013; Nangalia et al., 2013; Rumi et al., 2014a). JAK2 mutations occur in 95% of PV and in 50%–60% of ET and PMF. After JAK2 mutations, CALR mutations are the second most common driver mutations in ET and PMF, being reported primarily in the context of JAK2 and MPL wild-type PMF (25%–35%) and ET (20%–25%) (Rumi et al., 2014b; Tefferi et al., 2014). MPL mutation occurs in 5%–10% of ET and PMF.
CALR mutations activate the JAK/STAT pathway through MPL, causing a high platelet (PLT) count. Although approximately 50 different CALR mutations have been reported, the most common CALR mutations are type 1 with a 52-base pair deletion and type 2 with a 5-base pair insertion (Rumi et al., 2014b; Tefferi et al., 2014; Pietra et al., 2016; Loscocco et al., 2021; Guglielmelli et al., 2024; Loscocco et al., 2024). CALR type 1 mutation has been associated with an increased risk of myelofibrotic transformation in ET patients; on the other hand, CALR type 2 mutation is identified in ET with an indolent behavior and low thrombotic risk despite an elevated platelet (PTL) count (Rumi et al., 2014b; Tefferi et al., 2014; Pietra et al., 2016). CALR type 1 and CALR type 2 mutations show different morphological features and different outcomes. Both 2022 WHO classification and ICC classification take into consideration these molecular findings, refining the current diagnostic criteria of Ph-negative MPNs, but neither classification addresses the issue of MPNs, presenting more than one “driver” genetic alteration (Arber et al., 2022; Khoury et al., 2022).
BCR::ABL1 rearrangement and JAK2/MPL/CALR mutations were initially considered mutually exclusive genetic events. However, despite being a rare occurrence, a number of cases with concomitant Ph-positive and JAK2-mutated MPNs have been described with a frequency ranging from 0.2% to 2.5%, depending on different studies (Pieri et al., 2011; Martin-Cabrera et al., 2017; Soderquist et al., 2018).
The association of CALR mutations with BCR::ABL1 rearrangement is much rarer, being reported in isolated cases. In this article, we described a case from our daily practice with a long history of CALR-mutated PMF, subsequently developing CML with gaining of BCR::ABL1 rearrangement; meanwhile, through a literature search, we identified 23 cases harboring both CALR mutation and BCR::ABL1 rearrangement (Bonzheim et al., 2015; Cabagnols et al., 2015; Loghavi et al., 2015; Diamond et al., 2016; Seghatoleslami et al., 2016; Dogliotti et al., 2017; Gilles et al., 2017; Kandarpa et al., 2017; Blouet et al., 2018; Boddu et al., 2018; De Roeck et al., 2018; Klairmont et al., 2018; Lewandowski et al., 2018; Xia et al., 2019; Balducci et al., 2020; Da Costa et al., 2020; Guidotti et al., 2020; Liu et al., 2020; Yoon et al., 2020; Sobieralski et al., 2022; Huo et al., 2023).
Clinical manifestations, pathological features, clonal findings, and management of these cases with the concomitant CALR mutation and BCR::ABL1 are discussed.
In cases with discordant clinical and molecular features or uncommon BM histology, the co-occurrence of BCR::ABL1-positive CML with another Ph-negative MPN should be suspected.
2 Explicative case from routine daily practice
A 45-year-old man was noted to have a new onset thrombocytosis with a PTL count of 526 × 109/L and normochromic, normocytic mild anemia (Hb 10.5 g/dL), whereas the white blood cell (WBC) count (8.02 × 109/L) was within the normal limit with 63% neutrophils, 27% lymphocytes, and 8% monocytes. A moderate splenomegaly was present. Bone marrow (BM) biopsy showed a slightly hypercellular marrow with the prevalence of a normally maturing myeloid lineage; the erythroid lineage in different stages of maturation was reduced; and the megakaryocytic lineage was expanded with even dense clustering of variably sized elements, including megakaryocytes with hyperchromatic nuclei. CD34-positive hematopoietic precursors were not increased. Grade 0 reticulin fibrosis was present. Molecular studies showed wild-type JAK2 V617F and MPL genes, whereas CALR exon 9 mutation type 1 (60% allelic burden) was identified. No BCR::ABL1 rearrangement was found. Karyotype analysis was not available. The combination of clinical, pathologic, and molecular features was in keeping with the diagnosis of MPN and suggestive of PMF, in the pre-fibrotic stage. Anagrelide was initially administered, followed by hydroxyurea (HU) 3 years later with a good hematological control of the disease. After 11 years, a progressive increase in the WBC count was noted (57 × 109/L), with mild anemia (Hb 9.2 g/dL) and normal PTL count (177 × 109/L). Splenomegaly (18 cm diameter) was detected. BM biopsy showed a markedly hypercellular marrow (95% cellularity) with prevalent and normally maturing granulopoiesis, the erythroid lineage was reduced with features of dyserythropoiesis, and megakaryocytes were increased in number with large-sized elements along with small-sized cells with hyperchromatic nuclei and evident dense clustering (Figures 1, 2). Grade II reticulin fibrosis was present. Repeated molecular studies confirmed CALR exon 9 mutation (allelic burden 40%) and JAK2 V617F and MPL negativity, whereas the BCR::ABL1 rearrangement was identified. RT-PCR identified the BCR-ABL1 fusion transcript of the p210 variant (allelic burden 88%). Karyotype analysis detected a 46XY karyotype with t (9:22) (q34; q11) translocation. Altogether, the clinicopathologic and molecular findings supported the diagnosis of PMF (with CALR mutation) and subsequent occurrence of the BCR::ABL1 rearrangement, therefore, interpretable as the coexistence of a Ph-negative MPN with CML. Tyrosine-kinase therapy (TKI) with nilotinib (600 mg/die) associated with ruxolitinib was given. After introduction of TKI treatment, the patient achieved a deep molecular response (DMR) within 6 months, with negativity of BCR-ABL1 but persistence of CALR type 1 mutation (allelic burden 45%). The patient is still on combined treatment (ruxolitinib plus nilotinib) with DMR and good control of disease despite persistence of CALR mutation, 7 years after CML occurrence.
FIGURE 1

Medium power view of BM biopsy showing a hypercellular marrow with the prevalence of granulopoiesis and clustering of variably sized atypical MKs (hematoxylin and eosin, ×100 magnification).
FIGURE 2
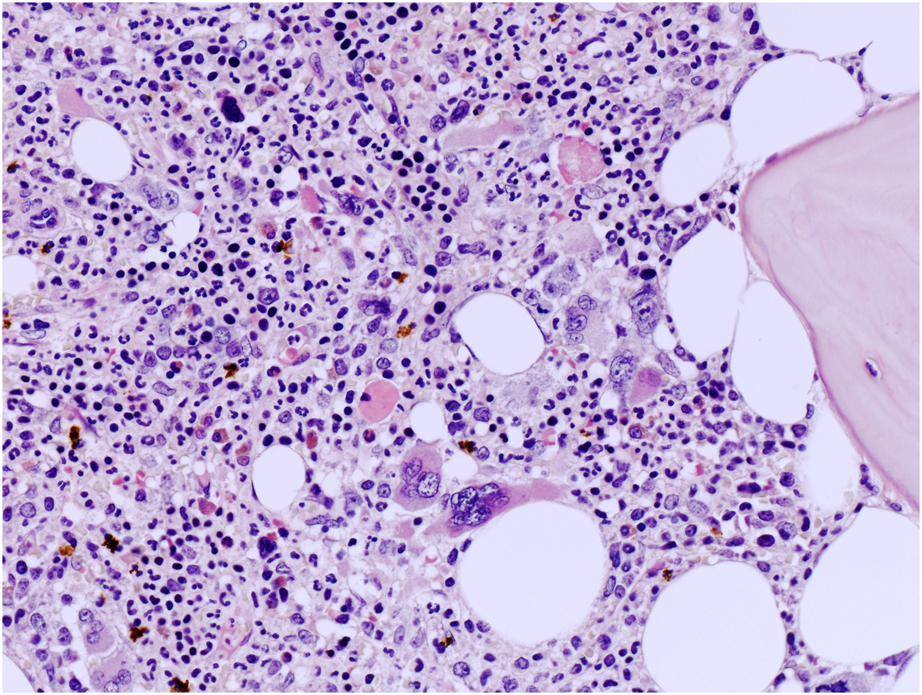
High power view of BM biopsy highlighting atypical MKs with non-classical CML features such as large MKs with bulbous nuclei (hematoxylin and eosin, ×200 magnification).
3 Systematic review of the literature: methods
We performed a systematic review adhering to the Preferred Reported Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines. The literature search was carried out through PubMed/MEDLINE, Embase, Scopus, Cochrane Library (Cochrane Database of Systematic Reviews), Cochrane Central Register of Controlled Trials (CENTRAL), and Web of Science (Science and Social Science Citation Index) databases, with the following non-MeSH/MeSH terms “CALR” AND BCR::ABL1 concomitant” [Mesh]. The search was performed from 2013 when CALR mutations were identified to be MPN-driven mutations to January 2024. The criteria for inclusion were as follows: 1) MPNs with concomitant CALR mutation and BCR::ABL1 translocation; 2) retrospective studies, case reports and/or case series, and literature review. The exclusion criteria were as follows: 1) studies not published in English; 2) lack of concomitant CALR mutation and BCR::ABL1 translocation. The papers were identified by three independent reviewers (M. Zanelli, VF, and GGL), first considering title, abstract, and key words and then reading the article full texts to evaluate if the articles met the inclusion criteria. From selected papers, the following information was collected: author’s surname, year of publication, patient’s age and sex, first disease diagnosis, second disease diagnosis, interval between the first and second diseases, BM histology during the disease course, type of CALR mutation, interaction between CALR and BCR::ABL1 clones, and treatment and prognosis. A third independent reviewer (AS) re-examined all collected results and resolved divergences.
4 Results of literature cases and the present case
4.1 Epidemiological and clinical data
Our literature search identified 21 articles reporting a total of 23 cases with concurrent BCR::ABL1 translocation and CALR mutation. The clinical, pathologic, and molecular characteristics, as well as treatment and outcome data, of the cases are summed up in Supplementary Table S1 (Supplementary Material).
Including our case, a total of 24 cases with concomitant BCR::ABL1 and CALR mutation have been described so far (Bonzheim et al., 2015; Cabagnols et al., 2015; Loghavi et al., 2015; Diamond et al., 2016; Seghatoleslami et al., 2016; Dogliotti et al., 2017; Gilles et al., 2017; Kandarpa et al., 2017; Blouet et al., 2018; Boddu et al., 2018; De Roeck et al., 2018; Klairmont et al., 2018; Lewandowski et al., 2018; Xia et al., 2019; Balducci et al., 2020; Da Costa et al., 2020; Guidotti et al., 2020; Liu et al., 2020; Yoon et al., 2020; Sobieralski et al., 2022; Huo et al., 2023).
Median age at the first diagnosis was 59.6 (range: 26–90), with a moderate prevalence in female individuals (F:14/24, 58.33%; M: 10/24, 41.66%).
In the majority of cases with concurrent BCR::ABL1 translocation and CALR mutation, CML was the first diagnosis, followed by Ph-negative MPN (13/24; 54.16%) (Cabagnols et al., 2015; Loghavi et al., 2015; Diamond et al., 2016; Dogliotti et al., 2017; Gilles et al., 2017; Kandarpa et al., 2017; Blouet et al., 2018; Lewandowski et al., 2018; Balducci et al., 2020; Da Costa et al., 2020; Guidotti et al., 2020; Yoon et al., 2020; Huo et al., 2023), and the Ph-negative MPNs were distributed as follows: four myeloproliferative neoplasm not otherwise specified (MPN, NOS); four PMF; and five ET.
In 9/24 cases (37.5%), CML was the second diagnosis preceded by Ph-negative MPNs (Bonzheim et al., 2015; Kandarpa et al., 2017; Boddu et al., 2018; De Roeck et al., 2018; Klairmont et al., 2018; Liu et al., 2020; Sobieralski et al., 2022), represented by four ET; three PMF; one MPN, NOS; and one post-essential thrombocythemia myelofibrosis (PET-MF).
In 2/24 cases (8.33%), Ph-negative MPN and CML occurred simultaneously (Seghatoleslami et al., 2016; Boddu et al., 2018), and the Ph-negative MPNs were one MPN, NOS and one PMF.
The median interval between the first and second diagnoses was 50 months (range: 3–154) in cases with CML preceding Ph-negative MPN and 141 months (range: 30–468) in cases with Ph-negative MPN preceding CML.
CML was found to be clinically in the chronic phase (CP) in all cases with the exception of one case with concomitant CML and MPN, NOS, in which CML was in the blast phase (BP) (Seghatoleslami et al., 2016).
In the group with CML preceding Ph-negative MPN, the clinical manifestations at the second disease occurrence were as follows: high PTL in 9/13 despite good molecular response of CML (Cabagnols et al., 2015; Dogliotti et al., 2017; Gilles et al., 2017; Lewandowski et al., 2018; Balducci et al., 2020; Da Costa et al., 2020; Guidotti et al., 2020; Yoon et al., 2020), increasing splenomegaly in 3/13 despite good molecular response of CML (Diamond et al., 2016; Kandarpa et al., 2017; Huo et al., 2023), anemia in 2/13 despite a good molecular response of CML (Cabagnols et al., 2015; Diamond et al., 2016), and appetite loss and fatigue in 1/13 despite a good molecular response of CML (Huo et al., 2023).
In the group with CML emerging as the second disease after Ph-negative MPN, the clinical manifestations at CML occurrence were as follows: high WBC in 9/9 (Bonzheim et al., 2015; Kandarpa et al., 2017; Boddu et al., 2018; De Roeck et al., 2018; Klairmont et al., 2018; Xia et al., 2019; Liu et al., 2020; Sobieralski et al., 2022), anemia in 3/9 (Kandarpa et al., 2017; Boddu et al., 2018; Xia et al., 2019), high PTL 1/9 (De Roeck et al., 2018), low PTL in 1/9 (Boddu et al., 2018), increasing splenomegaly in 5/9 (Kandarpa et al., 2017; Boddu et al., 2018; Liu et al., 2020; Sobieralski et al., 2022), hepatomegaly in 1/9 (Boddu et al., 2018), and fatigue and abdominal pain in 1/9 (De Roeck et al., 2018).
4.2 Histological features
In the group with CML preceding Ph-negative MPN (13/24 cases), BM histology at CML presentation in 10/13 cases with available BM data was as follows: CML (CP) histology in 2/10 (Kandarpa et al., 2017; Balducci et al., 2020), CML + fibrosis + hybrid MKs including both classical CML MKs with small and hypolobate nuclei and large hyperlobated forms in 3/10 (Loghavi et al., 2015; Gilles et al., 2017; Guidotti et al., 2020), CML + atypical MKs in 2/10 (Blouet et al., 2018; Da Costa et al., 2020), CML + fibrosis in 2/10 (Diamond et al., 2016; Yoon et al., 2020), and grade 2 reticulin fibrosis with no other BM data in 1/10 (Cabagnols et al., 2015). In this group, the emergence of the Ph-negative MPN phenotype usually occurred after TKI treatment and CML remission; the histology of BM at Ph-negative MPN emergence was available in 9/13 cases, and it was consistent with either PMF (4/9) (Loghavi et al., 2015; Diamond et al., 2016; Kandarpa et al., 2017; Huo et al., 2023), ET (4/9) (Dogliotti et al., 2017; Blouet et al., 2018; Balducci et al., 2020; Da Costa et al., 2020), or MPN, NOS (1/9) [ 25].
In the group with Ph-negative MPN preceding CML (9/24 cases), at presentation of first disease (Ph-negative MPN), BM histology was available in 6/9 cases, and it was consistent with either PMF (3/6) (Boddu et al., 2018; Sobieralski et al., 2022), ET (2/6) (Bonzheim et al., 2015; Liu et al., 2020), and MPN, NOS (1/6) (Klairmont et al., 2018). In this group, at the occurrence of CML, BM histological features were available in 7/9 cases as follows: CML (CP) histology in 1/7 (Bonzheim et al., 2015), CML + fibrosis in 3/7 (Klairmont et al., 2018; Liu et al., 2020; Sobieralski et al., 2022), and CML + fibrosis + hybrid MKs in 3/7 (Boddu et al., 2018; Xia et al., 2019).
Of 2/24 cases with concomitant CML and Ph-negative MPN, BM biopsy, performed only in 1/2 (CML + PMF), was obtained only at CML complete hematologic response and showed histological features consistent with PMF (Boddu et al., 2018).
4.3 Molecular data
In the group of CML preceding Ph-negative MPN (13/24 cases), data on the CALR type were available in 11/13 cases. CALR type 1 (52-bp deletion) was identified in 10/11 (Cabagnols et al., 2015; Loghavi et al., 2015; Diamond et al., 2016; Dogliotti et al., 2017; Gilles et al., 2017; Blouet et al., 2018; Balducci et al., 2020; Da Costa et al., 2020; Guidotti et al., 2020; Yoon et al., 2020) and CALR 34 bp deletion in 1/11 (Huo et al., 2023).
In the group of Ph-negative MPN preceding CML (9/24 cases), data on the CALR type were available in 8/9 cases, of which CALR type 1 was detected in 4/8 (Klairmont et al., 2018; Xia et al., 2019; Liu et al., 2020) and CALR type 2 (5-bp insertion) in 4/8 cases (Bonzheim et al., 2015; Boddu et al., 2018; De Roeck et al., 2018; Sobieralski et al., 2022). In the two cases of concomitant CML and Ph-negative MPN, CALR type 1 was found (Seghatoleslami et al., 2016; Boddu et al., 2018).
In the group of CML preceding Ph-negative MPN, CALR mutation and BCR::ABL1 rearrangement were identified simultaneously at CML diagnosis in 10/10 cases, in which CALR was evaluated at initial CML diagnosis (Cabagnols et al., 2015; Loghavi et al., 2015; Diamond et al., 2016; Dogliotti et al., 2017; Gilles et al., 2017; Blouet et al., 2018; Lewandowski et al., 2018; Balducci et al., 2020; Yoon et al., 2020; Huo et al., 2023). Data on the interaction between the BCR::ABL1 clone and CALR clone were available in 10/13 cases of this group; the Ph-positive clone resulted sensitive to TKI treatment with the progressive BCR::ABL1 decrease, whereas the CALR-mutant clone was persistent in all 10 cases with CALR allelic burden increasing at BCR::ABL1 decrease (Cabagnols et al., 2015; Loghavi et al., 2015; Diamond et al., 2016; Dogliotti et al., 2017; Gilles et al., 2017; Blouet et al., 2018; Lewandowski et al., 2018; Balducci et al., 2020; Yoon et al., 2020; Huo et al., 2023).
In the group of Ph-negative MPN preceding CML (9/24 cases), CALR was evaluated and found positive at initial Ph-negative MPN diagnosis only in 2/9 cases (Bonzheim et al., 2015), whereas in 7/9 cases, the CALR test was not performed at initial diagnosis because the test was not available. In all cases of this group, at CML diagnosis, both CALR mutation and BCR::ABL1 rearrangement were detected (Bonzheim et al., 2015; Kandarpa et al., 2017; Boddu et al., 2018; De Roeck et al., 2018; Klairmont et al., 2018; Xia et al., 2019; Liu et al., 2020; Sobieralski et al., 2022). The interaction between CALR and BCR::ABL1 clones was not available in 4/9 cases; the Ph-positive clone was sensitive to TKI and the CALR clone persistent in 4/9 cases (Bonzheim et al., 2015; Liu et al., 2020; Sobieralski et al., 2022), whereas in 1/9 cases, both CALR and BCR::ABL1 clones persisted despite treatment with TKI, HU, and ruxolitinib (Boddu et al., 2018).
Of the two cases with concomitant CML and Ph-negative MPN, the interaction between the CALR and BCR::ABL1 clone was available only in 1/2 cases, with the Ph-positive clone sensitive to TKI and the CALR-mutant clone persistent after DMR of CML (Boddu et al., 2018).
4.4 Treatment and prognosis
In the group of CML preceding Ph-negative MPN (13/24 cases), TKI treatment achieved DMR of CML in 10/13 cases (Loghavi et al., 2015; Diamond et al., 2016; Dogliotti et al., 2017; Kandarpa et al., 2017; Blouet et al., 2018; Balducci et al., 2020; Da Costa et al., 2020; Guidotti et al., 2020; Yoon et al., 2020; Huo et al., 2023), complete cytogenetic remission (CCyR) in 1/13 [ 23], and complete hematologic remission (CHR) in 1/13 (Gilles et al., 2017); however, in the 1/13 case, CML relapsed due to TKI stopping (Cabagnols et al., 2015). In 1/10 cases achieving DMR, TKI was stopped for gastric intolerance, and the patient was enrolled in a peptide CML vaccination protocol maintaining DMR of CML 12 years after TKI stopping (Dogliotti et al., 2017). Detailed data on different types of TKIs administered are present in Supplementary Table S1. In this group (CML preceding Ph-negative MPN), different treatments were administered in order to treat the Ph-negative MPN in 8/13 cases. In detail, HU in 1/8 (Yoon et al., 2020), interferon alpha (IFN) then replaced with HU and erythropoietin (EPO) in 1/8 (Cabagnols et al., 2015), HU followed by IFN + cytosine arabinoside (Ara-C) with slight PTL reduction in 1/8 (Lewandowski et al., 2018), HU followed by IFN in 1/8 (Dogliotti et al., 2017), hydroxycarbamide with a good control of ET in 1/8 (Blouet et al., 2018), hydroxycarbamide and acetylsalicylic acid (ASA) with good PTL control in 1/8 (Da Costa et al., 2020), HU with ASA with PTL normalization in 1/8 (Guidotti et al., 2020), and HU followed by anagrelide and subsequently by IFN with PTL normalization in 1/8 (Balducci et al., 2020). All patients of this group were alive at the last follow-up; OS was available in 12/13 cases with a median OS of 72 months (range 10–181).
In the group of Ph-negative MPN preceding CML (9/24 cases), TKI treatment achieved DMR in 4/7 cases with available follow-up data (De Roeck et al., 2018; Xia et al., 2019; Liu et al., 2020) and CHR in 2/7 cases (Bonzheim et al., 2015; Kandarpa et al., 2017), whereas in 1/7 cases, TKI did not achieve any response, and the patient underwent allogenic hematopoietic stem cell transplantation (allo-HSCT) with DMR (Boddu et al., 2018). In this group (Ph-negative MPN preceding CML), different therapies were administered for Ph-negative MPN as follows: IFN with no change in ET clinical features in 1/9 (Bonzheim et al., 2015); HU in 1/9 (Klairmont et al., 2018); anagrelide followed by ruxolitinib in 2/9 (Kandarpa et al., 2017); ASA in 1/9 (Xia et al., 2019); ruxolitinib with good control of ET in 1/9 (De Roeck et al., 2018); IFN + ASA with stable disease in 1/9 (Liu et al., 2020); ASA + HU, then ruxolitinib followed by spleen radiotherapy and allo-HSCT in 1/9 (Sobieralski et al., 2022); and HU, followed by ruxolitinib and subsequently by allo-HSCT in 1/9 with complete remission of both CML and Ph-negative MPN with undetectable genetic markers at 2 years from allo-HSCT (Boddu et al., 2018). All seven patients with available follow-up data were alive; median OS was 121 months (range 48–240).
Data on therapy and survival were available only in 1/2 cases with concomitant CML and Ph-negative MPN; in this case, TKI achieved DMR of CML, whereas no cytoreductive therapy was administered for Ph-negative MPN, and the patient was alive 24 months after diagnosis.
5 Discussion
The knowledge of the genetic basis of Ph-negative MPNs improved in the last few years because of the discovery of the main MPN driver mutations, including JAK2, MPL, and CALR mutations. The clinical manifestations and histological phenotype of MPNs mainly depend on their mutational status.
BCR::ABL1 rearrangement, at the basis of CML development, and JAK2 mutations were initially considered mutually exclusive genetic alterations. However, since JAK2 first discovery in 2005, an increasing number of patients with concomitant BCR::ABL1 rearrangement and JAK2 mutations have been reported, and in a recent comprehensive review of the literature, we identified 87 cases carrying both genetic abnormalities (Zanelli et al., 2024).
First described in 2013, CALR mutations have been detected in the majority of JAK2- and MPL-negative ET and PMF. CALR and BCR::ABL1 double-positive cases have been reported mainly as rare single-case descriptions. In the current review of the literature, we identified a total of 24 cases, including the present case, carrying the CALR mutation and BCR::ABL1 rearrangement.
The coexistence of CML and Ph-negative MPN may show three possible scenarios: CML preceding Ph-negative MPN; CML developing in patients with a previous history of Ph-negative MPN; and, finally, simultaneous presentation of CML and Ph-negative MPN.
In our recent review analyzing BCR::ABL1/JAK2 V617F double-positive cases, the majority of patients fell into the group of JAK2-mutated MPN preceding CML (49.42%) (Zanelli et al., 2024).
Unlike BCR::ABL1/JAK2 double-positive cases, the majority of cases carrying the CALR mutation and BCR::ABL1 rearrangement fell into the group of CML preceding Ph-negative MPN (54.16%), followed by Ph-negative MPN preceding CML (37.5%) and, finally, by concomitant CML and Ph-negative MPN (8.33%). However, the number of BCR::ABL1/CALR double-positive cases reported so far is too small to draw any conclusion about the different frequencies of the three scenarios between BCR::ABL1/JAK2 double-positive cases and patients carrying both BCR::ABL1 and CALR.
The coexistence of the BCR::ABL1 rearrangement and CALR mutation may change the clinical and laboratory manifestations of MPN and, for instance, in CML patients may be misinterpreted as failure of TKI therapy or disease transformation.
In CML patients, the persistence or occurrence of thrombocytosis, despite WBC decrease and good molecular BCR::ABL1 response under TKI, should alert clinicians to perform molecular screening for Ph-negative MPN, including JAK2, MPL, and CALR mutations. Similarly, cases of Ph-negative MPN developing CML-like manifestations (WBC increase or splenomegaly), even years after clinical stability, should lead to additional molecular investigations to rule out CML occurrence.
The pathologist may suspect the coexistence of Ph-negative MPN and Ph-positive CML from a close examination of BM histology. Non-clustering small MKs, the so-called dwarf MKs, are normally found in CML, whereas large and clustered MKs are common in Ph-negative MPNs.
Therefore, the identification of large MKs with hyperlobulated nuclei in BM of CML patients should prompt additional genetic testing including CALR mutations to exclude the coexistence of a Ph-negative MPN. Some cases of our literature review showed unusual MK morphology with composite features (hybrid MKs with both small and large forms), which should represent a clue for molecular testing.
Of note, even in cases with CML preceding Ph-negative MPN, at CML diagnosis, BM histology was rarely that of classical CML in CP, but more frequently, CML histology was associated with either fibrosis or MKs, showing no clear-cut CML features or both fibrosis and hybrid MKs.
In the majority of cases in the group with CML preceding Ph-negative MPN, CALR mutation and BCR::ABL1 translocation were retrospectively found to be coexistent at initial CML diagnosis, therefore explaining the abovementioned atypical histology at initial CML diagnosis.
The emergence of the CALR-mutated MPN phenotype often became clinically and histologically evident, following TKI therapy and CML remission as TKIs were generally ineffective for the CALR-mutated disease.
Data on the interaction between the BCR::ABL1 clone and CALR clone commonly showed an inverse relation as BCR::ABL1 decreased under TKI therapy, whereas the CALR-mutant clone persisted, often with a high allele burden, during the disease course despite successful TKI treatment of CML.
Similarly to BCR::ABL1/JAK2 double-positive cases, even in patients carrying both BCR::ABL1 and CALR, CML was easily managed with a good response to different types of TKIs. The CALR-mutated MPNs received different treatments (HU, anagrelide, IFN, and ruxolitinib) often as sequential therapies; however, data on the outcome are often incomplete and scarce; therefore, further studies are essential to establish the optimal management. In BCR::ABL1/JAK2 double-positive cases, interesting results have been reported with allo-HSCT, which may be a superior therapeutic option (Zanelli et al., 2024). This therapeutic strategy has been adopted in only two cases with concomitant BCR::ABL1 and CALR, and outcome data were available just in one case, which obtained complete remission of both CML and Ph-negative MPN with undetectable genetic markers (Boddu et al., 2018). Due to the limited number of cases, it is difficult to draw any definite conclusion on the optimal treatment modality for patients carrying both BCR::ABL1 and CALR, and data on a larger number of cases are essential to address this issue.
In conclusion, the combination of the BCR::ABL1 rearrangement and CALR mutation is rare but potentially underestimated due to low awareness of this entity, which can impact the therapeutic management and outcome of patients.
Statements
Author contributions
MZa: conceptualization, methodology, writing–original draft, and writing–review and editing. VF: conceptualization, methodology, and writing–original draft. GL: conceptualization, methodology, and writing–original draft. FS: data curation, methodology, and writing–original draft. GB: data curation, methodology, and writing–original draft. MZi: data curation and writing–original draft. AP: data curation and writing–original draft. SR: data curation and writing–original draft. EA: data curation and writing–original draft. GM: data curation and writing–original draft. SA: data curation and writing–original draft. FC: data curation and writing–original draft. PG: data curation and writing–original draft. FG: data curation and writing–original draft. RC: data curation and writing–original draft. NK: data curation and writing–original draft. AA: data curation, and writing–original draft. LC: data curation and writing–original draft. AC: data curation and writing–original draft. GO: formal analysis, investigation, supervision, and writing–original draft. SA: formal analysis, investigation, supervision, and writing–original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The study was partially supported by the Italian Ministry of Health- Ricerca Corrente Annual Program 2025.
Acknowledgments
The authors thank Dr. Francesca Sabrina Vinci, Dr. Giovanni Mattia, and Dr Virginia Dolcini of Grant Office & Research Administration, Azienda USL-IRCCS Reggio Emilia for their support. MZ is grateful to her husband for the informatics support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2024.1391078/full#supplementary-material
References
1
Arber D. A. Orazi A. Hasserjian R. P. Borowitz M. J. Calvo K. R. Kvasnicka H. M. et al (2022). International consensus classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood140, 1200–1228. 10.1182/blood.2022015850
2
Balducci E. Sanekli S. Hugues P. Soubeyrand M. Borie C. Fund X. et al (2020). Co-occurrence of BCR-ABL1 rearrangement and CALR mutation in a single leukemic stem cell: evidence that BCR-ABL1 oncogenic addiction prevails over CALR signaling. Leukemia Lymphoma61 (1), 209–212. 10.1080/10428194.2019.1658101
3
Blouet A. Rousselet M. C. Le Bris Y. Ribourtout B. Bouvier A. Cottin L. et al (2018). Imatinib treatment of chronic myeloid leukemia reveals a preexisting CALR-mutated essential thrombocythemia. HemasSphere2, e29. 10.1097/HS9.0000000000000029
4
Boddu P. Chihara D. Masarova L. Pemmaraju N. Patel K. P. Verstovsek S. (2018). The co-occurrence of driver mutations in chronic myeloproliferative neoplasms. Ann. Hematol.97, 2071–2080. 10.1007/s00277-018-3402-x
5
Bonzheim I. Mankel B. Klapthor P. Schmidt J. Hinrichsen T. Wachter O. et al (2015). CALR-mutated essential thrombocythemia evolving to chronic myeloid leukemia with coexistent CALR mutation and BCR-ABL translocation. Blood125 (14), 2309–2311. 10.1182/blood-2014-12-616847
6
Cabagnols X. Cayuela J. M. Vainchenker W. (2015). A CALR mutation preceding BCR-ABL1 in an atypical myeloproliferative neoplasm. NEJM372 (7), 688–690. 10.1056/NEJMc1413718
7
Cazzola M. Kralovics R. (2014). From Janus kinase 2 to calreticulin: the clinically relevant genomic landscape of myeloproliferative neoplasms. Blood123 (24), 3714–3719. 3719. 10.1182/blood-2014-03-530865
8
Da Costa V. E. F. de Oliveira R. D. Traina F. Chahud F. Palma L. C. de Figueredo-Pontes L. L. (2020). Co-occurrence of BCR-ABL1-positive chronic myeloid leukaemia and CALR-muated essential thrombocythaemia. BJH, e1–e30. 10.1111/bjh.16274
9
De Roeck L. Michaux L. Debackere K. Lierman E. Vandernberghe P. Devos T. (2018). Coexisting driver mutations in MPN: clinical and molecular characteristics of a series of 11 patients. Hematology23 (10), 785–792. 10.1080/10245332.2018.1498182
10
Diamond J. M. S. Medina de Almeida A. Belo HJLMR Pinheiro Gameiro de Costa G. de Sousa Ferreira Abecasis M. M. (2016). CALR-mutated primary myelofibrosis evolving to chronic myeloid leukemia with both CALR mutation and BCR-ABL1 fusion gene. Ann. Hematol.95, 2101–2104. 10.1007/s00277-016-2827-3
11
Dogliotti I. Fava C. Serra A. Gottardi E. Daraio F. Carnuccio F. et al (2017). CALR-positive myeloproliferative disorder in a patient with Ph-positive chronic myeloid leukemia in durable treatment-free remission: a case report. Stem Cell Investig.4, 57. 10.21037/sci.2017.06.02
12
Gilles S. R. Baughn L. B. Schomaker M. L. Courville E. L. Nelson A. C. Sachs Z. (2017). Buccal epithelial cells display somatic, bone marrow-derived CALR mutation. Blood Adv.1 (25), 2302–2306. 10.1182/bloodadvances.2017012229
13
Guglielmelli P. Szuber N. Gangat N. Capecchi G. Maccari P. Harnois M. et al (2024). CALR mutation burden in essential thrombocythemia and disease outcome. Blood, blood.2023023428. 10.1182/blood.2023023428
14
Guidotti F. Gardellini A. Feltri M. Zancanella M. Saccà V. Alberio F. et al (2020). Concurrent chronic myeloid leukemia and CALR-mutated chronic myeloproliferative neoplasm. Blood Cells, Mol. Dis.81, 102395. 10.1016/j.bcmd.2019.102395
15
Huo L. Xie J. Wang Q. Shen H. Ding Z. Wen L. et al (2023). Insights from a rare myeloproliferative neoplasm with coexisting BCR-ABL1 fusion gene, CALR and TET2 mutations treated with nilotinib and ruxolitinib. Clin. Case Rep.11, 06801. 10.1002/ccr3.6801
16
Kandarpa M. Wu Y. M. Robinson D. Burke P. W. Chinnaiyan A. M. Talpaz M. (2017). Clinical characteristics and whole exome/transcriptome sequencing of coexisting chronic myeloid leukemia and myelofibrosis. AJH92, 555–561. 10.1002/ajh.24728
17
Khoury J. D. Solary E. Abla O. Akkari Y. Alaggio R. Apperley J. F. et al (2022). The 5th edition of the world Health organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia36, 1703–1719. 10.1038/s41375-022-01613-1
18
Klairmont M. M. Cheng J. Schwartzberg L. Ho H. H. Gradowski J. F. (2018). Chronic myeloid leukemia, BCR-ABL1-positive with CALR and MPL mutation. Int. J. Lab. Hem., 1–2. 10.1111/ijlh.12792
19
Klampfl T. Gisslinger H. Harutyunyan A. S. Nivarthi H. Rumi E. Milosevic J. D. et al (2013). Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med.369 (25), 2379–2390. 10.1056/NEJMoa1311347
20
Lewandowski K. Gniot M. Wojtaszewska M. Kandula Z. Becht R. Paczkowska E. et al (2018). Coexistence of JAK2 or CALR mutation is a rare but clinically important event in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors. Int. J. Lab. Hem.40, 366–371. 10.1111/ijlh.12798
21
Liu C. Hu R. Du Z. Abecasis M. Wang C. (2020). Atypical myeloproliferative neoplasm with concurrent BCR-ABL1 fusion and CALR mutation. Medicine99, 5. 10.1097/MD.0000000000018811
22
Loghavi S. Pemmaraju N. Kanagal-Shamanna R. Mehrota M. Medeiros L. J. Luthra R. et al (2015). Insights from response to tyrosine kinase inhibitor therapy in a rare myeloproliferative neoplasm with CALR mutation and BCR-ABL1. Blood125 (21), 3360–3363. 10.1182/blood-2015-03-632893
23
Loscocco G. G. Gesullo F. Capecchi G. Atanasio A. Maccari C. Mannelli F. et al (2024). One thousand patients with essential thrombocythemia: the Florence-CRIMM experience. Blood Cancer J.14 (1), 10. 10.1038/s41408-023-00968-7
24
Loscocco G. G. Guglielmelli P. Gangat N. Rossi E. Mannarelli C. Betti S. et al (2021). Clinical and molecular predictors of fibrotic progression in essential thrombocythemia: a multicenter study involving 1607 patients. Am. J. Hematol.96 (11), 1472–1480. 10.1002/ajh.26332
25
Martin-Cabrera P. Haferlach C. Kern W. Schnitter S. Haferlach T. (2017). BCR-ABL1-positive and JAK2 V617F-positive clone in 23 patients with both aberrations reveal biologic and clinical importance. BJH176, 131–143. 10.1111/bjh.13932
26
Nangalia J. Massie C. E. Baxter E. J. Nice F. L. Gundem G. Wedge D. C. et al (2013). Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med.369 (25), 2391–2405. 10.1056/NEJMoa1312542
27
Pieri L. Spolverini A. Scappini B. Occhini U. Birtolo S. Bosi A. et al (2011). Concomitant occurrence of BCR-ABL and JAK2V617F mutation. Blood118 (12), 3445–3446. 10.1182/blood-2011-07-365007
28
Pietra D. Rumi E. Ferretti V. V. Di Buduo C. A. Milanesi C. Cavalloni C. et al (2016). Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia30, 431–438. 10.1038/leu.2015.277
29
Rumi E. Pietra D. Ferretti V. Klampfl T. Harutyunyan A. S. Milosevic J. D. et al (2014b). JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood123 (10), 1544–1551. 10.1182/blood-2013-11-539098
30
Rumi E. Pietra D. Ferretti V. Milosevic J. D. Casetti I. C. Bellini M. et al (2014a). CALR exon 9 mutations are somatically acquired events in familial cases of essential thrombocythemia or primary myelofibrosis. Blood123 (15), 2416–2419. 10.1182/blood-2014-01-550434
31
Seghatoleslami M. Ketabchi N. Ordo A. Asl J. M. Golchin N. Saki N. (2016). Coexistence of p190 BCR/ABL transcript and CALR 52-bp deletion in chronic myeloid leukemia blast crisis: a case report. Mediterr. J. Hematol. Infect. Dis.8 (1), e2016002. 10.4084/MJHID.2016.002
32
Sobieralski P. Bieniaszewska M. Leszczynska A. Zuk M. Wasag B. Zaucha J. M. (2022). Secondary chronic myeloid leukemia in a patient with CALR and ASXL1-mutated primary myelofibrosis. Int. J. Hematol.116, 442–445. 10.1007/s12185-022-03331-x
33
Soderquist C. R. Ewalt M. D. Czuchlewski D. R. Geyer J. T. Rogers H. J. His E. D. et al (2018). Myeloproliferative neoplasms with concurrent BCR-ABL1 translocation and JAK2V617F mutation: a multi-institutional study from the bone marrow pathology group. Mod. Path31 (5), 690–704. 10.1038/modpathol.2017.182
34
Tefferi A. Lasho T. L. Finke C. Belachew A. A. Wassie E. A. Ketterling R. P. et al (2014). Type 1 vs type 2 calreticulin mutations in primary myelofibrosis: differences in phenotype and prognostic impact. Leukemia28 (7), 1568–1570. 10.1038/leu.2014.83
35
WHO (2017). “WHO classification of tumours haematopoietic and lymphoid tissues, revised,” in WHO classification of tumours editorial board. 4th ed. (Lyon, France: IARC).
36
Xia D. His E. D. Dal Cin P. Hasserjian R. P. (2019). Composite chronic myeloid leukemia and essential thrombocythemia with BCR-ABL1 fusion and CALR mutation. AJH94, 504–505. 10.1002/ajh.25249
37
Yoon Sy Jeong Sy Kim C. Lee M. Y. Kim J. Kim K. H. et al (2020). Philadelphia+ chronic myeloid leukemia with CALR mutation: a case report and literature review. Cancer Res. Treat.52 (3), 987–991. 10.4143/crt2019.703
38
Zanelli M. Bisagni A. Sanguedolce F. Broggi G. Fragliasso V. Zizzo M. et al (2024). Co-occurrence of JAK2-V617F mutation and BCR::ABL1 translocation in chronic myeloproliferative neoplasms: a potentially confounding genetic combination. Front. Oncol.13, 1329298. 10.3389/fonc.2023.1329298
Summary
Keywords
BCR::ABL1, CALR, chronic myeloid leukemia, myeloproliferative neoplasm, primary myelofibrosis, essential thrombocythemia
Citation
Zanelli M, Fragliasso V, Loscocco GG, Sanguedolce F, Broggi G, Zizzo M, Palicelli A, Ricci S, Ambrogi E, Martino G, Aversa S, Coppa F, Gentile P, Gozzi F, Caltabiano R, Koufopoulos N, Asaturova A, Cimino L, Cavazza A, Orcioni GF and Ascani S (2024) Chronic myeloproliferative neoplasms with concomitant CALR mutation and BCR::ABL1 translocation: diagnostic and therapeutic implications of a rare hybrid disease. Front. Cell Dev. Biol. 12:1391078. doi: 10.3389/fcell.2024.1391078
Received
24 February 2024
Accepted
12 March 2024
Published
26 March 2024
Volume
12 - 2024
Edited by
Pier Paolo Piccaluga, University of Bologna, Italy
Reviewed by
Shaimaa Khattab, Alexandria University, Egypt
Francesca Palandri, University of Bologna, Italy
Updates
Copyright
© 2024 Zanelli, Fragliasso, Loscocco, Sanguedolce, Broggi, Zizzo, Palicelli, Ricci, Ambrogi, Martino, Aversa, Coppa, Gentile, Gozzi, Caltabiano, Koufopoulos, Asaturova, Cimino, Cavazza, Orcioni and Ascani.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magda Zanelli, magda.zanelli@ausl.re.it
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.