 Ting Huang
Ting Huang Chenqi Bei
Chenqi Bei Zhenhua Hu
Zhenhua Hu Yuanyuan Li
Yuanyuan Li- 1Zhongshan Institute for Drug Discovery, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Zhongshan, China
- 2School of Chinese Materia Medica, Nanjing University of Chinese Medicine, Nanjing, China
- 3Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China
- 4University of Chinese Academy of Sciences, Beijing, China
Chimeric Antigen Receptor (CAR) technology has revolutionized cellular immunotherapy, particularly with the success of CAR-T cells in treating hematologic malignancies. However, CAR-T cells have the limited efficacy of against solid tumors. To address these limitations, CAR-macrophages (CAR-Ms) leverage the innate properties of macrophages with the specificity and potency of CAR technology, offering a novel and promising approach to cancer immunotherapy. Preclinical studies have shown that CAR-Ms can effectively target and destroy tumor cells, even within challenging microenvironments, by exhibiting direct cytotoxicity and enhancing the recruitment and activation of other immune cells. Additionally, the favorable safety profile of macrophages and their persistence within solid tumors position CAR-Ms as potentially safer and more durable therapeutic options compared to CAR-T cells. This review explores recent advancements in CAR-Ms technology, including engineering strategies to optimize their anti-tumor efficacy and preclinical evidence supporting their use. We also discuss the challenges and future directions in developing CAR-Ms therapies, emphasizing their potential to revolutionize cellular immunotherapy. By harnessing the unique properties of macrophages, CAR-Ms offer a groundbreaking approach to overcoming the current limitations of CAR-T cell therapies, paving the way for more effective and sustainable cancer treatments.
1 Introduction
The landscape of cellular immunotherapy has been profoundly transformed by the advent of chimeric antigen receptor (CAR) technology (Biglari et al., 2006; Steinbach et al., 2022; Velasco-de Andres et al., 2023), which generates synthetic chimeric proteins that enable immune cells to recognize specific antigens on the surface of cancer cells, thereby triggering the immune cells’ ability to eliminate these cells (Kennedy and Salama, 2020; Lai et al., 2020). Particularly CAR-T cells have shown remarkable success in treating certain hematologic malignancies (Dana et al., 2021; Hong et al., 2020; Sterner and Sterner, 2021), but CAR-T cell therapies have limited efficacy against solid tumors (Halim and Maher, 2020; Kosti et al., 2018; Zhang et al., 2018; Brown and Adusumilli, 2016). As a result, there is a growing need to explore alternative immune cell types that can overcome these limitations and expand the therapeutic potential of CAR technology.
Macrophages, key players in the innate immune system (Cassetta and Pollard, 2018; Conte, 2022), are emerging as promising candidates for CAR-based therapies (DeNardo and Ruffell, 2019; Shin et al., 2023). Unlike T cells, macrophages possess inherent abilities to infiltrate solid tumors, modulate the tumor microenvironment, and sustain prolonged anti-tumor activity (Guerriero, 2018; Noy and Pollard, 2014; Sloas et al., 2021; Liu et al., 2022a). CAR-macrophages (CAR-Ms) have been engineered to combine these natural advantages with the specificity and potency of CAR technology, offering a novel approach to cancer immunotherapy (Unver, 2023). Preclinical studies have demonstrated the potential of CAR-Ms to effectively target and destroy tumor cells, even within the hostile microenvironment of solid tumors (Klichinsky et al., 2020; Chen et al., 2023). CAR-Ms not only exhibit direct cytotoxicity against cancer cells but also enhance the recruitment and activation of other immune cells (Su et al., 2022), thereby orchestrating a comprehensive anti-tumor response (Klichinsky et al., 2020). Furthermore, the safety profile of macrophages, coupled with their ability to persist and function within tumors, positions CAR-Ms as a potentially safer and more durable therapeutic option compared to CAR-T cells (Brudno and Kochenderfer, 2019). In this review, we provide a comprehensive and systematic overview of CAR-Ms, focusing on key aspects such as CAR structure, cell sources, and the progression of both preclinical and clinical studies. We also examine the challenges and future directions in the development of CAR-M therapies, highlighting their potential to revolutionize the field of cellular immunotherapy. This review aims to offer readers a concise and comprehensive overview of the rapidly evolving area of CAR-M research, helping them to grasp its current progress and future promise.
2 An overview of CAR-Ms
2.1 CAR structure and acting mechanism of CAR-Ms
The CAR structure in CAR-Ms shares similarities with that of CAR-T cells, comprising three core components: an extracellular domain, a transmembrane domain, and an intracellular domain (Figure 1) (Chen et al., 2024; Li J. et al., 2024; Li J. et al., 2022; Liu Y. et al., 2022; Wu et al., 2022; Yang et al., 2024). The extracellular domain, containing a single-chain variable fragment (scFv) that is primarily composed of the variable light chain and variable heavy chain, is responsible for recognizing antigens (Thakar et al., 2019; Zhang et al., 2022). CAR-Ms have been reported to target CD19, CD22, and HER2 on tumor cells for tumor clearance (Morrissey et al., 2018; Zhang et al., 2020; Zhang et al., 2019), and there are also reports indicating that they target the FAP protein for the treatment of liver fibrosis (Kim and Seki, 2023; Wegrzyniak et al., 2021; Yang AT. et al., 2023). The co-stimulatory and signal transduction domains are crucial for the proinflammatory and phagocytic functions of CAR-Ms. Several ligands from the immunoglobulin superfamily and the TNF receptor family, such as CD80, CD86, 4-1BBL, and OX40L, have been shown to enhance macrophage phagocytosis and cytokine secretion upon antigen binding (Stephan et al., 2007; Lei et al., 2024). CAR macrophages can utilize the same signaling domain as CAR-T cells, specifically CD3ζ, which contains the immunoreceptor tyrosine-based activation motif (ITAM) (Bettini et al., 2017). Studies on intracellular signaling pathways have shown that CAR macrophages containing CD3 are able to effectively exert their phagocytic function (Liu et al., 2022a). Additionally, a dual-signaling CAR incorporating CD3ζ and Toll-like receptor (TIR) domains has been designed to enhance target phagocytosis, promote antigen-dependent M1 polarization, and increase resistance to M2 polarization (Lei et al., 2024).
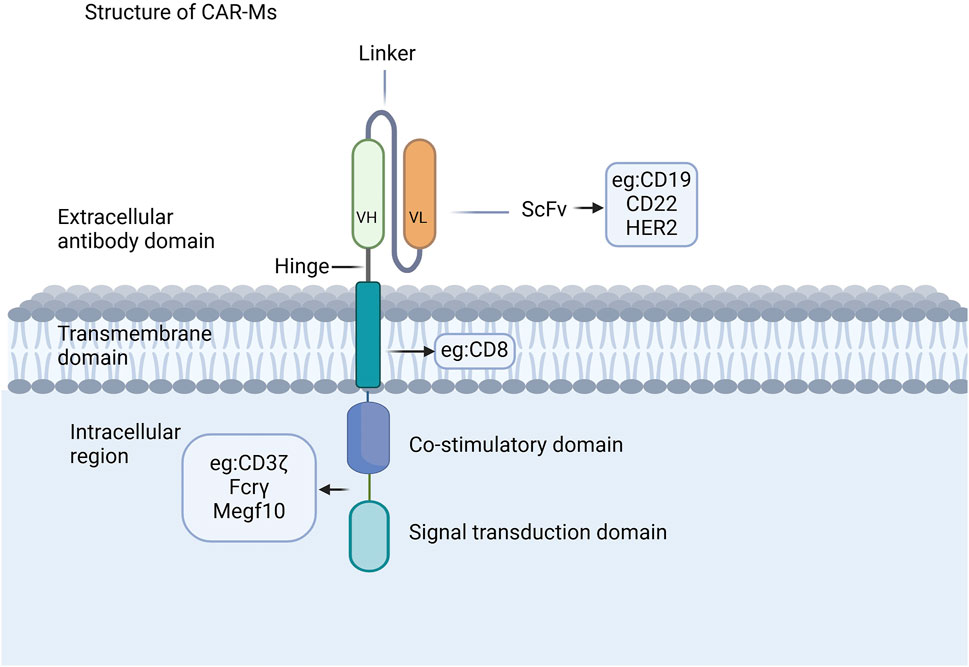
Figure 1. CAR structures of CAR-Ms. The CAR structure in CAR-Ms shares similarities with that of CAR-T cells, consisting of three core components: an extracellular domain, a transmembrane domain, and an intracellular domain.
Once introduced into the body, CAR-Ms can mediate phagocytosis through their chimeric antigen receptor, secrete various cytokines to combat the immunosuppressive microenvironment (Figure 2) (Sloas et al., 2021; Huo et al., 2023; Ji et al., 2024), and release matrix metalloproteinases to reshape the microenvironment’s structure (Yang et al., 2024; Villanueva, 2020; Kuznetsova et al., 2024; Li N. et al., 2024). As antigen-presenting cells, CAR-Ms can also enhance the cytotoxicity of T cells, thereby boosting immune efficacy (Shin et al., 2023; Hadiloo et al., 2023; Li SY. et al., 2023). Additionally, CAR-Ms can recruit the body’s own macrophages to further support immune functions and achieve therapeutic effects (Liang et al., 2023; Xu et al., 2022).
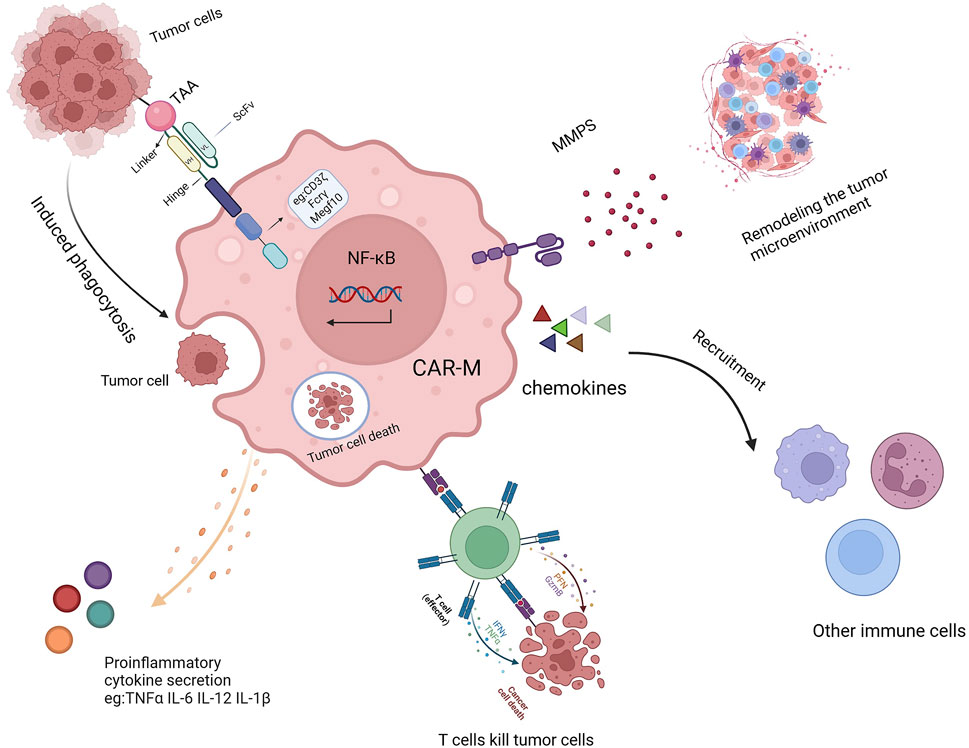
Figure 2. The mechanism of action of CAR-Ms includes TAA-induced phagocytosis, secretion of proinflammatory cytokines, activation of T cells, remodeling of the TME, and recruitment of other immune cells.
Macrophages exhibit significant heterogeneity and have the ability to swiftly modify their functions in response to signals from their local microenvironment. (Leung and Wong, 2021; Arora et al., 2021). They are generally categorized into two main types based on their phenotypes and functional roles: M1 macrophages, which are proinflammatory and classically activated, and M2 macrophages, which are anti-inflammatory and alternatively activated (Zheng et al., 2021). The polarization of macrophages into either the M1 or M2 subtype is a highly regulated process that involves various key signaling pathways, as well as transcriptional, epigenetic, and post-transcriptional regulatory mechanisms. M1 macrophages are induced by lipopolysaccharide (LPS) and interferon (IFN)-γ (Hu et al., 2023; Lewis et al., 2022). They secrete cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1, IL-6, IL-12, IL-23, and inducible nitric oxide synthase (iNOS) to promote a pro-inflammatory Th1 response (Italiani and Boraschi, 2014; Wang DR. et al., 2022; Anderson et al., 2021). Additionally, they secrete chemokines (CXCL9, CXCL10, CXCL11) to recruit Th1 cells to sites of inflammation (Patel et al., 2017). M2 macrophages, on the other hand, are induced by cytokines such as IL-4 and IL-13. They suppress inflammation by activating signal transducer and activator of transcription (STAT) six and secrete IL-10, arginase (ARG), and transforming growth factor (TGF)-β (Kadomoto et al., 2021; Li M. et al., 2023; Guo and Qian, 2024). Macrophages exhibit remarkable plasticity in vivo and play dual roles in various human diseases, contributing to both protective and pathogenic processes. Macrophages can be tailored for specific diseases by inducing them into targeted states of differentiation. (Murray et al., 2014).
2.2 Cell source of CAR-Ms
Macrophages have been recognized as effector cells that destroy cancer cells due to their phagocytosis ability, so they have received much attention in the field of tumor immunotherapy (Alvey and Discher, 2017). Next, we will discuss the different origins of Human CAR-Ms which can be derived from a variety of ways and which can be modified to express car molecules (Figure 3).
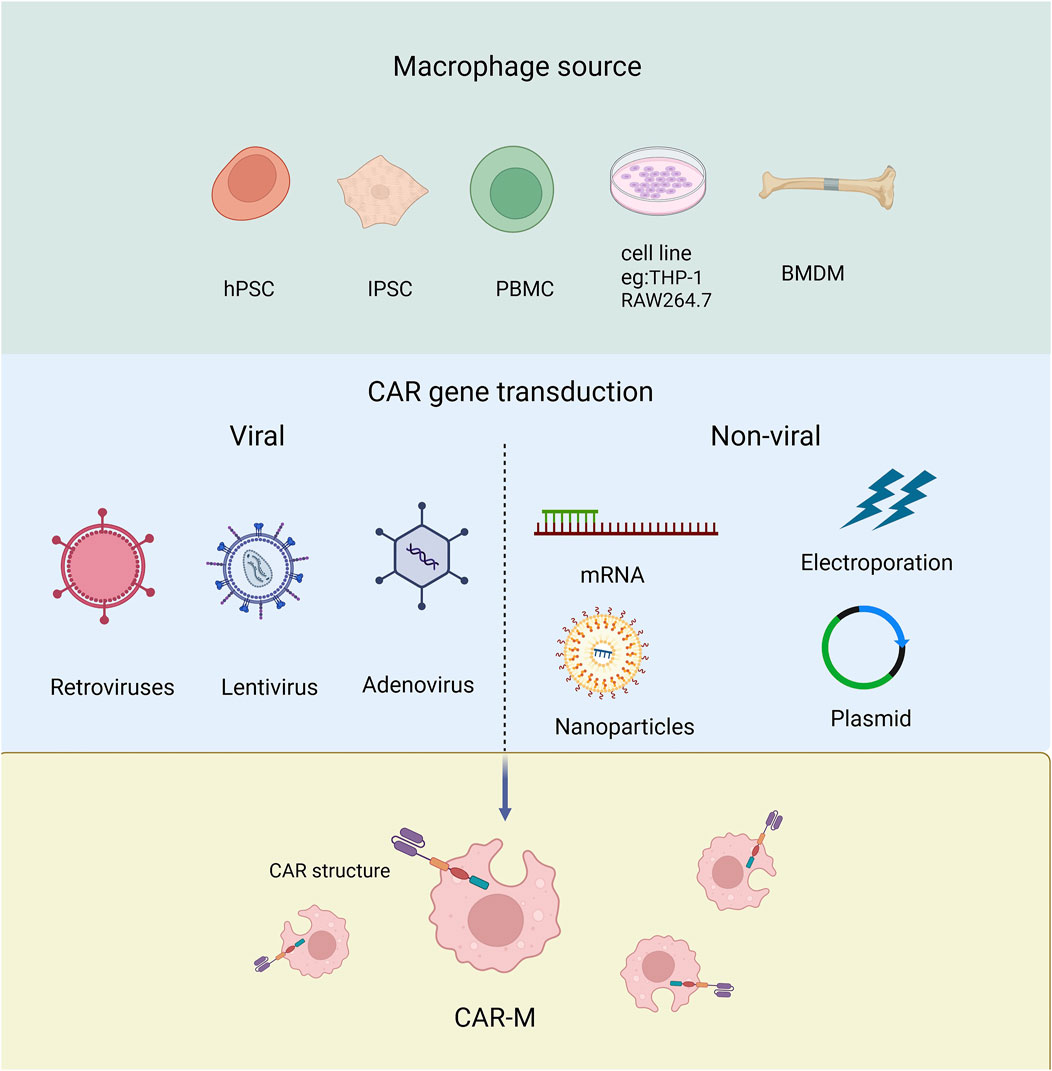
Figure 3. The source of macrophage used to produce CAR-Ms and the tools employed for transducing CAR genes.
Derived from cell line Human THP-1 cells can differentiate into CAR-macrophages after CAR engineering. The researchers attempted to evaluate the antigen-dependent phagocytotic ability of THP-1-derived macrophages, and the first generation of anti-CD19 CAR encoding CD3ζ intracellular segment was transduced to THP-1 cells. CD3ζ is homologous to the Fc co-gamma chain (FcεRI-γ), which is a specific classical signaling molecule for macrophages involved in antibody dependent cellular phagocytosis (ADCP). The results showed that CARs based on CD3ζ could enhance the anti-tumor phagocytosis activity of THP-1 macrophages (Sloas et al., 2021; Liu et al., 2022a; Su et al., 2022; Chocarro et al., 2022). Using RAW264.7 derived macrophages, the researchers designed CAR-Ms recognizing the tumor antigen HER2 (Morrissey et al., 2018).
Derived from monocytes Monocytes are widely distributed throughout the body, typically derived from peripheral blood or bone marrow (Biglari et al., 2006), and can be further differentiated into macrophages for the preparation of CAR-M. The peripheral blood monocytes (PBMC) with positive CD14 can be induced to macrophages by treating with granulocyte-macrophage colony-stimulating factor (GM-CSF) (Ball et al., 2022). While bone marrow-derived monocytes (BMMC) can be induced into mature macrophages by treating with macrophage colony-stimulating factor (M-CSF) (Dai et al., 2024). PBMCs are relatively easy to isolate and obtain in clinical, but have low differentiation efficiency than BMMC.
Pluripotent stem cells (PSCs) PSCs have the ability to differentiate into any type of cell in the body including macrophages. Embryoid bodies (EBs) represent the most prevalently employed approach for inducing PSCs to differentiate into macrophages. At present, the researchers have developed a highly efficient induction differentiation system, which can induce a single human PSC to produce about 6,000 macrophages within 2–3 weeks (Zhang et al., 2020; Netsrithong et al., 2023; Wang S. et al., 2022; Shen et al., 2024).
CAR-Ms sourced from different origins present distinct advantages and limitations. CAR-Ms derived from cell lines, such as THP-1, benefit from a stable genetic background, high operability, ease of culture, rapid proliferation, and convenient preservation. However, these immortalized macrophage cell lines are not suitable for clinical applications (Cousins et al., 2003; Mangan and Wahl, 1991; Mangan et al., 1991; Rogers et al., 2003; Gurunathan et al., 2021). On the other hand, CAR-Ms derived from monocytes retain their native form, offering better patient compatibility and specificity. Studies have shown that primary macrophages derived from PBMCs can produce higher levels of pro-inflammatory factors under M1 stimulation, but the availability of PBMCs is limited (Fuss et al., 2009; Schildberger et al., 2013). CAR-Ms derived from BMDMs are sourced from bone marrow and other tissues, which are relatively abundant, providing sufficient raw materials for CAR-M production. Additionally, PSCs offer a promising source for CAR-Ms, as they possess innate immune functions, including the expression and secretion of immune-related cytokines. The ease of amplifying PSCs, once the induction conditions and macrophage phenotypes are well established and characterized, makes them an attractive choice for producing CAR-Ms on a large scale (Zhang et al., 2020).
2.3 Preclinical and clinical studies of CAR-Ms
We conducted a search on PubMed and ClinicalTrials.gov using the keywords “chimeric antigen receptor macrophages” and summarized the preclinical and clinical studies on CAR-Ms in Table 1. The first report on CAR-Ms was published in 2017 by the team of Carl H. June and Saar Gill, pioneers in the field who initiated the first clinical trial of CAR-T cell therapy (Klichinsky et al., 2020). They explored the first-generation anti-CD19, anti-mesothelin, and anti-HER2 CARs in the THP-1 macrophage cell line and discovered that CAR-Ms could selectively phagocytose and eliminate cognate antigen-bearing tumor cells (Snyder and Gill, 2023). Subsequently, they introduced the anti-HER2 CAR into primary human macrophages, which demonstrated targeted phagocytosis and cytotoxicity against HER2-expressing ovarian and breast cancer cell lines (Klichinsky et al., 2020). In 2 mouse models of solid tumor xenotransplantation, a single infusion of human CAR-Ms was shown to reduce tumor burden and extend overall survival. Further studies in humanized mouse models revealed that CAR-Ms could induce a pro-inflammatory tumor microenvironment and enhance anti-tumor T cell activity. This groundbreaking work has since advanced to the clinical trial stage, as detailed in Table 1, representing the most advanced CAR-Ms cancer therapy to date.
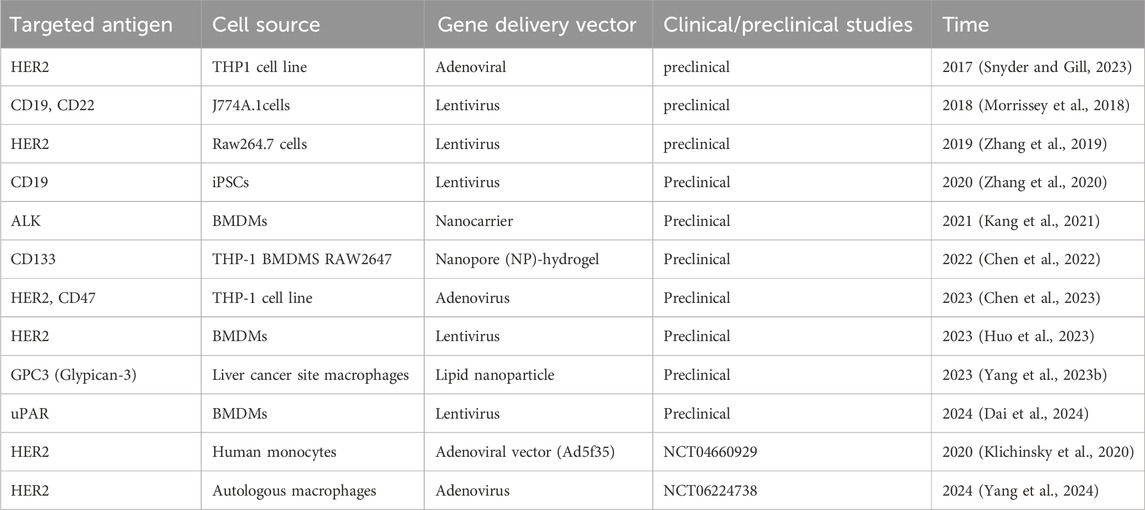
Table 1. Preclinical and clinical trials of CAR-Ms.
In 2018, Morrissey MA et al. created a new type of chimeric antigen receptor for phagocytosis, named CAR-P. By introducing CAR-P into macrophages, it was demonstrated that these modified cells could recognize and attack beads coated with proteins found on cancer cells. Additionally, they were able to limit the growth of live cancer cells in vitro by ‘biting’ and even ‘eating’ them (Morrissey et al., 2018).
In 2019, Zhang W et al. incorporated CD147 signaling molecule into the transmembrane region of HER2 CAR to reduce tumor collagen deposition and promote T-cell infiltration into tumors through effectively activating the expression of matrix metalloproteinases (MMPs) in macrophages after recognizing the antigen HER2. This work demonstrated that targeting the extracellular matrix (ECM) through engineered macrophages would be an effective treatment strategy for solid tumors (Zhang et al., 2019).
Zhang Jin’s team at Zhejiang University has made significant advancements in using induced pluripotent stem cells (iPSCs) to develop chimeric antigen receptor-expressing macrophages (CAR-iMac) for cancer immunotherapy. In November 2020, they were the first to report the successful application of CAR-iMac in both hematoma and solid tumor models in mice, demonstrating strong anti-cancer capabilities. In September 2023, they enhanced the efficacy of CAR-iMac against solid tumors by combining it with sialase and knocking down immune checkpoints Siglec-5 and Siglec-10 on macrophages (Wu et al., 2023). Additionally, they found that knocking out the Acod1 gene in CAR-iMacs led to a stronger pro-inflammatory response, improved phagocytosis, and enhanced tumor suppression in ovarian and pancreatic cancer models (Wang et al., 2023). Combining these modified CAR-iMacs with immune checkpoint inhibitors further improved tumor inhibition. Later that year, they developed a second-generation CAR-iMac with antigen-dependent polarization, enhancing cytotoxicity and antigen presentation, which offered a new method of tumor cell targeting called “cytoburial,” providing a solid theoretical foundation for CAR-iMac-based therapies in solid tumors (Lei et al., 2024).
In 2021, Kang’s team developed an innovative approach to cancer therapy by using nanoparticle carriers to transfect genes encoding CAR and IFN-γ into macrophages, creating MPEI/pCAR-IFN-γ. When injected directly into brain tumor-bearing mice (Neuro-2a), this treatment significantly extended the animals’ survival without causing notable changes in their body weight, indicating effective tumor inhibition with minimal side effects. This study marked a significant advance in cancer treatment by successfully converting macrophages into CAR-M1 type macrophages with potent anti-tumor activity, using a novel nanocomplex-mediated in vivo programming technique, offering new hope and challenges for cancer therapy (Kang et al., 2021).
In 2022, Professor Jiang Xinyi’s team from the School of Pharmacy at Shandong University, in collaboration with the School of Pharmacy at the University of Wisconsin-Madison, developed an injectable gene nanocarrier-hydrogel superstructure drug delivery system. In a mouse model of glioblastoma multiforme (GBM), this innovative system introduced a CAR gene targeting glioma stem cells (GSCs) into macrophage nuclei, resulting in the generation of GSC-specific CAR-Macrophages (CAR-Ms). These CAR-Ms were capable of specifically recognizing and phagocytizing GSCs, acting as antigen-presenting cells, and subsequently stimulating an adaptive anti-tumor immune response, ultimately forming immune memory (Chen et al., 2022).
In September 2023, the team of Tu Jiajie, Institute of Clinical Pharmacology, Anhui Medical University, constructed HER2-targeting CAR-Ms and CD47-targeting CAR-Ms, and co-cultured them with antigen-positive ovarian cancer cells in vitro. The results showed that HER2 CAR-Ms and CD47 CAR-Ms significantly inhibited tumorigenicity and tumor proliferation of ovarian cancer cell lines, confirming CAR-mediated phagocytosis of macrophages on ovarian cancer cells (Chen et al., 2023).
In 2023, Huo Y et al. conducted a study where macrophages were polarized in vitro using LPS combined with IFN-γ. The results showed a significant enhancement in the phagocytosis and killing ability of CAR-Macrophages (CAR-Ms) targeting cancer cells. Additionally, the expression of costimulatory molecules and proinflammatory cytokines increased markedly following polarization. By utilizing various in vivo isogenic tumor models, the researchers demonstrated that transfusions of these polarized M1 CAR-Ms effectively inhibited tumor progression, prolonged the survival of tumor-bearing mice, and enhanced overall cytotoxicity (Huo et al., 2023).
In 2023, by constructing an LNP that co-delivers CAR mRNA and mRNA encoding Siglec-G lacking ITIMs (Siglec-GΔITIMs), Yang Z et al. edited liver macrophages to generate phagocytic enhanced CAR macrophages in a mouse model of liver cancer. CAR-Ms generated by LNP and CD24-Siglec-G blockade can significantly improve the phagocytic function of liver macrophages, reduce tumor load and prolong survival time, providing an effective and flexible strategy for the treatment of HCC. This strategy avoids the complex procedure of making CAR-engineered cells in vitro, eliminates the potential side effects of systematic CAR-Ms transfusion, and has broad application prospects. Future studies will further evaluate the safety and efficacy of this strategy in clinical trials (Yang Z. et al., 2023).
In 2024, Wang Hua’s team at Anhui Medical University found that Mosaic antigen receptor-modified macrophages (CAR-Ms) can regulate the liver immune microenvironment to recruit and modify the activation of endogenous immune cells to drive the regression of fibrosis and produce specific anti-fibrotic T cell responses, providing the first preclinical evidence. Demonstrated that CAR-Ms targeting uPAR can enhance the immune response against liver fibrosis. Alleviating liver fibrosis and cirrhosis (Dai et al., 2024).
All the developments mentioned above are still in the pre-clinical experimental stage. For the clinical translation of CAR-Ms, careful attention must be given to their safety and efficacy. Although animal experiments have provided promising results, the safety and effectiveness of CAR-Ms in humans require further investigation (Chen et al., 2021). Notably, two CAR-Ms therapies have been registered with clinical trial numbers on the ClinicalTrials.gov website. (Tie et al., 2022; Schepisi et al., 2023). CT-0508 is an autologous cell therapy product consisting of pro-inflammatory macrophages derived from peripheral blood monocytes, developed by Carisma Therapeutics. It is the first CAR-M therapy to enter clinical trials, specifically a Phase I trial (NCT04660929) targeting HER2-overexpressing tumors. The HER2-directed CAR-Ms demonstrated a favorable safety profile and early signs of antitumor activity in patients with various solid tumors. Out of 14 patients treated, 28.6% achieved stable disease, all of whom had high HER2 expression (HER2+). Patients with lower HER2 expression (HER2-) experienced disease progression. No dose-limiting toxicities were reported, nor were severe cases of cytokine release syndrome (CRS) or neurotoxicity. Serious adverse events related to treatment were limited to grade 2 CRS and infusion reactions, which were manageable (Klichinsky et al., 2020; Li N. et al., 2024). Another early Phase I clinical trial (NCT06224738) was registered in March 2024 to evaluate human HER2-CAR-Ms therapy for HER2-positive advanced gastric cancer with peritoneal metastases. However, patient enrollment for the study has not yet been completed. (Yang et al., 2024).
Additionally, a Phase 2 randomized controlled clinical trial (ISRCTN 10368050) from the United Kingdom. Medicines and Healthcare products Regulatory Agency (MHRA) has evaluated the safety and feasibility of autologous macrophage from peripheral blood monocytes in patients with cirrhosis, providing valuable information and a theoretical foundation for subsequent efficacy studies (Brennan et al., 2021). This clinical trial evaluated the efficacy of RTX001 versus standard care in patients with compensated cirrhosis and a MELD score between 10 and 17. Patients were randomized into treatment groups receiving either one or three macrophage infusions, or standard care. The trial’s primary outcome was the change in MELD score over 90 days, with secondary outcomes including adverse events, fibrosis markers, and quality of life measures. Results showed a ΔΔMELD score of −0.87 in the treated group compared to the control group, approaching statistical significance (p = 0.06). The treated group also had fewer severe liver-related adverse events and deaths within 360 days of follow-up. However, there were no significant differences in non-invasive fibrosis markers or quality of life. The study concluded that macrophage therapy is safe and shows therapeutic potential in patients with cirrhosis, warranting further research (Brennan et al., 2021).
In conclusion, while the advancements in CAR-Ms therapy have shown significant promise in pre-clinical stages, translating these findings into clinical applications requires careful consideration of safety and efficacy in humans. The progression of CAR-Ms therapies into clinical trials, as evidenced by the FDA’s approval of candidates like CT-0508, marks a critical milestone in the field. These early-stage clinical trials have demonstrated encouraging results, particularly in targeting solid tumors, and have laid the groundwork for future exploration of CAR-Ms therapies. However, the continued development of CAR-Ms will necessitate further research to validate these therapies’ effectiveness and safety in diverse patient populations, ensuring that they can be reliably integrated into clinical practice.
3 CAR-M’s strengths, challenges and future direction
3.1 CAR-M’s strengths, and challenges
Compared with CAR-T and CAR-NK, CAR-Ms has its unique advantages as a new cellular immunotherapy (Table2), such as: 1). CAR-Ms can infiltrate tumor tissues in large quantities, reduce the proportion of TAMs, affect the phenotype of TAMs, and have a positive effect on tumor treatment (Su et al., 2022; Yoon et al., 2018). CAR-Ms possess the unique ability to reprogram the TME and sustain T cell infiltration. In a HER2-4T1 cold tumor model, the infusion of CAR-147 macrophages significantly inhibited tumor growth, leading to a fourfold increase in T cell infiltration compared to controls (Zhang et al., 2019). Similarly, Lei et al. demonstrated that combining anti-CD47 monoclonal antibodies with dual CD3ζ–TIR CAR macrophages resulted in superior tumor eradication in HEPG2 cells in vivo (Lei et al., 2024). In contrast, CAR-T therapies have shown limited success in solid tumor models, failing to produce satisfactory outcomes (Albelda, 2024). The effectiveness of CAR-M therapies against solid tumors is largely attributed to their ability to remodel the TME (Hadiloo et al., 2023). This remodeling is facilitated by the secretion of MMPs during antigen recognition, which degrade the ECM of the tumor. 2).In addition to phagocytosis of tumor cells, CAR-Ms can directly kill tumor cells expressing antigen by secreting cytokines (Keshavarz et al., 2022), promote antigen presentation and enhance T cell killing (Li W. et al., 2022; Uscanga-Palomeque et al., 2023). 3). Compared with CAR-T, CAR-Ms has limited time in circulation (Li SY. et al., 2023; Mercanti et al., 2023), moderate affinity of CAR-Ms can avoid unnecessary non-targeted toxicity, show little “off-target, non-tumor toxicity, and achieve better therapeutic effect (Su et al., 2022; Brudno and Kochenderfer, 2019). 4). CAR-Ms creates a pro-inflammatory environment in the tumor by secreting cytokines and chemokines and recruiting T cells and other white blood cells, reversing the inhibitory tumor microenvironment, and has also shown effective anti-tumor ability in animal experiments (Li W. et al., 2022). 5). CAR-Ms has the inherent tumor homing ability of myeloid cells, so it can enter solid tumors (Liu et al., 2022c).
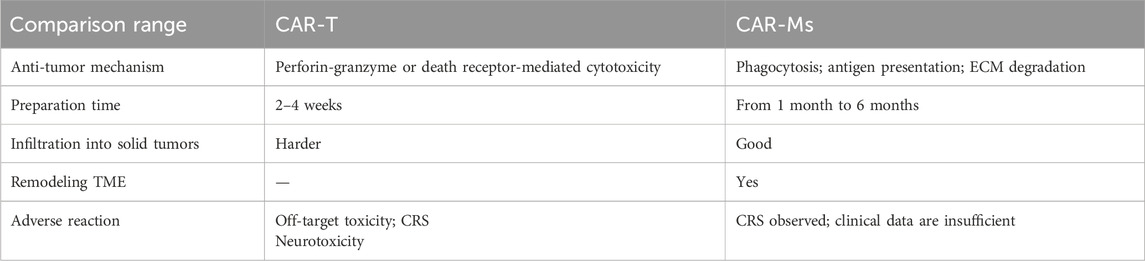
Table 2. Pros and Cons of CAR-T Cells vs CAR-Ms.
CAR-M therapy also faces some of the same challenges as CAR-T cell therapy, particularly regarding the time and cost of treatment (Pan et al., 2022). CAR-M therapy also faces some of the same challenges as CAR-T cell therapy, particularly regarding the time and cost of treatment (Kim and Bae, 2016). To address these issues, rapid CAR production processes, which enable CAR cell infusion within 24 h, and in vivo CAR induction strategies are being developed to shorten manufacturing timelines and reduce costs. Furthermore, Macrophages do not proliferate, and patients receive a limited number of macrophages, either in vitro or in vivo, which may affect the effectiveness of treatment. The ability of macrophage to remodel the TME and continuously stimulate T cell proliferation makes them a promising target for further enhancement. Strengthening these capabilities could significantly improve the effectiveness of CAR-M therapies. Additionally, the safety and efficacy of CAR-Ms in humans have yet to be confirmed by clinical trials (van der Heide et al., 2019). At present, CAR-Ms is mostly transfected by virus, which may lead to insertion mutation. The underlying mechanism of CAR-Ms resistance remains to be studied (Roth et al., 2018). (Chen et al., 2023; Zhang et al., 2023). The complex immune microenvironment should also be considered when applying CAR-Ms therapy (Nakamura and Smyth, 2020; Kochneva et al., 2020). Therefore, rational selection of existing therapies in combination with CAR-Ms may have a synergistic effect against tumor response.
3.2 Future direction of CAR-Ms therapy
3.2.1 Optimization of CAR-Ms function
Several areas warrant further exploration to enhance the efficacy of CAR-M therapy in the future. First, in terms of CAR molecule design, structural optimization—such as the integration of diverse domains or the introduction of tandem activation domains—could be explored to refine the architecture of CAR-Ms (Hadiloo et al., 2023). These modifications may improve the phagocytic efficacy of CAR-Ms against cancer cells. Additionally, genetic engineering approaches to enable CAR-Ms to express anti-tumor cytokines or immunostimulatory molecules could enhance antigen presentation and further boost therapeutic efficacy (Moreno-Lanceta et al., 2023). Moreover, safety considerations can be addressed by designing multi-antigen logic gates or drug-sensitive modules to modify CAR-Ms, potentially improving their safety profile (Savanur et al., 2021). Non-viral delivery systems for CAR-encoding DNA or mRNA can also be employed to program macrophages into CAR-Ms (Sloas et al., 2021). Beyond direct cytotoxicity, CAR-Ms have demonstrated other anti-tumor capabilities, such as secreting MMPs to degrade the extracellular matrix and inducing epitope spreading (Kang et al., 2021; Chen et al., 2022; Komohara et al., 2016; Paasch et al., 2022).
3.2.2 The future clinical applications of CAR-Ms on more diseases beyond cancers
Macrophages possess remarkable regenerative capacity, directly aiding tissue reconstruction in various injured organs, including the intestines, skin, liver, heart, kidneys, and lungs (Figure 4). Their phagocytic ability can be harnessed to clear cancer cells, neurodegenerative debris, and infectious agents (Na et al., 2023). For instance, the infusion of CAR-Ms is expected to efficiently clear accumulated neurotoxic materials in genetic brain diseases (Shibuya et al., 2022). Additionally, specific subtypes of CAR-Ms may reduce ECM content around heart injury sites, promoting regeneration (Henry et al., 2014; Podaru et al., 2019). CAR-Ms can also secrete CXCL1 and WNT, which coordinate the regeneration of damaged intestinal epithelial cells, offering a potential treatment for inflammatory bowel disease (IBD) (Na et al., 2021). Furthermore, CAR-Ms can help resolve the pathology of pulmonary alveolar proteinosis (PAP) by clearing excessive surfactant (Suzuki et al., 2014).

Figure 4. The future clinical applications of CAR-Ms extend to more diseases beyond cancers. Clear accumulated neurotoxic materials; Reduce extracellular matrix (ECM) contents around the heart injury site; Resolve the pathology of pulmonary alveolar proteinosis (PAP); Coordinate the regeneration of damaged intestinal epithelial cells; Resolve fibrosis and promote hepatocyte regeneration.
As potent pro-inflammatory regulators, CAR-M therapies are emerging as a promising area of research for infectious and autoimmune diseases. Macrophages play a crucial role in inflammatory responses, and CAR-Ms have the potential to modulate the inflammatory microenvironment by expressing anti-inflammatory factors or cytokine receptors, thereby controlling excessive inflammation (Zouali, 2024). For example, Perez Amill et al. highlighted the broad range of potential applications of CAR-Ms in targeted antigen recognition, with promising implications for autoimmune diseases (Perez-Amill et al., 2021).
3.2.3 CAR-Ms therapy in combination with other immunotherapeutic strategies
Although CAR-Ms have been proven effective in inhibiting tumors, the complexity of the tumor microenvironment and the challenges of cancer treatment have led researchers to explore combining CAR-Ms with other immunotherapies as a more comprehensive approach (Chen et al., 2021; Boshuizen and Peeper, 2020). For instance, blocking the CD47-SIRPα axis—the “do not eat me” signal—can enhance the phagocytic ability of CAR-Ms against cancer cells. Additionally, in patients with a high tumor burden, combining CAR-Ms with CAR-T cell therapy has emerged as a potential strategy. Since CAR-Ms are capable of suppressing cytokine release, their use alongside CAR-T therapy could reduce the risk of CRS and neurotoxicity, which are common complications associated with CAR-T treatments (Giavridis et al., 2018) (Shen et al., 2024). Liu et al. further investigated the synergistic potential of CAR-M and CAR-T cells in targeting tumor cells (Huang et al., 2024). They found that CAR-Ms and CAR-T cells demonstrated synergistic cytotoxicity against tumor cells in vitro. Inflammatory cytokines secreted by CAR-T cells induced M1 macrophage polarization, while increasing the expression of co-stimulatory ligands (CD86 and CD80) on CAR-Ms enhanced their cytotoxic effects. This study, for the first time, demonstrated the cooperative interaction between CAR-Ms and CAR-T cells in killing tumor cells, providing a novel avenue for combined immunotherapy approaches.
4 Prospects of CAR-Ms therapy
Any new invention must go through a long and arduous process from basic experiment to clinical application. Although there are still many difficulties to be overcome ahead, the future of CAR-Ms therapy holds significant promise for advancing medical treatments and addressing various diseases beyond its current applications. Real-world evidence from clinical practice will further inform best practices, optimize treatment protocols, and identify potential areas for improvement. Ongoing and future clinical trials will provide valuable insights into the safety, efficacy, and long-term effects of CAR-Ms therapy.
Author contributions
TH: Writing–original draft, Software, Methodology, Investigation. CB: Writing–original draft, Software, Resources. ZH: Writing–review and editing, Writing–original draft, Methodology, Conceptualization. YL: Writing–review and editing, Supervision, Project administration, Funding acquisition, Conceptualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study is supported by the National Natural Science Foundation of China 82104252, 82473985; Shanghai Pujiang Program 21PJ1415600, Guangdong Basic and Applied Basic Research Foundation 2022A1515010491; Zhongshan Municipal Bureau of Science and Technology.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Albelda, S. M. (2024). CAR T cell therapy for patients with solid tumours: key lessons to learn and unlearn. Nat. Rev. Clin. Oncol. 21 (1), 47–66. doi:10.1038/s41571-023-00832-4
Alvey, C., and Discher, D. E. (2017). Engineering macrophages to eat cancer: from “marker of self” CD47 and phagocytosis to differentiation. J. Leukoc. Biol. 102 (1), 31–40. doi:10.1189/jlb.4RI1216-516R
Anderson, N. R., Minutolo, N. G., Gill, S., and Klichinsky, M. (2021). Macrophage-based approaches for cancer immunotherapy. Cancer Res. 81 (5), 1201–1208. doi:10.1158/0008-5472.CAN-20-2990
Arora, S., Singh, P., Ahmad, S., Ahmad, T., Dohare, R., Almatroodi, S. A., et al. (2021). Comprehensive integrative analysis reveals the association of KLF4 with macrophage infiltration and polarization in lung cancer microenvironment. Cells 10 (8), 2091. doi:10.3390/cells10082091
Ball, M., Kremp, M., Qureshi, R., Sonawane, P., Schmierer, M., VanDuzer, J., et al. 315 Characterization of CT-0508, an anti-HER2 chimeric antigen receptor macrophage (CAR-M), manufactured from patients enrolled in the phase 1, first in human, clinical trial of CT-0508. 2022;10(Suppl. 2):A331-A.
Bettini, M. L., Chou, P. C., Guy, C. S., Lee, T., Vignali, K. M., and Vignali, D. A. A. (2017). Cutting edge: CD3 ITAM diversity is required for optimal TCR signaling and thymocyte development. J. Immunol. Baltim. Md 1950 199 (5), 1555–1560. doi:10.4049/jimmunol.1700069
Biglari, A., Southgate, T. D., Fairbairn, L. J., and Gilham, D. E. (2006). Human monocytes expressing a CEA-specific chimeric CD64 receptor specifically target CEA-expressing tumour cells in vitro and in vivo. Gene Ther. 13 (7), 602–610. doi:10.1038/sj.gt.3302706
Boshuizen, J., and Peeper, D. S. (2020). Rational cancer treatment combinations: an urgent clinical need. Mol. cell 78 (6), 1002–1018. doi:10.1016/j.molcel.2020.05.031
Brennan, P. N., MacMillan, M., Manship, T., Moroni, F., Glover, A., Graham, C., et al. (2021). Study protocol: a multicentre, open-label, parallel-group, phase 2, randomised controlled trial of autologous macrophage therapy for liver cirrhosis (MATCH). BMJ open 11 (11), e053190. doi:10.1136/bmjopen-2021-053190
Brown, C. E., and Adusumilli, P. S. (2016). Next frontiers in CAR T-cell therapy. Mol. Ther. Oncolytics 3, 16028. doi:10.1038/mto.2016.28
Brudno, J. N., and Kochenderfer, J. N. (2019). Recent advances in CAR T-cell toxicity: mechanisms, manifestations and management. Blood Rev. 34, 45–55. doi:10.1016/j.blre.2018.11.002
Cassetta, L., and Pollard, J. W. (2018). Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov. 17 (12), 887–904. doi:10.1038/nrd.2018.169
Chen, C., Jing, W., Chen, Y., Wang, G., Abdalla, M., Gao, L., et al. (2022). Intracavity generation of glioma stem cell-specific CAR macrophages primes locoregional immunity for postoperative glioblastoma therapy. Sci. Transl. Med. 14 (656), eabn1128. doi:10.1126/scitranslmed.abn1128
Chen, T., Wang, M., Chen, Y., and Liu, Y. (2024). Current challenges and therapeutic advances of CAR-T cell therapy for solid tumors. Cancer cell Int. 24 (1), 133. doi:10.1186/s12935-024-03315-3
Chen, Y., Yu, Z., Tan, X., Jiang, H., Xu, Z., Fang, Y., et al. (2021). CAR-macrophage: a new immunotherapy candidate against solid tumors. Biomed. Pharmacother. 139, 111605. doi:10.1016/j.biopha.2021.111605
Chen, Y., Zhu, X., Liu, H., Wang, C., Chen, Y., Wang, H., et al. (2023). The application of HER2 and CD47 CAR-macrophage in ovarian cancer. J. Transl. Med. 21 (1), 654. doi:10.1186/s12967-023-04479-8
Chocarro, L., Blanco, E., Fernandez-Rubio, L., Arasanz, H., Bocanegra, A., Echaide, M., et al. (2022). Cutting-edge CAR engineering: beyond T cells. Biomedicines 10 (12), 3035. doi:10.3390/biomedicines10123035
Conte, E. (2022). Targeting monocytes/macrophages in fibrosis and cancer diseases: therapeutic approaches. Pharmacol. Ther. 234, 108031. doi:10.1016/j.pharmthera.2021.108031
Cousins, R. J., Blanchard, R. K., Popp, M. P., Liu, L., Cao, J., Moore, J. B., et al. (2003). A global view of the selectivity of zinc deprivation and excess on genes expressed in human THP-1 mononuclear cells. Proc. Natl. Acad. Sci. U. S. A. 100 (12), 6952–6957. doi:10.1073/pnas.0732111100
Dai, H., Zhu, C., Huai, Q., Xu, W., Zhu, J., Zhang, X., et al. (2024). Chimeric antigen receptor-modified macrophages ameliorate liver fibrosis in preclinical models. J. hepatology 80 (6), 913–927. doi:10.1016/j.jhep.2024.01.034
Dana, H., Chalbatani, G. M., Jalali, S. A., Mirzaei, H. R., Grupp, S. A., Suarez, E. R., et al. (2021). CAR-T cells: early successes in blood cancer and challenges in solid tumors. Acta Pharm. Sin. B 11 (5), 1129–1147. doi:10.1016/j.apsb.2020.10.020
DeNardo, D. G., and Ruffell, B. (2019). Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 19 (6), 369–382. doi:10.1038/s41577-019-0127-6
Fuss, I. J., Kanof, M. E., Smith, P. D., and Zola, H. (2009). Isolation of whole mononuclear cells from peripheral blood and cord blood. Curr. Protoc. Immunol. 7, 7. doi:10.1002/0471142735.im0701s85
Giavridis, T., van der Stegen, S. J. C., Eyquem, J., Hamieh, M., Piersigilli, A., and Sadelain, M. (2018). CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 24 (6), 731–738. doi:10.1038/s41591-018-0041-7
Guerriero, J. L. (2018). Macrophages: the road less traveled, changing anticancer therapy. Trends Mol. Med. 24 (5), 472–489. doi:10.1016/j.molmed.2018.03.006
Guo, Q., and Qian, Z. M. (2024). Macrophage based drug delivery: key challenges and strategies. Bioact. Mater. 38, 55–72. doi:10.1016/j.bioactmat.2024.04.004
Gurunathan, S., Kang, M. H., Jeyaraj, M., and Kim, J. H. (2021). Palladium nanoparticle-induced oxidative stress, endoplasmic reticulum stress, apoptosis, and immunomodulation enhance the biogenesis and release of exosome in human leukemia monocytic cells (THP-1). Int. J. nanomedicine 16, 2849–2877. doi:10.2147/IJN.S305269
Hadiloo, K., Taremi, S., Heidari, M., and Esmaeilzadeh, A. (2023). The CAR macrophage cells, a novel generation of chimeric antigen-based approach against solid tumors. Biomark. Res. 11 (1), 103. doi:10.1186/s40364-023-00537-x
Halim, L., and Maher, J. (2020). CAR T-cell immunotherapy of B-cell malignancy: the story so far. Ther. Adv. Vaccines Immunother. 8, 2515135520927164. doi:10.1177/2515135520927164
Henry, T. D., Traverse, J. H., Hammon, B. L., East, C. A., Bruckner, B., Remmers, A. E., et al. (2014). Safety and efficacy of ixmyelocel-T: an expanded, autologous multi-cellular therapy, in dilated cardiomyopathy. Circulation Res. 115 (8), 730–737. doi:10.1161/CIRCRESAHA.115.304554
Hong, M., Clubb, J. D., and Chen, Y. Y. (2020). Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell 38 (4), 473–488. doi:10.1016/j.ccell.2020.07.005
Hu, M., Yao, Z., Xu, L., Peng, M., Deng, G., Liu, L., et al. (2023). M2 macrophage polarization in systemic sclerosis fibrosis: pathogenic mechanisms and therapeutic effects. Heliyon 9 (5), e16206. doi:10.1016/j.heliyon.2023.e16206
Huang, J., Yang, Q., Wang, W., and Huang, J. (2024). CAR products from novel sources: a new avenue for the breakthrough in cancer immunotherapy. Front. Immunol. 15, 1378739. doi:10.3389/fimmu.2024.1378739
Huo, Y., Zhang, H., Sa, L., Zheng, W., He, Y., Lyu, H., et al. (2023). M1 polarization enhances the antitumor activity of chimeric antigen receptor macrophages in solid tumors. J. Transl. Med. 21 (1), 225. doi:10.1186/s12967-023-04061-2
Italiani, P., and Boraschi, D. (2014). From monocytes to M1/M2 macrophages: phenotypical vs. Functional differentiation. Front. Immunol. 5, 514. doi:10.3389/fimmu.2014.00514
Ji, S., Shi, Y., and Yin, B. (2024). Macrophage barrier in the tumor microenvironment and potential clinical applications. Cell Commun. Signal. CCS 22 (1), 74. doi:10.1186/s12964-023-01424-6
Kadomoto, S., Izumi, K., and Mizokami, A. (2021). Macrophage polarity and disease control. Int. J. Mol. Sci. 23 (1), 144. doi:10.3390/ijms23010144
Kang, M., Lee, S. H., Kwon, M., Byun, J., Kim, D., Kim, C., et al. (2021). Nanocomplex-mediated in vivo programming to chimeric antigen receptor-M1 macrophages for cancer therapy. Adv. Mater. Deerf. Beach, Fla 33 (43), e2103258. doi:10.1002/adma.202103258
Kennedy, L. B., and Salama, A. K. S. (2020). A review of cancer immunotherapy toxicity. CA Cancer J. Clin. 70 (2), 86–104. doi:10.3322/caac.21596
Keshavarz, A., Salehi, A., Khosravi, S., Shariati, Y., Nasrabadi, N., Kahrizi, M. S., et al. (2022). Recent findings on chimeric antigen receptor (CAR)-engineered immune cell therapy in solid tumors and hematological malignancies. Stem Cell Res. Ther. 13 (1), 482. doi:10.1186/s13287-022-03163-w
Kim, J., and Bae, J. S. (2016). Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediat. Inflamm. 2016, 6058147. doi:10.1155/2016/6058147
Kim, J., and Seki, E. (2023). FAP: not just a biomarker but druggable target in liver fibrosis. Cell. Mol. gastroenterology hepatology 15 (4), 1018–1019. doi:10.1016/j.jcmgh.2022.12.018
Klichinsky, M., Ruella, M., Shestova, O., Lu, X. M., Best, A., Zeeman, M., et al. (2020). Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 38 (8), 947–953. doi:10.1038/s41587-020-0462-y
Kochneva, G. V., Sivolobova, G. F., Tkacheva, A. V., Gorchakov, A. A., and Kulemzin, S. V. (2020). Combination of oncolytic virotherapy and CAR T/NK cell therapy for the treatment of cancer. Mol. Biol. 54 (1), 3–16. doi:10.1134/S0026893320010100
Komohara, Y., Fujiwara, Y., Ohnishi, K., and Takeya, M. (2016). Tumor-associated macrophages: potential therapeutic targets for anti-cancer therapy. Adv. drug Deliv. Rev. 99 (Pt B), 180–185. doi:10.1016/j.addr.2015.11.009
Kosti, P., Maher, J., and Arnold, J. N. (2018). Perspectives on chimeric antigen receptor T-cell immunotherapy for solid tumors. Front. Immunol. 9, 1104. doi:10.3389/fimmu.2018.01104
Kuznetsova, A. V., Glukhova, X. A., Popova, O. P., Beletsky, I. P., and Ivanov, A. A. (2024). Contemporary approaches to immunotherapy of solid tumors. Cancers (Basel) 16 (12), 2270. doi:10.3390/cancers16122270
Lai, J., Mardiana, S., House, I. G., Sek, K., Henderson, M. A., Giuffrida, L., et al. (2020). Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat. Immunol. 21 (8), 914–926. doi:10.1038/s41590-020-0676-7
Lei, A., Yu, H., Lu, S., Lu, H., Ding, X., Tan, T., et al. (2024). A second-generation M1-polarized CAR macrophage with antitumor efficacy. Nat. Immunol. 25 (1), 102–116. doi:10.1038/s41590-023-01687-8
Leung, C. C. T., and Wong, C. K. C. (2021). Characterization of stanniocalcin-1 expression in macrophage differentiation. Transl. Oncol. 14 (1), 100881. doi:10.1016/j.tranon.2020.100881
Lewis, B. W., Patial, S., and Saini, Y. (2022). In vitro screening method for characterization of macrophage activation responses. Methods Protoc. 5 (5), 68. doi:10.3390/mps5050068
Li, J., Hu, H., Lian, K., Zhang, D., Hu, P., He, Z., et al. (2024a). CAR-NK cells in combination therapy against cancer: a potential paradigm. Heliyon 10 (5), e27196. doi:10.1016/j.heliyon.2024.e27196
Li, J., Tang, Y., and Huang, Z. (2022a). Efficacy and safety of chimeric antigen receptor (CAR)-T cell therapy in the treatment of relapsed and refractory multiple myeloma: a systematic-review and meta-analysis of clinical trials. Transl. cancer Res. 11 (3), 569–579. doi:10.21037/tcr-22-344
Li, M., Wang, M., Wen, Y., Zhang, H., Zhao, G. N., and Gao, Q. (2023b). Signaling pathways in macrophages: molecular mechanisms and therapeutic targets. MedComm 4 (5), e349. doi:10.1002/mco2.349
Li, N., Geng, S., Dong, Z. Z., Jin, Y., Ying, H., Li, H. W., et al. (2024b). A new era of cancer immunotherapy: combining revolutionary technologies for enhanced CAR-M therapy. Mol. Cancer 23 (1), 117. doi:10.1186/s12943-024-02032-9
Li, S. Y., Guo, Y. L., Tian, J. W., Zhang, H. J., Li, R. F., Gong, P., et al. (2023a). Anti-tumor strategies by harnessing the phagocytosis of macrophages. Cancers (Basel) 15 (10), 2717. doi:10.3390/cancers15102717
Li, W., Wang, F., Guo, R., Bian, Z., and Song, Y. (2022b). Targeting macrophages in hematological malignancies: recent advances and future directions. J. Hematol. Oncol. 15 (1), 110. doi:10.1186/s13045-022-01328-x
Liang, Y., Xu, Q., and Gao, Q. (2023). Advancing CAR-based immunotherapies in solid tumors: CAR-macrophages and neutrophils. Front. Immunol. 14, 1291619. doi:10.3389/fimmu.2023.1291619
Liu, M., Liu, J., Liang, Z., Dai, K., Gan, J., Wang, Q., et al. (2022a). CAR-macrophages and CAR-T cells synergistically kill tumor cells in vitro. Cells 11 (22), 3692. doi:10.3390/cells11223692
Liu, M., Liu, L., Song, Y., Li, W., and Xu, L. (2022c). Targeting macrophages: a novel treatment strategy in solid tumors. J. Transl. Med. 20 (1), 586. doi:10.1186/s12967-022-03813-w
Liu, Y., An, L., Huang, R., Xiong, J., Yang, H., Wang, X., et al. (2022b). Strategies to enhance CAR-T persistence. Biomark. Res. 10 (1), 86. doi:10.1186/s40364-022-00434-9
Mangan, D. F., and Wahl, S. M. (1991). Differential regulation of human monocyte programmed cell death (apoptosis) by chemotactic factors and pro-inflammatory cytokines. J. Immunol. Baltim. Md 1950 147 (10), 3408–3412. doi:10.4049/jimmunol.147.10.3408
Mangan, D. F., Welch, G. R., and Wahl, S. M. (1991). Lipopolysaccharide, tumor necrosis factor-alpha, and IL-1 beta prevent programmed cell death (apoptosis) in human peripheral blood monocytes. J. Immunol. Baltim. Md 1950 146 (5), 1541–1546. doi:10.4049/jimmunol.146.5.1541
Mercanti, L., Sindaco, M., Mazzone, M., Di Marcantonio, M. C., Piscione, M., Muraro, R., et al. (2023). PDAC, the influencer cancer: cross-talk with tumor microenvironment and connected potential therapy strategies. Cancers (Basel) 15 (11), 2923. doi:10.3390/cancers15112923
Moreno-Lanceta, A., Medrano-Bosch, M., Fundora, Y., Perramón, M., Aspas, J., Parra-Robert, M., et al. (2023). RNF41 orchestrates macrophage-driven fibrosis resolution and hepatic regeneration. Sci. Transl. Med. 15 (704), eabq6225. doi:10.1126/scitranslmed.abq6225
Morrissey, M. A., Williamson, A. P., Steinbach, A. M., Roberts, E. W., Kern, N., Headley, M. B., et al. (2018). Chimeric antigen receptors that trigger phagocytosis. eLife 7, e36688. doi:10.7554/eLife.36688
Murray, P. J., Allen, J. E., Biswas, S. K., Fisher, E. A., Gilroy, D. W., Goerdt, S., et al. (2014). Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41 (1), 14–20. doi:10.1016/j.immuni.2014.06.008
Na, Y. R., Jung, D., Stakenborg, M., Jang, H., Gu, G. J., Jeong, M. R., et al. (2021). Prostaglandin E(2) receptor PTGER4-expressing macrophages promote intestinal epithelial barrier regeneration upon inflammation. Gut 70 (12), 2249–2260. doi:10.1136/gutjnl-2020-322146
Na, Y. R., Kim, S. W., and Seok, S. H. (2023). A new era of macrophage-based cell therapy. Exp. and Mol. Med. 55 (9), 1945–1954. doi:10.1038/s12276-023-01068-z
Nakamura, K., and Smyth, M. J. (2020). Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell. and Mol. Immunol. 17 (1), 1–12. doi:10.1038/s41423-019-0306-1
Netsrithong, R., Garcia-Perez, L., and Themeli, M. (2023). Engineered T cells from induced pluripotent stem cells: from research towards clinical implementation. Front. Immunol. 14, 1325209. doi:10.3389/fimmu.2023.1325209
Noy, R., and Pollard, J. W. (2014). Tumor-associated macrophages: from mechanisms to therapy. Immunity 41 (1), 49–61. doi:10.1016/j.immuni.2014.06.010
Paasch, D., Meyer, J., Stamopoulou, A., Lenz, D., Kuehle, J., Kloos, D., et al. (2022). Ex vivo generation of CAR macrophages from hematopoietic stem and progenitor cells for use in cancer therapy. Cells 11 (6), 994. doi:10.3390/cells11060994
Pan, K., Farrukh, H., Chittepu, V., Xu, H., Pan, C. X., and Zhu, Z. (2022). CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J. Exp. Clin. Cancer Res. 41 (1), 119. doi:10.1186/s13046-022-02327-z
Patel, U., Rajasingh, S., Samanta, S., Cao, T., Dawn, B., and Rajasingh, J. (2017). Macrophage polarization in response to epigenetic modifiers during infection and inflammation. Drug Discov. Today 22 (1), 186–193. doi:10.1016/j.drudis.2016.08.006
Perez-Amill, L., Suñe, G., Antoñana-Vildosola, A., Castella, M., Najjar, A., Bonet, J., et al. (2021). Preclinical development of a humanized chimeric antigen receptor against B cell maturation antigen for multiple myeloma. Haematologica 106 (1), 173–184. doi:10.3324/haematol.2019.228577
Podaru, M. N., Fields, L., Kainuma, S., Ichihara, Y., Hussain, M., Ito, T., et al. (2019). Reparative macrophage transplantation for myocardial repair: a refinement of bone marrow mononuclear cell-based therapy. Basic Res. Cardiol. 114 (5), 34. doi:10.1007/s00395-019-0742-1
Rogers, P. D., Thornton, J., Barker, K. S., McDaniel, D. O., Sacks, G. S., Swiatlo, E., et al. (2003). Pneumolysin-dependent and -independent gene expression identified by cDNA microarray analysis of THP-1 human mononuclear cells stimulated by Streptococcus pneumoniae. Infect. Immun. 71 (4), 2087–2094. doi:10.1128/iai.71.4.2087-2094.2003
Roth, T. L., Puig-Saus, C., Yu, R., Shifrut, E., Carnevale, J., Li, P. J., et al. (2018). Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559 (7714), 405–409. doi:10.1038/s41586-018-0326-5
Savanur, M. A., Weinstein-Marom, H., and Gross, G. (2021). Implementing logic gates for safer immunotherapy of cancer. Front. Immunol. 12, 780399. doi:10.3389/fimmu.2021.780399
Schepisi, G., Gianni, C., Palleschi, M., Bleve, S., Casadei, C., Lolli, C., et al. (2023). The new frontier of immunotherapy: chimeric antigen receptor T (CAR-T) cell and macrophage (CAR-M) therapy against breast cancer. Cancers (Basel) 15 (5), 1597. doi:10.3390/cancers15051597
Schildberger, A., Rossmanith, E., Eichhorn, T., Strassl, K., and Weber, V. (2013). Monocytes, peripheral blood mononuclear cells, and THP-1 cells exhibit different cytokine expression patterns following stimulation with lipopolysaccharide. Mediat. Inflamm. 2013, 697972. doi:10.1155/2013/697972
Shen, J., Lyu, S., Xu, Y., Zhang, S., Li, L., Li, J., et al. (2024). Activating innate immune responses repolarizes hPSC-derived CAR macrophages to improve anti-tumor activity. Cell Stem Cell 31 (7), 1003–1019.e9. doi:10.1016/j.stem.2024.04.012
Shibuya, Y., Kumar, K. K., Mader, M. M., Yoo, Y., Ayala, L. A., Zhou, M., et al. (2022). Treatment of a genetic brain disease by CNS-wide microglia replacement. Sci. Transl. Med. 14 (636), eabl9945. doi:10.1126/scitranslmed.abl9945
Shin, M. H., Oh, E., Kim, Y., Nam, D. H., Jeon, S. Y., Yu, J. H., et al. (2023). Recent advances in CAR-based solid tumor immunotherapy. Cells 12 (12), 1606. doi:10.3390/cells12121606
Sloas, C., Gill, S., and Klichinsky, M. (2021). Engineered CAR-macrophages as adoptive immunotherapies for solid tumors. Front. Immunol. 12, 783305. doi:10.3389/fimmu.2021.783305
Snyder, C. M., and Gill, S. I. (2023). Good CARMA: turning bad tumor-resident myeloid cells good with chimeric antigen receptor macrophages. Immunol. Rev. 320 (1), 236–249. doi:10.1111/imr.13231
Steinbach, C., Merchant, A., Zaharie, A. T., Horak, P., Marhold, M., and Krainer, M. (2022). Current developments in cellular therapy for castration resistant prostate cancer: a systematic review of clinical studies. Cancers (Basel) 14 (22), 5719. doi:10.3390/cancers14225719
Stephan, M. T., Ponomarev, V., Brentjens, R. J., Chang, A. H., Dobrenkov, K. V., Heller, G., et al. (2007). T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat. Med. 13 (12), 1440–1449. doi:10.1038/nm1676
Sterner, R. C., and Sterner, R. M. (2021). CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 11 (4), 69. doi:10.1038/s41408-021-00459-7
Su, S., Lei, A., Wang, X., Lu, H., Wang, S., Yang, Y., et al. (2022). Induced CAR-macrophages as a novel therapeutic cell type for cancer immune cell therapies. Cells 11 (10), 1652. doi:10.3390/cells11101652
Suzuki, T., Arumugam, P., Sakagami, T., Lachmann, N., Chalk, C., Sallese, A., et al. (2014). Pulmonary macrophage transplantation therapy. Nature 514 (7523), 450–454. doi:10.1038/nature13807
Thakar, M. S., Kearl, T. J., and Malarkannan, S. (2019). Controlling cytokine release syndrome to harness the full potential of CAR-based cellular therapy. Front. Oncol. 9, 1529. doi:10.3389/fonc.2019.01529
Tie, Y., Tang, F., Wei, Y. Q., and Wei, X. W. (2022). Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J. Hematol. Oncol. 15 (1), 61. doi:10.1186/s13045-022-01282-8
Unver, N. (2023). Sophisticated genetically engineered macrophages, CAR-Macs, in hitting the bull's eye for solid cancer immunotherapy approaches. Clin. Exp. Med. 23 (7), 3171–3177. doi:10.1007/s10238-023-01106-0
Uscanga-Palomeque, A. C., Chávez-Escamilla, A. K., Alvizo-Báez, C. A., Saavedra-Alonso, S., Terrazas-Armendáriz, L. D., Tamez-Guerra, R. S., et al. (2023). CAR-T cell therapy: from the shop to cancer therapy. Int. J. Mol. Sci. 24 (21), 15688. doi:10.3390/ijms242115688
van der Heide, D., Weiskirchen, R., and Bansal, R. (2019). Therapeutic targeting of hepatic macrophages for the treatment of liver diseases. Front. Immunol. 10, 2852. doi:10.3389/fimmu.2019.02852
Velasco-de Andres, M., Munoz-Sanchez, G., Carrillo-Serradell, L., Gutierrez-Hernandez, M. D. M., Catala, C., Isamat, M., et al. (2023). Chimeric antigen receptor-based therapies beyond cancer. Eur. J. Immunol. 53 (3), e2250184. doi:10.1002/eji.202250184
Villanueva, M. T. (2020). Macrophages get a CAR. Nat. Rev. Drug Discov. 19 (5), 308. doi:10.1038/d41573-020-00053-9
Wang, D. R., Wu, X. L., and Sun, Y. L. (2022a). Therapeutic targets and biomarkers of tumor immunotherapy: response versus non-response. Signal Transduct. Target Ther. 7 (1), 331. doi:10.1038/s41392-022-01136-2
Wang, S., Yang, Y., Ma, P., Zha, Y., Zhang, J., Lei, A., et al. (2022b). CAR-macrophage: an extensive immune enhancer to fight cancer. EBioMedicine 76, 103873. doi:10.1016/j.ebiom.2022.103873
Wang, X., Su, S., Zhu, Y., Cheng, X., Cheng, C., Chen, L., et al. (2023). Metabolic Reprogramming via ACOD1 depletion enhances function of human induced pluripotent stem cell-derived CAR-macrophages in solid tumors. Nat. Commun. 14 (1), 5778. doi:10.1038/s41467-023-41470-9
Wegrzyniak, O., Rosestedt, M., and Eriksson, O. (2021). Recent progress in the molecular imaging of nonalcoholic fatty liver disease. Int. J. Mol. Sci. 22 (14), 7348. doi:10.3390/ijms22147348
Wu, J., Wang, X., Huang, Y., Zhang, Y., Su, S., Shou, H., et al. (2023). Targeted glycan degradation potentiates cellular immunotherapy for solid tumors. Proc. Natl. Acad. Sci. U. S. A. 120 (38), e2300366120. doi:10.1073/pnas.2300366120
Wu, M., Shi, Y., Zhu, L., Chen, L., Zhao, X., and Xu, C. (2022). Macrophages in glioblastoma development and therapy: a double-edged sword. Life Basel, Switz. 12 (8), 1225. doi:10.3390/life12081225
Xu, Y., Wang, X., Liu, L., Wang, J., Wu, J., and Sun, C. (2022). Role of macrophages in tumor progression and therapy (Review). Int. J. Oncol. 60 (5), 57. doi:10.3892/ijo.2022.5347
Yang, A. T., Kim, Y. O., Yan, X. Z., Abe, H., Aslam, M., Park, K. S., et al. (2023a). Fibroblast activation protein activates macrophages and promotes parenchymal liver inflammation and fibrosis. Cell. Mol. gastroenterology hepatology 15 (4), 841–867. doi:10.1016/j.jcmgh.2022.12.005
Yang, S., Wang, Y., Jia, J., Fang, Y., Yang, Y., Yuan, W., et al. (2024). Advances in engineered macrophages: a new frontier in cancer immunotherapy. Cell death and Dis. 15 (4), 238. doi:10.1038/s41419-024-06616-7
Yang, Z., Liu, Y., Zhao, K., Jing, W., Gao, L., Dong, X., et al. (2023b). Dual mRNA co-delivery for in situ generation of phagocytosis-enhanced CAR macrophages augments hepatocellular carcinoma immunotherapy. J. Control. release official J. Control. Release Soc. 360, 718–733. doi:10.1016/j.jconrel.2023.07.021
Yoon, D. H., Osborn, M. J., Tolar, J., and Kim, C. J. (2018). Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-Ts): combination or built-in CAR-T. Int. J. Mol. Sci. 19 (2), 340. doi:10.3390/ijms19020340
Zhang, E., Gu, J., and Xu, H. (2018). Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol. Cancer 17 (1), 7. doi:10.1186/s12943-018-0759-3
Zhang, H., Zhu, S., Deng, W., Li, R., Zhou, H., and Xiong, H. (2022). The landscape of chimeric antigen receptor T cell therapy in breast cancer: perspectives and outlook. Front. Immunol. 13, 887471. doi:10.3389/fimmu.2022.887471
Zhang, L., Tian, L., Dai, X., Yu, H., Wang, J., Lei, A., et al. (2020). Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. and Oncol. 13 (1), 153. doi:10.1186/s13045-020-00983-2
Zhang, W., Liu, L., Su, H., Liu, Q., Shen, J., Dai, H., et al. (2019). Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br. J. cancer 121 (10), 837–845. doi:10.1038/s41416-019-0578-3
Zhang, Y., Yang, J., Zhang, T., and Gu, H. (2023). Emerging advances in nanobiomaterials-assisted chimeric antigen receptor (CAR)-macrophages for tumor immunotherapy. Front. Bioeng. Biotechnol. 11, 1211687. doi:10.3389/fbioe.2023.1211687
Zheng, X., Jin, W., Wang, S., and Ding, H. (2021). Progression on the roles and mechanisms of tumor-infiltrating T lymphocytes in patients with hepatocellular carcinoma. Front. Immunol. 12, 729705. doi:10.3389/fimmu.2021.729705
Keywords: macrophage, chimeric antigen receptor, CAR-M therapy, tumor immunotherapy, solid tumor
Citation: Huang T, Bei C, Hu Z and Li Y (2024) CAR-macrophage: Breaking new ground in cellular immunotherapy. Front. Cell Dev. Biol. 12:1464218. doi: 10.3389/fcell.2024.1464218
Received: 16 July 2024; Accepted: 23 September 2024;
Published: 03 October 2024.
Edited by:
Samuel Ken-En Gan, Kean University-Wenzhou, ChinaReviewed by:
Chong Wu, School of Life Sciences, Sun Yat-sen University, ChinaXiaoming Feng, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Copyright © 2024 Huang, Bei, Hu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenhua Hu, YmFuZXlodWFAMTYzLmNvbQ==; Yuanyuan Li, bGl5dWFueXVhbkBzaW1tLmFjLmNu