 Jingxian Shi
Jingxian Shi Yuxi Jin
Yuxi Jin Sha Lin
Sha Lin Xing Li
Xing Li Donghui Zhang
Donghui Zhang Jinlin Wu
Jinlin Wu Yan Qi
Yan Qi Yifei Li
Yifei Li- 1Key Laboratory of Birth Defects and Related Diseases of Women and Children of MOE, Department of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu, Sichuan, China
- 2State Key Laboratory of Biocatalysis and Enzyme Engineering, School of Life Science, Hubei University, Wuhan, China
The initial contraction of the heart during the embryonic stage necessitates a substantial energy supply, predominantly derived from mitochondrial function. However, during embryonic heart development, mitochondria influence beyond energy supplementation. Increasing evidence suggests that mitochondrial permeability transition pore opening and closing, mitochondrial fusion and fission, mitophagy, reactive oxygen species production, apoptosis regulation, Ca2+ homeostasis, and cellular redox state also play critical roles in early cardiac development. Therefore, this review aims to describe the essential roles of mitochondrial non-energetic function embryonic cardiac development.
Introduction
During embryonic development in mammals, the heart is the first organ to develop and perform functionality (Bruneau, 2020; Buckingham et al., 2005). The primitive heart tube begins to beat for the first time on the twenty-second day of gestation (roughly equivalent to embryonic (E) day E8.5 in mice), allowing for active fetal circulation by the end of the fourth week of pregnancy (E9.5 in mice), culminating in the formation of a four-chamber heart by the seventh week of gestation (E13.5 in mice) (Tan and Lewandowski, 2020; Finnemore and Groves, 2015; Sánchez-Díaz et al., 2020). Cardiac development is a multi-step process that includes mesodermal differentiation, cardiac progenitor cell formation, and cardiac maturation (Zhu et al., 2020; Garry and Olson, 2006) (Figure 1). Recent studies have shown that the vast majority of cardiac cells are derived from Nkx2.5 cardiovascular progenitor cells (Stanley et al., 2002), the discovery of Nkx2.5 also ushered in a “golden age” of using molecular and genetic methods to study heart formation (Tanaka et al., 1999). Congenital heart defects resulting from abnormal heart development affect more than 1% of newborns (Wu et al., 2020). Accordingly, understanding and exploring the molecular and genetic mechanisms involved in the development of the heart embryo can provide important clues for better prevention and treatment of prevalent cardiac diseases.
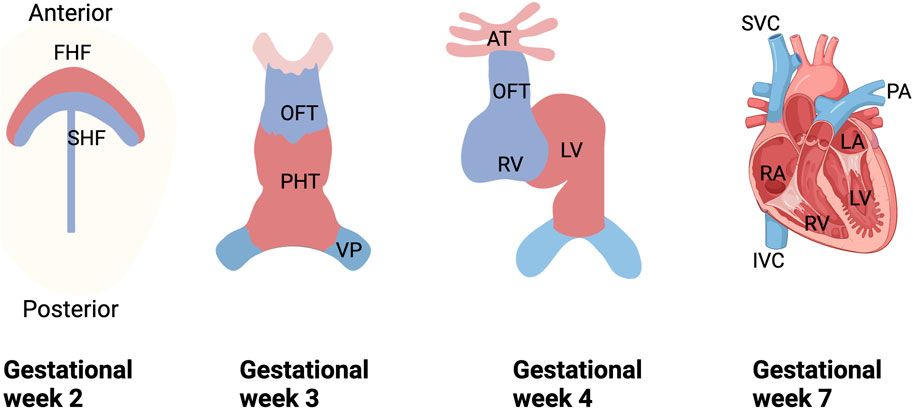
Figure 1. Schematic diagram of human embryonic heart development. During the 2nd week of gestation, cardiogenic mesodermal cells migrate towards the anterior side of the embryo to form FHF and SHF, which are designated to form specific regions of the PHT. During weeks 3 and 4 of gestation, FHF form the beating PHT and eventually give rise to the LV and portions of the right and left atria (RA and LA), and SHF are located posterior to the PHT and within the pharyngeal mesoderm to form the RV, portions of the atria and outflow tracts, and to form the base of the aorta and pulmonary arteries. And in the 3rd week of gestation, the cells of the venous pole form the superior and inferior vena cava (SVC and IVC), and ultimately, the four-chambered heart is formed by the end of the 7th week. FHF, first heart field; SHF, second heart field; PHT, primary heart tube; OFT, outflow tract; VP, venous pole; LV, left ventricle; RV, right ventricle; SVC, superior vena cava; IVC, inferior vena cava vein; PA, pulmonary artery.
Mitochondria, known as the power-factory of the cell, play a multifaceted role in the development and maturation of the embryonic heart (Gao and Zhang, 2018; Cheong et al., 2020). In almost all types of eukaryotic cells, mitochondria typically number in the hundreds within the cytoplasm of each cell, with each mitochondrion containing 2–10 copies of DNA (Harding, 1991; Andrieux et al., 2021). The specific number of mitochondria and their DNA copies generally correlate with the energy demands of the cell (Robin and Wong, 1988). As an organelle enveloped by a double phospholipid bilayer, the inner membrane of the mitochondrion encloses the matrix space and invaginates to form cristae (Vogel et al., 2006). This cristae structure maximizes the inner membrane’s surface area, providing ample attachment sites for various enzymes and electron transport chain complexes, thereby enhancing its ATP production capacity (Birkedal et al., 2022). The outer membrane, which encapsulates the inner membrane to form the intermembrane space, is highly permeable and creates a transitional space for the transport and storage of various metabolic substrates, facilitating enzymatic reactions occurring at the inner membrane (Gupta and Becker, 2021). As a semi-autonomous organelle, mitochondria contain approximately 1,500 proteins essential for their structure and function, with mitochondrial DNA (mtDNA) encoding less than 1% of these proteins, while the remainder are encoded by nuclear DNA (nDNA) (Nunnari and Suomalainen, 2012; Filograna et al., 2021; Vega et al., 2015). Unlike the long, linear structure of genomic DNA, mtDNA is a circular molecule consisting of about 16,000 base pairs (Chen Y. et al., 2014). Lacking introns, mtDNA has a high utilization rate and controls the expression of 37 genes, including 13 essential for the electron transport chain, 2 ribosomal RNAs, and 22 transfer RNAs (Chen Y. et al., 2014; Goffart et al., 2004; Bonekamp and Larsson, 2018; Becker et al., 2014). Mitochondria perform various cellular function, including ATP production, regulation of apoptosis, Ca2+ homeostasis, and their own homeostasis (Gao and Zhang, 2018; May-Panloup et al., 2021; Chandel, 2014; Shadel and Horvath, 2015; Gut and Verdin, 2013).
Nevertheless, there is accumulating evidence that mitochondria play a key role in the development of the embryonic heart, involving reactive oxygen species (ROS) production, cell apoptosis, mitochondrial fusion, and division. Accordingly, this paper focuses on the impact of non-energetic metabolism regarding mitochondria in embryonic cardiac development.
The connection between mitochondria and the embryonic heart
Mitochondria not only produce more than 90% of the ATP required for cardiomyocyte function via oxidative phosphorylation (OXPHOS), but also play a pivotal role in biosynthesis, apoptosis, calcium homeostasis, cell signaling pathways, and genetic expression (May-Panloup et al., 2021; Chandel, 2014; Shadel and Horvath, 2015; Gut and Verdin, 2013).
The mitochondria in mammals are semiautonomous organelle composed of nuclear DNA (nDNA) and mitochondrial DNA (mtDNA) encoding proteins (Pagliarini et al., 2008; Gao et al., 2023). Although mtDNA is critical for OXPHOS, its role in maintaining cardiac function goes beyond energy supply. Hundreds of different pathogenic mitochondrial genome (mtDNA) mutations have already been identified in humans, many of which are associated with cardiac deficiencies (Bray and Ballinger, 2017). The mtDNA single-strand breaks and mtDNA-depleted cells are susceptible to apoptosis (Yen et al., 2005; Tann et al., 2011). The research showed that in mouse models missing mitochondrial components (such as those necessary for mtDNA maintenance or gene expression), embryonic development typically stalled at embryonic (E) day E8.5 (roughly equivalent to E20 in humans) and seemed to lack all cardiac structures (Cerritelli et al., 2003; Cámara et al., 2011). Mitochondrial protein encoded by the nucleus, mitochondrial transcription factor A (TFAM), is a protein required for the transcription of mitochondrial DNA (mtDNA) (Ngo et al., 2014). In a model of Nkx2.5Cre, the detection of TFAM inactivation leads to mitochondrial dysfunction, which induces ROS production to activate the DNA damage response, ultimately leading to embryonic lethal cardiac dysplasia (Zhang et al., 2018). Deletion of cardiomyocyte-specific mitochondrial phosphatase (PTPMT1) in the TnT-Cre mouse model leads to abnormal cardiac development and embryonic lethality between E16.5 and E18.5 (Chen et al., 2021; Zhang et al., 2011). Furthermore, mutations in mtDNA encoding OXPHOS subunit complexes IV and V were found in congenital heart disease patients (Heidari et al., 2022). Cardiovascular involvement occurs early in more than one-third of children with primary mitochondrial disease, with a mean age of onset of 5.7 ± 7.8 years, associated with a poor prognosis (Brambilla et al., 2020; Holmgren et al., 2003).
During cardiac development, the maturation of mitochondrial structure and function is accompanied by an increase in mitochondrial number and the formation of lamellar cristae (David et al., 1981; Dorn et al., 2015). In vitro, early cardiomyocyte differentiation, mitochondria are relatively sparse, rounded, and dispersed throughout the cytoplasm, with increasing numbers of mitochondria as the development (Lyra-Leite et al., 2021; Kim et al., 1992). In the fetal mouse heart, mitochondria appear as elongated tubular organelles at embryonic (E) day E9.5, which are ovoid containing more endomembrane invaginations (cristae) at E13.5 (Maroli and Braun, 2021). The mitochondria permeability transition pores (mPTP) open at E9.5, but close mPTP at E13.5 (Murray et al., 2014). Moreover, mitochondria in fetal cardiomyocytes are fewer in number and more confined to the center of the cell, and energy metabolism in fetal myocardium is less dependent on oxidative phosphorylation and correlates with the mechanisms by which fetal myocardium resists hypoxia (Agata et al., 2019). All of these findings demonstrate that mitochondria are closely related to embryonic heart development.
Mitochondrial energy metabolism and embryonic heart development
Cardiac differentiation started in an early stage of embryonic progression (Kirby, 2002). During early cardiac development, energy metabolism transitions from reliance on anaerobic glycolysis to aerobic oxidative phosphorylation (Chung et al., 2007; Lopaschuk and Jaswal, 2010; Zhao et al., 2022) (Figure 2).
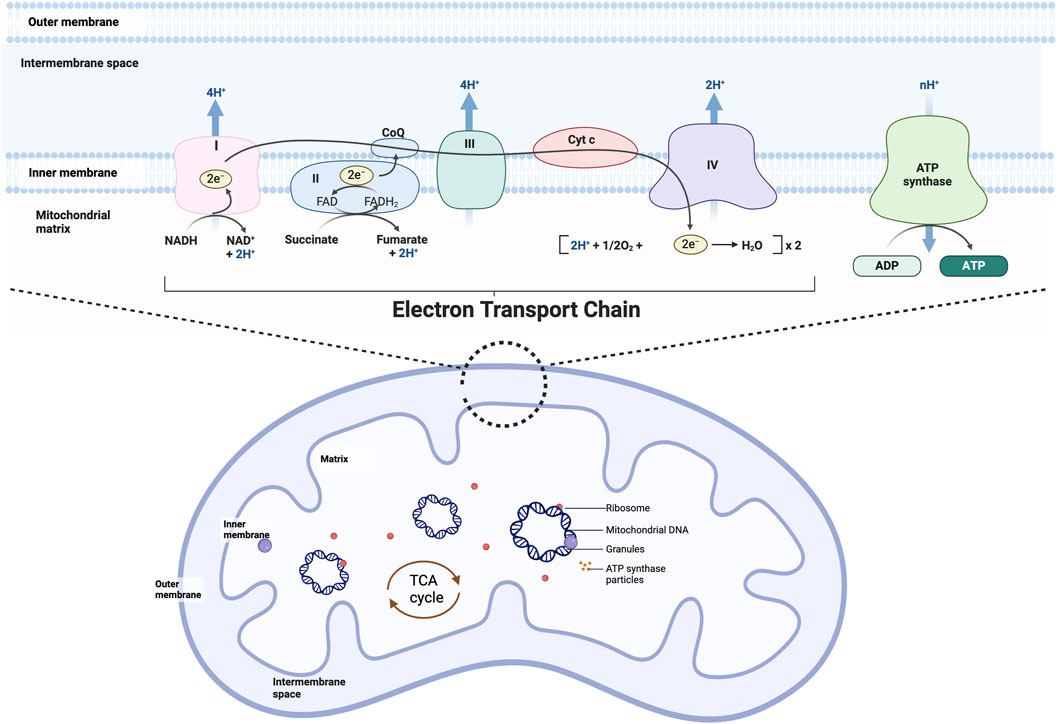
Figure 2. Mitochondrial structure and oxidative phosphorylation. Mitochondria can be divided from the outside to the inside into four functional regions: the outer mitochondrial membrane, the mitochondrial intermembrane space, the inner mitochondrial membrane, and the mitochondrial matrix, which generally also contains the mitochondria’s own DNA (mitochondrial DNA), RNA, and ribosomes (mitochondrial ribosomes). Mitochondrial oxidative phosphorylation is the movement of electrons through a complex (I-V) embedded in the inner mitochondrial membrane generates an electrical gradient through which protons are simultaneously pumped into the intermembrane space, producing a proton gradient. Together, the electrical gradient and the proton gradient generate electrochemical forces that lead to the synthesis of ATP by ATP-synthase.
Previous studies have shown that at the early stage of mouse heart development (E9.5), energy is primarily derived from glycolysis, and the activities of mitochondrial electron transport chain (ETC) complexes are at low levels (Tanimura and Shepard, 1970; Lai et al., 2008). Further measurements of oxygen concentration in the heart and other embryonic tissues indicate that oxygen concentration in cardiac tissue is only half that of other embryonic tissues, corroborating the low level of aerobic metabolism at this early stage of heart development (Burton, 2009). After E9.5, researchers observed a significant increase in the proportion of aerobic metabolism and mitochondrial ATP production, while glucose metabolism levels declined. As heart development progresses, from E8.5 to E14 in mice, the activities of, ETC complexes I, II, III, and V markedly increase, and mitochondria in the heart begin to actively participate in oxidative phosphorylation (Ingraham et al., 2009). Postnatally, the activity and quantity of ETC complexes continue to rise, with reliance on glycolysis and lactate metabolism decreasing until the development of mature cardiomyocytes, where fatty acid oxidation provides 90% of the energy (Lopaschuk and Jaswal, 2010; Zhao et al., 2022). These studies clearly demonstrate that the proportion of mitochondrial metabolism gradually increases during the development and maturation of the heart.
Mitochondrial non-energetic and embryonic heart development
The heart requires a constant supply of ATP from mitochondria for contraction and relaxation, but damaged mitochondria become a source of reactive ROS and cell death signals under pathological conditions (Saito et al., 2021; Saito and Sadoshima, 2015). More and more evidence suggests that mitochondria can influence cardiomyocyte differentiation and progression through more subtle means (Pohjoismäki and Goffart, 2017). For example, the regulation of reactive oxygen species (ROS) production and apoptosis, the opening and closing of the mitochondrial permeability transition pore, the dynamic balance between mitochondrial fusion and fission, Ca2+ homeostasis, and the regulation of the cellular redox state should not be neglected (Figure 3).
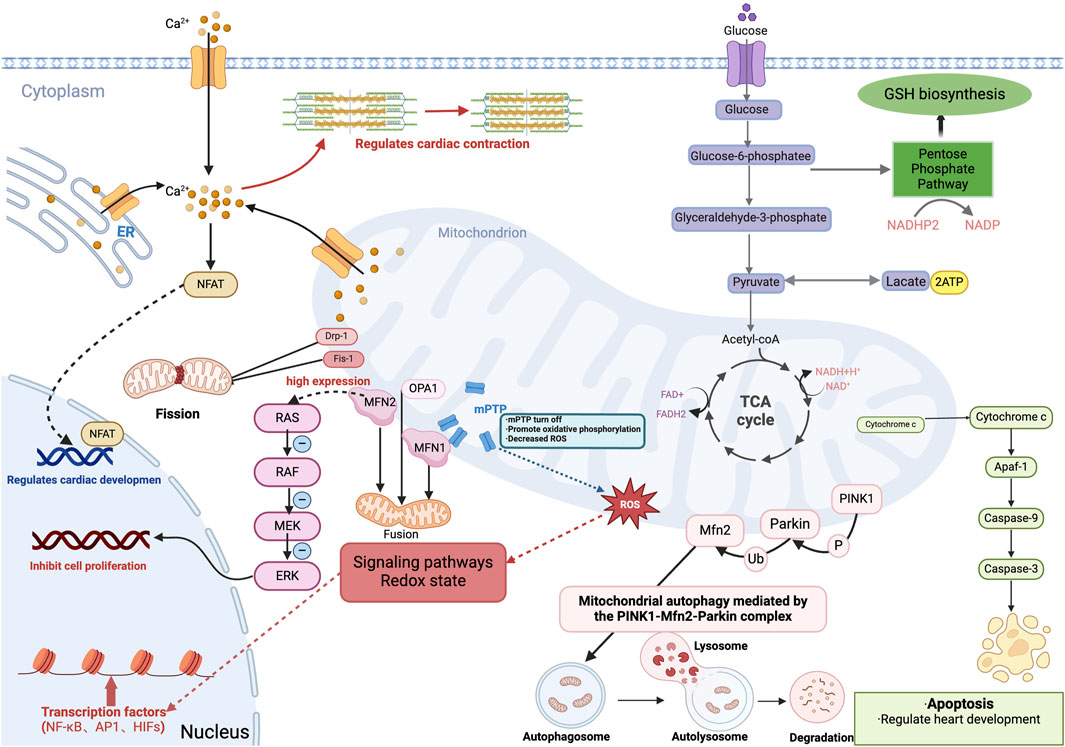
Figure 3. Role of mitochondria in embryonic heart development. Cardiomyocytes produce energy (ATP) by oxidative phosphorylation or anaerobic glycolysis, and during embryonic heart development, energy metabolism shifts from anaerobic glycolysis to aerobic oxidative phosphorylation involving mitochondria. And mitochondria produce intermediary metabolites and reactive oxygen species (ROS), which are involved in signaling pathways, redox homeostasis, and gene expression, and involve transcription factors including the κB family (NF-κB), activator protein-1 family (AP1) and hypoxia-inducible transcription factors (HIFs). Additionally, mitochondrial release of cytochrome c mediates apoptosis and regulates cardiac development during the embryonic period. Also, the closure of the mitochondrial permeability transition pore (mPTP) during the embryonic period facilitates oxidative phosphorylation, which reduces ROS levels in embryonic cardiomyocytes and promotes differentiation of embryonic cardiomyocytes. Mitochondrial quality control, including mitochondrial fusion, division, and mitochondrial autophagy is mediated by the PINK1-Mfn2-Parkin complex; Mitochondrial protein-2 (MFN2) regulates mitochondrial morphology and function by participating in mitochondrial fusion. While MFN2, which is highly expressed in embryonic stage, inhibits cardiomyocyte proliferation through the Ras-Raf-ERK signaling pathway. Mitochondria in the embryonic heart binds to ER and mediates the regulation of cardiac systolic-diastolic processes by Ca2+ concentration, and also activates the Ca +-dependent NFAT signaling pathway to regulate cardiac development. GSH, glutathione. ER, endoplasmic reticulum.
Pros and cons of embryonic ROS production
Most of the intracellular reactive oxygen species (ROS) are generated by mitochondrial metabolism (Birket et al., 2013). During electron transfer in mitochondria, premature oxidation of some electrons occurs, resulting in the production of ROS (Semenza, 2007; Zorov et al., 2014). The major ROS molecules in the body include superoxide anion (O2-), hydroxyl radical (•OH), and hydrogen peroxide (H2O2) (Shao et al., 2012). Initially, ROS, a byproduct of the electron transport chain, was traditionally thought to primarily damage DNA and cause strand breaks (Dennery, 2010). However, contemporary research increasingly suggests that ROS can regulate various biological processes (Sauer and Wartenberg, 2005). In the early development of the heart, ROS acts as a signaling molecule playing a crucial role (Thompson and Al-Hasan, 2012; Sauer et al., 1999).
There may be a delicate balance between oxygen levels and the production of ROS during the gestation period of embryonic growth. On the one hand, ROS can promote the differentiation efficiency of cardiomyocytes through multiple signaling pathways such as JNK, ERK1/2, p38, Ca2+, BMP, and increase the percentage of beating cardiomyocytes in embryoid bodies (Buggisch et al., 2007; Wei and Cong, 2018). High levels of ROS in embryonic stem cells are associated with an increase in the percentage of beating cardiomyocytes in embryos within the body (Sauer and Wartenberg, 2005). Moreover, hypoxia in the fetal rat heart induces epigenetic repression of the protein kinase C epsilon (PKCɛ) gene through ROS-mediated pathways, resulting in cardiac susceptibility to ischemic injury in the fetal heart (Hellgren et al., 2021; Patterson et al., 2012). While N-acetylcysteine as a ROS scavenger inhibits hypoxia-induced methylation of the SP1 binding site, decreases cardiac susceptibility to ischemic injury by increasing SP1 binding to the PKCɛ promoter (Patterson et al., 2012). As a result, ROS can enhance the differentiation of embryonic stem cells to cardiac cells.
On the other hand, high levels of ROS due to mitochondrial dysfunction may lead to severe congenital heart disease. High ROS levels lead to DNA damage and initiate a DNA damage response pathway that causes cardiomyocyte cell cycle arrest during embryonic development (Puente et al., 2014). The nucleus-encoded mitochondrial protein, TFAM (mitochondrial transcription factor A), is required for mtDNA transcription (Ngo et al., 2014). In the Nkx2.5-Cre model, TFAM deactivation leads to mitochondrial dysfunction, which induces ROS production to initiate a DNA damage response, ultimately contributing to embryonic lethal myocardial dysplasia (Zhang et al., 2018). The anticonvulsant drug valproic acid raises intracellular ROS levels in mouse embryoid somata, which can suppress cardiomyocyte polarization and lead to cardiac malformations (Na et al., 2003). The mitochondria-targeting antioxidant Mitoquinone (MitoQ), which consists of lipophilic triphenylphosphine (TPP) cations covalently attached to the antioxidant ubiquinone, prevents superoxide-induced lipid peroxidation and mitochondrial damage (Smith et al., 2008; Smith and Murphy, 2010). In chronically hypoxic chicken embryos, MitoQ treatment restores the mitochondrial respiratory control ratio, left ventricular structure, and contractile function of the heart by suppressing mitochondrial derived oxidative stress in embryonic heart (Botting et al., 2020). ROS undergo complex movements during embryonic cardiac development, whereby early ROS levels are increased to stimulate cardiomyocyte differentiation, whereas at later stages, ROS levels are reduced to allow cardiomyocyte maturation by shutting down mPTP (Hom et al., 2011).
Mitochondria participate in apoptosis
Mitochondria play a crucial role in regulating programmed cell death (apoptosis) (Jeong and Seol, 2008; Zhao et al., 2019; Fuchs and Steller, 2011). Mitochondria can mediate apoptosis by releasing cytochrome c (Cyt c), which binds to procaspase-9 and Apaf-1, subsequently activating procaspase-3 and ICAD downstream, leading to uncontrolled cleavage of nuclear DNA and initiating apoptosis (Green and Reed, 1998; Li et al., 2000; Lakhani et al., 2006). Studies have shown that mice lacking Apaf-1 typically exhibit severe developmental defects, and mutation at position 72 of cytochrome c results in similar phenotypes, underscoring the regulation of mitochondrial apoptotic pathways by cytochrome c (Nagasaka et al., 2010). Apoptosis also regulates multiple processes in cardiac development (Schaefer et al., 2004; Sharma et al., 2004; Maguire et al., 2017). Watanabe initially reported that cardiac outflow tract (OFT) shortening and rotation in chicken embryos occur through apoptosis of myocardial cells in the proximal OFT region (Watanabe et al., 1998). Studies in mouse embryonic hearts have confirmed the universality of this phenomenon, observing apoptosis initiation in the proximal OFT region around E12.5, peaking between E13.5 and E14.5, followed by a decline as development progresses (Barbosky et al., 2006). In addition to the OFT region, apoptosis has been detected in ventricular and endocardial cells during development, suggesting a potential role of apoptosis in ventricular morphogenesis (Watanabe et al., 2001).
Mitochondrial permeability transition pore opening and closing in cardiac development
The mitochondrial permeability transition pore (mPTP) is an electrical conduction channel in the inner mitochondrial membrane (Giorgio et al., 2018). Under physiological conditions, mPTP is turned off in mature cardiomyocytes (Beutner et al., 2017). Previous studies have demonstrated that the mPTP channel persists in mouse embryos from E9.5 to E13.5. Closure of the mPTP enhances mitochondrial membrane potential (Δψ) and facilitates OXPHOS, leading to decreased ROS levels in embryonic cardiomyocytes. These findings suggest that mPTP closure acts upstream in redox signaling, thereby stimulating cardiac maturation (Hom et al., 2011). In addition, it was found that heterogeneity in the degree of cardiomyocyte differentiation within the embryonic heart may then be associated with differential regulation of mPTP activity in the ventricular wall (Drenckhahn, 2011). Furthermore, to inhibit the mouse adenine nucleotide transporter 2 (ANT2) gene showed impaired cardiac development and depressed maturation of embryonic cardiomyocytes, which may be mainly related to altered regulation of mitochondrial mPTP (Kokoszka et al., 2016; Levy et al., 2000). In vitro, using cyclosporin A (CsA) promotes cardiomyocyte differentiation from pluripotent stem cells (PSCs) by inhibiting mPTP and activating mitochondrial oxidative metabolism (Cho et al., 2014). All these indicate that mPTP may be involved in regulating embryonic heart development and enhancing cardiomyocyte differentiation.
Fusion and fission of mitochondria in embryonic heart development
Mitochondria are a highly dynamic network system that is constantly remodeling mitochondria and maintaining their homeostasis through the processes of fusion and fission (Ma et al., 2020). Fusion is mediated by mitofusins-1 and mitofusins-2 (MFN1 and MFN2) on the outer membrane together with the inner membrane protein optic atrophy 1 (OPA1) (Adebayo et al., 2021). Fission is controlled by dynamin-related protein 1 (Drp1) and fission protein 1 (Fis1) (Wang et al., 2020). Both maintain dynamic balance, which is essential for the maintenance of normal mitochondrial morphology, distribution, and function (Mu et al., 2020; Shih and Robinson, 2018).
During the development and maturation of cardiomyocytes, there are significant changes in the number and size of mitochondria. To uncover the potential functions of mitochondrial fusion and fission in the process of cardiac development. Researchers utilized Nkx2.5-Cre to perform simultaneous deletion of Mfn1 and Mfn2 in embryonic hearts, resulting in embryonic lethality in experimental mice (Moses et al., 2001; Chen et al., 2011; Chen et al., 2003). Subsequent studies in embryonic stem cells demonstrated that blocking mitochondrial fusion diminishes the capacity of Ca2+ uptake into mitochondria and increases cytoplasmic Ca2+ concentration, leading to cellular demise (Kasahara et al., 2013). Conversely, employing Myh6-Cre to concurrently deactivate Mfn1 and Mfn2 in embryonic hearts from E15.5 to P0 showed no immediate cardiac dysfunction; however, these mice developed dilated cardiomyopathy starting 7 days after birth and all perished before reaching P16 (Papanicolaou et al., 2012). Another recent study also highlighted unbalanced mitochondrial dynamics and mitochondrial bioenergetics in bicuspid aortic valve (BAV) tissues together with reduced expression of Notch1 (Abudupataer et al., 2021). All these evidences support the important role of mitochondrial fusion in the developmental differentiation of cardiomyocytes (Tahrir et al., 2019; Dorn, 2016b; Stanley et al., 2005; Gong et al., 2015; Dorn, 2016a).
Besides, mutations in desmin during early embryonic development led to a significant increase in Drp1, subsequently causing notable cardiac dysfunction (Goldfarb and Dalakas, 2009; Alam et al., 2018). Further investigations using Myh6-Cre in mice to delete the mitochondrial fission protein Drp1 have demonstrated multiple cardiac defects, including reduced heart rate, abnormal electrocardiographic patterns, and decreased left ventricular contraction (Kageyama et al., 2014). All mutant mice succumbed between P9 and P11 (Kageyama et al., 2014). Specific deletion of Drp1 in mouse hearts at 4 weeks postnatally results in cardiac hypertrophy and death within 2 weeks (Ikeda et al., 2015). Conversely, deletion at 8 weeks reveals significant left ventricular enlargement and decreased ejection function, culminating in widespread heart failure 6–7 weeks post-deletion (Song et al., 2017; Song et al., 2015). These results underscore the essential role of mitochondrial fission in early cardiac development, with diminishing demand as maturity progresses (Dorn, 2015).
Furthermore, studies have shown that in addition to the involvement of MAFN2 in mitochondrial fusion to regulate mitochondrial morphology and function (de Brito and Scorrano, 2009; Santel, 2006), this gene has an important role in controlling cell proliferation and apoptosis (Guo et al., 2007). Maternal obesity and diabetes can increase the risk of cardiac disease in developing infants (Brite et al., 2014; Pauliks, 2015; Basu and Garg, 2018). In an animal study in rats, prenatal exposure to a high-fat diet led to an expression increase of the MFN2 protein, which affected mitochondrial viability in the developing fetus; moreover, the high expression of MFN2 depressed cell proliferation through the Ras-Raf-ERK signaling pathway, High expression of MFN2 during cardiac development may contribute to a decrease in the number of cardiomyocytes, secondary hypertrophy, as well as an impairment of diastolic and systolic functions (Chen K. H. et al., 2014; Chandhok et al., 2018; Larsen et al., 2019).
Mitophagy in cardiac development
Mitochondria as the powerhouse play a key role in the cell. As a defense mechanism, mitophagy selectively removes damaged and dysfunctional mitochondria from the cell in order to maintain mitochondrial mass and thus maintain mitochondrial physiological function (Campos et al., 2016). In mouse fetal cardiomyocytes, mitochondria were found to undergo mitochondrial autophagy mediated by PINK1-Mfn2-Parkin complexes (Tahrir et al., 2019). Through the phagocytic mechanism of PINK1-Mfn2-Parkin, the mitochondrial transition from fetal carbohydrate metabolism to normal fatty acid metabolism in postnatal life can be facilitated (Dorn, 2016b). In addition, in mouse embryonic cardiomyocytes, Mfn2 mutation leads to deletion of the PINK1 phosphorylation binding site, which inhibits mitochondrial maturation and leads to metabolic arrest, ultimately causing fatal cardiomyopathy (Gong et al., 2015; Dorn, 2016a). Accordingly, Parkin-mediated mitochondrial autophagy is essential for the maturation of normal perinatal cardiac mitochondrial metabolism.
Ca2+ homeostasis
Mitochondria possess several Ca2+ transport channels, including the Ca2+ uniporter complex, the mitochondrial Na+/Ca2+ exchanger, and the mitochondrial H+/Ca2+ exchanger, which, in conjunction with the endoplasmic reticulum, regulate the stability of mitochondrial Ca2+ levels (Kuster et al., 2010; Song et al., 2018; Berridge et al., 2000). Ca2+ plays a crucial role in cardiac development and maturation, acting as a second messenger involved in nearly all cellular biological processes during early development (Berridge et al., 2003; Créton et al., 1998). The critical role of increased Ca2+ concentration during embryonic development was first recognized during fertilization (Ducibella et al., 2006). Furthermore, mitochondria in the embryonic heart regulate cardiac development by activating the Ca2+-dependent NFAT signaling pathway (Guo et al., 2002; Cao and Chen, 2009). In cardiomyocytes, during cardiac contraction, intracellular Ca2+ accumulates, promoting the interaction between actin and myosin. During cardiac relaxation, intracellular Ca2+ decreases, leading to the decoupling of myofilaments and subsequent relaxation of cardiomyocytes (Bers and Despa, 2006; Aronsen et al., 2013). Therefore, mitochondria are essential for maintaining intracellular Ca2+ stability, which is vital for cardiac development and maturation.
Mitochondria regulate cellular redox state
The ROS levels are tightly regulated by the antioxidant system under physiological conditions (D’Autreaux and Toledano, 2007), In order to protect mitochondria from ROS, the GSH (γ-L-glutamyl-L-cysteinyl-glycine) pool of antioxidants is essential for the protection of mitochondrial function and cell survival (Quintana-Cabrera and Bolaños, 2013; Fahey et al., 1984). Studies have found that GPx-4 (glutathione peroxidase 4) deficient embryonic mice die at E7.5 (Yant et al., 2003). Prxs (peroxiredoxins) are a family of proteins that are very effective in scavenging peroxides (Cox et al., 2009). Whereas Prx III is an important H2O2-eliminating enzyme in mitochondria, intracellular ROS levels were significantly higher in macrophages from Prx III knockout mice, and Prx III depletion induced apoptosis susceptibility (Chang et al., 2004; Li et al., 2007).
In addition, mitochondria maintain cytosolic NAD+/NADH redox homeostasis through malate-aspartate shuttling and glycerophosphate shuttling activities (Broeks et al., 2021; Xiao et al., 2018). NAD+ adjusts the acetylation of mitochondrial proteins by acting as a coenzyme for protein deacetylases such as sirtuins (Chang and Guarente, 2014). Its deficiency decreases sirtuins activity, causing a decrease in placental function and resulting in increased oxidative stress, inflammation, altered placental metabolism, as well as decreased trophoblast viability (Kahmini et al., 2022; Teixeira et al., 2020; Alqarni et al., 2021). Complex I deficiency causes NADH accumulation and reduced NAD+/NADH ratio, depresses SIRT3 activity, resulting in hyperacetylation of mitochondrial proteins in cardiac tissues (Karamanlidis et al., 2013). Whereas mouse SIRT1 knocks die during fetal development or shortly after birth as well as exhibit severe developmental defects, including heart defects (Rodriguez et al., 2013). Consequently, the role of mitochondrial mechanisms in maintaining NAD+/NADH redox homeostasis in coordinating cardiac development cannot be ignored (Hocaoglu and Sieber, 2023).
Conclusion
Accumulating evidence indicates that mitochondria are not only the powerhouses driving the heart’s function, but also play a critical role in regulating mammalian embryonic heart development through non-energetic metabolic processes. This review highlights the regulatory roles of mitochondrial non-energetic metabolism in embryonic heart development, including the structure of the mPTP of mitochondria, fusion, fission and mitochondrial autophagy to maintain its homeostasis, as well as the regulation of ROS production, apoptosis, Ca2+, and cellular redox state, all of which reveal the roles of mitochondria and the potential mechanisms in embryonic heart development, aiming at the early detection, treatment of mitochondrial dysfunction-induced heart defects in the embryonic stage, to provide clues for the development of new clinical applications.
Author contributions
JS: Data curation, Formal Analysis, Investigation, Methodology, Resources, Validation, Visualization, Writing–original draft. YJ: Conceptualization, Data curation, Formal Analysis, Methodology, Resources, Validation, Writing–original draft. SL: Data curation, Formal Analysis, Visualization, Writing–original draft. XL: Resources, Validation, Visualization, Writing–original draft. DZ: Methodology, Project administration, Supervision, Validation, Visualization, Writing–original draft. JW: Conceptualization, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing–review and editing. YQ: Conceptualization, Data curation, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing–review and editing. YL: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was financially supported by the National Natural Science Foundation of China (82270249).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abudupataer, M., Zhu, S., Yan, S., Xu, K., Zhang, J., Luo, S., et al. (2021). Aorta smooth muscle-on-a-chip reveals impaired mitochondrial dynamics as a therapeutic target for aortic aneurysm in bicuspid aortic valve disease. Elife 10, e69310. doi:10.7554/eLife.69310
Adebayo, M., Singh, S., Singh, A. P., and Dasgupta, S. (2021). Mitochondrial fusion and fission: the fine-tune balance for cellular homeostasis. Faseb J. 35 (6), e21620. doi:10.1096/fj.202100067R
Agata, N., Kato, Y., Hamaguchi, S., Namekata, I., and Tanaka, H. (2019). Resistance of fetal Guinea pig ventricular myocardium to hypoxia: maintained intracellular ATP prevents the opening of ATP-sensitive potassium channels. Biol. Pharm. Bull. 42 (2), 268–272. doi:10.1248/bpb.b18-00762
Alam, S., Abdullah, C. S., Aishwarya, R., Miriyala, S., Panchatcharam, M., Peretik, J. M., et al. (2018). Aberrant mitochondrial fission is maladaptive in desmin mutation-induced cardiac proteotoxicity. J. Am. Heart Assoc. 7 (14), e009289. doi:10.1161/JAHA.118.009289
Alqarni, M. H., Foudah, A. I., Muharram, M. M., and Labrou, N. E. (2021). The pleiotropic function of human sirtuins as modulators of metabolic pathways and viral infections. Cells 10 (2), 460. doi:10.3390/cells10020460
Andrieux, P., Chevillard, C., Cunha-Neto, E., and Nunes, J. P. S. (2021). Mitochondria as a cellular hub in infection and inflammation. Int. J. Mol. Sci. 22 (21), 11338. doi:10.3390/ijms222111338
Aronsen, J. M., Swift, F., and Sejersted, O. M. (2013). Cardiac sodium transport and excitation-contraction coupling. J. Mol. Cell Cardiol. 61, 11–19. doi:10.1016/j.yjmcc.2013.06.003
Barbosky, L., Lawrence, D. K., Karunamuni, G., Wikenheiser, J. C., Doughman, Y. Q., Visconti, R. P., et al. (2006). Apoptosis in the developing mouse heart. Dev. Dyn. 235 (9), 2592–2602. doi:10.1002/dvdy.20885
Basu, M., and Garg, V. (2018). Maternal hyperglycemia and fetal cardiac development: clinical impact and underlying mechanisms. Birth Defects Res. 110 (20), 1504–1516. doi:10.1002/bdr2.1435
Becker, L., Kling, E., Schiller, E., Zeh, R., Schrewe, A., Hölter, S. M., et al. (2014). MTO1-deficient mouse model mirrors the human phenotype showing complex I defect and cardiomyopathy. PLoS One 9 (12), e114918. doi:10.1371/journal.pone.0114918
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4 (7), 517–529. doi:10.1038/nrm1155
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1 (1), 11–21. doi:10.1038/35036035
Bers, D. M., and Despa, S. (2006). Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J. Pharmacol. Sci. 100 (5), 315–322. doi:10.1254/jphs.cpj06001x
Beutner, G., Alavian, K. N., Jonas, E. A., and Porter, G. A. (2017). Erratum to: the mitochondrial permeability transition pore and ATP synthase. Handb. Exp. Pharmacol. 240, 489. doi:10.1007/164_2016_87
Birkedal, R., Laasmaa, M., Branovets, J., and Vendelin, M. (2022). Ontogeny of cardiomyocytes: ultrastructure optimization to meet the demand for tight communication in excitation-contraction coupling and energy transfer. Philos. Trans. R. Soc. Lond B Biol. Sci. 377 (1864), 20210321. doi:10.1098/rstb.2021.0321
Birket, M. J., Casini, S., Kosmidis, G., Elliott, D. A., Gerencser, A. A., Baartscheer, A., et al. (2013). PGC-1α and reactive oxygen species regulate human embryonic stem cell-derived cardiomyocyte function. Stem Cell Rep. 1 (6), 560–574. doi:10.1016/j.stemcr.2013.11.008
Bonekamp, N. A., and Larsson, N. G. (2018). SnapShot: mitochondrial nucleoid. Cell 172 (1-2), 388–388.e381. doi:10.1016/j.cell.2017.12.039
Botting, K. J., Skeffington, K. L., Niu, Y., Allison, B. J., Brain, K. L., Itani, N., et al. (2020). Translatable mitochondria-targeted protection against programmed cardiovascular dysfunction. Sci. Adv. 6 (34), eabb1929. doi:10.1126/sciadv.abb1929
Brambilla, A., Olivotto, I., Favilli, S., Spaziani, G., Passantino, S., Procopio, E., et al. (2020). Impact of cardiovascular involvement on the clinical course of paediatric mitochondrial disorders. Orphanet J. Rare Dis. 15 (1), 196. doi:10.1186/s13023-020-01466-w
Bray, A. W., and Ballinger, S. W. (2017). Mitochondrial DNA mutations and cardiovascular disease. Curr. Opin. Cardiol. 32 (3), 267–274. doi:10.1097/HCO.0000000000000383
Brite, J., Laughon, S. K., Troendle, J., and Mills, J. (2014). Maternal overweight and obesity and risk of congenital heart defects in offspring. Int. J. Obes. (Lond) 38 (6), 878–882. doi:10.1038/ijo.2013.244
Broeks, M. H., van Karnebeek, C. D. M., Wanders, R. J. A., Jans, J. J. M., and Verhoeven-Duif, N. M. (2021). Inborn disorders of the malate aspartate shuttle. J. Inherit. Metab. Dis. 44 (4), 792–808. doi:10.1002/jimd.12402
Bruneau, B. G. (2020). The developing heart: from the Wizard of Oz to congenital heart disease. Development 147 (21), dev194233. doi:10.1242/dev.194233
Buckingham, M., Meilhac, S., and Zaffran, S. (2005). Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 6 (11), 826–835. doi:10.1038/nrg1710
Buggisch, M., Ateghang, B., Ruhe, C., Strobel, C., Lange, S., Wartenberg, M., et al. (2007). Stimulation of ES-cell-derived cardiomyogenesis and neonatal cardiac cell proliferation by reactive oxygen species and NADPH oxidase. J. Cell Sci. 120 (Pt 5), 885–894. doi:10.1242/jcs.03386
Burton, G. J. (2009). Oxygen, the Janus gas; its effects on human placental development and function. J. Anat. 215 (1), 27–35. doi:10.1111/j.1469-7580.2008.00978.x
Cámara, Y., Asin-Cayuela, J., Park, C. B., Metodiev, M. D., Shi, Y., Ruzzenente, B., et al. (2011). MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab. 13 (5), 527–539. doi:10.1016/j.cmet.2011.04.002
Campos, J. C., Bozi, L. H., Bechara, L. R., Lima, V. M., and Ferreira, J. C. B. (2016). Mitochondrial quality control in cardiac diseases. Front. Physiol. 7, 479. doi:10.3389/fphys.2016.00479
Cao, X., and Chen, Y. (2009). Mitochondria and calcium signaling in embryonic development. Semin. Cell Dev. Biol. 20 (3), 337–345. doi:10.1016/j.semcdb.2008.12.014
Cerritelli, S. M., Frolova, E. G., Feng, C., Grinberg, A., Love, P. E., and Crouch, R. J. (2003). Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol. Cell 11 (3), 807–815. doi:10.1016/s1097-2765(03)00088-1
Chandel, N. S. (2014). Mitochondria as signaling organelles. BMC Biol. 12, 34. doi:10.1186/1741-7007-12-34
Chandhok, G., Lazarou, M., and Neumann, B. (2018). Structure, function, and regulation of mitofusin-2 in health and disease. Biol. Rev. Camb Philos. Soc. 93 (2), 933–949. doi:10.1111/brv.12378
Chang, H. C., and Guarente, L. (2014). SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 25 (3), 138–145. doi:10.1016/j.tem.2013.12.001
Chang, T. S., Cho, C. S., Park, S., Yu, S., Kang, S. W., and Rhee, S. G. (2004). Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J. Biol. Chem. 279 (40), 41975–41984. doi:10.1074/jbc.M407707200
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160 (2), 189–200. doi:10.1083/jcb.200211046
Chen, K. H., Dasgupta, A., Ding, J., Indig, F. E., Ghosh, P., and Longo, D. L. (2014). Role of mitofusin 2 (Mfn2) in controlling cellular proliferation. Faseb J. 28 (1), 382–394. doi:10.1096/fj.13-230037
Chen, Y., Liu, Y., and Dorn, G. W. (2011). Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 109 (12), 1327–1331. doi:10.1161/CIRCRESAHA.111.258723
Chen, Y., Sparks, M., Bhandari, P., Matkovich, S. J., and Dorn, G. W. (2014). Mitochondrial genome linearization is a causative factor for cardiomyopathy in mice and Drosophila. Antioxid. Redox Signal 21 (14), 1949–1959. doi:10.1089/ars.2013.5432
Chen, Z., Zhu, S., Wang, H., Wang, L., Zhang, J., Gu, Y., et al. (2021). PTPMT1 is required for embryonic cardiac cardiolipin biosynthesis to regulate mitochondrial morphogenesis and heart development. Circulation 144 (5), 403–406. doi:10.1161/CIRCULATIONAHA.121.054768
Cheong, A., Archambault, D., Degani, R., Iverson, E., Tremblay, K. D., and Mager, J. (2020). Nuclear-encoded mitochondrial ribosomal proteins are required to initiate gastrulation. Development 147 (10), dev188714. doi:10.1242/dev.188714
Cho, S. W., Park, J. S., Heo, H. J., Song, S., Kim, I., et al. (2014). Dual modulation of the mitochondrial permeability transition pore and redox signaling synergistically promotes cardiomyocyte differentiation from pluripotent stem cells. J. Am. Heart Assoc. 3 (2), e000693. doi:10.1161/JAHA.113.000693
Chung, S., Dzeja, P. P., Faustino, R. S., Perez-Terzic, C., Behfar, A., and Terzic, A. (2007). Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat. Clin. Pract. Cardiovasc Med. 4 (Suppl. 1), S60–S67. doi:10.1038/ncpcardio0766
Cox, A. G., Winterbourn, C. C., and Hampton, M. B. (2009). Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 425 (2), 313–325. doi:10.1042/BJ20091541
Créton, R., Speksnijder, J. E., and Jaffe, L. F. (1998). Patterns of free calcium in zebrafish embryos. J. Cell Sci. 111 (Pt 12), 1613–1622. doi:10.1242/jcs.111.12.1613
D’Autreaux, B., and Toledano, M. B. (2007). ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 8 (10), 813–824. doi:10.1038/nrm2256
David, H., Bózner, A., Meyer, R., and Wassilew, G. (1981). Pre- and postnatal development and ageing of the heart. Ultrastructural results and quantitative data. Exp. Pathol. Suppl. 7 (7), 1–176.
de Brito, O. M., and Scorrano, L. (2009). Mitofusin-2 regulates mitochondrial and endoplasmic reticulum morphology and tethering: the role of Ras. Mitochondrion 9 (3), 222–226. doi:10.1016/j.mito.2009.02.005
Dennery, P. A. (2010). Oxidative stress in development: nature or nurture? Free Radic. Biol. Med. 49 (7), 1147–1151. doi:10.1016/j.freeradbiomed.2010.07.011
Dorn, G. W. (2015). Gone fission…: diverse consequences of cardiac Drp1 deficiency. Circ. Res. 116 (2), 225–228. doi:10.1161/CIRCRESAHA.114.305672
Dorn, G. W. (2016a). Parkin-dependent mitophagy in the heart. J. Mol. Cell Cardiol. 95, 42–49. doi:10.1016/j.yjmcc.2015.11.023
Dorn, G. W. (2016b). Jurassic PARK2: You eat your mitochondria, and you are what your mitochondria eat. Autophagy 12 (3), 610–611. doi:10.1080/15548627.2016.1143210
Dorn, G. W., Vega, R. B., and Kelly, D. P. (2015). Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 29 (19), 1981–1991. doi:10.1101/gad.269894.115
Drenckhahn, J. D. (2011). Heart development: mitochondria in command of cardiomyocyte differentiation. Dev. Cell 21 (3), 392–393. doi:10.1016/j.devcel.2011.08.021
Ducibella, T., Schultz, R. M., and Ozil, J. P. (2006). Role of calcium signals in early development. Semin. Cell Dev. Biol. 17 (2), 324–332. doi:10.1016/j.semcdb.2006.02.010
Fahey, R. C., Newton, G. L., Arrick, B., Overdank-Bogart, T., and Aley, S. B. (1984). Entamoeba histolytica: a eukaryote without glutathione metabolism. Science 224 (4644), 70–72. doi:10.1126/science.6322306
Filograna, R., Mennuni, M., Alsina, D., and Larsson, N. G. (2021). Mitochondrial DNA copy number in human disease: the more the better? FEBS Lett. 595 (8), 976–1002. doi:10.1002/1873-3468.14021
Finnemore, A., and Groves, A. (2015). Physiology of the fetal and transitional circulation. Semin. Fetal Neonatal Med. 20 (4), 210–216. doi:10.1016/j.siny.2015.04.003
Fuchs, Y., and Steller, H. (2011). Programmed cell death in animal development and disease. Cell 147 (4), 742–758. doi:10.1016/j.cell.2011.10.033
Gao, F., Liang, T., Lu, Y. W., Fu, X., Dong, X., Pu, L., et al. (2023). A defect in mitochondrial protein translation influences mitonuclear communication in the heart. Nat. Commun. 14 (1), 1595. doi:10.1038/s41467-023-37291-5
Gao, F., and Zhang, J. (2018). Mitochondrial quality control and neurodegenerative diseases. Neuronal Signal 2 (4), Ns20180062. doi:10.1042/NS20180062
Garry, D. J., and Olson, E. N. (2006). A common progenitor at the heart of development. Cell 127 (6), 1101–1104. doi:10.1016/j.cell.2006.11.031
Giorgio, V., Guo, L., Bassot, C., Petronilli, V., and Bernardi, P. (2018). Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 70, 56–63. doi:10.1016/j.ceca.2017.05.004
Goffart, S., von Kleist-Retzow, J. C., and Wiesner, R. J. (2004). Regulation of mitochondrial proliferation in the heart: power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc Res. 64 (2), 198–207. doi:10.1016/j.cardiores.2004.06.030
Goldfarb, L. G., and Dalakas, M. C. (2009). Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J. Clin. Invest. 119 (7), 1806–1813. doi:10.1172/JCI38027
Gong, G., Song, M., Csordas, G., Kelly, D. P., Matkovich, S. J., and Dorn, G. W. (2015). Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350 (6265), aad2459. doi:10.1126/science.aad2459
Green, D. R., and Reed, J. C. (1998). Mitochondria and apoptosis. Science 281 (5381), 1309–1312. doi:10.1126/science.281.5381.1309
Guo, L., Nakamura, K., Lynch, J., Opas, M., Olson, E. N., Agellon, L. B., et al. (2002). Cardiac-specific expression of calcineurin reverses embryonic lethality in calreticulin-deficient mouse. J. Biol. Chem. 277 (52), 50776–50779. doi:10.1074/jbc.M209900200
Guo, X., Chen, K. H., Guo, Y., Liao, H., Tang, J., and Xiao, R. P. (2007). Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ. Res. 101 (11), 1113–1122. doi:10.1161/CIRCRESAHA.107.157644
Gupta, A., and Becker, T. (2021). Mechanisms and pathways of mitochondrial outer membrane protein biogenesis. Biochim. Biophys. Acta Bioenerg. 1862 (1), 148323. doi:10.1016/j.bbabio.2020.148323
Gut, P., and Verdin, E. (2013). The nexus of chromatin regulation and intermediary metabolism. Nature 502 (7472), 489–498. doi:10.1038/nature12752
Harding, A. E. (1991). Neurological disease and mitochondrial genes. Trends Neurosci. 14 (4), 132–138. doi:10.1016/0166-2236(91)90081-5
Heidari, M. M., Khatami, M., Kamalipour, A., Kalantari, M., Movahed, M., Emmamy, M. H., et al. (2022). Mitochondrial mutations in protein coding genes of respiratory chain including complexes IV, V, and mt-tRNA genes are associated risk factors for congenital heart disease. Excli J. 21, 1306–1330. doi:10.17179/excli2022-5298
Hellgren, K. T., Premanandhan, H., Quinn, C. J., Trafford, A. W., and Galli, G. L. J. (2021). Sex-dependent effects of developmental hypoxia on cardiac mitochondria from adult murine offspring. Free Radic. Biol. Med. 162, 490–499. doi:10.1016/j.freeradbiomed.2020.11.004
Hocaoglu, H., and Sieber, M. (2023). Mitochondrial respiratory quiescence: a new model for examining the role of mitochondrial metabolism in development. Semin. Cell Dev. Biol. 138, 94–103. doi:10.1016/j.semcdb.2022.03.040
Holmgren, D., Wåhlander, H., Eriksson, B. O., Oldfors, A., Holme, E., and Tulinius, M. (2003). Cardiomyopathy in children with mitochondrial disease; clinical course and cardiological findings. Eur. Heart J. 24 (3), 280–288. doi:10.1016/s0195-668x(02)00387-1
Hom, J. R., Quintanilla, R. A., Hoffman, D. L., de Mesy Bentley, K. L., Molkentin, J. D., Sheu, S. S., et al. (2011). The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Dev. Cell 21 (3), 469–478. doi:10.1016/j.devcel.2011.08.008
Ikeda, Y., Shirakabe, A., Maejima, Y., Zhai, P., Sciarretta, S., Toli, J., et al. (2015). Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 116 (2), 264–278. doi:10.1161/CIRCRESAHA.116.303356
Ingraham, C. A., Burwell, L. S., Skalska, J., Brookes, P. S., Howell, R. L., Sheu, S. S., et al. (2009). NDUFS4: creation of a mouse model mimicking a Complex I disorder. Mitochondrion 9 (3), 204–210. doi:10.1016/j.mito.2009.02.001
Jeong, S. Y., and Seol, D. W. (2008). The role of mitochondria in apoptosis. BMB Rep. 41 (1), 11–22. doi:10.5483/bmbrep.2008.41.1.011
Kageyama, Y., Hoshijima, M., Seo, K., Bedja, D., Sysa-Shah, P., Andrabi, S. A., et al. (2014). Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. Embo J. 33 (23), 2798–2813. doi:10.15252/embj.201488658
Kahmini, F. R., Ghaleh, H. D., and Shahgaldi, S. (2022). Sirtuins: subtle Regulators involved in Convoluted mechanisms of pregnancy. Cell Physiol. Biochem. 56 (6), 644–662. doi:10.33594/000000588
Karamanlidis, G., Lee, C. F., Garcia-Menendez, L., Kolwicz, S. C., Suthammarak, W., Gong, G., et al. (2013). Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 18 (2), 239–250. doi:10.1016/j.cmet.2013.07.002
Kasahara, A., Cipolat, S., Chen, Y., Dorn, G. W., and Scorrano, L. (2013). Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science 342 (6159), 734–737. doi:10.1126/science.1241359
Kim, H. D., Kim, D. J., Lee, I. J., Rah, B. J., Sawa, Y., and Schaper, J. (1992). Human fetal heart development after mid-term: morphometry and ultrastructural study. J. Mol. Cell Cardiol. 24 (9), 949–965. doi:10.1016/0022-2828(92)91862-y
Kirby, M. L. (2002). Molecular embryogenesis of the heart. Pediatr. Dev. Pathol. 5 (6), 516–543. doi:10.1007/s10024-002-0004-2
Kokoszka, J. E., Waymire, K. G., Flierl, A., Sweeney, K. M., Angelin, A., MacGregor, G. R., et al. (2016). Deficiency in the mouse mitochondrial adenine nucleotide translocator isoform 2 gene is associated with cardiac noncompaction. Biochim. Biophys. Acta 1857 (8), 1203–1212. doi:10.1016/j.bbabio.2016.03.026
Kuster, G. M., Lancel, S., Zhang, J., Communal, C., Trucillo, M. P., Lim, C. C., et al. (2010). Redox-mediated reciprocal regulation of SERCA and Na+-Ca2+ exchanger contributes to sarcoplasmic reticulum Ca2+ depletion in cardiac myocytes. Free Radic. Biol. Med. 48 (9), 1182–1187. doi:10.1016/j.freeradbiomed.2010.01.038
Lai, L., Leone, T. C., Zechner, C., Schaeffer, P. J., Kelly, S. M., Flanagan, D. P., et al. (2008). Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 22 (14), 1948–1961. doi:10.1101/gad.1661708
Lakhani, S. A., Masud, A., Kuida, K., Porter, G. A., Booth, C. J., Mehal, W. Z., et al. (2006). Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311 (5762), 847–851. doi:10.1126/science.1115035
Larsen, T. D., Sabey, K. H., Knutson, A. J., Gandy, T. C. T., Louwagie, E. J., Lauterboeck, L., et al. (2019). Diabetic pregnancy and Maternal high-fat diet Impair mitochondrial dynamism in the developing fetal rat heart by Sex-specific mechanisms. Int. J. Mol. Sci. 20 (12), 3090. doi:10.3390/ijms20123090
Levy, S. E., Chen, Y. S., Graham, B. H., and Wallace, D. C. (2000). Expression and sequence analysis of the mouse adenine nucleotide translocase 1 and 2 genes. Gene 254 (1-2), 57–66. doi:10.1016/s0378-1119(00)00252-3
Li, K., Li, Y., Shelton, J. M., Richardson, J. A., Spencer, E., Chen, Z. J., et al. (2000). Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell 101 (4), 389–399. doi:10.1016/s0092-8674(00)80849-1
Li, L., Shoji, W., Takano, H., Nishimura, N., Aoki, Y., Takahashi, R., et al. (2007). Increased susceptibility of MER5 (peroxiredoxin III) knockout mice to LPS-induced oxidative stress. Biochem. Biophys. Res. Commun. 355 (3), 715–721. doi:10.1016/j.bbrc.2007.02.022
Lopaschuk, G. D., and Jaswal, J. S. (2010). Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc Pharmacol. 56 (2), 130–140. doi:10.1097/FJC.0b013e3181e74a14
Lyra-Leite, D. M., Petersen, A. P., Ariyasinghe, N. R., and McCain, M. L. (2021). Mitochondrial architecture in cardiac myocytes depends on cell shape and matrix rigidity. J. Mol. Cell Cardiol. 150, 32–43. doi:10.1016/j.yjmcc.2020.10.004
Ma, Y., Wang, L., and Jia, R. (2020). The role of mitochondrial dynamics in human cancers. Am. J. Cancer Res. 10 (5), 1278–1293.
Maguire, J. J., Tyurina, Y. Y., Mohammadyani, D., Kapralov, A. A., Anthonymuthu, T. S., Qu, F., et al. (2017). Known unknowns of cardiolipin signaling: the best is yet to come. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862 (1), 8–24. doi:10.1016/j.bbalip.2016.08.001
Maroli, G., and Braun, T. (2021). The long and winding road of cardiomyocyte maturation. Cardiovasc Res. 117 (3), 712–726. doi:10.1093/cvr/cvaa159
May-Panloup, P., Boguenet, M., Hachem, H. E., Bouet, P. E., and Reynier, P. (2021). Embryo and its mitochondria. Antioxidants (Basel) 10 (2), 139. doi:10.3390/antiox10020139
Moses, K. A., DeMayo, F., Braun, R. M., Reecy, J. L., and Schwartz, R. J. (2001). Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis 31 (4), 176–180. doi:10.1002/gene.10022
Mu, J. K., Li, Y. Q., Shi, T. T., Yu, L. P., Yang, Y. Q., Gu, W., et al. (2020). Remedying the mitochondria to cure human diseases by Natural products. Oxid. Med. Cell Longev. 2020, 5232614. doi:10.1155/2020/5232614
Murray, T. V., Ahmad, A., and Brewer, A. C. (2014). Reactive oxygen at the heart of metabolism. Trends Cardiovasc Med. 24 (3), 113–120. doi:10.1016/j.tcm.2013.09.003
Na, L., Wartenberg, M., Nau, H., Hescheler, J., and Sauer, H. (2003). Anticonvulsant valproic acid inhibits cardiomyocyte differentiation of embryonic stem cells by increasing intracellular levels of reactive oxygen species. Birth Defects Res. A Clin. Mol. Teratol. 67 (3), 174–180. doi:10.1002/bdra.10030
Nagasaka, A., Kawane, K., Yoshida, H., and Nagata, S. (2010). Apaf-1-independent programmed cell death in mouse development. Cell Death Differ. 17 (6), 931–941. doi:10.1038/cdd.2009.186
Ngo, H. B., Lovely, G. A., Phillips, R., and Chan, D. C. (2014). Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat. Commun. 5, 3077. doi:10.1038/ncomms4077
Nunnari, J., and Suomalainen, A. (2012). Mitochondria: in sickness and in health. Cell 148 (6), 1145–1159. doi:10.1016/j.cell.2012.02.035
Pagliarini, D. J., Calvo, S. E., Chang, B., Sheth, S. A., Vafai, S. B., Ong, S. E., et al. (2008). A mitochondrial protein compendium elucidates complex I disease biology. Cell 134 (1), 112–123. doi:10.1016/j.cell.2008.06.016
Papanicolaou, K. N., Kikuchi, R., Ngoh, G. A., Coughlan, K. A., Dominguez, I., Stanley, W. C., et al. (2012). Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ. Res. 111 (8), 1012–1026. doi:10.1161/CIRCRESAHA.112.274142
Patterson, A. J., Xiao, D., Xiong, F., Dixon, B., and Zhang, L. (2012). Hypoxia-derived oxidative stress mediates epigenetic repression of PKCε gene in foetal rat hearts. Cardiovasc Res. 93 (2), 302–310. doi:10.1093/cvr/cvr322
Pauliks, L. B. (2015). The effect of pregestational diabetes on fetal heart function. Expert Rev. Cardiovasc Ther. 13 (1), 67–74. doi:10.1586/14779072.2015.988141
Pohjoismäki, J. L., and Goffart, S. (2017). The role of mitochondria in cardiac development and protection. Free Radic. Biol. Med. 106, 345–354. doi:10.1016/j.freeradbiomed.2017.02.032
Puente, B. N., Kimura, W., Muralidhar, S. A., Moon, J., Amatruda, J. F., Phelps, K. L., et al. (2014). The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157 (3), 565–579. doi:10.1016/j.cell.2014.03.032
Quintana-Cabrera, R., and Bolaños, J. P. (2013). Glutathione and γ-glutamylcysteine in the antioxidant and survival functions of mitochondria. Biochem. Soc. Trans. 41 (1), 106–110. doi:10.1042/BST20120252
Robin, E. D., and Wong, R. (1988). Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. J. Cell Physiol. 136 (3), 507–513. doi:10.1002/jcp.1041360316
Rodriguez, R. M., Fernandez, A. F., and Fraga, M. F. (2013). Role of sirtuins in stem cell differentiation. Genes Cancer 4 (3-4), 105–111. doi:10.1177/1947601913479798
Saito, T., Hamano, K., and Sadoshima, J. (2021). Molecular mechanisms and clinical implications of multiple forms of mitophagy in the heart. Cardiovasc Res. 117 (14), 2730–2741. doi:10.1093/cvr/cvaa340
Saito, T., and Sadoshima, J. (2015). Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ. Res. 116 (8), 1477–1490. doi:10.1161/CIRCRESAHA.116.303790
Sánchez-Díaz, M., Nicolás-Ávila, J., Cordero, M. D., and Hidalgo, A. (2020). Mitochondrial Adaptations in the Growing heart. Trends Endocrinol. Metab. 31 (4), 308–319. doi:10.1016/j.tem.2020.01.006
Santel, A. (2006). Get the balance right: mitofusins roles in health and disease. Biochim. Biophys. Acta 1763 (5-6), 490–499. doi:10.1016/j.bbamcr.2006.02.004
Sauer, H., Rahimi, G., Hescheler, J., and Wartenberg, M. (1999). Effects of electrical fields on cardiomyocyte differentiation of embryonic stem cells. J. Cell Biochem. 75 (4), 710–723. doi:10.1002/(sici)1097-4644(19991215)75:4<710::aid-jcb16>3.0.co;2-z
Sauer, H., and Wartenberg, M. (2005). Reactive oxygen species as signaling molecules in cardiovascular differentiation of embryonic stem cells and tumor-induced angiogenesis. Antioxid. Redox Signal 7 (11-12), 1423–1434. doi:10.1089/ars.2005.7.1423
Schaefer, K. S., Doughman, Y. Q., Fisher, S. A., and Watanabe, M. (2004). Dynamic patterns of apoptosis in the developing chicken heart. Dev. Dyn. 229 (3), 489–499. doi:10.1002/dvdy.10463
Semenza, G. L. (2007). Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem. J. 405 (1), 1–9. doi:10.1042/BJ20070389
Shadel, G. S., and Horvath, T. L. (2015). Mitochondrial ROS signaling in organismal homeostasis. Cell 163 (3), 560–569. doi:10.1016/j.cell.2015.10.001
Shao, D., Oka, S., Brady, C. D., Haendeler, J., Eaton, P., and Sadoshima, J. (2012). Redox modification of cell signaling in the cardiovascular system. J. Mol. Cell Cardiol. 52 (3), 550–558. doi:10.1016/j.yjmcc.2011.09.009
Sharma, P. R., Anderson, R. H., Copp, A. J., and Henderson, D. J. (2004). Spatiotemporal analysis of programmed cell death during mouse cardiac septation. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 277 (2), 355–369. doi:10.1002/ar.a.20006
Shih, E. K., and Robinson, M. B. (2018). Role of Astrocytic mitochondria in Limiting ischemic brain injury? Physiol. (Bethesda) 33 (2), 99–112. doi:10.1152/physiol.00038.2017
Smith, R. A., Adlam, V. J., Blaikie, F. H., Manas, A. R. B., Porteous, C. M., James, A. M., et al. (2008). Mitochondria-targeted antioxidants in the treatment of disease. Ann. N. Y. Acad. Sci. 1147, 105–111. doi:10.1196/annals.1427.003
Smith, R. A., and Murphy, M. P. (2010). Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 1201, 96–103. doi:10.1111/j.1749-6632.2010.05627.x
Song, J., Yang, R., Yang, J., and Zhou, L. (2018). Mitochondrial dysfunction-associated Arrhythmogenic substrates in diabetes Mellitus. Front. Physiol. 9, 1670. doi:10.3389/fphys.2018.01670
Song, M., Franco, A., Fleischer, J. A., Zhang, L., and Dorn, G. W. (2017). Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial Senescence. Cell Metab. 26 (6), 872–883. doi:10.1016/j.cmet.2017.09.023
Song, M., Mihara, K., Chen, Y., Scorrano, L., and Dorn, G. W. (2015). Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 21 (2), 273–286. doi:10.1016/j.cmet.2014.12.011
Stanley, E. G., Biben, C., Elefanty, A., Barnett, L., Koentgen, F., Robb, L., et al. (2002). Efficient Cre-mediated deletion in cardiac progenitor cells conferred by a 3'UTR-ires-Cre allele of the homeobox gene Nkx2-5. Int. J. Dev. Biol. 46 (4), 431–439.
Stanley, W. C., Recchia, F. A., and Lopaschuk, G. D. (2005). Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 85 (3), 1093–1129. doi:10.1152/physrev.00006.2004
Tahrir, F. G., Langford, D., Amini, S., Mohseni Ahooyi, T., and Khalili, K. (2019). Mitochondrial quality control in cardiac cells: mechanisms and role in cardiac cell injury and disease. J. Cell Physiol. 234 (6), 8122–8133. doi:10.1002/jcp.27597
Tan, C. M. J., and Lewandowski, A. J. (2020). The transitional heart: from early embryonic and fetal development to neonatal life. Fetal Diagn Ther. 47 (5), 373–386. doi:10.1159/000501906
Tanaka, M., Chen, Z., Bartunkova, S., Yamasaki, N., and Izumo, S. (1999). The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development 126 (6), 1269–1280. doi:10.1242/dev.126.6.1269
Tanimura, T., and Shepard, T. H. (1970). Glucose metabolism by rat embryos in vitro. Proc. Soc. Exp. Biol. Med. 135 (1), 51–54. doi:10.3181/00379727-135-34985
Tann, A. W., Boldogh, I., Meiss, G., Qian, W., Van Houten, B., Mitra, S., et al. (2011). Apoptosis induced by persistent single-strand breaks in mitochondrial genome: critical role of EXOG (5’-EXO/endonuclease) in their repair. J. Biol. Chem. 286 (37), 31975–31983. doi:10.1074/jbc.M110.215715
Teixeira, C. S. S., Cerqueira, N., Gomes, P., and Sousa, S. F. (2020). A molecular Perspective on sirtuin activity. Int. J. Mol. Sci. 21 (22), 8609. doi:10.3390/ijms21228609
Thompson, L. P., and Al-Hasan, Y. (2012). Impact of oxidative stress in fetal programming. J. Pregnancy 2012, 582748. doi:10.1155/2012/582748
Vega, R. B., Horton, J. L., and Kelly, D. P. (2015). Maintaining ancient organelles: mitochondrial biogenesis and maturation. Circ. Res. 116 (11), 1820–1834. doi:10.1161/CIRCRESAHA.116.305420
Vogel, F., Bornhövd, C., Neupert, W., and Reichert, A. S. (2006). Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell Biol. 175 (2), 237–247. doi:10.1083/jcb.200605138
Wang, Y., Dai, X., Li, H., Jiang, H., Zhou, J., Zhang, S., et al. (2020). The role of mitochondrial dynamics in disease. MedComm 4 (6), e462. doi:10.1002/mco2.462
Watanabe, M., Choudhry, A., Berlan, M., Singal, A., Siwik, E., Mohr, S., et al. (1998). Developmental remodeling and shortening of the cardiac outflow tract involves myocyte programmed cell death. Development 125 (19), 3809–3820. doi:10.1242/dev.125.19.3809
Watanabe, M., Jafri, A., and Fisher, S. A. (2001). Apoptosis is required for the proper formation of the ventriculo-arterial connections. Dev. Biol. 240 (1), 274–288. doi:10.1006/dbio.2001.0466
Wei, H., and Cong, X. (2018). The effect of reactive oxygen species on cardiomyocyte differentiation of pluripotent stem cells. Free Radic. Res. 52 (2), 150–158. doi:10.1080/10715762.2017.1420184
Wu, W., He, J., and Shao, X. (2020). Incidence and mortality trend of congenital heart disease at the global, regional, and national level, 1990-2017. Med. Baltim. 99 (23), e20593. doi:10.1097/MD.0000000000020593
Xiao, W., Wang, R. S., Handy, D. E., and Loscalzo, J. (2018). NAD(H) and NADP(H) redox Couples and cellular energy metabolism. Antioxid. Redox Signal 28 (3), 251–272. doi:10.1089/ars.2017.7216
Yant, L. J., Ran, Q., Rao, L., Van Remmen, H., Shibatani, T., Belter, J. G., et al. (2003). The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 34 (4), 496–502. doi:10.1016/s0891-5849(02)01360-6
Yen, H. C., Tang, Y. C., Chen, F. Y., Chen, S. W., and Majima, H. J. (2005). Enhancement of cisplatin-induced apoptosis and caspase 3 activation by depletion of mitochondrial DNA in a human osteosarcoma cell line. Ann. N. Y. Acad. Sci. 1042, 516–522. doi:10.1196/annals.1338.047
Zhang, D., Li, Y., Heims-Waldron, D., Bezzerides, V., Guatimosim, S., Guo, Y., et al. (2018). Mitochondrial cardiomyopathy caused by Elevated reactive oxygen species and impaired cardiomyocyte proliferation. Circ. Res. 122 (1), 74–87. doi:10.1161/CIRCRESAHA.117.311349
Zhang, J., Guan, Z., Murphy, A. N., Wiley, S. E., Perkins, G. A., Worby, C. A., et al. (2011). Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 13 (6), 690–700. doi:10.1016/j.cmet.2011.04.007
Zhao, K., Huang, X., Zhao, W., Lu, B., and Yang, Z. (2022). LONP1-mediated mitochondrial quality control safeguards metabolic shifts in heart development. Development 149 (6), dev200458. doi:10.1242/dev.200458
Zhao, Q., Sun, Q., Zhou, L., Liu, K., and Jiao, K. (2019). Complex regulation of mitochondrial function during cardiac development. J. Am. Heart Assoc. 8 (13), e012731. doi:10.1161/JAHA.119.012731
Zhu, X., Ding, S., Li, H., Zhang, Z., Xu, L., Wu, J., et al. (2020). Disruption of histamine/H(1)R signaling pathway represses cardiac differentiation and maturation of human induced pluripotent stem cells. Stem Cell Res. Ther. 11 (1), 27. doi:10.1186/s13287-020-1551-z
Keywords: mitochondria, cardiac development, embryo, non-energetic function, ROS
Citation: Shi J, Jin Y, Lin S, Li X, Zhang D, Wu J, Qi Y and Li Y (2024) Mitochondrial non-energetic function and embryonic cardiac development. Front. Cell Dev. Biol. 12:1475603. doi: 10.3389/fcell.2024.1475603
Received: 04 August 2024; Accepted: 20 September 2024;
Published: 07 October 2024.
Edited by:
Guojun Zhao, The Sixth Affiliated Hospital of Guangzhou Medical University, ChinaReviewed by:
Kameswara Rao Badri, Morehouse School of Medicine, United StatesYanguo Xin, Capital Medical University, China
Copyright © 2024 Shi, Jin, Lin, Li, Zhang, Wu, Qi and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinlin Wu, MzczMDUzNzg1QHFxLmNvbQ==; Yan Qi, Nzk0Nzc5Mzc0QHFxLmNvbQ==; Yifei Li, bGl5Zndjc2hAc2N1LmVkdS5jbg==
†These authors have contributed equally to this work