Abstract
Introduction:
Retinal dehydrogenase 12 (RDH12) is a photoreceptor NADPH-dependent retinal reductase enzyme, converting all-trans-retinal to all-trans-retinol. Heterozygous variants in RDH12 cause a rare autosomal dominant (AD) retinitis pigmentosa.
Methods:
As no disease models exist, we generated human induced pluripotent stem cell-derived retinal organoids (RO) from an RDH12-AD patient (with pathogenic variant c.759del, p.Phe254Leufs*24) alongside a healthy, unaffected control.
Results:
RDH12-AD RO exhibited correct localization of RDH12 to the photoreceptor inner segments up to week 44; however, transmission electron microscopy at week 37 showed that photoreceptors were less abundant and shorter in length. Visual cone function, retinol biosynthesis, and the vitamin A pathway were also highly disrupted at week 44.
Discussion:
Our study is the first to describe an RDH12-AD disease model with pathology at later stages of photoreceptor differentiation, in keeping with the milder disease course seen in humans. It provides insights into the etiology and possible targets for future therapeutic development.
1 Introduction
Inherited retinal diseases (IRDs) are a genetically and heterogeneous group of non-progressive and progressive sight loss disorders. Among the progressive group, the severity of the clinical phenotype can vary from a severe early onset Leber congenital amaurosis (LCA) to a mild adult-onset retinitis pigmentosa arising from variants in the same causative gene (Georgiou et al., 2021). RDH12 is a key example; ∼80 biallelic pathogenic variants account for up to 10% of LCA (Sarkar and Moosajee, 2019), but four rare cases of autosomal dominant (AD) late-onset retinitis pigmentosa (RP) have been reported (Fingert et al., 2008; Muthiah et al., 2022; Sarkar et al., 2020).
RDH12 encodes the retinal dehydrogenase 12 protein, a NADPH-dependent retinal reductase enzyme (Sarkar and Moosajee, 2019). Localized to the inner segment of photoreceptors (Kurth et al., 2007), RDH12 is involved in the visual cycle. In photoreceptor outer segments, rhodopsin interacts with 11-cis-retinal, and its photoactivation transforms it into all-trans-retinal. Most all-trans-retinal is reduced into all-trans-retinol by RDH8 in photoreceptor outer segments; the remaining is processed by RDH12 in the inner segment to protect cells from oxidative stress (Sarkar and Moosajee, 2019). RDH12 has also been reported to metabolize medium-chain aldehyde, especially nonanal (Belyaeva et al., 2005), to resist induced toxicity in HEK293 models (Lee et al., 2008).
While few in vitro (cell lines overexpression WT or mutated RDH12) and in vivo (mutant mouse and zebrafish) models exist for autosomal recessive RDH12-retinopathies, no animal model has been created for autosomal dominant RDH12-related retinitis pigmentosa. The heterozygous Rdh12 mouse model does not exhibit any retinal phenotype (Kurth et al., 2007; Maeda et al., 2006). Previous studies report retinal organoids derived from patient cells recapitulate the phenotype observed in patients and are a good model for understanding the pathophysiology associated with autosomal dominant retinal disorders (Buskin et al., 2018; Chirco et al., 2021; Gao et al., 2020; Kruczek et al., 2021; Rodrigues et al., 2022). Hence, in this study, we generated and characterized retinal organoid models differentiated from human induced pluripotent stem cells (HiPSC) derived from a patient with heterozygous pathogenic frameshift c.759del p.(Phe254Leufs*24) in RDH12, who was affected with dominant mild, late-onset RP (RDH12-AD) (Sarkar et al., 2021; Sarkar et al., 2020).
2 Materials and methods
2.1 Cell culture
HiPSCs were obtained from an RDH12-AD patient described by Sarkar et al. (2020) and an unrelated age- and gender-matched unaffected control (Méjécase et al., 2020; Sarkar et al., 2021). Three clones for each line were expanded, as previously reported (Méjécase et al., 2020; Sarkar et al., 2021). HiPSCs were differentiated into retinal organoids following a feeder-free xeno-free protocol (Reichman et al., 2017). Optic cups were picked between days 21 and 32 and cultured in 6-well plates with 10 ng/mL of human FGF2 for 7 days (Reichman et al., 2017). Each optic cup was selected and cultured in a 96-well plate, with the previously reported protocol to obtain laminated retinal organoids (Gonzalez-Cordero et al., 2017). Retinoic acid (0.5 µM) was maintained until the latest time point (week 44).
2.2 Retinal organoid characterization and electron microscopy analysis
Retinal organoids were cryoembedded at two time points (4× week-18 retinal organoids per line obtained from two differentiations and 5× week-44 retinal organoids per clone obtained from up to four differentiations) for immunostaining as previously reported (Reichman et al., 2014). Immunofluorescence staining was performed on 12-µm thick sections. Samples were incubated in a permeabilizing solution (0.5% Triton X-100 and 0.1% Tween 20 with PBS) for 1 h, in a blocking solution (PBS, 0.2% gelatin, and 0.25% Triton X-100) for 1 h at room temperature, and then in primary antibodies solutions diluted in blocking solution (Supplementary Table S1) overnight at 4°C. After washes in PBS with 0.1% Tween 20 solution, sections were incubated with secondary antibodies (Supplementary Table S1) diluted in blocking solution for 1 h at room temperature. Sections were mounted with ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific). CRX-positive cells with DAPI co-staining were manually counted in a stack of three slides using Fiji to determine the percentage representation. A t-test was performed with p ≤ 0.05 considered statistically significant.
A TUNEL assay was performed using the ApopTag Fluorescein In situ Apoptosis Detection Kit (Millipore) per the manufacturer’s instructions. The entire organoid section (19–28 images per stack) was imaged using an LSM710 confocal microscope at magnification ×40, and a maximum intensity projection Z stack was performed for each image (Lane et al., 2020). TUNEL-positive nuclei were counted manually on Fiji, and a t-test was performed, with p ≤ 0.05 considered statistically significant.
Week 44 retinal organoids (4× week-44 retinal organoids per line) were fixed in 4% paraformaldehyde overnight at 4°C, preserved in an increased sucrose gradient (10%–30%, in PBS1X) overnight for each glucose concentration at 4°C. Retinal organoids were then frozen in isopentane at −50°C. Samples and cryosections were kept at −80°C until use. RNAscope was performed to study RDH12 (1623951-C2), RBP4 (469781), RLBP1 (414221), and ARR3 (486461-C2) mRNA levels, following the manufacturer’s instructions (RNAscope Multiplex Fluorescent Reagent Kit v2), with a target retrieval step for 5 min and protease III steps for 30 min. The entire organoid section was imaged with a Zeiss Invert880 confocal microscope at magnification ×40, and a maximum intensity projection Z stack was performed for each image. Each dot corresponding to one mRNA molecule was counted with a 3D object counter plugin (Bolte and Cordelières, 2006) with Fiji. DAPI nuclei were counted manually to calculate the volume of dots per cell in each image. A t-test was performed with p ≤ 0.05 considered statistically significant.
Two retinal organoids per line (week 37) were fixed with Karnovsky’s fixative (2.5% glutaraldehyde, 1% PFA, 80 mM cacodylate buffer (pH 7.4), and 20 mM NaOH) for 1 h at room temperature in the dark. After washes in 0.2 M sodium cacodylate solution, samples were postfixed in 1% aqueous osmium tetroxide for 1–2 h at room temperature. Serial dehydration with increasing ethanol concentration (50%, 70%, 90%, and 100%) was performed, followed by a 100% propylene oxide bath. Samples were then embedded in 100% EPON resin. The samples were serially sectioned at the thickness of 70 nm using a UC7 ultramicrotome (Leica Microsystems), and sections were picked up on Formvar-coated 2-mm slot copper grids (Gilder Grids). The sections were post-stained with 2% UA and lead citrate, and sections containing the region of interest were imaged using a 120 kV JEM-1400Flash transmission electron microscope with a sCMOS Flash camera (JOEL, United Kingdom). For each organoid per line, three regions 60 µm apart were studied; photoreceptor segments, which could be small outer segment-like structures or inner segments, were counted and measured from the outer limiting membrane (Supplementary Figure S1). As the two structures can be difficult to distinguish due to the section and the angle of the organoid, they were not separated in our measurements. The measurement and counting were performed blind. As no significant difference was observed between the two researchers’ analyses for three blinded regions (Supplementary Figure S1), the remaining analysis (measurement and counting) was performed by only one researcher. A total of 2,097 and 1,238 segments were counted in the unaffected control and RDH12-AD samples. Observations can only be drawn from this as statistical analysis is not appropriate for n = 2 retinal organoids per group.
2.3 RNA-seq data analysis
At week 44 (day 303), 12 replicates (two organoids per replicate) from two differentiations of two RDH12-AD clones and eight replicates from three differentiations from one unaffected control clone were snap frozen. RNA was isolated and quantified, and cDNA was generated from pooled organoids (Qiagen, United Kingdom). cDNA libraries were sequenced on a HiSeq 2500 with paired-end reads of 150 bp using Illumina Stranded Total RNA with Ribo-Zero Plus (Illumina, United Kingdom). Raw reads were quality- and adapter-trimmed using Cutadapt (version 3.4) (Martin, 2011) before alignment. Reads were mapped, and subsequent gene levels were counted using RSEM 1.3.1 (Li and Dewey, 2011) and STAR 2.7.10a (Dobin et al., 2013) against the human genome GRCh38 using annotation release 95, both from Ensembl. Normalization of raw count data and differential expression analysis was performed with the DESeq2 package (version 1.38.3) (Love et al., 2014) within the R programming environment (version 4.2.2) (R Development Core Team, 2008). The pairwise comparison was performed with the contrast function, from which genes differentially expressed (adjusted p-value less than 0.05) between different conditions were determined. Gene lists were used to look for pathways and molecular functions with over-representation analysis using Reactome (Jassal et al., 2020) and Gene Ontology (GO) (Mi et al., 2019). Genes involved in RDH12 pathways were collected from the PathCards database (https://pathcards.genecards.org/, accessed 29 September 2023).
2.4 Statistics and reproducibility
All statistical analyses were performed using R for Statistics (https://www.r-project.org/, v4.4.3).
2.5 Data availability
The RNA-seq data generated by this study have been deposited in the NCBI Gene Expression Omnibus under the access code GSE271751, including unprocessed FASTQ files and associated gene count matrices.
3 Results
3.1 Retinal organoid characterization at different time points
HiPSC derived from an RDH12-AD patient and an unaffected control were differentiated into retinal organoids and characterized after 18 weeks and 44 weeks of differentiation (Figure 1). At week 18, both unaffected and RDH12-AD retinal organoids developed rod and cone photoreceptors, and there was no significant difference in either cell death (p = 0.42) or in the number of photoreceptor cells (CRX-positive cells, p = 0.62, Figure 1A). In both mutant and unaffected control retinal organoids, recoverin was localized in photoreceptor segments, with some recoverin-positive cells detected in the inner nuclear layer, corresponding to bipolar cells. At week 44, there was no significant difference in cell viability (p = 0.13), and both photoreceptor cells grew inner (recoverin-expressing) and outer segment-like structures (with L/M-opsin and rhodopsin expression) (Figure 1B). However, the percentage of photoreceptors (CRX-positive cells) in RDH12-AD retinal organoids was significantly decreased compared to the unaffected control at week 44 (p = 0.04, Figure 1B). RDH12 was detected in the photoreceptor inner segments in both the unaffected control and RDH12-AD retinal organoids at week 18 and week 44.
FIGURE 1
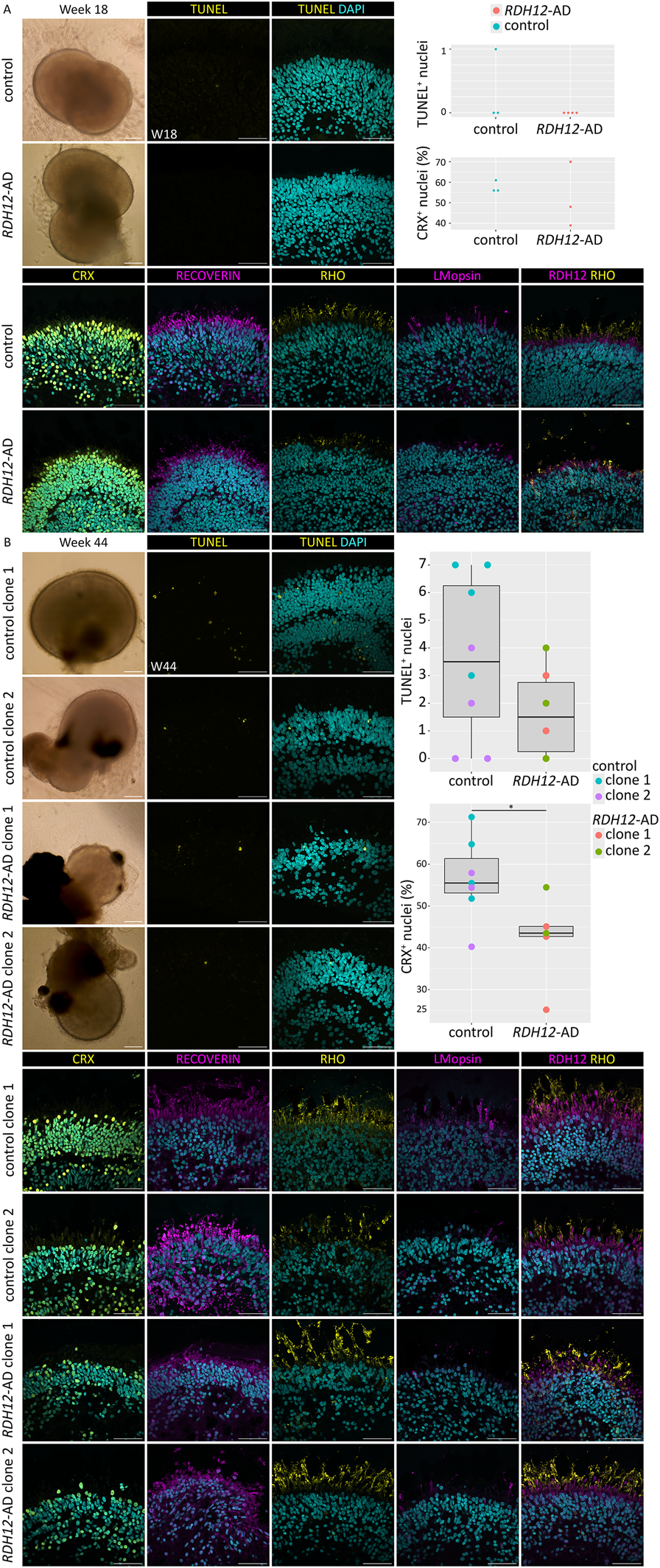
Immunofluorescence studies revealed that RDH12-AD retinal organoids developed fewer photoreceptors than unaffected controls. (A)RDH12-AD and control organoids developed rod and cone photoreceptors at week 18. Three unaffected controls and four RDH12-AD retinal organoids were studied. Analysis showed no significant difference in TUNEL+ nuclei in RDH12-AD organoids compared to control (t-test, p = 0.42) and no significant difference in the percentage of CRX-positive nuclei in RDH12-AD organoids compared to control (t-test, p = 0.62). (B) At week 44, photoreceptors displayed inner and outer segment-like structures in both control and mutant retinal organoids. Three RDH12-AD organoids per clone and five unaffected control organoids at week 44 were studied. Analysis showed no significant difference in TUNEL+ nuclei in RDH12-AD organoids compared to control (t-test, p = 0.13). RDH12-AD retinal organoids had significantly fewer photoreceptors (CRX-positive nuclei) than unaffected controls at week 44 (p = 0.04). CRX, RECOVERIN: photoreceptor precursors; RHO: rod; L/M-opsin: cone. The 200 µm scale bar belongs to brightfield images only; the 50 µm belongs to all the immunostaining images.
3.2 Photoreceptors are shorter and less abundant in RDH12-AD retinal organoids
Transmission electron microscopy of week 37 retinal organoids was undertaken to look for intracellular changes (Figure 2). Both unaffected control and RDH12-AD organoids developed segments around the organoid edges, with mitochondria and a clear, regular outer limiting membrane (Figure 2A). Small rudimentary outer segment-like structures and connecting cilia were observed in both WT and RDH12-AD organoids (Figure 2A). Blinded analyses of three regions per organoid showed unaffected control retinal organoids have an average number of 62 ± 5 photoreceptors per 100 µm whilst RDH12-AD have fewer photoreceptors (33 ± 4 photoreceptors per 100 µm) (Figure 2B). The average photoreceptor segment length was 10.09 ± 6.48 µm in unaffected controls (range between 0.25 µm and 44.02 µm) and reduced to 5.51 ± 3.42 µm in RDH12-AD retinal organoids (range between 0.36 µm and 26.77 µm) (Figure 2C).
FIGURE 2
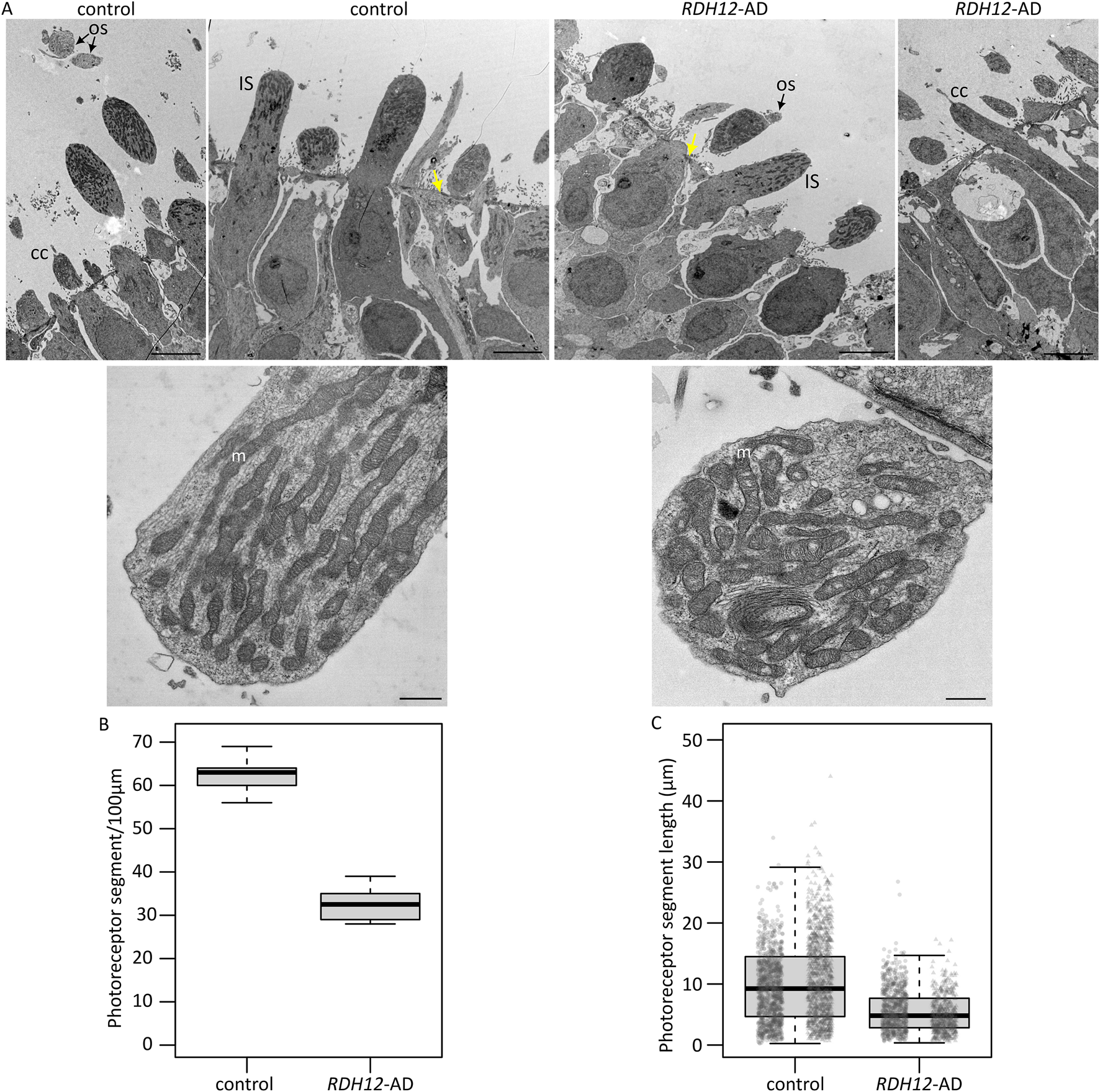
Ultrastructure of RDH12-AD mutants and unaffected control retinal organoids at week 37. (A) Both unaffected control and RDH12-AD retinal organoids developed photoreceptor outer segment (OS) and inner segment (IS)-like structures with mitochondria (m) and connecting cilium (CC). Yellow arrows indicate the outer limiting membrane. Scale bar: 5 µm (top) and 500 nm (bottom). (B)RDH12-AD retinal organoids had fewer photoreceptors per 100 µm than control organoids. (C) Photoreceptor segments were shorter in RDH12-AD retinal organoids than in unaffected controls. Data are represented as mean ± SEM. N = 3 regions from 2 organoids per line, from 1 differentiation batch, and a total of n = 2,097 and n = 1,238 photoreceptor segments, from the outer limiting membrane to tip end, were counted and measured in WT and RDH12-AD organoids, respectively.
3.3 Dominant disease-causing variant in RDH12 impairs cone functions
Bulk RNA sequencing was performed on mature RDH12 and unaffected control retinal organoids at week 44 to identify disease-causing pathways. In mature RDH12-AD retinal organoids, 2,446 genes were differentially expressed, with 1,424 upregulated and 1,022 downregulated, compared to the control expression (Figure 3A; Supplementary Table S2; Supplementary Figure S2). Gene ontology (GO) analysis identified GO overrepresentation of passive transmembrane transporter activity pathways (GO:0022803, GO:0005249, GO:0005261, GO:0046873, GO:0015103, GO:0015267, GO:0022836, GO:0005267, GO:0022843, GO:0005244, GO:0022832, GO:0015079, GO:0005216, GO:0022824, GO:0005230, GO:0022834, GO:0005231, GO:0005237, GO:0005245, GO:0005254, GO:0016917, GO:0004890, GO:0022835, GO:0015276, GO:0015085, GO:0005262, GO:0005253, GO:0015108, GO:1904315, GO:0099095, GO:0022851, and GO:0008331), neurotransmitter receptor activity (GO:0098960, GO:0099529, and GO:0008503), nucleoside-triphosphatase regulator activity (GO:0060589, GO:0030695, and GO:0005096), photoreceptor activity (GO:0009881 and GO:0008020), lipid phosphatase activity (GO:0042577 and GO:0008195), modified amino acid binding (GO:0072341 and GO:0001786), extracellular matrix structural constituent (GO:0005201 and GO:0030020), glycosaminoglycan binding (GO:0005539), transmembrane receptor protein tyrosine kinase activator activity (GO:0030297), ephrin receptor activity (GO:0005003), cGMP binding (GO:0030553), SNAP receptor activity (GO:0005484), cell–cell adhesion mediator activity (GO:0098632), coreceptor activity (GO:0015026), collagen binding (GO:0005518), actin binding (GO:0003779), cadherin binding (GO:0045296), integrin binding (GO:0005178), 1-phosphatidylinositol binding (GO:0005545), tubulin binding (GO:0015631 and GO:0043015), and microtubule binding (GO:0008017) in RDH12-AD (Figure 3B; Supplementary Table S3).
FIGURE 3
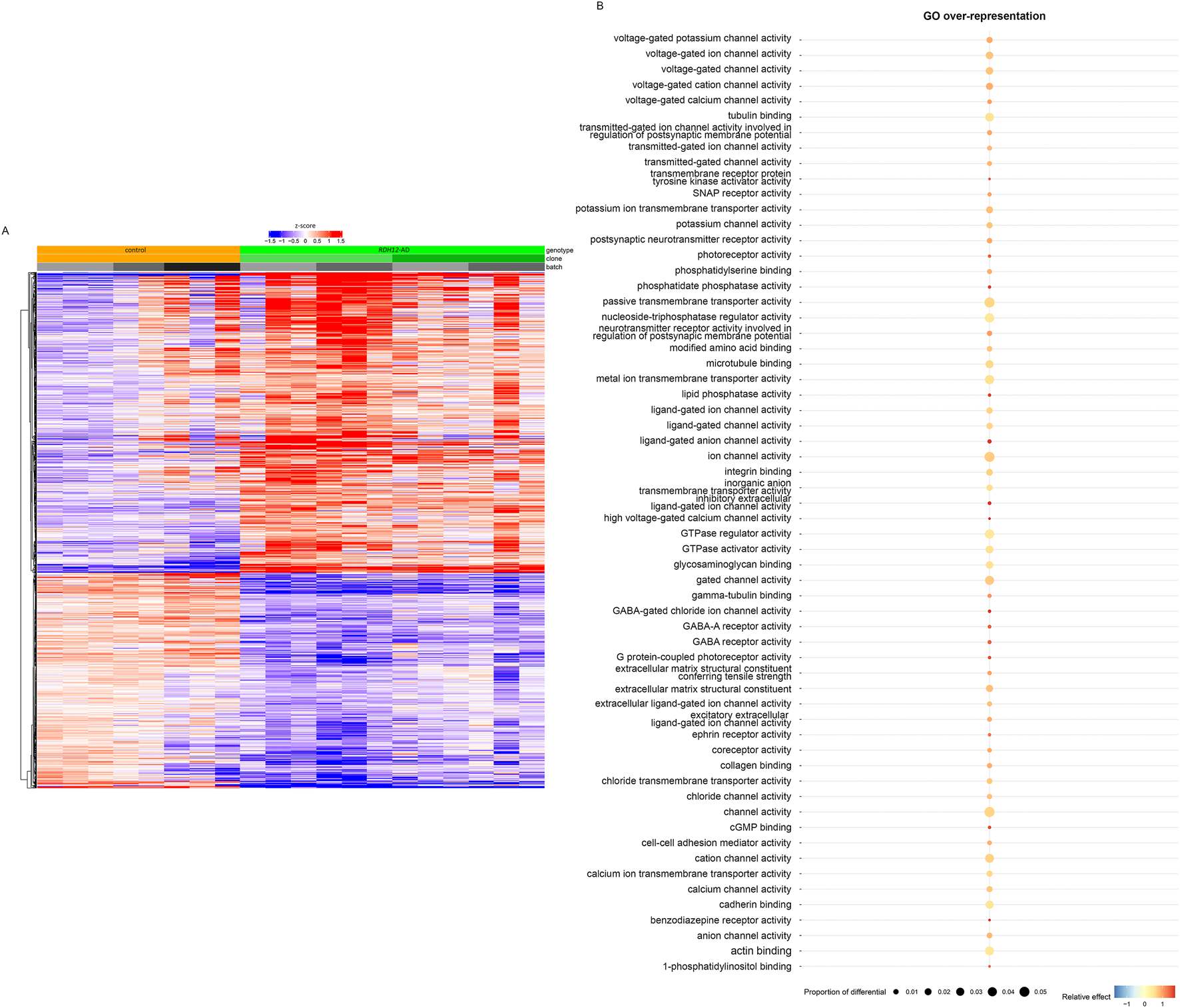
Transcriptomic analyses distinguished RDH12-AD mutant from unaffected control retinal organoids, highlighting molecular dysfunction (day 303, week 44). (A) Heatmap of differential genes from unaffected control (orange) and RDH12-AD (green) mature retinal organoids. Downregulated genes are indicated in blue, and upregulated genes are indicated in red. Both are described with standard deviations around the mean (z-score). (B) Gene ontology (GO) analysis identified overrepresented GO molecular function in RDH12-AD organoids, including passive transmembrane transporter activity pathways, neurotransmitter receptor activity, nucleoside-triphosphatase regulator activity, photoreceptor activity, lipid phosphatase activity, modified amino acid binding, extracellular matrix structural constituent, glycosaminoglycan binding, transmembrane receptor protein tyrosine kinase activator activity, ephrin receptor activity, cGMP binding, SNAP receptor activity, cell–cell adhesion mediator activity, coreceptor activity, collagen binding, actin binding, cadherin binding, integrin binding, 1-phosphatidylinositol binding, tubulin binding, and microtubule binding. Relative effect indicates under- (below 1) and overrepresented (above 1) pathways, supplemented with a statistically significant (hypergeometric) p-value. The proportion of differential indicates the fraction of the differentially expressed genes that belong to a specific pathway. Bulk RNA sequencing was performed on 12 replicates (two organoids per replicate) from two differentiations of two RDH12-AD clones and eight replicates from three differentiations from one control clone.
We examined different retinal cell markers (Kim et al., 2023) and found cone-specific markers were downregulated in RDH12-AD retinal organoids compared to the unaffected control: OPN1LW, TDRG1, GRK7, RAB41, MYL4, PDE6H, CNGB3, PDE6C, GNAT2, GUCA1C, GNGT2, ARR3, SLC24A2, KCNB2, and PEX5L (Supplementary Table S4; Figure 4A). However, only two stress and/or apoptosis markers were differentially expressed: MOAP1 (LFC −0.804, padj < 2.109 × 10−8) and AIFM2 (LFC 0.639, padj < 3.276 × 10−2) (Deng et al., 2018) (Supplementary Table S4).
FIGURE 4
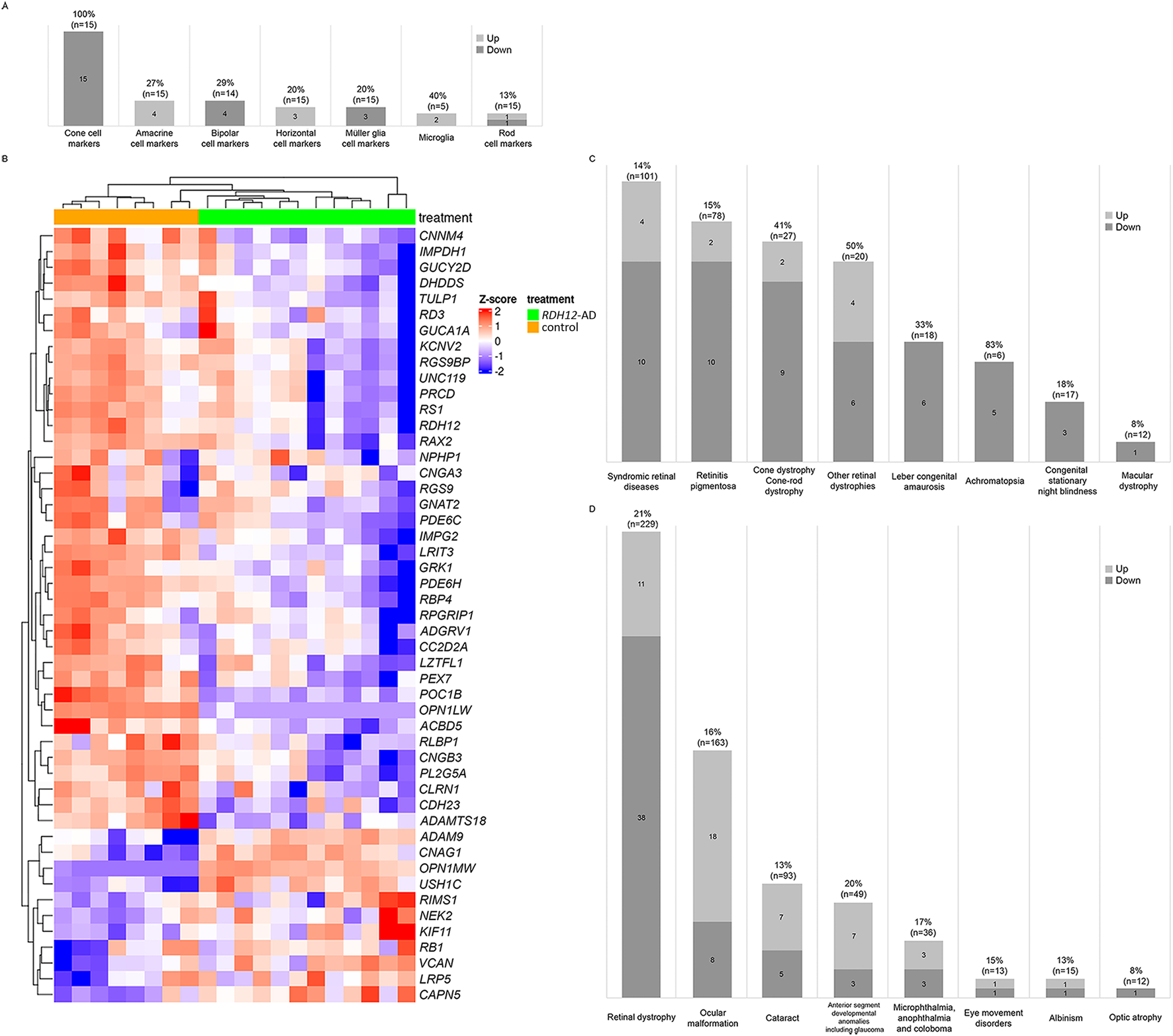
Retinal markers and genes associated with inherited eye diseases were dysregulated in RDH12-AD retinal organoids at day 303 (week 44). (A) Cone-specific cell markers were downregulated in RDH12-AD retinal organoids compared to unaffected controls. (B, C) Forty-nine unique genes associated with retinal dystrophies were significantly differentially expressed. (D) In total, 79 unique genes associated with inherited eye diseases were significantly dysregulated in RDH12-AD retinal organoids. Bulk RNA sequencing was performed on 12 replicates (two organoids per replicate) from two differentiations of two RDH12-AD clones and eight replicates from three differentiations from one control clone. Downregulated genes are indicated in blue, and upregulated genes are indicated in red. Both are described with standard deviations around the mean (z-score).
We compared the expression of genes previously reported to be associated with inherited eye diseases (Supplementary Table S5; Figure 4). Interestingly, 49 genes associated with retinal dystrophies were significantly differentially expressed in RDH12-AD (21%, n = 229), with 38 downregulated and 11 upregulated (Figures 4B–D; Supplementary Table S5). These genes are associated with syndromic retinal disorders (ACBD5, ADAMTS18, ADGRV1, CAPN5, CC2D2A, CDH23, CLRN1, KIF11, LZTFL1, NPHP1, PEX7, POC1B, USH1C, and VCAN), retinitis pigmentosa (ADAMTS18, CC2D2A, CLRN1, CNGA1, DHDDS, IMPDH1, IMPG2, NEK2, PRCD, RDH12, RLBP1, and TULP1), other retinal dystrophies (CAPN5, LRP5, OPN1LW, OPN1MW, PLA2G5, RB1, RBP4, RGS9, RGS9BP, and RS1), Leber congenital amaurosis (GUCY2D, IMPDH1, RD3, RDH12, RPGRIP1, and TULP1), cone dystrophy/cone-rod dystrophy (ADAM9, CNNM4, GUCA1A, GUCY2D, KCNV2, PDE6C, POC1B, RAX2, RIMS1, RPGRIP1, and UNC119), achromatopsia (CNGA3, CNGB3, GNAT2, PDE6C, and PDE6H), congenital stationary night blindness (LRIT3, GRK1, and RLBP1), and macular dystrophy (RAX2). Differential gene expression was also observed for the different groups of ocular diseases (Figure 4D; Supplementary Table S5): 26 genes are associated with ocular malformations (16%, n = 163), with 18 upregulated and eight downregulated; 12 are associated with cataracts (13%, n = 93), with seven upregulated and five downregulated; 10 are associated with anterior segment developmental anomalies including glaucoma (20%, n = 49), with seven upregulated and three downregulated; six associated with microphthalmia, anophthalmia, and coloboma (17%, n = 36), with three upregulated and three downregulated; two are associated with eye movement disorders (15%, n = 13), with one upregulated and one downregulated; two are associated with albinism (13%, n = 15), with one upregulated and one downregulated; and one gene associated with optic atrophy was downregulated (8%, n = 12).
RDH12 expression is downregulated in RDH12-AD retinal organoids compared to the unaffected control (LFC −1.158, padj < 4.633 × 10−3), and this was supported by RNA in situ hybridization (Supplementary Table S6; Supplementary Figure S3). RDH12 dysregulation may affect several pathways involving RDH12: the visual cycle in rod and cone photoreceptors, vitamin A and carotenoid metabolism, and retinol biosynthesis and metabolism (Sarkar and Moosajee, 2019). Interestingly, 45 unique genes involved in these pathways were dysregulated in RDH12-AD retinal organoids compared to the unaffected control (Figure 5A; Supplementary Table S6): of 18 genes involved in visual signal transduction in cones (78%, n = 23), 16 are downregulated, and two are upregulated; of 27 genes involved in visual phototransduction (25%, n = 110), 17 are downregulated, and 10 are upregulated; of 22 genes involved in the visual cycle in retinal rod (23%, n = 94), 17 are downregulated, and five are upregulated; of nine genes involved in vitamin A and carotenoid metabolism (21%, n = 42), seven are downregulated, and two are upregulated; two genes involved in retinol metabolism (5%, n = 38) are downregulated; and four genes involved in retinol biosynthesis are downregulated (31%, n = 13). Cone function appeared to be highly affected in RDH12-AD retinal organoids (Supplementary Table S6): GRK7 (LFC −1.974, padj < 2.061 × 10−10), OPN1MW3 (LFC −1.746, padj < 4.244 × 10−3), PDE6H (LFC −1.541, padj < 2.014 × 10−10), CNGB3 (LFC −1.514, padj < 4.937 × 10−5), PDE6C (LFC −1.509, padj < 1.042 × 10−12), GNAT2 (LFC −1.499, padj < 8.770 × 10−9), GNGT2 (LFC −1.447, padj < 1.042 × 10−5), ARR3 (LFC −1.438, padj < 1.528 × 10−6), SLC24A2 (LFC −1.403, padj < 1.079 × 10−3), GNB3 (LFC −1.202, padj < 6.145 × 10−4), RDH12 (LFC −1.158, padj < 4.633 × 10−3), GRK1 (LFC −1.132, padj < 6.192 × 10−5), RGS9BP (LFC −0.886, padj < 2.274 × 10−3), CNGA3 (LFC −0.824, padj < 2.127 × 10−2), GUCY2D (LFC −0.788, padj < 2.388 × 10−3), RGS9 (LFC −0.589, padj < 3.590 × 10−2); and OPN1MW (LFC 10.42, padj < 3.188 × 10−39), and OPN1MW2 (LFC 10.658, padj < 2.172 × 10−25). RNA in situ hybridization confirmed that RLBP1, a marker of the vitamin A pathway, was downregulated in the RDH12-AD retinal organoids compared to unaffected controls (p = 0.009, Supplementary Figure S3B). A trend toward reduction was seen with RBP4, which is involved in retinol biosynthesis (p = 0.121, Supplementary Figure S3C), and with ARR3, involved in cone phototransduction (p = 0.275, Supplementary Figure S3). The vitamin A pathway was downregulated in RDH12-AD retinal organoids (Figures 5B,C).
FIGURE 5
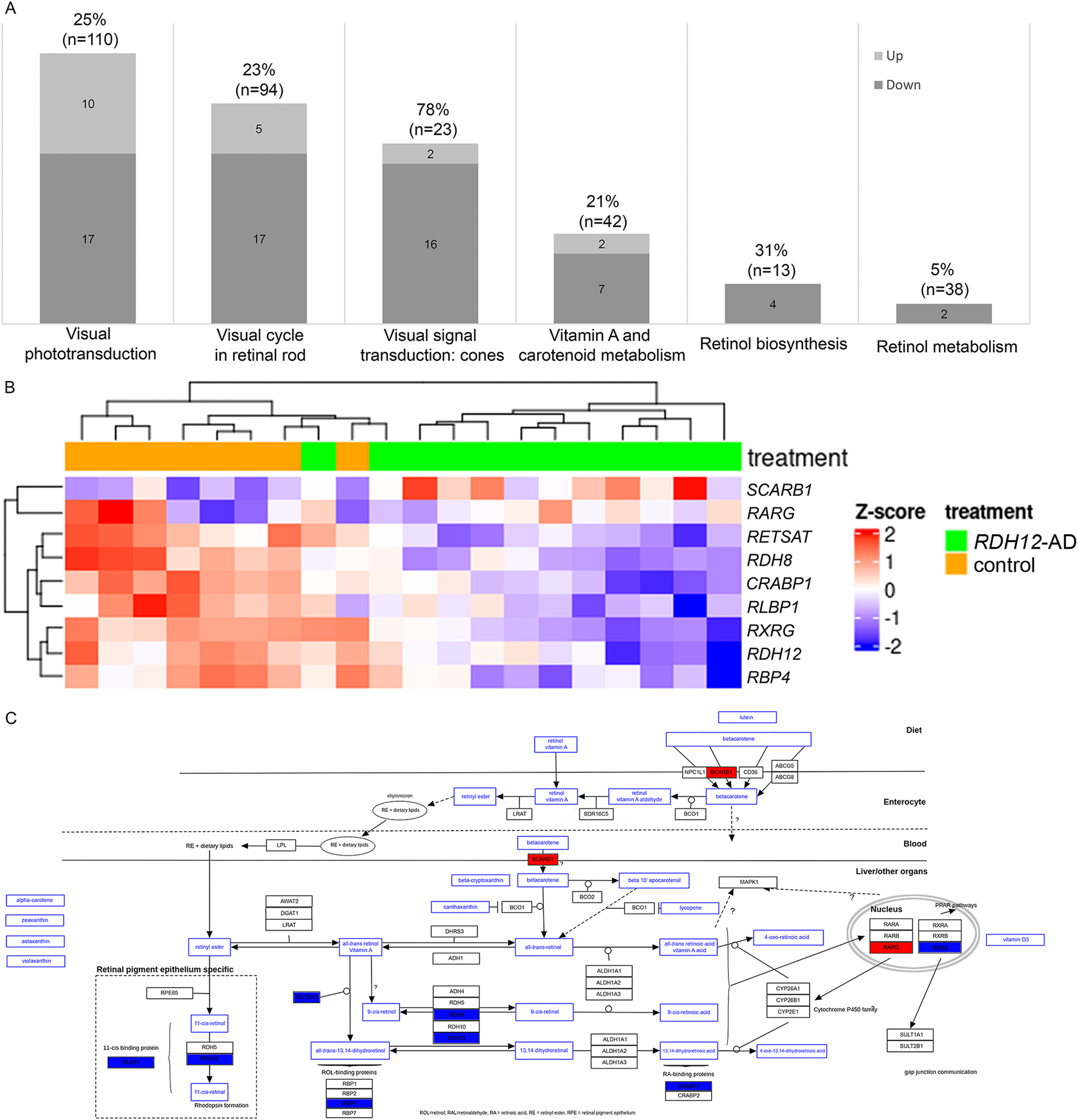
RDH12 pathways were differentially expressed in RDH12-AD retinal organoids at day 303 (week 44). (A) Forty-five genes involved in RDH12 pathways were significantly differentially expressed. Genes involved in RDH12 pathways were obtained from the PathCards database (https://pathcards.genecards.org/, accessed 29 September 2023). (B, C) The vitamin A pathway was affected in RDH12-AD retinal organoids at day 303 (W44). Downregulated genes are indicated in blue, and upregulated genes are indicated in red. Both are described with standard deviations around the mean (z-score). Bulk RNA sequencing was performed on 12 replicates (two organoids per replicate) from two differentiations of two RDH12-AD clones and eight replicates from three differentiations from one control clone. The scheme was adapted from the WikiPathways website (https://www.wikipathways.org/pathways/WP716.html, accessed on 11 November 2023) (Agrawal et al., 2024; Waagmeester et al., 2009).
4 Discussion
This study describes the first retinal organoid model for autosomal dominant RDH12-related disease, derived from a patient with a heterozygous frameshift variant c.759del p.(Phe254Leufs*24). This powerful tool unravels disease mechanisms associated with a milder and late-onset retinitis pigmentosa, providing further insights into RDH12 function in the retina.
Autosomal dominant RDH12 retinopathy is considered a milder form of IRD with relatively late onset in adulthood. It is characterized by nyctalopia and progressive visual field loss with associated bone spicules in the peripheral retina and retinal vessel attenuation. The central vision is affected much later in the disease, although the number of peripheral cone photoreceptors is reduced, and their outer segment is abnormal (Fingert et al., 2008; Muthiah et al., 2022; Sarkar et al., 2020). RDH12-AD retinal organoids model the mild phenotype seen in patients; they display a similar development process to unaffected control organoids with production of the outer nuclear layer with mature photoreceptors, albeit with shorter photoreceptor segments and reduced photoreceptor cell numbers. Variants in LRIT3, GRK1, and RLBP1 were previously reported to be associated with autosomal recessive or X-linked congenital stationary night blindness (Zeitz et al., 2015). Their effect was predicted to produce misfolded non-functional protein or decreased expression (Damodar et al., 2024; Poulter et al., 2021; Zeitz et al., 2013). Interestingly, these genes were downregulated in the RDH12-AD retinal organoids. Moreover, cone-specific markers associated with predominantly cone diseases (achromatopsia, cone/cone-rod dystrophy, and macular dystrophy) were downregulated, suggesting cones are preferentially affected in RDH12-AD models. In addition, 56 genes associated with ocular size, corneal defects, and cataracts were dysregulated, with 19 downregulated, suggesting a link to the hypermetropic refractive error commonly seen in RDH12 patients and associated complications seen with RP (Aleman et al., 2018). Further transcriptomics studies on IRD gene expression in different RP models may uncover common disease pathways.
Mature retinal organoids resemble more peripheral retina than macula (Cowan et al., 2020), with a well-delineated outer limiting membrane and photoreceptor segments exhibiting inner and outer segment characteristics, mitochondria, and discs (Zhong et al., 2014). Ultra-structural and immunostaining analysis of RDH12-AD retinal organoids revealed shorter photoreceptor segments and reduced cellular numbers than unaffected controls, consistent with shorter cone inner segments and a decreased cone density seen through adaptive optic scanning light ophthalmoscopy (AOSLO) in affected patients (Fingert et al., 2008; Muthiah et al., 2022; Sarkar et al., 2020). The number of photoreceptors at week 18 in RDH12-AD retinal organoids is similar to unaffected control organoids, showing cell death may have occurred between weeks 18 and 38, thus excluding any developmental defect in the RHD12-AD model. While cone defects are observed later in the disease, they cannot be excluded at earlier stages when the patient is potentially pre-symptomatic. Moreover, transmission electronic microscopy of retinal organoids cannot clarify which type of photoreceptors, rods or cones, are impacted. Shortened segments are an early sign of photoreceptor dysfunction and degeneration in RP patients (Milam et al., 1998) and in several mouse models (rds and rd; Travis, 1998). There was no change in the ultrastructure of cellular organelles, and immunostaining at various time points failed to detect a significant increase in cell death in RDH12-AD organoids. Moreover, in week 44 RDH12-AD retinal organoids, two pro-apoptotic genes, MOAP1 and AIFM2, were downregulated and upregulated, respectively (Su et al., 2022; Wu et al., 2002). The equilibrium of these genes might be altered in some cells, leading to their death and thus reducing the number of photoreceptors. Photoreceptor cell death may have occurred between weeks 18–38, time points not examined in this study; hence, further ultrastructure analyses at different time points during photoreceptor maturation would be needed to assess for longitudinal changes.
RDH12-AD retinal organoids are derived from patient cells, heterozygous for c.759del p.(Phe254Leufs*24) in RDH12 (Sarkar et al., 2021; Sarkar et al., 2020). This frameshift variant creates a premature termination codon (PTC) located 19 nucleotides upstream of the last exon–exon junction in exon 8, predicted to be resistant to nonsense-mediated decay (NMD) (Kurosaki et al., 2019). Transcriptomic analysis reveals that RDH12 expression decreased by 45% in RDH12-AD organoids compared to the unaffected control. The presence of mutated RDH12 mRNA in mutant retinal organoids suggests a partial degradation. The mutated RDH12 mRNA, which is insensitive to NMD, produces a 40-amino-acid shorter protein, with a 17-amino-acid sequence shared with the two other variants previously reported in autosomal dominant RDH12-retinal diseases (Fingert et al., 2008; Muthiah et al., 2022). The WT RDH12 allele localized to the photoreceptor inner segments (Kurth et al., 2007), including in RDH12-AD retinal organoids, was noted at week 18 and week 44. RDH12 was suggested to function as a dimer based on the ortholog photoreceptor retinal dehydrogenase in Drosophila melanogaster (Sarkar and Moosajee, 2019). The alternative C-terminus may function as a dominant-negative protein, sequestering WT protein to reduce its activity.
RDH12-AD-related RP is characterized by a late-onset milder phenotype as observed in patients affected with PDE6B-RP (Kim et al., 2020; Sarkar et al., 2020; Sarkar and Moosajee, 2019). RDH12 and PDE6B are both involved in the vision signal transduction pathway. PDE6B expression is unchanged in mature RDH12-AD organoids where RDH12 is downregulated, while RDH12 is upregulated in PDE6B mutant organoids with a missense variant leading to gene upregulation (Gao et al., 2020). Phototransduction in both PDE6B and RDH12-AD retinal organoid models reveal downregulation of ARR3, CNGA3, GNAT2, OPN1LW, PDE6C, PDE6H, RGS9BP, and RPGRIP1 (Gao et al., 2020). Further analysis comparing transcriptomic signatures in both RP models to investigate common pathways could be interesting. Transcriptomic analyses at week-44 RDH12-AD retinal organoids reveal 79 inherited eye disease-associated genes are differentially expressed, with 47 genes downregulated, including 49 associated with IRDs. None of these genes have previously been modeled with patient-derived iPSC to study their effect on the retina, but transcriptomic analysis of such retinal organoids may highlight common pathways that could be targeted for more agnostic therapeutic approaches. Of note, RDH12 expression is restricted to the retina, especially cone and rod photoreceptors (Cowan et al., 2020), but the visual cycle occurs in the retina and the RPE (Kiser et al., 2014). Amongst RPE proteins involved in the visual cycle, LRAT converts all-trans-retinol into all-trans-retinaldehyde, which is transformed into 11-cis-retinol by RPE65. RDH5 catalyzes 11-cis-retinal biosynthesis from 11-cis-retinol before retinoid transport back into photoreceptors to start a new phototransduction cycle. Mutations in LRAT or RPE65 are associated with Leber congenital amaurosis, and variants in RDH5 lead to fundus albipunctatus. Presently, differentiation and culture protocols do not effectively co-culture retinal organoids with RPE (Fathi et al., 2021), limiting the complete identification of affected pathways in AD RDH12-RP.
In summary, we have developed the first RDH12-AD retinal organoid model and highlighted photoreceptor dysfunction and vitamin A pathway defects. The genetic background is highly important in determining disease pathogenicity and severity; hence, isogenic controls would help validate these results (Riordan and Nadeau, 2017). Only four patients have been reported in the literature with AD RDH12-RP (Fingert et al., 2008; Muthiah et al., 2022; Sarkar et al., 2020); one unrelated family sharing the same variant studied here (Sarkar et al., 2020), and two other unrelated families, who were heterozygous for the c.776del p.(Glu260Argfs*18) or c.763del p.(Val255Serfs*23) variants in RDH12, respectively (Fingert et al., 2008; Muthiah et al., 2022). Retinal organoid models from each unrelated family or the introduction of the pathogenic variant on the same WT background using CRISPR-Cas9 gene editing would enable further characterization, electron microscopy, and transcriptomic analyses to advance our knowledge of the disease mechanisms associated with RDH12-RP.
Statements
Data availability statement
The RNA-seq data generated by this study have been deposited in the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under the access code GSE271751, including unprocessed FASTQ files and associated gene count matrices.
Ethics statement
The studies involving humans were approved by the 11/LO/243 NRES study of congenital eye diseases. The studies were conducted in accordance with local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
CM: Conceptualization, Investigation, Methodology, Visualization, Writing – original draft, Writing – review and editing. YZ: investigation, Visualization, Writing – review and editing. NO: data curation, Writing – review and editing. PS-B: Data curation, Visualization, Writing – review and editing. RC: Formal Analysis, Validation, Writing – review and editing. NN: Investigation, Writing – review and editing. HS: Methodology, Writing – review and editing. LT: Methodology, Writing – review and editing. MM: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by Retina UK GR594, Moorfields Eye Charity 569249.S-ACA.183882, and Wellcome Trust 205174/Z/16/Z.
Acknowledgments
The authors thank the RDH12 patients and their families. They gratefully acknowledge the National Institute for Health and Care Research (NIHR) Moorfields BRC; Dhani Tracey-White and Matt Hayes (UCL Institute of Ophthalmology, London) for sample preparation for transmission electron microscopy (TEM); Lucy Collison and the Electron Microscopy Science Technology Platform (STP) (The Francis Crick Institute, London) for the TEM imaging; Tom Burgoyne for his retinal TEM expertise (UCL Institute of Ophthalmology); and Camille Charoy and the Light Microscopy STP (The Francis Crick Institute) and the Bioinformatics and Biostatistics STP (The Francis Crick Institute) for their support and assistance in this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2025.1511066/full#supplementary-material
References
1
Agrawal A. Balcı H. Hanspers K. Coort S. L. Martens M. Slenter D. N. et al (2024). WikiPathways 2024: next generation pathway database. Nucleic Acids Res.52, D679–D689. 10.1093/nar/gkad960
2
Aleman T. S. Uyhazi K. E. Serrano L. W. Vasireddy V. Bowman S. J. Ammar M. J. et al (2018). RDH12 mutations cause a severe retinal degeneration with relatively spared rod function. Invest Ophthalmol. Vis. Sci.59, 5225–5236. 10.1167/iovs.18-24708
3
Belyaeva O. V. Korkina O. V. Stetsenko A. V. Kim T. Nelson P. S. Kedishvili N. Y. (2005). Biochemical properties of purified human retinol dehydrogenase 12 (RDH12): catalytic efficiency toward retinoids and C9 aldehydes and effects of cellular retinol-binding protein type I (CRBPI) and cellular retinaldehyde-binding protein (CRALBP) on the oxidation and reduction of retinoids. Biochemistry44, 7035–7047. 10.1021/bi050226k
4
Bolte S. Cordelières F. P. (2006). A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc.224, 213–232. 10.1111/j.1365-2818.2006.01706.x
5
Buskin A. Zhu L. Chichagova V. Basu B. Mozaffari-Jovin S. Dolan D. et al (2018). Disrupted alternative splicing for genes implicated in splicing and ciliogenesis causes PRPF31 retinitis pigmentosa. Nat. Commun.9, 4234. 10.1038/s41467-018-06448-y
6
Chirco K. R. Chew S. Moore A. T. Duncan J. L. Lamba D. A. (2021). Allele-specific gene editing to rescue dominant CRX-associated LCA7 phenotypes in a retinal organoid model. Stem Cell Rep.16, 2690–2702. 10.1016/j.stemcr.2021.09.007
7
Cowan C. S. Renner M. De Gennaro M. Gross-Scherf B. Goldblum D. Hou Y. et al (2020). Cell types of the human retina and its organoids at single-cell resolution. Cell182, 1623–1640. 10.1016/j.cell.2020.08.013
8
Damodar K. Dubois G. Guillou L. Mamaeva D. Pequignot M. Erkilic N. et al (2024). Dual CRALBP isoforms unveiled: iPSC-derived retinal modeling and AAV2/5-RLBP1 gene transfer raise considerations for effective therapy. Mol. Ther.32, 4319–4336. 10.1016/j.ymthe.2024.10.004
9
Deng W.-L. Gao M.-L. Lei X.-L. Lv J.-N. Zhao H. He K.-W. et al (2018). Gene correction reverses ciliopathy and photoreceptor loss in iPSC-derived retinal organoids from retinitis pigmentosa patients. Stem Cell Rep.10, 1267–1281. 10.1016/j.stemcr.2018.02.003
10
Dobin A. Davis C. A. Schlesinger F. Drenkow J. Zaleski C. Jha S. et al (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics29, 15–21. 10.1093/bioinformatics/bts635
11
Fathi M. Ross C. T. Hosseinzadeh Z. (2021). Functional 3-dimensional retinal organoids: technological progress and existing challenges. Front. Neurosci.15, 668857. 10.3389/fnins.2021.668857
12
Fingert J. H. Oh K. Chung M. Scheetz T. E. Andorf J. L. Johnson R. M. et al (2008). Association of a novel mutation in the retinol dehydrogenase 12 (RDH12) gene with autosomal dominant retinitis pigmentosa. Arch. Ophthalmol.126, 1301–1307. 10.1001/archopht.126.9.1301
13
Gao M.-L. Lei X.-L. Han F. He K.-W. Jin S.-Q. Zhang Y.-Y. et al (2020). Patient-specific retinal organoids recapitulate disease features of late-onset retinitis pigmentosa. Front. Cell Dev. Biol.8, 128. 10.3389/fcell.2020.00128
14
Georgiou M. Fujinami K. Michaelides M. (2021). Inherited retinal diseases: therapeutics, clinical trials and end points-A review. Clin. Exp. Ophthalmol.49, 270–288. 10.1111/ceo.13917
15
Gonzalez-Cordero A. Kruczek K. Naeem A. Fernando M. Kloc M. Ribeiro J. et al (2017). Recapitulation of human retinal development from human pluripotent stem cells generates transplantable populations of cone photoreceptors. Stem Cell Rep.9, 820–837. 10.1016/j.stemcr.2017.07.022
16
Jassal B. Matthews L. Viteri G. Gong C. Lorente P. Fabregat A. et al (2020). The reactome pathway knowledgebase. Nucleic Acids Res.48, D498–D503. 10.1093/nar/gkz1031
17
Kim H. J. O’Hara-Wright M. Kim D. Loi T. H. Lim B. Y. Jamieson R. V. et al (2023). Comprehensive characterization of fetal and mature retinal cell identity to assess the fidelity of retinal organoids. Stem Cell Rep.18, 175–189. 10.1016/j.stemcr.2022.12.002
18
Kim Y. N. Song J. S. Oh S. H. Kim Y. J. Yoon Y. H. Seo E.-J. et al (2020). Clinical characteristics and disease progression of retinitis pigmentosa associated with PDE6B mutations in Korean patients. Sci. Rep.10, 19540. 10.1038/s41598-020-75902-z
19
Kiser P. D. Golczak M. Palczewski K. (2014). Chemistry of the retinoid (visual) cycle. Chem. Rev.114, 194–232. 10.1021/cr400107q
20
Kruczek K. Qu Z. Gentry J. Fadl B. R. Gieser L. Hiriyanna S. et al (2021). Gene therapy of dominant CRX-leber congenital amaurosis using patient stem cell-derived retinal organoids. Stem Cell Rep.16, 252–263. 10.1016/j.stemcr.2020.12.018
21
Kurosaki T. Popp M. W. Maquat L. E. (2019). Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol.20, 406–420. 10.1038/s41580-019-0126-2
22
Kurth I. Thompson D. A. Rüther K. Feathers K. L. Chrispell J. D. Schroth J. et al (2007). Targeted disruption of the murine retinal dehydrogenase gene Rdh12 does not limit visual cycle function. Mol. Cell Biol.27, 1370–1379. 10.1128/MCB.01486-06
23
Lane A. Jovanovic K. Shortall C. Ottaviani D. Panes A. B. Schwarz N. et al (2020). Modeling and rescue of RP2 retinitis pigmentosa using iPSC-derived retinal organoids. Stem Cell Rep.15, 67–79. 10.1016/j.stemcr.2020.05.007
24
Lee S.-A. Belyaeva O. V. Kedishvili N. Y. (2008). Effect of lipid peroxidation products on the activity of human retinol dehydrogenase 12 (RDH12) and retinoid metabolism. Biochim. Biophys. Acta1782, 421–425. 10.1016/j.bbadis.2008.03.004
25
Li B. Dewey C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma.12, 323. 10.1186/1471-2105-12-323
26
Love M. I. Huber W. Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol.15, 550. 10.1186/s13059-014-0550-8
27
Maeda A. Maeda T. Imanishi Y. Sun W. Jastrzebska B. Hatala D. A. et al (2006). Retinol dehydrogenase (RDH12) protects photoreceptors from light-induced degeneration in mice. J. Biol. Chem.281, 37697–37704. 10.1074/jbc.M608375200
28
Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J.17, 10. 10.14806/ej.17.1.200
29
Méjécase C. Harding P. Sarkar H. Eintracht J. Lima Cunha D. Toualbi L. et al (2020). Generation of two human control iPS cell lines (UCLi016-A and UCLi017-A) from healthy donors with no known ocular conditions. Stem Cell Res.49, 102113. 10.1016/j.scr.2020.102113
30
Mi H. Muruganujan A. Ebert D. Huang X. Thomas P. D. (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res.47, D419–D426. 10.1093/nar/gky1038
31
Milam A. H. Li Z. Y. Fariss R. N. (1998). Histopathology of the human retina in retinitis pigmentosa. Prog. Retin Eye Res.17, 175–205. 10.1016/s1350-9462(97)00012-8
32
Muthiah M. N. Kalitzeos A. Oprych K. Singh N. Georgiou M. Wright G. A. et al (2022). Novel disease-causing variant in RDH12 presenting with autosomal dominant retinitis pigmentosa. Br. J. Ophthalmol.106, 1274–1281. 10.1136/bjophthalmol-2020-318034
33
Poulter J. A. Gravett M. S. C. Taylor R. L. Fujinami K. De Zaeytijd J. Bellingham J. et al (2021). New variants and in silico analyses in GRK1 associated Oguchi disease. Hum. Mutat.42, 164–176. 10.1002/humu.24140
34
R Development Core Team (2008). R a language and environment for statistical computing: reference index. Vienna: R Foundation for Statistical Computing.
35
Reichman S. Slembrouck A. Gagliardi G. Chaffiol A. Terray A. Nanteau C. et al (2017). Generation of storable retinal organoids and retinal pigmented epithelium from adherent human iPS cells in xeno-free and feeder-free conditions. Stem Cells35, 1176–1188. 10.1002/stem.2586
36
Reichman S. Terray A. Slembrouck A. Nanteau C. Orieux G. Habeler W. et al (2014). From confluent human iPS cells to self-forming neural retina and retinal pigmented epithelium. Proc. Natl. Acad. Sci. U. S. A.111, 8518–8523. 10.1073/pnas.1324212111
37
Riordan J. D. Nadeau J. H. (2017). From peas to disease: modifier genes, network resilience, and the genetics of Health. Am. J. Hum. Genet.101, 177–191. 10.1016/j.ajhg.2017.06.004
38
Rodrigues A. Slembrouck-Brec A. Nanteau C. Terray A. Tymoshenko Y. Zagar Y. et al (2022). Modeling PRPF31 retinitis pigmentosa using retinal pigment epithelium and organoids combined with gene augmentation rescue. NPJ Regen. Med.7, 39. 10.1038/s41536-022-00235-6
39
Sarkar H. Dubis A. M. Downes S. Moosajee M. (2020). Novel heterozygous deletion in retinol dehydrogenase 12 (RDH12) causes familial autosomal dominant retinitis pigmentosa. Front. Genet.11, 335. 10.3389/fgene.2020.00335
40
Sarkar H. Méjécase C. Harding P. Eintracht J. Toualbi L. Cunha D. L. et al (2021). Generation of two human iPSC lines from patients with autosomal dominant retinitis pigmentosa (UCLi014-A) and autosomal recessive Leber congenital amaurosis (UCLi015-A), associated with RDH12 variants. Stem Cell Res.54, 102449. 10.1016/j.scr.2021.102449
41
Sarkar H. Moosajee M. (2019). Retinol dehydrogenase 12 (RDH12): role in vision, retinal disease and future perspectives. Exp. Eye Res.188, 107793. 10.1016/j.exer.2019.107793
42
Su Y. Wang W. Meng X. (2022). Revealing the roles of MOAP1 in diseases: a review. Cells11, 889. 10.3390/cells11050889
43
Travis G. H. (1998). Mechanisms of cell death in the inherited retinal degenerations. Am. J. Hum. Genet.62, 503–508. 10.1086/301772
44
Waagmeester A. Pezik P. Coort S. Tourniaire F. Evelo C. Rebholz-Schuhmann D. (2009). Pathway enrichment based on text mining and its validation on carotenoid and vitamin A metabolism. OMICS13, 367–379. 10.1089/omi.2009.0029
45
Wu M. Xu L.-G. Li X. Zhai Z. Shu H.-B. (2002). AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J. Biol. Chem.277, 25617–25623. 10.1074/jbc.M202285200
46
Zeitz C. Jacobson S. G. Hamel C. P. Bujakowska K. Neuillé M. Orhan E. et al (2013). Whole-exome sequencing identifies LRIT3 mutations as a cause of autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet.92, 67–75. 10.1016/j.ajhg.2012.10.023
47
Zeitz C. Robson A. G. Audo I. (2015). Congenital stationary night blindness: an analysis and update of genotype–phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res.45, 58–110. 10.1016/j.preteyeres.2014.09.001
48
Zhong X. Gutierrez C. Xue T. Hampton C. Vergara M. N. Cao L.-H. et al (2014). Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun.5, 4047. 10.1038/ncomms5047
Summary
Keywords
retinal dehydrogenase 12, autosomal dominant, retinitis pigmentosa, retinal organoids, induced pluripotent stem cells, vitamin A
Citation
Méjécase C, Zhou Y, Owen N, Soro-Barrio P, Cheloni R, Nair N, Sarkar H, Toualbi L and Moosajee M (2025) Dominant RDH12-retinitis pigmentosa impairs photoreceptor development and implicates cone involvement in retinal organoids. Front. Cell Dev. Biol. 13:1511066. doi: 10.3389/fcell.2025.1511066
Received
14 October 2024
Accepted
31 March 2025
Published
29 April 2025
Volume
13 - 2025
Edited by
Kevin Francis, Sanford Research, United States
Reviewed by
Fei Liu, Chinese Academy of Sciences (CAS), China
Ruchira Singh, University of Rochester, United States
Kerstin Nagel-Wolfrum, Johannes Gutenberg University Mainz, Germany
Updates
Copyright
© 2025 Méjécase, Zhou, Owen, Soro-Barrio, Cheloni, Nair, Sarkar, Toualbi and Moosajee.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariya Moosajee, m.moosajee@ucl.ac.uk
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.