Abstract
Neurodegenerative diseases (NDs) such as Alzheimer’s, Parkinson’s and Huntington’s diseases as well as ataxias and fronto-temporal disorders are all characterized by the progressive accumulation of protein aggregates (amyloids) into inclusions bodies. In addition, recent experimental evidence is challenging the conventional view of the disease by revealing the ability of some of these disease-relevant proteins to be transferred between cells by means of extracellular vesicles (EVs), allowing the mutant protein to seed oligomers involving both the mutant and wild type forms of the protein. Abnormal secretion and levels of EVs are closely related to the pathogenesis of neurodegenerative diseases and contribute to disease progression. Numerous studies have proposed EVs as therapeutic targets or biomarkers for neurodegenerative diseases. In this review, we summarize and discuss the role of small heat shock proteins (sHSPs) and autophagy in cellular quality control and turn-over of the major aggregation-prone proteins associated to neurodegenerative disorders. We also highlight the advanced research progress on mechanisms regulating unconventional secretion, secretory autophagy and EVs biogenesis and their contribution in the pathological processes underlining these diseases. Finally, we outline the latest research on the roles of EVs in neurodegenerative diseases and their potential diagnostic and therapeutic significance for the treatment of these clinically relevant conditions.
1 Introduction
Small heat shock proteins (sHSPs) are a conserved family of molecular chaperones that play a critical role in maintaining protein homeostasis under stress conditions. Unlike other chaperones that actively refold misfolded proteins using ATP, sHSPs function in an ATP-independent manner, acting as molecular “holdases.” (Lambert et al., 1999; Arrigo, 2017; Boelens, 2020). They bind to partially unfolded or aggregation-prone proteins, preventing the formation of toxic aggregates and sequestering these proteins into soluble complexes. This action helps maintain cellular viability during stress and facilitates subsequent protein repair or degradation pathways. Protein aggregation is a hallmark of several neurodegenerative diseases, including Alzheimer’s, Parkinson’s, and Huntington’s diseases, where aberrant protein structures disrupt cellular homeostasis (Li D. Y. et al., 2022; Ecroyd et al., 2023).
Beyond their chaperone activity, recent studies have revealed additional roles for sHSPs at the intersection of protein aggregation, autophagy, and unconventional protein secretion pathways (Kwok et al., 2011; Li et al., 2017; Li et al., 2022 D. Y.; Secco et al., 2024).
Autophagy facilitates the degradation and recycling of damaged organelles, misfolded proteins, and other cytoplasmic components through lysosome-mediated degradation (Bento et al., 2016). This process plays a vital role in various physiological functions, including cellular adaptation to stress, immune responses, and the regulation of metabolic balance.
A common hallmark of numerous neurodegenerative diseases (NDs) is the accumulation of misfolded proteins along with the degeneration of specific neuronal populations. Autophagy is one of the major intracellular machineries for degrading aggregated proteins and maintaining cellular proteostasis (Menzies et al., 2017). Although there are currently no effective strategies that slow or prevent these NDs in humans, there is strong experimental evidence that the upregulation of intracellular clearance pathways (the autophagy-lysosome and ubiquitin-proteasome pathways) can clear aggregate-prone proteins, such as a-synuclein in experimental models. When the flux through these pathways is increased, the levels of aggregate-prone proteins can be reduced and this results in improved cell survival in both cell-based and animal models of NDs (Menzies et al., 2017).
Dysregulated autophagy plays a key role in most neurodegenerative diseases such as Parkinson disease, Huntington, Alzheimer and amyotrophic lateral sclerosis (Menzies et al., 2015). Polyubiquitinated proteins were found to be accumulated in autophagy-deficient neurons as inclusion bodies in Atg7-deficient mice that lack conventional autophagy (Komatsu et al., 2006). These findings have demonstrated the connection between autophagy and neurodegenerative disorders. Autophagy is essential for the survival of neural cells, and its impairment is involved in the pathogenesis of neurodegenerative disorders affecting ubiquitin-containing inclusion bodies formation (Komatsu et al., 2006).
Protein secretion can occur via the canonical ER-to-Golgi secretory pathway or by means or non-classical pathways. The canonical pathway involves signal peptide-containing proteins that are directed to the ER, undergo post-translational modifications in the Golgi, and are transported via coat protein complex II (COPII) and Rab proteins peptides (Guo et al., 2018; Ramazanov et al., 2021; Lippincott-Schwartz et al., 2000; Ramazanov et al., 2021). In contrast, unconventional protein secretion (also referred to as UPS) bypasses the Golgi and includes proteins lacking signal peptides or transmembrane proteins. UPS consists of four classes: direct translocation through plasma membrane pores, ABC transporter-mediated secretion, autophagosome/endosome-based secretion, and Golgi-independent secretion of membrane proteins. UPS is often triggered by cellular stress, activating alternative secretion mechanisms (Grieve and Rabouille, 2011; Dimou and Nickel, 2018; Rabouille 2017).
UPS, complementary to the canonical degradation, plays a main role in the pathophysiology of many neurodegenerative diseases (Buratta et al., 2020; Noh et al., 2022; Sepúlveda et al., 2022; Zubkova et al., 2024).
In view of its relevance for human diseases, the control of cellular proteostasis mediated by the chaperone and the autophagosome-lysosome systems has represented one of the most studied topics in the wide field of biomedical sciences. The involvement of sHSPs in several clinically relevant conditions, including neurodegenerative diseases, has brought a considerable interest toward these proteins, making them an attractive target for therapeutic intervention and has driven the development of a number of approaches to modulate their activity, some of which have progressed to clinical trials. On the same line, the observation that some of these proteins might undergo autophagy- or MVBs-dependent unconventional secretion and act extracellularly represents an attractive concept and has the likely potential to play an increasingly prominent role in future, from both the diagnostic and therapeutic point of view. Probably, unconventional secretion could be important to reduce the proteostatic stress induced by the intracellular accumulation of protein aggregates, but this may also lead to the transcellular propagation of pathological aggregates.
Extracellular vesicles (EVs) are small, membrane-bound particles secreted by cells into the extracellular environment. These vesicles play a crucial role in intercellular communication and have emerged as key mediators in various physiological and pathological processes (Abels and Breakefield, 2016; Dixson et al., 2023; Farhan et al., 2017; van Niel et al., 2022; Zubkova et al., 2024). EVs are classified into different subtypes, including exosomes (30–150 nm in diameter), microvesicles (100–1,000 nm), and apoptotic bodies, based on their size, biogenesis, and molecular composition. They carry a diverse cargo of bioactive molecules such as proteins, lipids, RNA, and DNA, reflecting the cell of origin and its state (Abels and Breakefield, 2016; van Niel et al., 2022; Dixson et al., 2023).
EVs have garnered significant attention for their roles in modulating immune responses, promoting tissue repair, and influencing cancer progression. They also serve as potential biomarkers for disease diagnosis and prognosis, owing to their presence in various biological fluids like blood, urine, saliva, and cerebrospinal fluid (CSF) (Hornung et al., 2020; van Niel et al., 2022; Younas et al., 2022).
Recent advancements in EV research highlight their therapeutic potential, particularly in drug delivery systems, regenerative medicine, and as targets for therapeutic interventions. As our understanding of EVs deepens, they offer exciting opportunities to unravel complex biological pathways and develop innovative clinical applications (Abels and Breakefield, 2016; Ramirez et al., 2018; Meldolesi, 2022; van Niel et al., 2022; Dixson et al., 2023; Li Z. et al., 2023).
For this reason, is important to characterize the molecular players of the biogenesis and the delivery of these vesicles. By doing so, it will be possible to identify novel druggable targets.
Here, we summarize and discuss the role of sHSPs and autophagy in cellular quality control and turn-over of the major aggregation-prone proteins associated to neurodegenerative disorders. We also highlight the advanced research progress on mechanisms regulating unconventional secretion, secretory autophagy and EVs biogenesis, outlining the latest research on the roles of EVs in neurodegenerative diseases and their potential diagnostic and therapeutic significance for the treatment of these clinically relevant conditions.
2 Role and functions of the small heat shock proteins
sHSPs do represent a fundamental class of molecular chaperones that play a vital role in protecting cells from various forms of stress, such as heat shock, oxidative stress, DNA damage and toxic agents (Boelens, 2020). These proteins, typically ranging from 12 to 43 kDa in size (as summarised in Table 1), are highly conserved across species and are characterized by a central α-crystallin domain (Delbecq and Klevit, 2013). Despite their relatively small size, sHSPs are essential for maintaining cellular health by ensuring the proper folding, stabilization, and function of other proteins within the cell.
TABLE 1
| Gene | Aliases for HSPBs gene and UniProt entry | Size and predicted M.W. | Quaternary structure (major interactors) | Genetic diseases association | References |
|---|---|---|---|---|---|
| HSPB1 | (Heat Shock 27kD Protein 1)-HSP27 [P04792] | 205aa 22.8 kDa | HSPB5, HSPB6, HSPB8, HSPBAP1 | Charcot Marie Tooth 2F HMND3 (autosomal distal hereditary motor neuronopathy 3) ALS and HSP (Hereditary Spastic Paraparesis) |
Almeida-Souza et al. (2010), Holmgren et al. (2013), Evgrafov et al. (2004), Capponi et al. (2016), Katz et al. (2020) |
| HSPB2 | Heat Shock Protein Family B (Small) Member 2 [Q16082] | 182aa 20.2 kDa | DMPK Kinase HSPB1, CRYAA, CRYAB, HSPB8, BAG3 |
None reported so far. One natural variant (position 111: Gly111Ser) described. HSPB2 facilitates neural regeneration upon traumatic brain injury | Huang et al. (2023) |
| HSPB3 | Heat Shock Protein Family B (Small) Member 3 [Q12988] | 150aa 16.9 kDa | ANP32A, HSPB2, HSPB8 | HMND4 (autosomal dominant pathology, phenotype similar to HMND3) | Kolb et al. (2010) |
| HSPB4 | Crystallin Alpha A, CRYAA, CRYA1 [P02489] | 173aa 19.9 kDa | CRYAA (1:3) (* lens-specific) CRYAB, HSPB1, HSPB2, BAG3 |
Cataract type 9 | Khoshaman et al. (2017) |
| HSPB5 | Crystallin Alpha B, CRYAB, CRYA2 [P02511] | 176aa 20.1 kDa | CRYAA (3:1), HSPB1, HSPB2, HSPBAP1, BAG3, HSPB6, HSPB8 | Myopathy myofibrillar 2 Cataract type 16 Dilatated cardiomyopathy |
Vicart et al. (1998), Berry et al. (2001), Fu and Liang (2003), Inagaki et al. (2006) |
| HSPB6 | Heat Shock Protein Family B (Small) Member 6 [Q14588] | 160aa 17.1 kDa | HSPB1, HSPB5, HSPB6, BAG3 | Dilated cardiomyopathy | Liu et al. (2018), Shatov and Gusev (2020) |
| HSPB7 | Heat Shock Protein Family B (Small) Member 7 (v2) [Q9UBY9] | 170aa 18.6 kDa | HSPB8, BAG3, APP C-term domain of actin-binding protein 280 |
Cardiomyopathy | Liao et al. (2017), Mercer et al. (2018) |
| HSPB8 | Heat Shock Protein Family B (Small) Member 8 [Q9UJY1] | 196aa 21.6 kDa | HSPB1, CRYAA, CRYAB, DNAJB6, BAG3 | HMND2 (autosomal distal hereditary motor neuronopathy 3) | Cortese et al. (2018), Echaniz-Laguna et al. (2017) |
| HSPB9 | Heat Shock Protein Family B (Small) Member 9 [Q9BQS6] | 159aa 17.5 kDa | HSPA1B, HSPA1L, HSPA2, HSPA6, HSPA9 (* testis-specific) | None reported so far One natural variant described (position 2: Gln2Pro) |
Kappé et al. (2001) |
| HSPB10 | Heat Shock Protein Family B ODF1/ODFP [Q14990] | 250aa 28.3 kDa | CCT2, CCT3, CCT4, CCT5, CCT6B, BAG4, TCP11, SPAG4, KLC3 (* testis-specific) | None reported so far Three natural variants described (position 216: Ser216Asn; position 243: Phe243Leu; deletion residues 219-227) |
Hofferbert et al. (1993) |
| HSPB11 | Heat Shock Protein Family B (Small) Member 11 [Q9Y547] | 144aa 16.3 kDa | IFT25 complex B, interacts with IFT22/IFT27/IFT88 (Intra-flagellar Transport Complex, membrane-associated) | initially classified as a member of the HSPB protein family; later shown not to be the case The impaired cooperation between IFT74/BBS22-IFT81 and IFT25-IFT27/BBS19 causes the Bardet-Biedl syndrome |
Kappé et al. (2001), Zhou et al. (2022) |
Human small heat shock proteins (HSPBs).
Source: Uniprot (https://www.uniprot.org) and BioGRID (https://thebiogrid.org).
Under normal conditions, proteins must fold into precise three-dimensional shapes to function correctly. However, environmental stresses, such as elevated temperatures, reactive oxygen species, or heavy metals, can cause proteins to misfold or aggregate, leading to cellular dysfunction and diseases (Li D. Y. et al., 2022; Ecroyd et al., 2023). sHSPs respond rapidly to these stresses, binding to unfolded or misfolded proteins, preventing them from forming toxic aggregates, and assisting in their recovery or degradation. While they do not actively refold proteins themselves, they work in tandem with larger heat shock proteins (such as HSP70 and HSP90), serving as the first line of defence in protein quality control (Peters et al., 2024).
Beyond their traditional role as stress responders, sHSPs are involved in several other cellular functions. For instance, they help regulate apoptosis (programmed cell death), maintain cytoskeletal integrity, and modulate immune responses. Furthermore, their roles in development, aging, and disease, including neurodegenerative disorders and cancer, highlight their broader physiological importance (Gu et al., 2023).
2.1 Phosphorylation and oligomerization
The activity of many sHSPs is regulated by phosphorylation, which alters their ability to bind to proteins or form large oligomeric structures (as depicted in Figures 1A,B). Phosphorylation typically occurs in response to stress and influences the dynamic behaviour of sHSPs, enabling them to switch between inactive and active states, as needed (Arrigo, 2017; Kostenko and Moens, 2009; Lambert et al., 1999).
FIGURE 1
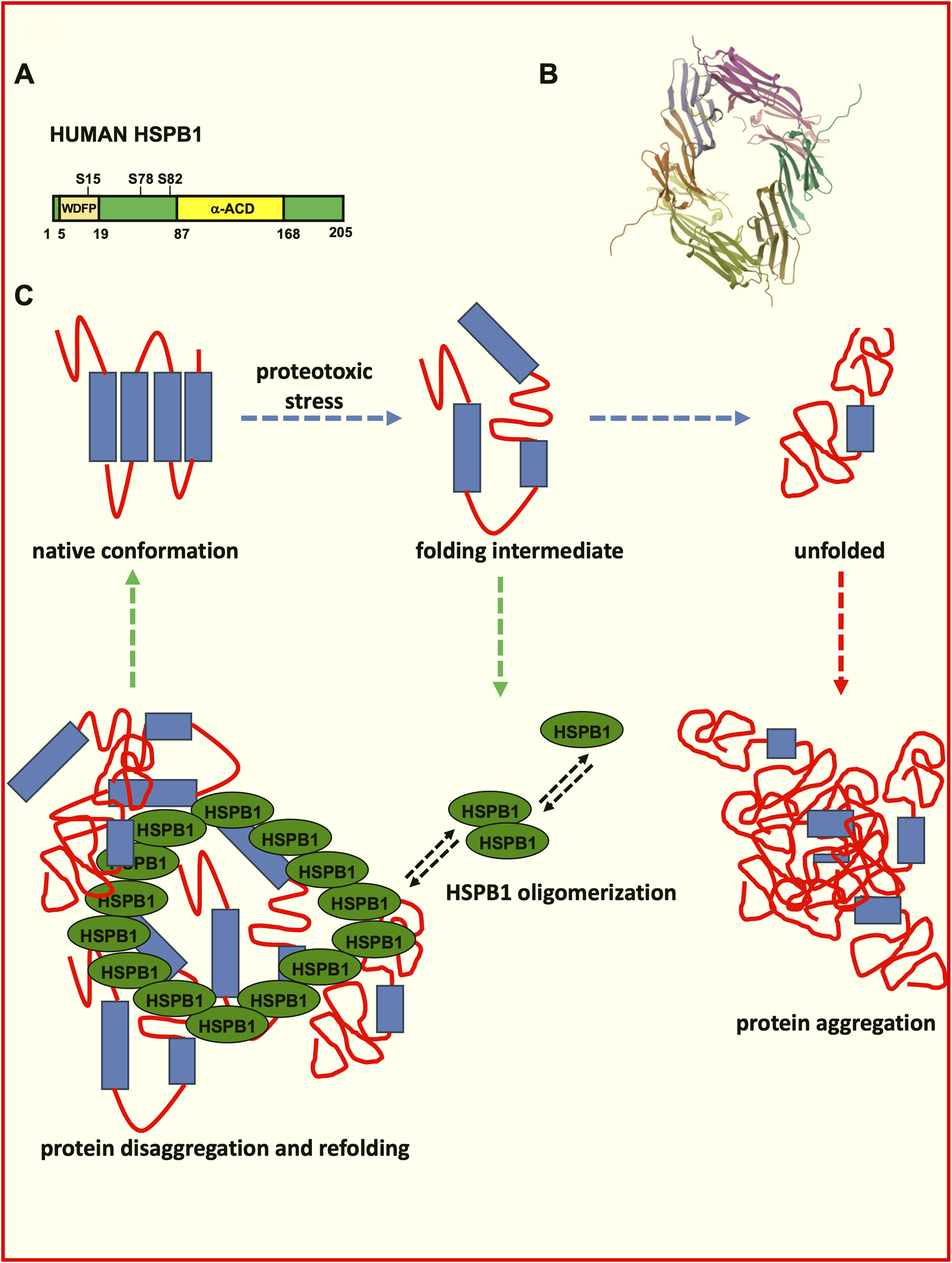
Role of HSPB1 in the control of protein folding and disaggregation. (A) Schematic representation of human HSPB1. The HSPB1 N-terminal domain phosphorylation influences oligomerization, F-actin stabilization and its chaperone activity. The ACD domain influences homo- and hetero-oligomerization, substrate binding and the chaperone activity. (B) Structure of the human Hsp27 (HspB1) alpha-crystallin domain oligomer in complex with a peptide mimic of its phosphorylated N-terminal region (structure deposited at the RCSB Protein Data Bank (PDB) (https://www.rcsb.org/structure/6GJH; DOI: https://doi.org/10.2210/pdb6GJH/pdb). Biological assembly 1 assigned by authors and generated by PISA (software); Biological Assembly Evidence: mass spectrometry; Global Symmetry: Asymmetric; Global Stoichiometry: Hetero 12-mer; Macromolecule Content: Total Structure Weight: 79.68 kDa; Atom Count: 6,007; Modelled Residue Count: 712; Unique protein chains: 4 (Collier et al., 2019). (C) Schematic depiction of the protein misfolding and aggregation and of the role exerted by HSPB1 in the disaggregation process.
Phosphorylation allows sHSPs to adjust their interactions with target proteins, cytoskeletal elements, and other chaperones, thereby optimizing their function during stress responses.
The α-crystallin domain (ACD) plays an important role in protein oligomerization, in fact, having conservative primary structures and being apparently similar, the ACDs of the different sHSPs differ in terms of their dimer stabilities (Figures 1A,B), which can influence the hetero-oligomerization preferences of sHSPs (Giese and Vierling, 2002; Shatov et al., 2023).
2.2 Molecular chaperoning function
The primary function of sHSPs is their role as molecular chaperones, which protect other proteins from damage under stress conditions. They prevent the aggregation of unfolded or misfolded proteins by binding to them, forming stable complexes that keep the damaged proteins soluble and non-toxic. Although sHSPs themselves do not refold proteins, they create a safe environment for proteins to be refolded by larger heat shock proteins, such as HSP70 or HSP90, once the stress subsides (Peters et al., 2024). Therefore, the chaperoning activity plays an important role in regulating protein homeostasis (proteostasis) within the cell (as depicted in Figure 1C). By preventing the formation of insoluble protein aggregates, sHSPs reduce cellular stress and prevent toxic accumulation of misfolded proteins that could otherwise impair cellular function or lead to cell death (Reinle et al., 2022).
Small heat shock proteins do usually display a cytosolic distribution; however, they can be imported into the mitochondrial intermembrane space, where they operate as molecular chaperones. In the mitochondrial intermembrane space, sHSPs prevent protein misfolding and aggregation, which is crucial for mitochondrial integrity and respiration. When sHSPs are depleted, mitochondria exhibit morphological changes, such as swelling and disrupted cristae, without significant impact on membrane potential. Mutations in HSPB1, a major sHSP, impair its mitochondrial import and functionality, suggesting that Charcot–Marie–Tooth-related mitochondrial dysfunction might stem from disrupted sHSP activity (Adriaenssens et al., 2023). In addition, chaperone activity can be increased by O-GlcNAc on some sHSPs, including HSPB1. O-GlcNAc modification of HSPB1 increases its binding to the Co-chaperone protein BAG3, which then promotes refolding of a model substrate by HSP70, suggesting that O-GlcNAc protects against protein aggregation (Javed et al., 2024).
HSPB8 selectively suppresses protein aggregation without affecting the native folding process. This anti-aggregation mechanism is distinct from previous models that rely on the stabilization of unfolded polypeptide chains or partially folded structures, as has been reported for other chaperones. HSPB8 selectively recognizes and binds to aggregated species formed at the early stages of aggregation, preventing them from growing into larger aggregated structures (Choudhary et al., 2023).
2.3 Response to environmental stress and cytoskeleton stabilization
sHSPs are rapidly upregulated in response to a variety of environmental stresses, with heat shock being one of the most studied triggers. When cells are exposed to elevated temperatures, proteins are at risk of denaturation and misfolding. sHSPs bind to these heat-damaged proteins, stabilizing them and preventing irreversible aggregation, which would otherwise result in cellular toxicity. sHSPs confer resistance to several kinds of stresses such as heat, cold, salinity, ethanol, acid, and chemicals (Carra et al., 2017; Waters and Vierling, 2020; Ma et al., 2021). sHSPs sequester denatured proteins in an ATP-independent manner (Rodriguez Ospina et al., 2022; Mogk et al., 2019; Reinle et al., 2022).
sHSPs also play a key role in defending cells against oxidative stress, where reactive oxygen species (ROS) cause protein oxidation and damage. By interacting with oxidized proteins, sHSPs protect them from aggregation and facilitate their repair. Levels of HSPB8, which is highly expressed in the brain, are known to be significantly elevated in cerebral injury models of cerebral oxidative stress, where HSPB8 protects the brain against mitochondrial damage (Wu et al., 2024).
An alternative scenario that can lead to the enhancement of the chaperone activity of sHSP is through the accumulation of substrate proteins (Freilich et al., 2018). Amyloid-forming Tau interacts with the HspB1 ACD, inducing sHSP de-oligomerization. This process exposes additional substrate-binding sites, leading to full sHsp activation and strong Tau binding (Freilich et al., 2018). This suggests that sHSPs can regulate their activity based on substrate availability, independent of external triggers (Freilich et al., 2018). The cytoskeleton, which includes structures like actin filaments and microtubules, is essential for maintaining cellular shape, facilitating movement, and ensuring proper cell division. sHSPs play a crucial role in maintaining the integrity of intermediary filaments (IFs) and actin filaments, particularly HSPB1 and αB-crystallin (HSPB5), are involved in maintaining and stabilizing the cytoskeleton under stress conditions (Liang and MacRae, 1997; Arrigo et al., 2007; Singh et al., 2007; Muranova et al., 2022). HSPB1 is known to bind directly to actin filaments, preventing their depolymerization during stress. This is critical for preserving the structural integrity of the cell during events such as heat shock or oxidative stress.
Charcot-Marie-Tooth (CMT) neuropathy is one of the most common inherited neuromuscular disorders, characterized by the degeneration of peripheral nerves that leads to muscle weakness in the hands and feet (Houlden et al., 2008; El Abassi et al., 2014). Mutations in the small heat shock protein HSPB1 (HSP27) have been identified as a cause of this disease. Additionally, several mutations affecting IFs, including those in the neurofilament light (NFL) protein, have been linked to the syndrome (Zhai et al., 2007). Notably, the HSPB1 (S135F) mutant exhibits stronger chaperone activity and a higher affinity for client proteins compared to wild-type HSPB1 (Almeida-Souza et al., 2011). This enhanced function results in increased affinity for tubulin and microtubules, ultimately improving the stabilization of the microtubule network in neurons isolated from a mouse model expressing the mutant HSPB1 (Almeida-Souza et al., 2011).
αB-crystallin is particularly important in muscle and neuronal cells, where it helps stabilize intermediate filaments, protecting cells from mechanical and environmental stress (Launay et al., 2006).
2.4 Regulation of apoptosis by sHSPs
Apoptosis, or programmed cell death, is a tightly regulated process that allows the body to remove damaged or unnecessary cells. sHSPs, particularly HSPB1, have been shown to act as anti-apoptotic proteins by inhibiting the apoptotic pathways (Chen et al., 2022; Takayama et al., 2003).
In the intrinsic apoptotic pathway, cytochrome c release from mitochondria is a crucial step leading to cell death. sHSPs can prevent this release, blocking the initiation of apoptosis.
sHSPs can also interact with caspases, the enzymes responsible for executing apoptosis, particularly by inhibiting caspase-3 activation. This further suppresses the cell death process, allowing the cell to survive under conditions where apoptosis might otherwise be triggered (Takayama et al., 2003). HSPB1 regulates the stability of BIM, a pro-apoptotic BH3-only protein, leading to protection against ER stress-induced apoptosis. BIM induction by ER stress is suppressed in rat PC12 cells overexpressing HSPB1. CMT-related HSPB1 mutants exhibited increased susceptibility to ER stress-induced cell death and high levels of BIM (Kennedy et al., 2017). HSPB1 can also protect neuronal cells from apoptosis triggered by factors like serum deprivation or nerve growth factor (NGF) withdrawal (Lewis et al., 1999; Wagstaff et al., 1999). Co-expression of HSPB1 with mutant huntingtin reduces cell death in HD models (Perrin et al., 2007; Wyttenbach et al., 2002). Overexpression of phospho-mimetic HSPB1 lost protective effects against poly(Q) overexpression (Wyttenbach et al., 2002). These data suggest a possible role for HSPB1 cellular protection upstream of mitochondrial cell death signaling. HSPB1 may increase aggregate solubilization shifting aggregate formation from large nuclear inclusions to smaller non-nuclear inclusions (Perrin et al., 2007).
Increased levels of HSPB1 protect against apoptosis induced by α-synuclein expression (Benn et al., 2002; Outeiro et al., 2006; Zourlidou et al., 2004). In Alzheimer’s disease (AD) models, HSPB1 bound to Aβ1-40 and decreased aggregation and cytotoxicity in cerebrovascular organotypic cultures (Wilhelmus et al., 2006). Small HSP proteins also decreased Aβ aggregation and toxicity in SH-SY5Y cells (Lee et al., 2006). αB-crystallin prevents the cell death induced Aβ (1-40) fibril formation in PC12 cells. In particular, αB-crystallin inhibited both Aβ (1-40) fibril formation and the associated cell toxicity (Dehle et al., 2010).
2.5 Role in disease protection
sHSPs are closely associated with protection against in several diseases and clinically relevant conditions, particularly those related to protein aggregation and cellular stress, such as neurodegenerative diseases, cancer, and cardiovascular conditions.
Diseases such as Alzheimer, Parkinson, and Huntington are characterized by the accumulation of misfolded proteins in the brain. sHSPs contribute in preventing the aggregation of these disease-related proteins, potentially slowing the progression of neurodegeneration (Escusa-Toret et al., 2013; Muranova et al., 2019; Chaplot et al., 2020; Vendredy et al., 2020).
HSPB1 interact with the autophagy cargo receptor p62/SQSTM1 and regulate the unconventional secretion of the HD-associated mutant HTT protein, but it also regulates synergistically the unconventional secretion of p62/SQSTM1. Importantly the HSPB1 over-expression can also facilitate transcellular spreading of HTT-loaded EVs (Bonavita et al., 2023).
Astrocytes secrete HSPB1 to mediate non–cell-autonomous protective functions. In the human AD brain, we demonstrate that HSPB1 levels increase in astrocytes clustering around amyloid plaques, as well as in the surrounding extracellular space. Furthermore, under conditions mimicking an inflammatory reactive response, astrocytes enhance HSPB1 secretion. Notably, both astrocytes and neurons can uptake astrocyte-secreted HSPB1, leading to a reduction in the inflammatory response in reactive astrocytes and a decrease in pathological tau inclusions (Fangjia et al., 2024).
Moreover, decrease of HSPB1 levels promotes cytoplasmic TDP-43 de-mixing and mis-localization. HSPB1 depletion is identified within ALS-patient spinal motor neurons containing aggregated TDP-43. These findings identify HSPB1 to be a regulator of cytoplasmic TDP-43 phase separation and aggregation (Lu et al., 2022). During ischemia-reperfusion injury, which is observed for instance upon heart attack or stroke, sHSPs protect heart and muscle cells by preventing protein denaturation and stabilizing cellular structures, thus reducing damage and enhancing recovery. HSPB1/Hsp27 is present in the EVs from plasma samples of rats after heart failure indicating a protective effect of Hsp27-overexpressed marrow-derived mesenchymal stem cells exosomes. The results of this study suggest that EVs and their cargo, specifically HSPB1/Hsp27, play a role in the development of heart failure in individuals with type 1 diabetes (Li C. et al., 2024).
HSPB3 protein is expressed in both the central and peripheral nervous system and its over-expression in motor neurons induces their survival after lesion-induced degeneration, indicating its potential role as a modulator of MN survival (La Padula et al., 2016).
Aβ has been identified as a key factor in the pathophysiology of age-related macular degeneration (AMD). Aβ fibrils treatment on wild-type rats and RPE cells induces αB-crystallin increase in retinal astrocytes. Such effect is in line with a role for αB-crystallin in regulating the function of the reactive astrocytes and could be a defence mechanism against apoptosis (Alge et al., 2002; Nagaraj et al., 2005; Ousman et al., 2007). Intestingly, treatment with exosomes derived from adipose tissue mesenchymal stem cells decrease the retinal levels of heat-shock proteins activated by pathogenic Aβ fibrils These results suggest the potential therapeutic target in Aβ-related retinal pathology (Qarawani et al., 2025). In the same way, the transcriptional activation of CRYAB/HSPB5, induced by the inhibition of the glutaminyl cyclase (QPCT) enzyme, affect mutant huntingtin aggregation and toxicity and rescue the HD phenotype (Jimenez-Sanchez et al., 2015).
HSPB6 has been originally identified as a small HSPB member present in skeletal muscle (Kato et al., 1994). Interestingly, the protein has emerged as a novel cardio-protector factor against stress-induced injury. A single point mutation of HSPB6 (HSPB6S10F) has been identified in patients affected by dilated cardiomyopathy (Liu et al., 2018). Interestingly, mutant HSPB6 displays a reduced interaction with the key autophagy regulator Beclin-1, which drives its ubiquitination and proteasomal-dependent degradation. As a result, autophagy flux was substantially inhibited and apoptosis was increased in HSPB6S10F-cells and mouse models (Liu et al., 2018). Recently, it has been also reported that HSPB6 shows a lipid-dependent chaperone activity and regulates aggregation of a-synuclein, a protein associated with Parkinson’s disease (Secco et al., 2024). The ability of HSPBs to interact with lipids and prevent a-synuclein lipid-induced aggregation might be influenced by their degree of disorder, as HSPB6 preferentially inhibit the aggregation of fibril-forming proteins that bind to lipids (Secco et al., 2024).
In neurons, chaperone-assisted selective autophagy promotes the removal of aggregating substrates. HSPB8 acts in a complex with HSPA, their cochaperone BAG3, and the E3 ubiquitin ligase STUB1. De novo frameshift (fs) mutations have been reported in HSPB8 (HSPB8_fs) (Tedesco et al., 2023). Interestingly, HSPB8_fs mutant variants are highly insoluble and tend to form protein aggregates in the cytoplasm. Such a co-partitioning process negatively affects the capability to remove aggregated proteins and can progressively hamper and eventually result in a general failure in the adaptive proteostatic response, indicating a pathogenic mechanism shared by the HSPB8_fs mutants in neuromuscular diseases (Tedesco et al., 2023).
HSPB8 functions as a chaperone interacting with the Bcl-2 associated athanogene 3 (BAG3) during autophagy and promotes the removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS) and other motor neuron diseases, such as TDP-43 (Crippa et al., 2010; Crippa et al., 2016). Consistent with this, HSPB8 mutations causing hereditary distal motor neuropathy have been shown to impair the delivery of autophagosomes to the lysosomal compartment (Kwok et al., 2011), whereas upon HSPB8 can efficiently promote the autophagosome-lysosome heterotypic fusion in neurons in a rat experimental model of diabetes (Li et al., 2017).
2.6 Role in autophagy and protein degradation
sHSPs can also influence cellular quality control systems such as autophagy, a process by which damaged proteins and organelles are degraded and recycled. By interacting with components of the autophagic machinery, sHSPs help direct misfolded or damaged proteins to be removed from the cell, further contributing to cellular proteostasis (Lambert et al., 1999; Arrigo, 2017; Boelens, 2020). Such a role is especially important during aging and in diseases where protein aggregation becomes prominent, as sHSPs help alleviate the burden of damaged proteins through autophagic pathways (Dugger and Dickson, 2017).
Autophagy plays a crucial role in ensuring cell survival under various stress conditions, including protein aggregate formation, nutrient and growth factor deprivation, ER stress, and pathogen infection (Moreau et al., 2010; Rosati et al., 2011). Impaired autophagy has been linked to neurodegeneration, lysosomal storage diseases, muscular dystrophies, cancer, and Crohn’s disease (Moreau et al., 2010). Beyond their chaperone activity, sHSPs offer neuroprotection by reducing oxidative stress and stimulating autophagy, both of which mitigate neuronal toxicity in HD (Wyttenbach et al., 2002; Carra et al., 2009).
The selective autophagy adaptor p62/SQSTM1 plays a crucial role in lysophagy by being recruited to damaged lysosomes and facilitating lysophagic flux. The Phox and Bem1p (PB1) domain of p62 is essential for its oligomerization and specific function in lysophagy. p62 assembles into condensates on damaged lysosomes, and these structures are finely regulated by the small heat shock protein HSP27. Phosphorylated in response to lysosomal damage, HSP27 maintains the liquidity of p62-containing condensates, enabling autophagosome formation. Mutations in p62/SQSTM1, identified in patients with amyotrophic lateral sclerosis (ALS), disrupt lysophagy, linking defects in this pathway to neurodegeneration. Hence, the HSP27-regulated formation of p62/SQSTM1 condensates support lysophagy by serving as platforms for autophagosome biogenesis at sites of lysosomal damage (Gallagher and Holzbaur, 2023).
Huntington’s disease (HD) is an inherited neurodegenerative disorder caused by expanded glutamine repeats in the huntingtin (Htt) protein, leading to abnormal folding and accumulation of mutant Htt. Hsp104 and HSPB1 rescue striatal dysfunction in primary neuronal cultures and HD rat models (Perrin et al., 2007). Polyglutamine (poly-Q) protein fibril aggregation leads to neuronal cell death in HD and spinocerebellar ataxias (SCAs) (Robertson et al., 2010). Proteins such as ataxin-1 (SCA1), ataxin-3 (SCA3), and huntingtin (HD) display a higher propension to aggregate because of a multi-domain misfolding mechanism, which includes poly-Q, poli-A traits and other aggregation-prone motifs or regions (Ellisdon et al., 2006; Thakur et al., 2009). For instance, ataxin-3 aggregation begins with the misfolding of its N-terminal Josephin domain, followed by poly-Q expansion-driven self-association (Ellisdon et al., 2006). αB-crystallin reduces SCA3 toxicity in a Drosophila model, with stronger effects when co-expressed with full-length ataxin-3 rather than a truncated variant lacking the Josephin domain (Bilen and Bonini, 2007). However, αB-crystallin does not prevent fibril formation in poly-Q proteins that aggregate solely through poly-Q expansion (Robertson et al., 2010). It does, however, inhibit the early Josephin domain-dependent stage of ataxin-3 aggregation (Robertson et al., 2010).
HSPB7 has also been identified as a potent suppressor of poly-Q protein aggregation and toxicity, likely through autophagy activation (Vos et al., 2010).
HSPB8 interacts with BAG3, a key activator of (macro) autophagy, to form a complex within cells (Carra et al., 2008). Over-expression of either BAG3 or HSPB8 enhances LC3-II formation, a critical marker of (macro) autophagy (Carra et al., 2008). Additionally, increased HSPB8 expression prevents aggregation of the mutant huntingtin protein (Htt43Q), a pathogenic factor in Huntington’s disease (Carra et al., 2005). The HSPB8-BAG3 complex, along with Hsp70, facilitates macroautophagic degradation of poly-Q protein aggregates, including the Htt-43Q variant (Carra et al., 2008).
HspB8 expression is upregulated in transgenic G93A-SOD1 and selectively removes the neurotoxic mutant SOD1 from motor neuronal cells, restoring normal proteasome activity. This action is specifically mediated by the activation of the autophagosome–lysosome pathway (Crippa et al., 2010).
Remarkably, post-mortem analyses of brain tissue from patients with Alzheimer disease, Parkinson disease, Huntington disease, and spinocerebellar ataxia type 3 (SCA3) have revealed increased HSPB8 and BAG3 expression in astrocytes within affected cerebral areas. This suggests that their upregulation enhances the ability of astrocytes to clear aggregated proteins and cellular debris from damaged neurons (Seidel et al., 2012). Mutations in HSPB8 associated with peripheral neuropathy (K141E and K141N) significantly reduce its efficiency in preventing ataxin-3 and P182L-HSPB1 aggregation, indicating that loss of HSPB8 function may accelerate the progression of these protein misfolding-associated diseases (Carra et al., 2010).
Interestingly, a number of recent studies have raised attention to the possibility of using sHPS as therapeutic target for these conditions. A high-through put screening to find small molecules capable of inducing HSPB8 in neurons for therapeutic purposes identified two compounds, colchicine and doxorubicin, that robustly upregulated HSPB8 expression. Both colchicine and doxorubicin increased the expression of TFEB and of the autophagy marker proteins p62/SQSTM1 and LC3-II. Both drugs counteracted the accumulation of TDP-43 and TDP-25 misfolded species responsible for motoneuronal death in ALS. Thus, analogues of colchicine and doxorubicin able to induce HSPB8 and with better safety and tolerability may result beneficial in neurodegenerative models (Crippa et al., 2016).
On the same line, a novel approach to potentiate a protein degradation system defined as “Chaperone-Assisted Selective Autophagy” (CASA) by using small molecules has been recently validated (Chierichetti et al., 2023). In this study, the chemical screening identified 3 compounds that prevented the formation of mutant SOD1 inclusions by stabilizing HSPB8. These compounds may represent valuable candidates to be tested in pre-clinical studies aimed at counteracting proteotoxicity observed in other neurodegenerative diseases caused by mutant aggregation-prone proteins (Chierichetti et al., 2023).
2.7 Extracellular secretion and function of sHSPs
Beyond their role as intracellular signalling molecules, evidence from the literature suggests that sHSPs are also released into the extracellular environment, where they contribute to intercellular communication and immunomodulation (Reddy et al., 2018; van Noort et al., 2012). Despite their presence outside the cell, the mechanisms underlying the secretion of sHSPs and their extracellular functions are less well characterized, if compared to larger HSPs (Reddy et al., 2018). Among the sHSP family members, HSPB1 is the most extensively studied, with elevated serum levels observed in patients with diabetic neuropathy (Gruden et al., 2008).
One proposed mechanism for HSPB1 secretion is via extracellular vesicle (EVs) formation (Nafar et al., 2016; Shi et al., 2019). Increasing evidence indicates that HSPB1 is associated with EVs. ACD domain of HSPB1 interact with specific lipid membrane components, including palmitoyl oleoyl phosphatidylserine, phosphatidylcholine, and phosphatidylglycerol in liposomes. As such, the association with lipid membranes may facilitate HSPB1 export (De Maio, 2011). HSPB1 has been detected in EVs isolated from B-lymphoblastoid cells, and it was shown that HSPB1 may reside inside the vesicles rather than on their surface (Rayner et al., 2009). In contrast, an ultrastructural study using transmission electron microscopy identified HSPB1 also on the EV membrane (Shi et al., 2019). Furthermore, exposure to amyloid-beta (Aβ) was shown to induce the release of membrane-bound HSPB1 from EVs in primary rat cortical astrocytes (De Maio, 2011). Collectively, the precise localization of sHSPs in EVs and whether they function solely as cargo or also as targeting molecules remains unclear.
Immunoprecipitation experiments have shown that extracellular HSPB1 can interact with extracellular Aβ, the primary component of amyloid plaques found in the brains of Alzheimer’s disease patients (Nafar et al., 2016). Additionally, HSPB1 has been found to bind α-synuclein fibrils associated with Parkinson’s disease and dementia, inhibiting their elongation and cytotoxicity. However, it remains unclear whether HSPB1 specifically binds extracellular α-synuclein (Cox et al., 2018).
While some studies suggest that extracellular HSPB1 is secreted and upregulated in cancer (Feng et al., 2005; Banerjee et al., 2011; Fanelli et al., 1998) and cardiovascular diseases (Martin-Ventura et al., 2004; Park et al., 2006), its defined extracellular roles in the context of neurodegenerative disorders are still not well understood. In contrast, the extracellular function of HSPB5 in neurodegenerative diseases has been more extensively studied. For instance, Zhu et al. demonstrated that extracellular HSPB5 protects astrocytes from cell death induced by staurosporine and C2 ceramide. Furthermore, they showed that extracellular HSPB5 enhances astrocyte viability through the PI3K/Akt/mTOR and ERK1-2/p38/JNK signalling pathways under serum-deprived conditions (Zhu et al., 2015). Another study found that extracellular HSPB5 suppresses astrocyte- and microglia-mediated inflammatory responses in both autocrine and paracrine manners. These findings highlight a novel role for extracellular HSPB5 in regulating neuroinflammation and suggest that targeting HSPB5-modulated neuroinflammation could be a potential therapeutic approach for MS (Guo et al., 2019).
Further supporting this, a Phase IIa clinical trial demonstrated that repeated administration of HSPB5 resulted in a progressive reduction in MS lesion activity (van Noort et al., 2015). Despite the established roles of HSPB1 and HSPB5, research on the secretion mechanisms and functions of other sHSP members in neurodegenerative diseases remains limited.
3 The autophagic machinery and regulation of the autophagic pathway
3.1 Core components of the autophagic machinery
Autophagy is a highly conserved cellular process that maintains cellular homeostasis by degrading and recycling damaged organelles and proteins. Autophagy, derived from the Greek words “auto” (self) and “phagy” (eating), describes a cellular mechanism for degrading and recycling cellular components (Bento et al., 2016). Autophagy has emerged as a vital component of cell survival, development, and homeostasis. Dysregulation of autophagy is implicated in various diseases, including neurodegeneration, cancer, and infectious disease (Bento et al., 2016).
Autophagy progresses through sequential stages: initiation, nucleation, elongation, closure, and degradation (Bento et al., 2016). The initiation of autophagy begins with the formation of the phagophore, a precursor membrane structure. The ULK1 complex, consisting of ULK1 (Unc-51-like kinase 1), ATG13, FIP200, and ATG101, is essential for this stage. Once the phagophore is formed, nucleation occurs, enabling the recruitment of essential proteins and lipids. The class III PI3K complex, consisting of VPS34, Beclin-1, ATG14, and p150, plays a pivotal role in this process. VPS34 generates phosphatidylinositol-3-phosphate (PI3P) at the phagophore, which acts as a recruitment site for downstream autophagic proteins. Beclin-1 is particularly significant in this complex, as its binding partners can either activate or inhibit autophagy, demonstrating its role as a regulatory hub. The elongation and closure of the autophagosome membrane require the conjugation systems ATG12-ATG5-ATG16L1 ternary complex. This system involves the conjugation of ATG12 to ATG5, which then interacts with ATG16L1 (Mizushima et al., 2011; Sica et al., 2015; Zhao et al., 2021). The complex attaches to the phagophore membrane, contributing to autophagosome elongation. ATG4 processes LC3 to its cytosolic form, LC3-I. Upon activation, LC3-I conjugates to phosphatidylethanolamine (PE) on the autophagosome membrane, forming LC3-II. This form of LC3 is essential for the expansion of the autophagosome and is widely used as a marker for autophagosome formation (Fujita et al., 2008; Lystad et al., 2019). Mature autophagosomes fuse with lysosomes to form autolysosomes, where lysosomal enzymes degrade the inner membrane and enclosed materials. Key players in this fusion process include SNARE proteins, Rab GTPases, and the homotypic fusion and protein sorting (HOPS) complex. Following fusion, the contents are broken down into macromolecules, which are eventually recycled back into the cytoplasm (Mizushima et al., 2010).
3.2 Regulation of the autophagic pathway
The regulation of autophagy is complex and tightly controlled by nutrient availability, energy levels and stress signals. Several major pathways and molecules influence autophagy regulation: The mTOR signalling pathway is one of the central regulators of autophagy. In nutrient-rich conditions, mTORC1 phosphorylates and inhibits the ULK1 complex, thereby suppressing autophagy. Under stress or nutrient deprivation, mTORC1 is inhibited, which relieves the suppression of ULK1, leading to autophagy activation (Nakatogawa, 2020). The mTOR pathway integrates signals from amino acids, growth factors and cellular energy status, making it a crucial regulator of autophagy (Kawabata and Yoshimori, 2016; Yu et al., 2018; Zhao et al., 2021).
AMP-activated protein kinase (AMPK) is an energy sensor that activates autophagy in response to low cellular ATP levels. AMPK directly phosphorylates ULK1, promoting autophagy initiation and can also inhibit mTORC1, thus enhancing autophagy indirectly. AMPK is activated in conditions of glucose deprivation and other forms of metabolic stress, highlighting its role as a cellular energy regulator (Kim et al., 2002; Kim et al., 2008; Saxton and Sabatini, 2017). In addition, several transcription factors, including TFEB, FOXO, NF-kB and p53 can regulate the expression of autophagy-related genes. Under nutrient starvation, TFEB translocates to the nucleus, up-regulating genes associated with autophagosome formation and lysosome biogenesis (Settembre et al., 2011). FOXO transcription factors can induce autophagy by up-regulating autophagy-related genes under conditions of oxidative stress or growth factor deprivation (Schäffner et al., 2018).
3.3 Functional links between HSPB1, actin cytoskeleton organization and regulation of autophagy
Historically, HSPB1 has been identified as a major regulator of actin cytoskeleton assembly (Collier and Schlesinger, 1986; Lavoie et al., 1995; Miron et al., 1988; Miron et al., 1991; Wettstein et al., 2012). In this context, HSPB1 might plays a role in the actin remodelling that is induced by mechanical stimulation. HspB1 physically associate with the actin cytoskeleton and contributes to the reinforcement of actin stress fibres upon mechanical stress. This phenomenon can be regulated by the p38MAPK-dependent phosphorylation in response to signalling and mechanical cues, reinforcing the role exerted by HSPB1 in actin cytoskeleton remodelling, cell growth control and cell migration (Clarke and Mearow, 2013; Hoffmann et al., 2019; Park et al., 2018).
It has been recently reported how HSPB1 is involved in the regulation of autophagy in astrocytes by means of ER stress and the purinergic receptor P2X7 (Kim J.E. et al., 2017; Kim J.E. et al., 2018).
In addition, most mutations in HSPB1 can cause axonal Charcot-Marie-Tooth neuropathy and/or distal hereditary motor neuropathy (Evgrafov et al., 2004; Echaniz-Laguna et al., 2017) and sporadic amyotrophic lateral sclerosis (ALS) (Capponi et al., 2016). As already mentioned, mutations in HSPB1 lead to impairment of autophagic flux (Haidar et al., 2019). In particular, the authors show how such an effect is mediated by the physical interaction with the autophagy receptor p62/SQSTM1 and the ability to form p62-bodies and phagophores, a function which is impaired by CMT-causing mutations. Noteworthy, the proteostatic activity of p62/SQSTM1 and its phosphorylation status have been recently shown to be regulated by the HSF1 stress response pathway (Watanabe et al., 2017), which is a master regulator of HSPB1 (De Thonel et al., 2012). Intriguingly, mutations in both HSPB1 and p62/SQSTM1 have been associated with ALS (Capponi et al., 2016; Teyssou et al., 2013).
Although autophagy is primarily considered a non-selective degradation process induced by nutrients deprivation, nutrient-independent basal autophagy can still impose intracellular quality control mechanisms by operating selective disposal of aberrant protein aggregates and damaged organelles, such as mitochondria (Bento et al., 2016). One of the molecular mechanisms that makes possible to switch between these two autophagic responses is linked to the re-organization of the actin cytoskeleton (Kast and Dominguez, 2017). One of the first evidence supporting a role for the actin cytoskeleton in regulating autophagy came from a study performed in yeast, where the authors showed how mutations in genes encoding subunits of the Arp2/3 complex interfered with Atg9 function. In particular, the actin nucleation by the Arp2/3 complex promotes the movement of Atg9-positive structures and the actin remodelling serves as a scaffold that is necessary for the function of autophagic membranes under non-limiting growth conditions (Reggiori et al., 2005; Monastyrska et al., 2008).
Indeed, despite the fact that autophagosome trafficking and fusion with the lysosomal compartment does largely rely on the microtubule-dependent transport system, a role for the actin cytoskeleton has been described for both the autophagosome biogenesis and maturation steps.
In particular, Lee et al. reported how the ubiquitin-binding histone deacetylase-6 (HDAC6) is an essential component for the regulation of basal autophagy that targets protein aggregates and damaged mitochondria (Lee JY et al., 2010). Surprisingly, HDAC6 is not required for autophagy activation, but it rather controls the autophagosome-lysosome heterotypic fusion, by recruiting a cortactin-dependent, actin-remodelling machinery, which in turn assembles an F-actin network that stimulates autophagosome-lysosome fusion and substrate degradation. Indeed, HDAC6 deficiency leads to autophagosome maturation failure, protein aggregate build-up, and neurodegeneration. Remarkably, at least in their experimental set-up, the authors reported how HDAC6 and F-actin assembly were completely dispensable for starvation-induced autophagy, (Lee JY et al., 2010). However, in another study it has been reported how the actin cytoskeleton participates in the early events of autophagosome formation, upon starvation induced autophagy (Aguilera et al., 2012). In particular, the authors showed that actin filaments colocalized with ATG14, BECN1/Beclin1 and PI3P-enriched structures, indicating that actin has a role at very early stages of autophagosome biogenesis, linked to the PI3P and omegasome formation early step. Consistent with the latter study, genetic or pharmacological perturbation to the actin cytoskeleton can impact the rate of clathrin-dependent endocytosis, which impacts autophagosome precursor formation and autophagosomes biogenesis (Moreau et al., 2015; Renna et al., 2013; Zavodszky et al., 2014).
4 Intersections between autophagy and conventional secretion pathways in cellular homeostasis
Autophagy and conventional secretion are fundamental cellular pathways that were once considered largely independent processes. Autophagy, an intracellular degradation and recycling pathway, is primarily responsible for removing damaged or superfluous organelles and proteins, contributing to cellular health and adaptation to stress. In contrast, conventional secretion functions in the transport of newly synthesized proteins and lipids from the endoplasmic reticulum (ER) and Golgi to the plasma membrane or extracellular space. Recent findings, however, demonstrate significant molecular and functional intersections between these pathways.
Both autophagy and conventional secretion are vital for maintaining cellular homeostasis. Autophagy serves as a cellular “cleanup” mechanism by degrading dysfunctional cellular components, while secretion ensures the transport of critical biomolecules outside the cell or to the plasma membrane. Traditional views held that these processes are separate, with autophagy focused on degradation and secretion dedicated to biosynthesis and transport. However, emerging research reveals shared regulators and molecular crosstalk, especially under stress conditions, highlighting an intricate balance between degradation and export in cellular health.
4.1 Mechanisms of autophagy and conventional secretion
Conventional secretion is characterized by a pathway originating from the ER, where proteins are synthesized, then processed and sorted in the Golgi apparatus (Figure 2). In the ER, newly synthesized proteins undergo initial folding and quality control. Then properly folded proteins are transported to the Golgi for further modification, such as glycosylation. Vesicles bud off from the trans-Golgi network and are directed to the plasma membrane or endosomal compartments. The distinct functions of autophagy and secretion may imply independence, but studies have revealed overlapping regulatory machinery and co-dependency under specific contexts (Bonifacino and Glick, 2004; Jena, 2007). Secreted proteins constitute approximately 9%–15% of the total human proteome and play crucial roles in cellular physiology and intercellular communication (Wu and Krijgsveld, 2024; Rabouille, 2017). Protein can be secreted through either classical or non-classical pathways. In the classical pathway, proteins are directed to the cell exterior by signal peptides (Guo et al., 2018). Signal peptide-containing proteins are specifically recognized in the cytoplasm by the signal recognition particle and localize to the ER. These peptides are typically located at the N-terminus of the protein and usually comprises a positively charged n-region, a hydrophobic h-region, and a signal peptidase recognition site (Guo et al., 2018). Secretory cargo is produced and assembled in the ER before being transported to the Golgi complex. In the Golgi, essential post-translational modifications, such as glycosylation, occur, regulating protein function and stability in the extracellular environment (Ramazanov et al., 2021; Lippincott-Schwartz et al., 2000). At the trans-Golgi network (TGN), proteins are sorted and packaged into post-Golgi carriers, which travel through the cytoplasm and their fusion with the plasma membrane is directed by cargo-containing coat protein complex II (COPII) and Rab proteins (Ramazanov et al., 2021). Most proteins follow this classical pathway, but proteins that do not contain identifiable signal peptides and are secreted by non-classical mechanisms that bypass the Golgi (Grieve and Rabouille, 2011; Dimou and Nickel 2018; Rabouille, 2017). This alternative route of secretion is defined unconventional protein secretion (UPS) and includes several mechanisms. Most of the proteins secreted by UPS lack targeting signal sequences, but also transmembrane proteins with or without signal sequences can be unconventionally secreted (Grieve and Rabouille 2011; Dimou and Nickel 2018; Rabouille 2017).
FIGURE 2
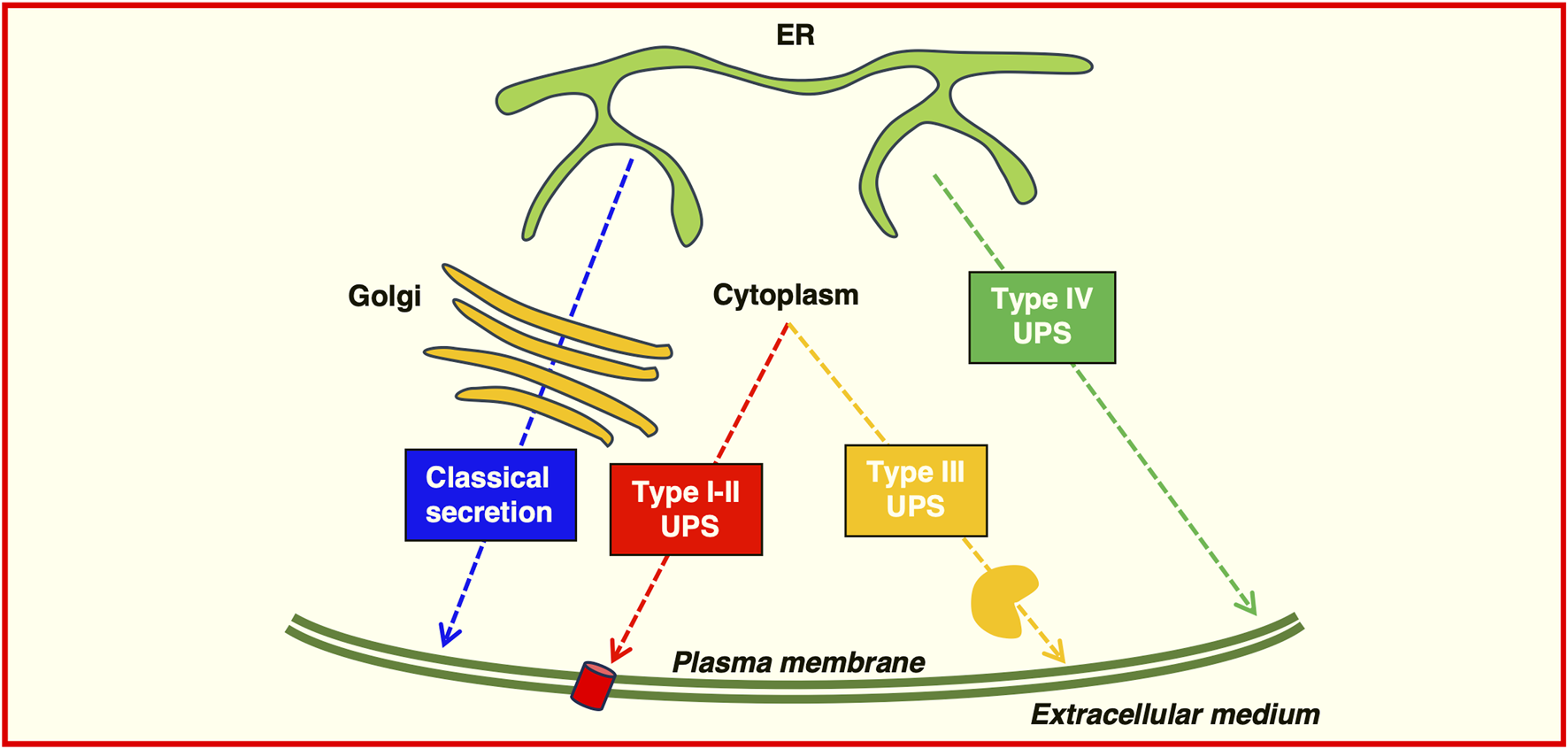
Possible routes of protein secretion. Proteins can be transported to the plasma membrane or secreted extracellularly by using the classical secretory pathway (blue) or the Golgi-bypass pathway (Type IV) unconventional protein secretion (green). Leaderless proteins can be transported using Type I-II unconventional protein secretion, translocating across the plasma membrane through a pore (red) or directly by membrane budding or shedding; Type III unconventional protein secretion requires a membrane-bound organelle carrier (yellow).
Four distinct classes of unconventional secretory mechanism have been identified (as depicted in Figure 3A).
FIGURE 3
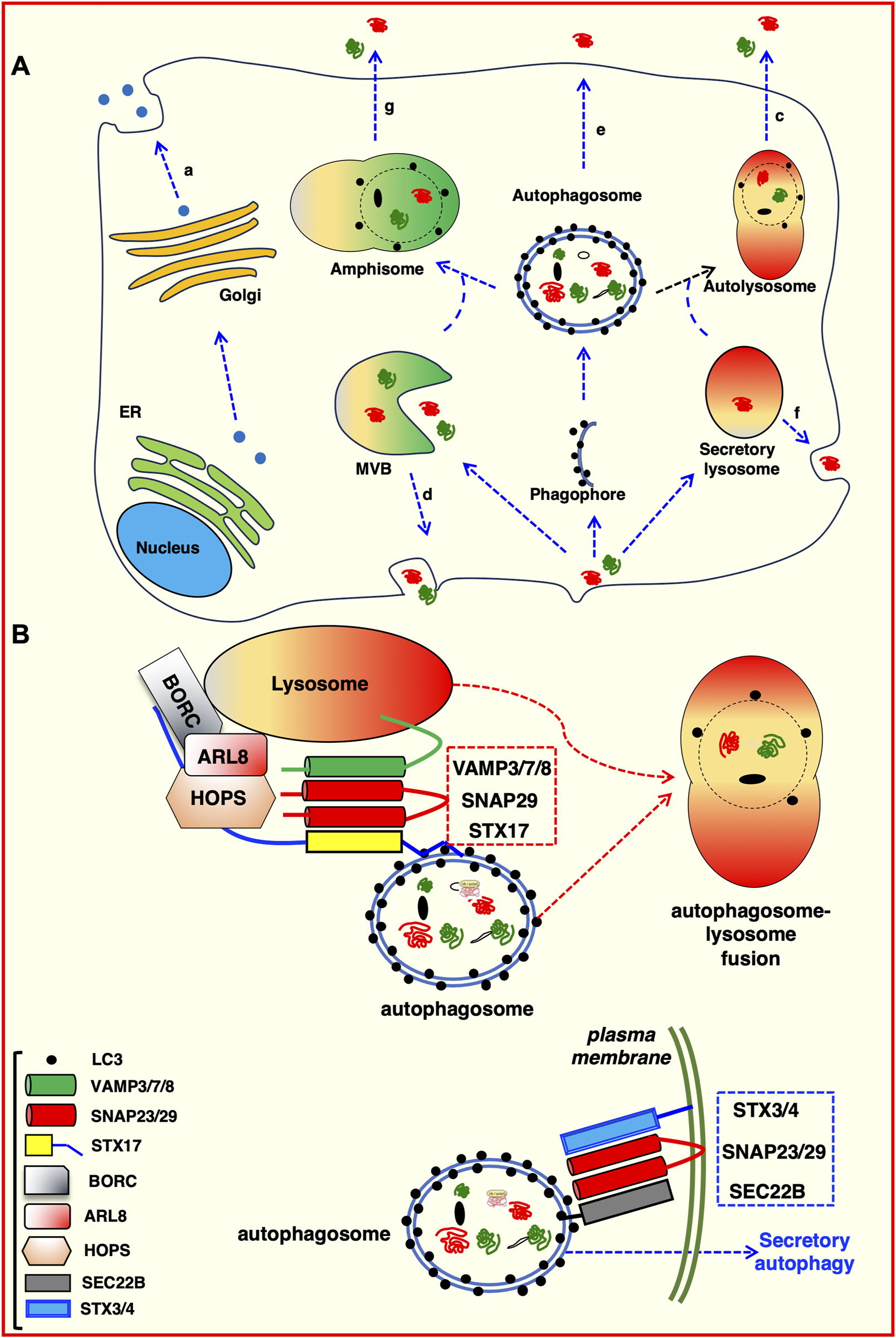
Intersections between autophagic pathway and unconventional protein secretion. (A) Proteins can be secreted using the canonical protein secretion pathway trough the Golgi to the plasma membrane (a). Alternatively, they can follow unconventional secretion pathways. For example: Secretory lysosome-mediated plasma membrane fusion (b); direct fusion of autolysosomes with the plasma membrane (c); exosomes generated in MVBs and released by fusion at the plasma membrane (d); direct fusion of either autophagosomes (e) or lysosomes (f) with the plasma membrane; amphisome-mediated secretion (g). (B) Molecular determinants of degradative and secretory autophagosomes. Trafficking and intracellular transport of autophagosomes depends on the proteins decorating the outer membrane. Both degradative and secretory routes are labelled with LC3 and require SNAREs–mediated processes (Wang et al., 2016). The STX17 SNARE (with the assistance of VAMP7 and SNAP29) directs heterotypic fusion of the degradative autophagosome with the lysosomal compartment (red dotted box), which allows the degradation of autophagosome content (Itakura et al., 2012; Jiang et al., 2014). Conversely, the SEC22B SNARE, in co-operation with STX3/4 and SNAP23/29 (blue dotted box), directs an autophagosome toward the plasma membrane for secretion (Kimura et al., 2017).
First class are the soluble proteins that can be secreted through direct protein translocation across pores in the plasma membrane despite the lack of a signal peptide or a transmembrane domain; second class are soluble proteins transported through ABC transporters after acylation. Third class are soluble proteins secreted by autophagosome/endosome-based secretion fusion with the plasma membrane. The fourth class are plasma membrane resident integral membrane proteins that bypass the Golgi (Grieve and Rabouille, 2011; Dimou and Nickel, 2018; Rabouille, 2017). These pathways share common features, in particular unconventional secretion is largely triggered by cellular stress such as nutrient stress, ER stress, mechanical stress, or inflammation. This stress may cause impairment in the functional integrity of the classical secretory pathway inducing the activation of an alternative secretion mechanism.
4.2 Common regulatory proteins
Over the last 2 decades, a number of studies have demonstrated the existence of multiple regulatory proteins and pathways shared between autophagy and secretion, suggesting that these two processes may rely on overlapping signalling networks. Among the others, the mammalian target of rapamycin (mTOR), a central regulator of cellular growth that inhibits autophagy under nutrient-rich conditions, can also influence the secretory pathway, particularly in controlling the biogenesis of secretory vesicles and their exocytosis (Koscielny et al., 2021). AMPK, a metabolic sensor that activates autophagy during energy stress, also modulates protein trafficking and secretion (O’donnell and Schmidt, 2019). In response to nutrient availability, mTORC1 phosphorylates GRASP55, inactivating the GRASP55-mediated unconventional secretion (Table 2). TASCC, a specialized cellular compartment, facilitates the biosynthesis of secretory proteins such as interleukin-6/8 (Table 2).
TABLE 2
| Protein | Function | References |
|---|---|---|
| GRASP55 | GRASP55 interacts with ATGs family’s members and is required for the unconventional protein secretion of interleukin 1b, F508 mutant CFTR. Bridges together LC3II and LAMP2 to facilitate the autophagosome-lysosome fusion. It stabilizes the translocon p23/TMED10, allowing the secretion of mutant HTT |
Kinseth et al. (2007); Dupont et al. (2011) Ge et al. (2013) Deretic et al. (2012) Ahat et al. (2022) |
| Acb1 | Acyl coenzyme A-binding protein 1 (Acb1) requires autophagy and the t-SNARE Sso1 for the fusion and release of the Acb1-containing vesicles into the extracellular space |
Bruns et al. (2011)
Duran et al. (2010) |
| Sec22B | The protein is usually involved in physiological cell trafficking between ER and Golgi. Furthermore it is involved in unconventional secretion, regulating the cargo loading in autophagosomes and promoting their secretion at the plasma membrane | Adnan et al. (2019) |
| p23 (TMED10) | Functions as a traslocon for cytosolic proteins to be translocate to the ERGIC, from which they are transferred in autophagosome | Zhang et al. (2020); Sun et al. (2024) |
| ATGs | In osteoclasts ATGs family’s members are essential to promote the secretory function of osteoclasts for bone resorption | DeSelm et al. (2011) |
| p62/SQSTM1 | Autophagy cargo receptor; forms a functional complex with HSPB1, that acts as a cargo selection platform allowing their EVs-dependent secretion | Bonavita et al. (2023) |
| mTORC1 | In response to nutrient availability, mTORC1 phosphorylates GRASP55, inactivating the GRASP55-mediated unconventional secretion | Nüchel et al. (2018) |
| SKP1 | Its phosphorylation states regulates either the degradation or the unconventional secretion of amphisomes-derived cargoes | Li et al. (2023b) |
| TPPP/p25⍺ | Inhibits deacetylase HDAC6, promoting anterograde transport of amphisome towards cell membrane | Ejlerskov et al. (2013) |
| TASCC | The TOR-autophagy spatial coupling compartment (TASCC), is a specialized cellular compartment, which comprises both signalling and degradative elements, induced in response to senescence; it facilitates the biosynthesis of secretory proteins such as interleukin-6/8 | Narita et al. (2011) |
| CFTR | The ΔF508 CFTR mutant is usually misfolded and degraded in proteasome. However, during stress conditions the mutant CFTR is transported to the cell surface, bypassing the Golgi |
Gee et al. (2017)
Noh et al. (2022) |
| IDE | The insuline degradative enzyme (IDE) is a natural efficient protease of amyloid beta peptide (Aβ). Despite the lack of a signal sequence for secretion, IDE can undergo autophagy-mediated unconventional secretion | Son et al. (2016) |
Autophagy related proteins and their functional intersection with unconventional and conventional secretion.
Soluble NSF Attachment Protein Receptors (SNAREs) and Rab GTPases are critical for vesicular trafficking in both autophagy and secretion. SNARE proteins such as syntaxin-17, which mediates the fusion of autophagosomes with lysosomes, also participate in the regulation of exocytosis in conventional secretion. Rab proteins, especially Rab8 and Rab11, play dual roles in autophagosome trafficking and secretion (Ding et al., 2019; Gyurkovska et al., 2023; Wu et al., 2023; Lin et al., 2024).
The ER and Golgi apparatus serve as points of convergence, influencing both autophagy and secretion through organelle crosstalk. For instance, the ER-to-Golgi Intermediate Compartment (ERGIC) provides membrane sources for autophagosome formation (Ge et al., 2013; Ge et al., 2014; Ge et al., 2017; Li S. et al., 2022). Importantly TMED10 functions as a translocator for leaderless cytosolic proteins, to be translocated to the ERGIC, from which they are transferred in autophagosomes (Zhang et al., 2020; Sun et al., 2024) (Table 2).
Furthermore, secretory vesicles derived from the Golgi can be often redirected to the autophagic pathway under stress conditions, highlighting a dynamic interchange (Cerrato et al., 2021). Sec22b a protein involved in trafficking between ER and Golgi, is also involved in unconventional secretion, regulating the cargo loading in autophagosomes and promoting their secretion at the plasma membrane (Adnan et al., 2019). GRASP55, a Golgi resident membrane protein, is required for the unconventional protein secretion of interleukin-1β (IL-1B), the ΔF508 mutant CFTR and is involved in the autophagosome-lysosome fusion bridging LC3 II and Lamp2 (Kinseth et al., 2007; Deretic et al., 2012) (Table 2).
5 Secretory autophagy as a form of unconventional protein secretion
Despite autophagy has been considered a cellular process primarily associated with the degradation and recycling of cellular components, especially under nutrient starvation, recent research reveals that autophagy also plays a role in unconventional protein secretion, termed “secretory autophagy”.
Secretory autophagy is a specialized form of autophagy involved in the transport and release of cellular cargo, particularly cytosolic proteins, to the extracellular environment (Figure 3A). Unlike canonical autophagy, which is primarily focused on the degradation of cytoplasmic components via lysosomal pathways, secretory autophagy mediates the selective export of certain proteins that lack a signal peptide and cannot access the traditional endoplasmic reticulum (ER)-Golgi secretory pathway. As such, this biological process has broad implications for cell signalling, immune responses and the cellular stress response. Cargo proteins selected for secretory autophagy often have specific structural motifs or post-translational modifications that mark them for this pathway. Proteins such as interleukin-1β (IL-1β) (Lopez-Castejon and Brough, 2011) and galectins (Bänfer et al., 2018; Davuluri et al., 2021; Popa et al., 2018), are known to be secreted through this route. These proteins generally lack signal peptides, which makes them incompatible with the conventional ER-Golgi pathway.
Some cytosolic proteins undergo specific modifications, such as phosphorylation (D’Agostino et al., 2019), ubiquitination or lipid modifications, which can make them recognizable by selective autophagy receptors (Hutchings et al., 2024).
Autophagy receptors, such as p62/SQSTM1, Optineurin, NBR1 and TOLLIP are known to mediate selective autophagic degradation. However, certain receptors can recognize and bind specific cargoes for secretory autophagy, tagging them for encapsulation within autophagosomes for eventual export rather than degradation. These receptors and adaptors can bind both the cargo and components of the autophagy machinery (e.g., LC3/GABARAP proteins) (Turco et al., 2019; Merkley et al., 2020; Wang et al., 2021; Gallagher and Holzbaur, 2023). Proteins bearing the LC3-interacting regions (LIR) may be selectively recognized by the autophagosome membrane. This binding allows LC3 or other autophagy-related (ATGs) proteins to facilitate cargo recruitment and packaging into autophagosomes.
As detailed in the next paragraph, secretory autophagy relies on specialized autophagosomes that differ in both composition and fate from those involved in canonical autophagy. The autophagosomes in secretory autophagy are often assembled in response to specific signalling cues, such as inflammatory signals or cellular stress. While many core autophagy proteins are involved (e.g., ULK1, Beclin-1, ATG5-ATG12), the regulation and membrane dynamics can vary to support cargo secretion (Table 2).
Secretory autophagy is classified as a form of unconventional protein secretion (UPS), which bypasses the classical endoplasmic reticulum (ER) and Golgi-dependent secretory pathways (Figure 3A). Secretory autophagy has been observed across a range of organisms and has been linked to several cellular processes, such as immune modulation, inter-cellular signalling, and tissue remodelling (Deretic et al., 2012). Secretory autophagy, like conventional autophagy, begins with the formation of autophagosomes. However, the selection of cargo differs markedly. Differently from canonical autophagy, where autophagosomes typically fuse with lysosomes for degradation, autophagosomes in secretory autophagy bypass lysosomal fusion. Instead, these autophagosomes may fuse with the plasma membrane, multivesicular bodies, or secretory vesicles, leading to the release of their contents into the extracellular space (D’Agostino et al., 2019; Padmanabhan and Manjithaya, 2020).
In secretory autophagy, cargo selection is often regulated by specific adaptor proteins or receptor-ligand interactions that recognize proteins destined for secretion. Notably, some secreted proteins, such as IL-1β and annexins, are selectively recruited to autophagosomes by specific mechanisms involving chaperones, lysosomal membrane proteins (e.g., LAMP2A), or components of the ESCRT (Endosomal Sorting Complex Required for Transport) machinery (Lopez-Castejon and Brough, 2011; Popa et al., 2018; Williams et al., 2023).
After cargo loading, autophagosomes destined for secretion bypass lysosomal degradation and instead fuse with the plasma membrane, releasing their contents directly into the extracellular space (Figure 3B). This pathway is modulated by proteins like RAB8A and RAB27A, which are involved in trafficking and membrane fusion. The precise regulation of vesicle fusion is critical to maintaining a balance between degradative autophagy and secretory autophagy, a balance that may be influenced by cellular stress levels and the specific needs of the organism (Fukuda, 2013; Chen et al., 2017). Several signalling pathways are known to modulate secretory autophagy, including the mTOR, AMPK, and ULK1 pathways. mTOR, a well-established regulator of conventional autophagy, also influences secretory autophagy, especially in response to nutrient availability. When nutrient levels are low, mTOR activity is suppressed, leading to the activation of autophagy. However, in cases where cellular stress triggers specific protein secretion, mTOR-independent pathways (e.g., the JNK pathway) can promote secretory autophagy (Cruz-Garcia et al., 2018). In addition, inflammatory signals, including TNF-α and IL-1β, have been shown to enhance secretory autophagy. Under such conditions, autophagy-related proteins ATG5 and ATG7, typically associated with the autophagosome formation pathway, are essential for secretory autophagy. In contrast, certain inflammatory mediators can bypass canonical autophagy proteins, suggesting alternative forms of autophagy-related secretion that still remain not completely understood (Cruz-Garcia et al., 2018; Gonzalez et al., 2020).
Secretory autophagy is implicated in the release of pro-inflammatory cytokines, particularly IL-1β and IL-18, both of which play crucial roles in immune responses. Unlike conventional secretion, secretory autophagy allows cells to release these cytokines in response to cellular stress, infections, or injury, even when the canonical secretion pathways are blocked. This function has been observed in macrophages and other immune cells that rely on secretory autophagy to initiate or propagate inflammatory responses (Chen et al., 2017; Martinelli et al., 2021; Weigert and Herhaus, 2023).
Secretory autophagy also plays a role in responding to cellular stress. For example, under oxidative stress or hypoxia, cells can release cytoplasmic components via secretory autophagy, thereby maintaining cellular homeostasis. By externalizing misfolded or aggregated proteins, cells protect themselves from cytotoxic effects, which is particularly relevant in neurodegenerative disorders, such as Alzheimer’s disease, where protein aggregates contribute to pathology (Li Q. et al., 2024).
Notably, disruptions in secretory autophagy are associated with various pathologies. Aberrant secretory autophagy has been implicated in chronic inflammatory diseases, such as rheumatoid arthritis, where excessive release of IL-1β contributes to sustained inflammation. Similarly, neurodegenerative diseases such as Parkinson’s and Huntington’s disease may involve altered autophagy pathways, leading to inappropriate release of neurotoxic proteins or failure to degrade misfolded proteins. In cancer, dysregulated secretory autophagy can enhance the aggressiveness of tumours, impacting metastasis and immune evasion (Wu et al., 2022; Debnath et al., 2023; Piletic et al., 2023).
5.1 Molecular mechanisms regulating secretory autophagy
The concept of autophagy-dependent secretion originally stems from observations of the unconventionally secreted protein Acb1 (Acyl-CoA binding protein, Dictyostelium homolog: AcbA). Under autophagy-enhancing conditions, such as low nitrogen concentrations, yeast cells secrete increased levels of Acb1 (Duran et al., 2010). Following individual knockdown of autophagy components, Atg5, Atg7, Atg8, and Atg12, Acb1 secretion significantly decreases, despite constant Acb1 concentrations within the cell. This foundational report suggested a role for the autophagosome in secretion of extracellular proteins (Duran et al., 2010). Additionally, Acb1 was determined to be secreted by means of a membrane-bound intermediate (Cabral et al., 2010). The connection of autophagic machinery with type III UPS arose from observations surrounding the secretion of Acb1. Acb1 secretion is dependent upon nutrient starvation and several autophagy-mediating proteins (Duran et al., 2010; Manjithaya et al., 2010). However, Acb1, and type III UPS as a whole, also has resemblances to endosome trafficking. Endosome sorting components, such as Stp22/Vps23, Grh1, and the endosome-specific t-SNARE Tlg2 are necessary for the secretion of Acb1 (Duran et al., 2010; Kinseth et al., 2007). Grh1 is trafficked to a unique, cup-shaped membrane upon nutrient starvation. This membrane is termed the compartment for unconventional protein secretion (CUPS). The CUPS contains both endosomal sorting components, such as Stp22, as well as autophagic machinery, such as Atg8 and Atg9 (Bruns et al., 2011). CUPS associations with autophagy include that it is induced by nutrient starvation (specifically glucose starvation), and that Atg8, Atg9, and a pool of PtdIns3P are necessary for its development. However, CUPS do not lead to an LC3+ bilayered membrane-bound intermediate that is degraded in the lysosome, indicating that although autophagic machinery and induction mechanisms play a role in CUPS development, CUPS and CUPS-dependent secretion are not involved in canonical autophagic flux (Bruns et al., 2011). However, the secretion of Acb1 can be triggered by rapamycin and nitrogen starvation (Manjithaya et al., 2010). Thus, although Acb1 secretion resembles endosomal/exosomal-mediated secretion, autophagy and the autophagic machinery are necessary for Acb1’s secretion, though the mechanism differs from degradative autophagy. As such, this also provides an example of pleiotropic roles exerted by the autophagic machinery in cellular secretion. Thus, the term autophagy-dependent secretion refers to the secretion of a factor that depends upon functional autophagic machinery, even if these machineries have roles outside of the canonical degradative mechanism of autophagic flux. This definition also accounts for the non-autophagy and alternative roles of autophagic machinery; for example, the involvement of Atg8 and Atg9 in CUPS formation (Bruns et al., 2011). One important potential bias deriving from this definition is that not all factors included by it are necessarily packaged and destined for secretion by a canonical, double-membrane, LC3+ autophagosome, but evidence does suggest that this occurs with some of the factors described, for instance IL-1β or TGF-B (Nüchel J et al., 2018). Indeed, most mechanistic work on autophagy-dependent secretion arises from studies analysing IL-1β secretion (Monteleone et al., 2015; Martín-Sánchez et al., 2016; Dupont et al., 2011). IL-1β secretion is enhanced following starvation of bone marrow-derived macrophages, similar to the enhancement of Acb1 secretion following nutrient starvation. Conversely, knockdown of ATG5 and colocalization with LC3 provides stronger evidence that autophagy mediates the secretion of IL-1β (Dupont et al., 2011). A modified autophagosome routes IL-1β for extracellular secretion. This is initially made possible by the cargo recruitment to the developing autophagosome. Mature IL-1β binds to TRIM16/ERBBP (tripartite motif containing 16) (Munding et al., 2006). This IL-1β -TRIM16 complex traffics to an autophagy sequestration membrane (Kimura et al., 2017), which corresponds to a previously described intermediate membrane compartment necessary for the conversion of LC3-I to LC3-II (Zhang et al., 2015). Without TRIM16, IL-1β cannot be conveyed to the sequestration membrane, or be found within the resulting autophagosome (Kimura et al., 2017). At the sequestration membrane, SEC22B (SEC22 homolog B, vesicle trafficking protein) binds the IL-1β -TRIM16 complex. SEC22B consists of a longin domain (involved in protein transport to the plasma membrane) and a SNARE motif (Moreau et al., 2013), with this SNARE motif critical to the vesicle fusion events involved in IL-1β secretion. Originally identified as part of the vesicle fusion machinery involved in COP-II coated vesicle fusion in the ER-Golgi intermediate compartment (Mancias and Goldberg, 2007), SEC22B is of particular importance to autophagy. Upon knockdown of SEC22B, LC3 lipidation is decreased (Ge et al., 2014). Paradoxically, SEC22B depletion leads to an increase in LC3-II levels by immunoblot, and LC3 puncta by immunofluorescence (Kimura et al., 2017; Renna et al., 2011), but no overall differences in autophagic flux. Reconciling this finding, SEC22B depletion blocks trafficking of lysosomal proteases to the lysosome, thereby rendering the lysosome ineffective (Renna et al., 2011). Upon SEC22B depletion, IL-1β secretion decreases (Kimura et al., 2017). Therefore, an autophagosome destined for secretion would have LC3-II, SEC22B, and TRIM16 on its cytosolic membrane. The modified autophagosome involved in IL-1β secretion displays characteristics similar to a degradative autophagosome, it does differ in a few key cytosolic membrane components to facilitate the trafficking to the plasma membrane (Figure 3B). In a way comparable to the canonical degradative autophagosome, a secretory autophagosome is characterised by the typical double membrane structure, labelled with LC3-II. Cargo recruitment in both secretion and degradation appear to rely on trafficking of cellular cargo to LC3-II. However, the destinations of the LC3+ double-membrane intermediate differ based upon the SNARE machinery coating the cytosolic membranes (Noh et al., 2022; Meldolesi, 2023; Wang et al., 2016). In a degradative autophagosome, STX17 allows for fusion with the lysosome (Itakura et al., 2012; Jiang et al., 2014). In a secretory autophagosome, SEC22B facilitates fusion with the plasma membrane (Figure 3B). Albeit subtle, these key differences in the cytosolic membrane proteins determine whether the contents are degraded or expelled. More specifically, in order to fuse with the plasma membrane, the secretory autophagosome undergoes a SNARE-mediated fusion event. The R-SNARE, SEC22B, on the secretory autophagosome binds to Qbc-SNAREs, SNAP23 and SNAP29 on the plasma membrane (Kimura et al., 2017). Together with STX3 (syntaxin-3) and STX4 (syntaxin-4) on the plasma membrane, these proteins mediate a SNARE complex allowing fusion of the secretory autophagosome with the plasma membrane (Kimura et al., 2017). The fusion of the secretory autophagosome with the plasma membrane facilitates secretion of IL-1β.
6 Secretory autophagy and extracellular release of intracellular organellar contents
Autophagy is considered the most important pathway for the degradation of damaged organelles, such as mitochondria, peroxisomes, or the endoplasmic reticulum (Gatica et al., 2018; Onishi et al., 2021). However, in recent years, several studies have revealed that dysfunctional organelles can also be secreted into the extracellular space through the fusion of autophagosomes with the plasma membrane, rather than being degraded. An important example is provided by mitochondria, whose clearance relies on the release of extracellular vesicles (EVs) in the absence of the mATG8-conjugation system (Tan et al., 2022) or in cases of lysosomal dysfunction (Liang et al., 2023). Specifically, when the mATG8-conjugation system is defective (ATG3-, ATG5-, and ATG7-knockout), PINK1-Parkin and the mitophagy cargo receptor NDP52 mediate the sorting of damaged mitochondria; upstream ATG proteins such as ATG9A, the ULK1 complex, and the PI3KC3-C1 complex regulate the formation of mitophagosomes, which later fuse with the plasma membrane (Tan et al., 2022). In addition to PINK1 and Parkin, other mitophagy receptors, such as optineurin (OPTN) and TAX1BP1, may play a role in recognizing damaged mitochondria and facilitating their sequestration within mitophagosomes. These receptors interact with LC3 and GABARAP family proteins via LIR (LC3-interacting region) motifs, highlighting their potential involvement in both degradation and secretion processes under specific conditions (Lazarou et al., 2015). From the signalling standpoint, the autophagic secretion of mitochondria can activate the cGAS-STING innate immune pathway in recipient HeLa cells, facilitating the induction of inflammation (Tan et al., 2022). The release of mitochondrial DNA (mtDNA) via extracellular vesicles has been shown to act as a potent trigger of cGAS-STING signalling pathway, emphasizing the dual role of these EVs in promoting either inflammation or tissue repair, depending on the cellular context (West and Shadel, 2017).
Additionally, a new type of extracellular vesicle containing damaged mitochondria has recently been identified: cardiac exophers, characterized by a variable diameter ranging from 3.5 to 0.1 µm and positive for the autophagosome marker LC3 (Nicolás-Ávila et al., 2020). Both cardiac exophers and mito-EVs formed following lysosomal dysfunction (Nicolás-Ávila et al., 2020; Liang et al., 2023) can be secreted by cardiomyocytes and internalized by surrounding macrophages to prevent inflammation in a process known as heterophagy (Nicolás-Ávila et al., 2022). Recent findings suggest that the heterophagy-mediated clearance of EVs by macrophages is critical for maintaining cardiac homeostasis, particularly under stress conditions such as ischemia or myocardial infarction. Secretory autophagy can also be exploited by cells during development, as in the case of human reticulocytes (Griffiths et al., 2012). During their maturation into erythrocytes, reticulocytes can reduce plasma membrane volume and eliminate excess organelles such as mitochondria, the Golgi apparatus, and the endoplasmic reticulum. Studies by Griffiths et al. revealed that vesicles containing glycophorin A (GPA), a surface protein used as a biomarker for the plasma membrane of mature reticulocytes, originate from the plasma membrane of reticulocytes. Specifically, GPA-positive vesicles can fuse with amphisomes, which are trafficking structures deriving by the fusion of autophagosomes with late endosomes and containing the organelles to be eliminated. TPPP/p25α promotes the anterograde transport of amphisomes towards cell membrane, by inhibiting HDAC6 (Ejlerskov et al., 2013). Phosphorylation of SPK1 regulates either the degradation or the unconventional secretion of amphisome-derived cargoes (Li J. et al., 2023) (Table 2). These vesicles can ultimately fuse with the plasma membrane, releasing mitochondria, the Golgi apparatus, and the endoplasmic reticulum into the extracellular space (Vats et al., 2025). Beyond developmental processes, the secretion of dysfunctional organelles via autophagy is increasingly recognized as a protective mechanism during stress adaptation. For instance, oxidative stress has been shown to enhance the release of mitochondrial EVs, contributing to inter-cellular communication and the modulation of the recipient cell’s metabolic state (Torralba et al., 2018). The identification of this pathway as a stress-response mechanism opens new avenues for therapeutic interventions targeting diseases characterized by defective organelle quality control, such as neurodegenerative disorders or metabolic syndromes.
7 Secretory autophagy of aggregation-prone proteins
One of the key hallmark of neurodegenerative diseases is the presence and progressive accumulation of misfolded proteins with a propensity to aggregate, causing cellular dysfunction, synaptic loss, and finally neuronal death (Soto and Pritzkow, 2018). The primary aggregation-prone proteins involved include amyloid-beta (Aβ) and tau in Alzheimer’s disease (AD), α-synuclein (α-Syn) in Parkinson’s disease (PD), and huntingtin (HTT) in Huntington’s disease (HD) (Jucker and Walker, 2018). Despite differences in their sequences, structures, localization and functions these proteins share a common mechanism of misfolding and aggregation, characterized by the formation of β-sheet-rich structures stabilized by hydrogen bonds and hydrophobic interactions. These fibrils can then recruit new monomers to accumulate into larger structures (Soto et al., 2006).
In this context, autophagy is essential to the pathogenesis of neurodegenerative diseases (Menzies et al., 2015). Autophagic degradation of cytosolic protein is the fate for most aggregation-prone proteins. However, under conditions of autophagic dysfunction, the clearance of misfolded proteins is impaired, leading to their accumulation and progressive neuronal damage (Son et al., 2012). Alzheimer’s disease is a late-onset neurological disorder that causes progressive memory loss and cognitive decline, characterized by the accumulation of Aβ plaques and hyperphosphorylated tau neurofibrillary tangles. Excessive levels of Aβ plaques can impair lysosomal trafficking and biogenesis (Tamminemi et al., 2017), whereas increased activity of p62/SQSTM1 or the transcription factor EB (TFEB) reduces plaque formation (Song et al., 2020). Hyperphosphorylated tau also interacts with autophagic receptors such as p62/SQSTM1 and OPTN (Xu et al., 2019). Parkinson’s disease is characterized by a progressive movement disorder caused by the accumulation of Lewy bodies, primarily composed of mutant α-synuclein, in dopaminergic neurons of the substantia nigra. Mutant α-synuclein interferes with autophagic degradation in multiple ways, such as inhibiting omegasome formation, by disrupting Rab1 function and misplacing ATG9A, a marker of autophagosomes (Winslow et al., 2010). It also impairs lysosomal protease cathepsin D activity (Hoffmann et al., 2017). Disruption of the autophagic degradation pathway is closely linked to modulation of exosome secretion. Several studies have reported that impaired “aggrephagy” is associated with increased release of extracellular vesicles containing aggregation-prone proteins. For instance, ATG5 silencing leads to increased α-synuclein secretion and reduced toxicity in human neurons, suggesting a protective mechanism against cellular damage (Fussi et al., 2018). A deficiency in PARK9 causes lysosomal dysfunction and a juvenile form of parkinsonism, the Kufor-Rakeb syndrome (Ramirez et al., 2006). In this case, the impaired lysosomal-dependent degradation leads to α-synuclein accumulation, whereas PARK9 over-expression reduces cellular toxicity and enhances the exosome-dependent secretion of the mutant protein (Tsunemi et al., 2014). However, α-synuclein release by exosomes has been shown to increase cell death in recipient human neuroglioma H4 cells, revealing for the first time the importance of exosomes in transmitting oligomers between neighbouring cells (Danzer et al., 2012). A subsequent study reported that exosomes provide an ideal microenvironment for α-synuclein aggregation, as they protect the protein from degradation and contain molecules that modulate aggregation. For example, phospholipids in exosomes inhibit aggregation, while gangliosides GM1 and GM3 promote nucleation (Marie et al., 2015). Conversely, specific gangliosides in exosomes facilitate Aβ aggregation. Inhibiting GM1 reduces Aβ aggregation and trafficking of its pathogenic form (Yuyama et al., 2008; Yuyama et al., 2012). The first evidence of exosome-dependent secretion of Alzheimer’s disease-related proteins came from the localization of the enzymes responsible for cleaving the amyloid precursor protein (APP). In particular, Rajendran et al. has reported that β-secretase cleaves APP in early endosomes, followed by APP trafficking to multi-vesicular bodies (MVBs). A portion of Aβ is released into the extracellular space via exosomes, contributing to the formation of amyloid plaques in the AD brain and therefore implicating the involvement of exosome-mediated secretion as a contributing factor in the spreading of protein (Rajendran et al., 2006). APP full-length, its metabolites, and cleaving enzymes have also been identified in exosomes, suggesting that processing may occur within the luminal compartment of these vesicles (Sharples et al., 2008). Tau can be also secreted via exosomes in Alzheimer’s disease. Notably, tau secretion by microglial cells is implicated in disease transmission to adjacent neurons, possibly through phagocytosis of tau-containing neurons followed by exosome-mediated release (Asai et al., 2015).
Huntington’s disease is an autosomal dominant disorder characterized by cognitive, behavioural and motor disturbances caused by an expansion of the CAG triplet (>35 repeats) in exon 1 of the HTT gene, leading to an abnormal polyglutamine tract. The pathogenesis of Huntington’s disease (HD) appears to be influenced by the expanded poly-Q segment. For example, it is well established that the nucleation time for amyloid fibrils inversely correlates with the length and concentration of the poly-Q repeat expansion (Pandey et al., 2018).
Similar to tau, mutant HTT interacts with p62/SQSTM1 and OPTN for its clearance (Fu et al., 2017). However, mutant HTT, along with the adaptor protein huntingtin-associated protein 1 (HAP1), has been shown to disrupt the retrograde transport of autophagosomes, thereby impairing their maturation and fusion with lysosomes (Wong and Holzbaur, 2014).
A number of studies have reported that mutant HTT exhibits prion-like behaviour; indeed, studies have shown that the mutant form can be transmitted to adjacent cells and induce aggregation of the wild-type protein (Babcock and Ganetzky, 2015). Masnata et al. reported that several cell lines can internalize mutant huntingtin fibrils, supporting the notion of disease spreading (Masnata et al., 2019). In this context, we have reported a novel role for the HSPB1-p62/SQSTM1 complex functions as a cargo-loading platform facilitating the unconventional secretion of mutant HTT via extracellular vesicles (EVs) (Bonavita et al., 2023). HSPB1 shows a stronger interaction with poly-Q expanded HTT compared to the wild-type protein, influencing its aggregation. In addition, HSPB1 levels are linked to the rate of mutant HTT secretion, which is regulated by the PI3K/AKT/mTOR signalling pathway. Finally, we demonstrate that these HTT-containing vesicles are biologically active, capable of being internalized by recipient cells, thereby contributing to the prion-like spreading properties of mutant HTT (Bonavita et al., 2023) (Figure 4). Interestingly, we have recently established a system to investigate the oligomerization of various disease-associated proteins by analysing sedimentation profiles obtained using a discontinuous sucrose gradient ultracentrifugation technique (Bonavita et al., 2024). This approach, applied specifically to mutant huntingtin, demonstrated the capability to separate protein oligomers based on their density, which correlates directly with their oligomerization state. The method described outlines an innovative protocol for characterizing the oligomerization profile of aggregation-prone proteins (Bonavita et al., 2024).
FIGURE 4
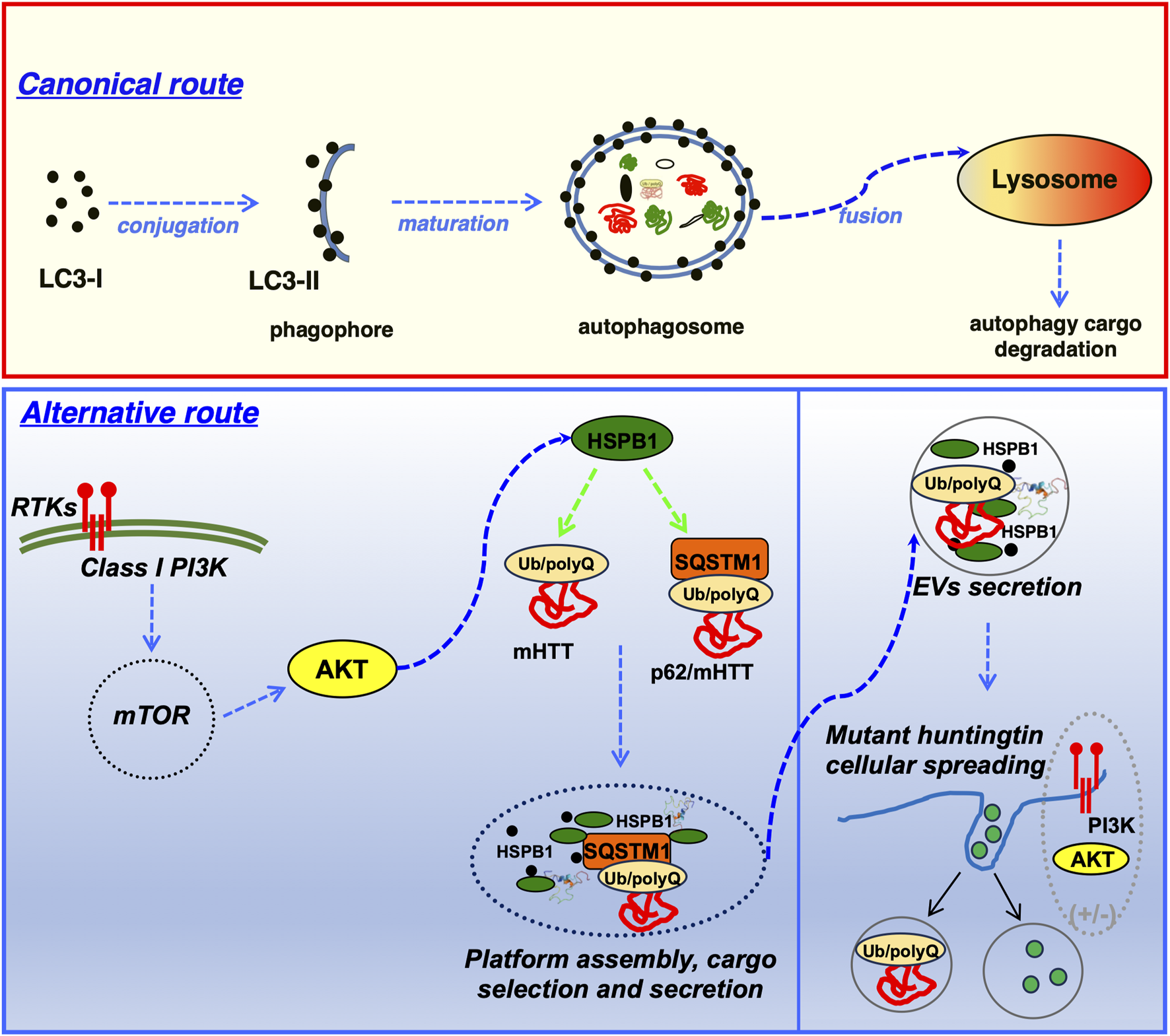
Schematic representation of the unconventional secretion and transcellular spreading of mutant huntingtin mediated by the HSPB1-p62/SQSTM1 functional complex. Top: most of the neurodegenerative diseases-associated (NDs) aggregation-prone proteins, including mutant huntingtin are engulfed into autophagosomes and ultimately delivered to the lysosomal compartment, where the degradation of the autophagy cargo occurs (canonical route). Bottom: however, under certain conditions, such as in response to variation of nutrient availability, cells can operate an alternative mechanism, where the HSPB1-p62/SQSTM1 functional complex acts as a cargo loading platform allowing the unconventional secretion of mutant HTT via extracellular vesicles (EVs). In addition, these HTT-containing vesicular structures are biologically active and able to be internalized by recipient cells, therefore providing an additional mechanism to explain the intercellular spreading of mutant HTT.
In Alzheimer disease, dystrophic neurites have an accumulation of autophagosomes (Nixon, 2007). These autophagosomes contain the bulk intracellular reservoir of b-amyloid (Yu et al., 2005). Although this finding led to the initial conclusion that the accumulation of b-amyloid and that autophagosome build-up resulted from a dysfunctional autolysosome, studies of Atg7 knockout transgenic mice have connected b-amyloid accumulation with a decreased secretory autophagy pathway. ATG7 knockout neurons have diminished b-amyloid secretion, and reconstitution of ATG7 can restore its secretion of β-amyloid (Nilsson et al., 2013). Furthermore, pharmacological induction of autophagy with rapamycin enhances amyloid beta secretion, whereas inhibition of autophagy with spautin-1 diminishes secretion (Nilsson et al., 2013). Thus, autophagy influences secretion of amyloid beta in Alzheimer disease.
In Parkinson disease, mutant a-synuclein aggregates accumulate within dopaminergic neurons (Luk et al., 2012). Both autophagy and the proteasome degrade SNCA. In neurons, TPPP/brain specific protein p25α (tubulin polymerization promoting protein), traffics SNCA to autophagic structures, while also preventing autophagosome-lysosome fusion (Ejlerskov et al., 2013). This promotes secretion of a-synuclein-containing autophagosomes. Conversely, autophagy inhibitors, such as 3-MA, attenuate a-synuclein release. Thus, autophagy-dependent secretion can facilitate the secretion of mutant a-synuclein in neurons (Ejlerskov et al., 2013).
Although the release of both a-synuclein and amyloid beta depend on autophagy, other routes have been described. Among the others, Misfolding-Associated Protein Secretion (MAPS) is one of the mechanisms that can contribute to the unconventional secretion of aggregation-prone proteins (Lee et al., 2016). In particular, the system relies on the ER-associated de-ubiquitinase USP19 (ubiquitin specific peptidase 19), which acts as a chaperone to enrich misfolded proteins at the ER surface (Lee et al., 2016).
Interestingly, two other chaperones, HSPA8/HSC70 (heat shock protein family A (HSP70) member 8) and DNAJC5 (DnaJ heat shock protein family (HSP40, member C5), function with USP19 to triage proteins in MAPS (Xu et al., 2018). Both SNCA and amyloid beta have been linked to a MAPS pathway of secretion (Fontaine et al., 2016; Lee et al., 2016). Further studies may provide a connection between MAPS and autophagy-dependent secretion.
Hence, the role of secretory autophagy in intercellular communication and propagation of aggregation-prone proteins is increasingly evident (summarised in Table 3). Understanding how proteins involved in neurodegenerative diseases are packaged into exosomes, the role of exosomal membrane composition in forming extracellular aggregates, and their uptake by recipient cells will be crucial for developing promising therapeutic strategies in the future.
TABLE 3
Main neurodegenerative diseases-associated proteins known to undergo unconventional secretion and intercellular spreading.
EVs, extracellular vesicle; ATG/LYSO, secretory autophagy/lysosomal exocytosis; TNT, tunnelling nanotubes.
8 The relevance of secretory autophagy in neurodegenerative, metabolic and endocrine diseases
Defects in autophagic processes, both in secretion and degradation, are pivotal in the aetiology of neurodegenerative and metabolic diseases, due to their critical role in cellular homeostasis. For instance, the Insulin-Degrading Enzyme (IDE) is a key regulator of peptide degradation, including insulin and β-amyloid (Aβ). In human astrocytes, IDE serves as the principal protease for Aβ degradation, and its dys-regulation is a hallmark of Alzheimer’s disease (AD) (Son et al., 2016) (Table 3). Notably, IDE secretion is modulated by autophagy through the LKB1-AMPK-mTOR signalling pathway, with drugs such as simvastatin enhancing autophagy and IDE secretion (Tamboli et al., 2010). Additionally, Aβ itself appears to promote IDE secretion via mechanisms involving autophagic proteins such as ATG7 and trafficking regulators such as GORASP (Golgi reassembly and stacking protein) and RAB8A, which collectively sustain an adequate autophagic flux that is essential for IDE secretion (Son et al., 2016). Studies in ATG7 knockout mouse models have shown reduced IDE levels in cerebrospinal fluid (CSF), linking defects in secretory autophagy to impaired Aβ metabolism. Beyond Aβ, IDE interacts with α-synuclein, a protein implicated in both neurodegenerative diseases and type 2 diabetes (T2DM). IDE activation by α-synuclein inhibits α-synuclein amyloid formation, offering protective effects. However, in T2DM models lacking IDE, high α-synuclein levels disrupt autophagic flux, increasing the risk of cellular damage (Sharma et al., 2015).
Islet Amyloid Polypeptide (IAPP), also known as amylin, is a 37-amino acid peptide co-secreted with insulin by pancreatic β-cells. Under physiological conditions, IAPP regulates processes such as gastric emptying and glucose homeostasis (Gonzalez et al., 2020). However, in T2DM, toxic oligomers of IAPP and amyloid deposits contribute to β-cell dysfunction and death (Hull et al., 2005). Autophagy in β-cells plays a central role in IAPP degradation, with stimulation of the AMPK pathway shown to reduce IAPP-induced oxidative stress and promote degradation (Gonzalez et al., 2020). Disruption of autophagic flux leads to the accumulation of toxic oligomers, as demonstrated in animal models expressing the human IAPP (hIAPP) variant. Deletion of key autophagic genes such as ATG7 results in amyloid accumulation, β-cell apoptosis, and exacerbation of disease pathology (Kim et al., 2014; Shigihara et al., 2014). Intracellular IAPP oligomers are particularly harmful, damaging mitochondrial and endoplasmic reticulum (ER) membranes and triggering oxidative stress (Kim et al., 2014). Moreover, IAPP exhibits a “prion-like” behaviour, propagating toxic aggregates across cells (Mukherjee, 2017). In metabolic contexts, adiponectin, a hormone derived from white adipose tissue highlights a distinct role for autophagy (Piletic et al., 2023). Adiponectin secretion is regulated by Beclin-1, a hyperactive autophagic protein that facilitates release via secretory vesicles rather than autophagosomes. This process relies on the exocyst assembly, promoting insulin sensitivity, glucose tolerance and lipid metabolism in non-adipose tissues. In this context, the upregulation of Beclin-1 activity has shown promise in improving metabolic parameters in obesity and T2DMmodels (Kuramoto et al., 2021). In summary, autophagic dysfunction significantly contributes to the pathogenesis of neurodegenerative, metabolic and endocrine disorders. Therapeutic strategies targeting autophagic flux hold promise for treating conditions such as AD, T2DM, and β-cell dysfunction. The intricate interplay between autophagy, degradation and secretion underscores the necessity of maintaining a functional autophagic system. Further studies will be required to unravel these mechanisms and harness autophagy to design novel therapeutic approaches.
9 Concluding remarks and future perspectives
One of the major features which is associated to neurodegenerative disorders with a simple Mendelian inheritance is the accumulation of misfolded proteins into insoluble aggregates (or inclusion bodies) in neurons, which is accompanied by the progressive neuronal loss in the affected regions of the central nervous system (CNS) (Dugger and Dickson, 2017). In view of the aging of the world population, the incidence and mortality associated with these disorders are rapidly increasing (Ferri et al., 2005). Although there are currently no effective strategies that slow or prevent these NDs in humans, there is strong experimental evidence that the upregulation of intracellular clearance pathways (the autophagy-lysosome and ubiquitin-proteasome pathways) can clear aggregate-prone proteins, such as a-synuclein in experimental models. When the flux through these pathways is increased, the levels of aggregate-prone proteins can be reduced and this results in improved cell survival in both cell-based and animal models of NDs (Menzies et al., 2017). More recently, a third strategy for clearing proteins from cells has been identified, which occurs via the unconventional secretion of proteins out of the cell. Unconventional secretion can occur via multiple routes, including the generation of extracellular vesicles (EVs) (Meldolesi, 2023; Noh et al., 2022; Zhang and Schekman, 2013; Zubkova et al., 2024). Interestingly, recent work has shown that these EVs can be taken up by neighbouring cells (van Niel et al., 2022). Hence, the contents/cargoes of these EVs can be transferred into recipient cells, thereby allowing cell-to-cell communication. Within the context of NDs, this spreading of cargoes could have two opposing consequences: on one hand, this could be a route for clearing misfolded proteins from neurons and the uptake of EVs by astrocytes or microglia may aid the removal of such aggregate-prone proteins. Alternatively, the spreading of misfolded and/or aggregate-prone proteins to surrounding neurons may lead the spreading and seeding of aggregates.
In this context, recent studies have suggested that the propagation and transmission of NDs-associated proteins such as α-synuclein (Gustafsson et al., 2018; Nakamura et al., 2024; Xie et al., 2022), SOD1 (Hutchings et al., 2024), Tau (Fowler et al., 2025; Lopez et al., 2025) and TDP-43 (Picard et al., 2025), can contribute to the pathogenesis of PD, AD and ALS/FTDs, respectively.
Furthermore, the identification of different populations of proteins within EVs and their aggregation state has the potential to serve as a biomarker for both disease progression and for identification of different forms of dementia. For instance, the autophagy cargo receptor p62/SQSTM1 represents a consistent component of pathological aggregates observed in ALS and FTDs (Kiernan et al., 2011; Burell et al., 2016). Mutations in p62/SQSTM1 have been associated with ALS and FTDs (Fecto et al., 2011; Le Ber et al., 2013; Teyssou et al., 2013). Interestingly, a number of these mutations are associated to impairment of autophagy, underlying the neurotoxicity and progressive neuronal loss observed in ALS and FTDs (Goode et al., 2016; Deng et al., 2019). Hence, its modulation to facilitate protein degradation has been proposed as a potential therapeutic target (Davidson et al., 2022). More recently, it has been suggested that p62/SQSTM1 might represent a valuable diagnostic and/or prognostic marker in these clinical conditions. For instance, the analysis of post-mortem tissues from patients affected by familial or sporadic forms of ALS has demonstrated the co-presence of SOD1, TDP-43 and p62/SQSTM-1 in motoneurons derived from the spinal cord (Trist et al., 2022). On the same line, a higher survival is associated with lower p62 levels in the spinal cord of patients affected by sporadic forms of ALS (Pinkerton et al., 2023). In addition, a positive correlation between the increased levels of p62/SQSTM1 has been reported in a study conducted on the cerebrospinal fluid of AD and FTD patients, confirming in a clinical context the important role exerted by the autophagic pathway in these neurodegenerative diseased (Rubino et al., 2022). As such, monitoring the levels of secreted/circulating p62/SQSTM1 might represent a potential in vivo biomarker to monitor autophagy in these clinical conditions.
Recent work in our lab has focussed on the small heat shock proteins (sHSPs), As key components of the chaperone system, sHSPs are constitutively expressed in several cell types and are involved in the maintenance of normal cellular protein homeostasis by regulating the proper folding of newly synthesized proteins, and the transport and turn-over of mature proteins. In addition, sHSPs can actively sequester proteins during initial unfolding and engage misfolded proteins, thereby preventing the formation of insoluble protein aggregates, while keeping them available for the ATPase-dependent chaperone complexes.
Therefore, the observations that some members of HSPBs family, such as HSPB1 and HSPB5/CRYAB might undergo unconventional secretion (D’Agostino et al., 2019; Bonavita et al., 2023; Bonavita et al., 2024; Van den Broek et al., 2021) could represent an alternative system by which cells are able to handle and reduce the accumulation of intracellular mutant, aggregate-prone or misfolded proteins. As such, the mechanism by which disease-relevant proteins (and their interacting proteins) can be transferred between cells by means of EVs deserves further investigations, as it could influence the pathogenesis and the onset of misfolding protein-associated diseases, and in perspective, represent a potential target to develop novel diagnostic tools and therapeutic strategies. Notably, a recent study has defined a novel non–cell-autonomous protective role played by the extracellular HSPB1 in neurodegeneration (Fangjia et al., 2024). In particular, in conditions that mimic an inflammatory response, such as in AD, human astrocytes can increase HSPB1secretion. The pool of HSPB1 secreted from astrocytes in human AD brain is able to cluster around amyloid plaques. Moreover, astrocytes and neurons can uptake astrocyte-secreted HSPB1, which is accompanied by an attenuation of the inflammatory response in reactive astrocytes and reduced pathological tau inclusions, findings highlighting the relevance of HSPB1 secretion for this protective mechanism, which would be typically regarded as intracellular (Fangjia et al., 2024). Such a phenomenon would be consistent with what has been reported for the chaperone DNAJB6, which can be loaded into extracellular vesicles (EVs) and is capable to decrease polyglutamine aggregation in in vitro and in vivo models of Huntington disease (Joshi et al., 2021; Joshi et al., 2022).
In addition, in a recent report the therapeutic potential of exosomes derived from adipose tissue mesenchymal stem cells in alleviating Aβ-induced retinal toxicity, which is a major factor contributing to age-related macular degeneration (AMD), has been evaluated both in vitro and in rat experimental model (Qarawani et al., 2025). Interestingly, the authors showed that such a protection is mediated by regulating the activity of HSPB5/CRYAB, offering a promising avenue for future AMD treatment strategies (Qarawani et al., 2025).
Indeed, in the past decade, exosomes and extracellular vesicles (EVs) have been reported as novel and important carrier system of signalling molecules. EVs are essential mediators of communication between cells. In particular, the EVs released from the brain and the spinal cord are able to transmit signalling information between neurons oligodendrocytes, microglia, as well as astrocytes in the CNS and contribute to their development and function, influencing the physiology of the recipient cells (Ramirez et al., 2018; Sharma et al., 2019; van Niel et al., 2022). In addition, EVs have been proposed to exert a protective role by expelling pathological molecules from cells (Howitt and Hill, 2016). In neurodegenerative diseases, misfolded proteins can be transferred to by EVs to perform pathological functions. A growing body of literature has highlighted an important role of EVs in the cell-to-cell transmission of pathogenic protein aggregates, thereby contributing to the pathological and clinical progression of neurodegenerative diseases (Hornung et al., 2020). In this context, growing evidence support the idea that EVs can function as reliable and robust biomarkers for neurodegenerative diseases, in comparison to the conventional patient-derived samples (Younas et al., 2022).
Interestingly, one of the outstanding and still not completely addressed questions in the field is how the selective cargo loading of these vesicles is regulated. Likewise, very little is known in regard to the mechanisms regulating the trafficking and the biological activity of the EVs internalised by recipient cells. Hence, large screening and database analyses are currently in progress in order to understand how these processes are regulated and what is their physiological relevance. In this scenario, a number of potential biomarkers have been identified by bioinformatics analyses and tested in experimental studies of EVs (Li J. et al., 2023). However, these studies were largely focused on the discovery of novel potential biomarkers and will require clinical validation. On the same line, additional studies will be needed to improve the reliability of EVs biomarkers to be used for clinical diagnostic purposes and to inform about the functional relationship existing between up- or downregulation of EVs biomarkers and disease progression. Nevertheless, the use of EVs as a potential biomarker and treatment for neurodegenerative diseases does represent an attractive concept that will certainly deserve future investigations.
Statements
Author contributions
RB: Conceptualization, Resources, Supervision, Visualization, Writing – original draft, Writing – review and editing. FV: Resources, Supervision, Visualization, Writing – original draft, Writing – review and editing. LV: Resources, Visualization, Writing – original draft. SW: Resources, Visualization, Writing – original draft. MC: Resources, Visualization, Writing – original draft. AF: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review and editing. MR: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This project was funded by the Italian Minister for Research and University (PRIN PNRR2022 research grant no.: P20224FYSY), assigned to MR and by a University of Cambridge Career Support Fund scheme, awarded to AF.
Acknowledgments
We are particularly grateful to Professor Stefano Bonatti and the other members of the laboratory for helpful comments. MR is grateful to the Department of Physiology, Development and Neuroscience (PDN, University of Cambridge, United Kingdom), for hosting him as a Visiting Academic Fellow.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Abels E. R. Breakefield X. O. (2016). Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell Mol. Neurobiol.36, 301–312. 10.1007/s10571-016-0366-z
2
Abounit S. Wu J. W. Duff K. Victoria G. S. Zurzolo C. (2016). Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion, 10 (5), 344–351. 10.1080/19336896.2016.1223003
3
Adnan M. Islam W. Zhang J. Zheng W. Lu G.-D. (2019). Diverse role of SNARE protein Sec22 in vesicle trafficking, membrane fusion, and autophagy. Cells8, 337. 10.3390/cells8040337
4
Adriaenssens E. Asselbergh B. Rivera-Mejías P. Bervoets S. Vendredy L. De Winter V. et al (2023). Small heat shock proteins operate as molecular chaperones in the mitochondrial intermembrane space. Nat. Cell Biol.25, 467–480. 10.1038/s41556-022-01074-9
5
Aguilera M. O. Berón W. Colombo M. I. (2012). The actin cytoskeleton participates in the early events of autophagosome formation upon starvation induced autophagy. Autophagy8 (11), 1590–1603. 10.4161/auto.21459
6
Ahat E. Bui S. Zhang J. da Veiga Leprevost F. Sharkey L. Reid W. et al (2022). GRASP55 regulates the unconventional secretion and aggregation of mutant huntingtin. J. Biological. Chem., 298 (8), 102219. 10.1016/j.jbc.2022.102219
7
Alge C. S. Priglinger S. G. Neubauer A. S. Kampik A. Zillig M. Bloemendal H. et al (2002). Retinal pigment epithelium is protected against apoptosis by alphaB-crystallin. Investigative Ophthalmol. and Vis. Sci.43 (11), 3575–3582.
8
Almeida-Souza L. Asselbergh B. d'Ydewalle C. Moonens K. Goethals S. de Winter V. et al (2011). Small heat-shock protein HSPB1 mutants stabilize microtubules in Charcot-Marie-Tooth neuropathy. J. Neurosci.31 (43), 15320–15328. 10.1523/JNEUROSCI.3266-11.2011
9
Almeida-Souza L. Goethals S. de Winter V. Dierick I. Gallardo R. Van Durme J. et al (2010). Increased monomerization of mutant HSPB1 leads to protein hyperactivity in Charcot-Marie-Tooth neuropathy. J. Biol. Chem.285 (17), 12778–12786. 10.1074/jbc.M109.082644
10
Arrigo A. P. (2017). Mammalian HspB1 (Hsp27) is a molecular sensor linked to the physiology and environment of the cell. Cell Stress Chaperones22, 517–529. 10.1007/s12192-017-0765-1
11
Arrigo A. P. Simon S. Gibert B. Kretz-Remy C. Nivon M. Czekalla A. et al (2007). Hsp27 (HspB1) and alphaB-crystallin (HspB5) as therapeutic targets. FEBS Lett.581, 3665–3674. 10.1016/j.febslet.2007.04.033
12
Asai H. Ikezu S. Tsunoda S. Medalla M. Luebke J. Haydar T. et al (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci.18, 1584–1593. 10.1038/nn.4132
13
Babcock D. T. Ganetzky B. (2015). Transcellular spreading of huntingtin aggregates in the Drosophila brain. Proc. Natl. Acad. Sci. U. S. A.112, E5427–E5433. 10.1073/pnas.1516217112
14
Banerjee S. Lin C. F. Skinner K. A. Schiffhauer L. M. Peacock J. Hicks D. G. et al (2011). Heat shock protein 27 differentiates tolerogenic macrophages that may support human breast cancer progression. Cancer Res.71 (2), 318–327. 10.1158/0008-5472.CAN-10-1778
15
Bänfer S. Schneider D. Dewes J. Strauss M. T. Freibert S. A. Heimerl T. et al (2018). Molecular mechanism to recruit galectin-3 into multivesicular bodies for polarized exosomal secretion. Proc. Natl. Acad. Sci. U. S. A.115, E4396–E4405. 10.1073/pnas.1718921115
16
Benn S. C. Perrelet D. Kato A. C. Scholz J. Decosterd I. Mannion R. J. et al (2002). Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival. Neuron36 (1), 45–56. 10.1016/s0896-6273(02)00941-8
17
Bento C. F. Renna M. Ghislat G. Puri C. Ashkenazi A. Vicinanza M. et al (2016). Mammalian autophagy: how does it work?Annu. Rev. Biochem.85, 685–713. 10.1146/annurev-biochem-060815-014556
18
Berry V. Francis P. Reddy M. A. Collyer D. Vithana E. MacKay I. et al (2001). Alpha-B crystallin gene (CRYAB) mutation causes dominant congenital posterior polar cataract in humans. Am. J. Hum. Genet.69 (5), 1141–1145. 10.1086/324158
19
Bilen J. Bonini N. M. (2007). Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet.3 (10), 1950–1964. PMID: 17953484; PMCID: PMC2041992. 10.1371/journal.pgen.0030177
20
Boelens W. C. (2020). Structural aspects of the human small heat shock proteins related to their functional activities. Cell Stress Chaperones25, 581–591. 10.1007/s12192-020-01093-1
21
Bonavita R. Di Martino R. Cortone G. Prodomo A. Di Gennaro M. Scerra G. et al (2024). A method for the analysis of the oligomerization profile of the Huntington’s disease-associated, aggregation-prone mutant huntingtin protein by isopycnic ultracentrifugation. Front. Mol. Biosci.11, 1420691. 10.3389/fmolb.2024.1420691
22
Bonavita R. Scerra G. Di Martino R. Nuzzo S. Polishchuk E. Di Gennaro M. et al (2023). The HSPB1-p62/SQSTM1 functional complex regulates the unconventional secretion and transcellular spreading of the HD-associated mutant huntingtin protein. Hum. Mol. Genet.32, 2269–2291. 10.1093/hmg/ddad047
23
Bonifacino J. S. Glick B. S. (2004). The mechanisms of vesicle budding and fusion. Cell116 (2), 153–166. 10.1016/S0092-8674(03)01079-1
24
Bruns C. McCaffery J. M. Curwin A. J. Duran J. M. Malhotra V. (2011). Biogenesis of a novel compartment for autophagosome-mediated unconventional protein secretion. J. Cell Biol.195, 979–992. 10.1083/jcb.201106098
25
Buratta S. Tancini B. Sagini K. Delo F. Chiaradia E. Urbanelli L. et al (2020). Lysosomal exocytosis, exosome release and secretory autophagy: the autophagic- and endo-lysosomal systems go extracellular. Int. J. Mol. Sci.21, 2576. 10.3390/ijms21072576
26
Burell J. R. Halliday G. M. Kril J. J. Ittner L. M. Götz J. Kiernan M. C. et al (2016). The frontotemporal dementia-motor neuron disease continuum. Lancet388, 919–931. 10.1016/S0140-6736(16)00737-6
27
Cabral M. Anjard C. Malhotra V. Loomis W. F. Kuspa A. (2010). Unconventional secretion of AcbA in dictyostelium discoideum through a vesicular intermediate. Eukaryot. Cell9, 1009–1017. 10.1128/EC.00337-09
28
Capponi S. Geuens T. Geroldi A. Origone P. Verdiani S. Cichero E. et al (2016). Molecular chaperones in the pathogenesis of amyotrophic lateral sclerosis: the role of HSPB1. Hum. Mutat.37, 1202–1208. 10.1002/humu.23062
29
Carra S. Alberti S. Arrigo P. A. Benesch J. L. Benjamin I. J. Boelens W. et al (2017). The growing world of small heat shock proteins: from structure to functions. Cell stress and chaperones22 (4), 601–611. 10.1007/s12192-017-0787-8
30
Carra S. Boncoraglio A. Kanon B. Brunsting J. F. Minoia M. Rana A. et al (2010). Identification of the Drosophila ortholog of HSPB8: implication of HSPB8 loss of function in protein folding diseases. J. Biol. Chem.285 (48), 37811–37822. 10.1074/jbc.M110.127498
31
Carra S. Seguin S. J. Lambert H. Landry J. (2008). HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J. Biol. Chem.283 (3), 1437–1444. 10.1074/jbc.M706304200
32
Carra S. Seguin S. J. Landry J. (2009). HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy4 (2), 237–239. 10.4161/auto.5407
33
Carra S. Sivilotti M. Chávez Zobel A. T. Lambert H. Landry J. (2005). HspB8, a small heat shock protein mutated in human neuromuscular disorders, has in vivo chaperone activity in cultured cells. Hum. Mol. Genet.14 (12), 1659–1669. 10.1093/hmg/ddi174
34
Cerrato G. Kepp O. Sauvat A. Kroemer G. (2021). A genome-wide RNA interference screen disentangles the Golgi tropism of LC3. Autophagy17, 820–822. 10.1080/15548627.2020.1861836
35
Chai X. Dage J. L. Citron M. (2012). Constitutive secretion of tau protein by an unconventional mechanism. Neurobio. Dis., 48 (3), 356–366. 10.1016/j.nbd.2012.05.021
36
Chaplot K. Jarvela T. S. Lindberg I. (2020). Secreted chaperones in neurodegeneration. Front. Aging Neurosci.12, 268. 10.3389/fnagi.2020.00268
37
Chen K. Hou C. Xu L. Peng H. He C. Liu J. et al (2022). HSPB1 regulates autophagy and apoptosis in vascular smooth muscle cells in arteriosclerosis obliterans. Cardiovasc Ther.2022, 3889419. 10.1155/2022/3889419
38
Chen Y.Da Fang Y. T. Cheng Y. L. Lin C. F. Hsu L. J. Wang S. Y. et al (2017). Exophagy of annexin A2 via RAB11, RAB8A and RAB27A in IFN-γ-stimulated lung epithelial cells. Sci. Rep.7, 5676. 10.1038/s41598-017-06076-4
39
Chierichetti M. Cerretani M. Ciammaichella A. Crippa V. Rusmini P. Ferrari V. et al (2023). Identification of HSPB8 modulators counteracting misfolded protein accumulation in neurodegenerative diseases. Life Sci.322, 121323. 10.1016/j.lfs.2022.121323
40
Choudhary D. Mediani L. Avellaneda M. J. Bjarnason S. Alberti S. Boczek E. E. et al (2023). Human small heat shock protein B8 inhibits protein aggregation without affecting the native folding process. J. Am. Chem. Soc.145, 15188–15196. 10.1021/jacs.3c02022
41
Clarke J. P. Mearow K. M. (2013). Cell stress promotes the association of phosphorylated HspB1 with F-actin. PLoS One8, e68978. 10.1371/journal.pone.0068978
42
Clayton A. Turkes A. Navabi H. Mason M. D. Tabi Z. (2005). Induction of heat shock proteins in B-cell exosomes. J. cell Sci.118 (Pt 16), 3631–3638. 10.1242/jcs.02494
43
Collier M. P. Alderson T. R. de Villiers C. P. Nicholls D. Gastall H. Y. Allison T. M. et al (2019). HspB1 phosphorylation regulates its intramolecular dynamics and mechanosensitive molecular chaperone interaction with filamin C. Sci. Adv.5 (5), eaav8421. 10.1126/sciadv.aav8421
44
Collier N. C. Schlesinger M. J. (1986). The dynamic state of heat shock proteins in chicken embryo fibroblasts. J. Cell Biol.103, 1495–1507. 10.1083/jcb.103.4.1495
45
Cortese A. Laurà M. Casali C. Nishino I. Hayashi Y. K. Magri S. et al (2018). Altered TDP-43-dependent splicing in HSPB8-related distal hereditary motor neuropathy and myofibrillar myopathy. Eur. J. neurology25 (1), 154–163. 10.1111/ene.13478
46
Costanzo M. Abounit S. Marzo L. Danckaert A. Chamoun Z. Roux P. et al (2013). Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J. Cell Sci., 126 (Pt 16), 3678–3685. 10.1242/jcs.126086
47
Cox D. Whiten D. R. Brown J. W. P. Horrocks M. H. San Gil R. Dobson C. M. et al (2018). The small heat shock protein Hsp27 binds α-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem.293 (12), 4486–4497. 10.1074/jbc.M117.813865
48
Crippa V. D’Agostino V. G. Cristofani R. Rusmini P. Cicardi M. E. Messi E. et al (2016). Transcriptional induction of the heat shock protein B8 mediates the clearance of misfolded proteins responsible for motor neuron diseases. Sci. Rep.6, 22827. 10.1038/srep22827
49
Crippa V. Sau D. Rusmini P. Boncoraglio A. Onesto E. Bolzoni E. et al (2010). The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum. Mol. Genet.19, 3440–3456. 10.1093/hmg/ddq257
50
Cruz-Garcia D. Malhotra V. Curwin A. J. (2018). Unconventional protein secretion triggered by nutrient starvation. Semin. Cell Dev. Biol.83, 22–28. 10.1016/j.semcdb.2018.02.021
51
D’Agostino M. Scerra G. Cannata Serio M. Caporaso M. G. Bonatti S. Renna M. (2019). Unconventional secretion of α-Crystallin B requires the Autophagic pathway and is controlled by phosphorylation of its serine 59 residue. Sci. Rep.9, 16892. 10.1038/s41598-019-53226-x
52
Danzer K. M. Kranich L. R. Ruf W. P. Cagsal-Getkin O. Winslow A. R. Zhu L. et al (2012). Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener.7, 42. 10.1186/1750-1326-7-42
53
Davidson J. M. Chung R. S. Lee A. (2022). The converging roles of sequestosome-1/p62 in the molecular pathways of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Neurobiol. Dis.166, 105653. 10.1016/j.nbd.2022.105653
54
Davuluri G. V. N. Chen C. C. Chiu Y. C. Tsai H. W. Chiu H. C. Chen Y. L. et al (2021). Autophagy drives galectin-1 secretion from tumor-associated macrophages facilitating hepatocellular carcinoma progression. Front. Cell Dev. Biol.9, 741820. 10.3389/fcell.2021.741820
55
Debnath J. Gammoh N. Ryan K. M. (2023). Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol.24, 560–575. 10.1038/s41580-023-00585-z
56
Dehle F. C. Ecroyd H. Musgrave I. F. Carver J. A. (2010). αB-Crystallin inhibits the cell toxicity associated with amyloid fibril formation by κ-casein and the amyloid-β peptide. Cell stress and chaperones15 (6), 1013–1026. 10.1007/s12192-010-0212-z
57
Delbecq S. P. Klevit R. E. (2013). One size does not fit all: the oligomeric states of αb crystallin. FEBS Lett.587, 1073–1080. 10.1016/j.febslet.2013.01.021
58
De Maio A. (2011). Extracellular heat shock proteins, cellular export vesicles, and the Stress Observation System: a form of communication during injury, infection, and cell damage. It is never known how far a controversial finding will go! Dedicated to Ferruccio Ritossa. Cell stress and chaperones16 (3), 235–249. 10.1007/s12192-010-0236-4
59
Deng Z. Wang Q. Purtell K. Wu S. Palomo G. M. Hu P. et al (2019). ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy16 (5), 917–931. 10.1080/15548627.2019.1644076
60
Deretic V. Jiang S. Dupont N. (2012). Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol.22, 397–406. 10.1016/j.tcb.2012.04.008
61
DeSelm C. J. Miller B. C. Zou W. Beatty W. L. van Meel E. Takahata Y. et al (2011). Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Develop. cell, 21 (5), 966–974. 10.1016/j.devcel.2011.08.016
62
De Thonel A. Le Mouël A. Mezger V. (2012). Transcriptional regulation of small HSP - HSF1 and beyond. Int. J. Biochem. Cell Biol.44, 1593–1612. 10.1016/j.biocel.2012.06.012
63
Dimou E. Nickel W. (2018). Unconventional mechanisms of eukaryotic protein secretion. Curr. Biol. CB28 (8), R406–R410. 10.1016/j.cub.2017.11.074
64
Ding X. Jiang X. Tian R. Zhao P. Li L. Wang X. et al (2019). RAB2 regulates the formation of autophagosome and autolysosome in mammalian cells. Autophagy15, 1774–1786. 10.1080/15548627.2019.1596478
65
Dixson A. C. Dawson T. R. Di Vizio D. Weaver A. M. (2023). Context-specific regulation of extracellular vesicle biogenesis and cargo selection. Nat. Rev. Mol. Cell Biol.24, 454–476. 10.1038/s41580-023-00576-0
66
Dugger B. N. Dickson D. W. (2017). Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol.9, a028035. 10.1101/cshperspect.a028035
67
Dupont N. Jiang S. Pilli M. Ornatowski W. Bhattacharya D. Deretic V. (2011). Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J.30, 4701–4711. 10.1038/emboj.2011.398
68
Duran J. M. Anjard C. Stefan C. Loomis W. F. Malhotra V. (2010). Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol.188, 527–536. 10.1083/jcb.200911154
69
Echaniz-Laguna A. Geuens T. Petiot P. Péréon Y. Adriaenssens E. Haidar M. et al (2017). Axonal neuropathies due to mutations in small heat shock proteins: clinical, genetic, and functional insights into novel mutations. Hum. Mutat.38, 556–568. 10.1002/humu.23189
70
Ecroyd H. Bartelt-Kirbach B. Ben-Zvi A. Bonavita R. Bushman Y. Casarotto E. et al (2023). The beauty and complexity of the small heat shock proteins: a report on the proceedings of the fourth workshop on small heat shock proteins. Cell Stress Chaperones28, 621–629. 10.1007/s12192-023-01360-x
71
Ejlerskov P. Rasmussen I. Nielsen T. T. Bergström A.-L. Tohyama Y. Jensen P. H. et al (2013). Tubulin polymerization-promoting protein (TPPP/p25α) promotes unconventional secretion of α-synuclein through exophagy by impairing autophagosome-lysosome fusion. J. Biol. Chem.288, 17313–17335. 10.1074/jbc.M112.401174
72
El-Abassi R. England J. D. Carter G. T. (2014). Charcot-Marie-Tooth disease: an overview of genotypes, phenotypes, and clinical management strategies. PM R6, 342–355. 10.1016/j.pmrj.2013.08.611
73
Ellisdon A. M. Thomas B. Bottomley S. P. (2006). The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J. Biol. Chem.281 (25), 16888–16896. 10.1074/jbc.M601470200
74
Emmanouilidou E. Melachroinou K. Roumeliotis T. Garbis S. D. Ntzouni M. Margaritis L. H. et al (2010). Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci: Off. J. Soc. Neurosci., 30 (20), 6838–6851. 10.1523/JNEUROSCI.5699-09.2010
75
Escusa-Toret S. Vonk W. I. M. Frydman J. (2013). Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat. Cell Biol.15, 1231–1243. 10.1038/ncb2838
76
Evgrafov O. V. Mersiyanova I. Irobi J. Van Den Bosch L. Dierick I. Leung C. L. et al (2004). Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet.36, 602–606. 10.1038/ng1354
77
Fanelli M. A. Cuello Carrion F. D. Dekker J. Schoemaker J. Ciocca D. R. (1998). Serological detection of heat shock protein hsp27 in normal and breast cancer patients. Cancer Epidemiol. Biomarkers Prev.7 (9), 791–795.
78
Fangjia Y. Beltran-Lobo P. Sung K. Goldrick C. Croft C. L. Nishimura A. et al (2024). Reactive astrocytes secrete the chaperone HSPB1 to mediate neuroprotection. Sci. Adv.10, eadk9884. 10.1126/sciadv.adk9884
79
Farhan H. Kundu M. Ferro-Novick S. (2017). The link between autophagy and secretion: a story of multitasking proteins. Mol. Biol. Cell28, 1161–1164. 10.1091/mbc.E16-11-0762
80
Fecto F. Yan J. Vemula S. P. Liu E. Yang Y. Chen W. et al (2011). SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol.68 (11), 1440–1446. 10.1001/archneurol.2011.250
81
Feng J. T. Liu Y. K. Song H. Y. Dai Z. Qin L. X. Almofti M. R. et al (2005). Heat-shock protein 27: a potential biomarker for hepatocellular carcinoma identified by serum proteome analysis. Proteomics5 (17), 4581–4588. 10.1002/pmic.200401309
82
Ferri C. P. Prince M. Brayne C. Brodaty H. Fratiglioni L. Ganguli M. et al (2005). Global prevalence of dementia: a Delphi consensus study. Lancet366, 2112–2117. 10.1016/S0140-6736(05)67889-0
83
Fevrier B. Vilette D. Archer F. Loew D. Faigle W. Vidal M. et al (2004). “Cells release prions in association with exosomes,” in Proceedings of the National Academy of Sciences of the United States of America, 101 (26), 9683–9688. 10.1073/pnas.0308413101
84
Fontaine S. N. Zheng D. Sabbagh J. J. Martin M. D. Chaput D. Darling A. et al (2016). DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J.35 (14), 1537–1549. 10.15252/embj.201593489
85
Fowler S. L. Behr T. S. Turkes E. O'Brien D. P. Cauhy P. M. Rawlinson I. et al (2025). Tau filaments are tethered within brain extracellular vesicles in Alzheimer’s disease. Nat. Neurosci.28, 40–48. 10.1038/s41593-024-01801-5
86
Freilich R. Arhar T. Abrams J. L. Gestwicki J. E. (2018). Protein-protein interactions in the molecular chaperone network. Accounts Chem. Res.51 (4), 940–949. 10.1021/acs.accounts.8b00036
87
Fu L. Liang J. J. (2003). Alteration of protein-protein interactions of congenital cataract crystallin mutants. Investigative Ophthalmol. and Vis. Sci.44 (3), 1155–1159. 10.1167/iovs.02-0950
88
Fu Y. Wu P. Pan Y. Sun X. Yang H. Difiglia M. et al (2017). A toxic mutant huntingtin species is resistant to selective autophagy. Nat. Chem. Biol.13, 1152–1154. 10.1038/nchembio.2461
89
Fujita N. Itoh T. Omori H. Fukuda M. Noda T. Yoshimori T. (2008). The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell19, 2092–2100. 10.1091/mbc.E07-12-1257
90
Fukuda M. (2013). Rab27 effectors, pleiotropic regulators in secretory pathways. Traffic14, 949–963. 10.1111/tra.12083
91
Fussi N. Höllerhage M. Chakroun T. Nykänen N. P. Rösler T. W. Koeglsperger T. et al (2018). Exosomal secretion of α-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis.9, 757. 10.1038/s41419-018-0816-2
92
Gallagher E. R. Holzbaur E. L. F. (2023). The selective autophagy adaptor p62/SQSTM1 forms phase condensates regulated by HSP27 that facilitate the clearance of damaged lysosomes via lysophagy. Cell Rep.42, 112037. 10.1016/j.celrep.2023.112037
93
Gatica D. Lahiri V. Klionsky D. J. (2018). Cargo recognition and degradation by selective autophagy. Nat. Cell Biol.20, 233–242. 10.1038/s41556-018-0037-z
94
Ge L. Melville D. Zhang M. Schekman R. (2013). The ER–Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife2, e00947. 10.7554/elife.00947
95
Ge L. Zhang M. Kenny S. J. Liu D. Maeda M. Saito K. et al (2017). Remodeling of ER ‐exit sites initiates a membrane supply pathway for autophagosome biogenesis. EMBO Rep.18, 1586–1603. 10.15252/embr.201744559
96
Ge L. Zhang M. Schekman R. (2014). Phosphatidylinositol 3-kinase and COPII generate LC3 lipidation vesicles from the ER-Golgi intermediate compartment. Elife3, 041355. 10.7554/eLife.04135
97
Giese K. C. Vierling E. (2002). Changes in oligomerization are essential for the chaperone activity of a small heat shock protein in vivo and in vitro. J. Biol. Chem.277, 46310–46318. 10.1074/jbc.M208926200
98
Gonzalez C. D. Resnik R. Vaccaro M. I. (2020). Secretory autophagy and its relevance in metabolic and degenerative disease. Front. Endocrinol. (Lausanne)11, 266. 10.3389/fendo.2020.00266
99
Goode A. Butler K. Long J. Cavey J. Scott D. Shaw B. et al (2016). Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy12 (7), 1094–1104. 10.1080/15548627.2016.1170257
100
Gousset K. Schiff E. Langevin C. Marijanovic Z. Caputo A. Browman D. T. et al (2009). Prions hijack tunnelling nanotubes for intercellular spread. Nat. cell Biol., 11 (3), 328–336. 10.1038/ncb1841
101
Grad L. I. Yerbury J. J. Turner B. J. Guest W. C. Pokrishevsky E. O'Neill M. A. et al (2014). “Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms,” in Proceedings of the National Academy of Sciences of the United States of America, 111 (9), 3620–3625. 10.1073/pnas.1312245111
102
Grieve A. G. Rabouille C. (2011). Golgi bypass: skirting around the heart of classical secretion. Cold Spring Harb. Perspect. Biol.3 (4), a005298. 10.1101/cshperspect.a005298
103
Griffiths R. E. Kupzig S. Cogan N. Mankelow T. J. Betin V. M. S. Trakarnsanga K. et al (2012). Maturing reticulocytes internalize plasma membrane in glycophorin A-containing vesicles that fuse with autophagosomes before exocytosis. Blood119, 6296–6306. 10.1182/blood-2011-09-376475
104
Gruden G. Bruno G. Chaturvedi N. Burt D. Schalkwijk C. Pinach S. et al (2008). Serum heat shock protein 27 and diabetes complications in the EURODIAB prospective complications study: a novel circulating marker for diabetic neuropathy. Diabetes57 (7), 1966–1970. 10.2337/db08-0009
105
Gu C. Fan X. Yu W. (2023). Functional diversity of mammalian small heat shock proteins: a review. Cells12, 1947. 10.3390/cells12151947
106
Guo H. Sun J. Li X. Xiong Y. Wang H. Shu H. et al (2018). Positive charge in the n-region of the signal peptide contributes to efficient post-translational translocation of small secretory preproteins. J. Biol. Chem.293 (6), 1899–1907. 10.1074/jbc.RA117.000922
107
Guo Y. S. Liang P. Z. Lu S. Z. Chen R. Yin Y. Q. Zhou J. W. (2019). Extracellular αB-crystallin modulates the inflammatory responses. Biochem. biophysical Res. Commun.508 (1), 282–288. 10.1016/j.bbrc.2018.11.024
108
Gustafsson G. Lööv C. Persson E. Lázaro D. F. Takeda S. Bergström J. et al (2018). Secretion and uptake of α-synuclein via extracellular vesicles in cultured cells. Cell. Mol. Neurobiol.38 (8), 1539–1550. 10.1007/s10571-018-0622-5
109
Gyurkovska V. Murtazina R. Zhao S. F. Shikano S. Okamoto Y. Segev N. (2023). Dual function of Rab1A in secretion and autophagy: hypervariable domain dependence. Life Sci. Alliance6, e202201810. 10.26508/lsa.202201810
110
Haidar M. Asselbergh B. Adriaenssens E. De Winter V. Timmermans J. P. Auer-Grumbach M. et al (2019). Neuropathy-causing mutations in HSPB1 impair autophagy by disturbing the formation of SQSTM1/p62 bodies. Autophagy15 (6), 1051–1068. 10.1080/15548627.2019.1569930
111
Hofferbert S. Burfeind P. Hoyer-Fender S. Lange R. Haidl G. Engel W. (1993). A homozygous deletion of 27 basepairs in the coding region of the human outer dense fiber protein gene does not result in a pathologic phenotype. Hum. Mol. Genet.2 (12), 2167–2170. 10.1093/hmg/2.12.2167
112
Hoffman L. Jensen C. C. Yoshigi M. Beckerle M. (2017). Mechanical signals activate p38 MAPK pathway-dependent reinforcement of actin via mechanosensitive HspB1. Mol. Biol. Cell28 (20), 2661–2675. 10.1091/mbc.E17-02-0087
113
Hoffmann A. C. Minakaki G. Menges S. Salvi R. Savitskiy S. Kazman A. et al (2019). Extracellular aggregated alpha synuclein primarily triggers lysosomal dysfunction in neural cells prevented by trehalose. Sci. Rep.9, 544. 10.1038/s41598-018-35811-8
114
Holmgren A. Bouhy D. De Winter V. Asselbergh B. Timmermans J. P. Irobi J. et al (2013). Charcot-Marie-Tooth causing HSPB1 mutations increase Cdk5-mediated phosphorylation of neurofilaments. Acta neuropathol.126 (1), 93–108. 10.1007/s00401-013-1133-6
115
Hornung S. Dutta S. Bitan G. (2020). CNS-derived blood exosomes as a promising source of biomarkers: opportunities and challenges. Front. Mol. Neurosci.13, 38. 10.3389/fnmol.2020.00038
116
Houlden H. Laura M. Wavrant-De Vrièze F. Blake J. Wood N. Reilly M. M. (2008). Mutations in the HSP27 (HSPB1) gene cause dominant, recessive, and sporadic distal HMN/CMT type 2. Neurology71 (21), 1660–1668. 10.1212/01.wnl.0000319696.14225.67
117
Howitt J. Hill A. F. (2016). Exosomes in the pathology of neurodegenerative diseases. J. Biol. Chem.291, 26589–26597. 10.1074/jbc.R116.757955
118
Huang Y. Meng S. Wu B. Shi H. Wang Y. Xiang J. et al (2023). HSPB2 facilitates neural regeneration through autophagy for sensorimotor recovery after traumatic brain injury. JCI insight8 (16), e168919. 10.1172/jci.insight.168919
119
Hull R. L. Shen Z.-P. Watts M. R. Kodama K. Carr D. B. Utzschneider K. M. et al (2005). Long-term treatment with rosiglitazone and metformin reduces the extent of, but does not prevent, islet amyloid deposition in mice expressing the gene for human islet amyloid polypeptide. Diabetes54 (7), 2235–2244. 10.2337/diabetes.54.7.2235
120
Hutchings A. J. Hambrecht B. Veh A. Giridhar N. J. Zare A. Angerer C. et al (2024). Plekhg5 controls the unconventional secretion of Sod1 by presynaptic secretory autophagy. Nat. Commun.15, 8622. 10.1038/s41467-024-52875-5
121
Iguchi Y. Eid L. Parent M. Soucy G. Bareil C. Riku Y. et al (2016). Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain: J. Neurol., 139 (Pt 12), 3187–3201. 10.1093/brain/aww237
122
Inagaki N. Hayashi T. Arimura T. Koga Y. Takahashi M. Shibata H. et al (2006). Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem. biophysical Res. Commun.342 (2), 379–386. 10.1016/j.bbrc.2006.01.154
123
Itakura E. Kishi-Itakura C. Mizushima N. (2012). The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell151 (6), 1256–1269. 10.1016/j.cell.2012.11.001
124
Javed A. Johnson O. T. Balana A. T. Volk R. F. Langen A. Ahn B. S. et al (2024). O-GlcNAc modification of HSP27 alters its protein interactions and promotes refolding of proteins through the BAG3/HSP70 co-chaperone. Protein Sci.33, e5173. 10.1002/pro.5173
125
Jena B. P. (2007). Secretion machinery at the cell plasma membrane. Curr. Opin. Struct. Biol.17, 437–443. 10.1016/j.sbi.2007.07.002
126
Jeon I. Cicchetti F. Cisbani G. Lee S. Li E. Bae J. et al (2016). Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta neuropathologica, 132 (4), 577–592. 10.1007/s00401-016-1582-9
127
Jiang P. Nishimura T. Sakamaki Y. Itakura E. Hatta T. Natsume T. et al (2014). The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell25 (8), 1327–1337. 10.1091/mbc.E13-08-0447
128
Jimenez-Sanchez M. Lam W. Hannus M. Sönnichsen B. Imarisio S. Fleming A. et al (2015). SiRNA screen identifies QPCT as a druggable target for Huntington’s disease. Nat. Chem. Biol.11, 347–354. 10.1038/nchembio.1790
129
Joshi B. S. Garcia Romeu H. Aliyandi A. de Vries M. P. Zuhorn I. S. (2022). DNAJB6-Containing extracellular vesicles as chaperone delivery systems: a proteomic analysis. Pharmaceutics14, 2485. 10.3390/pharmaceutics14112485
130
Joshi B. S. Youssef S. A. Bron R. de Bruin A. Kampinga H. H. Zuhorn I. S. (2021). DNAJB6b-enriched small extracellular vesicles decrease polyglutamine aggregation in in vitro and in vivo models of Huntington disease. iScience24, 103282. 10.1016/j.isci.2021.103282
131
Jucker M. Walker L. C. (2018). Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci.21, 1341–1349. 10.1038/s41593-018-0238-6
132
Kamelgarn M. Chen J. Kuang L. Arenas A. Zhai J. Zhu H. et al (2016). Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim. Biophys. Acta, 1862 (10), 2004–2014. 10.1016/j.bbadis.2016.07.015
133
Kappé G. Verschuure P. Philipsen R. L. Staalduinen A. A. Van de Boogaart P. Boelens W. C. et al (2001). Characterization of two novel human small heat shock proteins: protein kinase-related HspB8 and testis-specific HspB9. Biochimica biophysica acta1520 (1), 1–6. 10.1016/s0167-4781(01)00237-8
134
Kast D. J. Dominguez R. (2017). The cytoskeleton-autophagy connection. Curr. Biol.27 (8), R318–R326. 10.1016/j.cub.2017.02.061
135
Kato K. Goto S. Inaguma Y. Hasegawa K. Morishita R. Asano T. (1994). Purification and characterization of a 20-kDa protein that is highly homologous to αB crystallin. J. Biol. Chem.269, 15302–15309. 10.1016/s0021-9258(17)36606-1
136
Katz M. Davis M. Garton F. C. Henderson R. Bharti V. Wray N. et al (2020). Mutations in heat shock protein beta-1 (HSPB1) are associated with a range of clinical phenotypes related to different patterns of motor neuron dysfunction: a case series. J. Neurol. Sci.413, 116809. 10.1016/j.jns.2020.116809
137
Kawabata T. Yoshimori T. (2016). Beyond starvation: an update on the autophagic machinery and its functions. J. Mol. Cell Cardiol.95, 2–10. 10.1016/j.yjmcc.2015.12.005
138
Kennedy D. Mnich K. Oommen D. Chakravarthy R. Almeida-Souza L. Krols M. et al (2017). HSPB1 facilitates ERK-mediated phosphorylation and degradation of BIM to attenuate endoplasmic reticulum stress-induced apoptosis. Cell death and Dis.8 (8), e3026. 10.1038/cddis.2017.408
139
Khoshaman K. Yousefi R. Tamaddon A. M. Abolmaali S. S. Oryan A. Moosavi-Movahedi A. A. et al (2017). The impact of different mutations at Arg54 on structure, chaperone-like activity and oligomerization state of human αA-crystallin: the pathomechanism underlying congenital cataract-causing mutations R54L, R54P and R54C. Biochimica biophysica acta. Proteins proteomics1865 (5), 604–618. 10.1016/j.bbapap.2017.02.003
140
Kiernan M. C. Vucic S. Cheah B. C. Turner M. R. Eisen A. Hardiman O. et al (2011). Amyotrophic lateral sclerosis. Lancet377, 942–955. 10.1016/S0140-6736-(10)61156-7
141
Kim D.-H. Sarbassov D. D. Ali S. M. King J. E. Latek R. R. Erdjument-Bromage H. et al (2002). mTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex that Signals to the Cell Growth Machinery pathway regulates a variety of processes contributing to cell growth, including initiation of mRNA translation, ribosome synthesis, expression of metabolism-related genes, autophagy, and cytoskeletal reorganization recently reviewed by Schmelzle and Hall, 2000, and by Gin. Available online at: http://www.cell.com/cgi/content/full/110/.
142
Kim E. Goraksha-Hicks P. Li L. Neufeld T. P. Guan K. L. (2008). Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol.10, 935–945. 10.1038/ncb1753
143
Kim J. Cheon H. Jeong Y. T. Quan W. Kim K. H. Cho J. M. et al (2014). Amyloidogenic peptide oligomer accumulation in autophagy-deficient β cells induces diabetes. J. Clin. Investigation124, 3311–3324. 10.1172/JCI69625
144
Kim J. E. Hyun H. W. Min S. J. Kang T. C. (2017). Sustained HSP25 expression induces clasmatodendrosis via ER Stress in the rat hippocampus. Front. Cell. Neurosci.11, 47. 10.3389/fncel.2017.00047
145
Kim J. E. Ko A. R. Hyun H. W. Min S. J. Kang T. C. (2018). P2RX7-MAPK1/2-SP1 axis inhibits MTOR independent HSPB1-mediated astroglial autophagy. Cell Death Dis.9, 546. 10.1038/s41419-018-0586-x
146
Kimura T. Jia J. Kumar S. Choi S. W. Gu Y. Mudd M. et al (2017). Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J.36, 42–60. 10.15252/embj.201695081
147
Kinseth M. A. Anjard C. Fuller D. Guizzunti G. Loomis W. F. Malhotra V. (2007). The golgi-associated protein GRASP is required for unconventional protein secretion during development. Cell130, 524–534. 10.1016/j.cell.2007.06.029
148
Kolb S. J. Snyder P. J. Poi E. J. Renard E. A. Bartlett A. Gu S. et al (2010). Mutant small heat shock protein B3 causes motor neuropathy: utility of a candidate gene approach. Neurology74, 502–506. 10.1212/WNL.0b013e3181cef84a
149
Komatsu M. Waguri S. Chiba T. Murata S. Iwata J. Tanida I. et al (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature441 (7095), 880–884. 10.1038/nature04723
150
Koscielny A. Liszewska E. Machnicka K. Wezyk M. Kotulska K. Jaworski J. (2021). mTOR controls endoplasmic reticulum–Golgi apparatus trafficking of VSVg in specific cell types. Cell Mol. Biol. Lett.26, 18. 10.1186/s11658-021-00262-z
151
Kostenko S. Moens U. (2009). Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell Mol. Life Sci.66, 3289–3307. 10.1007/s00018-009-0086-3
152
Kuramoto K. Kim Y. J. Hong J. H. He C. (2021). The autophagy protein Becn1 improves insulin sensitivity by promoting adiponectin secretion via exocyst binding. Cell Rep.35, 109184. 10.1016/j.celrep.2021.109184
153
Kwok A. S. Phadwal K. Turner B. J. Oliver P. L. Raw A. Simon A. K. et al (2011). HspB8 mutation causing hereditary distal motor neuropathy impairs lysosomal delivery of autophagosomes. J. Neurochem.119, 1155–1161. 10.1111/j.1471-4159.2011.07521.x
154
Lambert H. Charette S. J. Bernier A. F. Guimond A. Landry J. (1999). HSP27 multimerization mediated by phosphorylation-sensitive intermolecular interactions at the amino terminus. J. Biol. Chem.274, 9378–9385. 10.1074/jbc.274.14.9378
155
La Padula V. Staszewski O. Nestel S. Busch H. Boerries M. Roussa E. et al (2016). HSPB3 protein is expressed in motoneurons and induces their survival after lesion-induced degeneration. Exp. Neurol.286, 40–49. 10.1016/j.expneurol.2016.08.014
156
Launay N. Goudeau B. Kato K. Vicart P. Lilienbaum A. (2006). Cell signaling pathways to alphaB-crystallin following stresses of the cytoskeleton. Exp. Cell Res.312, 3570–3584. 10.1016/j.yexcr.2006.07.025
157
Lavoie J. N. Lambert H. Hickey E. Weber L. A. Landry J. (1995). Modulation of cellular thermo-resistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol. Cell Biol.15, 505–516. 10.1128/MCB.15.1.505
158
Lazarou M. Sliter D. A. Kane L. A. Sarraf S. A. Wang C. Burman J. L. et al (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature524, 309–314. 10.1038/nature14893
159
Le Ber I. Camuzat A. Guerreiro R. Bouya-Ahmed K. Bras J. Nicolas G. et al (2013). SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol.70 (11), 1403–1410doi. 10.1001/jamaneurol.2013.3849
160
Lee J. G. Takahama S. Zhang G. Tomarev S. I. Ye Y. (2016). Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat. Cell Biol.18, 765–776. 10.1038/ncb3372
161
Lee J. Y. Koga H. Kawaguchi Y. Tang W. Wong E. Gao Y. S. et al (2010). HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J.29 (5), 969–980. 10.1038/emboj.2009.405
162
Lee S. Carson K. Rice-Ficht A. Good T. (2006). Small heat shock proteins differentially affect Abeta aggregation and toxicity. Biochem. biophysical Res. Commun.347 (2), 527–533. 10.1016/j.bbrc.2006.06.128
163
Lewis S. E. Mannion R. J. White F. A. Coggeshall R. E. Beggs S. Costigan M. et al (1999). A role for HSP27 in sensory neuron survival. J. Neurosci. official J. Soc. Neurosci.19 (20), 8945–8953. 10.1523/JNEUROSCI.19-20-08945.1999
164
Li C. Kuang J. Wang X. Zhang G. Hong M. Cheng G. et al (2024a). Exosomal Hsp27 protein are associated with heart failure in STZ-induced type 1 diabetic rats. Int. J. Biol. Macromol.277, 133772. 10.1016/j.ijbiomac.2024.133772
165
Li D. Y. Liang S. Wen J. H. Tang J. X. Deng S. L. Liu Y. X. (2022a). Extracellular HSPs: the potential target for human disease therapy. Molecules27, 2361. 10.3390/molecules27072361
166
Li J. Krause G. J. Gui Q. Kaushik S. Rona G. Zhang Q. et al (2023a). A noncanonical function of SKP1 regulates the switch between autophagy and unconventional secretion. Sci. Adv.9, eadh1134. 10.1126/sciadv.adh1134
167
Li Q. Peng G. Liu H. Wang L. Lu R. Li L. (2024b). Molecular mechanisms of secretory autophagy and its potential role in diseases. Life Sci.347, 122653. 10.1016/j.lfs.2024.122653
168
Li S. Yan R. Xu J. Zhao S. Ma X. Sun Q. et al (2022b). A new type of ERGIC–ERES membrane contact mediated by TMED9 and SEC12 is required for autophagosome biogenesis. Cell Res.32, 119–138. 10.1038/s41422-021-00563-0
169
Li X. C. Hu Q. K. Chen L. Liu S. Y. Su S. Tao H. et al (2017). HSPB8 promotes the fusion of autophagosome and lysosome during autophagy in diabetic neurons. Int. J. Med. Sci.14, 1335–1341. 10.7150/ijms.20653
170
Li Z. Wang X. Wang X. Yi X. Wong Y. K. Wu J. et al (2023b). Research progress on the role of extracellular vesicles in neurodegenerative diseases. Transl. Neurodegener.12, 43. 10.1186/s40035-023-00375-9
171
Liang P. MacRae T. H. (1997). Molecular chaperones and the cytoskeleton. J. cell Sci.110 (Pt 13), 1431–1440. 10.1242/jcs.110.13.1431
172
Liang W. Sagar S. Ravindran R. Najor R. H. Quiles J. M. Chi L. et al (2023). Mitochondria are secreted in extracellular vesicles when lysosomal function is impaired. Nat. Commun.14, 5031. 10.1038/s41467-023-40680-5
173
Liao W. C. Juo L. Y. Shih Y. L. Chen Y. H. Yan Y. T. (2017). HSPB7 prevents cardiac conduction system defect through maintaining intercalated disc integrity. PLoS Genet.13 (8), e1006984. 10.1371/journal.pgen.1006984
174
Lin P. W. Chu M. L. Liu Y. W. Chen Y. C. Shih Y. H. Lan S. H. et al (2024). Revealing potential Rab proteins participate in regulation of secretory autophagy machinery. Kaohsiung J. Med. Sci.40, 642–649. 10.1002/kjm2.12848
175
Lippincott-Schwartz J. Roberts T. H. Hirschberg K. (2000). Secretory protein trafficking and organelle dynamics in living cells. Annu. Rev. cell Dev. Biol.16, 557–589. 10.1146/annurev.cellbio.16.1.557
176
Liu G. S. Zhu H. Cai W. F. Wang X. Jiang M. Essandoh K. et al (2018). Regulation of BECN1-mediated autophagy by HSPB6: insights from a human HSPB6S10F mutant. Autophagy14, 80–97. 10.1080/15548627.2017.1392420
177
Lopez A. Siddiqi F. H. Villeneuve J. Ureshino R. P. Jeon H. Y. Koulousakis P. et al (2025). Carbonic anhydrase inhibition ameliorates tau toxicity via enhanced tau secretion. Nat. Chem. Biol.21 (4), 577–587. 10.1038/s41589-024-01762-7
178
Lopez-Castejon G. Brough D. (2011). Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev.22, 189–195. 10.1016/j.cytogfr.2011.10.001
179
Lu S. Hu J. Arogundade O. A. Goginashvili A. Vazquez-Sanchez S. Diedrich J. K. et al (2022). Heat-shock chaperone HSPB1 regulates cytoplasmic TDP-43 phase separation and liquid-to-gel transition. Nat. Cell Biol.24, 1378–1393. 10.1038/s41556-022-00988-8
180
Luk K. C. Kehm V. M. Zhang B. O'Brien P. Trojanowski J. Q. Lee V. M. (2012). Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med.209 (5), 975–986. 10.1084/jem.20112457
181
Lystad A. H. Carlsson S. R. de la Ballina L. R. Kauffman K. J. Nag S. Yoshimori T. et al (2019). Distinct functions of ATG16L1 isoforms in membrane binding and LC3B lipidation in autophagy-related processes. Nat. Cell Biol.21, 372–383. 10.1038/s41556-019-0274-9
182
Ma P. Li J. Qi L. Dong X. (2021). The archaeal small heat shock protein Hsp17.6 protects proteins from oxidative inactivation. Int. J. Mol. Sci.22 (5), 2591. 10.3390/ijms22052591
183
Mancias J. D. Goldberg J. (2007). The transport signal on Sec22 for packaging into COPII-coated vesicles is a conformational epitope. Mol. Cell26, 403–414. 10.1016/j.molcel.2007.03.017
184
Manjithaya R. Anjard C. Loomis W. F. Subramani S. (2010). Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J. Cell Biol.188, 537–546. 10.1083/jcb.200911149
185
Marie G. Dunning C. J. Gaspar R. Grey C. Brundin P. Sparr E. et al (2015). Acceleration of α-synuclein aggregation by exosomes. J. Biol. Chem.290, 2969–2982. 10.1074/jbc.M114.585703
186
Martinelli S. Anderzhanova E. A. Bajaj T. Wiechmann S. Dethloff F. Weckmann K. et al (2021). Stress-primed secretory autophagy promotes extracellular BDNF maturation by enhancing MMP9 secretion. Nat. Commun.12, 4643. 10.1038/s41467-021-24810-5
187
Martín-Sánchez F. Diamond C. Zeitler M. Gomez A. I. Baroja-Mazo A. Bagnall J. et al (2016). Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ.23, 1219–1231. 10.1038/cdd.2015.176
188
Martin-Ventura J. L. Duran M. C. Blanco-Colio L. M. Meilhac O. Leclercq A. Michel J. B. et al (2004). Identification by a differential proteomic approach of heat shock protein 27 as a potential marker of atherosclerosis. Circulation110 (15), 2216–2219. 10.1161/01.CIR.0000136814.87170.B1
189
Masnata M. Sciacca G. Maxan A. Bousset L. Denis H. L. Lauruol F. et al (2019). Demonstration of prion-like properties of mutant huntingtin fibrils in both in vitro and in vivo paradigms. Acta Neuropathol.137, 981–1001. 10.1007/s00401-019-01973-6
190
Meldolesi J. (2022). Unconventional protein secretion dependent on two extracellular vesicles: exosomes and ectosomes. Front. Cell Dev. Biol.10, 877344. 10.3389/fcell.2022.877344
191
Meldolesi J. (2023). Unconventional protein secretion (UPS): role in important diseases. Mol. Biomed.4, 2. 10.1186/s43556-022-00113-z
192
Menzies F. M. Fleming A. Caricasole A. Bento C. F. Andrews S. P. Ashkenazi A. et al (2017). Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron93 (5), 1015–1034. 10.1016/j.neuron.2017.01.022
193
Menzies F. M. Fleming A. Rubinsztein D. C. (2015). Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci.16, 345–357. 10.1038/nrn3961
194
Mercer E. J. Lin Y. F. Cohen-Gould L. Evans T. (2018). Hspb7 is a cardioprotective chaperone facilitating sarcomeric proteostasis. Dev. Biol.435 (1), 41–55. 10.1016/j.ydbio.2018.01.005
195
Merkley S. D. Goodfellow S. M. Guo Y. Wilton Z. E. R. Byrum J. R. Schwalm K. C. et al (2020). Non-autophagy role of Atg5 and NBR1 in unconventional secretion of IL-12 prevents gut dysbiosis and inflammation. 10.1101/2020.12.07.414227
196
Miron T. Vancompernolle K. Vandekerckhove J. Wilchek M. Geiger B. (1991). A 25-kD inhibitor of actin polymerization is a low molecular mass heat shock protein. J. Cell Biol.114 (2), 255–261. 10.1083/jcb.114.2.255
197
Miron T. Wilchek M. Geiger B. (1988). Characterization of an inhibitor of actin polymerization in vinculin-rich fraction of Turkey gizzard smooth muscle. Eur. J. Biochem.178 (2), 543–553. 10.1111/j.1432-1033.1988.tb14481.x
198
Mizushima N. Yoshimori T. Levine B. (2010). Methods in mammalian autophagy research. Cell140, 313–326. 10.1016/j.cell.2010.01.028
199
Mizushima N. Yoshimori T. Ohsumi Y. (2011). The role of atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol.27, 107–132. 10.1146/annurev-cellbio-092910-154005
200
Mogk A. Ruger-Herreros C. Bukau B. (2019). Cellular functions and mechanisms of action of small heat shock proteins. Annu. Rev. Microbiol.73, 89–110. 10.1146/annurev-micro-020518-115515
201
Monastyrska I. He C. Geng J. Hoppe A. D. Li Z. Klionsky D. J. (2008). Arp2 links autophagic machinery with the actin cytoskeleton. Mol. Biol. Cell19 (5), 1962–1975. 10.1091/mbc.e07-09-0892
202
Monteleone M. Stow J. L. Schroder K. (2015). Mechanisms of unconventional secretion of IL-1 family cytokines. Cytokine74, 213–218. 10.1016/j.cyto.2015.03.022
203
Moreau K. Ghislat G. Hochfeld W. Renna M. Zavodszky E. Runwal G. et al (2015). Transcriptional regulation of Annexin A2 promotes starvation-induced autophagy. Nat. Commun.6, 8045. 10.1038/ncomms9045
204
Moreau K. Luo S. Rubinsztein D. C. (2010). Cytoprotective roles for autophagy. Curr. Opin. cell Biol.22 (2), 206–211. 10.1016/j.ceb.2009.12.002
205
Moreau K. Renna M. Rubinsztein D. C. (2013). Connections between SNAREs and autophagy. Trends Biochem. Sci.38 (2), 57–63. 10.1016/j.tibs.2012.11.004
206
Mukherjee A. Morales-Scheihing D. Salvadores N. Moreno-Gonzalez I. Gonzalez C. Taylor-Presse K. et al (2017). Induction of IAPP amyloid deposition and associated diabetic abnormalities by a prion-like mechanism. J. Exp. Med.214, 2591–2610. 10.1084/jem.20161134
207
Munding C. Keller M. Niklaus G. Papin S. Tschopp J. Werner S. et al (2006). The estrogen-responsive B box protein: a novel enhancer of interleukin-1beta secretion. Cell Death Differ.13, 1938–1949. 10.1038/sj.cdd.4401896
208
Muranova L. K. Ryzhavskaya A. S. Sudnitsyna M. V. Shatov V. M. Gusev N. B. (2019). Small heat shock proteins and human neurodegenerative diseases. Biochem. Mosc.84, 1256–1267. 10.1134/S000629791911004X
209
Muranova L. K. Shatov V. M. Gusev N. B. (2022). Role of small heat shock proteins in the remodeling of actin microfilaments. Biochem. Mosc.87, 800–811. 10.1134/S0006297922080119
210
Nafar F. Williams J. B. Mearow K. M. (2016). Astrocytes release HspB1 in response to amyloid-β exposure in vitro. J. Alzheimer's Dis. JAD49 (1), 251–263. 10.3233/JAD-150317
211
Nagaraj R. H. Oya-Ito T. Bhat M. Liu B. (2005). Dicarbonyl stress and apoptosis of vascular cells: prevention by alphaB-crystallin. Ann. N. Y. Acad. Sci.1043, 158–165. 10.1196/annals.1333.020
212
Nakamura Y. Sawai T. Kakiuchi K. Arawaka S. (2024). Neuronal activity promotes secretory autophagy for the extracellular release of α-synuclein. J. Biol. Chem.300 (7), 107419. 10.1016/j.jbc.2024.107419
213
Nakatogawa H. (2020). Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol.21, 439–458. 10.1038/s41580-020-0241-0
214
Narita M. Young A. R. Arakawa S. Samarajiwa S. A. Nakashima T. Yoshida S. et al (2011). Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science (New York, N.Y.), 332 (6032), 966–970. 10.1126/science.1205407
215
Nicolás-Ávila J. A. Lechuga-Vieco A. V. Esteban-Martínez L. Sánchez-Díaz M. Díaz-García E. Santiago D. J. et al (2020). A network of macrophages supports mitochondrial homeostasis in the heart. Cell183, 94–109.e23. 10.1016/j.cell.2020.08.031
216
Nicolás-Ávila J. A. Pena-Couso L. Muñoz-Cánoves P. Hidalgo A. (2022). Macrophages, metabolism and heterophagy in the heart. Circ. Res.130, 418–431. 10.1161/CIRCRESAHA.121.319812
217
Nilsson P. Loganathan K. Sekiguchi M. Matsuba Y. Hui K. Tsubuki S. et al (2013). Aβ secretion and plaque formation depend on autophagy. Cell Rep.5 (1), 61–69. 10.1016/j.celrep.2013.08.042
218
Nixon R. A. (2007). Autophagy, amyloidogenesis and Alzheimer disease. J. cell Sci.120 (Pt 23), 4081–4091. 10.1242/jcs.019265
219
Noh S. H. Kim Y. J. Lee M. G. (2022). Autophagy-related pathways in vesicular unconventional protein secretion. Front. Cell Dev. Biol.10, 892450. 10.3389/fcell.2022.892450
220
Nüchel J. Ghatak S. Zuk A. V. Illerhaus A. Mörgelin M. Schönborn K. et al (2018). TGFB1 is secreted through an unconventional pathway dependent on the autophagic machinery and cytoskeletal regulators. Autophagy14, 465–486. 10.1080/15548627.2017.1422850
221
O’donnell A. F. Schmidt M. C. (2019). AMPK-mediated regulation of alpha-arrestins and protein trafficking. Int. J. Mol. Sci.20, 515. 10.3390/ijms20030515
222
Onishi M. Yamano K. Sato M. Matsuda N. Okamoto K. (2021). Molecular mechanisms and physiological functions of mitophagy. EMBO J.40, e104705. 10.15252/embj.2020104705
223
Ousman S. S. Tomooka B. H. van Noort J. M. Wawrousek E. F. O'Connor K. C. Hafler D. A. et al (2007). Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature, 448(7152), 474–479. 10.1038/nature05935
224
Outeiro T. F. Klucken J. Strathearn K. E. Liu F. Nguyen P. Rochet J. C. et al (2006). Small heat shock proteins protect against alpha-synuclein-induced toxicity and aggregation. Biochem. biophysical Res. Commun.351 (3), 631–638. 10.1016/j.bbrc.2006.10.085
225
Padmanabhan S. Manjithaya R. (2020). Facets of autophagy based unconventional protein secretion–the road less traveled. Front. Mol. Biosci.7, 586483. 10.3389/fmolb.2020.586483
226
Pandey N. K. Isas J. M. Rawat A. Lee R. V. Langen J. Pandey P. et al (2018). The 17-residue-long N terminus in huntingtin controls stepwise aggregation in solution and on membranes via different mechanisms. J. Biol. Chem.293, 2597–2605. 10.1074/jbc.M117.813667
227
Park A. M. Tsunoda I. Yoshie O. (2018). Heat shock protein 27 promotes cell cycle progression by down regulating E2F transcription factor 4 and retinoblastoma family protein p130. J. Biol. Chem.293 (41), 15815–15826. 10.1074/jbc.RA118.003310
228
Park H. K. Park E. C. Bae S. W. Park M. Y. Kim S. W. Yoo H. S. et al (2006). Expression of heat shock protein 27 in human atherosclerotic plaques and increased plasma level of heat shock protein 27 in patients with acute coronary syndrome. Circulation114 (9), 886–893. 10.1161/CIRCULATIONAHA.105.541219
229
Perrin V. Régulier E. Abbas-Terki T. Hassig R. Brouillet E. Aebischer P. et al (2007). Neuroprotection by Hsp104 and Hsp27 in lentiviral-based rat models of Huntington's disease. Mol. Ther. J. Am. Soc. Gene Ther.15 (5), 903–911. 10.1038/mt.sj.6300141
230
Peters C. Haslbeck M. Buchner J. (2024). Catchers of folding gone awry: a tale of small heat shock proteins. Trends Biochem. Sci.49, 1063–1078. 10.1016/j.tibs.2024.08.003
231
Picard F. Nonaka T. Belotti E. Osseni A. Errazuriz-Cerda E. Jost-Mousseau C. et al (2025). Enhanced secretion of the amyotrophic lateral sclerosis ALS-associated misfolded TDP-43 mediated by the ER-ubiquitin specific peptidase USP19. Cell. Mol. life Sci. CMLS82 (1), 76. 10.1007/s00018-025-05589-w
232
Piletic K. Alsaleh G. Simon A. K. (2023). Autophagy orchestrates the crosstalk between cells and organs. EMBO Rep.24, e57289. 10.15252/embr.202357289
233
Pinkerton M. Lourenco G. Torres Pacheco M. Halliday G. M. Kiernan M. C. Tan R. H. (2023). Survival in sporadic ALS is associated with lower p62 burden in the spinal cord. J. Neuropathol. Exp. Neurol.82 (9), 769–773. 10.1093/jnen/nlad051
234
Poehler A. M. Xiang W. Spitzer P. May V. E. Meixner H. Rockenstein E. et al (2014). Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy, 10 (12), 2171–2192. 10.4161/auto.36436
235
Popa S. J. Stewart S. E. Moreau K. (2018). Unconventional secretion of annexins and galectins. Semin. Cell Dev. Biol.83, 42–50. 10.1016/j.semcdb.2018.02.022
236
Qarawani A. Naaman E. Ben-Zvi Elimelech R. Harel M. Sigal-Dror S. Ben-Zur T. (2025). Mesenchymal stem cell-derived exosomes mitigate amyloid β-induced retinal toxicity: insights from rat model and cellular studies. J. Extracell. Biol.4, e70024. 10.1002/jex2.70024
237
Rabouille C. (2017). Pathways of unconventional protein secretion. Trends cell Biol.27 (3), 230–240. 10.1016/j.tcb.2016.11.007
238
Rajendran L. Honsho M. Zahn T. R. Keller P. Geiger K. D. Verkade P. et al (2006). Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U. S. A.103, 11172–11177. 10.1073/pnas.0603838103
239
Ramazanov B. R. Tran M. L. von Blume J. (2021). Sending out molecules from the TGN. Curr. Opin. cell Biol.71, 55–62. 10.1016/j.ceb.2021.02.005
240
Ramirez A. Heimbach A. Gründemann J. Stiller B. Hampshire D. Cid L. P. et al (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet.38, 1184–1191. 10.1038/ng1884
241
Ramirez S. H. Andrews A. M. Paul D. Pachter J. S. (2018). Extracellular vesicles: mediators and biomarkers of pathology along CNS barriers. Fluids Barriers CNS15, 19. 10.1186/s12987-018-0104-7
242
Rayner K. Sun J. Chen Y. X. McNulty M. Simard T. Zhao X. et al (2009). Heat shock protein 27 protects against atherogenesis via an estrogen-dependent mechanism: role of selective estrogen receptor beta modulation. Arteriosclerosis, thrombosis, Vasc. Biol.29 (11), 1751–1756. 10.1161/ATVBAHA.109.193656
243
Reddy V. S. Madala S. K. Trinath J. Reddy G. B. (2018). Extracellular small heat shock proteins: exosomal biogenesis and function. Cell stress and chaperones23 (3), 441–454. 10.1007/s12192-017-0856-z
244
Reggiori F. Monastyrska I. Shintani T. Klionsky D. J. (2005). The actin cytoskeleton is required for selective types of autophagy, but not nonspecific autophagy, in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell16 (12), 5843–5856. 10.1091/mbc.e05-07-0629
245
Reinle K. Mogk A. Bukau B. (2022). The diverse functions of small heat shock proteins in the proteostasis network: functions and mechanisms of sHsps. J. Mol. Biol.434, 167157. 10.1016/j.jmb.2021.167157
246
Ren P. H. Lauckner J. E. Kachirskaia I. Heuser J. E. Melki R. Kopito R. R. (2009). Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Bio., 11 (2), 219–225. 10.1038/ncb1830
247
Renna M. Bento C. F. Fleming A. Menzies F. M. Siddiqi F. H. Ravikumar B. et al (2013). IGF-1 receptor antagonism inhibits autophagy. Hum. Mol. Genet.22 (22), 4528–4544. 10.1093/hmg/ddt300
248
Renna M. Schaffner C. Winslow A. R. Menzies F. M. Peden A. A. Floto R. A. et al (2011). Autophagic substrate clearance requires activity of the syntaxin-5 SNARE complex. J. Cell Sci.124, 469–482. 10.1242/jcs.076489
249
Robertson A. L. Headey S. J. Saunders H. M. Ecroyd H. Scanlon M. J. Carver J. A. et al (2010). Small heat-shock proteins interact with a flanking domain to suppress polyglutamine aggregation. Proc. Natl. Acad. Sci. U. S. A.107 (23), 10424–10429. 10.1073/pnas.0914773107
250
Rodriguez Ospina S. Blazier D. M. Criado-Marrero M. Gould L. A. Gebru N. T. Beaulieu-Abdelahad D. et al (2022). Small heat shock protein 22 improves cognition and learning in the tauopathic brain. Int. J. Mol. Sci.23 (2), 851. 10.3390/ijms23020851
251
Rosati A. Graziano V. De Laurenzi V. Pascale M. Turco M. C. (2011). BAG3: a multifaceted protein that regulates major cell pathways. Cell death and Dis.2 (4), e141. 10.1038/cddis.2011.24
252
Rostami J. Holmqvist S. Lindström V. Sigvardson J. Westermark G. T. Ingelsson M. et al (2017). Human Astrocytes Transfer Aggregated Alpha-Synuclein via Tunneling Nanotubes. J. Neurosci: Off. J. Soc. Neurosci., 37 (49), 11835–11853. 10.1523/JNEUROSCI.0983-17.2017
253
Rubino E. Boschi S. Roveta F. Marcinnò A. Cermelli A. Borghese C. et al (2022). Concentrations in cerebrospinal fluid of patients with dementia: a potential autophagy biomarker in vivo?Brain Sci.12 (10), 1414. 10.3390/brainsci12101414
254
Saxton R. A. Sabatini D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell168, 960–976. 10.1016/j.cell.2017.02.004
255
Schäffner I. Minakaki G. Khan M. A. Balta E. A. Schlötzer-Schrehardt U. Schwarz T. J. et al (2018). FoxO function is essential for maintenance of autophagic flux and neuronal morphogenesis in adult neurogenesis. Neuron99, 1188–1203.e6. 10.1016/j.neuron.2018.08.017
256
Secco V. Tiago T. Staats R. Preet S. Chia S. Vendruscolo M. et al (2024). HSPB6: a lipid-dependent molecular chaperone inhibits α-synuclein aggregation. iScience27, 110657. 10.1016/j.isci.2024.110657
257
Seidel K. Vinet J. Dunnen W. F. Brunt E. R. Meister M. Boncoraglio A. et al (2012). The HSPB8-BAG3 chaperone complex is upregulated in astrocytes in the human brain affected by protein aggregation diseases. Neuropathology Appl. Neurobiol.38 (1), 39–53. 10.1111/j.1365-2990.2011.01198.x
258
Sepúlveda D. Cisternas-Olmedo M. Arcos J. Nassif M. Vidal R. L. (2022). Contribution of autophagy-lysosomal pathway in the exosomal secretion of alpha-synuclein and its impact in the progression of Parkinson’s disease. Front. Mol. Neurosci.15, 805087. 10.3389/fnmol.2022.805087
259
Settembre C. Di Malta C. Polito V. A. Arencibia M. G. Vetrini F. Erdin S. et al (2011). TFEB links autophagy to lysosomal biogenesis. Science332, 1429–1433. 10.1126/science.1204592
260
Sharma P. Mesci P. Carromeu C. McClatchy D. R. Schiapparelli L. Yates J. R. et al (2019). Exosomes regulate neurogenesis and circuit assembly. Proc. Natl. Acad. Sci. U. S. A.116, 16086–16094. 10.1073/pnas.1902513116
261
Sharma S. K. Chorell E. Wittung-Stafshede P. (2015). Insulin-degrading enzyme is activated by the C-terminus of α-synuclein. Biochem. Biophys. Res. Commun.466, 192–195. 10.1016/j.bbrc.2015.09.002
262
Sharples R. A. Vella L. J. Nisbet R. M. Naylor R. Perez K. Barnham K. J. et al (2008). Inhibition of γ‐secretase causes increased secretion of amyloid precursor protein C‐terminal fragments in association with exosomes. FASEB J.22, 1469–1478. 10.1096/fj.07-9357com
263
Shatov V. M. Gusev N. B. (2020). Physico-chemical properties of two point mutants of small heat shock protein HspB6 (Hsp20) with abrogated cardioprotection. Biochimie174, 126–135. 10.1016/j.biochi.2020.04.021
264
Shatov V. M. Muranova L. K. Zamotina M. A. Sluchanko N. N. Gusev N. B. (2023). α-Crystallin domains of five human small heat shock proteins (sHsps) differ in dimer stabilities and ability to incorporate themselves into oligomers of full-length sHsps. Int. J. Mol. Sci.24, 1085. 10.3390/ijms24021085
265
Shi C. Ulke-Lemée A. Deng J. Batulan Z. O'Brien E. R. (2019). Characterization of heat shock protein 27 in extracellular vesicles: a potential anti-inflammatory therapy. FASEB J. official Publ. Fed. Am. Soc. Exp. Biol.33 (2), 1617–1630. 10.1096/fj.201800987R
266
Shigihara N. Fukunaka A. Hara A. Komiya K. Honda A. Uchida T. et al (2014). Human IAPP-induced Pancreatic β cell toxicity and its regulation by autophagy. J. Clin. Investigation124, 3634–3644. 10.1172/JCI69866
267
Sica V. Galluzzi L. Bravo-San Pedro J. M. Izzo V. Maiuri M. C. Kroemer G. (2015). Organelle-specific initiation of autophagy. Mol. Cell59, 522–539. 10.1016/j.molcel.2015.07.021
268
Simón D. García-García E. Gómez-Ramos A. Falcón-Pérez J. M. Díaz-Hernández M. Hernández F. et al (2012). Tau overexpression results in its secretion via membrane vesicles. Neuro-Degener. Dis., 10 (1–4), 73–75. 10.1159/000334915
269
Singh B. N. Rao K. S. Ramakrishna T. Rangaraj N. Rao C. M. (2007). Association of alphaB-crystallin, a small heat shock protein, with actin: role in modulating actin filament dynamics in vivo. J. Mol. Biol.366, 756–767. 10.1016/j.jmb.2006.12.012
270
Son J. H. Shim J. H. Kim K. H. Ha J. Y. Han J. Y. (2012). Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med.44, 89–98. 10.3858/emm.2012.44.2.031
271
Son S. M. Cha M. Y. Choi H. Kang S. Choi H. Lee M. S. et al (2016). Insulin-degrading enzyme secretion from astrocytes is mediated by an autophagy-based unconventional secretory pathway in Alzheimer disease. Autophagy12, 784–800. 10.1080/15548627.2016.1159375
272
Song J. X. Malampati S. Zeng Y. Durairajan S. S. K. Yang C. B. Tong B. C. K. et al (2020). A small molecule transcription factor EB activator ameliorates beta-amyloid precursor protein and Tau pathology in Alzheimer’s disease models. Aging Cell19, e13069. 10.1111/acel.13069
273
Soto C. Estrada L. Castilla J. (2006). Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem. Sci.31, 150–155. 10.1016/j.tibs.2006.01.002
274
Soto C. Pritzkow S. (2018). Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci.21, 1332–1340. 10.1038/s41593-018-0235-9
275
Sun Y. Tao X. Han Y. Lin X. Tian R. Wang H. et al (2024). A dual role of ERGIC-localized Rabs in TMED10-mediated unconventional protein secretion. Nat. Cell Biol.26, 1077–1092. 10.1038/s41556-024-01445-4
276
Takayama S. Reed J. C. Homma S. (2003). Heat-shock proteins as regulators of apoptosis. Oncogene22, 9041–9047. 10.1038/sj.onc.1207114
277
Tamboli I. Y. Barth E. Christian L. Siepmann M. Kumar S. Singh S. et al (2010). Statins promote the degradation of extracellular amyloid {beta}-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion. J. Biol. Chem.285, 37405–37414. 10.1074/jbc.M110.149468
278
Tamminemi P. Ye X. Feng T. Aikal D. Cai Q. (2017). Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. Elife. 10.7554/eLife.21776.001
279
Tan H. W. S. Lu G. Dong H. Cho Y. L. Natalia A. Wang L. et al (2022). A degradative to secretory autophagy switch mediates mitochondria clearance in the absence of the mATG8-conjugation machinery. Nat. Commun.13, 3720. 10.1038/s41467-022-31213-7
280
Tanaka Y. Ito S. I. Honma Y. Hasegawa M. Kametani F. Suzuki G. et al (2023). Dysregulation of the progranulin-driven autophagy-lysosomal pathway mediates secretion of the nuclear protein TDP-43. J. Biol. Chem., 299 (11), 105272. 10.1016/j.jbc.2023.105272
281
Tardivel M. Bégard S. Bousset L. Dujardin S. Coens A. Melki R. et al (2016). Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun., 4 (1), 117. 10.1186/s40478-016-0386-4
282
Tedesco B. Vendredy L. Adriaenssens E. Cozzi M. Asselbergh B. Crippa V. et al (2023). HSPB8 frameshift mutant aggregates weaken chaperone-assisted selective autophagy in neuromyopathies. Autophagy19, 2217–2239. 10.1080/15548627.2023.2179780
283
Teyssou E. Takeda T. Lebon V. Boillée S. Doukouré B. Bataillon G. et al (2013). Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol.125, 511–522. 10.1007/s00401-013-1090-0
284
Thakur A. K. Jayaraman M. Mishra R. Thakur M. Chellgren V. M. Byeon I. J. et al (2009). Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. and Mol. Biol.16 (4), 380–389. 10.1038/nsmb.1570
285
Torralba D. Baixauli F. Villarroya-Beltri C. Fernández-Delgado I. Latorre-Pellicer A. Acín-Pérez R. et al (2018). Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat. Commun.9, 2658. 10.1038/s41467-018-05077-9
286
Trajkovic K. Jeong H. Krainc D. (2017). Mutant huntingtin is secreted via a late endosomal/lysosomal unconventional secretory pathway. J. Neurosci: Off. J. Soc. Neurosci., 37 (37), 9000–9012. 10.1523/JNEUROSCI.0118-17.2017
287
Trist B. G. Fifita J. A. Hogan A. Grima N. Smith B. Troakes C. et al (2022). Co-deposition of SOD1, TDP-43 and p62 proteinopathies in ALS: evidence for multifaceted pathways underlying neurodegeneration. Acta Neuropathol. Commun.10, 122. 10.1186/s40478-022-01421-9
288
Tsunemi T. Hamada K. Krainc D. (2014). ATP13A2/PARK9 regulates secretion of exosomes and α-Synuclein. J. Neurosci.34, 15281–15287. 10.1523/JNEUROSCI.1629-14.2014
289
Turco E. Witt M. Abert C. Bock-Bierbaum T. Su M. Y. Trapannone R. et al (2019). FIP200 claw domain binding to p62 promotes autophagosome formation at ubiquitin condensates. Mol. Cell74, 330–346.e11. 10.1016/j.molcel.2019.01.035
290
Van den Broek B. Wuyts C. Irobi J. (2021). Extracellular vesicle-associated small heat shock proteins as therapeutic agents in neurodegenerative diseases and beyond. Adv. drug Deliv. Rev.179, 114009. 10.1016/j.addr.2021.114009
291
van Niel G. Carter D. R. F. Clayton A. Lambert D. W. Raposo G. Vader P. (2022). Challenges and directions in studying cell–cell communication by extracellular vesicles. Nat. Rev. Mol. Cell Biol.23, 369–382. 10.1038/s41580-022-00460-3
292
van Noort J. M. Bsibsi M. Nacken P. Gerritsen W. H. Amor S. (2012). The link between small heat shock proteins and the immune system. Int. J. Biochem. and cell Biol.44 (10), 1670–1679. 10.1016/j.biocel.2011.12.010
293
van Noort J. M. Bsibsi M. Nacken P. J. Verbeek R. Venneker E. H. (2015). Therapeutic intervention in multiple sclerosis with alpha B-crystallin: a randomized controlled phase IIa trial. PloS one10 (11), e0143366. 10.1371/journal.pone.0143366
294
Vats S. Dionisio P. Pineau R. Schmitt A. Louet J. F. Tannour-Louet M. et al (2025). Role of late endosomal autophagy-dependent secretion of ER and mitochondrial components in cell fitness and communication. Cell Rep.10.2139/ssrn.5063619
295
Vendredy L. Adriaenssens E. Timmerman V. (2020). Small heat shock proteins in neurodegenerative diseases. Cell Stress Chaperones25, 679–699. 10.1007/s12192-020-01101-4
296
Vicart P. Caron A. Guicheney P. Li Z. Prévost M. C. Faure A. et al (1998). A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat. Genet.20 (1), 92–95. 10.1038/1765
297
Vos M. J. Zijlstra M. P. Kanon B. van Waarde-Verhagen M. A. Brunt E. R. Oosterveld-Hut H. M. et al (2010). HSPB7 is the most potent polyQ aggregation suppressor within the HSPB family of molecular chaperones. Hum. Mol. Genet.19 (23), 4677–4693. 10.1093/hmg/ddq398
298
Wagstaff M. J. Collaço-Moraes Y. Smith J. de Belleroche J. S. Coffin R. S. Latchman D. S. (1999). Protection of neuronal cells from apoptosis by Hsp27 delivered with a herpes simplex virus-based vector. J. Biol. Chem.274 (8), 5061–5069. 10.1074/jbc.274.8.5061
299
Wang Y. Li L. Hou C. Lai Y. Long J. Liu J. et al (2016). SNARE-mediated membrane fusion in autophagy. Semin. Cell Dev. Biol.60, 97–104. 10.1016/j.semcdb.2016.07.009
300
Wang Y. Zhang J. Liu X. Li Y. Sui J. Dong M. et al (2021). Molecular and structural mechanisms of ZZ domain‐mediated cargo selection by Nbr1. EMBO J.40, e107497. 10.15252/embj.2020107497
301
Watanabe Y. Tsujimura A. Taguchi K. Tanaka M. (2017). HSF1 stress response pathway regulates autophagy receptor SQSTM1/p62-associated proteostasis. Autophagy13, 133–148. 10.1080/15548627.2016.1248018
302
Waters E. R. Vierling E. (2020). Plant small heat shock proteins - evolutionary and functional diversity. New phytologist227 (1), 24–37. 10.1111/nph.16536
303
Weigert A. Herhaus L. (2023). Immune modulation through secretory autophagy. J. Cell Biochem.125, e30427. 10.1002/jcb.30427
304
West A. P. Shadel G. S. (2017). Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol.17, 363–375. 10.1038/nri.2017.21
305
Wettstein G. Bellaye P. S. Micheau O. Bonniaud P. (2012). Small heat shock proteins and the cytoskeleton: an essential interplay for cell integrity?Int. J. Biochem. and cell Biol.44 (10), 1680–1686. 10.1016/j.biocel.2012.05.024
306
Wilhelmus M. M. Boelens W. C. Otte-Höller I. Kamps B. de Waal R. M. Verbeek M. M. (2006). Small heat shock proteins inhibit amyloid-beta protein aggregation and cerebrovascular amyloid-beta protein toxicity. Brain Res.1089 (1), 67–78. 10.1016/j.brainres.2006.03.058
307
Williams J. K. Ngo J. M. Lehman I. M. Schekman R. (2023). Annexin A6 mediates calcium-dependent exosome secretion during plasma membrane repair. Elife12, e86556. 10.7554/eLife.86556
308
Winslow A. R. Chen C. W. Corrochano S. Acevedo-Arozena A. Gordon D. E. Peden A. A. et al (2010). α-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J. Cell Biol.190, 1023–1037. 10.1083/jcb.201003122
309
Wong Y. C. Holzbaur E. L. F. (2014). The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci.34, 1293–1305. 10.1523/JNEUROSCI.1870-13.2014
310
Wu S. Y. Chen J. W. Liu H. Y. Wang Y. C. Chu Y. S. Huang C. Y. et al (2022). Secretory autophagy promotes Rab37-mediated exocytosis of tissue inhibitor of metalloproteinase 1. J. Biomed. Sci.29, 103. 10.1186/s12929-022-00886-z
311
Wu S. Y. Wu H. T. Wang Y. C. Chang C. J. Shan Y. S. Wu S. R. et al (2023). Secretory autophagy promotes RAB37-mediated insulin secretion under glucose stimulation both in vitro and in vivo. Autophagy19, 1239–1257. 10.1080/15548627.2022.2123098
312
Wu W. Krijgsveld J. (2024). Secretome analysis: reading cellular sign language to understand intercellular communication. Mol. and Cell. proteomics MCP23 (1), 100692. 10.1016/j.mcpro.2023.100692
313
Wu Y. Xiong F. Ling J. (2024). The role of heat shock protein B8 in neuronal protection against oxidative stress and mitochondrial dysfunction: a literature review. Int. Immunopharmacol.140, 112836. 10.1016/j.intimp.2024.112836
314
Wyttenbach A. Sauvageot O. Carmichael J. Diaz-Latoud C. Arrigo A. P. Rubinsztein D. C. (2002). Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet.11 (9), 1137–1151. 10.1093/hmg/11.9.1137
315
Xie Y. X. Naseri N. N. Fels J. Kharel P. Na Y. Lane D. et al (2022). Lysosomal exocytosis releases pathogenic α-synuclein species from neurons in synucleinopathy models. Nat. Commun.13 (1), 4918. 10.1038/s41467-022-32625-1
316
Xu Y. Cui L. Dibello A. Wang L. Lee J. Saidi L. et al (2018). DNAJC5 facilitates USP19-dependent unconventional secretion of misfolded cytosolic proteins. Cell Discov.4, 11. 10.1038/s41421-018-0012-7
317
Xu Y. Zhang S. Zheng H. (2019). The cargo receptor SQSTM1 ameliorates neurofibrillary tangle pathology and spreading through selective targeting of pathological MAPT (microtubule associated protein tau). Autophagy15, 583–598. 10.1080/15548627.2018.1532258
318
Younas N. Fernandez Flores L. C. Hopfner F. Höglinger G. U. Zerr I. (2022). A new paradigm for diagnosis of neurodegenerative diseases: peripheral exosomes of brain origin. Transl. Neurodegener.11, 28. 10.1186/s40035-022-00301-5
319
Yu L. Chen Y. Tooze S. A. (2018). Autophagy pathway: cellular and molecular mechanisms. Autophagy14, 207–215. 10.1080/15548627.2017.1378838
320
Yu W. H. Cuervo A. M. Kumar A. Peterhoff C. M. Schmidt S. D. Lee J. H. et al (2005). Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J. cell Biol.171 (1), 87–98. 10.1083/jcb.200505082
321
Yuyama K. Sun H. Mitsutake S. Igarashi Y. (2012). Sphingolipid-modulated exosome secretion promotes clearance of amyloid-β by microglia. J. Biol. Chem.287, 10977–10989. 10.1074/jbc.M111.324616
322
Yuyama K. Yamamoto N. Yanagisawa K. (2008). Accelerated release of exosome-associated GM1 ganglioside (GM1) by endocytic pathway abnormality: another putative pathway for GM1-induced amyloid fibril formation. J. Neurochem.105, 217–224. 10.1111/j.1471-4159.2007.05128.x
323
Zavodszky E. Seaman M. N. Moreau K. Jimenez-Sanchez M. Breusegem S. Y. Harbour M. E. et al (2014). Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun.5, 3828. 10.1038/ncomms4828
324
Zhai J. Lin H. Julien J. P. Schlaepfer W. W. (2007). Disruption of neurofilament network with aggregation of light neurofilament protein: a common pathway leading to motor neuron degeneration due to Charcot-Marie-Tooth disease-linked mutations in NFL and HSPB1. Hum. Mol. Genet.16 (24), 3103–3116. 10.1093/hmg/ddm272
325
Zhang M. Kenny S. J. Ge L. Xu K. Schekman R. (2015). Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. eLife4, e11205. 10.7554/eLife.11205
326
Zhang M. Liu L. Lin X. Wang Y. Li Y. Guo Q. et al (2020). A translocation pathway for vesicle-mediated unconventional protein secretion. Cell181, 637–652.e15. 10.1016/j.cell.2020.03.031
327
Zhang M. Schekman R. (2013). Cell biology. Unconventional secretion, unconventional solutions. Science340 (6132), 559–561. 10.1126/science.1234740
328
Zhao Y. G. Codogno P. Zhang H. (2021). Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell Biol.22, 733–750. 10.1038/s41580-021-00392-4
329
Zhou Z. Qiu H. Castro-Araya R. F. Takei R. Nakayama K. Katoh Y. (2022). Impaired cooperation between IFT74/BBS22-IFT81 and IFT25-IFT27/BBS19 causes Bardet-Biedl syndrome. Hum. Mol. Genet.31 (10), 1681–1693. 10.1093/hmg/ddab354
330
Zhu Z. Li R. Stricker R. Reiser G. (2015). Extracellular α-crystallin protects astrocytes from cell death through activation of MAPK, PI3K/Akt signaling pathway and blockade of ROS release from mitochondria. Brain Res.1620, 17–28. 10.1016/j.brainres.2015.05.011
331
Zourlidou A. Payne Smith M. D. Latchman D. S. (2004). HSP27 but not HSP70 has a potent protective effect against alpha-synuclein-induced cell death in mammalian neuronal cells. J. Neurochem.88 (6), 1439–1448. 10.1046/j.1471-4159.2003.02273.x
332
Zubkova E. Kalinin A. Bolotskaya A. Beloglazova I. Menshikov M. (2024). Autophagy-dependent secretion: crosstalk between autophagy and exosome biogenesis. Curr. Issues Mol. Biol.46, 2209–2235. 10.3390/cimb46030142
Summary
Keywords
small heat shock proteins, protein misfolding, protein oligomerization and aggregation, autophagy, unconventional protein secretion, extracellular vesicles and exosomes, neurodegenerative diseases
Citation
Bonavita R, Vitale F, Verdicchio LV, Williams SV, Caporaso MG, Fleming A and Renna M (2025) Small HSPs at the crossroad between protein aggregation, autophagy and unconventional secretion: clinical implications and potential therapeutic opportunities in the context of neurodegenerative diseases. Front. Cell Dev. Biol. 13:1538377. doi: 10.3389/fcell.2025.1538377
Received
02 December 2024
Accepted
11 April 2025
Published
02 May 2025
Volume
13 - 2025
Edited by
Zoltán Wiener, Semmelweis University, Hungary
Reviewed by
Utpal Das, University of California, San Diego, United States
Karolina Pircs, Hungarian Centre of Excellence for Molecular Medicine (HCEMM), Hungary
Updates
Copyright
© 2025 Bonavita, Vitale, Verdicchio, Williams, Caporaso, Fleming and Renna.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angeleen Fleming, af425@cam.ac.uk; Maurizio Renna, maurizio.renna@unina.it
†These authors share first authorship
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.