 Fiona K. Leith1,2†
Fiona K. Leith1,2† Joey Lye1,2†
Joey Lye1,2† Derek S. Delaney1,3
Derek S. Delaney1,3 Samuel McLenachan4,5
Samuel McLenachan4,5 Fred K. Chen4,5,6,7,8
Fred K. Chen4,5,6,7,8 Marcus D. Atlas1,2
Marcus D. Atlas1,2 Elaine Y. M. Wong1,2,9*
Elaine Y. M. Wong1,2,9*- 1Hearing Therapeutics, Ear Science Institute Australia, Nedlands, WA, Australia
- 2Centre for Ear Sciences, Medical School, The University of Western Australia, Nedlands, WA, Australia
- 3Curtin Health Innovation Research Institute, Curtin University, Bentley, WA, Australia
- 4Ocular Tissue Engineering Laboratory, Lions Eye Institute Australia, Nedlands, WA, Australia
- 5Centre for Ophthalmology and Visual Sciences, The University of Western Australia, Nedlands, WA, Australia
- 6Department of Ophthalmology, Royal Perth Hospital, Perth, WA, Australia
- 7Ophthalmology, Department of Surgery, University of Melbourne, East Melbourne, VIC, Australia
- 8Centre for Eye Research Australia, Royal Victorian Eye and Ear Hospital, East Melbourne, VIC, Australia
- 9Curtin Medical School, Faculty of Health Sciences, Curtin University, Bentley, WA, Australia
Usher syndrome is a severely debilitating autosomal recessive disorder characterised by congenital or progressive hearing loss, gradual vision loss and in some subtypes, vestibular dysfunction. Much progress has been made in recent years in creating appropriate preclinical models for most subtypes of Usher syndrome to facilitate the development of novel therapies. In this review, we provide an update on new preclinical models of Usher syndrome, with a particular focus on induced pluripotent stem cells and new organoid models. An update on the status of novel therapies is provided, including the development of new genetic therapies using new preclinical models and those currently in clinical trials.
1 Introduction
Usher syndrome (USH) is a debilitating autosomal recessive disorder, representing almost 50% of genetic deaf-blindness and has a global prevalence of 4–17 per 100,000 people (Kimberling et al., 2010). The effect on patient morbidity is significant with some subtypes causing severe-to-profound bilateral sensorineural hearing loss (SNHL), profound vision loss due to retinitis pigmentosa (RP) and vestibular dysfunction. The healthcare burden of USH-related RP was predicted to cost up to USD $371–880 million annually in the United States and USD $45–185 million in Canada (Gong et al., 2021). USH is both clinically and genetically heterogenous and has been classified into three USH types based on age of onset, symptom progression and the causative gene. Eleven genes have been associated with USH: MYO7A, USH1C, CDH23, PCDH15, USH1G, CIB2, ESPN, USH2A, ADGRV1, WHRN and CLRN1, all of which are inherited in an autosomal recessive manner as shown in Figure 1 (Ahmed et al., 2018; Khateb et al., 2018; Whatley et al., 2020).
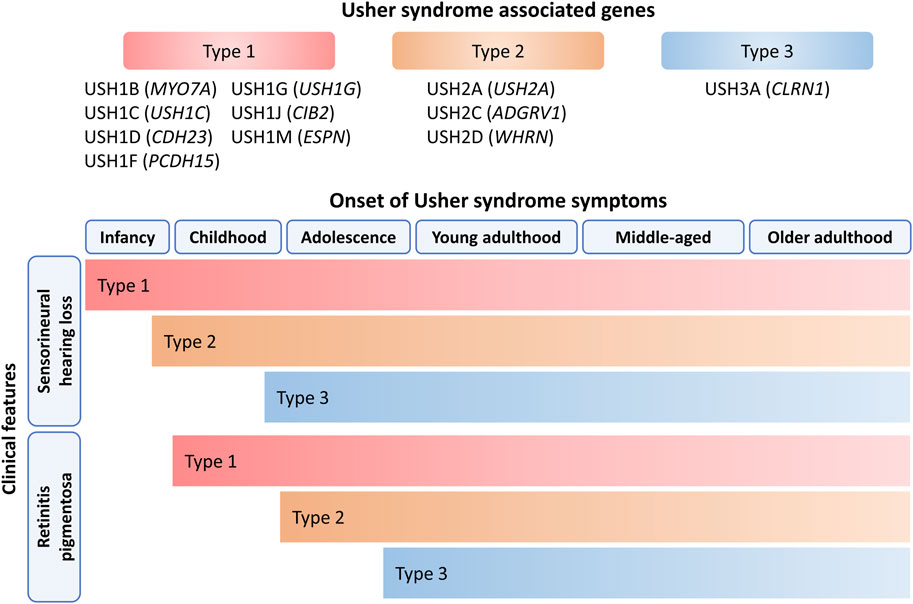
Figure 1. Onset of SNHL and RP symptoms in USH. The age, progression and severity of symptoms depends on the USH type which is determined by the underlying genetic cause. HL is the first clinical feature of USH and progressively deteriorates except for USH1 patients as they are born with severe-to-profound HL. USH2 patients are born with moderate-to-severe HL while USH3 patients are born with normal hearing and HL gradually becomes more severe over time. All USH patients are born with normal eyesight and retinal symptom onset is variable for each USH type. Vision loss typically begins during early childhood to adolescence for USH1 and USH2 patients and progressively worsens over several decades. The mean onset of retinal disease occurs during mid-adolescence for USH3 patients. The age groups were categorised as follows: infant (0–2 years old), childhood (2–11 years old), adolescence (12–18 years old), young adulthood (18–39 years old), middle-aged (40–59 years old) and older adulthood (60 years old and above).
2 Current management and treatment of Usher syndrome
There is no effective cure for any subtype of USH, merely ways to manage or mitigate symptoms. Hearing devices and cochlear implants are the standard of care for USH patients with residual hearing. Unfortunately, in cases of profound HL, cochlear implantation may not be sufficient in improving quality of life. Patients with USH who received late cochlear implantation have better hearing thresholds but improvement in speech recognition remains difficult to achieve (Davies et al., 2021). Most cases of USH are identified through hearing screening tests in newborns, due to early onset of hearing impairment for USH type 1 and 2 cases. Early intervention allows planning of long-term treatment and implementation of supportive strategies including learning speech signals and sign language at prelingual age and physiotherapy for balance issues for USH type 1 (USH1) patients.
There is no approved treatment to reverse the poor vision in dim light and slow down the progressive constricting visual field associated with USH-related RP (French et al., 2020; Zaw et al., 2022). Patients may present with central visual loss associated with cystoid macular oedema. These may respond to carbonic anhydrase inhibitor topically or systemically. However, intraocular injection of steroid implant (Ozurdex) and anti-vascular endothelial growth factor agents have been tried in those with oedema resistant to carbonic anhydrase inhibitors (Chen et al., 2022). Posterior subcapsular cataract is commonly seen in USH and standard cataract surgery in carefully selected patients may restore vision. However, post-operative complications such as cystoid macular oedema, posterior capsular opacification and zonular deficiency are more frequent in patients with RP (Nguyen et al., 2023b; Georgiou et al., 2024).
3 Usher syndrome type 1 (USH1)
USH1 is the most severe type and is characterised by severe-to-profound SNHL across all frequencies and vestibular dysfunction at birth followed by prepubertal onset of progressive RP. To date, various mutations in MYO7A, USH1C, CDH23, PCDH15, USH1G, CIB2 and ESPN have been shown to be causative of USH1 (Whatley et al., 2020; Fuster-García et al., 2021; Nisenbaum et al., 2022).
3.1 Usher syndrome type 1B (MYO7A)
MYO7A is located on chromosome 11q13.5 and has been linked to cause both USH1B and non-syndromic HL. The prevalence of MYO7A-associated HL has been reported across various populations with the lowest at 1.79% and the highest at 5.7% (Sloan-Heggen et al., 2016; Baux et al., 2017; Abu Rayyan et al., 2020). Recently, Ma et al. (2023) found that of 879 patients in Yunnan, China with HL onset before 6 years of age, 4.9% had a mutation in MYO7A. Another study by Watanabe et al. (2024) have identified 1.36% of HL patients within a Japanese cohort having MYO7A-associated HL, moreover, the prevalence of USH1B within autosomal recessive or sporadic HL patients was 0.32%.
The MYO7A protein is a member of the myosin family, essential for the development and maintenance of hair cell (HC) stereocilia in the cochlea. It is localised to the upper tip link density of HC stereocilia (Figure 2) where it complexes with harmonin, SANS and CDH23 to facilitate mechanotransduction and maintain tension of the tip link (Grati and Kachar, 2011; Li et al., 2020). During development it is thought that a complex of MYO7A and PCDH15 at the base of stereocilia play a role in setting stereocilia polarity and cohesion, while MYO7A plays a determining factor in stereocilia height (Senften et al., 2006; Prosser et al., 2008). MYO7A mutations result in HC with disorganised stereocilia in animals, with abnormalities in size and orientation. Several studies have also reported that Myo7a Ewaso mice, p.Ile487Asn, exhibit HC loss in the cochlea (Miller et al., 2012; Calabro et al., 2019).

Figure 2. Localisation of USH proteins in HCs. USH1 proteins are mainly localised in upper regions of HC. Myosin VIIA and harmonin contribute to the upper tip link density in HC, while protocadherin-23 and cadherin-23 form the lower and upper sections of tip links with associations to top connectors during development. SANS assembles with other USH1 proteins at stereocilia tips forming complexes, whereas CIB2 is concentrated on the apical surfaces of HC in addition to the tips of stereocilia. Meanwhile espin plays a role in stereocilia lengthening during development. USH2 proteins are primarily confined to the ankle link complex, with usherin being demonstrated to form the complex, ADGRV1 facilitating ankle link formation and whirlin playing a role in establishing the ankle link complex. Clarin-1, implicated in USH3, is localised to the cytosol and is concentrated on the apical and basal ends of HCs.
In the retina, MYO7A is mainly found in the apical region of retinal pigment epithelia (RPE) where it plays a key role in protein localisation of opsin and melanosomes, and transportation of phagosomes from the apical to basal RPE (Williams and Lopes, 2011). Within photoreceptors, MYO7A is localised to the ciliary and periciliary membranes (Liu et al., 1999; Gibbs et al., 2004). In an USH1B retinal organoid model, Leong et al. (2022) detected MYO7A expression in Muller and bipolar cells by single-cell RNA sequencing but were unable to detect protein localisation.
Tissue-specific functions of MYO7A are mediated by the expression of certain isoforms. There are currently two reported isoforms in the inner ear and two isoforms in the retina (Li et al., 2020; Gilmore et al., 2023). The canonical isoform was predominantly found in all inner hair cells (IHCs), whereas in outer hair cells (OHCs) its expression is highest in the apex and decreases towards the base of the cochlea (Li et al., 2020). The second isoform differs from the canonical isoform by the lack of 11 amino acids in the N-terminal extension of the motor head domain. While this shortened isoform is primarily expressed in OHCs and weakly expressed in IHCs, the expression in OHCs is thought to be in a tonotopic gradient inverse to that of the canonical isoform (Li et al., 2020). A recent study confirmed that the canonical isoform acts as a tip link motor to control tensioning of the mechanotransduction channel (Underhill et al., 2025).
3.2 Usher syndrome type 1C (USH1C)
Mutations in USH1C account for 2% of USH cases. USH1C is located on chromosome 11p15.1 and encodes the structural protein, harmonin. USH1C consists of 28 exons, with 20 that are conserved and 8 that are alternatively spliced to encode multiple isoforms categorised into three splice groups; a, b and c (Verpy et al., 2000). Mutations in domains shared across isoforms, N-harm, PDZ, or Coiled-coil (CC) domains, are more associated with USH1C (Grotz et al., 2022).
Harmonin is a PDZ-containing scaffold protein that is involved in the development, maintenance and excitation of sensory cells in the inner ear and retina by interacting with other USH genes. In the inner ear, harmonin isoform b is localised to the upper tip link density of the HC stereocilia where it interacts with SANS and MYO7A (Yan et al., 2022). Harmonin binds to CDH23 at the PDZ2 domain, the disruption of which deleteriously affects hair bundle development. Additionally, the CC and Proline-, Serine- and Threonine-rich (PST) domains with a known F-actin bundling function, when disrupted, did not affect development of the stereocilia bundle but did affect the transducer adaptability and displacement sensitivity (Grillet et al., 2009; Yan et al., 2022). Harmonin is additionally localised to the ribbon synapse of IHC and has been suggested to modulate Cav1.3 channels via ubiquitination (Gregory et al., 2011).
Several harmonin isoforms are present in the retina. Subclasses a and c are present in the inner and outer segments of photoreceptors as well as the synaptic terminal, while expression of subclass b has been reported in the outer segment (Reiners et al., 2003). Harmonin colocalises with CDH23 in the photoreceptor inner segment, and with CDH23, PCDH15 and MYO7A at the ribbon synapse where these proteins may contribute to cell adhesion and endo- or exocytosis. Previous studies have shown that the co-localisation and interaction of the USH1 proteins occurs at the ribbon synapses in the retina and cochlear HCs, however the role of USH1 proteins in the synaptic structure of retina and cochlea are still unclear (Reiners et al., 2005; Phillips et al., 2011; Miles et al., 2021). Harmonin is known to have a role in shaping cochlear stereocilia through β-catenin signalling during development of the inner ear (Boëda et al., 2002; Schäfer et al., 2023).
3.3 Usher syndrome type 1D (CDH23)
The CDH23 gene contains 69 exons that spans across 300 kb and it encodes for the cadherin 23 protein (CDH23) (Bork et al., 2001). Mutations in CDH23 results in USH1D and account for approximately 20% of USH1 cases making it the second most common cause of USH1 (Oshima et al., 2008). Cadherins are a family of calcium-dependent cell adhesion transmembrane proteins. Three subclasses of CDH23 isoforms are predominantly expressed in the inner ear and retina: A, B and C. Isoform A is the full-length protein consisting of an extracellular domain containing 27 extracellular cadherin (EC) repeats, a transmembrane domain and the cytoplasmic domain containing two PDZ-binding interfaces (Lagziel et al., 2005; Vanniya et al., 2018). Isoform A forms the stereocilia tip links with PCDH15 (Kazmierczak et al., 2007). Moreover, they are components of kinociliary links and are associated with transient lateral links that connects neighbouring stereocilia during development (Richardson and Petit, 2019). The specific role of isoforms B and C in the inner ear and retina is uncertain. Mutations in CDH23 cause disorganised HC stereocilia with misplaced kinocilia, resulting in HL and vestibular dysfunction (Palma et al., 2001). In the retina, CDH23 is found in the outer segment and calyceal processes of photoreceptors where, along with PCDH15, it is thought to have a role in the structural organisation of the outer segment (Schietroma et al., 2017).
3.4 Usher syndrome type 1F (PCDH15)
Protocadherin 15 (PCDH15) is the gene implicated in USH1F, which accounts for 11%–19% of USH1, however PCDH15 mutations are responsible for 50%–60% of USH1 cases within the Ashkenazi Jewish population (Yusuf et al., 2022). PCDH15 is 980 kb long and contains 33 exons, with mutations in this gene also being responsible for non-syndromic HL DFNB23 (Ahmed et al., 2001; Nisenbaum et al., 2022). In the inner ear, CDH23 and PCDH15 form the upper and lower sections of the tip link respectively and are components of transient links and kinocilial links (Kazmierczak et al., 2007; Richardson and Petit, 2019). At the lower tip link density of the stereocilia, PCDH15 binds to the mechanotransduction channel pore-forming subunits TMC1 and TMC2 (Maeda et al., 2014).
The PCDH15 protein has three isoforms: A B and C, of which isoform A is the longest, consisting of extracellular domains containing 11 EC repeats, a transmembrane domain and cytoplasmic domain. Isoform A has three subtypes (CD1, CD2 and CD3) which vary in cytoplasmic domain length (Pepermans et al., 2014). Ahmed et al. (2003) described isoform B containing a shortened extracellular domain with less EC repeats, a transmembrane domain and cytoplasmic domain. Isoform C lacks the transmembrane domain and cytoplasmic domain and is thought to be secreted, possibly acting as a ligand for a membrane receptor (Rouget-Quermalet et al., 2006). Studies by Pepermans et al. (2014) and Webb et al. (2011) reported that while Pcdh15 knockout mouse models for isoform CD1 and CD3 do not have HL, mice with isoform CD2 knocked out presented with functional and morphological HC defects. This indicates that the CD2 isoform is essential for hearing in humans which was shown in a study where the PCDH15 p.P1515Tfs*4 mutation only affected isoform CD2 in profoundly deaf children from two unrelated families (Pepermans et al., 2014). In the retina, PCDH15 is found in the outer segment and calyceal processes of photoreceptors (Schietroma et al., 2017).
3.5 Usher syndrome type 1G (SANS)
Mutations in USH1G account for up to 7% of USH1 cases (Nisenbaum et al., 2022). USH1G is located on chromosome 17q25.1 and encodes a scaffold protein containing ankyrin repeats and a sterile alpha motif (SAM) domain (SANS) protein which consists of three ankyrin repeats, a central domain followed by a sterile alpha motif domain and a class I PDZ-binding motif. Overlack et al. (2008) showed SANS was expressed in the photoreceptor cell layer and the inner and outer plexiform layer. SANS is localised within the ciliary apparatus and synapse of photoreceptor cells where it binds with usherin and whirlin and interconnects the complex with the microtubule cytoskeleton to facilitate transportation processes (Overlack et al., 2008; Sorusch et al., 2017). Recently, Yildirim et al. (2021) and Fritze et al. (2023) showed that SANS interacts with spliceosomal proteins such as SF3B1, PRPF6 and PRPF31 in Cajal bodies and nuclear speckles. This suggests SANS regulates pre-mRNA splicing of USH genes and other genes related to cellular proliferation.
3.6 Usher syndrome type 1J (CIB2)
CIB2 encodes for calcium and integrin binding protein 2 (CIB2), a DNA-dependent protein kinase interacting protein also associated with the non-syndromic HL DFNB48. Three isoforms of CIB2 have been identified in humans (Riazuddin et al., 2012). In the inner ear, CIB2 is localised to the tip of stereocilia and apical surfaces of HCs (Michel et al., 2017). Moreover, Riazuddin et al. (2012) demonstrated that CIB2 interacts with MYO7A and whirlin, and hypothesised several functions: to regulate calcium during mechanotransduction, photoreceptor maintenance and homeostasis. In Cib2 knockout mice, mechanotransduction in cochlear HC but not vestibular HC was reduced due to CIB3 acting as a redundancy for CIB2 function (Michel et al., 2017; Wang et al., 2017; Liang et al., 2021). In the retina, CIB2 is expressed by RPE, photoreceptors and certain ganglion cells. Sethna et al. (2021) demonstrated that loss of CIB2 in the mice RPE resulted in a phenotype similar to that of age-related macular degeneration and dysregulation of phagolysosomal processing.
Some studies have expressed scepticism of CIB2 as being causative of USH1. Booth et al. (2018) found no patients with RP in their investigation of six families across three ethnicities with a mutation in CIB2 during ophthalmological evaluation. Similarly, murine models differ in phenotype, a lack of behaviour characterising vestibular dysfunction and changes in stereocilia morphology inconsistent with other USH mouse models. Moreover, a meta-analysis of 11 next-generation sequencing studies of patients with USH and 21 next-generation sequencing studies of patients with isolated deafness found no mutations of CIB2 in subjects with HL and visual impairment (Jouret et al., 2019).
3.7 Usher syndrome type 1M (ESPN)
ESPN was recently associated with USH1M in a study where a consanguineous Pakistani family with prelingual SNHL, vestibular dysfunction and progressive vision impairment were identified to have an in-frame deletion within the gene (Ahmed et al., 2018). ESPN is also associated with the human deafness locus DFNB36 (Naz et al., 2004). ESPN encodes for the actin-bundling, cytoskeletal regulatory protein espin. Espin has a critical role in the inner ear for stereocilia lengthening during development (Donaudy et al., 2006). In the retina, espin is expressed in the outer limiting membrane, localised to the inner segment and the calyceal processes (Wang et al., 2011a). It is also thought to be present in the microvilli of Muller cells (Sekerková et al., 2004). Espin and the USH2 protein whirlin have been found to colocalise and interact at the ankle link of cochlear stereocilia and at the periciliary membrane complex of photoreceptors (Wang et al., 2011a).
4 Usher syndrome type 2 (USH2)
USH type 2 (USH2) is the most common type of USH and accounts for more than 60% of USH cases (Castiglione and Möller, 2022). USH2 patients are born with moderate-to-severe SNHL with progressive vision loss in the second decade of life and very rarely present with balance issues (Magliulo et al., 2017; Wafa et al., 2021). USH2 is characterised by congenital SNHL with a down-sloping audiometric configuration in which HL at low frequencies is typically mild-to-moderate and gradually becomes more severe at higher frequencies (Abadie et al., 2012). USH2 patients have variable onset of RP, which typically develops during adolescence and progressively worsens, though patients aged 5 years with RP have also been described (Sadeghi et al., 2006).
4.1 Usher syndrome type 2A (USH2A)
Mutations in the USH2A gene were predicted to be involved in 55%–90% of USH2 cases (Millán et al., 2011; Jouret et al., 2019). USH2A is located on chromosome 1q41 and spans over ∼800 kb. Usherin is a 5,202 amino acid transmembrane protein encoded by 72 exons (Eudy et al., 1998). There are two usherin isoforms: the short extracellular and long transmembrane isoform. The short isoform is encoded by 21 exons and contains a signal peptide, a laminin-globular-like domain, a laminin N-terminus domain, 10 laminin-epidermal growth factor domains and four fibronectin type III (FN3) domains. The long isoform is encoded by another 51 exons with two additional laminin globular domains and 28 FN3 domains in its N-terminus, followed by a transmembrane domain and an intracellular PDZ-binding motif (PBM) domain at the C-terminal tail.
In the developing cochlea, usherin is expressed in the ankle link (Figure 2) and spiral ganglion cells. Usherin forms the ankle link complex, a transient structure that connects and supports the growing stereocilia in immature HCs (Liu et al., 2007; Zou et al., 2015; Wang et al., 2023). In the retina, usherin is expressed in the connecting cilium and was recently proposed to facilitate ciliary trafficking of intracellular protein components in the inner segment of photoreceptors (Toms et al., 2020; Crane et al., 2023; Tebbe et al., 2023).
4.2 Usher syndrome type 2C (ADGRV1)
ADGRV1 is the fourth most commonly mutated gene in USH and is located on chromosome 5q14.3-21.3 (Jouret et al., 2019). ADGRV1 contains 90 exons, spanning over 600 kb of genomic DNA and encodes for Adhesion G protein-coupled Receptor V1 (ADGRV1; also known as Very Large G protein-coupled Receptor-1). There are three major alternatively spliced transcripts identified in humans, each encoding different isoforms of various lengths termed ADGRV1a, ADGRV1b and ADGRV1c. ADGRV1b is the largest full-length isoform, containing all 6,306 amino acids and is the predominant form in the inner ear and retina. At the N-terminal, its extracellular portion consists of 35 sodium-calcium exchangers (Calxβ) domains, one laminin-G/pentraxin domain and seven epilepsy-associated repeat (EAR) domains located in between the 22-23rd Calxβ domain. At the intracellular carboxyl end, there is a G-protein-coupled proteolytic site (GPS) domain, seven-transmembrane (7TM) domain and a PBM domain. ADGRV1a (1967 amino acids) and ADGRV1c (2296 amino acids) are shorter isoforms without the signal transduction 7TM domain due to partial deletion of exon 31.
Like other USH2 genes, the ADGRV1 protein is essential for ankle link formation during cochlear HC development (McGee et al., 2006; Liu et al., 2007). In the developing retina, ADGRV1 is highly expressed in neural retinal precursors and in mature RPE (McMillan et al., 2002). A recent study revealed ADGRV1 and CIB2 co-localised in the synaptic and ciliary region of photoreceptor cells. Linnert et al. (2023) demonstrated their interaction with ciliary proteins, such as the TRiC/CCT chaperonin complex, to facilitate cargo transportation from the inner to the outer segment of photoreceptors. Along with other recent studies, ADGRV1 is involved in focal adhesions for mechanosensing during cell migration, as demonstrated by the absence of ADGRV1 resulting in autophagy (Kusuluri et al., 2021; Linnert et al., 2023).
4.3 Usher syndrome type 2D (WHRN)
USH2D is caused by pathogenic WHRN variants and is the least prevalent USH2 subtype. The WHRN gene is located on chromosome 9q32-34 and contains 12 exons that encode whirlin, a 907 amino acid protein. The full-length whirlin isoform is highly expressed in cochlear HCs and retinal photoreceptors, while the shorter isoform is only present in HCs and is controlled by a different promoter (Mburu et al., 2003). The long whirlin isoform consists of an Ala/Gly/Ser-rich region at the N-terminal end, followed by a harmonin-homology domain (HHD1), two PDZ domains (PDZ1, PDZ2), a second HHD (HHD2), a proline-rich region, a third PDZ domain (PDZ3) and a PBM domain at its C-terminal end. The PDZ1 domain of the long isoform has been shown to interact with the C-terminal tails of cadherin 23 and protocadherin 15 (Michel et al., 2020). The short isoform consists of HHD1, PDZ1, PDZ2 and HHD2, and was proposed to be involved in polymerisation and stabilisation of actin filaments at the tips of the tallest stereocilia with stereociliary components Eps8 and Myosin XVa for stereocilia elongation (Manor et al., 2011).
Similar to other USH2 proteins, the PDZ1 and PDZ2 domain of whirlin interacts with the PBM domain of usherin and ADGRV1 to establish the ankle link complex, where whirlin acts as a scaffold to connect the neighbouring stereocilia with other USH2 proteins (van Wijk et al., 2006; Chen et al., 2014; Guan et al., 2023). Moreover, whirlin has been shown to interact with Esp8 and myosin XVa in cochlear HCs, in which they are essential for regulating stereocilia growth during development (Mburu et al., 2006; Wang et al., 2011a). In the retina, whirlin recruits other USH2 proteins to the periciliary membrane complex in the photoreceptors (Zou et al., 2011).
5 Usher syndrome type 3 (USH3)
USH type 3 (USH3) is the rarest form of USH and exhibits the most phenotypic heterogeneity. USH3 accounts for 1%–6% of worldwide USH cases, however it is significantly more prevalent among Finnish and Ashkenazi Jewish populations. In these populations, USH3 accounts for up to 40% of total USH cases (Ness et al., 2003; Plantinga et al., 2005; Herrera et al., 2008). As for its clinical presentation, USH3 typically features a later onset of the classic USH symptoms compared with USH1 and USH2. HL in USH3 patients is progressive, being mostly diagnosed by the age of 10, though onset has been observed to occur as late as 35 (Ness et al., 2003; Plantinga et al., 2005). As for visual function, RP generally occurs from late adolescence to the fourth decade of life and is also progressive, with patients experiencing total or near-total blindness past the age of 50 (Ness et al., 2003; Herrera et al., 2008; Yoshimura et al., 2015). Vestibular dysfunction is variable, occurring in approximately half of USH3 patients (Sadeghi et al., 2005; Wafa et al., 2021).
CLRN1 is the gene implicated in USH3 and encodes clarin-1, a membrane protein generally involved in organisation of cilia and F-actin in the cytoskeleton (Adato et al., 2002; Herrera et al., 2008; Tian et al., 2009; Ratnam et al., 2013). Clrn1 expression has been demonstrated to occur in HCs in the apical region (Figure 2) and at the base near the ribbon synapse in mice, zebrafish and non-human primate models (Zallocchi et al., 2009; Ogun and Zallocchi, 2014). In the retina it is expressed by Muller cells, making it unique among the USH proteins as it is not expressed by photoreceptors (Xu et al., 2020). Mutations in CLRN1 generally affect the clarin-1 protein by impeding its trafficking to the plasma membrane (Isosomppi et al., 2009; Ogun and Zallocchi, 2014; Gopal et al., 2015; Geng et al., 2017). The most common mutation in North American and Ashkenazi Jewish populations is the c.143T>C mutation causing a N48K substitution in the clarin-1 protein (Herrera et al., 2008; Ratnam et al., 2013). This has been demonstrated to affect glycosylation of the final protein, resulting in reduced trafficking to HC apices and reduced stability (Gopal et al., 2015).
6 USH-related gene modifier (PDZD7)
PDZ-domain containing 7 (PDZD7) is a large structural protein and paralog of WHRN. The PDZD7 gene spans 23.3 kb on chromosome 10 and comprises 16 exons, while the protein consists of three PDZ-like domains and a HHD which is found between the second and third PDZ domains. PDZD7 forms part of the transient ankle link complex at the base of hair bundles with usherin, ADGRV1 and whirlin (Grati et al., 2012; Chen et al., 2014; Du et al., 2020; Guan et al., 2023). It has also been observed to interact with myosin VIIa and other proteins forming the stereocilia (Morgan et al., 2016). PDZD7 has two isoforms: a short isoform exclusively expressed in the cytoplasm of HCs; and a long isoform, which is also localised to cochlear hair bundles and is part of ankle link complexes (Du et al., 2020). PDZD7 protein expression in the retina peaks prenatally and is almost undetectable in the mature retina (Zou et al., 2013; Yang et al., 2014). Zou et al. (2013) generated mice with digenic heterozygous mutations in PDZD7 with USH2A, ADGRV1, WHRN or SANS and did not observe any effect on hearing function. This was despite demonstrating mice harbouring homozygous PDZD7 mutations disrupting the localisation of USH2 protein complex to the hair bundle (Zou et al., 2013). These mice exhibited profound HL and malformed hair bundles without vestibular dysfunction or RP.
Other studies examining the physical interaction between PDZD7 and USH proteins have suggested that mutations in PDZD7 could modify the phenotype of USH2 patients due to the role of PDZD7 in bridging the ankle link proteins (Grati et al., 2012; Chen et al., 2014; Morgan et al., 2016; Lin et al., 2021). However, a few studies have reported non-syndromic HL caused solely by mutations in PDZD7, which have undermined its association with USH in recent years (Booth et al., 2015; Guan et al., 2018; Fahimi et al., 2021).
7 Digenic inheritance
Possible digenic inheritance has been reported in some families with mutations in USH genes. Three USH1 patients from a study by Zheng et al. (2004) harboured pathogenic monoallelic mutations for CDH23 and PCDH15; these patients exhibit congenital profound deafness and vestibular dysfunction, accompanied with progressive vision loss. Through genetic screening, family members of the affected USH1 patients were used to confirm the variant origin and each parent was heterozygous for either CDH23 or PCDH15. Yoshimura et al. (2014) also reported digenic inheritance caused by USH1 mutations in MYO7A and PCDH15 in the Japanese population. Other combinations of pathogenic mutations were identified for biallelic mutations in MYO7A and monoallelic mutation in CDH23, attributing to more severe USH phenotype where earlier onset of RP symptoms such as night blindness was described (Yoshimura et al., 2014). Another study showed digenic inheritance of PCDH15 and USH1G which have been identified in five family members from a Pakistani consanguineous family (Schrauwen et al., 2018). These individuals were presented with non-syndromic HL and no visual or vestibular abnormalities, similar to double heterozygous Pcdh15+/av−3J and Ush1g+/js mice thus suggesting true digenism for PCDH15-USH1G (Zheng et al., 2012; Schrauwen et al., 2018).
Digenic inheritance has also been described for USH2, where a single pathogenic variant was identified in either ADGRV1 or PDZD7 presented with mild disease phenotype (Ebermann et al., 2010). Another patient with a homozygous truncation mutation in USH2A and a heterozygous frameshift mutation in PDZD7 was observed to have earlier onset and more severe RP compared to her sister who carried the homozygous USH2A mutation but not the PDZD7 mutation (Ebermann et al., 2010). However, this digenic form of USH2 has not been recapitulated in mice models with heterozygous mutations in PDZD7 and either USH2 genes or SANS (Zou et al., 2013).
8 Emerging USH genes
HARS has previously been implicated in USH3 and encodes a class IIa aminoacyl tRNA synthetase that loads tRNA with histidine (Abbott et al., 2017). Although HARS is ubiquitously expressed throughout the body, for unknown reasons pathological HARS variants are exclusively associated with rare neuropathies. A study by Puffenberger et al. (2012) was the first to identify the HARS c.1361A>C (p.Y454S) homozygous variant and associate it to USH3. The HARS variant was identified in an Amish Plain population, in which the patients exhibited severe retinal dystrophy and cone dysfunction from birth and later onset HL in childhood. The disorder was present in 1.5% of this population. This variant is also linked to damaged afferent sensory cells, predisposing affected individuals to episodic psychosis and sudden death through unknown mechanisms (Puffenberger et al., 2012). While the variable deaf-blindness severity and onset observed with this HARS variant could warrant its association with USH3, the additional symptoms observed in affected individuals complicate the designation of HARS as a bona fide USH gene. The number of reports on this HARS variant are also limited.
Recently several clinical reports of patients with ARSG mutations in have been proposed as Usher syndrome type 4 (USH4). These patients were presented with a combination of late-onset RP and SNHL (Khateb et al., 2018; Abad-Morales et al., 2020; Fowler et al., 2021; Peter et al., 2021; Velde et al., 2022; Bauwens et al., 2025). In the animal study, there is a clear phenotypic change in Arsg−/− mice which they were characterised with progressive retinal degeneration at 1–6 months of age (Kruszewski et al., 2016). There are limited reports on ARSG in the inner ear, though Girotto et al. (2014) revealed restricted ARSG expression at the apical side of inner and outer HCs of P5 mice cochlea. Despite the hints of ARSG reported in current literature, its precise functional role in the inner ear and retinal sensory cells remains too early to be a causative USH gene.
9 Preclinical animal models
There are various animal models that exist for the study of USH. Up until recently, animal models were limited to mice and some zebrafish models (Williams, 2008). The current available animal models for USH are too numerous to discuss individually and have been listed in Table 1. The earlier USH mouse models were characterised by their tendency to run in circles and head tossing, which arise from vestibular dysfunction caused by their mutation. They are even named as such, some examples include shaker-1 (USH1B), deaf circler (USH1C), waltzer (USH1D), Ames waltzer (USH1F), Jackson shaker (USH1G) and whirler (USH2D). Aside from the latter example, most mouse models for USH2 do not present with vestibular dysfunction, reflecting the human disease.
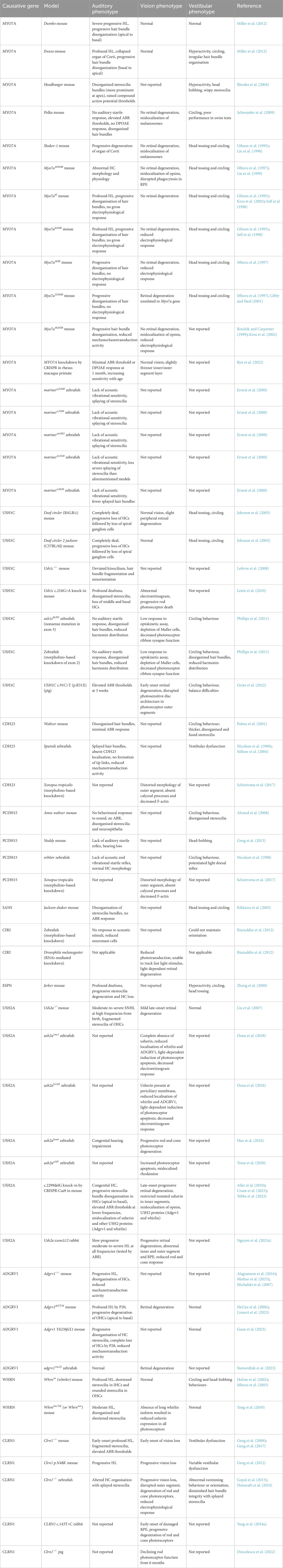
Table 1. Preclinical animal models used for the study of USH disease mechanisms and therapies. Only the homozygotic phenotypes are reported here.
Mouse models have been useful for modelling the phenotype of various USH gene mutations in vivo, as mutations induced in different regions of the same gene can yield varying effects on mouse phenotype as in humans. Miller et al. (2012) reported different phenotypes in Ewaso (p.I487N) and Dumbo (p.F947I) mice, where the former model had profound HL with circling behaviour indicative of vestibular dysfunction and the latter mice had progressive HL without a vestibular phenotype. The Ewaso model would thus be a more appropriate model for USH1B, while the Dumbo model appears to match the phenotype for DFNB2, a non-syndromic hearing disorder. Moreover, the c.2839T>A mutation in the Dumbo model affects the linker region of MYO7A, while the c.1460T>A Ewaso mutation affects the head domain. Mutations in the head domain have shown a deafness and vestibular dysfunction phenotype like the shaker-1 (c.1505G>C) and Headbanger (c.531A>T) models, and likely have the greatest effect on phenotype due to the head domain being essential for myosin protein function (Gibson et al., 1995; Rhodes et al., 2004).
Despite their utility for studying the hearing and vestibular phenotype of USH, the majority of mouse models do not show an abnormal vision phenotype (Williams, 2008). The shaker-1 mouse for example, exhibits progressive HL, degeneration of the organ of Corti and behaviours indicative of vestibular dysfunction. However, their vision is only mildly impaired, though this was found to be exacerbated by light exposure (Peng et al., 2011). Some theories have suggested functional redundancy, alternative splicing, the higher ratio of rod to cone photoreceptors in mice and slightly differing localisation of USH proteins in the human and mouse retina (Sahly et al., 2012). A more faithful phenotype of disease progression was observed in Ush2a−/− mice, which exhibit moderate-to-severe SNHL, stereocilia fragmentation and late-onset, mild retinal degeneration (Liu et al., 2007). There are other rare cases where the mouse phenotype may not exactly align with the human condition. For example, Whrnwi (whirler) mice also visibly display vestibular dysfunction through circling behaviour, yet USH2 patients rarely have issues with balance (Holme et al., 2002; Mburu et al., 2003; Mustapha et al., 2007).
Significant progress has been achieved in the characterisation of new zebrafish models. Zebrafish are particularly useful due to their transparent bodies, large eyes and additional presence of HCs along a longitudinal strip of their bodies known as the lateral line. Additionally, more zebrafish models of USH have retinal degeneration phenotypes than their mouse counterparts. USH models created in other animals including rabbits and pigs have also been reported recently (Table 1). These were created in the absence of a retinal phenotype for a given subtype in mice and zebrafish and are advantageous in their closer anatomical and physiological resemblance to humans in the case of the pig (Nguyen et al., 2023a). A non-human primate model of USH1B generated by CRISPR-Cas9 was recently reported, however editing efficiency was insufficient to create a disease phenotype in the animals (Ryu et al., 2022).
Despite the obvious anatomical, physiological and genetic differences between human and animal model in disease progression, animal models remain important for elucidating the disease mechanism of USH subtypes and generating novel treatments. The more translationally relevant animal models will have a similar mutation to the orthologous human gene and expected phenotype. Moreover, the spatiotemporal expression of some USH genes have been comprehensively mapped in mice, with one study by Kolla et al. (2020) showing the expression of genes such as Myo7a, Pcdh15 and Cdh23 at various timepoints between E14 and P7 in mice. This will allow for the better design of gene therapies that can be transferred or redesigned for human use. Moreover, animals remain essential for standard preclinical studies of drug delivery, dosage and safety for any new therapy of USH.
10 Precision medicine for Usher syndrome
Precision medicine involves the use of patient data to refine therapeutic options and decisions to suit an individual patient’s condition. Comprehensive data such as; genetics, omics, lifestyle, demographics, physiological measurements and comorbid conditions of the patient, combined with clinical and therapeutic outcomes are shared and databased. This information can be used to refine diagnostic and therapeutic choices of future patients to allow faster intervention and improve clinical and therapeutic outcomes (König et al., 2017; Kosorok and Laber, 2019).
The application of precision medicine could improve diagnosis and treatment outcomes for SNHL (Ginsburg and Phillips, 2018; Wafi and Mirnezami, 2018). Genetic screening could be coupled with existing newborn hearing screening programs to improve diagnostic efficiency, improve detection of genetic SNHL and monitor progression of hearing impairment in USH individuals (Wang et al., 2011b; Wu et al., 2017; Shearer et al., 2019). A necessity of precision medicine is the compilation and computation of a vast amount of patient and population data for which suitable infrastructure is required (Ginsburg and Phillips, 2018; Wafi and Mirnezami, 2018). The Usher Syndrome Coalition is an organisation working to support individuals and families living with USH (Usher Syndrome Coalition, 2025). This along with biobanks such as the Australasian Hearing Registry and Biobank, and the Western Australian Retinal Degeneration study biobank aim to improve understanding of genotype-phenotype correlation; improve communication between the scientific community, patients and their families and; facilitate researcher access to patient samples and clinical data. Advancing understanding of gene therapy strategies, population genetics and the structure and functions of the USH genes will aid in developing methods for restoring function in USH proteins to treat those affected by it (Redfield et al., 2025).
10.1 Preclinical Usher patient-specific induced pluripotent stem cell and organoid models
Since human induced pluripotent stem cells (iPSCs) were first generated by the Yamanaka group, iPSCs have become an invaluable tool for modelling many human diseases and as a potential source for cell therapy (Takahashi and Yamanaka, 2006). iPSCs can be generated from non-invasively collected somatic cell types including dermal fibroblasts, peripheral blood mononuclear cells (PBMCs), keratinocytes and urine-derived cells (Aasen et al., 2008; Loh et al., 2009; Zhou et al., 2011; McLenachan et al., 2019; Zaw et al., 2021; Wong et al., 2024).
Recently, protocols for the differentiation of inner ear and retinal organoids from iPSCs have emerged (Eiraku et al., 2011; Zhong et al., 2014; Koehler et al., 2017; Jeong et al., 2018). Over time protocols have been adapted to specifically manipulate signalling pathways, such as Fgf, BMP, TGFβ, Wnt and Sonic Hedgehog at various timepoints during organoid growth (Zhou et al., 2015; Lahlou et al., 2018). This is done to mimic the early growth of the human inner ear and provides the opportunity to study the role of genes and diseases during development. These protocols could finally allow investigation of USH disease pathophysiology in a human model. These organoid models have been characterised to grow cochlear, vestibular and retinal cell types (Finkbeiner et al., 2022; Doda et al., 2023; Moore et al., 2023; Steinhart et al., 2023; Tresenrider et al., 2023). Moreover, protein interactions key to USH mechanisms could be modelled. The reported iPSC lines generated from USH1B and USH2A patients with mutations in MYO7A and USH2A have been summarised in Table 2 iPSC lines for other USH subtypes are yet to be reported.
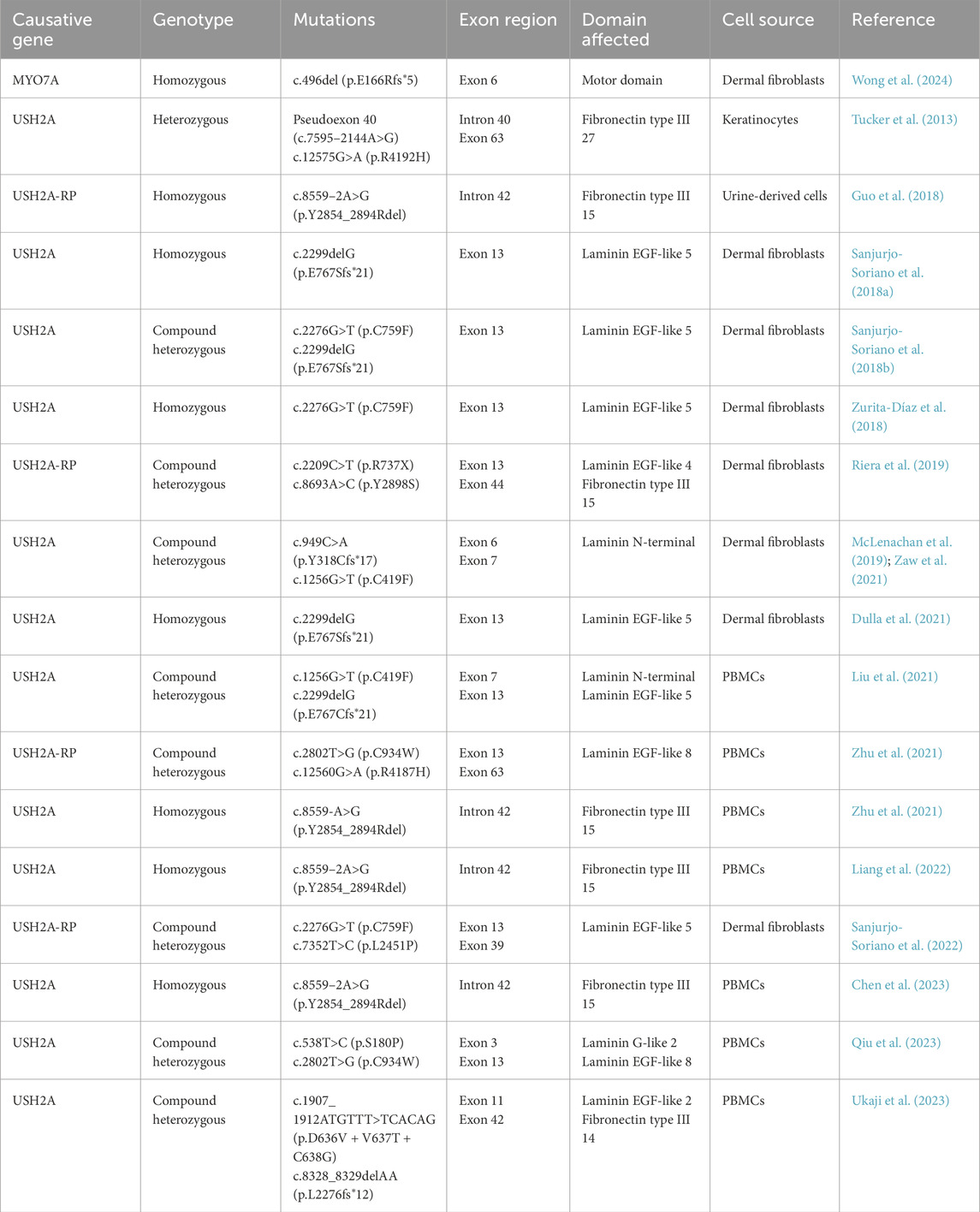
Table 2. Reported iPSC lines generated from patient tissues.
10.1.1 Usher syndrome type 1B
Recently, Gilmore et al. (2023) studied the expression of MYO7A isoforms in retinal organoids, which were generated from a donor with no known retinal disease and a commercially available episomal human iPSC line (Buchholz et al., 2013; Zhong et al., 2014; Hazim et al., 2017; Aparicio-Domingo et al., 2023). The organoids exhibited similar RPE features as human RPE, including critical protein expression, tight junction formation and phagocytosis of photoreceptor outer segments. Three-dimensional modelling of the two MYO7A isoforms showed differences in amino acid sequences at the tip of a loop of the FERM1 domain, which may result in changes in protein interaction. Interestingly, while both isoforms were expressed in human retinal organoids, mouse and pig RPE and neural retina, the proportion of the isoforms varied between these studied species. For instance, the human retinal organoid and pig models express similar levels of MYO7A short isoform at 82%–90%, while the long isoform is predominantly found in the mouse retina (Gilmore et al., 2023). This study demonstrates how iPSC can be used in situations where; animal models are known to differ from human genetics or physiology; to better understand the precise differences between human and animal disease models; and when human tissue types are inaccessible or scarce.
Leong et al. (2022) observed changes in retinal phenotype in iPSC-derived USH1B retinal organoids from three patients with MYO7A mutations. The organoids were found to be at a developmental stage equivalent to human foetal retina. The organoids did not show any cellular degradation but displayed heightened expression of genes related to adaptive stress response and apoptosis. Rod photoreceptors displayed upregulated expression of a pro-apoptotic factor (BNIP3), antioxidant enzymes (PRDX1, PRDX2, and PRDX5), and a free radical scavenging enzyme (SOD1). Apoptosis and stress response-related processes were also differentially expressed in Muller cell and bipolar cells. This stress response may be a primary factor in retinal degeneration caused by mutations in MYO7A, but upregulation of apoptotic pathways may indicate that Muller and bipolar cells should be further investigated for their role in USH1B pathophysiology (Leong et al., 2022).
10.1.2 Usher syndrome type 2A
In a study by Tucker et al. (2013), keratinocytes from a 62-years old USH2A-RP patient carrying missense mutation c.12575G>A and deep-intronic mutation intron 40 (pseudoexon 40) were reprogrammed into iPSCs and then differentiated into rod photoreceptor precursor cells (PPCs) (Tucker et al., 2013). These retinal-like cells developed eyecup-like structures with a layer of RPE and non-pigmented neural retina, sharing structural features with human retinal precursor cells. Surprisingly, Tucker et al. (2013) did not report any notable morphological changes between USH2A and normal retinal-like cells.
Recently, Guo et al. (2019) generated three-dimensional retinal organoids from USH2A-RP patient iPSC with compound heterozygous mutations c.8559–2A>G and c.9127-9129delTCC in USH2A. The authors described USH2A patient retinal organoids as being morphologically smaller and had abnormal retinal formation compared to control organoids. Layers of neural retina and RPE were observed in organoids by day 34 of culture with normal RPE-like cells displaying a cobblestone-like morphology with pigmentation. Conversely, USH2A patient retinal organoids lacked pigmentation due to absence of melanin and showed signs of cellular degeneration due to RPE atrophy. Indeed, expression of apoptotic genes were also significantly higher than controls on day 34 (Guo et al., 2019). Moreover, gene expression of retinal developmental markers were significantly reduced at day 18 in USH2A retinal organoids compared to control. Moreover, the patient organoids had decreased expression of ankle link complex-related genes, including PDZD7 (Guo et al., 2019).
Similarly, two USH2A patient cell lines with either one or two copies of the c.8559–2A>G mutation in USH2A were used to generate retinal organoids in a microfluidic system (Su et al., 2022). By day 18 of differentiation, expression level of pro-apoptotic protein BAX was significantly elevated in USH2A retinal organoids while anti-apoptotic protein BCL2 expression had decreased when compared to normal control organoids. These patient retinal organoids also have reduced laminin and collagen type IV expression affecting integrin expression and thereby downregulating PI3K-Akt signalling, an essential pathway for regulating cell proliferation and growth. Furthermore, expression of cytoskeleton organisation proteins were disrupted in USH2A retinal organoids, suggesting mutations in USH2A are linked to impaired extracellular functions causing cell apoptosis in the retina (Su et al., 2022).
11 Developing therapies
Developing therapies to date have largely targeted either hearing or vision loss rather than both senses, with recent clinical trials mostly focused on treatment of RP. This could be due to HL preceding the onset of RP, giving a wider treatment window for vision loss than hearing. Another consideration is drug delivery, as the retina is easier to access than the inner ear. Drug delivery to the inner ear would require cochleostomy for direct application to the cochlea or transtympanic injection to apply the drug to the round window niche. The latter method however, assumes passive diffusion of the drug across the round window membrane and this is governed by numerous chemical properties of the drug (Hao and Li, 2019; Delaney et al., 2023).
In this section, we will discuss gene augmentation, gene editing, drug therapy and cell therapy development for various USH subtypes (Figure 3) which are summarised in Tables 3–5.
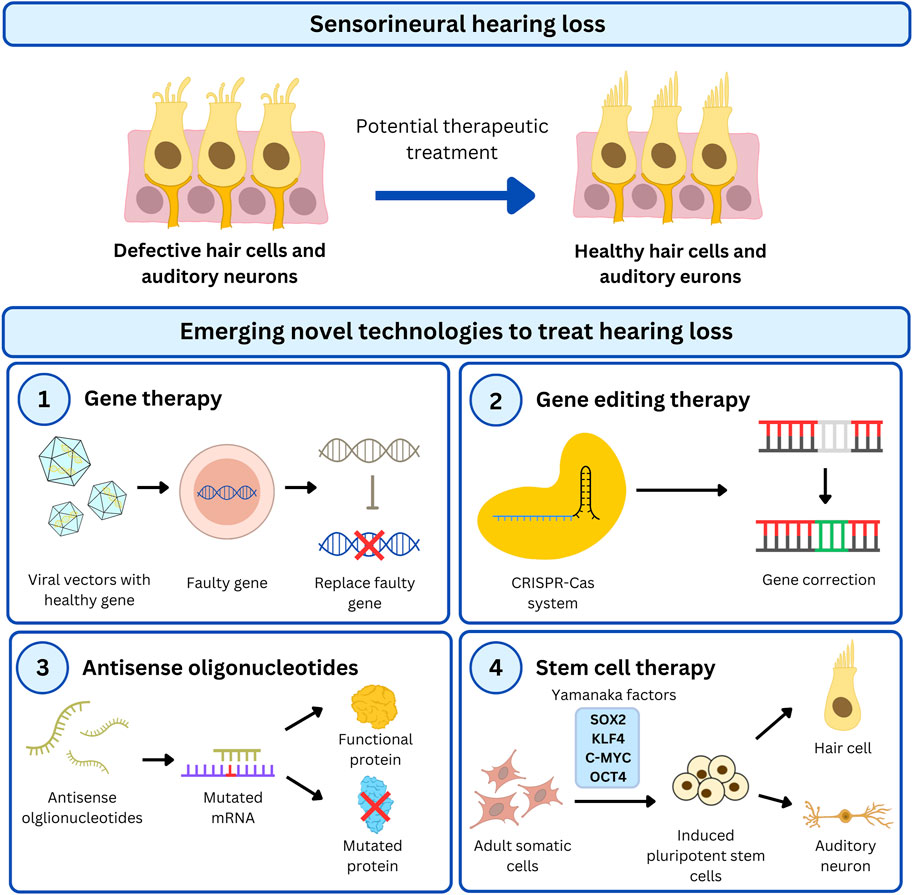
Figure 3. Overview of emerging therapeutic strategies to treat HL in USH patients. USH genes are a major cause of untreatable genetic HL and has recently been targeted by cutting-edge technologies to restore functional HCs in the cochlea. There are different approaches used in preclinical studies to restore hearing which includes gene- and cell-based therapy. Gene therapy is a popular treatment option to correct faulty genes by (1) introducing healthy gene transcripts via viral vectors, (2) precisely correct the genome using the CRISPR-Cas system, or (3) modify protein function by modulating gene expression with antisense oligonucleotides. Additionally, stem cells can be genetically modified with gene-targeted therapy for (4) cell therapy to replace compromised cells.
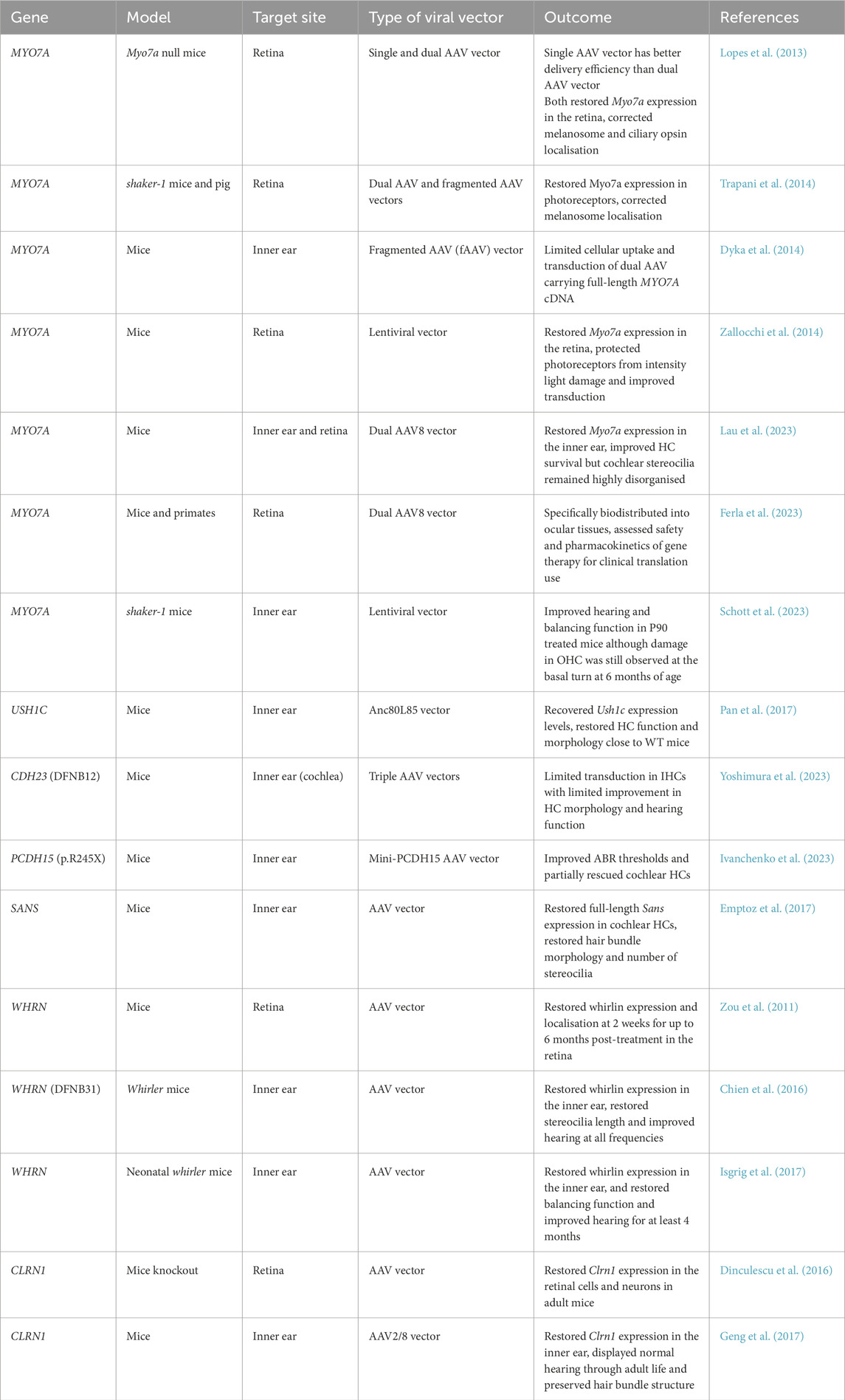
Table 3. Gene augmentation therapy for USH genes in preclinical models.
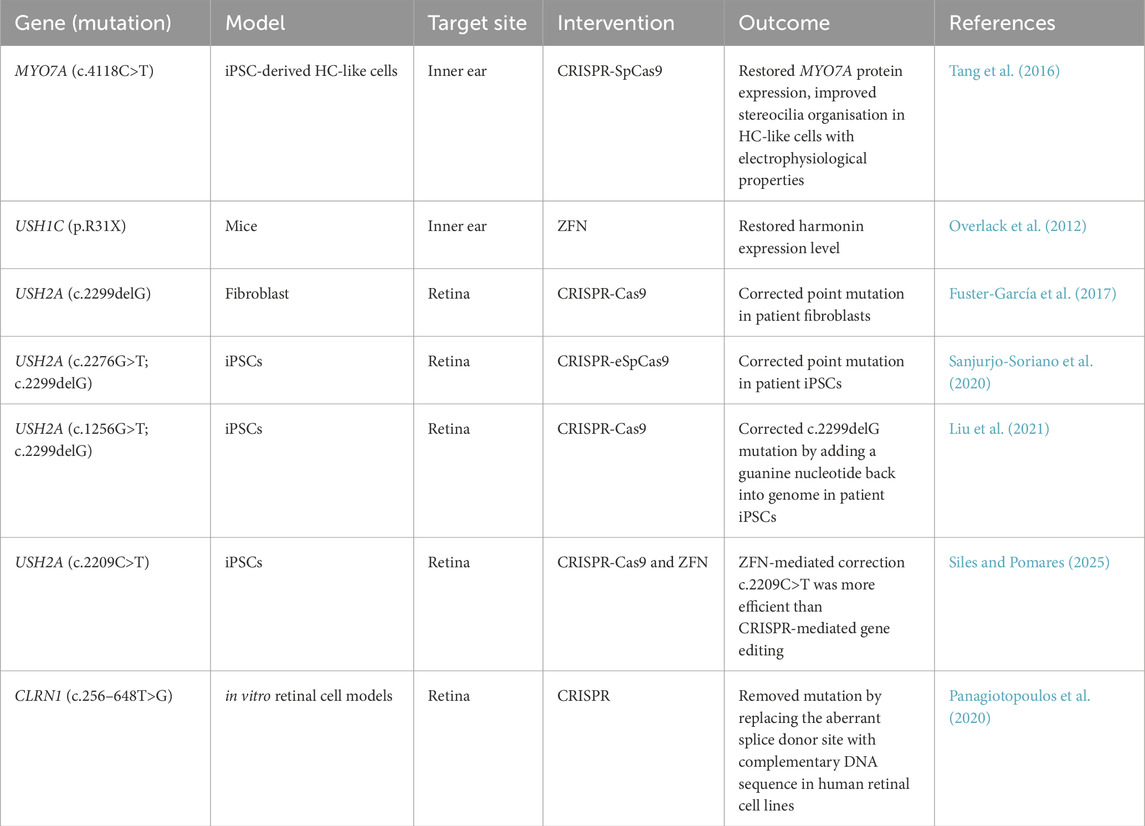
Table 4. Gene editing therapy for USH in preclinical models.
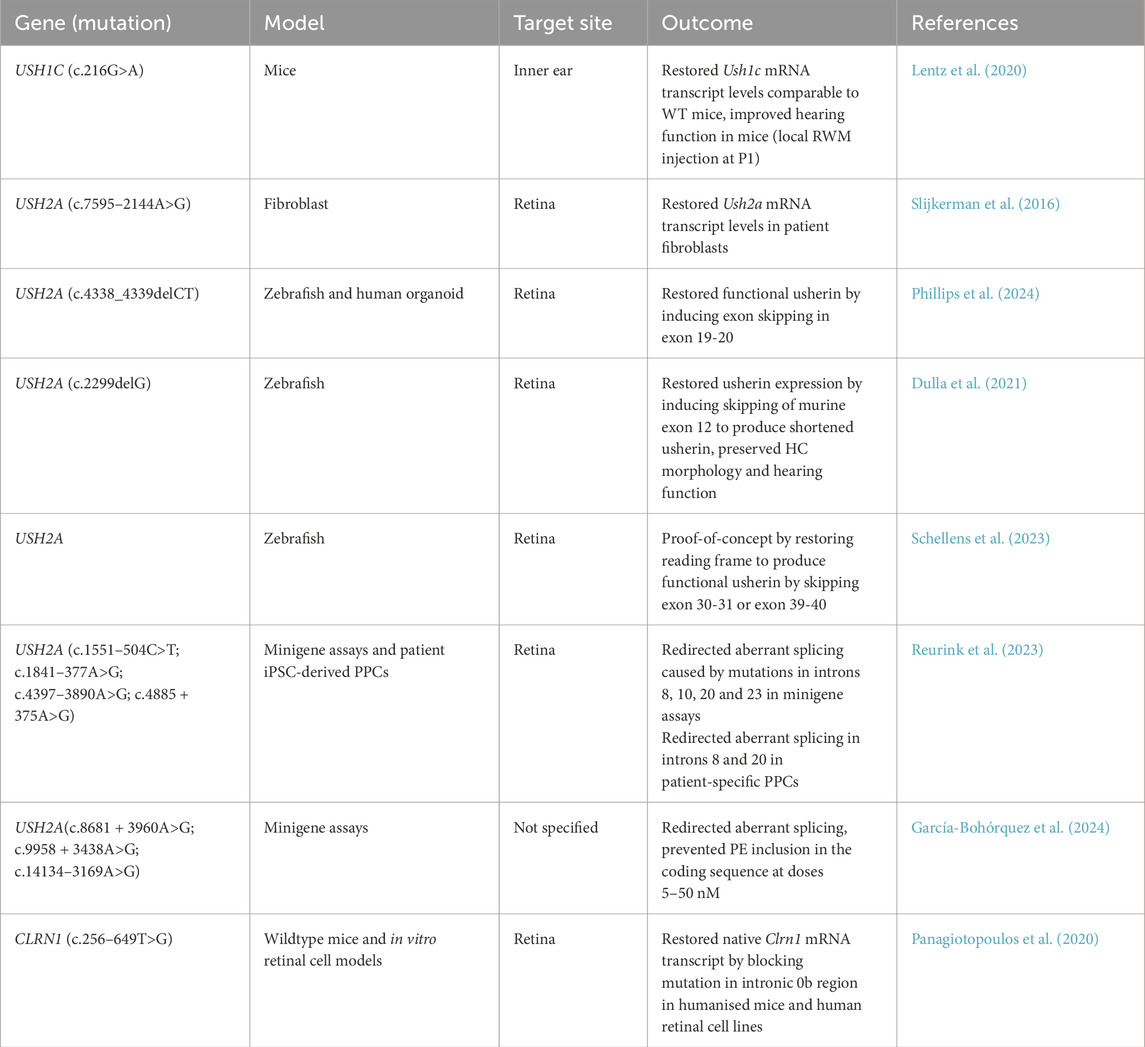
Table 5. Antisense oligonucleotide-based therapy for USH in preclinical models.
11.1 Gene replacement therapies
Gene replacement therapy, or gene augmentation, is the delivery of a functional copy of the affected gene, which can be delivered by viral or non-viral methods. Recent progress in gene augmentation has shown promising outcomes in preclinical studies which has been summarised in Table 3, with some advancing into clinical trials (Table 6). Adeno-associated viruses (AAV) are widely used as a delivery vector as they are not associated with any human disease and are essentially inert without cargo. The use of viral vectors can be limited by the packaging capacity of the vector. AAV vectors are generally constrained to 5 kb such that genes that exceed this capacity, such as several USH genes, require alternative methods to allow their delivery. Despite the capacity limit AAVs have been used to deliver USH genes, for example, rAAV2/8 has been used to deliver Sans and Anc80L65 for Ush1c early postnatal knockout mice which improved hearing and restored vestibular function (Emptoz et al., 2017; Pan et al., 2017).
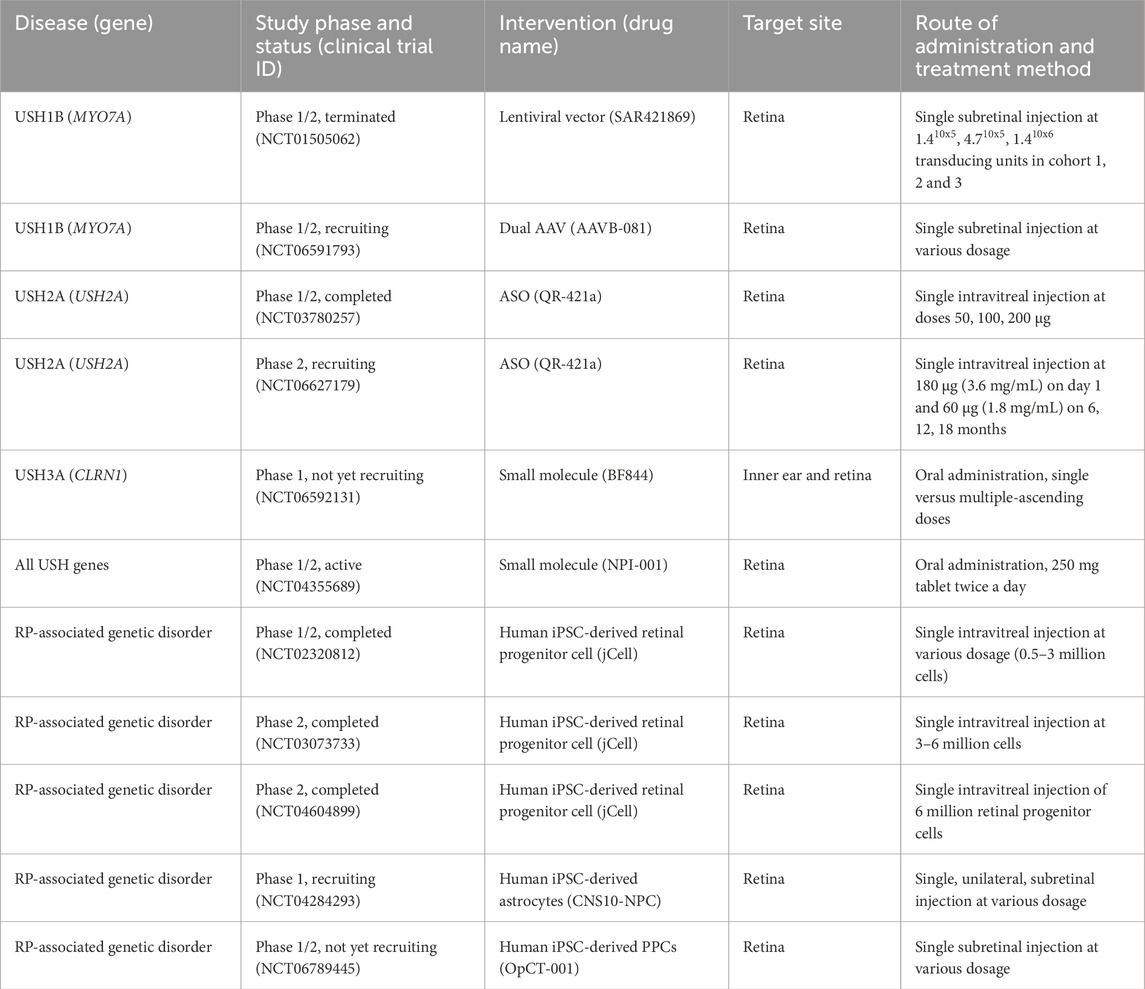
Table 6. Current clinical trial status of gene and cell therapy interventions for USH-associated disorders.
AAVs are thought to provide transgene expression even in non-replicating cells as they persist as extrachromosomal DNA. Moreover, this lack of integration has the advantage as it minimises safety concerns of insertion into the genome causing deleterious effects due to disruption of the genes at the insertion site. However, the stability of expression is likely to be impacted by any immune response mounted against the viral vector or delivered transgene (Li and Samulski, 2020). Inhibition of transduction and expression of the delivery product and damage due to inflammatory responses are all factors affected by the immune response to gene therapy treatments that must be considered. However, there is continued research into ways to mitigate the immune response elicited by viral vector delivery such as modification of viral vector such as the virus capsids to evade immune response and immunomodulation (Shirley et al., 2020).
The use of specific serotypes and promoters have been investigated to achieve gene delivery specific to targeted cell types, for example, AAV serotype 2/8 has been used to target IHCs and AAV5 is known to transfect photoreceptors and RPE (Zou et al., 2011; Chien et al., 2016; Isgrig et al., 2017). Zou et al. (2011) further used the human rhodopsin kinase promoter to specify expression to photoreceptor cells and attempt to mimic the low abundance expression level of wildtype whirlin. The use of serotypes known to selectively transduce specific cell types and promoters that mimic endogenous expression of the delivered gene can reduce off-target effect of gene therapy and concerns of overexpression of the delivered genes improving safety of future prospective treatments.
One strategy to address genes that are too large for delivery in a standard AAV method includes fragmented genome (fAAV) delivery. An oversized gene is packaged into a single AAV vector resulting in fragmentation of the gene which is then reassembled via recombination of the full-length cDNA upon delivery to the cell. While delivery of full-sized MYO7A has been achieved by fAAV overall this approach has been limited by low vector titers and transduction efficiency (Lopes et al., 2013; Akil, 2020). Additionally, the randomised manner of gene fragmentation on insertion could lead to a heterozygous payload across the vector preparation.
Multi-vectors would avoid this complication as the predetermined sectioning of the gene split across multiple AAV vectors would ensure a consistent payload across the vector preparation. Once the AAV vectors are transduced, the gene segments are joined by one of several strategies; overlapping, trans-splicing or a hybrid of the two approaches. The use of multiple vectors may hamper transduction with dual and triple vector approaches reporting reduced efficiency compared to single AAV delivery (Colella et al., 2014; Carvalho et al., 2017; Maddalena et al., 2018; Yoshimura et al., 2023).
A promising dual vector approach was recently tested for USH1B, with preclinical animal studies in both the inner ear and retina. Dual vectors are able to mediate expression of full-length MYO7A with efficiency equivalent or surpassing fAAV (Dyka et al., 2014). Lau et al. (2023) tested dual vector AAV8 delivery of MYO7A to the inner ear of shaker-1 mice. Although cochlear HC morphology and auditory function were not rescued, improved vestibular HC morphology, vestibular sensory-evoked potential threshold and reduced circling behavior were observed. This dual vector approach may be viable for the treatment of vestibular function in USH1B. Ferla et al. (2023) assessed pharmacokinetics and safety of good-manufacturing-practice-like AAV8.MYO7A dual vector of low and high doses delivered subretinally to shaker-1 mice and non-human primates. Administration resulted in expression of MYO7A protein and improved melanosome localisation in shaker-1 mice. In non-human primates, biodistribution of the vector was localised to the retinal and ocular tissues with minimal detection, aside from in serum and the lymphatic system with no detectable spread to the gonads. Overall data reported no major adverse effects (Ferla et al., 2023). These preclinical studies using the dual vector approach have culminated in a Phase I/II clinical trial (NCT06591793) for USH1B-related RP. This clinical trial is sponsored by AAVangard Bio and this study aims to evaluate the safety and efficiency of a single subretinal administration in 15 patients over 61 months.
Another strategy to compensate for large gene delivery is mini-genes, in which versions of the gene are designed without non-essential regions to enable delivery in a single AAV vector. For example, a mini-PCDH15 gene delivery to Pcdh15R245X mice, which carry the orthologous nonsense mutation in USH1F patients, showed improved ABR thresholds and partial rescue of HCs after treatment (Ivanchenko et al., 2023). However, the mini-gene approach is only suitable to genes for which the protein has non-essential sections that can be removed without compromising function.
Other viral vectors in use for gene replacement therapy include lentiviruses, which have a larger carrying capacity of approximately 10 kb. Following the preclinical study for testing and safety evaluation of EIAV-based lentiviral delivery of MYO7A in shaker-1 mice and rhesus macaques, this gene therapy progressed into clinical trials (Zallocchi et al., 2014). The UshStat clinical trial (NCT01505062) study was to assess the safety and tolerability of UshStat when subretinally injected at ascending doses. This study was terminated by Sanofi due to factors not related to safety. There is an active long-term study (NCT02065011) to assess safety, tolerability and biological activity in the 9 participants. In another lentiviral vector-based gene therapy, Schott et al. (2023) demonstrated partial recovery of hearing and full recovery of vestibular function in homozygous shaker-1 mice. Moreover, lentiviral vectors carrying the full-length MYO7A cDNA were able to fully restore auditory and vestibular function in heterozygous shaker-1 mice. This suggests this treatment may be more effective for other forms of MYO7A-associated HL such as DFNB2. While lentiviral vectors are ideal for its larger packaging capacity, low-eliciting immune response and long-term expression, there is a lingering question as to their safety. Lentiviruses can integrate randomly into the host genome and potentially result in gene disruption or mutagenesis.
11.2 Gene editing
Gene editing involves correction of mutations in a site-specific manner that retains the endogenous regulation of the gene of interest and have been applied in USH models (Table 4). Older editing strategies include transcription activator-like effector nucleases (TALENs) and Zinc-finger nucleases (ZFNs). ZFNs were previously used to correct a Ush1c nonsense mutation, p.R31X, and recover harmonin expression (Overlack et al., 2012). Clustered regularly interspaced palindromic repeats (CRISPR) approaches are considered simpler, cheaper and more efficient and are currently being more widely used than ZFN and TALENs.
Gene editing has similar limitations to gene augmentation as the inner ear remains difficult to access. Additionally, a common delivery method of CRISPR is by viral vector, resulting in complications due to tissue targeting and specificity, efficiency of delivery and immune response. As such advancements in strategies to improve the safety and efficiency of viral vector delivery will benefit both gene editing and gene augmentation.
11.2.1 MYO7A gene editing
Tang et al. (2016) generated three iPSC cell lines from; a deaf patient with compound heterozygous MYO7A c.1184G>A and c.4118C>T mutations, his asymptomatic father and a normal donor. The c.4118C>T mutation was corrected using Streptococcus pyogenes Cas9 (SpCas9) in iPSC and was differentiated into HC-like cells. Interestingly, stereocilia of these HC-like cells did not conform to the classic staircase-like pattern of mammalian stereocilia, which was proposed to be due to decreased Wnt signalling. When compared with differentiated control lines, MYO7A mutant stereocilia were disorganized and lacked bonding with neighbouring stereocilia, with significant changes to HC electrophysiology. The CRISPR-corrected HC-like cells expressed similar levels of ATOH1, POU4F3, MYO7A, and ESPN compared to normal donor HC-like cells. The corrected iPSC produced a full-sized MYO7A protein when analysed by immunoblotting, indicating genetic correction mitigated truncation of the protein. Morphology of the stereocilia showed organisation and bonding comparable to controls. Electrophysiology showed inward and outward currents consistent with HC (Tang et al., 2016).
11.2.2 USH2A gene editing
The CRISPR/Cas9 system was used to correct c.2299delG USH2A mutation in fibroblasts with no off-target effects detected (Fuster-García et al., 2017). This was followed by using CRISPR to correct mutations in patient-derived iPSCs wherein Sanjurjo-Soriano et al. (2020) utilised iPSCs from patients with USH2A mutations and corrected these mutations using CRISPR editing. Dermal fibroblasts from a homozygous USH2A patient harbouring c.2299delG (Sanjurjo-Soriano et al., 2018a) and a RP-associated patient with compound heterozygous mutations c.2276G>T and c.2299delG in USH2A (Sanjurjo-Soriano et al., 2018b) were used to generate iPSCs. Both USH2A iPSC lines were corrected using the enhanced specificity SpCas9 (eSpCas9) with high targeting efficiency and did not induce off-target effects. These CRISPR-corrected iPSC lines maintained pluripotency as they expressed similar levels of OCT3/4, SOX2 and NANOG compared to their untreated parental iPSC lines.
Liu et al. (2021) collected PBMCs from an USH2A patient harbouring compound heterozygous variants c.1256G>T and c.2299delG to generate iPSCs. Using these iPSCs, they employed CRISPR-Cas9 with a homology repair template to introduce the missing guanine back into the sequence, which they confirmed by DNA sequencing after treatment. The corrected USH2A patient iPSCs had similar pluripotency characteristics and showed the ability to differentiate into the three primary germline layers, suggesting that CRISPR-mediated genome editing did not affect iPSC characteristics.
Recently, gene correction using CRISPR or TALENs technology was compared in iPSCs derived from patients with different genetic forms of inherited retinal disorder (IRD), including USH2A-associated RP. Although CRISPR-mediated gene editing showed superior homology-directed repair correction in other IRD patient iPSCs, TALEN-mediated gene editing efficiency was moderate but higher than CRISPR for correcting USH2A c.2209C>T mutation (Siles and Pomares, 2025).
Currently, there are no gene editing-based therapies in clinical trials. However, a CRISPR-meditated therapy for targeting USH2A mutations in exon 13, EDIT-102, is being developed by Allergan and Editas Medicine. EDIT-102 comprises the same proprietary enzyme, AAV vector, promoters, and route of delivery as EDIT-101. Currently, EDIT-101 is undergoing the Brilliance clinical trial (NCT03872479) for Leber congenital amaurosis to correct an intronic mutation in CEP290, after achieving rapid and sustained editing of somatic non-human primate cells at a level that met the target therapeutic threshold (Maeder et al., 2019).
11.3 Drug therapies
USH3A has been proposed to be one of the easier USH subtypes to target for therapeutic intervention. This is owing to its prolonged latency compared to other subtypes and that the causative gene does not encode a structural protein of HCs or photoreceptors. Theoretically therefore, USH3A should have a wider therapeutic window for intervention. Currently, the clinical trial of the antioxidant N-acetylcysteine amide, NPI-001 (NCT04355689) for general USH treatment could be of particular interest for treating USH3A, as animal models have shown improved hearing after antioxidant treatment (Gopal et al., 2019). N-acetylcysteine is currently undergoing a Phase III clinical trial with Johns Hopkins University (NCT05537220) for RP, irrespective of genetic source with 438 participants over 45 months. This follows previous clinical trials in which 6 months of treatment was safe and well-tolerated with improvement of best-corrected visual acuity and macular sensitivity (Campochiaro et al., 2020). Another small molecule drug currently in preclinical development, BF844, has been demonstrated to stabilize the CLRN1N48K mutation and attenuate progressive HL in mouse models (Alagramam et al., 2016). The drug is currently undergoing a Phase I clinical trial (NCT06592131) to evaluate pharmacokinetics.
As mentioned previously, upregulation of genes related to apoptosis has been observed in retinal organoids derived from USH patients (Guo et al., 2019; Leong et al., 2022; Su et al., 2022). One explanation for the activation of the apoptosis pathway is the over-accumulation of USH proteins in the endoplasmic reticulum. For instance, MYO7A, harmonin and cadherin 23 are known to localise and assemble a protein complex in the endoplasmic reticulum before being trafficked to the stereocilia (Blanco-Sánchez et al., 2014). A small molecule, Salubrinal, acts to prevent dephosphorylation of eukaryotic translation initiation factor 2 alpha (eIF2α) which has a cytoprotective effect during endoplasmic reticulum stress when phosphorylated (Matsuoka and Komoike, 2015). Salubrinal-treated erlong mice, harbouring missense Cdh23 mutations, had improved ABR thresholds and DPOAE amplitudes and OHC survival after treatment (Hu et al., 2016). These encouraging results indicate endoplasmic reticulum stress may be an interesting novel target to delay HC death for some USH types, and extend the treatment window for other therapeutic strategies that require intact HCs.
11.3.1 Antisense oligonucleotide therapies
Antisense oligonucleotide (ASO) therapy is an alternative method to treat inherited disorders like USH by using a synthetic 21–25 single-stranded oligodeoxynucleotide that complementarily bind to the target mRNA to regulate gene expression (Rinaldi and Wood, 2018). ASO-based drugs have proven to be a promising therapeutic approach to target specific genetic diseases such as Duchenne muscular dystrophy, which restored functional mRNA transcripts of the DMD gene (Lauffer et al., 2024). Fortunately, development of ASO treatments is rapidly advancing in preclinical studies for USH genes with promising outcome as summarised in Table 5.
11.3.1.1 USH1C antisense therapies
Preclinical applications of drug candidate ASO-29 to target Acadian mutation c.216G>A in exon 3 of USH1C for the inner ear have showed promising results. Mice harbouring murine equivalent c.214G>A mutation treated at different dosage and inner ear HC developmental stages have various levels of hearing restoration (Lentz et al., 2013; Lentz et al., 2020; Wang et al., 2020). In Lentz et al. (2020) study, ASO-29 was directly administered in P1 Ush1c216AA mice via local round window membrane injection. Treated mice had significantly improved hearing thresholds and cochlear hair bundle morphology. Currently, the Lentz group are actively conducting natural history studies of USH1C patients to investigate the disease progression and identify suitable participants for future clinical trials (Lentz et al., 2023). Their earlier study also enlightened another possibility for treating visual impairment caused by USH1C c.216G>A, where significant improvement in visual function and retinal structure was shown in neonatal mice when ASO was locally administered to the eye (Amato et al., 2015). In 2019, the Lentz and Koenekopp groups collaborated to investigate potential ASO treatment to target vision loss caused by USH1C c.216G>A, where in their most recent work, they reported mRNA transcript levels of corrected Ush1c significantly increased in treated mutant mice by modifying the chemical moiety of the parent drug ASO-29 (Alapure et al., 2023; Alapure et al., 2024).
11.3.1.2 USH2A antisense therapies
There are an increasing number of newly identified deep-intronic variants in USH2A, often creating pseudoexon inclusions, premature stop codons or altering the protein function (García-Bohórquez et al., 2024). A deep-intronic c.7595–2144A>G mutation in USH2A was first reported in 2012, which results in an insertion of a 152 bp pseudoexon 40 (PE40) into the mature mRNA transcript (Vaché et al., 2012). Fibroblasts from a heterozygous patient with USH2A c.7595–2144A>G mutation were treated with ASO to induce splicing correction (Slijkerman et al., 2016). Two different ASOs targeting the PE40 mutation promoted splicing correction in USH2A mRNA transcripts which were confirmed by Sanger sequencing. Slijkerman et al. (2016) also transfected both ASOs into patient fibroblasts which achieved higher splicing correction, suggesting a cocktail of ASOs can improve therapeutic outcome. Recently, ASOs targeting deep-intronic mutations in patient iPSC-derived PPCs have shown to be an effective method to correct abberant splicing mutations c.1551–504C>T (PE8) and c.4397–3890A>G (PE20) in USH2A (Reurink et al., 2023). Moreover, splicing redirection by ASOs have also been reported for three novel deep-intronic variants in minigene splicing assays (García-Bohórquez et al., 2024).
Multiple exon skipping was recently demonstrated for founder mutation c.4338_4339delCT in exon 20 of USH2A, which accounts for 55.6% of USH2 cases in the Quebec French-Canadian population (Ebermann et al., 2009; Phillips et al., 2024). The designed ASO induced in-frame deletion by skipping exons 19 and 20 which together encode for a single fibronectin domain and had showed successful exon skipping potential in a zebrafish, and in human iPSC-derived retinal and inner ear organoid models (Phillips et al., 2024). This effective strategy not only preserved usherin protein folding properties based on an in silico analysis but allowing broader application for treating patients with pathogenic variants found in either exon 19 and/or exon 20 (Schellens et al., 2023; Phillips et al., 2024). In a similar study, Schellens et al. (2023) showed simultaneous exon skipping of exons 30-31, or exons 39-40 in USH2A, which both encodes for one of the FN3 domains, did not affect its protein function in the zebrafish photoreceptor cells.
In a proof-of-concept study, deletion of murine equivalent exon 12 in Ush2a (Ush2aΔex12) resulted in a shortened usherin protein. Despite this, Ush2aΔex12 mice demonstrated improved hearing function along with preserved HC morphology (Pendse et al., 2019). Exon skipping of the most prevalent c.2299delG mutation in exon 13 of USH2A was demonstrated by Dulla et al. (2021) using the ASO, QR-421a. This method of splicing correction also does not disrupt the reading frame for translating USH2A, thus producing a shortened usherin protein with residual function. Indeed, QR-421a corrected the ush2a transcript reading frame in ush2armc1 zebrafish; the treatment successfully restored some level of usherin protein expression in mutant photoreceptors (Dulla et al., 2021). Moreover, PPCs derived from USH2A patient iPSCs carrying homozygous c.2299delG mutation were treated with QR-421a at concentrations 1–10 µM for 28 days using gymnotic delivery. Exon 13 skipping in USH2A was highest at 62% for 10 µM concentration whilst no correction was observed in untreated or control group, indicating high target sequence specificity of QR-421a.
Due to the recent success of Ultevursen (QR-421a) in restoring functional ush2a in the zebrafish model, the Stellar trial, a Phase II clinical trial (NCT03780257) was conducted over 24 months with 20 subjects. The study reported that a single intravitreal injection of QR-421a was well-tolerated with stabilisation of visual acuity and improvement in retinal sensitivity and structure (Audo et al., 2022). Since the positive outcome of Stellar trial, the Phase II LUNA trial (NCT06627179) is currently recruiting USH2A-RP patients with mutations in exon 13 to determine the safety and tolerability of QR-421a over the course of 24 months. USH2A-RP patients will initially receive an intravitreal injection of QR-421a at 160 µg and is further administered at 80 µg on 6, 12 and 18 months. This ongoing administration allows the production of partially functional protein by repeatedly inducing skipping of exon 13 in USH2A at the mRNA level (Boros et al., 2022; Komaki et al., 2025). For instance, Nusinersen is the only FDA-approved ASO drug for treating spinal muscular atrophy and studies have shown patients require regular administration every 4–6 months at lowered concentration of Nusinersen to maintain levels of corrected protein expression (De Vivo et al., 2019). To mitigate this issue, Ou et al. (2024) introduced an AAV-based gene therapy approach called RM-101 containing SmOPT snRNA and have showed high efficiency of skipping exon 13 in USH2A. Moreover, humanised mice treated with RM-101 have sustained USH2AΔex13 transcript expression and did not show any retinal abnormalities, offering a different perspective to treat USH2A-associated RP.
12 Cell therapies
Cell therapy is an approach whereby healthy and functional cells can be transplanted into an individual to replace damaged or lost cells. The application of cell therapy has been thoroughly investigated for regenerative medicine or treating neurodegenerative conditions like USH. Stem cells are widely used due to their self-renewal ability and potency to differentiate into any cell type in the body. There are currently two types of stem cells used for inner ear and retinal cell therapy including embryonic stem cells (ESCs) and iPSCs. Currently, various stem cell products are being developed and tested in clinical trials for RP treatment which has been summarised in Table 6.
jCell is the first allogeneic iPSC-derived retinal progenitor cell product developed by jCyte, which has neuroprotective mechanisms by releasing neurotrophic factors (Yang et al., 2024b). Early results from Phase I/II clinical trial (NCT02320812) have shown jCell to be safe and well-tolerated at various dosages. In the latest Phase IIb study (NCT04604899), participants from the previous Phase II trial (NCT03073733) were re-injected with jCell on the same treated eye and had sustained improvement in visual acuity, contrast sensitivity and kinetic visual fields. Following the success of this clinical trial, jCyte reported they anticipate proceeding into Phase III for jCell.
In 2024, an iPSC-derived neural progenitor cell (NPC) product called CNS10-NPC entered Phase I clinical trials (NCT04284293) for RP. CNS10-NPC was shown to significantly delay photoreceptor degeneration through the promotion of antioxidant effects and release of trophic factors through various signalling pathways in RCS rats modelling retinal degeneration (Lu et al., 2023). Moreover, CNS10-NPC were able to mature into astrocytes and did not show tumorigenicity, making this a promising treatment for RP (Lu et al., 2023). Similarly, the US FDA recently approved fast track designation for OpCT-001, an allogeneic iPSC-derived PPC product developed by BlueRock Therapeutics to treat primary photoreceptor diseases including RP. Currently, a Phase I clinical trial (NCT06789445) is undergoing patient recruitment to investigate safety, tolerability and efficacy of OpCT-001 for 52 weeks.
Rincell-1 by Rinri Therapeutics, is expected to enter clinical trials in 2025. Rincell-1 is a human ESC-derived otic neural progenitor (ONP) product used for regenerating auditory neurons. In their preliminary study, transplantation of human ESC-derived ONP cells into the cochlea of a gerbil model of auditory neuropathy was able to re-establish the synaptic connection with native cochlear HCs, improving hearing ability (Chen et al., 2012). Additionally, Rinri Therapeutics have two stem cell products in preclinical development for hearing restoration, including iPSC-derived ONP (Rincell-2) and otic epithelial progenitor (Rincell-3). While ANP1 (ReSonance) is another neural cell-based product developed by Lineage Cell Therapeutics for treating auditory neuronal disorders, this form of cell therapy does not meet the clinical needs of USH patients. For instance, majority of USH proteins are expressed in HCs and stereocilia, and in USH patients with severely impaired cochlear HCs and auditory nerves, replacing neural cells alone would be insufficient to restore hearing (Sekiya and Holley, 2021).
To date, the only stem cell-based therapy in clinical trial for hearing impairment is the use of autologous mesenchymal stem cells to regenerate HCs in children with acquired SNHL (Baumgartner et al., 2018). Although their results demonstrated the patients had hearing improvement, mesenchymal stem cells have limited differentiation potential and are unable to differentiate into the necessary cell types such as HCs that are missing in USH patients (Pittenger et al., 2019). While these stem cells have restricted ability to only differentiate into neural stem cells, USH patients with preserved HCs could benefit from its immunomodulatory and regenerative properties as mesenchymal stem cells can secrete growth factors and cytokines to preserve HCs as shown in rodent cochlea (Kada et al., 2020; Tsai et al., 2022). Transplanting stem cells that can simultaneously differentiate into both HCs and auditory nerves may be a feasible method to reverse HL in USH patients. Several groups have recently shown iPSCs can differentiate into cochlear sensory epithelium, containing both HCs and neural cells in inner ear organoids, which could potentially provide a renewable source of HCs (Koehler et al., 2017; Moore et al., 2023).
13 Conclusion and future therapeutics
Mutations in USH-associated genes affect the sensory cells of the inner ear and retina which do not replenish once damaged. Moreover, USH is still considered an incurable disease, despite being the most common genetic disorder of deaf-blindness. Our understanding of the role that USH proteins play in the inner ear and retina has advanced due to the continued progress in optimising animal models. The USH proteins form an important interactome in the sensory cells of the mammalian inner ear and retina, as they support both the development of sensory cells and their maintenance after birth. Genetic screening and identification of USH mutations combined with patient-derived organoid models will improve our ability to link the functions of USH protein variants with their pathological mechanisms and help identify new therapeutic targets.
The recent success of the clinical trial that delivered a functional copy of the OTOF gene with an adeno-associated virus provides hope of further progress in treating genetic HL via gene therapy. OTOF gene therapy has been administered in 11 children and improved ABR, speech perception and sound localisation with no dose-limiting toxicity or serious adverse events were reported (Lv et al., 2024; Wang et al., 2024). While these patients will require follow-up to assess long-term effects, it is outstanding progress. It is thought adults may also benefit from similar gene therapy (Qi et al., 2025). One reason that the OTOF treatment may be efficient at restoring hearing, is the preservation of the architecture of the organ of Corti and HCs. This is of particular note when considering the treatment of USH3 due to the late onset of the subtype. Genetic screening to identify these patients presents the opportunity to treat their USH with gene replacement or editing therapies before symptoms become too severe.
Cell therapy is a promising strategy to replace defective cochlear HCs and retinal photoreceptor with healthy ones for USH patients, however, there are concerns regarding the safety of stem cells. Manipulating stem cell differentiation is difficult and could potentially lead to undesirable cell types and teratomas (Gunewardene et al., 2014; Isoda et al., 2023). Promisingly, results from clinical trials for retinal disorders using stem cell therapy have yet to report adverse events (Radu et al., 2024). Moreover, most stem cell products in clinical trials are harvested from allogeneic sources and are a popular choice as a functional copy of the gene is readily available. One drawback of allogeneic stem cells is the risk of immune rejection, limiting effective tissue transplantation. Alternatively, autologous stem cells derived from patient’s own cells can mitigate tissue rejection issues, but in USH cases, the dependent on gene therapy will still be necessary.
While we have discussed the new therapeutic strategies individually, there is promising work in other fields where technologies are combined, which could be applicable to USH. Current studies have already shown ways to improve cell transplantation such as knocking out the human leukocyte antigen with CRISPR-Cas gene editing technology (Amiri et al., 2021). Furthermore, transplanting genetically corrected patient iPSCs have already been reported in various inherited disorders including Huntington’s disease and Parkinson’s disease (Cho et al., 2019; Moriarty et al., 2022). Genetically modified stem cells have already been tested in deaf-blind animal models and have been demonstrated to evade immune rejection and successfully improve the engraftment rate of transplanted cells, though a method for transplanting cells in their precise locations, particularly in the organ of Corti has yet to be demonstrated (da Silva et al., 2023; Ishida et al., 2024).
The therapeutic strategies of gene augmentation, gene editing, drug therapy and cell therapy discussed in this review are promising approaches for the treatment of USH. The current and future progress in preclinical and clinical trials will continue to refine these strategies and their delivery, efficacy and safety. The global burden of these sensory deficits has over 1.4 billion people suffering from HL and 1.3 billion from vision loss, with this expected to rise to 2.5 billion people projected to have HL by 2050 (GBD, 2017 Disease and Injury Incidence and Prevalence Collaborators, 2018; World Health Organisation, 2025). The novel therapies discussed in this review and the deeper understanding of the inner ear and retina gained from recent organoid and animal models could be applied to address other causes of hearing and vision loss.
Author contributions
FL: Writing – original draft, Writing – review and editing. JL: Writing – original draft, Writing – review and editing. DD: Writing – original draft, Writing – review and editing. SM: Writing – review and editing, Writing – original draft. FC: Writing – review and editing, Writing – original draft. MA: Writing – review and editing, Writing – original draft. EW: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing, Data curation, Formal Analysis.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research has been supported by the Western Australian Future Health Research and Innovation Fund Grant ID FHRIFES2024/2 and Near-miss Awards: Ideas Grants (WANMA): WANMA/Ideas2023-24/X. Australian government National Health and Medical Research Council (NHMRC) Ideas Grant: GNT1188694 and Medical Research Future Fund (MRFF) Stem Cell Therapies Mission Grant: MRF2017281, Commonwealth Scientific and Industrial Research Organisation (CSIRO) ON Prime Innovation Program: P12-1426, The Ian Potter Foundation, Passe and Williams Foundation, Perron Charitable Foundation, MedChem Australia, Channel 7 Telethon grant. FL and JL were supported by the PhD scholarship provided by Ear Science Institute Australia.
Acknowledgments
The authors thank Peter Millington and David Sly for their additional review and proof reading of this manuscript. The authors thank Josh Gacer for figure editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aasen, T., Raya, A., Barrero, M. J., Garreta, E., Consiglio, A., Gonzalez, F., et al. (2008). Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 26 (11), 1276–1284. doi:10.1038/nbt.1503
Abadie, C., Blanchet, C., Baux, D., Larrieu, L., Besnard, T., Ravel, P., et al. (2012). Audiological findings in 100 USH2 patients. Clin. Genet. 82 (5), 433–438. doi:10.1111/j.1399-0004.2011.01772.x
Abad-Morales, V., Navarro, R., Burés-Jelstrup, A., and Pomares, E. (2020). Identification of a novel homozygous ARSG mutation as the second cause of Usher syndrome type 4. Am. J. Ophthalmol. Case Rep. 19, 100736. doi:10.1016/j.ajoc.2020.100736
Abbott, J. A., Guth, E., Kim, C., Regan, C., Siu, V. M., Rupar, C. A., et al. (2017). The usher syndrome type IIIB histidyl-tRNA synthetase mutation confers temperature sensitivity. Biochemistry 56 (28), 3619–3631. doi:10.1021/acs.biochem.7b00114
Abu Rayyan, A., Kamal, L., Casadei, S., Brownstein, Z., Zahdeh, F., Shahin, H., et al. (2020). Genomic analysis of inherited hearing loss in the Palestinian population. Proc. Natl. Acad. Sci. 117 (33), 20070–20076. doi:10.1073/pnas.2009628117
Adato, A., Vreugde, S., Joensuu, T., Avidan, N., Hamalainen, R., Belenkiy, O., et al. (2002). USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur. J. Hum. Genet. 10 (6), 339–350. doi:10.1038/sj.ejhg.5200831
Ahmed, Z. M., Jaworek, T. J., Sarangdhar, G. N., Zheng, L., Gul, K., Khan, S. N., et al. (2018). Inframe deletion of human ESPN is associated with deafness, vestibulopathy and vision impairment. J. Med. Genet. 55 (7), 479–488. doi:10.1136/jmedgenet-2017-105221
Ahmed, Z. M., Kjellstrom, S., Haywood-Watson, R. J., Bush, R. A., Hampton, L. L., Battey, J. F., et al. (2008). Double homozygous waltzer and Ames waltzer mice provide no evidence of retinal degeneration. Mol. Vis. 14, 2227–2236.
Ahmed, Z. M., Riazuddin, S., Ahmad, J., Bernstein, S. L., Guo, Y., Sabar, M. F., et al. (2003). PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum. Mol. Genet. 12 (24), 3215–3223. doi:10.1093/hmg/ddg358
Ahmed, Z. M., Riazuddin, S., Bernstein, S. L., Ahmed, Z., Khan, S., Griffith, A. J., et al. (2001). Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am. J. Hum. Genet. 69 (1), 25–34. doi:10.1086/321277
Akil, O. (2020). Dual and triple AAV delivery of large therapeutic gene sequences into the inner ear. Hear. Res. 394, 107912. doi:10.1016/j.heares.2020.107912
Alagramam, K. N., Gopal, S. R., Geng, R., Chen, D.H.-C., Nemet, I., Lee, R., et al. (2016). A small molecule mitigates hearing loss in a mouse model of Usher syndrome III. Nat. Chem. Biol. 12 (6), 444–451. doi:10.1038/nchembio.2069
Alapure, B. V., Robillard, K., Hathaway, M., and Lentz, J. J. (2024). Optimized ASO chemistry significantly improves Ush1c gene splicing and visual function in Ush1c-216AA mice. Investigative Ophthalmol. Vis. Sci. 65 (7), 2194.
Alapure, B. V., Robillard, K. N., and Lentz, J. J. (2023). Combination ASO therapy improves gene expression compared with single ASO therapy in Ush1c mice. Investigative Ophthalmol. Vis. Sci. 64 (8), 3870.
Aller, E., Larrieu, L., Jaijo, T., Baux, D., Espinós, C., González-Candelas, F., et al. (2010). The USH2A c.2299delG mutation: dating its common origin in a Southern European population. Eur. J. Hum. Genet. 18 (7), 788–793. doi:10.1038/ejhg.2010.14
Amato, R. J., Rosencrans, R., Jodelka, F. M., Hinrich, A. J., Bazan, N. G., Rigo, F., et al. (2015). Early effects of antisense oligonucleotide treatment on photoreceptor function and retinal structure in a mouse model of Usher Syndrome. Investigative Ophthalmol. Vis. Sci. 56 (7), 5452.
Amiri, F., Ranjbar, M., Pirouzfar, M., Nourigorji, M., and Dianatpour, M. (2021). HLA-A gene knockout using CRISPR/Cas9 system toward overcoming transplantation concerns. Egypt. J. Med. Hum. Genet. 22, 37–38. doi:10.1186/s43042-021-00155-y
Aparicio-Domingo, S., Flores-Bellver, M., Cobb, H., Li, K. V., Conrad, B., Chen, C., et al. (2023). “Generation of three-dimensional retinal tissue with physiologically competent, light-sensitive photoreceptors from human-induced pluripotent stem cells,” in Brain organoid research. Editor J. Gopalakrishnan (New York, NY: Springer US), 99–119.
Audo, I., Birch, D. G., Thiran Jayasundera, K., Meunier, I., Huckfeldt, R. M., Koenekoop, R. K., et al. (2022). QR-421a RNA therapy in retinitis pigmentosa due to mutations in USH2A: Stellar trial Phase [AP1] 1b/2 interim results. Acta Ophthalmol. 100. doi:10.1111/j.1755-3768.2022.205
Baumgartner, L. S., Moore, E., Shook, D., Messina, S., Day, M. C., Green, J., et al. (2018). Safety of autologous umbilical cord blood therapy for acquired sensorineural hearing loss in children. J. Audiology Otology 22 (4), 209–222. doi:10.7874/jao.2018.00115
Bauwens, M., De Man, V., Audo, I., Balikova, I., Zein, W. M., Smirnov, V., et al. (2025). Expanding the genetic landscape of Usher syndrome type IV caused by pathogenic ARSG variants. Clin. Genet. 107 (1), 44–55. doi:10.1111/cge.14614
Baux, D., Vaché, C., Blanchet, C., Willems, M., Baudoin, C., Moclyn, M., et al. (2017). Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci. Rep. 7 (1), 16783. doi:10.1038/s41598-017-16846-9
Blanco-Sánchez, B., Clément, A., Fierro Jr, J., Washbourne, P., and Westerfield, M. (2014). Complexes of Usher proteins preassemble at the endoplasmic reticulum and are required for trafficking and ER homeostasis. Dis. Models Mech. 7 (5), 547–559. doi:10.1242/dmm.014068
Boëda, B., El-Amraoui, A., Bahloul, A., Goodyear, R., Daviet, L., Blanchard, S., et al. (2002). Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 21 (24), 6689–6699. doi:10.1093/emboj/cdf689
Booth, K. T., Azaiez, H., Kahrizi, K., Simpson, A. C., Tollefson, W. T. A., Sloan, C. M., et al. (2015). PDZD7 and hearing loss: more than just a modifier. Am. J. Med. Genet. Part A 167 (12), 2957–2965. doi:10.1002/ajmg.a.37274
Booth, K. T., Kahrizi, K., Babanejad, M., Daghagh, H., Bademci, G., Arzhangi, S., et al. (2018). Variants in CIB2 cause DFNB48 and not USH1J. Clin. Genet. 93 (4), 812–821. doi:10.1111/cge.13170
Bork, J. M., Peters, L. M., Riazuddin, S., Bernstein, S. L., Ahmed, Z. M., Ness, S. L., et al. (2001). Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 68 (1), 26–37. doi:10.1086/316954
Boros, B. D., Schoch, K. M., Kreple, C. J., and Miller, T. M. (2022). Antisense oligonucleotides for the study and treatment of ALS. Neurotherapeutics 19 (4), 1145–1158. doi:10.1007/s13311-022-01247-2
Buchholz, D. E., Pennington, B. O., Croze, R. H., Hinman, C. R., Coffey, P. J., and Clegg, D. O. (2013). Rapid and efficient directed differentiation of human pluripotent stem cells into retinal pigmented epithelium. Stem Cells Transl. Med. 2 (5), 384–393. doi:10.5966/sctm.2012-0163
Calabro, K. R., Boye, S. L., Choudhury, S., Fajardo, D., Peterson, J. J., Li, W., et al. (2019). A novel mouse model of MYO7A USH1B reveals auditory and visual system haploinsufficiencies. Front. Neurosci. 13, 1255. doi:10.3389/fnins.2019.01255
Campochiaro, P. A., Iftikhar, M., Hafiz, G., Akhlaq, A., Tsai, G., Wehling, D., et al. (2020). Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. J. Clin. Investigation 130 (3), 1527–1541. doi:10.1172/JCI132990
Carvalho, L. S., Turunen, H. T., Wassmer, S. J., Luna-Velez, M. V., Xiao, R., Bennett, J., et al. (2017). Evaluating efficiencies of dual AAV approaches for retinal targeting. Front. Neurosci. 11, 503. doi:10.3389/fnins.2017.00503
Castiglione, A., and Möller, C. (2022). Usher syndrome. Audiology Res. 12 (1), 42–65. doi:10.3390/audiolres12010005
Chang, S.-Y., Kim, E., Carpena, N. T., Lee, J.-H., Kim, D. H., and Lee, M. Y. (2023). Photobiomodulation can enhance stem cell viability in cochlea with auditory neuropathy but does not restore hearing. Stem Cells Int. 2023 (1), 6845571. doi:10.1155/2023/6845571
Chen, C., Liu, X., and Peng, X. (2022). Management of cystoid macular edema in retinitis pigmentosa: a systematic review and meta-analysis. Front. Med. 9, 895208. doi:10.3389/fmed.2022.895208
Chen, L., Wang, J., Yang, T., Xie, L., Cui, Z., Yu, Q., et al. (2023). Establishment of iPS cell line (KLRMMEi003-A) from a patient with Usher syndrome due to USH2A mutation. Stem Cell Res. 68, 103055. doi:10.1016/j.scr.2023.103055
Chen, Q., Zou, J., Shen, Z., Zhang, W., and Yang, J. (2014). Whirlin and PDZ domain-containing 7 (PDZD7) proteins are both required to form the quaternary protein complex associated with usher syndrome type 2. J. Biol. Chem. 289 (52), 36070–36088. doi:10.1074/jbc.M114.610535
Chen, W., Jongkamonwiwat, N., Abbas, L., Eshtan, S. J., Johnson, S. L., Kuhn, S., et al. (2012). Restoration of auditory evoked responses by human ES-cell-derived otic progenitors. Nature 490 (7419), 278–282. doi:10.1038/nature11415
Chien, W. W., Isgrig, K., Roy, S., Belyantseva, I. A., Drummond, M. C., May, L. A., et al. (2016). Gene therapy restores hair cell stereocilia morphology in inner ears of deaf whirler mice. Mol. Ther. 24 (1), 17–25. doi:10.1038/mt.2015.150
Cho, I. K., Hunter, C. E., Ye, S., Pongos, A. L., and Chan, A. W. S. (2019). Combination of stem cell and gene therapy ameliorates symptoms in Huntington's disease mice. NPJ Regen. Med. 4, 7. doi:10.1038/s41536-019-0066-7
Colella, P., Trapani, I., Cesi, G., Sommella, A., Manfredi, A., Puppo, A., et al. (2014). Efficient gene delivery to the cone-enriched pig retina by dual AAV vectors. Gene Ther. 21 (4), 450–456. doi:10.1038/gt.2014.8
Crane, R., Tebbe, L., Mwoyosvi, M. L., Al-Ubaidi, M. R., and Naash, M. I. (2023). Expression of the human usherin c.2299delG mutation leads to early-onset auditory loss and stereocilia disorganization. Commun. Biol. 6 (1), 933. doi:10.1038/s42003-023-05296-x
da Silva, L. H. A., Heuer, R. A., Roque, C. B., McGuire, T. L., Hosoya, T., Kimura, H., et al. (2023). Enhanced survival of hypoimmunogenic otic progenitors following intracochlear xenotransplantation: repercussions for stem cell therapy in hearing loss models. Stem Cell Res. Ther. 14, 83. doi:10.1186/s13287-023-03304-9
Davies, C., Bergman, J., Misztal, C., Ramchandran, R., Mittal, J., Bulut, E., et al. (2021). The outcomes of cochlear implantation in usher syndrome: a systematic review. J. Clin. Med. 10 (13), 2915. doi:10.3390/jcm10132915
Delaney, D. S., Liew, L. J., Lye, J., Atlas, M. D., and Wong, E. Y. (2023). Overcoming barriers: a review on innovations in drug delivery to the middle and inner ear. Front. Pharmacol. 14, 1207141. doi:10.3389/fphar.2023.1207141
De Vivo, D. C., Bertini, E., Swoboda, K. J., Hwu, W.-L., Crawford, T. O., Finkel, R. S., et al. (2019). Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul. Disord. 29 (11), 842–856. doi:10.1016/j.nmd.2019.09.007
Dinculescu, A., McCall, M. A., Gregg, R. G., Jalligampala, A., Noel, J. M., Whiting, R., et al. (2022). The generation and characterization of a large animal model of USH3 disease. Investigative Ophthalmol. Vis. Sci. 63 (7), 1926–A0072.
Dinculescu, A., Stupay, R. M., Deng, W.-T., Dyka, F. M., Min, S.-H., Boye, S. L., et al. (2016). AAV-mediated clarin-1 expression in the mouse retina: implications for USH3A gene therapy. PLOS ONE 11 (2), e0148874. doi:10.1371/journal.pone.0148874
Doda, D., Alonso Jimenez, S., Rehrauer, H., Carreño, J. F., Valsamides, V., Di Santo, S., et al. (2023). Human pluripotent stem cell-derived inner ear organoids recapitulate otic development in vitro. Development 150 (19), dev201865. doi:10.1242/dev.201865
Dona, M., Slijkerman, R., Lerner, K., Broekman, S., Wegner, J., Howat, T., et al. (2018). Usherin defects lead to early-onset retinal dysfunction in zebrafish. Exp. eye Res. 173, 148–159. doi:10.1016/j.exer.2018.05.015
Donaudy, F., Zheng, L., Ficarella, R., Ballana, E., Carella, M., Melchionda, S., et al. (2006). Espin gene (ESPN) mutations associated with autosomal dominant hearing loss cause defects in microvillar elongation or organisation. J. Med. Genet. 43 (2), 157–161. doi:10.1136/jmg.2005.032086
Du, H., Zou, L., Ren, R., Li, N., Li, J., Wang, Y., et al. (2020). Lack of PDZD7 long isoform disrupts ankle-link complex and causes hearing loss in mice. FASEB J. 34 (1), 1136–1149. doi:10.1096/fj.201901657RR
Dulla, K., Slijkerman, R., van Diepen, H. C., Albert, S., Dona, M., Beumer, W., et al. (2021). Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol. Ther. 29 (8), 2441–2455. doi:10.1016/j.ymthe.2021.04.024
Dyka, F. M., Boye, S. L., Chiodo, V. A., Hauswirth, W. W., and Boye, S. E. (2014). Dual adeno-associated virus vectors result in efficient in vitro and in vivo expression of an oversized gene, MYO7A. Hum. Gene Ther. Methods 25 (2), 166–177. doi:10.1089/hgtb.2013.212
Ebermann, I., Koenekoop, R. K., Lopez, I., Bou-Khzam, L., Pigeon, R., and Bolz, H. J. (2009). An USH2A founder mutation is the major cause of Usher syndrome type 2 in Canadians of French origin and confirms common roots of Quebecois and Acadians. Eur. J. Hum. Genet. EJHG 17 (1), 80–84. doi:10.1038/ejhg.2008.143
Ebermann, I., Phillips, J. B., Liebau, M. C., Koenekoop, R. K., Schermer, B., Lopez, I., et al. (2010). PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J. Clin. Investigation 120 (6), 1812–1823. doi:10.1172/JCI39715
Eiraku, M., Takata, N., Ishibashi, H., Kawada, M., Sakakura, E., Okuda, S., et al. (2011). Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472 (7341), 51–56. doi:10.1038/nature09941
Emptoz, A., Michel, V., Lelli, A., Akil, O., Boutet de Monvel, J., Lahlou, G., et al. (2017). Local gene therapy durably restores vestibular function in a mouse model of Usher syndrome type 1G. Proc. Natl. Acad. Sci. 114 (36), 9695–9700. doi:10.1073/pnas.1708894114
Ernest, S., Rauch, G.-J., Haffter, P., Geisler, R., Petit, C., and Nicolson, T. (2000). Mariner is defective in myosin VIIA: a zebrafish model for human hereditary deafness. Hum. Mol. Genet. 9 (14), 2189–2196. doi:10.1093/hmg/9.14.2189
Eudy, J. D., Weston, M. D., Yao, S., Hoover, D. M., Rehm, H. L., Ma-Edmonds, M., et al. (1998). Mutation of a gene encoding a protein with extracellular matrix motifs in usher syndrome type IIa. Science 280 (5370), 1753–1757. doi:10.1126/science.280.5370.1753
Fahimi, H., Behroozi, S., Noavar, S., and Parvini, F. (2021). A novel recessive PDZD7 bi-allelic mutation in an Iranian family with non-syndromic hearing loss. BMC Med. Genomics 14 (1), 37. doi:10.1186/s12920-021-00884-4
Ferla, R., Dell’Aquila, F., Doria, M., Ferraiuolo, M., Noto, A., Grazioli, F., et al. (2023). Efficacy, pharmacokinetics, and safety in the mouse and primate retina of dual AAV vectors for Usher syndrome type 1B. Mol. Ther. Methods Clin. Dev. 28, 396–411. doi:10.1016/j.omtm.2023.02.002
Finkbeiner, C., Ortuño-Lizarán, I., Sridhar, A., Hooper, M., Petter, S., and Reh, T. A. (2022). Single-cell ATAC-seq of fetal human retina and stem-cell-derived retinal organoids shows changing chromatin landscapes during cell fate acquisition. Cell Rep. 38 (4), 110294. doi:10.1016/j.celrep.2021.110294
Fowler, N. H., El-Rashedy, M. I., Chishti, E. A., Vander Kooi, C. W., and Maldonado, R. S. (2021). Multimodal imaging and genetic findings in a case of ARSG-related atypical Usher syndrome. Ophthalmic Genet. 42 (3), 338–343. doi:10.1080/13816810.2021.1891552
French, L. S., Mellough, C. B., Chen, F. K., and Carvalho, L. S. (2020). A review of gene, drug and cell-based therapies for usher syndrome. Front. Cell. Neurosci. 14, 183. doi:10.3389/fncel.2020.00183
Fritze, J. S., Stiehler, F. F., and Wolfrum, U. (2023). Pathogenic variants in USH1G/SANS alter protein interaction with pre-RNA processing factors PRPF6 and PRPF31 of the spliceosome. Int. J. Mol. Sci. 24 (24), 17608. doi:10.3390/ijms242417608
Fuster-García, C., García-Bohórquez, B., Rodríguez-Muñoz, A., Aller, E., Jaijo, T., Millán, J. M., et al. (2021). Usher syndrome: genetics of a human ciliopathy. Int. J. Mol. Sci. 22 (13), 6723. doi:10.3390/ijms22136723
Fuster-García, C., García-García, G., González-Romero, E., Jaijo, T., Sequedo, M. D., Ayuso, C., et al. (2017). USH2A gene editing using the CRISPR system. Mol. Ther. Nucleic Acids 8, 529–541. doi:10.1016/j.omtn.2017.08.003
García-Bohórquez, B., Barberán-Martínez, P., Aller, E., Jaijo, T., Mínguez, P., Rodilla, C., et al. (2024). Exploring non-coding variants and evaluation of antisense oligonucleotides for splicing redirection in Usher syndrome. Mol. Ther. Nucleic Acids 35 (4), 102374. doi:10.1016/j.omtn.2024.102374
GBD 2017 Disease and Injury Incidence and Prevalence Collaborators (2018). Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet London, Engl. 392 (10159), 1789–1858. doi:10.1016/S0140-6736(18)32279-7
Geng, R., Geller, S. F., Hayashi, T., Ray, C. A., Reh, T. A., Bermingham-McDonogh, O., et al. (2009). Usher syndrome IIIA gene clarin-1 is essential for hair cell function and associated neural activation. Hum. Mol. Genet. 18 (15), 2748–2760. doi:10.1093/hmg/ddp210
Geng, R., Melki, S., Chen, D.H.-C., Tian, G., Furness, D. N., Oshima-Takago, T., et al. (2012). The mechanosensory structure of the hair cell requires clarin-1, a protein encoded by usher syndrome III causative gene. J. Neurosci. 32 (28), 9485–9498. doi:10.1523/jneurosci.0311-12.2012
Geng, R., Omar, A., Gopal, S. R., Chen, D. H. C., Stepanyan, R., Basch, M. L., et al. (2017). Modeling and preventing progressive hearing loss in usher syndrome III. Sci. Rep. 7 (1), 13480. doi:10.1038/s41598-017-13620-9
Geng, R., Sotomayor, M., Kinder, K. J., Gopal, S. R., Gerka-Stuyt, J., Chen, D.H.-C., et al. (2013). Noddy, a mouse harboring a missense mutation in protocadherin-15, reveals the impact of disrupting a critical interaction site between tip-link cadherins in inner ear hair cells. J. Neurosci. 33 (10), 4395–4404. doi:10.1523/JNEUROSCI.4514-12.2013
Georgiou, M., Shakarchi, A. F., Elhusseiny, A. M., Michaelides, M., and Sallam, A. B. (2024). Cataract surgery outcomes in retinitis pigmentosa A comparative clinical database study. Am. J. Ophthalmol. 262, 34–39. doi:10.1016/j.ajo.2024.01.037
Gibbs, D., Azarian, S. M., Lillo, C., Kitamoto, J., Klomp, A. E., Steel, K. P., et al. (2004). Role of myosin VIIa and Rab27a in the motility and localization of RPE melanosomes. J. Cell Sci. 117 (26), 6473–6483. doi:10.1242/jcs.01580
Gibson, F., Walsh, J., Mburu, P., Varela, A., Brown, K. A., Antonio, M., et al. (1995). A type VII myosin encoded by the mouse deafness gene shaker-1. Nature 374 (6517), 62–64. doi:10.1038/374062a0
Gilmore, W. B., Hultgren, N. W., Chadha, A., Barocio, S. B., Zhang, J., Kutsyr, O., et al. (2023). Expression of two major isoforms of MYO7A in the retina: considerations for gene therapy of Usher syndrome type 1B. Vis. Res. 212, 108311. doi:10.1016/j.visres.2023.108311
Ginsburg, G. S., and Phillips, K. A. (2018). Precision medicine: from science to value. Health Aff. 37 (5), 694–701. doi:10.1377/hlthaff.2017.1624
Girotto, G., Vuckovic, D., Buniello, A., Lorente-Cánovas, B., Lewis, M., Gasparini, P., et al. (2014). Expression and replication studies to identify new candidate genes involved in normal hearing function. PLOS ONE 9 (1), e85352. doi:10.1371/journal.pone.0085352
Gong, J., Cheung, S., Fasso-Opie, A., Galvin, O., Moniz, L. S., Earle, D., et al. (2021). The impact of inherited retinal diseases in the United States of America (US) and Canada from a cost-of-illness perspective. Clin. Ophthalmol. 15, 2855–2866. doi:10.2147/opth.s313719
Gopal, S. R., Chen, D.H.-C., Chou, S.-W., Zang, J., Neuhauss, S. C. F., Stepanyan, R., et al. (2015). Zebrafish models for the mechanosensory hair cell dysfunction in usher syndrome 3 reveal that clarin-1 is an essential hair bundle protein. J. Neurosci. 35 (28), 10188–10201. doi:10.1523/jneurosci.1096-15.2015
Gopal, S. R., Lee, Y. T., Stepanyan, R., McDermott, B. M., and Alagramam, K. N. (2019). Unconventional secretory pathway activation restores hair cell mechanotransduction in an USH3A model. Proc. Natl. Acad. Sci. U. S. A. 116 (22), 11000–11009. doi:10.1073/pnas.1817500116
Grati, M. h., and Kachar, B. (2011). Myosin VIIa and sans localization at stereocilia upper tip-link density implicates these Usher syndrome proteins in mechanotransduction. Proc. Natl. Acad. Sci. 108 (28), 11476–11481. doi:10.1073/pnas.1104161108
Grati, M. h., Shin, J.-B., Weston, M. D., Green, J., Bhat, M. A., Gillespie, P. G., et al. (2012). Localization of PDZD7 to the stereocilia ankle-link associates this scaffolding protein with the usher syndrome protein network. J. Neurosci. 32 (41), 14288–14293. doi:10.1523/jneurosci.3071-12.2012
Gregory, F. D., Bryan, K. E., Pangršič, T., Calin-Jageman, I. E., Moser, T., and Lee, A. (2011). Harmonin inhibits presynaptic Cav1.3 Ca²⁺ channels in mouse inner hair cells. Nat. Neurosci. 14 (9), 1109–1111. doi:10.1038/nn.2895
Grillet, N., Xiong, W., Reynolds, A., Kazmierczak, P., Sato, T., Lillo, C., et al. (2009). Harmonin mutations cause mechanotransduction defects in cochlear hair cells. Neuron 62 (3), 375–387. doi:10.1016/j.neuron.2009.04.006
Grotz, S., Schäfer, J., Wunderlich, K. A., Ellederova, Z., Auch, H., Bähr, A., et al. (2022). Early disruption of photoreceptor cell architecture and loss of vision in a humanized pig model of usher syndromes. EMBO Mol. Med. 14 (4), e14817. doi:10.15252/emmm.202114817
Guan, J., Wang, H., Lan, L., Wang, L., Yang, J., Xie, L., et al. (2018). Novel recessive PDZD7 biallelic mutations in two Chinese families with non-syndromic hearing loss. Am. J. Med. Genet. Part A 176 (1), 99–106. doi:10.1002/ajmg.a.38477
Guan, Y., Du, H.-B., Yang, Z., Wang, Y.-Z., Ren, R., Liu, W.-W., et al. (2023). Deafness-associated ADGRV1 mutation impairs USH2A stability through improper phosphorylation of WHRN and WDSUB1 recruitment. Adv. Sci. 10 (16), 2205993. doi:10.1002/advs.202205993
Gunewardene, N., Bergen, N. V., Crombie, D., Needham, K., Dottori, M., and Nayagam, B. A. (2014). Directing human induced pluripotent stem cells into a neurosensory lineage for auditory neuron replacement. BioResearch Open Access 3 (4), 162–175. doi:10.1089/biores.2014.0019
Guo, Y., Wang, P., Ma, J. H., Cui, Z., Yu, Q., Liu, S., et al. (2019). Modeling retinitis pigmentosa: retinal organoids generated from the iPSCs of a patient with the USH2A mutation show early developmental abnormalities. Front. Cell. Neurosci. 13, 361. doi:10.3389/fncel.2019.00361
Guo, Y., Zeng, Q., Liu, S., Yu, Q., Wang, P., Ma, H., et al. (2018). Generation of an iPS cell line via a non-integrative method using urine-derived cells from a patient with USH2A-associated retinitis pigmentosa. Stem Cell Res. 29, 139–142. doi:10.1016/j.scr.2018.03.022
Han, S., Wang, Q., Cheng, M., Hu, Y., Liu, P., Hou, W., et al. (2024). The effects of ush2a gene knockout on vesicle transport in photoreceptors. Gene 892, 147885. doi:10.1016/j.gene.2023.147885
Hao, J., and Li, S. K. (2019). Inner ear drug delivery: recent advances, challenges, and perspective. Eur. J. Pharm. Sci. 126, 82–92. doi:10.1016/j.ejps.2018.05.020
Hazim, R. A., Karumbayaram, S., Jiang, M., Dimashkie, A., Lopes, V. S., Li, D., et al. (2017). Differentiation of RPE cells from integration-free iPS cells and their cell biological characterization. Stem Cell Res. Ther. 8 (1), 217. doi:10.1186/s13287-017-0652-9
Herrera, W., Aleman, T. S., Cideciyan, A. V., Roman, A. J., Banin, E., Ben-Yosef, T., et al. (2008). Retinal disease in usher syndrome III caused by mutations in the clarin-1 gene. Investigative Ophthalmol. Vis. Sci. 49 (6), 2651–2660. doi:10.1167/iovs.07-1505
Holme, R. H., Kiernan, B. W., Brown, S. D., and Steel, K. P. (2002). Elongation of hair cell stereocilia is defective in the mouse mutant whirler. J. Comp. Neurology 450 (1), 94–102. doi:10.1002/cne.10301
Hu, J., Li, B., Apisa, L., Yu, H., Entenman, S., Xu, M., et al. (2016). ER stress inhibitor attenuates hearing loss and hair cell death in Cdh23erl/erl mutant mice. Cell Death Dis. 7 (11), e2485. doi:10.1038/cddis.2016.386
Isgrig, K., Shteamer, J. W., Belyantseva, I. A., Drummond, M. C., Fitzgerald, T. S., Vijayakumar, S., et al. (2017). Gene therapy restores balance and auditory functions in a mouse model of Usher syndrome. Mol. Ther. 25 (3), 780–791. doi:10.1016/j.ymthe.2017.01.007
Ishida, M., Masuda, T., Sakai, N., Nakai-Futatsugi, Y., Kamao, H., Shiina, T., et al. (2024). Graft survival of major histocompatibility complex deficient stem cell-derived retinal cells. Commun. Med. 4 (1), 187. doi:10.1038/s43856-024-00617-5
Isoda, M., Sanosaka, T., Tomooka, R., Mabuchi, Y., Shinozaki, M., Andoh-Noda, T., et al. (2023). Mesenchymal properties of iPSC-derived neural progenitors that generate undesired grafts after transplantation. Commun. Biol. 6 (1), 611. doi:10.1038/s42003-023-04995-9
Isosomppi, J., Västinsalo, H., Geller, S. F., Heon, E., Flannery, J. G., and Sankila, E.-M. (2009). Disease-causing mutations in the CLRN1 gene alter normal CLRN1 protein trafficking to the plasma membrane. Mol. Vis. 15, 1806–1818.
Ivanchenko, M. V., Hathaway, D. M., Klein, A. J., Pan, B., Strelkova, O., De-la-Torre, P., et al. (2023). Mini-PCDH15 gene therapy rescues hearing in a mouse model of Usher syndrome type 1F. Nat. Commun. 14 (1), 2400. doi:10.1038/s41467-023-38038-y
Jeong, M., O'Reilly, M., Kirkwood, N. K., Al-Aama, J., Lako, M., Kros, C. J., et al. (2018). Generating inner ear organoids containing putative cochlear hair cells from human pluripotent stem cells. Cell Death Dis. 9 (9), 922. doi:10.1038/s41419-018-0967-1
Johnson, K. R., Gagnon, L. H., Webb, L. S., Peters, L. L., Hawes, N. L., Chang, B., et al. (2003). Mouse models of USH1C and DFNB18: phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene. Hum. Mol. Genet. 12 (23), 3075–3086. doi:10.1093/hmg/ddg332
Jouret, G., Poirsier, C., Spodenkiewicz, M., Jaquin, C., Gouy, E., Arndt, C., et al. (2019). Genetics of usher syndrome: new insights from a meta-analysis. Otol. Neurotol. 40 (1), 121–129. doi:10.1097/mao.0000000000002054
Kada, S., Hamaguchi, K., Ito, J., Omori, K., and Nakagawa, T. (2020). Bone marrow stromal cells accelerate hearing recovery via regeneration or maintenance of cochlear fibrocytes in mouse spiral ligaments. Anatomical Rec. 303 (3), 478–486. doi:10.1002/ar.24063
Kazmierczak, P., Sakaguchi, H., Tokita, J., Wilson-Kubalek, E. M., Milligan, R. A., Müller, U., et al. (2007). Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature 449 (7158), 87–91. doi:10.1038/nature06091
Khateb, S., Kowalewski, B., Bedoni, N., Damme, M., Pollack, N., Saada, A., et al. (2018). A homozygous founder missense variant in arylsulfatase G abolishes its enzymatic activity causing atypical Usher syndrome in humans. Genet. Med. 20 (9), 1004–1012. doi:10.1038/gim.2017.227
Kikkawa, Y., Shitara, H., Wakana, S., Kohara, Y., Takada, T., Okamoto, M., et al. (2003). Mutations in a new scaffold protein Sans cause deafness in Jackson shaker mice. Hum. Mol. Genet. 12 (5), 453–461. doi:10.1093/hmg/ddg042
Kimberling, W. J., Hildebrand, M. S., Shearer, A. E., Jensen, M. L., Halder, J. A., Trzupek, K., et al. (2010). Frequency of Usher syndrome in two pediatric populations: implications for genetic screening of deaf and hard of hearing children. Genet. Med. 12 (8), 512–516. doi:10.1097/GIM.0b013e3181e5afb8
Koehler, K. R., Nie, J., Longworth-Mills, E., Liu, X.-P., Lee, J., Holt, J. R., et al. (2017). Generation of inner ear organoids containing functional hair cells from human pluripotent stem cells. Nat. Biotechnol. 35 (6), 583–589. doi:10.1038/nbt.3840
Kolla, L., Kelly, M. C., Mann, Z. F., Anaya-Rocha, A., Ellis, K., Lemons, A., et al. (2020). Characterization of the development of the mouse cochlear epithelium at the single cell level. Nat. Commun. 11 (1), 2389. doi:10.1038/s41467-020-16113-y
Komaki, H., Takeshita, E., Kunitake, K., Ishizuka, T., Shimizu-Motohashi, Y., Ishiyama, A., et al. (2025). Phase 1/2 trial of brogidirsen: dual-targeting antisense oligonucleotides for exon 44 skipping in Duchenne muscular dystrophy. Cell Rep. Med. 6 (1), 101901. doi:10.1016/j.xcrm.2024.101901
König, I. R., Fuchs, O., Hansen, G., von Mutius, E., and Kopp, M. V. (2017). What is precision medicine? Eur. Respir. J. 50 (4), 1700391. doi:10.1183/13993003.00391-2017
Kosorok, M. R., and Laber, E. B. (2019). Precision medicine. Annu. Rev. statistics Its Appl. 6 (1), 263–286. doi:10.1146/annurev-statistics-030718-105251
Kros, C., Marcotti, W., Van Netten, S., Self, T., Libby, R., Brown, S., et al. (2002). Reduced climbing and increased slipping adaptation in cochlear hair cells of mice with Myo7a mutations. Nat. Neurosci. 5 (1), 41–47. doi:10.1038/nn784
Kruszewski, K., Lüllmann-Rauch, R., Dierks, T., Bartsch, U., and Damme, M. (2016). Degeneration of photoreceptor cells in arylsulfatase G-deficient mice. Investigative Ophthalmol. Vis. Sci. 57 (3), 1120–1131. doi:10.1167/iovs.15-17645
Kusuluri, D. K., Güler, B. E., Knapp, B., Horn, N., Boldt, K., Ueffing, M., et al. (2021). Adhesion G protein-coupled receptor VLGR1/ADGRV1 regulates cell spreading and migration by mechanosensing at focal adhesions. iScience 24 (4), 102283. doi:10.1016/j.isci.2021.102283
Lagziel, A., Ahmed, Z. M., Schultz, J. M., Morell, R. J., Belyantseva, I. A., and Friedman, T. B. (2005). Spatiotemporal pattern and isoforms of cadherin 23 in wild type and waltzer mice during inner ear hair cell development. Dev. Biol. 280 (2), 295–306. doi:10.1016/j.ydbio.2005.01.015
Lahlou, H., Nivet, E., Lopez-Juarez, A., Fontbonne, A., Assou, S., and Zine, A. (2018). Enriched differentiation of human otic sensory progenitor cells derived from induced pluripotent stem cells. Front. Mol. Neurosci. 11, 452. doi:10.3389/fnmol.2018.00452
Lau, S. C., Grati, M., Isgrig, K., Sinan, M., Calabro, K. R., Zhu, J., et al. (2023). Dual-AAV vector-mediated expression of MYO7A improves vestibular function in a mouse model of Usher syndrome 1B. Mol. Ther. Methods Clin. Dev. 30, 534–545. doi:10.1016/j.omtm.2023.08.012
Lauffer, M. C., van Roon-Mom, W., Aartsma-Rus, A., and Collaborative, N. (2024). Possibilities and limitations of antisense oligonucleotide therapies for the treatment of monogenic disorders. Commun. Med. 4 (1), 6. doi:10.1038/s43856-023-00419-1
Lefevre, G., Michel, V., Weil, D., Lepelletier, L., Bizard, E., Wolfrum, U., et al. (2008). A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development 135 (8), 1427–1437. doi:10.1242/dev.012922
Lentz, J. J., Gordon, W. C., Farris, H. E., MacDonald, G. H., Cunningham, D. E., Robbins, C. A., et al. (2010). Deafness and retinal degeneration in a novel USH1C knock-in mouse model. Dev. Neurobiol. 70 (4), 253–267. doi:10.1002/dneu.20771
Lentz, J. J., Jodelka, F. M., Hinrich, A. J., McCaffrey, K. E., Farris, H. E., Spalitta, M. J., et al. (2013). Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat. Med. 19 (3), 345–350. doi:10.1038/nm.3106
Lentz, J. J., Kristaponyte, I., Rauterkus, G., Kim, D., Klumpp, M., Crabtree, J., et al. (2023). USH1C vision and balance natural history studies and approach to sharing clinical data. Investigative Ophthalmol. Vis. Sci. 64 (8), 3096.
Lentz, J. J., Pan, B., Ponnath, A., Tran, C. M., Nist-Lund, C., Galvin, A., et al. (2020). Direct delivery of antisense oligonucleotides to the middle and inner ear improves hearing and balance in Usher mice. Mol. Ther. 28 (12), 2662–2676. doi:10.1016/j.ymthe.2020.08.002
Leong, Y. C., Di Foggia, V., Pramod, H., Bitner-Glindzicz, M., Patel, A., and Sowden, J. C. (2022). Molecular pathology of Usher 1B patient-derived retinal organoids at single cell resolution. Stem Cell Rep. 17 (11), 2421–2437. doi:10.1016/j.stemcr.2022.09.006
Li, C., and Samulski, R. J. (2020). Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 21 (4), 255–272. doi:10.1038/s41576-019-0205-4
Li, S., Mecca, A., Kim, J., Caprara, G. A., Wagner, E. L., Du, T.-T., et al. (2020). Myosin-VIIa is expressed in multiple isoforms and essential for tensioning the hair cell mechanotransduction complex. Nat. Commun. 11 (1), 2066. doi:10.1038/s41467-020-15936-z
Liang, L., Xue, Y., Su, C., Wang, J., Chen, L., Su, T., et al. (2022). Establishment of iPS cell line (KLRMMEi002-A) by reprogramming peripheral blood mononuclear cells from a patient with USH2A-associated Usher syndrome. Stem Cell Res. 60, 102699. doi:10.1016/j.scr.2022.102699
Liang, X., Qiu, X., Dionne, G., Cunningham, C. L., Pucak, M. L., Peng, G., et al. (2021). CIB2 and CIB3 are auxiliary subunits of the mechanotransduction channel of hair cells. Neuron 109 (13), 2131–2149.e15. doi:10.1016/j.neuron.2021.05.007
Libby, R., and Steel, K. (2001). Electroretinographic anomalies in mice with mutations in Myo7a, the gene involved in human Usher syndrome type 1B. Investigative Ophthalmol. Vis. Sci. 42 (3), 770–778.
Lin, L., Wang, H., Ren, D., Xia, Y., He, G., and Lu, Q. (2021). Structure and membrane targeting of the PDZD7 harmonin homology domain (HHD) associated with hearing loss. Front. Cell Dev. Biol. 9, 642666. doi:10.3389/fcell.2021.642666
Linnert, J., Güler, B. E., Krzysko, J., and Wolfrum, U. (2023). The adhesion G protein-coupled receptor VLGR1/ADGRV1 controls autophagy. Basic Clin. Pharmacol. Toxicol. 133 (4), 313–330. doi:10.1111/bcpt.13869
Liu, X., Bulgakov, O. V., Darrow, K. N., Pawlyk, B., Adamian, M., Liberman, M. C., et al. (2007). Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl. Acad. Sci. 104 (11), 4413–4418. doi:10.1073/pnas.0610950104
Liu, X., Lillywhite, J., Zhu, W., Huang, Z., Clark, A. M., Gosstola, N., et al. (2021). Generation and genetic correction of USH2A c.2299delG mutation in patient-derived induced pluripotent stem cells. Genes 12 (6), 805. doi:10.3390/genes12060805
Liu, X., Ondek, B., and Williams, D. S. (1998). Mutant myosin VIIa causes defective melanosome distribution in the RPE of shaker-1 mice. Nat. Genet. 19 (2), 117–118. doi:10.1038/470
Liu, X., Udovichenko, I. P., Brown, S. D. M., Steel, K. P., and Williams, D. S. (1999). Myosin VIIa participates in opsin transport through the photoreceptor cilium. J. Neurosci. 19 (15), 6267–6274. doi:10.1523/jneurosci.19-15-06267.1999
Loh, Y.-H., Agarwal, S., Park, I.-H., Urbach, A., Huo, H., Heffner, G. C., et al. (2009). Generation of induced pluripotent stem cells from human blood. Blood, J. Am. Soc. Hematol. 113 (22), 5476–5479. doi:10.1182/blood-2009-02-204800
Lopes, V. S., Boye, S. E., Louie, C. M., Boye, S., Dyka, F., Chiodo, V., et al. (2013). Retinal gene therapy with a large MYO7A cDNA using adeno-associated virus. Gene Ther. 20 (8), 824–833. doi:10.1038/gt.2013.3
Lu, B., Avalos, P., Svendsen, S., Zhang, C., Nocito, L., Jones, M. K., et al. (2023). GMP-grade human neural progenitors delivered subretinally protect vision in rat model of retinal degeneration and survive in minipigs. J. Transl. Med. 21 (1), 650. doi:10.1186/s12967-023-04501-z
Lv, J., Wang, H., Cheng, X., Chen, Y., Wang, D., Zhang, L., et al. (2024). AAV1-hOTOF gene therapy for autosomal recessive deafness 9: a single-arm trial. Lancet 403 (10441), 2317–2325. doi:10.1016/S0140-6736(23)02874-X
Ma, J., Ma, X., Lin, K., Huang, R., Bi, X., Ming, C., et al. (2023). Genetic screening of a Chinese cohort of children with hearing loss using a next-generation sequencing panel. Hum. Genomics 17 (1), 1. doi:10.1186/s40246-022-00449-1
Maddalena, A., Tornabene, P., Tiberi, P., Minopoli, R., Manfredi, A., Mutarelli, M., et al. (2018). Triple vectors expand AAV transfer capacity in the retina. Mol. Ther. 26 (2), 524–541. doi:10.1016/j.ymthe.2017.11.019
Maeda, R., Kindt, K. S., Mo, W., Morgan, C. P., Erickson, T., Zhao, H., et al. (2014). Tip-link protein protocadherin 15 interacts with transmembrane channel-like proteins TMC1 and TMC2. Proc. Natl. Acad. Sci. 111 (35), 12907–12912. doi:10.1073/pnas.1402152111
Maeder, M. L., Stefanidakis, M., Wilson, C. J., Baral, R., Barrera, L. A., Bounoutas, G. S., et al. (2019). Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 25 (2), 229–233. doi:10.1038/s41591-018-0327-9
Magliulo, G., Iannella, G., Gagliardi, S., Iozzo, N., Plateroti, R., Mariottini, A., et al. (2017). Usher’s syndrome type II: a comparative study of genetic mutations and vestibular system evaluation. Otolaryngology–Head Neck Surg. 157 (5), 853–860. doi:10.1177/0194599817715235
Manor, U., Disanza, A., Grati, M. H., Andrade, L., Lin, H., Di Fiore, P. P., et al. (2011). Regulation of stereocilia length by myosin XVa and whirlin depends on the actin-regulatory protein Eps8. Curr. Biol. CB 21 (2), 167–172. doi:10.1016/j.cub.2010.12.046
Mathur, P. D., Zou, J., Neiswanger, G., Zhu, D., Wang, Y., Almishaal, A. A., et al. (2023). Adenylyl cyclase 6 plays a minor role in the mouse inner ear and retina. Sci. Rep. 13 (1), 7075. doi:10.1038/s41598-023-34361-y
Matsuoka, M., and Komoike, Y. (2015). Experimental evidence shows salubrinal, an eIF2α dephosphorylation inhibitor, reduces xenotoxicant-induced cellular damage. Int. J. Mol. Sci. 16 (7), 16275–16287. doi:10.3390/ijms160716275
Mburu, P., Kikkawa, Y., Townsend, S., Romero, R., Yonekawa, H., and Brown, S. D. (2006). Whirlin complexes with p55 at the stereocilia tip during hair cell development. Proc. Natl. Acad. Sci. U. S. A. 103 (29), 10973–10978. doi:10.1073/pnas.0600923103
Mburu, P., Liu, X. Z., Walsh, J., Saw, D., Cope, M., Gibson, F., et al. (1997). Mutation analysis of the mouse myosin VIIA deafness gene. Genes Funct. 1 (3), 191–203. doi:10.1046/j.1365-4624.1997.00020.x
Mburu, P., Mustapha, M., Varela, A., Weil, D., El-Amraoui, A., Holme, R. H., et al. (2003). Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat. Genet. 34 (4), 421–428. doi:10.1038/ng1208
McGee, J., Goodyear, R. J., McMillan, D. R., Stauffer, E. A., Holt, J. R., Locke, K. G., et al. (2006). The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J. Neurosci. 26 (24), 6543–6553. doi:10.1523/JNEUROSCI.0693-06.2006
McLenachan, S., Wong, E. Y. M., Zhang, X., Leith, F., Moon, S. Y., Zhang, D., et al. (2019). Generation of two induced pluripotent stem cell lines from a patient with compound heterozygous mutations in the USH2A gene. Stem Cell Res. 36, 101420. doi:10.1016/j.scr.2019.101420
McMillan, D. R., Kayes-Wandover, K. M., Richardson, J. A., and White, P. C. (2002). Very large G protein-coupled receptor-1, the largest known cell surface protein, is highly expressed in the developing central nervous system. J. Biol. Chem. 277 (1), 785–792. doi:10.1074/jbc.M108929200
Michalski, N., Michel, V., Bahloul, A., Lefèvre, G., Barral, J., Yagi, H., et al. (2007). Molecular characterization of the ankle-link complex in cochlear hair cells and its role in the hair bundle functioning. J. Neurosci. 27 (24), 6478–6488. doi:10.1523/jneurosci.0342-07.2007
Michel, V., Booth, K. T., Patni, P., Cortese, M., Azaiez, H., Bahloul, A., et al. (2017). CIB2, defective in isolated deafness, is key for auditory hair cell mechanotransduction and survival. EMBO Mol. Med. 9 (12), 1711–1731. doi:10.15252/emmm.201708087
Michel, V., Pepermans, E., Boutet de Monvel, J., England, P., Nouaille, S., Aghaie, A., et al. (2020). Interaction of protocadherin-15 with the scaffold protein whirlin supports its anchoring of hair-bundle lateral links in cochlear hair cells. Sci. Rep. 10 (1), 16430. doi:10.1038/s41598-020-73158-1
Miles, A., Blair, C., Emili, A., and Tropepe, V. (2021). Usher syndrome type 1-associated gene, pcdh15b, is required for photoreceptor structural integrity in zebrafish. Dis. Models Mech. 14 (12), dmm048965. doi:10.1242/dmm.048965
Millán, J. M., Aller, E., Jaijo, T., Blanco-Kelly, F., Gimenez-Pardo, A., and Ayuso, C. (2011). An update on the genetics of usher syndrome. J. Ophthalmol. 2011 (1), 417217. doi:10.1155/2011/417217
Miller, K. A., Williams, L. H., Rose, E., Kuiper, M., Dahl, H.-H. M., and Manji, S. S. M. (2012). Inner ear morphology is perturbed in two novel mouse models of recessive deafness. PLOS ONE 7 (12), e51284. doi:10.1371/journal.pone.0051284
Moore, S. T., Nakamura, T., Nie, J., Solivais, A. J., Aristizábal-Ramírez, I., Ueda, Y., et al. (2023). Generating high-fidelity cochlear organoids from human pluripotent stem cells. Cell Stem Cell 30 (7), 950–961.e7. doi:10.1016/j.stem.2023.06.006
Morgan, C. P., Krey, J. F., Grati, M. h., Zhao, B., Fallen, S., Kannan-Sundhari, A., et al. (2016). PDZD7-MYO7A complex identified in enriched stereocilia membranes. eLife 5, e18312. doi:10.7554/eLife.18312
Moriarty, N., Gantner, C. W., Hunt, C. P., Ermine, C. M., Frausin, S., Viventi, S., et al. (2022). A combined cell and gene therapy approach for homotopic reconstruction of midbrain dopamine pathways using human pluripotent stem cells. Cell Stem Cell 29 (3), 434–448.e5. doi:10.1016/j.stem.2022.01.013
Mustapha, M., Beyer, L. A., Izumikawa, M., Swiderski, D. L., Dolan, D. F., Raphael, Y., et al. (2007). Whirler mutant hair cells have less severe pathology than shaker 2 or double mutants. J. Assoc. Res. Otolaryngology 8, 329–337. doi:10.1007/s10162-007-0083-x
Naz, S., Griffith, A., Riazuddin, S., Hampton, L., Battey Jr, J., Khan, S., et al. (2004). Mutations of ESPN cause autosomal recessive deafness and vestibular dysfunction. J. Med. Genet. 41 (8), 591–595. doi:10.1136/jmg.2004.018523
Ness, S. L., Ben-Yosef, T., Bar-Lev, A., Madeo, A. C., Brewer, C. C., Avraham, K. B., et al. (2003). Genetic homogeneity and phenotypic variability among Ashkenazi Jews with Usher syndrome type III. J. Med. Genet. 40 (10), 767–772. doi:10.1136/jmg.40.10.767
Nguyen, V. P., Song, J., Prieskorn, D., Zou, J., Li, Y., Dolan, D., et al. (2023a). USH2A gene mutations in rabbits lead to progressive retinal degeneration and hearing loss. Transl. Vis. Sci. Technol. 12 (2), 26. doi:10.1167/tvst.12.2.26
Nguyen, X.-T.-A., Thiadens, A. A., Fiocco, M., Tan, W., McKibbin, M., Klaver, C. C., et al. (2023b). Outcome of cataract surgery in patients with retinitis pigmentosa. Am. J. Ophthalmol. 246, 1–9. doi:10.1016/j.ajo.2022.10.001
Nicolson, T., Rüsch, A., Friedrich, R. W., Granato, M., Ruppersberg, J. P., and Nüsslein-Volhard, C. (1998). Genetic analysis of vertebrate sensory hair cell mechanosensation: the zebrafish circler mutants. Neuron 20 (2), 271–283. doi:10.1016/S0896-6273(00)80455-9
Nisenbaum, E., Thielhelm, T. P., Nourbakhsh, A., Yan, D., Blanton, S. H., Shu, Y., et al. (2022). Review of genotype-phenotype correlations in usher syndrome. Ear Hear. 43 (1), 1–8. doi:10.1097/aud.0000000000001066
Nonarath, H. J. T., Simpson, S. L., Slobodianuk, T. L., Collery, R. F., Dinculescu, A., and Link, B. A. (2024). The USH3A causative gene clarin1 functions in Müller glia to maintain retinal photoreceptors. bioRxiv. doi:10.1101/2024.02.29.582878
Ogun, O., and Zallocchi, M. (2014). Clarin-1 acts as a modulator of mechanotransduction activity and presynaptic ribbon assembly. J. Cell Biol. 207 (3), 375–391. doi:10.1083/jcb.201404016
Oshima, A., Jaijo, T., Aller, E., Millan, J. M., Carney, C., Usami, S., et al. (2008). Mutation profile of the CDH23 gene in 56 probands with Usher syndrome type I. Hum. Mutat. 29 (6), E37–E46. doi:10.1002/humu.20761
Ou, J., Tu, J., Zhang, H., Wu, K., Li, Y., Xu, H., et al. (2024). Pre-clinical study of RM-101, a novel AAV-based gene therapy for USH2A-related Retinitis Pigmentosa. Investigative Ophthalmol. Vis. Sci. 65 (7), 5337.
Overlack, N., Goldmann, T., Wolfrum, U., and Nagel-Wolfrum, K. (2012). Gene repair of an Usher syndrome causing mutation by zinc-finger nuclease mediated homologous recombination. Investigative Ophthalmol. Vis. Sci. 53 (7), 4140–4146. doi:10.1167/iovs.12-9812
Overlack, N., Maerker, T., Latz, M., Nagel-Wolfrum, K., and Wolfrum, U. (2008). SANS (USH1G) expression in developing and mature mammalian retina. Vis. Res. 48 (3), 400–412. doi:10.1016/j.visres.2007.08.021
Palma, F. D., Holme, R. H., Bryda, E. C., Belyantseva, I. A., Pellegrino, R., Kachar, B., et al. (2001). Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat. Genet. 27 (1), 103–107. doi:10.1038/83660
Pan, B., Askew, C., Galvin, A., Heman-Ackah, S., Asai, Y., Indzhykulian, A. A., et al. (2017). Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol. 35 (3), 264–272. doi:10.1038/nbt.3801
Panagiotopoulos, A.-L., Karguth, N., Pavlou, M., Böhm, S., Gasparoni, G., Walter, J., et al. (2020). Antisense oligonucleotide-and CRISPR-Cas9-mediated rescue of mRNA splicing for a deep intronic CLRN1 mutation. Mol. Therapy-Nucleic Acids 21, 1050–1061. doi:10.1016/j.omtn.2020.07.036
Pendse, N. D., Lamas, V., Pawlyk, B. S., Maeder, M. L., Chen, Z.-Y., Pierce, E. A., et al. (2019). In vivo assessment of potential therapeutic approaches for USH2A-associated diseases. Adv. Exp. Med. Biol. 1185, 91–96. doi:10.1007/978-3-030-27378-1_15
Peng, Y.-W., Zallocchi, M., Wang, W.-M., Delimont, D., and Cosgrove, D. (2011). Moderate light-induced degeneration of rod photoreceptors with delayed transducin translocation in shaker1 mice. Investigative Ophthalmol. Vis. Sci. 52 (9), 6421–6427. doi:10.1167/iovs.10-6557
Pepermans, E., Michel, V., Goodyear, R., Bonnet, C., Abdi, S., Dupont, T., et al. (2014). The CD2 isoform of protocadherin-15 is an essential component of the tip-link complex in mature auditory hair cells. EMBO Mol. Med. 6 (7), 984–992. doi:10.15252/emmm.201403976
Peter, V. G., Quinodoz, M., Sadio, S., Held, S., Rodrigues, M., Soares, M., et al. (2021). New clinical and molecular evidence linking mutations in ARSG to Usher syndrome type IV. Hum. Mutat. 42 (3), 261–271. doi:10.1002/humu.24150
Phillips, J. B., Blanco-Sanchez, B., Lentz, J. J., Tallafuss, A., Khanobdee, K., Sampath, S., et al. (2011). Harmonin (Ush1c) is required in zebrafish Müller glial cells for photoreceptor synaptic development and function. Dis. Models Mech. 4 (6), 786–800. doi:10.1242/dmm.006429
Phillips, J. B., Mauriac, S. A., Huang, Y.-H., Wegner, J., Westerfield, M., Yu, T., et al. (2024). Testing a dual-exon skip treatment for usher syndrome type 2A. Investigative Ophthalmol. Vis. Sci. 65 (7), 5329.
Pittenger, M. F., Discher, D. E., Péault, B. M., Phinney, D. G., Hare, J. M., and Caplan, A. I. (2019). Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regen. Med. 4 (1), 22. doi:10.1038/s41536-019-0083-6
Plantinga, R. F., Kleemola, L., Huygen, P. L. M., Joensuu, T., Sankila, E.-M., Pennings, R. J. E., et al. (2005). Serial audiometry and speech recognition findings in Finnish usher syndrome type III patients. Audiology Neurotol. 10 (2), 79–89. doi:10.1159/000083363
Prosser, H. M., Rzadzinska, A. K., Steel, K. P., and Bradley, A. (2008). Mosaic complementation demonstrates a regulatory role for myosin VIIa in actin dynamics of stereocilia. Mol. Cell. Biol. 28 (5), 1702–1712. doi:10.1128/MCB.01282-07
Puffenberger, E. G., Jinks, R. N., Sougnez, C., Cibulskis, K., Willert, R. A., Achilly, N. P., et al. (2012). Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLOS ONE 7 (1), e28936. doi:10.1371/journal.pone.0028936
Qi, J., Xu, L., Zeng, F.-G., and Chai, R. (2025). OTOF-related gene therapy: a new way but a long road ahead. Lancet 405 (10481), 777–779. doi:10.1016/S0140-6736(25)00248-X
Qiu, S., Zhang, X., Zhang, L., Liu, Z., Wang, L., Jin, Z.-B., et al. (2023). Generation of the induced pluripotent stem cell line SFMUi001-A from a patient with usher syndrome type 2 caused by biallelic variants in the USH2A gene. Stem Cell Res. 69, 103101. doi:10.1016/j.scr.2023.103101
Qiu, Y., Kenana, R., Beharry, A., Wilhelm, S. D. P., Hsu, S. Y., Siu, V. M., et al. (2022). Histidine supplementation can escalate or rescue HARS deficiency in a Charcot–Marie–Tooth disease model. Hum. Mol. Genet. 32 (5), 810–824. doi:10.1093/hmg/ddac239
Radu, M., Brănişteanu, D. C., Pirvulescu, R. A., Dumitrescu, O. M., Ionescu, M. A., and Zemba, M. (2024). Exploring stem-cell-based therapies for retinal regeneration. Life 14 (6), 668. doi:10.3390/life14060668
Ratnam, K., Västinsalo, H., Roorda, A., Sankila, E.-M. K., and Duncan, J. L. (2013). Cone structure in patients with usher syndrome type III and mutations in the clarin 1 gene. JAMA Ophthalmol. 131 (1), 67–74. doi:10.1001/2013.jamaophthalmol.2
Redfield, S. E., Mauriac, S. A., Géléoc, G. S., and Shearer, A. E. (2025). A genomic analysis of usher syndrome: population-scale prevalence and therapeutic targets. Am. J. Med. Genet. Part C Seminars Med. Genet., 2025.02.27.25323008. doi:10.1101/2025.02.27.25323008
Reiners, J., Marker, T., Jurgens, K., Reidel, B., and Wolfrum, U. (2005). Photoreceptor expression of the Usher syndrome type 1 protein protocadherin 15 (USH1F) and its interaction with the scaffold protein harmonin (USH1C). Mol. Vis. 12 (11), 347–355.
Reiners, J., Reidel, B., El-Amraoui, A., Boeda, B., Huber, I., Petit, C., et al. (2003). Differential distribution of harmonin isoforms and their possible role in usher-1 protein complexes in mammalian photoreceptor cells. Investigative Ophthalmol. Vis. Sci. 44 (11), 5006–5015. doi:10.1167/iovs.03-0483
Reurink, J., Weisschuh, N., Garanto, A., Dockery, A., van den Born, L. I., Fajardy, I., et al. (2023). Whole genome sequencing for USH2A-associated disease reveals several pathogenic deep-intronic variants that are amenable to splice correction. HGG Adv. 4 (2), 100181. doi:10.1016/j.xhgg.2023.100181
Rhodes, C. R., Hertzano, R., Fuchs, H., Bell, R. E., de Angelis, M. H., Steel, K. P., et al. (2004). A Myo7a mutation cosegregates with stereocilia defects and low-frequency hearing impairment. Mamm. Genome 15 (9), 686–697. doi:10.1007/s00335-004-2344-x
Riazuddin, S., Belyantseva, I. A., Giese, A. P. J., Lee, K., Indzhykulian, A. A., Nandamuri, S. P., et al. (2012). Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat. Genet. 44 (11), 1265–1271. doi:10.1038/ng.2426
Richardson, G. P., and Petit, C. (2019). Hair-bundle links: genetics as the gateway to function. Cold Spring Harb. Perspect. Med. 9 (12), a033142. doi:10.1101/cshperspect.a033142
Riera, M., Patel, A., Corcostegui, B., Chang, S., Corneo, B., Sparrow, J. R., et al. (2019). Generation of an induced pluripotent stem cell line (FRIMOi002-A) from a retinitis pigmentosa patient carrying compound heterozygous mutations in USH2A gene. Stem Cell Res. 35, 101386. doi:10.1016/j.scr.2019.101386
Rinaldi, C., and Wood, M. J. (2018). Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 14 (1), 9–21. doi:10.1038/nrneurol.2017.148
Rinchik, E. M., and Carpenter, D. A. (1999). N-ethyl-N-nitrosourea mutagenesis of a 6-to 11-cM subregion of the Fah–Hbb interval of mouse chromosome 7: completed testing of 4557 gametes and deletion mapping and complementation analysis of 31 mutations. Genetics 152 (1), 373–383. doi:10.1093/genetics/152.1.373
Rouget-Quermalet, V., Giustiniani, J., Marie-Cardine, A., Beaud, G., Besnard, F., Loyaux, D., et al. (2006). Protocadherin 15 (PCDH15): a new secreted isoform and a potential marker for NK/T cell lymphomas. Oncogene 25 (19), 2807–2811. doi:10.1038/sj.onc.1209301
Ryu, J., Statz, J. P., Chan, W., Burch, F. C., Brigande, J. V., Kempton, B., et al. (2022). CRISPR/Cas9 editing of the MYO7A gene in rhesus macaque embryos to generate a primate model of Usher syndrome type 1B. Sci. Rep. 12 (1), 10036. doi:10.1038/s41598-022-13689-x
Sadeghi, A. M., Eriksson, K., Kimberling, W. J., Sjöström, A., and Möller, C. (2006). Longterm visual prognosis in Usher syndrome types 1 and 2. Acta Ophthalmol. Scand. 84 (4), 537–544. doi:10.1111/j.1600-0420.2006.00675.x
Sadeghi, M., Cohn, E. S., Kimberling, W. J., Tranebjærg, L., and Möller, C. (2005). Audiological and vestibular features in affected subjects with USH3: a genotype/phenotype correlation. Int. J. Audiology 44 (5), 307–316. doi:10.1080/14992020500060610
Sahly, I., Dufour, E., Schietroma, C., Michel, V., Bahloul, A., Perfettini, I., et al. (2012). Localization of Usher 1 proteins to the photoreceptor calyceal processes, which are absent from mice. J. Cell Biol. 199 (2), 381–399. doi:10.1083/jcb.201202012
Sanjurjo-Soriano, C., Erkilic, N., Baux, D., Mamaeva, D., Hamel, C. P., Meunier, I., et al. (2020). Genome editing in patient iPSCs corrects the most prevalent USH2A mutations and reveals intriguing mutant mRNA expression profiles. Mol. Ther. Methods Clin. Dev. 17, 156–173. doi:10.1016/j.omtm.2019.11.016
Sanjurjo-Soriano, C., Erkilic, N., Manes, G., Dubois, G., Hamel, C. P., Meunier, I., et al. (2018a). Generation of a human iPSC line, INMi002-A, carrying the most prevalent USH2A variant associated with Usher syndrome type 2. Stem Cell Res. 33, 247–250. doi:10.1016/j.scr.2018.11.007
Sanjurjo-Soriano, C., Erkilic, N., Manes, G., Dubois, G., Hamel, C. P., Meunier, I., et al. (2018b). Generation of an iPSC line, INMi001-A, carrying the two most common USH2A mutations from a compound heterozygote with non-syndromic retinitis pigmentosa. Stem Cell Res. 33, 228–232. doi:10.1016/j.scr.2018.11.004
Sanjurjo-Soriano, C., Erkilic, N., Vache, C., Dubois, G., Roux, A.-F., Meunier, I., et al. (2022). Generation of a human iPSC line, INMi005-A, from a patient with non-syndromic USH2A-associated retinitis pigmentosa. Stem Cell Res. 60, 102738. doi:10.1016/j.scr.2022.102738
Schäfer, J., Wenck, N., Janik, K., Linnert, J., Stingl, K., Kohl, S., et al. (2023). The Usher syndrome 1C protein harmonin regulates canonical Wnt signaling. Front. Cell Dev. Biol. 11, 1130058. doi:10.3389/fcell.2023.1130058
Schellens, R. T., Broekman, S., Peters, T., Graave, P., Malinar, L., Venselaar, H., et al. (2023). A protein domain-oriented approach to expand the opportunities of therapeutic exon skipping for USH2A-associated retinitis pigmentosa. Mol. Ther. Nucleic Acids 32, 980–994. doi:10.1016/j.omtn.2023.05.020
Schietroma, C., Parain, K., Estivalet, A., Aghaie, A., Boutet de Monvel, J., Picaud, S., et al. (2017). Usher syndrome type 1–associated cadherins shape the photoreceptor outer segment. J. Cell Biol. 216 (6), 1849–1864. doi:10.1083/jcb.201612030
Schott, J. W., Huang, P., Morgan, M., Nelson-Brantley, J., Koehler, A., Renslo, B., et al. (2023). Third-generation lentiviral gene therapy rescues function in a mouse model of Usher 1B. Mol. Ther. 31 (12), 3502–3519. doi:10.1016/j.ymthe.2023.10.018
Schrauwen, I., Chakchouk, I., Acharya, A., Liaqat, K., and Bamshad, M. J. (2018). Novel digenic inheritance of PCDH15 and USH1G underlies profound non-syndromic hearing impairment. BMC Med. Genet. 19 (1), 122. doi:10.1186/s12881-018-0618-5
Schwander, M., Lopes, V., Sczaniecka, A., Gibbs, D., Lillo, C., Delano, D., et al. (2009). A novel allele of myosin VIIa reveals a critical function for the C-terminal FERM domain for melanosome transport in retinal pigment epithelial cells. J. Neurosci. 29 (50), 15810–15818. doi:10.1523/jneurosci.4876-09.2009
Sekerková, G., Zheng, L., Loomis, P. A., Changyaleket, B., Whitlon, D. S., Mugnaini, E., et al. (2004). Espins are multifunctional actin cytoskeletal regulatory proteins in the microvilli of chemosensory and mechanosensory cells. J. Neurosci. 24 (23), 5445–5456. doi:10.1523/jneurosci.1279-04.2004
Sekiya, T., and Holley, M. C. (2021). Cell transplantation to restore lost auditory nerve function is a realistic clinical opportunity. Cell Transplant. 30, 09636897211035076. doi:10.1177/09636897211035076
Self, T., Mahony, M., Fleming, J., Walsh, J., Brown, S. D., and Steel, K. P. (1998). Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development 125 (4), 557–566. doi:10.1242/dev.125.4.557
Senften, M., Schwander, M., Kazmierczak, P., Lillo, C., Shin, J.-B., Hasson, T., et al. (2006). Physical and functional interaction between protocadherin 15 and myosin VIIa in mechanosensory hair cells. J. Neurosci. 26 (7), 2060–2071. doi:10.1523/jneurosci.4251-05.2006
Sethna, S., Scott, P. A., Giese, A. P. J., Duncan, T., Jian, X., Riazuddin, S., et al. (2021). CIB2 regulates mTORC1 signaling and is essential for autophagy and visual function. Nat. Commun. 12 (1), 3906. doi:10.1038/s41467-021-24056-1
Shearer, A. E., Shen, J., Amr, S., Morton, C. C., and Smith, R. J.Newborn Hearing Screening Working Group of the National Coordinating Center for the Regional Genetics Networks (2019). A proposal for comprehensive newborn hearing screening to improve identification of deaf and hard-of-hearing children. Genet. Med. 21 (11), 2614–2630. doi:10.1038/s41436-019-0563-5
Shirley, J. L., De Jong, Y. P., Terhorst, C., and Herzog, R. W. (2020). Immune responses to viral gene therapy vectors. Mol. Ther. 28 (3), 709–722. doi:10.1016/j.ymthe.2020.01.001
Siles, L., and Pomares, E. (2025). Rescue of the disease-associated phenotype in CRISPR-corrected hiPSCs as a therapeutic approach for inherited retinal dystrophies. Mol. Ther. Nucleic Acids 36, 102482. doi:10.1016/j.omtn.2025.102482
Slijkerman, R. W. N., Vaché, C., Dona, M., García-García, G., Claustres, M., Hetterschijt, L., et al. (2016). Antisense oligonucleotide-based splice correction for USH2A-associated retinal degeneration caused by a frequent deep-intronic mutation. Mol. Ther. - Nucleic Acids 5, e381. doi:10.1038/mtna.2016.89
Sloan-Heggen, C. M., Bierer, A. O., Shearer, A. E., Kolbe, D. L., Nishimura, C. J., Frees, K. L., et al. (2016). Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 135, 441–450. doi:10.1007/s00439-016-1648-8
Söllner, C., Rauch, G.-J., Siemens, J., Geisler, R., Schuster, S. C., Tübingen Screen Consortium, t., et al. (2004). Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature 428 (6986), 955–959. doi:10.1038/nature02484
Sorusch, N., Bauß, K., Plutniok, J., Samanta, A., Knapp, B., Nagel-Wolfrum, K., et al. (2017). Characterization of the ternary Usher syndrome SANS/ush2a/whirlin protein complex. Hum. Mol. Genet. 26 (6), 1157–1172. doi:10.1093/hmg/ddx027
Steinhart, M. R., van der Valk, W. H., Osorio, D., Serdy, S. A., Zhang, J., Nist-Lund, C., et al. (2023). Mapping oto-pharyngeal development in a human inner ear organoid model. Development 150 (19), dev201871. doi:10.1242/dev.201871
Stemerdink, M., Broekman, S., Peters, T., Kremer, H., de Vrieze, E., and van Wijk, E. (2023). Generation and characterization of a zebrafish model for ADGRV1-associated retinal dysfunction using CRISPR/Cas9 genome editing technology. Cells 12 (12), 1598. doi:10.3390/cells12121598
Su, T., Liang, L., Zhang, L., Wang, J., Chen, L., Su, C., et al. (2022). Retinal organoids and microfluidic chip-based approaches to explore the retinitis pigmentosa with USH2A mutations. Front. Bioeng. Biotechnol. 10, 939774. doi:10.3389/fbioe.2022.939774
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126 (4), 663–676. doi:10.1016/j.cell.2006.07.024
Tang, Z.-H., Chen, J.-R., Zheng, J., Shi, H.-S., Ding, J., Qian, X.-D., et al. (2016). Genetic correction of induced pluripotent stem cells from a deaf patient with MYO7A mutation results in morphologic and functional recovery of the derived hair cell-like cells. Stem Cells Transl. Med. 5 (5), 561–571. doi:10.5966/sctm.2015-0252
Tebbe, L., Mwoyosvi, M. L., Crane, R., Makia, M. S., Kakakhel, M., Cosgrove, D., et al. (2023). The usherin mutation c.2299delG leads to its mislocalization and disrupts interactions with whirlin and VLGR1. Nat. Commun. 14 (1), 972. doi:10.1038/s41467-023-36431-1
Tian, G., Zhou, Y., Hajkova, D., Miyagi, M., Dinculescu, A., Hauswirth, W. W., et al. (2009). Clarin-1, encoded by the Usher Syndrome III causative gene, forms a membranous microdomain: possible role of clarin-1 in organizing the actin cytoskeleton. J. Biol. Chem. 284 (28), 18980–18993. doi:10.1074/jbc.M109.003160
Toms, M., Dubis, A. M., de Vrieze, E., Tracey-White, D., Mitsios, A., Hayes, M., et al. (2020). Clinical and preclinical therapeutic outcome metrics for USH2A-related disease. Hum. Mol. Genet. 29 (11), 1882–1899. doi:10.1093/hmg/ddaa004
Trapani, I., Colella, P., Sommella, A., Iodice, C., Cesi, G., De Simone, S., et al. (2014). Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol. Med. 6 (2), 194–211. doi:10.1002/emmm.201302948
Tresenrider, A., Sridhar, A., Eldred, K. C., Cuschieri, S., Hoffer, D., Trapnell, C., et al. (2023). Single-cell sequencing of individual retinal organoids reveals determinants of cell-fate heterogeneity. Cell Rep. Methods 3 (8), 100548. doi:10.1016/j.crmeth.2023.100548
Tsai, S.C.-S., Lin, F.C.-F., Chang, K.-H., Li, M.-C., Chou, R.-H., Huang, M.-Y., et al. (2022). The intravenous administration of skin-derived mesenchymal stem cells ameliorates hearing loss and preserves cochlear hair cells in cisplatin-injected mice: SMSCs ameliorate hearing loss and preserve outer hair cells in mice. Hear. Res. 413, 108254. doi:10.1016/j.heares.2021.108254
Tucker, B. A., Mullins, R. F., Streb, L. M., Anfinson, K., Eyestone, M. E., Kaalberg, E., et al. (2013). Patient-specific iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa. eLife 2, e00824. doi:10.7554/eLife.00824
Ukaji, T., Takahashi-Shibata, M., Arai, D., Tsutsumi, H., Tajima, S., Akamatsu, W., et al. (2023). Generation and characterization of a human iPSC line (JUFMDOi007-A) from a patient with Usher syndrome due to mutation in USH2A. Stem Cell Res. 69, 103100. doi:10.1016/j.scr.2023.103100
Underhill, A., Webb, S., Grandi, F. C., Jeng, J.-Y., de Monvel, J. B., Plion, B., et al. (2025). MYO7A is required for the functional integrity of the mechanoelectrical transduction complex in hair cells of the adult cochlea. Proc. Natl. Acad. Sci. 122 (1), e2414707122. doi:10.1073/pnas.2414707122
Usher Syndrome Coalition (2025). Join the USH trust. Available online at: https://www.usher-syndrome.org/what-is-usher-syndrome/join-the-ush-trust/join-the-ush-trust.html (Accessed March 25, 2025).
Vaché, C., Besnard, T., Le Berre, P., García-García, G., Baux, D., Larrieu, L., et al. (2012). Usher syndrome type 2 caused by activation of an USH2A pseudoexon: implications for diagnosis and therapy. Hum. Mutat. 33 (1), 104–108. doi:10.1002/humu.21634
Vanniya, S. P., Srisailapathy, C. S., and Mohanram, R. K. (2018). The tip link protein Cadherin-23: from hearing loss to cancer. Pharmacol. Res. 130, 25–35. doi:10.1016/j.phrs.2018.01.026
van Wijk, E., van der Zwaag, B., Peters, T., Zimmermann, U., Te Brinke, H., Kersten, F. F., et al. (2006). The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum. Mol. Genet. 15 (5), 751–765. doi:10.1093/hmg/ddi490
Velde, H. M., Reurink, J., Held, S., Li, C. H. Z., Yzer, S., Oostrik, J., et al. (2022). Usher syndrome type IV: clinically and molecularly confirmed by novel ARSG variants. Hum. Genet. 141 (11), 1723–1738. doi:10.1007/s00439-022-02441-0
Verpy, E., Leibovici, M., Zwaenepoel, I., Liu, X.-Z., Gal, A., Salem, N., et al. (2000). A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat. Genet. 26 (1), 51–55. doi:10.1038/79171
Wafa, T. T., Faridi, R., King, K. A., Zalewski, C., Yousaf, R., Schultz, J. M., et al. (2021). Vestibular phenotype-genotype correlation in a cohort of 90 patients with Usher syndrome. Clin. Genet. 99 (2), 226–235. doi:10.1111/cge.13868
Wafi, A., and Mirnezami, R. (2018). Translational–omics: future potential and current challenges in precision medicine. Methods 151, 3–11. doi:10.1016/j.ymeth.2018.05.009
Wang, H., Chen, Y., Lv, J., Cheng, X., Cao, Q., Wang, D., et al. (2024). Bilateral gene therapy in children with autosomal recessive deafness 9: single-arm trial results. Nat. Med. 30 (7), 1898–1904. doi:10.1038/s41591-024-03023-5
Wang, H., Du, H., Ren, R., Du, T., Lin, L., Feng, Z., et al. (2023). Temporal and spatial assembly of inner ear hair cell ankle link condensate through phase separation. Nat. Commun. 14 (1), 1657. doi:10.1038/s41467-023-37267-5
Wang, L., Kempton, J. B., Jiang, H., Jodelka, F. M., Brigande, A. M., Dumont, R. A., et al. (2020). Fetal antisense oligonucleotide therapy for congenital deafness and vestibular dysfunction. Nucleic Acids Res. 48 (9), 5065–5080. doi:10.1093/nar/gkaa194
Wang, L., Zou, J., Shen, Z., Song, E., and Yang, J. (2011a). Whirlin interacts with espin and modulates its actin-regulatory function: an insight into the mechanism of Usher syndrome type II. Hum. Mol. Genet. 21 (3), 692–710. doi:10.1093/hmg/ddr503
Wang, Q.-J., Zhao, Y.-L., Rao, S.-Q., Guo, Y.-F., He, Y., Lan, L., et al. (2011b). Newborn hearing concurrent gene screening can improve care for hearing loss: a study on 14,913 Chinese newborns. Int. J. Pediatr. Otorhinolaryngology 75 (4), 535–542. doi:10.1016/j.ijporl.2011.01.016
Wang, Y., Li, J., Yao, X., Li, W., Du, H., Tang, M., et al. (2017). Loss of CIB2 causes profound hearing loss and abolishes mechanoelectrical transduction in mice. Front. Mol. Neurosci. 10, 401. doi:10.3389/fnmol.2017.00401
Watanabe, K., Nishio, S., Usami, S., Kumai, T., Katada, A., Ogasawara, N., et al. (2024). The prevalence and clinical features of MYO7A-related hearing loss including DFNA11, DFNB2 and USH1B. Sci. Rep. 14 (1), 8326. doi:10.1038/s41598-024-57415-1
Webb, S. W., Grillet, N., Andrade, L. R., Xiong, W., Swarthout, L., Della Santina, C. C., et al. (2011). Regulation of PCDH15 function in mechanosensory hair cells by alternative splicing of the cytoplasmic domain. Development 138 (8), 1607–1617. doi:10.1242/dev.060061
Whatley, M., Francis, A., Ng, Z. Y., Khoh, X. E., Atlas, M. D., Dilley, R. J., et al. (2020). Usher syndrome: genetics and molecular links of hearing loss and directions for therapy. Front. Genet. 11, 565216. doi:10.3389/fgene.2020.565216
Wilhelm, S. D. P., Kenana, R., Qiu, Y., O’Donoghue, P., and Heinemann, I. U. (2023). Towards a cure for HARS disease. Genes 14 (2), 254. doi:10.3390/genes14020254
Williams, D. S. (2008). Usher syndrome: animal models, retinal function of Usher proteins, and prospects for gene therapy. Vis. Res. 48 (3), 433–441. doi:10.1016/j.visres.2007.08.015
Williams, D. S., and Lopes, V. S. (2011). The many different cellular functions of MYO7A in the retina. Biochem. Soc. Trans. 39 (5), 1207–1210. doi:10.1042/BST0391207
Wong, E. Y. M., Khoh, X. E., Chen, S.-C., Lye, J., Leith, F. K., Zhang, D., et al. (2024). Generation of two induced pluripotent stem cell lines from an Usher syndrome type 1B patient with the homozygous c.496del MYO7A variant. Stem Cell Res. 79, 103492. doi:10.1016/j.scr.2024.103492
World Health Organisation (2025). Deafness and hearing loss. Available online at: https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (Accessed March 31, 2025).
Wu, C.-C., Tsai, C.-H., Hung, C.-C., Lin, Y.-H., Lin, Y.-H., Huang, F.-L., et al. (2017). Newborn genetic screening for hearing impairment: a population-based longitudinal study. Genet. Med. 19 (1), 6–12. doi:10.1038/gim.2016.66
Xu, L., Bolch, S. N., Santiago, C. P., Dyka, F. M., Akil, O., Lobanova, E. S., et al. (2020). Clarin-1 expression in adult mouse and human retina highlights a role of Müller glia in Usher syndrome. J. Pathology 250 (2), 195–204. doi:10.1002/path.5360
Yan, W., Chen, G., and Li, J. (2022). Structure of the Harmonin PDZ2 and coiled-coil domains in a complex with CDHR2 tail and its implications. FASEB J. 36 (7), e22425. doi:10.1096/fj.202200403RR
Yang, D., Nguyen, V. P., Wei, Z., Iannuzzi, M., Bisgaier, C., Chen, Y. E., et al. (2024a). Rabbit models of usher syndrome type 3A. Investigative Ophthalmol. Vis. Sci. 65 (7), 3105.
Yang, J., Lewis, G. P., Hsiang, C.-H., Menges, S., Luna, G., Cho, W., et al. (2024b). Amelioration of photoreceptor degeneration by intravitreal transplantation of retinal progenitor cells in rats. Int. J. Mol. Sci. 25 (15), 8060. doi:10.3390/ijms25158060
Yang, J., Liu, X., Zhao, Y., Adamian, M., Pawlyk, B., Sun, X., et al. (2010). Ablation of whirlin long isoform disrupts the USH2 protein complex and causes vision and hearing loss. PLoS Genet. 6 (5), e1000955. doi:10.1371/journal.pgen.1000955
Yang, J., Zou, J., and Zheng, T. (2014). A study of PDZD7 in the mouse retina. Investigative Ophthalmol. Vis. Sci. 55 (13), 4363.
Yildirim, A., Mozaffari-Jovin, S., Wallisch, A.-K., Schäfer, J., Ludwig, S. E. J., Urlaub, H., et al. (2021). SANS (USH1G) regulates pre-mRNA splicing by mediating the intra-nuclear transfer of tri-snRNP complexes. Nucleic Acids Res. 49 (10), 5845–5866. doi:10.1093/nar/gkab386
Yoshimura, H., Iwasaki, S., Nishio, S.-y., Kumakawa, K., Tono, T., Kobayashi, Y., et al. (2014). Massively parallel DNA sequencing facilitates diagnosis of patients with usher syndrome type 1. PLOS ONE 9 (3), e90688. doi:10.1371/journal.pone.0090688
Yoshimura, H., Oshikawa, C., Nakayama, J., Moteki, H., and Usami, S.-i. (2015). Identification of a novel CLRN1 gene mutation in usher syndrome type 3:two case reports. Ann. Otology, Rhinology Laryngology 124 (1_Suppl. l), 94S–99S. doi:10.1177/0003489415574069
Yoshimura, H., Yokota, S., and Takumi, Y. (2023). Treatment following triple-AAV delivery in mature murine model of human CDH23-associated hearing loss. Curr. Issues Mol. Biol. 45 (12), 9413–9421. doi:10.3390/cimb45120590
Yusuf, I. H., Garrett, A. M., MacLaren, R. E., and Issa, P. C. (2022). Retinal cadherins and the retinal cadherinopathies: current concepts and future directions. Prog. Retin. Eye Res. 90, 101038. doi:10.1016/j.preteyeres.2021.101038
Zallocchi, M., Binley, K., Lad, Y., Ellis, S., Widdowson, P., Iqball, S., et al. (2014). EIAV-based retinal gene therapy in the shaker1 mouse model for usher syndrome type 1B: development of UshStat. PloS one 9 (4), e94272. doi:10.1371/journal.pone.0094272
Zallocchi, M., Meehan, D. T., Delimont, D., Askew, C., Garige, S., Gratton, M. A., et al. (2009). Localization and expression of clarin-1, the Clrn1 gene product, in auditory hair cells and photoreceptors. Hear. Res. 255 (1), 109–120. doi:10.1016/j.heares.2009.06.006
Zaw, K., Carvalho, L. S., Aung-Htut, M. T., Fletcher, S., Wilton, S. D., Chen, F. K., et al. (2022). Pathogenesis and treatment of usher syndrome type IIA. Asia-Pacific J. Ophthalmol. (Philadelphia, Pa.) 11 (4), 369–379. doi:10.1097/APO.0000000000000546
Zaw, K., Wong, E. Y. M., Zhang, X., Zhang, D., Chen, S.-C., Thompson, J. A., et al. (2021). Generation of three induced pluripotent stem cell lines from a patient with Usher syndrome caused by biallelic c.949C > A and c.1256G > T mutations in the USH2A gene. Stem Cell Res. 50, 102129. doi:10.1016/j.scr.2020.102129
Zheng, L., Sekerková, G., Vranich, K., Tilney, L., Mugnaini, E., and Bartles, J. (2000). The deaf jerker mouse has a mutation in the gene encoding the espin actin-bundling proteins of hair cell stereocilia and lacks espins. Cell 102 (3), 377–385. doi:10.1016/s0092-8674(00)00042-8
Zheng, Q. Y., Scarborough, J. D., Zheng, Y., Yu, H., Choi, D., and Gillespie, P. G. (2012). Digenic inheritance of deafness caused by 8J allele of myosin-VIIA and mutations in other Usher I genes. Hum. Mol. Genet. 21 (11), 2588–2598. doi:10.1093/hmg/dds084
Zheng, Q. Y., Yan, D., Ouyang, X. M., Du, L. L., Yu, H., Chang, B., et al. (2004). Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans. Hum. Mol. Genet. 14 (1), 103–111. doi:10.1093/hmg/ddi010
Zhong, X., Gutierrez, C., Xue, T., Hampton, C., Vergara, M. N., Cao, L.-H., et al. (2014). Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 5 (1), 4047. doi:10.1038/ncomms5047
Zhou, S., Flamier, A., Abdouh, M., Tétreault, N., Barabino, A., Wadhwa, S., et al. (2015). Differentiation of human embryonic stem cells into cone photoreceptors through simultaneous inhibition of BMP, TGFβ and Wnt signaling. Development 142 (19), 3294–3306. doi:10.1242/dev.125385
Zhou, T., Benda, C., Duzinger, S., Huang, Y., Li, X., Li, Y., et al. (2011). Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. JASN 22 (7), 1221–1228. doi:10.1681/ASN.2011010106
Zhu, T., Wu, S., Sun, Z., Wei, X., Han, X., Zou, X., et al. (2021). Generation of two human induced pluripotent stem cell lines from patients with biallelic USH2A variants. Stem Cell Res. 55, 102502. doi:10.1016/j.scr.2021.102502
Zou, J., Luo, L., Shen, Z., Chiodo, V. A., Ambati, B. K., Hauswirth, W. W., et al. (2011). Whirlin replacement restores the formation of the USH2 protein complex in whirlin knockout photoreceptors. Investigative Ophthalmol. Vis. Sci. 52 (5), 2343–2351. doi:10.1167/iovs.10-6141
Zou, J., Mathur, P. D., Zheng, T., Wang, Y., Almishaal, A., Park, A. H., et al. (2015). Individual USH2 proteins make distinct contributions to the ankle link complex during development of the mouse cochlear stereociliary bundle. Hum. Mol. Genet. 24 (24), 6944–6957. doi:10.1093/hmg/ddv398
Zou, J., Zheng, T., Ren, C., Askew, C., Liu, X.-P., Pan, B., et al. (2013). Deletion of PDZD7 disrupts the Usher syndrome type 2 protein complex in cochlear hair cells and causes hearing loss in mice. Hum. Mol. Genet. 23 (9), 2374–2390. doi:10.1093/hmg/ddt629
Zurita-Díaz, F., Ortuño-Costela, M. C., Moreno-Izquierdo, A., Galbis, L., Millán, J. M., Ayuso, C., et al. (2018). Establishment of a human iPSC line, IISHDOi004-A, from a patient with Usher syndrome associated with the mutation c.2276G>T; p.Cys759Phe in the USH2A gene. Stem Cell Res. 31, 152–156. doi:10.1016/j.scr.2018.08.002
Keywords: Usher syndrome, hearing loss, inner ear, hair cell, gene therapy
Citation: Leith FK, Lye J, Delaney DS, McLenachan S, Chen FK, Atlas MD and Wong EYM (2025) Current approaches for Usher syndrome disease models and developing therapies. Front. Cell Dev. Biol. 13:1547523. doi: 10.3389/fcell.2025.1547523
Received: 18 December 2024; Accepted: 29 May 2025;
Published: 20 June 2025.
Edited by:
Erkan Kiris, Middle East Technical University, TürkiyeReviewed by:
Armel Hervé Nwabo Kamdje, University of Garoua, CameroonKshitiz Raj Shrestha, Independent Researcher, Kathmandu, Nepal
Alessandro Castiglione, Örebro University Hospital, Sweden
Copyright © 2025 Leith, Lye, Delaney, McLenachan, Chen, Atlas and Wong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elaine Y. M. Wong, ZWxhaW5lLndvbmdAZWFyc2NpZW5jZS5vcmcuYXU=
†These authors have contributed equally to this work