 Valentina Rapozzi
Valentina Rapozzi Clara Comuzzi
Clara Comuzzi Eros Di Giorgio
Eros Di Giorgio Luigi E. Xodo
Luigi E. Xodo- 1Department of Medicine, Laboratory of Biochemistry, University of Udine, Udine, Italy
- 2Department of Agricultural Food Environmental and Animal Science, University of Udine, Udine, Italy
Cancer cells are subject to metabolic reprogramming, which leads to a sustained production of reactive oxygen species (ROS). Increased oxidative stress contributes to genomic instability and promotes malignant transformation. To counteract excessive ROS levels, cells activate nuclear factor erythroid 2–related factor 2 (NRF2), a key regulator of redox homeostasis that coordinates the transcription of a wide range of antioxidant and cytoprotective genes. This review examines the metabolic adaptations controlled by the KRAS–NRF2 axis under oxidative stress conditions. In addition, we highlight a novel function of NRF2 in regulating the expression of NOS2 by binding to a DNA enhancer element, thereby modulating the production of reactive nitrogen species (RNS). Finally, we discuss novel molecular strategies aimed at disrupting adaptive antioxidant responses in cancer cells and provide insights into combinatorial therapeutic approaches targeting redox balance in cancer.
1 Introduction
Pancreatic cancer is an extremely aggressive and deadly type of cancer that originates in the pancreas: an organ that lies behind the stomach and plays a crucial role in digestion and blood sugar regulation (Park et al., 2021). Among the different types of pancreatic cancer, pancreatic ductal adenocarcinoma (PDAC) is the most common, originating from the exocrine cells responsible for the production of digestive enzymes (Back et al., 2022). PDAC accounts for about 90% of all pancreatic cancers (exocrine and neuroendocrine cancers) and develops from the epithelial cells lining the ducts that transport digestive enzymes to the small intestine. PDAC is characterized by rapid progression and a high propensity to metastasize to nearby organs such as the liver and lungs (Ho et al., 2021) and is often only diagnosed at an advanced stage as it is asymptomatic in early stages. This late detection combined with resistance to conventional therapies contributes to the poor prognosis, with a 5-year survival rate of only ∼10%. It is predicted that this disease will be the second most common cause of cancer-related death in Western countries by 2030 (He et al., 2024). Somatic mutations play a key role in the development of PDAC, and KRAS is the most commonly mutated gene and found in over 90% of cases (Lennerz and Stenzinger, 2015). Mutations in KRAS lead to uncontrolled cell growth and contribute significantly to the progression of pancreatic cancer. Other frequently mutated genes are TP53 (∼50–75% of cases), CDKN2A (p16), SMAD4 (in ∼50% of cases), BRCA2 and ATM (Hahn et al., 1996; Lai et al., 2021; Schutte et al., 1997; Matrone et al., 2004).
A characteristic feature of PDAC cells is their increased oxidative stress compared to normal cells (Hayes et al., 2020). This results from an imbalance between the production of reactive oxygen species (ROS) and nitrogen oxide species (RNS) and the detoxification capacity of the cells. In cancer cells, mitochondrial dysfunction in the electron transport chain (ETC.) is the main cause that leads to an increase in ROS production (Hardie et al., 2017; Sarwar et al., 2022). Oncogenes such as KRAS and MYC also drive metabolic changes that further increase ROS levels. In addition, hypoxic regions in tumours are exposed to metabolic changes that also increase ROS production. ROS levels are mainly regulated by NRF2, the main transcription factor that controls the cell’s antioxidant defence system (Ma, 2013). Dysregulation of NRF2 in cancer cells can create a delicate balance where ROS levels remain high enough to support tumour growth and survival but still within the cell’s tolerance threshold (Jung et al., 2018; Osman et al., 2023; Wu et al., 2020) This review examines the intricate interplay between ROS, oncogenic KRAS and NRF2 in cancer, with a focus on metabolic reprogramming that occurs under oxidative stress conditions. This may provide valuable insights for the rational design of new therapeutic strategies for this disease, which tends to be refractory to conventional treatments.
2 Sources of oxidative and nitrosative stress
2.1 Sources of ROS in cancer cells
The mitochondrial, ETC., and nicotinamide dinucleotide phosphate (NADPH) oxidases (NOX) are the main sources of ROS in the cell (Figures 1A,B) (Skonieczna et al., 2017). In addition, cytochrome P450 and xanthine oxidase, that transfer electrons from NADPH to O2 via FAD and heme cofactors, produce superoxide (•O2−) and hydrogen peroxide (H2O2) (Parvez et al., 2018). In the ETC, coenzyme Q (CoQ) plays a central role in the generation of the superoxide anion (•O2−). This occurs because during the transport of electrons from NADH/FADH2 to oxygen, some of them escape when the semiquinone CoQ• accidently transfers its electron to O2 and forms •O2−, a reactive but relatively short-lived oxygen radical (Sohal and Forster, 2007). ROS are also generated by mitochondrial dysfunction induced by mutated KRAS. For example, KRAS G12V disrupts mitochondrial function, reducing oxygen consumption and increasing ROS production (Hu et al., 2012). Downregulation of NDUFAF1, a factor for mitochondrial complex I assembly, has been linked to reduced mitochondrial respiration in KRAS-related cancers (Wang et al., 2015). Mitochondrial production of •O2− is estimated to be <0.2% of the total O2 consumed by the organelle (St-Pierre et al., 2002). Superoxide is rapidly detoxified by mitochondrial superoxide dismutase (SOD) to hydrogen peroxide (2 •O2−+2H+ → H2O2 + O2) or can cross the mitochondrial membrane via VDAC, a voltage-dependent anion channel (Tikunov et al., 2010; Madesh and Hajnóczky G., 2001). Hydrogen peroxide easily enters the cytosol via the membrane aquaporin (Tafani et al., 2016), but can also be converted into H2O and O2 by catalase (2 H2O2 → 2 H2O+ O2). The superoxide anion can also react through non-enzymatic Haber-Weiss and Fenton reactions to form hydroxyl radicals, the most reactive ROS (Kehrer JP, 2000; Thomas et al., 2009) (Figure 1C). It has been demonstrated that hypoxic conditions exacerbate oxidative stress mainly by disrupting mitochondrial function and increasing ROS production (D’Aiuto et al., 2022). The exact mechanisms by which hypoxia increases ROS levels are still unclear, but there is evidence that hypoxia can increase ROS production by impairing complexes I, II and III of the, ETC (Kondoh et al., 2013; Wang et al., 2007). In addition, cancer cells often exhibit impaired antioxidant defence mechanisms, e.g., reduced glutathione levels or decreased activity of SOD and catalase, making them more susceptible to oxidative damage (Niu et al., 2021). The neutrophils and macrophages present in the tumour microenvironment are also a source of ROS (Wu et al., 2020; Wang et al., 2021).
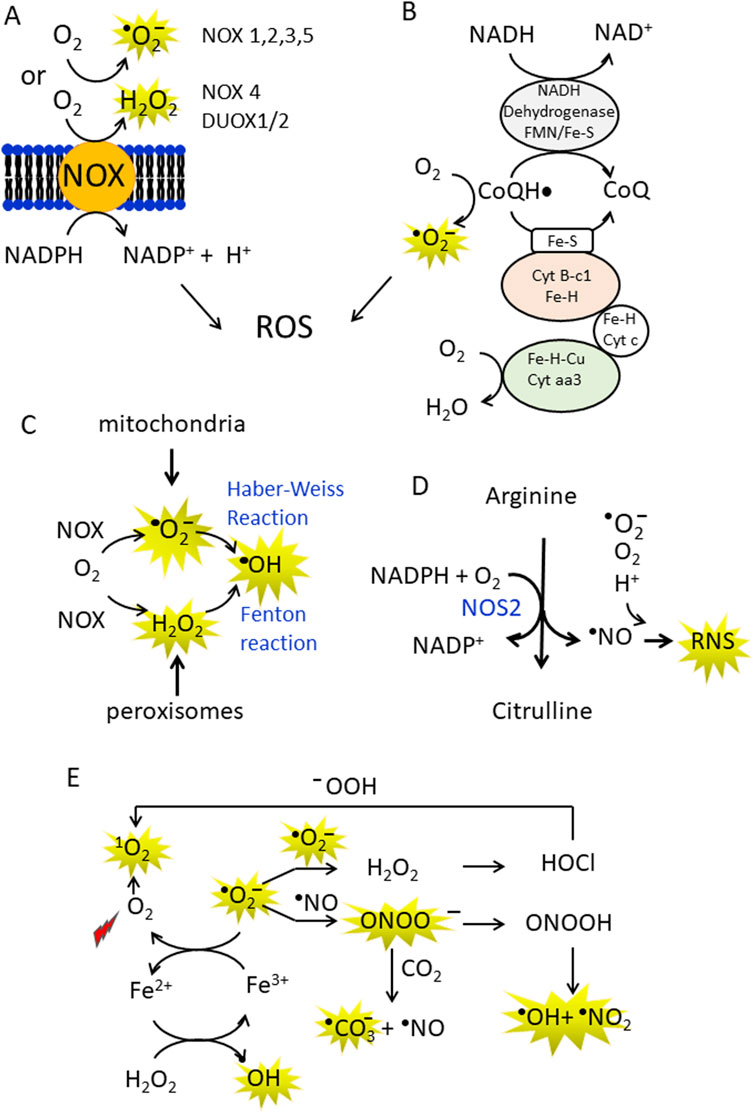
Figure 1. (A,B) Production of superoxide anions (•O2¯) and hydrogen peroxide (H2O2) by NADPH oxidases (NOX enzymes) and CoQ in the mitochondrial electron transport chain (ETC.). In the ETC some electrons are inadvertently transferred from CoQH• to O2 in the mitochondria to generate •O2¯; (C) •O2¯ can be non-enzymatically converted to the more reactive hydroxyl radical (•OH) in the Haber-Weiss reaction. In addition, metal ions such as Fe2+, Cu+ can act as single electron donors in the Fenton reaction to give •OH; (D) Arginine is the substrate of NOS2, which converts arginine to citrulline, releasing •NO and RNS; (E) A conversion between ROS and RNS occurs in the cell. At high concentrations, •NO can combine non-enzymatically with •O2¯ and form peroxynitrite (ONOO¯). Peroxynitrite is a strong oxidising agent that is stable and can diffuse through membranes and interact with proteins (methionine and -SH groups). It can also split into the hydroxyl radical and nitrogen dioxide (NO2). In addition, ONOO¯ can interact with CO2 to form the carbonate anion and •NO. The superoxide anion can be converted into H2O2and O2 by spontaneous or enzymatically controlled dismutation. Hydrogen peroxide via Fenton reaction is transformed in •OH or to hypochlorous acid (HClO) by myeloperoxidase. HClO produces singlet oxygen, a strong oxidising agent, in the presence of hydroperoxides.
2.2 Cancer cells produce nitric oxide
Cancer cells produce nitric oxide (•NO) and reactive nitrogen species RNS (Xu et al., 2002). The main source of •NO in the cells are the nitric oxide synthases (Föstermann and Sessa, 2012) (Figure 1D). Of the three known isoforms, inducible NOS (iNOS or NOS2) can increase the aggressiveness of pancreatic cancer cells (Wang et al., 2016). NOS2 contributes to higher •NO production under inflammatory and hypoxic conditions (Franco et al., 2004). Elevated •NO levels are often associated with tumour progression, metastasis and chemoresistance. These levels are generally observed when iNOS is highly expressed due to inflammatory signalling (Fukumura et al., 2006). An important aspect of ROS/RNS is that they interconvert each other (Figure 1E). The superoxide anion produced by the NOX enzymes and the ETC can rapidly dismutate spontaneously or enzymatically to H2O2 and O2. The spontaneous dismutation has a rate constant of 105 M-1s-1 (Sheng et al., 2014), but the enzymatic reaction is four orders of magnitude higher and therefore extremely efficient, with a kinetic constant comparable to the diffusion rate. However, superoxide can react with •NO at a high rate (1010 M-1s-1) to generate peroxynitrite ONOO¯, a powerful oxidising agent (Fridovich, 1983). Although this RNS species intrinsically decays to •OH and •NO2 (Radi, 2013a; Radi, 2013b), it can modify proteins, oxidise thiols and react with fatty acids to generate reactive electrophilic species (Bartesaghi and Radi, 2018). The peroxynitrite species can undergo a one-electron reaction with CO2 to produce the carbonate •CO3− and •NO2 radicals (Denicola et al., 1996). Despite the carbonate radical is less oxidising than •OH, it reacts with amino acids with a high second-order rate constant (106–108 M-1s-1) (Bonini and Augusto, 2001). Finally, H2O2 is converted to HOCl in neutrophils by myeloperoxidase. HOCl can react with the peroxide ion to form singlet oxygen 1O2 (Onyango AN, 2016). In cancer cells, both ROS and RNS are important activators of cell signalling and in the next paragraph we will take a closer look at cell signalling mediated by ROS.
3 ROS-mediated cellular signalling in cancer cells
3.1 Enhanced levels of H2O2 stimulate cell proliferation
Due to its relatively longer half-life and higher diffusivity compared to superoxide and the hydroxyl radical (10−3 s for H2O2 compared to 10−9 s for •OH and 10−6 s for •O2−) (Rubio and Ceron, 2021), H2O2 is the most important ROS in cell signalling. Since H2O2 is generated from various sources, including •O2−dismutation, NOX and xanthine oxidase enzymes and fatty acid oxidation in peroxisomes, it is the most abundant ROS in cells, including pancreatic cancer cells (Liu et al., 2023). There is clear evidence that the increase of ROS in cancer cells stimulates cell growth and survival (Hayes et al., 2020; Storz, 2016). This occurs by inhibiting the activity of protein tyrosine phosphatases (PTPs), the phosphatase and tensin homologue (PTEN) and MAPK phosphatases, thereby activating the MAPK/ERK, PI3P/PKB/AkT and PKD1/NFkB signalling pathways (Moloney and Cotter, 2018; Song et al., 2005) (Figure 2A). These PTPs phosphatases (PTP1B, PTPN2, PTPN11 and PTEN) are characterised by an active site containing a thiolate group susceptible to oxidation. Elevated H2O2 concentrations, as in cancer cells, oxidise the cysteine thiolate to sulfenate (SOH), sulfinate (SO2H) or sulfonate (SO3H), depending on the H2O2 content and duration of the ROS exposure (Figure 2B). The oxidative modification of the cysteine residue in the catalytic site of the phosphatases leads to their inactivation and thus to an increase in MAPK/ERK, PI3P/PKB/AKT and PKD/NFkB signaling, which promotes cell growth and survival. In the oxidised state at sulfenate, the catalytic site can be further inactivated by forming a disulfide bridge with either reduced glutathione (GSH) or with another cysteine in the catalytic site, leading to an increase in growth pathways (Hayes et al., 2020). These modifications are reversible as the disulfide can be reversed by the antioxidant systems thioredoxin (TXN1)/thioredoxin reductase (TXNRD1) or sufiredoxin 1 (SRXN1) (Hayes et al., 2020). This leads to activation of phosphatases and blocking of the MAPK/ERK, PI3P/PKB/AKT and PKD/NFkB signalling (Figure 2B). However, it should be noted that increased ROS levels are beneficial to cells if the risk of ROS-induced death is controlled by antioxidant systems. In other words, the ROS content should not be too high, otherwise the cells will die by apoptosis or other types of death instead of proliferating. Therefore, tumour cells upregulate antioxidant transcription factors and reprogram the metabolism to increase the levels of NADPH and GSH. That ROS in non-toxic concentrations are beneficial for cancer growth was shown in the study by Song et al. (2018), which demonstrated that suppression of ROS by N-acetylcysteine (NAC) reduced lung carcinoma in a KRAS G12D-driven mouse model.
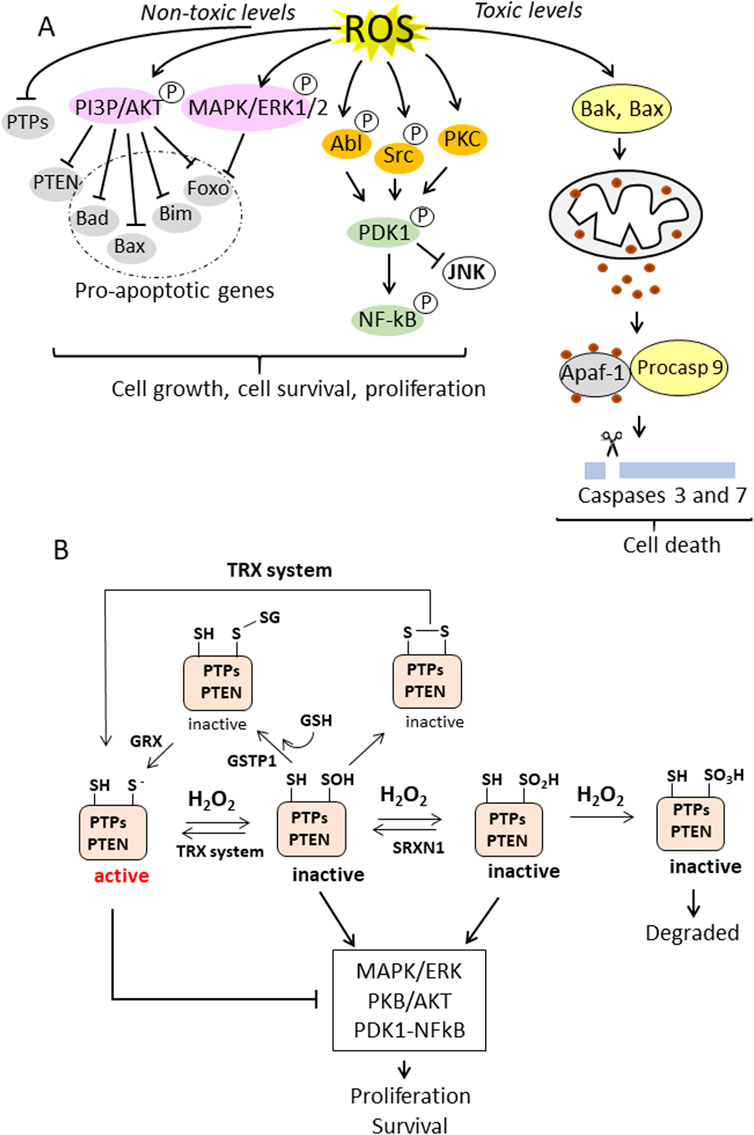
Figure 2. (A) ROS-induced cellular signalling in cancer cells. Non-toxic levels of ROS induce the phosphorylation and activation of PI3P/AKT and MAPK/ERK1/2 and the simultaneous inactivation of protein tyrosine phosphatases (PTPs) and lipid phosphatase, resulting in the inhibition of proapoptotic genes and the stimulation of cell growth and survival. ROS also activates the PDK1/NF-kB signalling pathway, leading to survival and proliferation. In contrast, overproduction of ROS leads to toxic oxidative stress, which activates Bak (Bcl-2 homologue antagonist/killer) and Bax (Bcl-2-associated X protein), which are pro-apoptotic members of the Bcl-2 protein family that regulate apoptosis and in particular the intrinsic (mitochrondrial) apoptosis pathway. Bax and Bak form pores in the outer mitochrondrial membrane that allow the release of cytochrome c into the cytoplasm. Cytochrome c induces the formation of the apoptosome, which activates caspase 9, which in turn activates executioner caspases 3 and 7; (B) PTPs and PTEN have a Cys residue in the active site in a thiolate state, which is susceptible to oxidation. The thiolate can be oxidised to sulfenate (-SOH), sulfinate (-SO2H) or sulfonate (-SO3H) depending on the H2O2 concentration. The Cys in the active site can also form disulfides, either with GSH, a reaction catalysed by GSTP1, or with another thiol in the active site, forming a disulphide bridge. These oxidative modifications inactivate the phosphatases and thereby enhance the MAPK/ERK and PKB/AKT pathways. The oxidative inactivation to sulfenate or sulfinate can be reversed by the antioxidant systems TXN1 or SRXN1. This restores phosphatase activity and promotes suppression of the MAPK/ERK and PKB/AKT signalling pathways. Similarly, Cys in the active site that have formed S-glutathionylation or have formed a disulfide bridge can be reversed by the enzyme glutaredoxin (GRX) (catalyses the reversible reduction of glutathione-protein mixed disulfide) or the TRX system. The oxidation of Cys in the active centre to a sulfonate state is irreversible (right) and the altered protein is degraded.
3.2 High levels of ROS activates apoptosis
Overproduction of ROS is toxic to the cell and induces cell death by apoptosis (Zhang et al., 2016). Superoxide •O2−produced in the, ETC., can impair mitochondrial functions (Kondoh et al., 2013) and cause mitochondrial membrane damage with leakage of cytochrome c from the intermembrane space. In the cytoplasm, cytochrome c induces the activation of caspase 9 and executioner caspases 3 and 7 (Figure 2A) (Walsh et al., 2008). These caspases cleave PARP-1 into two fragments of 24 and 89 kDa, which are no longer able to perform DNA repair functions. These conditions favour DNA fragmentation, chromatin condensation and membrane blebbing, the hallmarks of apoptotic cells. (Mills et al., 1998).
High levels of ROS can also activate the extrinsic apoptotic pathway, known as the death receptor pathway, which is triggered by the binding of signals from outside the cell such as tumour necrosis factor-alpha (TNF-α) or Fas ligand (FasL) to their respective death receptors (Fulda and Debatin, 2006). After TNF-α and FasL bind to the death receptors, procaspase-8 (or procaspase-10, depending on the cell type) is recruited to the death receptor. The recruited procaspase-8 is autocatalytically cleaved and converted to active caspase-8, which initiates the downstream cascade by direct cleavage and activation of executioner caspases (Kantari and Walczak, 2011).
In the next section, we will discuss how the KRAS-NRF2 axis controls ROS homeostasis and induces a metabolic shift in cancer cells.
4 The interplay between ROS, KRAS, and NRF2 in redox homeostasis and metabolic reprogramming in pancreatic cancer
4.1 NRF2 controls redox homeostasis
Cancer cells preferentially convert glucose into lactate even when sufficient oxygen is available: a phenomenon known as the Warburg effect (Vander Heiden et al., 2009). This metabolic shift is in stark contrast to normal cells, which under aerobic conditions mainly use oxidative phosphorylation (OXPHOS) to maximise ATP production. Since aerobic glycolysis yields only 2 ATP per glucose molecule compared to the 36 ATP generated by full glucose oxidation via glycolysis, tricarboxylic acid cycle and OXPHOS, it is upregulated in cancer cells to provide biomass for rapid proliferation and adapt to hypoxic conditions (Gatenby and Gillies, 2004; Zhou et al., 2022).
There is increasing evidence that metabolism in pancreatic cancer cells is controlled by the oncogenic KRAS, which activates several downstream signalling pathways. In particular, the PI3K/AKT signalling pathway increases glucose uptake (Hong et al., 2016; Fontana et al., 2024) and glycolysis (Hu et al., 2016), while the MAPK/ERK signalling pathway stimulates cell proliferation (Drosten and Barbacid, 2020) and modulates the expression of metabolic enzymes (Papa et al., 2019). In early tumour stages, the blood supply is reduced, leading to hypoxic conditions. Hypoxia inducible factor 1 (H1F-1a) is upregulated by the KRAS signalling pathway, allowing cancer cells to adapt to hypoxia. HIF-1α increases anaerobic glycolysis by stimulating the expression of glycolytic genes (Zhu et al., 2024). High glycolytic flux also drives the pentose phosphate pathway (PPP), which is critical for the production of ribose-5-phosphate (a precursor for nucleotide biosynthesis) and NADPH. NADPH maintains redox balance and serves as coenzyme for NOX enzymes, particularly NOX4, which is overexpressed in pancreatic cancer cells and is a major source of oxidative stress (•O2−) (Ju et al., 2017). The control of redox homeostasis is critical for cancer cell survival and is primarily regulated by the nuclear factor erythroid 2-related factor 2 (NRF2) (Ngo and Duennwald, 2022). NRF2 activates the expression of various antioxidant genes − including superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx) and haemoxygenase-1 (H O -1) − and prevents excessive accumulation of ROS in the cell. Low or moderate ROS levels act as signalling molecules that regulate cell growth, differentiation and survival, while excessive ROS damage DNA, proteins and lipids and impair cell viability (Srinivas et al., 2019) (Figure 2A). NRF2, a member of the NFE2 family of transcription factors, plays a crucial role in redox homeostasis. Under non-stressed conditions, NRF2 is sequestered in the cytoplasm by its inhibitor KEAP1. KEAP1 promotes the ubiquitination of NRF2 and subsequent proteasomal degradation. When cellular ROS increase, as in the case of cancer cells, KEAP1, which contains several cysteine residues that are essential for its interaction with NRF2, is oxidised. The oxidation of the cysteines alters the conformation of KEAP1 and reduces its affinity for NRF2 (Wakahayashi et al., 2004). This allows NRF2 to escape proteasomal degradation and migrate to the nucleus, where it acts as a transcription factor by binding to the antioxidant response element (ARE), thereby activating the expression of antioxidant genes. When the oxidative stress decreases, KEAP1 is restored to its reduced state and resumes its function as a target for NRF2 degradation. This dynamic regulation ensures that NRF2 activation is tightly controlled and only occurs during cellular stress. Mutations in KEAP1 have been identified in patients with PDAC (Lister et al., 2011). Loss of KEAP1 function leads to abnormal activation of NRF2, which promotes the progression of PDAC. Conversely, depletion of NRF2 has been shown to inhibit tumour progression in mouse models of PDAC and non-small cell lung cancer (Romero et al., 2017).
4.2 NRF2 causes a metabolic reprogramming in PDAC
NRF2 plays a central role also in metabolic reprogramming by activating glycolysis, PPP, glutathione synthesis, long-chain fatty acid and glutamine metabolism (Di Giorgio et al., 2023; Mitsuishi et al., 2012). Similarly, KRAS increases the expression of enzymes involved in glucose and glutamine metabolism in PDAC (Ying et al., 2012; Gaglio et al., 2011). This functional synergy between NRF2 and KRAS supports the hypothesis that these oncogenes jointly reprogramme the metabolism of cancer cells. Both genes are upregulated in pancreatic cancer cells (Figure 3A). Analysis of 175 tumour tissues from The Cancer Genome Atlas (TCGA) revealed a positive correlation between mRNA levels of KRAS and NRF2. Furthermore, PDAC patients with high expression of KRAS and NRF2 had significantly poorer survival than patients with lower expression (Di Giorgio et al., 2023) (Figure 3B). These results emphasise a functional link between KRAS and NRF2. Direct evidence for the regulation of NRF2 by KRAS comes from studies showing that oncogenic KRAS stimulates NRF2 expression (Ferino et al., 2020a; Tao et al., 2019; Yang et al., 2021). Overexpression of KRAS G12D or KRAS G12V in Panc-1 cells leads to increased NRF2 levels, while silencing KRAS with specific siRNA leads to reduced NRF2 expression (Di Giorgio et al., 2023). Another important observation is that both KRAS and NRF2 expression levels are modulated by H2O2, further linking their regulation to oxidative stress (Figure 3C). All these data are consistent with the hypothesis that the regulation of metabolism and oxidative stress management in pancreatic cancer cells is related to the coordinated activity of KRAS and NRF2. This concept is illustrated in the diagram in Figure 3D. Mutant KRAS G12D, which occurs in ∼90% of PDAC, causes a metabolic switch in favour of aerobic glycolysis (Warburg effect), the pentose phosphate pathway (PPP) and increased glutamine metabolism (Ying et al., 2012). This reprogramming activates NOX enzymes, disrupts mitochondrial function and increases the production of reactive oxygen species (ROS), which promotes cancer growth. However, an excess of ROS can be cytotoxic, so KRAS upregulates NRF2 via the PI3K/AKT and MAPK/ERK pathways. Under oxidative stress, NRF2 activates the expression of antioxidant genes and maintains ROS at levels that promote tumour proliferation. NRF2 also contributes to KRAS-driven metabolic reprogramming by promoting anabolic metabolism (Di Giorgio et al., 2023; Mitsuishi et al., 2012). For a comprehensive description of the metabolic reprogramming induced by the KRAS-NRF2 axis in conjunction with hypoxia we refer to reference Di Giorgio et al., 2023.
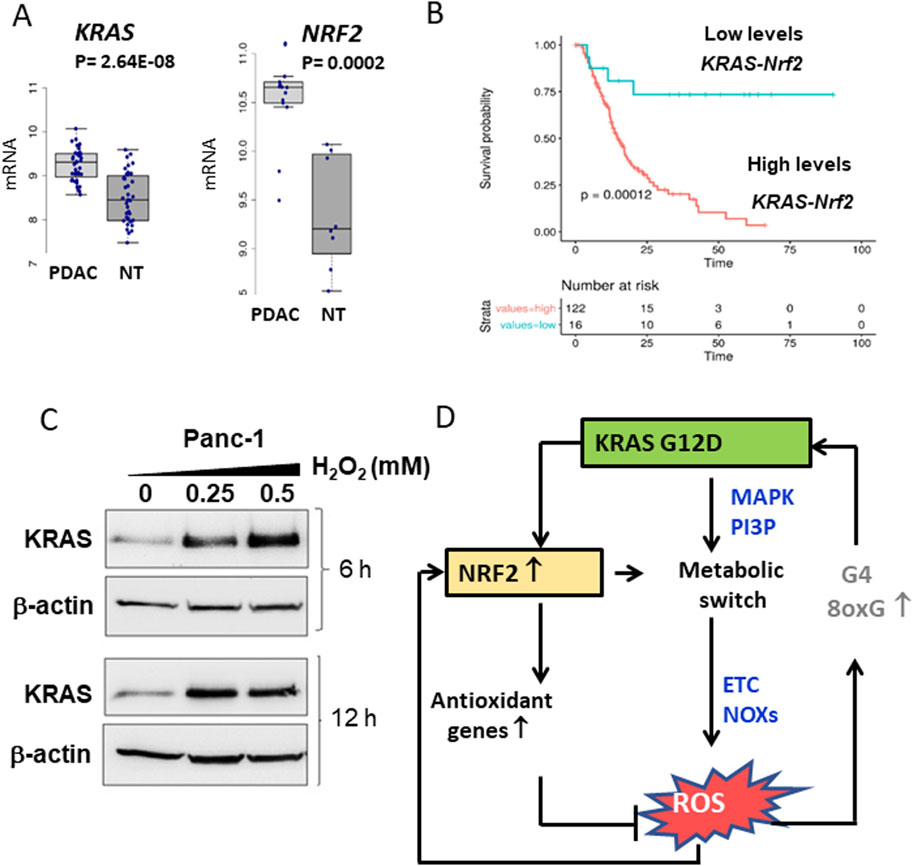
Figure 3. (A) Differential expression of KRAS and NRF2 between normal and tumor tissues in PDAC patients. Data obtained from GSE15471; (B) Kaplan-Meir plots show that patients with high levels of KRAS and NRF2 expression exhibit a lower survival probability than patients with low KRAS and NRF2 expression; (C) Levels of KRAS and actin in Panc-1 cells treated with increasing amounts of H2O2; (D) Scheme showing the relationships between KRAS, NRF2 and ROS in pancreatic cancer cells. Panels A,B,C adapted with permission (iScience 2023, 26, 108566).
4.3 Mechanism by which ROS upregulate KRAS
Interestingly, ROS themselves stimulate the expression of both KRAS and NRF2. In the case of KRAS, the mechanism of ROS-mediated upregulation involves a G-rich promoter sequence upstream of the transcription start site (TSS). This region contains two G-quadruplex (G4) motifs that form folded secondary structures that recruit transcription factors (MAZ, PARP1 and hnRNPA1) (Cogoi et al., 2008). These G4 motifs are highly susceptible to oxidative modifications, in particular by the oxidation of guanine to 8-oxoguanine (Cogoi et al., 2018). Oxidation of the G-quadruplex structure facilitates the recruitment of transcription factors and increases KRAS transcription under oxidative stress. Taken together, these mechanisms reveal an intricate feedback loop in which KRAS, NRF2 and ROS jointly regulate oxidative stress and drive metabolic reprogramming in PDAC cells. This interplay between KRAS, NRF2 and ROS is shown schematically in Figure 3D. KRAS G12D stimulates PDAC growth by promoting a metabolic switch that drives anabolic processes and ROS production. To counteract the potentially cytotoxic ROS levels, KRAS activates NRF2, which not only attenuates oxidative stress but also promotes the metabolic shifts required for tumour growth. This dynamic interplay highlights the coordinated activity of KRAS and NRF2 in PDAC pathophysiology and emphasises their potential as therapeutic targets.
4.4 The suppression of NRF2 switches PDAC cells to aerobic metabolism
Recent research from our laboratory has further investigated the influence of KRAS and NRF2 on the modulation of PDAC metabolism (Di Giorgio et al., 2023; 2024). NRF2 was stably knocked down with CRISPR/Cas9 and two PDAC cell lines, Panc-1 and MIA-PaCa-2, without NRF2 (labelled NRF2−/−) were obtained. Using these edited cell lines, the cellular response was investigated under conditions in which the coordinated action of KRAS and NRF2 is disrupted (Di Giorgio et al., 2023). RNA-seq analysis on Panc-1 NRF2−/− cells (GEO: GSE217965) revealed 2,554 differentially expressed genes (DEGs) in NRF2−/− cells compared to wild-type cells, with 1,888 downregulated DEGs and 666 upregulated DEGs, based on a threshold of |log2 FC| ≥ 1 and P < 0.05 (Di Giorgio et al., 2023) (Figure 4A). The functional enrichment analysis of DEGs with ClusterProfiler showed a significant decrease in glycolysis, the pentose phosphate pathway (PPP), the glutathione cycle and long-chain fatty acid metabolism as well as a simultaneous deep reactivation of arginine/proline and medium-chain fatty acid metabolism in NRF2−/− Panc-1 cells compared to WT cells (Di Giorgio et al., 2023) (Figure 4B). These results suggest that NRF2 plays an important role in the switch of pancreatic cancer cells to aerobic glycolysis for ATP and biomass production as well as in the activation of PPP and glutathione cycle to maintain redox balance (Figure 4C). This is consistent with gene microarray data from two GEO datasets showing upregulation of glycolysis, PPP and GSH signalling pathways in NRF2-active oesophageal cells (Fu et al., 2019). Furthermore, silencing of NRF2 by siRNA in A549 lung cancer cells similarly reduced PPP enzymes and glutathione synthesis (Mitsuishi et al., 2012; Lu, 2009). Taken together, these data support the notion that NRF2 co-operates with KRAS G12D to shift glucose metabolism in PDAC towards aerobic glycolysis. Furthermore, recent work from our laboratory showed that the KRAS-NRF2 axis regulates the expression of NOS2 and the production of NO and RNS in PDAC: an argument that will be discussed in the next section (Di Giorgio et al., 2024).
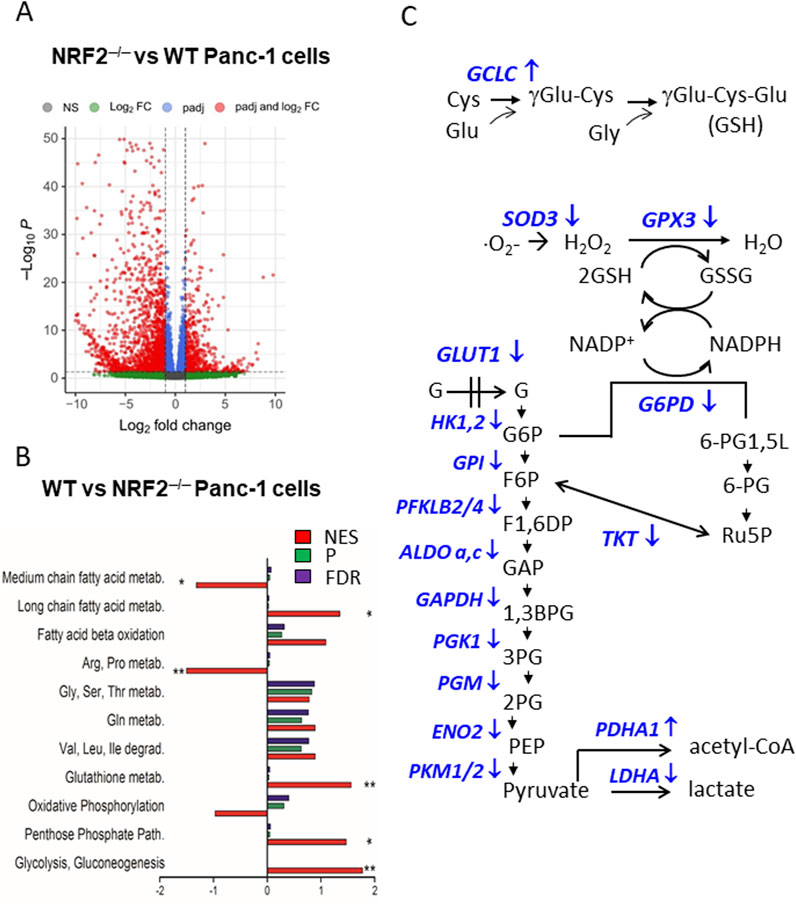
Figure 4. (A) Volcano plot of DEGs in Panc-1 NRF2−/− cells compared to WT cells; (B) Functional enrichment analysis of WT Panc-1 cells compared to NRF2−/− Panc-1 cells. This analysis shows that in cells where NRF2 was deleted (NRF2−/−), glycolysis, PPP, glutathione metabolism and long-chain fatty acid metabolism are inhibited, while arginine/proline and medium fatty acid metabolism are activated. NES = normal enrichment score; P = p-value; FDR = false discovery rate; (C) Glycolytic, PPP and glutathione metabolism enzymes are downregulated in NRF2−/− cells (indicated with ↓). Panels A and B adapted with permission (iScience 2023, 26, 108566).
5 NRF2 regulates NOS2 expression in pancreatic cancer cells
5.1 Suppression of NRF2 upregulates NOS2 in PDAC
The functional enrichment analysis of DEGs in NRF2−/− Panc-1 cells compared to the WT cells showed a reactivation of the arginine/proline metabolism (Figure 4B). As arginine is a nonessential amino acid that serves as a substrate for nitric oxide synthase (NOS) enzymes, DEG analysis suggests that NRF2, and consequently KRAS, should control the expression of NOS2 and nitric oxide (NO) production in PDAC cells. Gene Ontology (GO) analysis of DEGs in Panc-1 NRF2−/−cells compared to WT cells revealed four enriched terms involving NOS2, one of which is “HIF-1 signalling pathway (group P = 4.36 × 10−3). Considering its role in metabolic adaptation to oxygen and oxidative stress, analysis of the HIF signalling pathway showed that 5 genes were upregulated and 15 were downregulated, with NOS2 being strongly upregulated (log2 FC = 6.84, P = 5.53 × 10−6) (Di Giorgio et al., 2024) (Figure 5A). GENEMANIA analysis of upregulated DEGs in the KEGG.HIF pathway (NOS2, PRKCB, IGF1R, CYBB, and AKT3) suggests interactions with genes encoding subunits of the enzyme complex that converts GTP to cGMP, a key second messenger in the NO-mediated signalling pathway. The downregulated DEGs in the HIF-1 pathway encode enzymes involved in anaerobic processes—glycolysis, PPP, and the glutathione cycle—suggesting that NRF2 suppression renders Panc-1 cells dependent on aerobic metabolism. An important observation is that glycolytic Panc-1 cells, which rely primarily on glucose for ATP production and biomass synthesis, have low levels of NOS2 and NO, indicating that their metabolism is substantially dependent on arginine. In contrast, NRF2−/− Panc-1 cells have approximately 4-fold higher levels of NOS2. This increase is related to their dependence on aerobic metabolism and altered arginine utilisation. Measurements of •NO levels using DAF-FM DA, a non-fluorescent molecule that reacts with •NO to produce a fluorescent benzotriazole, confirm that NRF2−/− Panc-1 cells indeed contain more •NO than wild-type (WT) cells (Di Giorgio et al., 2024). When NRF2 expression is restored in NRF2−/− Panc-1 cells, NO levels decrease to the level of WT cells. This rescue experiment demonstrates that the regulation of NOS2 and •NO in pancreatic cancer cells is controlled by NRF2, or more comprehensively, the KRAS-NRF2 axis. Further evidence for NOS2 activity in NRF2−/− Panc-1 cells is provided by the citrulline/arginine ratio, which is 6-fold higher in NRF2−/− cells compared to WT cells (Di Giorgio et al., 2024). Restoring NRF2 expression lowers this ratio to WT levels. Similarly, treatment with the NOS2 inhibitor 1400W equalizes the citrulline/arginine ratio between WT and NRF2−/− cells, highlighting that NOS2 activity is dependent on NRF2 (Di Giorgio et al., 2024).
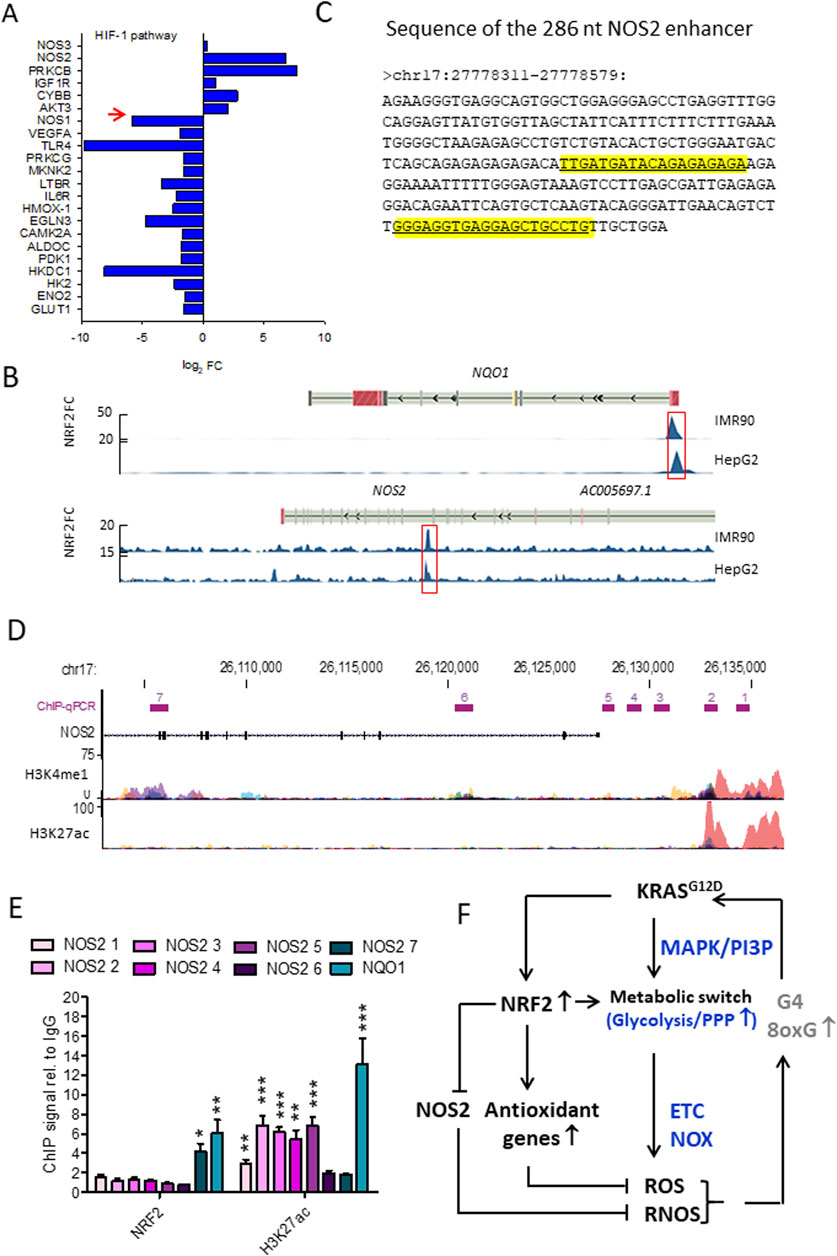
Figure 5. (A) Gene expression of the HIF pathway in NRF2−/− cells. Note that NOS2 is highly expressed in NRF2−/− cells while NOS1 is downregulated. The opposite holds for WT Panc-1 cells; (B) NRF2 ChIP-seq signals expressed as fold-change (FC) respect to Input in correspondence of NQO1 and NOS2 genomic loci. Data were retrieved from Encode and the significantly enriched peaks are highlighted; (C) Sequence of the NOS2 enhancer, the two predicted NRF2 binding sites are highlighted in yellow; (D) H3K4me1 and H3K27ac ChIP-seq signals expressed as fold-change (FC) compared to Input in correspondence of NOS2. The amplified regions investigated in qPCR are indicated (from 1 to 7), H3K4me1 and H3K27ac levels were obtained from Encode (cell lines: GM12878, H1-hESC, HSMM, HUVEC, K562, NHEK, NHLF); (E) ChIP signals relative to IgG obtained with anti-NRF2 and anti-H3K27ac antibodies in WT Panc-1 cells relative to the indicated genomic loci; (F) Interplay between KRAS, NRF2 and NOS2 in the control of oxidative and nitrosative homeostasis in PDAC. Panels A,B,C,D,E adapted with permission (BBA MCR 2024, 1871, 119106).
5.2 NRF2 binds to NOS2 enhancer and inhibits transcription
To investigate the mechanism by which NRF2 regulates NOS2, NRF2 ChIP-seq data from IMR90 (ENCSR197WGI) and HepG2 (ENCSR488EES) cells, available through ENCODE (Encyclopedia of DNA Elements), revealed that NRF2 interacts with a distal enhancer located 22 kb downstream of the NOS2 transcription start site (Figure 5B). Enhancers are short DNA sequences that increases the transcription of target genes. Unlike promoters, which are positioned directly adjacent to the genes they regulate, enhancers can influence gene expression from a great distance (Panigrahi and O’Malley, 2021). They recruit transcription factors and cofactors that facilitate the assembly of the transcriptional machinery at the promoter of the target gene. The NOS2 enhancer is 269 nt long and harbors two putative antioxidant response elements (AREs) recognized by NRF2 (Figure 5C) (Di Giorgio et al., 2024). Evidence that NRF2 binds to NOS2 in Panc-1 cells was obtained by ChIP experiments using antibodies anti-NRF2 and anti-H3K27ac in which 7 sites of NOS2 in genomic regions where histone H3 is epigenetically modified were amplified (Di Giorgio et al., 2024). The results showed that NRF2 binds to a genomic region of NOS2 (site 7, Figure 5D) where histone H3 is epigenetically modified by methylation and not acetylation (Figures 5D,E). It should be remembered that histone H3 acetylation at lysine 27 (H3K27ac) is a well-characterized histone modification linked to active gene transcription and is often used as a marker to distinguish active enhancers from poised or inactive ones (Creyghton et al., 2010). This modification is catalysed by histone acetyltransferases (HATs), which transfer an acetyl group to lysine, which loses its positive charge and weakens the interaction between histones and DNA. The relaxed chromatin structure makes the DNA more accessible to transcription factors and other components of the transcription machinery, which promotes gene activation. When H3K27ac marks enhancer regions, it signifies an active state, as opposed to enhancers marked only by H3 methylation H3K4me1, which can be other in an active or in a poised/inactive state (Creyghton et al., 2010). The presence of H3K27ac at enhancers often facilitates the recruitment of coactivators, transcription factors, and mediator complex components, which collectively enhance transcriptional activation of nearby genes by stabilizing RNA polymerase II at the promoter region (Kang et al., 2021; Yao et al., 2020). The DNA region within the NOS2 locus to which NRF2 binds to is marked by a lack of H3K27 acetylation (H3K27ac-) and a presence of H3K4 monomethylation (H3K4me1+) in wild-type Panc-1 cells (Figure 5D, site 7). This epigenetic profile indicates a repressed chromatin state with little or no transcriptional activity. At the same time, the presence of H3K4me1+, which indicates monomethylation at lysine 4 of histone H3, is usually associated with potential enhancer activity rather than active transcription. Taken together, these markers—H3K27ac- and H3K4me1+— represent a poised, not fully active state of the NOS2 enhancer in WT Panc-1 cells. In summary, NRF2 inhibits the transcription of NOS2. The interplay between KRAS G12D, NRF2 and NOS2 in the control of oxidative and nitrosative homeostasis in pancreatic cancer cells is summarised in Figure 5F. Given that NO and RNS are important contributors to the pathogenesis and progression of PDAC, in the next section, we discuss the effects of NO and RNS on pancreatic cancer cells.
6 Effect of nitric oxide on PDAC cells
Enhanced levels of NO/RNS have been observed in PDAC (Wang and Xie, 2010; Wang et al., 2016). NO acts as a signalling molecule in various physiological and pathological processes (Lowenstein and Padalko, 2004). NOS2 expression is triggered by inflammatory cytokines (TNF-α and IFN-γ) and hypoxic conditions in the tumour microenvironment. NO plays a dual role in tumour biology, influencing tumour growth, angiogenesis, metastasis and immune responses.
6.1 Dual role of NO in cancer cells
At low concentrations, •NO promotes cancer cell survival and proliferation by activating the PI3K/AKT and MAPK/ERK1/2 signalling pathways (Ding et al., 2021). It also (i) stimulates the expression of vascular endothelial growth factor (VEGF), promoting angiogenesis and tumour growth (Dulak et al., 2000); (ii) modulates cell adhesion molecules and metalloproteases, enhancing cancer cell migration and invasion (Carreau et al., 2011); (iii) facilitates immune evasion by inhibiting cytotoxic T-cells and natural killer (NK) cells (Sato et al., 2007; Cifone et al., 2001). At high concentrations, •NO has cytotoxic effects through the formation of RNS such as peroxynitrite, which triggers DNA damage and apoptosis in cancer cells (Virag et al., 2003). At high concentrations, NO can induce cell cycle arrest and inhibit proliferation of pancreatic cancer cells. To prevent the accumulation of toxic levels of ROS and RNS, PDAC cells upregulate NRF2, the master regulator of cellular redox homeostasis (Lister et al., 2011). NRF2 activation lowers ROS levels and, as recently reported, inhibits NOS2, the primary source of •NO and RNS in cells (Di Giorgio et al., 2024). The upregulation of NRF2 helps cancer cells maintain redox balance, which supports their survival and continued proliferation even under oxidative stress conditions.
6.2 Canonical and non-cnonical NO signalling pathways and protein S-nitrosylation
Nitric oxide signalling pathways are divided into canonical and non-canonical pathways based on their mechanisms and targets. The canonical •NO signalling involves the activation of soluble guanylyl cyclase (sGC) and the production of cyclic guanosine monophosphate (cGMP), which acts as a second messenger and activates downstream targets such as protein kinase G (PKG), phosphodiesterases (PDEs) and ion channels (Francis et al., 2010). At low NO levels (<100 nM), the cGMP/PKB pathway can protect against apoptosis and promote cell survival (Yasuhiro et al., 2006). At higher concentrations (>400 nM), NO activates non-canonical signalling by direct interaction with proteins leading to apoptosis. S-nitrosylation refers to a reversible modification in which •NO binds covalently to a cysteine thiol and forms a nitrosothiol -S-N=O group (Figure 6A) (Hess et al., 2005; Jaffrey et al, 2001). Extensive S-nitrosylation of proteins has been associated with various diseases, including pancreatic cancer (Tan et al., 2019). This post-translational modification can affect function, stability, localisation and interaction of proteins and often contributes to carcinogenesis by altering important signalling pathways (Plenchette et al., 2015). Since excessive S-nitrosylation can be detrimental, cells have developed systems to denitrosylate proteins. Thioredoxin and glutathione, for example, remove •NO groups from proteins, restoring free thiols and maintaining redox homeostasis (Sengupta and Holmgren, 2013). The biological role of S-nitrosylated proteins in PDAC, adjacent non-cancerous tissue, and Panc-1 cells was analysed separately (Tan et al., 2019). Functional analysis in non-cancerous tissues revealed that S-nitrosylated proteins are primaraly associated with basic biological processes, including primary cell metabolism, regulation of biological quality, response to stress and stimuli, catabolic activities, oxidation-reduction processes, secondary metabolism and initiation of translation. However, in PDAC tissues and Panc-1 cells, S-nitrosylated proteins were specifically enriched in pathways associated with tumourigenesis, such as cell cycle regulation, cell division, cell motility and actin filament-based processes. In addition, gene ontology annotations of cellular components revealed that S-nitrosylated proteins in these three samples are distributed in different subcellular compartments: cytoplasm, nuclear components, ribonucleoprotein complexes and ribosomes. These results emphasise the extensive involvement of S-nitrosylation of proteins in the pathogenesis of PDAC.
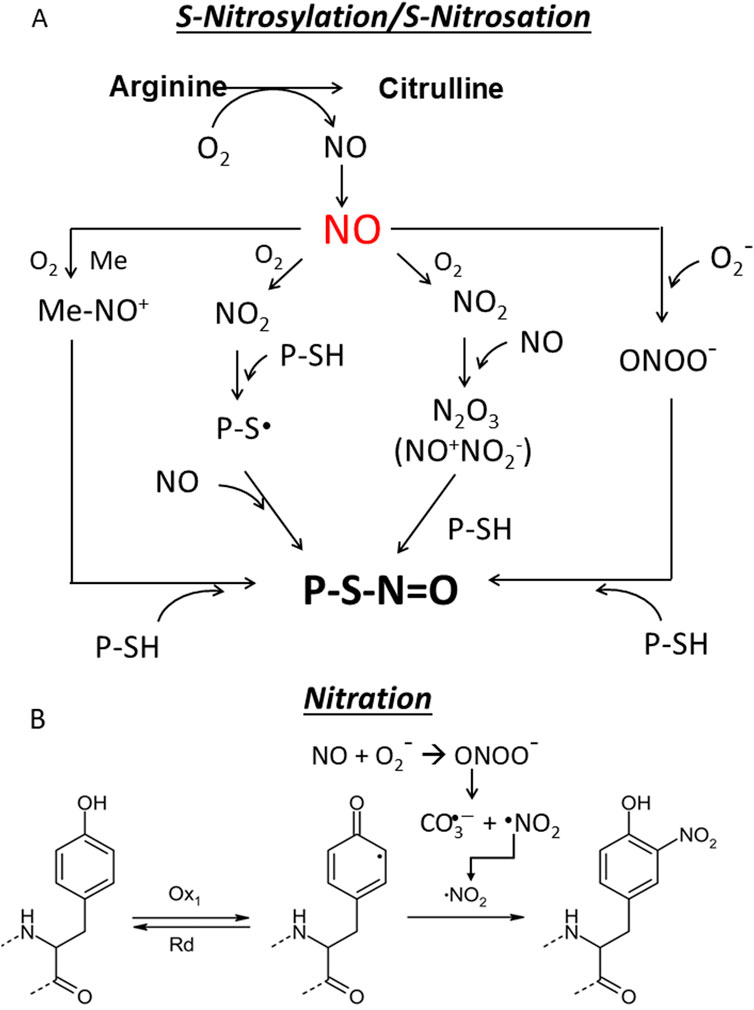
Figure 6. (A) S-nitrosylation/S-nitrosation. This cellular modification can occur in different ways. First, NO can form with O2 dinitrogen trioxide N2O3 which isomerises to nitrosonium nitrite (NO+NO2−) (Zakharov and Zakharov, 2009) whose nitrosonium NO+ reacts with a protein thiol (P-SH) to produce a nitrosothiol (P-S-N=O). In another pathway NO is oxidized to NO2 which reacts with a thiol group to give a thiol radical (-S∙) that with NO provides a nitrosothiol. The third pathway is mediated by metal which generates with NO a nitrosonium (NO+) (Mn+1 + NO + O2→ Mn-NO+), that adds to the thiol group to form a nitrosothiol (Aboalroub and Al Azzam, 2024; Ye et al., 2022; Reactive cysteines of proteins and glutathione (GSH) can undergo S-nitrosation by the peroxynitrite ONOO¯ to form S-nitrosothiol derivatives, P-SNOs and S-nitrosoglutathione (GSNO), respectively. Not all Cys are susceptible to S-nitrosylation. Those that are subject to S-nitrosylation lie within a consensus sequence that includes amino acids that create a hydrophobic environment. S-nitrosylation depends on several factors, including the acid/base and hydrophobic residues in the vicinity of the cysteine and the accessibility of the solvent (Marino and Gladyshev, 2010; Doulias et al., 2010). Notably, a hydrophobic environment attracts hydrophobic gases like NO and O2 and strongly enhances the rate of S-nitrosylation (Möller et al., 2007). S-nitrosation refers to a chemical process that occurs under physiological and pathological conditions in which peroxinitrite ONOO− reacts non-enzymatically with the thiol group to form a nitrosothiol (P-S-N=O); (B) Nitration. The one-electron oxidation of tyrosine produces a tyrosine radical Tyr•, which is converted into 3-nitrotyrosine by reaction with •NO2. As the pK value of the −OH group of Tyr is 10.3, it is 100% protonated at physiological pH, while the pK value drops to 7.3 during nitration and the -OH group is almost 50% deprotonated. This can strongly influence the structure of the nitrated protein.
6.3 S-nitrosation and nitration of proteins
S-nitrosation is a chemical process occurring under physiological and pathological conditions where peroxinitrite ONOO− reacts non-enzymatically with the thiol group, forming a nitrosothiol (-S-N=O) (Figure 6A). It is often used interchangeably with S-nitrosylation. This chemical reaction occurs on proteins and non-protein molecules such as glutathione GSH which is transformed into S-nitrosoglutathione (GSNO): a molecule acting as a NO reservoir in the cell (Ye et al., 2022). S-nitrosation of cysteines in the catalytic site of enzymes is a biologically important modification because it can alter metabolic pathays (Bruegger et al., 2018).
The term nitration refers to the addition of a nitro group (-NO2) to a tyrosine residue or other aromatic amino acids in proteins to form nitrotyrosine. Like S-nitrosation, it requires a peroxynitrite (ONOO-), which is formed by the reaction of NO with superoxide anion (•O2−). It is often associated with oxidative and nitrosative stress (Radi, 2013a) (Figure 6B). Nitration can irreversibly alter the structure and function of proteins and dysregulate signalling pathways (Bandookwala and Sengupta, 2020). Nitration is used as a biomarker for oxidative damage and inflammation in diseases such as atherosclerosis and neurodegenerative diseases. The characteristics of •NO signalling pathways are summarised in Table1.
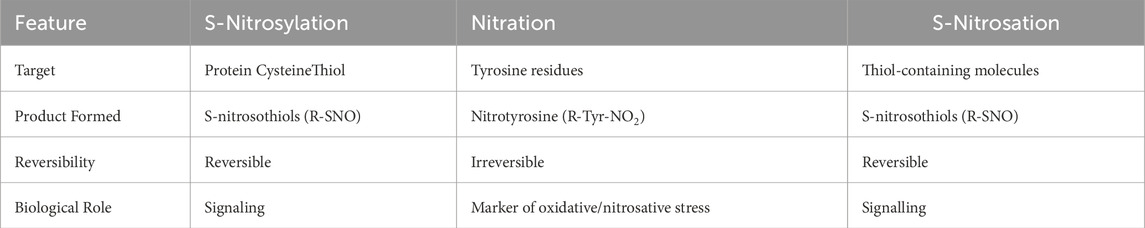
Table 1. Nitric oxide signaling.
The level of S-nitrosylated proteins in cells is primarily regulated by enzymatic processes that ensure that S-nitrosylation occurs in a controlled manner to maintain homeostasis and signaling. The main source of S-nitrosylation is •NO, but also S-nitrosoglutathione (GSNO), a low molecular weight S-nitrosothiol, can serve as •NO donor for protein S-nitrosylation. GSNO acts as a stable •NO reservoir and ensures the availability of NO for S-nitrosylation under physiological conditions (Broniowska et al., 2013). Key enzymes such as nitrosylases and thioredoxin/thioredoxin reductase (Trx/TrxR) and S-nitrosoglutathione reductase (GSNOR) precisely regulate the level of S-nitrosylated proteins in the cells (Anand and Stamler, 2012; Sengupta and Holmgren, 2012). Trx/TrxR reverses S-nitrosylation in proteins by catalysing denitrosylation, while GSNOR degrades GSNO and thus reduces the pool of available •NO donors. This dynamic balance between S-nitrosylation and denitrosylation allows cells to fine-tune the levels of S-nitrosylated proteins in response to physiological and pathological conditions.
6.4 NO donors in cancer therapy
Given that •NO at high concentrations arrests the cell cycle and inhibits proliferation, the use of •NO donors has gained increasing interest in therapeutic applications. The therapeutic potential of •NO was first explored many years ago, particularly in the treatment of pulmonary hypertension (Abman, 2013). However, due to the challenges associated with handling this gaseous molecule, •NO donors present an attractive alternative. These compounds can generate •NO in a controlled manner and have shown promise in cancer therapy. Wink and colleagues (Thomas et al., 2009) investigated the dose-dependent effects of •NO on human breast cancer MCF7 cells. At low concentrations (1–30 nM), •NO activates cyclic guanosine monophosphate (cGMP); at 30–100 nM, •NO phosphorylates AKT; and at 100–300 nM, •NO stabilizes hypoxia-inducible factor 1-alpha (HIF1α). At these relatively low concentrations, •NO promotes proliferation and survival. However, at concentrations exceeding 1 μM, •NO induces nitrosative stress, leading to cytostatic and apoptotic effects. Several NO donors have been proposed for therapeutic applications, offering a range of properties and effectiveness, including N-nitroso compounds as diazeniumdiolates, (NONOates); 3-morpholinosydnonimide (SIN-1) generating peroxynitrite and polymeric N-nitrosamines (Huerta, 2015; Mondal et al., 2024). For a comprehensive review of their characteristics, readers are referred to specific studies (Bhowmik and Roy, 2024; Huang et al., 2017; Huerta et al., 2008; Huerta S, 2015).
7 ChiP-seq analysis in 3D Panc-1 spheroids confirm NOS2 regulation by NRF2
7.1 NRF2 controls NOS2 in Panc-1 sheroids
The regulation of NOS2 expression by NRF2 was also investigated in Panc-1 spheroids, which are 3D cell models that better mimic the structural and biological features of tumours than 2D culture cells (Di Giorgio et al., 2024). H3K27ac-ChIP-seq analyses of WT and NRF2−/− Panc-1 spheroids revealed significant acetylation in over 13,500 genes. Specifically, 569 genes showed acetylation in NRF2−/− spheroids, while 734 genes showed acetylation in WT NRF2+/+ spheroids. Genes with increased acetylation in NRF2−/− spheroids are primarily associated with signalling pathways involving arginine metabolism, epithelial-mesenchymal transition (EMT) and •NO signalling (Di Giorgio et al., 2024). The regulatory role of NRF2 on NOS2 through the binding to a DNA enhancer was confirmed by mapping the H3K27ac signal at the NOS2 locus, which showed a marked increase in acetylation at the distal enhancer region in NRF2−/− spheroids compared to WT spheroids. The increased acetylation extended to a regulatory element located 5 kb upstream of the NOS2 transcription start site (Di Giorgio et al., 2024). These findings reinforce the conclusion that NRF2 directly represses NOS2 expression by modulating the chromatin state at its enhancer region in 3D spheroid model.
In addition to NRF2, hypoxia is a critical regulator of NOS2 transcription. Suppression of NRF2 permits the release of the distal enhancer, facilitating the recruitment of transcriptional activators. The chromatin at the NOS2 locus is characterized by the presence of H3K4me1—a mark of poised enhancers—indicating partial accessibility. Under hypoxic conditions, histone H3 becomes further acetylated at lysine 27 (H3K27ac), promoting chromatin relaxation and enhancer activation. Concurrently, levels of HIF-1 increase, leading to its binding at hypoxia response elements (HREs) within the NOS2 locus, further enhancing transcription. As shown in Figure 7, a three-step mechanism for the full activation of NOS2 has been proposed: (i) release of the enhancer from NRF2 inhibition; (ii) H3K27 acetylation leading to chromatin decompaction at the NOS2 locus; and (iii) binding of activating protein to the enhancer and HIF-1 to the NOS2 promoter (Matrone et al., 2004), forming the transcriptional complex.
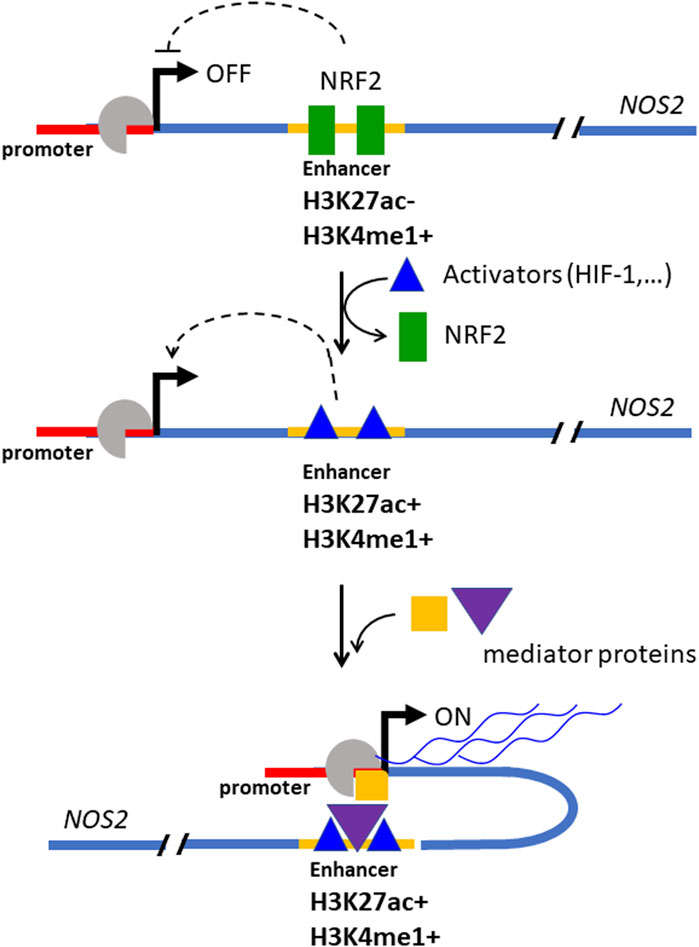
Figure 7. Mechanism by which NRF2 suppresses the expression of NOS2 in Panc-1 cells. At elevated concentrations, NRF2 binds to a distal enhancer, inactivating it. The enhancer bound by NRF2 is locked to the activators protein and the expression of NOS2 is inhibited (in this state the DNA at the NOS2 locus is marked by H3K27ac- and H3K5me1+). When the NRF2 level decreases, the enhancer is unlocked, free of NRF2, and is bound by activator proteins that promote transcription (in this state the DNA at the NOS2 locus is marked by H3K27ac+ and H3K5me1+). (“Med” stands for Mediator, a protein complex involved in gene expression in eukaryotic cells; “ac” = activator protein, TSS = transcription start site. Adapted with permission (BBA MCR 2024, 1871, 119106).
7.2 Epigenetic mechanisms regulationg NOS2
Epigenetic mechanisms that regulate NOS2 expression have been previously documented. For example, De Andrés et al. (2013) demonstrated that abnormal NOS2 expression in osteoarthritic human chondrocytes is associated with DNA methylation changes in NOS2 promoter and enhancer regions. Specifically, these authors reported demethylation of an NF-κB enhancer located −5.8 kb upstream of the transcription start site of NOS2 gene. These authors demonstrated that demethylation of this enhancer is essential for NOS2 transactivation. A comparable epigenetic regulatory mechanism was observed in human macrophages, where NOS2 expression differs significantly from that in mouse macrophages (Ross et al., 2014). While mouse macrophages express high levels of NOS2, human macrophages produce low levels of NOS2 and •NO due to extensive DNA methylation near the transcription start site of human NOS2. In contrast, the mouse NOS2 gene has low methylation in the 5′-flanking CpG regions. Analyses of chromatin accessibility and histone modifications showed that human macrophages have a closed chromatin conformation at the NOS2 locus, whereas mouse macrophages have an open chromatin state that facilitates NOS2 expression. This work emphasises how epigenetic changes control species-specific gene expression (Ross et al., 2014). Dreger et al. (Dreger et al., 2016) further investigated NOS2 regulation by showing that cytokines strongly induce NOS2 expression in rodent endothelial cells but not in human endothelial cells. NOS2 was identified in human umbilical vein endothelial cells as a potential target of the histone methyltransferase EZH2, which catalyses the tri-methylation of histone H3 at lysine 27 (H3K27me3). The EZH2-mediated modification represents an epigenetic mechanism of gene silencing.
Another notable mechanism affecting chromatin accessibility at the NOS2 promoter involves the NLRC4/caspase-1 axis in human macrophages (Buzzo et al., 2017). Caspase-1 cleaves PARP-1, a protein traditionally associated with apoptosis but also known to alter chromatin states essential for gene expression (Hottiger, 2015). Normally, PARP-1 maintains chromatin in a condensed state and suppresses gene expression. However, under stress conditions, PARP-1 is displaced from chromatin by caspase-1-mediated cleavage, leading to localised decondensation and allowing transcription factors to access DNA and promote gene expression (Erener et al., 2012).
Collectively, these studies highlight the multifaceted role of epigenetic mechanisms—including DNA methylation, histone modifications and chromatin remodelling—in regulating NOS2 expression in different cell types and species. In the following section, we report that NOS2 also plays a critical role for growth in NRF2−/− Panc-1 spheroids.
8 PDAC-spheroids formation depends on NOS2: therapeutic implications
8.1 NOS2 strongly affect the growth of NRF2−/− spheroids
The role of NOS2 in the proliferation of Panc-1 cells was examined by comparing NRF2−/− Panc-1 cells, which exhibit elevated NOS2 levels, to wild-type (WT) Panc-1 cells, which have lower NOS2 expression (Di Giorgio et al., 2024). To effectively suppress NOS2, esiNOS2—a pool of small interfering RNAs (siRNAs) targeting NOS2—was applied to both cell types. The impact of NOS2 knockdown on 3D spheroid formation was evaluated under conditions of both normal and suppressed NOS2 expression. Strikingly, NOS2 silencing had minimal impact on spheroid formation in WT Panc-1 cells, but resulted in more than a 50% reduction in spheroid formation in NRF2−/− Panc-1 cells (Di Giorgio et al., 2024). These results underscore the metabolic dependence of NRF2−/− Panc-1 cells on NOS2, and suggest that NOS2 is a potential therapeutic target. •NO, at non-toxic levels (<100 nM), is a key regulator of cellular metabolism (López-Sánchez et al., 2020; Zhou et al., 2019). In Panc-1 cells, •NO stabilizes HIF-1α, a transcription factor that upregulates glycolytic enzymes such as hexokinase (HK), phosphofructokinase (PFK), and pyruvate kinase (PK) under hypoxic conditions (Di Giorgio et al., 2024; Semenza, 2013). This promotes glycolysis as a compensatory mechanism for reduced mitochondrial respiration. Additionally, •NO activates signalling pathways that support cell growth and survival (vide infra), suggesting that NOS2 inhibition—and consequently reduced •NO production—may hinder PDAC progression. This notion is supported by recent findings from Reddy et al. (2024), who demonstrated that metaplastic breast cancer—a chemoresistant subtype with few treatment options—becomes sensitized to PI3K inhibitors when co-treated with NOS inhibitor L-NMMA. Building on the data presented here, a promising therapeutic strategy for PDAC may lie in targeting both NOS2 and the KRAS–NRF2 axis, as elaborated in the following section.
8.2 Combination therapeutic strategies for PDAC
The observation that the inhibition of NOS2 (and the arrest of •NO production) blocks the growth of NRF2−/− Panc-1 spheroids can be used in combined therapeutic approaches. This is supported by several important considerations: (i) KRAS is essential for the growth of PDAC. Its inhibition suppresses cell proliferation and induces apoptosis and ferroptosis (Miglietta et al., 2017; Ferino et al., 2020b; Di Giorgio et al., 2022); (ii) KRAS regulates NRF2. KRAS overexpression upregulating NRF2 and KRAS inhibition downregulates NRF2; (iii) Therapeutic suppression of KRAS results in the downregulation of NRF2 and consequently upregulation of NOS2. This leads to a metabolic switch from anaerobic glycolysis to aerobic metabolism. PDAC cells resist to the KRAS-targeted therapy by redirecting energy production to aerobic pathways in which arginine plays a crucial role, supporting the synthesis of phosphocreatine (an ATP buffer), polyamines and the production of NO/RNS through NOS2 (Di Giorgio et al., 2023). This metabolic adaptation was also evident in analysis of RNA-seq data from organoids derived from PDAC patients treated with FOLFIRINOX, a combination of chemotherapeutic agents (folinic acid, fluorouracil, irinotecan hydrochloride, oxaliplatin) used to treat tumours with KRAS mutations, including PDAC. A comparative analysis showed that genes that were suppressed in FOLFIRINOX organoids were also significantly suppressed in NRF2−/− Panc-1 cells. The most strongly repressed genes belong to the KEGG categories “glycolysis” and “genes upregulated by oncogenic KRAS” (Di Giorgio et al., 2023). This suggests that the suppression of the KRAS-NRF2 axis and the simultaneous activation of arginine-based metabolic pathways is also observed in PDAC patients undergoing FOLFIRINOX therapy.
These observations suggest that combination therapies targeting both KRAS and NOS2 may outperform KRAS-targeted monotherapies. While KRAS inhibition blocks tumour growth, resistance may develop as cells adapt metabolically (they undergo a metabolic shift from anaerobic to aerobic arginine-dependent metabolism). The resistance is accompanied by increased NOS2 and •NO production, which promote tumour growth and reduce the effect of anti-KRAS drugs. So, a dual therapeutic approach combining KRAS inhibitors with NOS2 inhibitors could enhance the anti-tumour response. Another interesting combination could include KRAS inhibitors and arginine antagonists such as homoarginine, which reduces •NO production by NOS2. This strategy could further improve the efficacy of targeted KRAS therapies. Future research should focus on exploring the metabolic consequences of NOS2 inhibition in PDAC cells, particularly in combination with KRAS-targeted treatments.
9 Conclusion
Over the last 2 decades, considerable progress has been made in understanding the effects of ROS/RNS on cancer biology, but many aspects remain unresolved. Since KRAS mutations (mainly G12D and G12V) are present in over 90% of PDAC cases, this oncogene is a major trigger of the disease and a primary target for rational drug development. Recent advances have yielded promising drugs targeting specific KRAS mutations, including KRAS G12C inhibitors (such as AMG510 and MRTX849) (Canon et al., 2019; Mahadevan et al., 2023) and inhibitors for KRAS G12D and KRAS G12V. However, despite significant efforts, the efficacy of these KRAS-targeting strategies remains a challenge, which is why KRAS was considered an undruggable target for many years.
More effective therapies can be developed by understanding the metabolic response of tumour cells to treatments. Pancreatic cancer cells, for example, are often found in a hypoxic microenvironment characterised by a specific genetic signature: overexpression and constitutive activation of KRAS, overexpression of NRF2 and HIF-1, and restricted expression of NOS2 (Di Giorgio et al., 2024). PDAC cells with this genetic profile are highly dependent on glucose for ATP production and biomass for proliferation. They produce ROS, mainly through NOX enzymes and electron leakage from the ETC, but remain below the toxic threshold thanks to the robust antioxidant defence of NRF2, which prevents ROS overload of cancer cells. In addition to controlling the cellular redox system, an increased NRF2 level favours binding to the NOS2 enhancer. This leads to inhibition of NOS2 expression and relatively low levels of •NO and RNS in PDAC. As mentioned above, this carefully regulated balance of ROS and NO/RNS supports both cell proliferation and survival. In addition, the hypoxic conditions that characterise PDAC stimulate the expression of HIF-1 and thus the synthesis of glycolytic and PPP enzymes, adapting the cells to anaerobic metabolism (Di Giorgio et al., 2023).
When KRAS is targeted, the treatment not only downregulates or inhibits KRAS, but also reduces NRF2 expression. This leads to a profound metabolic reprogramming in which the cancer cells switch from an anaerobic to an aerobic metabolism, with arginine playing a crucial role in this transition (Di Giorgio et al., 2023). In addition, reduced NRF2 levels weaken the antioxidant defence, leading to an increase in ROS and favouring the dissociation of NRF2 from the NOS2 enhancer and thus the transcription of NOS2. This leads to an increase in the concentration of •NO. At high concentrations, •NO has a negative effect on metabolism, but at lower concentrations •NO stimulates metabolic activity. This suggests that NOS2 and its substrate arginine are critical components for cancer cell survival and adaptation during anti-KRAS therapies. Although FOLFIRINOX does not directly target KRAS, this combination chemotherapy protocol which is primarily used to treat PDAC, also activates arginine metabolism, highlighting the role of metabolic reprogramming in the development of therapy resistance in pancreatic cancer. These results indicate that KRAS and NOS2 are promising candidates for a synthetic lethality approach. Combination therapies with small molecules or inhibitors targeting both KRAS and NOS2 may prove more effective than KRAS-targeted monotherapies alone. Future research will focus on exploring such combination strategies to improve treatment outcomes in pancreatic cancer.
Author contributions
VR: Funding acquisition, Writing – review and editing. CC: Writing – review and editing, Formal Analysis, Investigation. ED: Formal Analysis, Methodology, Writing – review and editing. LX: Conceptualization, Funding acquisition, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work has been carried out with the financial support of the “Ministero dell’Università e Ricerca Scientifica”, PRIN 2022 to LX and VR.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fcell.2025.1661525.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abman, S. H. (2013). Inhaled nitric oxide for the treatment of pulmonary arterial hypertension. Handb. Exp. Pharmacol. 218, 257–276. doi:10.1007/978-3-642-38664-0_11
Aboalroub, A. A., and Al Azzam, K. M. (2024). Protein S-nitrosylation: a chemical modification with ubiquitous biological activities. Protein J. 43, 639–655. doi:10.1007/s10930-024-10223-y
Anand, P., and Stamler, J. S. (2012). Enzymatic mechanisms regulating protein S-nitrosylation: implications in health and disease. J. Mol. Med. Berl. 90 (3), 233–244. doi:10.1007/s00109-012-0878-z
Back, E., Coolens, K., Van den Bossche, J. L., Houbracken, I., Espinet, E., and Rooman, I. (2022). On the origin of pancreatic cancer: molecular tumor subtypes in perspective of exocrine cell plasticity. Cell. Mol. Gastroenterol. Hepatol. 13 (4), 1243–1253. doi:10.1016/j.jcmgh.2021.11.010ì
Bandookwala, M., and Sengupta, P. (2020). 3-Nitrotyrosine: a versatile oxidative stress biomarker for major neurodegenerative diseases. Int. J. Neurosci. 130 (10), 1047–1062. doi:10.1080/00207454.2020.1713776
Bartesaghi, S., and Radi, R. (2018). Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 14, 618–625. doi:10.1016/j.redox.2017.09.009
Bhowmik, R., and Roy, M. (2024). Recent advances on the development of NO-releasing molecules (NORMs) for biomedical applications. Eur. J. Med. Chem. 268, 116217. doi:10.1016/j.ejmech.2024.116217
Bonini, M. G., and Augusto, O. (2001). Carbon dioxide stimulates the production of thiyl, sulfinyl, and disulfide radical anion from thiol oxidation by peroxynitrite. J. Biol. Chem. 276, 9749–9754. doi:10.1074/jbc.M008456200
Broniowska, K. A., Diers, A. R., and Hogg, N. (2013). S-nitrosoglutathione. Biochim. Biophys. Acta. 1830 (5), 3173–3181. doi:10.1016/j.bbagen.2013.02.004
Bruegger, J. J., Smith, B. C., Wynia-Smith, S. L., and Marletta, M. A. (2018). Comparative and integrative metabolomics reveal that S-nitrosation inhibits physiologically relevant metabolic enzymes. J. Biol. Chem. 293, 6282–6296. doi:10.1074/jbc.M117.817700
Buzzo, C. L., Medina, T., Branco, L. M., Lage, S. L., Ferreira, L. C., Amarante-Mendes, G. P., et al. (2017). Epigenetic regulation of nitric oxide synthase 2, inducible (Nos2) by NLRC4 inflammasomes involves PARP1 cleavage. Sci. Rep. 7, 41686–41698. doi:10.1038/srep41686
Canon, J., Rex, K., Saiki, A. Y., Mohr, C., Cooke, K., Bagal, D., et al. (2019). The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223. doi:10.1038/s41586-019-1694-1
Carreau, A., Kieda, C., and Grillon, C. (2011). Nitric oxide modulates the expression of endothelial cell adhesion molecules involved in angiogenesis and leukocyte recruitment. Exp. Cell. Res. 317 (1), 29–41. doi:10.1016/j.yexcr.2010.08.011
Cifone, M. G., Ulisse, S., and Santoni, A. (2001). Natural killer cells and nitric oxide. Int. Immunopharmacol. 1 (8), 1513–1524. doi:10.1016/s1567-5769(01)00095-9
Cogoi, S., Ferino, A., Miglietta, G., Pedersen, E. B., and Xodo, L. E. (2018). The regulatory G4 motif of the Kirsten ras (KRAS) gene is sensitive to guanine oxidation: implications on transcription. Nucleic Acids Res. 46 (2), 661–676. doi:10.1093/nar/gkx1142
Cogoi, S., Paramasivam, M., Spolaore, B., and Xodo, L. E. (2008). Structural polymorphism within a regulatory element of the human KRAS promoter: formation of G4-DNA recognized by nuclear proteins. Nucleic Acids Res. 36 (11), 3765–3780. doi:10.1093/nar/gkn120
Creyghton, M., Cheng, A. W., Welsteadet, G. G., Kooistra, T., Carey, B. W., Steine, E. J., et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. PNAS 107, 21931–21936. doi:10.1073/pnas.1016071107
D’Aiuto, N., Hochmann, J., Millán, M., Di Paolo, A., Bologna-Molina, R., Sotelo Silveira, J., et al. (2022). Hypoxia, acidification and oxidative stress in cells cultured at large distances from an oxygen source. Sci. doi:10.1038/s41598-022-26205-y
De Andrés, M. C., Imagawa, K., Hashimoto, K., Gonzalez, A., Roach, H. I., Goldring, M. B., et al. (2013). Loss of methylation in CpG sites in the NF-κB enhancer elements of inducible nitric oxide synthase is responsible for gene induction in human articular chondrocytes. Arthritis Rheum. 65, 732–742. doi:10.1002/art.37806
Denicola, A., Freeman, B. A., Trujillo, M., and Radi, R. (1996). Peroxynitrite reaction with carbon dioxide/bicarbonate. Kinetics and influence on peroxynitrite-mediated oxidations. Arch. Biochem. Biophys. 333 (1), 49–58. doi:10.1006/abbi.1996.0363
Di Giorgio, E., Choudhary, H., Ferino, A., Cortolezzis, Y., Dalla, E., D’Este, F., et al. (2023). Suppression of the KRAS-NRF2 axis shifts arginine into the phosphocreatine energy system in pancreatic cancer cells. iScience 26 (12), 108566. doi:10.1016/j.isci.2023.108566
Di Giorgio, E., Cortolezzis, Y., Gualandi, N., Agostini, F., Rapozzi, V., and Xodo, L. E. (2024). NRF2 interacts with distal enhancer and inhibits nitric oxide synthase 2 expression in KRAS-driven pancreatic cancer cells. Biochim. Biophys. Acta Mol. Cell. Res. 1871 (1), 119606. doi:10.1016/j.bbamcr.2023.119606
Di Giorgio, E., Ferino, A., Choudhary, H., Löffler, P. M. G., D'Este, F., Rapozzi, V., et al. (2022). Photosensitization of pancreatic cancer cells by cationic alkyl-porphyrins in free form or engrafted into POPC liposomes: the relationship between delivery mode and mechanism of cell death. J. Photochem. Photobiol. B 231, 112449–112464. doi:10.1016/j.jphotobiol.2022.112449
Ding, Z., Ogata, D., Roszik, J., Qin, Y., Kim, S. H., Tetzlaff, M. T., et al. (2021). iNOS associates with poor survival in melanoma: a role for nitric oxide in the PI3K-AKT pathway stimulation and PTEN S-nitrosylation. Front. Oncol. 11, 631766. doi:10.3389/fonc.2021.631766
Doulias, P. T., Greene, J. L., Greco, T. M., Tenopoulou, M., Seeholzer, S. H., Dunbrack, R. L., et al. (2010). Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc. Natl. Acad. Sci. U. S. A. 107, 16958–16963. doi:10.1073/pnas.1008036107
Dreger, H., Ludwig, A., Weller, A., Baumann, G., Stangl, V., and Stangl, K. (2016). Epigenetic suppression of iNOS expression in human endothelial cells: a potential role of Ezh2-mediated H3K27me3. Genomics 107, 145–149. doi:10.1016/j.ygeno.2016.02.002
Drosten, M., and Barbacid, M. (2020). Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell 37 (4), 543–550. doi:10.1016/j.ccell.2020.03.013
Dulak, J., Józkowicz, A., Dembinska-Kiec, A., Guevara, I., Zdzienicka, A., Zmudzinska-Grochot, D., et al. (2000). Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 20 (3), 659–666. doi:10.1161/01.atv.20.3.659
Erener, S., Pétrilli, V., Kassner, I., Minotti, R., Castillo, R., Santoro, R., et al. (2012). Inflammasome-activated caspase 7 cleaves PARP1 to enhance the expression of a subset of NF-κB target genes. Mol. Cell. 46, 200–211. doi:10.1016/j.molcel.2012.02.016
Ferino, A., Nicoletto, G., D'Este, F., Zorzet, S., Lago, S., Richter, S. N., et al. (2020a). Photodynamic therapy for ras-driven cancers: targeting G-quadruplex RNA structures with bifunctional alkyl-modified porphyrins. J. Med. Chem. 63, 1245–1260. doi:10.1021/acs.jmedchem.9b01577
Ferino, A., Rapozzi, V., and Xodo, L. E. (2020b). The ROS-KRAS-Nrf2 axis in the control of the redox homeostasis and the intersection with survival-apoptosis pathways: implications for photodynamic therapy. J. Photochem. Photobiol. B 202, 111672–111681. doi:10.1016/j.jphotobiol.2019.111672
Fontana, F., Giannitti, G., Marchesi, S., and Limonta, P. (2024). The PI3K/akt pathway and glucose metabolism: a dangerous liaison in cancer. Int. J. Biol. Sci. 20 (8), 3113–3125. doi:10.7150/ijbs.89942
Förstermann, U., and Sessa, W. C. (2012). Nitric oxide synthases: regulation and function. Eur. Heart J. 33 (7), 829–837. 837a-837d. doi:10.1093/eurheartj/ehr304
Francis, S. H., Busch, J. L., Corbin, J. D., and Sibley, D. (2010). cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 62 (3), 525–563. doi:10.1124/pr.110.002907
Franco, L., Doria, D., Bertazzoni, E., Benini, A., and Bassi, C. (2004). Increased expression of inducible nitric oxide synthase and cyclooxygenase-2 in pancreatic cancer. Prostagl. Other Lipid Mediat 73 (1-2), 51–58. doi:10.1016/j.prostaglandins.2003.12.001
Fridovich, I. (1983). Superoxide radical: an endogenous toxicant. Annu. Rev. Pharmacol. Toxicol. 23, 239–257. doi:10.1146/annurev.pa.23.040183.001323
Fu, J., Xiong, Z., Huang, C., Li, J., Yang, W., Han, Y., et al. (2019). Hyperactivity of the transcription factor Nrf2 causes metabolic reprogramming in mouse esophagus. J. Biol. Chem. 294, 327–340. doi:10.1074/jbc.RA118.005963
Fukumura, D., Kashiwagi, S., and Jain, R. K. (2006). The role of nitric oxide in tumour progression. Nat. Rev. Cancer. 6 (7), 521–534. doi:10.1038/nrc1910
Fulda, S., and Debatin, K. M. (2006). Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25 (34), 4798–4811. doi:10.1038/sj.onc.1209608
Gaglio, D., Metallo, C. M., Gameiro, P. A., Hiller, K., Danna, L. S., Balestrieri, C., et al. (2011). Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 7, 523. doi:10.1038/msb.2011.56
Gatenby, R., and Gillies, R. (2004). Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer. 4, 891–899. doi:10.1038/nrc1478
Hahn, S. A., Schutte, M., Hoque, A. T., Moskaluk, C. A., da Costa, L. T., Rozenblum, E., et al. (1996). DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 27, 350–353. doi:10.1126/science.271.5247.350
Hardie, R. A., van Dam, E., Cowley, M., Han, T. L., Balaban, S., Pajic, M., et al. (2017). Mitochondrial mutations and metabolic adaptation in pancreatic cancer. Cancer Metab. 5, 2. doi:10.1186/s40170-017-0164-1
Hayes, J. D., Dinkova-Kostova, A. T., and Tew, K. T. (2020). Oxidative stress in cancer. Cancer Cell 38, 167–197. doi:10.1016/j.ccell.2020.06.001
He, R., Jiang, W., Wang, C., Li, X., and Zhou, W. (2024). Global burden of pancreatic cancer attributable to metabolic risks from 1990 to 2019, with projections of mortality to 2030. BMC Public Health 24, 456. doi:10.1186/s12889-024-17875-6
Hess, D. T., Matsumoto, A., Kim, S. O., Marshall, H. E., and Stamler, J. S. (2005). Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166. doi:10.1038/nrm1569
Ho, W. J., Erbe, R., Danilova, L., Bigelow, E., Phyo, Z., Stein-O’Brien, G., et al. (2021). Multi-omic profiling of lung and liver tumor microenvironments of metastatic pancreatic cancer reveals site-specific immune regulatory pathways. Genome Biol. 22, 154. doi:10.1186/s13059-021-02363-6
Hong, S. Y., Yu, F.-X., Luo, Y., and Hagen, T. (2016). Oncogenic activation of the PI3K/Akt pathway promotes cellular glucose uptake by downregulating the expression of thioredoxin-interacting protein. Cell. Signal. 28, 377–383. doi:10.1016/j.cellsig.2016.01.011
Hottiger, M. O. (2015). Poly(ADP-ribose) polymerase inhibitor therapeutic effect: are we just scratching the surface? Expert. Opin. Ther. Targets. 19, 1149–1152. doi:10.1517/14728222.2015.1073262
Hu, H., Juvekar, A., Lyssiotis, C. A., Lien, E. C., Albeck, J. G., Oh, D., et al. (2016). Phosphoinositide 3-kinase regulates glycolysis through mobilization of aldolase from the actin cytoskeleton. Cell 164, 433–446. doi:10.1016/j.cell.2015.12.042
Hu, Y., Lu, W., Chen, G., Wang, P., Chen, Z., Zhou, Y., et al. (2012). K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 22, 399–412. doi:10.1038/cr.2011.145
Huang, Z., Fu, J., and Zhang, Y. (2017). Nitric oxide donor-based cancer therapy: advances and prospects. J. Med. Chem. 60 (18), 7617–7635. doi:10.1021/acs.jmedchem.6b01672
Huerta, S. (2015). Nitric oxide for cancer therapy. Future Sci. OA 1 (1), FSO44. doi:10.4155/fso.15.44
Huerta, S., Chilka, S., and Bonavida, B. (2008). Nitric oxide donors: novel cancer therapeutics (review). Int. J. Oncol. 33 (5), 909–927. doi:10.4155/fso.15.44
Jaffrey, S. R., Erdjument-Bromage, H., Ferris, C. D., Tempst, P., and Snyder, S. H. (2001). Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 3 (2), 193–197. doi:10.1038/35055104
Ju, H. Q., Ying, H., Tian, T., Ling, J., Fu, J., Lu, Y., et al. (2017). Mutant Kras- and p16-regulated NOX4 activation overcomes metabolic checkpoints in development of pancreatic ductal adenocarcinoma. Nat. Commun. 8, 14437. doi:10.1038/ncomms14437
Jung, B. J., Yoo, H. S., Shin, S., Park, Y. J., and Jeon, S. M. (2018). Dysregulation of NRF2 in cancer: from molecular mechanisms to therapeutic opportunities. Biomol. Ther. Seoul. 26 (1), 57–68. doi:10.4062/biomolther.2017.195
Kang, Y., Kim, Y. W., Kang, J., and Kim, A. (2021). Histone H3K4me1 and H3K27ac play roles in nucleosome eviction and eRNA transcription, respectively, at enhancers. FASEB J. 35, e21781–e21795. doi:10.1096/fj.202100488R
Kantari, C., and Walczak, H. (2011). Caspase-8 and Bid: caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta. 1813, 558–563. doi:10.1016/j.bbamcr.2011.01.026
Kehrer, J. P. (2000). The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 149 (1), 43–50. doi:10.1016/s0300-483x(00)00231-6
Kondoh, M., Ohga, N., Akiyama, K., Hida, Y., Maishi, N., Towfik, M. T., et al. (2013). Hypoxia-induced reactive oxygen species cause chromosomal abnormalities in endothelial cells in the tumor microenvironment. PLoS ONE 8 (11), e80349. doi:10.1371/journal.pone.0080349
Lai, E., Ziranu, P., Dubois, M., Pretta, A., Tolu, S., Camera, S., et al. (2021). BRCA-mutant pancreatic ductal adenocarcinoma. Br. J. Cancer. 125, 1321–1332. doi:10.1038/s41416-021-01469-9
Lennerz, J. K., and Stenzinger, A. (2015). Allelic ratio of KRAS mutations in pancreatic cancer. Oncologist 20 (4), e8–e9. doi:10.1634/theoncologist.2014-0408
Lister, A., Nedjadi, T., Kitteringham, N. R., Campbell, F., Costello, E., Lloyd, B., et al. (2011). Nrf2 is overexpressed in pancreatic cancer: implications for cell proliferation and therapy. Mol. Cancer. 10, 37. doi:10.1186/1476-4598-10-37
Liu, Q., Ge, W., Martínez-Jarquín, S., He, Y., Wu, R., Stoffel, M., et al. (2023). Mass spectrometry reveals high levels of hydrogen peroxide in pancreatic cancer cells. Angew. Chem. Int. Ed. Engl. 62 (19), e202213703. doi:10.1002/anie.202213703
López-Sánchez, L. M., Aranda, E., and Rodríguez-Ariza, A. (2020). Nitric oxide and tumor metabolic reprogramming. Biochem. Pharmacol. 176, 113769. doi:10.1016/j.bcp.2019.113769
Lowenstein, C. J., and Padalko, E. (2004). iNOS (NOS2) at a glance. J. Cell Sci. 117, 2865–2867. doi:10.1242/jcs.01166
Lu, S. C. (2009). Regulation of glutathione synthesis. Mol. Asp. Med. 30, 42–59. doi:10.1016/j.mam.2008.05.005
Ma, Q. (2013). Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 53, 401–426. doi:10.1146/annurev-pharmtox-011112-140320
Madesh, M., and Hajnóczky, G. (2001). VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 155, 1003–1015. doi:10.1083/jcb.200105057
Mahadevan, K. K., McAndrews, K. M., LeBleu, V. S., Yang, S., Lyu, H., Li, B., et al. (2023). KRASG12D inhibition reprograms the microenvironment of early and advanced pancreatic cancer to promote FAS-mediated killing by CD8+ T cells. Cancer Cell 41, 1606–1620.e8. doi:10.1016/j.ccell.2023.07.002
Marino, S. M., and Gladyshev, V. N. (2010). Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J. Mol. Biol. 404 (5), 902–906. doi:10.1016/j.jmb.2010.09.027
Matrone, C., Pignataro, G., Molinaro, P., Irace, C., Scorziello, A., Di Renzo, G. F., et al. (2004). HIF-1alpha reveals a binding activity to the promoter of iNOS gene after permanent middle cerebral artery occlusion. J. Neurochem. 90, 368–378. doi:10.1111/j.1471-4159.2004.02483.x
Miglietta, G., Cogoi, S., Marinello, J., Capranico, G., Tikhomirov, A. S., Shchekotikhin, A., et al. (2017). RNA G-quadruplexes in kirsten ras (KRAS) oncogene as targets for small molecules inhibiting translation. J. Med. Chem. 60, 9448–9461. doi:10.1021/acs.jmedchem.7b00622
Mills, J. C., Stone, N. L., Erhardt, J., and Pittman, R. N. (1998). Apoptotic membrane blebbing is regulated by myosin light chain phosphorylation. J. Cell Biol. 140 (3), 627–636. doi:10.1083/jcb.140.3.627
Mitsuishi, Y., Taguchi, K., Kawatani, Y., Shibata, T., Nukiwa, T., Aburatani, H., et al. (2012). Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22 (1), 66–79. doi:10.1016/j.ccr.2012.05.016
Möller, M. N., Li, Q., Lancaster, J. R., and Denicola, A. (2007). Acceleration of nitric oxide autoxidation and nitrosation by membranes. IUBMB Life 59 (4-5), 243–248. doi:10.1080/15216540701311147
Moloney, J. N., and Cotter, T. G. (2018). ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 80, 50–64. doi:10.1016/j.semcdb.2017.05.023
Mondal, A., Paul, S., and De, P. (2024). Recent advancements in polymeric N-Nitrosamine-Based nitric oxide (NO) donors and their therapeutic applications. Biomacromolecules 25, 5592–5608. doi:10.1021/acs.biomac.4c00685
Ngo, V., and Duennwald, M. L. (2022). Nrf2 and oxidative stress: a general overview of mechanisms and implications in human disease. Antioxidants (Basel) 11 (12), 2345. doi:10.3390/antiox11122345
Niu, B., Liao, K., Zhou, Y., Wen, T., Quan, G., Pan, X., et al. (2021). Application of glutathione depletion in cancer therapy: enhanced ROS-based therapy, ferroptosis, and chemotherapy. Biomaterials 277, 121110. doi:10.1016/j.biomaterials.2021.121110
Onyango, A. N. (2016). Endogenous generation of singlet oxygen and ozone in human and animal tissues: mechanisms, biological significance, and influence of dietary components. Oxid. Med. Cell Longev. 2016, 2398573–22. doi:10.1155/2016/2398573
Osman, A. A., Aslan, E., Bartels, M., Michikawa, C., Lindemann, A., Tomczak, K., et al. (2023). Dysregulation and epigenetic reprogramming of NRF2 SignallingAxis promote acquisition of cisplatin resistance and metastasis in head and neck squamous cell carcinoma. Clin. Cancer Res. 29 (7), 1344–1359. doi:10.1158/1078-0432.CCR-22-2747
Panigrahi, A., and O’Malley, B. W. (2021). Mechanisms of enhancer action: the known and the unknown. Genome Biol. 22 (1), 108. doi:10.1186/s13059-021-02322-1
Papa, S., Choy, P. M., and Bubici, C. (2019). The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 38 (13), 2223–2240. doi:10.1038/s41388-018-0582-8
Park, W., Chawla, A., and O'Reilly, E. M. (2021). Pancreatic cancer: a review. JAMA 326 (9), 851–862. doi:10.1001/jama.2021.13027
Parvez, S., Long, M. J. C., Poganik, J. R., and Aye, Y. (2018). Redox signaling by reactive electrophiles and oxidants. Chem. Rev. 118, 8798–8888. doi:10.1021/acs.chemrev.7b00698
Plenchette, S., Romagny, S., Laurens, V., and Bettaieb, A. (2015). S-Nitrosylation in TNF superfamily signaling pathway: implication in cancer. Redox Biol. 6, 507–515. doi:10.1016/j.redox.2015.08.019
Radi, R. (2013a). Peroxynitrite, a stealthy biological oxidant. J. Biol. Chem. 288 (37), 26464–26472. doi:10.1074/jbc.R113.472936
Radi, R. (2013b). Protein tyrosine nitration: biochemical mechanisms and structural basis of functional effects. Acc. Chem. Res. 46 (2), 550–559. doi:10.1021/ar300234c
Reddy, T., Puri, A., Guzman-Rojas, L., Thomas, C., Qian, W., Zhou, J., et al. (2024). NOS inhibition sensitizes metaplastic breast cancer to PI3K inhibition and taxane therapy via c-JUN repression. Nat. Commun. 15 (1), 10737. doi:10.1038/s41467-024-54651-x
Romero, R., Sayin, V. I., Davidson, S. M., Bauer, M. R., Singh, S. X., LeBoeuf, S. E., et al. (2017). Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 23, 1362–1368. doi:10.1038/nm.4407
Ross, T. J., Kremens, K., Powers, L. S., Brink, B., Knutson, T., Domann, F. E., et al. (2014). Epigenetic silencing of the human NOS2 gene: rethinking the role of nitric oxide in human macrophage inflammatory responses. J. Immunol. 192, 2326–2338. doi:10.4049/jimmunol.1301758
Rubio, C. P., and Ceron, J. J. (2021). Spectrophotometric assays for evaluation of Reactive Oxygen Species (ROS) in serum: general concepts and applications in dogs and humans. BMC Vet. Res. 17, 226. doi:10.1186/s12917-021-02924-8
Sarwar, A., Zhu, M., Su, Q., Zhu, Z., Yang, T., Chen, Y., et al. (2022). Targeting mitochondrial dysfunctions in pancreatic cancer evokes new therapeutic opportunities. Crit. Rev. Oncol. Hematol. 180, 103858. doi:10.1016/j.critrevonc.2022.103858
Sato, K., Ozaki, K., Oh, I., Meguro, A., Hatanaka, K., Nagai, T., et al. (2007). Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood 109 (1), 228–234. doi:10.1182/blood-2006-02-002246
Schutte, M., Hruban, R. H., Geradts, J., Maynard, R., Hilgers, W., Rabindran, S. K., et al. (1997). Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 57, 3126–3130.
Semenza, G. L. (2013). HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Invest. 123 (9), 3664–3671. doi:10.1172/JCI67230
Sengupta, R., and Holmgren, A. (2012). The role of thioredoxin in the regulation of cellular processes by S-nitrosylation. Biochim. Biophys. Acta. 1820 (6), 689–700. doi:10.1016/j.bbagen.2011.08.012
Sengupta, R., and Holmgren, A. (2013). Thioredoxin and thioredoxin reductase in relation to reversible S -nitrosylation. Antioxid. Redox Signal. 18 (3), 259–269. doi:10.1089/ars.2012.4716
Sheng, Y., Abreu, I. A., Cabelli, D. E., Maroney, M. J., Miller, A. F., Teixeira, M., et al. (2014). Superoxide dismutases and superoxide reductases. Chem. Rev. 114, 3854–3918. doi:10.1021/cr4005296
Skonieczna, M., Hejmo, T., Poterala-Hejmo, A., Cieslar-Pobuda, A., and Buldak, R. J. (2017). NADPH oxidases: insights into selected functions and mechanisms of action in cancer and stem cells. Oxid. Med. Cell Longev. 2017, 9420539. doi:10.1155/2017/9420539
Sohal, R. S., and Forster, M. J. (2007). Coenzyme Q, oxidative stress and aging. Mitochondrion 7, S103–S111. doi:10.1016/j.mito.2007.03.006
Song, G., Ouyang, G., and Bao, S. (2005). The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 9, 59–71. doi:10.1111/j.1582-4934.2005.tb00337.x
Song, N. Y., Zhu, F., Wang, Z., Willette-Brown, J., Xi, S., Sun, Z., et al. (2018). IKKα inactivation promotes Kras-initiated lung adenocarcinoma development through disrupting major redox regulatory pathways. Proc. Natl. Acad. Sci. U. S. A. 115 (4), E812-E821–E821. doi:10.1073/pnas.1717520115
Srinivas, U. S., Tan, B. W. Q., Vellayappan, B. A., and Jeyasekharan, A. D. (2019). ROS and the DNA damage response in cancer. Redox Biol. 25, 101084. doi:10.1016/j.redox.2018.101084
Storz, P. (2016). KRas, ROS and the initiation of pancreatic cancer. Small GTPases 8 (1), 38–42. doi:10.1080/21541248.2016.1192714
St-Pierre, J., Buckingham, J. A., Roebuck, S. J., and Brand, M. D. (2002). Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 277, 44784–44790. doi:10.1074/jbc.M207217200
Tafani, M., Sansone, L., Limana, F., Arcangeli, T., De Santis, E., et al. (2016). The interplay of reactive oxygen species, hypoxia, inflammation, and sirtuins in cancer initiation and progression. Oxid. Med. Cell Longev. 2016, 3907147. doi:10.1155/2016/3907147
Tan, C., Li, Y., Huang, X., Wei, M., Huang, Y., Tang, Z., et al. (2019). Extensive protein S-nitrosylation associated with human pancreatic ductal adenocarcinoma pathogenesis. Cell Death and Dis. 10, 914. doi:10.1038/s41419-019-2144-6
Tao, S., Wang, S., Moghaddam, S. J., Ooi, A., Chapman, E., Wong, P. K., et al. (2019). Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 74, 7430–7441. doi:10.1158/0008-5472.CAN-14-1439
Thomas, C., Mackey, M. M., Diaz, A. A., and Cox, D. P. (2009). Hydroxyl radical is produced via the Fenton reaction in submitochondrial particles under oxidative stress: implications for diseases associated with iron accumulation. Redox Rep. 14 (3), 102–108. doi:10.1179/135100009X392566
Tikunov, A., Johnson, C. B., Pediaditakis, P., Markevich, N., Macdonald, J. M., Lemasters, J. J., et al. (2010). Closure of VDAC causes oxidative stress and accelerates the Ca(2+)-induced mitochondrial permeability transition in rat liver mitochondria. Arch. Biochem. Biophys. 495 (2), 174–181. doi:10.1016/j.abb.2010.01.008
Vander Heiden, M. G., Cantley, L. C., and Thompson, C. B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324 (5930), 1029–1033. doi:10.1126/science.1160809
Virag, L., Szabó, E., Gergely, P., and Szabó, C. (2003). Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicol. Lett. 140-141, 113–124. doi:10.1016/s0378-4274(02)00508-8
Wakabayashi, N., Dinkova-Kostova, A. T., Holtzclaw, W. D., Kang, M., Kobayash, A., Yamamoto, M., et al. (2004). Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. U.S.A. 101 (7), 2040–2045. doi:10.1073/pnas.0307301101
Walsh, J. G., Cullen, S. P., Sheridan, C., Lüthi, A. U., Gerner, C., and Martin, S. J. (2008). Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc. Natl. Acad. Sci. U. S. A. 105 (35), 12815–12819. doi:10.1073/pnas.0707715105
Wang, J., He, P., Gaida, M., Yang, S., Schetter, A. J., Gaedcke, J., et al. (2016). Inducible nitric oxide synthase enhances disease aggressiveness in pancreatic cancer. Oncotarget 7 (33), 52993–53004. doi:10.18632/oncotarget.10323
Wang, L., Kuang, Z., Zhang, D., Gao, Y., Ying, M., and Wang, T. (2021). Reactive oxygen species in immune cells: a new antitumor target. Biomed. Pharmacother. 133, 110978. doi:10.1016/j.biopha.2020.110978
Wang, L., and Xie, K. (2010). Nitric oxide and pancreatic cancer pathogenesis, prevention, and treatment. Curr. Pharm. Des. 16 (4), 421–427. doi:10.2174/138161210790232194
Wang, P., Song, M., Zeng, Z. L., Zhu, C. F., Lu, W. H., Yang, J., et al. (2015). Identification of NDUFAF1 in mediating K-Ras induced mitochondrial dysfunction by a proteomic screening approach. Oncotarget 6 (6), 3947–3962. doi:10.18632/oncotarget.2968
Wang, Q.-S., Zheng, Y.-M., Dong, L., Ho, Y.-S., Guo, Z., and Wang, Y.-X. (2007). Role of mitochondrial reactive oxygen species in hypoxia-dependent increase in intracellular calcium in pulmonary artery myocytes. Free Radic. Biol. Med. 42 (5), 642–653. doi:10.1016/j.freeradbiomed.2006.12.008
Wu, L., Saxena, S., and Singh, R. K. (2020). Neutrophils in the tumor microenvironment. Adv. Exp. Med. Biol. 1224, 1–20. doi:10.1007/978-3-030-35723-8_1
Xu, W., Liu, L., Loizidou, M., Ahmed, M., and Charles, I. G. (2002). The role of nitric oxide in cancer. Cell Res. 12, 311–320. doi:10.1038/sj.cr.7290133
Yang, L., Shen, C., Estrada-Bernal, A., Robb, R., Chatterjee, M., Sebastian, N., et al. (2021). Oncogenic KRAS drives radioresistance through upregulation of NRF2-53BP1-mediated non-homologous end-joining repair. Nucleic Acids Res. 49, 11067–11082. doi:10.1093/nar/gkab871
Yao, J., Chen, J., Li, L. Y., and Wu, M. (2020). Epigenetic plasticity of enhancers in cancer. Transcription 11, 26–36. doi:10.1080/21541264.2020.1713682
Yasuhiro, Y., Akiko, Y., and Sadaaki, K.-A. (2006). Nitric oxide/cGMP signaling pathway protects RAW264 cells against nitric oxide-induced apoptosis by inhibiting the activation of p38 mitogen-activated protein kinase. J. Pharmacol. Sci. 101 (2), 126–134. doi:10.1254/jphs.FPJ06001X
Ye, H., Wu, J., Liang, Z., Zhang, Y., and Huang, Z. (2022). Protein S-nitrosation: biochemistry, identification, molecular mechanisms and therapeutic applications. J. Med. Chem. 65, 5902–5925. doi:10.1021/acs.jmedchem.1c02194
Ying, H., Kimmelman, A. C., Lyssiotis, C. A., Hua, S., Chu, G. C., Fletcher-Sananikone, E., et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670. doi:10.1016/j.cell.2012.01.058
Zakharov, I. I., and Zakharova, O. I. (2009). Nitrosonium nitrite isomer of N2O3: quantum-chemical data. J. Struct. Chem. 50, 212–218. doi:10.1007/s10947-009-0031-1
Zhang, L., Li, J., Zong, L., Chen, X., Chen, K., Jiang, Z., et al. (2016). Reactive oxygen species and targeted therapy for pancreatic cancer. Oxid. Med. Cell Longev. 2016, 1616781. doi:10.1155/2016/1616781
Zhou, D., Duan, Z., Li, Z., Ge, F., Wei, R., and Kong, L. (2022). The significance of glycolysis in tumor progression and its relationship with the tumor microenvironment. Front. Pharmacol. 13, 1091779. doi:10.3389/fphar.2022.1091779
Zhou, H. L., Zhang, R., Anand, P., Stomberski, C. T., Qian, Z., Hausladen, A., et al. (2019). Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature 565, 96–100. doi:10.1038/s41586-018-0749-z
Zhu, B., Cheng, L., Huang, B., Liu, R., and Ren, B. (2024). Central role of hypoxia-inducible factor-1α in metabolic reprogramming of cancer cells: a review. Med. Baltim. 103 (44), e40273. doi:10.1097/MD.0000000000040273
Glossary
mRNA Messanger RNA
ATP Adenosin triphosphate
PDAC Pancreatic ductal adenocarcinoma
NADPH Nicotinamide adenine dinucleotide phosphate
NRF2 Nuclear factor erythroid 2-related factor 2
NOS1 Neuronal nitric oxide synthase (also called nNOS)
NOS2 Inducible nitric oxide synthase (also called iNOS)
NOS3 Endothelial nitric oxide synthase (also called eNOS)
•NO Nitric oxide
ONOO− Peroxynitrite
ROS Reactive oxygen species
RNS Reactive oxygen nitrogen species
H3K27ac histone 3 lysine 27 acetylation
H3K4me1 Histone3 lysine 4 mono-methylation
HIF-1α Hypoxia-inducible factor 1
DEG Differentially expressed gene
KRAS Kirsten ras gene
OXPHOS Oxidative phosphorylation
DAF-FM DA 4-Amino-5-methylamino-2′,7′-difluorofluorescein diacetate
ENCODE Encyclopedia of DNA Elements
FAD Flavin adenine dinucleotide
FMN Flavin mononucleotide
GMPc Cyclic guanosine monophosphate
sGC Soluble Guanylyl cyclase
GTP Guanosine triphospohate
IL-1 Interleukin-1
IL-1R1 Interleukin-1 receptor type I
INF-γ Type II interferon
JAKs Janus family kinases
LPS Lipopolysaccharides
NF-kB nuclear factor kappa-light-chain-enhancer of activated B cells
Keywords: KRAS, NRF2, NOS2, metabolic reprogramming, PDAC, ROS, RNS
Citation: Rapozzi V, Comuzzi C, Di Giorgio E and Xodo LE (2025) KRAS and NRF2 drive metabolic reprogramming in pancreatic cancer cells: the influence of oxidative and nitrosative stress. Front. Cell Dev. Biol. 13:1547582. doi: 10.3389/fcell.2025.1547582
Received: 18 December 2024; Accepted: 26 May 2025;
Published: 11 June 2025; Corrected: 07 August 2025.
Edited by:
Jidong Yan, Xi’an Jiaotong University, ChinaReviewed by:
Alessio Lepore, University of Leeds, United KingdomYifan Zhang, Shanghai Jiao Tong University, China
Copyright © 2025 Rapozzi, Comuzzi, Di Giorgio and Xodo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luigi E. Xodo, bHVpZ2kueG9kb0B1bml1ZC5pdA==