Abstract
Podocytes are highly specialized, terminally differentiated cells in the glomerulus of the kidney and these cells play a central role in blood filtration. In this review, we comprehensively describe the cell biology of podocytes under healthy conditions and in glomerular disorders wherein podocyte injury is a major pathological mechanism. First, the molecular mechanisms that maintain podocyte actin cytoskeleton structure, permanent cell cycle exit, and metabolism under healthy conditions are described. Secondly, the mechanisms of podocyte injury, including genetic alterations and external insults that ultimately disrupt podocyte actin cytoskeleton dynamics or interrupt podocyte quiescence and mitochondrial metabolism are discussed. This understanding forms the basis of described potential therapeutic agents that act by modulating dysregulated podocyte cytoskeleton organization, prevent or reverse cell cycle re-entry, and re-establish normal mitochondrial energy production. Lastly, the application of modern techniques such as single cell RNA sequencing, super resolution microscopy, atomic force microscopy, and glomerular organoids is improving the resolution of mechanistic podocytopathy knowledge. Taken together, our review provides critical insights into the cellular and molecular mechanisms leading to podocyte loss, necessary for the advancement of therapeutic development in glomerular diseases.
1 Introduction
Chronic kidney disease (CKD) is a major global health concern affecting 1 in 10 individuals, representing over 850 million people worldwide and resulting in more than 1 million deaths per year (Jha et al., 2013; Verberne et al., 2019; Jager et al., 2019). Care for patients with CKD presents a significant healthcare burden, with hospitalization costs of over 100 billion USD per year (Jha et al., 2013; Verberne et al., 2019; Jager et al., 2019).
Glomerular diseases are a major cause of CKD. Glomeruli (Figure 1) are the blood-filtering units of the kidney comprised of four specialized epithelial cells consisting of podocytes, endothelial cells, mesangial cells, and parietal epithelial cells. Podocytes (Figure 1) are highly specialized, terminally differentiated cells that serve a crucial role in glomerular filtration function. Podocyte loss defines many forms of glomerular disease, which are challenging to treat given the highly specialized, differentiated state of these cells. In this review, we focus on podocytes and the pathologic molecular mechanisms that define their role in disease. Ultimately this knowledge is critical to identify molecular targets and pathways for therapeutic advancement.
FIGURE 1
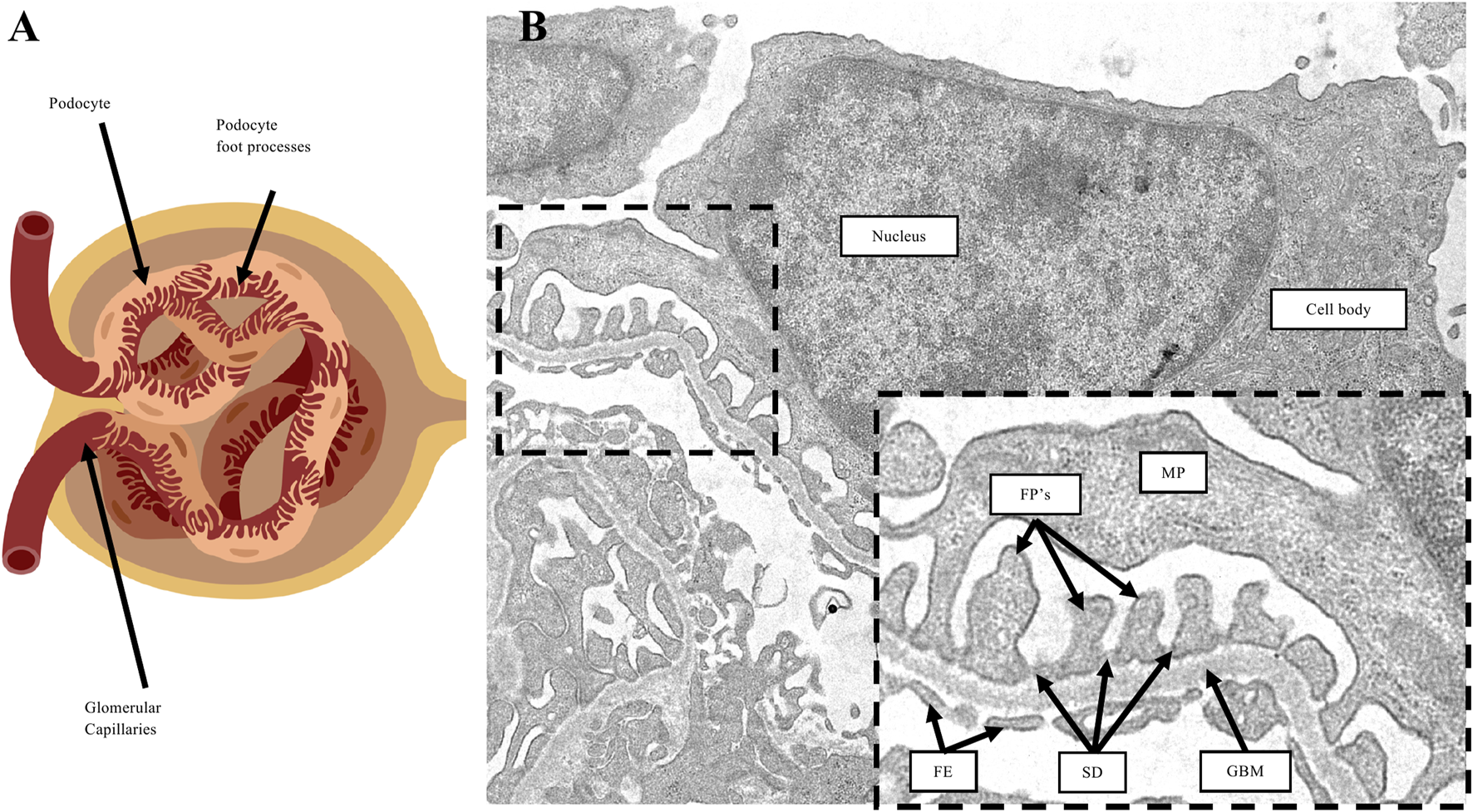
Podocyte morphology. (A) Diagram representation of a glomerulus. Podocytes are specialized epithelial cells comprised of cellular projections called foot processes which wrap around the surface of glomerular capillaries. (B) Transmission electron microscopy image of a podocyte. The cell body of podocytes branch into major processes (MP), which in turn form the foot processes (FP’s) that are in direct contact with the underlying glomerular basement membrane (GBM). On the capillary lumen side are fenestrated endothelium (FE). Adjacent foot processes form a space in-between called the slit diaphragm (SD).
1.1 Glomerular diseases characterized by podocyte loss in kidney disease patients
Glomerular diseases represent approximately 85% of CKD in the USA and Western Europe (Grahammer and Huber, 2016). Disruption of the glomerular filtration barrier at the capillary wall leads to nephrotic syndrome characterized by >3 g/day proteinuria, hypoalbuminemia, hyperlipidemia, and edema (Ding and Saleem, 2012; Andolino and Reid-Adam, 2015). Broadly, there are 3 major histopathologic lesions observed in nephrotic syndrome which are descriptively labeled as minimal change disease, focal segmental glomerulosclerosis (FSGS) and membranous nephropathy (Vivarelli et al., 2017; Bose et al., 2014; Ronco et al., 2021). Podocyte abnormalities is a hallmark feature in minimal change disease and FSGS, manifested as disruption of the highly organized cell-cell junction known as the slit diaphragm, referred to as foot process effacement. However, podocyte injury–whether central or secondary as a result of the complex cellular interrelationships within the glomerulus–is a major contributor to the development and progression of glomerular diseases (Kriz, 2002). Podocyte damage, along with its potential for recovery or lack thereof, is a defining event in the chronic kidney disease pathway (Kriz, 2002). Given that podocytes have limited replicative capacity, the primary response to its loss is podocyte hypertrophy (Wiggins et al., 2005). When this compensatory response is exceeded (≥20% podocyte loss in vivo), then there is mechanical strain, detachment and exposure of the glomerular basement membrane precipitating a sequence of events culminating in glomerular scarring (Wharram et al., 2005; Kretzler et al., 1994; Kriz, 2003).
1.2 Experimental models of podocyte loss
Clinically relevant experimental models of podocytopathies have proven useful to delineate cellular and molecular mechanisms governing glomerular diseases. Among the most commonly employed rodent models are the use of podocytotoxic agents such as in adriamycin nephropathy (Lee and Harris, 2011), puromycin aminonucleoside (Marshall et al., 2006), long term stimulation by FGF-2 (Kriz et al., 1995), Masugi nephritis (induction of glomerulonephritis using anti-kidney serum) (Shirato et al., 1996), and Thy-1.1 glomerulonephritis (induction of mesangial proliferative glomerulonephritis using an anti-Thy1.1 monoclonal antibody) (Kriz et al., 2003); as well as different genetic models such as knockout or loss-of-function mutations in key podocyte proteins like nephrin, podocin, and podocalyxin (Verma et al., 2018; Refaeli et al., 2019; Fan et al., 2009; Gigante et al., 2009). Drosophila nephrocytes have also been used as podocyte models, particularly to study the glomerular filtration barrier ultrastructure, due to their shared functional, morphological and molecular features with vertebrate podocytes (Helmstadter et al., 2017; van de Leemput and Han, 2024; Zhang and Chen, 2014). While no model perfectly recapitulates human nephrotic syndrome, these models mimic certain aspects of podocyte damage and symptoms observed in patients, including the formation of glomerular segmental scars, podocyte foot process effacement, podocyte loss and albuminuria.
1.3 Podocytes maintain a highly organized actin cytoskeleton and interdigitating foot processes to form slit diaphragms, which are crucial for their filtration function
Podocytes are highly specialized, terminally differentiated visceral epithelial cells that have a complex cell structure comprised of a cell body, major processes and foot processes (Figure 1). Lasagni et al. (2013), Lennon et al. (2014), Faul et al. (2007) The foot processes are cellular extensions of podocytes that wrap around the outer surface of glomerular capillaries, forming interdigitations with foot processes of adjacent podocytes and creating filtration slits called the slit diaphragm. The slit diaphragm is a specialized cell-cell junction consisting of various structural proteins including nephrin, podocin and TRPC6, and plays a role in preventing the leak of plasma proteins into the urine (Wiggins et al., 2005; Fan et al., 2009; Gigante et al., 2009). The basal surfaces of foot processes are in direct contact with the glomerular basement membrane, comprised of extracellular matrix proteins, which underlie fenestrated endothelium (Figure 1). Lasagni et al. (2013), Lennon et al. (2014), Faul et al. (2007) At the molecular level, foot processes are comprised of a highly organized actin-based contractile apparatus, including an actin filament network, actin-associated proteins, membrane-associated proteins that anchor actin filaments to the cell membrane of the foot process, and membrane-associated proteins that anchor actin filaments to the glomerular basement membrane (Figure 2A). Lasagni et al. (2013), Lennon et al. (2014), Faul et al. (2007) RHO-family GTPases such as RHOA, RAC1, and CDC42 are known regulators of podocyte actin cytoskeletal dynamics by activating the formation of actin stress fibers (Mouawad et al., 2013; Matsuda et al., 2021; Boi et al., 2023). RHO GTPases respond to external stimuli, including mechanical stretch induced by changes in glomerular capillary blood flow, by cycling between an active GTP-bound and inactive GDP-bound conformation that activates or deactivates, respectively, their downstream effectors. This coordination results in modulating podocyte morphology, actin cytoskeleton dynamics, and slit diaphragm integrity, critical for maintaining glomerular filtration function (Mouawad et al., 2013; Matsuda et al., 2021; Boi et al., 2023).
FIGURE 2
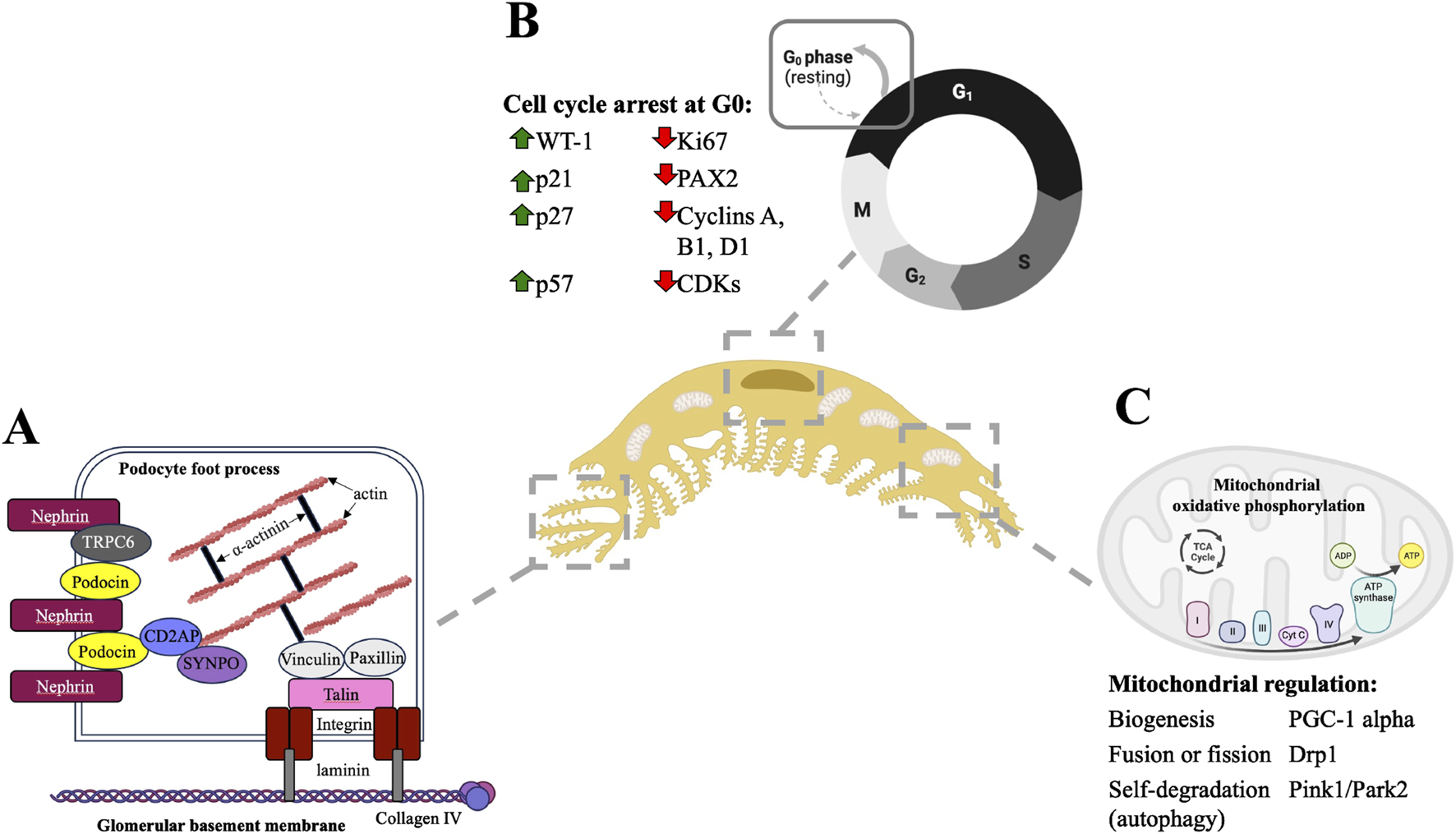
Podocytes are postmitotic cells comprised of a highly organized actin cytoskeleton, and these two cellular properties are supported by mitochondrial processes. (A) Podocyte foot processes are comprised of parallel actin filaments which are anchored to the cell membrane and to the underlying glomerular basement membrane by various actin-binding and actin-associated proteins (A) adapted from Lasagni et al., 2013; Curr Mol Med). (B) Podocytes undergo cell cycle arrest at the G0 resting phase. Within the nucleus, this terminally differentiated state is maintained through increased expression of cell cycle checkpoint proteins such as cyclin dependent kinase inhibitors and simultaneous decreased expression of proteins that promote cell cycle entry and proliferation. (C) The podocyte metabolic demands are largely due to the maintenance of their highly organized cellular structure and terminally differentiated state. Podocyte mitochondria have several adaptations to meet dynamic changes in metabolic demands, including biogenesis, fusion and fission to increase mitochondrial numbers and self-degradation through autophagy to remove damaged mitochondria and prevent excessive oxidative stress.
1.4 Podocytes are terminally differentiated post-mitotic cells
The highly organized podocyte actin cytoskeleton architecture is associated with their terminally differentiated state (Figure 2B). Lasagni et al. (2013), Thomasova and Anders (2015), Liapis et al. (2013) Mature podocytes show quiescence and permanent arrest in the G0/resting phase of the cell cycle (Liapis et al., 2013). Similarly, other highly specialized cell types such as neurons and cardiomyocytes maintain a permanent exit from the cell cycle, coupled with a highly structured actin cytoskeleton (Thomasova and Anders, 2015). Since actin cytoskeleton dynamics and rearrangement underlie cellular changes including de-differentiation, migration and division, by nature any rearrangement in the podocyte actin cytoskeleton due to kidney injury or other external insults has potential to disrupt its core cell structure and function. Any attempt for podocyte self-renewal through the proliferation and replication would require significant actin cytoskeleton rearrangement, causing ultrastructural changes in their foot processes and could introduce ‘leaks’ in the glomerular filtration barrier. Under baseline conditions, mature podocytes are felt to have a very limited capability for cell division and poses a challenge for self-regeneration when lost through injury (Lasagni et al., 2013; Thomasova and Anders, 2015; Liapis et al., 2013). Podocytes gradually acquire their post-mitotic, terminally differentiated state as nephrogenesis progresses during kidney development. In the earlier stages, precursor cells that generate podocytes initially express high levels of proliferation markers such as Ki67, cyclins A, B1 and D1 and cyclin dependent kinases; while downregulating expression of cell cycle checkpoint proteins and cyclin dependent kinase inhibitors such as p21, p27 and p57 (Thomasova and Anders, 2015). Transcription factors like PAX2 that regulate mesenchymal-to-epithelial transition are also initially expressed at high levels but become downregulated as epithelial differentiation occurs (Quaggin, 2002; Cunanan et al., 2024). These cellular changes allow podocyte precursor cells to retain motility, facilitating cellular and morphological changes during development. At later stages of nephrogenesis, precursor cells begin to express markers of adult podocytes including Wilms Tumour-1 (WT-1) and podocalyxin, increase expression of cell cycle checkpoint proteins, and conversely downregulate cell proliferation proteins, leading to cell cycle exit and differentiation (Figure 2B). Thomasova and Anders (2015) The differentiation of podocyte progenitors are also coupled with the transition from a simple to highly organized actin cytoskeleton (Lasagni et al., 2013). Adult podocytes maintain a post-mitotic state through unique mechanisms: increased expression of cyclin-dependent kinase inhibitors (Combs et al., 1998; Shankland et al., 2000; Shankland et al., 2017; Hiromura et al., 2001; Barisoni et al., 2000; Cormack-Aboud et al., 2009), DNA regulatory mechanisms leading to cell cycle arrest (Shankland et al., 2017; Shankland et al., 1999), expression of podocyte histone deacetylases which modulate DNA damage responses (Medina et al., 2023), lack of ability to establish an effective mitotic spindle which is required to complete cytokinesis (Lasagni et al., 2010), and abnormal M-type cyclin activity during mitosis (Shankland et al., 2017; Marshall and Shankland, 2007).
1.5 Podocytes require energy to maintain actin cytoskeleton dynamics, the slit diaphragm and their differentiated state
Podocytes require energy to maintain actin cytoskeleton dynamics, the slit diaphragm, and their differentiated state. While studies have shown mitochondria contribute to podocyte energy metabolism, there are also reports that have shown podocytes primarily rely on anaerobic glycolysis instead of mitochondrial metabolism to maintain glomerular filtration (Brinkkoetter et al., 2019). Other studies have also established that podocytes are not as reliant on mitochondrial oxidative phosphorylation as other renal cells, such as proximal tubules, which have higher metabolic demands and are more susceptible to ischemic injury (Gobe et al., 1999; Wei et al., 2010). Nonetheless, transmission electron microscopy reveals a concentration of mitochondria within the podocyte cell body and foot processes, suggesting some involvement in cellular function and homeostasis (Imasawa and Rossignol, 2013; Abe et al., 2010). To adapt to changes in energy demand, podocyte mitochondria can undergo biogenesis, fusion or fission, mediated by proteins such as PGC-1 alpha and DRP1, to modulate mitochondrial dynamics as needed (Figure 2C). Additionally, podocytes maintain mitochondrial control through autophagy, a process mediated by proteins such as PINK1 and PARK2, which facilitates the degradation of damaged mitochondria to preserve cellular integrity (Handschin and Spiegelman, 2006; Fontecha-Barriuso et al., 2020; Elgass et al., 2013; Chen et al., 2020; Galluzzi et al., 2017; Jiang et al., 2020).
1.6 Podocyte loss results from disruptions in essential functions, including actin cytoskeleton integrity, slit diaphragm structure, cell differentiation and metabolism
1.6.1 Actin cytoskeleton and slit diaphragm
Podocytes are continuously exposed to significant mechanical stress generated by the flow of glomerular ultrafiltrate (Faul et al., 2007; Blaine and Dylewski, 2020). Podocytes respond by modifying their foot process actin cytoskeleton. Expectedly, disruptions in actin cytoskeleton structure have been linked to podocyte loss in glomerular disease. One of the major causes of podocyte disruption are pathogenic variants in genes that encode actin binding and slit diaphragm proteins (Faul et al., 2007; Blaine and Dylewski, 2020). Some genes that contribute or regulate the actin cytoskeleton are ACTN4, ARHGAP24 and INF2, pathogenic variants in which are causative for adult-onset FSGS (Akilesh et al., 2011; Schell and Huber, 2012; Brown et al., 2010; Sun et al., 2021; Kaplan et al., 2000). Mice deficient in or with a mutant form of alpha-actinin-4 developed podocyte damage and glomerular disease (Kos et al., 2003; Michaud et al., 2003; Yao et al., 2004). ARHGAP24 inhibits cell spreading and motility in podocytes by regulating RHOA/RAC1 signalling (Matsuda et al., 2021); consequently, Arghap24 knockdown in mouse podocyte cell cultures demonstrated abnormally increased motility associated with actin cytoskeleton disruption (Akilesh et al., 2011; Schell and Huber, 2012). INF2 regulates podocyte actin dynamics and slit diaphragm protein trafficking by opposing RHO-mediated actin polymerization and rearrangement (Brown et al., 2010; Sun et al., 2013). As a result, kidney biopsy tissue obtained from a patient with a pathogenic variant in INF2 showed abnormal aggregation of podocyte actin filaments associated with podocyte foot process effacement (Brown et al., 2010). Cultured human podocytes with INF2 knockdown showed a deregulated podocyte actin cytoskeleton and disrupted trafficking of slit diaphragm proteins (Sun et al., 2013). Variants in nephrin (NPHS1) and podocin (NPHS2), both encoding slit diaphragm transmembrane proteins, are also causative for Mendelian forms of nephrotic syndrome (Verma et al., 2018; Kestila et al., 1998; Ruotsalainen et al., 2000; Welsh and Saleem, 2010; Boute et al., 2000).
Experimentally induced podocyte injury through toxins or external insults also results in disrupted actin cytoskeleton organization. Exposure of podocytes to puromycin aminonucleoside causes altered intracellular localization of slit diaphragm proteins nephrin and podocin with disruptions in scaffolding protein CD2AP, resulting in decreased binding and depolymerization of actin filaments (Saleem et al., 2002). Adriamycin injured mice with knockout of synaptopodin (Synpo), which encodes an actin-associated protein, showed worsened podocyte injury and glomerular disease due to loss of actin stress fibers, impaired cell migration, mislocalization and decreased expression of alpha-actinin-4, (ACTN4) and altered activity levels of RHOA and RAC1 (Ning et al., 2020). In another model, exposure of podocytes to palmitic acid led to disruptions in actin cytoskeleton integrity, as well as decreased levels of actin-associated proteins including nephrin, vimentin, and the α3 chain of type IV collagen (CO4A3), overall culminating in cell loss (Martinez-Garcia et al., 2015). Furthermore, mouse podocytes grown in a high glucose environment led to abnormally increased activation of FYN/ROCK signalling, resulting in the phosphorylation of paxillin, interrupting the link between the foot process actin cytoskeleton and the glomerular basement membrane. These changes led to decreased levels of synaptopodin, marked actin cytoskeleton rearrangement and increased cell motility which exacerbated podocyte loss and glomerular dysfunction (Lv et al., 2016). HIV-1 protein NEF also promotes actin cytoskeleton changes in podocytes due to the ability of NEF to alter the activity levels of actin cytoskeleton regulators RHOA and RAC1 (Lu et al., 2008). These cellular and molecular effects led to podocyte damage, albuminuria and glomerular disease consistent with pathological findings in HIV-associated nephropathy (HIVAN) (Lu et al., 2008).
Taken together, these findings demonstrate that actin cytoskeleton integrity is crucial for healthy podocyte function. Regardless of underlying cause, alterations in normal podocyte actin cytoskeleton and foot process dynamics are strongly linked to overall loss (Faul et al., 2007; Blaine and Dylewski, 2020).
1.6.2 Differentiated state and cell cycle abnormalities
Podocyte cell cycle re-entry has been observed in human glomerular diseases such as IgA nephropathy, collapsing FSGS, HIVAN, lupus nephritis, and in experimental models of podocyte injury including adriamycin nephropathy (Figure 3), aminoglycoside-induced nephrosis, and TGF-beta overexpression in cultured mouse podocytes (Thomasova and Anders, 2015; Liapis et al., 2013; Barisoni et al., 2000; Barisoni et al., 2007). In these disease conditions, podocytes show a maladaptive attempt for cell cycle re-entry indicated by abnormal levels of cell cycle regulator proteins cyclins and cyclin-dependent kinase inhibitors, binucleation, micronuclei formation, and abnormally-formed mitotic spindles (Thomasova and Anders, 2015; Liapis et al., 2013; Barisoni et al., 2000; Barisoni et al., 2007). For example, in patient biopsies with HIVAN and collapsing FSGS, the loss of expression of cell cycle inhibitor proteins p27, p57 and cyclin D1, coupled with the abnormal re-expression of proliferation markers Ki67 and cyclin A, was observed specifically in podocytes that are found in areas of glomerular collapse (Barisoni et al., 2000). Increased levels of mitotic arrest deficient 2-like protein 2 (MAD2B), which regulates metaphase-to-anaphase transition, was also observed in FSGS patients and adriamycin treated mice and associated with podocyte cell cycle re-entry (Bao et al., 2021). These findings thus suggest that abnormal podocyte cell cycle re-entry plays a role in glomerular disease progression.
FIGURE 3
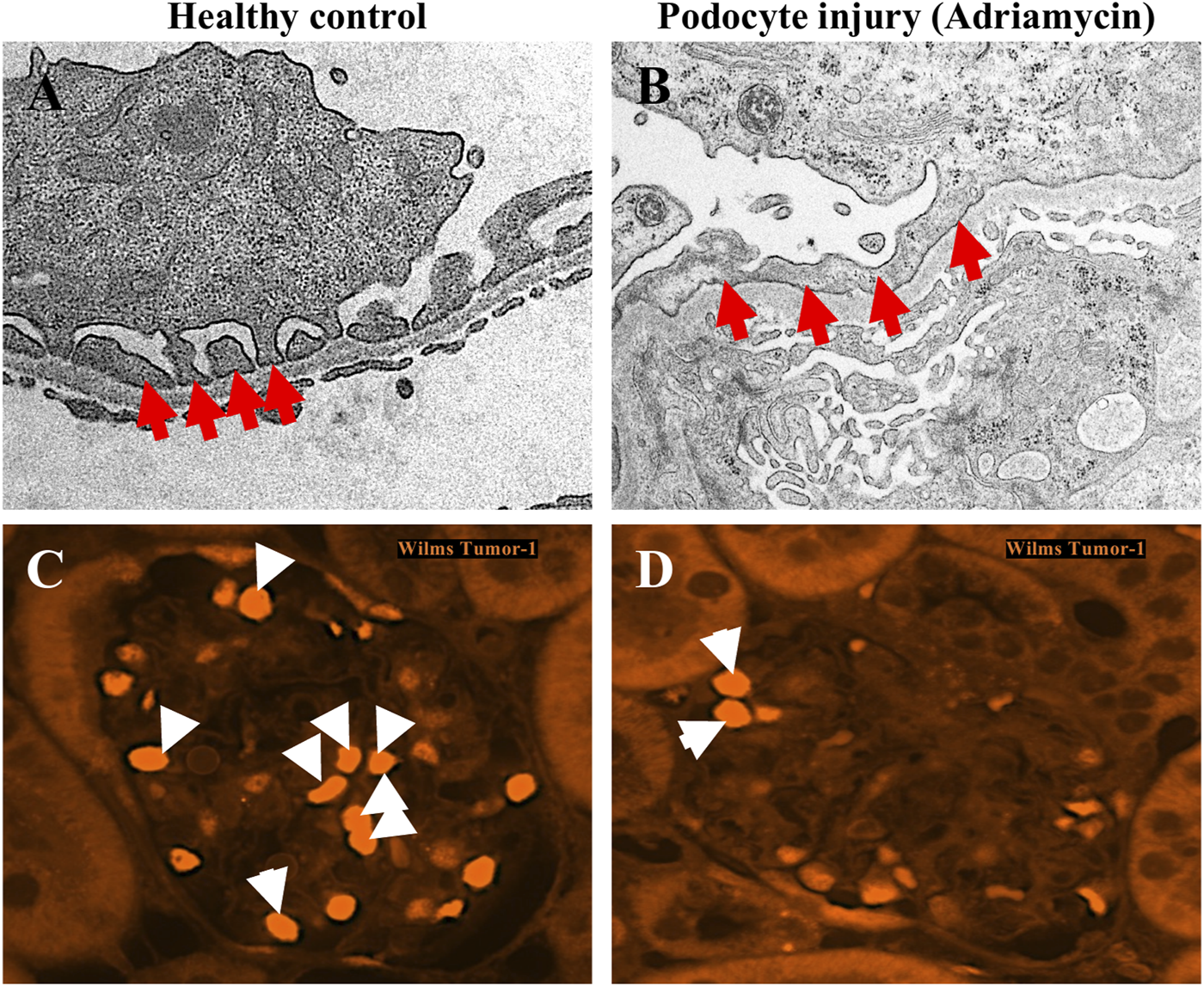
Podocyte foot process effacement and podocyte loss resulting from adriamycin injury. (A,B) Transmission electron microscopy images of podocytes from a healthy mouse glomerulus shows normal podocyte foot process morphology, characterized as interdigitating finger-like projections (red arrows) from the cytoplasm of podocytes which are in direct contact with the underlying glomerular basement membrane (A). In mice with podocyte injury due to adriamycin, the characteristic interdigitating pattern of podocyte foot processes is lost (red arrows), and instead are observed as a continuous, flattened structure lining the glomerular basement membrane. (C,D) Immunofluorescence staining for podocyte nuclear marker Wilms Tumour-1 (WT-1) demonstrates decreased number of podocytes (white arrows) in adriamycin-injured mice (D) compared to healthy controls (C).
Under normal conditions, podocytes maintain cell quiescence but there are a number of pathologic or stress-inducing stimuli that can trigger them to undergo cell cycle re-entry including lupus, high glucose, ionizing radiation, fibroblast growth factors, and drugs that can interfere with cytoskeletal assembly stability (Kriz et al., 1995; Hagen et al., 2016; Vakifahmetoglu et al., 2008; Surova and Zhivotovsky, 2013; Xie et al., 2019). Under glomerular disease conditions characterized by podocyte loss, it is possible that the remaining podocytes attempt self-renewal to a certain extent (Cunanan et al., 2024). When attempting cell cycle re-entry, podocytes are forced to override certain checkpoints such as G2/M arrest (Lasagni et al., 2013; Thomasova and Anders, 2015; Liapis et al., 2013). This enables podocytes to undergo DNA synthesis and chromosome segregation, but the lack of sufficient actin dynamics renders them unable to complete cytokinesis efficiently. This generates podocytes with chromosomal irregularities and multinucleated cells that render them genetically unstable, leading to necrosis, apoptosis, podocyte detachment and loss, ultimately causing proteinuria and glomerulosclerosis, a phenomenon referred to as “mitotic catastrophe” (Lasagni et al., 2013; Thomasova and Anders, 2015; Liapis et al., 2013; Tian X. et al., 2023). In support of this, studies have found abnormal levels of binucleated podocytes in urine samples from patients with FSGS and IgA nephropathy (Hara et al., 2001; Lemley et al., 2002). In addition, mouse podocyte culture demonstrated binucleation and cell cycle re-entry upon stimulation with basic fibroblast growth factor. After puromycin aminonucleoside injury, the binucleated and proliferating podocytes were more susceptible to cell death compared to unstimulated podocytes (Hagen et al., 2016). As an alternative mechanism in response to pathological stimuli, podocytes can maintain a state of arrest in cell cycle checkpoints but instead attempt to undergo cell hypertrophy to compensate for podocyte loss (Lasagni et al., 2013; Thomasova and Anders, 2015; Liapis et al., 2013). In a case of human advanced diabetic glomerulosclerosis, most podocytes showed hypertrophy and partial detachment from the glomerular basement membrane (Liapis et al., 2013). In either case, podocyte cell cycle re-entry and growth arrest leading to hypertrophy both generate abnormal cells which ultimately cannot fully recapitulate the structure and function of normal podocytes (Lasagni et al., 2013; Thomasova and Anders, 2015; Liapis et al., 2013).
Overall, these findings demonstrate that podocytes retain a post-mitotic state through tightly controlled cellular and molecular mechanisms. Under conditions of significant stress, podocytes may be forced to undergo mitosis, but this results in nuclear irregularities that eventually lead to loss.
1.6.3 Metabolism
Genetic abnormalities in key metabolic regulators, which lead to mitochondrial dysfunction and injury in podocytes, have been reported in both patients and rodent models of glomerular disease. Pathogenic variants in COQ2 (coenzyme Q2) and COQ6 (coenzyme Q6), which encode coenzymes required for the biosynthesis of the essential electron carrier ubiquinone in the mitochondrial transport chain, have been reported in familial FSGS (Diomedi-Camassei et al., 2007; Heeringa et al., 2011). Patients with COQ2 mutations have increased numbers of dysmorphic mitochondria in podocytes, associated with decreased activities of electron transport chain complexes II and III and decreased ATP generation (Diomedi-Camassei et al., 2007). Podocytes also displayed severe foot process effacement and significant hypertrophy (Diomedi-Camassei et al., 2007). Furthermore, knockdown of COQ6 in mouse podocyte cell cultures and zebrafish embryos resulted in apoptosis (Heeringa et al., 2011). Genetic defects in PDSS2 (decaprenyl diphosphate synthase-2), encoding another enzyme involved in ubiquinone biosynthesis, were also found in patients with nephrotic syndrome (Lopez et al., 2006). Podocyte specific Pdss2 knockout in mice resulted in foot process effacement and proteinuria (Peng et al., 2008). Podocyte-specific knockout of Mtorc1 (mammalian target of rapamycin complex 1) in mice, which regulates mitochondrial activity and biogenesis, resulted in proteinuria and glomerular scarring (Godel et al., 2011). Similarly, podocyte-specific knockout of Atg5 (autophagy related-5), a ubiquitin ligase involved in autophagy, resulted in increased oxidative stress, podocyte injury, glomerulosclerosis and proteinuria (Hartleben et al., 2010). Lastly, mice with a loss-of-function variant in Crif1 (CR6-interacting factor-1), encoding a protein that participates in oxidative phosphorylation by regulating the insertion of polypeptides into the inner mitochondrial membrane, developed podocyte injury, glomerular sclerosis and marked albuminuria (Na et al., 2021). Studies using cultured mouse podocytes also showed loss of CRIF1 protein resulted in disrupted oxidative phosphorylation and abnormal F-actin aggregation, demonstrating that normal oxidative phosphorylation is necessary to maintain podocyte actin cytoskeleton and glomerular filtration barrier integrity (Na et al., 2021).
External insults can also lead to changes in expression levels of mitochondrial proteins and compromise function. For example, adriamycin-induced podocyte damage in rats, as well as in mouse podocyte cell cultures, resulted in decreased levels of PGC-1 alpha, a key regulator of mitochondrial biogenesis (Zhu et al., 2014). In these models, decreased PGC-1 alpha levels led to impaired oxidative phosphorylation, increased susceptibility to oxidative stress and apoptosis (Zhu et al., 2014). Similarly, mice with aldosterone-induced podocyte injury demonstrated increased renal oxidative stress and mitochondrial dysfunction, evident in reduced mitochondrial DNA copy number, mitochondrial membrane potential, and ATP production (Su et al., 2013). Proteinuria and podocyte injury were observed in these mice several days after the mitochondrial defects took place, demonstrating that mitochondrial dysfunction is an early event in podocyte injury (Su et al., 2013). Lastly, human podocytes exposed to a high glucose growth medium triggered an abnormal accumulation of pyruvate, the end-product of glycolysis (Feng et al., 2019). This resulted in interruption of downstream metabolic processes, oxidative phosphorylation and mitochondrial fragmentation leading to podocyte cell death (Feng et al., 2019).
Altogether, these findings demonstrate that podocytes have metabolic demands which are involved in maintaining podocyte structure and function. Mutations in key mitochondrial or metabolic regulators and various external insults can disrupt podocyte mitochondrial function and lead to decreased energy production, representing a mechanism that could contribute to podocyte loss.
1.7 Experimental approaches to mitigate podocyte loss
An understanding of the mechanisms that maintain podocyte function and those that contribute to injury provide insights for therapeutic targeting in chronic kidney disease. Several studies have demonstrated that a variety of molecules and compounds can act on podocytes to stabilize their actin cytoskeleton, regulate homeostasis to maintain their post-mitotic state, and regulate mitochondrial dynamics and metabolism (Figure 4).
FIGURE 4
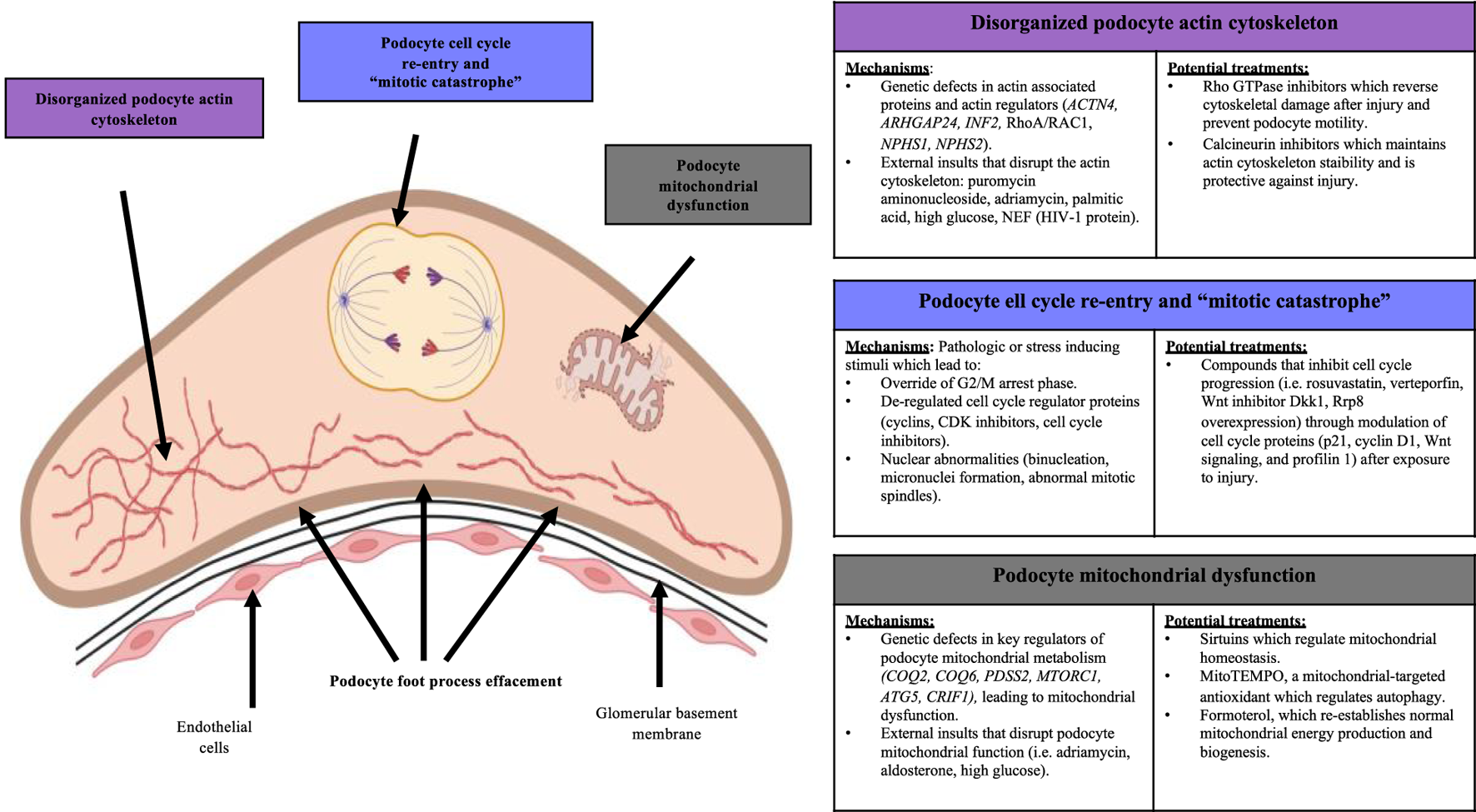
Mechanisms of podocyte loss and potential treatments. Disorganization of the podocyte actin cytoskeleton, cell cycle re-entry and mitotic catastrophe, and mitochondrial dysfunction are three cellular mechanisms that underlie podocyte loss. These mechanisms are also modifiable by potential treatments that can modulate the severity of podocyte injury.
Mechanisms leading to podocyte actin cytoskeleton derangements can be reversed through certain drugs and molecules. Elevated levels of RHO GTPases and their regulators RHOA, RAC1 and ROCK contribute to podocyte dysfunction through disrupting the actin cytoskeleton resulting in abnormally increased motility (Akilesh et al., 2011; Babelova et al., 2013; Tian F. et al., 2023). These observations have led to the development of various agents that can inhibit RHO GTPases and their regulators. Mouse podocyte cultures exposed to mechanical strain simulating glomerular hypertension showed increased activity of RHOA/RAC1, actin cytoskeleton re-organization, de-differentiation of cells towards a mesenchymal phenotype, increased levels of fibrotic markers alpha-SMA (alpha-smooth muscle actin) and fibronectin, and decreased levels of podocyte proteins WT-1 and nephrin (Babelova et al., 2013). Inhibition of RAC1 and ROCK through EHT1846 and SAR407881, respectively, led to re-organization of the mouse podocyte actin cytoskeleton, reduction of mesenchymal de-differentiation, reduced fibrosis markers, and rescued WT-1 and nephrin levels similar to healthy controls (Babelova et al., 2013). Similar findings were reported in mice with experimental lupus nephritis where treatment with RHO GTPase inhibitor Fasudil prevented cytoskeletal breakage in podocytes (Tian F. et al., 2023), and in cultured human podocytes grown with damaging agents where glucocorticoid treatment markedly reduced RAC1 overactivity and cell motility (McCaffrey et al., 2017). Synaptopodin is also a regulator of RHO GTPases in podocytes. Dephosphorylation of synaptopodin by calcineurin, leading to synaptopodin degradation, was markedly increased in mice with lipopolysaccharide-induced podocyte injury (Faul et al., 2008). This also resulted in de-stabilization of the podocyte actin cytoskeleton. Cyclosporine A is a well-established treatment for FSGS and is known for its role in stabilizing the podocyte cytoskeleton (Cattran et al., 1999). Similarly, mice treated with cyclosporine A, a calcineurin inhibitor, prevented synaptopodin degradation and maintained actin cytoskeleton stability, demonstrating a protective effect against lipopolysaccharide injury (Faul et al., 2008). Mycophenolate Mofetil, an immunosuppressive drug, has been shown to stabilize the podocyte actin cytoskeleton in mice with podocyte injury from nephrotoxic serum. Mechanisms involved modulating calcium signaling, leading to reduced proteinuria and improved foot process architecture (Hackl et al., 2023). Altogether, these findings demonstrate that modulating podocyte actin cytoskeleton dynamics represents a therapeutic pathway.
Maladaptive podocyte mitosis can also be targeted to prevent podocyte loss. For example, the cell cycle protein p21 has a pro-survival effect in podocytes through modulating intrinsic DNA repair processes and inhibiting cell cycle (Cormack-Aboud et al., 2009). Mouse podocytes grown with rosuvastatin prior to induction of injury with puromycin aminonucleoside resulted in increased p21 levels and reduced apoptosis (Cormack-Aboud et al., 2009). Abnormally increased levels of YES-associated protein (YAP) was found in cultured mouse podocytes after adriamycin injury, accompanied by cell cycle re-entry and mesenchymal transition shown by increased cyclin D1 levels and downregulation of podocyte proteins (Xie et al., 2019). By contrast, treatment of podocytes with YAP inhibitor verteporfin prior to adriamycin injury resulted in decreased cell cycle re-entry and decreased mesenchymal transformation by reversing overexpression of cyclin D1 (Xie et al., 2019). Increased levels of telomerase reverse transcriptase (TERT) accompanied by abnormally increased activation of WNT signalling were observed in human and mouse kidneys with HIVAN (Shkreli et al., 2011). These were associated with cell cycle re-entry and loss of differentiated phenotypes, similar to immature podocyte progenitor cells. However, silencing TERT expression and treatment with WNT inhibitor DKK1 in mice reverted the podocytes to a differentiated state and reduced glomerular disease (Shkreli et al., 2011). Lastly, profilin 1 is a critical regulator of podocyte quiescence and its disrupted expression was accompanied by mitotic catastrophe leading to podocyte loss. Overexpression of ribosomal RNA processing 8 (RRP8) in podocyte cell cultures obtained from mice with profilin knockout partially reversed and prevented abnormal cell cycle re-entry (Tian X. et al., 2023). Thus, these studies demonstrate that targeting abnormal cell cycle re-entry in podocytes can re-establish podocyte quiescence and reduce the severity of glomerular injury.
Restoring podocyte mitochondrial homeostasis can also mitigate podocyte loss. Decreased levels of sirtuins have been associated with podocyte injury in rodents due to aging-associated podocyte loss (Liu T. et al., 2022; Chen et al., 2023). Sirtuins protect podocytes by deacetylating various mitochondrial targets including PGC-1 alpha, NF‐κB, FOXO, HIF-1 alpha, leading to the modulation of mitochondrial homeostasis. Compounds that increase sirtuin activity include natural products such as resveratrol, baicalin and grape seed extract, and synthetic compounds such as metformin, olmesartan, and SRT1720, all of which have been reported to reduce the severity of podocyte injury in experimental models (Liu T. et al., 2022; Chen et al., 2023). Decreased levels of PINK1/Parkin pathway-mediated mitochondrial autophagy was observed in Sprague-Dawley rats with experimental membranous glomerulonephritis, resulting in increased podocyte loss due to dysregulated mitochondrial function (Liu B. et al., 2022). This was reversed by treatment with MitoTEMPO, a mitochondria-targeted antioxidant, which increased PINK1/Parkin activity, resulting in modulated mitochondrial autophagy and reduced susceptibility to podocyte injury (Liu B. et al., 2022). Decreased levels of progranulin, a secreted glycoprotein that plays a role in mitochondrial autophagy, was associated with exacerbated mitochondrial damage and podocyte apoptosis in diabetic nephropathy (Zhou et al., 2019). In vitro treatment with progranulin on human podocytes cultured in high glucose medium attenuated the SIRT1-PCG-1 alpha/FOXI signaling pathway, resulting in re-establishment of mitochondrial membrane potential, improved mitochondrial biogenesis and decreased podocyte injury (Zhou et al., 2019). Similarly, placenta-derived mesenchymal stem cells were found to reduce the severity of high glucose induced podocyte injury by stimulating increased levels of mitochondrial autophagy mediated by PINK1/Parkin and the SIRT1-PCG-1 alpha pathways (Han et al., 2023). Lastly, human podocyte cultures and mice exposed to podocyte injury showed accelerated recovery when treated with formoterol, a β2-adrenergic receptor agonist (Arif et al., 2019). Formoterol was associated with increased PGC-1 levels, which resulted in re-establishment of normal mitochondrial structure, oxygen consumption rate, ATP generation, and enhanced podocyte mitochondrial biogenesis (Arif et al., 2019). Taken together, these studies demonstrate that targeting mitochondrial homeostasis in podocytes can attenuate injury and loss.
1.8 Future perspectives
Recent advances in single-cell transcriptomics, high-resolution imaging, and organoid models have significantly improved our understanding of podocyte structure and function in healthy and diseased states. Recent single-cell RNA sequencing (scRNA-seq) studies have identified key transcriptional programs and molecular signatures of podocyte injury in models of diabetic kidney disease, immune-mediated glomerulopathy, podocyte drug toxicities, and genetic podocytopathies (Table 1). Fu et al. (2016), Tsai et al. (2023), Chung et al. (2020), Bais et al. (2021) Gene ontology and pathway enrichment analyses reveal podocyte-specific biological and molecular processes that take place during podocyte injury and loss, all of which fall under the umbrella of dysregulated actin cytoskeleton, cell cycle, and metabolism. For example, in mouse models of diabetic kidney disease, pathway analysis of dysregulated genes revealed involvement in actin cytoskeleton organization, RNA processing and endoplasmic reticulum function, autophagy, altered cell survival, alterations in glucose and lipid metabolism, induction of apoptosis, and increased expression of extracellular matrix and matrix-modifying proteins in podocytes (Fu et al., 2016; Tsai et al., 2023; Chung et al., 2020). Furthermore, scRNA-seq has uncovered unique podocyte subpopulations with distinct responses to stress, suggesting heterogeneity in podocyte resilience and repair potential (Clark et al., 2022). As scRNA-seq datasets continue to grow, integrating transcriptomic findings with functional assays and spatial multi-omics will be essential to refine our understanding of podocyte pathology. Next, super resolution microscopy techniques including STED (stimulated emission depletion microscopy), STORM (stochastic optical reconstruction microscopy) and 3D-SIM (3 dimensional structured illumination microscopy) can reach resolutions down to 80–20 nm and have been used in obtaining nanoscale localization of podocyte slit diaphragm proteins and glomerular basement membrane proteins, nanoscopic resolution of actin cytoskeleton organization in podocytes under baseline and pathological conditions, and a higher resolution identification of foot process effacement in patient biopsies (Angelotti et al., 2021; Siegerist et al., 2018; Unnersjo-Jess, 2023). Atomic force microscopy has also been used to examine the mechanical properties of podocytes and the underlying glomerular basement membrane in healthy and diseased states (Siegerist et al., 2018; Wyss et al., 2011; Reynolds, 2020). Lastly, studies using glomerular organoids derived from human cells (i.e., induced pluripotent stem cells/iPSCs obtained from glomerular disease patients) have also provided accessible approaches to perform in vitro modelling of human podocyte injury and loss (Hale et al., 2018; Hale and Little, 2023; Lasse et al., 2023). These glomerular organoids have a more comparable morphology to human glomeruli in vivo and contain podocytes that recapitulate in vivo structure and apicobasal polarity, in comparison to cultured primary human and rodent podocytes. As a result, these glomerular models have helped advance the understanding of the podocyte transcriptome in patients with glomerular disease. Glomerular organoids have also served as valuable tools for drug screening to determine the efficacy of compounds that can mitigate abnormalities in podocyte structure and function (Hale et al., 2018; Hale and Little, 2023; Lasse et al., 2023).
TABLE 1
| Glomerular disease | Disease model | Pathways involved |
|---|---|---|
| Diabetic Nephropathy | Type I diabetes (streptozotocin injection with eNOS knockout) | Upregulated: cell death, actin organization |
| Downregulated: RNA processing, endoplasmic reticulum (ER) function (mTOR signaling, autophagy, ER stress response) | ||
| Type II diabetes (db/db leptin receptor-deficient mice) | Dysregulated: autophagy, cell survival, cell death | |
| Type II diabetes (ob/ob leptin-deficient mice) | Dysregulated: glucose and lipid metabolism | |
| Upregulated: apoptosis, matrix and matrix-modifying proteins | ||
| Lupus Nephritis/Goodpasture Disease | Nephrotoxic serum injection | Upregulated: cytoskeleton regulation, cell adhesion, inflammatory response, Hippo signaling, TGF-β signaling, NF-κB signaling, FoxO signaling |
| Downregulated: podocyte differentiation | ||
| Podocyte Drug Toxicity | Adriamycin injection | Upregulated: p53 signaling |
| Downregulated: podocyte markers | ||
| Podocyte-Specific Genetic Disease | CD2AP knockout | Upregulated: apoptosis, matrix proteins |
| Doxycycline-induced CTCF knockout in podocytes | Dysregulated: mitochondrial function, cytoskeletal organization, cell-matrix adhesion | |
| Upregulated: Col4a5 (potential cell-matrix adhesion) | ||
| Downregulated: Vegfa (autocrine pro-survival ligand) |
Summary of findings from single cell RNA sequencing studies, demonstrating abnormalities in the podocyte transcriptome under disease conditions.
While the past 2 decades have largely expanded our knowledge of podocyte biology under health and disease, there are several withstanding challenges and gaps in knowledge in therapeutic development for podocyte loss. Despite promising pre-clinical data for a variety of compounds, there has been a lack of significant clinical translation (Meliambro et al., 2024). The current standard of care for podocytopathies largely rely on repurposed immunosuppressive treatments, with a relatively limited understanding of underlying mechanisms (Meliambro et al., 2024; Schonenberger et al., 2011; Cambier et al., 2018; Mirioglu et al., 2024). A major reason for the lack of therapeutic development for podocytopathies is the challenge of regenerating podocytes. In this review, we discussed mechanisms underlying the highly specialized, terminally differentiated nature of podocytes. These findings have led to the widely accepted notion that podocytes have a limited capacity for regeneration and loss is mostly irreversible. There is promise, however, with recent studies suggesting that there is a limited capacity for podocyte regeneration by other resident glomerular cell populations including parietal epithelial cells, which are proposed to have stem cell-like properties (Cunanan et al., 2024; Romagnani and Remuzzi, 2013; Ronconi et al., 2009; Lasagni et al., 2015; Wanner et al., 2014; Berger et al., 2014; Peired et al., 2014; Sagrinati et al., 2006; Kaverina et al., 2019), progenitor “renin lineage” cells (Pippin et al., 2013; Pippin et al., 2015; Eng et al., 2018), and bone marrow-derived mesenchymal stem cells (Zoja et al., 2012; Khalilpourfarshbafi et al., 2017; Wang et al., 2013). However, the concept of stem cell-based regeneration of podocytes from these cells is widely debated. Some studies demonstrate stem cell-like properties for potential podocyte progenitors (Romagnani and Remuzzi, 2013; Ronconi et al., 2009; Lasagni et al., 2015; Wanner et al., 2014; Berger et al., 2014; Peired et al., 2014; Sagrinati et al., 2006; Kaverina et al., 2019), while in contrast other studies support the contribution of these cells to pathogenic proliferation and the activation of pro-fibrotic pathways (Dijkman et al., 2005; Kuppe et al., 2019; Hakroush et al., 2014; Smeets et al., 2004; El Agha et al., 2017; Qin et al., 2023). These seemingly contrary studies have significant differences in their methodologies that could reconcile disparate results.
Another significant challenge in the treatment of glomerular diseases is the development of effective podocyte-directed drug delivery systems. Despite promising data from in vitro and animal studies showing therapeutic agents that can act on injured podocytes to re-establish their normal structure and function, our understanding of podocyte-targeted delivery systems is relatively lacking. For example, there are genes and signalling pathways, such as the Hippo/YAP signaling pathway and Tyro3, that are critical for podocyte survival but non-targeted delivery to other cell types can confer an oncogenic risk (Meliambro et al., 2024; Campbell et al., 2013; Zhong et al., 2023; Smart et al., 2018). Nanoparticle-based delivery systems can help reduce these off-target effects by protecting drugs from degradation and enabling controlled release. Nanoparticles can also be modified to carry ligands that specifically bind to podocyte receptors, such as nephrin and podocin, to improve podocyte delivery and uptake of drugs (Meliambro et al., 2024; Campbell et al., 2013; Zhong et al., 2023; Smart et al., 2018). Gene therapy, another promising approach, have been used to correct genetic mutations in podocytopathies (Mason et al., 2022; Daga et al., 2024; Ding et al., 2023). For example, adeno-associated virus (AAV) mediated delivery has been used in combination with CRISPR-Cas9 gene editing techniques to correct mutations in COL4A3 and COL4A5 in cultured podocytes from patients Alport syndrome, as well as to rescue NPHS2 mutations in cultured podocytes from nephrotic syndrome patients (Mason et al., 2022; Daga et al., 2024; Ding et al., 2023). While nanoparticle and AAV-mediated drug delivery to injured podocytes show evidence reduced injury in these pre-clinical studies, both techniques also have significant challenges and limitations. Nanoparticles have instability issues such as aggregation, premature drug release, and degradation at inappropriate times, as well as difficulty in achieving consistent drug encapsulation, controlled release profiles, and proper biodistribution (Geszke-Moritz and Moritz, 2024). Gene therapy techniques such as AAV and CRISPR-Cas9 also have limitations, such as AAV’s limited packaging capacity (4.7 kb), immune responses to Cas9 proteins, and off-target effects that can cause unintended genetic alterations (Wang et al., 2024). While smaller Cas9 variants, dual AAV systems, and self-complementary AAV improve delivery and efficiency, they are limited by reduced cleavage activity, the need for simultaneous vector delivery, and higher risks at increased doses (Wang et al., 2024).
Overall, the translation of these findings from research to clinical practice require further rigorous investigation to determine patient safety and efficacy. Despite these withstanding challenges, our current knowledge of podocyte biology together with the identification of novel targets to attenuate podocyte injury, certainly provide promising insights to help overcome the lack of therapeutic development in this area.
2 Conclusion
The studies highlighted in this review demonstrate the challenge in therapeutic development for diseases of the podocyte, which are highly specialized in their morphology and function. The highly organized podocyte actin cytoskeleton is difficult to re-establish in the event of podocyte loss. Further, podocytes retain a quiescent, terminally differentiated cell state, making it unlikely for them to undergo cell division and self-renewal in the event of injury. Lastly, various insults leading to increased mitochondrial demands and stress also make podocytes vulnerable to injury. Overall, an understanding of the various cellular and molecular mechanisms that retain this intricate podocyte structure and function under healthy conditions, as well as the pathogenic mechanisms that lead to disruptions in the actin cytoskeleton, cell cycle arrest, and mitochondrial function has led to the identification of potential therapeutic targets that have the potential to mitigate the severity of glomerular diseases characterized by podocyte loss.
Statements
Author contributions
JC: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review and editing. DZ: Writing – original draft. AJP: Writing – review and editing. MB: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Joanna Cunanan is a recipient of the Frederick Banting and Charles Best Canada Graduate Scholarships–Doctoral Award from the Canadian Institutes of Health Research (CIHR), and the Michael Smith Foreign Study Supplements from CIHR. Moumita Barua is the Principal Investigator for a research grant entitled ‘PAX2 Mediates Glomerular Repair’, funded by CIHR.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Abe Y. Sakairi T. Kajiyama H. Shrivastav S. Beeson C. Kopp J. B. (2010). Bioenergetic characterization of mouse podocytes. Am. J. Physiol. Cell Physiol.299 (2), C464–C476. 10.1152/ajpcell.00563.2009
2
Akilesh S. Suleiman H. Yu H. Stander M. C. Lavin P. Gbadegesin R. et al (2011). Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J. Clin. Invest121 (10), 4127–4137. 10.1172/JCI46458
3
Andolino T. P. Reid-Adam J. (2015). Nephrotic syndrome. Pediatr. Rev.36 (3), 117–125. 10.1542/pir.36-3-117
4
Angelotti M. L. Antonelli G. Conte C. Romagnani P. (2021). Imaging the kidney: from light to super-resolution microscopy. Nephrol. Dial. Transpl.36 (1), 19–28. 10.1093/ndt/gfz136
5
Arif E. Solanki A. K. Srivastava P. Rahman B. Fitzgibbon W. R. Deng P. et al (2019). Mitochondrial biogenesis induced by the β2-adrenergic receptor agonist formoterol accelerates podocyte recovery from glomerular injury. Kidney Int.96 (3), 656–673. 10.1016/j.kint.2019.03.023
6
Babelova A. Jansen F. Sander K. Lohn M. Schafer L. Fork C. et al (2013). Activation of Rac-1 and RhoA contributes to podocyte injury in chronic kidney disease. PLoS One8 (11), e80328. 10.1371/journal.pone.0080328
7
Bais A. S. Cerqueira D. M. Clugston A. Bodnar A. J. Ho J. Kostka D. (2021). Single-cell RNA sequencing reveals differential cell cycle activity in key cell populations during nephrogenesis. Sci. Rep.11 (1), 22434. 10.1038/s41598-021-01790-6
8
Bao D. Su H. Lei C. T. Tang H. Ye C. Xiong W. et al (2021). MAD2B-mediated cell cycle reentry of podocytes is involved in the pathogenesis of FSGS. Int. J. Biol. Sci.17 (15), 4396–4408. 10.7150/ijbs.62238
9
Barisoni L. Mokrzycki M. Sablay L. Nagata M. Yamase H. Mundel P. (2000). Podocyte cell cycle regulation and proliferation in collapsing glomerulopathies. Kidney Int.58 (1), 137–143. 10.1046/j.1523-1755.2000.00149.x
10
Barisoni L. Schnaper H. W. Kopp J. B. (2007). A proposed taxonomy for the podocytopathies: a reassessment of the primary nephrotic diseases. Clin. J. Am. Soc. Nephrol.2 (3), 529–542. 10.2215/CJN.04121206
11
Berger K. Schulte K. Boor P. Kuppe C. van Kuppevelt T. H. Floege J. et al (2014). The regenerative potential of parietal epithelial cells in adult mice. J. Am. Soc. Nephrol.25 (4), 693–705. 10.1681/ASN.2013050481
12
Blaine J. Dylewski J. (2020). Regulation of the actin cytoskeleton in podocytes. Cells9 (7), 1700. 10.3390/cells9071700
13
Boi R. Bergwall L. Ebefors K. Bergo M. O. Nystrom J. Buvall L. (2023). Podocyte geranylgeranyl transferase type-I is essential for maintenance of the glomerular filtration barrier. J. Am. Soc. Nephrol.34 (4), 641–655. 10.1681/ASN.0000000000000062
14
Bose B. Cattran D. Toronto Glomerulonephritis R. (2014). Glomerular diseases: FSGS. Clin. J. Am. Soc. Nephrol.9 (3), 626–632. 10.2215/CJN.05810513
15
Boute N. Gribouval O. Roselli S. Benessy F. Lee H. Fuchshuber A. et al (2000). NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat. Genet.24 (4), 349–354. 10.1038/74166
16
Brinkkoetter P. T. Bork T. Salou S. Liang W. Mizi A. Ozel C. et al (2019). Anaerobic glycolysis maintains the glomerular filtration barrier independent of mitochondrial metabolism and dynamics. Cell Rep.27 (5), 1551–1566. 10.1016/j.celrep.2019.04.012
17
Brown E. J. Schlondorff J. S. Becker D. J. Tsukaguchi H. Tonna S. J. Uscinski A. L. et al (2010). Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat. Genet.42 (1), 72–76. 10.1038/ng.505
18
Cambier A. Rabant M. Peuchmaur M. Hertig A. Deschenes G. Couchoud C. et al (2018). Immunosuppressive treatment in children with IgA nephropathy and the clinical value of podocytopathic features. Kidney Int. Rep.3 (4), 916–925. 10.1016/j.ekir.2018.03.013
19
Campbell K. N. Wong J. S. Gupta R. Asanuma K. Sudol M. He J. C. et al (2013). Yes-associated protein (YAP) promotes cell survival by inhibiting proapoptotic dendrin signaling. J. Biol. Chem.288 (24), 17057–17062. 10.1074/jbc.C113.457390
20
Cattran D. C. Appel G. B. Hebert L. A. Hunsicker L. G. Pohl M. A. Hoy W. E. et al (1999). A randomized trial of cyclosporine in patients with steroid-resistant focal segmental glomerulosclerosis. North America Nephrotic Syndrome Study Group. Kidney Int.56 (6), 2220–2226. 10.1046/j.1523-1755.1999.00778.x
21
Chen H. H. Zhang Y. X. Lv J. L. Liu Y. Y. Guo J. Y. Zhao L. et al (2023). Role of sirtuins in metabolic disease-related renal injury. Biomed. Pharmacother.161, 114417. 10.1016/j.biopha.2023.114417
22
Chen Z. Ma Y. Yang Q. Hu J. Feng J. Liang W. et al (2020). AKAP1 mediates high glucose-induced mitochondrial fission through the phosphorylation of Drp1 in podocytes. J. Cell Physiol.235 (10), 7433–7448. 10.1002/jcp.29646
23
Chung J. J. Goldstein L. Chen Y. J. Lee J. Webster J. D. Roose-Girma M. et al (2020). Single-cell transcriptome profiling of the kidney glomerulus identifies key cell types and reactions to injury. J. Am. Soc. Nephrol.31 (10), 2341–2354. 10.1681/ASN.2020020220
24
Clark A. R. Marshall J. Zhou Y. Montesinos M. S. Chen H. Nguyen L. et al (2022). Single-cell transcriptomics reveal disrupted kidney filter cell-cell interactions after early and selective podocyte injury. Am. J. Pathol.192 (2), 281–294. 10.1016/j.ajpath.2021.11.004
25
Combs H. L. Shankland S. J. Setzer S. V. Hudkins K. L. Alpers C. E. (1998). Expression of the cyclin kinase inhibitor, p27kip1, in developing and mature human kidney. Kidney Int.53 (4), 892–896. 10.1111/j.1523-1755.1998.00842.x
26
Cormack-Aboud F. C. Brinkkoetter P. T. Pippin J. W. Shankland S. J. Durvasula R. V. (2009). Rosuvastatin protects against podocyte apoptosis in vitro. Nephrol. Dial. Transpl.24 (2), 404–412. 10.1093/ndt/gfn528
27
Cunanan J. Rajyam S. S. Sharif B. Udwan K. Rana A. De Gregorio V. et al (2024). Mice with a Pax2 missense variant display impaired glomerular repair. Am. J. Physiol. Ren. Physiol.326, F704–F726. 10.1152/ajprenal.00259.2023
28
Daga S. Donati F. Capitani K. Croci S. Tita R. Giliberti A. et al (2024). Correction: new frontiers to cure Alport syndrome: COL4A3 and COL4A5 gene editing in podocyte-lineage cells. Eur. J. Hum. Genet.32 (1), 131. 10.1038/s41431-023-01287-y
29
Dijkman H. Smeets B. van der Laak J. Steenbergen E. Wetzels J. (2005). The parietal epithelial cell is crucially involved in human idiopathic focal segmental glomerulosclerosis. Kidney Int.68 (4), 1562–1572. 10.1111/j.1523-1755.2005.00568.x
30
Ding W. Y. Kuzmuk V. Hunter S. Lay A. Hayes B. Beesley M. et al (2023). Adeno-associated virus gene therapy prevents progression of kidney disease in genetic models of nephrotic syndrome. Sci. Transl. Med.15 (708), eabc8226. 10.1126/scitranslmed.abc8226
31
Ding W. Y. Saleem M. A. (2012). Current concepts of the podocyte in nephrotic syndrome. Kidney Res. Clin. Pract.31 (2), 87–93. 10.1016/j.krcp.2012.04.323
32
Diomedi-Camassei F. Di Giandomenico S. Santorelli F. M. Caridi G. Piemonte F. Montini G. et al (2007). COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J. Am. Soc. Nephrol.18 (10), 2773–2780. 10.1681/ASN.2006080833
33
El Agha E. Kramann R. Schneider R. K. Li X. Seeger W. Humphreys B. D. et al (2017). Mesenchymal stem cells in fibrotic disease. Cell Stem Cell21 (2), 166–177. 10.1016/j.stem.2017.07.011
34
Elgass K. Pakay J. Ryan M. T. Palmer C. S. (2013). Recent advances into the understanding of mitochondrial fission. Biochim. Biophys. Acta1833 (1), 150–161. 10.1016/j.bbamcr.2012.05.002
35
Eng D. G. Kaverina N. V. Schneider R. R. S. Freedman B. S. Gross K. W. Miner J. H. et al (2018). Detection of renin lineage cell transdifferentiation to podocytes in the kidney glomerulus with dual lineage tracing. Kidney Int.93 (5), 1240–1246. 10.1016/j.kint.2018.01.014
36
Fan Q. Zhang H. Ding J. Liu S. Miao J. Xing Y. et al (2009). R168H and V165X mutant podocin might induce different degrees of podocyte injury via different molecular mechanisms. Genes cells.14 (9), 1079–1090. 10.1111/j.1365-2443.2009.01336.x
37
Faul C. Asanuma K. Yanagida-Asanuma E. Kim K. Mundel P. (2007). Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol.17 (9), 428–437. 10.1016/j.tcb.2007.06.006
38
Faul C. Donnelly M. Merscher-Gomez S. Chang Y. H. Franz S. Delfgaauw J. et al (2008). The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat. Med.14 (9), 931–938. 10.1038/nm.1857
39
Feng J. Ma Y. Chen Z. Hu J. Yang Q. Ding G. (2019). Mitochondrial pyruvate carrier 2 mediates mitochondrial dysfunction and apoptosis in high glucose-treated podocytes. Life Sci.237, 116941. 10.1016/j.lfs.2019.116941
40
Fontecha-Barriuso M. Martin-Sanchez D. Martinez-Moreno J. M. Monsalve M. Ramos A. M. Sanchez-Nino M. D. et al (2020). The role of PGC-1α and mitochondrial biogenesis in kidney diseases. Biomolecules10 (2), 347. 10.3390/biom10020347
41
Fu J. Wei C. Lee K. Zhang W. He W. Chuang P. et al (2016). Comparison of glomerular and podocyte mRNA profiles in streptozotocin-induced diabetes. J. Am. Soc. Nephrol.27 (4), 1006–1014. 10.1681/ASN.2015040421
42
Galluzzi L. Baehrecke E. H. Ballabio A. Boya P. Bravo-San Pedro J. M. Cecconi F. et al (2017). Molecular definitions of autophagy and related processes. EMBO J.36 (13), 1811–1836. 10.15252/embj.201796697
43
Geszke-Moritz M. Moritz M. (2024). Biodegradable polymeric nanoparticle-based drug delivery systems: comprehensive overview, perspectives and challenges. Polym. (Basel)16 (17), 2536. 10.3390/polym16172536
44
Gigante M. Pontrelli P. Montemurno E. Roca L. Aucella F. Penza R. et al (2009). CD2AP mutations are associated with sporadic nephrotic syndrome and focal segmental glomerulosclerosis (FSGS). Nephrol. Dial. Transpl.24 (6), 1858–1864. 10.1093/ndt/gfn712
45
Gobe G. Willgoss D. Hogg N. Schoch E. Endre Z. (1999). Cell survival or death in renal tubular epithelium after ischemia-reperfusion injury. Kidney Int.56 (4), 1299–1304. 10.1046/j.1523-1755.1999.00701.x
46
Godel M. Hartleben B. Herbach N. Liu S. Zschiedrich S. Lu S. et al (2011). Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Invest121 (6), 2197–2209. 10.1172/JCI44774
47
Grahammer F. Huber T. B. (2016). Aberrant podocyte cell cycle in glomerular disease. Cell Cycle15 (17), 2237–2238. 10.1080/15384101.2016.1205413
48
Hackl A. Nusken E. Voggel J. Abo Zed S. E. D. Binz-Lotter J. Unnersjo-Jess D. et al (2023). The effect of mycophenolate mofetil on podocytes in nephrotoxic serum nephritis. Sci. Rep.13 (1), 14167. 10.1038/s41598-023-41222-1
49
Hagen M. Pfister E. Kosel A. Shankland S. Pippin J. Amann K. et al (2016). Cell cycle re-entry sensitizes podocytes to injury induced death. Cell Cycle15 (14), 1929–1937. 10.1080/15384101.2016.1191710
50
Hakroush S. Cebulla A. Schaldecker T. Behr D. Mundel P. Weins A. (2014). Extensive podocyte loss triggers a rapid parietal epithelial cell response. J. Am. Soc. Nephrol.25 (5), 927–938. 10.1681/ASN.2013070687
51
Hale L. J. Howden S. E. Phipson B. Lonsdale A. Er P. X. Ghobrial I. et al (2018). 3D organoid-derived human glomeruli for personalised podocyte disease modelling and drug screening. Nat. Commun.9 (1), 5167. 10.1038/s41467-018-07594-z
52
Hale L. J. Little M. H. (2023). Generating human glomeruli from pluripotent stem cells for disease modelling and drug screening. Methods Mol. Biol.2664, 55–68. 10.1007/978-1-0716-3179-9_5
53
Han X. Wang J. Li R. Huang M. Yue G. Guan L. et al (2023). Placental mesenchymal stem cells alleviate podocyte injury in diabetic kidney disease by modulating mitophagy via the SIRT1-PGC-1alpha-TFAM pathway. Int. J. Mol. Sci.24 (5), 4696. 10.3390/ijms24054696
54
Handschin C. Spiegelman B. M. (2006). Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev.27 (7), 728–735. 10.1210/er.2006-0037
55
Hara M. Yanagihara T. Kihara I. (2001). Urinary podocytes in primary focal segmental glomerulosclerosis. Nephron.89 (3), 342–347. 10.1159/000046097
56
Hartleben B. Godel M. Meyer-Schwesinger C. Liu S. Ulrich T. Kobler S. et al (2010). Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Invest120 (4), 1084–1096. 10.1172/JCI39492
57
Heeringa S. F. Chernin G. Chaki M. Zhou W. Sloan A. J. Ji Z. et al (2011). COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Invest121 (5), 2013–2024. 10.1172/JCI45693
58
Helmstadter M. Huber T. B. Hermle T. (2017). Using the Drosophila nephrocyte to model podocyte function and disease. Front. Pediatr.5, 262. 10.3389/fped.2017.00262
59
Hiromura K. Haseley L. A. Zhang P. Monkawa T. Durvasula R. Petermann A. T. et al (2001). Podocyte expression of the CDK-inhibitor p57 during development and disease. Kidney Int.60 (6), 2235–2246. 10.1046/j.1523-1755.2001.00057.x
60
Imasawa T. Rossignol R. (2013). Podocyte energy metabolism and glomerular diseases. Int. J. Biochem. Cell Biol.45 (9), 2109–2118. 10.1016/j.biocel.2013.06.013
61
Jager K. J. Kovesdy C. Langham R. Rosenberg M. Jha V. Zoccali C. (2019). A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Kidney Int.96 (5), 1048–1050. 10.1016/j.kint.2019.07.012
62
Jha V. Garcia-Garcia G. Iseki K. Li Z. Naicker S. Plattner B. et al (2013). Chronic kidney disease: global dimension and perspectives. Lancet382 (9888), 260–272. 10.1016/S0140-6736(13)60687-X
63
Jiang X. S. Chen X. M. Hua W. He J. L. Liu T. Li X. J. et al (2020). PINK1/Parkin mediated mitophagy ameliorates palmitic acid-induced apoptosis through reducing mitochondrial ROS production in podocytes. Biochem. Biophys. Res. Commun.525 (4), 954–961. 10.1016/j.bbrc.2020.02.170
64
Kaplan J. M. Kim S. H. North K. N. Rennke H. Correia L. A. Tong H. Q. et al (2000). Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet.24 (3), 251–256. 10.1038/73456
65
Kaverina N. V. Eng D. G. Freedman B. S. Kutz J. N. Chozinski T. J. Vaughan J. C. et al (2019). Dual lineage tracing shows that glomerular parietal epithelial cells can transdifferentiate toward the adult podocyte fate. Kidney Int.96 (3), 597–611. 10.1016/j.kint.2019.03.014
66
Kestila M. Lenkkeri U. Mannikko M. Lamerdin J. McCready P. Putaala H. et al (1998). Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol. Cell.1 (4), 575–582. 10.1016/s1097-2765(00)80057-x
67
Khalilpourfarshbafi M. Hajiaghaalipour F. Selvarajan K. K. Adam A. (2017). Mesenchymal stem cell-based therapies against podocyte damage in diabetic nephropathy. Tissue Eng. Regen. Med.14 (3), 201–210. 10.1007/s13770-017-0026-5
68
Kos C. H. Le T. C. Sinha S. Henderson J. M. Kim S. H. Sugimoto H. et al (2003). Mice deficient in alpha-actinin-4 have severe glomerular disease. J. Clin. Invest111 (11), 1683–1690. 10.1172/JCI17988
69
Kretzler M. Koeppen-Hagemann I. Kriz W. (1994). Podocyte damage is a critical step in the development of glomerulosclerosis in the uninephrectomised-desoxycorticosterone hypertensive rat. Virchows Arch.425 (2), 181–193. 10.1007/BF00230355
70
Kriz W. (2002). Podocyte is the major culprit accounting for the progression of chronic renal disease. Microsc. Res. Tech.57 (4), 189–195. 10.1002/jemt.10072
71
Kriz W. (2003). The pathogenesis of 'classic' focal segmental glomerulosclerosis-lessons from rat models. Nephrol. Dial. Transpl.18 (Suppl. 6), vi39–44. 10.1093/ndt/gfg1064
72
Kriz W. Hahnel B. Hosser H. Ostendorf T. Gaertner S. Kranzlin B. et al (2003). Pathways to recovery and loss of nephrons in anti-Thy-1 nephritis. J. Am. Soc. Nephrol.14 (7), 1904–1926. 10.1097/01.asn.0000070073.79690.57
73
Kriz W. Hahnel B. Rosener S. Elger M. (1995). Long-term treatment of rats with FGF-2 results in focal segmental glomerulosclerosis. Kidney Int.48 (5), 1435–1450. 10.1038/ki.1995.433
74
Kuppe C. Leuchtle K. Wagner A. Kabgani N. Saritas T. Puelles V. G. et al (2019). Novel parietal epithelial cell subpopulations contribute to focal segmental glomerulosclerosis and glomerular tip lesions. Kidney Int.96 (1), 80–93. 10.1016/j.kint.2019.01.037
75
Lasagni L. Angelotti M. L. Ronconi E. Lombardi D. Nardi S. Peired A. et al (2015). Podocyte regeneration driven by renal progenitors determines glomerular disease remission and can Be pharmacologically enhanced. Stem Cell Rep.5 (2), 248–263. 10.1016/j.stemcr.2015.07.003
76
Lasagni L. Ballerini L. Angelotti M. L. Parente E. Sagrinati C. Mazzinghi B. et al (2010). Notch activation differentially regulates renal progenitors proliferation and differentiation toward the podocyte lineage in glomerular disorders. Stem Cells28 (9), 1674–1685. 10.1002/stem.492
77
Lasagni L. Lazzeri E. Shankland S. J. Anders H. J. Romagnani P. (2013). Podocyte mitosis - a catastrophe. Curr. Mol. Med.13 (1), 13–23. 10.2174/1566524011307010013
78
Lasse M. El Saghir J. Berthier C. C. Eddy S. Fischer M. Laufer S. D. et al (2023). An integrated organoid omics map extends modeling potential of kidney disease. Nat. Commun.14 (1), 4903. 10.1038/s41467-023-39740-7
79
Lee V. W. Harris D. C. (2011). Adriamycin nephropathy: a model of focal segmental glomerulosclerosis. Nephrol. Carlt.16 (1), 30–38. 10.1111/j.1440-1797.2010.01383.x
80
Lemley K. V. Lafayette R. A. Safai M. Derby G. Blouch K. Squarer A. et al (2002). Podocytopenia and disease severity in IgA nephropathy. Kidney Int.61 (4), 1475–1485. 10.1046/j.1523-1755.2002.00269.x
81
Lennon R. Randles M. J. Humphries M. J. (2014). The importance of podocyte adhesion for a healthy glomerulus. Front. Endocrinol. (Lausanne)5, 160. 10.3389/fendo.2014.00160
82
Liapis H. Romagnani P. Anders H. J. (2013). New insights into the pathology of podocyte loss: mitotic catastrophe. Am. J. Pathol.183 (5), 1364–1374. 10.1016/j.ajpath.2013.06.033
83
Liu B. Wang D. Cao Y. Wu J. Zhou Y. Wu W. et al (2022b). MitoTEMPO protects against podocyte injury by inhibiting NLRP3 inflammasome via PINK1/Parkin pathway-mediated mitophagy. Eur. J. Pharmacol.929, 175136. 10.1016/j.ejphar.2022.175136
84
Liu T. Yang L. Mao H. Ma F. Wang Y. Li S. et al (2022a). Sirtuins as novel pharmacological targets in podocyte injury and related glomerular diseases. Biomed. Pharmacother.155, 113620. 10.1016/j.biopha.2022.113620
85
Lopez L. C. Schuelke M. Quinzii C. M. Kanki T. Rodenburg R. J. Naini A. et al (2006). Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet.79 (6), 1125–1129. 10.1086/510023
86
Lu T. C. He J. C. Wang Z. H. Feng X. Fukumi-Tominaga T. Chen N. et al (2008). HIV-1 Nef disrupts the podocyte actin cytoskeleton by interacting with diaphanous interacting protein. J. Biol. Chem.283 (13), 8173–8182. 10.1074/jbc.M708920200
87
Lv Z. Hu M. Ren X. Fan M. Zhen J. Chen L. et al (2016). Fyn mediates high glucose-induced actin cytoskeleton reorganization of podocytes via promoting ROCK activation in vitro. J. Diabetes Res.2016, 5671803. 10.1155/2016/5671803
88
Marshall C. B. Pippin J. W. Krofft R. D. Shankland S. J. (2006). Puromycin aminonucleoside induces oxidant-dependent DNA damage in podocytes in vitro and in vivo. Kidney Int.70 (11), 1962–1973. 10.1038/sj.ki.5001965
89
Marshall C. B. Shankland S. J. (2007). Cell cycle regulatory proteins in podocyte health and disease. Nephron Exp. Nephrol.106 (2), e51–e59. 10.1159/000101793
90
Martinez-Garcia C. Izquierdo-Lahuerta A. Vivas Y. Velasco I. Yeo T. K. Chen S. et al (2015). Renal lipotoxicity-associated inflammation and insulin resistance affects actin cytoskeleton organization in podocytes. PLoS One10 (11), e0142291. 10.1371/journal.pone.0142291
91
Mason W. J. Jafree D. J. Pomeranz G. Kolatsi-Joannou M. Rottner A. K. Pacheco S. et al (2022). Systemic gene therapy with thymosin β4 alleviates glomerular injury in mice. Sci. Rep.12 (1), 12172. 10.1038/s41598-022-16287-z
92
Matsuda J. Asano-Matsuda K. Kitzler T. M. Takano T. (2021). Rho GTPase regulatory proteins in podocytes. Kidney Int.99 (2), 336–345. 10.1016/j.kint.2020.08.035
93
McCaffrey J. C. Webb N. J. Poolman T. M. Fresquet M. Moxey C. Zeef L. A. H. et al (2017). Glucocorticoid therapy regulates podocyte motility by inhibition of Rac1. Sci. Rep.7 (1), 6725. 10.1038/s41598-017-06810-y
94
Medina R. P. X. Cross E. Liu C. Pedigo C. E. Tian X. Gutierrez-Calabres E. et al (2023). Cell cycle and senescence regulation by podocyte histone deacetylase 1 and 2. J. Am. Soc. Nephrol.34 (3), 433–450. 10.1681/ASN.2022050598
95
Meliambro K. He J. C. Campbell K. N. (2024). Podocyte-targeted therapies - progress and future directions. Nat. Rev. Nephrol.20, 643–658. 10.1038/s41581-024-00843-z
96
Michaud J. L. Lemieux L. I. Dube M. Vanderhyden B. C. Robertson S. J. Kennedy C. R. (2003). Focal and segmental glomerulosclerosis in mice with podocyte-specific expression of mutant alpha-actinin-4. J. Am. Soc. Nephrol.14 (5), 1200–1211. 10.1097/01.asn.0000059864.88610.5e
97
Mirioglu S. Daniel-Fischer L. Berke I. Ahmad S. H. Bajema I. M. Bruchfeld A. et al (2024). Management of adult patients with podocytopathies: an update from the ERA Immunonephrology Working Group. Nephrol. Dial. Transpl.39 (4), 569–580. 10.1093/ndt/gfae025
98
Mouawad F. Tsui H. Takano T. (2013). Role of Rho-GTPases and their regulatory proteins in glomerular podocyte function. Can. J. Physiol. Pharmacol.91 (10), 773–782. 10.1139/cjpp-2013-0135
99
Na K. R. Jeong J. Y. Shin J. A. Chang Y. K. Suh K. S. Lee K. W. et al (2021). Mitochondrial dysfunction in podocytes caused by CRIF1 deficiency leads to progressive albuminuria and glomerular sclerosis in mice. Int. J. Mol. Sci.22 (9), 4827. 10.3390/ijms22094827
100
Ning L. Suleiman H. Y. Miner J. H. (2020). Synaptopodin is dispensable for normal podocyte homeostasis but is protective in the context of acute podocyte injury. J. Am. Soc. Nephrol.31 (12), 2815–2832. 10.1681/ASN.2020050572
101
Peired A. Lazzeri E. Lasagni L. Romagnani P. (2014). Glomerular regeneration: when can the kidney regenerate from injury and what turns failure into success?Nephron Exp. Nephrol.126 (2), 70. 10.1159/000360669
102
Peng M. Falk M. J. Haase V. H. King R. Polyak E. Selak M. et al (2008). Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet.4 (4), e1000061. 10.1371/journal.pgen.1000061
103
Pippin J. W. Kaverina N. V. Eng D. G. Krofft R. D. Glenn S. T. Duffield J. S. et al (2015). Cells of renin lineage are adult pluripotent progenitors in experimental glomerular disease. Am. J. Physiol. Ren. Physiol.309 (4), F341–F358. 10.1152/ajprenal.00438.2014
104
Pippin J. W. Sparks M. A. Glenn S. T. Buitrago S. Coffman T. M. Duffield J. S. et al (2013). Cells of renin lineage are progenitors of podocytes and parietal epithelial cells in experimental glomerular disease. Am. J. Pathol.183 (2), 542–557. 10.1016/j.ajpath.2013.04.024
105
Qin L. Liu N. Bao C. L. Yang D. Z. Ma G. X. Yi W. H. et al (2023). Mesenchymal stem cells in fibrotic diseases-the two sides of the same coin. Acta Pharmacol. Sin.44 (2), 268–287. 10.1038/s41401-022-00952-0
106
Quaggin S. E. (2002). Transcriptional regulation of podocyte specification and differentiation. Microsc. Res. Tech.57 (4), 208–211. 10.1002/jemt.10076
107
Refaeli I. Hughes M. R. McNagny K. M. (2019). The first identified heterozygous nonsense mutations in podocalyxin offer new perspectives on the biology of podocytopathies. Clin. Sci. (Lond).133 (3), 443–447. 10.1042/CS20181067
108
Reynolds P. A. (2020). The mechanobiology of kidney podocytes in health and disease. Clin. Sci. (Lond).134 (11), 1245–1253. 10.1042/CS20190764
109
Romagnani P. Remuzzi G. (2013). Renal progenitors in non-diabetic and diabetic nephropathies. Trends Endocrinol. Metab.24 (1), 13–20. 10.1016/j.tem.2012.09.002
110
Ronco P. Beck L. Debiec H. Fervenza F. C. Hou F. F. Jha V. et al (2021). Membranous nephropathy. Nat. Rev. Dis. Prim.7 (1), 69. 10.1038/s41572-021-00303-z
111
Ronconi E. Sagrinati C. Angelotti M. L. Lazzeri E. Mazzinghi B. Ballerini L. et al (2009). Regeneration of glomerular podocytes by human renal progenitors. J. Am. Soc. Nephrol.20 (2), 322–332. 10.1681/ASN.2008070709
112
Ruotsalainen V. Patrakka J. Tissari P. Reponen P. Hess M. Kestila M. et al (2000). Role of nephrin in cell junction formation in human nephrogenesis. Am. J. Pathol.157 (6), 1905–1916. 10.1016/S0002-9440(10)64829-8
113
Sagrinati C. Netti G. S. Mazzinghi B. Lazzeri E. Liotta F. Frosali F. et al (2006). Isolation and characterization of multipotent progenitor cells from the Bowman's capsule of adult human kidneys. J. Am. Soc. Nephrol.17 (9), 2443–2456. 10.1681/ASN.2006010089
114
Saleem M. A. Ni L. Witherden I. Tryggvason K. Ruotsalainen V. Mundel P. et al (2002). Co-localization of nephrin, podocin, and the actin cytoskeleton: evidence for a role in podocyte foot process formation. Am. J. Pathol.161 (4), 1459–1466. 10.1016/S0002-9440(10)64421-5
115
Schell C. Huber T. B. (2012). New players in the pathogenesis of focal segmental glomerulosclerosis. Nephrol. Dial. Transpl.27 (9), 3406–3412. 10.1093/ndt/gfs273
116
Schonenberger E. Ehrich J. H. Haller H. Schiffer M. (2011). The podocyte as a direct target of immunosuppressive agents. Nephrol. Dial. Transpl.26 (1), 18–24. 10.1093/ndt/gfq617
117
Shankland S. J. Eitner F. Hudkins K. L. Goodpaster T. D'Agati V. Alpers C. E. (2000). Differential expression of cyclin-dependent kinase inhibitors in human glomerular disease: role in podocyte proliferation and maturation. Kidney Int.58 (2), 674–683. 10.1046/j.1523-1755.2000.00213.x
118
Shankland S. J. Freedman B. S. Pippin J. W. (2017). Can podocytes be regenerated in adults?Curr. Opin. Nephrol. Hypertens.26 (3), 154–164. 10.1097/MNH.0000000000000311
119
Shankland S. J. Pippin J. W. Couser W. G. (1999). Complement (C5b-9) induces glomerular epithelial cell DNA synthesis but not proliferation in vitro. Kidney Int.56 (2), 538–548. 10.1046/j.1523-1755.1999.00560.x
120
Shirato I. Sakai T. Kimura K. Tomino Y. Kriz W. (1996). Cytoskeletal changes in podocytes associated with foot process effacement in Masugi nephritis. Am. J. Pathol.148 (4), 1283–1296.
121
Shkreli M. Sarin K. Y. Pech M. F. Papeta N. Chang W. Brockman S. A. et al (2011). Reversible cell-cycle entry in adult kidney podocytes through regulated control of telomerase and Wnt signaling. Nat. Med.18 (1), 111–119. 10.1038/nm.2550
122
Siegerist F. Endlich K. Endlich N. (2018). Novel microscopic techniques for podocyte research. Front. Endocrinol. (Lausanne)9, 379. 10.3389/fendo.2018.00379
123
Smart S. K. Vasileiadi E. Wang X. DeRyckere D. Graham D. K. (2018). The emerging role of TYRO3 as a therapeutic target in cancer. Cancers (Basel)10 (12), 474. 10.3390/cancers10120474
124
Smeets B. Te Loeke N. A. Dijkman H. B. Steenbergen M. L. Lensen J. F. Begieneman M. P. et al (2004). The parietal epithelial cell: a key player in the pathogenesis of focal segmental glomerulosclerosis in Thy-1.1 transgenic mice. J. Am. Soc. Nephrol.15 (4), 928–939. 10.1097/01.asn.0000120559.09189.82
125
Su M. Dhoopun A. R. Yuan Y. Huang S. Zhu C. Ding G. et al (2013). Mitochondrial dysfunction is an early event in aldosterone-induced podocyte injury. Am. J. Physiol. Ren. Physiol.305 (4), F520–F531. 10.1152/ajprenal.00570.2012
126
Sun H. Perez-Gill C. Schlondorff J. S. Subramanian B. Pollak M. R. (2021). Dysregulated dynein-mediated trafficking of nephrin causes INF2-related podocytopathy. J. Am. Soc. Nephrol.32 (2), 307–322. 10.1681/ASN.2020081109
127
Sun H. Schlondorff J. Higgs H. N. Pollak M. R. (2013). Inverted formin 2 regulates actin dynamics by antagonizing Rho/diaphanous-related formin signaling. J. Am. Soc. Nephrol.24 (6), 917–929. 10.1681/ASN.2012080834
128
Surova O. Zhivotovsky B. (2013). Various modes of cell death induced by DNA damage. Oncogene32 (33), 3789–3797. 10.1038/onc.2012.556
129
Thomasova D. Anders H. J. (2015). Cell cycle control in the kidney. Nephrol. Dial. Transpl.30 (10), 1622–1630. 10.1093/ndt/gfu395
130
Tian F. Huang S. Xu W. Xie G. Gan Y. Huang F. et al (2023b). Fasudil compensates podocyte injury via CaMK4/Rho GTPases signal and actin cytoskeleton-dependent activation of YAP in MRL/lpr mice. Int. Immunopharmacol.119, 110199. 10.1016/j.intimp.2023.110199
131
Tian X. Pedigo C. E. Li K. Ma X. Bunda P. Pell J. et al (2023a). Profilin1 is required for prevention of mitotic catastrophe in murine and human glomerular diseases. J. Clin. Invest133 (24), e171237. 10.1172/JCI171237
132
Tsai Y. C. Kuo M. C. Huang J. C. Chang W. A. Wu L. Y. Huang Y. C. et al (2023). Single-cell transcriptomic profiles in the pathophysiology within the microenvironment of early diabetic kidney disease. Cell Death Dis.14 (7), 442. 10.1038/s41419-023-05947-1
133
Unnersjo-Jess D. (2023). Quantitative imaging of podocyte foot processes in the kidney using confocal and STED microscopy. Methods Mol. Biol.2664, 185–199. 10.1007/978-1-0716-3179-9_14
134
Vakifahmetoglu H. Olsson M. Zhivotovsky B. (2008). Death through a tragedy: mitotic catastrophe. Cell Death Differ.15 (7), 1153–1162. 10.1038/cdd.2008.47
135
van de Leemput J. Han Z. (2024). Drosophila nephrocyte as a model for studying glomerular basement membrane physiology. J. Am. Soc. Nephrol.35 (9), 1141–1142. 10.1681/ASN.0000000000000461
136
Verberne W. R. Das-Gupta Z. Allegretti A. S. Bart H. A. J. van Biesen W. Garcia-Garcia G. et al (2019). Development of an international standard set of value-based outcome measures for patients with chronic kidney disease: a report of the international consortium for health outcomes measurement (ichom) CKD working group. Am. J. Kidney Dis.73 (3), 372–384. 10.1053/j.ajkd.2018.10.007
137
Verma R. Venkatareddy M. Kalinowski A. Li T. Kukla J. Mollin A. et al (2018). Nephrin is necessary for podocyte recovery following injury in an adult mature glomerulus. PLoS One13 (6), e0198013. 10.1371/journal.pone.0198013
138
Vivarelli M. Massella L. Ruggiero B. Emma F. (2017). Minimal change disease. Clin. J. Am. Soc. Nephrol.12 (2), 332–345. 10.2215/CJN.05000516
139
Wang S. Li Y. Zhao J. Zhang J. Huang Y. (2013). Mesenchymal stem cells ameliorate podocyte injury and proteinuria in a type 1 diabetic nephropathy rat model. Biol. Blood Marrow Transpl.19 (4), 538–546. 10.1016/j.bbmt.2013.01.001
140
Wang Y. Jiang H. Li M. Xu Z. Xu H. Chen Y. et al (2024). Delivery of CRISPR/Cas9 system by AAV as vectors for gene therapy. Gene927, 148733. 10.1016/j.gene.2024.148733
141
Wanner N. Hartleben B. Herbach N. Goedel M. Stickel N. Zeiser R. et al (2014). Unraveling the role of podocyte turnover in glomerular aging and injury. J. Am. Soc. Nephrol.25 (4), 707–716. 10.1681/ASN.2013050452
142
Wei Q. Bhatt K. He H. Z. Mi Q. S. Haase V. H. Dong Z. (2010). Targeted deletion of Dicer from proximal tubules protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol.21 (5), 756–761. 10.1681/ASN.2009070718
143
Welsh G. I. Saleem M. A. (2010). Nephrin-signature molecule of the glomerular podocyte?J. Pathol.220 (3), 328–337. 10.1002/path.2661
144
Wharram B. L. Goyal M. Wiggins J. E. Sanden S. K. Hussain S. Filipiak W. E. et al (2005). Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J. Am. Soc. Nephrol.16 (10), 2941–2952. 10.1681/ASN.2005010055
145
Wiggins J. E. Goyal M. Sanden S. K. Wharram B. L. Shedden K. A. Misek D. E. et al (2005). Podocyte hypertrophy, “adaptation,” and “decompensation” associated with glomerular enlargement and glomerulosclerosis in the aging rat: prevention by calorie restriction. J. Am. Soc. Nephrol.16 (10), 2953–2966. 10.1681/ASN.2005050488
146
Wyss H. M. Henderson J. M. Byfield F. J. Bruggeman L. A. Ding Y. Huang C. et al (2011). Biophysical properties of normal and diseased renal glomeruli. Am. J. Physiol. Cell Physiol.300 (3), C397–C405. 10.1152/ajpcell.00438.2010
147
Xie K. Xu C. Zhang M. Wang M. Min L. Qian C. et al (2019). Yes-associated protein regulates podocyte cell cycle re-entry and dedifferentiation in adriamycin-induced nephropathy. Cell Death Dis.10 (12), 915. 10.1038/s41419-019-2139-3
148
Yao J. Le T. C. Kos C. H. Henderson J. M. Allen P. G. Denker B. M. et al (2004). Alpha-actinin-4-mediated FSGS: an inherited kidney disease caused by an aggregated and rapidly degraded cytoskeletal protein. PLoS Biol.2 (6), e167. 10.1371/journal.pbio.0020167
149
Zhang F. Chen X. (2014). The Drosophila nephrocyte has a glomerular filtration system. Nat. Rev. Nephrol.10 (9), 491. 10.1038/nrneph.2012.290-c1
150
Zhong F. Cai H. Fu J. Sun Z. Li Z. Bauman D. et al (2023). TYRO3 agonist as therapy for glomerular disease. JCI Insight8 (1), e165207. 10.1172/jci.insight.165207
151
Zhou D. Zhou M. Wang Z. Fu Y. Jia M. Wang X. et al (2019). PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis.10 (7), 524. 10.1038/s41419-019-1754-3
152
Zhu C. Xuan X. Che R. Ding G. Zhao M. Bai M. et al (2014). Dysfunction of the PGC-1α-mitochondria axis confers adriamycin-induced podocyte injury. Am. J. Physiol. Ren. Physiol.306 (12), F1410–F1417. 10.1152/ajprenal.00622.2013
153
Zoja C. Garcia P. B. Rota C. Conti S. Gagliardini E. Corna D. et al (2012). Mesenchymal stem cell therapy promotes renal repair by limiting glomerular podocyte and progenitor cell dysfunction in adriamycin-induced nephropathy. Am. J. Physiol. Ren. Physiol.303 (9), F1370–F1381. 10.1152/ajprenal.00057.2012
Summary
Keywords
podocytes, glomerular disease, actin cytoskeleton, slit diaphragm, differentiated, metabolism, therapeutic targets
Citation
Cunanan J, Zhang D, Peired AJ and Barua M (2025) Podocytes in health and glomerular disease. Front. Cell Dev. Biol. 13:1564847. doi: 10.3389/fcell.2025.1564847
Received
22 January 2025
Accepted
08 April 2025
Published
24 April 2025
Volume
13 - 2025
Edited by
Simon Rousseau, McGill University, Canada
Reviewed by
Francesca Di Sole, Des Moines University, United States
Agnes Hackl, University Hospital of Cologne, Germany
Updates
Copyright
© 2025 Cunanan, Zhang, Peired and Barua.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Moumita Barua, moumita.barua@uhn.ca
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.