 John H. Rinald1
John H. Rinald1 Carol M. Troy2,3,4*
Carol M. Troy2,3,4*- 1Neurobiology and Behavior PhD Program, Columbia University, New York, NY, United States
- 2Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, NY, United States
- 3Department of Neurology, Vagelos College of Physicians and Surgeons, Columbia University, New York, NY, United States
- 4The Taub Institute for Research on Alzheimer's Disease and the Aging Brain, Vagelos College of Physicians and Surgeons, Columbia University, New York, NY, United States
Hemorrhagic stroke is a debilitating neurological disease, affecting millions worldwide. Characterized by bleeding in the brain, it is caused by a breakdown of the blood-brain barrier (BBB) and causes damage through the presence of iron in the brain, immune activation and increased intracranial pressure. The goal of this mini-review is to explore the signaling pathways that lead to cell death that are a part of disease progression in hemorrhagic stroke. This mini-review will highlight clinical observations and data, while also incorporating findings using preclinical disease models. There are important roles for apoptosis, necroptosis, necrosis, autophagy, ferroptosis, and pyroptosis in hemorrhagic stroke. Recent work has highlighted the interplay between these phenomena, providing key regulators as potential therapeutic targets, including reactive oxygen species, iron metabolism, and caspases. Therapeutic strategies that can delay or counteract the cytotoxic effects of hemorrhage can improve clinical outcomes in hemorrhagic stroke patients.
1 Introduction
Stroke is the third leading cause of death and disability globally (Feigin et al., 2022). There are two categories of stroke: ischemic, in which blood flow through blood vessels is occluded, and hemorrhagic, in which a blood vessel ruptures, and blood spills into the neural parenchyma (Kuriakose and Xiao, 2020). Hemorrhagic stroke can be further classified by the location of the hemorrhage into subarachnoid hemorrhage, and intracerebral hemorrhage. Subarachnoid hemorrhage and intracerebral hemorrhage have very different clinical presentations, and different treatment options (Coelho et al., 2016; An et al., 2017). Subarachnoid hemorrhage typically presents as a sudden onset severe headache. Intracerebral hemorrhage manifests differently depending on the affected brain region, e.g., thalamic bleeds typically cause unilateral motor impacts. However, while the clinical manifestations of these diseases are different, the cellular and molecular pathology is comparable. In this mini-review, we will be discussing the cellular effects of hemorrhagic stroke, specifically exploring the pathological mechanisms of cell death that are involved in hemorrhagic stroke.
Hemorrhagic stroke significantly impacts neurological function by destroying areas of the brain and preventing the normal function of neural behavioral circuits. The pathology of hemorrhagic stroke changes as the disease progresses past the initial time of injury (Magid-Bernstein et al., 2022). Primary injury occurs within the first 2 days, and is largely mediated by the mass effect of the hematoma in the neural parenchyma (Zazulia et al., 1999). Inside the hemorrhagic core, there is a high concentration of toxic blood components due to the lysis of hemoglobin-filled erythrocytes (Fang et al., 2022). This can cause neuronal death through, for example, an increase in reactive oxygen species (ROS) due to increased local iron concentrations (Koduri et al., 2022). Additionally, the increase of intracranial pressure from the mass of the blood can cause cell death through restricting flow of blood to other regions of the brain (Zazulia et al., 1999). Secondary injury occurs over a longer time period, and is largely understood to be the consequences of several toxic pathways, including: inflammation, excitotoxicity, oxidative stress, and hemolysis (Ziai, 2013; Shoamanesh and Katsanos, 2021). These pathways can actually be observed within the first hour after stroke, but continue to have deleterious effects even after hematoma clearance (Bautista et al., 2021). Both the primary and secondary injuries cause irreversible neuronal death, which causes the neurological effects of stroke.
In hemorrhagic stroke, there are multiple modes of cell death active at any time, depicted in Figure 1. For example, in the regions closest to the injury, tissue is quickly liquified via necrosis, whereas in the peri-hematoma regions the majority of the dying cells appear apoptotic, rather than necrotic at several timepoints after injury (Qureshi et al., 2003). This initial finding that both apoptosis and necrosis play roles in hemorrhagic stroke has expanded to the discoveries that many other types of cell death are also present in hemorrhagic stroke (Zhang et al., 2022). By studying the plurality of cell death modes in hemorrhagic stroke, we can better understand the molecular pathways that are responsible for the devastating consequences of this disease and develop more specific disease-modifying strategies.
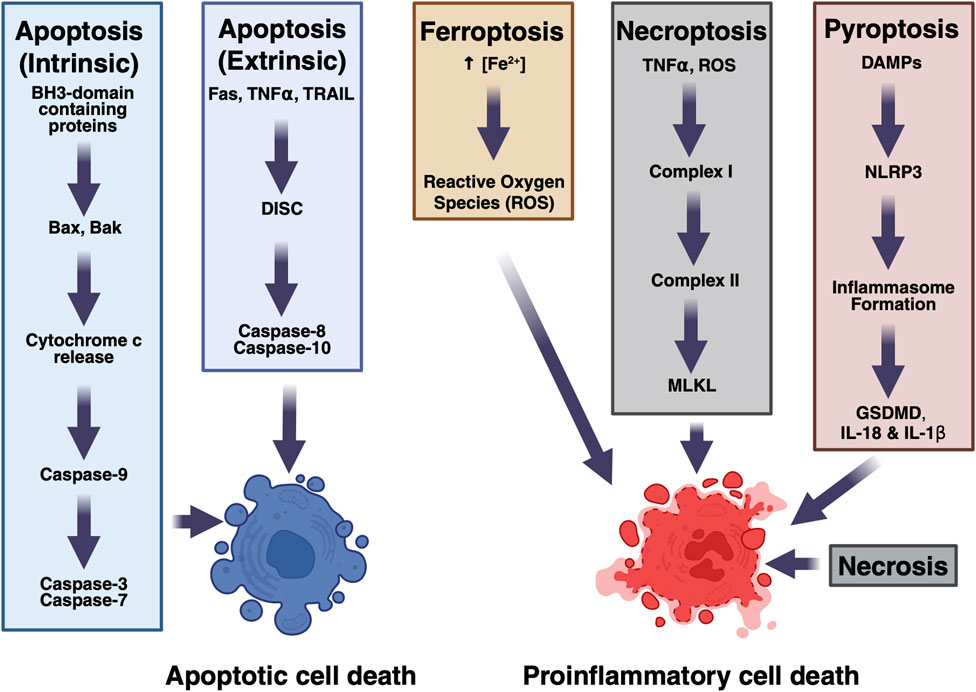
Figure 1. Mechanisms of cell death in hemorrhagic stroke. Multiple pathways contribute to cell death.
2 Apoptosis
Apoptosis was the first active form of cell death discovered. Apoptosis requires energy, and is pathologically characterized by cell shrinkage, chromatin condensation and the formation of apoptotic bodies (Coleman et al., 2001). Although apoptosis is often called programmed cell death, programmed cell death is normal death by apoptosis that occurs during development and maintenance of homeostasis; apoptosis is also responsible for cell death during disease (Elmore, 2007).
Depending on the stimulation, cells can either undergo intrinsic or extrinsic apoptosis (Green, 2000). These pathways have been well described previously (Qureshi et al., 2003; Green, 2000; Xu and Shi, 2007; Moujalled et al., 2021). Briefly, intrinsic apoptosis, also known as the mitochondrial apoptotic pathway, can be a response to various intracellular stressors, including DNA damage, hypoxia, or oxidative stress (Westphal et al., 2014). These pathways activate p53, leading to the expression of BH3-domain containing proteins including Bim and Bid, which bind to and activate Bax and Bak. Bax and Bak form pores in the outer mitochondrial membrane, releasing cytochrome c from the mitochondria, causing the ATP-dependent formation of the apoptosome, a large multiunit complex of APAF-1 and caspase-9. The apoptosome provides a platform for caspase-9 activation, and the activated protease then cleaves executioner caspases, caspase-3 and -7, which will enact irreversible cell death. Extrinsic apoptosis is a response to extracellular pro-death signals. Three major classes of pro-death receptor-ligand pairs have been well described: Fas and Fas ligand, TNFα and the TNF receptor, and the death receptors DR4 and DR5 and TRAIL (Xu and Shi, 2007). Activation of these pathways triggers the formation of the death-induced signaling complex (DISC), to act as a platform for activation of caspase-8 or -10, which can then go on to cleave and activate effector caspases. In hemorrhagic stroke tissue, this extrinsic pathway is likely triggered by intracellular contents from other dying cells.
Apoptosis is observed as a large portion of cell death in hemorrhagic stroke. A study in 2003 by Qureshi et al. showed that in the perihematomal region in clinical cases of intracerebral hemorrhage, a majority of the cells were positive for terminal deoxynucleotidyl transferase-mediated 5-triphosphate nick-end (TUNEL) labeling (Qureshi et al., 2003). This study likely represents a plurality of modes of cell death, rather than just apoptosis, as the authors initially claimed, as TUNEL-positivity can be indicative of more than apoptosis (Kelly et al., 2003; Grasl-Kraupp et al., 1995). Another study has found that this proportion of apoptotic cells increases to a peak in the first 12–24 h in the perihematomal tissue after intracerebral hemorrhage, measured as an increase in TUNEL positivity and Bax protein and mRNA expression (Guo et al., 2006). This indicates that a large portion of cells that are near the most affected areas are undergoing apoptotic death, presumably stimulated by pro-death signals received from the brain parenchyma. Additionally, in a dog model of subarachnoid hemorrhage, caspase inhibitors were able to protect against some of the behavioral effects, indicating that caspase-3 activity is an important pathological mechanism of hemorrhage (Zhou et al., 2004). Altogether, apoptosis is likely to be present in both subarachnoid and intracerebral hemorrhage tissue, and blocking apoptosis may be a plausible therapeutic route.
3 Necrosis
Necrosis is a form of uncontrolled, pro-inflammatory cell death that occurs under damaging conditions. Necrosis is characterized pathologically by membrane breakdown and cytoplasmic swelling, without the chromatin condensation that is characteristic of apoptosis (Khalid and Azimpouran, 2025). Necrosis is triggered by cell-external factors, such as mechanical breakdown of tissue or intense ischemia. Necrotic cells release intracellular contents, which act as pro-inflammatory damage-associated molecular patterns (DAMPs), triggering an immune response. Necrosis is understood as an unregulated, energy-independent, and non-cell-initiated form of death, but there are ways that a cell can induce its own pro-inflammatory necrotic fate through necroptosis or pyroptosis, as described later in this paper.
Necrosis is responsible for a large amount of cell death during the primary and short-term secondary injury after a hemorrhagic stroke. The mechanical force generated from increased intracranial pressure causes cells to rupture and become necrotic. It has been shown in samples taken from patients with intracerebral hemorrhage that around 25% of cells in the perihematomal region are necrotic at 24 h post-stroke (Qureshi et al., 2003). Since necrosis is a physical consequence of traumatic environmental changes that occur during the primary injury during a hemorrhagic stroke, modification with therapies presents a significant challenge. There are interventions that can block oxidative stress-induced death by scavenging reactive oxygen species, which have shown some beneficial preclinical effects in ischemic injury, but have not been tested for application in hemorrhagic stroke (Hwang et al., 2018).
4 Necroptosis
Necroptosis is an induced form of necrosis. This is a pro-inflammatory death in response to either an external factor, such as ligands for Toll-like receptors, including the TNFα receptor, or an abundance of intracellular reactive oxygen species (ROS) (Dhuriya and Sharma, 2018).
A cell can induce its own necrotic death in response to extrinsic factors secreted by the cell, most importantly in response to the stimulation of its death receptors, including by tumor necrosis factor 1 (TNFα). TNFα binds to the TNFα receptor in the plasma membrane, causing a conformation shift in the receptor’s luminal domain, which starts the platform for the membrane-associated Complex I. Complex I is made up of TRADD, FADD, RIPK1, TRAF2/TRAF5, and cIAP1/cIAP2, and causes a variety of post-translational modifications of TRADD, TFNR1, and RIP1, including the ubiquitination of RIP1. A portion of Complex I containing RIP1, TRADD, FADD, and TRAF2 then detaches from the membrane, where it forms Complex II by incorporating caspase-8 and RIPK3. Activated RIPK3 can then phosphorylate the pseudokinase MLKL, which enacts necroptosis through permeabilization of various intracellular membranes (Martens et al., 2021). Thus, phosphorylated MLKL is a bona fide marker for necroptotic cells.
Whether this cell proceeds to a necroptotic fate, or apoptosis is triggered, is dependent on caspase-8 (and caspase-10 in humans). If procaspase-8 is present in the cell when RIPK1 is released from the membrane, caspase-8 will form a complex with it and FADD and cleave itself. This activated form of caspase-8 will then enact apoptosis, suppressing necroptosis. This then positions the presence of caspase-8 as the switch between a cell’s necroptotic and apoptotic fates. Since caspase-8 is differentially expressed in different cell populations, whether they face apoptosis or necroptosis is partially determined by the presence of caspase-8 (Velier et al., 1999). For example, different subpopulations of pyramidal neurons differentially express capsase-8, and that affects their longevity in a rodent model of ischemic stroke.
Necroptotic death has been shown to be an abundant form of death in response to blood and blood oxidation products, such as heme. A dog model of subarachnoid hemorrhage has shown that TNFR1 is present in dying cells after injury (Zhou et al., 2004), indicating that the signaling mechanism to initiate necroptosis is present. Additionally, Necrostatin-1, an inhibitor of RIPK1, has been shown to reduce neurovascular injury after collagenase-induced hemorrhage in rodents (Chang et al., 2014). Taken together, these findings demonstrate that necroptosis may be contributing to the pathology of hemorrhagic stroke in this model, and presents the necroptotic pathway as a potential therapeutic target. This implicates necroptosis as a key pathological mechanism of cell death in hemorrhagic stroke.
5 Pyroptosis
Pyroptosis is another pro-inflammatory method of cell death, characterized by the release of IL-1β and IL-18 from the dying cell. When DAMPs bind to their conjugate receptors, including NLRP3, DNA receptors, and the pyrin receptor, they trigger the formation of the inflammasome, a large protein complex, which serves as an activating platform for caspase-1 (Bertheloot et al., 2021). Caspase-1 cleaves pro-IL-1β and pro-IL-18 into their mature forms (IL-1β and IL-18 respectively), as well as cleaving Gasdermin D (GSDMD) into a form that can form pores in the plasma membrane. This breakdown of the plasma membrane causes the death of the cell, as well as the release of the newly synthesized pro-inflammatory cytokines. This triggers an immune response through the direct sensing of these cytokines by immune cells.
The activity of caspase-1, an essential step for pyroptosis, has been shown to be required for the breakdown of the BBB during collagenase-induced hemorrhage in mice (Lin et al., 2018). Indeed, an inhibitor of caspase-1, AC-YVAD-CMK, has been shown to inhibit pyroptosis and protect against motor deficits in this model. However, it is important to know that this model of stroke likely overrepresents immune activation, as collagenase itself causes immune cell activation, independent of its effect on the BBB (Singh and Bhattacharyya, 2014).
In perihematomal tissue from patients with ICH, single cell sequencing analysis has shown that a transcriptionally unique population of IL-1β+ microglia undergoes pyroptosis within the first 2 days after injury (Zhang et al., 2024). These pro-inflammatory dying microglia exist in a balance with another population of microglia, identified as expressing P2RY12. As a therapeutic target, this molecular distinction of at least two microglial subpopulations is important, since strategies manipulating only pro-inflammatory cells could be beneficial (Scott et al., 2021).
6 Ferroptosis
Ferroptosis is an iron-dependent method of cell death (Dixon et al., 2012). In intracerebral hemorrhage, lysed red blood cells release hemoglobin into the neural parenchyma. The iron bound to this hemoglobin leads to the generation of reactive oxygen species, in a mitochondria-dependent manner. An increase in the local concentration of iron also disrupts normal iron metabolism (Koduri et al., 2022). Ferroptosis exists as a unique death pathway as it does not use energy and does not involve caspases or lysosomes (Dixon et al., 2012; Wang et al., 2002; Zille et al., 2017).
Ferroptosis has been shown to be active in oligodendrocyte precursor cells in a mouse model of intraventricular hemorrhage (Shen et al., 2022). Additionally, it has been confirmed that inhibition of ferroptosis with ferrostatin-1 protects against neuronal death in the collagenase model of intracerebral hemorrhage in mice (Zille et al., 2017; Li et al., 2017). This effect may be through the rescue of cells that would otherwise die due to iron overload, or it could be through an indirect, synergistic effect on inflammation, as ferroptosis has been shown to be immunogenic (Zhang et al., 2018).
7 Autophagic cell death
Autophagy is a common mechanism for cell death and clearance after damage. Autophagy is triggered by activation of the mTOR pathway (Zhang et al., 2022). When an organelle is recognized for autophagy, a second membrane is formed around the organellar membrane, and that then fuses with the lysosome, acidifying the autophagosome (Towers and Thorburn, 2016). This begins the breakdown of the vesicle contents. While autophagic materials are usually used for the clearance of defective organelles or cytoplasmic components, excessive autophagy can cause cell death via a pathway that is distinct from apoptosis or necroptosis (Denton and Kumar, 2019).
Autophagy and phagocytosis work through similar mechanisms, and are thus inextricably linked. There is evidence that increased autophagy can modulate microglial receptiveness to inflammatory signaling, and defective autophagy may impair phagocytic function (Chen et al., 2022). Thus, microglial autophagy is an essential player in the secondary injury from hemorrhagic stroke.
The presence of autophagic cell death has been verified in a rat model of subarachnoid hemorrhage by electron microscopic identification of autophagosomes and autolysosomes (Lee et al., 2009). Additionally, Beclin-1, a marker of autophagic tissue, has been observed in a collagenase mouse model of intracerebral hemorrhage (Shen et al., 2016). This same study also found that there is an increase in NF-κβ signaling that can be reduced by blocking autophagy, indicating that this pathway contributes to inflammation and immune response. Inhibition of autophagy has also been shown to be beneficial by preventing ferroptotic brain injury (Chen et al., 2012). While autophagy is essential for clearance of debris by macrophages, this indicates that autophagy may also be playing a deleterious role in hemorrhagic stroke pathology, and therefore could serve as a therapeutic target, with careful consideration of its beneficial role. Currently, therapeutics targeting autophagy are being investigated in many diseases, including neurodegenerative and infectious (Towers and Thorburn, 2016).
8 Therapeutic considerations and conclusion
Under the conditions of hemorrhagic stroke, it is likely that all these death pathways are occurring at some level. By understanding the contribution and impact that each of these distinct molecular death pathways has on the pathology of hemorrhagic stroke, it may be possible to design specific therapeutics that can target only the negative consequences of hemorrhagic stroke.
Investigations into inhibitors of cell death pathways have provided promising preclinical results but have not yet translated into successful clinical trials. In a screen of many inhibitors of different cell death pathways, inhibitors of ferroptosis demonstrated the most consistent protection against hemin toxicity in cultured cortical neurons (Zille et al., 2017). Ischemia, however, has been shown in vivo to cause sequential waves of necroptosis and apoptosis (Naito et al., 2020). Therefore, therapeutics that can address a single pathology of hemorrhagic stroke may not be able to ameliorate the effects of a stroke, due to the multiplicity of cell death pathways that have been shown to be active. Uncovering interplay between these pathways will also be essential for developing effective treatments.
To effectively evaluate the efficacy of therapeutics, there must be testing in a multimodal and human-relevant disease model (Table 1). Subarachnoid hemorrhage can be effectively replicated in rodents through endovascular perforation, in which blood vessels are ruptured from the inside (Schüller et al., 2013). Intracerebral hemorrhage is typically either modeled through intracerebral injection of autologous blood, to recreate hemotoxic effects, or collagenase, which degrades extracellular matrix including the tight junctions that hold together blood vessels (Thangameeran et al., 2022; MacLellan et al., 2010). Both models are able to model some portion of intracerebral hemorrhage, but neither fully recapitulate the vasogenic nature of the injury. To understand how different methods of cell death interact in a disease state, a better animal model of intracerebral hemorrhage is critical.
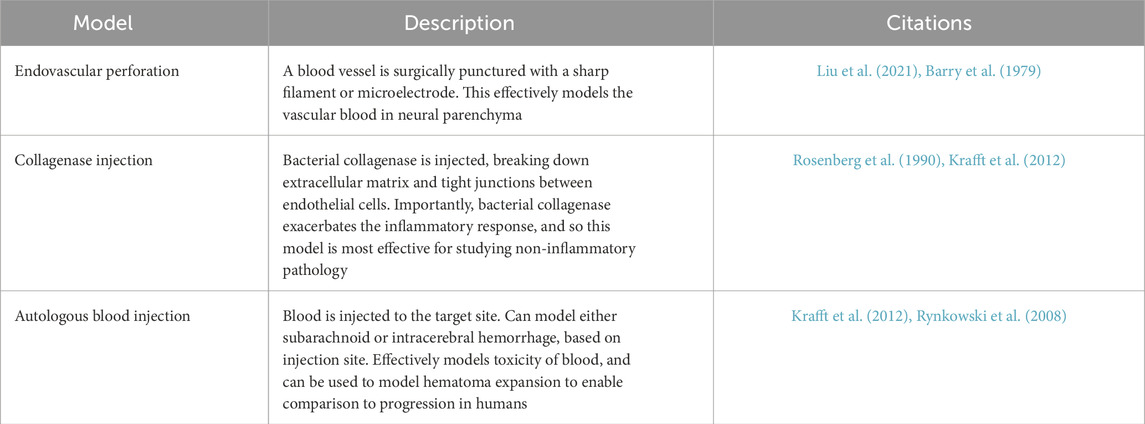
Table 1. Common animal models of hemorrhagic stroke.
Additionally, stem cell transplantation has also been investigated, based on the findings that the certain populations of stem cell can provide an anti-inflammatory and pro-growth environment through secreted factors (Stonesifer et al., 2017). As mentioned earlier, investigations in a mouse model of intraventricular hemorrhage has shown that oligodendrocyte precursor cells die by ferroptosis, thus the protection or replacement of these cells may protect white matter connections in brain tissue (Shen et al., 2022).
Ultimately, hemorrhagic stroke is a disease with a complex pathology which involves multiple mechanisms for cell death. With continued research, we can develop a better understanding of disease progression, and we may be able to develop treatments that can be powerful strategies to prevent brain cell death in hemorrhagic stroke.
Author contributions
JHR: Conceptualization, Writing – original draft, Writing – review and editing. CMT: Conceptualization, Writing – review and editing, Funding acquisition, Resources, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by a Sponsored Research Agreement from IPCure to CMT, by pilot funding from The Taub Institute Grants for EmergingResearch (TIGER) to CMT and by pilot funding from The Carol and Gene Ludwig Center for Neurodegeneration to CMT.
The authors declare that this study received funding from Cure/Deerfield to CMT. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Acknowledgments
Figure created in BioRender. JR, J. (2025) https://BioRender.com/v64r183.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
An, S. J., Kim, T. J., and Yoon, B.-W. (2017). Epidemiology, risk factors, and clinical features of intracerebral hemorrhage: an update. J. Stroke 19, 3–10. doi:10.5853/jos.2016.00864
Barry, K. J., Gogjian, M. A., and Stein, B. M. (1979). Small animal model for investigation of subarachnoid hemorrhage and cerebral vasospasm. Stroke 10, 538–541. doi:10.1161/01.str.10.5.538
Bautista, W., Adelson, P. D., Bicher, N., Themistocleous, M., Tsivgoulis, G., and Chang, J. J. (2021). Secondary mechanisms of injury and viable pathophysiological targets in intracerebral hemorrhage. Ther. Adv. Neurol. Disord. 14, 17562864211049208. doi:10.1177/17562864211049208
Bertheloot, D., Latz, E., and Franklin, B. S. (2021). Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell. Mol. Immunol. 18, 1106–1121. doi:10.1038/s41423-020-00630-3
Chang, P., Dong, W., Zhang, M., Wang, Z., Wang, Y., Wang, T., et al. (2014). Anti-necroptosis chemical necrostatin-1 can also suppress apoptotic and autophagic pathway to exert neuroprotective effect in mice intracerebral hemorrhage model. J. Mol. Neurosci. 52, 242–249. doi:10.1007/s12031-013-0132-3
Chen, C.-W., Chen, T. Y., Tsai, K. L., Lin, C. L., Yokoyama, K. K., Lee, W. S., et al. (2012). Inhibition of autophagy as a therapeutic strategy of iron-induced brain injury after hemorrhage. Autophagy 8, 1510–1520. doi:10.4161/auto.21289
Chen, M., Zhang, H., Chu, Y. H., Tang, Y., Pang, X. W., Qin, C., et al. (2022). Microglial autophagy in cerebrovascular diseases. Front. Aging Neurosci. 14, 1023679. doi:10.3389/fnagi.2022.1023679
Coelho, L. G. B. S. A., Costa, J. M. D., and Silva, E. I. P. A. (2016). Non-aneurysmal spontaneous subarachnoid hemorrhage: perimesencephalic versus non-perimesencephalic. Rev. Bras. Ter. Intensiva 28, 141–146. doi:10.5935/0103-507X.20160028
Coleman, M. L., Sahai, E. A., Yeo, M., Bosch, M., Dewar, A., and Olson, M. F. (2001). Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell. Biol. 3, 339–345. doi:10.1038/35070009
Denton, D., and Kumar, S. (2019). Autophagy-dependent cell death. Cell. Death Differ. 26, 605–616. doi:10.1038/s41418-018-0252-y
Dhuriya, Y. K., and Sharma, D. (2018). Necroptosis: a regulated inflammatory mode of cell death. J. Neuroinflammation 15, 199. doi:10.1186/s12974-018-1235-0
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 149, 1060–1072. doi:10.1016/j.cell.2012.03.042
Elmore, S. (2007). Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516. doi:10.1080/01926230701320337
Fang, M., Xia, F., Chen, Y., Shen, Y., Ma, L., You, C., et al. (2022). Role of eryptosis in hemorrhagic stroke. Front. Mol. Neurosci. 15, 932931. doi:10.3389/fnmol.2022.932931
Feigin, V. L., Brainin, M., Norrving, B., Martins, S., Sacco, R. L., Hacke, W., et al. (2022). World stroke organization (WSO): global stroke fact sheet 2022. Int. J. Stroke 17, 18–29. doi:10.1177/17474930211065917
Grasl-Kraupp, B., Ruttkay-Nedecky, B., Koudelka, H., Bukowska, K., Bursch, W., and Schulte-Hermann, R. (1995). In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology 21, 1465–1468. doi:10.1002/hep.1840210534
Green, D. R. (2000). Apoptotic pathways: paper wraps stone blunts scissors. Cell. 102, 1–4. doi:10.1016/s0092-8674(00)00003-9
Guo, F., Li, X. j., Chen, L. y., Yang, H., Dai, H. y., Wei, Y. s., et al. (2006). Study of relationship between inflammatory response and apoptosis in perihematoma region in patients with intracerebral hemorrhage. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue 18, 290–293.
Hwang, I.-C., Kim, J. Y., Kim, J. H., Lee, J. E., Seo, J. Y., Lee, J. W., et al. (2018). Therapeutic potential of a novel necrosis inhibitor, 7-amino-indole, in myocardial ischemia–reperfusion injury. Hypertension 71, 1143–1155. doi:10.1161/HYPERTENSIONAHA.117.09405
Kelly, K. J., Sandoval, R. M., Dunn, K. W., Molitoris, B. A., and Dagher, P. C. (2003). A novel method to determine specificity and sensitivity of the TUNEL reaction in the quantitation of apoptosis. Am. J. Physiol. Cell. Physiol. 284, C1309–C1318. doi:10.1152/ajpcell.00353.2002
Khalid, N., and Azimpouran, M. (2025). Necrosis. In StatPearls. Treasure Island (FL): StatPearls Publishing.
Koduri, S., Keep, R. F., Xi, G., Chaudhary, N., and Pandey, A. S. (2022). The role of iron in hemorrhagic stroke. Stroke Vasc. Interventional Neurology 2, e000419. doi:10.1161/svin.122.000419
Krafft, P. R., Rolland, W. B., Duris, K., Lekic, T., Campbell, A., Tang, J., et al. (2012). Modeling intracerebral hemorrhage in mice: injection of autologous blood or bacterial collagenase. J. Vis. Exp. 4289, e4289. doi:10.3791/4289
Kuriakose, D., and Xiao, Z. (2020). Pathophysiology and treatment of stroke: present status and future perspectives. Int. J. Mol. Sci. 21, 7609. doi:10.3390/ijms21207609
Lee, J.-Y., He, Y., Sagher, O., Keep, R., Hua, Y., and Xi, G. (2009). Activated autophagy pathway in experimental subarachnoid hemorrhage. Brain Res. 1287, 126–135. doi:10.1016/j.brainres.2009.06.028
Li, Q., Han, X., Lan, X., Gao, Y., Wan, J., Durham, F., et al. (2017). Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2, e90777. doi:10.1172/jci.insight.90777
Lin, X., Ye, H., Siaw-Debrah, F., Pan, S., He, Z., Ni, H., et al. (2018). AC-YVAD-CMK inhibits pyroptosis and improves functional outcome after intracerebral hemorrhage. Biomed. Res. Int. 2018, 3706047. doi:10.1155/2018/3706047
Liu, S., Tielking, K., von Wedel, D., Nieminen-Kelhä, M., Mueller, S., Boehm-Sturm, P., et al. (2021). Endovascular perforation model for subarachnoid hemorrhage combined with magnetic resonance imaging (MRI). J. Vis. Exp. (JoVE), e63150. doi:10.3791/63150
MacLellan, C. L., Silasi, G., Auriat, A. M., and Colbourne, F. (2010). Rodent models of intracerebral hemorrhage. Stroke 41, S95–S98. doi:10.1161/STROKEAHA.110.594457
Magid-Bernstein, J., Girard, R., Polster, S., Srinath, A., Romanos, S., Awad, I. A., et al. (2022). Cerebral hemorrhage: pathophysiology, treatment, and future directions. Circulation Res. 130, 1204–1229. doi:10.1161/CIRCRESAHA.121.319949
Martens, S., Bridelance, J., Roelandt, R., Vandenabeele, P., and Takahashi, N. (2021). MLKL in cancer: more than a necroptosis regulator. Cell. Death Differ. 28, 1757–1772. doi:10.1038/s41418-021-00785-0
Moujalled, D., Strasser, A., and Liddell, J. R. (2021). Molecular mechanisms of cell death in neurological diseases. Cell. Death Differ. 28, 2029–2044. doi:10.1038/s41418-021-00814-y
Naito, M. G., Xu, D., Amin, P., Lee, J., Wang, H., Li, W., et al. (2020). Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke. Proc. Natl. Acad. Sci. U. S. A. 117, 4959–4970. doi:10.1073/pnas.1916427117
Qureshi, A. I., Suri, M. F. K., Ostrow, P. T., Kim, S. H., Ali, Z., Shatla, A. A., et al. (2003). Apoptosis as a form of cell death in intracerebral hemorrhage. Neurosurgery 52, 1041–1048. doi:10.1227/01.neu.0000057694.96978.bc
Rosenberg, G. A., Mun-Bryce, S., Wesley, M., and Kornfeld, M. (1990). Collagenase-induced intracerebral hemorrhage in rats. Stroke 21, 801–807. doi:10.1161/01.str.21.5.801
Rynkowski, M. A., Kim, G. H., Komotar, R. J., Otten, M. L., Ducruet, A. F., Zacharia, B. E., et al. (2008). A mouse model of intracerebral hemorrhage using autologous blood infusion. Nat. Protoc. 3, 122–128. doi:10.1038/nprot.2007.513
Schüller, K., Bühler, D., and Plesnila, N. (2013). A murine model of subarachnoid hemorrhage. J. Vis. Exp. 50845, e50845. doi:10.3791/50845
Scott, M. C., Bedi, S. S., Olson, S. D., Sears, C. M., and Cox, C. S. (2021). Microglia as therapeutic targets after neurological injury: strategy for Cell Therapy. Expert Opin. Ther. Targets 25, 365–380. doi:10.1080/14728222.2021.1934447
Shen, D., Wu, W., Liu, J., Lan, T., Xiao, Z., Gai, K., et al. (2022). Ferroptosis in oligodendrocyte progenitor cells mediates white matter injury after hemorrhagic stroke. Cell. Death Dis. 13, 259–314. doi:10.1038/s41419-022-04712-0
Shen, X., Dong, W., Wu, Q., Gao, Y., Luo, C., et al. (2016). Autophagy regulates intracerebral hemorrhage induced neural damage via apoptosis and NF-κB pathway. Neurochem. Int. 96, 100–112. doi:10.1016/j.neuint.2016.03.004
Shoamanesh, A., and Katsanos, A. H. (2021). Combatting secondary injury from intracerebral hemorrhage with supplemental antioxidant therapy. Stroke 52, 1182–1184. doi:10.1161/STROKEAHA.121.033849
Singh, N., and Bhattacharyya, D. (2014). Collagenases in an ether extract of bacterial metabolites used as an immunostimulator induces TNF-α and IFN-γ. Int. Immunopharmacol. 23, 211–221. doi:10.1016/j.intimp.2014.08.026
Stonesifer, C., Corey, S., Ghanekar, S., Diamandis, Z., Acosta, S. A., and Borlongan, C. V. (2017). Stem cell therapy for abrogating stroke-induced neuroinflammation and relevant secondary cell death mechanisms. Prog. Neurobiol. 158, 94–131. doi:10.1016/j.pneurobio.2017.07.004
Thangameeran, S. I. M., Pang, C. Y., Lee, C. H., Tsai, S. T., Hu, W. F., and Liew, H. K. (2022). Experimental animal models and evaluation techniques in intracerebral hemorrhage. Tzu Chi Med. J. 35, 1–10. doi:10.4103/tcmj.tcmj_119_22
Towers, C. G., and Thorburn, A. (2016). Therapeutic targeting of autophagy. EBioMedicine 14, 15–23. doi:10.1016/j.ebiom.2016.10.034
Velier, J. J., Ellison, J. A., Kikly, K. K., Spera, P. A., Barone, F. C., and Feuerstein, G. Z. (1999). Caspase-8 and caspase-3 are expressed by different populations of cortical neurons undergoing delayed cell death after focal stroke in the rat. J. Neurosci. 19, 5932–5941. doi:10.1523/JNEUROSCI.19-14-05932.1999
Wang, X., Mori, T., Sumii, T., and Lo, E. H. (2002). Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress. Stroke 33, 1882–1888. doi:10.1161/01.str.0000020121.41527.5d
Westphal, D., Kluck, R. M., and Dewson, G. (2014). Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell. Death Differ. 21, 196–205. doi:10.1038/cdd.2013.139
Xu, G., and Shi, Y. (2007). Apoptosis signaling pathways and lymphocyte homeostasis. Cell. Res. 17, 759–771. doi:10.1038/cr.2007.52
Zazulia, A. R., Diringer, M. N., Derdeyn, C. P., and Powers, W. J. (1999). Progression of mass effect after intracerebral hemorrhage. Stroke 30, 1167–1173. doi:10.1161/01.str.30.6.1167
Zhang, P., Gao, C., Guo, Q., Yang, D., Zhang, G., Lu, H., et al. (2024). Single-cell RNA sequencing reveals the evolution of the immune landscape during perihematomal edema progression after intracerebral hemorrhage. J. Neuroinflammation 21, 140. doi:10.1186/s12974-024-03113-8
Zhang, Y., Khan, S., Liu, Y., Zhang, R., Li, H., Wu, G., et al. (2022). Modes of brain cell death following intracerebral hemorrhage. Front. Cell. Neurosci. 16, 799753. doi:10.3389/fncel.2022.799753
Zhang, Z., Wu, Y., Yuan, S., Zhang, P., Zhang, J., Li, H., et al. (2018). Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res. 1701, 112–125. doi:10.1016/j.brainres.2018.09.012
Zhou, C., Yamaguchi, M., Kusaka, G., Schonholz, C., Nanda, A., and Zhang, J. H. (2004). Caspase inhibitors prevent endothelial apoptosis and cerebral vasospasm in dog model of experimental subarachnoid hemorrhage. J. Cereb. Blood Flow. Metab. 24, 419–431. doi:10.1097/00004647-200404000-00007
Ziai, W. C. (2013). Hematology and inflammatory signaling of intracerebral hemorrhage. Stroke 44, S74–S78. doi:10.1161/STROKEAHA.111.000662
Keywords: hemorrhagic stroke, cell death, apoptosis, inflammation, neurovascular disease, caspases
Citation: Rinald JH and Troy CM (2025) A review of cell death pathways in hemorrhagic stroke. Front. Cell Dev. Biol. 13:1570569. doi: 10.3389/fcell.2025.1570569
Received: 03 February 2025; Accepted: 15 April 2025;
Published: 28 April 2025.
Edited by:
Amit Singh, University of Dayton, United StatesReviewed by:
Ling Lin, The Pennsylvania State University, United StatesCopyright © 2025 Rinald and Troy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carol M. Troy, Y210MkBjdW1jLmNvbHVtYmlhLmVkdQ==