Abstract
Precursor mRNA (pre-mRNA) must undergo splicing to remove intron sequences and join exons. This splicing process is catalysed by an RNA/protein complex called the spliceosome. At the centre of the catalytic spliceosome is the U5 small nuclear ribonucleoprotein (snRNP). Pathogenic variants in U5 snRNP core proteins are associated with various diseases commonly known as spliceosomopathies. Variants in TXNL4A and EFTUD2 manifest in craniofacial malformations while variants in PRPF8 and SNRNP200 manifest in retinitis pigmentosa. This perspective highlights research addressing how these specific manifestations come about as the spliceosome is required in all cells and at all developmental stages. Cell and animal models can replicate the human clinical specificity providing explanations for the specificity of the disorders. We propose that future research could benefit from models originating from patient-derived induced pluripotent stem cells (iPSCs) and isogenic controls to compare the coding and non-coding transcriptomic perturbations. Analysis of spliceosomal protein complexes and their interactome could also uncover novel insights on molecular pathogenesis. Finally, as studies highlight changes in metabolic processes, metabolomic studies could become a new venture in studying the consequences of U5 snRNP variants.
1 Introduction
Human genes, like other eukaryotes, contain translated exons and untranslated introns. To correctly produce the final messenger RNA and protein products, the precursor messenger RNA (pre-mRNA), as the initial product of transcription, must undergo splicing. The molecular mechanism of the splicing process has been reviewed elsewhere (Wan et al., 2020; Wilkinson et al., 2020). The RNA splicing process is catalysed by a complex RNA/protein machinery, the spliceosome. The spliceosome involves five small nuclear ribonucleoproteins (snRNPs) composed of small nuclear RNAs (snRNAs) and their respective associated proteins (Beusch and Madhani, 2024). In humans, the majority of introns (more than 99.5%) are spliced by the major spliceosome while the rest are catalysed by the minor spliceosome. The U5 snRNP is involved in the catalytic process of both spliceosomes (Akinyi and Frilander, 2021).
The U5 snRNP consists of the U5 snRNA, seven Sm proteins, and eight core proteins (Wood et al., 2021). The core U5 snRNP is assembled in the cytoplasm from an Sm protein ring and U5 snRNA and then imported to the nucleus. In nuclear Cajal bodies, the core U5 snRNP interacts with an RNA-free heterotetrameric complex consisting PRPF8, SNRNP200, EFTUD2, and SNRNP40. Afterwards, PRPF6, DDX23, CD2BP2, and TXNL4A join the complex. The final U5 snRNP then interacts with the U4/U6 di-snRNP to form U4/U6.U5 tri-snRNP (Klimešová et al., 2021; Riabov Bassat et al., 2024). In the central region of the tri-snRNP, PRPF8 serves as scaffold. It interacts and positions the U5 snRNA with exons. PRPF8 also interacts with EFTUD2 and regulates SNRNP200 ATPase activity (Kastner et al., 2019). TXNL4A is also located centrally in the tri-snRNP. For the formation of the catalytic spliceosome, TXNL4A must depart the spliceosome (Schreib et al., 2018).
Pathogenic variants affecting spliceosome components manifest in a wide range of diseases or disorders commonly known as spliceosomopathies (Love et al., 2023). Variants in TXNL4A and EFTUD2 manifest in similar craniofacial malformations while variants in PRPF8, SNRNP200, and PRPF6 result in retinal degeneration. These genetic disorders have been previously reviewed elsewhere (Beauchamp et al., 2020; Griffin and Saint-Jeannet, 2020; Maxwell et al., 2021; Wood et al., 2021). In this perspective, we would like to highlight research that has been conducted to address the specific disease manifestations as well as raise insights for future research. Due to space constraints, this perspective will not focus on PRPF6. For consistency, in this article, we use TXNL4A, EFTUD2, PRPF8, and SNRNP200 to refer to the human and animal proteins and Dib1, Snu114, Prp8, and Brr2 to its respective orthologue in yeast.
2 U5 snRNP components with craniofacial manifestation
2.1 TXNL4A
TXNL4A, orthologous to yeast Dib1, is one of the core proteins of the U5 snRNP (Liu et al., 2006). It stabilises U4/U6.U5 tri-snRNP complex, preventing premature spliceosome activation (Schreib et al., 2018). Variants in this gene cause Burn-McKeown syndrome (BMKS). The syndrome is a rare autosomal-recessive disorder with choanal atresia, craniofacial anomalies including lower eyelid coloboma and cleft lip, and other extra-craniofacial manifestations. Almost all patients have normal intellectual development with only one reported having intellectual disability (Wieczorek et al., 2014; Wood et al., 2022). Clinical manifestations arise from compound heterozygosity of a loss-of-function allele and either type 1 or type 2 promoter deletion or homozygosity of either type of promoter deletion (Wieczorek et al., 2014; Goos et al., 2017; Wood et al., 2022). Promoter deletion reduces TXNL4A levels shown by luciferase assays (Wieczorek et al., 2014; Wood et al., 2022).
Using frog Xenopus, Park et al. (2022) showed spatiotemporal specificity of Txnl4a during embryo development. While Txnl4a is expressed at all developmental stages, the gene is enriched at the anterior neural plate and the neural-crest-forming regions during the neurula stage. Knockdown of Txnl4a decreases the expression of several neural crest genes and negatively affects neural crest formation and survival, thus impacting craniofacial development (Park et al., 2022). As shown in mouse embryonic stem cells, the molecular pathogenesis involves mis-splicing of Mdm2, upregulation of p53, and downregulation of several glycolytic genes (Varineau and Calo, 2024). Utilising BMKS patient-derived induced pluripotent stem cells (iPSCs), Wood et al. (2020) found that patient cells have delayed proliferation and differentiation into neural crest cells (NCCs). The NCCs had delayed epithelial-to-mesenchymal transition which might be caused by downregulated WNT signalling via mis-splicing of TCF7L2. Both in vitro and animal model data indicate that neural crest developmental defect causes the craniofacial malformations in BMKS patients.
2.2 EFTUD2
EFTUD2 and its yeast orthologue Snu114 are GTP-bound proteins in the core of the U5 snRNP providing a scaffold for spliceosome complex assembly and disassembly (Jia et al., 2020). Variants that inactivate one allele causing haploinsufficiency in EFTUD2 result in Mandibulofacial dysostosis Guion-Almeida type (MFDGA). Patients are characterized by malar and mandibular hypoplasia as the core craniofacial malformations as well as microcephaly, external ear malformations, and intellectual disability (Abell et al., 2021). The clinical phenotype hints at cell-type specific effects of EFTUD2 variants. To study the molecular mechanism of EFTUD2 variant, Wood et al. (2019) introduced a knockdown mutation in HEK293 human cell line. The knockdown caused changes not only in the expression level but also in the mis-splicing of genes associated with biological processes involved in craniofacial development. In a zebrafish model (Lei et al., 2017), apoptosis occurs in the head and spinal cord’s neuronal progenitors of the Eftud2 mutant leading to smaller heads and abnormal brain development. Non-neuronal organ patterning and development had little or no changes. In a mouse model, similar tissue specificity of Eftud2 expression was also observed. Eftud2 was expressed in precursors of the brain, face, and head which are the affected organs of MFDGA patients. However, there was a lack of craniofacial malformation and p53 activation in the heterozygous mouse model. Additionally, homozygous mutants were lethal (Beauchamp et al., 2019). To address the lethality issue, neural crest cell-specific Eftud2 homozygous mutants were developed using a Wnt1-Cre transgene (Beauchamp et al., 2021). The resulting mutants developed brain and craniofacial malformation, replicating the human phenotype.
EFTUD2 molecular pathogenesis in neural crest cells is proposed to be mediated by p53 upregulation. In HEK293 cells, p53 was identified as a top upstream regulator of genes with increased exon skipping caused by EFTUD2 knockdown (Wood et al., 2019). The p53 signalling pathway is also activated in neural cell progenitors by aberrant splicing in Eftud2-mutant zebrafish (Lei et al., 2017). The involvement of p53 was also supported by the improvement of craniofacial development by a p53 inhibitor in the mouse model (Beauchamp et al., 2021). Specifically, p53 pathway upregulation seems to be induced by exon 3 skipping in Mdm2 (Beauchamp et al., 2021; Varineau and Calo, 2024). However, craniofacial malformations from Eftud2 insufficiency can also be triggered by p53-independent pathways (Beauchamp et al., 2022). As EFTUD2 knockdown elicits endoplasmic reticulum stress and unfolded protein responses in a human cell line (Wood et al., 2019), these pathways might contribute to the abnormal cellular processes leading to apoptosis of neural crest cells. While the cell line and animal models have provided additional insights on MFDGA pathogenesis, patient-derived iPSCs should be generated and differentiated into neural crest cells and other relevant cell types to better understand the molecular mechanisms of EFTUD2 variants.
3 U5 snRNP components with retinal manifestation
3.1 PRPF8
PRPF8, the human orthologue of yeast Prp8, occupies the catalytic centre of the spliceosome (Grainger and Beggs, 2005). Its N-terminal portion interacts with EFTUD2 (Jia et al., 2020) while its C-terminal Jab1/MPN domain regulates SNRNP200 ATPase activity (Mayerle and Guthrie, 2016). Variants in PRPF8 cause retinitis pigmentosa type 13 (RP-13). This progressive retinal degeneration is inherited in an autosomal dominant manner (McKie et al., 2001). Specific manifestation of PRPF8 variants in the retina might result from the increased splicing activity in retina. Compared to other cell types, retina highly expresses snRNAs and spliced transcripts of housekeeping/constitutive mRNAs (Tanackovic et al., 2011). The worm C. elegans model of PRPF8 knockdown also revealed that defect phenotype occurs in a cell-type with high transcriptional activity (Rubio-Pena et al., 2015). Using fibroblasts from RP patients, Atkinson et al. (2024) generated iPSCs and used CRISPR-Cas9 correcting the variant in PRPF8 to generate isogenic normal controls. Both iPSCs were differentiated into retinal pigment epithelium (RPE), retinal organoids, as well as kidney organoids. The study supported the tissue/organ-specific defects in RP where impaired alternative splice site selection and enhanced usage of cryptic/aberrant splice sites occur in RPE and retinal organoids compared to kidney organoids. The specificity might stem from special nuclear clusters of transcription and splicing machineries in human photoreceptor cells. RPE and retinal organoids derived from RP patient’s iPSCs had dispersed nuclear speckles, while kidney organoids from the same iPSCs had nuclear speckles structurally similar to controls (Atkinson et al., 2024). While this subcellular organisation facilitates higher efficiency in human photoreceptor cells, this organisation introduces a vulnerability to pathogenesis observed in RP.
Drosophila models expressing variant Prpf8 had increased apoptosis in developing eye primordium leading to smaller adult eyes. Interestingly, this apoptosis was only observed when the variant proteins were expressed early in the eye primordium but not when expressed late in the differentiated photoreceptors (Stanković et al., 2020). In line with the fly model, mouse models with heterozygous and homozygous Prpf8 variants also had no photoreceptor degeneration. However, the retinal pigment epithelium (RPE) had degenerative ultrastructural changes that were more severe in the homozygous model (Graziotto et al., 2011). Using primary RPE culture from the homozygous variant model, Farkas et al. (2014) showed functional disturbance of adhesion and phagocytosis in the variant RPE’s photoreceptor outer segments. The animal models show that the RP manifestation is not only organ-specific, but also cell-type specific.
3.2 SNRNP200
SNRNP200, and its yeast counterpart Brr2, are helicases crucial for the snRNA unwinding process during spliceosome activation (Ledoux and Guthrie, 2016). Variants in SNRNP200 cause retinitis pigmentosa type 33 (RP-33). This inherited retinal degeneration seems to have a dual inheritance pattern. Initially identified as autosomal dominant, there have been several reports indicating autosomal recessive inheritance pattern (Holtes et al., 2025). While the variants specifically affect the retina, SNRNP200 is ubiquitously expressed in human, mice, and zebrafish (Zhao et al., 2009; Zhang T. et al., 2021). Both SNRNP200 knockdown and SNRNP200 variant expression resulted in systemic deformities in zebrafish embryos. The zebrafish model developed normal ocular size and cone photoreceptor morphology, but had significantly decreased rhodopsin (Liu et al., 2015; Zhang T. et al., 2021). Interestingly, while knockdown increased larvae deformities with moderate death, variant protein significantly increased larval death (Zhang T. et al., 2021). This observation supports the dominant-negative pattern of SNRNP200, as observed in humans.
As with other spliceosomal defects, the molecular pathogenesis might involve disturbance in cell cycle/growth. Expression of variant SNRNP200 in HEK293 cells resulted in observable cell dysmorphology (Ehsani et al., 2013). Cell cycle is also affected, with increased cell populations in G2/M phase compared to the control. On the other hand, knocking down SNRNP200 resulted in decreased G1 phase and increased S phase but unchanged G2/M phase. This observation hints at a different mechanism between dominant-negative variant and knockdown. One putative mechanism is through more marked reduction of alpha-tubulin expression in cell lines expressing variant protein (Ehsani et al., 2013). Patient-derived iPSCs could be used to further study the SNRNP200 variants mechanism in human cells and tissues (Zhang D. et al., 2021).
4 Discussion
Even though splicing is a ubiquitous process, different variants in the pre-mRNA splicing machineries result in different pathologies. Here we discuss two contrasting phenotypes from variants in U5 snRNP proteins. TXNL4A and EFTUD2 manifest in craniofacial deformation which might arise from similar defects in cranial neural crest cell formation (Park et al., 2022). On the other hand, PRPF8 and SNRNP200 have an even more specific phenotype in the form of retinal dysfunction. Even within the same type of defect, the phenotypes are still distinct. BMKS is distinguishable from MFDGA based on microcephaly and developmental delay in MFDGA. Distinctive facial phenotypes such as lower eyelid coloboma in BMKS and malar hypoplasia in MFDGA also help to distinguish these craniofacial disorders (Lüdecke and Wieczorek, 2016; Abell et al., 2021). Differences between BMKS and MFDGA clinical phenotypes might be explained by unique gene expression changes (Park et al., 2022). Interestingly, TXNL4A and EFTUD2 knockdowns shared common molecular mechanism. The splicing perturbation in the Mdm2 gene leads to p53 upregulation followed by glycolytic transcript downregulation (Varineau and Calo, 2024). In contrast, different variants of the same gene might result in different transcriptional changes. In Drosophila, different Prpf8 point-mutations had dissimilar differentially expressed genes with only limited overlap (Stanković et al., 2020). As more patients are identified and analysed, we might see distinct manifestations that might confront the cell specificity hypothesis. For example, a study reported retinal dystrophy in an MFDGA patient with a novel EFTUD2 variant, where the condition does not typically involve eye anomalies (Deml et al., 2015).
The variants in U5 snRNP components usually manifest in cell types and developmental stages that are relatively inaccessible in humans. Therefore, in vitro and animal models have been utilised to obtain insights on the effects of U5 snRNP component variants (Figure 1). Variants of interest can be introduced into established human cell lines. For example, Wood et al. (2019) utilised CRISPR-Cas modification to introduce EFTUD2 haploinsufficiency in human embryonic kidney HEK293 cells. Moreover, patient-derived cells can be induced for pluripotency and differentiated into target cells, even organoids. Several studies have compared patient-derived iPSCs with healthy control-derived iPSCs, but this approach must account for different genetic background between patient and control cells. To address this issue, Atkinson et al. (2024) developed an isogenic normal control where the disease-causing variant is corrected by CRISPR-Cas9.
FIGURE 1
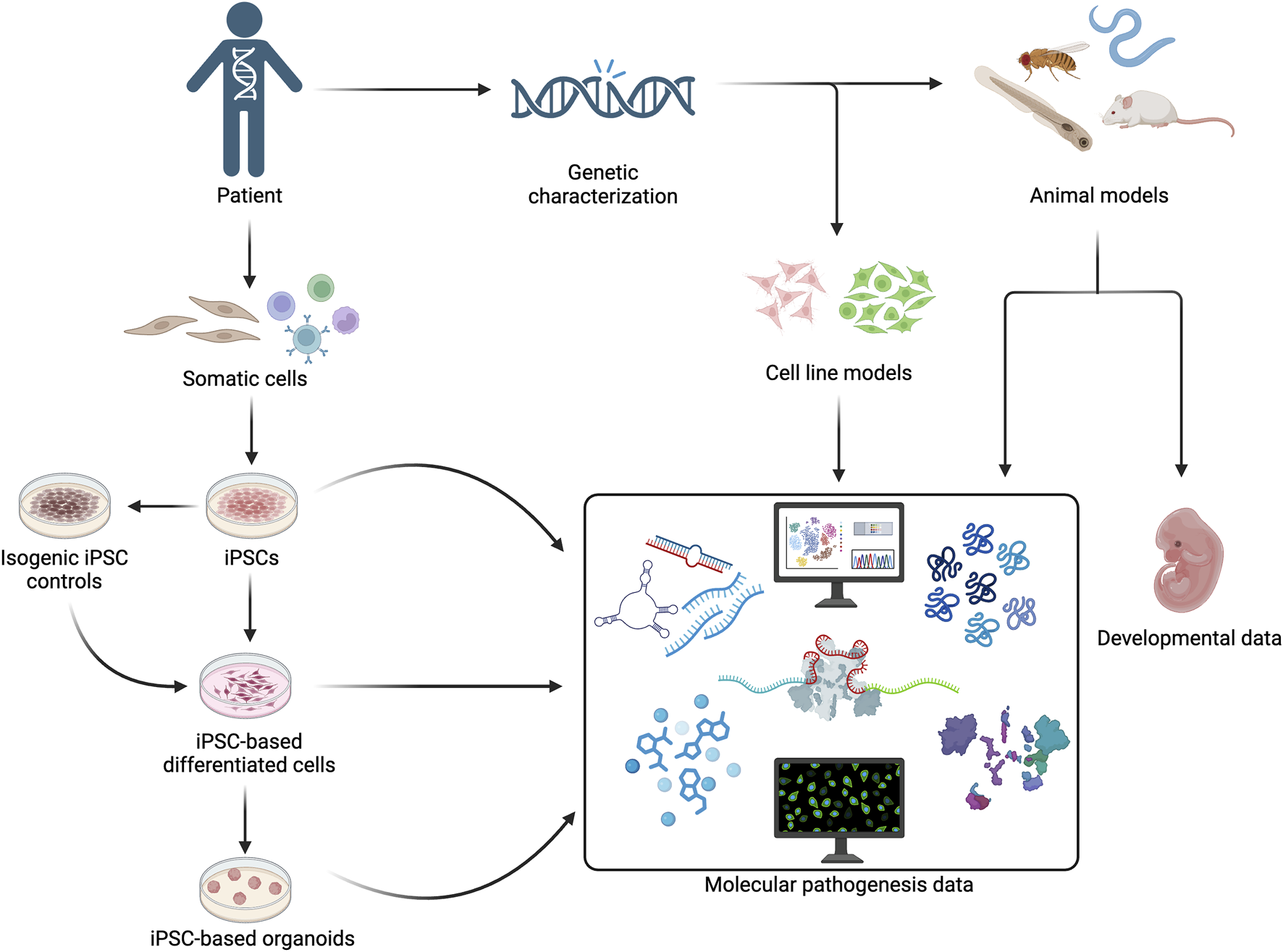
Patient-derived cells, human cell lines, and animal models can be utilised to obtain molecular pathogenesis and developmental manifestation addressing the tissue-specificity of U5 snRNP protein gene variants.
Most spliceosomopathies are studied in vitro by differentiating iPSC into the (putative) affected cell types. While this approach seems the most sensible approach, it has not fully addressed the specificity question. If variants in PRPF8 manifest in retina as a result of its high splicing activity, why do variants in TXNL4A or EFTUD2 not manifest in the retina as well? Atkinson et al. (2024) has tried to address the specificity by analysing PRPF8 variants in unaffected tissue/organs. They suggested that different subcellular organization, such as of nuclear speckles, in different cell types could serve as the basis of cell specificity. However, future research could compare the manifestation of TXNL4A and EFTUD2 variants in retinal cell/organ or PRPF8 and SNRNP200 variants in neural crest cell to gain more insights on the mechanisms. Additionally, since spliceosomopathies could affect several organs/systems, analysing the manifestation in more cell types could uncover the mechanism(s) behind the specificity. For example, osteoblast and chondrocyte cells could be utilized to study skeletal dysplasia in MFDGA (Wu et al., 2019). While RP does not usually have skeletal manifestation, SNRNP200 knockdown inhibits osteo-/dentinogenic differentiation of stem cells from the apical papilla (Su et al., 2020). These studies show the benefit of expanding the cell type landscape in increasing our understanding on spliceosomopathies.
Spliceosomopathies manifest early in development. Since it is controversial to study the disease in developing human embryos, animal models have provided a way to study the effect of spliceosome gene variants in early development. Lower animal models such as C. elegans and Drosophila as well as higher animal models such as zebrafish, frog, and mice have been developed. A common challenge is the lethality of knockdowns or homozygous mutants. Several methods have been utilized to overcome this phenomenon such as using Wnt1-Cre transgene to develop cell-type specific mutants (Beauchamp et al., 2021) or utilising different promoter systems to express the target protein at different developmental stages (Stanković et al., 2020). Using those methods, animal models could be designed to observe the variant effects not only on specific cell types but also at specific developmental stages.
Once the disease models have been developed, various molecular analysis could be conducted (Figure 1). Transcriptomic and proteomic changes are measured in various studies with spliceosome dysregulation. While comparison between variant and healthy controls has been the norm, one might also add another layer of analysis such as different transcriptional and proteomic changes resulting from different variants of the same gene. For example, in Drosophila, the expression of more toxic PRPF8 variants had only limited overlapping differentially expressed genes compared to a variant associated with mild or no human symptoms. Apoptosis and adult eye defect were induced by the more toxic variants but not by a milder one (Stanković et al., 2020). The information might explain the manifestation variability between various U5 snRNP variants. Comparing the molecular pathogenesis of different SNRNP200 variants might answer the question of its dual inheritance pattern (Holtes et al., 2025). Another parameter of interest is the non-coding RNAs. It has been shown with PRPF8 that variant protein causes misexpression of circRNAs and degeneration of cerebellar granule cells in a mouse model (Krausová et al., 2023). The effect of other U5 snRNP variants on non-coding RNA should also be studied. SNRNP200 plays a role in plant miRNA biogenesis (Li et al., 2024), therefore variants affecting SNRNP200 activity might result in changes of miRNA expression. As mutating Dicer protein responsible for miRNA biogenesis results in neural crest cells defects and craniofacial anomalies (Huang et al., 2010), understanding the role of TXNL4A and EFTUD2 variants on miRNA biogenesis in neural crest cells could open a new molecular mechanism in craniofacial malformations.
U5 snRNP variants affect spliceosome assembly or catalytic capacities. As shown in yeast, both Dib1 and Snu114 variants reduce the U5 and/or U4/U6.U5 tri-snRNP spliceosome complex assembly (Brenner and Guthrie, 2006; Wieczorek et al., 2014). The molecular mechanism behind this effect could be the decrease of functional proteins caused by loss of function, reduced expression from mutated promoter, or mis-splicing of the gene transcript (Wieczorek et al., 2014; Thomas et al., 2020; Wood et al., 2022). Meanwhile, variant PRPF8 and SNRNP200 proteins are well-incorporated into the U4.U6/U5 tri-snRNP complex (Tanackovic et al., 2011; Cvačková et al., 2014; Atkinson et al., 2024). However, the variants cause functional defects. PRPF8 variants cause defects in the second catalytic step of splicing via deregulation of SNRNP200 helicase activity (Mozaffari-Jovin et al., 2013; Mozaffari-Jovin et al., 2014; Mayerle and Guthrie, 2016). SNRNP200 variants with reduced ATPase activity had lower RNA duplex unwinding activity, which is crucial to unwind the U4/U6 duplex (Ledoux and Guthrie, 2016). In yeast, defective Brr2 unwinding activity inhibits the formation of catalytic spliceosomes (Zhao et al., 2009). Variants in the Brr2 RNA-binding pocket also seems to shorten the Brr2 interaction time with pre-mRNA (Cvačková et al., 2014).
Transcriptomic and proteomic analysis also hint at the effects of variants in an individual U5 snRNP to different snRNP components. EFTUD2 knockdown decreases TXNL4A expression in HEK293 cells (Wood et al., 2019). SNRNP200 knockdown in zebrafish upregulates several spliceosome components, such as PRPF3, PRPF6, PRPF8, and PRPF31 (Liu et al., 2015). Since the spliceosome operates as a complex, a complexome type of analysis could be utilised to study the effect of variants on the complex dynamics (Wittig and Malacarne, 2021). Interactome analysis could also enrich the mechanistic insight behind variant U5 snRNPs pathogenesis. For example, a study showed that human cohesin protein interacts with EFTUD2 and SNRNP200, among several components of U4/U6.U5 tri-snRNP, which might explain cell cycle defect resulting from U5 snRNP variants (Kim et al., 2019). Since U5 snRNP is assembled in both the cytoplasm and nucleus (Matera and Wang, 2014), analysis on subcellular fractions should also be conducted. Study from another protein complex, the C-terminal to LisH (CTLH) E3 ligase complex, showed distinct assembly and interactomes in the cytoplasm and nucleus (Onea et al., 2022). Recent advances in spatial transcriptomics and spatial proteomics could also become useful tools to understand the contrasting craniofacial and retinal manifestations (Montgomery et al., 2022; Nalbach et al., 2023).
Finally, it has been shown that splicing perturbation caused by splicing factor knockdowns results in glycolytic transcript downregulation leading to cellular metabolic changes (Varineau and Calo, 2024). As glycolysis is important for neural crest migration (Bhattacharya et al., 2020) and survival of rod cells (Petit et al., 2018), its downregulation could also explain the molecular mechanism of U5 snRNP variants in the respective affected cells. Therefore, metabolomic studies could provide a novel point of view on the pathogenesis in spliceosomopathies.
The strategies discussed in this perspective are not only applicable for the four snRNP components described. The iPSC-derived RPE cells have been utilised to study the molecular pathogenesis of PRPF6 in retinitis pigmentosa 60 (Liang et al., 2022). For other less characterised U5 snRNP components, studies could be initiated by collecting patient cohorts through GeneMatcher (Sobreira et al., 2015). This approach has identified an association of DDX23 variants to a neurodevelopmental disorder (Burns et al., 2021). In mice, CD2BP2 and SNRNP40 knockout have been associated with podocyte and immune cell function, respectively (Albert et al., 2015; Zhang et al., 2019). Future studies could utilize GeneMatcher to identify and characterize patients with CD2BP2 or SNRNP40 variants. Once human association is found, further dissection of molecular and developmental pathogenesis could be investigated utilizing approaches outlined in this perspective.
Statements
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
RK: Conceptualization, Writing – original draft, Writing – review and editing. RO: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
RK study at the University of Manchester is funded by the Indonesia Endowment Fund for Education Agency.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Abell K. Hopkin R. J. Bender P. L. Jackson F. Smallwood K. Sullivan B. et al (2021). Mandibulofacial dysostosis with microcephaly: an expansion of the phenotype via parental survey. Am. J. Med. Genet. A185 (2), 413–423. 10.1002/ajmg.a.61977
2
Akinyi M. V. Frilander M. J. (2021). At the intersection of major and minor spliceosomes: crosstalk mechanisms and their impact on gene expression. Front. Genet.12, 700744. 10.3389/fgene.2021.700744
3
Albert G. I. Schell C. Kirschner K. M. Schäfer S. Naumann R. Müller A. et al (2015). The GYF domain protein CD2BP2 is critical for embryogenesis and podocyte function. J. Mol. Cell. Biol.7 (5), 402–414. 10.1093/jmcb/mjv039
4
Atkinson R. Georgiou M. Yang C. Szymanska K. Lahat A. Vasconcelos E. J. R. et al (2024). PRPF8-mediated dysregulation of hBrr2 helicase disrupts human spliceosome kinetics and 5′-splice-site selection causing tissue-specific defects. Nat. Commun.15 (1), 3138. 10.1038/s41467-024-47253-0
5
Beauchamp M. C. Alam S. S. Kumar S. Jerome-Majewska L. A. (2020). Spliceosomopathies and neurocristopathies: two sides of the same coin?Dev. Dyn.249 (8), 924–945. 10.1002/dvdy.183
6
Beauchamp M. C. Boucher A. Dong Y. Aber R. Jerome-Majewska L. A. (2022). Craniofacial defects in embryos with homozygous deletion of Eftud2 in their neural crest cells are not rescued by Trp53 deletion. Int. J. Mol. Sci.23 (16), 9033. 10.3390/ijms23169033
7
Beauchamp M. C. Djedid A. Bareke E. Merkuri F. Aber R. Tam A. S. et al (2021). Mutation in Eftud2 causes craniofacial defects in mice via mis-splicing of Mdm2 and increased P53. Hum. Mol. Genet.30 (9), 739–757. 10.1093/hmg/ddab051
8
Beauchamp M. C. Djedid A. Daupin K. Clokie K. Kumar S. Majewski J. et al (2019). Loss of function mutation of Eftud2, the gene responsible for mandibulofacial dysostosis with microcephaly (MFDM), leads to pre-implantation arrest in mouse. PLoS One14 (7), e0219280. 10.1371/journal.pone.0219280
9
Beusch I. Madhani H. D. (2024). Understanding the dynamic design of the spliceosome. Trends Biochem. Sci.49 (7), 583–595. 10.1016/j.tibs.2024.03.012
10
Bhattacharya D. Azambuja A. P. Simoes-Costa M. (2020). Metabolic reprogramming promotes neural crest migration via Yap/Tead signaling. Dev. Cell.53 (2), 199–211. 10.1016/j.devcel.2020.03.005
11
Brenner T. J. Guthrie C. (2006). Assembly of Snu114 into U5 snRNP requires Prp8 and a functional GTPase domain. RNA12 (5), 862–871. 10.1261/rna.2319806
12
Burns W. Bird L. M. Heron D. Keren B. Ramachandra D. Thiffault I. et al (2021). Syndromic neurodevelopmental disorder associated with de novo variants in DDX23. Am. J. Med. Genet. A185 (10), 2863–2872. 10.1002/ajmg.a.62359
13
Cvačková Z. Matějů D. Staněk D. (2014). Retinitis pigmentosa mutations of SNRNP200 enhance cryptic splice-site recognition. Hum. Mutat.35 (3), 308–317. 10.1002/humu.22481
14
Deml B. Reis L. M. Muheisen S. Bick D. Semina E. V. (2015). EFTUD2 deficiency in vertebrates: identification of a novel human mutation and generation of a zebrafish model. Birth Defects Res. A Clin. Mol. Teratol.103 (7), 630–640. 10.1002/bdra.23397
15
Ehsani A. Alluin J. V. Rossi J. J. (2013). Cell cycle abnormalities associated with differential perturbations of the human U5 snRNP associated U5-200kD RNA helicase. PLoS One8 (4), e62125. 10.1371/journal.pone.0062125
16
Farkas M. H. Lew D. S. Sousa M. E. Bujakowska K. Chatagnon J. Bhattacharya S. S. et al (2014). Mutations in pre-mRNA processing factors 3, 8, and 31 cause dysfunction of the retinal pigment epithelium. Am. J. Pathol.184 (10), 2641–2652. 10.1016/j.ajpath.2014.06.026
17
Goos J. A. C. Swagemakers S. M. A. Twigg S. R. F. van Dooren M. F. Hoogeboom A. J. M. Beetz C. et al (2017). Identification of causative variants in TXNL4A in Burn-McKeown syndrome and isolated choanal atresia. Eur. J. Hum. Genet.25 (10), 1126–1133. 10.1038/ejhg.2017.107
18
Grainger R. J. Beggs J. D. (2005). Prp8 protein: at the heart of the spliceosome. RNA11 (5), 533–557. 10.1261/rna.2220705
19
Graziotto J. J. Farkas M. H. Bujakowska K. Deramaudt B. M. Zhang Q. Nandrot E. F. et al (2011). Three gene-targeted mouse models of RNA splicing factor RP show late-onset RPE and retinal degeneration. Invest. Ophthalmol. Vis. Sci.52 (1), 190–198. 10.1167/iovs.10-5194
20
Griffin C. Saint-Jeannet J. P. (2020). Spliceosomopathies: diseases and mechanisms. Dev. Dyn.249 (9), 1038–1046. 10.1002/dvdy.214
21
Holtes L. K. de Bruijn S. E. Cremers F. P. M. Roosing S. (2025). Dual inheritance patterns: a spectrum of non-syndromic inherited retinal disease phenotypes with varying molecular mechanisms. Prog. Retin. Eye Res.104, 101308. 10.1016/j.preteyeres.2024.101308
22
Huang Z. P. Chen J. F. Regan J. N. Maguire C. T. Tang R. H. Dong X. R. et al (2010). Loss of microRNAs in neural crest leads to cardiovascular syndromes resembling human congenital heart defects. Arterioscler. Thromb. Vasc. Biol.30 (12), 2575–2586. 10.1161/ATVBAHA.110.213306
23
Jia J. Ganichkin O. M. Preußner M. Absmeier E. Alings C. Loll B. et al (2020). A Snu114-GTP-Prp8 module forms a relay station for efficient splicing in yeast. Nucleic Acids Res.48 (8), 4572–4584. 10.1093/nar/gkaa182
24
Kastner B. Will C. L. Stark H. Lührmann R. (2019). Structural insights into nuclear pre-mRNA splicing in higher eukaryotes. Cold Spring Harb. Perspect. Biol.11 (11), a032417. 10.1101/cshperspect.a032417
25
Kim J. S. He X. Liu J. Duan Z. Kim T. Gerard J. et al (2019). Systematic proteomics of endogenous human cohesin reveals an interaction with diverse splicing factors and RNA-binding proteins required for mitotic progression. J. Biol. Chem.294 (22), 8760–8772. 10.1074/jbc.RA119.007832
26
Klimešová K. Vojáčková J. Radivojević N. Vandermoere F. Bertrand E. Verheggen C. et al (2021). TSSC4 is a component of U5 snRNP that promotes tri-snRNP formation. Nat. Commun.12 (1), 3646. 10.1038/s41467-021-23934-y
27
Krausová M. Kreplová M. Banik P. Cvačková Z. Kubovčiak J. Modrák M. et al (2023). Retinitis pigmentosa-associated mutations in mouse Prpf8 cause misexpression of circRNAs and degeneration of cerebellar granule cells. Life Sci. Alliance6 (6), e202201855. 10.26508/lsa.202201855
28
Ledoux S. Guthrie C. (2016). Retinitis pigmentosa mutations in bad response to refrigeration 2 (Brr2) impair ATPase and helicase activity. J. Biol. Chem.291 (23), 11954–11965. 10.1074/jbc.M115.710848
29
Lei L. Yan S. Y. Yang R. Chen J. Y. Li Y. Bu Y. et al (2017). Spliceosomal protein eftud2 mutation leads to p53-dependent apoptosis in zebrafish neural progenitors. Nucleic Acids Res.45 (6), 3422–3436. 10.1093/nar/gkw1043
30
Li X. Zhong S. Li C. Yan X. Zhu J. Li Y. et al (2024). RNA helicase Brr2a promotes miRNA biogenesis by properly remodelling secondary structure of pri-miRNAs. Nat. Plants10 (10), 1532–1547. 10.1038/s41477-024-01788-8
31
Liang Y. Tan F. Sun X. Cui Z. Gu J. Mao S. et al (2022). Aberrant retinal pigment epithelial cells derived from induced pluripotent stem cells of a retinitis pigmentosa patient with the PRPF6 mutation. Int. J. Mol. Sci.23 (16), 9049. 10.3390/ijms23169049
32
Liu S. Rauhut R. Vornlocher H. P. Lührmann R. (2006). The network of protein-protein interactions within the human U4/U6.U5 tri-snRNP. RNA12 (7), 1418–1430. 10.1261/rna.55406
33
Liu Y. Chen X. Qin B. Zhao K. Zhao Q. Staley J. P. et al (2015). Knocking down Snrnp200 initiates demorphogenesis of rod photoreceptors in zebrafish. J. Ophthalmol.2015, 816329. 10.1155/2015/816329
34
Love S. L. Emerson J. D. Koide K. Hoskins A. A. (2023). Pre-mRNA splicing-associated diseases and therapies. RNA Biol.20 (1), 525–538. 10.1080/15476286.2023.2239601
35
Lüdecke H. J. Wieczorek D. (2016). “TXNL4A-related craniofacial disorders,” in GeneReviews®. ed. AdamM. P.FeldmanJ.MirzaaG. M. (Seattle, WA: University of Washington), 1993–2025. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK373577/ (accessed March 18, 2025).
36
Matera A. G. Wang Z. (2014). A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol.15 (2), 108–121. 10.1038/nrm3742
37
Maxwell D. W. O’Keefe R. T. Roy S. Hentges K. E. (2021). The role of splicing factors in retinitis pigmentosa: links to cilia. Biochem. Soc. Trans.49 (3), 1221–1231. 10.1042/BST20200798
38
Mayerle M. Guthrie C. (2016). Prp8 retinitis pigmentosa mutants cause defects in the transition between the catalytic steps of splicing. RNA22 (5), 793–809. 10.1261/rna.055459.115
39
McKie A. B. McHale J. C. Keen T. J. Tarttelin E. E. Goliath R. van Lith-Verhoeven J. J. et al (2001). Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum. Mol. Genet.10 (15), 1555–1562. 10.1093/hmg/10.15.1555
40
Montgomery S. B. Bernstein J. A. Wheeler M. T. (2022). Toward transcriptomics as a primary tool for rare disease investigation. Cold Spring Harb. Mol. Case Stud.8 (2), a006198. 10.1101/mcs.a006198
41
Mozaffari-Jovin S. Wandersleben T. Santos K. F. Will C. L. Lührmann R. Wahl M. C. (2013). Inhibition of RNA helicase Brr2 by the C-terminal tail of the spliceosomal protein Prp8. Science341 (6141), 80–84. 10.1126/science.1237515
42
Mozaffari-Jovin S. Wandersleben T. Santos K. F. Will C. L. Lührmann R. Wahl M. C. (2014). Novel regulatory principles of the spliceosomal Brr2 RNA helicase and links to retinal disease in humans. RNA Biol.11 (4), 298–312. 10.4161/rna.28353
43
Nalbach K. Schifferer M. Bhattacharya D. Ho-Xuan H. Tseng W. C. Williams L. A. et al (2023). Spatial proteomics reveals secretory pathway disturbances caused by neuropathy-associated TECPR2. Nat. Commun.14 (1), 870. 10.1038/s41467-023-36553-6
44
Onea G. Maitland M. E. R. Wang X. Lajoie G. A. Schild-Poulter C. (2022). Distinct nuclear and cytoplasmic assemblies and interactomes of the mammalian CTLH E3 ligase complex. J. Cell Sci.135 (14), jcs259638. 10.1242/jcs.259638
45
Park B. Y. Tachi-Duprat M. Ihewulezi C. Devotta A. Saint-Jeannet J. P. (2022). The core splicing factors EFTUD2, SNRPB and TXNL4A are essential for neural crest and craniofacial development. J. Dev. Biol.10 (3), 29. 10.3390/jdb10030029
46
Petit L. Ma S. Cipi J. Cheng S. Y. Zieger M. Hay N. et al (2018). Aerobic glycolysis is essential for normal rod function and controls secondary cone death in retinitis pigmentosa. Cell Rep.23 (9), 2629–2642. 10.1016/j.celrep.2018.04.111
47
Riabov Bassat D. Visanpattanasin S. Vorländer M. K. Fin L. Phillips A. W. Plaschka C. (2024). Structural basis of human U5 snRNP late biogenesis and recycling. Nat. Struct. Mol. Biol.31 (5), 747–751. 10.1038/s41594-024-01243-4
48
Rubio-Peña K. Fontrodona L. Aristizábal-Corrales D. Torres S. Cornes E. García-Rodríguez F. J. et al (2015). Modeling of autosomal-dominant retinitis pigmentosa in Caenorhabditis elegans uncovers a nexus between global impaired functioning of certain splicing factors and cell type-specific apoptosis. RNA21 (12), 2119–2131. 10.1261/rna.053397.115
49
Schreib C. C. Bowman E. K. Hernandez C. A. Lucas A. L. Potts C. H. S. Maeder C. (2018). Functional and biochemical characterization of Dib1’s role in pre-messenger RNA splicing. J. Mol. Biol.430 (11), 1640–1651. 10.1016/j.jmb.2018.04.027
50
Sobreira N. Schiettecatte F. Valle D. Hamosh A. (2015). GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat.36 (10), 928–930. 10.1002/humu.22844
51
Stanković D. Claudius A. K. Schertel T. Bresser T. Uhlirova M. (2020). A Drosophila model to study retinitis pigmentosa pathology associated with mutations in the core splicing factor Prp8. Dis. Model. Mech.13 (6), dmm043174. 10.1242/dmm.043174
52
Su X. Yang H. Shi R. Zhang C. Liu H. Fan Z. et al (2020). Depletion of SNRNP200 inhibits the osteo-/dentinogenic differentiation and cell proliferation potential of stem cells from the apical papilla. BMC Dev. Biol.20 (1), 22. 10.1186/s12861-020-00228-y
53
Tanackovic G. Ransijn A. Thibault P. Abou Elela S. Klinck R. Berson E. L. et al (2011). PRPF mutations are associated with generalized defects in spliceosome formation and pre-mRNA splicing in patients with retinitis pigmentosa. Hum. Mol. Genet.20 (11), 2116–2130. 10.1093/hmg/ddr094
54
Thomas H. B. Wood K. A. Buczek W. A. Gordon C. T. Pingault V. Attié-Bitach T. et al (2020). EFTUD2 missense variants disrupt protein function and splicing in mandibulofacial dysostosis Guion-Almeida type. Hum. Mutat.41 (8), 1372–1382. 10.1002/humu.24027
55
Varineau J. E. Calo E. (2024). A common cellular response to broad splicing perturbations is characterized by metabolic transcript downregulation driven by the Mdm2-p53 axis. Dis. Model Mech.17 (2), dmm050356. 10.1242/dmm.050356
56
Wan R. Bai R. Zhan X. Shi Y. (2020). How is precursor messenger RNA spliced by the spliceosome?Annu. Rev. Biochem.89, 333–358. 10.1146/annurev-biochem-013118-111024
57
Wieczorek D. Newman W. G. Wieland T. Berulava T. Kaffe M. Falkenstein D. et al (2014). Compound heterozygosity of low-frequency promoter deletions and rare loss-of-function mutations in TXNL4A causes Burn-McKeown syndrome. Am. J. Hum. Genet.95 (6), 698–707. 10.1016/j.ajhg.2014.10.014
58
Wilkinson M. E. Charenton C. Nagai K. (2020). RNA splicing by the spliceosome. Annu. Rev. Biochem.89, 359–388. 10.1146/annurev-biochem-091719-064225
59
Wittig I. Malacarne P. F. (2021). Complexome profiling: assembly and remodeling of protein complexes. Int. J. Mol. Sci.22, 7809. 10.3390/ijms22157809
60
Wood K. A. Eadsforth M. A. Newman W. G. O’Keefe R. T. (2021). The role of the U5 snRNP in genetic disorders and cancer. Front. Genet.12, 636620. 10.3389/fgene.2021.636620
61
Wood K. A. Ellingford J. M. Thomas H. B. Hobson E. Douzgou S. Beaman G. M. et al (2022). Expanding the genotypic spectrum of TXNL4A variants in Burn-McKeown syndrome. Clin. Genet.101(2), 255–259. 10.1111/cge.14082
62
Wood K. A. Rowlands C. F. Qureshi W. M. S. Thomas H. B. Buczek W. A. Briggs T. A. et al (2019). Disease modeling of core pre-mRNA splicing factor haploinsufficiency. Hum. Mol. Genet.28 (22), 3704–3723. 10.1093/hmg/ddz169
63
Wood K. A. Rowlands C. F. Thomas H. B. Woods S. O'Flaherty J. Douzgou S. et al (2020). Modelling the developmental spliceosomal craniofacial disorder Burn-McKeown syndrome using induced pluripotent stem cells. PLoS One15 (7), e0233582. 10.1371/journal.pone.0233582
64
Wu J. Yang Y. He Y. Li Q. Wang X. Sun C. et al (2019). EFTUD2 gene deficiency disrupts osteoblast maturation and inhibits chondrocyte differentiation via activation of the p53 signaling pathway. Hum. Genomics.13 (1), 63. 10.1186/s40246-019-0238-y
65
Zhang D. McLenachan S. Chen S. C. Zaw K. Alziyadat Y. Zhang X. et al (2021). Generation of two induced pluripotent stem cell lines from a patient with recessive inherited retinal disease caused by compound heterozygous mutations in SNRNP200. Stem Cell Res.51, 102154. 10.1016/j.scr.2020.102154
66
Zhang D. Yue T. Choi J. H. Nair-Gill E. Zhong X. Wang K. W. et al (2019). Syndromic immune disorder caused by a viable hypomorphic allele of spliceosome component Snrnp40. Nat. Immunol.20 (10), 1322–1334. 10.1038/s41590-019-0464-4
67
Zhang T. Bai J. Zhang X. Zheng X. Lu N. Liang Z. et al (2021). SNRNP200 mutations cause autosomal dominant retinitis pigmentosa. Front. Med. (Lausanne).7, 588991. 10.3389/fmed.2020.588991
68
Zhao C. Bellur D. L. Lu S. Zhao F. Grassi M. A. Bowne S. J. et al (2009). Autosomal-dominant retinitis pigmentosa caused by a mutation in SNRNP200, a gene required for unwinding of U4/U6 snRNAs. Am. J. Hum. Genet.85 (5), 617–627. 10.1016/j.ajhg.2009.09.020
Summary
Keywords
craniofacial malformation, pre-mRNA splicing, retinitis pigmentosa, spliceosomopathies, tissue specificity, U5 snRNP
Citation
Kemal RA and O’Keefe RT (2025) Addressing the tissue specificity of U5 snRNP spliceosomopathies. Front. Cell Dev. Biol. 13:1572188. doi: 10.3389/fcell.2025.1572188
Received
06 February 2025
Accepted
20 March 2025
Published
08 April 2025
Volume
13 - 2025
Edited by
Loydie Jerome-Majewksa, McGill University Health Centre, Canada
Reviewed by
Katherine Ann Fantauzzo, University of Colorado Anschutz Medical Campus, United States
Updates
Copyright
© 2025 Kemal and O’Keefe.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raymond T. O’Keefe, rokeefe@manchester.ac.uk
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.