Abstract
Late endosomes/lysosomes (LE/Lys) and lysosome related organelles (LROs) move dynamically through cells which involves many levels of regulation. To reach their destination, they need to connect to the motor proteins dynein-dynactin, kinesin or myosin for long-range bidirectional transport along microtubules and short-range movement along actin filaments. This connection depends on various factors at the microtubule, including the MAP- and tubulin-code, as well as adaptors, Rab GTPases and effector proteins marking the LE/Lys and LRO membranes. Mutations affecting this transport results in defective LE/Lys or LRO cargo delivery often resulting in skin, neurological and/or immunological diseases. How LE/Lys and LRO transport is orchestrated and how it fails in disease states, will be discussed.
Introduction
Motor protein controlled vesicular transport is essential for maintaining cellular homeostasis. Without motor protein support, vesicles will not move in cells. Various types of vesicles are transported through the cellular space to deliver their cargo in response to signaling and cellular demand. These include compartments of the endolysosomal system (early endosomes and late endosome/lysosomes (LE/Lys)), Golgi-derived vesicles, ER-derived vesicles, peroxisomes, autophagosomes and lipid droplets. Furthermore, specialized cell types contain dedicated lysosome-related organelles (LROs) packed with specific cargo often destined for secretion, such as melanosomes in melanocytes, lytic granules (LGs) in cytotoxic T-cells (CTLs) and Natural Killer (NK)-cells and secretory vesicles in neurons (Delevoye et al., 2019; Marks et al., 2013; Raposo et al., 2007). To reach their destination, vesicles need to be actively transported. They can be transported in a fast, bidirectional manner along microtubules whereas short-range transport occurs along actin filaments. Microtubule-based transport is facilitated by two groups of motor proteins: the dynein-dynactin complex for minus-end directed (inward) movement, and the members of the kinesin (KIF) family moving cargo in the opposite direction (plus-end directed/outwards) (Figure 1) (Endow et al., 2010; Hook and Vallee, 2006; Bonifacino and Neefjes, 2017). The family of myosin motor proteins mediates transport along actin filaments (Bonifacino and Neefjes, 2017).
FIGURE 1
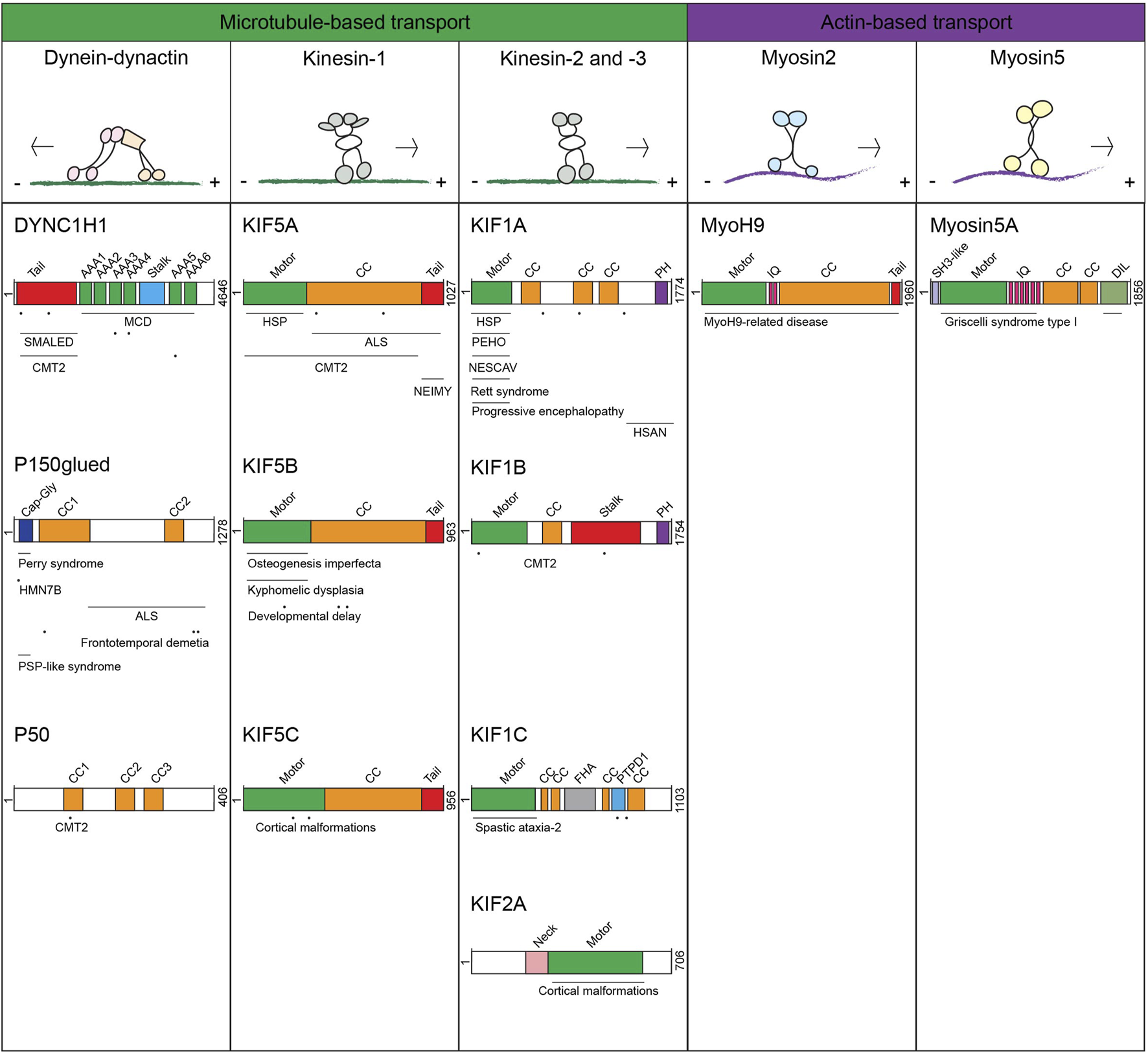
Overview of different motor proteins transporting late endosomes, lysosomes and lysosome-related organelles and mapping of mutations associated with motor protein-associated diseases Upper panel: motor proteins transporting late endosomes, lysosomes and lysosome-related organelles with their track and direction of movement. Lower panel: schematic representation of the protein domains and associated diseases per proteins. Horizontal lines above the mentioned disease indicate multiple mutations have been found in this (these) domain(s), dots indicate specific locations of mutations that have been reported.
The GTPases dance
Motor proteins require specific adaptors to attach to their cargoes. These adaptors usually involve small GTPases from the Rab, Arf and Arf-like (Arl)-family and interacting co-factors, adaptor and effector proteins at the target-membrane, or phosphoinositides (Stenmark, 2009; Donaldson and Honda, 2005; Balla, 2013; Li and Marlin, 2015; Posor et al., 2022). Approximately 60 different Rab and around 20 Arf/Arl small GTPases extensively label different organelles in mammalian cells (Pasqualato et al., 2002; Homma et al., 2021). Various lipids and/or small GTPases define the target membrane and thus the motor-type and the resulting transport. Small GTPases act as molecular switches that are activated when loaded with GTP and inactivated by GTP hydrolysis, a process accelerated by specific GAP proteins. Rab/Arf/Arl GTPases bind target vesicles in their activated GTP-bound state and then recruit effector proteins to mediate motor protein binding for cargo transport initiation. For example, LE/Lys are marked by Rab7, which recruits the effector proteins RILP or FYCO1 for dynein-dynactin or kinesin-motor dependent transport, respectively. The Rab7-positive LE/Lys can further mature into a LE/Lys marked by Arl8b. Arl8b provides a platform to recruit the GAP of Rab7 that then is removed. This yields a Rab7-Arl8b handover mechanism and illustrates how vesicle maturation is molecularly controlled (Jongsma et al., 2020). Arl8b recruits its own effectors, RUFY3/4 and JIP4 (recruiting dynein) or SKIP (recruiting kinesin), for bidirectional transport (Rosa-Ferreira and Munro, 2011; Keren-Kaplan et al., 2022; Kumar et al., 2022). Recently, the GEF DENND6A was shown to bind Arl8b, whereafter it activates another GTPase, Rab34, leading to recruitment of effector protein RILP and then the dynein-dynactin motor resulting in vesicle transport in the retrograde direction (Kumar et al., 2024). Thus multiple GTPases with different effectors binding oppositely directed motor proteins are recruited during the life- and maturation-time of LE/Lys.
LROs are like LE/Lys marked by different Rabs and effectors recruiting fusion machinery and motor proteins that are often uniquely expressed in the specialized cell types. For example, in melanocytes, melanin-containing melanosomes change their membrane composition during maturation. Melanosomes are then transported bidirectionally along microtubules by Rab36-RILP-Mreg-p150glued complex formation resulting in dynein-dynactin mediated retrograde transport (Ohbayashi et al., 2012; Matsui et al., 2012) and Rab1A-SKIP-kinesin-1 (KIF5B) complex formation for anterograde transport (Ishida et al., 2015), albeit this is not confirmed by others (Robinson et al., 2017). At the end of the microtubules, melanosomes have to pass the cortical actin cytoskeleton under the cell surface to secrete their melanin content. Mature melanocytes acquire the small GTPase Rab27a that binds effector protein melanophilin and subsequently the actin-based motor Myosin5a (Wu et al., 2006; Park et al., 2019). As a result, the mature melanosomes accumulate below the cell membrane for secretion, which likely involves actin disassembly and formation of a functional SNARE complex (Van Den Bossche et al., 2006). The situation is similar in neurons and immune cells. NK-cells and CTLs contain LGs packed with cytotoxic enzymes that are released after activation in a more-or-less closed synapse with a target cell to-be-killed. LGs are transported towards the microtubule-organizing center (MTOC) (which reorientates towards the formed immune synapse) in a Rab7-RILP-dynein-dynactin dependent manner while Rab27a-Slp3-kinesin-1 (KIF5B) complex formation (in CTLs) as well as Arl8b-SKIP-kinesin-1 (KIF5B) complex formation (in NK-cells) mediates LGs anterograde transport (Tuli et al., 2013; Kurowska et al., 2012). The MTOC locates the LGs close to the membrane after activation of the NK-cell or CTL, allowing swift delivery of content (Daniele et al., 2011). Yet, the LGs have to pass the actin cytoskeleton involving the UNC45A-Myosin2a complex (Iizuka et al., 2015). Transport is more complicated in neurons, for the simple reason that microtubule-based transport goes along long distances to deliver the vesicles from the cell body to the axon terminus (Guedes-Dias and Holzbaur, 2019). Anterograde and retrograde transport is strictly coordinated in these axons by adaptor proteins that bind dynein-dynactin and kinesin-1 (KIF5B) motor proteins. For example, Alzheimer’s β-amyloid precursor protein (APP)-positive neuronal vesicles bind to JIP1 (Matsuda et al., 2001; Scheinfeld et al., 2002), which interacts with both dynein-dynactin and FEZ1-KIF5B depending on its phosphorylation state (Fu and Holzbaur, 2013; Blasius et al., 2007), while GABA(A)R-marked neuronal vesicles recruit HAP1-huntingtin (htt) to coordinate the activity of dynein-dynactin and kinesin-1 (KIF5B) motor proteins (Twelvetrees et al., 2010; Caviston et al., 2007; McGuire et al., 2006). With the long distances that need to be travelled in neurons, it is not surprising that mutations in protein complexes transporting these vesicles are usually first recognized in diseases with a neurological basis (see later). A list of diseases related to mutations in transport machineries is given in (Table 1).
TABLE 1
| Protein | Function | Associated diseases | References |
|---|---|---|---|
| APP | Adaptor protein | Alzheimer’s disease | Kang et al. (1987), Goate et al. (1991) |
| BICD2 | Adaptor protein | Spinal muscular atrophy with lower extremity dominance | Oates et al. (2013), Peeters et al. (2013), Neveling et al. (2013) |
| BLOC1S3 (BLOS3) | BLOC1 complex | Hermansky-Pudlak syndrome 8 | Morgan et al. (2006) |
| BLOC1S5 (muted) | BLOC1 complex | Hermansky-Pudlak syndrome 11 | Pennamen et al. (2020) |
| BLOC1S6 (Pallidin) | BLOC1 complex | Hermansky-Pudlak syndrome 9 | Badolato et al. (2012) |
| BLOC1S8 (dysbindin) | BLOC1 complex | Hermansky-Pudlak syndrome 7 | Li et al. (2003) |
| Schizophrenia | Straub et al. (2002) | ||
| CEP169 | MT-associated | Autism | Chahrour et al. (2012) |
| CLN3 | Rab7 interactor | Neuronal ceroid lipofuscinosis (Batten disease) | Isolation of a novel gene underlying Batten disease (1995) |
| CLN5 | Rab7 interactor | Neuronal ceroid lipofuscinosis | Savukoski et al. (1998) |
| Dynein heavy chain | Dynein-dynactin motor | Spinal muscular atrophy with lower extremity dominance | Harms et al. (2012) |
| Malformations in cortical development | Willemsen et al. (2012) | ||
| Charcot Marie Tooth disease type 2 | Weedon et al. (2011) | ||
| EB2 | MT-associated | congenital symmetric circumferential skin creases-2 | Isrie et al. (2015) |
| FYCO1 | Adaptor protein | Cataract | Chen et al. (2011) |
| HPS1 | BLOC3 complex/GEF | Hermansky-Pudlak syndrome 1 | Fukai et al. (1995); Wildenberg et al. (1995); Oh et al. (1996) |
| HPS2/adaptin/AP3B1 | AP3 complex | Hermansky-Pudlak syndrome 2 | Dell'Angelica et al. (1999) |
| HPS3 | BLOC2 complex | Hermansky-Pudlak syndrome 3 | Anikster et al. (2001) |
| HPS4 | BLOC3 complex | Hermansky-Pudlak syndrome 4 | Suzuki et al. (2002) |
| Huntingtin | Adaptor protein | Huntington’s disease | Marcy et al. (1993) |
| JIP3 | Adaptor protein | Neurodevelopmental disorder with or without variable brain abnormalities | Iwasawa et al. (2019) |
| KIF1A | Kinesin-3 motor | Hereditary Spastic Paraplegia | Erlich et al. (2011) |
| Hereditary sensory and autonomic neuropathy | Riviere et al. (2011) | ||
| NESCAV syndrome | Hamdan et al. (2011) | ||
| PEHO syndrome | Langlois et al. (2016) | ||
| Rett syndrome | Wang et al. (2019) | ||
| progressive encephalopathy and brain atrophy | Esmaeeli et al. (2015) | ||
| KIF1B | Kinesin-3 motor | Charcot Marie Tooth disease type 2 | Zhao et al. (2001) |
| KIF1C | Kinesin-3 motor | autosomal recessive spastic ataxia-2 | Dor et al. (2014) |
| KIF2A | Kinesin-2 motor | Cortical malformations | Poirier et al. (2013) |
| KIF5A | Kinesin-1 motor | Hereditary Spastic Paraplegia | Reid et al. (2002) |
| Amyotrophic Lateral Sclerosis | Nicolas et al. (2018) | ||
| Charcot Marie Tooth disease type 2 | Crimella et al. (2012); Brenner et al. (2018) | ||
| Neonatal Intractable MYoclonus | Duis et al. (2016) | ||
| KIF5B | Kinesin-1 motor | Osteogenesis imperfecta | Marom et al. (2023) |
| kyphomelic dysplasia | Itai et al. (2022) | ||
| Developmental delay with variable symptoms including myopathic features | Flex et al. (2023) | ||
| KIF5C | Kinesin-1 motor | Cortical malformations | Poirier et al. (2013) |
| LIS1 | MT-associated | Lissencephaly | Reiner et al. (1993) |
| LYST | Adaptor protein | Chediak-Higashi syndrome | Barbosa et al. (1996) |
| MAP6 | MT-associated | schizophrenia | Merenlender-Wagner et al. (2014); Shimizu et al. (2006) |
| MID1 | MT-associated | Opitz syndrome | Quaderi et al. (1997) |
| MLPH | Myosin effector | Griscelli syndrome type 3 | Menasche et al. (2003) |
| MyoH9 | Myosin motor | MYOH9-related disease | Kunishima et al. (1999); Seri et al. (2000) |
| Myosin 5a | Myosin motor | Griscelli syndrome type 1 | Pastural et al. (1997) |
| Myosin 7a | Myosin motor | Usher syndrome type 1B | Weil et al. (1995) |
| NPC-1 | Cholesterol transporter | Niemann Pick Type C1-disease | Carstea et al. (1997) |
| NPC-2 | Cholesterol transporter | Niemann Pick Type C2-disease | Naureckiene et al. (2000) |
| P50/dynamitin | Dynein/dynactin motor | Charcot Marie Tooth disease type 2 | Braathen et al. (2016) |
| P150glued | Dynein-dynactin motor | Perry syndrome | Farrer et al. (2009) |
| hereditary motor neuronopathy 7B | Puls et al. (2003) | ||
| Frontotemporal dementia | Munch et al. (2005) | ||
| progressive supranuclear palsy-like syndrome | Stockmann et al. (2013) | ||
| Amyotrophic Lateral Sclerosis | Munch et al. (2004) | ||
| Rab7a | Small GTPase | Alzheimer’s disease | Vardarajan et al. (2012) |
| Charcot Marie Tooth disease type 2 | Verhoeven et al. (2003) | ||
| RAB27a | Small GTPase | Griscelli syndrome type 2 | Menasche et al. (2000) |
| Rab32 | GEF | Parkinsons | Gustavsson et al. (2024); Hop et al. (2024) |
| SNX1 | Retromer complex | Alzheimer’s disease | Vardarajan et al. (2012) |
| Tau | MT-associated | Alzheimer’s disease | Grundke-Iqbal et al. (1986); Conrad et al. (2002) |
| Frontotemporal dementia | Joachim et al. (1987); Hutton et al. (1998) | ||
| progressive supranuclear palsy | Bancher et al. (1987); Conrad et al. (1997)) | ||
| Parkinson’s disease | Joachim et al. (1987); Martin et al. (2001) | ||
| VAPB | MCS | Amyotrophic Lateral Sclerosis | Nishimura et al. (2004) |
| VPS11 | HOPS complex | Hypomyelinating Leukodystrophy 12 | Edvardson et al. (2015) |
| Dystonia 32 | Monfrini et al. (2021) | ||
| VPS16 | HOPS complex | Dystonia 30 | Cai et al. (2016); Steel et al. (2020)) |
| VPS33a | HOPS complex | mucopolysaccharidosis-plus syndrome | Dursun et al. (2017) |
| VPS35 | Retromer complex | Parkinson’s disease | Vilarino-Guell et al. (2011); Zimprich et al. (2011) |
| VPS39 | HOPS complex | Schizophrenia | Xu et al. (2012) |
| VPS41 | HOPS complex | autosomal recessive spinocerebellar ataxia-29 | Steel et al. (2020) |
overview of transport-related proteins of which mutations are associated with disease.
The microtubule highway and traffic control
Fast vesicle transport occurs along microtubules by microtubule-based motor proteins. These microtubules are not empty roads but covered by various microtubule-associated proteins (MAPs). The MAP-family consists of different proteins including microtubule stabilizing proteins and proteins controlling motor protein transport (Jijumon et al., 2022; Bodakuntla et al., 2019; Goodson and Jonasson, 2018; Matteoni and Kreis, 1987) (summarized in (Table 2)). MAP2, MAP4, MAP7 and MAP9 can inhibit or activate dynein-dynactin and kinesin motor proteins (Jongsma et al., 2023; Monroy et al., 2020; Monroy et al., 2018; Hagiwara et al., 1994; Paschal et al., 1989; Hooikaas et al., 2019; Chaudhary et al., 2019; Barlan et al., 2013; Semenova et al., 2014; Ferro et al., 2022), while other MAPs are involved in motor protein recruitment to the microtubule. These include the atypical dynein heavy chain (dynein HC) activating MAP LIS1, as well as the E3 ligase MID1 and the plus-end proteins EB1 and CEP169 that recruit activated dynein HC to the microtubule growing plus-end (Splinter et al., 2012; Elshenawy et al., 2020; Singh et al., 2024; Gillies et al., 2022; Baumbach et al., 2017; Karasmanis et al., 2023; Dixit et al., 2008a; Duellberg et al., 2014; Jongsma et al., 2024). The dynein HC has to assemble with the dynactin complex to form a functional dynein-dynactin motor, which is recruited to the microtubule plus end by factors including EB1 and CLIP170 (Jongsma et al., 2024; Bjelic et al., 2012; Watson and Stephens, 2006). Consequently, the dynein-dynactin motor is assembled at the plus-end and then moves inward to catch associated LE/Lys for minus end transport. LRO transport is also regulated by MAPs. For example, MAP4 mediates switching between kinesin and dynein-dynactin dependent transport in melanocytes. When dephosphorylated, MAP4 binds to the microtubule surface where it recruits kinesin-2 towards the melanosome, whereas phosphorylated MAP4 is cytosolic and unable to recruit kinesin-2, thereby favoring dynein-mediated transport (Semenova et al., 2014). The neuronal MAP tau stabilizes and bundles axonal microtubules (Chung et al., 2016). In addition, tau inhibits kinesin-1 and kinesin-3 microtubule binding and motility without effecting kinesin-2 and dynein-mediated transport of LE/Lys and LROs (Monroy et al., 2020; Monroy et al., 2018; Chaudhary et al., 2018). Yet, the control of cargo transport along microtubules is even more complicated by various tubulin isotypes and post-translational modifications that also control microtubule stability and motor protein activation (McKenna et al., 2023). On these modification and protein littered roads, vesicles and their associated microtubule-based motor proteins have to find their ways while moving in a bidirectional and stop-and-go manner.
TABLE 2
Overview of microtubule-associated proteins regulating motor proteins and microtubule dynamics.
Although microtubules allow transport over long distances in cells, the microtubule highways fail to deliver cargo to the plasma membrane, as they do not reach the cell surface. Where microtubules end, the cortical actin network takes over. To move vesicles along actin cables requires a different set of motor proteins, the myosin motor proteins. At the growing plus-end of microtubules, the EB1 protein can orchestrate the hand-over of melanosomes from microtubule-based motors to the actin-associated motor Myosin5a, which is an important step preceding plasma membrane fusion and release of melanin. EB1 binds Melanophilin-Myosin5a allowing transfer of the Melanophilin-Myosin5a complex to GTPase Rab27a as present at the mature melanosomes membrane, initiating actin-binding before membrane fusion (Wu et al., 2005). Similarly, Myosin5a facilitates the delivery of neuronal vesicles (Lise et al., 2006; Takamori et al., 2006; Correia et al., 2008; Roder et al., 2010; Roder et al., 2008), whereas LGs are transported by UNC45-Myosin2 (Iizuka et al., 2015; Krzewski et al., 2006). When the plasma membrane is reached, this activates SNARE-complex assembly and fusion. The road to plasma membrane delivery is complicated and dynamic but it works!
LE/Lys and LRO transport is controlled by a series of molecules acting at different levels. It is therefore not too surprising that mutations in the different proteins involved can result in disease states. We will dissect the different steps in this pathway and describe the different mutations found in the mammalian system and their phenotypes. We will start at the microtubules and then work our way up to the LE/Lys and LROs by discussing the motor proteins and adaptor/effector proteins bound at the various cargo membranes. In assembly, they illustrate the relevance of proper control of lysosomal transport processes for a healthy state.
At the microtubule highways
Intracellular long-range motor-mediated transport is possible through a functional highway-system, the microtubule network, build-up from various tubulin subunits. These microtubules, their modifications and their associated proteins are in control of LE/Lys and LRO transport.
Diseases associated with microtubules and their associated proteins
The basis of microtubules is formed by different isotypes of α- and β-tubulin dimers. Missense and splice-site mutations in genes encoding for tubulin subunits result in various diseases including lissencephaly brain and ocular cranial nerve disorders, illustrating the importance for proper maintenance of these trafficking-roads (Tischfield et al., 2011). Besides the tubulin isotypes, there are many microtubule-associated post-translational modifications including acetylation, ubiquitination, sumoylation, detyrosination, glutamylation and phosphorylation (McKenna et al., 2023). This so-called tubulin-code controls microtubule dynamics and stability, can act at the growing plus-end and controls vesicle transport and issues like neuronal growth, differentiation and axonal regeneration (Lu et al., 2024). The various enzymes involved in the different post-translational modifications are then expected to yield neuronal diseases when mutated. Indeed, mutations in DYRK1A, which phosphorylates Ser172 on β -tubulin, have been associated with intellectual developmental disorder (van Bon et al., 2011; O'Roak et al., 2012; Courcet et al., 2012). However, as DYRK1A is involved in the phosphorylation of many targets, including transcription factors such as CREB, FKHR, GLI1, NFAT, and STAT3, it is unlikely that the disease is solely caused by the loss of β-tubulin phosphorylation. Mutations in the tubulin-glycosylating enzyme TTLL10 have been identified in patients with severe bleeding disorder, where it is suggested to play a crucial role in the microtubule dynamics involved in platelet production (Khan et al., 2022). In addition to the tubulin PTMs, there are many MAPs decorating the tubulin subunits, together forming the MAP-code (Monroy et al., 2020), ochestrating many different functions, including the control of dynein and kinesin motor protein-mediated transport. A well known MAP affecting transport is tau, encoded by the MAPT gene, which is specifically expressed in neuronal cells. Aggregates containing hyperphosphorylated Tau are linked to multiple neurodegenerative diseases including Alzheimer’s Disease (Grundke-Iqbal et al., 1986). In some studies, certain MAPT variants associated with an increased risk of tauopathies, likely due to increased MAPT expression (Tanahashi et al., 2004; Laws et al., 2007; Myers et al., 2007; Russ et al., 2001; Baker et al., 2000; Bullido et al., 2000; Clark et al., 2003; Kwok et al., 2004). When binding the microtubule surface, tau obstructs vesicle transport by acting as an obstacle for kinesin and dynein motor proteins (Vershinin et al., 2008; Ebneth et al., 1998), which can then induce neurodegeneration (Chaudhary et al., 2018). Another neuronal specific MAP suggested to be associated to disease is MAP6. MAP6 mutant mice are a model for schizophrenia and show a reduced presynaptic glutamate vesicle density (Andrieux et al., 2002; Daoust et al., 2014; Gimenez et al., 2017; Merenlender-Wagner et al., 2014). Furthermore, MAP6 expression was upregulated in post-mortem brains of schizophrenia patients, along with two SNPs that showed an association with schizophrenia (Shimizu et al., 2006).
At the microtubule plus-end, various MAPs regulate the dynamic growth and shrinkage (catastrophy) of the microtubule. This process is mainly regulated by the end-binding family proteins, EB1-3. EB1, MID1 and CEP169 (encoded by MAPRE1, MID1 and NCKAP5L genes) are essential to locate the activated (Lis1 containing) dynein HC at the growing microtubule plus-end (Jongsma et al., 2024). So far, altered expression of EB1 has been observed in pediatric ependymoma (underexpression) (Suarez-Merino et al., 2005) and esophageal squamous cell carcinoma (overexpression) (Wang et al., 2005), as well as a lymphoblastic leukemia patient showing a fusion of EB1 and MLL (Mixed-Lineage Leukemia) (Fu et al., 2005). Mutations in the MID1 gene have been linked to Opitz Syndrome, a disease caused by defects in cell migration resulting in maldeveloped midline structures (Schweiger and Schneider, 2003; Aranda-Orgilles et al., 2008). MID1 is known to stabilize microtubules (Schweiger et al., 1999), yet MID1 can also ubiquitinate phosphatase 2A (PP2A) leading to its degradation (Trockenbacher et al., 2001). Mutated MID1 therefore leads to increased PP2A levels which alters cytoskeletal remodeling, intracellular transport and cell migration (Perea-Cabrera et al., 2023). Next, dysfunctional centrosomal protein CEP169 has been associated to Autism spectrum disorder (ASD) (Chahrour et al., 2012). How mutations in CEP169 contribute to ASD is currently not understood, but it might be related to its function in synaptic plasticity, as CEP169 was found to be upregulated in response to neuronal activity (Chahrour et al., 2012), similarly to other genes associated with autism (Walsh et al., 2008; Ramocki and Zoghbi, 2008).
The MAP-code controls both the microtubule highway and the activation of kinesin and dynein-dynactin motor proteins. It is therefore not surprising that many diseases associated to LE/Lys and LRO transport are the result of mutations in these proteins. How about the motor proteins, which need to walk along these roads?
Dynein-Dynactin mediated transport
Dynein-dynactin is a large, multi-subunit motor protein complex interacting with cargo adaptors and effector proteins to transport a various cargoes towards the microtubule minus-end, including vesicles, mitochondria and mRNA (Wilkie and Davis, 2001; Pilling et al., 2006; Jordens et al., 2001; Gross et al., 2002; Driskell et al., 2007). It consists of two multi-subunit complexes, the dynein motor and its cofactor dynactin, which assemble as an active motor at the microtubule plus-end after recruitment by various MAPs (Jongsma et al., 2024; Bjelic et al., 2012; Watson and Stephens, 2006). Dynein is formed by two dynein heavy chains (HCs) (DYNC1H1) activated by Lis1 (Elshenawy et al., 2020; Htet et al., 2020), an intermediate chain (IC, DYNC1I1 or DYNC1I2), a light-intermediate chain (LIC, DYNC1LI1 or DYNC1LI2), and three light chains (LCs, DYNLT1, DYNLL1 and DYNLRB1) (recently reviewed by (Rao and Gennerich, 2024). The dynactin complex contains 23 proteins, including the microtubule binding subunit p150glued, built around a short filament of actin related protein-1 (Arp1) (Urnavicius et al., 2015). At the microtubule plus-end, two dynein dimers can assemble with one dynactin complex and (when available) a cargo adaptor (Splinter et al., 2012; Urnavicius et al., 2015; Schlager et al., 2014; McKenney et al., 2014). Since cells express just a single type of dynein HC motor that facilitates minus-end directed transport in the cytoplasm (Roberts et al., 2013), the formation with various IC, LIC and cargo adaptor proteins allows the specificity to regulate transport of distinct cargoes. Dynactin enhances the processivity of the dynein motor (King and Schroer, 2000). Mutations in the dynein-dynactin subunits should then affect transport of lysosomes and related organelles translating in disease phenotypes.
Diseases related to defects in the dynein motor
Mutations in the human DYNC1H1 gene (encoding the 500 kDa dynein HC) have been associated to multiple neurological diseases (Marzo et al., 2019; Hoang et al., 2017; Becker et al., 2020) (an overview of the motor proteins with associated diseases and location of mutations can be found in (Figure 1)). More than 40 heterozygous missense mutations in the DYNC1H1 gene are identified in patients with malformations in cortical development (MCD, mutations mostly found in dynein HC motor-domain) and spinal muscular atrophy with lower extremity dominance (SMALED, mutations found in dynein HC tail-domain) (Becker et al., 2020; Willemsen et al., 2012; Weedon et al., 2011; Vissers et al., 2010; Tsurusaki et al., 2012; Strickland et al., 2015; Scoto et al., 2015; Schiavo et al., 2013; Poirier et al., 2013; Niu et al., 2015; Mei et al., 2023; Lipka et al., 2013; Harms et al., 2012; Gelineau-Morel et al., 2016; Fiorillo et al., 2014). These are neuromuscular disorders caused by defects in neuronal proliferation and migration resulting in intellectual disabilities and epilepsy (MCD) or spinal cord motor neuron loss affecting lower limp function (SMALED). A study investigating the effect of 14 MCD or SMALED-associated dynein HC mutations on dynein-dynactin function showed that most human disease-associated mutations reduced dynein-dynactin-BICD2 complex motility (Hoang et al., 2017). Peripheral neurons with long axons, requiring long distance transport, are likely the first cells affected by the reduced processivity of the dynein-dynactin-cargo complex, explaining why specifically lower extremities of the body are affected in SMALED. Stronger effects on dynein-dynactin motility also hamper retrograde transport in neurons with shorter axons, which contributes to the cortical malformations found in MCD. Another neurological disease is caused by mutations in the dynein HC tail-domain, essential for dynein HC dimerization, and is called Charcot Marie Tooth disease type 2 (CMT2), a progressive disease characterized by muscle weakness (Weedon et al., 2011). A mouse model mimicking human CMT2-associated mutations showed reduced innervation and lower synaptic vesicle density at the gastrocnemius neuromuscular junctions, likely caused by defective dynein transport of the synaptic vesicles (Weedon et al., 2011; Nandini et al., 2019).
Diseases related to defects in the dynactin complex
Although consisting of 23 subunits, most dynactin mutations associated to neurodegenerative diseases are observed in the DCTN1 gene encoding for the dynactin subunit p150glued. Mutations localizing to the CAP-gly domain in or close to the GKNDG-motif, which controls p150glued binding to microtubules, have been identified in Perry syndrome and hereditary motor neuronopathy 7B (HMN7B) patients (Waterman-Storer et al., 1995; Schroer, 2004; Li et al., 2002). Perry syndrome is a neurodegenerative disease characterized by parkinsonism, psychiatric symptoms and TDP-43 (transactive-response DNA-binding protein of 43 kDa) aggregation in the brain. Perry syndrome-associated p150glued mutants are able to dimerize and associate to the dynein IC and did not affect axonal transport, yet dynactin recruitment to microtubules is limited as is the inability to accumulate dynactin at neurite tips (Moughamian and Holzbaur, 2012). Although these P150 mutations only mildly affect microtubule binding, even such subtle effects on dynein motor transport can result in disrupted lysosome and autophagosome retrograde transport leading to accumulation and aggregation of pathological proteins (Perlson et al., 2010), including TDP-43 (Xia et al., 2016). But not all mutations in the p150glued CAP-Gly domain lead to similar phenotypes. The Gly59Ser substitution in p150glued has been diagnosed in just a few families as HMN7B disease (Puls et al., 2003). Whereas there is no evidence for motor neuron pathology in Perry syndrome, HMN7B patients show chronic motor neuron denervation and have an earlier onset of disease. The HMN7B Gly59Ser mutation is located at the center of the CAP-Gly domain while Perry Syndrome-related mutations are located at the protein surface (Moughamian and Holzbaur, 2012). This mutation is not directly involved in p150glued binding to the microtubule, but since Serine-residues are greater in size than Glycine-residues the substitution will cause steric hindrance and disturb proper folding of the CAP-Gly domain resulting in mildly affected microtubule binding. This mutation not only decreases the interaction with dynein HC but also hinders dynactin recruitment to the microtubule resulting in reduced minus-end directed cargo transport. Of note, mutant p150glued can form toxic aggregates that also contribute to death of neurons (Moughamian and Holzbaur, 2012). In addition, some rare mutations in the DCTN1 gene have been associated to other neurodegenerative diseases, such as frontotemporal dementia, progressive supranuclear palsy-like syndrome and the motor neuron disease Amyotrophic Lateral Sclerosis (ALS), which results in loss of muscle control (Konno et al., 2017). Various p150glued mutations are found in ALS patients (Met571Thr, Arg785Trp, Arg1101Lys and Thr1249Ile) that reside outside the CAP-Gly domain and do not affect microtubule binding and do not result in p150glued aggregation (Dixit et al., 2008a; Stockmann et al., 2013). They affect p150glued binding to the dynein HC motor (Munch et al., 2004). These mutations may form a genomic risk factor for developing ALS. However, as ALS is a multifactorial disease caused by a combination of mutations and environmental factors, ALS patients with p150glued mutations should also include other ALS-associated genes (Cady et al., 2015), as not all family members carrying these DCTN1 gene mutations developed ALS. Mutations in the p50/dynamitin subunit have been identified in patients with CMT2 (Braathen et al., 2016). The His113Tyr mutation is located in the first coiled-coil motif, which is predicted to mediate self‐association and stabilization of the dynactin complex (Jacquot et al., 2010; Maier et al., 2008). Mutations in other dynactin subunits are more rare but do occur.
Diseases related to defects in dynein-dynactin activating adaptor proteins
The dynein-dynactin motor does not function alone, there are multiple dynein-activating adaptor proteins, including BICD-family, Hook-family and JIP-family proteins that bind the dynein-dynactin complex to enhance motor-complex stability and allow the motor to connect to specific cargo (Olenick and Holzbaur, 2019). Activating adaptors contain a N-terminal dynein-dynactin binding domain which often includes a domain interacting with the dynein LIC while its C-terminus is involved in cargo-binding (Reck-Peterson et al., 2018). Most adaptor proteins enhance dynein-dynactin motility, yet some, like JIP-family adaptors, TRAK1/2 and HAP1, act as motility switches by coordinating both dynein-dynactin and kinesin motors (Fu and Holzbaur, 2013; Twelvetrees et al., 2010; McGuire et al., 2006; Engelender et al., 1997; Li et al., 1998; van Spronsen et al., 2013). Some neuronal diseases have been associated to mutations in these adaptor proteins. For example, mutations in the BICD2 gene are associated with SMALED (Fiorillo et al., 2016; Oates et al., 2013; Peeters et al., 2013; Picher-Martel et al., 2020; Ravenscroft et al., 2016), while a study in C. Elegans showed that mutations in the neuronal expressed MAPK8IP3 gene (encoding the homologue of mammalian JIP3) results in disturbed LE/Lys trafficking associated to NEDBA (Neurodevelopmental Disorder with or Without Variable Brain Abnormalities) (Arimoto et al., 2011; Edwards et al., 2013). Also, mutations in the dynein activator LIS1 gene lead to the neuronal migration disease Lissencephaly causing severe brain malformations (Dobyns et al., 1993; Guerrini and Parrini, 2010). Possibly, dysfunctional Lis1 affects neuronal migration as a result of defective nuclear movement. During neuronal migration, the nucleus couples to the centrosome, a process depending on dynein-mediated nuclear translocation. Depletion of LIS1 was found to hamper nucleus-centrosome coupling, which could be rescued by overexpression of wild-type Lis1 but not Lis1 constructs harboring Lissencephaly-associated patient mutations (Tanaka et al., 2004). Similarly, defects in neuronal migration are also observed when the dynein motor is inactivated, suggesting that Lis1 is required to activate the dynein motor for correct nuclear translocation (Tanaka et al., 2004).
It may be surprising that mutations with relatively small effects on dynein-dynactin motor transport already result in various neurological diseases. However, full inhibition of dynein motor activity will be lethal and it is likely that small effects are tolerated at the cost of diseases in the system most sensitive to alteration in microtubule based transport, the neurological system.
The other direction: kinesin-mediated transport
The kinesin-superfamily (KIFs) consists of 14 subfamilies, which includes 45 different kinesin HCs (Lawrence et al., 2004; Hirokawa et al., 2009). In general, kinesins contain an N-terminal motor domain, followed by a family-specific neck region that determines the generated force, a coiled-coil domain for dimerization and a C-terminal tail defining cargo specificity. Two kinesin HCs form a dimer and assemble with various co-factors, including light chains for kinesin-1, allowing interaction with specific cargoes (Verhey and Hammond, 2009; Dimitrova-Paternoga et al., 2021). To become an active motor, kinesins require MAPs or other co-factors to be released from their autoinhibited state, as shown for MAP7-family members activating kinesin-1 member KIF5B (Hooikaas et al., 2019), and Kinesin-associated protein 3 (KAP3) activating the kinesin-2 heterodimers KIF3A–KIF3B and KIF3A–KIF3C by forming active heterotrimeric complexes (Sonar et al., 2020; Cole et al., 1993; Garbouchian et al., 2022; Yamazaki et al., 1995), thus supporting microtubule binding and subsequent cargo transport by these kinesin motors towards the microtubule plus-end.
Diseases related to defects in the kinesin-motors
The kinesin motors reported to transport LE/Lys and most LROs are KIF1A/B (kinesin-3) and KIF5A/B/C (kinesin-1). While KIF1B and KIF5B are ubiquitously expressed, KIF1A, KIF5A and KIF5C are mainly expressed in neuronal cells (Niclas et al., 1994; Nangaku et al., 1994; Kanai et al., 2000; Aizawa et al., 1992). Of these, especially mutations in kinesin-1 family member KIF5A have been associated with neuronal diseases (Cozzi et al., 2024). The type of disease depends on the location of the KIF5A mutation. Hereditary Spastic Paraplegia (HSP) is caused by mutations in the KIF5A N-terminal motor domain. HSP-associated Arg17Gln and Arg280Cys mutant KIF5A showed reduced motility and microtubule binding capacity and destabilized the protein (Cozzi et al., 2024; Goizet et al., 2009; Ebbing et al., 2008; Fichera et al., 2004; Liu et al., 2014; Reid et al., 2002; Blair et al., 2006; Crimella et al., 2012). CMT2 is linked to KIF5A mutations in both the N-terminal motor domain and stalk region leading to truncated motor proteins without tail-domain (Cozzi et al., 2024; Crimella et al., 2012). ALS is associated to mutations in the KIF5A C-terminal cargo-binding tail disturbing cargo-binding and then cargo localization (Cozzi et al., 2024; Nicolas et al., 2018; Brenner et al., 2018). Another C-terminal KIF5A mutation, Cys975Valfs*73, elongates the KIF5A tail-domain. The extended tail region reduces solubility of the protein that then aggregates. This also reduces the number of active KIF5A motors, causing NEonatal Intractable MYoclonus (NEIMY) (Cozzi et al., 2024; Rydzanicz et al., 2017). The wide variety of diseases linked to KIF5A shows the importance of KIF5A-mediated transport in neuronal cells. However, also mutations in KIF1A (kinesin-3) and KIF5C (kinesin-1) yield defects in brain development leading to brain malformation (Poirier et al., 2013; Esmaeeli et al., 2015; Klebe et al., 2012; Lee et al., 2015; Michels et al., 2017; Riviere et al., 2011) as are mutations in KIF1B (kinesin-3), which have been linked to CMT2 (Zhao et al., 2001). The identified mutations in KIF1A, KIF1B and KIF5C cluster in the motor domains and result in reduced ATP hydrolysis capacity and consequently, reduced motility of the motors (Esmaeeli et al., 2015; Zhao et al., 2001). Mutations in the motor domain of KIF5B have also been observed, and are associated with skeletal dysplasias (Itai et al., 2022; Marom et al., 2023). The KIF5B Leu498Pro and Leu537Pro mutations were found in patients with developmental delay translating in variable symptoms including myopathic features, and localize to the KIF5B coiled-coil domains and likely affects KIF5B dimerization (Flex et al., 2023).
Since LE/Lys and LROs move along microtubules in a bidirectional manner involving dynein-dynactin as well as kinesin motor proteins, it would have been surprising if only one of these motors would associate to neurological diseases. Indeed, mutations in both motor classes are ultimately involved in a plethora of neurological disorders as they are active in the same process; long distance transport of LE/Lys and LROs. But there is a third class of motor proteins using another highway, the actin-based myosin motos. What about these?
Myosin-mediated transport along actin highways
When LE/Lys and LROs move to the cell surface along microtubules, they will reach the actin cytoskeleton. Also this transport is polarized and requires myosin motors to move towards the actin plus-end, with the exception of Myosin6 that moves in the opposite direction (Wells et al., 1999). The Myosin-family can be divided into at least 20 subclasses (Odronitz and Kollmar, 2007; Sebe-Pedros et al., 2014; Richards and Cavalier-Smith, 2005; Foth et al., 2006), that participate in various trafficking and anchoring events at many cellular locations. Specificity of cargo binding occurs through the divergent myosin tail-regions while their N-terminal catalytic-domains are conserved between subclasses (Thompson and Langford, 2002).
Diseases related to defects in myosin-motors
Since the last, but equally essential part in vesicle transport towards the plasma membrane involves myosin motors that control the actin based transport step, it is predictable malfunction at this transport step should also yield disease. Since multiple myosin motors (Myosin1 (Donaudy et al., 2003; Zadro et al., 2009), Myosin2 (Kunishima and Saito, 2010), Myosin3 (Walsh et al., 2002), Myosin6 (Melchionda et al., 2001), Myosin7 (Liu et al., 1997) and Myosin15 (Wang et al., 1998)) contribute to the structure of stereocilia essential for hearing (Nambiar et al., 2010), the most common abnormality related to mutations/dysfunction of these motor proteins is deafness. This is illustrated by Myosin7-related Usher syndrome (Kremer et al., 2006). Mutations in myosin motors are also associated to many other diseases, including the LRO-transport related diseases Griscelli syndrome Type 1 (GS1) and MYH9 (Myosin9)-related disease (Van Gele et al., 2009). GS1 is caused by mutations in the MYO5A gene, encoding Myosin5 HC, the motor that links Rab27A-marked melanosomes to actin filaments preceding plasma membrane fusion for melanin release. Similarly, it is involved in the release of LROs in neuronal cells (Lise et al., 2006; Takamori et al., 2006; Correia et al., 2008; Roder et al., 2010; Roder et al., 2008). Because of its function in both melanocytes and neurons, GS1 patients suffer from neurological abnormalities next to the well described pigmentation abnormalities. MYO5A mutations identified in GS1 patients include mutations in the motor domain, among which the nonsense mutation Arg779X resulting in truncated Myosin5 lacking its calmodulin, neck and tail region leading to complete loss-of-function (Pastural et al., 1997). In other patients, a 47 base pair insertion was identified at the start of the Myosin5 tail-domain leading to a truncated Myosin5 lacking its cargo binding tail (Pastural et al., 2000). Depending on cell type, MYO5A transcripts are alternatively spliced leading to various Myosin5 isoforms. While brain cells only express a shorter Myosin5 isoform, melanocytes mostly express Myosin5 with a longer tail domain (including the F-exon) essential for binding to melanophillin. Therefore, GS1 patients with F-exon deletions display pigmentation abnormalities while neuronal cells and function are unaffected (Menasche et al., 2003). MYH9-RD is a collection of disorders caused by mutations in the MYH9 gene, encoding the Myosin9 subunit of Myosin2A, which includes May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome. Three forms of Myosin2 (Myosin 2A, Myosin2B and Myosin2C) exist and perform several function in various cell types usually in combinations. Yet, certain blood cells, including platelets and leukocytes, only express Myosin2A explaining the bleeding problems and immune abnormalities observed in MYH9-RD patients. More than 45 mutations in MYH9 gene have been associated to MYH9-RDs. The severity of symptoms varies with mutations inside the motor domain leading to more severe disorders than mutations in the tail-domain (Althaus and Greinacher, 2009; Sanborn et al., 2011). For example, mutations in the Myosin9 head- and N-terminal S2-domain (Ser96Leu and Thr1155Ile) strongly affected NK-cell cytotoxicity, whereas mutations more C-terminal (Arg1400Trp and Asp1424Asn) yielded milder effects. However, NK-cell cytotoxicity of a patient with a 5779delC mutation (leading to a Myosin9 truncation at residue 1942) showed again strongly diminished NK-cell cytotoxicity. This truncation lacks an important phosphorylation site essential for the interaction between Myosin2A and LGs leading to defective LGs transport (Sanborn et al., 2011). Myosin motors control the actin based step in intracellular transport of LE/Lys and LROs. Mutations then result in various genetic diseases. Also these actin-based motor proteins need to find the correct vesicles by interacting with defined adaptors, which can also be subject to mutations and disease phenotypes.
Motor adaptors at the lysosomal surface and diseases
The LE/Lys outer membrane is decorated with proteins regulating LE/Lys trafficking and functions including protein sorting, fusion with other organelles and protein degradation. The small GTPase Rab7 marks these organelles and controls HOmotypic fusion and vacuole Protein Sorting (HOPS) complex and motor protein recruitment (Jordens et al., 2001; Johansson et al., 2007; Pankiv et al., 2010; van der Kant et al., 2013; Raiborg et al., 2015). Defects in these proteins are mainly associated to neurodegenerative diseases. This includes defects in the retromer-complex associated to Parkinsons disease (Harrington et al., 2012; Zimprich, 2011; Vilarino-Guell et al., 2011), mutations in HOPS-complex subunits associated to Schizophrenia (Xu et al., 2012), Rab7 mutations linked to Alzheimers disease and CMT2 (Romano et al., 2022; Meggouh et al., 2006; Vardarajan et al., 2012; Verhoeven et al., 2003; Houlden et al., 2004; Wang et al., 2014) and VAPB mutations involved in ALS development (Vardarajan et al., 2012). These neurological problems are due to the absence of substrate degrading enzymes resulting in LE/Lys misfunctioning and accumulation of undigested and accumulated material inside the neuronal cell. For example, Alzheimers disease is described to accumulate Tau-containing neurofilaments and Beta-amyloid plaques formed from APP (Amyloid Precursor Protein) (Grundke-Iqbal et al., 1986; Glenner and Wong, 1984; Delacourte and Defossez, 1986; Kosik et al., 1986; Wood et al., 1986; Nukina and Ihara, 1986; Kang et al., 1987). In addition, missense mutations affecting Rab7a, the GTPase important for both dynein-dynactin and kinesin-mediated LE/Lys transport, leads to decreased presence of autolysosomes suggesting limited clearance of intracellular protein aggregates which may explain its association to CMT2 (Romano et al., 2022; Meggouh et al., 2006; Verhoeven et al., 2003; Houlden et al., 2004; Wang et al., 2014). Also, mutations in the proteins CLN3 (leading to a truncated protein) and CLN5 (leading to a misfolded protein) disturb LE/Lys trafficking and sorting machineries. CLN3 is important for recruiting Rab7 and failure affects Rab7-associated functions including motor recruitment, whereas CLN5 interacts with the retromer complex for protein sorting. Insufficient recruitment of either the motor proteins or sorting complexes leads to the accumulation of ceroid lipofuscin and consequently the neurodegenerative disorder Neuronal Ceroid Lipofuscinois (Uusi-Rauva et al., 2012; Mamo et al., 2012). Another group of LE/Lys related diseases are Lysosomal storage disorders (LSDs), including Niemann Pick disease Type-C, also resulting from accumulated undigested material inside the lysosome. Gene mutations associated to LSDs are mutations in NPC1 and NPC2 (mostly single amino acid mutations reducing or eliminating their cholesterol transport activities), resulting in accumulation of cholesterol in LE/Lys, which is sensed by the cholesterol-sensor ORP1L (Rocha et al., 2009; Chang et al., 2005). As a result, ORP1L fails to remove the dynein-dynactin motor from Rab7-RILP resulting in net minus-end transport and accumulation of LE/Lys close to the nucleus around the centriole/MTOC (Rocha et al., 2009). Also, mutations in the effector proteins for Rab7 and other LE/Lys GTPases may result in disease. Indeed, mutations in the Rab7 effector FYCO1 are associated with Cataract (Barashkov et al., 2021; Ullah et al., 2023; Aprahamian et al., 2021; Chen et al., 2011; Iqbal et al., 2020; Mei et al., 2022; Saleem et al., 2022; Shirzadeh et al., 2022).
The dual-adaptor protein huntingtin is mutated in Huntington’s disease (Marcy et al., 1993). Wild-type huntingtin (no capital) is involved in various cellular processes related to not only endosomal transport but also transcriptional regulation and synaptic functioning (Barron et al., 2021; Benn et al., 2008; Dunah et al., 2002). Huntingtin may act as a switch between dynein-dynactin and kinesin-mediated LE/Lys transport. Via the huntingtin-interacting protein HAP1 (Harjes and Wanker, 2003; Caviston and Holzbaur, 2009; Li and Li, 2004; Truant et al., 2006), both the kinesin-1 and dynein/dynactin motor complex can be recruited to the same LE/Lys vesicle (Twelvetrees et al., 2010; Caviston et al., 2007; McGuire et al., 2006; Engelender et al., 1997; Li et al., 1998). Depending on its phosphorylation status, huntingtin can regulate switches in transport direction. Whereas kinesin-1 interacts with phosphorylated huntingtin for plus-end directed transport, kinesin-1 is removed upon huntingtin dephosphorylation thereby favoring dynein-mediated transport in the opposite direction (Colin et al., 2008). In Huntington’s disease patients, the huntingtin gene Htt contains a prolonged CAG repeat (Gil and Rego, 2008). Because huntingtin performs multiple roles in the cell, this mutation is suggested to have multiple pathogenic mechanism, including aggregate formation, altered gene expression, mitochondrial dysfunction and impaired autophagy (reviewed in (Jimenez-Sanchez et al., 2017; Tong et al., 2024)). Focusing on the effect on endosomal transport, the expanded CAG repeat was found to limit transport of BDNF-containing neuronal vesicles by affecting dynein/dynactin motor formation as well as disruption of their association to microtubules (Gauthier et al., 2004).
It is likely that other transport-associated proteins whose function cannot be easily compensated also cause diseases in neurological, immunological or other systems relying on proper LE/Lys or LRO transport. Indeed, novel disease causing mutations in the vesicle transport machinery are still being identified. The functioning of LROs similarly depends on trafficking factors. How do mutations in proteins specifically involved in LRO dynamics associate to different diseases?
The motor adaptors at the LRO membrane and diseases
Although LROs share many transport characteristics with LE/Lys, their specialized character provides them with their own set of transport-related membrane proteins. Consequently, when these proteins are dysfunctional, specialized cargo secretion will be affected only in these specialized cells. For example, transport related defects in melanocytes results in pigmentation defects resulting in albinism. This is observed in Griscelli syndrome (GS) two and 3, Chediak-Higashi syndrome (CHS) and Hermansky-Pudlak syndrome (HPS) (Dell'Angelica et al., 2000). GS2 and GS3 are caused by mutations in the RAB27A gene or MLPH gene (encoding for the Rab27a effector Melanophilin), respectively. While GS2 and GS3 patients both show pigmentation defects due to abnormal melanin secretion, only GS2 patients have additional immune defects, such as hemophagocytic lymphohistiocystosis. This difference in symptoms is caused by the unique function of melanophilin in melanosome secretion as part of the Rab27A-Melanophilin-Myosin5 complex, whereas Rab27A also mediates LGs transport in CTLs as part of the RAB27A-Slp3a-kinesin-1 (KIF5B) complex (Kurowska et al., 2012). Several mutations in the LYST gene, encoding for LYSosomal Trafficking regulator, have been identified in CHS patients (Karim et al., 2002; Dufourcq-Lagelouse et al., 1999; Barbosa et al., 1997; Barbosa et al., 1996; Karim et al., 1997; Morimoto et al., 2024). Although the function of LYST is not exactly known, cells of these patients expressing a short truncated LYST version show an increased LE/Lys and LRO size resulting in an altered structure and function (Ji et al., 2016). Enlarged melanosomes containing accumulated melanin are found in these patients as well as enlarged platelet dense bodies resulting in defective platelet-function and neurological abnormalities (Ji et al., 2016). Another melanosome-associated gene mutated in HPS is the HPS1 gene, encoding for HPS1, a subunit of the Biogenesis of Lysosomal-related Organelles Complex (BLOC)-3 complex. BLOC-3 is a GEF for Rab32 and Rab38. Mutated HPS1 fails to activate the GTPases Rab32 and Rab38, then inhibiting melanin release (Gerondopoulos et al., 2012). Rab32 defects are also related to Parkinson Disease (Gustavsson et al., 2024; Hop et al., 2024). As described above, some LRO membrane proteins have specific roles in controlling their intracellular transport in certain cell types, while others fulfill general functions in LRO and LE/Lys transport. When mutated, they affect one, two or multiple cell types. Different combinations of neurological defects, immune and blood deficiencies as well as pigmentation abnormalities are therefore often seen in patients with mutations in these LRO transport regulating genes.
Conclusion
The transport of LE/Lys and LROs involves many large and small intracellular structures that in assembly coordinate proper cargo transport (Figure 2). Its importance is best illustrated by the fact that many proteins involved in this process cause neurological or other disorders when mutated. These mutations can occur at the level of the highways (microtubules or actin), the signboards on these highways (post-translational mutations, MAPs), lorries (the motor proteins) and their cargo (the adaptor/effector proteins linking motor proteins to their cargo). We begin to understand the system and the reason for the associated diseases. The next challenge will be ways to correct the traffic jams and often lethal accidents.
FIGURE 2
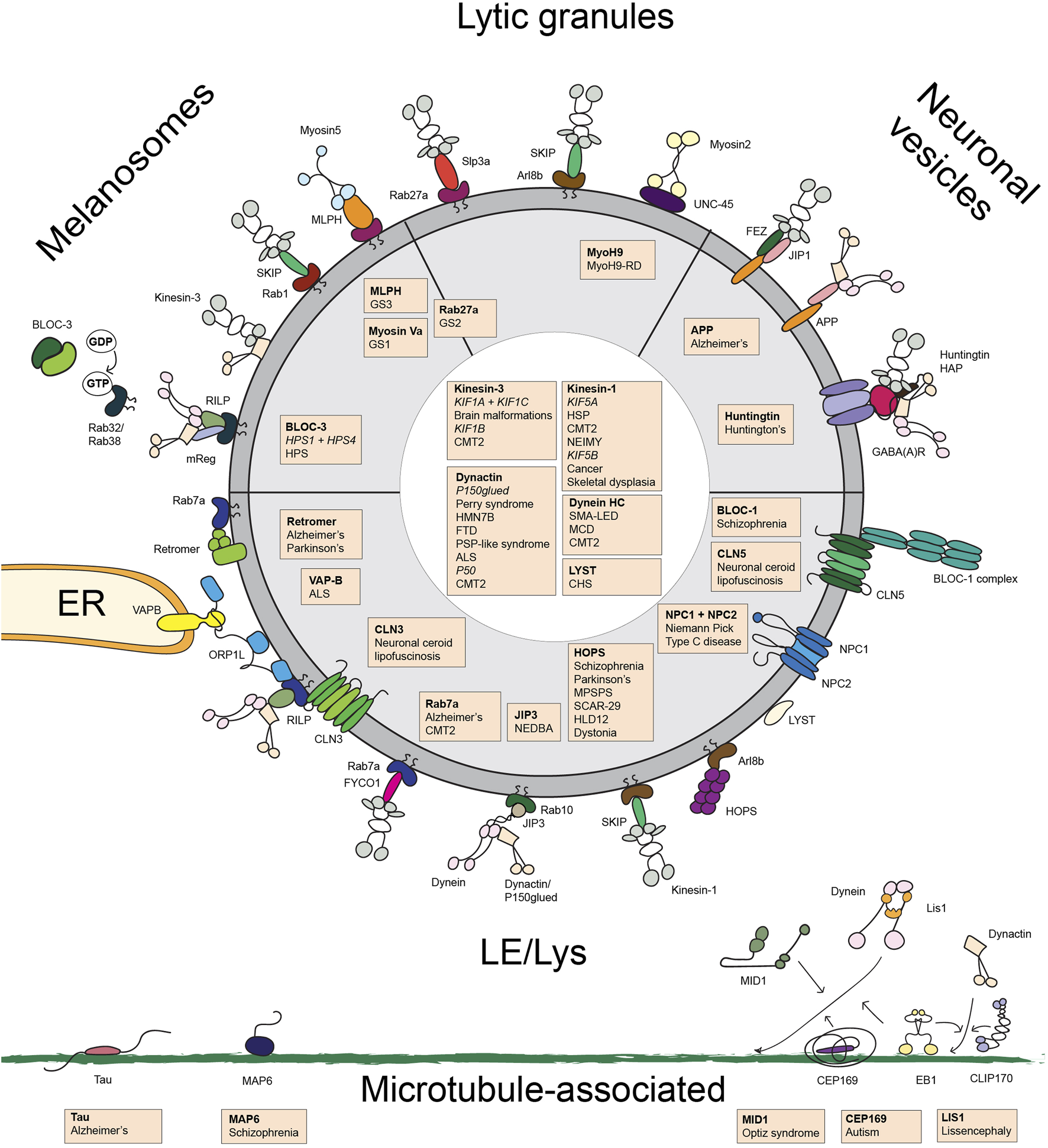
Overview of the transport machinery controlling the transport of late endosomes/lysosomes, melanosomes, lytic granules and and neuronal vesicles. Schematic overview of the proteins involved in late endosome/lysosome and lysosome-related organelle transport. Proteins of which mutations are associated with disease are indicated in orange boxes with a list of the associated disease. Abbreviations used: ALS, amyotrophic lateral sclerosis; BLOC, Biogenesis of Lysosomal-related Organelles Complex; CHS, Chediak-Higashi syndrome; CMT2, Charcot Marie Tooth disease type 2; FTD, frontotemporal dementia; GS, Griscelli syndrome; HLD12, Hypomyelinating Leukodystrophy 12; HMN7B, hereditary motor neuronopathy 7B; HOPS, HOmotypic fusion and vacuole Protein Sorting; HSD hereditary spastic paraplegia; HSP, Hereditary Spastic Paraplegia; MCD, malformations in cortical development; MLPH, melanophilin; MPSPS, mucopolysaccharidosis-plus syndrome; MyoH9-RD, myosinH9-related disease; NEDBA, neurodevelopmental disorder with or without variable brain abnormalities; NEIMY, Neonatal Intractable Myoclonus; NPC, Niemann Pick Type C; SCAR-29, autosomal recessive spinocerebellar ataxia-29; SMALED, spinal muscular atrophy with lower extremity dominance.
Statements
Author contributions
NB: Conceptualization, Writing – original draft, Writing – review and editing, Visualization. MJ: Conceptualization, Supervision, Writing – original draft, Writing – review and editing. JN: Funding acquisition, Supervision, Writing – review and editing, Writing – original draft, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by an ERC ERCOPE grant (50438) and an NWO BBoL GLCCER grant (737.016.002) awarded to JN.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ALS, amyotrophic lateral sclerosis; APP, β-amyloid precursor protein; Arl, Arf-like; Arp1, actin-related protein-1; ASD, autism spectrum disorder; BLOC, Biogenesis of Lysosomal-related Organelles Complex; CMT2, Charcot Marie Tooth disease type 2; CSCSC2, congenital symmetric circumferential skin creases-2; CTL, cytotoxic T cell; HC, heavy chain; HMN7B, hereditary motor neuronopathy 7B; HPS, Hermansky-Pudlak syndrome; HOPS, HOmotypic fusesion and vacuole Protein Sorting; HSD, hereditary spastic paraplegia; IC, intermediate chain; KAP3, kinesin associated protein 3; LC, light chain; LE/Lys, late endosome/lysosome; LG, lytic granule; LIC, light intermediate chain; LRO, lysosome related organelle; MAP, microtubule-associated protein; MCD, malformations in cortical development; MTOC, microtubule-organizing center; MYH9, Myosin9; NEDBA, neurodevelopmental disorder with or without variable brain abnormalities; NK-cell, natural killer cell; PP2A, phosphatase 2A; SMALED, spinal muscular atrophy with lower extremity dominance; TDP-43, transactive-response DNA-binding protein of 43 kDa.
References
1
Aizawa H. Emori Y. Mori A. Murofushi H. Sakai H. Suzuki K. (1991). Functional analyses of the domain structure of microtubule-associated protein-4 (MAP-U). J. Biol. Chem.266 (15), 9841–9846. 10.1016/s0021-9258(18)92896-6
2
Aizawa H. Sekine Y. Takemura R. Zhang Z. Nangaku M. Hirokawa N. (1992). Kinesin family in murine central nervous system. J. Cell Biol.119 (5), 1287–1296. 10.1083/jcb.119.5.1287
3
Akhmanova A. Steinmetz M. O. (2008). Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol.9 (4), 309–322. 10.1038/nrm2369
4
Althaus K. Greinacher A. (2009). MYH9-related platelet disorders. Semin. Thromb. Hemost.35 (2), 189–203. 10.1055/s-0029-1220327
5
Andrieux A. Salin P. A. Vernet M. Kujala P. Baratier J. Gory-Fauré S. et al (2002). The suppression of brain cold-stable microtubules in mice induces synaptic defects associated with neuroleptic-sensitive behavioral disorders. Gene Dev.16 (18), 2350–2364. 10.1101/gad.223302
6
Anikster Y. Huizing M. White J. Shevchenko Y. O. Fitzpatrick D. L. Touchman J. W. et al (2001). Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat. Genet.28 (4), 376–380. 10.1038/ng576
7
Aprahamian R. Yammine T. Salem N. Souaid M. Mansour H. Farra C. (2021). Identification of a novel nonsense variant in FYCO1 gene associated with infantile cataract and cortical atrophy. Ophthalmic Genet.42 (6), 744–746. 10.1080/13816810.2021.1955277
8
Aranda-Orgilles B. Aigner J. Kunath M. Lurz R. Schneider R. Schweiger S. (2008). Active transport of the ubiquitin ligase MID1 along the microtubules is regulated by protein phosphatase 2A. PLoS One3 (10), e3507. 10.1371/journal.pone.0003507
9
Arimoto M. Koushika S. P. Choudhary B. C. Li C. Matsumoto K. Hisamoto N. (2011). The Caenorhabditis elegans JIP3 protein UNC-16 functions as an adaptor to link kinesin-1 with cytoplasmic dynein. J. Neurosci.31 (6), 2216–2224. 10.1523/JNEUROSCI.2653-10.2011
10
Badolato R. Prandini A. Caracciolo S. Colombo F. Tabellini G. Giacomelli M. et al (2012). Exome sequencing reveals a pallidin mutation in a Hermansky-Pudlak-like primary immunodeficiency syndrome. Blood119 (13), 3185–3187. 10.1182/blood-2012-01-404350
11
Baker M. Graff-Radford D. Wavrant DeVrieze F. Graff-Radford N. Petersen R. C. Kokmen E. et al (2000). No association between TAU haplotype and Alzheimer's disease in population or clinic based series or in familial disease. Neurosci. Lett.285 (2), 147–149. 10.1016/s0304-3940(00)01057-0
12
Balla T. (2013). Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev.93 (3), 1019–1137. 10.1152/physrev.00028.2012
13
Bancher C. Lassmann H. Budka H. Grundke-Iqbal I. Iqbal K. Wiche G. et al (1987). Neurofibrillary tangles in Alzheimer's disease and progressive supranuclear palsy: antigenic similarities and differences. Microtubule-associated protein tau antigenicity is prominent in all types of tangles. Acta Neuropathol.74 (1), 39–46. 10.1007/BF00688336
14
Barashkov N. A. Konovalov F. A. Borisova T. V. Teryutin F. M. Solovyev A. V. Pshennikova V. G. et al (2021). Autosomal recessive cataract (CTRCT18) in the Yakut population isolate of Eastern Siberia: a novel founder variant in the FYCO1 gene. Eur. J. Hum. Genet.29 (6), 965–976. 10.1038/s41431-021-00833-w
15
Barbosa M. D. Nguyen Q. A. Tchernev V. T. Ashley J. A. Detter J. C. Blaydes S. M. et al (1996). Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature382 (6588), 262–265. 10.1038/382262a0
16
Barbosa M. D. Barrat F. J. Tchernev V. T. Nguyen Q. A. Mishra V. S. Colman S. D. et al (1997). Identification of mutations in two major mRNA isoforms of the Chediak-Higashi syndrome gene in human and mouse. Hum. Mol. Genet.6 (7), 1091–1098. 10.1093/hmg/6.7.1091
17
Barlan K. Lu W. Gelfand V. I. (2013). The microtubule-binding protein ensconsin is an essential cofactor of kinesin-1. Curr. Biol.23 (4), 317–322. 10.1016/j.cub.2013.01.008
18
Barron J. C. Hurley E. P. Parsons M. P. (2021). Huntingtin and the synapse. Front. Cell Neurosci.15, 689332. 10.3389/fncel.2021.689332
19
Baumbach J. Murthy A. McClintock M. A. Dix C. I. Zalyte R. Hoang H. T. et al (2017). Lissencephaly-1 is a context-dependent regulator of the human dynein complex. Elife6, e21768. 10.7554/eLife.21768
20
Becker L. L. Dafsari H. S. Schallner J. Abdin D. Seifert M. Petit F. et al (2020). The clinical-phenotype continuum in DYNC1H1-related disorders-genomic profiling and proposal for a novel classification. J. Hum. Genet.65 (11), 1003–1017. 10.1038/s10038-020-0803-1
21
Benn C. L. Sun T. Sadri-Vakili G. McFarland K. N. DiRocco D. P. Yohrling G. J. et al (2008). Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J. Neurosci.28 (42), 10720–10733. 10.1523/JNEUROSCI.2126-08.2008
22
Berti C. Fontanella B. Ferrentino R. Meroni G. (2004). Mig12, a novel Opitz syndrome gene product partner, is expressed in the embryonic ventral midline and co-operates with Mid1 to bundle and stabilize microtubules. BMC Cell Biol.5, 9. 10.1186/1471-2121-5-9
23
Bieling P. Laan L. Schek H. Munteanu E. L. Sandblad L. Dogterom M. et al (2007). Reconstitution of a microtubule plus-end tracking system in vitro. Nature450 (7172), 1100–1105. 10.1038/nature06386
24
Bieling P. Kandels-Lewis S. Telley I. A. van Dijk J. Janke C. Surrey T. (2008). CLIP-170 tracks growing microtubule ends by dynamically recognizing composite EB1/tubulin-binding sites. J. Cell Biol.183 (7), 1223–1233. 10.1083/jcb.200809190
25
Bjelic S. De Groot C. O. Scharer M. A. Jaussi R. Bargsten K. Salzmann M. et al (2012). Interaction of mammalian end binding proteins with CAP-Gly domains of CLIP-170 and p150(glued). J. Struct. Biol.177 (1), 160–167. 10.1016/j.jsb.2011.11.010
26
Blair M. A. Ma S. Hedera P. (2006). Mutation in KIF5A can also cause adult-onset hereditary spastic paraplegia. Neurogenetics7 (1), 47–50. 10.1007/s10048-005-0027-8
27
Blasius T. L. Cai D. Jih G. T. Toret C. P. Verhey K. J. (2007). Two binding partners cooperate to activate the molecular motor Kinesin-1. J. Cell Biol.176 (1), 11–17. 10.1083/jcb.200605099
28
Bodakuntla S. Jijumon A. S. Villablanca C. Gonzalez-Billault C. Janke C. (2019). Microtubule-associated proteins: structuring the cytoskeleton. Trends Cell Biol.29 (10), 804–819. 10.1016/j.tcb.2019.07.004
29
Bonifacino J. S. Neefjes J. (2017). Moving and positioning the endolysosomal system. Curr. Opin. Cell Biol.47, 1–8. 10.1016/j.ceb.2017.01.008
30
Braathen G. J. Hoyer H. Busk O. L. Tveten K. Skjelbred C. F. Russell M. B. (2016). Variants in the genes DCTN2, DNAH10, LRIG3, and MYO1A are associated with intermediate Charcot-Marie-Tooth disease in a Norwegian family. Acta Neurol. Scand.134 (1), 67–75. 10.1111/ane.12515
31
Brenner D. Yilmaz R. Muller K. Grehl T. Petri S. Meyer T. et al (2018). Hot-spot KIF5A mutations cause familial ALS. Brain141 (3), 688–697. 10.1093/brain/awx370
32
Bullido M. J. Aldudo J. Frank A. Coria F. Avila J. Valdivieso F. (2000). A polymorphism in the tau gene associated with risk for Alzheimer's disease. Neurosci. Lett.278 (1-2), 49–52. 10.1016/s0304-3940(99)00893-9
33
Cady J. Allred P. Bali T. Pestronk A. Goate A. Miller T. M. et al (2015). Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann. Neurol.77 (1), 100–113. 10.1002/ana.24306
34
Cai X. Chen X. Wu S. Liu W. Zhang X. Zhang D. et al (2016). Homozygous mutation of VPS16 gene is responsible for an autosomal recessive adolescent-onset primary dystonia. Sci. Rep.6, 25834. 10.1038/srep25834
35
Carstea E. D. Morris J. A. Coleman K. G. Loftus S. K. Zhang D. Cummings C. et al (1997). Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science277 (5323), 228–231. 10.1126/science.277.5323.228
36
Caviston J. P. Holzbaur E. L. (2009). Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol.19 (4), 147–155. 10.1016/j.tcb.2009.01.005
37
Caviston J. P. Ross J. L. Antony S. M. Tokito M. Holzbaur E. L. (2007). Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. U. S. A.104 (24), 10045–10050. 10.1073/pnas.0610628104
38
Chahrour M. H. Yu T. W. Lim E. T. Ataman B. Coulter M. E. Hill R. S. et al (2012). Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet.8 (4), e1002635. 10.1371/journal.pgen.1002635
39
Chang T. Y. Reid P. C. Sugii S. Ohgami N. Cruz J. C. Chang C. C. (2005). Niemann-Pick type C disease and intracellular cholesterol trafficking. J. Biol. Chem.280 (22), 20917–20920. 10.1074/jbc.R400040200
40
Chaudhary A. R. Berger F. Berger C. L. Hendricks A. G. (2018). Tau directs intracellular trafficking by regulating the forces exerted by kinesin and dynein teams. Traffic19 (2), 111–121. 10.1111/tra.12537
41
Chaudhary A. R. Lu H. Krementsova E. B. Bookwalter C. S. Trybus K. M. Hendricks A. G. (2019). MAP7 regulates organelle transport by recruiting kinesin-1 to microtubules. J. Biol. Chem.294 (26), 10160–10171. 10.1074/jbc.RA119.008052
42
Chen J. Ma Z. Jiao X. Fariss R. Kantorow W. L. Kantorow M. et al (2011). Mutations in FYCO1 cause autosomal-recessive congenital cataracts. Am. J. Hum. Genet.88 (6), 827–838. 10.1016/j.ajhg.2011.05.008
43
Chung P. J. Song C. Deek J. Miller H. P. Li Y. Choi M. C. et al (2016). Tau mediates microtubule bundle architectures mimicking fascicles of microtubules found in the axon initial segment. Nat. Commun.7, 12278. 10.1038/ncomms12278
44
Clark L. N. Levy G. Tang M. X. Mejia-Santana H. Ciappa A. Tycko B. et al (2003). The Saitohin 'Q7R' polymorphism and tau haplotype in multi-ethnic Alzheimer disease and Parkinson's disease cohorts. Neurosci. Lett.347 (1), 17–20. 10.1016/s0304-3940(03)00635-9
45
Cole D. G. Chinn S. W. Wedaman K. P. Hall K. Vuong T. Scholey J. M. (1993). Novel heterotrimeric kinesin-related protein purified from sea urchin eggs. Nature366 (6452), 268–270. 10.1038/366268a0
46
Colin E. Zala D. Liot G. Rangone H. Borrell-Pages M. Li X. J. et al (2008). Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J.27 (15), 2124–2134. 10.1038/emboj.2008.133
47
Conrad C. Andreadis A. Trojanowski J. Q. Dickson D. W. Kang D. Chen X. et al (1997). Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann. Neurol.41 (2), 277–281. 10.1002/ana.410410222
48
Conrad C. Vianna C. Freeman M. Davies P. (2002). A polymorphic gene nested within an intron of the tau gene: implications for Alzheimer's disease. Proc. Natl. Acad. Sci. U. S. A.99 (11), 7751–7756. 10.1073/pnas.112194599
49
Correia S. S. Bassani S. Brown T. C. Lise M. F. Backos D. S. El-Husseini A. et al (2008). Motor protein-dependent transport of AMPA receptors into spines during long-term potentiation. Nat. Neurosci.11 (4), 457–466. 10.1038/nn2063
50
Courcet J. B. Faivre L. Malzac P. Masurel-Paulet A. Lopez E. Callier P. et al (2012). The DYRK1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy. J. Med. Genet.49 (12), 731–736. 10.1136/jmedgenet-2012-101251
51
Cozzi M. Magri S. Tedesco B. Patelli G. Ferrari V. Casarotto E. et al (2024). Altered molecular and cellular mechanisms in KIF5A-associated neurodegenerative or neurodevelopmental disorders. Cell Death Dis.15 (9), 692. 10.1038/s41419-024-07096-5
52
Crimella C. Baschirotto C. Arnoldi A. Tonelli A. Tenderini E. Airoldi G. et al (2012). Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin. Genet.82 (2), 157–164. 10.1111/j.1399-0004.2011.01717.x
53
Cuveillier C. Delaroche J. Seggio M. Gory-Faure S. Bosc C. Denarier E. et al (2020). MAP6 is an intraluminal protein that induces neuronal microtubules to coil. Sci. Adv.6 (14), eaaz4344. 10.1126/sciadv.aaz4344
54
Daniele T. Hackmann Y. Ritter A. T. Wenham M. Booth S. Bossi G. et al (2011). A role for Rab7 in the movement of secretory granules in cytotoxic T lymphocytes. Traffic12 (7), 902–911. 10.1111/j.1600-0854.2011.01194.x
55
Daoust A. Bohic S. Saoudi Y. Debacker C. Gory-Faure S. Andrieux A. et al (2014). Neuronal transport defects of the MAP6 KO mouse - a model of schizophrenia - and alleviation by Epothilone D treatment, as observed using MEMRI. Neuroimage.96, 133–142. 10.1016/j.neuroimage.2014.03.071
56
Delacourte A. Defossez A. (1986). Alzheimer's disease: tau proteins, the promoting factors of microtubule assembly, are major components of paired helical filaments. J. Neurol. Sci.76 (2-3), 173–186. 10.1016/0022-510x(86)90167-x
57
Delevoye C. Marks M. S. Raposo G. (2019). Lysosome-related organelles as functional adaptations of the endolysosomal system. Curr. Opin. Cell Biol.59, 147–158. 10.1016/j.ceb.2019.05.003
58
Dell'Angelica E. C. Shotelersuk V. Aguilar R. C. Gahl W. A. Bonifacino J. S. (1999). Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol. Cell3 (1), 11–21. 10.1016/s1097-2765(00)80170-7
59
Dell'Angelica E. C. Aguilar R. C. Wolins N. Hazelwood S. Gahl W. A. Bonifacino J. S. (2000). Molecular characterization of the protein encoded by the Hermansky-Pudlak syndrome type 1 gene. J. Biol. Chem.275 (2), 1300–1306. 10.1074/jbc.275.2.1300
60
Dimitrova-Paternoga L. Jagtap P. K. A. Cyrklaff A. Vaishali L. K. Sehr P. et al (2021). Molecular basis of mRNA transport by a kinesin-1-atypical tropomyosin complex. Genes Dev.35 (13-14), 976–991. 10.1101/gad.348443.121
61
Dixit R. Levy J. R. Tokito M. Ligon L. A. Holzbaur E. L. (2008a). Regulation of dynactin through the differential expression of p150Glued isoforms. J. Biol. Chem.283 (48), 33611–33619. 10.1074/jbc.M804840200
62
Dixit R. Ross J. L. Goldman Y. E. Holzbaur E. L. (2008b). Differential regulation of dynein and kinesin motor proteins by tau. Science319 (5866), 1086–1089. 10.1126/science.1152993
63
Dobyns W. B. Reiner O. Carrozzo R. Ledbetter D. H. (1993). Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA270 (23), 2838–2842. 10.1001/jama.270.23.2838
64
Donaldson J. G. Honda A. (2005). Localization and function of Arf family GTPases. Biochem. Soc. Trans.33 (Pt 4), 639–642. 10.1042/BST0330639
65
Donaudy F. Ferrara A. Esposito L. Hertzano R. Ben-David O. Bell R. E. et al (2003). Multiple mutations of MYO1A, a cochlear-expressed gene, in sensorineural hearing loss. Am. J. Hum. Genet.72 (6), 1571–1577. 10.1086/375654
66
Dor T. Cinnamon Y. Raymond L. Shaag A. Bouslam N. Bouhouche A. et al (2014). KIF1C mutations in two families with hereditary spastic paraparesis and cerebellar dysfunction. J. Med. Genet.51 (2), 137–142. 10.1136/jmedgenet-2013-102012
67
Driskell O. J. Mironov A. Allan V. J. Woodman P. G. (2007). Dynein is required for receptor sorting and the morphogenesis of early endosomes. Nat. Cell Biol.9 (1), 113–120. 10.1038/ncb1525
68
Duellberg C. Trokter M. Jha R. Sen I. Steinmetz M. O. Surrey T. (2014). Reconstitution of a hierarchical +TIP interaction network controlling microtubule end tracking of dynein. Nat. Cell Biol.16 (8), 804–811. 10.1038/ncb2999
69
Dufourcq-Lagelouse R. Lambert N. Duval M. Viot G. Vilmer E. Fischer A. et al (1999). Chediak-Higashi syndrome associated with maternal uniparental isodisomy of chromosome 1. Eur. J. Hum. Genet.7 (6), 633–637. 10.1038/sj.ejhg.5200355
70
Duis J. Dean S. Applegate C. Harper A. Xiao R. He W. et al (2016). KIF5A mutations cause an infantile onset phenotype including severe myoclonus with evidence of mitochondrial dysfunction. Ann. Neurol.80 (4), 633–637. 10.1002/ana.24744
71
Dunah A. W. Jeong H. Griffin A. Kim Y. M. Standaert D. G. Hersch S. M. et al (2002). Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science296 (5576), 2238–2243. 10.1126/science.1072613
72
Dursun A. Yalnizoglu D. Gerdan O. F. Yucel-Yilmaz D. Sagiroglu M. S. Yuksel B. et al (2017). A probable new syndrome with the storage disease phenotype caused by the VPS33A gene mutation. Clin. Dysmorphol.26 (1), 1–12. 10.1097/MCD.0000000000000149
73
Dye R. B. Fink S. P. Williams R. C. Jr (1993). Taxol-induced flexibility of microtubules and its reversal by MAP-2 and Tau. J. Biol. Chem.268 (10), 6847–6850. 10.1016/s0021-9258(18)53113-6
74
Ebbing B. Mann K. Starosta A. Jaud J. Schols L. Schule R. et al (2008). Effect of spastic paraplegia mutations in KIF5A kinesin on transport activity. Hum. Mol. Genet.17 (9), 1245–1252. 10.1093/hmg/ddn014
75
Ebneth A. Godemann R. Stamer K. Illenberger S. Trinczek B. Mandelkow E. (1998). Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J. Cell Biol.143 (3), 777–794. 10.1083/jcb.143.3.777
76
Edvardson S. Gerhard F. Jalas C. Lachmann J. Golan D. Saada A. et al (2015). Hypomyelination and developmental delay associated with VPS11 mutation in Ashkenazi-Jewish patients. J. Med. Genet.52 (11), 749–753. 10.1136/jmedgenet-2015-103239
77
Edwards S. L. Yu S. C. Hoover C. M. Phillips B. C. Richmond J. E. Miller K. G. (2013). An organelle gatekeeper function for Caenorhabditis elegans UNC-16 (JIP3) at the axon initial segment. Genetics194 (1), 143–161. 10.1534/genetics.112.147348
78
Elshenawy M. M. Kusakci E. Volz S. Baumbach J. Bullock S. L. Yildiz A. (2020). Lis1 activates dynein motility by modulating its pairing with dynactin. Nat. Cell Biol.22 (5), 570–578. 10.1038/s41556-020-0501-4
79
Endow S. A. Kull F. J. Liu H. (2010). Kinesins at a glance. J. Cell Sci.123 (Pt 20), 3420–3424. 10.1242/jcs.064113
80
Engelender S. Sharp A. H. Colomer V. Tokito M. K. Lanahan A. Worley P. et al (1997). Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum. Mol. Genet.6 (13), 2205–2212. 10.1093/hmg/6.13.2205
81
Erlich Y. Edvardson S. Hodges E. Zenvirt S. Thekkat P. Shaag A. et al (2011). Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis. Genome Res.21 (5), 658–664. 10.1101/gr.117143.110
82
Esmaeeli N. S. Madou M. R. Sirajuddin M. Fregeau B. McKnight D. Lexa K. et al (2015). De novo mutations in KIF1A cause progressive encephalopathy and brain atrophy. Ann. Clin. Transl. Neurol.2 (6), 623–635. 10.1002/acn3.198
83
Farrer M. J. Hulihan M. M. Kachergus J. M. Dachsel J. C. Stoessl A. J. Grantier L. L. et al (2009). DCTN1 mutations in Perry syndrome. Nat. Genet.41 (2), 163–165. 10.1038/ng.293
84
Felgner H. Frank R. Biernat J. Mandelkow E. M. Mandelkow E. Ludin B. et al (1997). Domains of neuronal microtubule-associated proteins and flexural rigidity of microtubules. J. Cell Biol.138 (5), 1067–1075. 10.1083/jcb.138.5.1067
85
Ferro L. S. Fang Q. Eshun-Wilson L. Fernandes J. Jack A. Farrell D. P. et al (2022). Structural and functional insight into regulation of kinesin-1 by microtubule-associated protein MAP7. Science375 (6578), 326–331. 10.1126/science.abf6154
86
Fichera M. Lo Giudice M. Falco M. Sturnio M. Amata S. Calabrese O. et al (2004). Evidence of kinesin heavy chain (KIF5A) involvement in pure hereditary spastic paraplegia. Neurology63 (6), 1108–1110. 10.1212/01.wnl.0000138731.60693.d2
87
Fiorillo C. Moro F. Yi J. Weil S. Brisca G. Astrea G. et al (2014). Novel dynein DYNC1H1 neck and motor domain mutations link distal spinal muscular atrophy and abnormal cortical development. Hum. Mutat.35 (3), 298–302. 10.1002/humu.22491
88
Fiorillo C. Moro F. Brisca G. Accogli A. Trucco F. Trovato R. et al (2016). Beyond spinal muscular atrophy with lower extremity dominance: cerebellar hypoplasia associated with a novel mutation in BICD2. Eur. J. Neurol.23 (4), e19–e21. 10.1111/ene.12914
89
Flex E. Albadri S. Radio F. C. Cecchetti S. Lauri A. Priolo M. et al (2023). Dominantly acting KIF5B variants with pleiotropic cellular consequences cause variable clinical phenotypes. Hum. Mol. Genet.32 (3), 473–488. 10.1093/hmg/ddac213
90
Foth B. J. Goedecke M. C. Soldati D. (2006). New insights into myosin evolution and classification. Proc. Natl. Acad. Sci. U. S. A.103 (10), 3681–3686. 10.1073/pnas.0506307103
91
Fu J. F. Hsu H. C. Shih L. Y. (2005). MLL is fused to EB1 (MAPRE1), which encodes a microtubule-associated protein, in a patient with acute lymphoblastic leukemia.Genes Chromosomes Cancer43 (2), 206–210. 10.1002/gcc.20174
92
Fu M. M. Holzbaur E. L. (2013). JIP1 regulates the directionality of APP axonal transport by coordinating kinesin and dynein motors. J. Cell Biol.202 (3), 495–508. 10.1083/jcb.201302078
93
Fukai K. Oh J. Frenk E. Almodovar C. Spritz R. A. (1995). Linkage disequilibrium mapping of the gene for Hermansky-Pudlak syndrome to chromosome 10q23.1-q23.3. Hum. Mol. Genet.4 (9), 1665–1669. 10.1093/hmg/4.9.1665
94
Garbouchian A. Montgomery A. C. Gilbert S. P. Bentley M. (2022). KAP is the neuronal organelle adaptor for Kinesin-2 KIF3AB and KIF3AC. Mol. Biol. Cell33 (14), ar133. 10.1091/mbc.E22-08-0336
95
Gauthier L. R. Charrin B. C. Borrell-Pages M. Dompierre J. P. Rangone H. Cordelieres F. P. et al (2004). Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell118 (1), 127–138. 10.1016/j.cell.2004.06.018
96
Gelineau-Morel R. Lukacs M. Weaver K. N. Hufnagel R. B. Gilbert D. L. Stottmann R. W. (2016). Congenital cataracts and gut dysmotility in a DYNC1H1 dyneinopathy patient. Genes (Basel)7 (10), 85. 10.3390/genes7100085
97
Gerondopoulos A. Langemeyer L. Liang J. R. Linford A. Barr F. A. (2012). BLOC-3 mutated in Hermansky-Pudlak syndrome is a Rab32/38 guanine nucleotide exchange factor. Curr. Biol.22 (22), 2135–2139. 10.1016/j.cub.2012.09.020
98
Gil J. M. Rego A. C. (2008). Mechanisms of neurodegeneration in Huntington's disease. Eur. J. Neurosci.27 (11), 2803–2820. 10.1111/j.1460-9568.2008.06310.x
99
Gillies J. P. Reimer J. M. Karasmanis E. P. Lahiri I. Htet Z. M. Leschziner A. E. et al (2022). Structural basis for cytoplasmic dynein-1 regulation by Lis1. Elife11, e71229. 10.7554/eLife.71229
100
Gimenez U. Boulan B. Mauconduit F. Taurel F. Leclercq M. Denarier E. et al (2017). 3D imaging of the brain morphology and connectivity defects in a model of psychiatric disorders: MAP6-KO mice. Sci. Rep.7 (1), 10308. 10.1038/s41598-017-10544-2
101
Glenner G. G. Wong C. W. (1984). Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun.120 (3), 885–890. 10.1016/s0006-291x(84)80190-4
102
Goate A. Chartier-Harlin M. C. Mullan M. Brown J. Crawford F. Fidani L. et al (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature349 (6311), 704–706. 10.1038/349704a0
103
Goizet C. Boukhris A. Mundwiller E. Tallaksen C. Forlani S. Toutain A. et al (2009). Complicated forms of autosomal dominant hereditary spastic paraplegia are frequent in SPG10. Hum. Mutat.30 (2), E376–E385. 10.1002/humu.20920
104
Goodson H. V. Jonasson E. M. (2018). Microtubules and microtubule-associated proteins. Cold Spring Harb. Perspect. Biol.10 (6), a022608. 10.1101/cshperspect.a022608
105
Gross S. P. Tuma M. C. Deacon S. W. Serpinskaya A. S. Reilein A. R. Gelfand V. I. (2002). Interactions and regulation of molecular motors in Xenopus melanophores. J. Cell Biol.156 (5), 855–865. 10.1083/jcb.200105055
106
Grundke-Iqbal I. Iqbal K. Quinlan M. Tung Y. C. Zaidi M. S. Wisniewski H. M. (1986). Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem.261 (13), 6084–6089. 10.1016/s0021-9258(17)38495-8
107
Guedes-Dias P. Holzbaur E. L. F. (2019). Axonal transport: driving synaptic function. Science.366 (6462), eaaw9997. 10.1126/science.aaw9997
108
Guerrini R. Parrini E. (2010). Neuronal migration disorders. Neurobiol. Dis.38 (2), 154–166. 10.1016/j.nbd.2009.02.008
109
Gumy L. F. Katrukha E. A. Grigoriev I. Jaarsma D. Kapitein L. C. Akhmanova A. et al (2017). MAP2 defines a pre-axonal filtering zone to regulate KIF1- versus KIF5-dependent cargo transport in sensory neurons. Neuron94 (2), 347–362. 10.1016/j.neuron.2017.03.046
110
Gustavsson E. K. Follett J. Trinh J. Barodia S. K. Real R. Liu Z. et al (2024). RAB32 Ser71Arg in autosomal dominant Parkinson's disease: linkage, association, and functional analyses. Lancet Neurol.23 (6), 603–614. 10.1016/S1474-4422(24)00121-2
111
Hagiwara H. Yorifuji H. Sato-Yoshitake R. Hirokawa N. (1994). Competition between motor molecules (kinesin and cytoplasmic dynein) and fibrous microtubule-associated proteins in binding to microtubules. J. Biol. Chem.269 (5), 3581–3589. 10.1016/s0021-9258(17)41903-x
112
Hamdan F. F. Gauthier J. Araki Y. Lin D. T. Yoshizawa Y. Higashi K. et al (2011). Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am. J. Hum. Genet.88 (3), 306–316. 10.1016/j.ajhg.2011.02.001
113
Harjes P. Wanker E. E. (2003). The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem. Sci.28 (8), 425–433. 10.1016/S0968-0004(03)00168-3
114
Harms M. B. Ori-McKenney K. M. Scoto M. Tuck E. P. Bell S. Ma D. et al (2012). Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology78 (22), 1714–1720. 10.1212/WNL.0b013e3182556c05
115
Harrington A. J. Yacoubian T. A. Slone S. R. Caldwell K. A. Caldwell G. A. (2012). Functional analysis of VPS41-mediated neuroprotection in Caenorhabditis elegans and mammalian models of Parkinson's disease. J. Neurosci.32 (6), 2142–2153. 10.1523/JNEUROSCI.2606-11.2012
116
Hirokawa N. Noda Y. Tanaka Y. Niwa S. (2009). Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol.10 (10), 682–696. 10.1038/nrm2774
117
Hoang H. T. Schlager M. A. Carter A. P. Bullock S. L. (2017). DYNC1H1 mutations associated with neurological diseases compromise processivity of dynein-dynactin-cargo adaptor complexes. Proc. Natl. Acad. Sci. U. S. A.114 (9), E1597-E1606–E606. 10.1073/pnas.1620141114
118
Homma Y. Hiragi S. Fukuda M. (2021). Rab family of small GTPases: an updated view on their regulation and functions. FEBS J.288 (1), 36–55. 10.1111/febs.15453
119
Hooikaas P. J. Martin M. Muhlethaler T. Kuijntjes G. J. Peeters C. A. E. Katrukha E. A. et al (2019). MAP7 family proteins regulate kinesin-1 recruitment and activation. J. Cell Biol.218 (4), 1298–1318. 10.1083/jcb.201808065
120
Hook P. Vallee R. B. (2006). The dynein family at a glance. J. Cell Sci.119 (Pt 21), 4369–4371. 10.1242/jcs.03176
121
Hop P. J. Lai D. Keagle P. J. Baron D. M. Kenna B. J. Kooyman M. et al (2024). Systematic rare variant analyses identify RAB32 as a susceptibility gene for familial Parkinson's disease. Nat. Genet.56 (7), 1371–1376. 10.1038/s41588-024-01787-7
122
Houlden H. King R. H. Muddle J. R. Warner T. T. Reilly M. M. Orrell R. W. et al (2004). A novel RAB7 mutation associated with ulcero-mutilating neuropathy. Ann. Neurol.56 (4), 586–590. 10.1002/ana.20281
123
Htet Z. M. Gillies J. P. Baker R. W. Leschziner A. E. DeSantis M. E. Reck-Peterson S. L. (2020). LIS1 promotes the formation of activated cytoplasmic dynein-1 complexes. Nat. Cell Biol.22 (5), 518–525. 10.1038/s41556-020-0506-z
124
Hutton M. Lendon C. L. Rizzu P. Baker M. Froelich S. Houlden H. et al (1998). Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature393 (6686), 702–705. 10.1038/31508
125
Iizuka Y. Cichocki F. Sieben A. Sforza F. Karim R. Coughlin K. et al (2015). UNC-45A is a nonmuscle myosin IIA chaperone required for NK cell cytotoxicity via control of lytic granule secretion. J. Immunol.195 (10), 4760–4770. 10.4049/jimmunol.1500979
126
Iqbal H. Khan S. Y. Zhou L. Irum B. Ali M. Ahmed M. R. et al (2020). Mutations in FYCO1 identified in families with congenital cataracts. Mol. Vis.26, 334–344.
127
Ishida M. Ohbayashi N. Fukuda M. (2015). Rab1A regulates anterograde melanosome transport by recruiting kinesin-1 to melanosomes through interaction with SKIP. Sci. Rep.5, 8238. 10.1038/srep08238
128
Isolation of a novel gene underlying Batten disease (1995). Isolation of a novel gene underlying batten disease, CLN3. The international batten disease consortium. Cell82 (6), 949–957. 10.1016/0092-8674(95)90274-0
129
Isrie M. Breuss M. Tian G. Hansen A. H. Cristofoli F. Morandell J. et al (2015). Mutations in either TUBB or MAPRE2 cause circumferential skin creases kunze type. Am. J. Hum. Genet.97 (6), 790–800. 10.1016/j.ajhg.2015.10.014
130
Itai T. Wang Z. Nishimura G. Ohashi H. Guo L. Wakano Y. et al (2022). De novo heterozygous variants in KIF5B cause kyphomelic dysplasia. Clin. Genet.102 (1), 3–11. 10.1111/cge.14133
131
Itoh T. J. Hisanaga S. Hosoi T. Kishimoto T. Hotani H. (1997). Phosphorylation states of microtubule-associated protein 2 (MAP2) determine the regulatory role of MAP2 in microtubule dynamics. Biochemistry36 (41), 12574–12582. 10.1021/bi962606z
132
Iwasawa S. Yanagi K. Kikuchi A. Kobayashi Y. Haginoya K. Matsumoto H. et al (2019). Recurrent de novo MAPK8IP3 variants cause neurological phenotypes. Ann. Neurol.85 (6), 927–933. 10.1002/ana.25481
133
Jacquot G. Maidou-Peindara P. Benichou S. (2010). Molecular and functional basis for the scaffolding role of the p50/dynamitin subunit of the microtubule-associated dynactin complex. J. Biol. Chem.285 (30), 23019–23031. 10.1074/jbc.M110.100602
134
Ji X. Chang B. Naggert J. K. Nishina P. M. (2016). Lysosomal trafficking regulator (LYST). Adv. Exp. Med. Biol.854, 745–750. 10.1007/978-3-319-17121-0_99
135
Jijumon A. S. Bodakuntla S. Genova M. Bangera M. Sackett V. Besse L. et al (2022). Lysate-based pipeline to characterize microtubule-associated proteins uncovers unique microtubule behaviours. Nat. Cell Biol.24 (2), 253–267. 10.1038/s41556-021-00825-4
136
Jimenez-Sanchez M. Licitra F. Underwood B. R. Rubinsztein D. C. (2017). Huntington's disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb. Perspect. Med.7 (7), a024240. 10.1101/cshperspect.a024240
137
Joachim C. L. Morris J. H. Kosik K. S. Selkoe D. J. (1987). Tau antisera recognize neurofibrillary tangles in a range of neurodegenerative disorders. Ann. Neurol.22 (4), 514–520. 10.1002/ana.410220411
138
Johansson M. Rocha N. Zwart W. Jordens I. Janssen L. Kuijl C. et al (2007). Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor betalll spectrin. J. Cell Biol.176 (4), 459–471. 10.1083/jcb.200606077
139
Jongsma M. L. Bakker J. Cabukusta B. Liv N. van Elsland D. Fermie J. et al (2020). SKIP-HOPS recruits TBC1D15 for a Rab7-to-Arl8b identity switch to control late endosome transport. EMBO J.39 (6), e102301. 10.15252/embj.2019102301
140
Jongsma M. L. M. Bakker N. Neefjes J. (2023). Choreographing the motor-driven endosomal dance. J. Cell Sci.136 (5), jcs259689. 10.1242/jcs.259689
141
Jongsma M. L. M. Bakker N. Voortman L. M. Koning R. I. Bos E. Akkermans J. et al (2024). Systems mapping of bidirectional endosomal transport through the crowded cell. Curr. Biol.34 (19), 4476–4494.e11. 10.1016/j.cub.2024.08.026
142
Jordens I. Fernandez-Borja M. Marsman M. Dusseljee S. Janssen L. Calafat J. et al (2001). The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol.11 (21), 1680–1685. 10.1016/s0960-9822(01)00531-0
143
Kanai Y. Okada Y. Tanaka Y. Harada A. Terada S. Hirokawa N. (2000). KIF5C, a novel neuronal kinesin enriched in motor neurons. J. Neurosci.20 (17), 6374–6384. 10.1523/JNEUROSCI.20-17-06374.2000
144
Kang J. Lemaire H. G. Unterbeck A. Salbaum J. M. Masters C. L. Grzeschik K. H. et al (1987). The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature325 (6106), 733–736. 10.1038/325733a0
145
Karasmanis E. P. Reimer J. M. Kendrick A. A. Nguyen K. H. V. Rodriguez J. A. Truong J. B. et al (2023). Lis1 relieves cytoplasmic dynein-1 autoinhibition by acting as a molecular wedge. Nat. Struct. Mol. Biol.30 (9), 1357–1364. 10.1038/s41594-023-01069-6
146
Karim M. A. Nagle D. L. Kandil H. H. Burger J. Moore K. J. Spritz R. A. (1997). Mutations in the Chediak-Higashi syndrome gene (CHS1) indicate requirement for the complete 3801 amino acid CHS protein. Hum. Mol. Genet.6 (7), 1087–1089. 10.1093/hmg/6.7.1087
147
Karim M. A. Suzuki K. Fukai K. Oh J. Nagle D. L. Moore K. J. et al (2002). Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am. J. Med. Genet.108 (1), 16–22. 10.1002/ajmg.10184.abs
148
Keren-Kaplan T. Saric A. Ghosh S. Williamson C. D. Jia R. Li Y. et al (2022). RUFY3 and RUFY4 are ARL8 effectors that promote coupling of endolysosomes to dynein-dynactin. Nat. Commun.13 (1), 1506. 10.1038/s41467-022-28952-y
149
Khan A. O. Slater A. Maclachlan A. Nicolson P. L. R. Pike J. A. Reyat J. S. et al (2022). Post-translational polymodification of β1-tubulin regulates motor protein localisation in platelet production and function. Haematologica107 (1), 243–259. 10.3324/haematol.2020.270793
150
Kikuchi K. Sakamoto Y. Uezu A. Yamamoto H. Ishiguro K. I. Shimamura K. et al (2022). Map7D2 and Map7D1 facilitate microtubule stabilization through distinct mechanisms in neuronal cells. Life Sci. Alliance5 (8), e202201390. 10.26508/lsa.202201390
151
King S. J. Schroer T. A. (2000). Dynactin increases the processivity of the cytoplasmic dynein motor. Nat. Cell Biol.2 (1), 20–24. 10.1038/71338
152
Klebe S. Lossos A. Azzedine H. Mundwiller E. Sheffer R. Gaussen M. et al (2012). KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: distinct phenotypes according to the nature of the mutations. Eur. J. Hum. Genet.20 (6), 645–649. 10.1038/ejhg.2011.261
153
Konno T. Ross O. A. Teive H. A. G. Slawek J. Dickson D. W. Wszolek Z. K. (2017). DCTN1-related neurodegeneration: Perry syndrome and beyond. Park. Relat. Disord.41, 14–24. 10.1016/j.parkreldis.2017.06.004
154
Kosik K. S. Joachim C. L. Selkoe D. J. (1986). Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A.83 (11), 4044–4048. 10.1073/pnas.83.11.4044
155
Kremer H. van Wijk E. Marker T. Wolfrum U. Roepman R. (2006). Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum. Mol. Genet.15 (Spec No 2), R262–R270. 10.1093/hmg/ddl205
156
Krzewski K. Chen X. Orange J. S. Strominger J. L. (2006). Formation of a WIP-WASp-actin-and myosin IIA-containing multiprotein complex in activated NK cells and its alteration by KIR inhibitory signaling. J. Cell Biol.173 (1), 121–132. 10.1083/jcb.200509076
157
Kumar G. Chawla P. Dhiman N. Chadha S. Sharma S. Sethi K. et al (2022). RUFY3 links Arl8b and JIP4-Dynein complex to regulate lysosome size and positioning. Nat. Commun.13 (1), 1540. 10.1038/s41467-022-29077-y
158
Kumar R. Khan M. Francis V. Aguila A. Kulasekaran G. Banks E. et al (2024). DENND6A links Arl8b to a Rab34/RILP/dynein complex, regulating lysosomal positioning and autophagy. Nat. Commun.15 (1), 919. 10.1038/s41467-024-44957-1
159
Kunishima S. Saito H. (2010). Advances in the understanding of MYH9 disorders. Curr. Opin. Hematol.17 (5), 405–410. 10.1097/MOH.0b013e32833c069c
160
Kunishima S. Kojima T. Tanaka T. Kamiya T. Ozawa K. Nakamura Y. et al (1999). Mapping of a gene for May-Hegglin anomaly to chromosome 22q. Hum. Genet.105 (5), 379–383. 10.1007/s004390051119
161
Kurowska M. Goudin N. Nehme N. T. Court M. Garin J. Fischer A. et al (2012). Terminal transport of lytic granules to the immune synapse is mediated by the kinesin-1/Slp3/Rab27a complex. Blood119 (17), 3879–3889. 10.1182/blood-2011-09-382556
162
Kwok J. B. Teber E. T. Loy C. Hallupp M. Nicholson G. Mellick G. D. et al (2004). Tau haplotypes regulate transcription and are associated with Parkinson's disease. Ann. Neurol.55 (3), 329–334. 10.1002/ana.10826
163
Langlois S. Tarailo-Graovac M. Sayson B. Drogemoller B. Swenerton A. Ross C. J. et al (2016). De novo dominant variants affecting the motor domain of KIF1A are a cause of PEHO syndrome. Eur. J. Hum. Genet.24 (6), 949–953. 10.1038/ejhg.2015.217
164
Lawrence C. J. Dawe R. K. Christie K. R. Cleveland D. W. Dawson S. C. Endow S. A. et al (2004). A standardized kinesin nomenclature. J. Cell Biol.167 (1), 19–22. 10.1083/jcb.200408113
165
Laws S. M. Friedrich P. Diehl-Schmid J. Muller J. Eisele T. Bauml J. et al (2007). Fine mapping of the MAPT locus using quantitative trait analysis identifies possible causal variants in Alzheimer's disease. Mol. Psychiatry12 (5), 510–517. 10.1038/sj.mp.4001935
166
Lee J. R. Srour M. Kim D. Hamdan F. F. Lim S. H. Brunel-Guitton C. et al (2015). De novo mutations in the motor domain of KIF1A cause cognitive impairment, spastic paraparesis, axonal neuropathy, and cerebellar atrophy. Hum. Mutat.36 (1), 69–78. 10.1002/humu.22709
167
Li S. H. Li X. J. (2004). Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet.20 (3), 146–154. 10.1016/j.tig.2004.01.008
168
Li G. Marlin M. C. (2015). Rab family of GTPases. Methods Mol. Biol.1298, 1–15. 10.1007/978-1-4939-2569-8_1
169
Li S. H. Gutekunst C. A. Hersch S. M. Li X. J. (1998). Interaction of huntingtin-associated protein with dynactin P150Glued. J. Neurosci.18 (4), 1261–1269. 10.1523/JNEUROSCI.18-04-01261.1998
170
Li S. Finley J. Liu Z. J. Qiu S. H. Chen H. Luan C. H. et al (2002). Crystal structure of the cytoskeleton-associated protein glycine-rich (CAP-Gly) domain. J. Biol. Chem.277 (50), 48596–48601. 10.1074/jbc.M208512200
171
Li W. Zhang Q. Oiso N. Novak E. K. Gautam R. O'Brien E. P. et al (2003). Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat. Genet.35 (1), 84–89. 10.1038/ng1229
172
Lipka J. Kuijpers M. Jaworski J. Hoogenraad C. C. (2013). Mutations in cytoplasmic dynein and its regulators cause malformations of cortical development and neurodegenerative diseases. Biochem. Soc. Trans.41 (6), 1605–1612. 10.1042/BST20130188
173
Lise M. F. Wong T. P. Trinh A. Hines R. M. Liu L. Kang R. et al (2006). Involvement of myosin Vb in glutamate receptor trafficking. J. Biol. Chem.281 (6), 3669–3678. 10.1074/jbc.M511725200
174
Liu X. Vansant G. Udovichenko I. P. Wolfrum U. Williams D. S. (1997). Myosin VIIa, the product of the Usher 1B syndrome gene, is concentrated in the connecting cilia of photoreceptor cells. Cell Motil. Cytoskelet.37 (3), 240–252. 10.1002/(SICI)1097-0169(1997)37:3<240::AID-CM6>3.0.CO;2-A
175
Liu Y. T. Laura M. Hersheson J. Horga A. Jaunmuktane Z. Brandner S. et al (2014). Extended phenotypic spectrum of KIF5A mutations: from spastic paraplegia to axonal neuropathy. Neurology83 (7), 612–619. 10.1212/WNL.0000000000000691
176
Lu Y. M. Yan S. Ti S. C. Zheng C. (2024). Editing of endogenous tubulins reveals varying effects of tubulin posttranslational modifications on axonal growth and regeneration. Elife13. 10.7554/eLife.94583
177
Maier K. C. Godfrey J. E. Echeverri C. J. Cheong F. K. Schroer T. A. (2008). Dynamitin mutagenesis reveals protein-protein interactions important for dynactin structure. Traffic9 (4), 481–491. 10.1111/j.1600-0854.2008.00702.x
178
Mamo A. Jules F. Dumaresq-Doiron K. Costantino S. Lefrancois S. (2012). The role of ceroid lipofuscinosis neuronal protein 5 (CLN5) in endosomal sorting. Mol. Cell Biol.32 (10), 1855–1866. 10.1128/MCB.06726-11
179
Marcy E. Christine M. Mabel P. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell72 (6), 971–983. 10.1016/0092-8674(93)90585-e
180
Marks M. S. Heijnen H. F. Raposo G. (2013). Lysosome-related organelles: unusual compartments become mainstream. Curr. Opin. Cell Biol.25 (4), 495–505. 10.1016/j.ceb.2013.04.008
181
Marom R. Zhang B. Washington M. E. Song I. W. Burrage L. C. Rossi V. C. et al (2023). Dominant negative variants in KIF5B cause osteogenesis imperfecta via down regulation of mTOR signaling. PLoS Genet.19 (11), e1011005. 10.1371/journal.pgen.1011005
182
Martin E. R. Scott W. K. Nance M. A. Watts R. L. Hubble J. P. Koller W. C. et al (2001). Association of single-nucleotide polymorphisms of the tau gene with late-onset Parkinson disease. JAMA286 (18), 2245–2250. 10.1001/jama.286.18.2245
183
Marzo M. G. Griswold J. M. Ruff K. M. Buchmeier R. E. Fees C. P. Markus S. M. (2019). Molecular basis for dyneinopathies reveals insight into dynein regulation and dysfunction. Elife8, e47246. 10.7554/eLife.47246
184
Masson D. Kreis T. E. (1993). Identification and molecular characterization of E-MAP-115, a novel microtubule-associated protein predominantly expressed in epithelial cells. J. Cell Biol.123 (2), 357–371. 10.1083/jcb.123.2.357
185
Matsuda S. Yasukawa T. Homma Y. Ito Y. Niikura T. Hiraki T. et al (2001). c-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain-1 scaffolds Alzheimer's amyloid precursor protein with JNK. J. Neurosci.21 (17), 6597–6607. 10.1523/JNEUROSCI.21-17-06597.2001
186
Matsui T. Ohbayashi N. Fukuda M. (2012). The Rab interacting lysosomal protein (RILP) homology domain functions as a novel effector domain for small GTPase Rab36: Rab36 regulates retrograde melanosome transport in melanocytes. J. Biol. Chem.287 (34), 28619–28631. 10.1074/jbc.M112.370544
187
Matteoni R. Kreis T. E. (1987). Translocation and clustering of endosomes and lysosomes depends on microtubules. J. Cell Biol.105 (3), 1253–1265. 10.1083/jcb.105.3.1253
188
Maurer S. P. Fourniol F. J. Bohner G. Moores C. A. Surrey T. (2012). EBs recognize a nucleotide-dependent structural cap at growing microtubule ends. Cell149 (2), 371–382. 10.1016/j.cell.2012.02.049
189
Maurer S. P. Cade N. I. Bohner G. Gustafsson N. Boutant E. Surrey T. (2014). EB1 accelerates two conformational transitions important for microtubule maturation and dynamics. Curr. Biol.24 (4), 372–384. 10.1016/j.cub.2013.12.042
190
McGuire J. R. Rong J. Li S. H. Li X. J. (2006). Interaction of Huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J. Biol. Chem.281 (6), 3552–3559. 10.1074/jbc.M509806200
191
McKenna E. D. Sarbanes S. L. Cummings S. W. Roll-Mecak A. (2023). The tubulin code, from molecules to health and disease. Annu. Rev. Cell Dev. Biol.39, 331–361. 10.1146/annurev-cellbio-030123-032748
192
McKenney R. J. Huynh W. Tanenbaum M. E. Bhabha G. Vale R. D. (2014). Activation of cytoplasmic dynein motility by dynactin-cargo adapter complexes. Science345 (6194), 337–341. 10.1126/science.1254198
193
McVicker D. P. Chrin L. R. Berger C. L. (2011). The nucleotide-binding state of microtubules modulates kinesin processivity and the ability of Tau to inhibit kinesin-mediated transport. J. Biol. Chem.286 (50), 42873–42880. 10.1074/jbc.M111.292987
194
Meggouh F. Bienfait H. M. Weterman M. A. de Visser M. Baas F. (2006). Charcot-Marie-Tooth disease due to a de novo mutation of the RAB7 gene. Neurology67 (8), 1476–1478. 10.1212/01.wnl.0000240068.21499.f5
195
Mei S. Lin J. Liu Z. Li C. (2022). A novel mutation in the FYCO1 gene causing congenital cataract: case study of a Chinese family. Dis. Markers2022, 5838104. 10.1155/2022/5838104
196
Mei Y. Jiang Y. Zhang Z. Zhang H. (2023). Muscle and bone characteristics of a Chinese family with spinal muscular atrophy, lower extremity predominant 1 (SMALED1) caused by a novel missense DYNC1H1 mutation. BMC Med. Genomics16 (1), 47. 10.1186/s12920-023-01472-4
197
Melchionda S. Ahituv N. Bisceglia L. Sobe T. Glaser F. Rabionet R. et al (2001). MYO6, the human homologue of the gene responsible for deafness in Snell's waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am. J. Hum. Genet.69 (3), 635–640. 10.1086/323156
198
Menasche G. Pastural E. Feldmann J. Certain S. Ersoy F. Dupuis S. et al (2000). Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat. Genet.25 (2), 173–176. 10.1038/76024
199
Menasche G. Ho C. H. Sanal O. Feldmann J. Tezcan I. Ersoy F. et al (2003). Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J. Clin. Invest112 (3), 450–456. 10.1172/JCI18264
200
Merenlender-Wagner A. Shemer Z. Touloumi O. Lagoudaki R. Giladi E. Andrieux A. et al (2014). New horizons in schizophrenia treatment: autophagy protection is coupled with behavioral improvements in a mouse model of schizophrenia. Autophagy10 (12), 2324–2332. 10.4161/15548627.2014.984274
201
Michels S. Foss K. Park K. Golden-Grant K. Saneto R. Lopez J. et al (2017). Mutations of KIF5C cause a neurodevelopmental disorder of infantile-onset epilepsy, absent language, and distinctive malformations of cortical development. Am. J. Med. Genet. A173 (12), 3127–3131. 10.1002/ajmg.a.38496
202
Monfrini E. Cogiamanian F. Salani S. Straniero L. Fagiolari G. Garbellini M. et al (2021). A novel homozygous VPS11 variant may cause generalized dystonia. Ann. Neurol.89 (4), 834–839. 10.1002/ana.26021
203
Monroy B. Y. Sawyer D. L. Ackermann B. E. Borden M. M. Tan T. C. Ori-McKenney K. M. (2018). Competition between microtubule-associated proteins directs motor transport. Nat. Commun.9 (1), 1487. 10.1038/s41467-018-03909-2
204
Monroy B. Y. Tan T. C. Oclaman J. M. Han J. S. Simo S. Niwa S. et al (2020). A combinatorial MAP code dictates polarized microtubule transport. Dev. Cell53 (1), 60–72. 10.1016/j.devcel.2020.01.029
205
Morgan N. V. Pasha S. Johnson C. A. Ainsworth J. R. Eady R. A. Dawood B. et al (2006). A germline mutation in BLOC1S3/reduced pigmentation causes a novel variant of Hermansky-Pudlak syndrome (HPS8). Am. J. Hum. Genet.78 (1), 160–166. 10.1086/499338
206
Mori Y. Inoue Y. Tanaka S. Doda S. Yamanaka S. Fukuchi H. et al (2015b). Cep169, a novel microtubule plus-end-tracking centrosomal protein, binds to CDK5RAP2 and regulates microtubule stability. PLoS One10 (10), e0140968. 10.1371/journal.pone.0140968
207
Mori Y. Taniyama Y. Tanaka S. Fukuchi H. Terada Y. (2015c). Microtubule-bundling activity of the centrosomal protein, Cep169, and its binding to microtubules. Biochem. Biophys. Res. Commun.467 (4), 754–759. 10.1016/j.bbrc.2015.10.069
208
Morimoto M. Nicoli E. R. Kuptanon C. Roney J. C. Serra-Vinardell J. Sharma P. et al (2024). Spectrum of LYST mutations in Chediak-Higashi syndrome: a report of novel variants and a comprehensive review of the literature. J. Med. Genet.61 (3), 212–223. 10.1136/jmg-2023-109420
209
Moughamian A. J. Holzbaur E. L. (2012). Dynactin is required for transport initiation from the distal axon. Neuron74 (2), 331–343. 10.1016/j.neuron.2012.02.025
210
Moughamian A. J. Osborn G. E. Lazarus J. E. Maday S. Holzbaur E. L. (2013). Ordered recruitment of dynactin to the microtubule plus-end is required for efficient initiation of retrograde axonal transport. J. Neurosci.33 (32), 13190–13203. 10.1523/JNEUROSCI.0935-13.2013
211
Munch C. Sedlmeier R. Meyer T. Homberg V. Sperfeld A. D. Kurt A. et al (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology63 (4), 724–726. 10.1212/01.wnl.0000134608.83927.b1
212
Munch C. Rosenbohm A. Sperfeld A. D. Uttner I. Reske S. Krause B. J. et al (2005). Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann. Neurol.58 (5), 777–780. 10.1002/ana.20631
213
Myers A. J. Pittman A. M. Zhao A. S. Rohrer K. Kaleem M. Marlowe L. et al (2007). The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol. Dis.25 (3), 561–570. 10.1016/j.nbd.2006.10.018
214
Nakagawa H. Koyama K. Murata Y. Morito M. Akiyama T. Nakamura Y. (2000). EB3, a novel member of the EB1 family preferentially expressed in the central nervous system, binds to a CNS-specific APC homologue. Oncogene19 (2), 210–216. 10.1038/sj.onc.1203308
215
Nambiar R. McConnell R. E. Tyska M. J. (2010). Myosin motor function: the ins and outs of actin-based membrane protrusions. Cell Mol. Life Sci.67 (8), 1239–1254. 10.1007/s00018-009-0254-5
216
Nandini S. Conley Calderon J. L. Sabblah T. T. Love R. King L. E. King S. J. (2019). Mice with an autosomal dominant Charcot-Marie-Tooth type 2O disease mutation in both dynein alleles display severe moto-sensory phenotypes. Sci. Rep.9 (1), 11979. 10.1038/s41598-019-48431-7
217
Nangaku M. Sato-Yoshitake R. Okada Y. Noda Y. Takemura R. Yamazaki H. et al (1994). KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell79 (7), 1209–1220. 10.1016/0092-8674(94)90012-4
218
Naureckiene S. Sleat D. E. Lackland H. Fensom A. Vanier M. T. Wattiaux R. et al (2000). Identification of HE1 as the second gene of Niemann-Pick C disease. Science290 (5500), 2298–2301. 10.1126/science.290.5500.2298
219
Neveling K. Martinez-Carrera L. A. Holker I. Heister A. Verrips A. Hosseini-Barkooie S. M. et al (2013). Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy. Am. J. Hum. Genet.92 (6), 946–954. 10.1016/j.ajhg.2013.04.011
220
Niclas J. Navone F. Hom-Booher N. Vale R. D. (1994). Cloning and localization of a conventional kinesin motor expressed exclusively in neurons. Neuron12 (5), 1059–1072. 10.1016/0896-6273(94)90314-x
221
Nicolas A. Kenna K. P. Renton A. E. Ticozzi N. Faghri F. Chia R. et al (2018). Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron97 (6), 1267–1288. 10.1016/j.neuron.2018.02.027
222
Nishimura A. L. Mitne-Neto M. Silva H. C. Richieri-Costa A. Middleton S. Cascio D. et al (2004). A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet.75 (5), 822–831. 10.1086/425287
223
Niu Q. Wang X. Shi M. Jin Q. (2015). A novel DYNC1H1 mutation causing spinal muscular atrophy with lower extremity predominance. Neurol. Genet.1 (2), e20. 10.1212/NXG.0000000000000017
224
Nukina N. Ihara Y. (1986). One of the antigenic determinants of paired helical filaments is related to tau protein. J. Biochem.99 (5), 1541–1544. 10.1093/oxfordjournals.jbchem.a135625
225
Oates E. C. Rossor A. M. Hafezparast M. Gonzalez M. Speziani F. MacArthur D. G. et al (2013). Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. Am. J. Hum. Genet.92 (6), 965–973. 10.1016/j.ajhg.2013.04.018
226
Odronitz F. Kollmar M. (2007). Drawing the tree of eukaryotic life based on the analysis of 2,269 manually annotated myosins from 328 species. Genome Biol.8 (9), R196. 10.1186/gb-2007-8-9-r196
227
Oh J. Bailin T. Fukai K. Feng G. H. Ho L. Mao J. I. et al (1996). Positional cloning of a gene for Hermansky-Pudlak syndrome, a disorder of cytoplasmic organelles. Nat. Genet.14 (3), 300–306. 10.1038/ng1196-300
228
Ohbayashi N. Maruta Y. Ishida M. Fukuda M. (2012). Melanoregulin regulates retrograde melanosome transport through interaction with the RILP-p150Glued complex in melanocytes. J. Cell Sci.125 (Pt 6), 1508–1518. 10.1242/jcs.094185
229
Olenick M. A. Holzbaur E. L. F. (2019). Dynein activators and adaptors at a glance. J. Cell Sci.132 (6), jcs227132. 10.1242/jcs.227132
230
O'Roak B. J. Vives L. Fu W. Egertson J. D. Stanaway I. B. Phelps I. G. et al (2012). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science338 (6114), 1619–1622. 10.1126/science.1227764
231
Pankiv S. Alemu E. A. Brech A. Bruun J. A. Lamark T. Overvatn A. et al (2010). FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol.188 (2), 253–269. 10.1083/jcb.200907015
232
Park J. I. Lee J. E. Myung C. H. Jo C. S. Jang H. S. Hwang J. S. (2019). The absence of Rab27a accelerates the degradation of Melanophilin. Exp. Dermatol28 (1), 90–93. 10.1111/exd.13840
233
Paschal B. M. Obar R. A. Vallee R. B. (1989). Interaction of brain cytoplasmic dynein and MAP2 with a common sequence at the C terminus of tubulin. Nature342 (6249), 569–572. 10.1038/342569a0
234
Pasqualato S. Renault L. Cherfils J. (2002). Arf, Arl, Arp and Sar proteins: a family of GTP-binding proteins with a structural device for 'front-back' communication. EMBO Rep.3 (11), 1035–1041. 10.1093/embo-reports/kvf221
235
Pastural E. Barrat F. J. Dufourcq-Lagelouse R. Certain S. Sanal O. Jabado N. et al (1997). Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene. Nat. Genet.16 (3), 289–292. 10.1038/ng0797-289
236
Pastural E. Ersoy F. Yalman N. Wulffraat N. Grillo E. Ozkinay F. et al (2000). Two genes are responsible for Griscelli syndrome at the same 15q21 locus. Genomics63 (3), 299–306. 10.1006/geno.1999.6081
237
Peeters K. Litvinenko I. Asselbergh B. Almeida-Souza L. Chamova T. Geuens T. et al (2013). Molecular defects in the motor adaptor BICD2 cause proximal spinal muscular atrophy with autosomal-dominant inheritance. Am. J. Hum. Genet.92 (6), 955–964. 10.1016/j.ajhg.2013.04.013
238
Pennamen P. Le L. Tingaud-Sequeira A. Fiore M. Bauters A. Van Duong Beatrice N. et al (2020). BLOC1S5 pathogenic variants cause a new type of Hermansky-Pudlak syndrome. Genet. Med.22 (10), 1613–1622. 10.1038/s41436-020-0867-5
239
Perea-Cabrera M. Granados-Riveron J. T. Segura-Stanford B. Moreno-Vargas L. M. Prada-Gracia D. Moran-Espinosa M. C. et al (2023). Opitz GBBB syndrome with total anomalous pulmonary venous connection: a new MID1 gene variant. Mol. Genet. Genomic Med.11 (9), e2234. 10.1002/mgg3.2234
240
Perlson E. Maday S. Fu M. M. Moughamian A. J. Holzbaur E. L. (2010). Retrograde axonal transport: pathways to cell death?Trends Neurosci.33 (7), 335–344. 10.1016/j.tins.2010.03.006
241
Permana S. Hisanaga S. Nagatomo Y. Iida J. Hotani H. Itoh T. J. (2005). Truncation of the projection domain of MAP4 (microtubule-associated protein 4) leads to attenuation of microtubule dynamic instability. Cell Struct. Funct.29 (5-6), 147–157. 10.1247/csf.29.147
242
Picher-Martel V. Morin C. Brunet D. Dionne A. (2020). SMALED2 with BICD2 gene mutations: report of two cases and portrayal of a classical phenotype. Neuromuscul. Disord.30 (8), 669–673. 10.1016/j.nmd.2020.05.009
243
Pilling A. D. Horiuchi D. Lively C. M. Saxton W. M. (2006). Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell17 (4), 2057–2068. 10.1091/mbc.e05-06-0526
244
Poirier K. Lebrun N. Broix L. Tian G. Saillour Y. Boscheron C. et al (2013). Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet.45 (6), 639–647. 10.1038/ng.2613
245
Posor Y. Jang W. Haucke V. (2022). Phosphoinositides as membrane organizers. Nat. Rev. Mol. Cell Biol.23 (12), 797–816. 10.1038/s41580-022-00490-x
246
Puls I. Jonnakuty C. LaMonte B. H. Holzbaur E. L. Tokito M. Mann E. et al (2003). Mutant dynactin in motor neuron disease. Nat. Genet.33 (4), 455–456. 10.1038/ng1123
247
Quaderi N. A. Schweiger S. Gaudenz K. Franco B. Rugarli E. I. Berger W. et al (1997). Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat. Genet.17 (3), 285–291. 10.1038/ng1197-285
248
Raiborg C. Wenzel E. M. Pedersen N. M. Olsvik H. Schink K. O. Schultz S. W. et al (2015). Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature520 (7546), 234–238. 10.1038/nature14359
249
Ramocki M. B. Zoghbi H. Y. (2008). Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature455 (7215), 912–918. 10.1038/nature07457
250
Rao L. Gennerich A. (2024). Structure and function of dynein's non-catalytic subunits. Cells13 (4), 330. 10.3390/cells13040330
251
Raposo G. Marks M. S. Cutler D. F. (2007). Lysosome-related organelles: driving post-Golgi compartments into specialisation. Curr. Opin. Cell Biol.19 (4), 394–401. 10.1016/j.ceb.2007.05.001
252
Ravenscroft G. Di Donato N. Hahn G. Davis M. R. Craven P. D. Poke G. et al (2016). Recurrent de novo BICD2 mutation associated with arthrogryposis multiplex congenita and bilateral perisylvian polymicrogyria. Neuromuscul. Disord.26 (11), 744–748. 10.1016/j.nmd.2016.09.009
253
Reck-Peterson S. L. Redwine W. B. Vale R. D. Carter A. P. (2018). The cytoplasmic dynein transport machinery and its many cargoes. Nat. Rev. Mol. Cell Biol.19 (6), 382–398. 10.1038/s41580-018-0004-3
254
Reid E. Kloos M. Ashley-Koch A. Hughes L. Bevan S. Svenson I. K. et al (2002). A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am. J. Hum. Genet.71 (5), 1189–1194. 10.1086/344210
255
Reiner O. Carrozzo R. Shen Y. Wehnert M. Faustinella F. Dobyns W. B. et al (1993). Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature364 (6439), 717–721. 10.1038/364717a0
256
Richards T. A. Cavalier-Smith T. (2005). Myosin domain evolution and the primary divergence of eukaryotes. Nature436 (7054), 1113–1118. 10.1038/nature03949
257
Riviere J. B. Ramalingam S. Lavastre V. Shekarabi M. Holbert S. Lafontaine J. et al (2011). KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am. J. Hum. Genet.89 (2), 219–230. 10.1016/j.ajhg.2011.06.013
258
Roberts A. J. Kon T. Knight P. J. Sutoh K. Burgess S. A. (2013). Functions and mechanics of dynein motor proteins. Nat. Rev. Mol. Cell Biol.14 (11), 713–726. 10.1038/nrm3667
259
Robinson C. L. Evans R. D. Briggs D. A. Ramalho J. S. Hume A. N. (2017). Inefficient recruitment of kinesin-1 to melanosomes precludes it from facilitating their transport. J. Cell Sci.130 (12), 2056–2065. 10.1242/jcs.186064
260
Rocha N. Kuijl C. van der Kant R. Janssen L. Houben D. Janssen H. et al (2009). Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J. Cell Biol.185 (7), 1209–1225. 10.1083/jcb.200811005
261
Roder I. V. Petersen Y. Choi K. R. Witzemann V. Hammer J. A. 3rd Rudolf R. (2008). Role of Myosin Va in the plasticity of the vertebrate neuromuscular junction in vivo. PLoS One3 (12), e3871. 10.1371/journal.pone.0003871
262
Roder I. V. Choi K. R. Reischl M. Petersen Y. Diefenbacher M. E. Zaccolo M. et al (2010). Myosin Va cooperates with PKA RIalpha to mediate maintenance of the endplate in vivo. Proc. Natl. Acad. Sci. U. S. A.107 (5), 2031–2036. 10.1073/pnas.0914087107
263
Romano R. Del Fiore V. S. Saveri P. Palama I. E. Pisciotta C. Pareyson D. et al (2022). Autophagy and lysosomal functionality in CMT2B fibroblasts carrying the RAB7(K126R) mutation. Cells11 (3), 496. 10.3390/cells11030496
264
Rosa-Ferreira C. Munro S. (2011). Arl8 and SKIP act together to link lysosomes to kinesin-1. Dev. Cell21 (6), 1171–1178. 10.1016/j.devcel.2011.10.007
265
Russ C. Powell J. F. Zhao J. Baker M. Hutton M. Crawford F. et al (2001). The microtubule associated protein Tau gene and Alzheimer's disease--an association study and meta-analysis. Neurosci. Lett.314 (1-2), 92–96. 10.1016/s0304-3940(01)02289-3
266
Rydzanicz M. Jagla M. Kosinska J. Tomasik T. Sobczak A. Pollak A. et al (2017). KIF5A de novo mutation associated with myoclonic seizures and neonatal onset progressive leukoencephalopathy. Clin. Genet.91 (5), 769–773. 10.1111/cge.12831
267
Saffin J. M. Venoux M. Prigent C. Espeut J. Poulat F. Giorgi D. et al (2005). ASAP, a human microtubule-associated protein required for bipolar spindle assembly and cytokinesis. Proc. Natl. Acad. Sci. U. S. A.102 (32), 11302–11307. 10.1073/pnas.0500964102
268
Saleem R. S. Siddiqui S. N. Irshad S. Ashraf N. M. Hamid A. Khan M. A. U. et al (2022). Targeted gene sequencing of FYCO1 identified a novel mutation in a Pakistani family for autosomal recessive congenital cataract. Mol. Genet. Genomic Med.10 (8), e1985. 10.1002/mgg3.1985
269
Samora C. P. Mogessie B. Conway L. Ross J. L. Straube A. McAinsh A. D. (2011). MAP4 and CLASP1 operate as a safety mechanism to maintain a stable spindle position in mitosis. Nat. Cell Biol.13 (9), 1040–1050. 10.1038/ncb2297
270
Sanborn K. B. Mace E. M. Rak G. D. Difeo A. Martignetti J. A. Pecci A. et al (2011). Phosphorylation of the myosin IIA tailpiece regulates single myosin IIA molecule association with lytic granules to promote NK-cell cytotoxicity. Blood118 (22), 5862–5871. 10.1182/blood-2011-03-344846
271
Savukoski M. Klockars T. Holmberg V. Santavuori P. Lander E. S. Peltonen L. (1998). CLN5, a novel gene encoding a putative transmembrane protein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis. Nat. Genet.19 (3), 286–288. 10.1038/975
272
Scheinfeld M. H. Roncarati R. Vito P. Lopez P. A. Abdallah M. D'Adamio L. (2002). Jun NH2-terminal kinase (JNK) interacting protein 1 (JIP1) binds the cytoplasmic domain of the Alzheimer's beta-amyloid precursor protein (APP). J. Biol. Chem.277 (5), 3767–3775. 10.1074/jbc.M108357200
273
Schiavo G. Greensmith L. Hafezparast M. Fisher E. M. (2013). Cytoplasmic dynein heavy chain: the servant of many masters. Trends Neurosci.36 (11), 641–651. 10.1016/j.tins.2013.08.001
274
Schlager M. A. Hoang H. T. Urnavicius L. Bullock S. L. Carter A. P. (2014). In vitro reconstitution of a highly processive recombinant human dynein complex. EMBO J.33 (17), 1855–1868. 10.15252/embj.201488792
275
Schroer T. A. (2004). Dynactin. Annu. Rev. Cell Dev. Biol.20, 759–779. 10.1146/annurev.cellbio.20.012103.094623
276
Schweiger S. Schneider R. (2003). The MID1/PP2A complex: a key to the pathogenesis of Opitz BBB/G syndrome. Bioessays25 (4), 356–366. 10.1002/bies.10256
277
Schweiger S. Foerster J. Lehmann T. Suckow V. Muller Y. A. Walter G. et al (1999). The Opitz syndrome gene product, MID1, associates with microtubules. Proc. Natl. Acad. Sci. U. S. A.96 (6), 2794–2799. 10.1073/pnas.96.6.2794
278
Scoto M. Rossor A. M. Harms M. B. Cirak S. Calissano M. Robb S. et al (2015). Novel mutations expand the clinical spectrum of DYNC1H1-associated spinal muscular atrophy. Neurology84 (7), 668–679. 10.1212/WNL.0000000000001269
279
Sebe-Pedros A. Grau-Bove X. Richards T. A. Ruiz-Trillo I. (2014). Evolution and classification of myosins, a paneukaryotic whole-genome approach. Genome Biol. Evol.6 (2), 290–305. 10.1093/gbe/evu013
280
Semenova I. Ikeda K. Resaul K. Kraikivski P. Aguiar M. Gygi S. et al (2014). Regulation of microtubule-based transport by MAP4. Mol. Biol. Cell25 (20), 3119–3132. 10.1091/mbc.E14-01-0022
281
Seri M. Cusano R. Gangarossa S. Caridi G. Bordo D. Lo Nigro C. et al (2000). Mutations in MYH9 result in the may-hegglin anomaly, and fechtner and sebastian syndromes. The may-heggllin/fechtner syndrome consortium. Nat. Genet.26 (1), 103–105. 10.1038/79063
282
Shimizu H. Iwayama Y. Yamada K. Toyota T. Minabe Y. Nakamura K. et al (2006). Genetic and expression analyses of the STOP (MAP6) gene in schizophrenia. Schizophr. Res.84 (2-3), 244–252. 10.1016/j.schres.2006.03.017
283
Shirzadeh E. Piryaei F. Naddaf H. Barabadi Z. (2022). Two new variants in FYCO1 are responsible for autosomal recessive congenital cataract in Iranian population. Cell J.24 (9), 546–551. 10.22074/cellj.2022.8116
284
Singh K. Lau C. K. Manigrasso G. Gama J. B. Gassmann R. Carter A. P. (2024). Molecular mechanism of dynein-dynactin complex assembly by LIS1. Science383 (6690), eadk8544. 10.1126/science.adk8544
285
Sonar P. Youyen W. Cleetus A. Wisanpitayakorn P. Mousavi S. I. Stepp W. L. et al (2020). Kinesin-2 from C. Reinhardtii is an atypically fast and auto-inhibited motor that is activated by heterotrimerization for intraflagellar transport. Curr. Biol.30 (6), 1160–1166. 10.1016/j.cub.2020.01.046
286
Splinter D. Razafsky D. S. Schlager M. A. Serra-Marques A. Grigoriev I. Demmers J. et al (2012). BICD2, dynactin, and LIS1 cooperate in regulating dynein recruitment to cellular structures. Mol. Biol. Cell23 (21), 4226–4241. 10.1091/mbc.E12-03-0210
287
Steel D. Zech M. Zhao C. Barwick K. E. S. Burke D. Demailly D. et al (2020). Loss-of-Function variants in HOPS complex genes VPS16 and VPS41 cause early onset dystonia associated with lysosomal abnormalities. Ann. Neurol.88 (5), 867–877. 10.1002/ana.25879
288
Stenmark H. (2009). Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol.10 (8), 513–525. 10.1038/nrm2728
289
Stockmann M. Meyer-Ohlendorf M. Achberger K. Putz S. Demestre M. Yin H. et al (2013). The dynactin p150 subunit: cell biology studies of sequence changes found in ALS/MND and Parkinsonian syndromes. J. Neural Transm. (Vienna)120 (5), 785–798. 10.1007/s00702-012-0910-z
290
Straub R. E. Jiang Y. MacLean C. J. Ma Y. Webb B. T. Myakishev M. V. et al (2002). Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am. J. Hum. Genet.71 (2), 337–348. 10.1086/341750
291
Strickland A. V. Schabhuttl M. Offenbacher H. Synofzik M. Hauser N. S. Brunner-Krainz M. et al (2015). Mutation screen reveals novel variants and expands the phenotypes associated with DYNC1H1. J. Neurol.262 (9), 2124–2134. 10.1007/s00415-015-7727-2
292
Suarez-Merino B. Hubank M. Revesz T. Harkness W. Hayward R. Thompson D. et al (2005). Microarray analysis of pediatric ependymoma identifies a cluster of 112 candidate genes including four transcripts at 22q12.1-q13.3.Neuro Oncol.7 (1), 20–31. 10.1215/S1152851704000596
293
Sun X. Shi X. Liu M. Li D. Zhang L. Liu X. et al (2011). Mdp3 is a novel microtubule-binding protein that regulates microtubule assembly and stability. Cell Cycle10 (22), 3929–3937. 10.4161/cc.10.22.18106
294
Suzuki T. Li W. Zhang Q. Karim A. Novak E. K. Sviderskaya E. V. et al (2002). Hermansky-Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light-ear gene. Nat. Genet.30 (3), 321–324. 10.1038/ng835
295
Takamori S. Holt M. Stenius K. Lemke E. A. Gronborg M. Riedel D. et al (2006). Molecular anatomy of a trafficking organelle. Cell127 (4), 831–846. 10.1016/j.cell.2006.10.030
296
Tala S. X. Chen J. Zhang L. Liu N. Zhou J. et al (2014). Microtubule stabilization by Mdp3 is partially attributed to its modulation of HDAC6 in addition to its association with tubulin and microtubules. PLoS One9 (3), e90932. 10.1371/journal.pone.0090932
297
Tan R. Lam A. J. Tan T. Han J. Nowakowski D. W. Vershinin M. et al (2019). Microtubules gate tau condensation to spatially regulate microtubule functions. Nat. Cell Biol.21 (9), 1078–1085. 10.1038/s41556-019-0375-5
298
Tanahashi H. Asada T. Tabira T. (2004). Association between tau polymorphism and male early-onset alzheimer's disease. Neuroreport15 (1), 175–179. 10.1097/00001756-200401190-00034
299
Tanaka T. Serneo F. F. Higgins C. Gambello M. J. Wynshaw-Boris A. Gleeson J. G. (2004). Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J. Cell Biol.165 (5), 709–721. 10.1083/jcb.200309025
300
Thompson R. F. Langford G. M. (2002). Myosin superfamily evolutionary history. Anat. Rec.268 (3), 276–289. 10.1002/ar.10160
301
Tischfield M. A. Cederquist G. Y. Gupta M. L. Jr. Engle E. C. (2011). Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr. Opin. Genet. Dev.21 (3), 286–294. 10.1016/j.gde.2011.01.003
302
Tokuraku K. Noguchi T. Q. Nishie M. Matsushima K. Kotani S. (2007). An isoform of microtubule-associated protein 4 inhibits kinesin-driven microtubule gliding. J. Biochem.141 (4), 585–591. 10.1093/jb/mvm063
303
Tong H. Yang T. Xu S. Li X. Liu L. Zhou G. et al (2024). Huntington's disease: complex pathogenesis and therapeutic strategies. Int. J. Mol. Sci.25 (7), 3845. 10.3390/ijms25073845
304
Trockenbacher A. Suckow V. Foerster J. Winter J. Krauss S. Ropers H. H. et al (2001). MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat. Genet.29 (3), 287–294. 10.1038/ng762
305
Truant R. Atwal R. Burtnik A. (2006). Hypothesis: huntingtin may function in membrane association and vesicular trafficking. Biochem. Cell Biol.84 (6), 912–917. 10.1139/o06-181
306
Tsurusaki Y. Saitoh S. Tomizawa K. Sudo A. Asahina N. Shiraishi H. et al (2012). A DYNC1H1 mutation causes a dominant spinal muscular atrophy with lower extremity predominance. Neurogenetics13 (4), 327–332. 10.1007/s10048-012-0337-6
307
Tuli A. Thiery J. James A. M. Michelet X. Sharma M. Garg S. et al (2013). Arf-like GTPase Arl8b regulates lytic granule polarization and natural killer cell-mediated cytotoxicity. Mol. Biol. Cell24 (23), 3721–3735. 10.1091/mbc.E13-05-0259
308
Twelvetrees A. E. Yuen E. Y. Arancibia-Carcamo I. L. MacAskill A. F. Rostaing P. Lumb M. J. et al (2010). Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant huntingtin. Neuron65 (1), 53–65. 10.1016/j.neuron.2009.12.007
309
Tymanskyj S. R. Ma L. (2019). MAP7 prevents axonal branch retraction by creating a stable microtubule boundary to rescue polymerization. J. Neurosci.39 (36), 7118–7131. 10.1523/JNEUROSCI.0775-19.2019
310
Ullah M. I. Rehman Z. Dad R. Alsrhani A. Shakil M. Ghanem H. B. et al (2023). Identification and functional characterization of mutation in FYCO1 in families with congenital cataract. Life (Basel)13 (8), 1788. 10.3390/life13081788
311
Urnavicius L. Zhang K. Diamant A. G. Motz C. Schlager M. A. Yu M. et al (2015). The structure of the dynactin complex and its interaction with dynein. Science347 (6229), 1441–1446. 10.1126/science.aaa4080
312
Uusi-Rauva K. Kyttala A. van der Kant R. Vesa J. Tanhuanpaa K. Neefjes J. et al (2012). Neuronal ceroid lipofuscinosis protein CLN3 interacts with motor proteins and modifies location of late endosomal compartments. Cell Mol. Life Sci.69 (12), 2075–2089. 10.1007/s00018-011-0913-1
313
van Bon B. W. Hoischen A. Hehir-Kwa J. de Brouwer A. P. Ruivenkamp C. Gijsbers A. C. et al (2011). Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly. Clin. Genet.79 (3), 296–299. 10.1111/j.1399-0004.2010.01544.x
314
Van Den Bossche K. Naeyaert J. M. Lambert J. (2006). The quest for the mechanism of melanin transfer. Traffic7 (7), 769–778. 10.1111/j.1600-0854.2006.00425.x
315
van der Kant R. Fish A. Janssen L. Janssen H. Krom S. Ho N. et al (2013). Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L. J. Cell Sci.126 (Pt 15), 3462–3474. 10.1242/jcs.129270
316
Van Gele M. Dynoodt P. Lambert J. (2009). Griscelli syndrome: a model system to study vesicular trafficking. Pigment. Cell Melanoma Res.22 (3), 268–282. 10.1111/j.1755-148X.2009.00558.x
317
van Spronsen M. Mikhaylova M. Lipka J. Schlager M. A. van den Heuvel D. J. Kuijpers M. et al (2013). TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron77 (3), 485–502. 10.1016/j.neuron.2012.11.027
318
Vardarajan B. N. Bruesegem S. Y. Harbour M. E. Inzelberg R. Friedland R. St George-Hyslop P. et al (2012). Identification of Alzheimer disease-associated variants in genes that regulate retromer function. Neurobiol. Aging33 (9), 2231 e15–2231. 10.1016/j.neurobiolaging.2012.04.020
319
Verhey K. J. Hammond J. W. (2009). Traffic control: regulation of kinesin motors. Nat. Rev. Mol. Cell Biol.10 (11), 765–777. 10.1038/nrm2782
320
Verhoeven K. De Jonghe P. Coen K. Verpoorten N. Auer-Grumbach M. Kwon J. M. et al (2003). Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet.72 (3), 722–727. 10.1086/367847
321
Vershinin M. Carter B. C. Razafsky D. S. King S. J. Gross S. P. (2007). Multiple-motor based transport and its regulation by Tau. Proc. Natl. Acad. Sci. U. S. A.104 (1), 87–92. 10.1073/pnas.0607919104
322
Vershinin M. Xu J. Razafsky D. S. King S. J. Gross S. P. (2008). Tuning microtubule-based transport through filamentous MAPs: the problem of dynein. Traffic9 (6), 882–892. 10.1111/j.1600-0854.2008.00741.x
323
Vilarino-Guell C. Wider C. Ross O. A. Dachsel J. C. Kachergus J. M. Lincoln S. J. et al (2011). VPS35 mutations in Parkinson disease. Am. J. Hum. Genet.89 (1), 162–167. 10.1016/j.ajhg.2011.06.001
324
Vissers L. E. de Ligt J. Gilissen C. Janssen I. Steehouwer M. de Vries P. et al (2010). A de novo paradigm for mental retardation. Nat. Genet.42 (12), 1109–1112. 10.1038/ng.712
325
Walsh T. Walsh V. Vreugde S. Hertzano R. Shahin H. Haika S. et al (2002). From flies' eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc. Natl. Acad. Sci. U. S. A.99 (11), 7518–7523. 10.1073/pnas.102091699
326
Walsh C. A. Morrow E. M. Rubenstein J. L. (2008). Autism and brain development. Cell135 (3), 396–400. 10.1016/j.cell.2008.10.015
327
Wang A. Liang Y. Fridell R. A. Probst F. J. Wilcox E. R. Touchman J. W. et al (1998). Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science280 (5368), 1447–1451. 10.1126/science.280.5368.1447
328
Wang X. Han C. Liu W. Wang P. Zhang X. (2014). A novel RAB7 mutation in a Chinese family with Charcot-Marie-Tooth type 2B disease. Gene534 (2), 431–434. 10.1016/j.gene.2013.10.023
329
Wang J. Zhang Q. Chen Y. Yu S. Wu X. Bao X. (2019). Rett and Rett-like syndrome: expanding the genetic spectrum to KIF1A and GRIN1 gene. Mol. Genet. Genomic Med.7 (11), e968. 10.1002/mgg3.968
330
Wang Y. Zhou X. Zhu H. Liu S. Zhou C. Zhang G. et al (2005). Overexpression of EB1 in human esophageal squamous cell carcinoma (ESCC) may promote cellular growth by activating beta-catenin/TCF pathway.Oncogene24 (44), 6637–6645. 10.1038/sj.onc.1208819
331
Wang S. Huang J. Li C. Zhao L. Wong C. C. Zhai J. et al (2020). MAP9 loss triggers chromosomal instability, initiates colorectal tumorigenesis, and is associated with poor survival of patients with colorectal cancer. Clin. Cancer Res.26 (3), 746–757. 10.1158/1078-0432.CCR-19-1611
332
Waterman-Storer C. M. Karki S. Holzbaur E. L. (1995). The p150Glued component of the dynactin complex binds to both microtubules and the actin-related protein centractin (Arp-1). Proc. Natl. Acad. Sci. U. S. A.92 (5), 1634–1638. 10.1073/pnas.92.5.1634
333
Watson P. Stephens D. J. (2006). Microtubule plus-end loading of p150(Glued) is mediated by EB1 and CLIP-170 but is not required for intracellular membrane traffic in mammalian cells. J. Cell Sci.119 (Pt 13), 2758–2767. 10.1242/jcs.02999
334
Weedon M. N. Hastings R. Caswell R. Xie W. Paszkiewicz K. Antoniadi T. et al (2011). Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet.89 (2), 308–312. 10.1016/j.ajhg.2011.07.002
335
Weil D. Blanchard S. Kaplan J. Guilford P. Gibson F. Walsh J. et al (1995). Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature374 (6517), 60–61. 10.1038/374060a0
336
Wells A. L. Lin A. W. Chen L. Q. Safer D. Cain S. M. Hasson T. et al (1999). Myosin VI is an actin-based motor that moves backwards. Nature401 (6752), 505–508. 10.1038/46835
337
Wildenberg S. C. Oetting W. S. Almodovar C. Krumwiede M. White J. G. King R. A. (1995). A gene causing Hermansky-Pudlak syndrome in a Puerto Rican population maps to chromosome 10q2. Am. J. Hum. Genet.57 (4), 755–765.
338
Wilkie G. S. Davis I. (2001). Drosophila wingless and pair-rule transcripts localize apically by dynein-mediated transport of RNA particles. Cell105 (2), 209–219. 10.1016/s0092-8674(01)00312-9
339
Willemsen M. H. Vissers L. E. Willemsen M. A. van Bon B. W. Kroes T. de Ligt J. et al (2012). Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J. Med. Genet.49 (3), 179–183. 10.1136/jmedgenet-2011-100542
340
Wood J. G. Mirra S. S. Pollock N. J. Binder L. I. (1986). Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc. Natl. Acad. Sci. U. S. A.83 (11), 4040–4043. 10.1073/pnas.83.11.4040
341
Wu X. S. Tsan G. L. Hammer J. A. 3rd (2005). Melanophilin and myosin Va track the microtubule plus end on EB1. J. Cell Biol.171 (2), 201–207. 10.1083/jcb.200503028
342
Wu X. Sakamoto T. Zhang F. Sellers J. R. Hammer J. A. 3rd (2006). In vitro reconstitution of a transport complex containing Rab27a, melanophilin and myosin Va. FEBS Lett.580 (25), 5863–5868. 10.1016/j.febslet.2006.09.047
343
Xia Q. Wang H. Hao Z. Fu C. Hu Q. Gao F. et al (2016). TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J.35 (2), 121–142. 10.15252/embj.201591998
344
Xu B. Ionita-Laza I. Roos J. L. Boone B. Woodrick S. Sun Y. et al (2012). De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat. Genet.44 (12), 1365–1369. 10.1038/ng.2446
345
Yadav S. Verma P. J. Panda D. (2014). C-terminal region of MAP7 domain containing protein 3 (MAP7D3) promotes microtubule polymerization by binding at the C-terminal tail of tubulin. PLoS One9 (6), e99539. 10.1371/journal.pone.0099539
346
Yamazaki H. Nakata T. Okada Y. Hirokawa N. (1995). KIF3A/B: a heterodimeric kinesin superfamily protein that works as a microtubule plus end-directed motor for membrane organelle transport. J. Cell Biol.130 (6), 1387–1399. 10.1083/jcb.130.6.1387
347
Yang C. Wu J. de Heus C. Grigoriev I. Liv N. Yao Y. et al (2017). EB1 and EB3 regulate microtubule minus end organization and Golgi morphology. J. Cell Biol.216 (10), 3179–3198. 10.1083/jcb.201701024
348
Zadro C. Alemanno M. S. Bellacchio E. Ficarella R. Donaudy F. Melchionda S. et al (2009). Are MYO1C and MYO1F associated with hearing loss?Biochim. Biophys. Acta1792 (1), 27–32. 10.1016/j.bbadis.2008.10.017
349
Zhao C. Takita J. Tanaka Y. Setou M. Nakagawa T. Takeda S. et al (2001). Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell105 (5), 587–597. 10.1016/s0092-8674(01)00363-4
350
Zhong F. J. Li Y. M. Xu C. Sun B. Wang J. L. Yang L. Y. (2021). EB2 promotes hepatocellular carcinoma proliferation and metastasis via MAPK/ERK pathway by modulating microtubule dynamics. Clin. Sci. (Lond).135 (7), 847–864. 10.1042/CS20201500
351
Zimprich A. Benet-Pages A. Struhal W. Graf E. Eck S. H. Offman M. N. et al (2011). A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet.89 (1), 168–175. 10.1016/j.ajhg.2011.06.008
352
Zimprich A. (2011). Genetics of Parkinson's disease and essential tremor. Curr. Opin. Neurol.24 (4), 318–323. 10.1097/WCO.0b013e3283484b87
Summary
Keywords
lysosomes, lysosome-related organelles (LROs), transport, kinesin, dynein, microtubules, disease
Citation
Bakker N, Jongsma MLM and Neefjes J (2025) Engine breakdown of lysosomes and related organelles and the resulting physiology. Front. Cell Dev. Biol. 13:1575571. doi: 10.3389/fcell.2025.1575571
Received
12 February 2025
Accepted
29 May 2025
Published
16 June 2025
Volume
13 - 2025
Edited by
Cédric Delevoye, INSERM U1151 Institut Necker Enfants Malades, France
Reviewed by
Laura Elizabeth Swan, University of Liverpool, United Kingdom
Silvia Jansen, Washington University in St. Louis, United States
Updates
Copyright
© 2025 Bakker, Jongsma and Neefjes.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacques Neefjes, j.j.c.neefjes@lumc.nl
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.