 Zhaohua Cai
Zhaohua Cai Ben He
Ben He- Department of Cardiology, Shanghai Chest Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
1 Introduction
Arteriomegaly refers to a diffuse arterial ectasia, which may manifest independently or concomitantly with aneurysmal pathology. This complex vasculopathy is characterized by pronounced dilation, excessive tortuosity, segmental elongation, and luminal irregularities accompanied by hemodynamic flow deceleration. While initially conceptualized by Leriche in 1942 using the descriptive term “dolicho et mega artere” (denoting elongated and enlarged arteries) (R, 1942), the entity remained poorly characterized until Thomas described the detailed angiographic findings and established the current nomenclature arteriomegaly in 1971 (Thomas, 1971). Pathoanatomically, arteriomegaly diverges from classical aneurysms, which are strictly defined as focal, persistent dilatations exceeding 1.5 times the reference arterial diameter. Emerging evidence reveals significant pathophysiological correlations between arteriomegaly and aneurysm development, particularly in the abdominal aorta and peripheral arterial systems, suggesting shared mechanisms underlying both pathological processes.
Clinical investigations have increasingly demonstrated the coexistence of systemic arteriomegaly in abdominal aortic aneurysm (AAA) pathogenesis. Tilson et al. initially documented concurrent suprarenal aortic and iliac arterial dilatation in AAA cohorts (Tilson and Dang, 1981), a finding later expanded by Baxter et al. who identified pan-aortic dilation (encompassing ascending/descending thoracic, supraceliac, and suprarenal segments) in patients with infrarenal AAA (Baxter et al., 1994). Notably, the arterial remodeling extends beyond the aortic territory in AAA patients. Comparative ultrasonographic analyses by Ward et al. revealed significant diameter increases in carotid, brachial, femoral, and popliteal arteries among AAA patients versus controls (Ward, 1992). This pattern of peripheral vascular involvement is further corroborated by multiple studies demonstrating statistically significant common carotid enlargement in AAA patients (Makita et al., 2000; Iwamoto et al., 2004; van Laake et al., 2005; Nordon et al., 2009). Furthermore, generalized arteriomegaly frequently coexists with multisegmental aneurysms in popliteal artery aneurysm cases (Yamamoto et al., 2002; Widmer et al., 2008). These multisystem manifestations collectively redefine aneurysmal disease as a pan-vascular disorder rather than localized pathology.
The progressive nature of arteriomegaly is underscored by its strong temporal association with multifocal aneurysm development (Thomas, 1971; Carlson et al., 1975; Hollier et al., 1983; Chan and Thomas, 1990; Barandiaran et al., 2012). Current epidemiological data reveal that 30.3%–80.6% of arteriomegaly patients develop multiple or diffuse aneurysms in arteriomegalic populations (Table 1), with longitudinal studies demonstrating critical progression patterns (Thomas, 1971; Carlson et al., 1975; Hollier et al., 1983; Chan and Thomas, 1990; Barandiaran et al., 2012). A pivotal 76-month cohort study documented baseline aneurysm prevalence of 53.7% at diagnosis, escalating to 80.6% during surveillance through de novo formation across distinct arterial territories. This characteristic pattern of temporal-spatial progression—marked by sequential aneurysm development in previously unaffected vascular regions—provides compelling evidence for redefining arteriomegaly as a progressive pan-arteriopathy (Barandiaran et al., 2012). Such insights fundamentally alter therapeutic paradigms, necessitating a shift from localized, lesion-specific interventions to systemic strategies targeting the underlying pan-vascular pathophysiology.
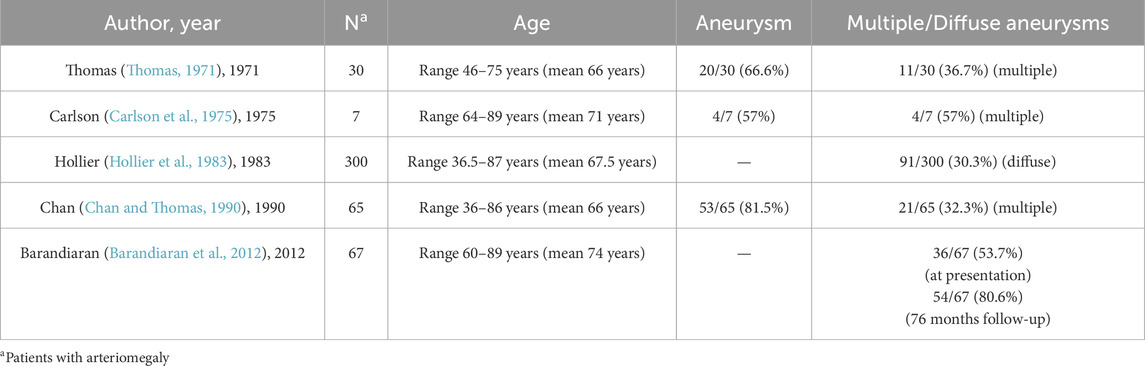
Table 1. Demographic characteristics and aneurysm incidence in published cases of patients with arteriomegaly.
Nevertheless, the precise etiopathogenetic mechanisms governing the co-evolution of generalized arteriomegaly and focal aneurysm formation remain elusive, constrained by two research limitations. First, the current experimental paradigm suffers from a critical absence of genetically faithful animal models spontaneously recapitulating the full spectrum of human arteriomegaly-aneurysm progression. Second, human translational studies are fundamentally limited to histopathological analysis of end-stage surgical specimens, which by their terminal nature inherently obscure inciting molecular events. This dual barrier impedes delineation of molecular drivers underlying macroscopic vascular changes, underscoring the urgent need for preclinical models that recapitulate generalized arteriomegaly and aneurysm co-evolution.
2 Discovery of the murine model of generalized arteriomegaly and aortic/arterial aneurysm formation
Our laboratory engineered a tamoxifen-inducible smooth muscle cell (SMC)-specific Lkb1 knockout mouse model through Myh11-CreERT2-mediated recombination in Lkb1flox/flox mice (Cai et al., 2024). Remarkably, Lkb1flox/flox;Myh11-CreERT2 mice invariably succumbed to fatal aortic/arterial rupture within 8 months post-tamoxifen induction (Cai et al., 2024). Serial in vivo micro-ultrasound imaging over 4.5 months demonstrated progressive lumen expansion in ascending aorta, abdominal aorta, and carotid arteries in Lkb1flox/flox;Myh11-CreERT2 mice versus control mice, establishing this model as manifesting generalized arteriomegaly (Cai et al., 2024). Systematic histomorphometric analysis of aortic segments and peripheral arteries in Lkb1flox/flox;Myh11-CreERT2 mice revealed concordant pathological dilation patterns. Specifically, hematoxylin-eosin staining quantitatively confirmed progressive luminal enlargement in carotid arteries, femoral arteries, and aortic regions, consistent precisely with ultrasound metrics (Cai et al., 2024). Notably, as time developed, Lkb1flox/flox;Myh11-CreERT2 mice exhibited aneurysm formation in abdominal aorta, renal artery, iliac artery, femoral artery, and/or popliteal artery leading to arterial rupture (Cai et al., 2024), which is highly reminiscent of human aneurysm of the aorta-iliac-femoral tree (Norman and Powell, 2010).
In our subsequent multimodal imaging investigation, we implemented longitudinal computed tomography angiography (CTA) and high-resolution magnetic resonance imaging (MRI) to delineate the spatiotemporal progression of aneurysmal disease in the Lkb1flox/flox;Myh11-CreERT2 model (Cai et al., 2025). Quantitative imaging protocols revealed several distinct pathological phases in Lkb1flox/flox;Myh11-CreERT2 mice: 1) systemic arteriomegaly with significant increases in cross-sectional diameters across aortic segments and multiple medium arteries (including left common carotid artery, celiac trunk, renal artery, common iliac artery, and femoral artery), 2) multiterritorial aneurysm development involving renal, iliac, caudal, and femoral arteries and abdominal aortas, and 3) terminal rupture events. This comprehensive phenotyping establishes that postnatal Lkb1 ablation in mature vascular smooth muscle cell (VSMC) induces a progressive vascular pathology: initial generalized arteriomegaly progressing to multifocal aneurysm formation, ultimately culminating in fatal rupture, recapitulating key aspects of human multiterritorial aneurysmal diseases. The anatomical distribution (aorto-iliac-femoral predominance) and temporal progression pattern (diffuse dilation preceding focal aneurysms) mirror clinical observations in aneurysmal patients.
Notably, conditional Lkb1 ablation in mature VSMC using the tamoxifen-inducible Myh11-CreERT2 system initiated a sequential phenotypic transition characterized by progressive transdifferentiation. Initially, contractile VSMCs adopted an early modulated phenotype, followed by their transformation into fibroblast-like, chondrocyte-like (cartilage-forming), and ultimately osteocyte-like (calcification-prone) cells (Cai et al., 2024). Furthermore, Lkb1 deficiency triggered profound extracellular matrix (ECM) remodeling, including excessive deposition of collagen-rich and proteoglycan-rich matrix, as well as elastic lamina fragmentation and disruption (Cai et al., 2024). Collectively, these findings delineate a causal cascade wherein Lkb1 loss drives VSMC lineage reprogramming, which in turn precipitates ECM compositional and structural dysregulation. This multistep pathological cascade ultimately compromises vascular wall integrity, serving as a pivotal driver of arteriomegaly and aneurysm pathogenesis.
3 Clinical implications
3.1 Redefining arteriomegaly-aneurysm continuum
The Lkb1flox/flox;Myh11-CreERT2 murine model recapitulates a biphasic disease continuum: (1) an initial systemic pan-arterial ectasia phase marked by diffuse vascular dilatation, progressing to (2) focal aneurysm formation—a pathological trajectory that precisely mirrors the clinical progression from arteriomegaly to aneurysm formation in humans. While no existing murine models fully replicate this continuum, certain genetically engineered strains exhibit analogous vascular dysfunction (Quelquejay et al., 2024; Arévalo et al., 2023). Our findings resolve a longstanding nosological debate by demonstrating that generalized arteriomegaly and focal aneurysms represent either distinct phenotypic manifestations (systemic vs. localized) or sequential stages of a unified arteriopathy characterized by progressive loss of vascular wall integrity. This pathochronic relationship is corroborated by clinical epidemiology showing arteriomegaly typically manifests decades prior to aneurysmal presentation (Hollier et al., 1983). Importantly, longitudinal clinical studies by Barandiaran et al. revealed that 58% of arteriomegaly patients without baseline aneurysms developed multifocal lesions across the aorto-iliac-femoral axis during 76-month median follow-up (Barandiaran et al., 2012). These collective findings establish AAA as a representative manifestation of systemic arterial wall dysregulation rather than an isolated vascular pathology.
3.2 Mechanistic insight into aneurysm drivers
The Lkb1flox/flox;Myh11-CreERT2 murine model reveals an intrinsic cell-autonomous mechanism governing aneurysmal pathogenesis. Our findings establish that sequential VSMC transformation and maladaptive extracellular matrix (ECM) remodeling constitute the principal pathogenic drivers of generalized arteriomegaly and loss of vessel structural integrity. The initiation of VSMC transformation triggers ECM remodeling characterized by excessive collagen deposition, proteoglycan accumulation, and elastic lamina degradation, creating a feedforward loop that collectively compromises vascular wall integrity. The pathophysiological progression in Lkb1flox/flox;Myh11-CreERT2 model aligns with clinical observations of panvascular ECM abnormalities in AAA patients (Baxter et al., 1994; van Laake et al., 2005). While prior research emphasizes matrix metalloproteinase (MMP)-mediated proteolysis in the pathogenesis of aneurysm formation (Baxter et al., 1994), our findings position VSMC phenotypic determination as the upstream regulator initiating ECM dysregulation. This mechanistic paradigm reorients therapeutic strategies to preserving VSMC contractile identity, offering novel intervention opportunities during early disease progression.
3.3 Genetic contribution to aortic aneurysm
Despite the absence of definitively identified genetic loci in clinical cohorts to date, accumulating evidence underscores a pronounced familial predisposition to arteriomegaly and aneurysmal disease (Lawrence et al., 1998). These observations strongly suggest an underlying heritable component, even as the precise molecular drivers remain unresolved. Our recent study (Cai et al., 2024) provides mechanistic insights into this phenomenon, proposing that genetic mutations disrupting VSMC identity and homeostasis constitute a pivotal pathogenic axis in generalized arteriomegaly and aneurysm formation. Specifically, we hypothesize that VSMC transformation or “loss of fate” compromises arterial wall integrity, rendering vessels susceptible to pathological dilatation and aneurysm formation. This model aligns with the systemic nature of arteriomegaly, implicating pan-vascular defects in VSMC biology rather than localized environmental stressors. While candidate genes have yet to be validated in human AAA populations, our findings bridge clinical epidemiology (familial clustering) and cellular pathophysiology, advocating for a paradigm where inherited or de novo genetic alterations disrupt vascular homeostasis through impaired VSMC-dependent remodeling processes.
4 Conclusion and perspectives
Generalized arteriomegaly and localized aneurysm formation represent sequential pathological stages within a unified vascular disease continuum, both stemming from systemic dysregulation of arterial wall homeostasis. The tamoxifen-inducible Lkb1flox/flox;Myh11-CreERT2 murine model emerges as a superior preclinical platform for investigating this disease progression, as it faithfully recapitulates both the anatomical characteristics and pathophysiological mechanisms observed in human aneurysm formation. This genetically engineered model system provides unprecedented opportunities for early intervention strategy development through three unique capacities: 1) Spatiotemporal recapitulation of human disease progression—from diffuse arteriomegaly to aortic/iliac aneurysm rupture, 2) Identifying critical molecular pathways governing VSMC cell fate and vascular matrix remodeling, and 3) Enabling stage-specific therapeutic testing during the pre-aneurysmal window. By bridging molecular insights with clinical progression patterns, this strategic transition from reactive aneurysm repair to early intervention during the arteriomegaly phase provides innovative approaches to prevent progressive vascular damage.
Author contributions
ZC: Conceptualization, Funding acquisition, Writing – original draft. BH: Conceptualization, Funding acquisition, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (Grant Number: 82300489, 82130012, and 81830010), the Shanghai Pujiang Program (No. 23PJD084), and the Nurture projects for basic research of Shanghai Chest Hospital (Grant Number: 2022YNJCQ03).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Arévalo, M. M., Ritsvall, O., Bastrup, J. A., Celik, S., Jakobsson, G., Daoud, F., et al. (2023). Vascular smooth muscle-specific YAP/TAZ deletion triggers aneurysm development in mouse aorta. JCI Insight 8, e170845. doi:10.1172/jci.insight.170845
Barandiaran, J. V., Hall, T. C., Glaves, I., El-Barghouti, N., and Perry, E. P. (2012). An observational study into the management of arteriomegaly: a call for a revised classification system. Ann. R. Coll. Surg. Engl. 94, 250–255. doi:10.1308/003588412X13171221498505
Baxter, B. T., Davis, V. A., Minion, D. J., Wang, Y. P., Lynch, T. G., and McManus, B. M. (1994). Abdominal aortic aneurysms are associated with altered matrix proteins of the nonaneurysmal aortic segments. J. Vasc. Surg. 19, 797–803. ; discussion 803. doi:10.1016/s0741-5214(94)70004-4
Cai, Z., Fang, L., Wei, Z., Liang, M., Jiang, Y., Huang, Y., et al. (2025). Abdominal aortic aneurysm is a systemic and generalized arteriomegaly in mice and humans. Sci. Rep. 15, 8477. doi:10.1038/s41598-025-92599-0
Cai, Z., Satyanarayana, G., Song, P., Zhao, F., You, S., Liu, Z., et al. (2024). Regulation of Ptbp1-controlled alternative splicing of pyruvate kinase muscle by liver kinase B1 governs vascular smooth muscle cell plasticity in vivo. Cardiovasc Res. 120, 1780–1793. doi:10.1093/cvr/cvae187
Carlson, D. H., Gryska, P., Seletz, J., and Armstrong, S. (1975). Arteriomegaly. Am. J. Roentgenol. radium Ther. Nucl. Med. 125, 553–558. doi:10.2214/ajr.125.3.553
Chan, O., and Thomas, M. L. (1990). The incidence of popliteal aneurysms in patients with arteriomegaly. Clin. Radiol. 41, 185–189. doi:10.1016/s0009-9260(05)80965-1
Hollier, L. H., Stanson, A. W., Gloviczki, P., Pairolero, P. C., Joyce, J. W., Bernatz, P. E., et al. (1983). Arteriomegaly: classification and morbid implications of diffuse aneurysmal disease. Surgery 93, 700–708.
Iwamoto, T., Kimura, A., Nakai, T., Kanaya, K., and Ishimaru, S. (2004). Implications of carotid arteriomegaly in patients with aortic aneurysm. J. Atheroscler. thrombosis 11, 348–353. doi:10.5551/jat.11.348
Lawrence, P. F., Wallis, C., Dobrin, P. B., Bhirangi, K., Gugliuzza, N., Galt, S., et al. (1998). Peripheral aneurysms and arteriomegaly: is there a familial pattern? J. Vasc. Surg. 28, 599–605. doi:10.1016/s0741-5214(98)70082-5
Makita, S., Ohira, A., Tachieda, R., Itoh, S., Moriai, Y., Niinuma, H., et al. (2000). Dilation and reduced distensibility of carotid artery in patients with abdominal aortic aneurysms. Am. heart J. 140, 297–302. doi:10.1067/mhj.2000.108000
Nordon, I., Brar, R., Taylor, J., Hinchliffe, R., Loftus, I. M., and Thompson, M. M. (2009). Evidence from cross-sectional imaging indicates abdominal but not thoracic aortic aneurysms are local manifestations of a systemic dilating diathesis. J. Vasc. Surg. 50, 171–176 e1. doi:10.1016/j.jvs.2009.03.007
Norman, P. E., and Powell, J. T. (2010). Site specificity of aneurysmal disease. Circulation 121, 560–568. doi:10.1161/CIRCULATIONAHA.109.880724
Quelquejay, H., Al-Rifai, R., Silvestro, M., Vandestienne, M., Ferreira, I., Mirault, T., et al. (2024). L-Wnk1 deletion in smooth muscle cells causes aortitis and inflammatory shift. Circ. Res. 135, 488–502. doi:10.1161/CIRCRESAHA.124.324366
R, L. (1942). Dilatation pathologique des artères endehors des anéurysmes vie tissulaire des artères. Prèsse Med. 50, 641–642.
Tilson, M. D., and Dang, C. (1981). Generalized arteriomegaly. A possible predisposition to the formation of abdominal aortic aneurysms. Arch. Surg. 116, 1030–1032. doi:10.1001/archsurg.1981.01380200038007
van Laake, L. W., Vainas, T., Dammers, R., Kitslaar, P. J., Hoeks, A. P., and Schurink, G. W. (2005). Systemic dilation diathesis in patients with abdominal aortic aneurysms: a role for matrix metalloproteinase-9? Eur. J. Vasc. endovascular Surg. official J. Eur. Soc. Vasc. Surg. 29, 371–377. doi:10.1016/j.ejvs.2005.01.009
Ward, A. S. (1992). Aortic aneurysmal disease. A generalized dilating diathesis. Arch. Surg. 127, 990–991. doi:10.1001/archsurg.1992.01420080124021
Widmer, M. K., Blatter, S., Schmidli, J., Baumgartner, I., Gahl, B., Carrel, T., et al. (2008). Generalized dilating diathesis in patients with popliteal arterial aneurysm. VASA Z. fur Gefasskrankh. 37, 157–163. doi:10.1024/0301-1526.37.2.157
Keywords: abdominal aortic aneurysm, arteriomegaly, dilation, LKB1 (STK11), mouse model
Citation: Cai Z and He B (2025) Generalized arteriomegaly as a precursor to multifocal aneurysmal disease: implications for early intervention. Front. Cell Dev. Biol. 13:1592225. doi: 10.3389/fcell.2025.1592225
Received: 12 March 2025; Accepted: 13 May 2025;
Published: 20 May 2025.
Edited by:
Zhaoyu Liu, Sun Yat-sen Memorial Hospital, ChinaReviewed by:
Fujie Zhao, Emory University, United StatesCopyright © 2025 Cai and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhaohua Cai, emNhaTVAeWFob28uY29t; Ben He, aGViZW5Ac2hjaGVzdC5vcmc=