 Meilin Ji
Meilin Ji Yaopeng Guo3†
Yaopeng Guo3† Qingshi Chen
Qingshi Chen- 1The Second Clinical Medical College, Fujian Medical University, Quanzhou, China
- 2The Second Affiliated Hospital of Fujian Medical University, Quanzhou, China
- 3Department of Endocrinology and Metabolism, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, China
Obstructive sleep apnea (OSA) is a sleep-related respiratory disorder. Although recent studies have shown that OSA may be an alterable risk factor for metabolic syndrome (MS), the precise mechanism remains unknown. This study was designed with the purpose of identifying differentially expressed microRNAs (DEmiRs) in OSA-induced brown adipose tissue (BAT) injury. In this study, mouse models of chronic intermittent hypoxia (CIH)-related BAT injury were established using APOE mice. The microRNAs (miRNAs) expression profiles of the CIH-caused BAT injury were analyzed by the miRNA sequencing technology. The miRNA-seq data were analyzed using Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. An analysis of real-time quantitative PCR (RT-qPCR) confirmed the presence of several typical miRNAs. Ultimately, we constructed a network to illustrate the correlation between the miRNAs and target genes. In the CIH-induced BAT damage mouse models, 7 miRNAs experienced an upregulation, and 16 miRNAs underwent a downregulation. Six DEmiRs were confirmed using RT-qPCR. Additionally, GO and KEGG analyses were adopted to annotate the potential biological role of miRNAs. As a final step, we construct a miRNA–mRNA network for predicting miRNAs target genes. In conclusion, we first discovered that OSA-induced BAT dysfunction is associated with abnormal miRNA expression. This study exhibited a novel understanding of the potential molecular mechanism of OSA-related MS.
Introduction
As a highly prevalent disorder, obstructive sleep apnea (OSA) is defined by the repeated narrowing or closure of the airway during sleep. The airway collapse leads to increased chronic intermittent hypoxia (CIH), sleep fragmentation, and negative intrathoracic pressure. Considerable evidence has been proved to support an independent correlation of OSA with comorbidities including metabolic dysfunction (Almendros et al., 2022), atherosclerotic disease (Sapiña-Beltrán et al., 2022), and pulmonary hypertension (Sharma et al., 2021). Multiple experimental studies have proved that OSA is closely correlated with metabolic syndrome (MS) (Cui et al., 2018; Dahan et al., 2022; Huang et al., 2022). While these clinical links are well-documented, the search for biological mediators has identified brown adipose tissue (BAT)-a key thermogenic organ involved in energy expenditure-as a potential target for OSA-related metabolic dysfunction. Brown adipocytes are considered an effective target for treating OSA-related metabolic disorders (Dong et al., 2018). Brown adipocytes uniquely express uncoupling protein 1 (UCP1), enabling nonshivering thermogenesis through mitochondrial uncoupling. This process consumes glucose and lipids, making BAT a critical regulator of systemic metabolism. Dysfunctional BAT is implicated in insulin resistance and dyslipidemia---hallmarks of metabolic syndrome (Dong et al., 2018; Saito and Okamatsu-Ogura, 2023). Thus, elucidating BAT pathology in OSA may reveal actionable therapeutic targets. Despite this potential, the molecular mechanisms linking OSA-induced hypoxia to BAT dysfunction remain poorly characterized. In particular, the role of epigenomic regulators like microRNAs (miRNAs) in this context is entirely unexplored.
miRNAs are small non-coding RNAs that fine-tune gene expression by targeting mRNAs for degradation or translational repression. Critically, miRNAs such as miR-669a-5p directly regulate UCP1 expression and adipocyte browning (Tan et al., 2022). Hypoxia-responsive miRNAs (e.g., miR-210) further modulate metabolic adaptation in adipose tissue (Caca et al., 2025). The importance of miRNAs has been noted in prior studies, which get involved in innumerable biological processes, such as cell proliferation, differentiation, apoptosis, and tumorigenesis (Naqvi et al., 2022; Tealdi et al., 2022; Zeng et al., 2022; Zhu et al., 2023). Previous studies have certificated that miRNAs also play an essential part in BAT as transcriptional regulators and biomarkers (Goody and Pfeifer, 2019). For instance, miR-34C-5P was proven to regulate DPYSL4 expression and have an influence on insulin β-cells, the inflammatory response and glucose oxidative catabolism (Qiu et al., 2023). Elevated levels of miR-210, induced by both hypoxia and adrenergic stimulation, played a key role in regulating the differentiation and thermogenic function of brown adipocytes (Caca et al., 2025). Nevertheless, no studies have investigated miRNA networks in OSA-mediated BAT dysfunction.
This study pioneers the profiling of miRNA alterations in BAT during OSA progression. Using an ApoE mouse model of CIH, we integrated miRNA sequencing, bioinformatics, and experimental validation to identify dysregulated miRNAs and their mechanistic roles in BAT dysfunction. Finally, a DemiRs and target mRNA regulatory network was construed. Our study may pave a novel strategy for understanding molecular mechanisms of OSA-associated MS.
Materials and methods
Animals
APOE-deficient mice were chosen for their heightened susceptibility to metabolic dysfunction, including insulin resistance and dyslipidemia, which mirrors OSA-associated metabolic syndrome in humans. Male APOE mice were obtained from Guangdong Yaokang Biological Technology Co., Ltd. Approval was granted to all experiments by the Institutional Animal Care and Use Committee of the Second Affiliated Hospital of Fujian Medical University.
Chronic intermittent hypoxia system
The CIH protocol was adapted from established models of OSA (Chen et al., 2025; Zhang et al., 2025). APOE male mice were assigned to CIH or normal in random and exposed for 8 weeks in an environmental chamber applying a gas control system. To replicate CIH that experienced by patients with OSA, for 8 weeks, mice in the CIH group were kept in a device that produced 60 hypoxic episodes each hour (40 s of room air exposure followed by 20 s of 5% oxygen) for 8 hours each day. The normal group was placed in the same apparatus, and regular air was supplied to the apparatus.
miRNAs-seq and bioinformatics analysis
To obtain miRNAs and mRNA expression profiles, total RNA extraction from BAT was carried out. BAT samples were homogenized in 1 mL TRI Reagent. Phase separation was achieved by adding 200 μL chloroform, followed by centrifugation for 15 min at 4°C. The aqueous phase was transferred, and RNA was precipitated with 500 μL isopropanol, washed twice with 75% ethanol, and dissolved in RNase-free water. RNA concentration and purity were verified via NanoDrop. RNA integrity was assessed using Agilent Bioanalyzer 2100, and only samples with an RNA Integrity Number (RIN) >7.0 were used for sequencing. Total RNA (1 μg) was reverse-transcribed into cDNA using the PrimeScript™ RT Reagent Kit with gDNA Eraser, following the manufacturer’s protocol. Reactions included: 37°C for 15 min, 85°C for 5 s, and 4°C hold. The small RNA library was established with the aid of the NEB Multiplex Small RNA Library Prep Set for Illumina, according to the manufacturer’s instructions. Differentially expressed genes and DEmiRs were detected by software edgeR (v3.40.2) with thresholds: |log2FC| > 1.0 and adjusted p value <0.05,and then were visualized on a volcano plot. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses on the parental genes of the miRNAs were conducted to analyze the functions of the DEmiRs. Targeted mRNAs of the DEmiRs were analyzed using the GO and KEGG databases to show the vital functions and pathways.
Real-time quantitative PCR assay (RT-qPCR)
The extraction of all RNA from BAT was performed using TRI Reagent (Sigma: T9424) in accordance with the manufacturer’s protocol and reverse transcription was undertaken using a QuantStudio5 Real-time PCR System. RT-qPCR was performed using 2×PCR master mix (Arraystar: AS-MR-006-5) on a QuantStudio5 System (Applied Biosystems). Reaction conditions: 95°C for 10 min; 40 cycles of 95°C for 10 s and 60°C for 60 s. Each reaction contained 5 μL 2×Master Mix, 1.0 μL primers (10 μM), 2 μL cDNA, and 2.0 μL nuclease-free water. For RT-qPCR, data were normalized to the endogenous control U6 snRNA using the 2−ΔΔCT method. Each experiment was carried out three times. The primer sequences can be seen in Table 1.
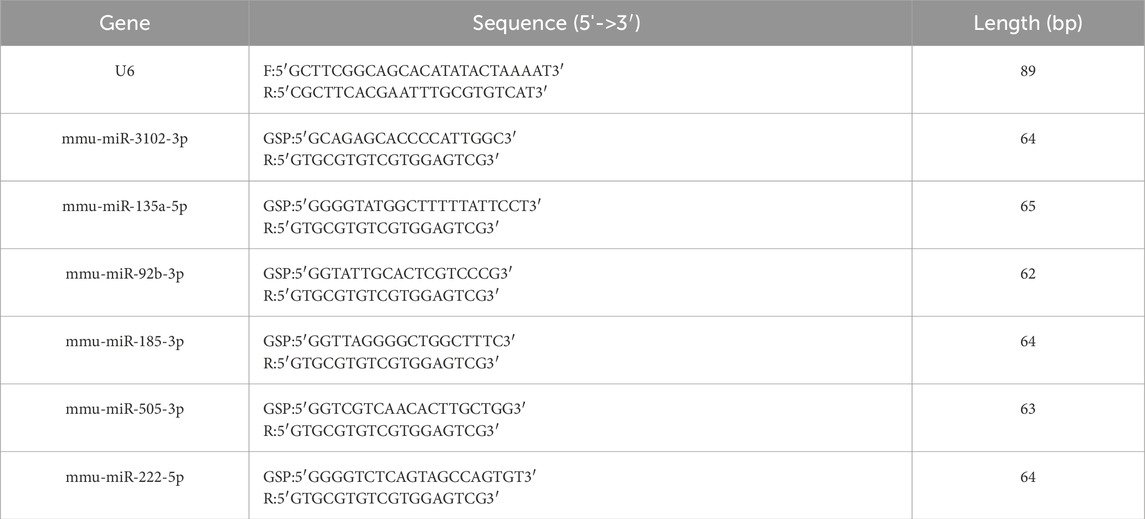
Table 1. RT-qPCR primers.
miRNAs targets prediction
Remarkably enriched KEGG pathways (P < 0.05) were identified for the predicted target genes using the Kyoto Encyclopedia of Genes and Genome. Besides, combining the predicted target genes with the use of miRDB (v6.0) and Targeted Scan (v7.0) and the DEmiRs for these miRNAs, a core miRNA target network was built by using the Cytoscape software (v3.9.1).
Statistical analysis
Every figure was expressed as the mean ± standard deviation. Unpaired Student’s t-tests were used to compare CIH groups with normal groups. In order to qualify as a notable difference, the P value had to be less than 0.05. Meanwhile, we used FDR (False Discovery Rate) correction via the Benjamini–Hochberg method to account for multiple testing. Differential expression was defined as adjusted p-value <0.05 and |log2FC| > 1.0.
Results
Differentially expressed miRNAs
A total of 23 DEmiRs were found in the BAT of the CIH group compared with the normal group. In CIH-induced BAT injury models, 7 miRNAs were significantly upregulated (e.g., mmu-miR-3102-3p: log2FC = 4.171, adjusted p value = 0.010), and 16 miRNAs were downregulated (e.g., mmu-miR-185-3p: log2FC = −3.145, adjusted p value = 0.029). A comprehensive list of all differentially expressed miRNAs was provided in Supplementary Table S1. Results of cluster analysis and volcano plot analysis of all DEmiRs were displayed respectively in Figures 1A,B. The gene expression variation between the CIH and control group is presented in Supplementary Figure S1A. The chromosomal distribution of these differentially expressed genes (DEGs) is shown in Supplementary Figure S1B.
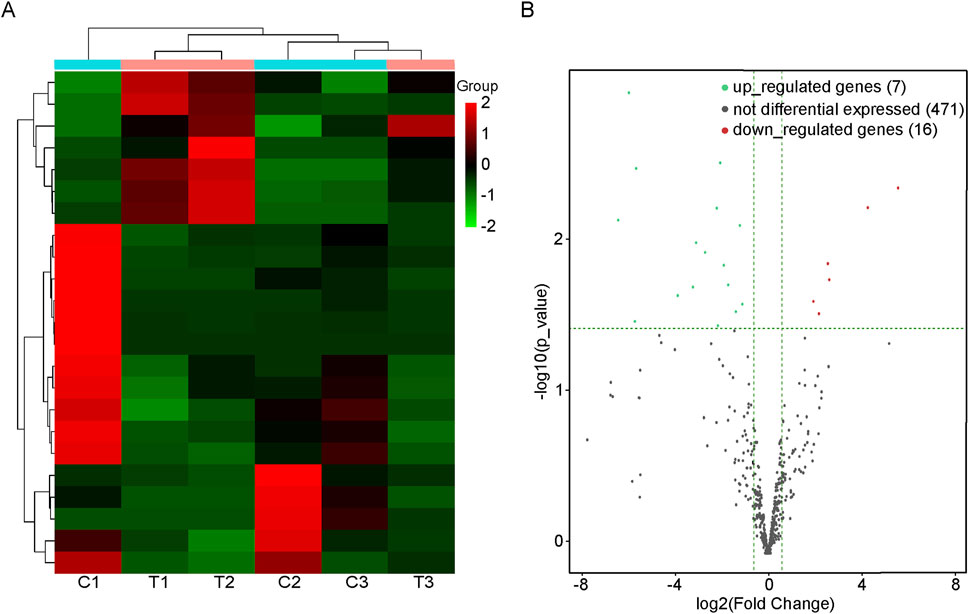
Figure 1. DEmiRs in BAT of mice with or without OSA. (A) Heat map of the DEmiRs in mice with or without OSA. C1, C2, and C3 were control groups; T1, T2, and T3 were OSA groups. (B) The volcano plot reveals the DEmiRs between the OSA mice and the control mice. Those dots that are red represent miRNAs that are upregulated, while those that are green represent miRNAs that are downregulated.
Enrichment analysis for the target genes of demiRs
To gain more information about the potential mechanisms of DEmiRs, GO and KEGG pathway enrichment analyses were conducted on target genes of all 23 DEmiRs. Results of the GO analysis of DEmiRs target genes are displayed in Figure 2. In order to analyze predicted genes, the corresponding GO annotations were examined, which include information about cellular components (CC), biological processes (BP), and molecular functions (MF). The analysis demonstrated that these genes were implicated in metabolic process, such as regulation of cellular metabolic process, regulation of nitrogen compound metabolic process, organic substance metabolic process, and regulation of RNA metabolic process. KEGG Pathway analysis was used to identify enriched pathways based on representative profiles of miRNA targeted genes. The analysis revealed that some of these genes were involved in signal transduction pathways (Figure 3), including focal adhesion kinase, FoxO signaling pathway, Sulfur metabolism, Cholinergic synapse and Choline metabolism in cancer.
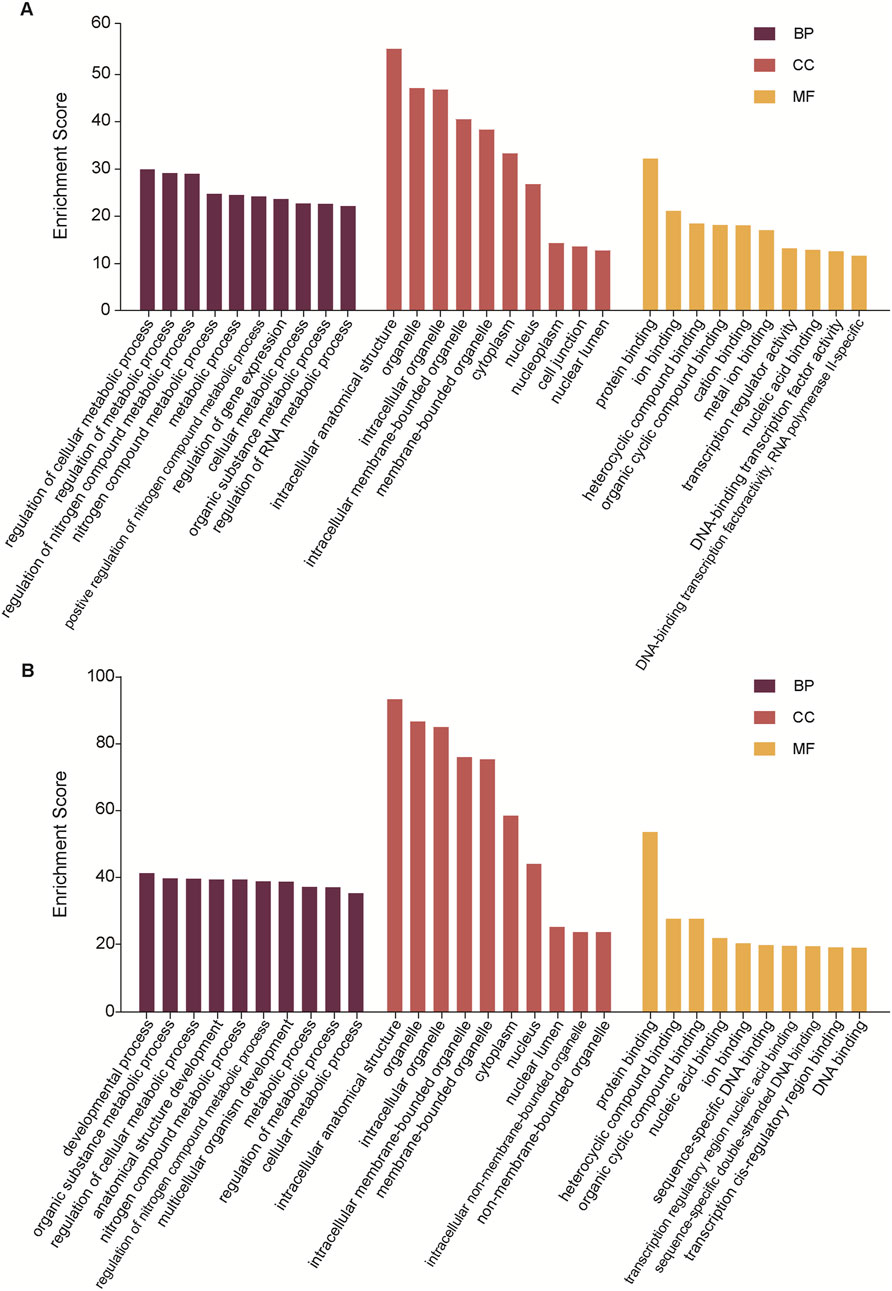
Figure 2. GO analysis. (A) The GO terms for the DemiRs that are upregulated. (B) The GO terms for the DemiRs that are downregulated.
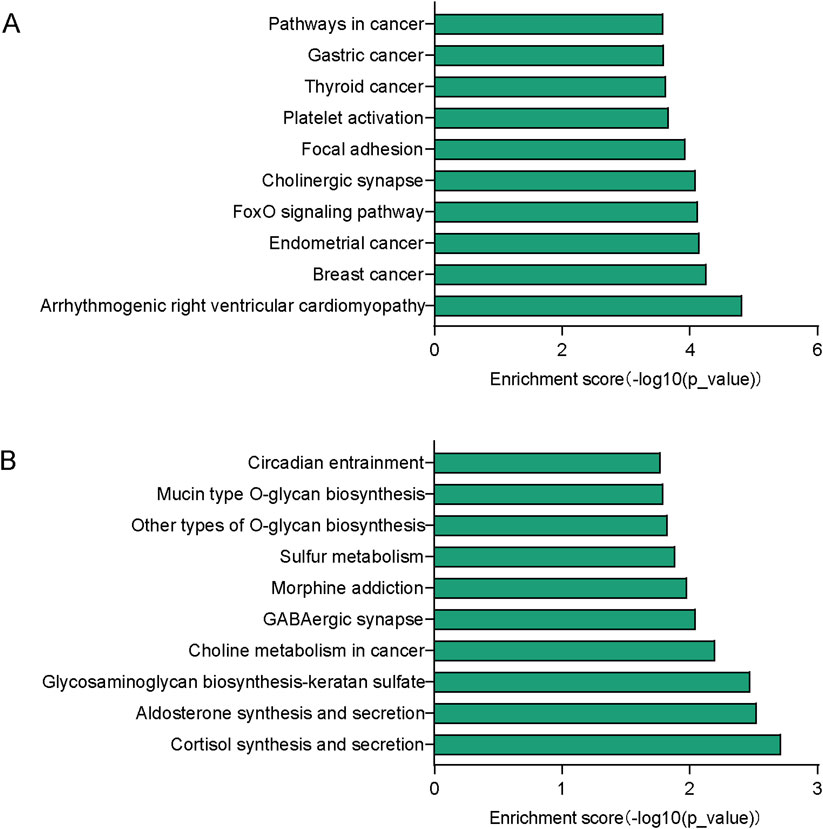
Figure 3. KEGG pathway analysis. (A) Top 10 significant pathways associated with upregulated genes. (B) Top 10 significant pathways related to downregulated genes.
RT-qPCR validation of DEmiRs
Then the accurateness and reliability of the sequencing analysis were validated using RT-qPCR. Based on their magnitude of expression change, established roles in metabolic pathways, and practical constraints, we chose three upregulated miRNAs (mmu-miR-3102-3p, mmu-miR-135a-5p, mmu-miR-92b-3p) and three downregulated miRNAs (mmu-miR-185-3p, mmu-miR-505-3p, mmu-miR-222-5p). As shown in Figure 4, expression profiles of these selected miRNAs show a similar trend with the results from our RNA-Seq analysis, which means the reliability of our miRNAs-seq data was high.
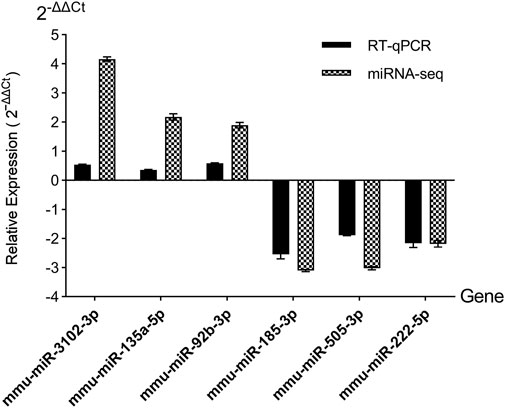
Figure 4. RT-qPCR validation of candidate DEmiRs. miRNAs-seq, miRNAs sequencing. Relative expression was calculated using the 2−ΔΔCT method, normalized to U6 snRNA.
miRNA/mRNA interaction network analysis
Based on RT-qPCR validation and relevance to OSA-associated metabolic syndrome, five miRNAs were selected for miRNA-mRNA network analysis (Figure 5). The integrated miRNA-mRNA network was built based on the dependence of these miRNAs on their targeted genes. This network provides a comprehensive understanding of the interaction between miRNAs and their targeted genes. By revealing their targeted mRNAs, we can investigate the fundamental mechanism of these DemiRs.
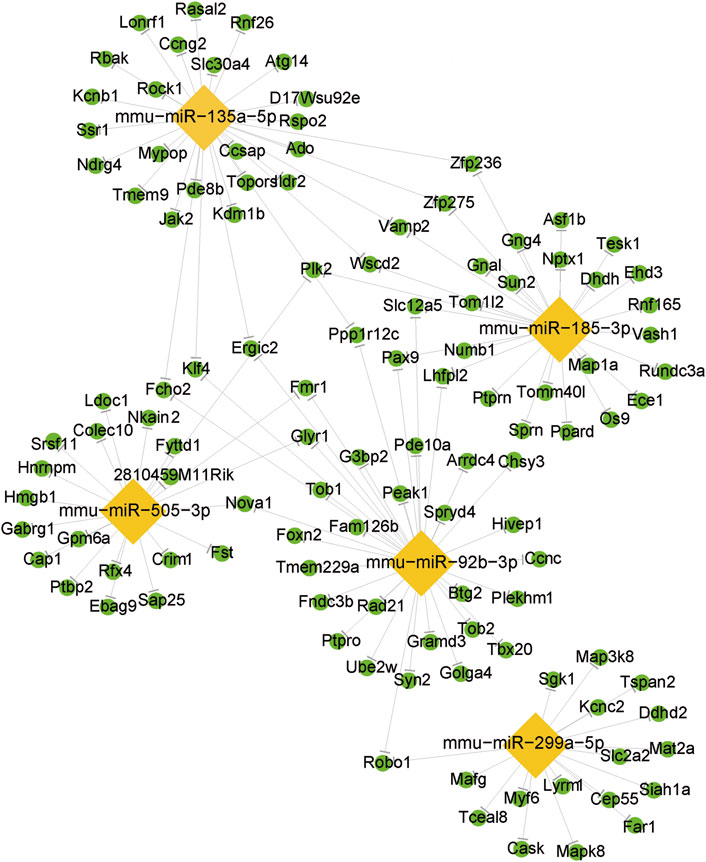
Figure 5. The differentially expressed miRNA/mRNA interaction network. The lozenge represents the DEmiRs. The circle represents the target mRNAs.
Discussion
More and more studies indicate that there exist a complicated and bidirectional association between OSA and MS (Framnes and Arble, 2018; Karuga et al., 2023; Martins and Conde, 2021). OSA leads to and exacerbates metabolic disorders, and cardiovascular dysfunction. CIH, the hallmark feature of OSA, triggers cyclic oxygen deprivation/reoxygenation, directly inducing BAT dysfunction through three key mechanisms: insulin resistance, sympathetic activation, or inflammation. It was reported that CIH-driven hepatic inflammation, mitochondrial dysfunction, and oxidative stress critically contributed to the development of MS associated with OSA (Fernandes et al., 2023). MS is also considered an emerging major risk factor for the development of OSA. According to a report, the incidence of OSA is high among those with MS, as well as MS prevalence among those with OSA, which ranges from 60% to 70% (Giampá et al., 2023). Short-chain fatty acids from gut microbiota might influence MS in OSA via immune system regulation, as suggested by studies linking gut microbiota to OSA-associated hypertension (Zhang et al., 2022). This feature can trigger intestinal oxidative stress and inflammation, ultimately culminating in OSA-related MS (Shobatake et al., 2022). However, the definite underlying mechanisms of OSA-related MS remain incompletely understood.
The discovery of miRNAs in diseases has opened new vistas for researchers in recent years. In addition to post-transcriptional regulation of gene expression, miRNAs have a range of physiological effects (Zhang et al., 2020). Furthermore, miRNAs are being identified as crucial regulators of numerous pathological and physiological processes and have been reported to have an impact on various diseases. However, the precise contribution of miRNAs to the pathogenesis of OSA-related MS remains unclear. Thus, we constructed CIH-induced BAT injury models to uncover the underlying mechanisms of miRNAs in OSA-related MS.
The aim of this study was to analyze the abnormal miRNAs profiles using RNA-seq in the CIH mouse models. The findings of this study revealed that a total of 23 DEmiRs were identified and selected. Of them, 7 miRNAs were upregulated. In addition to the upregulated miRNAs, we obtained 16 downregulated miRNAs. Notably, downregulated miR-505-3p, previously linked to macrophage dysfunction in hypercholesterolemia (Escate et al., 2018), may exacerbate inflammation in OSA-related BAT injury. Conversely, upregulated miR-135a-5p, which targets insulin signaling pathways (Teichenne et al., 2015), could drive metabolic dysregulation. These findings extend prior work on OSA-gut microbiota-metabolism interactions (Xue et al., 2021) by implicating BAT-specific miRNA networks. The RT-qPCR results were in accord with the miRNAs sequencing data. We also discovered that a diverse range of biological and pathological processes have also been linked to these identified miRNAs in this study. For example, the presence of miR-16-2-3p in diabetes is linked to coronary microvascular dysfunction by controlling the breakdown of fatty acids in endothelial cells (Liu et al., 2024). Nevertheless, further investigation of these DEmiRs is needed to understand the role they play in the progression of CIH-related MS.
It was necessary to identify potential target genes for these DEmiRs and to determine likely signaling pathways. Our research revealed that these DEmiRs may potentially regulate 107 pathways. For the upregulated DEmiRs, the arrhythmogenic right ventricular cardiomyopathy pathway was the most enriched one, which indicates that OSA has a great impact on cardiovascular disease (CVD). Recent reports have stated that MS is a clustering of metabolic complications that increase the risk of CVD (Kuckuck et al., 2024). Among the downregulated DemiRs cortisol synthesis and secretion pathway was the most enriched. Previous studies showed that repeated physiological stress such as hypoxia and fragmentation of sleep can affect cortisol secretion (Mohammadi et al., 2020). Meanwhile, the development of MS may be influenced by stress and its related hormones, such as cortisol, the glucocorticoid hormone (Kuckuck et al., 2024). However, the precise underlying mechanism between OSA and MS remains uncertain.
Then we identified the crucial miRNAs and mRNAs based on the quantity of their neighbors or connections. By conducting an intricate investigation of the miRNA-mRNA network, we found that a single miRNA exerted control over a sequence of target genes, while many miRNAs regulated several target genes. Prior to studying miRNA-regulated genes, we should also pay attention to mRNAs and genes regulated by several miRNAs. On the other hand, miRNAs regulating a larger number of mRNAs/genes have higher possibilities to influence a pathway and perform their biological functions. While the diverse nature of miRNA-mRNA interactions is well-documented, our network analysis identified miR-135a-5p as a high-connectivity hub (regulating three targets in arrhythmogenic pathways, Figure 4). This positions it as a candidate for future mechanistic studies of OSA-related cardiovascular complications, such as via in vivo knockout models. We emphasize that these findings are exploratory and require functional validation to validate the specific roles of the miRNA-mRNA network in OSA-related MS, further research is needed.
There are multiple constraints that need to be acknowledged in this study. First, there was no investigation of the function and relevance of identified DemiRs. Second, the study did not include female mice, hence the potential impact of gender could not be ruled out. Third, while validating all 23 DEmiRs experimentally is ideal, this was not feasible due to sample limitations. Future studies will expand validation. Fourth, our study relies exclusively on CIH to model OSA. While CIH effectively induces hypoxia-reoxygenation cycles analogous to OSA, it does not replicate sleep fragmentation or neurocognitive arousals-critical elements of clinical OSA pathophysiology. Recent studies demonstrate that combined models (e.g., CIH + sleep disruption) better mimic multisystemic OSA manifestations. For instance, Yin et al. (Wang et al., 2022) showed that adding sleep fragmentation to CIH exacerbates metabolic dysfunction beyond hypoxia alone. This may limit direct translation to human OSA complexity. Fifth, the RT-qPCR validation is limited to 6 miRNAs. Luciferase reporter assays and functional studies in vitro/in vivo will be essential to confirm causal relationships between key DEmiRs and BAT dysfunction. Last but not least, the limited number of samples makes the generalized results difficult to achieve.
Conclusion
To summarize, an abundance of DEmiRs was found in BAT of mouse models with CIH and a functional interaction network was identified between them. These findings broadened our present understanding of the molecular etiology of OSA-related MS, which could make a contribution to the development of novel therapeutic strategies for this disease.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was approved by the Institutional Animal Care and Use Committee of the Second Affiliated Hospital of Fujian Medical University (No. 2022-15). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
MJ: Writing – review and editing, Writing – original draft. YG: Writing – original draft. JZ: Writing – original draft. SL: Writing – review and editing. LL: Writing – original draft. QC: Writing – review and editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Joint Funds for the Innovation of Science and Technology, Fujian province (Grant number: 2021Y9027), Fujian Medical University Student Innovation and Entrepreneurship Training Program Funding Project (Grant number: C2024051 and JC2023180), Quanzhou Science and Technology Projects (Grant number: 2022C038R), Natural Science Foundation of Fujian Province (Grant number: 2024J01662), and Medical Innovation Project of Fujian Provincial Health Science and Technology Program (Grant number: 2023CXA034).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2025.1598018/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | (A) DEGs are presented on a scatter plot. Respectively, Green and red dots indicated decreased and increased expressed genes in BAT. (B) The distribution plot indicates the distribution of DEGs in chromosomes. Green and red indicated downregulated and upregulated genes in the BAT of the CIH mouse model, respectively.
References
Almendros, I., Basoglu, Ö. K., Conde, S. V., Liguori, C., and Saaresranta, T. (2022). Metabolic dysfunction in osa: is there something new under the sun? J. Sleep. Res. 31 (1), e13418. doi:10.1111/jsr.13418
Caca, J., Bartelt, A., and Egea, V. (2025). Hypoxia regulates brown adipocyte differentiation and stimulates mir-210 by hif-1α. Int. J. Mol. Sci. 26 (1), 117. doi:10.3390/ijms26010117
Chen, Q., Lai, H., Chen, Y., Peng, Z., Wu, S., and Liu, D. (2025). Characterization of circrna expression profiles and functional roles in a mouse model of liver injury induced by osa. Sci. Rep.-UK 15 (1), 15615. doi:10.1038/s41598-025-99612-6
Cui, F., Guan, Y., Guo, J., Tian, Y., Hu, H., Zhang, X., et al. (2018). Chronic intermittent hypobaric hypoxia protects vascular endothelium by ameliorating autophagy in metabolic syndrome rats. Life Sci. 205, 145–154. doi:10.1016/j.lfs.2018.05.008
Dahan, T., Nassar, S., Yajuk, O., Steinberg, E., Benny, O., Abudi, N., et al. (2022). Chronic intermittent hypoxia during sleep causes browning of interscapular adipose tissue accompanied by local insulin resistance in mice. Int. J. Mol. Sci. 23 (24), 15462. doi:10.3390/ijms232415462
Dong, M., Lin, J., Lim, W., Jin, W., and Lee, H. J. (2018). Role of brown adipose tissue in metabolic syndrome, aging, and cancer cachexia. Front. Med. 12 (2), 130–138. doi:10.1007/s11684-017-0555-2
Escate, R., Mata, P., Cepeda, J. M., Padreó, T., and Badimon, L. (2018). Mir-505-3p controls chemokine receptor up-regulation in macrophages: role in familial hypercholesterolemia. FASEB J. 32 (2), 601–612. doi:10.1096/fj.201700476R
Fernandes, J. L., Martins, F. O., Olea, E., Prieto-Lloret, J., Braga, P. C., Sacramento, J. F., et al. (2023). Chronic intermittent hypoxia-induced dysmetabolism is associated with hepatic oxidative stress, mitochondrial dysfunction and inflammation. Antioxidants (Basel) 12 (11), 1910. doi:10.3390/antiox12111910
Framnes, S. N., and Arble, D. M. (2018). The bidirectional relationship between obstructive sleep apnea and metabolic disease. Front. Endocrinol. (Lausanne) 9, 440. doi:10.3389/fendo.2018.00440
Giampá, S. Q. C., Lorenzi Filho, G., and Drager, L. F. (2023). Obstructive sleep apnea and metabolic syndrome. Obesity 31 (4), 900–911. doi:10.1002/oby.23679
Goody, D., and Pfeifer, A. (2019). Micrornas in brown and beige fat. Biochimica Biophysica Acta (BBA) - Mol. Cell Biol. Lipids 1864 (1), 29–36. doi:10.1016/j.bbalip.2018.05.003
Huang, X., Huang, X., Guo, H., Li, J., Zhou, C., Huang, Y., et al. (2022). Intermittent hypoxia-induced mettl3 downregulation facilitates mgll-mediated lipolysis of adipocytes in osas. Cell Death Discov. 8 (1), 352. doi:10.1038/s41420-022-01149-4
Karuga, F. F., Jaromirska, J., Malicki, M., Sochal, M., Szmyd, B., Białasiewicz, P., et al. (2023). The role of micrornas in pathophysiology and diagnostics of metabolic complications in obstructive sleep apnea patients. Front. Mol. Neurosci. 16, 1208886. doi:10.3389/fnmol.2023.1208886
Kuckuck, S., Lengton, R., Boon, M. R., Boersma, E., Penninx, B. W. J. H., Kavousi, M., et al. (2024). Long-term glucocorticoids in relation to the metabolic syndrome and cardiovascular disease: a systematic review and meta-analysis. J. Intern. Med. 295 (1), 2–19. doi:10.1111/joim.13739
Liu, Y., Zhong, C., Chen, S., Xue, Y., Wei, Z., Dong, L., et al. (2024). Circulating exosomal mir-16-2-3p is associated with coronary microvascular dysfunction in diabetes through regulating the fatty acid degradation of endothelial cells. Cardiovasc. Diabetol. 23 (1), 60. doi:10.1186/s12933-024-02142-0
Martins, F. O., and Conde, S. V. (2021). Gender differences in the context of obstructive sleep apnea and metabolic diseases. Front. physiology 12, 792633. doi:10.3389/fphys.2021.792633
Mohammadi, H., Rezaei, M., Sharafkhaneh, A., Khazaie, H., and Ghadami, M. R. (2020). Serum testosterone/cortisol ratio in people with obstructive sleep apnea. J. Clin. Lab. Anal. 34 (1), e23011. doi:10.1002/jcla.23011
Naqvi, R. A., Datta, M., Khan, S. H., and Naqvi, A. R. (2022). Regulatory roles of microrna in shaping t cell function, differentiation and polarization. Semin. Cell Dev. Biol. 124, 34–47. doi:10.1016/j.semcdb.2021.08.003
Qiu, L., Sheng, P., and Wang, X. (2023). Identification of metabolic syndrome-related mirna–mrna regulatory networks and key genes based on bioinformatics analysis. Biochem. Genet. 61 (1), 428–447. doi:10.1007/s10528-022-10257-w
Saito, M., and Okamatsu-Ogura, Y. (2023). Thermogenic brown fat in humans: implications in energy homeostasis, obesity and metabolic disorders. World J. Mens. Health 41 (3), 489–507. doi:10.5534/wjmh.220224
Sapiña-Beltrán, E., Gracia-Lavedan, E., Torres, G., Gaeta, A. M., Paredes, J., Mayoral, A., et al. (2022). Prevalence of obstructive sleep apnoea and its association with atherosclerotic plaques in a cohort of subjects with mild–moderate cardiovascular risk. Arch. Bronconeumología 58 (6), 490–497. doi:10.1016/j.arbres.2021.01.026
Sharma, S., Stansbury, R., Hackett, B., and Fox, H. (2021). Sleep apnea and pulmonary hypertension: a riddle waiting to be solved. Pharmacol. Ther. 227, 107935. doi:10.1016/j.pharmthera.2021.107935
Shobatake, R., Ota, H., Takahashi, N., Ueno, S., Sugie, K., and Takasawa, S. (2022). The impact of intermittent hypoxia on metabolism and cognition. Int. J. Mol. Sci. 23 (21), 12957. doi:10.3390/ijms232112957
Tan, X., Zhu, T., Zhang, L., Fu, L., Hu, Y., Li, H., et al. (2022). Mir-669a-5p promotes adipogenic differentiation and induces browning in preadipocytes. Adipocyte 11 (1), 120–132. doi:10.1080/21623945.2022.2030570
Tealdi, S., Ferro, E., Campa, C. C., and Bosia, C. (2022). Microrna-mediated encoding and decoding of time-dependent signals in tumorigenesis. Biomolecules 12 (2), 213. doi:10.3390/biom12020213
Teichenne, J., Morro, M., Casellas, A., Jimenez, V., Tellez, N., Leger, A., et al. (2015). Identification of mirnas involved in reprogramming acinar cells into insulin producing cells. PLoS One 10 (12), e0145116. doi:10.1371/journal.pone.0145116
Wang, F., Zou, J., Xu, H., Huang, W., Zhang, X., Wei, Z., et al. (2022). Effects of chronic intermittent hypoxia and chronic sleep fragmentation on gut microbiome, serum metabolome, liver and adipose tissue morphology. Front. Endocrinol. (Lausanne) 13, 820939. doi:10.3389/fendo.2022.820939
Xue, J., Allaband, C., Zhou, D., Poulsen, O., Martino, C., Jiang, L., et al. (2021). Influence of intermittent hypoxia/hypercapnia on atherosclerosis, gut microbiome, and metabolome. Front. physiology 12, 663950. doi:10.3389/fphys.2021.663950
Zeng, H., Huang, M., and Gong, X. (2022). Microrna-124-3p promotes apoptosis and autophagy of glioma cells by down-regulating crebrf. Neurol. Res. 44 (12), 1094–1103. doi:10.1080/01616412.2022.2112374
Zhang, X., Lin, X., Wu, X., Zeng, Y., Chen, X., Luo, X., et al. (2020). Differential expression of micrornas in xenografted lewis lung carcinomas subjected to intermittent hypoxia: a next-generation sequence analysis. Transl. cancer Res. 9 (7), 4354–4365. doi:10.21037/tcr-19-2913
Zhang, L., Ko, C., and Zeng, Y. (2022). Immunoregulatory effect of short-chain fatty acids from gut microbiota on obstructive sleep apnea-associated hypertension. Nat. Sci. sleep 14, 393–405. doi:10.2147/NSS.S354742
Zhang, J., Guo, Y., Ji, M., Lin, S., Liu, D., and Chen, Q. (2025). A comprehensive analysis of microrna alteration in an apoe(−/−) mice model of white adipose tissue injury induced by chronic intermittent hypoxia. Front. Genet. 16, 1474223. doi:10.3389/fgene.2025.1474223
Keywords: obstructive sleep apnea, brown adipose tissue, microRNA, metabolic syndrome, bioinformatic analysis
Citation: Ji M, Guo Y, Zhang J, Lin S, Li L and Chen Q (2025) Differential expression profiles and bioinformatics analysis of microRNAs in brown adipose tissue dysfunction induced by chronic intermittent hypoxia in obstructive sleep apnea. Front. Cell Dev. Biol. 13:1598018. doi: 10.3389/fcell.2025.1598018
Received: 22 March 2025; Accepted: 07 August 2025;
Published: 15 August 2025.
Edited by:
Yu-Jen Chen, National Taiwan University, TaiwanReviewed by:
Sruti Chandra, Tulane University, United StatesCarmen Lambert, Health Research Institute of Asturias (ISPA), Spain
Xujun Feng, The First Affiliated Hospital of Nanchang University, China
Copyright © 2025 Ji, Guo, Zhang, Lin, Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingshi Chen, Y2hlbnFpbmdzaGkxOTg2QDEyNi5jb20=; Liangyi Li, MTM5MDUwODgwNDRAMTYzLmNvbQ==
†These authors have contributed equally to this work