 Xu Zhang1,2†
Xu Zhang1,2† Hegui Wang
Hegui Wang- 1Department of Cardiology, Yijishan Hospital of Wannan Medical College, Wuhu, Anhui, China
- 2Department of Cardiology, Liyang Branch Hospital, The First Affiliated Hospital of Nanjing Medical University, Changzhou, Jiangsu, China
- 3Department of Cardiology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, China
- 4Department of Surgery, Fengcheng Hospital of Fengxian District, Shanghai, China
Cardiovascular diseases are a major global health concern, leading to high morbidity, mortality, and disability rates. Scaffold proteins, particularly A-kinase anchoring proteins (AKAPs), play a crucial role in signal transduction within the cardiovascular system. This review provides a comprehensive analysis of AKAPs’ involvement in the pathogenesis of cardiovascular diseases, emphasizing their key function in coordinating diverse signaling molecules, directing them to specific cellular microdomains, and minimizing signal interference. Disruptions in these interactions are linked to several cardiovascular disorders, such as cardiac hypertrophy, myocardial apoptosis, heart failure, arrhythmias, dysfunction in myocardial contraction and relaxation, and hypertension. Our goal was to explore the therapeutic potential of targeting the AKAP signaling pathway and offer new perspectives for the development and application of cardiovascular drugs that modulate AKAP signaling complexes.
1 Background
Cardiovascular diseases represent a major global health challenge, contributing considerably to both mortality and morbidity (Ong et al., 2018; Luo et al., 2021). A-kinase anchoring proteins (AKAPs) act as molecular scaffolds for a family of functionally related proteins that interact with various signaling molecules, including cyclic adenosine monophosphate (cAMP)-dependent protein kinases (PKAs). Furthermore, AKAPs associate with G-protein-coupled receptors, GTPases, kinases, phosphatases, phosphodiesterases, and cytoskeletal components, all of which are localized within distinct cytoskeletal microdomains. This strategic localization ensures that neighboring signaling complexes are insulated from other pathways, preserving specificity and minimizing potential crosstalk (Scott et al., 2013; Maric et al., 2021). The assembly of these multivalent signaling complexes allows AKAPs to integrate and coordinate signals from multiple pathways, enabling the precise regulation of complex cellular responses.
AKAPs are widely expressed in the heart and are critical to cardiac function. Notable examples include AKAP1 (D-AKAP1/AKAP121/AKAP149), AKAP5 (AKAP79/150/75), AKAP6 (mAKAPβ), AKAP7 (AKAP15/18), AKAP9 (Yotiao/AKAP350/450), AKAP10 (D-AKAP2), AKAP12 (Gravin), and AKAP13 (AKAP-Lbc) (Marin, 2020). A key feature of AKAPs is the structurally conserved domain responsible for PKA binding. This domain comprises a 14–18-residue amphipathic α-helix, which selectively interacts with the regulatory subunit of PKA, guiding its localization to specific subcellular regions where substrates are present (Per et al., 2012). Each AKAP, however, possesses a distinct subcellular targeting structure, with similar regions confined to homologous areas that bind to the RII dimer. PKA consists of two isoform types, I and II, each containing two regulatory subunits (RIα/β, RIIα/β) and two catalytic subunits (Cα, Cβ, or Cγ). According to the different regulatory subunits (RⅠ/RⅡ), PKA can be divided into two isoforms (PKAⅠ/PKAⅡ) (Christian et al., 2011; Liu et al., 2022; Bedioune et al., 2024). AKAPs are essential for compartmentalizing intracellular signal regulation, enabling precise control of site-specific signaling pathways. This regulation occurs through interactions with the regulatory subunits of PKA, particularly the RII subunits, which guide PKA to specific subcellular microdomains and modulate its substrate activity.
Some dual-specific AKAPs, or d-AKAPs, can simultaneously bind to both RI and RII (Table 1). RII subunits are typically localized to specific cellular sites, such as the plasma membrane, mitochondria, cytoskeleton, and centrosomes, whereas RI subunits tend to be more diffusely distributed (Kinderman et al., 2006; Omar and Scott, 2020). cAMP, as the second messenger activating PKA, is crucial for maintaining cellular physiological activities. In cardiomyocytes, the levels of cAMP are dynamically regulated by the balance between adenylate cyclases (ACs) and phosphodiesterases (PDEs). Upon activation of β-adrenergic receptors (β-ARs), the α subunit of G-proteins (Gas) is released within the target cells, which activates ACs, converting ATP to cAMP. This results in a rapid increase in intracellular cAMP levels and subsequent activation of PKA. In contrast, PDEs hydrolyze cAMP into 5′-AMP, reducing cAMP levels and consequently decreasing PKA activity (Liu et al., 2022; Zhang et al., 2024). The cAMP signaling pathway is compartmentalized. First, this second messenger is not uniformly distributed within the cell, and the spatial regulation of PDEs leads to varying cAMP concentrations across different subcellular compartments. PDEs regulate the localization, duration, and amplitude of cAMP signals within subcellular domains, controlling its diffusion to neighboring compartments, thereby preventing unnecessary PKA activation (Musheshe et al., 2018). Furthermore, AKAPs determine the subcellular localization of cAMP effectors by binding with PKA regulatory subunits and anchoring PKA to specific substrates. AKAPs can also interact with PDEs and phosphatases, providing local elements for signal termination. The spatial arrangement of these regulatory factors, effectors, and targets gives rise to the specific signaling of cAMP and governs the compartmentalized signaling mechanisms of AKAP complexes (Musheshe et al., 2018; Tomek and Zaccolo, 2023; Ripoll et al., 2024).
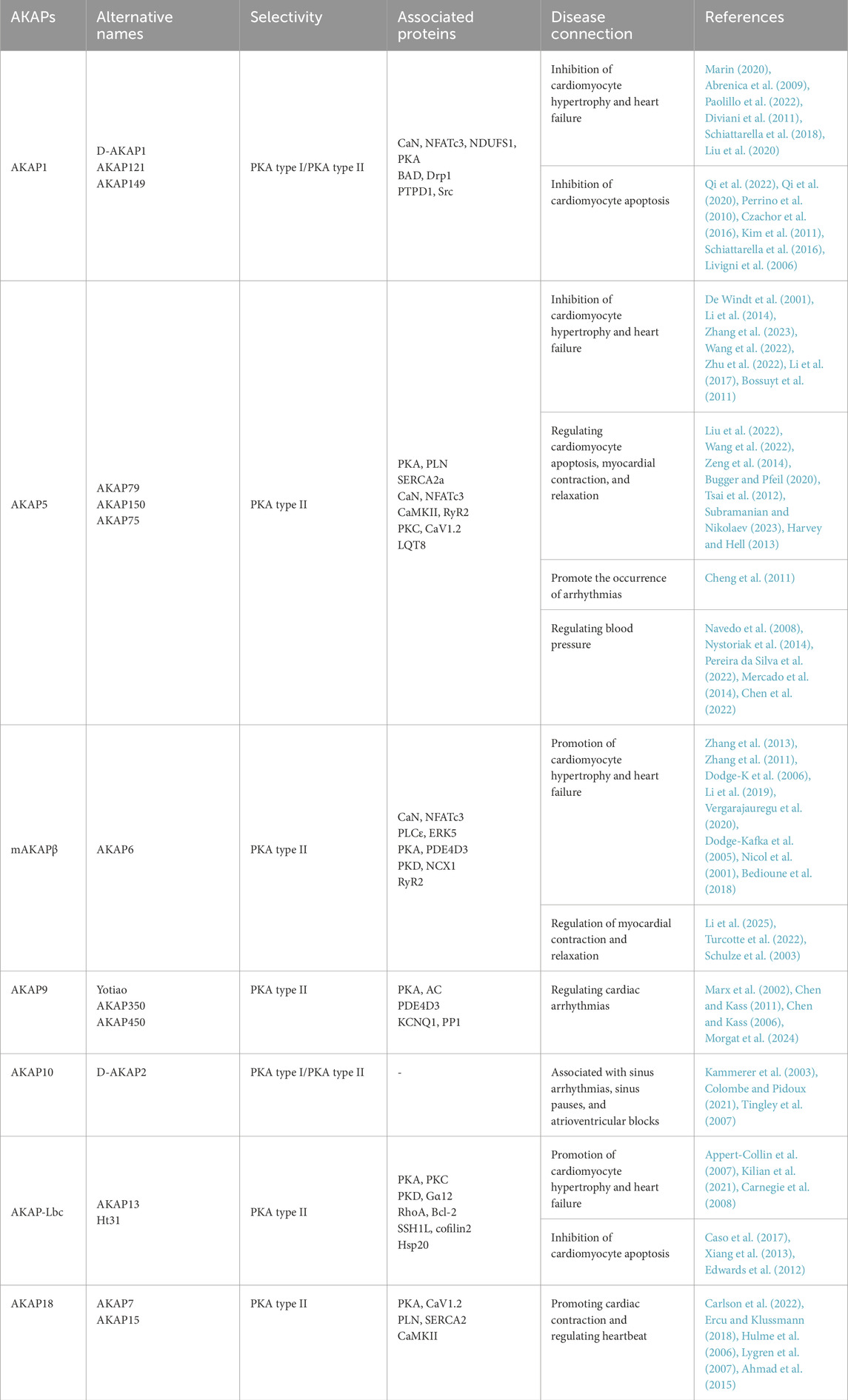
Table 1. Summary of AKAP complex features.
In cardiomyocytes, AKAP complexes play a critical role in maintaining and coordinating key cardiac functions by localizing to specific cellular sites. They regulate various physiological processes, including calcium cycling, excitation–contraction coupling, energy metabolism, transcriptional regulation, mitochondrial integrity, and action potential duration. Moreover, AKAPs play a crucial role in signaling pathways associated with pathophysiological conditions such as arrhythmias, cardiomyocyte hypertrophy, fibrosis, heart failure, and adaptive responses to hypoxia (Maric et al., 2021; Czepiel et al., 2022; Zhang et al.; Li et al., 2020; Delaunay et al., 2019; Carlson et al., 2022).
2 The role of AKAPs in cardiac hypertrophy development
Pathological cardiac hypertrophy is characterized by heart dilation, triggered by various adverse factors. The mechanisms underlying this condition are complex and multifaceted. Notably, AKAPs play a critical role in the signaling pathways involved in cardiac hypertrophy (Figure 1). Specifically, AKAP1 acts as a negative regulator of cardiomyocyte hypertrophy through the calcineurin (CaN)/NFAT transcription factor 3 (NFATc3) pathway. In pathological conditions such as hypertension or chronic isoproterenol exposure, desensitization of β-adrenergic receptors (β-ARs) in cardiomyocytes leads to a decrease in AKAP1 gene transcription. As a result, the impaired AKAP1 protein is released into the cytoplasm by forming a complex with CaN, which dephosphorylates NFATc3, thereby triggering the gene expression associated with cardiac hypertrophy (Abrenica et al., 2009; Paolillo et al., 2022). Phosphorylated NFATc3 is predominantly inactive and localized to the cell membrane. However, genetic deletion of AKAP1 (AKAP1−/−) results in the release of active CaN. The availability of free CaN at the cell membrane promotes the dephosphorylation of NFATc3, facilitating its translocation from the membrane to the nucleus. This translocation activates cardiac hypertrophic genes, contributing to the progression of heart failure. Thus, AKAP1 may serve as a critical inhibitor of pathological cardiac hypertrophy (Paolillo et al., 2022; Diviani et al., 2011; Schiattarella et al., 2018).
AKAP5 shares significant homology with the CaN binding site in AKAP1 (Marin, 2020), and the AKAP5–CaN complex and its downstream effectors are crucial in the expression of pathological hypertrophic genes. Research has demonstrated that ΔAKAP5, which incorporates the CaN inhibitory structural domain of AKAP5, inhibits isoproterenol (ISO)-induced cardiac hypertrophy in genetically modified mice (De Windt et al., 2001). Li et al. found that carvedilol effectively reverses cardiac hypertrophy in AKAP5-deficient mice by normalizing the activity of cardiac CaN and calcium/calmodulin-dependent protein kinase II (CaMKII) (Li et al., 2014).
Following these findings, our study demonstrated that hypoxia/reoxygenation (H/R) reduces AKAP5 expression in cardiomyocytes. Concurrently, the activation of CaN and CaMKII promotes cardiomyocyte hypertrophy by modulating downstream molecular complexes (Zhang et al., 2023; Wang et al., 2022; Zhu et al., 2022; Dondi et al., 2024). The phosphorylation of phospholamban (PLN) by PKA, which is anchored by AKAP5, is essential for regulating intracellular calcium cycling. The phosphorylation or ablation of PLN, resulting in its dissociation from sarcoplasmic reticulum calcium ATPase 2a (SERCA2a), enhances calcium recycling through SERCA2a, thus reducing hypertrophic responses and arrhythmias in cardiomyocytes (Cheng et al., 2019; M et al., 2016; Bai et al., 2013; Fang et al., 2018; Koch et al., 2021).
Additionally, mAKAP plays a critical role in the development of cardiac hypertrophy. It functions as a docking platform for various signaling proteins, including CaN/NFATc3, phospholipase C epsilon (PLCε), and the kinase ERK5, all of which are directly anchored to the nuclear membrane of hypertrophic genes (Zhang et al., 2011; Dodge-K et al., 2006; Li et al., 2019). In addition to the classical hypertrophic pathway involving CaN/NFATc3, mAKAPβ anchors the PLCε complex at the nuclear membrane, facilitating the hydrolysis of phosphatidylinositol 4-phosphate (PI4P). The resulting hydrolysate, diacylglycerol (DAG), activates the hypertrophic pathway by promoting the activation of nuclear protein kinase D (PKD) and inhibiting myocardial hypertrophy considerably when mAKAPβ is depleted (Zhang et al., 2013; Vergarajauregu et al., 2020). Furthermore, mAKAP facilitates the phosphorylation of phosphodiesterase (PDE) 4D3 by anchoring PKA, which reduces local cAMP levels. This reduction weakens the inhibition of ERK5 activity mediated by Epac1, enhancing myocardial hypertrophy induced by leukemia inhibitory factor (LIF) (Dodge-Kafka et al., 2005; Nicol et al., 2001; Bedioune et al., 2018).
AKAP–Lbc, a scaffolding protein associated with protein kinase A (PKA) and protein kinase C (PKC), facilitates the activation of protein kinase D (PKD). This activation results in the phosphorylation of histone deacetylase 5 (HDAC5) and its translocation from the nucleus, thus enhancing the MEF2-mediated transcription of hypertrophic genes (Taglieri et al., 2014; He et al., 2020). Additionally, the Gα12–AKAP–Lbc–RhoA signaling pathway is likely involved in α1-adrenergic receptor (α1-AR)-induced hypertrophy of cardiomyocytes (Appert-Collin et al., 2007; Kilian et al., 2021).
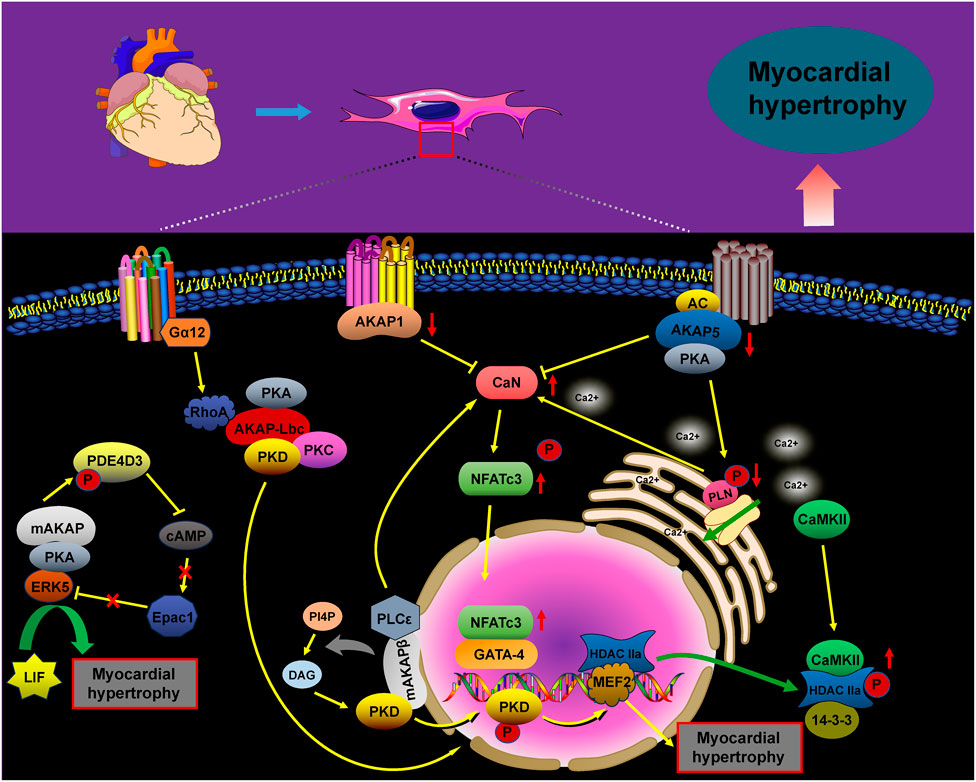
Figure 1. AKAP-mediated signaling in cardiomyocyte hypertrophy. The signaling pathways of AKAPs involved in cardiomyocyte hypertrophy, including the AKAP1/5/mAKAP/CaN/NFATc3, AKAP5/PKA/PLN, mAKAP/PLCε/PKD, mAKAP/PKA/PDE4D3, and Gα12/AKAP–Lbc/RhoA/PKD pathways, promote cardiomyocyte hypertrophy upon activation.
3 The role of AKAPs in cardiomyocyte apoptosis
Myocardial ischemia is a complex pathological condition resulting from reduced localized blood flow to tissues or organs. Although it prevents tissue necrosis, myocardial ischemia–reperfusion can induce metabolic disorders and cardiac dysfunction. Apoptosis, in particular, is a key pathological process (Chen et al., 2025; Han et al., 2024). AKAP complexes play a crucial role in regulating myocardial cell apoptosis (Figure 2).
Under physiological conditions, AKAP5 anchors PKA, facilitating the phosphorylation of ryanodine receptor 2 (RyR2) and PLN, which is crucial for maintaining intracellular calcium (Ca2+) cycling homeostasis. In contrast, mice deficient in AKAP5 exhibit reduced phosphorylation of RyR2 and PLN, leading to compromised Ca2+ cycling in response to adrenergic stimulation or pressure overload. This deficiency results in impaired myocardial diastolic and systolic function, along with significant cardiomyocyte apoptosis (Li et al., 2017). Our study showed a significant reduction in AKAP5 protein expression and a concomitant increase in apoptosis in H9C2 cells subjected to hypoxia/reoxygenation. Notably, activation of the PLN/SERCA2a signaling pathway following AKAP5 upregulation was linked to decreased apoptosis. Thus, AKAP5 may influence cardiomyocyte apoptosis through the PKA/PLN/SERCA complex (Wang et al., 2022). Additionally, under hyperglycemic conditions, AKAP5 and cPKC signaling complexes enhance anchoring at the plasma membrane and activate cPKC, promoting the phosphorylation of p47(phox) and the production of reactive oxygen species (ROS). This cascade leads to impaired myocardial diastolic function, apoptosis, and oxidative stress following hyperglycemic exposure (Zeng et al., 2014; Bugger and Pfeil, 2020; Tsai et al., 2012).
NADH-ubiquinone oxidoreductase subunit S1 (NDUFS1) protects against myocardial infarction and hypoxia-induced apoptosis associated with ROS in mitochondria. However, the deletion of AKAP1 impairs the mitochondrial translocation of NDUFS1, resulting in mitochondrial dysfunction, suppression of oxidative phosphorylation (OXPHOS), and increased mitochondrial ROS production. This cascade exacerbates cardiac myocyte apoptosis and contributes to the pathogenesis of diabetic cardiomyopathy (Qi et al., 2022; Qi et al., 2020). Furthermore, the downregulation of AKAP1 in response to pressure overload exacerbates mitochondrial dysfunction, increases ROS production, and promotes cardiomyocyte death (Perrino et al., 2010; Czachor et al., 2016). Research has shown that AKAP1 anchors protein kinase A (PKA) to various mitochondrial substrates, such as NDUFS4, enhancing the activity of the mitochondrial respiratory complex. Additionally, PKA exerts an anti-apoptotic effect by phosphorylating and inactivating the pro-apoptotic protein BAD, thereby preventing its association with Bcl-2 (Perrino et al., 2010; Harada et al., 1999; Haushalter et al., 2019; Affaitati et al., 2003; Gao et al., 2022). Kim et al. demonstrated that AKAP1, located on the mitochondrial membrane, plays a crucial role in inhibiting mitochondrial fission. This inhibition is facilitated by providing docking sites for PKA and dynamin-related protein 1 (Drp1), enabling the phosphorylation of Drp1 by PKA. Under ischemic hypoxic conditions, the expression and activity of Siah2 are upregulated, leading to the dysregulation of AKAP1. This dysregulation results in decreased Drp1 phosphorylation and increased interaction between Drp1 and Fis1, ultimately promoting mitochondrial fission and apoptosis in cardiomyocytes (Kim et al., 2011).
The observed reduction in AKAP1 levels under ischemic–hypoxic conditions may be linked to increased degradation of AKAP1, mediated by Siah2 (Schiattarella et al., 2016). Additionally, AKAP1 can direct src tyrosine kinase to the mitochondria through the action of protein tyrosine phosphatase (PTPD1). This targeting facilitates src-dependent tyrosine phosphorylation of mitochondrial substrates, thereby enhancing mitochondrial respiration and ATP synthesis (Perrino et al., 2010; Livigni et al., 2006). Mitochondrial dysfunction and apoptosis resulting from AKAP1 deficiency may be attributed to altered interactions between AKAP1, PKA, and src.
The literature indicates that AKAP–Lbc serves as a molecular scaffold, coordinating protective signaling pathways against doxorubicin (DOX)-induced cardiac cytotoxicity (Caso et al., 2017). Activation of PKD1, anchored by AKAP–Lbc, promotes the transcription of the anti-apoptotic protein Bcl-2 and inhibits the phosphatase SSH1L. This inhibition prevents the dephosphorylation of the actin-binding protein cofilin2, thus inhibiting the translocation of Bax to the mitochondria. As a result, this mitigates mitochondrial dysfunction, cytochrome C (CytC) release, and apoptotic cell death (Caso et al., 2017; Xiang et al., 2013). Additionally, localized increases in cyclic adenosine monophosphate (cAMP) due to β-adrenergic receptor stimulation activate PKA, anchored by AKAP–Lbc. This activation enhances the phosphorylation of the 20-kDa heat shock protein (Hsp20) at the Ser16 site, preventing cardiomyocyte apoptosis (Edwards et al., 2012).

Figure 2. AKAP signaling in cardiomyocyte apoptosis. The signaling pathways involving AKAPs are crucial for cardiomyocyte apoptosis. Key pathways include AKAP5/PKA/PLN/RyR2, AKAP5/PKC/p47(phox), AKAP1/NDUFS1/OXPHOS, AKAP1/PKA/BAD, AKAP1/PKA/Drp1/Fis1, AKAP1/PTPD1/src, AKAP-Lbc/PKD1/SSH1L/Cofilin2, and AKAP-Lbc/PKD1/Bcl-2. Activation of these pathways promotes cardiomyocyte apoptosis.
4 The role of AKAPs in cardiac arrhythmias
In the cardiac system, the AKAP macromolecular complex coordinates the phosphorylation of various channel proteins, including RyR2 calcium channels, L-type calcium channels, and potassium channels (IKs). Mutations in these channels have been linked to inherited arrhythmia syndromes, such as long QT syndrome (LQT) and catecholaminergic polymorphic ventricular tachycardia (Chen et al., 2007; Hegyi et al., 2021; Wu and Larsson, 2020). The sympathetic nervous system tightly regulates the activation of IKs, with AKAPs playing a crucial role in cardiac repolarization. Studies have shown that AKAP9 (Yotiao) forms a macromolecular complex with the α-subunit of the IK potassium channel (KCNQ1), the regulatory subunit of PKA type II (RII), and protein phosphatase 1 (PP1). Additionally, AKAP9 can activate PKA by modulating cAMP levels through interactions with adenylyl cyclase (AC) and phosphodiesterase 4D3 (PDE4D3). Activated PKA then facilitates the phosphorylation of serine 43 (S43) on AKAP9 and promotes the phosphorylation of serine 27 (S27) in the amino-terminal region of KCNQ1. The removal of phosphorylation at the AKAP9 S43 site considerably impairs PKA-induced voltage-dependent activation of IKs, altering these dynamics. Conversely, AKAP9’s binding to PP1 leads to the dephosphorylation of KCNQ1, and mutations within this complex have been implicated in type 1 long QT syndrome (LQT1), a potentially fatal inherited arrhythmia syndrome (Marx et al., 2002; Chen and Kass, 2011; Chen and Kass, 2006; Morgat et al., 2024) (Figure 3).
Timothy syndrome (TS), also referred to as long QT syndrome type 8 (LQT8), is a rare pediatric disorder caused by the G406R mutation in the CaV1.2 channel. This mutation disrupts the normal inactivation of the channel, resulting in a prolonged influx of Ca2+ during the action potential (AP). This predisposes individuals to life-threatening arrhythmias (Drum et al., 2014; Pitt et al., 2021). CaV1.2 channels are crucial for excitation–contraction coupling in the heart, as they considerably influence the AP waveform. They play an essential role in the heart’s excitation–contraction mechanism. In the case of TS (LQT8), the interaction between the anchoring protein AKAP5 and the CaV1.2–LQT8 channel forms a complex that enhances calcium influx, prolongs the AP duration, and promotes arrhythmogenesis. This occurs through stabilization of the open conformation and facilitation of the gating of the CaV1.2–LQT8 channel. Interestingly, AKAP5 ablation has been shown to rectify the pathological gating of the CaV1.2–LQT8 channel, helping mitigate the development of arrhythmias (Cheng et al., 2011).
AKAP10 is a bispecific A-kinase anchoring protein primarily localized in the mitochondria, cytoplasm, and plasma membrane, where it plays a crucial role in regulating heart rate in both mice and humans. A functional single nucleotide polymorphism (SNP) in AKAP10 has been identified, involving the substitution of isoleucine (Ile) at position 646 with valine (Val). Individuals carrying this SNP exhibit an increased heart rate, reduced heart rate variability, and an higher risk of cardiac arrest and sudden death (Kammerer et al., 2003; Colombe and Pidoux, 2021; Tingley et al., 2007). Tingley et al. demonstrated that mutations in AKAP10 increased the sensitivity of cardiomyocytes to cholinergic signaling, contributing to arrhythmia development. Mice with AKAP10 mutations exhibited significant sinus arrhythmias, sinus pauses, and atrioventricular blocks. These mice experienced sinus pauses with junctional escape beats 40 times more frequently and atrioventricular block 15 times more frequently than wild-type (WT) mice (Tingley et al., 2007). Additionally, a correlation was proposed between the 1936A > G AKAP10 variant and the corrected QT interval (QTc) in a cohort of European-descent newborns (Loniewska et al., 2015).
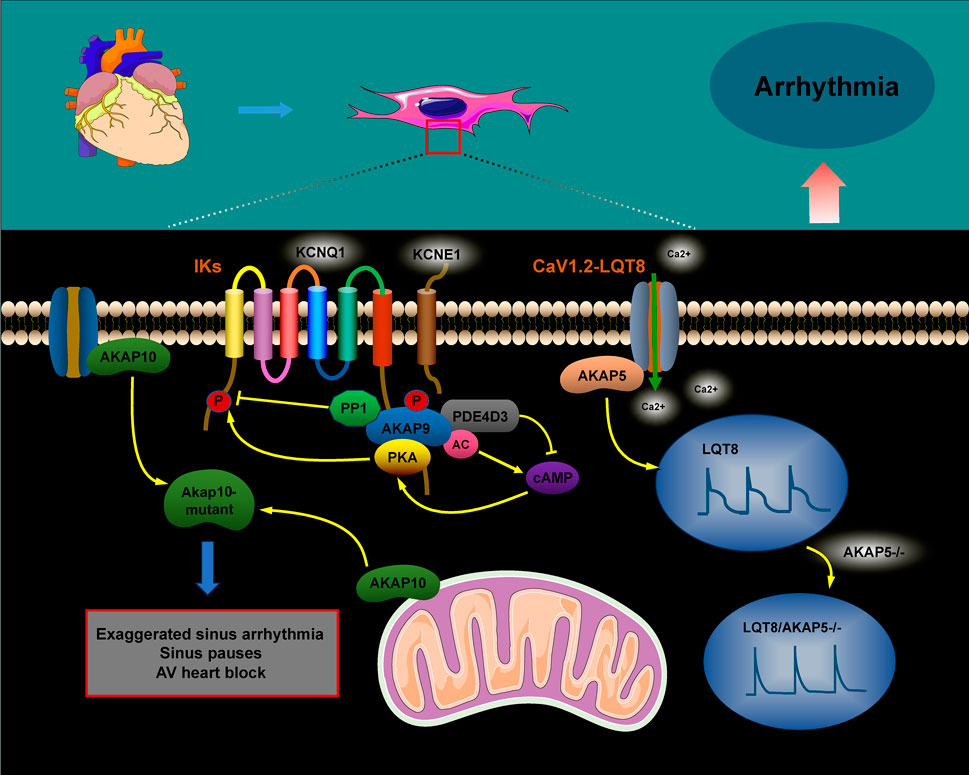
Figure 3. The role of AKAPs in signaling pathways during cardiac arrhythmias. The AKAP9/AC/PDE4D3/PKA and AKAP9/PP1 complexes regulate KCNQ1 phosphorylation, thus controlling the activity and current kinetics of IKs. Mutations in KCNQ1 within this complex are associated with LQT1. AKAP5 enhances calcium influx, prolongs action potential duration, and promotes arrhythmogenesis by increasing the coupling and gating of the CaV1.2–LQT8 channel, while AKAP5 ablation corrects the CaV1.2–LQT8-related arrhythmias. AKAP10 mutations can result in sinus arrhythmia, sinus arrest, and atrioventricular conduction blocks.
5 The role of AKAPs in heart failure
Heart failure is a progressive, often fatal condition marked by a decline in the heart’s ability to pump blood efficiently. Factors such as myocardial infarction, hypertension, congenital heart disease, and chronic activation of neurohumoral factors and cytokines contribute to its onset. The progression of heart failure is further aggravated by cardiomyocyte hypertrophy, apoptosis, and fibrotic remodeling. The AKAP complex plays a significant role in the pathophysiology of heart failure (Figure 4), with protein kinase B (Akt), a crucial signaling molecule involved in cardiomyocyte growth and diastolic function, influencing its progression through various signaling pathways. Research suggests that Akt can promote the translocation of glucose transporter 4 (GLUT4) to the plasma membrane by activating the substrate AS160. This action enhances glucose uptake, helping to prevent diabetic cardiomyopathy and heart failure (Hou et al., 2019; Shu et al., 2021). In AKAP1 knockout mice subjected to transverse aortic constriction (TAC), the absence of Akt activation accelerated cardiomyocyte death and worsened the progression of heart failure (Marin, 2020; Schiattarella et al., 2018). Abnormalities in mitochondrial metabolism, impaired oxidative phosphorylation (OXPHOS), excessive ROS production, and dysregulated mitochondrial dynamics have all been implicated in the development of heart failure (Wu et al., 2022). Specifically, downregulation of AKAP1 reduces PKA localization to the mitochondria, resulting in decreased inhibitory phosphorylation of Drp1 at serine 637. This promotes mitochondrial fission, leading to increased cytoplasmic ROS production, which contributes to cardiac hypertrophy and the progression of heart failure (Liu et al., 2020).
Abnormal changes in cardiomyocyte properties are strongly associated with heart failure. These alterations include the reactivation of fetal gene programs, disruptions in calcium handling and energy metabolism, abnormal protein synthesis, and sarcomere reorganization. Such modifications impair myocardial contractility, promote cardiomyocyte apoptosis, and exacerbate the progression of heart failure. A-kinase anchoring proteins play a critical role in coordinating the signaling pathways involved in these processes (Cibi et al., 2020; Subramanian and Nikolaev, 2023). Heart failure is marked by increased activation of the CaN–NFAT signaling pathway. AKAP5 plays a cardioprotective role by modulating the pathological signaling triggered by β-adrenergic receptors (β-ARs) and CaN. Notably, the absence of AKAP5 in murine models leads to a considerable rise in CaN and CaMKII activity, which is closely associated with the onset of age-related cardiac hypertrophy, ventricular dilation, and the progression of heart failure (Li et al., 2014; Li et al., 2017; Bossuyt et al., 2011). The mAKAPβ/PLCε/PKD and AKAP-Lbc/PKD/HDAC5 pathways play a critical role in regulating myocardial hypertrophy, considerably contributing to the progression of heart failure (Zhang et al., 2013; Vergarajauregu et al., 2020; He et al., 2020; Appert-Collin et al., 2007; Kilian et al., 2021; Carnegie et al., 2008).
Figure 4. Role of AKAP signaling in heart failure. The AKAP signaling pathways involved in heart failure, including the AKAP1/Akt/AS160, AKAP1/PKA/Drp1/Fis1, AKAP1/NDUFS1/OXPHOS, AKAP5/CaN/NFATc3, AKAP5/CaMKII/HDAC, AKAP-Lbc/PKD/HDAC, and mAKAPβ/PLCε/PKD pathways, contribute to heart failure progression through their activation.
6 The role of AKAPs in myocardial contraction and relaxation
AKAPs are integral to the signaling pathways of various hormones and neurotransmitters within the heart, facilitating excitatory–contractile coupling and intracellular calcium cycling in cardiomyocytes in response to β-AR stimulation. In this signaling pathway, AKAPs anchor PKA and regulate the phosphorylation of key substrate proteins, including L-type calcium channels (LTCC, Cav1.2), ryanodine receptors (RYRs), PLN, troponin I (cTnI), and cardiac myosin-binding protein C (cMyBP-C) (Carlson et al., 2022; Wang et al., 2022; Li et al., 2017; Rababa’h et al., 2014; Pallien and Klussmann, 2020).
During the excitation–contraction coupling (ECC) process, the transient opening of L-type calcium channels (LTCCs) in the transverse tubules and surface sarcoplasmic membrane causes a localized increase in intracellular calcium concentration ([Ca2+]i). This increase activates ryanodine receptor 2 (RyR2) in the sarcoplasmic reticulum (SR) via calcium-induced calcium release (CICR). Consequently, a calcium transient occurs, leading to an overall increase in [Ca2+]i and subsequent contraction of the cardiomyocyte. Following this, LTCC and RyR2 rapidly inactivate through a calcium-dependent mechanism, halting further calcium release from the SR. This process enables the sarcoplasmic/endoplasmic reticulum calcium ATPase 2a (SERCA2a) to recycle the released calcium before the next heartbeat. SERCA2a plays a critical role in regulating myocardial calcium cycling by facilitating the reuptake of considerable amounts of cytoplasmic calcium into the SR (Rhana et al., 2024; Jiang et al., 2025).
PLN negatively regulates SERCA2a activity by binding to it and reducing its calcium affinity (MacLennan and Kranias, 2003; Weber et al., 2021; Ren et al., 2024). Phosphorylation of PLN, mediated by AKAPs in conjunction with PKA, causes PLN to dissociate from SERCA2a. This process improves the calcium recycling by SERCA2a into the endoplasmic reticulum, reducing cytoplasmic calcium accumulation and promoting cardiomyocyte diastole. The phosphorylation of these calcium-related proteins is regulated by multiple AKAPs (Subramanian and Nikolaev, 2023; Szentesi et al., 2004; Papa et al., 2022) (Figure 5).
AKAP5 macromolecular complexes consist of β-ARs, PKA, CaN, PLN, RyR2, and others. PKA-mediated phosphorylation regulates the activities of CaV1.2, PLN, and RyR2, which are associated with CaV3. AKAP5 anchors PKA to CaV1.2, PLN, and RyR2, promoting Ca2+ release through the activation of CaV1.2. This activation in turn stimulates RyR2 and PLN, playing a crucial role in myocardial contraction and relaxation (Liu et al., 2022; Harvey and Hell, 2013; Xiao et al., 2005). Studies have shown that genetically engineered mice lacking AKAP5 exhibit reduced calcium release from RyR2 channels and impaired calcium recycling by PLN/SERCA2a. This dysfunction results from diminished phosphorylation of RyR2 and PLN by the AKAP5/PKA complex, disrupting calcium cycling in cardiomyocytes and leading to abnormal myocardial contraction and relaxation (Li et al., 2017; Subramanian and Nikolaev, 2023).
AKAP18α is a membrane-associated scaffolding protein that enhances calcium currents by facilitating PKA-dependent phosphorylation of serine 1928 in CaV1.2 channels, thus promoting cardiac contraction (Ercu and Klussmann, 2018; Hulme et al., 2006). In rat hearts, AKAP18δ forms a supramolecular complex with PKA, PLN, and SERCA2, anchoring PKA to phosphorylate PLN in response to adrenergic stimulation. This process regulates SERCA2-mediated Ca2+ reuptake into the SR (Lygren et al., 2007). AKAP18δ is likely crucial for regulating the heartbeat by modulating the Ca2+ frequency-dependent activation of CaMKII at the SERCA2–PLN complex and RyR channels (Carlson et al., 2022). In the human heart, AKAP18γ promotes PKA-mediated phosphorylation of PLN, leading to its dissociation from SERCA2, which activates ATPase and enhances Ca2+ reuptake into the SR (Ahmad et al., 2015). The muscle-selective A-kinase anchoring protein, mAKAPβ, interacts with RyR2 at the SR, promoting PKA-mediated phosphorylation of the receptor. This modification enhances channel opening, facilitating the release of Ca2+ from the SR into the cytoplasm (Li et al., 2025; Turcotte et al., 2022). Additionally, mAKAPβ interacts with the sodium/calcium exchanger protein NCX1 at the sarcolemmal membrane, facilitating PKA-dependent activation of NCX1. This leads to an increased Ca2+ efflux (Schulze et al., 2003).
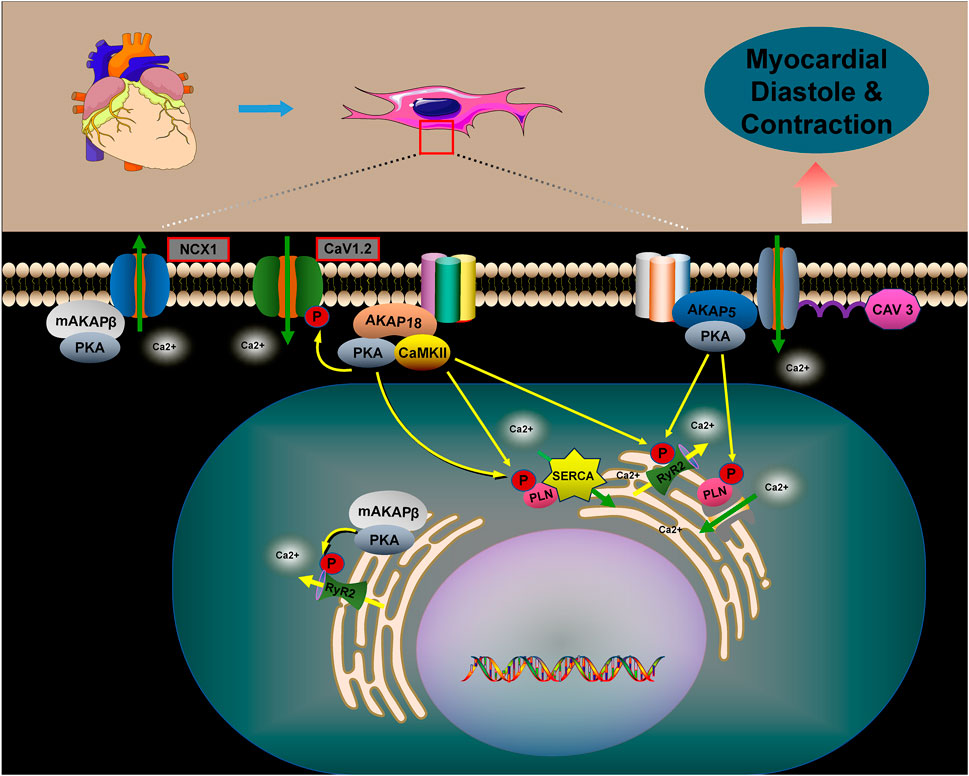
Figure 5. The role of AKAPs in regulating myocardial contraction and relaxation. The role of AKAPs in regulating myocardial contraction and relaxation is pivotal. Specifically, AKAP5, AKAP18, and mAKAPβ regulate cellular calcium cycling, cardiac contraction, and relaxation by forming complexes with PKA, CaV1.2, PLN, and RyR2. In the absence of AKAPs, disruptions in calcium cycling occur within cardiomyocytes, leading to abnormalities in myocardial contraction and relaxation. Furthermore, mAKAPβ plays a crucial role by interacting with NCX1, thereby promoting PKA-dependent activation of NCX1. This interaction facilitates the efflux of Ca2+, thus influencing myocardial contraction and relaxation.
7 The role of AKAPs in hypertension pathogenesis
Hypertension is a major risk factor for the development of cardiovascular, cerebrovascular, and renal diseases. Its etiology is multifactorial, with vascular dysfunction playing a central role (Navedo et al., 2008). Changes in vasoconstrictive properties and increased arterial remodeling contribute to the progression of conditions like chronic hypertension, atherosclerosis, and heart failure (Touyz et al., 2018). Research has demonstrated that AKAPs are vital in regulating vascular integrity and peripheral arterial vasoconstriction by integrating and processing various signal transduction pathways, which are essential for maintaining blood pressure homeostasis (Nystoriak et al., 2017; Prada et al., 2020; Ottolini et al., 2020) (Figure 6).
AKAP5 is a key scaffolding protein involved in regulating blood pressure. It facilitates calcium influx through PKC activation of the voltage-dependent calcium channel CaV1.2, promoting cellular contraction and increasing vascular tone. In contrast, the absence of AKAP5 disrupts the PKC-mediated targeting of CaV1.2, leading to reduced calcium release and decreased vascular tone (Navedo et al., 2008). Under hyperglycemic conditions, AKAP5 anchors PKC to phosphorylate the Cav1.2 channel, enhancing calcium ion influx and activating the CaN/NFATc3 complex. This results in reduced expression of the β1 subunit of the large-conductance calcium-activated K+ channel (BKCa), inhibiting K+ efflux, promoting vasoconstriction, and raising blood pressure (Nystoriak et al., 2014; Pereira da Silva et al., 2022). However, evidence suggests that the AKAP5–PKC complex also interacts with transient receptor potential vanilloid 4 (TRPV4) calcium channels, facilitating the coupling of RyRs to BKCa channels. This interaction helps inhibit the increase in blood pressure (Mercado et al., 2014; Chen et al., 2022).
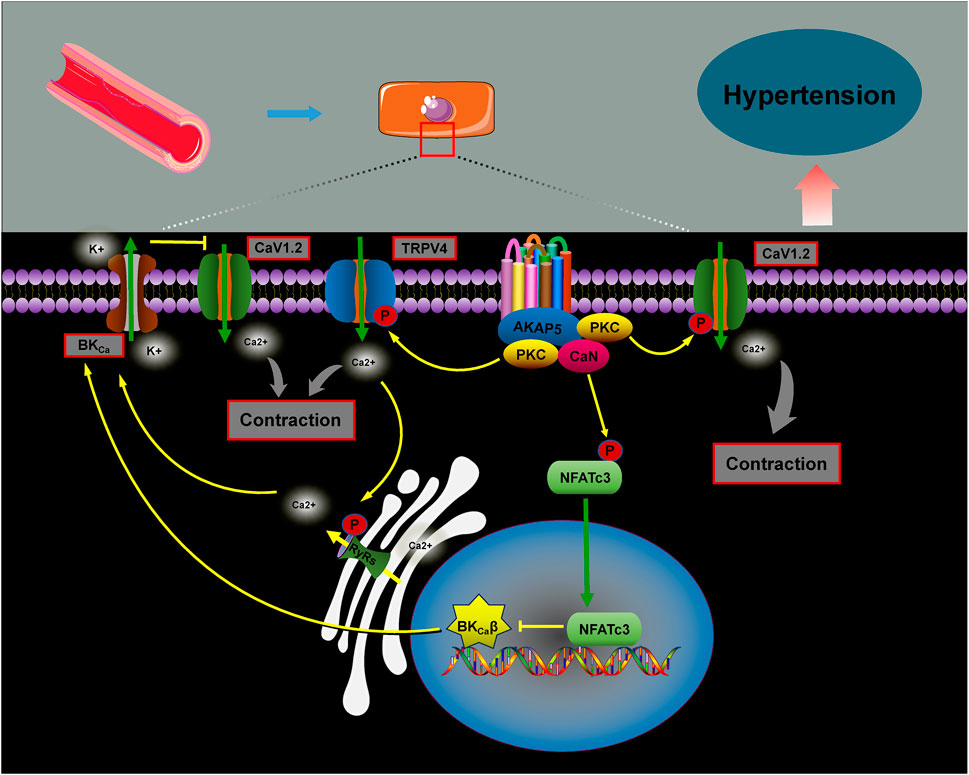
Figure 6. The role of AKAPs in blood pressure regulation. AKAP5 facilitates calcium influx by anchoring PKC to phosphorylate Cav1.2 channels, which activates the CaN/NFATc3 complex. This reduces the expression of the β1 subunit of BKCa, inhibiting potassium efflux, promoting vascular contraction, and raising blood pressure. Additionally, studies show that the AKAP5–PKC complex interacts with the TRPV4 calcium channel, promoting RyRs–BKCa coupling and activating the BKCa channel, thereby suppressing blood pressure elevation.
8 Conclusion
AKAPs are ubiquitously expressed in the cardiovascular system, anchoring various signaling molecules to multiprotein complexes. As dynamic hubs for multiple signaling pathways, AKAPs are essential for maintaining the homeostasis and functionality of the cardiovascular system. Disruptions in the interactions between AKAPs and their associated molecules are closely linked to the pathophysiology of cardiovascular diseases, including heart failure, cardiomyocyte apoptosis and hypertrophy, myocardial contractile and diastolic dysfunction, arrhythmias, and hypertension. Preliminary drug development and therapeutic strategies have begun to target AKAPs as potential interventions for cardiovascular diseases. One such agent, St-Ht31, is a peptide inhibitor of AKAPs that disrupts the interaction between AKAPs and PKA by mimicking the RIIα binding domain of PKA. This disruption results in decreased phosphorylation of the PKA substrate, RyR2 (Marx et al., 2000; Soni et al., 2014; McConnell et al., 2009). Hyperphosphorylation of RyR2 has been implicated in the pathogenesis of various cardiac dysfunctions, including myocardial systolic and diastolic dysfunction, arrhythmias, and heart failure (Marx et al., 2000; Belevych et al., 2013; Do and Knollmann, 2025; Shan et al., 2010). Despite its widespread use in fundamental research, the peptide inhibitor St-Ht31 has multiple drawbacks (Troger et al., 2012). St-Ht31 lacks specificity, targeting multiple AKAP isoforms and potentially interfering with distinct AKAP-mediated signaling pathways, which complicates the interpretation of its inhibitory effects. Moreover, it has shortcomings such as limited cellular uptake and a short biological half-life. Thus, further investigation is necessary to fully assess St-Ht31’s impact on cardiac conditions. In contrast, the small molecule inhibitor FMP-API-1 is better suited for cellular and animal studies due to its stability. FMP-API-1 has shown promise in disrupting the AKAP–PKA interaction and enhancing myocardial contractility in rats. However, the enhanced myocardial contractility observed with FMP-API-1 may also result from its activation of PKA (Christian et al., 2011; Troger et al., 2012). Recent advances in gene regulation technologies, such as CRISPR-Cas9 gene editing, RNA interference, and viral vector-mediated gene therapy, have shown considerable promise in cardiovascular disease research by enabling precise targeting of AKAPs (Carlson et al., 2022; Caso et al., 2017; Ibarrola et al., 2018; Guo et al., 2015; Mayers et al., 2010; You et al., 2022; Xu et al., 2025). The crucial role of AKAPs in cardiovascular diseases has been consistently validated. Ongoing and future research into AKAP complexes has the potential to provide novel insights that could overcome the limitations of conventional therapies and inform the development of molecularly targeted drugs for the treatment of cardiovascular diseases.
Author contributions
XZ: Funding acquisition, Writing – original draft, Writing – review and editing, Resources, Visualization. FZ: Software, Validation, Writing – original draft. ZX: Resources, Visualization, Writing – original draft. HW: Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Anhui Province University Science Research Projects (YJS20210553), and the Wuhu Science and Technology Program, Anhui Province (2023jc26).
Acknowledgments
We thank LetPub (www.letpub.com.cn) for its linguistic assistance during the preparation of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abrenica, B., AlShaaban, M., and Czubryt, M. P. (2009). The A-kinase anchor protein AKAP121 is a negative regulator of cardiomyocyte hypertrophy. J. Mol. Cell Cardiol. 46 (5), 674–681. doi:10.1016/j.yjmcc.2009.01.018
Affaitati, A., Cardone, L., de Cristofaro, T., Carlucci, A., Ginsberg, M. D., Varrone, S., et al. (2003). Essential role of A-kinase anchor protein 121 for cAMP signaling to mitochondria. J. Biol. Chem. 278 (6), 4286–4294. doi:10.1074/jbc.M209941200
Ahmad, F., Shen, W., Vandeput, F., Szabo-Fresnais, N., Krall, J., Degerman, E., et al. (2015). Regulation of sarcoplasmic reticulum Ca2+ ATPase 2 (SERCA2) activity by phosphodiesterase 3A (PDE3A) in human myocardium: phosphorylation-dependent interaction of PDE3A1 with SERCA2. J. Biol. Chem. 290 (11), 6763–6776. doi:10.1074/jbc.M115.638585
Appert-Collin, A., Cotecchia, S., Nenniger-Tosato, M., Pedrazzini, T., and Diviani, D. (2007). The A-kinase anchoring protein (AKAP)-Lbc-signaling complex mediates alpha1 adrenergic receptor-induced cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 104 (24), 10140–10145. doi:10.1073/pnas.0701099104
Bai, Y., Jones, P. P., Guo, J., Zhong, X., Clark, R. B., Zhou, Q., et al. (2013). Phospholamban knockout breaks arrhythmogenic Ca2+ waves and suppresses catecholaminergic polymorphic ventricular tachycardia in mice. Circ. Res. 113 (5), 517–526. doi:10.1161/CIRCRESAHA.113.301678
Bedioune, I., Gandon-Renard, M., Dessillons, M., Barthou, A., Varin, A., Mika, D., et al. (2024). Essential role of the RIα subunit of cAMP-dependent protein kinase in regulating cardiac contractility and heart failure development. Circulation 150 (25), 2031–2045. doi:10.1161/CIRCULATIONAHA.124.068858
Bedioune, I., Lefebvre, F., Lechene, P., Varin, A., Domergue, V., Kapiloff, M. S., et al. (2018). PDE4 and mAKAPβ are nodal organizers of β2-ARs nuclear PKA signalling in cardiac myocytes. Cardiovasc Res. 114 (11), 1499–1511. doi:10.1093/cvr/cvy110
Belevych, A. E., Radwanski, P. B., Carnes, C. A., and Gyorke, S. (2013). 'Ryanopathy': causes and manifestations of RyR2 dysfunction in heart failure. Cardiovasc Res. 98 (2), 240–247. doi:10.1093/cvr/cvt024
Bossuyt, J., Chang, C. W., Helmstadter, K., Kunkel, M. T., Newton, A. C., Campbell, K. S., et al. (2011). Spatiotemporally distinct protein kinase D activation in adult cardiomyocytes in response to phenylephrine and endothelin. J. Biol. Chem. 286 (38), 33390–33400. doi:10.1074/jbc.M111.246447
Bugger, H., and Pfeil, K. (2020). Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 1866 (7), 165768. doi:10.1016/j.bbadis.2020.165768
Carlson, C. R., Aronsen, J. M., Bergan-Dahl, A., Moutty, M. C., Lunde, M., Lunde, P. K., et al. (2022). AKAP18δ anchors and regulates CaMKII activity at phospholamban-SERCA2 and RYR. Circ. Res. 130 (1), 27–44. doi:10.1161/CIRCRESAHA.120.317976
Carnegie, G. K., Soughayer, J., Smith, F. D., Pedroja, B. S., Zhang, F., Diviani, D., et al. (2008). AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol. Cell 32 (2), 169–179. doi:10.1016/j.molcel.2008.08.030
Caso, S., Maric, D., Arambasic, M., Cotecchia, S., and Diviani, D. (2017). AKAP-Lbc mediates protection against doxorubicin-induced cardiomyocyte toxicity. Biochim. Biophys. Acta Mol. Cell Res. 1864 (12), 2336–2346. doi:10.1016/j.bbamcr.2017.09.007
Chen, L., and Kass, R. S. (2006). Dual roles of the A kinase-anchoring protein Yotiao in the modulation of a cardiac potassium channel: a passive adaptor versus an active regulator. Eur. J. Cell Biol. 85 (7), 623–626. doi:10.1016/j.ejcb.2006.03.002
Chen, L., and Kass, R. S. (2011). A-kinase anchoring protein 9 and IKs channel regulation. J. Cardiovasc Pharmacol. 58 (5), 459–13. doi:10.1097/FJC.0b013e318232c80c
Chen, L., Marquardt, M. L., Tester, D. J., Sampson, K. J., Ackerman, M. J., and Kass, R. S. (2007). Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc. Natl. Acad. Sci. U. S. A. 104 (52), 20990–20995. doi:10.1073/pnas.0710527105
Chen, Q., Zheng, A., Xu, X., Shi, Z., Yang, M., Sun, S., et al. (2025). Nrf3-Mediated mitochondrial superoxide promotes cardiomyocyte apoptosis and impairs cardiac functions by suppressing Pitx2. Circulation 151 (14), 1024–1046. doi:10.1161/CIRCULATIONAHA.124.070286
Chen, Y. L., Daneva, Z., Kuppusamy, M., Ottolini, M., Baker, T. M., Klimentova, E., et al. (2022). Novel smooth muscle Ca(2+)-signaling nanodomains in blood pressure regulation. Circulation 146 (7), 548–564. doi:10.1161/CIRCULATIONAHA.121.058607
Cheng, E. P., Yuan, C., Navedo, M. F., Dixon, R. E., Nieves-Cintron, M., Scott, J. D., et al. (2011). Restoration of normal L-type Ca2+ channel function during Timothy syndrome by ablation of an anchoring protein. Circ. Res. 109 (3), 255–261. doi:10.1161/CIRCRESAHA.111.248252
Cheng, K. C., Chang, W. T., Kuo, F. Y., Chen, Z. C., Li, Y., and Cheng, J. T. (2019). TGR5 activation ameliorates hyperglycemia-induced cardiac hypertrophy in H9c2 cells. Sci. Rep. 9 (1), 3633. doi:10.1038/s41598-019-40002-0
Christian, F., Szaszak, M., Friedl, S., Drewianka, S., Lorenz, D., Goncalves, A., et al. (2011). Small molecule AKAP-protein kinase A (PKA) interaction disruptors that activate PKA interfere with compartmentalized cAMP signaling in cardiac myocytes. J. Biol. Chem. 286 (11), 9079–9096. doi:10.1074/jbc.M110.160614
Cibi, D. M., Bi-Lin, K. W., Shekeran, S. G., Sandireddy, R., Tee, N., Singh, A., et al. (2020). Prdm16 deficiency leads to age-dependent cardiac hypertrophy, adverse remodeling, mitochondrial dysfunction, and heart failure. Cell Rep. 33 (3), 108288. doi:10.1016/j.celrep.2020.108288
Colombe, A. S., and Pidoux, G. (2021). Cardiac cAMP-PKA signaling compartmentalization in myocardial infarction. Cells 10 (4), 922. doi:10.3390/cells10040922
Czachor, A., Failla, A., Lockey, R., and Kolliputi, N. (2016). Pivotal role of AKAP121 in mitochondrial physiology. Am. J. Physiol. Cell Physiol. 310 (8), C625–C628. doi:10.1152/ajpcell.00292.2015
Czepiel, M., Diviani, D., Jazwa-Kusior, A., Tkacz, K., Rolski, F., Smolenski, R. T., et al. (2022). Angiotensin II receptor 1 controls profibrotic Wnt/beta-catenin signalling in experimental autoimmune myocarditis. Cardiovasc Res. 118 (2), 573–584. doi:10.1093/cvr/cvab039
Delaunay, M., Osman, H., Kaiser, S., and Diviani, D. (2019). The role of cyclic AMP signaling in cardiac fibrosis. Cells 9 (1), 69. doi:10.3390/cells9010069
De Windt, L. J., Lim, H. W., Bueno, O. F., Liang, Q., Delling, U., Braz, J. C., et al. (2001). Targeted inhibition of calcineurin attenuates cardiac hypertrophy in vivo. Proc. Natl. Acad. Sci. U. S. A. 98 (6), 3322–3327. doi:10.1073/pnas.031371998
Diviani, D., Dodge-Kafka, K. L., Li, J., and Kapiloff, M. S. (2011). A-kinase anchoring proteins: scaffolding proteins in the heart. Am. J. Physiol. Heart Circ. Physiol. 301 (5), H1742–H1753. doi:10.1152/ajpheart.00569.2011
Do, T. Q., and Knollmann, B. C. (2025). Inhibitors of intracellular RyR2 calcium release channels as therapeutic agents in arrhythmogenic heart diseases. Annu. Rev. Pharmacol. Toxicol. 65 (1), 443–463. doi:10.1146/annurev-pharmtox-061724-080739
Dodge-Kafka, K. L., and Kapiloff, M. S. (2006). The mAKAP signaling complex: integration of cAMP, calcium, and MAP kinase signaling pathways. Eur. J. Cell Biol. 85 (7), 593–602. doi:10.1016/j.ejcb.2006.01.007
Dodge-Kafka, K. L., Soughayer, J., Pare, G. C., Carlisle Michel, J. J., Langeberg, L. K., Kapiloff, M. S., et al. (2005). The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437 (7058), 574–578. doi:10.1038/nature03966
Dondi, C., Vogler, G., Gupta, A., Walls, S. M., Kervadec, A., Marchant, J., et al. (2024). The nutrient sensor CRTC and Sarcalumenin/thinman represent an alternate pathway in cardiac hypertrophy. Cell Rep. 43 (8), 114549. doi:10.1016/j.celrep.2024.114549
Drum, B. M., Dixon, R. E., Yuan, C., Cheng, E. P., and Santana, L. F. (2014). Cellular mechanisms of ventricular arrhythmias in a mouse model of Timothy syndrome (long QT syndrome 8). J. Mol. Cell Cardiol. 66, 63–71. doi:10.1016/j.yjmcc.2013.10.021
Edwards, H. V., Scott, J. D., and Baillie, G. S. (2012). The A-kinase-anchoring protein AKAP-Lbc facilitates cardioprotective PKA phosphorylation of Hsp20 on Ser(16). Biochem. J. 446 (3), 437–443. doi:10.1042/BJ20120570
Ercu, M., and Klussmann, E. (2018). Roles of A-kinase anchoring proteins and phosphodiesterases in the cardiovascular system. J. Cardiovasc Dev. Dis. 5 (1), 14. doi:10.3390/jcdd5010014
Fang, H. Y., Hung, M. Y., Lin, Y. M., Pandey, S., Chang, C. C., Lin, K. H., et al. (2018). 17β-Estradiol and/or estrogen receptor alpha signaling blocks protein phosphatase 1 mediated ISO induced cardiac hypertrophy. PLoS One 13 (5), e0196569. doi:10.1371/journal.pone.0196569
Gao, R., Guo, W., Fan, T., Pang, J., Hou, Y., Feng, X., et al. (2022). Phosphodiesterase 4D contributes to angiotensin II-induced abdominal aortic aneurysm through smooth muscle cell apoptosis. Exp. Mol. Med. 54 (8), 1201–1213. doi:10.1038/s12276-022-00815-y
Guo, H., Liu, B., Hou, L., The, E., Li, G., Wang, D., et al. (2015). The role of mAKAPβ in the process of cardiomyocyte hypertrophy induced by angiotensin II. Int. J. Mol. Med. 35 (5), 1159–1168. doi:10.3892/ijmm.2015.2119
Han, X., Zhang, G., Pang, M., Hu, C., Xu, T., Wu, Y., et al. (2024). Taohong siwu decoction suppresses oxidative stress-induced myocardial apoptosis post-myocardial infarction by inhibiting PTEN pathway. Phytomedicine 135, 155388. doi:10.1016/j.phymed.2024.155388
Harada, H., Becknell, B., Wilm, M., Mann, M., Huang, L. J., Taylor, S. S., et al. (1999). Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol. Cell 3 (4), 413–422. doi:10.1016/s1097-2765(00)80469-4
Harvey, R. D., and Hell, J. W. (2013). CaV1.2 signaling complexes in the heart. J. Mol. Cell Cardiol. 58, 143–152. doi:10.1016/j.yjmcc.2012.12.006
Haushalter, K. J., Schilling, J. M., Song, Y., Sastri, M., Perkins, G. A., Strack, S., et al. (2019). Cardiac ischemia-reperfusion injury induces ROS-dependent loss of PKA regulatory subunit RIalpha. Am. J. Physiol. Heart Circ. Physiol. 317 (6), H1231–H1242. doi:10.1152/ajpheart.00237.2019
He, T., Huang, J., Chen, L., Han, G., Stanmore, D., Krebs-Haupenthal, J., et al. (2020). Cyclic AMP represses pathological MEF2 activation by myocyte-specific hypo-phosphorylation of HDAC5. J. Mol. Cell Cardiol. 145, 88–98. doi:10.1016/j.yjmcc.2020.05.018
Hegyi, B., Polonen, R. P., Hellgren, K. T., Ko, C. Y., Ginsburg, K. S., Bossuyt, J., et al. (2021). Cardiomyocyte Na(+) and Ca(2+) mishandling drives vicious cycle involving CaMKII, ROS, and ryanodine receptors. Basic Res. Cardiol. 116 (1), 58. doi:10.1007/s00395-021-00900-9
Hou, N., Mai, Y., Qiu, X., Yuan, W., Li, Y., Luo, C., et al. (2019). Carvacrol attenuates diabetic cardiomyopathy by modulating the PI3K/AKT/GLUT4 pathway in diabetic mice. Front. Pharmacol. 10, 998. doi:10.3389/fphar.2019.00998
Hulme, J. T., Westenbroek, R. E., Scheuer, T., and Catterall, W. A. (2006). Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac CaV1.2 channels during beta1-adrenergic regulation. Proc. Natl. Acad. Sci. U. S. A. 103 (44), 16574–16579. doi:10.1073/pnas.0607294103
Ibarrola, J., Sadaba, R., Martinez-Martinez, E., Garcia-Pena, A., Arrieta, V., Alvarez, V., et al. (2018). Aldosterone impairs mitochondrial function in human cardiac fibroblasts via A-kinase anchor protein 12. Sci. Rep. 8 (1), 6801. doi:10.1038/s41598-018-25068-6
Jiang, P., Huang, F., Chen, L., Zhou, H., Deng, Y., Li, L., et al. (2025). Intercellular NETwork-facilitated sarcoplasmic reticulum targeting for myocardial ischemia-reperfusion injury treatment. Sci. Adv. 11 (7), eadr4333. doi:10.1126/sciadv.adr4333
Kammerer, S., Burns-Hamuro, L. L., Ma, Y., Hamon, S. C., Canaves, J. M., Shi, M. M., et al. (2003). Amino acid variant in the kinase binding domain of dual-specific A kinase-anchoring protein 2: a disease susceptibility polymorphism. Proc. Natl. Acad. Sci. U. S. A. 100 (7), 4066–4071. doi:10.1073/pnas.2628028100
Kilian, L. S., Voran, J., Frank, D., and Rangrez, A. Y. (2021). RhoA: a dubious molecule in cardiac pathophysiology. J. Biomed. Sci. 28 (1), 33. doi:10.1186/s12929-021-00730-w
Kim, H., Scimia, M. C., Wilkinson, D., Trelles, R. D., Wood, M. R., Bowtell, D., et al. (2011). Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol. Cell 44 (4), 532–544. doi:10.1016/j.molcel.2011.08.045
Kinderman, F. S., Kim, C., von Daake, S., Ma, Y., Pham, B. Q., Spraggon, G., et al. (2006). A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol. Cell 24 (3), 397–408. doi:10.1016/j.molcel.2006.09.015
Koch, D., Alexandrovich, A., Funk, F., Kho, A. L., Schmitt, J. P., and Gautel, M. (2021). Molecular noise filtering in the beta-adrenergic signaling network by phospholamban pentamers. Cell Rep. 36 (4), 109448. doi:10.1016/j.celrep.2021.109448
Li, A., Shen, Y., Li, Z., and Li, L. (2025). Wnt/β-catenin pathway induces cardiac dysfunction via AKAP6-mediated RyR2 phosphorylation and sarcoplasmic reticulum calcium leakage. J. Mol. Cell Biol., mjaf002. doi:10.1093/jmcb/mjaf002
Li, J., Aponte Paris, S., Thakur, H., Kapiloff, M. S., and Dodge-Kafka, K. L. (2019). Muscle A-kinase-anchoring protein-beta-bound calcineurin toggles active and repressive transcriptional complexes of myocyte enhancer factor 2D. J. Biol. Chem. 294 (7), 2543–2554. doi:10.1074/jbc.RA118.005465
Li, J., Tan, Y., Passariello, C. L., Martinez, E. C., Kritzer, M. D., Li, X., et al. (2020). Signalosome-regulated serum response factor phosphorylation determining myocyte growth in width versus length as a therapeutic target for heart failure. Circulation 142 (22), 2138–2154. doi:10.1161/CIRCULATIONAHA.119.044805
Li, L., Li, J., Drum, B. M., Chen, Y., Yin, H., Guo, X., et al. (2017). Loss of AKAP150 promotes pathological remodelling and heart failure propensity by disrupting calcium cycling and contractile reserve. Cardiovasc Res. 113 (2), 147–159. doi:10.1093/cvr/cvw221
Li, X., Matta, S. M., Sullivan, R. D., and Bahouth, S. W. (2014). Carvedilol reverses cardiac insufficiency in AKAP5 knockout mice by normalizing the activities of calcineurin and CaMKII. Cardiovasc Res. 104 (2), 270–279. doi:10.1093/cvr/cvu209
Liu, Y., Chen, J., Fontes, S. K., Bautista, E. N., and Cheng, Z. (2022). Physiological and pathological roles of protein kinase A in the heart. Cardiovasc Res. 118 (2), 386–398. doi:10.1093/cvr/cvab008
Liu, Y., Merrill, R. A., and Strack, S. (2020). A-kinase anchoring protein 1: emerging roles in regulating mitochondrial form and function in health and disease. Cells 9 (2), 298. doi:10.3390/cells9020298
Livigni, A., Scorziello, A., Agnese, S., Adornetto, A., Carlucci, A., Garbi, C., et al. (2006). Mitochondrial AKAP121 links cAMP and src signaling to oxidative metabolism. Mol. Biol. Cell 17 (1), 263–271. doi:10.1091/mbc.e05-09-0827
Loniewska, B., Kaczmarczyk, M., Clark, J. S., Goracy, I., Horodnicka-Jozwa, A., and Ciechanowicz, A. (2015). Association of functional genetic variants of A-kinase anchoring protein 10 with QT interval length in full-term Polish newborns. Arch. Med. Sci. 11 (1), 149–154. doi:10.5114/aoms.2013.34172
Luo, Y., Jiang, N., May, H. I., Luo, X., Ferdous, A., Schiattarella, G. G., et al. (2021). Cooperative binding of ETS2 and NFAT links erk1/2 and calcineurin signaling in the pathogenesis of cardiac hypertrophy. Circulation 144 (1), 34–51. doi:10.1161/CIRCULATIONAHA.120.052384
Lygren, B., Carlson, C. R., Santamaria, K., Lissandron, V., McSorley, T., Litzenberg, J., et al. (2007). AKAP complex regulates Ca2+ re-uptake into heart sarcoplasmic reticulum. EMBO Rep. 8 (11), 1061–1067. doi:10.1038/sj.embor.7401081
MacLennan, D. H., and Kranias, E. G. (2003). Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 4 (7), 566–577. doi:10.1038/nrm1151
Maric, D., Paterek, A., Delaunay, M., Lopez, I. P., Arambasic, M., and Diviani, D. (2021). A-kinase anchoring protein 2 promotes protection against myocardial infarction. Cells 10 (11), 2861. doi:10.3390/cells10112861
Marin, W. (2020). A-kinase anchoring protein 1 (AKAP1) and its role in some cardiovascular diseases. J. Mol. Cell Cardiol. 138, 99–109. doi:10.1016/j.yjmcc.2019.11.154
Marx, S. O., Kurokawa, J., Reiken, S., Motoike, H., D'Armiento, J., Marks, A. R., et al. (2002). Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 295 (5554), 496–499. doi:10.1126/science.1066843
Marx, S. O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N., et al. (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101 (4), 365–376. doi:10.1016/s0092-8674(00)80847-8
Mayers, C. M., Wadell, J., McLean, K., Venere, M., Malik, M., Shibata, T., et al. (2010). The Rho guanine nucleotide exchange factor AKAP13 (BRX) is essential for cardiac development in mice. J. Biol. Chem. 285 (16), 12344–12354. doi:10.1074/jbc.M110.106856
Mazzocchi, G., Sommese, L., Palomeque, J., Felice, J. I., Di Carlo, M. N., Fainstein, D., et al. (2016). Phospholamban ablation rescues the enhanced propensity to arrhythmias of mice with CaMKII-constitutive phosphorylation of RyR2 at site S2814. J. Physiol. 594 (11), 3005–3030. doi:10.1113/JP271622
McConnell, B. K., Popovic, Z., Mal, N., Lee, K., Bautista, J., Forudi, F., et al. (2009). Disruption of protein kinase A interaction with A-kinase-anchoring proteins in the heart in vivo: effects on cardiac contractility, protein kinase A phosphorylation, and troponin I proteolysis. J. Biol. Chem. 284 (3), 1583–1592. doi:10.1074/jbc.M806321200
Mercado, J., Baylie, R., Navedo, M. F., Yuan, C., Scott, J. D., Nelson, M. T., et al. (2014). Local control of TRPV4 channels by AKAP150-targeted PKC in arterial smooth muscle. J. Gen. Physiol. 143 (5), 559–575. doi:10.1085/jgp.201311050
Morgat, C., Fressart, V., Porretta, A. P., Neyroud, N., Messali, A., Temmar, Y., et al. (2024). Genetic characterization of KCNQ1 variants improves risk stratification in type 1 long QT syndrome patients. Europace 26 (6), euae136. doi:10.1093/europace/euae136
Musheshe, N., Schmidt, M., and Zaccolo, M. (2018). cAMP: from long-range second messenger to nanodomain signalling. Trends Pharmacol. Sci. 39 (2), 209–222. doi:10.1016/j.tips.2017.11.006
Navedo, M. F., Nieves-Cintron, M., Amberg, G. C., Yuan, C., Votaw, V. S., Lederer, W. J., et al. (2008). AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin II-induced hypertension. Circ. Res. 102 (2), e1–e11. doi:10.1161/CIRCRESAHA.107.167809
Nicol, R. L., Frey, N., Pearson, G., Cobb, M., Richardson, J., and Olson, E. N. (2001). Activated MEK5 induces serial assembly of sarcomeres and eccentric cardiac hypertrophy. EMBO J. 20 (11), 2757–2767. doi:10.1093/emboj/20.11.2757
Nystoriak, M. A., Nieves-Cintron, M., Nygren, P. J., Hinke, S. A., Nichols, C. B., Chen, C. Y., et al. (2014). AKAP150 contributes to enhanced vascular tone by facilitating large-conductance Ca2+-activated K+ channel remodeling in hyperglycemia and diabetes mellitus. Circ. Res. 114 (4), 607–615. doi:10.1161/CIRCRESAHA.114.302168
Nystoriak, M. A., Nieves-Cintron, M., Patriarchi, T., Buonarati, O. R., Prada, M. P., Morotti, S., et al. (2017). Ser1928 phosphorylation by PKA stimulates the L-type Ca2+ channel CaV1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci. Signal. 10 (463), eaaf9647. doi:10.1126/scisignal.aaf9647
Omar, M. H., and Scott, J. D. (2020). AKAP signaling islands: venues for precision pharmacology. Trends Pharmacol. Sci. 41 (12), 933–946. doi:10.1016/j.tips.2020.09.007
Ong, S. B., Hernandez-Resendiz, S., Crespo-Avilan, G. E., Mukhametshina, R. T., Kwek, X. Y., Cabrera-Fuentes, H. A., et al. (2018). Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol. Ther. 186, 73–87. doi:10.1016/j.pharmthera.2018.01.001
Ottolini, M., Hong, K., Cope, E. L., Daneva, Z., DeLalio, L. J., Sokolowski, J. D., et al. (2020). Local peroxynitrite impairs endothelial transient receptor potential vanilloid 4 channels and elevates blood pressure in obesity. Circulation 141 (16), 1318–1333. doi:10.1161/CIRCULATIONAHA.119.043385
Pallien, T., and Klussmann, E. (2020). New aspects in cardiac L-type Ca2+ channel regulation. Biochem. Soc. Trans. 48 (1), 39–49. doi:10.1042/BST20190229
Paolillo, R., D'Apice, S., Schiattarella, G. G., Ameri, P., Borzacchiello, D., Catalucci, D., et al. (2022). Mitochondrial a kinase anchor proteins in cardiovascular health and disease: a review article on behalf of the working group on cellular and molecular biology of the heart of the Italian society of cardiology. Int. J. Mol. Sci. 23 (14), 7691. doi:10.3390/ijms23147691
Papa, A., Kushner, J., and Marx, S. O. (2022). Adrenergic regulation of calcium channels in the heart. Annu. Rev. Physiol. 84, 285–306. doi:10.1146/annurev-physiol-060121-041653
Pereira da Silva, E. A., Martin-Aragon Baudel, M., Navedo, M. F., and Nieves-Cintron, M. (2022). Ion channel molecular complexes in vascular smooth muscle. Front. Physiol. 13, 999369. doi:10.3389/fphys.2022.999369
Perino, A., Ghigo, A., Scott, J. D., and Hirsch, E. (2012). Anchoring proteins as regulators of signaling pathways. Circ. Res. 111 (4), 482–492. doi:10.1161/CIRCRESAHA.111.262899
Perrino, C., Feliciello, A., Schiattarella, G. G., Esposito, G., Guerriero, R., Zaccaro, L., et al. (2010). AKAP121 downregulation impairs protective cAMP signals, promotes mitochondrial dysfunction, and increases oxidative stress. Cardiovasc Res. 88 (1), 101–110. doi:10.1093/cvr/cvq155
Pitt, G. S., Matsui, M., and Cao, C. (2021). Voltage-gated calcium channels in nonexcitable tissues. Annu. Rev. Physiol. 83, 183–203. doi:10.1146/annurev-physiol-031620-091043
Prada, M. P., Syed, A. U., Reddy, G. R., Martin-Aragon Baudel, M., Flores-Tamez, V. A., Sasse, K. C., et al. (2020). AKAP5 complex facilitates purinergic modulation of vascular L-type Ca(2+) channel Ca(V)1.2. Nat. Commun. 11 (1), 5303. doi:10.1038/s41467-020-18947-y
Qi, B., He, L., Zhao, Y., Zhang, L., He, Y., Li, J., et al. (2020). Akap1 deficiency exacerbates diabetic cardiomyopathy in mice by NDUFS1-mediated mitochondrial dysfunction and apoptosis. Diabetologia 63 (5), 1072–1087. doi:10.1007/s00125-020-05103-w
Qi, B., Song, L., Hu, L., Guo, D., Ren, G., Peng, T., et al. (2022). Cardiac-specific overexpression of Ndufs1 ameliorates cardiac dysfunction after myocardial infarction by alleviating mitochondrial dysfunction and apoptosis. Exp. Mol. Med. 54 (7), 946–960. doi:10.1038/s12276-022-00800-5
Rababa'h, A., Singh, S., Suryavanshi, S. V., Altarabsheh, S. E., Deo, S. V., and McConnell, B. K. (2014). Compartmentalization role of A-kinase anchoring proteins (AKAPs) in mediating protein kinase A (PKA) signaling and cardiomyocyte hypertrophy. Int. J. Mol. Sci. 16 (1), 218–229. doi:10.3390/ijms16010218
Ren, A. J., Wei, C., Liu, Y. J., Liu, M., Wang, P., Fan, J., et al. (2024). ZBTB20 regulates SERCA2a activity and myocardial contractility through phospholamban. Circ. Res. 134 (3), 252–265. doi:10.1161/CIRCRESAHA.123.323798
Rhana, P., Matsumoto, C., Fong, Z., Costa, A. D., Del Villar, S. G., Dixon, R. E., et al. (2024). Fueling the heartbeat: dynamic regulation of intracellular ATP during excitation-contraction coupling in ventricular myocytes. Proc. Natl. Acad. Sci. U. S. A. 121 (25), e2318535121. doi:10.1073/pnas.2318535121
Ripoll, L., Li, Y., Dessauer, C. W., and von Zastrow, M. (2024). Spatial organization of adenylyl cyclase and its impact on dopamine signaling in neurons. Nat. Commun. 15 (1), 8297. doi:10.1038/s41467-024-52575-0
Schiattarella, G. G., Boccella, N., Paolillo, R., Cattaneo, F., Trimarco, V., Franzone, A., et al. (2018). Loss of Akap1 exacerbates pressure overload-induced cardiac hypertrophy and heart failure. Front. Physiol. 9, 558. doi:10.3389/fphys.2018.00558
Schiattarella, G. G., Cattaneo, F., Pironti, G., Magliulo, F., Carotenuto, G., Pirozzi, M., et al. (2016). Akap1 deficiency promotes mitochondrial aberrations and exacerbates cardiac injury following permanent coronary ligation via enhanced mitophagy and apoptosis. PLoS One 11 (5), e0154076. doi:10.1371/journal.pone.0154076
Schulze, D. H., Muqhal, M., Lederer, W. J., and Ruknudin, A. M. (2003). Sodium/calcium exchanger (NCX1) macromolecular complex. J. Biol. Chem. 278 (31), 28849–28855. doi:10.1074/jbc.M300754200
Scott, J. D., Dessauer, C. W., and Tasken, K. (2013). Creating order from chaos: cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol. 53, 187–210. doi:10.1146/annurev-pharmtox-011112-140204
Shan, J., Betzenhauser, M. J., Kushnir, A., Reiken, S., Meli, A. C., Wronska, A., et al. (2010). Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J. Clin. Invest. 120 (12), 4375–4387. doi:10.1172/JCI37649
Shu, H., Hang, W., Peng, Y., Nie, J., Wu, L., Zhang, W., et al. (2021). Trimetazidine attenuates heart failure by improving myocardial metabolism via AMPK. Front. Pharmacol. 12, 707399. doi:10.3389/fphar.2021.707399
Soni, S., Scholten, A., Vos, M. A., and van Veen, T. A. (2014). Anchored protein kinase A signalling in cardiac cellular electrophysiology. J. Cell Mol. Med. 18 (11), 2135–2146. doi:10.1111/jcmm.12365
Subramanian, H., and Nikolaev, V. O. (2023). A-kinase anchoring proteins in cardiac myocytes and their roles in regulating calcium cycling. Cells 12 (3), 436. doi:10.3390/cells12030436
Szentesi, P., Pignier, C., Egger, M., Kranias, E. G., and Niggli, E. (2004). Sarcoplasmic reticulum Ca2+ refilling controls recovery from Ca2+-induced Ca2+ release refractoriness in heart muscle. Circ. Res. 95 (8), 807–813. doi:10.1161/01.RES.0000146029.80463.7d
Taglieri, D. M., Johnson, K. R., Burmeister, B. T., Monasky, M. M., Spindler, M. J., DeSantiago, J., et al. (2014). The C-terminus of the long AKAP13 isoform (AKAP-Lbc) is critical for development of compensatory cardiac hypertrophy. J. Mol. Cell Cardiol. 66, 27–40. doi:10.1016/j.yjmcc.2013.10.010
Tingley, W. G., Pawlikowska, L., Zaroff, J. G., Kim, T., Nguyen, T., Young, S. G., et al. (2007). Gene-trapped mouse embryonic stem cell-derived cardiac myocytes and human genetics implicate AKAP10 in heart rhythm regulation. Proc. Natl. Acad. Sci. U. S. A. 104 (20), 8461–8466. doi:10.1073/pnas.0610393104
Tomek, J., and Zaccolo, M. (2023). Compartmentalized cAMP signalling and control of cardiac rhythm. Philos. Trans. R. Soc. Lond B Biol. Sci. 378 (1879), 20220172. doi:10.1098/rstb.2022.0172
Touyz, R. M., Alves-Lopes, R., Rios, F. J., Camargo, L. L., Anagnostopoulou, A., Arner, A., et al. (2018). Vascular smooth muscle contraction in hypertension. Cardiovasc Res. 114 (4), 529–539. doi:10.1093/cvr/cvy023
Troger, J., Moutty, M. C., Skroblin, P., and Klussmann, E. (2012). A-kinase anchoring proteins as potential drug targets. Br. J. Pharmacol. 166 (2), 420–433. doi:10.1111/j.1476-5381.2011.01796.x
Tsai, K. H., Wang, W. J., Lin, C. W., Pai, P., Lai, T. Y., Tsai, C. Y., et al. (2012). NADPH oxidase-derived superoxide anion-induced apoptosis is mediated via the JNK-dependent activation of NF-κB in cardiomyocytes exposed to high glucose. J. Cell Physiol. 227 (4), 1347–1357. doi:10.1002/jcp.22847
Turcotte, M. G., Thakur, H., Kapiloff, M. S., and Dodge-Kafka, K. L. (2022). A perinuclear calcium compartment regulates cardiac myocyte hypertrophy. J. Mol. Cell Cardiol. 172, 26–40. doi:10.1016/j.yjmcc.2022.07.007
Vergarajauregui, S., Becker, R., Steffen, U., Sharkova, M., Esser, T., Petzold, J., et al. (2020). AKAP6 orchestrates the nuclear envelope microtubule-organizing center by linking golgi and nucleus via AKAP9. Elife 9, e61669. doi:10.7554/eLife.61669
Wang, Z., Zhang, X., Zhu, F., Zhou, S., Wang, Q., and Wang, H. (2022). A-kinase anchoring protein 5 anchors protein kinase A to mediate PLN/SERCA to reduce cardiomyocyte apoptosis induced by hypoxia and reoxygenation. Biochem. Cell Biol. 100 (2), 162–170. doi:10.1139/bcb-2021-0466
Weber, D. K., Reddy, U. V., Wang, S., Larsen, E. K., Gopinath, T., Gustavsson, M. B., et al. (2021). Structural basis for allosteric control of the SERCA-Phospholamban membrane complex by Ca(2+) and phosphorylation. Elife 10, e66226. doi:10.7554/eLife.66226
Wu, C., Zhang, Z., Zhang, W., and Liu, X. (2022). Mitochondrial dysfunction and mitochondrial therapies in heart failure. Pharmacol. Res. 175, 106038. doi:10.1016/j.phrs.2021.106038
Wu, X., and Larsson, H. P. (2020). Insights into cardiac IKs (KCNQ1/KCNE1) channels regulation. Int. J. Mol. Sci. 21 (24), 9440. doi:10.3390/ijms21249440
Xiang, S. Y., Ouyang, K., Yung, B. S., Miyamoto, S., Smrcka, A. V., Chen, J., et al. (2013). PLCε, PKD1, and SSH1L transduce RhoA signaling to protect mitochondria from oxidative stress in the heart. Sci. Signal 6 (306), ra108. doi:10.1126/scisignal.2004405
Xiao, B., Jiang, M. T., Zhao, M., Yang, D., Sutherland, C., Lai, F. A., et al. (2005). Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ. Res. 96 (8), 847–855. doi:10.1161/01.RES.0000163276.26083.e8
Xu, Z., Hussain, A., Jiang, H., Zhang, H., Raza, Y., Mustafa, G., et al. (2025). AKAP14 is dispensable for mouse fertility. Cells Dev., 204032. doi:10.1016/j.cdev.2025.204032
You, Y., Hersh, S. W., Aslebagh, R., Shaffer, S. A., Ikezu, S., Mez, J., et al. (2022). Alzheimer's disease associated AKAP9 I2558M mutation alters posttranslational modification and interactome of tau and cellular functions in CRISPR-edited human neuronal cells. Aging Cell 21 (6), e13617. doi:10.1111/acel.13617
Zeng, C., Wang, J., Li, N., Shen, M., Wang, D., Yu, Q., et al. (2014). AKAP150 mobilizes cPKC-dependent cardiac glucotoxicity. Am. J. Physiol. Endocrinol. Metab. 307 (4), E384–E397. doi:10.1152/ajpendo.00175.2014
Zhang, H., Liu, Y., Liu, J., Chen, J., Wang, J., Hua, H., et al. (2024). cAMP-PKA/EPAC signaling and cancer: the interplay in tumor microenvironment. J. Hematol. Oncol. 17 (1), 5. doi:10.1186/s13045-024-01524-x
Zhang, L., Malik, S., Kelley, G. G., Kapiloff, M. S., and Smrcka, A. V. (2011). Phospholipase C epsilon scaffolds to muscle-specific A kinase anchoring protein (mAKAPbeta) and integrates multiple hypertrophic stimuli in cardiac myocytes. J. Biol. Chem. 286 (26), 23012–23021. doi:10.1074/jbc.M111.231993
Zhang, L., Malik, S., Pang, J., Wang, H., Park, K. M., Yule, D. I., et al. (2013). Phospholipase Cε hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell 153 (1), 216–227. doi:10.1016/j.cell.2013.02.047
Zhang, X., Wang, Q., Wang, Z., Zhang, H., Zhu, F., Ma, J., et al. (2023). Interaction between A-kinase anchoring protein 5 and protein kinase A mediates CaMKII/HDAC signaling to inhibit cardiomyocyte hypertrophy after hypoxic reoxygenation. Cell Signal 103, 110569. doi:10.1016/j.cellsig.2022.110569
Zhu, F., Yuan, C., Zhang, X., Wang, Z., Wang, Q., and Wang, H. (2022). A-kinase anchoring protein 5-ancored calcineurin regulates the remodeling of H9c2 cardiomyocytes exposed to hypoxia and reoxygenation. Biomed. Pharmacother. 155, 113689. doi:10.1016/j.biopha.2022.113689
Glossary
AC adenylate cyclase
ADP adenosine diphosphate
AKAP A-kinase anchoring protein
Akt protein kinase B
AS160 Akt substrate of 160 kDa
ATP adenosine triphosphate
BAD Bcl-2 antagonist of cell death
Bax Bcl-2-associated X protein
Bcl-2 anti-apoptotic B cell leukemia/lymphoma 2
BKCa large conductance calcium-activated potassium channel
CaMKII Ca2+/calmodulin-dependent protein kinase type II
cAMP cyclic-3′,5′-adenosine monophosphate
CaN calcineurin
CaV1.2 voltage-gated calcium channels
Cav3 caveolin 3
DAG diacylglycerol
Drp1 dynamin-related protein 1
Epac1 exchange protein activated by cAMP 1
ERK5 extracellular signal-regulated kinase 5
Fis1 mitochondrial fission 1 protein
GLUT4 glucose transporter type-4
Gα12 G protein subunit alpha 12
HDAC histone deacetylase
IKs slow component of the delayed rectifier K+ current
KCNQ1 potassium voltage-gated channel subfamily Q member 1
LIF leukemia inhibitory factor
LQT8 type 8 long QT syndrome
mAKAP muscle-specific A kinase anchoring protein
MEF2 myocyte enhancer factor 2
NCX1 sodium/calcium exchanger
NDUFS1 ubiquinone oxidoreductase core subunit S1
NFAT nuclear factor of activated T cells
NFkB nuclear factor kappa B
OXPHOS oxidative phosphorylation
p47(phox) a component of NADPH oxidase
PDE4D3 phosphodiesterase-4D3
PI4P phosphatidylinositol 4-phosphate
PKA protein kinase A
PKC protein kinase C
PKD protein kinase D
PLCε phospholipase Cε
PLN phospholamban
PP1 Ser/Thr protein phosphatase 1
PTPD1 protein-tyrosine phosphatase D1
RhoA Ras homolog gene family member A
ROS reactive oxygen species
RyR2 ryanodine receptor 2
RyRs ryanodine receptor
SERCA sarco/endoplasmic reticulum Ca2+-ATPase
Siah2 seven in absentia homolog 2
Src a non-receptor tyrosine kinase protein
SSH1L slingshot-1L
TRPV4 transient receptor potential vanilloid 4
Keywords: A-kinase anchoring proteins, scaffold proteins, cellular microdomains, cardiovascular diseases, cardiovascular drugs
Citation: Zhang X, Zhu F, Xiao Z and Wang H (2025) The role of A-kinase anchoring proteins in cardiovascular diseases and recent advances. Front. Cell Dev. Biol. 13:1611583. doi: 10.3389/fcell.2025.1611583
Received: 14 April 2025; Accepted: 03 June 2025;
Published: 17 June 2025.
Edited by:
Martin Werner Berchtold, University of Copenhagen, DenmarkReviewed by:
Naveed Aslam, BioSystOmics, United StatesVirginia Actis Dato, University of California, San Diego, United States
Copyright © 2025 Zhang, Zhu, Xiao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hegui Wang, d2FuZ2hlZ3VpQHdubWMuZWR1LmNu
†These authors share first authorship