 Casey Griffin
Casey Griffin- Department of Molecular Pathobiology, College of Dentistry, New York University, New York, United States
Craniofacial spliceosomopathies are syndromes resulting from mutations in components of the spliceosome, presenting with facial dysostosis in combination with other phenotypes. An outstanding question in the field is how mutations in the ubiquitously expressed spliceosome lead to such cell- and tissue-specific disorders. To understand the etiology of these diseases and decipher the underlying mechanisms, scientists have turned to modeling these disorders in the laboratory. In vivo modeling of these disorders includes the use of mice, zebrafish, and frogs, whereas in vitro modeling typically uses embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). The goal with these models is to recapitulate the human disorders in a manner that is conducive to scientific exploration. In this review, we briefly describe the major craniofacial spliceosomopathies and discuss recent advances using model systems that have helped understand the root cause of these conditions.
Introduction
The spliceosome is a complex of RNA and proteins that functions to process pre-messenger RNA (pre-mRNA) into mRNA by identifying introns, splicing them out, and joining the exons. The steps of pre-mRNA splicing are as follows: (1) 5′ intron recognition, (2) 3′ intron recognition, (3) pre-catalytic spliceosome recruitment, (4) catalytic activation, and (5) exon joining (Will and Lührmann, 2011; Griffin and Saint-Jeannet, 2020). The major spliceosome is made up of five U subunits: U1, U2, U4, U5, and U6, which are each composed of small nuclear RNAs (snRNAs) associated with small nuclear ribonucleoproteins (snRNPs) and other proteins (Will and Lührmann, 2011). In the minor spliceosome, which is involved in the recognition of rare introns (Verma et al., 2018), the U2 subunit is replaced by the U12 snRNA.
Mutations in any of the components of the spliceosome can give rise to diseases known as spliceosomopathies. Although the spliceosome is active in all cells of the body to process pre-mRNA, most spliceosomopathies are cell- or tissue-specific in their manifestation and, as such, represent a conundrum in the field to understand the mechanism underlying these pathologies. The four major classes of spliceosomopathies are retinitis pigmentosa, myelodysplastic syndromes, cancers, and craniofacial spliceosomopathies (Griffin and Saint-Jeannet, 2020). Retinitis pigmentosa is a genetic disorder characterized by the deterioration of the photoreceptors of the retina, which can result in blindness (Hamel, 2006). Myelodysplastic syndromes are disorders in which there is defective hematopoiesis, affecting one or more hematopoietic lineages (Catenacci and Schiller, 2005). Many cancers can be caused by mutations in spliceosome components; aberrant splicing events have been linked to cancer proliferation, invasion, and metastasis (Mrid et al., 2025; Cao and Li, 2024; Bak-Gordon and Manley, 2025; Hermán-Sánchez et al., 2024; Stanley and Abdel-Wahab, 2022, and many more). Craniofacial spliceosomopathies are disorders in which mutations of the spliceosome cause defects in the skeletal elements of the craniofacial complex, more specifically, the neural crest-derived skeletal elements of the face (Lehalle et al., 2015).
The neural crest is an embryonic cell population that derives from the neural plate border as epithelial cells, undergoes an epithelial-to-mesenchymal transition, and then migrates through the pharyngeal arches to give rise to a variety of cell types, including the craniofacial skeleton. Although craniofacial spliceosomopathies cover a wide range of phenotypes and manifestations, they all share defects in the neural crest-derived structures of the face. In this review, we focus on craniofacial spliceosomopathies and the models that have been developed to study them, with the goal of discovering why mutations in the ubiquitously active spliceosomal complex give rise to such phenotypically specific disorders.
Craniofacial spliceosomopathies
Although craniofacial spliceosomopathies are rare diseases, they belong to the category of facial dysostoses, which represent one-third of all live births with congenital anomalies (Trainor and Andrews, 2013). The craniofacial component of these diseases often occurs in combination with other phenotypes (Figure 1; Table 1). Many of these disorders are due to loss-of-function mutations in genes that encode for spliceosomal proteins, making many of the patients haploinsufficient. The common features of these disorders are malformations of the derivates of the first and second pharyngeal arches, which occur during embryogenesis (Trainor and Andrews, 2013). These defects are typically considered maxillary, malar, and mandibular hypoplasia, cleft palate, and outer and/or middle ear defects. In particular, the skeletal defects seen in these disorders are developmental in nature and are mostly due to impairment of the neural crest. The majority of these disorders are non-lethal, with the current treatment involving reconstructive surgeries to ease pain and improve cosmetics, usually beginning at birth and continuing throughout adolescence and adulthood. Diagnostic criteria for these disorders include clinical assessment and genetic testing.
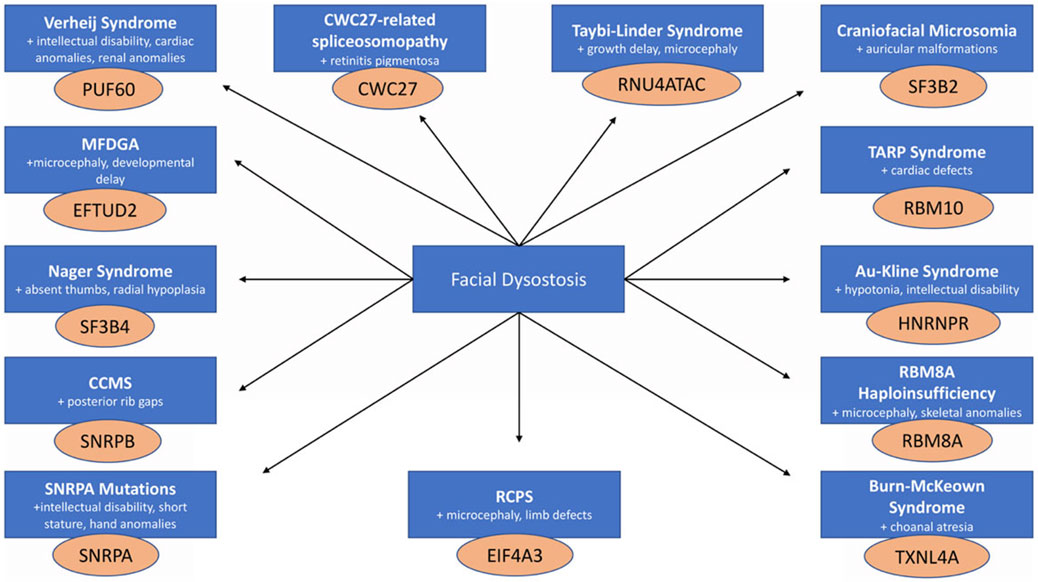
Figure 1. Craniofacial spliceosomopathies are all characterized by facial dysostoses that present in combination with a diverse array of phenotypes arising from mutations in genes that encode proteins of the spliceosome.
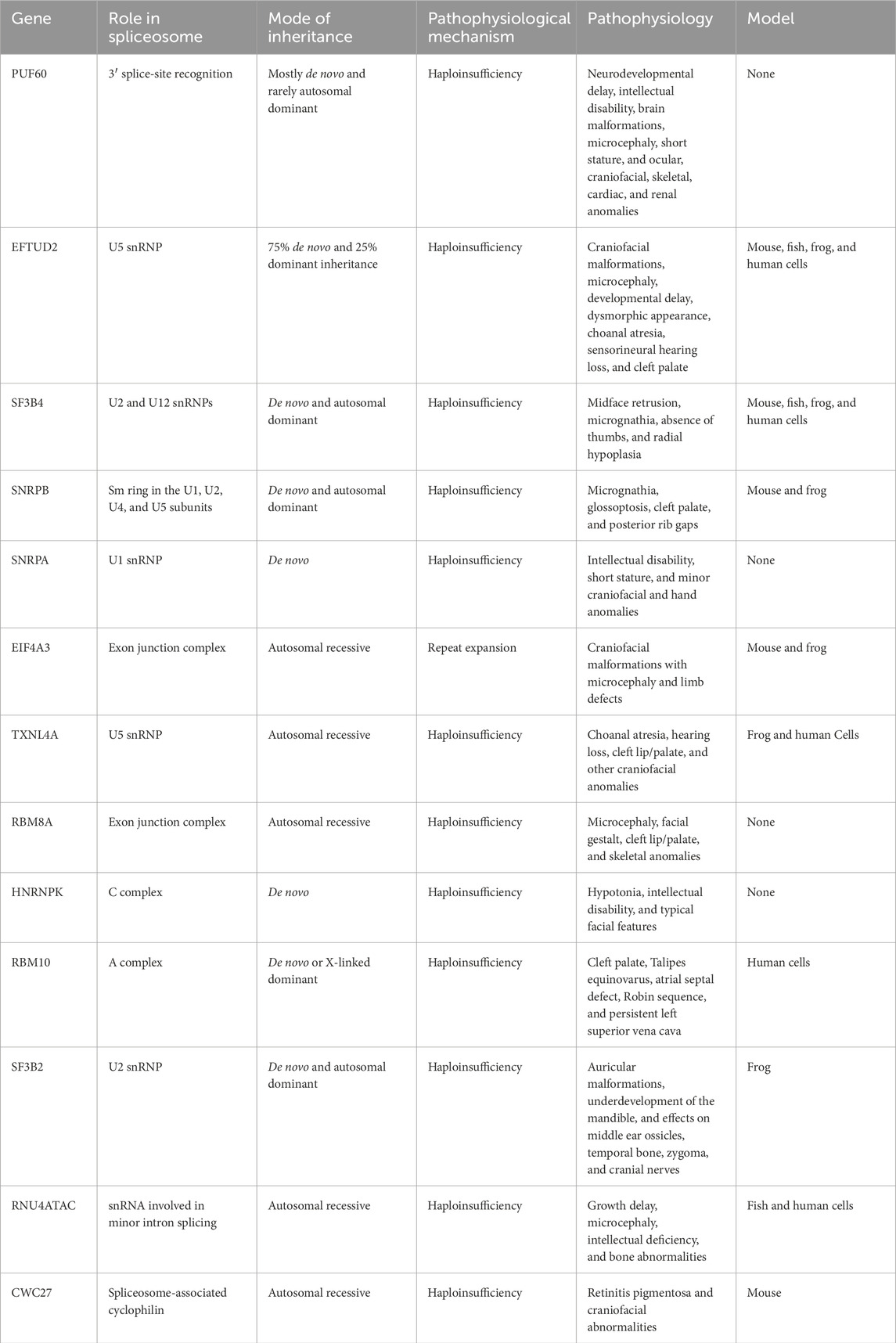
Table 1. List of genes causing craniofacial spliceosomopathies, characteristics of these genes, and types of models available.
Verheij syndrome
Verheij syndrome (OMIM #615583) involves a spectrum of phenotypes, including neurodevelopmental delay, intellectual disability, brain malformations, microcephaly, short stature, and ocular, craniofacial, skeletal, cardiac, and renal anomalies (Fennell et al., 2022). This syndrome is caused by a deletion in the 8q24.3 region, where the PUF60 gene is located (Verheij et al., 2009; Miao et al., 2024). PUF60 is involved in 3′ splice-site recognition, interacting with U2AF in RNA binding and splicing activation (Hoogenboom et al., 2024; Hastings et al., 2007). The majority of cases of Verheij syndrome are due to de novo mutations; however, rare cases show an autosomal dominant inheritance pattern (Verheij et al., 2009; Sivasubramanian and Ayyavoo, 2024). Haploinsufficiency of PUF60, due to deletions spanning from 78 kb to 1 Mb, has been found to be the driver of Verheij syndrome, with the copy number variants (CNVs) affecting the dose of multiple genes depending on the size of the deletion (Hoogenboom et al., 2024; Dauber et al., 2013).
Mandibulofacial dysostosis, Guion-Almeida type
Mutations in EFTUD2, part of the U5 snRNP of the spliceosome, have been identified as the cause of mandibulofacial dysostosis, Guion-Almeida type (MFDGA; OMIM #610536) (Lines et al., 2012; Beauchamp and Jerome-Majewska, 2024). MFDGA is characterized by craniofacial malformations, microcephaly, developmental delay, and dysmorphic appearance but may also include choanal atresia, sensorineural hearing loss, and cleft palate (Guion-Almeida et al., 2006; Wieczorek et al., 2009). The frequency of MFDGA is unknown, with approximately 100 affected individuals identified so far, harboring 86 distinct EFTUD2 mutations (Beauchamp et al., 2020). Most mutations are stop-gain and splicing mutations, with roughly 75% of patients harboring de novo mutations, whereas dominant inheritance is observed in the remaining patients (Huang et al., 2016).
Nager and Rodriguez syndromes
Nager syndrome (OMIM #154400) is a type of acrofacial dysostosis characterized by midface retrusion, micrognathia, absence of thumbs, and radial hypoplasia (Bernier et al., 2012; Czeschik et al., 2013; Petit et al., 2014). Sixty percent of patients with Nager syndrome have mutations in SF3B4, with haploinsufficiency of SF3B4 being the underlying cause of the disorder (Bernier et al., 2012). Rodriguez syndrome (OMIM #201170) is also caused by mutations in SF3B4 (Drivas et al., 2019). The patients have similar features as Nager syndrome patients; however, the phenotype is typically more severe and involves lower limb and cardiac defects (Rodríguez et al., 1990). SF3B4 encodes for SAP49 and is part of the U2 and U12 snRNPs (Will and Lührmann, 2011), functioning in 3′ branchpoint sequence recognition. The frequency of Nager syndrome is unknown, with approximately 100 cases found worldwide.
Cerebro-costo-mandibular syndrome
Mutations in SNRPB cause cerebro-costo-mandibular syndrome (CCMS; OMIM #117650), which is a disorder that includes micrognathia, glossoptosis, cleft palate, and posterior rib gaps (Lynch et al., 2014). SNRPB is part of the Sm ring that is the scaffold for snRNPs in the U1, U2, U4, and U5 subunits (Schwer et al., 2016; Zahoor et al., 2024). Mutations tend to be heterozygous regulatory mutations, with high frequency of mutation in the premature termination codon-containing exons. CCMS is a rare disease, with approximately 80 reported cases (Bacrot et al., 2015).
Mutations in SNRPA
Mutations in gene SNRPA (OMIM #182285) lead to a yet unnamed syndrome that includes intellectual disability, short stature, and minor craniofacial and hand anomalies (Rangel-Sosa et al., 2018). The mutations are homozygous missense variants in SNRPA, which encodes for an snRNP in the U1 subunit of the spliceosome (Nelissen et al., 1991). Mutations tend to be localized to the first 10–89 amino acids, which is the domain associated with RNA binding (Rangel-Sosa et al., 2018; Jessen et al., 1991).
Richieri-Costa–Pereira syndrome
Richieri-Costa–Pereira syndrome (RCPS; OMIM #268305) is a type of acrofacial dysostosis, in which patients exhibit craniofacial malformations with microcephaly and limb defects (Bertola et al., 2018; Favaro et al., 2014; Hsia et al., 2018). This disorder is due to decreased expression levels of EIF4A3, a member of the exon junction complex in the spliceosome (Le Hir et al., 2016). The decreased levels of this gene are attributed to increased repeats in the 5′UTR of the gene. RCPS is a rare disorder, with less than 50 published cases (Pardo et al., 2021), with a possible phenotypic spectrum related to the number of repeats in the gene (Bertola et al., 2018).
Burn–McKeown syndrome
Mutations in TXNL4A cause a disorder known as Burn–McKeown syndrome (BMKS; OMIM #608572). This condition is characterized by choanal atresia, hearing loss, cleft lip/palate, and other craniofacial anomalies (Wieczorek et al., 2014). TXNL4A encodes a component of the spliceosome U5 snRNP, and the mutations in patients lead to reduced expression and ultimately reduced assembly of the snRNP complex. This disease has been found in 20 individuals with biallelic pathogenic variants (Lüdecke and Wieczorek, 2022). Most patients have a loss-of-function deletion in the promoter region of the gene; however, patients have been identified with intronic deletions (Wood et al., 2022).
RBM8A haploinsufficiency/1q21.1 deletion syndrome
Mutations in RBM8A, a member of the exon junction complex, resulting in haploinsufficiency lead to a disorder characterized by microcephaly, facial gestalt, cleft lip/palate, and skeletal anomalies (OMIM #274000) (Gamba et al., 2016; Mao et al., 2015). These mutations also involve microdeletions of the 1q21.1 chromosome, resulting in variable syndromic phenotypes (Upadhyai et al., 2020). Occurrence of CNVs in 1q21.1 is rare, with less than 40 reports in the literature (Brunetti-Pierri, et al., 2008).
Au–Kline syndrome
Au–Kline syndrome (OMIM #616580) is a developmental disorder characterized by hypotonia, intellectual disability, and typical facial features (Au et al., 2019). This disorder is due to variants in HNRNPK, which is part of the spliceosome C complex, resulting in impairment of Hox gene expression (Duijkers et al., 2019). Loss-of-function mutations are also associated with a specific DNA methylation signature (Choufani et al., 2022). All currently known patients have de novo mutations, with some including missense variants and others being deletions of 9q21.32, encompassing HNRNPK (Au et al., 2018).
TARP syndrome
Mutations in RBM10 cause an X-linked form of cleft palate known as TARP syndrome (Talipes equinovarus, Atrial septal defect, Robin sequence, and Persistent left superior vena cava; OMIM #311900) (Johnston et al., 2010; Gripp et al., 2011). RBM10 is an RNA-binding protein that plays a role in the A complex of the spliceosome, regulating alternative splicing. TARP syndrome is a very rare disorder, with approximately 30 cases reported (Omorodion et al., 2023). Mutations tend to be loss-of-function, occurring either de novo or via X-linked dominant inheritance, in which male children are affected and mothers may present some mosaicism (Johnston et al., 2013).
Craniofacial microsomia/oculo-auriculo-vertebral spectrum/ Goldenhar syndrome
Craniofacial microsomia (OMIM #164210) is a disorder that includes auricular malformations and underdevelopment of the mandible but may also affect the middle ear ossicles, temporal bone, zygoma, and cranial nerves (Beleza-Meireles et al., 2014; Keogh et al., 2007; Timberlake et al., 2021). The most prevalent genetic cause of craniofacial microsomia is haploinsufficiency of SF3B2, a component of the U2 small nuclear ribonucleoprotein complex. Loss-of-function mutations in SF3B2 account for 3% of sporadic cases and 25% of familial cases, with mutations spread across the entirety of the gene (Timberlake et al., 2021). Craniofacial microsomia occurs in between 1 in 5,600 and 1 in 26,550 births, but mild cases are often difficult to diagnose (Gougoutas et al., 2007).
Taybi–Linder, Roifman, and Lowry–Wood syndromes
Taybi–Linder syndrome (TALS; OMIM #210710), or microcephalic osteodysplastic primordial dwarfism type I (MOPD1), is characterized by severe growth delay, microcephaly, intellectual deficiency, bone abnormalities, and other factors ultimately resulting in early mortality (Hagiwara et al., 2021). Roifman syndrome (OMIM #616651) is a disorder characterized by growth retardation, cognitive delay, and spondyloepiphyseal dysplasia (Merico et al., 2015). Lowry–Wood syndrome (OMIM #226960) is a similar disorder characterized by multiple epiphyseal dysplasia, microcephaly, and intellectual disability (Farach et al., 2018). All three of these disorders are attributed to mutations in RNU4ATAC, a small nuclear RNA essential for minor intron splicing (Edery et al., 2011).
CWC27-related spliceosomopathy
The CWC27-related spliceosomopathy (OMIM #250410) is also known as retinitis pigmentosa with or without skeletal anomalies. Although this is mainly categorized as a retina disorder, when the patients have skeletal anomalies, they include craniofacial abnormalities, classifying this disorder also as a craniofacial spliceosomopathy (Xu et al., 2017). Variants of CWC27 are diverse and may result from missense mutations, nonsense mutations, splice-site variants, small insertions, small deletions, and gross deletions (Li et al., 2024).
Craniofacial spliceosomopathies represent a broad array of phenotypes affecting many parts of the body; however, they all have one common denominator: defects in the neural crest-derived craniofacial skeleton, and can be categorized as facial dysostoses (Figure 1). Although the splicing factors affected under these conditions are found across the spliceosome and carry distinct functions (Griffin and Saint-Jeannet, 2020), they all cause a similar craniofacial phenotype, pointing at a possible common root cause and driving the need to further investigate the underlying mechanisms of these disorders.
Modeling craniofacial spliceosomopathies
The etiology of congenital diseases can be studied through in vivo or in vitro modeling, which are expected to closely duplicate these human conditions (Figure 2). Preferred in vivo models include mouse (Mus musculus), zebrafish (Danio rerio), and frog (Xenopus laevis or Xenopus tropicalis), with tools such as CRISPR/Cas9, Cre/lox, TALENs, and mutagenesis screens to target specific genes and/or mutations. Morpholino antisense oligonucleotides are also used in fish and frogs for gene knockdowns (Figure 2). For in vitro modeling, mouse or human embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs) from patient samples are commonly used. Gene function can be manipulated by small interfering RNA (siRNA), short hairpin RNA (shRNA), or CRISPR/Cas9 to engineer disease-causing mutations, allowing for the investigation of the consequences of these mutations on cellular processes such as proliferation, apoptosis, migration, and differentiation (Figure 2). In this section, current models available for studying craniofacial spliceosomopathies are summarized (Table 2), with an emphasis on how these tools can be used to understand the underlying mechanisms of these diseases.
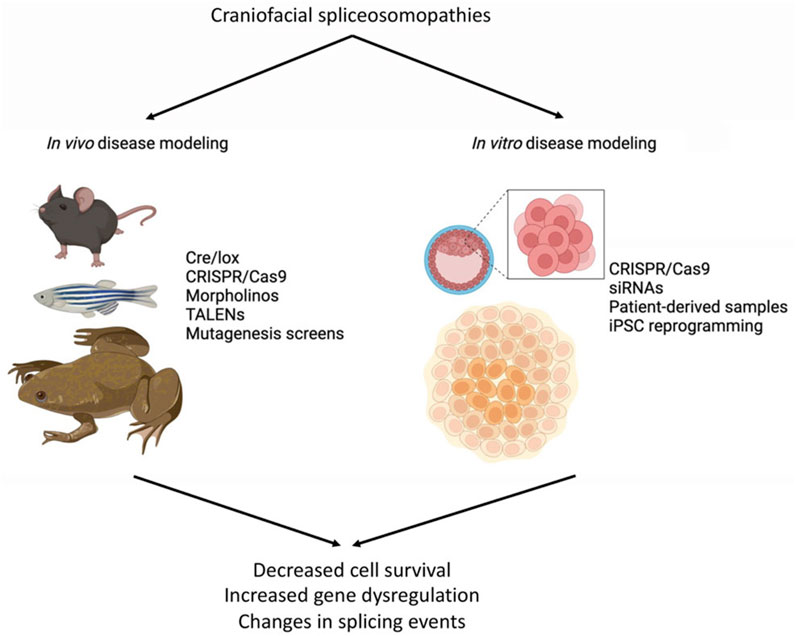
Figure 2. Tools available for in vivo and in vitro disease modeling.
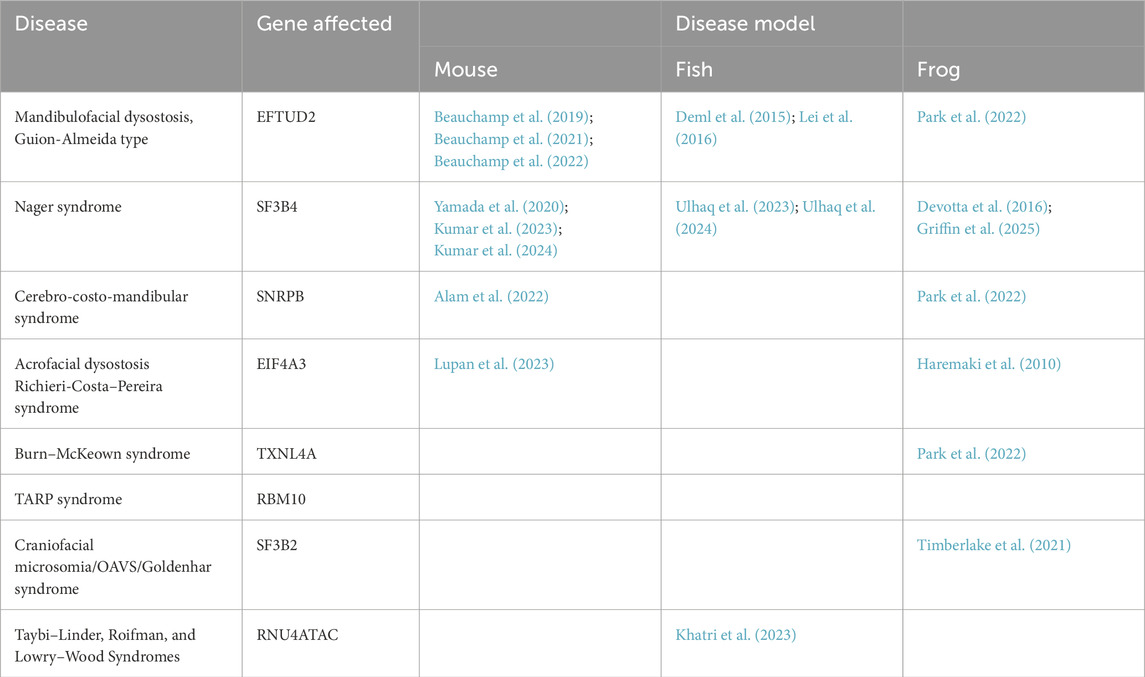
Table 2. List of craniofacial spliceosomopathies and the models available to study them.
Mouse models
Mouse models are a hallmark of disease modeling and can be a powerful system for understanding disease mechanisms and phenotypes, when the model actually represents the disease in a clinically relevant manner.
Conditional knockout of Snrpb in the brain and neural crest lineages using the Wnt1-Cre2 driver was used to model CCMS. The heterozygous mutant embryos (Snrpbncc+/−) recapitulate the disease, showing craniofacial hypoplasia with decreased differentiation of craniofacial cartilage and bone, and reduced postnatal survival (Alam et al., 2022). Although most neural crest cells form in the head and migrate into the pharyngeal arches in the mutants, a subset of neural crest cells undergo apoptosis, indicating that increased neural crest cell death accounts for aspects of this disease. Snrpbncc+/− embryos at E9.0 also had many significantly altered splicing events compared to the wild type, with the most abundant being skipped exons and retained introns. Among these, 13 transcripts required for craniofacial development, including Rere, Dyrk2, and Pou2f1, were identified as having increased exon skipping, potentially contributing to the craniofacial defects observed in the mutant embryos (Alam et al., 2022).
The Sf3b4ncc/ncc and Sf3b4ncc/− mice with loss of Sf3b4 in neural crest cells is another useful model for Nager and Rodriguez syndromes (Kumar et al., 2024). This conditional knockout was able to recapitulate the craniofacial and cardiac phenotype observed in patients. Similarly, Eftud2ncc−/− mouse was generated to recapitulate MFDGA in vivo. Although these mice exhibited craniofacial malformations, they did not survive until birth; however, this model was still used to understand the connection between Eftud2 and the P53 pathway (Beauchamp et al., 2021; Beauchamp et al., 2022). In particular, exon skipping and increased levels of an alternatively spliced form of Mdm2, a p53 pathway gene, were found in mouse embryos. Treatment of mutant embryos with an inhibitor of p53 (pifithrin-a) ameliorated the craniofacial abnormalities found in the untreated embryos, connecting increased p53 activity to the mechanism of MFDGA.
The mouse, however, is not always the best system for modeling craniofacial spliceosomopathies. Frequently, homozygous knockouts of splicing factors are embryonic lethal, whereas heterozygous knockouts show no craniofacial phenotype. Such is the case for the Eftud2 CRISPR/Cas9 knockout mouse, in which there is no survival post-implantation, with the heterozygotes failing to model MFDGA (Beauchamp et al., 2019). Similarly, the Sf3b4 heterozygous knockout mouse does not show any craniofacial phenotype and instead shows defects in the axial skeleton and the forebrain (Yamada et al., 2020), accompanied by mis-splicing of chromatin remodelers and dysregulation of Hox gene expression (Kumar et al., 2023). In the case of CWC27, mutant mice show the retinal degeneration phenotype of the associated disorder, but no craniofacial malformations are described in either of two mutant models—Cwc27K338fs/K338fs and Cwc27Tm1a/K338fs (gene trapping of exon 3 and CRISPR/Cas9-mediated frameshift compound heterozygote) (Bertrand et al., 2022; Lu et al., 2023).
The best approach to study spliceosomopathies in mice appears to be the use of conditional knockouts, as demonstrated with Eftud2 and Sf3b4 using the Wnt1-Cre2 driver. However, this limits the tissues in which the defects can be examined when multiple tissues are affected. For example, conditional knockout of Eif4a3 in the radial glial cells allows for the examination of the microcephaly phenotype of patients but disregards any analysis of the craniofacial malformations observed in RCPS (Lupan et al., 2023). Therefore, considering other models in addition to the mouse may be beneficial.
Zebrafish models
Zebrafish is a popular vertebrate model system, recognized for the ease at making transgenic animals and imaging analysis due to the transparency of the embryos. Mutant embryos can be generated in a number of ways in zebrafish, with tools lending themselves to the specific attributes of a gene or disease. For example, morpholino antisense oligonucleotides can be used to target specific genes and knockdown their function. Such is the case with RNU4ATAC, in which morpholino-mediated knockdown resulted in defects in primary cilia, such as decreased number and function, thereby recapitulating the phenotype of TALS-patient fibroblasts (Khatri et al., 2023). TALEN-mediated disruption has also been used to induce mutations in zebrafish. This was done for a truncation mutation in the eftud2 gene to mimic a mutation found in a MFDGA patient. Mutants displayed a small head and small eye, identifying novel eye phenotypes possibly associated with MFDGA (Deml et al., 2015). A separate mutant construct of eftud2 known as the fn10a mutant has been generated from a mutagenesis screen, and this mutant is useful for studying the impact on neurogenesis (Lei et al., 2016). In this model, neural progenitors experience increased apoptosis and mitosis, coupled with splicing deficiencies including increased retained intron and exon skipping in genes enriched for several KEGG pathways such as “cell cycle,” “p53 signaling pathway,” and “spliceosome.” However, this mutant lacks the craniofacial phenotype of MFDGA.
Unfortunately, similar to the mouse, zebrafish models might lack certain characteristics of craniofacial spliceosomopathies observed in patients. For example, sf3b4−/− mutant zebrafish show no craniofacial malformations but instead exhibit features of retinitis pigmentosa (Ulhaq et al., 2023; Ulhaq et al., 2024). Although retinitis pigmentosa is a spliceosomopathy (Griffin and Saint-Jeannet, 2020), it has not clinically been attributed to mutations in Sf3b4, and Nager syndrome patients do not show clinical signs of retinitis pigmentosa. Further studies will define the clinical relevance of this model.
Frog models
Xenopus is an excellent model system for studying developmental disorders because of the ease at which key developmental processes can be observed and manipulated. A tool broadly used in X. laevis is morpholino antisense oligonucleotides to knockdown gene function during development. For example, a comparative study has been performed by knocking down individual splicing factors—namely, eftud2, snrpb, and txnl4a, which have been linked to MFDGA, CCMS, and BMKS, respectively—and analyzing the consequences on neural crest and craniofacial development. The main results indicate that neural crest progenitor formation is similarly affected in each knockdown through a mechanism that involves increased apoptosis and results in hypoplastic craniofacial cartilages (Park et al., 2022). A study using a morpholino against eif4a3 found that loss of this protein function disrupts derivatives of the neural plate and neural plate border, including some neural crest derivatives (although the craniofacial structure is unaffected) (Haremaki et al., 2010). Similar morpholino studies to interfere with sf3b4 or sf3b2 function have shown shared mechanisms underlying these phenotypes, characterized by impaired neural crest formation, coupled with an increase in apoptosis in the head region and reduced craniofacial cartilages, perhaps hinting at a common root cause to some, if not all craniofacial spliceosomopathies (Devotta et al., 2016; Timberlake et al., 2021).
The X. laevis allotetraploid genome and the relatively long generation time (10–12 months) render genetic analysis challenging in this organism. In recent years, the related species X. tropicalis has become more broadly used. It offers the same embryological advantages as its allotetraploid counterpart, with a shorter generation time (5–7 months) and a diploid genome. Xenopus tropicalis has been used to generate CRISPR/Cas9 knockout mutant lines, which enable a more consistent knockout than morpholinos, reducing off-target effects and allowing for large-scale genomic analysis. Recently, an Sf3b4 knockout mutant line was generated, and it was found that the homozygous null embryos showed reduced neural crest cell migration, increased apoptosis in the head region, and decreased craniofacial cartilage precursors (the heterozygous embryos were comparable to the wild type). These phenotypes were reflected in dysregulated genes as revealed by bulk RNA-sequencing, with downregulated genes categorized into GO terms such as “neural crest cell migration,” “extracellular matrix organization,” and “negative regulation of extrinsic apoptotic signaling.” These dysregulated genes were preceded in developmental time by mis-splicing events, predominately increased abnormal skipped exons, for genes categorized into GO terms such as “RNA splicing,” “regulation of embryonic development,” and “regulation of apoptotic process” (Griffin et al., 2025). Further studies will use this information to identify the gene networks and pathways affected under this craniofacial condition. These studies show that frogs provide a unique system for studying craniofacial spliceosomopathies within the context of development.
Animal models are extremely powerful tools to investigate disease mechanisms. However, there are also downsides to working with these models. Most animal models require a complete gene dosage reduction to recapitulate the disease, taking away from the ability to exactly replicate the human conditions, which are often haploinsufficient. Moreover, the reliance on conditional mutations to model a desired phenotype restricts the ability to interrogate the role of a gene in a broad range of cell and tissue types. Finally, animal models are inherently different than humans and, therefore, may introduce variables that are not directly relevant to the human diseases. Therefore, turning to in vitro modeling may help support and expand the findings of in vivo studies.
In vitro modeling
In vitro modeling of craniofacial spliceosomopathies is an emerging field, with only a small number of disorders examined so far. The use of relevant cell lines, whether primary or engineered, especially those of human origin, holds great potential for elucidating the underlying mechanisms of these diseases in terms of gene function.
For example, the use of mouse mandibular MEPA (mouse embryonic pharyngeal arch) cell lines to examine the role of RBM10 in TARP syndrome has allowed for the identification of RBM10-binding sites in the genome and the elucidation of its role in regulating alternative splicing (Rodor et al., 2016). This group also used the MEPA cells to generate an RBM10 CRISPR/Cas9 knockout cell line to characterize the phenotype at the cellular level, which highlights that loss of RBM10 leads to proliferation defects and changes in the differentiation potential of mutant cells. Human cell line HEK293 has been used to generate an EFTUD2 CRISPR/Cas9 knockout cell line, with a heterozygous loss-of-function mutation that is a null allele equivalent to MFDGA patient mutations (Wood et al., 2019). This cell line was used to identify diminished proliferation, increased sensitivity to endoplasmic reticulum (ER) stress, and mis-expression of ER stress response genes as the potential underlying mechanisms of MFDGA. Another study used primary cells—fibroblasts from TALS patients—to understand the role of RNU4ATAC in the disorder and compare function to the in vivo phenotypes (Khatri et al., 2023). The work indicates alterations in primary cilium function in these cells, which reflected phenotypes observed in vivo, thereby demonstrating the strength of this in vitro model in recapitulating some aspects of the disease.
The use of human embryonic stem cells (hESCs) and iPSCs to model diseases has grown exponentially over the recent years. These cells offer the ability to use human samples to investigate the manifestation of disorders in specific cell types or in tissues in the form of organoids. Recently, hESCs have been used to investigate the underlying mechanism of Nager syndrome. Taking advantage of a well-described protocol to derive neural crest cells from hESCs (Bajpai et al., 2010), it is possible to specifically investigate the function of SF3B4 in differentiating neural crest cells. siRNA-mediated knockdown of SF3B4 revealed a requirement for SF3B4 in neural crest cell production, survival, and differentiation (Griffin and Saint-Jeannet, 2025), showing some parallels with the corresponding animal models (Devotta et al., 2016; Kumar et al., 2024; Griffin et al., 2025). Similarly, BMKS patient iPSCs have been used to investigate the differentiation potential and behavior of neural crest cells with reduced TXNL4A expression (Wood et al., 2020). TXNL4-deficient cells exhibited defective differentiation into neural crest cells, with significant differences in neural border and neural crest marker genes, a delay in the epithelial-to-mesenchymal transition, and dampened response to WNT signaling, an important regulator of craniofacial development (Wood et al., 2020). RCPS patient-derived iPSCs have been used to generate cortical organoids to study neurogenesis with EIF4A3 haploinsufficiency (Lupan et al., 2023). Coupled with in vivo mouse work, it was determined that EIF4A3 mediates neurogenesis by controlling mitosis and cell survival; with reduction in EIF4A3, there is extensive cell death and impaired neurogenesis.
In vitro modeling of diseases allows for the use of human samples to interrogate the mechanisms of the disorders in the context of the patient mutations and/or specific cell types that are affected. In the case of craniofacial spliceosomopathies, hESCs and iPSCs can be differentiated into neural crest cells and their derivatives, allowing for examination of disease-causing mutations in the cells that are primarily affected in these patients. However, there are also some limitations to in vitro disease modeling, such as the limited number of cell types differentiating in a dish that does not fully capture the complexity of tissues in vivo; future technological advances such as 3D organoids may help alleviate some of these shortcomings. Another limitation is the difficulty of obtaining samples from patients with such rare diseases.
Conclusions and perspectives
In this paper, we summarize and discuss the models developed to understand the etiology of several craniofacial spliceosomopathies. Although these models have started to narrow down some of the key mechanisms underlying these diseases, which include increased apoptosis and dysregulated gene expression and splicing events, important gaps remain to be addressed to better understand what makes neural crest cells a preferred target in these pathologies. It is also important to point out that several craniofacial spliceosomopathies have not yet benefited from in vivo or in vitro modeling, which includes Verheij syndrome (PUF60), mutations in SNRPA, RBM8A haploinsufficiency, and Au–Kline syndrome (HNRNPK). It is essential that this group of diseases be studied at a global level to obtain a comprehensive understanding of the underlying pathomechanisms.
It is intriguing that most models of craniofacial spliceosomopathies in vivo do not fully replicate the human diseases in their presentation. Furthermore, animal models typically require homozygosity to develop the phenotype, whereas the majority of craniofacial spliceosomopathies are found to be heterozygous mutations in patients. Perhaps, this is due to some compensatory underlying mechanisms in these organisms’ spliceosomes that have been lost or are lacking in humans. Either way, it makes studying these diseases more challenging because the systems must be manipulated in ways that may affect downstream mechanistic studies.
Important efforts are currently underway in the field to develop in vitro models using patient-derived cells or genome-edited cells that reflect patient mutations. These cells can be obtained from patients directly and studied as primary cells or be reprogrammed into iPSCs. Alternatively, hESCs or iPSCs can be edited with CRISPR/Cas9 to induce patient mutations. The derived stem cells can then be differentiated into neural crest cells using defined protocols, testing the impact of the mutations on neural crest generation, differentiation potential, and survival, with the added potential for transcriptomic and proteomic analyses. Eventually, neural crest cells will need to be incorporated into 3D organoids with other cell types to reproduce the in vivo patient environment more closely.
Overall, disease modeling is an important tool to understand the etiology of understudied disorders. Although all model systems have limitations, it is critical to use them in combination to develop the most comprehensive understanding of the mechanisms underlying these conditions.
Author contributions
CG: Writing – review and editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by a grant from the National Institute of Health to CG (K99-DE034476).
Acknowledgments
The author would like to thank Dr. Jean-Pierre Saint-Jeannet for critical reading of the manuscript and helpful discussions. The author is grateful to the members of the Saint-Jeannet laboratory past and present for their support.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alam, S. S., Kumar, S., Beauchamp, M. C., Bareke, E., Bourcher, A., Nzirorera, N., et al. (2022). Snrpb is required in murine neural crest cells for proper splicing and craniofacial morphogenesis. Dis. Model Mech. 15 (6), dmm049544. doi:10.1242/dmm.049544
Au, P. Y. B., Goedhart, C., Ferguson, M., Breckpot, J., Devriendt, K., Wierenga, K., et al. (2018). Phenotypic spectrum of Au-Kline syndrome: a report of six new cases and review of the literature. Eur. J. Hum. Genet. 26 (9), 1272–1281. doi:10.1038/s41431-018-0187-2
Au, P. Y. B., McNiven, V., Phillips, L., Innes, A. M., and Kline, A. D. (2019). Au-Kline syndrome. GeneReviews PMID, 30998304.
Bacrot, S., Doyard, M., Huber, C., Alibeu, O., Feldhahn, N., Lehalle, D., et al. (2015). Mutations in SNRPB, encoding components of the core splicing machinery, cause cerebro-costo-mandibular syndrome. Hum. Mutat. 36 (2), 187–190. doi:10.1002/humu.22729
Bajpai, R., Chen, D. A., Rada-Iglesias, A., Zhang, J., Xiong, Y., Helms, J., et al. (2010). CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463 (7283), 958–962. doi:10.1038/nature08733
Bak-Gordon, P., and Manley, J. L. (2025). SF3B1: from core splicing factor to oncogenic driver. RNA 31 (3), 314–332. doi:10.1261/rna.080368.124
Beauchamp, M. C., and Jerome-Majewska, L. A. (2024). A protective role for EFTUD2 in the brain. Neuron 112 (20), 3378–3380. doi:10.1016/j.neuron.2024.10.003
Beauchamp, M. C., Djedid, A., Daupin, K., Clokie, K., Kumar, S., Majewski, J., et al. (2019). Loss of function mutation of Eftud2, the gene responsible for mandibulofacial dysostosis with microcephaly (MFDM), leads to pre-implantation arrest in mouse. PLoS One 14 (7), e0219280. doi:10.1371/journal.pone.0219280
Beauchamp, M. C., Alam, S. S., Kumar, S., and Jerome-Majewska, L. A. (2020). Spliceosomapthies and neurocristopathies: two sides of the same coin? Dev. Dyn. 249 (8), 924–945. doi:10.1002/dvdy.183
Beauchamp, M. C., Djedid, A., Bareke, E., Merkuri, F., Aber, R., Tam, A. S., et al. (2021). Mutation in Eftud2 causes craniofacial defects in mice via mis-splicing of Mdm2 and increased P53. Hum. Mol. Genet. 30 (9), 739–757. doi:10.1093/hmg/ddab051
Beauchamp, M. C., Boucher, A., Dong, Y., Aber, R., and Jerome-Majewska, L. A. (2022). Craniofacial defects in embryos with homozygous deletion of Eftud2 in their neural crest cells are not rescued by Trp53 deletion. Int. J. Mol. Sci. 23 (16), 9033. doi:10.3390/ijms23169033
Beleza-Meireles, A., Clayton-Smith, J., Saraiva, J. M., and Tassabehji, M. (2014). Oculo-auriculo-vertebral spectrum: a review of the literature and genetic update. J. Med. Genet. 52 (10), 635–645. doi:10.1136/jmedgenet-2014-102476
Bernier, F. P., Caluseriu, O., Ng, S., Schwartzentruber, J., Buckingham, K. J., Innes, A. M., et al. (2012). Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am. J. Hum. Genet. 90 (5), 925–933. doi:10.1016/j.ajhg.2012.04.004
Bertola, D. R., Hsia, G., Alvizi, L., Gardham, A., Wakeling, E. L., Yamamoto, G. L., et al. (2018). Richieri-costa-Pereira syndrome: expanding its phenotypic and genotypic spectrum. Clin. Genet. 93 (4), 800–811. doi:10.1111/cge.13169
Bertrand, R. E., Wang, J., Li, Y., Cheng, X., Wang, K., Stoilov, P., et al. (2022). Cwc27, associated with retinal degeneration, functions as a splicing factor in vivo. Hum. Mol. Genet. 31 (8), 1278–1292. doi:10.1093/hmg/ddab319
Brunetti-Pierri, N., Berg, J. S., Scaglia, F., Belmont, J., Bacino, C. A., Sahoo, T., et al. (2008). Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 40 (12), 1466–1471. doi:10.1038/ng.279
Cao, Z., and Li, Y. (2024). Mechanisms of RNA alternative splicing dysregulation in triple-negative breast cancer. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 49 (7), 1143–1154. doi:10.11817/j.issn.1672-7347.2024.240434
Catenacci, D. V. T., and Schiller, G. J. (2005). Myelodysplasic syndromes: a comprehensive review. Blood Rev. 19 (6), 301–319. doi:10.1016/j.blre.2005.01.004
Choufani, S., McNiven, V., Cytrynbaum, C., Jangjoo, M., Adam, M. P., Bjornsson, H. T., et al. (2022). An HNRNPK-specific DNA methylation signature makes sense of missense variants and expands the phenotypic spectrum of Au-Kline syndrome. Am. J. Hum. Genet. 109 (10), 1867–1884. doi:10.1016/j.ajhg.2022.08.014
Czeschik, J. C., Voigt, C., Alanay, Y., Albrecht, B., Avci, S., Fitzpatrick, D., et al. (2013). Clinical and mutation data in 12 patients with the clinical diagnosis of Nager syndrome. Hum. Genet. 132 (8), 885–898. doi:10.1007/s00439-013-1295-2
Dauber, A., Golzio, C., Guenot, C., Jodelka, F. M., Kibaek, M., Kjaergaard, S., et al. (2013). SCRIB and PUF60 are primary drivers of the multisystemic phenotypes of the 8q24.3 copy-number variant. Am. J. Hum. Genet. 93 (5), 798–811. doi:10.1016/j.ajhg.2013.09.010
Deml, B., Reis, L. M., Muheisen, S., Bick, D., and Semina, E. V. (2015). EFTUD2 deficiency in vertebrates: identification of a novel human mutation and generation of a zebrafish model. Birth Defects Res. A Clin. Mol. Teratol. 103 (7), 630–640. doi:10.1002/bdra.23397
Devotta, A., Juraver-Geslin, H., Gonzalez, J. A., Hong, C. S., and Saint-Jeannet, J. P. (2016). Sf3b4-depleted Xenopus embryos: a model to study the pathogenesis of craniofacial defects in Nager syndrome. Dev. Biol. 415 (2), 371–382. doi:10.1016/j.ydbio.2016.02.010
Drivas, T. G., Taylor, J. A., and Zackai, E. H. (2019). The final demise of Rodriguez lethal acrofacial dysostosis: a case report and review of the literature. Am. J. Med. Genet. A 179 (6), 1063–1068. doi:10.1002/ajmg.a.61121
Duijkers, F. A., McDonald, A., Janssens, G. E., Lezzerini, M., Jongejan, A., van Koningsbruggen, S., et al. (2019). HNRNPR variants that impair homeobox gene expression drive developmental disorders in humans. Am. J. Hum. Genet. 104 (6), 1040–1059. doi:10.1016/j.ajhg.2019.03.024
Edery, P., Marcaillou, C., Sahbatou, M., Labalme, A., Chastang, J., Touraine, R., et al. (2011). Association of TALS developmental disorder with defect in minor splicing component U4atac snRNA. Science 332 (6026), 240–243. doi:10.1126/science.1202205
Farach, L. S., Little, M. E., Duker, A. L., Logan, C. V., Jackson, A., Hecht, J. T., et al. (2018). The expanding phenotype of RNU4ATAC pathogenic variants to Lowry Wood syndrome. Am. J. Med. Genet. A 176 (2), 465–469. doi:10.1002/ajmg.a.38581
Favaro, F. P., Alvizi, L., Zechi-Ceide, R. M., Bertola, D., Felix, T. M., de Souza, J., et al. (2014). “A noncoding expansion in EIF4A3 causes Richieri-Costa-Pereira syndrome,” in A craniofacial disorder associated with limb defects.
Fennell, A. P., Baxter, A. E., Berkovic, S. F., Ellaway, C. J., Forwood, C., Hildebrand, M. S., et al. (2022). The diverse pleiotropic effects of spliceosomal protein PUF60: a case series of Verheij syndrome. Am. J. Med. Genet. 188A, 3432–3447. doi:10.1002/ajmg.a.62950
Gamba, B. F., Zechi-Ceide, R. M., Kokitsu-Nakata, N. M., Vendramini-Pittoli, S., Rosenberg, C., Krepischi Santos, A. C. V., et al. (2016). Interstitial 1q21.1 microdeletion is associated with severe skeletal anomalies, dysmorphic face and moderate intellectual disability. Mol. Syndromol. 7 (6), 344–348. doi:10.1159/000450971
Gougoutas, A. J., Singh, D. J., Low, D. W., and Bartlett, S. P. (2007). Hemifacial microsomia: clinical features and pictographic representations of the OMENS classification system. Plast. Reconstr. Surg. 120 (7), 112e–120e. doi:10.1097/01.prs.0000287383.35963.5e
Griffin, C., and Saint-Jeannet, J. P. (2020). Spliceosomopathies: diseases and mechanisms. Dev. Dyn. 249 (9), 1038–1046. doi:10.1002/dvdy.214
Griffin, C., and Saint-Jeannet, J. P. (2025). Human stem cell model of neural crest cell differentiation reveals a requirement of SF3B4 in survival, maintenance, and differentiation. Dev. Dyn., dvdy.70009. doi:10.1002/dvdy.70009
Griffin, C., Coppenrath, K., Khan, D., Lin, Z., Horb, M., and Saint-Jeannet, J. P. (2025). Sf3b4 mutation in Xenopus tropicalis causes RNA splicing defects followed by massive gene dysregulation that disrupt cranial neural crest development.
Gripp, K. W., Hopkins, E., Johnston, J. J., Krause, C., Dobyns, W. B., and Biesecker, L. G. (2011). Long-term survival in TARP syndrome and confirmation of RBM10 as the disease-causing gene. Am. J. Med. Genet. A 155A (10), 2516–2520. doi:10.1002/ajmg.a.34190
Guion-Almeida, M. L., Zechi-Ceide, R. M., Vendramini, S., and Tabith Júnior, A. (2006). A new syndrome with growth and mental retardation, mandibulofacial dysostosis, microcephaly, and cleft palate. Clin. Dysmorphol. 15, 171–174. doi:10.1097/01.mcd.0000220603.09661.7e
Hagiwara, H., Matsumoto, H., Uematsu, K., Zaha, K., Sekinaka, Y., Miyake, N., et al. (2021). Immunodeficiency in a patient with microcephalic osteodysplastic primordial dwarfism type I as compared to Roifman syndrome. Brain Dev. 43 (2), 337–342. doi:10.1016/j.braindev.2020.09.007
Haremaki, T., Sridharan, J., Dvora, S., and Weinstain, D. C. (2010). Regulation of vertebrate embryogenesis by the exon junction complex core component Eif4a3. Dev. Dyn. 239 (7), 1977–1987. doi:10.1002/dvdy.22330
Hastings, M. L., Allemand, E., Duelli, D. M., Myers, M. P., and Krainer, A. R. (2007). Control of pre-mRNA splicing by the general splicing factors PUF60 and U2AF(65). PLoS One 2 (6), e538. doi:10.1371/journal.pone.0000538
Hermán-Sánchez, N., G-García, M. E., Jiménez-Vacas, J. M., Yubero-Serrano, E. M., López-Sánchez, L. M., Romero-Martín, S., et al. (2024). The splicing machinery is dysregulated and represents a therapeutic vulnerability in breast cancer. Cell Mol. Life Sci. 82 (1), 18. doi:10.1007/s00018-024-05515-6
Hoogenboom, A., Falix, F. A., van der Laan, L., Kerkhof, J., Alders, M., Sadikovic, B., et al. (2024). Novel PUF60 variant suggesting an interaction between Verheij and Cornelia de Lange syndrome: phenotype description and review of the literature. Eur. J. Hum. Genet. 32 (4), 435–439. doi:10.1038/s41431-023-01527-1
Hsia, G. S. P., Musso, C. M., Alvizi, L., Brito, L. A., Kobayashi, G. S., Pavanello, R. C. M., et al. (2018). Complexity of the 5’ untranslated region of EIF4A3, a critical factor for craniofacial and neural development. Front. Genet. 9, 149. doi:10.3389/fgene.2018.00149
Huang, L., Vanstone, M. R., Hartley, T., Osmond, M., Barrowman, N., Allanson, J., et al. (2016). Mandibulofacial dysostosis with microcephaly: mutation and database update. Hum. Mutat. 37 (2), 148–154. doi:10.1002/humu.22924
Jessen, T. H., Oubridge, C., Teo, C. H., Pritchard, C., and Nagai, K. (1991). Identification of molecular contacts between the U1 A small nuclear ribonucleoprotein and U1 RNA. EMBO J. 10 (11), 3447–3456. doi:10.1002/j.1460-2075.1991.tb04909.x
Johnston, J. J., Teer, J. K., Cherukuri, P. F., Hansen, N. F., Loftus, S. K., Chong, K., et al. (2010). Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am. J. Hum. Genet. 86 (5), 743–748. doi:10.1016/j.ajhg.2010.04.007
Johnston, J. J., Sapp, J. C., Curry, C., Horton, M., Leon, E., Cusmano-Ozog, K., et al. (2013). Expansion of the TARP syndrome phenotype associated with de novo mutations and mosaicism. Am. J. Med. Genet. A 164A (1), 120–128. doi:10.1002/ajmg.a.36212
Keogh, I. J., Troulis, M. J., Monroy, A. A., Eavey, R. D., and Kaban, L. B. (2007). Isolated microtia as a marker for unsuspected hemifacial microsomia. Arch. Otolaryngol. Head. Neck Surg. 133 (10), 997–1001. doi:10.1001/archotol.133.10.997
Khatri, D., Putoux, A., Cologne, A., Kaltenbach, S., Besson, A., Bertiaux, E., et al. (2023). Deficiency of the minor spliceosome component U4atac snRNA secondarily results in ciliary defects in human and zebrafish. Proc. Natl. Acad. Sci. U S A 120 (9), e2102569120. doi:10.1073/pnas.2102569120
Kumar, S., Alam, S. S., Bareke, E., Beauchamp, M. C., Dong, Y., Chan, W., et al. (2023). Sf3b4 regulates chromatin remodeler splicing and hox expression. Differentiation 131, 59–73. doi:10.1016/j.diff.2023.04.004
Kumar, S., Bareke, E., Lee, J., Carlson, E., Merkuri, F., Schwager, E. E., et al. (2024). Etiology of craniofacial and cardiac malformations in a mouse model of SF3B4-related syndromes. Proc. Natl. Acad. Sci. U S A 121 (39), e2405523121. doi:10.1073/pnas.2405523121
Le Hir, H., Saulière, J., and Wang, Z. (2016). The exon junction complex as a node of post-transcriptional networks. Nat. Rev. Mol. Cell Biol. 17 (1), 41–54. doi:10.1038/nrm.2015.7
Lehalle, D., Wieczorek, D., Zechi-Ceide, R. M., Passos-Bueno, M. R., Lyonnet, S., Amiel, J., et al. (2015). A review of craniofacial disorders caused by spliceosomal defects. Clin. Genet. 88 (5), 405–415. doi:10.1111/cge.12596
Lei, L., Yan, S. Y., Yang, R., Chen, J. Y., Li, Y., Bu, Y., et al. (2016). Spliceosomal protein eftud2 mutation leads to p53-dependent apoptosis in zebrafish neural progenitors. Nucleic Acids Res. 45 (6), 3422–3436. doi:10.1093/nar/gkw1043
Li, H., Zheng, K., and Xie, M. (2024). A novel small deletion in CWC27 gene associated with CWC27-related spliceosomeopathy. Ophthalmic Genet. 45 (5), 537–541. doi:10.1080/13816810.2024.2368791
Lines, M. A., Huang, L., Schwartzentruber, J., Douglas, S. L., Lynch, D. C., Beaulieu, C., et al. (2012). Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am. J. Hum. Genet. 90 (2), 369–377. doi:10.1016/j.ajhg.2011.12.023
Lu, J., Zheng, K. Q., Bertrand, R. E., Quinlan, J., Ferdous, S., Srinivasan, T., et al. (2023). Gene augmentation therapy to rescue degenerative photoreceptors in a Cwc27 mutant mouse model. Exp. Eye Res. 234, 109596. doi:10.1016/j.exer.2023.109596
Lüdecke, H. J., and Wieczorek, D. (2022). TXNL4A-related craniofacial disorders. Seattle, Washington: GeneReviews.
Lupan, B. M., Solecki, R. A., Musso, C. M., Alsina, F. C., and Silver, D. L. (2023). The exon junction complex component EIF4A3 is essential for mouse and human cortical progenitor mitosis and neurogenesis. Development 150 (10), dev201619. doi:10.1242/dev.201619
Lynch, D. C., Revil, T., Schwartzentruber, J., Bhoj, E. J., Innes, A. M., Lamont, R. E., et al. (2014). Disrupted auto-regulation of the spliceosomal gene SNRPB causes cerebro-costo-mandibular syndrome. Nat. Commun. 5, 4483. doi:10.1038/ncomms5483
Mao, H., Pilaz, L. J., McMahon, J. J., Golzio, C., Wu, D., Shi, L., et al. (2015). Rbm8a haploinsufficiency disrupts embryonic cortical development resulting in microcephaly. J. Neurosci. 35 (18), 7003–7018. doi:10.1523/JNEUROSCI.0018-15.2015
Merico, D., Roifman, M., Braunschweig, U., Yuen, R. K. C., Alexandrova, R., Bates, A., et al. (2015). Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman Syndrome by disrupting minor intron splicing. Nat. Commun. 6, 8718. doi:10.1038/ncomms9718
Miao, M., Wang, J., Guo, C., Su, X., Sun, L., and Lu, S. (2024). Identification of a novel de novo PUF60 variant causing Verheij syndrome in a fetus. Gene 897, 148092. doi:10.1016/j.gene.2023.148092
Mrid, R. B., El Guendouzi, S., Mineo, M., and El Fatimy, R. (2025). The emerging roles of aberrant alternative splicing in glioma. Cell Death Discov. 11 (1), 50. doi:10.1038/s41420-025-02323-0
Nelissen, R. L., Sillekens, P. T., Beijer, R. P., van Kessel, A. H. G., and van Venrooij, W. J. (1991). Structure, chromosomal localization and evolutionary conservation of the gene encoding human U1 snRNP-specific A protein. Gene 102 (2), 189–196. doi:10.1016/0378-1119(91)90077-o
Omorodion, J., Tannenbaum, L., O’Neill, J. P., Cummings, C., and Wojcik, M. H. (2023). Vitelline vascular remnant causing intestinal obstruction in a patient with TARP syndrome. Birth Defects Res. 115 (13), 1216–1221. doi:10.1002/bdr2.2212
Pardo, M. P., Dos Santos, G. L., Carvalho, I. M. M., and Tjioe, K. C. (2021). Craniofacial features in Richieri-Costa-Pereira syndrome. Cleft Palate Craniofac J. 58 (11), 1370–1375. doi:10.1177/1055665620987749
Park, B. Y., Tachi-Duprat, M., Ihewulezi, C., Devotta, A., and Saint-Jeannet, J. P. (2022). The core splicing factors EFTUD2, SNRPB and TXNL4A are essential for neural crest and craniofacial development. J. Dev. Biol. 10 (3), 29. doi:10.3390/jdb10030029
Petit, F., Escande, F., Jourdain, A. S., Porchet, N., Amiel, J., Doray, B., et al. (2014). Nager syndrome: confirmation of SF3B4 haploinsufficiency as the major cause. Clin. Genet. 86 (3), 246–251. doi:10.1111/cge.12259
Rangel-Sosa, M. M., Figuera-Villanueva, L. E., González-Ramos, I. A., Pérez-Páramo, Y. X., Martínez-Jacobo, L. A., Arnaud-López, L., et al. (2018). Exome sequencing reveals three homozygous missense variants in SNRPA in two sisters with syndromic intellectual disability. Clin. Genet. 93 (6), 1229–1233. doi:10.1111/cge.13235
Rodor, J., FitzPatrick, D. R., Eyras, E., and Cáceres, J. F. (2016). The RNA-binding landscape of RBM10 and its role in alternative splicing regulation in models of mouse early development. RNA Biol. 14 (1), 45–57. doi:10.1080/15476286.2016.1247148
Rodríguez, J. I., Palacios, J., and Urioste, M. (1990). New acrofacial dysostosis syndrome in 3 sibs. Am. J. Med. Genet. 35 (4), 484–489. doi:10.1002/ajmg.1320350408
Schwer, B., Kruchten, J., and Shuman, S. (2016). Structure-function analysis and genetic interactions of the SmG, SmE, and SmF subunits of the yeast Sm protein ring. RNA 22 (9), 1320–1328. doi:10.1261/rna.057448.116
Sivasubramanian, D., and Ayyavoo, A. (2024). A case report of Verheij Syndrome. Cureus 16 (8), e66692. doi:10.7759/cureus.66692
Stanley, R. F., and Abdel-Wahab, O. (2022). Dysregulation and therapeutic targeting of RNA splicing in cancer. Nat. Cancer 3 (5), 536–546. doi:10.1038/s43018-022-00384-z
Timberlake, A. T., Griffin, C., Heike, C. L., Hing, A. V., Cunningham, M. L., Chitayat, D., et al. (2021). Haploinsufficiency of SF3B2 causes craniofacial microsomia. Nat. Commun. 12 (1), 4680. doi:10.1038/s41467-021-24852-9
Trainor, P. A., and Andrews, B. T. (2013). Facial dysostoses: etiology, pathogenesis and management. Am. J. Med. Genet. C Semin. Med. Genet. 163C (4), 283–294. doi:10.1002/ajmg.c.31375
Ulhaq, Z. S., Okamoto, K., Ogino, Y., and Tse, W. K. F. (2023). Dysregulation of spliceosomes complex induces retinitis pigmentosa-like characteristics in sf3b4-depleted zebrafish. Am. J. Pathol. 193 (9), 1223–1233. doi:10.1016/j.ajpath.2023.05.008
Ulhaq, Z. S., Ogino, Y., and Tse, W. K. F. (2024). Transcriptome alterations in sf3b4-depleted zebrafish: insights into cataract formation in retinitis pigmentosa model. Exp. Eye Res. 240, 109819. doi:10.1016/j.exer.2024.109819
Upadhyai, P., Amiri, E. F., Guleria, V. S., Bielas, S. L., Girisha, K. M., and Shukla, A. (2020). Recurrent 1q21.1 deletion syndrome: report on variable expression, nonpenetrance and review of literature. Clin. Dysmorphol. 29 (3), 127–131. doi:10.1097/MCD.0000000000000327
Verheij, J. B. G. M., de Munnik, S. A., Dijkhuizen, T., de Leeuw, N., Olde Weghuis, D., van den Hoek, G. J., et al. (2009). An 8.35 Mb overlapping interstitial deletion of 8q24 in two patients with coloboma, congenital heart defect, limb abnormalities, psychomotor retardation and convulsions. Eur. J. Med. Genet. 52 (5), 353–357. doi:10.1016/j.ejmg.2009.05.006
Verma, B., Akinyi, M. V., Norppa, A. J., and Frilander, M. J. (2018). Minor spliceosome and disease. Semin. Cell Dev. Biol. 79, 103–112. doi:10.1016/j.semcdb.2017.09.036
Wieczorek, D., Gener, B., González, M. J., Seland, S., Fischer, S., Hehr, U., et al. (2009). Microcephaly, microtia, preauricular tags, choanal atresia and developmental delay in three unrelated patients: a mandibulofacial dysostosis distinct from Treacher Collins syndrome. Am. J. Med. Genet. 149A, 837–843. doi:10.1002/ajmg.a.32747
Wieczorek, D., Newman, W. G., Wieland, T., Berulava, T., Kaffe, M., Falkenstein, D., et al. (2014). Compound heterozygosity of low-frequency promoter deletions and rare loss-of-function mutations in TXNL4A causes Burn-McKeown syndrome. Am. J. Hum. Genet. 95 (6), 698–707. doi:10.1016/j.ajhg.2014.10.014
Will, C. L., and Lührmann, R. (2011). Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 3 (7), a003707. doi:10.1101/cshperspect.a003707
Wood, K. A., Rowlands, C. F., Qureshi, W. M. S., Thomas, H. B., Buczek, W. A., Briggs, T. A., et al. (2019). Disease modeling of core pre-mRNA splicing factor haploinsufficiency. Hum. Mol. Genet. 28 (22), 3704–3723. doi:10.1093/hmg/ddz169
Wood, K. A., Rowlands, C. F., Thomas, H. B., Woods, S., O’Flaherty, J., Douzgou, S., et al. (2020). Modelling the developmental spliceosomal craniofacial disorder Burn-McKeown syndrome using induced pluripotent stem cells. PLoS One 15 (7), e0233582. doi:10.1371/journal.pone.0233582
Wood, K. A., Ellingford, J. M., Thomas, H. B., Genomics, U. K. R. C., Douzgou, S., Beaman, G. M., et al. (2022). Expanding the genotypic spectrum of TXNL4A variants in Burn-McKeown syndrome. Clin. Genet. 101 (2), 255–259. doi:10.1111/cge.14082
Xu, M., Xie, Y. A., Abouzeid, H., Gordon, C. T., Fiorentino, A., Sun, Z., et al. (2017). Mutations in the spliceosome component CWC27 cause retinal degeneration with or without additional developmental anomalies. Am. J. Hum. Genet. 100 (4), 592–604. doi:10.1016/j.ajhg.2017.02.008
Yamada, T., Takechi, M., Yokoyama, N., Hiraoka, Y., Ishikubo, H., Usami, T., et al. (2020). Heterozygous mutation of the splicing factor Sf3b4 affects development of the axial skeleton and forebrain in mouse. Dev. Dyn. 249 (5), 622–635. doi:10.1002/dvdy.148
Keywords: craniofacial spliceosomopathies, neural crest, spliceosome, in vitro, in vivo
Citation: Griffin C (2025) Modeling craniofacial spliceosomopathies: a pathway toward deciphering disease mechanisms. Front. Cell Dev. Biol. 13:1624043. doi: 10.3389/fcell.2025.1624043
Received: 06 May 2025; Accepted: 04 August 2025;
Published: 26 September 2025.
Edited by:
Loydie Jerome-Majewksa, McGill University Health Centre, CanadaReviewed by:
Stefan Pinter, University of Connecticut Health Center, United StatesIsabelle Perrault, INSERM U1163 Institut Imagine, France
Nobue Itasaki, University of Bristol, United Kingdom
Copyright © 2025 Griffin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Casey Griffin, Y3JnMjY5QG55dS5lZHU=