 Yu Pan1,2†Lin Chen3†
Yu Pan1,2†Lin Chen3† Yan Chen3†Elizabeth Rosalind Thomas4Shiying Zhou1
Yan Chen3†Elizabeth Rosalind Thomas4Shiying Zhou1 You Yang1Kezhi Liu5*
You Yang1Kezhi Liu5* Jianming Wu1*
Jianming Wu1* Xiang Li1,5,6*
Xiang Li1,5,6*- 1Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Southwest Medical University, Luzhou, China
- 2School of Nursing, Southwest Medical University, Luzhou, China
- 3Department of Dermatology, Department of Orthopedics and Joint Surgery, The Affiliated Hospital, Southwest Medical University, Luzhou, China
- 4Department of Medical Microbiology, PGIMER, Chandigarh, India
- 5Zigong Institute of Brain Science, Zigong mental health Center, The Zigong Affiliated of Hospital of Southwest Medical University, Zigong, Sichuan, China
- 6Health Science Center, Xi’an Jiaotong University, Xi’an, China
Diabetic foot ulcers (DFUs) are a serious complication of diabetes, characterized by delayed wound healing, recurrent infection, and risk of amputation. Mitochondrial dysfunction has emerged as a central pathological mechanism underlying impaired wound healing. Persistent hyperglycemia triggers a cascade of mitochondrial abnormalities like disrupted calcium homeostasis, excessive ROS production, impaired autophagy, increased apoptosis, and imbalanced mitochondrial dynamics. These alterations hinder ATP production, damage repair cells and delays tissue regeneration. This review comprehensively explores the mechanism of action of oxidative stress, mitochondrial apoptosis, autophagy dysfunction, calcium imbalance and ferroptosis on DFU pathogenesis. It also highlights promising mitochondrial targeted therapies. As mitochondria regulates key cellular processes, targeting mitochondrial dysfunction represents a novel and promising strategy. Future research should focus on integrated approaches to restore mitochondrial homeostasis in diabetic wound healing.
1 Introduction
Diabetes mellitus is a serious and chronic metabolic disease, characterized primarily by hyperglycemia. Its global incidence continues to rise, posing significant public health challenges. Among the numerous complications associated with diabetes, diabetic foot ulcers (DFUs) is one of the most severe and prevalent issue, affecting approximately 18.6 million individuals worldwide annually (Armstrong et al., 2023). The pathogenesis of DFUs is multifactorial, involving a complex interplay of pathological factors that ultimately result in cellular dysfunction and impaired wound healing. A critical aspect of diabetic ulcers is the altered wound microenvironment, particularly abnormalities in the extracellular matrix, which directly impair wound repair mechanisms (Santarella et al., 2020; Chang and Nguyen, 2021). These alterations manifest through disturbances in the immune microenvironment (Zhao et al., 2023; Mohsin et al., 2024), imbalances in cytokines (Zubair and Ahmad, 2019), growth factors (Yan et al., 2018; Zhang et al., 2023b), and dysregulated protease activity (McCarty et al., 2012; Gao et al., 2015; Chen et al., 2023b). Collectively, these factors disrupt the normal cellular functions, making wound healing exceedingly challenging. In addition, prolonged hyperglycemia contributes to serious neurological and vascular complications, diminishing sensory acuity and increasing vulnerability to skin injuries. Even minor lesions may serve as entry points for pathogenic microorganisms, gradually developing into chronic, hard-to-heal ulcers within the glucose-rich microenvironment of diabetic patients (Uberoi et al., 2024; Yang et al., 2024). Such ulcers significantly reduce patients’ quality of life, escalating healthcare costs, and can lead to severe outcomes such as infections, amputations (Schwarz et al., 2013), and even death (Senneville et al., 2024). Therefore, early diagnosis, timely intervention, and exploration of innovative treatment options are essential to improve outcome for patients with diabetic ulcers (McDermott et al., 2023).
Mitochondria, a primary site of cellular energy production and metabolism, play a pivotal role in maintaining cellular homeostasis (Spinelli and Haigis, 2018). Glucose undergoes glycolysis in the cytoplasm, generating pyruvate, which is converted to acetyl coenzyme A and enters the mitochondrial matrix to fuel oxidative phosphorylation. This process produces essential molecules like nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2), which drives the electron transport chain, culminating in ATP generation (Walsh et al., 2018). Beyond energy metabolism, mitochondria are integral to diverse cellular processes, including intracellular calcium homeostasis (Cartes-Saavedra et al., 2025), reactive oxygen species (ROS) production (Zorov et al., 2014; Rizwan et al., 2020), regulation of intracellular protein folding (Shin et al., 2021), apoptosis, immune response modulation (Zecchini et al., 2023; Trinchese et al., 2024), mitochondrial quality control (Al Ojaimi et al., 2022), and mitophagy (Lorentzen et al., 2025).
Despite advances in conventional treatments, diabetic wounds often exhibit suboptimal healing. Targeting mitochondrial biological functions present a promising therapeutic approach. By improving cellular energy metabolism, reducing oxidative stress, promoting angiogenesis, inhibiting apoptosis, and modulating immune responses, mitochondrial-targeted interventions can accelerate tissue repair and wound healing (Lin M. et al., 2023; Qi et al., 2024). Consequently, improving mitochondrial function represents an urgent and promising strategy for the treatment of diabetic wounds.
Recent studies increasingly highlight the critical role of mitochondria in the pathogenesis and progression of diabetic ulcers (Jiang et al., 2023). This review summarizes the current status of mitochondrial function in diabetic ulcer healing and explores potential therapeutic approaches, providing a foundation for the development of improved clinical treatment strategies.
1.1 Effects of mitochondrial oxidative stress on diabetic wound healing
Under normal physiological conditions, wound healing consists of four successive stages: hemostasis, inflammation, proliferation, and remodeling (Lindley et al., 2016). However, in diabetic patients, this process is often disrupted by cellular dysfunction (Liu C. et al., 2023) and prolonged inflammation (Xia et al., 2023) that prevents progression to subsequent healing stages (Yang et al., 2023; Xiong et al., 2025). A prolonged hyperglycemic state in diabetic patients significantly increases the production of ROS (Yaribeygi et al., 2019b; Wan et al., 2022), including superoxide anion, hydrogen peroxide and hydroxyl radicals (Staveness et al., 2016). Under normal physiological conditions, ROS act as essential signaling molecules, regulates pathogen defense, autophagy and cell proliferation (Carrasco et al., 2015; Sahin et al., 2019; Li W. et al., 2024; Zhao et al., 2024).
However, in diabetic patients, the overproduction of ROS triggers oxidative stress, causing damage to cell membranes, proteins, and DNA, thereby impairing wound healing (Deng et al., 2021; Hong et al., 2023). Additionally, mitochondrial antioxidant defense systems - such as manganese superoxide dismutase (MnSOD), glutathione peroxidase (GPX), and glutathione reductase - exhibit diminished activity in diabetic patients (Oyewole and Birch-Machin, 2015; Li F. et al., 2024). This imbalance between ROS production and antioxidant capacity exacerbates tissue injury, disrupts cellular redox homeostasis, and further impairs healing (Yaribeygi et al., 2019a; Zhu et al., 2024). Notably, reduced peroxidase III expression in diabetic wounds is associated with mitochondrial membrane potential (ΔΨm), a key factor in triggering apoptotic signaling (Wolf et al., 2010; Wang X. et al., 2020; Wang et al., 2022). Consequently, mitochondrial dysfunction and ROS are recognized not only as markers but also as drivers of impaired healing in diabetic wounds (Wang X. et al., 2020).
High levels of ROS also damages extracellular matrix (ECM) proteins, leading to non-enzymatic glycosylation due to excess glucose (Du et al., 2020; Sabbatinelli et al., 2022). This process generates intermediates that result in the formation of advanced glycation end-products (AGEs), which interacts with their corresponding receptors (RAGE), further accelerating glycosylation (Zheng et al., 2022) and exacerbating vascular and neural toxicity, while impairing the functions of macrophages (Geng et al., 2023), fibroblasts, and vascular endothelial cells (Mao et al., 2022) - all of which are critical for wound healing (Li et al., 2022b; Fu et al., 2023). Moreover, diabetic patients also exhibit reduced antioxidant enzyme activity in the ECM, making their wounds highly sensitive to oxidative stress, particularly during the tissue remodeling phase (Kunkemoeller and Kyriakides, 2017; Elbatreek et al., 2019). The activation of the AGE-RAGE signaling pathway is a key component in driving the vicious cycle of oxidative stress (Shi et al., 2013). The binding of AGE-RAGE activates NADPH oxidase (NOX), leading to the generation of large amounts of cytoplasmic ROS (Chen et al., 2018), and NOX-derived ROS contributes to mitochondrial dysfunction, secondary ROS generation, mtDNA damage, and impaired antioxidant defenses. Continuous activation of the AGE-RAGE pathway leads to the accumulation of ROS, which impedes the healing of diabetic wounds (Piperi et al., 2015; Bai et al., 2022; Zhang et al., 2025) (Figure 1).
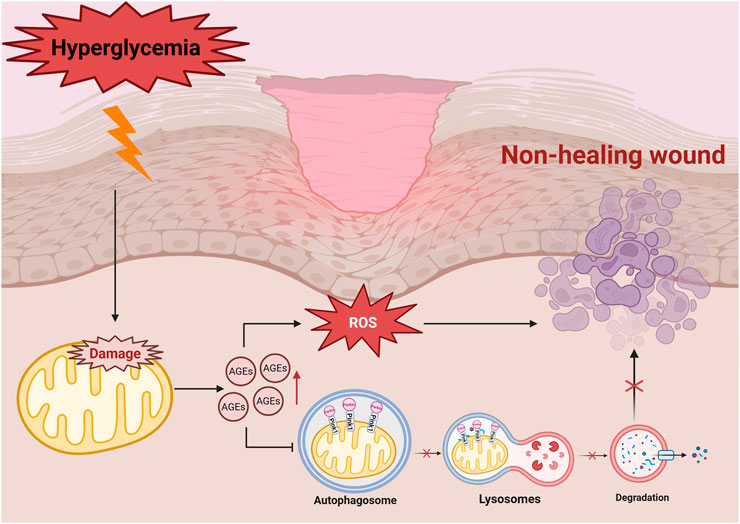
Figure 1. Hyperglycemia-affected cell with high ROS production triggers mitochondrial dysfunction, exacerbates oxidative stress, promotes the formation of AGEs, and further activates signalling pathways, such as NF-κB and PKC, which ultimately leads to delayed wound healing. Hyperglycemia induces the accumulation of AGEs, leading to increased levels of ROS, which in turn downregulates mitochondrial autophagy proteins such as PINK1/Parkin. As the autophagosome cannot fuse with the lysosome properly, mitochondrial degradation is blocked, which ultimately leads to non-healing wound in diabetes mellitus. Damaged mitochondria are not effectively cleared, which further exacerbates oxidative stress and disrupts cellular metabolism.
In addition, oxidative signals activates multiple signaling pathways, such as protein kinase C (PKC), nuclear factor-κB (NF-κB), mitogen-activated protein kinase (MAPK), and c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) (Choudhury et al., 2015; Qin et al., 2019; Meng W. et al., 2025). Activating these signaling pathways regulates matrix metalloproteinase expression, and combined with high levels of redox reactions, disrupts ECM remodeling, delays wound healing, and promotes inflammation and apoptosis (Kowluru et al., 2016; Zhu et al., 2022). It is worth noting that damage-associated molecular patterns (DAMPs) (e.g., mtDNA, ATP) released by mitochondrial dysfunction can trigger sterile inflammation through TLRs, NLRP3 inflammasomes, and cGAS-STING pathways, hindering diabetic wound healing (Grazioli and Pugin, 2018). Improving mitochondrial function by suppressing mtDNA leakage and promoting ATP production reduce inflammatory responses and accelerate wound healing (He et al., 2024; Mao et al., 2025).
A prolonged hyperglycemic state significantly increases electron leakage in complexes I and III of the electron transport chain (ETC), generating large amounts of superoxide anions (O2−) (Kowluru et al., 2016; Huo et al., 2023). These anions get converted into hydroxyl radicals (OH) and hydrogen peroxide (H2O2), which damage the mitochondrial structures, reducing ATP production, decreasing ΔΨm, and induces mitochondrial DNA (mtDNA) mutations (Yuzefovych et al., 2012; Li et al., 2022a; Shen et al., 2023). Studies have shown that introducing the 8-oxo guanine DNA glycosylase 1 gene into mitochondria using adenoviral vector technology effectively mitigates mtDNA damage, thereby improving ROS and promoting wound-healing in diabetic rat models (Yuzefovych et al., 2012).
Furthermore, Rizwan et al. found that excess ROS in hyperglycemic environments damages mtDNA in keratinocytes (Rizwan et al., 2020), triggering inflammation and apoptosis via the cGAS-STING-IRF3 pathway. Targeting mtDNA protection offers a great potential strategy for diabetic wound healing. Chronic hyperglycemia also suppresses vascular regeneration, prolongs epithelial migration, exacerbates inflammatory cell infiltration, and hinders granulation tissue formation, all of which contribute to poor wound healing (Peplow and Baxter, 2012; Luo et al., 2023). Prolonged exposure of the body to high glucose levels activates hypoxia-induced pathways, further perpetuating inflammation and tissue damage (Gerber and Rutter, 2017; Huang et al., 2024). However, Shi et al. revealed that bone marrow mesenchymal stem cells (BMSCs) under hypoxic conditions secretes TGF-β1, promotes autophagy, reduces inflammation, and enhancing epidermal cell proliferation and migration through the HIF-1α/TGF-β1/SMAD signaling pathway. This accelerated wound healing in diabetic ulcers (Shi et al., 2022). These findings suggest that hypoxia, despite its detrimental role in diabetic wounds, may have context-dependent therapeutic potential warranting further investigation.
ROS exhibit a dual role in wound healing; while moderate ROS levels stimulate early inflammatory responses, recruit immune cells and promote angiogenesis and epithelial migration (Sies and Jones, 2020), excessive ROS induces oxidative stress, damages cellular structures (Li M. et al., 2024), and activates pro-inflammatory pathways, such as NF-κB, impeding healing (Ji et al., 2024).
Novel approaches, like the glucose-responsive hydrogel GHM3, show promise. GHM3 reduces glucose levels in the wound microenvironment, scavenges ROS, improving inflammation and accelerating wound healing (Qi et al., 2023).
In summary, mitochondrial dysfunction and oxidative stress play crucial roles in the pathophysiology of diabetic wound healing. Strategies to inhibit ROS overproduction, maintain mitochondrial function, and enhance antioxidant defenses are essential for improving wound healing in diabetic patients.
1.2 Effects of mitophagy on diabetic wound healing
Mitophagy is a self-regulatory mechanism that maintains cellular homeostasis by selectively removing damaged or dysfunctional mitochondria (Jiang et al., 2023). It plays a crucial role in regulating cellular metabolism and stress responses. In diabetic patients, the hyperglycemic microenvironment and the subsequent accumulation of AGEs impair cellular mitophagy, delay wound healing and disrupts cellular metabolism (Han et al., 2017; Wu et al., 2024). Studies have shown that mitophagy - related proteins, such as PINK1, Parkin, Beclin1 and LC3-II/LC3-I are significantly downregulated during the wound infection stage, exacerbating mitochondrial dysfunction as the condition progresses (Xiang et al., 2022; Deng et al., 2024).
Angiogenesis is critical for wound healing during the proliferation phase, and mitophagy promotes vascular endothelial cell survival and proliferation, facilitating the formation of new blood vessels (Zhu et al., 2018; Fan et al., 2023). Laughlin et al. demonstrated that enhancing the level of mitophagy in keratinocytes counteracted the negative effects of AGEs, promoting differentiation, proliferation, and epithelialization in diabetic ulcers (Laughlin et al., 2020). However, in diabetic patients, oxidative stress and hyperglycemia inhibits mitophagy, impairs angiogenesis and hinders wound repair. Collectively, mitophagy dysfunction emerges as a key factor contributing to impaired wound healing in diabetes.
Mitophagy reduces ROS accumulation by efficiently removing damaged mitochondria, thus lowering oxidative stress and maintaining normal cellular metabolism–crucial for wound healing (Tan et al., 2022; Zhao et al., 2022). Interestingly, ROS also play a dual role in mitophagy: excessive oxidative stress impairs mitophagy, while low levels activate mitophagy as a protective response (Liu J. et al., 2023).
The PINK1/Parkin pathway is a fundamental regulatory mechanism in mitophagy (Figure 1) (Wang H. et al., 2024). In healthy cells, PTEN-induced kinase 1 (PINK1) is imported into the mitochondria via the translocase complexes (TOM and TIM) and is rapidly degraded (Eldeeb et al., 2024). However, under oxidative stress, mitochondrial depolarisation prevents PINK1 degradation, enabling it to recruit Parkin to label damaged mitochondria for autophagic clearance (Shu et al., 2021). The damaged mitochondria are degraded by lysosomal enzymes into essential biomolecules such as amino acids and lipids, which are reused for cell regeneration and tissue repair (Zhang et al., 2022). In diabetic wounds, impaired mitophagy reduces the metabolic recycling capacity. Further research is needed to elucidate its exact role in delayed diabetic wound healing (Figure 1).
Mitophagy reduces the release of apoptotic factors, thereby preserving tissue integrity and cellular function. Studies show that mitophagy related proteins, including PINK1, Parkin, LC3-I and Beclin1, are downregulated in vascular endothelial cells under hyperglycemic conditions (Xi et al., 2021; Xiang et al., 2022), resulting in mitochondrial damage, increased apoptosis, and reduced endothelial cell activity and migration (Figure 1). Su et al. demonstrated that denatured collagen enhances autophagy and inhibits fibroblast apoptosis, facilitating wound repair (Su et al., 2022). In fibroblasts, denatured collagen reduces the activation of the apoptotic marker, caspase-3 and increases the expression of autophagic markers like Beclin-1 and LC3, highlighting its potential in promoting diabetic wound healing.
Chen et al. further confirmed the critical role of mitophagy in diabetic wound healing (Chen et al., 2015), showing that high glucose levels inhibit autophagy in endothelial progenitor cells (EPCs), increasing apoptosis and impairing its function. It is well known that mechanistic target of rapamycin (mTOR) plays an important role in the regulation of autophagy. Inhibiting mTOR signaling pathway activity reverses AGEs-induced autophagy impairment in endothelial progenitor cells (EPCs), thereby accelerating wound healing in diabetes (Jin et al., 2018). These findings underscore the importance of regulating mitochondrial mitophagy to reduce mitochondrial damage, inhibit apoptosis, and improve diabetic wound healing.
The imbalance between autophagy and apoptosis is a major contributor to delayed healing in diabetic wounds (Ko et al., 2020). Regulating autophagy pathways and addressing mitochondrial dysfunction could significantly enhance therapeutic strategies. Future research should focus on elucidating the precise mechanisms of autophagy inhibition in diabetic wounds, identifying novel targets and developing treatments to restore cellular homeostasis and accelerate wound healing.
1.3 Mitochondrial fission and fusion in diabetic wound healing
Mitochondrial quality control is a dynamic process through which mitochondria regulate their morphology, size, number, and function to maintain cellular homeostasis, respond to oxidative stress, and regulate energy metabolism (Jiang et al., 2022). Mitochondrial fission and fusion - the two key dynamics of mitochondrial quality control - enable mitochondria to adapt to cellular demands, ensuring the proper balance required for cellular function and environmental adaptation (Liu B. H. et al., 2024). Disruption of this balance is implicated in various pathological conditions, including diabetic wound healing (Zheng et al., 2021).
Mitochondrial quality control processes are precisely regulated by a specific set of regulatory proteins. During mitochondrial fission, the primary regulatory proteins include dynamin-related protein 1 (Drp1) and its receptor proteins - mitochondrial fission protein 1 (Fis1), mitochondrial fission factor (MFF) and MiD49/MiD51 (Konig et al., 2021). Drp1, a GTPase, is recruited to the outer mitochondrial membrane where it forms oligomeric structures with these receptor proteins (Gao and Hu, 2021), thus playing a central role in mitochondrial constriction and division. This process ensures proper mitochondrial distribution during cell division and the removal of damaged mitochondria (Kleele et al., 2021).
In diabetic patients, chronic hyperglycemia and impaired insulin signaling increases cellular energy demand (Tseng et al., 2024). To compensate, mitochondrial fission proteins are upregulated, while mitochondrial fusion proteins are downregulated. However, excessive activation of Drp1 leads to overactive mitochondrial fission, producing dysfunctional mitochondria that produces large amounts of ROS (Hao et al., 2019). This exacerbates intracellular oxidative stress, creating a vicious cycle of mitochondrial damage and cellular dysfunction (Ruegsegger et al., 2018) (Figure 2). Zhang et al. demonstrated that high glucose conditions lead to rapid mitochondrial fragmentation and increased expression of fission-related proteins, such as Drp1 and Fis1, disrupting mitochondrial morphology and exacerbating ROS production (Zhang et al., 2023a). Shi et al. showed that inhibiting the ROCK1/Drp1 mediated mitochondrial fission pathway reduced mitochondrial ROS (mtROS) production, restored blood flow, promoted capillary formation, and accelerated wound healing in diabetic mice (Shi et al., 2018).
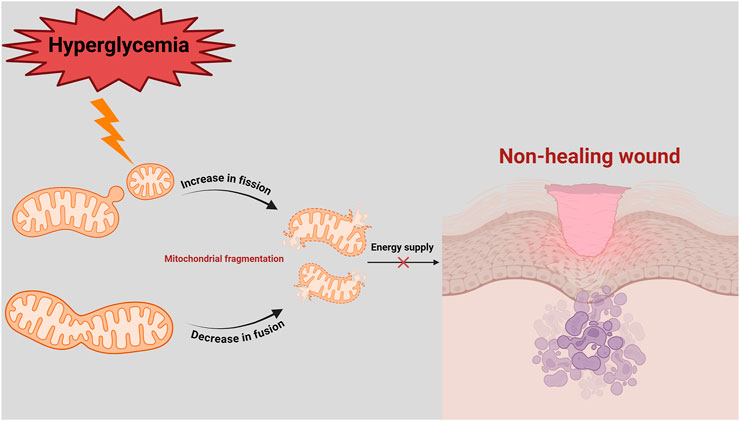
Figure 2. Imbalance of mitochondrial fission and fusion in delayed diabetic wound healing. In a hyperglycaemic environment, mitochondrial fission is increased, while mitochondrial fusion is decreased. This leads to mitochondrial fragmentation, energy deficit (decreased ATP) and ROS production. Since diabetic wounds have a high energy demand, inadequate ATP supply and imbalance in mitochondrial quality control lead to prolonged non-healing of diabetic wounds.
Conversely, mitochondrial fusion involves the progressive merging of two or more mitochondria into a continuous network, including both inner and outer mitochondrial membranes. This process is mediated primarily by mitochondrial fusion proteins such as optic atrophy 1 (OPA1), mitofusin1 (Mfn1) and mitofusin2 (Mfn2) (Hu et al., 2021). Mfn1 and Mfn2 mediates the fusion of outer mitochondrial membrane by forming homodimers or heterodimers (e.g., Mfn1-Mfn1, Mfn2-Mfn2, or Mfn1-Mfn2), while OPA1 mediates inner membrane fusion (Gordaliza-Alaguero et al., 2024). Mitochondrial fusion enhances bioenergetic capacity, supports mitochondrial genome intergrity, and enables cells to respond more effectively to injury and stress (Tabara et al., 2024). Lu et al. found that mesenchymal stem cells’ extracellular vesicles (MSC-EVs) promote mitochondrial fusion, reduce ROS and mtDNA release, and inhibit ferroptosis in endothelial cells, thus improving angiogenesis and wound healing in diabetic mice (Lu et al., 2024).
In diabetic patients, the balance between mitochondrial fission and fusion is often disrupted, as evidenced with increased fission and decreased fusion observed under hyperglycemic stress (Tatmatsu-Rocha et al., 2018). This imbalance contributes to mitochondrial fragmentation, functional impairment, and oxidative stress, all of which exacerbate endothelial cell dysfunction, apoptosis, and impaired wound healing (Sun et al., 2016). Zheng et al. reported that high-glucose induced dysregulation of mitochondrial dynamics disrupts vascular endothelial function, with upregulated fission proteins (Drp1 and Fis1) and downregulated fusion proteins (Mfn1, Mfn2, and OPA1), contributing to mitochondrial dysfunction and increased oxidative stress (Zheng et al., 2021) (Figure 2).
Mitochondrial fusion is particularly crucial for repairing damaged mitochondria and maintaining energy metabolism (Quintana-Cabrera et al., 2021; Guo et al., 2023). Disruption of fusion proteins impairs ATP production, directly affecting energy intensive processes such as cell migration, proliferation, and tissue regeneration, which are essential for wound healing (Amini et al., 2018; Sun et al., 2022). Wang et al. showed that overexpression of NDUFB5 promotes mitochondrial fusion, restores mitochondrial oxidative phosphorylation, and accelerates diabetic wound healing by improving mitochondrial function and reducing ROS production (Wang T. et al., 2024). Similarly, Chen et al. found that the S1PR2 antagonists modulate the RhoA/ROCK1/Drp1 signaling pathway, reversing high glucose-induced mitochondrial fission, improving endothelial cell migration, and inhibiting apoptosis (Chen et al., 2019).
Therefore, dysregulation of mitochondrial dynamics plays an essential pathological role in diabetic wound healing (Dai et al., 2022). An in-depth research is needed to elucidate the molecular mechanisms governing mitochondrial fission and fusion in diabetic wounds. Understanding these pathways can pave the way for new treatments that address mitochondrial dysfunction, providing innovative therapeutic avenues for managing diabetes-related complications.
1.4 Mitochondrial apoptosis in diabetic wound healing
Apoptosis, a genetically controlled, process of programmed cell death, plays a critical role in tissue homeostasis by rapidly removing excess or damaged cells. This process involves two major pathways: the mitochondria-mediated intrinsic pathway and the death receptor-mediated extrinsic pathway (Son and Lee, 2021). Among these, the mitochondria-mediated pathway is the predominant intrinsic apoptotic mechanism (Brenner and Mak, 2009). In DFUs, the activation of mitochondrial apoptosis disrupts physiological processes such as cell proliferation, angiogenesis, and reconstruction of extracellular matrix (Nagarjuna Reddy et al., 2022). The apoptosis of key cells, including fibroblasts, keratinocytes, and vascular endothelial cells, delays wound healing and hampers tissue regeneration (Kim and Park, 2019; Liang et al., 2019; Liu H. et al., 2024), highlighting the critical role of apoptotic mechanisms in wound healing.
Mitochondrial apoptosis is initiated by multiple pro-apoptotic signals, including oxidative stress triggered by hyperglycemia, sustained inflammation, DNA damage, and severe hypoxia (Kaina, 2003; Chen et al., 2024). These pro-apoptotic signals act synergistically through BH3 domain - containing proteins, such as Bim and Bid, which activates key pro-apoptotic effectors like Bax and Bak (Ruhl et al., 2025). When activated, Bax and Bak translocate to the outer mitochondrial membrane, resulting in mitochondrial outer membrane permeabilisation (MOMP) (Riley et al., 2018). This disrupts membrane integrity, decreases mitochondrial membrane potential, and facilitates the release of pro-apoptotic factors such as cytochrome C (Cyt C) (Cheng and Ferrell, 2018).
In DFUs, hyperglycemia disrupts the balance between pro-apoptotic (e.g., Bax) and anti-apoptotic (e.g., Bcl-2) proteins, triggering mitochondrial apoptosis (Wu et al., 2025). Studies on diabetic wounds have show significant reduced expression of Bcl-2, and weakened anti-apoptotic defenses, leading to cell apoptosis (Ye et al., 2025) (Figure 3). This phenomenon exacerbates the loss of fibroblasts and vascular endothelial cells, impairs tissue regeneration and delays wound healing (Justynski et al., 2023). Moreover, AGEs activate apoptotic signaling pathways through interaction with their receptor, RAGE. This interaction enhances the production of ROS and upregulates the expression of Bax, caspase-9, and cytochrome c, ultimately activating apoptotic markers such as caspase-3 and PARP. Consequently, endothelial progenitor cell apoptosis occurs, impairing tissue repair and further delaying wound healing (Jin et al., 2018; Li et al., 2018). Similarly, Ren et al. found that hyperglycemia increases the expression of cleaved Bax and Caspase-3 in human microvascular endothelial cells (HMEC-1), promoting apoptosis, oxidative stress and inflammation. However, increased expression of angiotensin-converting enzyme 2 (ACE2) was found to attenuate hyperglycemia-triggered apoptosis by inhibiting the JAK2/STAT3 signaling pathway, improving cell viability, and decreasing mitochondrial apoptotic protein expression (Ren et al., 2022). This suggests that ACE2 could be a potential therapeutic target for improving vascular endothelial cell dysfunction and promoting wound healing in diabetic patients.
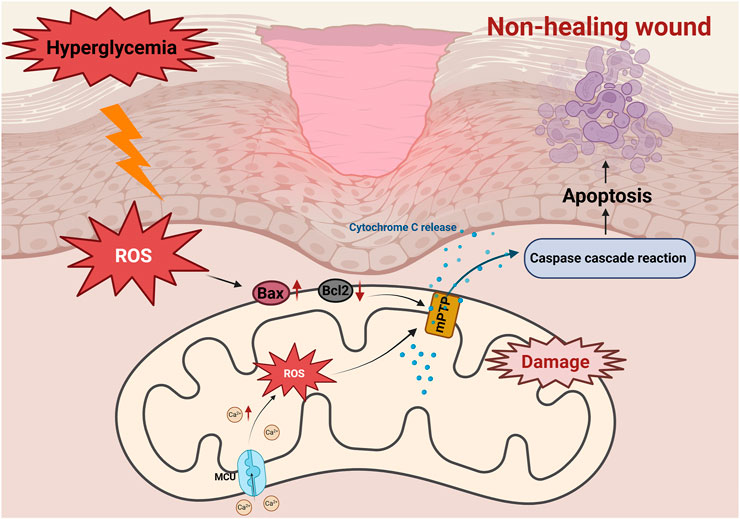
Figure 3. Role of mitochondrial apoptosis in non-healing diabetic wounds. Activation of the AGEs/RAGE axis induces overproduction of ROS, leading to upregulation of Bax and downregulation of Bcl-2. The subsequent opening of the mitochondrial membrane permeability transition pore (mPTP) triggers a decrease in ΔΨm, which leads to the release of Cyt C and activation of the caspase cascade reaction, ultimately inducing apoptosis of fibroblasts, exacerbating tissue repair disorders and delaying healing of diabetic wounds. Hyperglycemia triggers mitochondrial calcium (Ca2+) overload through MCU channels. Excess Ca2+ triggers ROS accumulation, disrupts mitochondrial membrane potential (ΔΨm), and causes Cyt C release, activates caspase cascade reaction, which ultimately induces apoptosis and leads to delayed diabetic wound healing.
Changes in mitochondrial membrane permeability is pivotal in mitochondria-mediated apoptosis, directly determining cell fate (Tomasina et al., 2022). Increased mitochondrial membrane permeability facilitates the rapid release of Cyt C, a key mediator in the mitochondrial respiratory chain. Cyt C disrupts electron transfer, impairs respiratory chain function, and leads to excessive production of superoxide ions. This triggers oxidative stress, exacerbates the inflammatory response and further delays wound healing in diabetic patients (Figure 3).
In addition, extrinsic apoptosis plays an important role in wound healing in diabetes. TNF-α inhibition promoted wound healing in diabetic mice and inhibited fibroblast apoptosis (Siqueira et al., 2010). In diabetic wound, TNF superfamily, member 6 (FasL) expression induces keratinocyte apoptosis, leading to delayed healing (Liang et al., 2019; Wang et al., 2023). Modulating mitochondrial apoptotic proteins or directly targeting mitochondrial function presents a promising intervention strategy for diabetic wound management. However, the precise mechanisms by which mitochondrial apoptosis influences diabetic wound healing remains incompletely understood. Further research is needed to explore the molecular pathways involved and develop targeted pharmacological interventions. Advancing our understanding in this area could open new avenues for the treatment of diabetes-induced complications and accelerate wound repair.
1.5 Mitochondrial calcium homeostasis and its role in diabetic wound healing
Mitochondria, often referred as the “powerhouse” of the cell, serves as an important regulatory centre for intracellular calcium signaling (De Stefani et al., 2016). In 1960, De Luca et al. first discovered that mitochondrial calcium uptake is mediated by the mitochondrial calcium uniporter (MCU), a channel located across the inner mitochondrial membrane. The MCU complex consists of channel subunits (MCU and MCUb), regulatory subunits (EMRE, MICU1, MICU2, and MCUR1), and additional proteins associated with calcium transport. Calcium ion (Ca2+) translocation is regulated by the interaction between the channel subunits and the regulatory proteins (Xue et al., 2022).
The mitochondrial Ca2+ uptake is driven by the electrochemical gradient established during oxidative phosphorylation, where the proton concentration gradient across the inner mitochondrial membrane fuels ATP synthesis (Patron et al., 2022; Szabo and Szewczyk, 2023). This Ca2+ influx is essential for regulating aerobic metabolism and maintaining redox homeostasis (Wescott et al., 2019). However, disrupted mitochondrial calcium homeostasis triggers oxidative stress, overproduction of ROS, mitochondrial depolarization, and apoptosis (Meng et al., 2023; Weiser et al., 2023).
In the resting state, the cytoplasmic Ca2+ concentration is low, and regulatory proteins such as MICU1 and MICU2 prevent Ca2+ from entering the mitochondria through MCU (Liu et al., 2016). Upon stimulated, cytoplasmic Ca2+ levels rises and activates MICU1, enabling mitochondrial Ca2+influx; closure of MCU, mediated by MCUR1, restores balance (Dong et al., 2017). After sufficient calcium uptake, MCUR1 assists in closing the MCU channel, preventing calcium overload. This tightly regulated mechanism ensures that the mitochondria is protected from oxidative damage caused by excess calcium while maintaining their ability to respond to cytoplasmic calcium signals, thereby preserving normal physiological functions of the cells (Wang C. et al., 2020; Garbincius and Elrod, 2022).
Chronic hyperglycemic induces persistent oxidative stress, which disrupts mitochondrial calcium homeostasis (Gerber and Rutter, 2017). Normally Ca2+ helps regulate NADPH production to counteract oxidative stress (Park et al., 2022). However, in diabetes, mitochondrial dysfunction leads to calcium dysregulation, impairing the mitochondrial electron transport chain and decreasing the membrane potential (Belosludtsev et al., 2019; Dia et al., 2020), which exacerbates ROS generation and delays wound healing.
Mitochondrial calcium plays a dual role: in moderate amounts, it activates enzymes in the tricarboxylic acid (TCA) cycle, promoting ATP production. However, ROS-induced damage to the mitochondrial membrane causes excess calcium influx into the mitochondria, disrupts this balance. Chen et al. showed that MCU’s mRNA and its regulatory protein MCUR1 were upregulated in high-glucose environments, leading to increased mitochondrial calcium levels and ROS production. This, in turn, triggers endothelial cell dysfunction, apoptosis and impaired wound healing (Chen et al., 2017). Regulating MCU expression improves Ca2+ homeostasis, thereby protecting the biological function of dermal fibroblasts in wound healing (Wang M. et al., 2024).
Calcium overload opens the mitochondrial permeability transition pore (mPTP), a critical event in mitochondrial apoptosis (Dubois et al., 2024). The mPTP opening leads to a loss of mitochondrial membrane potential, mitochondrial swelling, and the release of pro-apoptotic factors such as Cyt C. These events exacerbate apoptosis, further impairing diabetic wound healing (Figure 3).
Therefore, hyperglycemia-induced oxidative stress and dysregulation of mitochondrial calcium homeostasis form a self-reinforcing loop. Oxidative stress damages mitochondrial membrane and impairs MCU function, leading to Ca2+ overload (Zhang et al., 2024). The imbalance increases ROS production, aggravating oxidative stress (Ly et al., 2017). This vicious cycle ultimately delays wound healing in diabetic patients. Although these mechanisms are theoretically supported, the exact processes remains to be determined.
Future studies should aim to further investigate the molecular pathways involved and develop new strategies to effectively regulate mitochondrial calcium homeostasis as a means to treat diabetic wounds.
1.6 Ferroptosis and its role in diabetic wound healing
Mitochondrial dysfunction is an important factor contributing to delayed wound healing in diabetic ulcers. Studies have shown that mitochondrial dysfunction is strongly associated with ferroptosis, a hallmark of diabetes-related complications (He et al., 2022). Ferroptosis, a Fe2+-dependent type of programmed cell death, differs from traditional modes of cell death such as apoptosis, necrosis, and autophagy. It is characterized by excessive lipid peroxidation. Mitochondria, enriched in Fe2+, serves as the primary site of ROS production, which enhances cellular sensitivity to ferroptosis through a variety of mechanisms.
In diabetic conditions, chronic hyperglycemia induces persistent oxidative stress, leading to excessive ROS production and Fe2+ accumulation (Wei et al., 2020; Feng et al., 2021). This activates lipid peroxidation, impairs cellular functions, and triggers ferroptosis, ultimately delaying wound healing (Feng et al., 2022) (Figure 4). However, the mechanisms linking mitochondrial dysfunction to ferroptosis in diabetic wounds remains incompletely understood, warranting further studies.
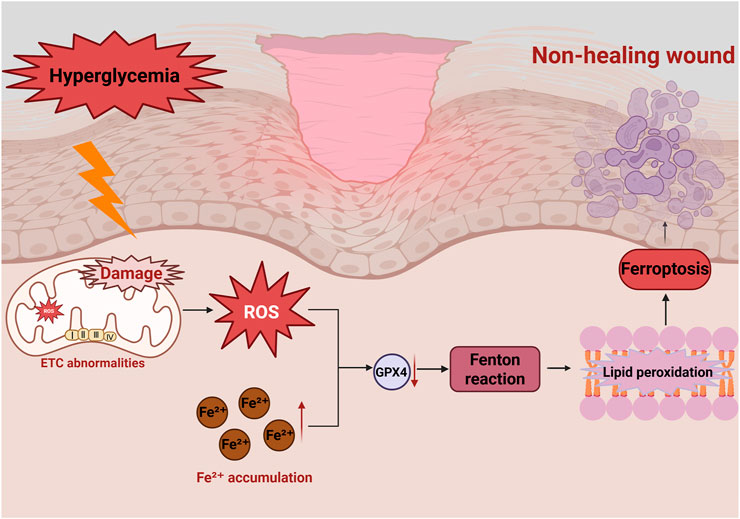
Figure 4. Role of ferroptosis in diabetic wounds in non-healing diabetic wounds. Hyperglycemia induces mitochondrial dysfunction, leading to excessive ROS production, reduced GPX4 activity and ETC., abnormalities. Excessive Fe2+ accumulation induces the Fenton reaction to produce lipid peroxides, which results in ferroptosis. This damage repair cells and impede wound healing.
The critical ferroptosis inhibitors are glutathione peroxidase 4 (GPX4) and glutathione (GSH) (Xi et al., 2025). Their depletion enhances lipid peroxide accumulation, exacerbating ferroptosis, leading to cellular damage and delayed tissue repair (Li et al., 2021).
Hyperglycemia-induced mitochondrial dysfunction and endoplasmic reticulum stress can result in Fe2+ accumulation and excessive ROS production, further driving ferroptosis (Ma et al., 2020). Cui et al. found that elevated Fe2+ levels and ROS production damage mitochondria, impairing the proliferation and migration of critical skin repair cells such as human dermal fibroblasts (HDFs) and endothelial cells (Cui et al., 2023), significantly hindering diabetic wound healing. Studies have shown that administration of the Deferoxamine (DFO) improves ferroptosis in human umbilical vein endothelial cells (HUVECs) induced by high glucose (Chen et al., 2023a).
Xiong et al. (2024) developed a novel therapeutic approach using a PF-PEG@ASIV-EXO hydrogel, which inhibited ferroptosis pathways to promote wound healing. The hydrogel improved mitochondrial function, inhibited ferroptosis, and promoted angiogenesis by increasing the expression of SLC7A11, GPX4, mitochondrial GSH and superoxide dismutase (SOD), while decreasing the expression of the Acyl-CoA synthetase long chain family member 4 (ACSL4) – accelerating wound healing. This finding underscores the therapeutic potential of ferroptosis inhibitors in diabetic wound therapy.
Thus, hyperglycemia-induced ferroptosis contributes significantly to diabetic ulcer pathology by altering mitochondrial function, increasing oxidative stress and promoting lipid peroxidation (Meng X. et al., 2025). While the interplay between mitochondrial dysfunction and ferroptosis in diabetic ulcers is still unclear, targeting ferroptosis-related pathways holds significant therapeutic promise. Ferroptosis inhibitors, along with strategies to enhance mitochondrial health and regulate oxidative stress, could accelerate the healing of diabetic ulcers.
Thus, the modulation of ferroptosis pathways not only provides insights into pathological mechanism of diabetic ulcers but also offers a foundation for developing novel therapeutic strategy for future clinical interventions (Han et al., 2025).
2 Conclusion
Sustained hyperglycemia-induced mitochondrial dysfunction–characterized by disrupted calcium homeostasis, excessive ROS production, impaired mitophagy, increased apoptosis and ferroptosis, and altered mitochondrial dynamics–is a central pathological mechanism hindering diabetic wound healing (Figure 5). These dysfunctions impair cellular energy metabolism and compromise the activity of critical repair cells, leading to delayed tissue regeneration. Consequently, therapeutic strategies aimed at restoring mitochondrial function–particularly by modulating calcium signaling, mitochondrial dynamics, mitophagy, and ferroptosis–hold significant promise.
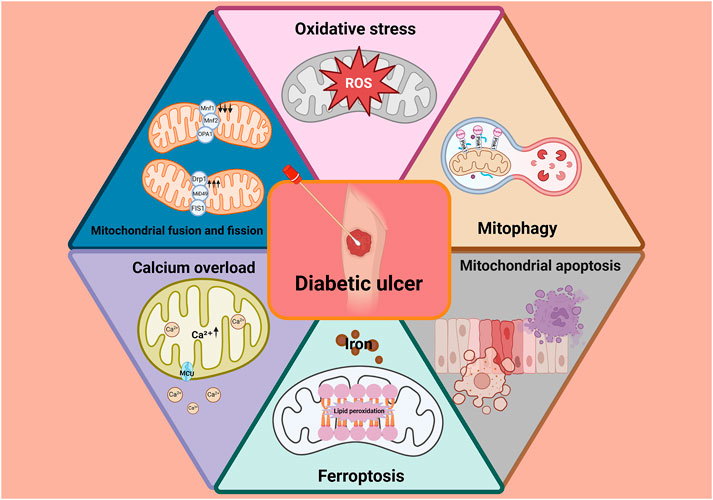
Figure 5. Sustained hyperglycemia-induced mitochondrial dysfunction–characterized by disrupted calcium homeostasis, excessive ROS production, impaired mitophagy, increased apoptosis and ferroptosis, and altered mitochondrial dynamics–is a central pathological mechanism hindering diabetic wound healing.
Many drugs have shown to have better therapeutic effects on diabetes (Lin Y. et al., 2023; Oladoja et al., 2023). Notably, mitochondrial modulators such as metformin have demonstrated beneficial effects in diabetic wound models. Metformin inhibits excessive mitochondrial fission, reduces oxidative stress, and enhances mitophagy, collectively promoting wound repair. Table 1 summarizes pharmacological agents that target mitochondrial pathways for diabetic wound treatment.
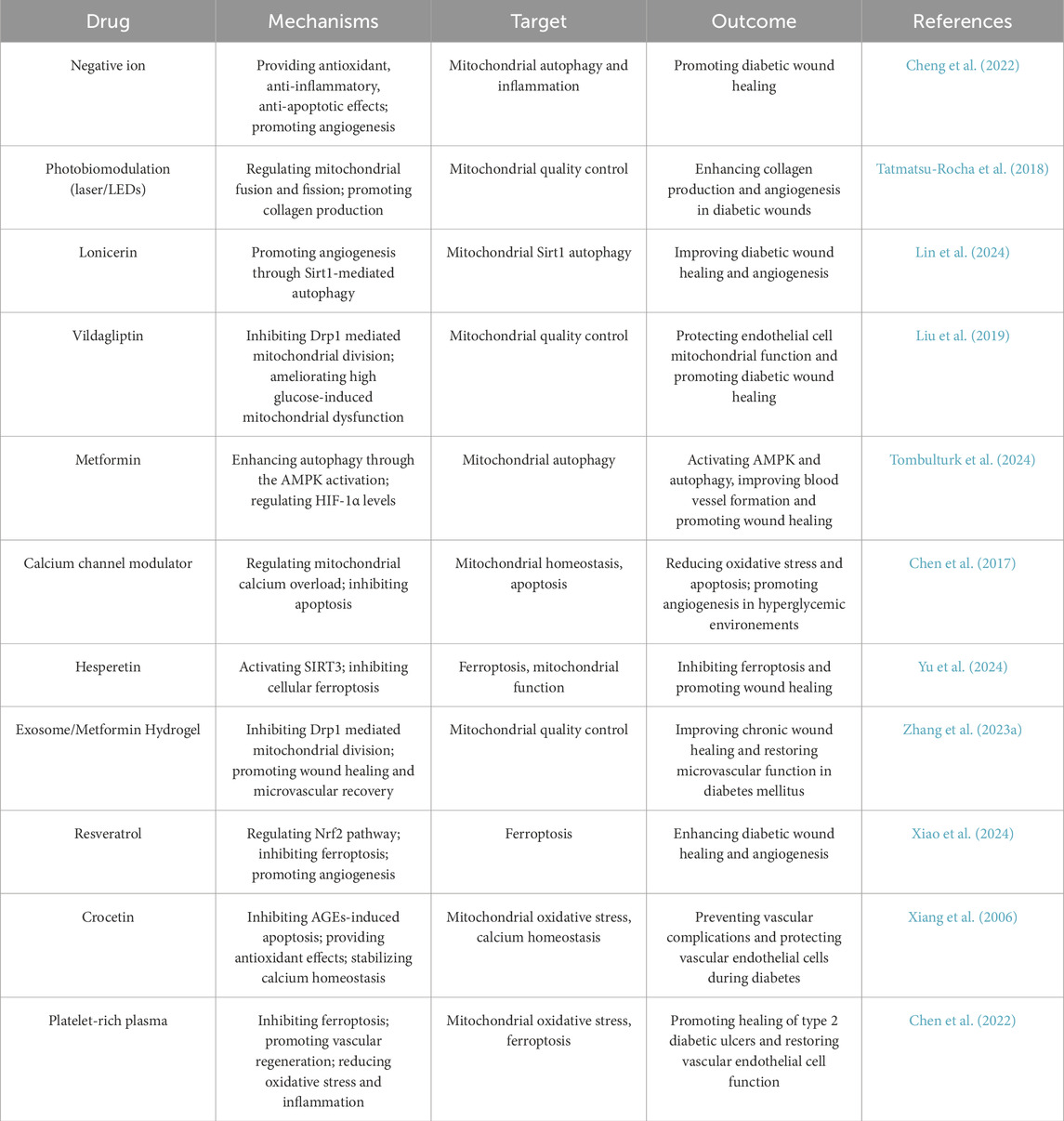
Table 1. Drugs targeting mitochondria improve diabetic wound healing.
Future research should focus on developing integrated therapeutic approaches that comprehensively regulate mitochondrial biological functions. Such strategies offer a targeted and effective path toward improved clinical outcomes in diabetic wound healing.
Author contributions
YP: Writing – original draft. LC: Writing – original draft. YC: Writing – original draft. ET: Writing – review and editing, Writing – original draft. SZ: Writing – original draft. YY: Writing – original draft. KL: Writing – review and editing, Funding acquisition. JW: Writing – review and editing, Funding acquisition. XL: Funding acquisition, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was financially supported by Sichuan Science and Technology Program (No. 2024NSFSC0610 and 2022YFS0622). This study was financially supported by National Natural Science Foundation of China (Grant No. 82374073) and Science and Technology Planning Project of Luzhou, Sichuan Province, China (Grant No. 2024LZXNYDJ008). This study was financially supported by the Applied Basic research program of Southwest Medical University (2024ZKY107).
Acknowledgments
The authors acknowledges the use of Biorender that is used to create the figure.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Al Ojaimi, M., Salah, A., and El-Hattab, A. W. (2022). Mitochondrial fission and fusion: molecular mechanisms, biological functions, and related disorders. Membr. (Basel) 12 (9), 893. doi:10.3390/membranes12090893
Amini, P., Stojkov, D., Felser, A., Jackson, C. B., Courage, C., Schaller, A., et al. (2018). Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat. Commun. 9 (1), 2958. doi:10.1038/s41467-018-05387-y
Armstrong, D. G., Tan, T. W., Boulton, A. J. M., and Bus, S. A. (2023). Diabetic foot ulcers: a review. JAMA 330 (1), 62–75. doi:10.1001/jama.2023.10578
Bai, R., Zhang, T., Gao, Y., Shu, T., Zhou, Y., Wang, F., et al. (2022). Rab31, a receptor of advanced glycation end products (RAGE) interacting protein, inhibits AGE induced pancreatic beta-cell apoptosis through the pAKT/BCL2 pathway. Endocr. J. 69 (8), 1015–1026. doi:10.1507/endocrj.EJ21-0594
Belosludtsev, K. N., Talanov, E. Y., Starinets, V. S., Agafonov, A. V., Dubinin, M. V., and Belosludtseva, N. V. (2019). Transport of Ca(2+) and Ca(2+)-Dependent permeability transition in Rat liver Mitochondria under the streptozotocin-induced type I diabetes. Cells 8 (9), 1014. doi:10.3390/cells8091014
Brenner, D., and Mak, T. W. (2009). Mitochondrial cell death effectors. Curr. Opin. Cell Biol. 21 (6), 871–877. doi:10.1016/j.ceb.2009.09.004
Carrasco, E., Calvo, M. I., Blazquez-Castro, A., Vecchio, D., Zamarron, A., de Almeida, I. J. D., et al. (2015). Photoactivation of ROS production in situ transiently activates cell proliferation in mouse skin and in the hair follicle stem cell niche promoting hair growth and wound healing. J. Invest. Dermatol 135 (11), 2611–2622. doi:10.1038/jid.2015.248
Cartes-Saavedra, B., Ghosh, A., and Hajnoczky, G. (2025). The roles of mitochondria in global and local intracellular calcium signalling. Nat. Rev. Mol. Cell Biol. 26 (6), 456–475. doi:10.1038/s41580-024-00820-1
Chang, M., and Nguyen, T. T. (2021). Strategy for treatment of infected diabetic foot ulcers. Acc. Chem. Res. 54 (5), 1080–1093. doi:10.1021/acs.accounts.0c00864
Chen, W., Wu, Y., Li, L., Yang, M., Shen, L., Liu, G., et al. (2015). Adenosine accelerates the healing of diabetic ischemic ulcers by improving autophagy of endothelial progenitor cells grown on a biomaterial. Sci. Rep. 5, 11594. doi:10.1038/srep11594
Chen, W., Yang, J., Chen, S., Xiang, H., Liu, H., Lin, D., et al. (2017). Importance of mitochondrial calcium uniporter in high glucose-induced endothelial cell dysfunction. Diab Vasc. Dis. Res. 14 (6), 494–501. doi:10.1177/1479164117723270
Chen, Y. H., Chen, Z. W., Li, H. M., Yan, X. F., and Feng, B. (2018). AGE/RAGE-Induced EMP release via the NOX-Derived ROS pathway. J. Diabetes Res. 2018, 6823058. doi:10.1155/2018/6823058
Chen, W., Xiang, H., Chen, R., Yang, J., Yang, X., Zhou, J., et al. (2019). S1PR2 antagonist ameliorate high glucose-induced fission and dysfunction of mitochondria in HRGECs via regulating ROCK1. BMC Nephrol. 20 (1), 135. doi:10.1186/s12882-019-1323-0
Chen, L., Wu, D., Zhou, L., and Ye, Y. (2022). Platelet-rich plasma promotes diabetic ulcer repair through inhibition of ferroptosis. Ann. Transl. Med. 10 (20), 1121. doi:10.21037/atm-22-4654
Chen, J., Li, X., Liu, H., Zhong, D., Yin, K., Li, Y., et al. (2023a). Bone marrow stromal cell-derived exosomal circular RNA improves diabetic foot ulcer wound healing by activating the nuclear factor erythroid 2-related factor 2 pathway and inhibiting ferroptosis. Diabet. Med. 40 (7), e15031. doi:10.1111/dme.15031
Chen, J., Qin, S., Liu, S., Zhong, K., Jing, Y., Wu, X., et al. (2023b). Targeting matrix metalloproteases in diabetic wound healing. Front. Immunol. 14, 1089001. doi:10.3389/fimmu.2023.1089001
Chen, S., Sun, D., Zhang, S., Xu, L., Wang, N., Li, H., et al. (2024). TIN2 modulates FOXO1 mitochondrial shuttling to enhance oxidative stress-induced apoptosis in retinal pigment epithelium under hyperglycemia. Cell Death Differ. 31 (11), 1487–1505. doi:10.1038/s41418-024-01349-8
Cheng, X., and Ferrell, J. E. (2018). Apoptosis propagates through the cytoplasm as trigger waves. Science 361 (6402), 607–612. doi:10.1126/science.aah4065
Cheng, Y. H., Li, H. K., Yao, C. A., Huang, J. Y., Sung, Y. T., Chung, S. D., et al. (2022). Negative air ions through the action of antioxidation, anti-inflammation, anti-apoptosis and angiogenesis ameliorate lipopolysaccharide induced acute lung injury and promote diabetic wound healing in rat. PLoS One 17 (10), e0275748. doi:10.1371/journal.pone.0275748
Choudhury, S., Ghosh, S., Gupta, P., Mukherjee, S., and Chattopadhyay, S. (2015). Inflammation-induced ROS generation causes pancreatic cell death through modulation of Nrf2/NF-κB and SAPK/JNK pathway. Free Radic. Res. 49 (11), 1371–1383. doi:10.3109/10715762.2015.1075016
Cui, S., Liu, X., Liu, Y., Hu, W., Ma, K., Huang, Q., et al. (2023). Autophagosomes defeat ferroptosis by decreasing generation and increasing discharge of free Fe(2+) in skin repair cells to accelerate diabetic wound healing. Adv. Sci. (Weinh) 10 (25), e2300414. doi:10.1002/advs.202300414
Dai, X., Wang, K., Fan, J., Liu, H., Fan, X., Lin, Q., et al. (2022). Nrf2 transcriptional upregulation of IDH2 to tune mitochondrial dynamics and rescue angiogenic function of diabetic EPCs. Redox Biol. 56, 102449. doi:10.1016/j.redox.2022.102449
De Stefani, D., Rizzuto, R., and Pozzan, T. (2016). Enjoy the trip: calcium in Mitochondria back and forth. Annu. Rev. Biochem. 85, 161–192. doi:10.1146/annurev-biochem-060614-034216
Deng, L., Du, C., Song, P., Chen, T., Rui, S., Armstrong, D. G., et al. (2021). The role of oxidative stress and antioxidants in diabetic wound healing. Oxid. Med. Cell Longev. 2021, 8852759. doi:10.1155/2021/8852759
Deng, L., Wang, G., and Ju, S. (2024). Correlation between inflammatory factors, autophagy protein levels, and infection in granulation tissue of diabetic foot ulcer. Immun. Inflamm. Dis. 12 (4), e1233. doi:10.1002/iid3.1233
Dia, M., Gomez, L., Thibault, H., Tessier, N., Leon, C., Chouabe, C., et al. (2020). Reduced reticulum-mitochondria Ca(2+) transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy. Basic Res. Cardiol. 115 (6), 74. doi:10.1007/s00395-020-00835-7
Dong, Z., Shanmughapriya, S., Tomar, D., Siddiqui, N., Lynch, S., Nemani, N., et al. (2017). Mitochondrial Ca(2+) uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol. Cell 65 (6), 1014–1028. doi:10.1016/j.molcel.2017.01.032
Du, L., Wang, L., Wang, B., Wang, J., Hao, M., Chen, Y. B., et al. (2020). A novel compound AB38b attenuates oxidative stress and ECM protein accumulation in kidneys of diabetic mice through modulation of Keap1/Nrf2 signaling. Acta Pharmacol. Sin. 41 (3), 358–372. doi:10.1038/s41401-019-0297-6
Dubois, M., Boulghobra, D., Rochebloine, G., Pallot, F., Yehya, M., Bornard, I., et al. (2024). Hyperglycemia triggers RyR2-dependent alterations of mitochondrial calcium homeostasis in response to cardiac ischemia-reperfusion: key role of DRP1 activation. Redox Biol. 70, 103044. doi:10.1016/j.redox.2024.103044
Elbatreek, M. H., Pachado, M. P., Cuadrado, A., Jandeleit-Dahm, K., and Schmidt, H. (2019). Reactive oxygen comes of Age: Mechanism-Based therapy of diabetic end-organ damage. Trends Endocrinol. Metab. 30 (5), 312–327. doi:10.1016/j.tem.2019.02.006
Eldeeb, M. A., Bayne, A. N., Fallahi, A., Goiran, T., MacDougall, E. J., Soumbasis, A., et al. (2024). Tom20 gates PINK1 activity and mediates its tethering of the TOM and TIM23 translocases upon mitochondrial stress. Proc. Natl. Acad. Sci. U. S. A. 121 (10), e2313540121. doi:10.1073/pnas.2313540121
Fan, L., Zhang, X., Huang, Y., Zhang, B., Li, W., Shi, Q., et al. (2023). Homoplantaginin attenuates high glucose-induced vascular endothelial cell apoptosis through promoting autophagy via the AMPK/TFEB pathway. Phytother. Res. 37 (7), 3025–3041. doi:10.1002/ptr.7797
Feng, X., Wang, S., Sun, Z., Dong, H., Yu, H., Huang, M., et al. (2021). Ferroptosis enhanced diabetic renal tubular injury via HIF-1α/HO-1 pathway in db/db mice. Front. Endocrinol. (Lausanne) 12, 626390. doi:10.3389/fendo.2021.626390
Feng, J., Wang, J., Wang, Y., Huang, X., Shao, T., Deng, X., et al. (2022). Oxidative stress and lipid peroxidation: prospective associations between ferroptosis and delayed wound healing in diabetic ulcers. Front. Cell Dev. Biol. 10, 898657. doi:10.3389/fcell.2022.898657
Fu, Y. J., Shi, Y. F., Wang, L. Y., Zhao, Y. F., Wang, R. K., Li, K., et al. (2023). All-Natural immunomodulatory bioadhesive hydrogel promotes angiogenesis and diabetic wound healing by regulating macrophage heterogeneity. Adv. Sci. (Weinh) 10 (13), e2206771. doi:10.1002/advs.202206771
Gao, S., and Hu, J. (2021). Mitochondrial fusion: the machineries In and out. Trends Cell Biol. 31 (1), 62–74. doi:10.1016/j.tcb.2020.09.008
Gao, M., Nguyen, T. T., Suckow, M. A., Wolter, W. R., Gooyit, M., Mobashery, S., et al. (2015). Acceleration of diabetic wound healing using a novel protease–anti-protease combination therapy. Proc. Natl. Acad. Sci. 112 (49), 15226–15231. doi:10.1073/pnas.1517847112
Garbincius, J. F., and Elrod, J. W. (2022). Mitochondrial calcium exchange in physiology and disease. Physiol. Rev. 102 (2), 893–992. doi:10.1152/physrev.00041.2020
Geng, K., Ma, X., Jiang, Z., Gu, J., Huang, W., Wang, W., et al. (2023). WDR74 facilitates TGF-β/Smad pathway activation to promote M2 macrophage polarization and diabetic foot ulcer wound healing in mice. Cell Biol. Toxicol. 39 (4), 1577–1591. doi:10.1007/s10565-022-09748-8
Gerber, P. A., and Rutter, G. A. (2017). The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxid. Redox Signal 26 (10), 501–518. doi:10.1089/ars.2016.6755
Gordaliza-Alaguero, I., Sanchez-Fernandez-de-Landa, P., Radivojevikj, D., Villarreal, L., Arauz-Garofalo, G., Gay, M., et al. (2024). Endogenous interactomes of MFN1 and MFN2 provide novel insights into interorganelle communication and autophagy. Autophagy 21, 957–978. doi:10.1080/15548627.2024.2440843
Grazioli, S., and Pugin, J. (2018). Mitochondrial damage-associated molecular patterns: from inflammatory signaling to human diseases. Front. Immunol. 9, 832. doi:10.3389/fimmu.2018.00832
Guo, Y., Zhang, H., Yan, C., Shen, B., Zhang, Y., Guo, X., et al. (2023). Small molecule agonist of mitochondrial fusion repairs mitochondrial dysfunction. Nat. Chem. Biol. 19 (4), 468–477. doi:10.1038/s41589-022-01224-y
Han, Y., Sun, T., Tao, R., Han, Y., and Liu, J. (2017). Clinical application prospect of umbilical cord-derived mesenchymal stem cells on clearance of advanced glycation end products through autophagy on diabetic wound. Eur. J. Med. Res. 22 (1), 11. doi:10.1186/s40001-017-0253-1
Han, Q., Gu, Y., and Qian, Y. (2025). Study on the mechanism of activating SIRT1/Nrf2/p62 pathway to mediate autophagy-dependent ferroptosis to promote healing of diabetic foot ulcers. Naunyn Schmiedeb. Arch. Pharmacol. 398 (3), 3015–3025. doi:10.1007/s00210-024-03400-4
Hao, Y., Liu, H. M., Wei, X., Gong, X., Lu, Z. Y., and Huang, Z. H. (2019). Diallyl trisulfide attenuates hyperglycemia-induced endothelial apoptosis by inhibition of Drp1-mediated mitochondrial fission. Acta Diabetol. 56 (11), 1177–1189. doi:10.1007/s00592-019-01366-x
He, J., Li, Z., Xia, P., Shi, A., FuChen, X., Zhang, J., et al. (2022). Ferroptosis and ferritinophagy in diabetes complications. Mol. Metab. 60, 101470. doi:10.1016/j.molmet.2022.101470
He, S., Li, Z., Wang, L., Yao, N., Wen, H., Yuan, H., et al. (2024). A nanoenzyme-modified hydrogel targets macrophage reprogramming-angiogenesis crosstalk to boost diabetic wound repair. Bioact. Mater 35, 17–30. doi:10.1016/j.bioactmat.2024.01.005
Hong, Y., Li, J., Zhong, Y., Yang, S., Pei, L., Huang, Z., et al. (2023). Elabela inhibits TRAF1/NF-κB induced oxidative DNA damage to promote diabetic foot ulcer wound healing. iScience 26 (9), 107601. doi:10.1016/j.isci.2023.107601
Hu, Y., Chen, H., Zhang, L., Lin, X., Li, X., Zhuang, H., et al. (2021). The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy 17 (5), 1142–1156. doi:10.1080/15548627.2020.1749490
Huang, Y., Xing, H., Naud, S., and Kyriakides, T. R. (2024). Targeting hypoxia and thrombospondin-2 in diabetic wound healing. FASEB J. 38 (19), e70091. doi:10.1096/fj.202302429RRR
Huo, S., Wang, Q., Shi, W., Peng, L., Jiang, Y., Zhu, M., et al. (2023). ATF3/SPI1/SLC31A1 Signaling Promotes cuproptosis induced by advanced glycosylation end products in diabetic myocardial injury. Int. J. Mol. Sci. 24 (2), 1667. doi:10.3390/ijms24021667
Ji, X., Zhou, J., Zhou, Z., Liu, Z., Yan, L., Li, Y., et al. (2024). Recovering skin-nerve interaction by nanoscale metal-organic framework for diabetic ulcers healing. Bioact. Mater 42, 112–123. doi:10.1016/j.bioactmat.2024.08.024
Jiang, Y., Krantz, S., Qin, X., Li, S., Gunasekara, H., Kim, Y. M., et al. (2022). Caveolin-1 controls mitochondrial damage and ROS production by regulating fission - fusion dynamics and mitophagy. Redox Biol. 52, 102304. doi:10.1016/j.redox.2022.102304
Jiang, G., Jiang, T., Chen, J., Yao, H., Mao, R., Yang, X., et al. (2023). Mitochondrial dysfunction and oxidative stress in diabetic wound. J. Biochem. Mol. Toxicol. 37 (7), e23407. doi:10.1002/jbt.23407
Jin, H., Zhang, Z., Wang, C., Tang, Q., Wang, J., Bai, X., et al. (2018). Melatonin protects endothelial progenitor cells against AGE-Induced apoptosis via autophagy flux stimulation and promotes wound healing in diabetic mice. Exp. Mol. Med. 50 (11), 1–15. doi:10.1038/s12276-018-0177-z
Justynski, O., Bridges, K., Krause, W., Forni, M. F., Phan, Q. M., Sandoval-Schaefer, T., et al. (2023). Apoptosis recognition receptors regulate skin tissue repair in mice. Elife 12, e86269. doi:10.7554/eLife.86269
Kaina, B. (2003). DNA damage-triggered apoptosis: critical role of DNA repair, double-strand breaks, cell proliferation and signaling. Biochem. Pharmacol. 66 (8), 1547–1554. doi:10.1016/s0006-2952(03)00510-0
Kim, S. H., and Park, J. W. (2019). IDH2 deficiency impairs cutaneous wound healing via ROS-Dependent apoptosis. Biochim. Biophys. Acta Mol. Basis Dis. 1865 (11), 165523. doi:10.1016/j.bbadis.2019.07.017
Kleele, T., Rey, T., Winter, J., Zaganelli, S., Mahecic, D., Perreten Lambert, H., et al. (2021). Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 593 (7859), 435–439. doi:10.1038/s41586-021-03510-6
Ko, Y. S., Jin, H., Park, S. W., and Kim, H. J. (2020). Salvianolic acid B protects against oxLDL-induced endothelial dysfunction under high-glucose conditions by downregulating ROCK1-mediated mitophagy and apoptosis. Biochem. Pharmacol. 174, 113815. doi:10.1016/j.bcp.2020.113815
Konig, T., Nolte, H., Aaltonen, M. J., Tatsuta, T., Krols, M., Stroh, T., et al. (2021). MIROs and DRP1 drive mitochondrial-derived vesicle biogenesis and promote quality control. Nat. Cell Biol. 23 (12), 1271–1286. doi:10.1038/s41556-021-00798-4
Kowluru, R. A., Shan, Y., and Mishra, M. (2016). Dynamic DNA methylation of matrix metalloproteinase-9 in the development of diabetic retinopathy. Lab. Invest. 96 (10), 1040–1049. doi:10.1038/labinvest.2016.78
Kunkemoeller, B., and Kyriakides, T. R. (2017). Redox signaling in diabetic wound healing regulates extracellular matrix deposition. Antioxid. Redox Signal 27 (12), 823–838. doi:10.1089/ars.2017.7263
Laughlin, T., Tan, Y., Jarrold, B., Chen, J., Li, L., Fang, B., et al. (2020). Autophagy activators stimulate the removal of advanced glycation end products in human keratinocytes. J. Eur. Acad. Dermatol Venereol. 34 (Suppl. 3), 12–18. doi:10.1111/jdv.16453
Li, Q., Xia, S., Yin, Y., Guo, Y., Chen, F., and Jin, P. (2018). miR-5591-5p regulates the effect of ADSCs in repairing diabetic wound via targeting AGEs/AGER/JNK signaling axis. Cell Death Dis. 9 (5), 566. doi:10.1038/s41419-018-0615-9
Li, S., Li, Y., Wu, Z., Wu, Z., and Fang, H. (2021). Diabetic ferroptosis plays an important role in triggering on inflammation in diabetic wound. Am. J. Physiol. Endocrinol. Metab. 321 (4), E509–E520. doi:10.1152/ajpendo.00042.2021
Li, S., Deng, J., Sun, D., Chen, S., Yao, X., Wang, N., et al. (2022a). FBXW7 alleviates hyperglycemia-induced endothelial oxidative stress injury via ROS and PARP inhibition. Redox Biol. 58, 102530. doi:10.1016/j.redox.2022.102530
Li, S., Ding, X., Zhang, H., Ding, Y., and Tan, Q. (2022b). IL-25 improves diabetic wound healing through stimulating M2 macrophage polarization and fibroblast activation. Int. Immunopharmacol. 106, 108605. doi:10.1016/j.intimp.2022.108605
Li, F., Mao, Z., Du, Y., Cui, Y., Yang, S., Huang, K., et al. (2024a). Mesoporous MOFs with ROS scavenging capacity for the alleviation of inflammation through inhibiting stimulator of interferon genes to promote diabetic wound healing. J. Nanobiotechnology 22 (1), 246. doi:10.1186/s12951-024-02423-6
Li, M., Dong, Y., Shang, Y., Liu, J., Wang, Y., Zhang, D., et al. (2024b). Metformin syncs CeO(2) to recover Intra- and extra-cellular ROS homeostasis in diabetic wound healing. Small 20 (52), e2407802. doi:10.1002/smll.202407802
Li, W., Zhu, H., Chen, J., Ru, B., Peng, Q., Miao, J., et al. (2024c). PsAF5 functions as an essential adapter for PsPHB2-mediated mitophagy under ROS stress in Phytophthora sojae. Nat. Commun. 15 (1), 1967. doi:10.1038/s41467-024-46290-z
Liang, Y., Yang, C., Lin, Y., Parviz, Y., Sun, K., Wang, W., et al. (2019). Matrix metalloproteinase 9 induces keratinocyte apoptosis through FasL/Fas pathway in diabetic wound. Apoptosis 24 (7-8), 542–551. doi:10.1007/s10495-019-01536-w
Lin, Z., Li, L. Y., Chen, L., Jin, C., Li, Y., Yang, L., et al. (2024). Lonicerin promotes wound healing in diabetic rats by enhancing blood vessel regeneration through Sirt1-mediated autophagy. Acta Pharmacol. Sin. 45 (4), 815–830. doi:10.1038/s41401-023-01193-5
Lin, M., Wang, L., Wan, L. H., Xu, J. D., Li, Y., Cao, L. Y., et al. (2023). The antidiabetic effect and mechanism of JinXiaoXiaoKe decoction in type 2 diabetic goto–kakizaki rats. Clin. Complementary Med. Pharmacol. 3 (1), 100049. doi:10.1016/j.ccmp.2022.100049
Lin, Y., Wei, Y., Wei, Y., Yu, H., Zhang, W., Li, C., et al. (2023). Dexmedetomidine alleviates oxidative stress and mitochondrial dysfunction in diabetic peripheral neuropathy via the microRNA-34a/SIRT2/S1PR1 axis. Int. Immunopharmacol. 117, 109910. doi:10.1016/j.intimp.2023.109910
Lindley, L. E., Stojadinovic, O., Pastar, I., and Tomic-Canic, M. (2016). Biology and biomarkers for wound healing. Plast. Reconstr. Surg. 138 (Suppl. l), 18S–28S. doi:10.1097/PRS.0000000000002682
Liu, J. C., Liu, J., Holmstrom, K. M., Menazza, S., Parks, R. J., Fergusson, M. M., et al. (2016). MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep. 16 (6), 1561–1573. doi:10.1016/j.celrep.2016.07.011
Liu, H., Xiang, H., Zhao, S., Sang, H., Lv, F., Chen, R., et al. (2019). Vildagliptin improves high glucose-induced endothelial mitochondrial dysfunction via inhibiting mitochondrial fission. J. Cell Mol. Med. 23 (2), 798–810. doi:10.1111/jcmm.13975
Liu, C., Yalavarthi, S., Tambralli, A., Zeng, L., Rysenga, C. E., Alizadeh, N., et al. (2023a). Inhibition of neutrophil extracellular trap formation alleviates vascular dysfunction in type 1 diabetic mice. Sci. Adv. 9 (43), eadj1019. doi:10.1126/sciadv.adj1019
Liu, J., Lu, S., Zheng, L., Guo, Q., Cao, L., Xiao, Y., et al. (2023b). ATM-CHK2-TRIM32 axis regulates ATG7 ubiquitination to initiate autophagy under oxidative stress. Cell Rep. 42 (11), 113402. doi:10.1016/j.celrep.2023.113402
Liu, B. H., Xu, C. Z., Liu, Y., Lu, Z. L., Fu, T. L., Li, G. R., et al. (2024a). Mitochondrial quality control in human health and disease. Mil. Med. Res. 11 (1), 32. doi:10.1186/s40779-024-00536-5
Liu, H., Yao, Q., Wang, X., Xie, H., Yang, C., Gao, H., et al. (2024b). The research progress of crosstalk mechanism of autophagy and apoptosis in diabetic vascular endothelial injury. Biomed. Pharmacother. 170, 116072. doi:10.1016/j.biopha.2023.116072
Lorentzen, K. C., Prescott, A. R., and Ganley, I. G. (2025). Artificial targeting of autophagy components to mitochondria reveals both conventional and unconventional mitophagy pathways. Autophagy 21 (2), 315–337. doi:10.1080/15548627.2024.2395149
Lu, W., Li, X., Wang, Z., Zhao, C., Li, Q., Zhang, L., et al. (2024). Mesenchymal stem cell-derived extracellular vesicles accelerate diabetic wound healing by inhibiting NET-Induced ferroptosis of endothelial cells. Int. J. Biol. Sci. 20 (9), 3515–3529. doi:10.7150/ijbs.97150
Luo, L., An, Y., Geng, K., Wan, S., Zhang, F., Tan, X., et al. (2023). High glucose-induced endothelial STING activation inhibits diabetic wound healing through impairment of angiogenesis. Biochem. Biophys. Res. Commun. 668, 82–89. doi:10.1016/j.bbrc.2023.05.081
Ly, L. D., Xu, S., Choi, S. K., Ha, C. M., Thoudam, T., Cha, S. K., et al. (2017). Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 49 (2), e291. doi:10.1038/emm.2016.157
Ma, H., Wang, X., Zhang, W., Li, H., Zhao, W., Sun, J., et al. (2020). Melatonin suppresses ferroptosis induced by high glucose via activation of the Nrf2/HO-1 signaling pathway in type 2 diabetic osteoporosis. Oxid. Med. Cell Longev. 2020, 9067610. doi:10.1155/2020/9067610
Mao, W., Fan, Y., Wang, X., Feng, G., You, Y., Li, H., et al. (2022). Phloretin ameliorates diabetes-induced endothelial injury through AMPK-Dependent anti-EndMT pathway. Pharmacol. Res. 179, 106205. doi:10.1016/j.phrs.2022.106205
Mao, J., Xia, W., Wu, Y., Li, M., Zhao, Y., Zhai, P., et al. (2025). Biosynthesis of lysosomally escaped apoptotic bodies inhibits inflammasome synthesis in macrophages. Res. (Wash D C) 8, 0581. doi:10.34133/research.0581
McCarty, S. M., Cochrane, C. A., Clegg, P. D., and Percival, S. L. (2012). The role of endogenous and exogenous enzymes in chronic wounds: a focus on the implications of aberrant levels of both host and bacterial proteases in wound healing. Wound Repair Regen. 20 (2), 125–136. doi:10.1111/j.1524-475X.2012.00763.x
McDermott, K., Fang, M., Boulton, A. J. M., Selvin, E., and Hicks, C. W. (2023). Etiology, epidemiology, and disparities in the burden of diabetic foot ulcers. Diabetes Care 46 (1), 209–221. doi:10.2337/dci22-0043
Meng, M., Jiang, Y., Wang, Y., Huo, R., Ma, N., Shen, X., et al. (2023). β-carotene targets IP3R/GRP75/VDAC1-MCU axis to renovate LPS-induced mitochondrial oxidative damage by regulating STIM1. Free Radic. Biol. Med. 205, 25–46. doi:10.1016/j.freeradbiomed.2023.05.021
Meng, W., Chen, X., Chen, Y., Li, M., Zhang, L., Luo, Q., et al. (2025a). Self-Cascade of ROS/Glucose-Scavenging immunomodulatory hydrogels for programmed therapeutics of infected diabetic ulcers via Nrf2/NF-κB pathway. Small 21 (7), e2411189. doi:10.1002/smll.202411189
Meng, X., Pu, Z., He, J., Li, Q., and Xie, Y. (2025b). Metrnl ameliorates ferroptosis in model of diabetic foot ulcer through the inhibition of mitochondrial damage via LKB1/AMPK signaling. Exp. Clin. Endocrinol. Diabetes 133 (3), 120–132. doi:10.1055/a-2502-8712
Mohsin, F., Javaid, S., Tariq, M., and Mustafa, M. (2024). Molecular immunological mechanisms of impaired wound healing in diabetic foot ulcers (DFU), current therapeutic strategies and future directions. Int. Immunopharmacol. 139, 112713. doi:10.1016/j.intimp.2024.112713
Nagarjuna Reddy, V., Nyamathulla, S., Abdul Kadir Pahirulzaman, K., Mokhtar, S. I., Giribabu, N., and Pasupuleti, V. R. (2022). Gallocatechin-silver nanoparticles embedded in cotton gauze patches accelerated wound healing in diabetic rats by promoting proliferation and inhibiting apoptosis through the Wnt/β-catenin signaling pathway. PLoS One 17 (6), e0268505. doi:10.1371/journal.pone.0268505
Oladoja, F. A., Irokosu, E. S., Ayoola, M. D., Elijah, O. O., Akanji, M. A., Beatrice, O. T., et al. (2023). Evaluation of the antidiabetic activity and toxicological properties of Hippocratea Velutina (Afzel.). Clin. Complementary Med. Pharmacol. 3 (2), 100080. doi:10.1016/j.ccmp.2023.100080
Oyewole, A. O., and Birch-Machin, M. A. (2015). Mitochondria-targeted antioxidants. FASEB J. 29 (12), 4766–4771. doi:10.1096/fj.15-275404
Park, J. M., Do, V. Q., Seo, Y. S., Kim, H. J., Nam, J. H., Yin, M. Z., et al. (2022). NADPH oxidase 1 mediates acute blood pressure response to angiotensin II by contributing to calcium influx in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 42 (5), e117–e130. doi:10.1161/ATVBAHA.121.317239
Patron, M., Tarasenko, D., Nolte, H., Kroczek, L., Ghosh, M., Ohba, Y., et al. (2022). Regulation of mitochondrial proteostasis by the proton gradient. EMBO J. 41 (16), e110476. doi:10.15252/embj.2021110476
Peplow, P. V., and Baxter, G. D. (2012). Gene expression and release of growth factors during delayed wound healing: a review of studies in diabetic animals and possible combined laser phototherapy and growth factor treatment to enhance healing. Photomed. Laser Surg. 30 (11), 617–636. doi:10.1089/pho.2012.3312
Piperi, C., Goumenos, A., Adamopoulos, C., and Papavassiliou, A. G. (2015). AGE/RAGE signalling regulation by miRNAs: associations with diabetic complications and therapeutic potential. Int. J. Biochem. Cell Biol. 60, 197–201. doi:10.1016/j.biocel.2015.01.009
Qi, X., Cai, E., Xiang, Y., Zhang, C., Ge, X., Wang, J., et al. (2023). An immunomodulatory hydrogel by hyperthermia-assisted self-cascade glucose depletion and ROS scavenging for diabetic foot ulcer wound therapeutics. Adv. Mater 35 (48), e2306632. doi:10.1002/adma.202306632
Qi, X., Liu, C., Si, J., Yin, B., Huang, J., Wang, X., et al. (2024). A bioenergetically-active ploy (glycerol sebacate)-based multiblock hydrogel improved diabetic wound healing through revitalizing mitochondrial metabolism. Cell Prolif. 57 (7), e13613. doi:10.1111/cpr.13613
Qin, J., Peng, Z., Yuan, Q., Li, Q., Peng, Y., Wen, R., et al. (2019). AKF-PD alleviates diabetic nephropathy via blocking the RAGE/AGEs/NOX and PKC/NOX Pathways. Sci. Rep. 9 (1), 4407. doi:10.1038/s41598-018-36344-w
Quintana-Cabrera, R., Manjarres-Raza, I., Vicente-Gutierrez, C., Corrado, M., Bolanos, J. P., and Scorrano, L. (2021). Opa1 relies on cristae preservation and ATP synthase to curtail reactive oxygen species accumulation in mitochondria. Redox Biol. 41, 101944. doi:10.1016/j.redox.2021.101944
Ren, Y., Xie, W., Yang, S., Jiang, Y., Wu, D., Zhang, H., et al. (2022). Angiotensin-converting enzyme 2 inhibits inflammation and apoptosis in high glucose-stimulated microvascular endothelial cell damage by regulating the JAK2/STAT3 signaling pathway. Bioengineered 13 (4), 10802–10810. doi:10.1080/21655979.2022.2065760
Riley, J. S., Quarato, G., Cloix, C., Lopez, J., O'Prey, J., Pearson, M., et al. (2018). Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 37 (17), e99238. doi:10.15252/embj.201899238
Rizwan, H., Pal, S., Sabnam, S., and Pal, A. (2020). High glucose augments ROS generation regulates mitochondrial dysfunction and apoptosis via stress signalling cascades in keratinocytes. Life Sci. 241, 117148. doi:10.1016/j.lfs.2019.117148
Ruegsegger, G. N., Creo, A. L., Cortes, T. M., Dasari, S., and Nair, K. S. (2018). Altered mitochondrial function in insulin-deficient and insulin-resistant states. J. Clin. Invest. 128 (9), 3671–3681. doi:10.1172/JCI120843
Ruhl, S., Li, Z., Srivastava, S., Mari, L., Guy, C. S., Yang, M., et al. (2025). Inhibition of BAK-mediated apoptosis by the BH3-only protein BNIP5. Cell Death Differ. 32 (2), 320–336. doi:10.1038/s41418-024-01386-3
Sabbatinelli, J., Castiglione, S., Macri, F., Giuliani, A., Ramini, D., Vinci, M. C., et al. (2022). Circulating levels of AGEs and soluble RAGE isoforms are associated with all-cause mortality and development of cardiovascular complications in type 2 diabetes: a retrospective cohort study. Cardiovasc Diabetol. 21 (1), 95. doi:10.1186/s12933-022-01535-3
Sahin, N., Orhan, C., Erten, F., Tuzcu, M., Defo Deeh, P. B., Ozercan, I. H., et al. (2019). Effects of allyl isothiocyanate on insulin resistance, oxidative stress status, and transcription factors in high-fat diet/streptozotocin-induced type 2 diabetes mellitus in rats. J. Biochem. Mol. Toxicol. 33 (7), e22328. doi:10.1002/jbt.22328
Santarella, F., Sridharan, R., Marinkovic, M., Do Amaral, R., Cavanagh, B., Smith, A., et al. (2020). Scaffolds functionalized with matrix from induced pluripotent stem cell fibroblasts for diabetic wound healing. Adv. Healthc. Mater 9 (16), e2000307. doi:10.1002/adhm.202000307
Schwarz, P. E., Gallein, G., Ebermann, D., Muller, A., Lindner, A., Rothe, U., et al. (2013). Global Diabetes Survey--an annual report on quality of diabetes care. Diabetes Res. Clin. Pract. 100 (1), 11–18. doi:10.1016/j.diabres.2012.11.008
Senneville, E., Albalawi, Z., van Asten, S. A., Abbas, Z. G., Allison, G., Aragon-Sanchez, J., et al. (2024). IWGDF/IDSA guidelines on the diagnosis and treatment of diabetes-related foot infections (IWGDF/IDSA 2023). Diabetes Metab. Res. Rev. 40 (3), e3687. doi:10.1002/dmrr.3687
Shen, Q., Fang, J., Guo, H., Su, X., Zhu, B., Yao, X., et al. (2023). Astragaloside IV attenuates podocyte apoptosis through ameliorating mitochondrial dysfunction by up-regulated Nrf2-ARE/TFAM signaling in diabetic kidney disease. Free Radic. Biol. Med. 203, 45–57. doi:10.1016/j.freeradbiomed.2023.03.022
Shi, L., Chen, H., Yu, X., and Wu, X. (2013). Advanced glycation end products delay corneal epithelial wound healing through reactive oxygen species generation. Mol. Cell Biochem. 383 (1-2), 253–259. doi:10.1007/s11010-013-1773-9
Shi, Y., Fan, S., Wang, D., Huyan, T., Chen, J., Chen, J., et al. (2018). FOXO1 inhibition potentiates endothelial angiogenic functions in diabetes via suppression of ROCK1/Drp1-mediated mitochondrial fission. Biochim. Biophys. Acta Mol. Basis Dis. 1864 (7), 2481–2494. doi:10.1016/j.bbadis.2018.04.005
Shi, Y., Wang, S., Zhang, W., Zhu, Y., Fan, Z., Huang, Y., et al. (2022). Bone marrow mesenchymal stem cells facilitate diabetic wound healing through the restoration of epidermal cell autophagy via the HIF-1α/TGF-β1/SMAD pathway. Stem Cell Res. Ther. 13 (1), 314. doi:10.1186/s13287-022-02996-9
Shin, C. S., Meng, S., Garbis, S. D., Moradian, A., Taylor, R. W., Sweredoski, M. J., et al. (2021). LONP1 and mtHSP70 cooperate to promote mitochondrial protein folding. Nat. Commun. 12 (1), 265. doi:10.1038/s41467-020-20597-z
Shu, L., Hu, C., Xu, M., Yu, J., He, H., Lin, J., et al. (2021). ATAD3B is a mitophagy receptor mediating clearance of oxidative stress-induced damaged mitochondrial DNA. EMBO J. 40 (8), e106283. doi:10.15252/embj.2020106283
Sies, H., and Jones, D. P. (2020). Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 21 (7), 363–383. doi:10.1038/s41580-020-0230-3
Siqueira, M. F., Li, J., Chehab, L., Desta, T., Chino, T., Krothpali, N., et al. (2010). Impaired wound healing in mouse models of diabetes is mediated by TNF-alpha dysregulation and associated with enhanced activation of forkhead box O1 (FOXO1). Diabetologia 53 (2), 378–388. doi:10.1007/s00125-009-1529-y
Son, J. M., and Lee, C. (2021). Aging: all roads lead to mitochondria. Semin. Cell Dev. Biol. 116, 160–168. doi:10.1016/j.semcdb.2021.02.006
Spinelli, J. B., and Haigis, M. C. (2018). The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 20 (7), 745–754. doi:10.1038/s41556-018-0124-1
Staveness, D., Bosque, I., and Stephenson, C. R. (2016). Free radical chemistry enabled by visible light-induced electron transfer. Acc. Chem. Res. 49 (10), 2295–2306. doi:10.1021/acs.accounts.6b00270
Su, Y., Li, M., Wang, X., Wang, Z., and Yi, L. (2022). Denatured collagen could increase the autophagy level and inhibit apoptosis of fibroblasts to help cell survival and influence wound healing. Int. J. Low. Extrem Wounds 21 (1), 92–99. doi:10.1177/1534734620925942
Sun, A., Wang, Y., Liu, J., Yu, X., Sun, Y., Yang, F., et al. (2016). Exogenous H2S modulates mitochondrial fusion-fission to inhibit vascular smooth muscle cell proliferation in a hyperglycemic state. Cell Biosci. 6, 36. doi:10.1186/s13578-016-0102-x
Sun, Q., Jia, H., Cheng, S., Wang, Y., and Wang, J. (2022). Metformin alleviates epirubicin-induced endothelial impairment by restoring mitochondrial homeostasis. Int. J. Mol. Sci. 24 (1), 343. doi:10.3390/ijms24010343
Szabo, I., and Szewczyk, A. (2023). Mitochondrial ion channels. Annu. Rev. Biophys. 52, 229–254. doi:10.1146/annurev-biophys-092622-094853
Tabara, L. C., Burr, S. P., Frison, M., Chowdhury, S. R., Paupe, V., Nie, Y., et al. (2024). MTFP1 controls mitochondrial fusion to regulate inner membrane quality control and maintain mtDNA levels. Cell 187 (14), 3619–3637.e27. doi:10.1016/j.cell.2024.05.017
Tan, H. W. S., Lu, G., Dong, H., Cho, Y. L., Natalia, A., Wang, L., et al. (2022). A degradative to secretory autophagy switch mediates mitochondria clearance in the absence of the mATG8-conjugation machinery. Nat. Commun. 13 (1), 3720. doi:10.1038/s41467-022-31213-7
Tatmatsu-Rocha, J. C., Tim, C. R., Avo, L., Bernardes-Filho, R., Brassolatti, P., Kido, H. W., et al. (2018). Mitochondrial dynamics (fission and fusion) and collagen production in a rat model of diabetic wound healing treated by photobiomodulation: comparison of 904 nm laser and 850 nm light-emitting diode (LED). J. Photochem Photobiol. B 187, 41–47. doi:10.1016/j.jphotobiol.2018.07.032
Tomasina, F., Martinez, J., Zeida, A., Chiribao, M. L., Demicheli, V., Correa, A., et al. (2022). De novo sequencing and construction of a unique antibody for the recognition of alternative conformations of cytochrome c in cells. Proc. Natl. Acad. Sci. U. S. A. 119 (47), e2213432119. doi:10.1073/pnas.2213432119
Tombulturk, F. K., Soydas, T., and Kanigur-Sultuybek, G. (2024). Metformin as a modulator of autophagy and hypoxia responses in the enhancement of wound healing in diabetic rats. Inflammation 48, 1391–1402. doi:10.1007/s10753-024-02129-9
Trinchese, G., Cimmino, F., Catapano, A., Cavaliere, G., and Mollica, M. P. (2024). Mitochondria: the gatekeepers between metabolism and immunity. Front. Immunol. 15, 1334006. doi:10.3389/fimmu.2024.1334006
Tseng, W. W., Chu, C. H., Lee, Y. J., Zhao, S., Chang, C., Ho, Y. P., et al. (2024). Metabolic regulation of mitochondrial morphologies in pancreatic beta cells: coupling of bioenergetics and mitochondrial dynamics. Commun. Biol. 7 (1), 1267. doi:10.1038/s42003-024-06955-3
Uberoi, A., McCready-Vangi, A., and Grice, E. A. (2024). The wound microbiota: microbial mechanisms of impaired wound healing and infection. Nat. Rev. Microbiol. 22 (8), 507–521. doi:10.1038/s41579-024-01035-z
Walsh, C. T., Tu, B. P., and Tang, Y. (2018). Eight kinetically stable but thermodynamically activated molecules that power cell metabolism. Chem. Rev. 118 (4), 1460–1494. doi:10.1021/acs.chemrev.7b00510
Wan, L., Bai, X., Zhou, Q., Chen, C., Wang, H., Liu, T., et al. (2022). The advanced glycation end-products (AGEs)/ROS/NLRP3 inflammasome axis contributes to delayed diabetic corneal wound healing and nerve regeneration. Int. J. Biol. Sci. 18 (2), 809–825. doi:10.7150/ijbs.63219
Wang, C., Jacewicz, A., Delgado, B. D., Baradaran, R., and Long, S. B. (2020a). Structures reveal gatekeeping of the mitochondrial Ca(2+) uniporter by MICU1-MICU2. Elife 9, e59991. doi:10.7554/eLife.59991
Wang, X., Shen, K., Wang, J., Liu, K., Wu, G., Li, Y., et al. (2020b). Hypoxic preconditioning combined with curcumin promotes cell survival and mitochondrial quality of bone marrow mesenchymal stem cells, and accelerates cutaneous wound healing via PGC-1α/SIRT3/HIF-1α signaling. Free Radic. Biol. Med. 159, 164–176. doi:10.1016/j.freeradbiomed.2020.07.023
Wang, G., Wang, Y., Yang, Q., Xu, C., Zheng, Y., Wang, L., et al. (2022). Metformin prevents methylglyoxal-induced apoptosis by suppressing oxidative stress in vitro and in vivo. Cell Death Dis. 13 (1), 29. doi:10.1038/s41419-021-04478-x
Wang, J. W., Zhu, Y. Z., Ouyang, J. Y., Nie, J. Y., Wang, Z. H., Wu, S., et al. (2023). Adipose-Derived stem cell extracellular vesicles improve wound closure and angiogenesis in diabetic mice. Plast. Reconstr. Surg. 151 (2), 331–342. doi:10.1097/PRS.0000000000009840
Wang, H., Luo, W., Chen, H., Cai, Z., and Xu, G. (2024a). Mitochondrial dynamics and mitochondrial autophagy: molecular structure, orchestrating mechanism and related disorders. Mitochondrion 75, 101847. doi:10.1016/j.mito.2024.101847
Wang, M., Yang, D., Li, L., Wu, P., Sun, Y., Zhang, X., et al. (2024b). A dual role of mesenchymal stem cell derived small extracellular vesicles on TRPC6 protein and Mitochondria to promote diabetic wound healing. ACS Nano 18 (6), 4871–4885. doi:10.1021/acsnano.3c09814
Wang, T., Li, X., Tao, Y., Wang, X., Li, L., and Liu, J. (2024c). METTL3-mediated NDUFB5 m6A modification promotes cell migration and mitochondrial respiration to promote the wound healing of diabetic foot ulcer. J. Transl. Med. 22 (1), 643. doi:10.1186/s12967-024-05463-6
Wei, S., Qiu, T., Yao, X., Wang, N., Jiang, L., Jia, X., et al. (2020). Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS-autophagy-lysosomal pathway. J. Hazard Mater 384, 121390. doi:10.1016/j.jhazmat.2019.121390
Weiser, A., Hermant, A., Bermont, F., Sizzano, F., Karaz, S., Alvarez-Illera, P., et al. (2023). The mitochondrial calcium uniporter (MCU) activates mitochondrial respiration and enhances mobility by regulating mitochondrial redox state. Redox Biol. 64, 102759. doi:10.1016/j.redox.2023.102759
Wescott, A. P., Kao, J. P. Y., Lederer, W. J., and Boyman, L. (2019). Voltage-energized calcium-sensitive ATP production by mitochondria. Nat. Metab. 1 (10), 975–984. doi:10.1038/s42255-019-0126-8
Wolf, G., Aumann, N., Michalska, M., Bast, A., Sonnemann, J., Beck, J. F., et al. (2010). Peroxiredoxin III protects pancreatic ß cells from apoptosis. J. Endocrinol. 207 (2), 163–175. doi:10.1677/JOE-09-0455
Wu, D., Huang, W., Zhang, J., He, L., Chen, S., Zhu, S., et al. (2024). Downregulation of VEGFA accelerates AGEs-mediated nucleus pulposus degeneration through inhibiting protective mitophagy in high glucose environments. Int. J. Biol. Macromol. 262 (Pt 1), 129950. doi:10.1016/j.ijbiomac.2024.129950
Wu, X., Gu, R., Tang, M., Mu, X., He, W., and Nie, X. (2025). Elucidating the dual roles of apoptosis and necroptosis in diabetic wound healing: implications for therapeutic intervention. Burns Trauma 13, tkae061. doi:10.1093/burnst/tkae061
Xi, J., Rong, Y., Zhao, Z., Huang, Y., Wang, P., Luan, H., et al. (2021). Scutellarin ameliorates high glucose-induced vascular endothelial cells injury by activating PINK1/Parkin-mediated mitophagy. J. Ethnopharmacol. 271, 113855. doi:10.1016/j.jep.2021.113855
Xi, L., Du, J., Lu, Y., Xue, W., Xia, Y., Chen, T., et al. (2025). Dulaglutide accelerates diabetic wound healing by suppressing Nrf2-dependent ferroptosis in diabetic mice. Peptides 185, 171366. doi:10.1016/j.peptides.2025.171366
Xia, W., Liu, Y., Jiang, X., Li, M., Zheng, S., Zhang, Z., et al. (2023). Lean adipose tissue macrophage derived exosome confers immunoregulation to improve wound healing in diabetes. J. Nanobiotechnology 21 (1), 128. doi:10.1186/s12951-023-01869-4
Xiang, M., Yang, M., Zhou, C., Liu, J., Li, W., and Qian, Z. (2006). Crocetin prevents AGEs-induced vascular endothelial cell apoptosis. Pharmacol. Res. 54 (4), 268–274. doi:10.1016/j.phrs.2006.06.010
Xiang, J., Zhang, C., Di, T., Chen, L., Zhao, W., Wei, L., et al. (2022). Salvianolic acid B alleviates diabetic endothelial and mitochondrial dysfunction by down-regulating apoptosis and mitophagy of endothelial cells. Bioengineered 13 (2), 3486–3502. doi:10.1080/21655979.2022.2026552
Xiao, K., Wang, S., Li, G., Chen, W., Chen, B., and Li, X. (2024). Resveratrol promotes diabetic wound healing by inhibiting ferroptosis in vascular endothelial cells. Burns 50 (9), 107198. doi:10.1016/j.burns.2024.07.002
Xiong, W., Zhang, X., Hu, J., Zou, X., Huang, H., Qu, W., et al. (2024). PF-PEG@ASIV-EXO hydrogel accelerates diabetic wound healing by ferroptosis resistance and promoting angiogenesis. ACS Biomater. Sci. Eng. 10 (10), 6263–6285. doi:10.1021/acsbiomaterials.4c00692
Xiong, Y., Knoedler, S., Alfertshofer, M., Kim, B. S., Jiang, D., Liu, G., et al. (2025). Mechanisms and therapeutic opportunities in metabolic aberrations of diabetic wounds: a narrative review. Cell Death Dis. 16 (1), 341. doi:10.1038/s41419-025-07583-3
Xue, K., Wu, D., Wang, Y., Zhao, Y., Shen, H., Yao, J., et al. (2022). The mitochondrial calcium uniporter engages UCP1 to form a thermoporter that promotes thermogenesis. Cell Metab. 34 (9), 1325–1341.e6. doi:10.1016/j.cmet.2022.07.011
Yan, J., Tie, G., Wang, S., Tutto, A., DeMarco, N., Khair, L., et al. (2018). Diabetes impairs wound healing by Dnmt1-dependent dysregulation of hematopoietic stem cells differentiation towards macrophages. Nat. Commun. 9 (1), 33. doi:10.1038/s41467-017-02425-z
Yang, S., Wang, S., Chen, L., Wang, Z., Chen, J., Ni, Q., et al. (2023). Neutrophil extracellular traps delay diabetic wound healing by inducing endothelial-to-mesenchymal transition via the Hippo pathway. Int. J. Biol. Sci. 19 (1), 347–361. doi:10.7150/ijbs.78046
Yang, X., Che, T., Tian, S., Zhang, Y., Zheng, Y., Zhang, Y., et al. (2024). A living microecological hydrogel with Microbiota remodeling and immune reinstatement for diabetic wound healing. Adv. Healthc. Mater 13 (23), e2400856. doi:10.1002/adhm.202400856
Yaribeygi, H., Atkin, S. L., and Sahebkar, A. (2019a). Mitochondrial dysfunction in diabetes and the regulatory roles of antidiabetic agents on the mitochondrial function. J. Cell Physiol. 234 (6), 8402–8410. doi:10.1002/jcp.27754
Yaribeygi, H., Farrokhi, F. R., Butler, A. E., and Sahebkar, A. (2019b). Insulin resistance: review of the underlying molecular mechanisms. J. Cell Physiol. 234 (6), 8152–8161. doi:10.1002/jcp.27603
Ye, P., Wu, X., Gu, R., Zhu, H., Chen, J., Dai, Y., et al. (2025). ACNO hydrogel enhances diabetic wound healing by modulating the Bcl-2/Bax/Caspase-3/PARP pathway. Int. Immunopharmacol. 147, 113997. doi:10.1016/j.intimp.2024.113997
Yu, X., Liu, Z., Yu, Y., Qian, C., Lin, Y., Jin, S., et al. (2024). Hesperetin promotes diabetic wound healing by inhibiting ferroptosis through the activation of SIRT3. Phytother. Res. 38 (3), 1478–1493. doi:10.1002/ptr.8121
Yuzefovych, L. V., Solodushko, V. A., Wilson, G. L., and Rachek, L. I. (2012). Protection from palmitate-induced mitochondrial DNA damage prevents from mitochondrial oxidative stress, mitochondrial dysfunction, apoptosis, and impaired insulin signaling in rat L6 skeletal muscle cells. Endocrinology 153 (1), 92–100. doi:10.1210/en.2011-1442
Zecchini, V., Paupe, V., Herranz-Montoya, I., Janssen, J., Wortel, I. M. N., Morris, J. L., et al. (2023). Fumarate induces vesicular release of mtDNA to drive innate immunity. Nature 615 (7952), 499–506. doi:10.1038/s41586-023-05770-w
Zhang, T., Liu, Q., Gao, W., Sehgal, S. A., and Wu, H. (2022). The multifaceted regulation of mitophagy by endogenous metabolites. Autophagy 18 (6), 1216–1239. doi:10.1080/15548627.2021.1975914
Zhang, Y., Li, M., Wang, Y., Han, F., Shen, K., Luo, L., et al. (2023a). Exosome/metformin-loaded self-healing conductive hydrogel rescues microvascular dysfunction and promotes chronic diabetic wound healing by inhibiting mitochondrial fission. Bioact. Mater 26, 323–336. doi:10.1016/j.bioactmat.2023.01.020
Zhang, Y., Zhang, Y. Y., Pan, Z. W., Li, Q. Q., Sun, L. H., Li, X., et al. (2023b). GDF11 promotes wound healing in diabetic mice via stimulating HIF-1ɑ-VEGF/SDF-1ɑ-mediated endothelial progenitor cell mobilization and neovascularization. Acta Pharmacol. Sin. 44 (5), 999–1013. doi:10.1038/s41401-022-01013-2
Zhang, X., Leng, S., Liu, X., Hu, X., Liu, Y., Li, X., et al. (2024). Ion channel Piezo1 activation aggravates the endothelial dysfunction under a high glucose environment. Cardiovasc Diabetol. 23 (1), 150. doi:10.1186/s12933-024-02238-7
Zhang, S., Zhao, X., Zhang, W., Wei, X., Chen, X. L., and Wang, X. (2025). Zn-DHM nanozymes regulate metabolic and immune homeostasis for early diabetic wound therapy. Bioact. Mater 49, 63–84. doi:10.1016/j.bioactmat.2025.02.041
Zhao, L., Zhang, C. L., He, L., Chen, Q., Liu, L., Kang, L., et al. (2022). Restoration of Autophagic Flux improves endothelial function in diabetes through lowering mitochondrial ROS-Mediated eNOS monomerization. Diabetes 71 (5), 1099–1114. doi:10.2337/db21-0660
Zhao, Y., Wang, D., Qian, T., Zhang, J., Li, Z., Gong, Q., et al. (2023). Biomimetic nanozyme-decorated hydrogels with H(2)O(2)-Activated oxygenation for modulating immune microenvironment in diabetic wound. ACS Nano 17 (17), 16854–16869. doi:10.1021/acsnano.3c03761
Zhao, M., Kang, M., Wang, J., Yang, R., Zhong, X., Xie, Q., et al. (2024). Stem cell-derived nanovesicles embedded in dual-layered hydrogel for programmed ROS regulation and comprehensive tissue regeneration in burn wound healing. Adv. Mater 36 (32), e2401369. doi:10.1002/adma.202401369
Zheng, Y., Luo, A., and Liu, X. (2021). The imbalance of mitochondrial Fusion/Fission drives high-glucose-induced vascular injury. Biomolecules 11 (12), 1779. doi:10.3390/biom11121779
Zheng, W., Li, H., Go, Y., Chan, X. H. F., Huang, Q., and Wu, J. (2022). Research advances on the damage mechanism of skin glycation and related inhibitors. Nutrients 14 (21), 4588. doi:10.3390/nu14214588
Zhu, W., Yuan, Y., Liao, G., Li, L., Liu, J., Chen, Y., et al. (2018). Mesenchymal stem cells ameliorate hyperglycemia-induced endothelial injury through modulation of mitophagy. Cell Death Dis. 9 (8), 837. doi:10.1038/s41419-018-0861-x
Zhu, P., Chen, C., Wu, D., Chen, G., Tan, R., and Ran, J. (2022). AGEs-induced MMP-9 activation mediated by Notch1 signaling is involved in impaired wound healing in diabetic rats. Diabetes Res. Clin. Pract. 186, 109831. doi:10.1016/j.diabres.2022.109831
Zhu, Z., Ding, J., Qin, M., Wang, L., Jiang, D., Zhao, J., et al. (2024). Enhanced.OH-Scavenging activity of Cu-CeO(x) nanozyme via resurrecting macrophage Nrf2 transcriptional activity facilitates diabetic wound healing. Adv. Healthc. Mater 13 (12), e2303229. doi:10.1002/adhm.202303229
Zorov, D. B., Juhaszova, M., and Sollott, S. J. (2014). Mitochondrial reactive oxygen species (ROS) and ROS-Induced ROS release. Physiol. Rev. 94 (3), 909–950. doi:10.1152/physrev.00026.2013
Keywords: diabetes, mitochondria, trauma, apoptosis, ROS
Citation: Pan Y, Chen L, Chen Y, Thomas ER, Zhou S, Yang Y, Liu K, Wu J and Li X (2025) Mitochondrial dysfunction in diabetic ulcers: pathophysiological mechanisms and targeted therapeutic strategies. Front. Cell Dev. Biol. 13:1625474. doi: 10.3389/fcell.2025.1625474
Received: 09 May 2025; Accepted: 05 August 2025;
Published: 21 August 2025.
Edited by:
Natascia Tiso, University of Padua, ItalyReviewed by:
Sergey Yaklichkin, Memorial Sloan Kettering Cancer Center, United StatesAnant Kumar, Council of Scientific and Industrial Research (CSIR), India
Copyright © 2025 Pan, Chen, Chen, Thomas, Zhou, Yang, Liu, Wu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang Li, bGl4MjAwOUAxMjYuY29t; Jianming Wu, amlhbm1pbmd3dUBzd211LmVkdS5jbg==; Kezhi Liu, bGt6QHN3bXUuZWR1LmNu
†These authors have contributed equally to this work