Abstract
Phospholamban (PLN) is a key regulator of cardiac muscle contractility and has become a central focus in the study of cardiac disease. Variants in the PLN gene have been identified in patients with a wide range of phenotypes, including hypertrophic, dilated, and arrhythmogenic cardiomyopathies. The growing number of identified variants highlights the previously underappreciated role of PLN in cardiac pathophysiology. This review offers a comprehensive examination of the genetic landscape of PLN and evaluates the mechanistic effects of specific variants on cardiac function, aiming to uncover potential genotype-phenotype correlations. The rapidly expanding body of knowledge in this area is driving the development of advanced diagnostic and prognostic tools, as well as highly targeted therapeutic strategies. These advances underscore the importance of recognizing PLN’s role in cardiac disease and the value of genetic testing for accurate diagnosis, prognosis, effective management, and early risk prediction for family members.
1 Introduction
Cardiac muscle contraction and relaxation rely on tightly regulated calcium cycling. Contraction is initiated by Ca2+ influx through transmembrane voltage-gated calcium channels, which subsequently induces a sharp release of Ca2+ from the sarcoplasmic reticulum (SR) via ryanodine receptor activation. This rise in cytosolic Ca2+ enables its binding to troponin C, which displaces tropomyosin from actin filaments and promotes actin-myosin interaction and contraction (Bers, 2002). Relaxation is achieved primarily through the reuptake of Ca2+ into the SR by the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase isoform 2a (SERCA2a), whose activity is controlled by phospholamban (PLN) (MacLennan and Kranias, 2003).
The PLN gene (OMIM: 172405) encodes a highly conserved protein that is primarily expressed in cardiac muscle and to a lesser degree in skeletal and smooth muscles (Fujii et al., 1991; Tada and Toyofuku, 1998; Tada et al., 1998). PLN is expressed early in development, indicating its essential role in cardiac physiology (Ganim et al., 1992; Simmerman and Jones, 1998). At the transcriptional level, PLN expression is known to be regulated by various transcription factors, including thyroid hormone receptors, glucocorticoid nuclear receptor, nuclear factor YA (NF-YA) and NF-YB, and the zinc finger protein ZBTB20 (Yabuki et al., 1998; Haghighi et al., 2008; Belakavadi et al., 2010; Ren et al., 2024), while post-transcriptionally its mRNA is stabilized by the RNA-binding protein Human Antigen R (HuR) (Hu et al., 2020). PLN is a 52-amino-acid transmembrane protein, localizing at the SR membrane and regulating SERCA2a activity (Tada and Toyofuku, 1998; Tada et al., 1998). Although information regarding trafficking of the protein to the SR is limited, the di-arginine motif in the cytoplasmic domain of PLN was shown to be required for retrograde trafficking between the Golgi to ER/SR compartments (Sharma et al., 2010) (Figure 1).
FIGURE 1
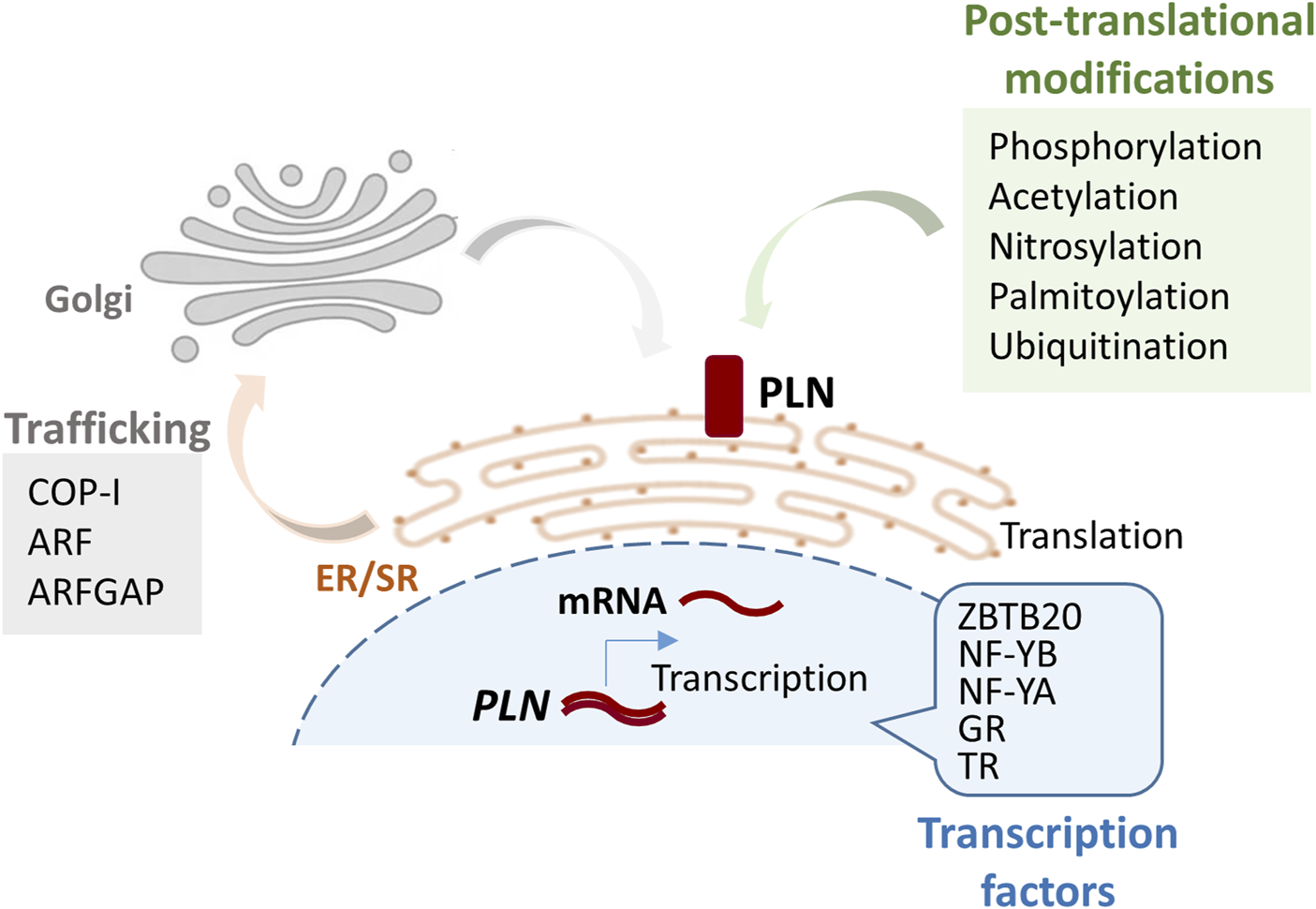
Schematic figure illustrating the pathway from PLN gene to protein and its localization at the SR membrane. Key factors and modifications involved in each step of the process are indicated. Post-translational modifications occur at lysine 3 (ubiquitination), cysteine 36 (palmitoylation), cysteines 41 and 46 (nitrosylation). GR, glucocorticoid nuclear receptor; TR, thyroid hormone receptor; COP-I, Coat Protein Complex I; ARF, ADP-ribosylation factor; ARFGAP, ADP-ribosylation factor GTPase-activating protein 1.
At the functional level, PLN regulates SERCA2a in a phosphorylation-dependent manner (MacLennan and Kranias, 2003; Kranias and Hajjar, 2017). When dephosphorylated, PLN inhibits SERCA2a by reducing its affinity for Ca2+, thereby delaying SR Ca2+ reuptake and relaxation. PLN phosphorylation at serine 16 (Ser16) by cAMP-dependent protein kinase (PKA) and at threonine 17 (Thr17) by Ca2+-calmodulin-dependent protein kinase (CaMKII) relieves this inhibition, leading to enhanced SR Ca2+ uptake and cardiac relaxation, particularly during β-adrenergic stimulation (MacLennan and Kranias, 2003). PLN acetylation, S-palmitoylation, and nitrosylation have emerged as additional post-translational modifications that modulate PLN function by influencing its membrane association, oligomerization, or interaction with SERCA2a, while ubiquitination regulates PLN protein levels (Froehlich et al., 2008; Irie et al., 2015; Zhou et al., 2015; Nakagawa et al., 2016; Keceli et al., 2019; Rogers et al., 2023; Mushala et al., 2024). Cytosolic Ca2+ levels also influence PLN-SERCA2a interactions, with elevated Ca2+ promoting PLN dissociation and SERCA2a activation (MacLennan and Kranias, 2003; Kranias and Hajjar, 2017). Structural studies have provided evidence to suggest that PLN adopts multiple conformational states which affect its inhibitory function on SERCA2a (Gustavsson et al., 2011; Gustavsson et al., 2013; Raguimova et al., 2020; Weber et al., 2021; Weber et al., 2024). In contrast to the initial, more simplistic, view that SERCA2a represents the sole binding partner of PLN, a more complex regulatory network has now been established through the identification of multiple and multifunctional binding proteins, signifying an essential contribution of PLN in cardiac function through multiple pathways (Vafiadaki et al., 2009; Haghighi et al., 2014; Kranias and Hajjar, 2017; Mattiazzi and Kranias, 2024).
The fundamental role of PLN in the heart is further corroborated by the detrimental effects PLN variants can have. To date, multiple pathogenic PLN variants have been identified in patients with a range of clinical cardiac phenotypes that include arrhythmogenic (ACM), dilated (DCM) and hypertrophic cardiomyopathy (HCM) (Kranias and Hajjar, 2017; Vafiadaki et al., 2023; Mattiazzi and Kranias, 2024).
According to the latest international guidelines and statements on genetic testing for cardiac disease, of the American Heart Association (AHA), the European Society of Cardiology (ESC), and the European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) (Musunuru et al., 2020; Wilde et al., 2022; Arbelo et al., 2023), PLN is in the recommended shortlist of genes to be tested in patients with a confirmed or suspected diagnosis of inherited HCM, DCM or ACM, or in individuals at high risk due to a previously identified pathogenic variant in their family.
The implementation of these guidelines is gradually leading to a marked increase in the number of novel PLN variants identified. Herein, we provide a comprehensive and critical overview of all human PLN variants known to date, including extensively characterized ones and/or described in the literature, as well as those reported solely across publicly available databases. The expanding research evidence on their respective pathogenetic mechanisms, is fueling the intense exploration of targeted precision medicine therapies, while genotype-phenotype correlations are geared towards guiding the development of diagnostically and prognostically valuable tools. Our study reveals a previously unanticipated extent of PLN contribution to cardiac disease worldwide, highlighting the need for heightened clinical awareness on the value of PLN genetic testing, as well as the interpretation and clinical integration of these results towards enhanced diagnosis, prognosis and clinical management of cardiomyopathy patients and their family members.
2 Published pathogenic PLN variants
The discovery of the first pathogenic PLN variants in 2003 (Haghighi et al., 2003; Schmitt et al., 2003), sparked interest in this gene and initiated a long path of research and discovery. Approximately half of the published pathogenic PLN variants have been characterized at the molecular/cellular/functional level, albeit the extent of characterization varies. The findings offer a deep understanding of the complex molecular mechanisms, implicated in PLN disease pathogenesis, and have led to the more effective treatment and clinical management of PLN cardiomyopathy patients (Hof et al., 2019). The key conclusions from these pathogenic PLN variants and their implications, are critically discussed by genomic location (coding or promoter region) and type of variation (deletion, missense, nonsense or insertion) (Table 1).
TABLE 1
| Type of variant | PLN variant | Classification | State | Clinical phenotype | Effect on protein | Reference |
|---|---|---|---|---|---|---|
| Deletion | c.40_42delAGA, p.Arg14del | Pathogenic | Heterozygote | Asymptomatic, ACM or DCM with arrhythmias | PLN structural changes, protein interaction aberrations, Ca2+ handling dysregulation, mitochondrial dysfunction, UPR activation, autophagy defects, SR disorganization and protein aggregation | Haghighi et al. (2003), van der Zwaag et al. (2012), Hof et al. (2019), Haghighi et al. (2006), Karakikes et al. (2015), Eijgenraam et al. (2020), Badone et al. (2021), Cuello et al. (2021), Feyen et al. (2021), Grote Beverborg et al. (2021), Haghighi et al. (2021), Raad et al. (2021), Dave et al. (2022), Vafiadaki et al. (2022), Cleary et al. (2023), Rogalska et al. (2023), Stege et al. (2023), Cleary et al. (2024), Foo et al. (2024), Maniezzi et al. (2024), Vafiadaki et al. (2024) |
| c.95_98delTTAT, p.Phe32SerfsTer7 | Pathogenic | Heterozygote | HCM | Predicted to result in nonsense-mediated mRNA decay but no evidence provided | Du et al. (2024) | |
| Missense | c.23C>T, p.Thr8Ile | N/A | Not specified | DCM | Not known | Sousa et al. (2019) |
| c.25C>T, p.Arg9Cys | Pathogenic | Heterozygote | DCM | Loss of function, increased propensity for oligomerization, decreased binding to SERCA2, decreased SERCA2 inhibition, aberrant Ca2+ handling, lack of phosphorylation, structural changes | Schmitt et al. (2003), Schmitt et al. (2009), Ha et al. (2011), Ceholski et al. (2012b), Abrol et al. (2015), Truszkowska et al. (2015), Fish et al. (2016), Nelson et al. (2018), Vicente et al. (2022), Weber et al. (2024) | |
| c.26G>T, p.Arg9Leu | Likely Pathogenic | Heterozygote | DCM | Loss of function, lack of phosphorylation, structural changes | Medeiros et al. (2011), Ceholski et al. (2012a), Ceholski et al. (2012b), Truszkowska et al. (2015), Weber et al. (2024) | |
| c.26G>A, p.Arg9His | Likely Pathogenic/VUS | Heterozygote | DCM | Loss of function, lack of phosphorylation | Medeiros et al. (2011), Ceholski et al. (2012a), Sousa et al. (2019), Weber et al. (2024) | |
| c.43G>A, p.Ala15Thr | Likely Pathogenic/VUS | Heterozygote | DCM | Gain of function, increased SERCA inhibition, reduced phosphorylation and dephosphorylation at Ser16 | Pugh et al. (2014), Armanious et al. (2024) | |
| c.46T>G, p.Ser16Pro | N/A | Not specified | HCM | Not known | Walsh et al. (2017b) | |
| c.50C>A, p.Thr17Asn | N/A | Heterozygote | DCM | Not known | Mollanoori et al. (2018) | |
| c.53T>C; p.Ile18Thr | VUS | Not specified | HCM | Partial loss of function, reduced inhibition of SERCA, lack of PKA phosphorylation | Burns et al. (2017), Lopes et al. (2015), Armanious et al. (2024) | |
| c.61C>A, p.Pro21Thr | VUS | Heterozygote | DCM | Gain of function, increased SERCA inhibition, reduced phosphorylation and dephosphorylation at Ser16, increased helical structure | Pugh et al. (2014), Armanious et al. (2024) | |
| c.73C>T, p.Arg25Cys | Likely Pathogenic/VUS | Heterozygote | DCM, HCM and Bruganda syndrome | Gain of function, SERCA2 super-inhibition, elevated diastolic Ca2+, increased RyR Ser2814 phosphorylation | Behr et al. (2015), Liu et al. (2015), Lopes et al. (2015) | |
| c.121T>A, p.Cys41Ser | VUS | Not specified | HCM | Not known | Walsh et al. (2017b) | |
| c.145G>A, p.Val49Met | VUS | Heterozygote | HCM | Not known | Xu et al. (2015) | |
| c.152T>C, p.Leu51Pro | VUS | Not specified | HCM | Not known | Walsh et al. (2017b), Lopes et al. (2015) | |
| Nonsense | c.4G>T, p.Glu2Ter | Pathogenic | Homozygote | DCM and severe heart failure | Loss of function, no PLN protein expression | Li et al. (2019) |
| c.116T>G, p.Leu39Ter | Pathogenic/Likely Pathogenic | Heterozygote Homozygote | Asymptomatic hypertrophy, HCM or severe DCM with cardiac arrhythmias | Loss of function, no PLN protein expression, no SERCA2 inhibition | Haghighi et al. (2003), Landstrom et al. (2011), Medeiros et al. (2011), Sanoudou et al. (2015), Walsh et al. (2017a), Walsh et al. (2017b), Mazzone et al. (2024) | |
| Insertion | c.9_10insA, p.Val4SerFsTer15 | Pathogenic | Heterozygote | Wolf-Parkinson-White syndrome with unconfirmed diagnosis of cardiomyopathy | Frameshift and premature termination, produces protein of 17 amino acids but with only the initial 3 residues corresponding to PLN, no evidence provided | Truszkowska et al. (2015) |
| c.37_38insA, p.Arg13LysfsTer7 | N/A | Heterozygote | DCM | Frameshift alterations and premature termination after 7 amino acids but no evidence provided | Pugh et al. (2014) | |
| c.61_62insCT, p.Pro21LeufsTer19 | N/A | Heterozygote | HCM | Frameshift alterations and premature termination after 19 amino acids but no evidence provided | Lopes et al. (2015) | |
| c.63_64dupTC, p.Gln22LeufsTer19 | Pathogenic/Likely pathogenic/VUS | Not specified | HCM | Frameshift alterations and premature termination after 19 amino acids but no evidence provided | Walsh et al. (2017b) | |
| c.138dupT, p.Ile47TyrfsTer14 | N/A | Not specified | HCM | Changes the last 6 amino acids of PLN and extends the protein by 8 residues but no evidence provided | Walsh et al. (2017b) | |
| Promoter | −77 A>G | N/A | Heterozygote | HCM | Increased promoter activity | Minamisawa et al. (2003) |
| −42 C>G | N/A | Heterozygote | HCM and healthy | Decreased promoter activity | Medin et al. (2007) | |
| −36 A>C | N/A | Heterozygote | DCM and healthy | Increased promoter activity | Haghighi et al. (2008) |
PLN variants published to date and key phenotypic features. Classification is based on ClinVar’s 29 April 2025 release. VUS: variant of unknown significance, N/A: not available.
2.1 Pathogenic coding PLN variants
The majority of published pathogenic PLN variants lie in the coding region, and they include missense and nonsense substitutions, as well as small deletions or insertions. While missense substitutions represent the most frequent type of variant, only a few have been well characterized.
2.1.1 Pathogenic deletion variants
The PLN deletion c.40_42delAGA, p. Arg14del (or R14del) (OMIM: 172405.0003) has been the most extensively studied variant across clinical, cellular and molecular levels (Doevendans et al., 2019). It was originally identified in a large Greek family with DCM and arrhythmia symptoms (Haghighi et al., 2006) and has now been reported in >1,500 patients worldwide, with the vast majority being in the Netherlands (Hof et al., 2019). PLN-Arg14del has only been found in the heterozygous state, with patients exhibiting a spectrum of highly variable phenotypes that range from asymptomatic to ACM or DCM that may progress to heart failure or sudden cardiac death (Hof et al., 2019). PLN-Arg14del represents a prime example of the significant clinical impact that the in-depth characterization of a genetic variant can have.
Clinical research on PLN-Arg14del has demonstrated that these patients exhibit certain distinct clinical characteristics including low-voltage electrocardiograms (ECGs), negative T waves in left precordial leads and a high prevalence of malignant ventricular arrhythmias (van der Zwaag et al., 2012; van Rijsingen et al., 2014; Hof et al., 2019). These findings have been used for the development of multiple impactful clinical diagnostic and prognostic tools. Deep learning models have been developed that significantly improve ECG-based PLN-Arg14del automated detection of this rare group of patients (Lopes et al., 2021; Bleijendaal et al., 2021). At the prognostic level, multivariant clinical prediction algorithms have been aimed at identifying PLN-Arg14del patients at high risk of malignant ventricular arrhythmia, who should therefore be considered for primary prevention implantable cardioverter defibrillator (ICD) implantation (Verstraelen et al., 2021). The delineation of PLN-Arg14del as a distinct disease entity has increased the rate of PLN-Arg14del patient identification, leading to better clinical management. Furthermore, it enabled the formation of an international patient community and the establishment of the patient-led PLN Foundation (Kranias et al., 2018; Doevendans et al., 2019), which has since been actively facilitating and promoting PLN-Arg14del research towards finding a cure.
At the basic and translational research level, a multitude of in vitro and in vivo tools have been employed to decipher the PLN-Arg14del driven pathogenesis, including animal models ranging from zebrafish to mouse and patient-derived induced pluripotent stem cell cardiomyocytes (iPSC-CMs) (Haghighi et al., 2006; Karakikes et al., 2015; Eijgenraam et al., 2020; Badone et al., 2021; Cuello et al., 2021; Feyen et al., 2021; Grote Beverborg et al., 2021; Haghighi et al., 2021; Raad et al., 2021; Dave et al., 2022; Vafiadaki et al., 2022; Cleary et al., 2023; Rogalska et al., 2023; Stege et al., 2023; Cleary et al., 2024; Foo et al., 2024; Maniezzi et al., 2024; Vafiadaki et al., 2024). The results pinpoint defects in molecular mechanisms, including PLN structural changes, protein interaction aberrations, Ca2+ handling dysregulation, mitochondrial dysfunction, unfolded protein response (UPR) activation, autophagy defects, SR disorganization and protein aggregation [reviewed in (Vafiadaki et al., 2023; Stege et al., 2024; Deimen et al., 2022)]. Standard heart failure therapy involving eplerenone or metoprolol was shown to be ineffective in PLN-Arg14del heterozygous mice (Eijgenraam et al., 2020), underscoring the need for the development of novel therapeutic strategies targeting PLN-Arg14del. Excitingly, within 20 years since the discovery of this rare variant, a multitude of novel therapeutic approaches are being developed, including gene therapy, gene editing (CRISPR/Cas9 and TALENs), antisense oligonucleotides, and small molecules (Karakikes et al., 2015; Feyen et al., 2021; Grote Beverborg et al., 2021; Dave et al., 2022; Eijgenraam et al., 2022).
Recently, another pathogenic (ACMG criteria based classification: PVS1, PS2, PM2) deletion variant was reported in PLN (c.95_98del, p. Phe32SerfsTer7) in a heterozygous patient with complex clinical features due to dual diagnosis of cardiomyopathy and Perrault syndrome (Du et al., 2024). Notably, along with this PLN variant, a pathogenic variant was detected in Required for Meiotic Nuclear Division 1 homolog (RMND1) which might be responsible for the Perrault syndrome (Du et al., 2024). PLN-Phe32SerfsTer7 was predicted to disrupt the translational reading frame, leading to nonsense-mediated mRNA decay. However, there are no data available regarding the impact of this mutation on PLN protein expression levels, or its potential functional consequences in the cardiomyocytes.
2.1.2 Pathogenic missense variants
The missense variant c.25C>T, p. Arg9Cys (OMIM: 172405.0001) (Schmitt et al., 2003) along with the nonsense variant c.116T>G, p. Leu39Term (OMIM: 172405.0002) [(Haghighi et al., 2003); see below] were the first PLN variants to be discovered in DCM patients, linking PLN genetic changes to cardiac disease. The familial PLN-Arg9Cys has only been encountered in heterozygosity and exhibits autosomal dominant inheritance (Schmitt et al., 2003; Truszkowska et al., 2015; Fish et al., 2016). Detailed analysis at the molecular, cellular and animal model levels has shown that PLN-Arg9Cys represents a loss-of-function variant, with a dominant-negative effect on wild-type PLN. It interferes with PKA-mediated PLN phosphorylation and consequently disrupts the regulatory function of PLN resulting in sustained SERCA2 inhibition, development of DCM, heart failure and premature death.
Recent studies on iPSC-CMs, recapitulated disease mechanisms associated with PLN-Arg9Cys, exhibiting aberrant Ca2+ handling, blunted β-adrenergic signaling, hypertrophy and fibrosis-related changes along with alterations in cellular metabolism and proteostasis disruption (Ceholski et al., 2018; Yu et al., 2024). Interestingly, exposing these cells to functional challenges exacerbated the PLN-Arg9Cys defects, leading to autophagic overload, structural remodeling and functional deficiencies (Yu et al., 2024). These research insights are guiding efforts towards the development of targeted therapeutic approaches for PLN-Arg9Cys carriers. Autophagy inducing agents tested on patient-derived PLN-Arg9Cys iPSC-CMs gave promising results, rescuing, at least in part, functional defects including structural remodeling, Ca2+ handling and contractile function (Yu et al., 2024).
In the same nucleotide position of PLN two more variants, c.26G>T, p. Arg9Leu and c.26G>A, p. Arg9His, have been identified, in heterozygosity, following mutation screening of Polish, Brazilian and Portuguese DCM patient cohorts (Medeiros et al., 2011; Truszkowska et al., 2015; Sousa et al., 2019). Similar to PLN-Arg9Cys, the Arg9Leu and Arg9His variant proteins do not get phosphorylated, most likely due to structural alterations in the region around the PKA phosphorylation site (Medeiros et al., 2011; Ceholski et al., 2012a). Interestingly however, only PLN-Arg9Leu failed to inhibit SERCA activity as, similarly with PLN-Arg9Cys, this variant results in altered hydrophobicity that impacts its function (Ceholski et al., 2012a; Ceholski et al., 2012b). In contrast, the inhibitory effect of Arg9His was indistinguishable from the wild-type PLN probably due to similar physicochemical properties of the substituted amino acid (Ceholski et al., 2012a).
A different heterozygous substitution, PLN c.73C>T, p. Arg25Cys, has been identified by multiple studies in patients presenting DCM and cardiac arrhythmias, HCM or Brugada syndrome patients with QRS duration (Behr et al., 2015; Liu et al., 2015; Lopes et al., 2015). Functional analysis by adenoviral overexpression in adult rat cardiomyocytes demonstrated that the PLN-Arg25Cys protein causes SERCA2 super-inhibition, resulting in suppressed Ca2+ kinetic parameters and depressed myocyte contractility (Liu et al., 2015). The molecular mechanisms associated with this included increased interaction of PLN-Arg25Cys with SERCA2a, most likely due to structural alterations (Liu et al., 2015; Nelson et al., 2018), leading to elevated diastolic Ca2+ and consequent activation of CaM kinase II (CaMKII). Although the latter did not affect PLN phosphorylation, it caused enhanced RyR phosphorylation at the Ser2814 site with consequent increases in SR Ca2+ leak which promoted arrhythmogenesis under stress conditions (Liu et al., 2015). These findings provided initial evidence to support the hypothesis that the increased PLN inhibitory function may impact both SR Ca2+ uptake and Ca2+ release, expanding our understanding of cardiomyocyte Ca2+ homeostasis regulatory mechanisms (Liu et al., 2015; Wagner et al., 2015).
Over the years, an increasing number of heterozygous missense PLN variants have been detected including c.23C>T, pThr8Ile; c.43G>A, p. Ala15Thr; c.46T>G, p. Ser16Pro; c.50C>A, pThr17Asn; c.53T>C; p. Ile18Thr; c.61C>A, p. Pro21Thr; c.121T>A, Cys41Ser; c.145G>A, p. Val49Met and c.152T>C, Leu51Pro (Pugh et al., 2014; Lopes et al., 2015; Xu et al., 2015; Burns et al., 2017; Walsh et al., 2017b; Mollanoori et al., 2018; Sousa et al., 2019). Among them, structural and functional evaluation has been pursued only for Ala15Thr, Pro21Thr, and Ile18Thr, albeit to a limited extent. Both former variants exhibited increased inhibition of SERCA activity and reduced rates of both PLN phosphorylation and dephosphorylation, whereas the latter variant caused reduced SERCA inhibition and no PKA phosphorylation (Armanious et al., 2024). While no structural changes were observed for Ala15Thr and Ile18Thr, the PLN Pro21Thr variant had increased helical structure, an alteration that could be correlated with its gain-of-function effect on SERCA activity (Armanious et al., 2024).
Overall, missense variants represent the most populated category of PLN variants. Their downstream molecular implications vary considerably, while in many instances they remain unknown. As the patient population carrying these variants increases, genotype-phenotype studies will be valuable to unveil diagnostically, prognostically and/or therapeutically relevant correlations.
2.1.3 Pathogenic nonsense variants
The PLN nonsense variant c.116T>G, p. Leu39Term (also encountered in the literature as Leu39stop and Leu39X) was initially reported in a Greek family with DCM (Haghighi et al., 2003) but has since been identified in additional populations, with patients exhibiting clinical phenotypes ranging from asymptomatic hypertrophy to HCM or severe DCM with cardiac arrhythmias (Chiu et al., 2007; Landstrom et al., 2011; Medeiros et al., 2011; Alfares et al., 2015; Sanoudou et al., 2015; Walsh et al., 2017a; Walsh et al., 2017b; Mazzone et al., 2024). This familial PLN variant has been encountered in both heterozygous and homozygous state, with the existence of genetic modifiers potentially contributing in this wide phenotypic variability (Sanoudou et al., 2015). Examination of explanted cardiac tissue from a homozygote patient revealed more than 50% reduction in PLN mRNA levels and no detectable PLN protein, indicating that PLN-Leu39Ter is a null variant (Haghighi et al., 2003). These findings were corroborated by cellular studies following overexpression of recombinant PLN-Leu39Ter in HEK293 and adult rat cardiomyocytes which determined absence of stable PLN-Leu39Ter protein expression and consequent lack of SERCA2a activity inhibition (Haghighi et al., 2003). Recently, the absence of PLN-Leu39Ter protein was demonstrated to be due to its rapid proteosomal degradation following its translation (Rohner et al., 2021).
Another nonsense PLN variant, c.4G>T, p. Glu2Ter, has been identified in a 36 year old patient with DCM and severe heart failure (Li et al., 2019). This variant exhibited autosomal recessive inheritance. The mutation was present in homozygous state in the identified patient, while heterozygous carriers of the family had normal cardiac function. The p. Glu2Ter variant did not affect PLN expression at the mRNA level but it reduced PLN protein levels to about 50% in unaffected heterozygotes and abolished PLN protein expression in the homozygous patient (Li et al., 2019). Absence of the PLN protein was therefore proposed to be causative of the severe cardiac phenotype observed in this patient.
2.1.4 Pathogenic insertion variants
Several PLN insertion variants have been reported so far, however, their functional significance has yet to been explored experimentally. All variants pertain a single or double base insertion that causes open reading frameshift changes with consequent inclusion of premature stop codon and protein termination. Among them, the single base insertion c.37_38insA, p. Arg13LysfsTer7, which causes frameshift alterations and premature termination after 7 amino acids, was identified in a patient with clinical diagnosis and familial history of DCM (Pugh et al., 2014). The single base insertion c.138dup, p. Ile47TyrfsTer14 was identified in an HCM patient (Walsh et al., 2017b). This variant occurs at the C-terminal end of the protein and would be expected to change the last 6 amino acids, removing the termination codon and extending the protein by 8 residues. Given that this insertion occurs within the transmembrane domain of PLN, a region that is critical both for its interaction with SERCA2 and its SR membrane localization (Kimura et al., 1996; Asahi et al., 1999; Abrol et al., 2014), expression of this protein variant would be expected to severely disrupt PLN function. Another single base insertion (c.9_10insA, p. Val4SerFsTer15) has been reported in a case with Wolf-Parkinson-White syndrome but with unconfirmed diagnosis of cardiomyopathy. As this insertion occurs close to the amino terminal sequence of the protein, it is expected to cause a frameshift and premature termination after 15 amino acids, leading to a protein of just 17 amino acids in length but with only the initial 3 residues corresponding to PLN (Truszkowska et al., 2015). Apart from these single base insertions, two cases of two base pair insertions (c.63_64dup, p. Gln22LeufsTer19 and c.61_62insCT, p. Pro21LeufsTer19) have been reported in patients with HCM (Lopes et al., 2015; Walsh et al., 2017b). Both variants were predicted to cause frameshift alterations and introduction of a premature termination after 19 amino acids. Collectively, while several insertion variants have been identified, it remains to be determined if PLN protein can be expressed in their presence, and if so, to delineate the impact on PLN function.
2.2 Non-coding pathogenic PLN variants
Only three variants in the promoter region of PLN have been published to date (−77 A>G, OMIM: 172405.0004, −42 C>G, OMIM: 172405.0005 and −36 A>C, OMIM: 172405.0006) (Minamisawa et al., 2003; Medin et al., 2007; Haghighi et al., 2008). They were all found in heterozygous state in patients with DCM (Haghighi et al., 2008) or HCM (Minamisawa et al., 2003; Medin et al., 2007). In vitro analysis in cell culture systems determined that the −77 A>G and −36 A>C variants increase PLN promoter activity by 50% and 24% respectively, while the presence of −42 C>G had the opposite effect causing nearly 50% decrease in promoter activity (Minamisawa et al., 2003; Medin et al., 2007; Haghighi et al., 2008). These changes may be due to alterations in transcription factor binding on the variant sequence, as indicated in the case of −36 A>C which resulted in enhanced binding of the glucocorticoid nuclear receptor (Haghighi et al., 2008). However, both the −42 C>G and the −36 A>C variants have also been detected in unaffected (up to the time of genetic testing) individuals (Medin et al., 2007). As a consequence, although one could speculate a tentative effect on PLN expression levels, the precise contribution of these non-coding variants in disease pathogenesis is currently unclear (Santos et al., 2009; Hirtle-Lewis et al., 2013).
3 PLN variants reported in genetic databases
Beyond the published PLN variants, the increasing awareness and application of cardiogenetic testing is leading to a rapidly expanding list of PLN variants in public repositories. Diving into this information can offer valuable insights. The ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/; search performed on 11th March 2025) has 145 entries of short (<50bps) PLN variants. Among them, the vast majority (n = 82) is listed as variants of unknown significance (VUS), 12 as pathogenic/likely pathogenic (P/LP) and 10 as conflicting (for 6 of which the conflict was between P/LP and VUS). In this list of PLN ClinVar P/LP/VUS/conflicting variants, the majority (n = 47) involves missense alterations, 12 frameshift and 3 nonsense. Interestingly, 29 UTR alterations are also listed, all of which are currently categorized as VUS (with the exception of 1 conflicting: VUS vs. benign), and 27 of which were detected in patients with either DCM or HCM. In some of these cases, the inheritance pattern of DCM in the family is described as dominant. The majority (n = 24) of the 29 PLN UTR alterations involved the 3′ UTR sequence, however, there is currently no experimental evidence regarding their contribution in disease pathogenesis. Although our ability to determine the clinical impact of variants in the 3′ UTRs of genes remains poor, it is well established that they comprise the bulk of noncoding sequences present in exomes, and are important for regulation of messenger RNA (mRNA) processing, stability, translation and subcellular localization (Mayr, 2019). Investigating the implications of these UTR variants at the molecular and clinical level could impact the design and interpretation of PLN genetic test results.
In addition to these short (<50 bps) PLN variants, copy number variants (CNVs) are also listed in the database (n = 12), including both copy number loss (n = 7) and copy number gain (n = 5). However, as these type of variants encompass large genomic regions (>1 kb), affecting multiple genes, their effect would be non-PLN specific and therefore difficult to interpret at the clinical level.
Similarly to the information available in ClinVar, gnomAD (https://gnomad.broadinstitute.org/) lists a large number of PLN variants beyond what is described in the literature (search performed on 11th March 2025). In specific, 193 variants are listed, of which only 47 are reported as overlapping with the PLN ClinVar variants. Notably, among the 193 variants, 65 involve missense changes, 10 frameshift, and strikingly, 95 involve UTR changes. Unlike the ClinVar data, most of the UTR variants in gnomAD are located in the 5′ UTR region (n = 56). Although the role of PLN UTR variants remains hypothetical and without experimental validation, such 5′ UTR variants are increasingly associated with cardiovascular disease in literature reports and investigated as therapeutic targets (Liang et al., 2017; Roberts et al., 2020; Soukarieh et al., 2022).
The predicted impact of many of these novel coding variants on PLN protein structure will require molecular and cellular approaches similar to those employed to date for published PLN variants. However, the even greater number of UTR variants, points to the need for additional research avenues that will delve into the under-recognized class of non-coding UTR variants, and characterize their tentative role in PLN cardiomyopathies’ pathogenesis, as well as their diagnostic value and therapeutic potential (Whiffin et al., 2020).
4 Discussion
The present study has compiled information from all published PLN variants as well as in-depth analysis of the reported PLN variants in the ClinVar and gnomAD genetic databases, revealing the complex genetic contribution of PLN to cardiac disease. Among the rapidly increasing number of published PLN variants, the vast majority are missense, while nonsense variants appear to be rare (Figure 2). Several small insertions have been detected, all of which result in frameshift alterations that cause premature termination of protein. On the contrary, the small PLN deletions described cause either premature termination due to open reading frame alterations or single amino acid residue deletion, as in the case of PLN-Arg14del. The same trend of variant type frequencies is observed in the P/LP/VUS PLN variants submitted to the ClinVar and gnomAD databases.
FIGURE 2
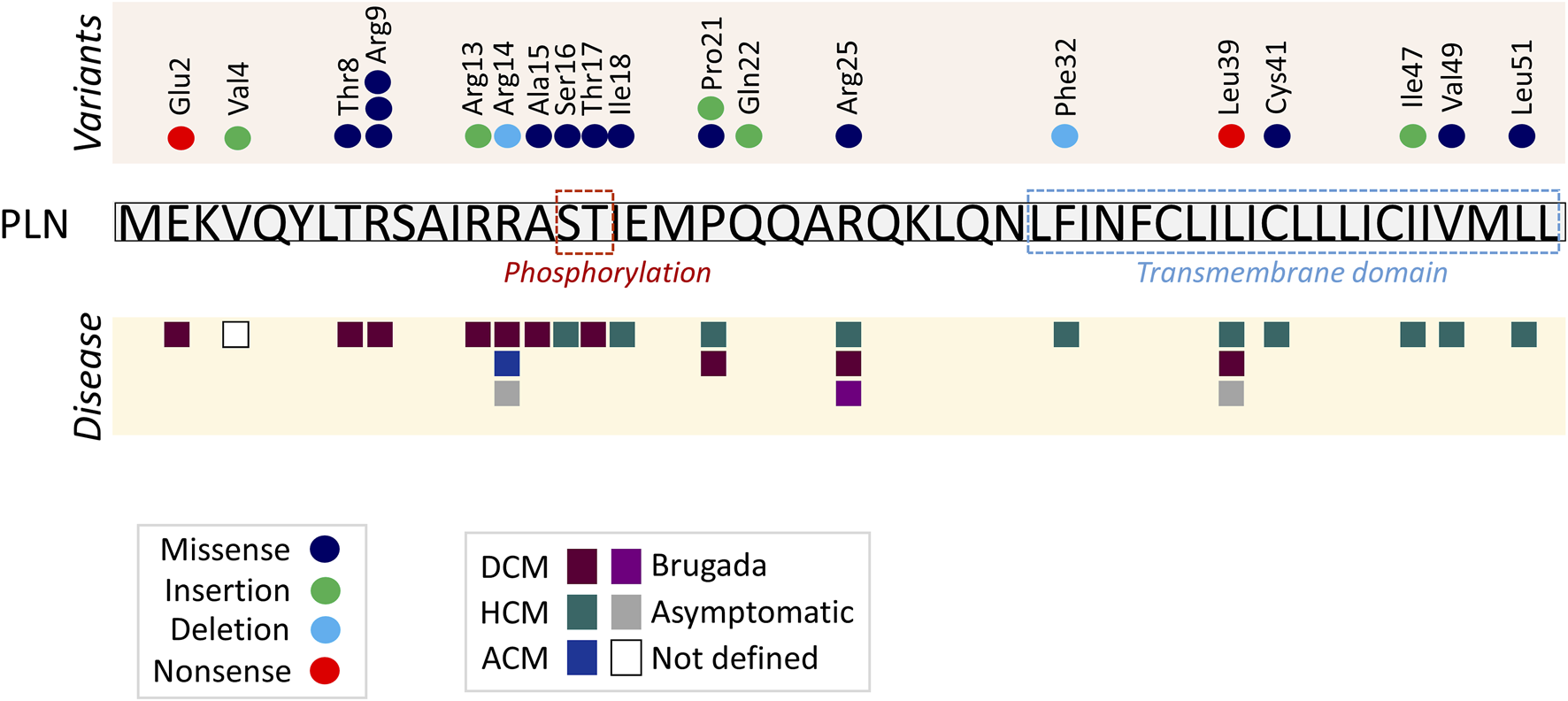
Diagrammatic representation of published PLN variants and associated clinical phenotypes. Each circle represents a different published PLN variant in the specific amino acid, and each square a different phenotype. Protein phosphorylation residues and transmembrane domain are indicated in dashed line boxes. PLN cytoplasmic residues 2–18 interact with a cytoplasmic fragment of SERCA2 while residues 20–30 and 31–52 bind to different transmembrane regions of SERCA2 (Kimura et al., 1996; Kimura et al., 1998; MacLennan et al., 1998; Asahi et al., 1999; Asahi et al., 2003; Toyoshima et al., 2003).
The majority of published pathogenic variants are located within the N-terminal region of PLN (amino acids 1–30; n = 16), and fewer at the transmembrane domain (amino acids 31–52; n = 6) (Figure 2). A similar trend, yet less prominent, is observed among the P/LP/VUS/Conflicting ClinVar (40 N-terminal vs. 27 transmembrane) and the non-synonymous gnomAD (45 N-terminal vs. 31 transmembrane) PLN variants.
Furthermore, although there does not appear to be a distinct “variant hotspot,” specific residues may be more prone to mutagenesis. For example, multiple different alterations have been observed at a single amino acid position (Arg9), namely, Arg9Cys, Arg9Leu and Arg9His. Intriguingly, despite the fact that substitution occurs at the same position, not all variants have the same impact on the protein. This may be directly associated with physicochemical properties of the substituted amino acid and/or structural changes ensued. In specific, hydrophobic substitutions, such as Arg9Cys and Arg9Leu, increase propensity of oligomerization and eliminate SERCA inhibition while the aromatic substitution Arg9His does not impact SERCA activity (Ceholski et al., 2012a; Ceholski et al., 2012b; Abrol et al., 2015). According to a recent NMR spectroscopy study, the extent of PLN inhibition on SERCA activity correlates with the tilt angle of PLN’s transmembrane domain and PLN-Arg9Cys, Arg9Leu and Arg9His variants were shown to impact this on varying degrees (Weber et al., 2024). Nevertheless, unresponsiveness to β-adrenergic stimulation due to lack of phosphorylation of all three variants represents a common mechanism contributing to disease (Ceholski et al., 2012a; Ceholski et al., 2012b; Abrol et al., 2015). Similarly to the published Arg9 variants, three or more different variants have been submitted to ClinVar and gnomAD for the PLN amino acid positions Ile12, Arg25, Gln29, Ile40, Ile47, Met50 and Leu51, all of which are missense or frameshift.
The fact that PLN variants lead to multiple forms of cardiomyopathies, along with the various clinical manifestations associated with single variants, such as Arg14del, Arg25Cys and Leu39Ter, suggest the absence of a clear genotype-phenotype correlation. However, close examination of the location of the variants suggests a potential topological correlation with the type of disease ensued. In specific, variants causing DCM are only occurring in the N-terminal and cytoplasmicc region of PLN, while HCM-causing variants are primarily found towards the C-terminal and within the transmembrane domain of PLN (Figure 2). An exception to this, are variants on residues Pro21, Arg25 and Leu39 that have been described in both DCM and HCM patients. In these cases, genetic or non-genetic factors, as discussed below, may act as modifiers of disease. While the contribution of such modifiers cannot be excluded, additional contributing mechanisms may underlie the intriguing topological distinction between the type of disease (DCM vs. HCM). In particular, the location of each variant may have a differential impact on PLN functional properties. This may include alterations in PLN structure, phosphorylation and/or protein interactions (Figure 3). Indeed, based on evidence from PLN-Arg9Cys, PLN-Arg14del and PLN-Arg25Cys, structural alterations are caused by these three variants (Hughes and Middleton, 2014; Liu et al., 2015; Vostrikov et al., 2015; Nelson et al., 2018; Vafiadaki et al., 2022). While their interaction to SERCA2 is impaired by all (Abrol et al., 2015; Liu et al., 2015; Vafiadaki et al., 2022), only PLN-Arg9Cys and PLN-Arg14del have been reported to exhibit additional aberrant interactions to proteins implicated in regulation of SERCA2 or PLN phosphorylation (Rigatti et al., 2015; Vafiadaki et al., 2022). In support of this, phosphorylation of PLN-Arg9Cys and PLN-Arg14del is abrogated while no change has been observed in the phosphorylation status of PLN-Arg25Cys (Schmitt et al., 2003; Ceholski et al., 2012b; Liu et al., 2015; Vafiadaki et al., 2022). These findings suggest that the presence of multiple, simultaneous alterations in critical PLN functional parameters may contribute towards development of a more severe spectrum of disease (i.e., DCM) in PLN-Arg9Cys and PLN-Arg14del, while a milder phenotype (i.e., HCM) is associated with PLN-Arg25Cys. It remains to be determined whether this differential topological effect on disease manifestation may be applicable to other PLN variants.
FIGURE 3
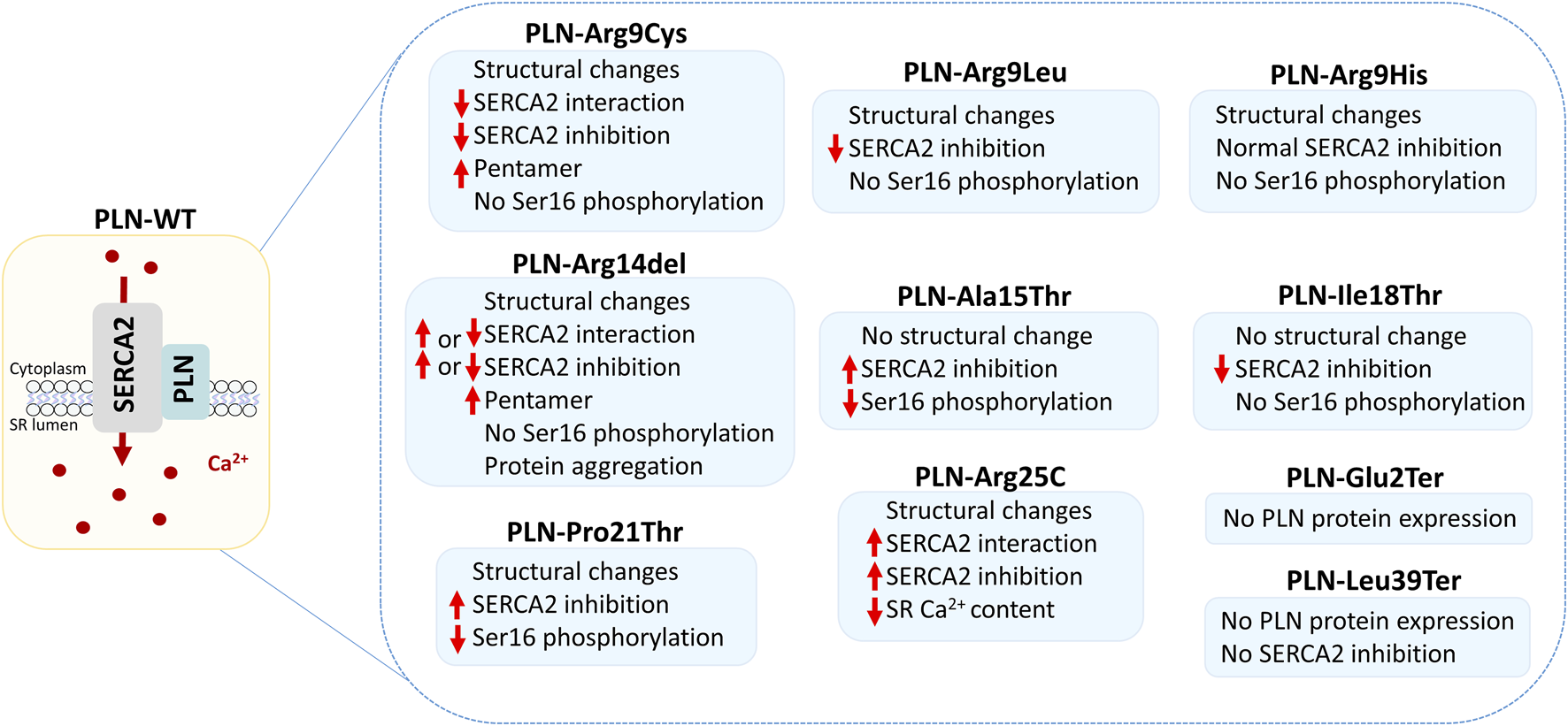
Functional effects of PLN variants. The schematic depicts the PLN and SERCA2 protein complex at SR membrane, where it regulates Ca2+ homeostasis and cardiac function. Alterations mediated by the different PLN variants are shown, based on currently available experimental evidence.
4.1 The pathogenic PLN variants involve predominantly loss of protein function
While the impact of published variants on PLN function has been characterized in only a small subset, it appears that the majority of pathogenic variants result in loss-of-function (Table 2). This observation, along with the fact that they are mainly found in heterozygous state, suggests a PLN dosage effect, with reduction in PLN function being central to disease pathogenesis as the presence of the wild-type PLN allele does not appear to suffice for normal cardiac function. A representative example is the particularly severe phenotype in the case of the dominant-negative variant Arg9Cys, with progressive heart failure requiring heart transplantation in early adult life, which has been described across unrelated families (Schmitt et al., 2003; Truszkowska et al., 2015; Fish et al., 2016). An exception to this loss-of-function effect appears to be the nonsense variant Glu2Ter, which has been observed in homozygosity in a 36 years old individual with severe DCM and heart failure, whereas heterozygous carriers were reported to have normal cardiac function (Li et al., 2019). It remains to be determined whether a single copy of this variant can lead to late onset disease or whether other mechanisms, such as incomplete penetrance, may be contributing to this apparent lack of cardiac phenotype. Another intriguing variant is Leu39Ter, which associates with a dosage-dependent effect correlating to disease severity. Heterozygotes exhibit HCM due to partial PLN functionality, while homozygotes develop severe DCM at a young age due to complete loss of PLN function (Landstrom et al., 2011).
TABLE 2
Functional effects and experimental evidence on currently characterized PLN variants.
To date, the only reported gain-of-function variants comprise Ala15Thr, Pro21Thr and Arg25Cys, which have all been shown to result in increased SERCA inhibition. It is of interest that they are closely located along the length of the protein, with both Pro21Thr and Arg25Cys causing an increase in PLN helical structure that possibly indicates a common pathogenic mechanism (Liu et al., 2015; Armanious et al., 2024). PLN-Arg14del is another putative gain-of-function variant that was initially proposed to cause SERCA super-inhibition although subsequent studies challenged this observation (Haghighi et al., 2006; Badone et al., 2021; Maniezzi et al., 2024). Thus, the impact of PLN-Arg14del on SERCA activity remains to be clarified.
4.2 The role of genetic modifiers in PLN cardiomyopathies
The phenotype of carriers with pathogenic PLN variants is highly variable, with evidence of both incomplete penetrance and variable expressivity. Patients may present with DCM, HCM or ARVC, while the age of onset and the disease severity ranges from asymptomatic all the way to sudden cardiac death (SCD) or end-stage heart failure (HF) at a young age (Table 3). The phenotypic variability is evident even among carriers of the same PLN variant, such as PLN-Arg14del (Hof et al., 2019). Although the potential role of non-genetic parameters including exercise, chronic stress, personality traits, lifestyle factors and major disease manifestations has been assessed, no significant association has been established to date (van Drie et al., 2023; van Lint et al., 2023; Mahmoud et al., 2025). Meanwhile, the implication of genetic modifiers has long been suspected, and evidence to support this is being explored.
TABLE 3
| Type of variant | PLN variant | Domain location | Clinical phenotype | Age of onset | Penetrance | Number of reported cases | Reference |
|---|---|---|---|---|---|---|---|
| Deletion | c.40_42delAGA, p.Arg14del | Cytoplasmic | Asymptomatic, ACM or DCM with arrhythmias | 30–60 years, arrhythmias may occur at earlier age | Incomplete | >1,500 | Haghighi et al. (2003), van der Zwaag et al. (2012), van Rijsingen et al. (2014), Cheung et al. (2019), Hof et al. (2019), Jiang et al. (2020), Tabata et al. (2021), Verstraelen et al. (2025) |
| c.95_98delTTAT, p.Phe32SerfsTer7 | Cytoplasmic | HCM | 14 years | Unknown | 1 | Du et al. (2024) | |
| Missense | c.23C>T, p.Thr8Ile | Cytoplasmic | DCM | Unknown | Unknown | Not specified | Sousa et al. (2019) |
| c.25C>T, p.Arg9Cys | Cytoplasmic | DCM | 20–35 years | Complete | 10 | Schmitt et al. (2003), Truszkowska et al. (2015), Fish et al. (2016) | |
| c.26G>T, p.Arg9Leu | Cytoplasmic | DCM | 30–50 years | Unknown | 4 | Medeiros et al. (2011), Truszkowska et al. (2015) | |
| c.26G>A, p.Arg9His | Cytoplasmic | DCM | 43 years | Incomplete | >7 (Not specified in Sousa et al.) | Medeiros et al. (2011), Sousa et al. (2019) | |
| c.43G>A, p.Ala15Thr | Cytoplasmic | DCM | 4 years | Unknown | 1 | Pugh et al. (2014) | |
| c.46T>G, p.Ser16Pro | Cytoplasmic | HCM | Unknown | Unknown | 1 | Walsh et al. (2017b) | |
| c.50C>A, pThr17Asn | Cytoplasmic | DCM | 44 years | Unknown | 1 | Mollanoori et al. (2018) | |
| c.53T>C; p.Ile18Thr | Cytoplasmic | HCM | Unknown | Unknown | >1 (Not specified in Lopes et al.) | Burns et al. (2017), Lopes et al. (2015) | |
| c.61C>A, p.Pro21Thr | Cytoplasmic | DCM | 60 years | Unknown | >1 (Not specified in Sousa et al.) | Pugh et al. (2014), Sousa et al. (2019) | |
| c.73C>T, p.Arg25Cys | Cytoplasmic | DCM, HCM and Bruganda syndrome | 45 years | >5 (Not specified in Lopes et al.) | Behr et al. (2015), Liu et al. (2015), Lopes et al. (2015) | ||
| c.121T>A, p.Cys41Ser | Transmembrane | HCM | Unknown | Unknown | 1 | Walsh et al. (2017b) | |
| c.145G>A, p.Val49Met | Transmembrane | HCM | Unknown | Unknown | 1 | Xu et al. (2015) | |
| c.152T>C, p.Leu51Pro | Transmembrane | HCM | Unknown | Unknown | >1 (Not specified in Lopes et al.) | Walsh et al. (2017b), Lopes et al. (2015) | |
| Nonsense | c.4G>T, p.Glu2Ter | Cytoplasmic | DCM and severe heart failure, arrhythmias | 36 years | Unknown | 6 | Li et al. (2019) |
| c.116T>G, p.Leu39Ter | Transmembrane | Asymptomatic hypertrophy, HCM or severe DCM with cardiac arrhythmias | 17–51 years | Incomplete | 28 | Haghighi et al. (2003), Landstrom et al. (2011), Medeiros et al. (2011), Sanoudou et al. (2015), Walsh et al. (2017a), Walsh et al. (2017b), Mazzone et al. (2024), Burns et al. (2017) | |
| Insertion | c.9_10insA, p.Val4SerFsTer15 | Cytoplasmic | Wolf-Parkinson-White syndrome with unconfirmed diagnosis of cardiomyopathy | 31 years | Unknown | 1 | Truszkowska et al. (2015) |
| c.37_38insA, p.Arg13LysfsTer7 | Cytoplasmic | DCM | 39 years | Family history of DCM | 1 | Pugh et al. (2014) | |
| c.61_62insCT, p.Pro21LeufsTer19 | Cytoplasmic | HCM | Unknown | Unknown | Not specified | Lopes et al. (2015) | |
| c.63_64dupTC, p.Gln22LeufsTer19 | Cytoplasmic | HCM | Unknown | Unknown | 1 | Walsh et al. (2017b) | |
| c.138dupT, p.Ile47TyrfsTer14 | Transmembrane | HCM | Unknown | Unknown | 1 | Walsh et al. (2017b) | |
| Promoter | −77 A>G | Promoter | HCM | 56 years | Unknown | 1 | Minamisawa et al. (2003) |
| −42 C>G | Promoter | HCM and healthy | 67 years | Incomplete | 3 | Medin et al. (2007) | |
| −36 A>C | Promoter | DCM and healthy | 18–44 years | Incomplete | 24 | Medin et al. (2007), Haghighi et al. (2008) |
PLN variants, clinical characteristics and cases reported to date.
A tentative effect of individual modifier genes has also been proposed in relation to different PLN variants. For example, although PLN-Leu39Ter heterozygosity has been reported to lead to HCM, in the presence of three additional genetic variants in DCM/arrhythmia associated genes, a PLN-Leu39Ter heterozygote patient presented with DCM and sustained ventricular tachycardia (Sanoudou et al., 2015). Similarly, PLN-Arg25Cys has been identified in patients presenting with HCM or Brugada. However in the presence of both PLN-Arg25Cys and a LMNA variant severe DCM was observed, with the patient requiring heart transplantation (Behr et al., 2015; Liu et al., 2015; Lopes et al., 2015).
These observations in PLN cardiomyopathy patients are well-aligned with increasing evidence supporting the coexistence of multiple genetic variants in a proportion of DCM patients (Haas et al., 2015; Pankuweit and Richter, 2015). In specific, it was recently proposed that the overall genetic landscape underlying DCM and HCM is significantly different, with increased coexistence of genetic variants correlating with disease severity and predisposition to DCM, compared to HCM (Puckelwartz et al., 2021). For example, BAG3 Cys151Arg was proposed to serve as an important genetic modifier variant in DCM, by modulating risk on the DCM-HCM spectrum, and impacting DCM risk in carriers of pathogenic truncating titin variants (Park et al., 2024). Similarly, a variant of unknown significance in the myosin heavy chain 7 gene (MYH7 p. Ile1927Phe) was shown to contribute to the development of severe HCM in the presence of truncating MyBPC3 variants (Escriba et al., 2023).
5 Conclusion and perspective
PLN has a well-established and central role in cardiac function and disease. Pathogenic PLN variants, span missense, nonsense, small deletions, and insertions that are associated with a range of cardiomyopathy phenotypes. Although only some of these variants have been functionally characterized, their analysis has contributed to elucidation of key pathogenic mechanisms, such as altered Ca2+ handling, structural disruption, and aberrant protein interactions. In public genetic databases many more novel PLN variants have been submitted, including predicted P/LP, even though experimental validation is pending. The increasing use of genetic testing as part of the Cardiology Clinics’ routine is anticipated to unveil a greater number and frequency of PLN variants. Importantly, pathogenic PLN variants are associated with a spectrum of cardiomyopathy-related phenotypes. Detailed mapping of these variants, careful characterization of their biological role, and investigation of genotype-phenotype correlations, has the potential to transform healthcare for patients with PLN variants, by enhancing predictive, diagnostic, and prognostic accuracy, as well as the precision of future therapeutic approaches. Towards this, the development of targeted interventions, guided by the unique properties of specific PLN variants, represents a promising avenue to address the unmet clinical needs of the patients and ultimately mitigate PLN-related cardiac diseases. For Cardiology clinics to reap the full potential of these fascinating developments, close interaction with genetics experts and genetic counselors is recommended (Musunuru et al., 2020; Wilde et al., 2022; Morales et al., 2023; Goehringer et al., 2025; Sanoudou et al., 2025). Meanwhile, as current understanding of genetic, epigenetic and epitranscriptomic variation evolves, and the artificial intelligence milieu matures, more sophisticated all-encompassing clinical prediction/classification/prognostic/prevention tools are foreseen (Kalozoumi et al., 2012; Leptidis et al., 2022; Sel et al., 2024).
Statements
Author contributions
EV: Data curation, Writing – review and editing, Conceptualization, Writing – original draft. IC: Formal Analysis, Writing – review and editing. KS: Writing – review and editing, Formal Analysis. AGE: Writing – review and editing. EK: Writing – review and editing. DS: Writing – original draft, Funding acquisition, Conceptualization, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by CURE-PLaN, a grant from the Leducq Foundation for Cardiovascular Research (18CVD01).
Conflict of interest
AGE is co-founder and CSO of GENOSOPHY S.A., spin-off company of the National and Kapodistrian University of Athens.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Abrol N. de Tombe P. P. Robia S. L. (2015). Acute inotropic and lusitropic effects of cardiomyopathic R9C mutation of phospholamban. J. Biol. Chem.290 (11), 7130–7140. 10.1074/jbc.M114.630319
2
Abrol N. Smolin N. Armanious G. Ceholski D. K. Trieber C. A. Young H. S. et al (2014). Phospholamban C-terminal residues are critical determinants of the structure and function of the calcium ATPase regulatory complex. J. Biol. Chem.289 (37), 25855–25866. 10.1074/jbc.M114.562579
3
Alfares A. A. Kelly M. A. McDermott G. Funke B. H. Lebo M. S. Baxter S. B. et al (2015). Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet. Med.17 (11), 880–888. 10.1038/gim.2014.205
4
Arbelo E. Protonotarios A. Gimeno J. R. Arbustini E. Barriales-Villa R. Basso C. et al (2023). 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J.44 (37), 3503–3626. 10.1093/eurheartj/ehad194
5
Armanious G. P. Lemieux M. J. Espinoza-Fonseca L. M. Young H. S. (2024). Missense variants in phospholamban and cardiac myosin binding protein identified in patients with a family history and clinical diagnosis of dilated cardiomyopathy. Biochim. Biophys. Acta Mol. Cell Res.1871 (4), 119699. 10.1016/j.bbamcr.2024.119699
6
Asahi M. Kimura Y. Kurzydlowski K. Tada M. MacLennan D. H. (1999). Transmembrane helix M6 in sarco(endo)plasmic reticulum Ca(2+)-ATPase forms a functional interaction site with phospholamban. Evidence for physical interactions at other sites. J. Biol. Chem.274 (46), 32855–32862. 10.1074/jbc.274.46.32855
7
Asahi M. Nakayama H. Tada M. Otsu K. (2003). Regulation of sarco(endo)plasmic reticulum Ca2+ adenosine triphosphatase by phospholamban and sarcolipin: implication for cardiac hypertrophy and failure. Trends Cardiovasc Med.13 (4), 152–157. 10.1016/s1050-1738(03)00037-9
8
Badone B. Ronchi C. Lodola F. Knaust A. E. Hansen A. Eschenhagen T. et al (2021). Characterization of the PLN p.Arg14del Mutation in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Int. J. Mol. Sci.22 (24), 13500. 10.3390/ijms222413500
9
Behr E. R. Savio-Galimberti E. Barc J. Holst A. G. Petropoulou E. Prins B. P. et al (2015). Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc Res.106 (3), 520–529. 10.1093/cvr/cvv042
10
Belakavadi M. Saunders J. Weisleder N. Raghava P. S. Fondell J. D. (2010). Repression of cardiac phospholamban gene expression is mediated by thyroid hormone receptor-{alpha}1 and involves targeted covalent histone modifications. Endocrinology151 (6), 2946–2956. 10.1210/en.2009-1241
11
Bers D. M. (2002). Cardiac excitation-contraction coupling. Nature415 (6868), 198–205. 10.1038/415198a
12
Bleijendaal H. Ramos L. A. Lopes R. R. Verstraelen T. E. Baalman S. W. E. Oudkerk Pool M. D. et al (2021). Computer versus cardiologist: Is a machine learning algorithm able to outperform an expert in diagnosing a phospholamban p.Arg14del mutation on the electrocardiogram?Heart rhythm.18 (1), 79–87. 10.1016/j.hrthm.2020.08.021
13
Burns C. Bagnall R. D. Lam L. Semsarian C. Ingles J. (2017). Multiple gene variants in hypertrophic cardiomyopathy in the era of next-generation sequencing. Circ. Cardiovasc Genet.10 (4), e001666. 10.1161/CIRCGENETICS.116.001666
14
Ceholski D. K. Trieber C. A. Holmes C. F. Young H. S. (2012a). Lethal, hereditary mutants of phospholamban elude phosphorylation by protein kinase A. J. Biol. Chem.287 (32), 26596–26605. 10.1074/jbc.M112.382713
15
Ceholski D. K. Trieber C. A. Young H. S. (2012b). Hydrophobic imbalance in the cytoplasmic domain of phospholamban is a determinant for lethal dilated cardiomyopathy. J. Biol. Chem.287 (20), 16521–16529. 10.1074/jbc.M112.360859
16
Ceholski D. K. Turnbull I. C. Kong C. W. Koplev S. Mayourian J. Gorski P. A. et al (2018). Functional and transcriptomic insights into pathogenesis of R9C phospholamban mutation using human induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell Cardiol.119, 147–154. 10.1016/j.yjmcc.2018.05.007
17
Cheung C. C. Healey J. S. Hamilton R. Spears D. Gollob M. H. Mellor G. et al (2019). Phospholamban cardiomyopathy: a Canadian perspective on a unique population. Neth Heart J.27 (4), 208–213. 10.1007/s12471-019-1247-0
18
Chiu C. Tebo M. Ingles J. Yeates L. Arthur J. W. Lind J. M. et al (2007). Genetic screening of calcium regulation genes in familial hypertrophic cardiomyopathy. J. Mol. Cell Cardiol.43 (3), 337–343. 10.1016/j.yjmcc.2007.06.009
19
Cleary S. R. Seflova J. Cho E. E. Bisht K. Khandelia H. Espinoza-Fonseca L. M. et al (2024). Phospholamban inhibits the cardiac calcium pump by interrupting an allosteric activation pathway. J. Biol. Chem.300 (5), 107267. 10.1016/j.jbc.2024.107267
20
Cleary S. R. Teng A. C. T. Kongmeneck A. D. Fang X. Phillips T. A. Cho E. E. et al (2023). Dilated cardiomyopathy variant R14del increases phospholamban pentamer stability, blunting dynamic regulation of cardiac calcium handling. bioRxiv. 10.1101/2023.05.26.542463
21
Cuello F. Knaust A. E. Saleem U. Loos M. Raabe J. Mosqueira D. et al (2021). Impairment of the ER/mitochondria compartment in human cardiomyocytes with PLN p.Arg14del mutation. EMBO Mol. Med.13 (6), e13074. 10.15252/emmm.202013074
22
Dave J. Raad N. Mittal N. Zhang L. Fargnoli A. Oh J. G. et al (2022). Gene editing reverses arrhythmia susceptibility in humanized PLN-R14del mice: modelling a European cardiomyopathy with global impact. Cardiovasc Res.118 (15), 3140–3150. 10.1093/cvr/cvac021
23
Deiman F. E. Bomer N. van der Meer P. Grote Beverborg N. (2022). Review: precision medicine approaches for genetic cardiomyopathy: targeting phospholamban R14del. Curr. Heart Fail. Rep. 10.1007/s11897-022-00558-x
24
Doevendans P. A. Glijnis P. C. Kranias E. G. (2019). Leducq transatlantic network of excellence to cure phospholamban-induced cardiomyopathy (CURE-PLaN). Circ. Res.125 (7), 720–724. 10.1161/CIRCRESAHA.119.315077
25
Du X. Barnett C. L. Widmeyer K. M. Wang X. Brightman D. S. Noonan C. W. et al (2024). RMND1 and PLN variants are the underlying cause of Perrault-like syndrome and cardiac anomalies in a patient. Clin. Case Rep.12 (11), e9537. 10.1002/ccr3.9537
26
Eijgenraam T. R. Boukens B. J. Boogerd C. J. Schouten E. M. van de Kolk C. W. A. Stege N. M. et al (2020). The phospholamban p.(Arg14del) pathogenic variant leads to cardiomyopathy with heart failure and is unreponsive to standard heart failure therapy. Sci. Rep.10 (1), 9819. 10.1038/s41598-020-66656-9
27
Eijgenraam T. R. Stege N. M. Oliveira Nunes Teixeira V. de Brouwer R. Schouten E. M. Grote Beverborg N. et al (2022). Antisense Therapy Attenuates Phospholamban p.(Arg14del) Cardiomyopathy in Mice and Reverses Protein Aggregation. Int. J. Mol. Sci.23 (5), 2427. 10.3390/ijms23052427
28
Escriba R. Larranaga-Moreira J. M. Richaud-Patin Y. Pourchet L. Lazis I. Jimenez-Delgado S. et al (2023). iPSC-based modeling of variable clinical presentation in hypertrophic cardiomyopathy. Circ. Res.133 (2), 108–119. 10.1161/CIRCRESAHA.122.321951
29
Feyen D. A. M. Perea-Gil I. Maas R. G. C. Harakalova M. Gavidia A. A. Arthur Ataam J. et al (2021). Unfolded Protein Response as a Compensatory Mechanism and Potential Therapeutic Target in PLN R14del Cardiomyopathy. Circulation144 (5), 382–392. 10.1161/CIRCULATIONAHA.120.049844
30
Fish M. Shaboodien G. Kraus S. Sliwa K. Seidman C. E. Burke M. A. et al (2016). Mutation analysis of the phospholamban gene in 315 South Africans with dilated, hypertrophic, peripartum and arrhythmogenic right ventricular cardiomyopathies. Sci. Rep.6, 22235. 10.1038/srep22235
31
Foo B. Amedei H. Kaur S. Jaawan S. Boshnakovska A. Gall T. et al (2024). Unbiased complexome profiling and global proteomics analysis reveals mitochondrial impairment and potential changes at the intercalated disk in presymptomatic R14Δ/+ mice hearts. PLoS One19 (10), e0311203. 10.1371/journal.pone.0311203
32
Froehlich J. P. Mahaney J. E. Keceli G. Pavlos C. M. Goldstein R. Redwood A. J. et al (2008). Phospholamban thiols play a central role in activation of the cardiac muscle sarcoplasmic reticulum calcium pump by nitroxyl. Biochemistry47 (50), 13150–13152. 10.1021/bi801925p
33
Fujii J. Zarain-Herzberg A. Willard H. F. Tada M. MacLennan D. H. (1991). Structure of the rabbit phospholamban gene, cloning of the human cDNA, and assignment of the gene to human chromosome 6. J. Biol. Chem.266 (18), 11669–11675. 10.1016/s0021-9258(18)99009-5
34
Ganim J. R. Luo W. Ponniah S. Grupp I. Kim H. W. Ferguson D. G. et al (1992). Mouse phospholamban gene expression during development in vivo and in vitro. Circ. Res.71 (5), 1021–1030. 10.1161/01.res.71.5.1021
35
Goehringer J. Sanoudou D. Morales A. (2025). “Genetic counseling for cardiovascular disease: Part A – pre-test approaches and considerations,” in Genetic counselling - navigating the future. Editor SeifiM. (London, United Kingdom: IntechOpen). 10.5772/intechopen.1007908
36
Grote Beverborg N. Spater D. Knoll R. Hidalgo A. Yeh S. T. Elbeck Z. et al (2021). Phospholamban antisense oligonucleotides improve cardiac function in murine cardiomyopathy. Nat. Commun.12 (1), 5180. 10.1038/s41467-021-25439-0
37
Gustavsson M. Traaseth N. J. Karim C. B. Lockamy E. L. Thomas D. D. Veglia G. (2011). Lipid-mediated folding/unfolding of phospholamban as a regulatory mechanism for the sarcoplasmic reticulum Ca2+-ATPase. J. Mol. Biol.408 (4), 755–765. 10.1016/j.jmb.2011.03.015
38
Gustavsson M. Verardi R. Mullen D. G. Mote K. R. Traaseth N. J. Gopinath T. et al (2013). Allosteric regulation of SERCA by phosphorylation-mediated conformational shift of phospholamban. Proc. Natl. Acad. Sci. U. S. A.110 (43), 17338–17343. 10.1073/pnas.1303006110
39
Ha K. N. Masterson L. R. Hou Z. Verardi R. Walsh N. Veglia G. et al (2011). Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase A. Proc. Natl. Acad. Sci. U. S. A.108 (7), 2735–2740. 10.1073/pnas.1013987108
40
Haas J. Frese K. S. Peil B. Kloos W. Keller A. Nietsch R. et al (2015). Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J.36 (18), 1123–135a. 10.1093/eurheartj/ehu301
41
Haghighi K. Bidwell P. Kranias E. G. (2014). Phospholamban interactome in cardiac contractility and survival: a new vision of an old friend. J. Mol. Cell Cardiol.77, 160–167. 10.1016/j.yjmcc.2014.10.005
42
Haghighi K. Chen G. Sato Y. Fan G. C. He S. Kolokathis F. et al (2008). A human phospholamban promoter polymorphism in dilated cardiomyopathy alters transcriptional regulation by glucocorticoids. Hum. Mutat.29 (5), 640–647. 10.1002/humu.20692
43
Haghighi K. Gardner G. Vafiadaki E. Kumar M. Green L. C. Ma J. et al (2021). Impaired right ventricular calcium cycling is an early risk factor in r14del-phospholamban arrhythmias. J. Pers. Med.11 (6), 502. 10.3390/jpm11060502
44
Haghighi K. Kolokathis F. Gramolini A. O. Waggoner J. R. Pater L. Lynch R. A. et al (2006). A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A.103 (5), 1388–1393. 10.1073/pnas.0510519103
45
Haghighi K. Kolokathis F. Pater L. Lynch R. A. Asahi M. Gramolini A. O. et al (2003). Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J. Clin. Invest111 (6), 869–876. 10.1172/JCI17892
46
Hirtle-Lewis M. Desbiens K. Ruel I. Rudzicz N. Genest J. Engert J. C. et al (2013). The genetics of dilated cardiomyopathy: a prioritized candidate gene study of LMNA, TNNT2, TCAP, and PLN. Clin. Cardiol.36 (10), 628–633. 10.1002/clc.22193
47
Hof I. E. van der Heijden J. F. Kranias E. G. Sanoudou D. de Boer R. A. van Tintelen J. P. et al (2019). Prevalence and cardiac phenotype of patients with a phospholamban mutation. Neth Heart J.27 (2), 64–69. 10.1007/s12471-018-1211-4
48
Hu H. Jiang M. Cao Y. Zhang Z. Jiang B. Tian F. et al (2020). HuR regulates phospholamban expression in isoproterenol-induced cardiac remodelling. Cardiovasc Res.116 (5), 944–955. 10.1093/cvr/cvz205
49
Hughes E. Middleton D. A. (2014). Comparison of the structure and function of phospholamban and the arginine-14 deficient mutant associated with dilated cardiomyopathy. PLoS One9 (9), e106746. 10.1371/journal.pone.0106746
50
Irie T. Sips P. Y. Kai S. Kida K. Ikeda K. Hirai S. et al (2015). S-nitrosylation of calcium-handling proteins in cardiac adrenergic signaling and hypertrophy. Circ. Res.117 (9), 793–803. 10.1161/CIRCRESAHA.115.307157
51
Jiang X. Xu Y. Sun J. Wang L. Guo X. Chen Y. (2020). The phenotypic characteristic observed by cardiac magnetic resonance in a PLN-R14del family. Sci. Rep.10 (1), 16478. 10.1038/s41598-020-73359-8
52
Kalozoumi G. Tzimas C. Sanoudou D. (2012). The expanding role of epigenetics. Glob. Cardiol. Sci. Pract.2012 (1), 7. 10.5339/gcsp.2012.7
53
Karakikes I. Stillitano F. Nonnenmacher M. Tzimas C. Sanoudou D. Termglinchan V. et al (2015). Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun.6, 6955. 10.1038/ncomms7955
54
Keceli G. Majumdar A. Thorpe C. N. Jun S. Tocchetti C. G. Lee D. I. et al (2019). Nitroxyl (HNO) targets phospholamban cysteines 41 and 46 to enhance cardiac function. J. Gen. Physiol.151 (6), 758–770. 10.1085/jgp.201812208
55
Kimura Y. Asahi M. Kurzydlowski K. Tada M. MacLennan D. H. (1998). Phospholamban domain Ib mutations influence functional interactions with the Ca2+-ATPase isoform of cardiac sarcoplasmic reticulum. J. Biol. Chem.273 (23), 14238–14241. 10.1074/jbc.273.23.14238
56
Kimura Y. Kurzydlowski K. Tada M. MacLennan D. H. (1996). Phospholamban regulates the Ca2+-ATPase through intramembrane interactions. J. Biol. Chem.271 (36), 21726–21731. 10.1074/jbc.271.36.21726
57
Kranias E. G. Doevendans P. A. Glijnis P. C. Hajjar R. J. (2018). PLN foundation. Circulation Res.123 (12), 1276–1278. 10.1161/Circresaha.118.314014
58
Kranias E. G. Hajjar R. J. (2017). The phospholamban journey 4 decades after setting out for ithaka. Circ. Res.120 (5), 781–783. 10.1161/CIRCRESAHA.116.310007
59
Landstrom A. P. Adekola B. A. Bos J. M. Ommen S. R. Ackerman M. J. (2011). PLN-encoded phospholamban mutation in a large cohort of hypertrophic cardiomyopathy cases: summary of the literature and implications for genetic testing. Am. Heart J.161 (1), 165–171. 10.1016/j.ahj.2010.08.001
60
Leptidis S. Papakonstantinou E. Diakou K. I. Pierouli K. Mitsis T. Dragoumani K. et al (2022). Epitranscriptomics of cardiovascular diseases (Review). Int. J. Mol. Med.49 (1), 9. (Review). 10.3892/ijmm.2021.5064
61
Li Z. Chen P. Xu J. Yu B. Li X. Wang D. W. et al (2019). A PLN nonsense variant causes severe dilated cardiomyopathy in a novel autosomal recessive inheritance mode. Int. J. Cardiol.279, 122–125. 10.1016/j.ijcard.2018.12.075
62
Liang X. H. Sun H. Shen W. Wang S. Yao J. Migawa M. T. et al (2017). Antisense oligonucleotides targeting translation inhibitory elements in 5' UTRs can selectively increase protein levels. Nucleic Acids Res.45 (16), 9528–9546. 10.1093/nar/gkx632
63
Liu G. S. Morales A. Vafiadaki E. Lam C. K. Cai W. F. Haghighi K. et al (2015). A novel human R25C-phospholamban mutation is associated with super-inhibition of calcium cycling and ventricular arrhythmia. Cardiovasc Res.107 (1), 164–174. 10.1093/cvr/cvv127
64
Lopes L. R. Syrris P. Guttmann O. P. O'Mahony C. Tang H. C. Dalageorgou C. et al (2015). Novel genotype-phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart101 (4), 294–301. 10.1136/heartjnl-2014-306387
65
Lopes R. R. Bleijendaal H. Ramos L. A. Verstraelen T. E. Amin A. S. Wilde A. A. M. et al (2021). Improving electrocardiogram-based detection of rare genetic heart disease using transfer learning: An application to phospholamban p.Arg14del mutation carriers. Comput. Biol. Med.131, 104262. 10.1016/j.compbiomed.2021.104262
66
MacLennan D. H. Kimura Y. Toyofuku T. (1998). Sites of regulatory interaction between calcium ATPases and phospholamban. Ann. N. Y. Acad. Sci.853, 31–42. 10.1111/j.1749-6632.1998.tb08254.x
67
MacLennan D. H. Kranias E. G. (2003). Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol.4 (7), 566–577. 10.1038/nrm1151
68
Mahmoud B. van der Heide M. Y. C. Cox M. Verstraelen T. E. de Brouwer R. van Drie E. et al (2025). The role of Comorbidities and lifestyle factors in disease progression of phospholamban cardiomyopathy. Eur. J. Prev. Cardiol., zwaf084. 10.1093/eurjpc/zwaf084
69
Maniezzi C. Eskandr M. Florindi C. Ferrandi M. Barassi P. Sacco E. et al (2024). Early consequences of the phospholamban mutation PLN-R14del(+/-) in a transgenic mouse model. Acta Physiol. (Oxf)240 (3), e14082. 10.1111/apha.14082
70
Mattiazzi A. Kranias E. G. (2024). Unleashing the Power of genetics: PLN Ablation, Phospholambanopathies and evolving challenges. Circ. Res.134 (2), 138–142. 10.1161/CIRCRESAHA.123.323053
71
Mayr C. (2019). What are 3' UTRs doing?Cold Spring Harb. Perspect. Biol.11 (10), a034728. 10.1101/cshperspect.a034728
72
Mazzone F. Giovannico L. Fischetti G. Parigino D. Guaricci A. I. Forleo C. et al (2024). Homozygous phospholamban mutation causing dilated cardiomyopathy in a young man: from cardiogenic Shock to Tennis Tournaments. Clin. Transpl.38 (11), e70031. 10.1111/ctr.70031
73
Medeiros A. Biagi D. G. Sobreira T. J. de Oliveira P. S. Negrao C. E. Mansur A. J. et al (2011). Mutations in the human phospholamban gene in patients with heart failure. Am. Heart J.162 (6), 1088–1095. 10.1016/j.ahj.2011.07.028
74
Medin M. Hermida-Prieto M. Monserrat L. Laredo R. Rodriguez-Rey J. C. Fernandez X. et al (2007). Mutational screening of phospholamban gene in hypertrophic and idiopathic dilated cardiomyopathy and functional study of the PLN -42 C>G mutation. Eur. J. Heart Fail9 (1), 37–43. 10.1016/j.ejheart.2006.04.007
75
Minamisawa S. Sato Y. Tatsuguchi Y. Fujino T. Imamura S. Uetsuka Y. et al (2003). Mutation of the phospholamban promoter associated with hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun.304 (1), 1–4. 10.1016/s0006-291x(03)00526-6
76
Mollanoori H. Naderi N. Amin A. Hassani B. Shahraki H. Teimourian s. (2018). A novel human T17N-phospholamban variation in idiopathic dilated cardiomyopathy. Gene Rep.12, 122–127. 10.1016/j.genrep.2018.06.014
77
Morales A. Goehringer J. Sanoudou D. (2023). Evolving cardiovascular genetic counseling needs in the era of precision medicine. Front. Cardiovasc Med.10, 1161029. 10.3389/fcvm.2023.1161029
78
Mushala B. Cho E. Stoner M. Gibson G. Sembrat J. Bugga P. et al (2024). Abstract Mo069: phospholamban acetylation Enhances cardiomyocyte calcium cycling under conditions of high-Fat Feeding. Circulation Res.135 (Suppl. 1), AMo069. 10.1161/res.135.suppl_1.Mo069
79
Musunuru K. Hershberger R. E. Day S. M. Klinedinst N. J. Landstrom A. P. Parikh V. N. et al (2020). Genetic testing for inherited cardiovascular diseases: a Scientific statement from the American heart association. Circ. Genom Precis. Med.13 (4), e000067. 10.1161/HCG.0000000000000067
80
Nakagawa T. Yokoe S. Asahi M. (2016). Phospholamban degradation is induced by phosphorylation-mediated ubiquitination and inhibited by interaction with cardiac type Sarco(endo)plasmic reticulum Ca(2+)-ATPase. Biochem. Biophys. Res. Commun.472 (3), 523–530. 10.1016/j.bbrc.2016.03.009
81
Nelson S. E. D. Ha K. N. Gopinath T. Exline M. H. Mascioni A. Thomas D. D. et al (2018). Effects of the Arg9Cys and Arg25Cys mutations on phospholamban's conformational equilibrium in membrane bilayers. Biochim. Biophys. Acta Biomembr.1860 (6), 1335–1341. 10.1016/j.bbamem.2018.02.030
82
Pankuweit S. Richter A. (2015). Clinical genetics of dilated cardiomyopathy: on the way to personalized medicine?Eur. Heart J.36 (18), 1074–1077. 10.1093/eurheartj/ehu402
83
Park J. Levin M. G. Zhang D. Reza N. Mead J. O. Carruth E. D. et al (2024). Bidirectional risk modulator and modifier variant of dilated and hypertrophic cardiomyopathy in BAG3. JAMA Cardiol.9 (12), 1124–1133. 10.1001/jamacardio.2024.3547
84
Puckelwartz M. J. Pesce L. L. Dellefave-Castillo L. M. Wheeler M. T. Pottinger T. D. Robinson A. C. et al (2021). Genomic Context Differs between human dilated cardiomyopathy and hypertrophic cardiomyopathy. J. Am. Heart Assoc.10 (7), e019944. 10.1161/JAHA.120.019944
85
Pugh T. J. Kelly M. A. Gowrisankar S. Hynes E. Seidman M. A. Baxter S. M. et al (2014). The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med.16 (8), 601–608. 10.1038/gim.2013.204
86
Raad N. Bittihn P. Cacheux M. Jeong D. Ilkan Z. Ceholski D. et al (2021). Arrhythmia Mechanism and Dynamics in a Humanized Mouse Model of Inherited Cardiomyopathy Caused by Phospholamban R14del Mutation. Circulation144 (6), 441–454. 10.1161/CIRCULATIONAHA.119.043502
87
Raguimova O. N. Aguayo-Ortiz R. Robia S. L. Espinoza-Fonseca L. M. (2020). Dynamics-driven Allostery underlies Ca(2+)-mediated release of SERCA inhibition by phospholamban. Biophys. J.119 (9), 1917–1926. 10.1016/j.bpj.2020.09.014
88
Ren A. J. Wei C. Liu Y. J. Liu M. Wang P. Fan J. et al (2024). ZBTB20 regulates SERCA2a activity and Myocardial contractility through phospholamban. Circ. Res.134 (3), 252–265. 10.1161/CIRCRESAHA.123.323798
89
Rigatti M. Le A. V. Gerber C. Moraru I. I. Dodge-Kafka K. L. (2015). Phosphorylation state-dependent interaction between AKAP7δ/γ and phospholamban increases phospholamban phosphorylation. Cell Signal27 (9), 1807–1815. 10.1016/j.cellsig.2015.05.016
90
Roberts T. C. Langer R. Wood M. J. A. (2020). Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov.19 (10), 673–694. 10.1038/s41573-020-0075-7
91
Rogalska M. Vafiadaki E. Erpapazoglou Z. Haghighi K. Green L. Mantzoros C. S. et al (2023). Isoform changes of action potential regulators in the ventricles of arrhythmogenic phospholamban-R14del humanized mouse hearts. Metabolism138, 155344. 10.1016/j.metabol.2022.155344
92
Rogers H. T. Roberts D. S. Larson E. J. Melby J. A. Rossler K. J. Carr A. V. et al (2023). Comprehensive characterization of Endogenous phospholamban Proteoforms enabled by Photocleavable Surfactant and Top-down proteomics. Anal. Chem.95 (35), 13091–13100. 10.1021/acs.analchem.3c01618
93
Rohner E. Witman N. Sohlmer J. De Genst E. Louch W. E. Sahara M. et al (2021). An mRNA assay system demonstrates proteasomal-specific degradation contributes to cardiomyopathic phospholamban null mutation. Mol. Med.27 (1), 102. 10.1186/s10020-021-00362-8
94
Sanoudou D. Goehringer J. Morales A. (2025). “Genetic counseling for cardiovascular disease: Part B – post-test approaches and Considerations,” in Genetic counseling for cardiovascular disease: Part B – post-test approaches and considerations. Editor SeifiM. (London, United Kingdom: IntechOpen). 10.5772/intechopen.1007942
95
Sanoudou D. Kolokathis F. Arvanitis D. Al-Shafai K. Krishnamoorthy N. Buchan R. J. et al (2015). Genetic modifiers to the PLN L39X mutation in a patient with DCM and sustained ventricular tachycardia?Glob. Cardiol. Sci. Pract.2015 (2), 29. 10.5339/gcsp.2015.29
96
Santos D. G. Medeiros A. Brum P. C. Mill J. G. Mansur A. J. Krieger J. E. et al (2009). No evidence for an association between the -36A>C phospholamban gene polymorphism and a worse prognosis in heart failure. BMC Cardiovasc Disord.9, 33. 10.1186/1471-2261-9-33
97
Schmitt J. P. Ahmad F. Lorenz K. Hein L. Schulz S. Asahi M. et al (2009). Alterations of phospholamban function can exhibit cardiotoxic effects independent of excessive sarcoplasmic reticulum Ca2+-ATPase inhibition. Circulation119 (3), 436–444. 10.1161/CIRCULATIONAHA.108.783506
98
Schmitt J. P. Kamisago M. Asahi M. Li G. H. Ahmad F. Mende U. et al (2003). Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science299 (5611), 1410–1413. 10.1126/science.1081578
99
Sel K. Osman D. Zare F. Masoumi Shahrbabak S. Brattain L. Hahn J. O. et al (2024). Building digital twins for cardiovascular health: from principles to clinical impact. J. Am. Heart Assoc.13 (19), e031981. 10.1161/JAHA.123.031981
100
Sharma P. Ignatchenko V. Grace K. Ursprung C. Kislinger T. Gramolini A. O. (2010). Endoplasmic reticulum protein targeting of phospholamban: a common role for an N-terminal di-arginine motif in ER retention?PLoS One5 (7), e11496. 10.1371/journal.pone.0011496
101
Simmerman H. K. Jones L. R. (1998). Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol. Rev.78 (4), 921–947. 10.1152/physrev.1998.78.4.921
102
Soukarieh O. Meguerditchian C. Proust C. Aissi D. Eyries M. Goyenvalle A. et al (2022). Common and rare 5'UTR variants altering upstream open reading frames in cardiovascular genomics. Front. Cardiovasc Med.9, 841032. 10.3389/fcvm.2022.841032
103
Sousa A. Canedo P. Azevedo O. Lopes L. Pinho T. Baixia M. et al (2019). Molecular characterization of Portuguese patients with dilated cardiomyopathy. Rev. Port. Cardiol. Engl. Ed.38 (2), 129–139. 10.1016/j.repc.2018.10.010
104
Stege N. M. de Boer R. A. Makarewich C. A. van der Meer P. Sillje H. H. W. (2024). Reassessing the Mechanisms of PLN-R14del Cardiomyopathy: From Calcium Dysregulation to S/ER Malformation. JACC Basic Transl. Sci.9 (8), 1041–1052. 10.1016/j.jacbts.2024.02.017
105
Stege N. M. Eijgenraam T. R. Oliveira Nunes Teixeira V. Feringa A. M. Schouten E. M. Kuster D. W. D. et al (2023). DWORF Extends Life Span in a PLN-R14del Cardiomyopathy Mouse Model by Reducing Abnormal Sarcoplasmic Reticulum Clusters. Circ. Res.133 (12), 1006–1021. 10.1161/CIRCRESAHA.123.323304
106
Tabata T. Kuramoto Y. Ohtani T. Miyawaki H. Miyashita Y. Sera F. et al (2021). Phospholamban p.Arg14del Cardiomyopathy: A Japanese Case Series. Intern Med.61 (13), 1987–1993. 10.2169/internalmedicine.8594-21
107
Tada M. Toyofuku T. (1998). Molecular regulation of phospholamban function and expression. Trends Cardiovasc Med.8 (8), 330–340. 10.1016/s1050-1738(98)00032-2
108
Tada M. Yabuki M. Toyofuku T. (1998). Molecular regulation of phospholamban function and gene expression. Ann. N. Y. Acad. Sci.853, 116–129. 10.1111/j.1749-6632.1998.tb08261.x
109
Toyoshima C. Asahi M. Sugita Y. Khanna R. Tsuda T. MacLennan D. H. (2003). Modeling of the inhibitory interaction of phospholamban with the Ca2+ ATPase. Proc. Natl. Acad. Sci. U. S. A.100 (2), 467–472. 10.1073/pnas.0237326100
110
Truszkowska G. T. Bilinska Z. T. Kosinska J. Sleszycka J. Rydzanicz M. Sobieszczanska-Malek M. et al (2015). A study in Polish patients with cardiomyopathy emphasizes pathogenicity of phospholamban (PLN) mutations at amino acid position 9 and low penetrance of heterozygous null PLN mutations. BMC Med. Genet.16, 21. 10.1186/s12881-015-0167-0
111
Vafiadaki E. Glijnis P. C. Doevendans P. A. Kranias E. G. Sanoudou D. (2023). Phospholamban R14del disease: the past, the present and the future. Front. Cardiovasc Med.10, 1162205. 10.3389/fcvm.2023.1162205
112
Vafiadaki E. Haghighi K. Arvanitis D. A. Kranias E. G. Sanoudou D. (2022). Aberrant PLN-R14del Protein Interactions Intensify SERCA2a Inhibition, Driving Impaired Ca(2+) Handling and Arrhythmogenesis. Int. J. Mol. Sci.23 (13), 6947. 10.3390/ijms23136947
113
Vafiadaki E. Kranias E. G. Eliopoulos A. G. Sanoudou D. (2024). The phospholamban R14del generates pathogenic aggregates by impairing autophagosome-lysosome fusion. Cell Mol. Life Sci.81 (1), 450. 10.1007/s00018-024-05471-1
114
Vafiadaki E. Papalouka V. Arvanitis D. A. Kranias E. G. Sanoudou D. (2009). The role of SERCA2a/PLN complex, Ca(2+) homeostasis, and anti-apoptotic proteins in determining cell fate. Pflugers Arch.457 (3), 687–700. 10.1007/s00424-008-0506-5
115
van der Zwaag P. A. van Rijsingen I. A. Asimaki A. Jongbloed J. D. van Veldhuisen D. J. Wiesfeld A. C. et al (2012). Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail14 (11), 1199–1207. 10.1093/eurjhf/hfs119
116
van Drie E. Taal S. E. L. Schmidt A. F. Verstraelen T. E. de Brouwer R. Schoormans D. et al (2023). Influence of stressful life events and personality traits on PLN cardiomyopathy severity: an exploratory study. Europace26 (1), euad368. 10.1093/europace/euad368
117
van Lint F. H. M. Hassanzada F. Verstraelen T. E. Wang W. Bosman L. P. van der Zwaag P. A. et al (2023). Exercise does not influence development of phenotype in PLN p.(Arg14del) cardiomyopathy. Neth Heart J.31 (7-8), 291–299. 10.1007/s12471-023-01800-4
118
van Rijsingen I. A. van der Zwaag P. A. Groeneweg J. A. Nannenberg E. A. Jongbloed J. D. Zwinderman A. H. et al (2014). Outcome in phospholamban R14del carriers: results of a large multicentre cohort study. Circ. Cardiovasc Genet.7 (4), 455–465. 10.1161/CIRCGENETICS.113.000374
119
Verstraelen T. E. van Lint F. H. M. Bosman L. P. de Brouwer R. Proost V. M. Abeln B. G. S. et al (2021). Prediction of ventricular arrhythmia in phospholamban p.Arg14del mutation carriers-reaching the frontiers of individual risk prediction. Eur. Heart J.42 (29), 2842–2850. 10.1093/eurheartj/ehab294
120
Verstraelen T. E. van Lint F. H. M. de Brouwer R. Proost V. M. van Drie E. Bosman L. P. et al (2025). Age-related penetrance of phospholamban p.Arg14del cardiomyopathy. Eur. J. Heart Fail. 10.1002/ejhf.3672
121
Vicente M. Salgado-Almario J. Valiente-Gabioud A. A. Collins M. M. Vincent P. Domingo B. et al (2022). Early calcium and cardiac contraction defects in a model of phospholamban R9C mutation in zebrafish. J. Mol. Cell Cardiol.173, 127–140. 10.1016/j.yjmcc.2022.10.005
122
Vostrikov V. V. Soller K. J. Ha K. N. Gopinath T. Veglia G. (2015). Effects of naturally occurring arginine 14 deletion on phospholamban conformational dynamics and membrane interactions. Biochim. Biophys. Acta1848 (1 Pt B), 315–322. 10.1016/j.bbamem.2014.09.007
123
Wagner M. Weber S. El-Armouche A. (2015). Linking superinhibitory PLN mutations to CaMKII activation: a new arrhythmogenic mechanism in genetic DCM?Cardiovasc Res.107 (1), 5–6. 10.1093/cvr/cvv163
124
Walsh R. Buchan R. Wilk A. John S. Felkin L. E. Thomson K. L. et al (2017a). Defining the genetic architecture of hypertrophic cardiomyopathy: re-evaluating the role of non-sarcomeric genes. Eur. Heart J.38 (46), 3461–3468. 10.1093/eurheartj/ehw603
125
Walsh R. Thomson K. L. Ware J. S. Funke B. H. Woodley J. McGuire K. J. et al (2017b). Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med.19 (2), 192–203. 10.1038/gim.2016.90
126
Weber D. K. Reddy U. V. Robia S. L. Veglia G. (2024). Pathological mutations in the phospholamban cytoplasmic region affect its topology and dynamics modulating the extent of SERCA inhibition. Biochim. Biophys. Acta Biomembr.1866 (7), 184370. 10.1016/j.bbamem.2024.184370
127
Weber D. K. Reddy U. V. Wang S. Larsen E. K. Gopinath T. Gustavsson M. B. et al (2021). Structural basis for allosteric control of the SERCA-Phospholamban membrane complex by Ca(2+) and phosphorylation. Elife10, e66226. 10.7554/eLife.66226
128
Whiffin N. Karczewski K. J. Zhang X. Chothani S. Smith M. J. Evans D. G. et al (2020). Characterising the loss-of-function impact of 5' untranslated region variants in 15,708 individuals. Nat. Commun.11 (1), 2523. 10.1038/s41467-019-10717-9
129
Wilde A. A. M. Semsarian C. Marquez M. F. Shamloo A. S. Ackerman M. J. Ashley E. A. et al (2022). European heart Rhythm association (EHRA)/Heart Rhythm society (HRS)/Asia pacific heart Rhythm society (APHRS)/Latin American heart Rhythm society (LAHRS) expert consensus statement on the state of genetic testing for cardiac diseases. Europace24 (8), 1307–1367. 10.1093/europace/euac030
130
Xu J. Li Z. Ren X. Dong M. Li J. Shi X. et al (2015). Investigation of pathogenic genes in Chinese sporadic hypertrophic cardiomyopathy patients by whole exome sequencing. Sci. Rep.5, 16609. 10.1038/srep16609
131
Yabuki M. Toyofuku T. Otsu K. Nishida M. Kuzuya T. Hori M. et al (1998). Involvement of NF-Y in transcriptional regulation of the phospholamban gene. Eur. J. Biochem.258 (2), 744–751. 10.1046/j.1432-1327.1998.2580744.x
132
Yu Q. Barndt R. J. Shen Y. Sallam K. Tang Y. Chan S. Y. et al (2024). Mitigation of stress-induced structural remodeling and functional deficiency in iPSC-CMs with PLN R9C mutation by promoting autophagy. bioRxiv. 10.1101/2024.04.17.589921
133
Zhou T. Li J. Zhao P. Liu H. Jia D. Jia H. et al (2015). Palmitoyl acyltransferase Aph2 in cardiac function and the development of cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A.112 (51), 15666–15671. 10.1073/pnas.1518368112
Summary
Keywords
phospholamban, genetic variants, cardiomyopathies, precision medicine, genetic testing, genetic counseling
Citation
Vafiadaki E, Chaudhari I, Soliman KM, Eliopoulos AG, Kranias EG and Sanoudou D (2025) Genetic landscape of phospholamban cardiomyopathies. Front. Cell Dev. Biol. 13:1626242. doi: 10.3389/fcell.2025.1626242
Received
10 May 2025
Accepted
26 May 2025
Published
10 June 2025
Volume
13 - 2025
Edited by
Natascia Tiso, University of Padua, Italy
Reviewed by
Rudy Celeghin, University of Padua, Italy
Andreea Sorina Afana, University of Medicine and Pharmacy of Craiova, Romania
Frederik Deiman, University Medical Center Groningen, Netherlands
Updates
Copyright
© 2025 Vafiadaki, Chaudhari, Soliman, Eliopoulos, Kranias and Sanoudou.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth Vafiadaki, lvafiadaki@bioacademy.gr; Despina Sanoudou, dsanoudou@med.uoa.gr
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.