 Laura Venegas
Laura Venegas Stephanie Lheureux
Stephanie Lheureux- Division of Medical Oncology and Hematology, Princess Margaret Cancer Centre, University Health Network, Toronto, ON, Canada
High-grade serous ovarian cancer (HGSOC) is the most lethal gynecological malignancy. Therapeutic options remain limited for patients lacking predictive biomarkers, particularly those with BRCA wild-type tumors or those who have acquired resistance to both PARP inhibitors and platinum-based chemotherapy. Replication stress, TP53 mutations, and genomic instability characterize HGSOC. The cellular response to replication stress is primarily mediated by checkpoint kinases; however, this mechanism is frequently impaired in tumor cells. Consequently, cancer cells become increasingly dependent on the replication stress response (RSR) pathway for survival, and susceptible to therapies targeting the ATR-CHK1-WEE1 axis—a key regulator of genomic integrity. Inhibition of these checkpoint kinases can disrupt cell cycle control, inducing mitotic catastrophe and subsequent cancer cell death. Another defining feature of HGSOC is its immunosuppressive tumor microenvironment (TME), which has limited the efficacy of immune checkpoint inhibitors. Emerging evidence suggests that inhibition of the RSR pathway may not only exploit intrinsic tumor vulnerabilities but also modulate the TME to enhance anti-tumor immune responses. This provides rationale for combination approaches integrating RSR pathway inhibitors with innovative immune checkpoint blockade (ICB). This review examines the mechanistic rationale and therapeutic potential of such combinations, drawing on both preclinical and clinical data.
1 Introduction
DNA damage induces replicative stress, a critical cellular alteration that can arise from exogenous agents—such as cytotoxic chemotherapies (e.g., gemcitabine, 5-fluorouracil, cisplatin)—or endogenous factors, including misincorporation of ribonucleotides or mutations in tumor suppressor genes (Zeman and Cimprich, 2014). The cellular response to replicative stress is a regulated mechanism that ensures accurate DNA replication and genome integrity (Saxena and Zou, 2022). In tumor cells, this response becomes essential for survival; when compromised, tumor cell proliferation is impaired (Schoonen et al., 2019).
Ovarian cancer is the most lethal gynecologic malignancy, responsible for approximately 207,000 deaths worldwide each year (Huang et al., 2022). HGSOC is the most common histology subtype and is characterized by genomic instability, universal TP53 mutations, and profound copy number changes (Hillmann et al., 2025; Cancer Genome Atlas Research Network, 2011). The loss of the tumor suppressor gene p53 promotes a sequential pattern of genomic instability as tumors evolve. This progression begins with the accumulation of deletions, particularly in p53, and copy number alterations, followed by genome doubling and subclonal expansion, leading to intratumoral heterogeneity that contributes to poor prognosis and treatment resistance (Baslan et al., 2022). In a preclinical model using cell lines derived from non-ciliated fallopian tube epithelial cells, TP53 mutation appears to act as an initiating event, while subsequent BRCA1 loss further increases chromosomal instability (CIN) (Bronder et al., 2021). These molecular alterations coincide with progressive changes in the TME, transitioning from immune surveillance in early serous tubal intraepithelial carcinomas (STICs) to immune suppression in advanced STICs and cancer (Kader et al., 2024). The loss of p53 also upregulates repetitive elements, triggering an antiviral immune response known as viral mimicry; however, in premalignant lesions, this response becomes progressively suppressed, contributing to the development of immune tolerance (Ishak et al., 2025). Another contributor to the progressive cascade of events is the amplification of Cyclin E1 (CCNE1), which accelerates the transition into synthesis phase (S phase), increases cellular proliferation, and exacerbates replication stress (Aziz et al., 2019).
A major therapeutic discovery in HGSOC has been the introduction of Poly(ADP-ribose) polymerase 1/2 inhibitors (PARPi), which have shown clinical benefit predominantly in patients with defects in DNA damage repair pathways based on the concept of synthetic lethality (Farmer et al., 2005; Konstantinopoulos et al., 2015). More recently, the inhibition of cell cycle–regulating kinases has emerged as an interesting treatment strategy. These agents are currently under investigation and have demonstrated encouraging activity, particularly in a selective group of patients, including CCNE1 amplified tumors (Xu et al., 2024). However, patients with no identified biomarker, such as BRCA mutation, homologous recombination deficiency (HRD) phenotype, or CCNE1 amplification, face a biological challenge with limited therapeutic options, representing a significant unmet need (Wang YW. et al., 2025). This underscores the importance of identifying novel target therapies or rational combination strategies for this population beyond genomic alterations. Efforts to improve clinical outcomes using anti-PD(L)1 therapies—either as monotherapies or in combination with PARPi or chemotherapy—have mainly failed, demonstrating limited efficacy across multiple clinical trials. (Ghisoni et al., 2024). Emerging evidence suggests that modulation of the TME and inhibition of kinases involved in the replicative stress process could enhance therapeutic efficacy (Hardaker et al., 2024a). However, a deeper mechanistic understanding of these interactions is still needed. This review explores the interaction between replicative stress and the TME and summarizes current preclinical and clinical evidence supporting the combination of cell cycle checkpoint inhibitors with anti-PD(L)1 therapy in HGSOC. Our literature review is narrative in nature rather than a systematic review, we included preclinical original research, and clinical trials relevant for the topic, non-english publications or non-peer-reviewed materials were excluded.
2 Intercommunication between replication stress and the immune microenvironment
DNA replication, under normal conditions, occurs in an organized and coordinated manner, ensuring that DNA is replicated only once and is equally distributed to the daughter cells (Sørensen and Syljuåsen, 2012). However, various factors can disrupt this delicate process, leading to replication stress. Some causes of replication stress include the release of reactive oxygen species (ROS), incorrect incorporation of ribonucleotides, alterations in DNA structure, or collisions between the transcription and replication machinery (Zeman and Cimprich, 2014). In response to this stress, a cascade of proteins is activated (Figure 1), starting with the replication protein A (RPA), the initial sensor that binds to single-stranded DNA (ssDNA) at the stalled replication fork and recruits ATR kinase. Subsequently, ATR kinase collaborates with Interacting Protein (ATRIP), activated by Topoisomerase II Binding Protein 1 (TopBP1). Once activated, ATR phosphorylates Checkpoint kinase 1 (Chk1), which induces cell cycle arrest at the S-G2 phase, providing time for DNA repair mechanisms to act, including homologous recombination (HRR) and non-homologous end joining (NHEJ) pathways. In addition, Chk1 regulates the G2-M transition by reducing cyclin-dependent kinase 2 (CDK2), slowing replication in the S phase. Chk1 also phosphorylates and activates WEE1, which negatively regulates cyclin-dependent kinase 1 (CDK1), also known as CDC2, resulting in cell cycle arrest, which is essential for entry into mitosis. WEE1 also stabilizes the replication fork by inhibiting nucleases and preventing DNA degradation (Domínguez-Kelly et al., 2011). Other participants in the DNA damage response (DDR) include BRCA2; Its function is to protect the replication fork from degradation by MRE11 nuclease (Oh and Symington, 2018).
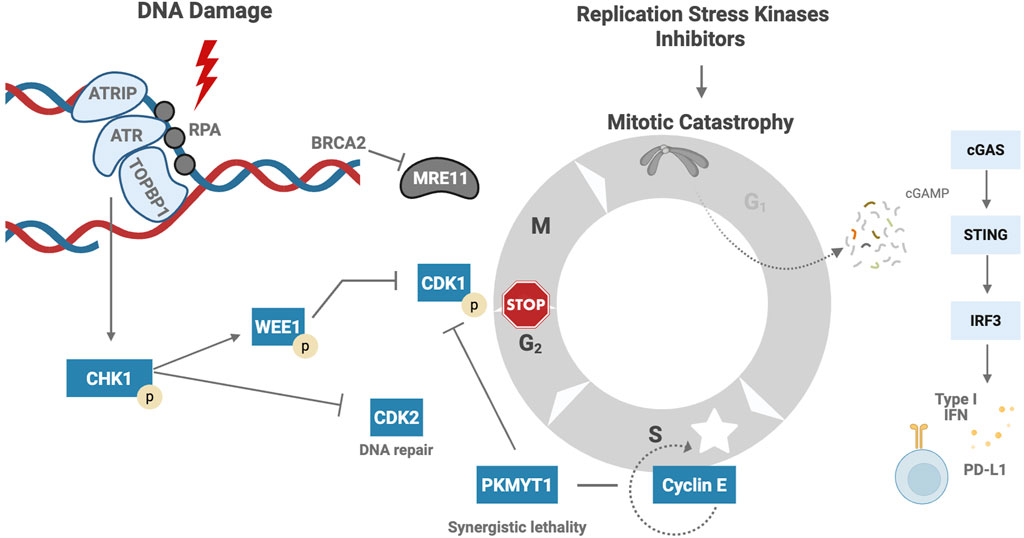
Figure 1. Cellular Response to Replication Stress and Immune Activation. RPA binds to ssDNA at the stalled fork and recruits ATR kinase through its partner ATRIP. ATR activation is facilitated by TopBP1 and leads to Chk1 phosphorylation (Saldivar et al., 2017). Activated Chk1 induces cell cycle arrest at the S-G2 phase by downregulating CDK2 and delaying S-phase progression. Chk1 also activates WEE1, which inhibits CDK1, enforcing G2-M arrest and stabilizing the replication fork by preventing nuclease-mediated degradation (Saxena and Zou, 2022). BRCA2 plays a critical role in protecting stalled forks from degradation by the MRE11 nuclease (Oh and Symington, 2018). In the absence of efficient checkpoint signaling, cells may enter mitosis prematurely, resulting in micronuclei formation. DNA fragments within the cytoplasm activate the cGAS-STING pathway, leading to transcription of type I interferon genes and PDL1 overexpression (Tong et al., 2024; Sato et al., 2017). ssDNA – Single-stranded DNA, ATR – Ataxia telangiectasia and Rad3-related protein, ATRIP – ATR Interacting Protein, TopBP1 – Topoisomerase II Binding Protein 1, Chk1 – Checkpoint kinase 1, CDK2 – Cyclin-dependent kinase 2, S-G2 phase – Synthesis to Gap 2 phase of the cell cycle, WEE1 – WEE1 G2 checkpoint kinase, CDK1 – Cyclin-dependent kinase 1, BRCA2 – Breast Cancer gene 2, MRE11 – Meiotic recombination 11 homolog, cGAS – Cyclic GMP-AMP synthase, STING – Stimulator of interferon genes, PDL1 (or PD-L1) – Programmed death-ligand 1. Created in BioRender. venegas, l. (2025) https://BioRender.com/58mfsso.
In cancer, replication stress is particularly prevalent due to the loss of function of tumor suppressor genes like TP53, RB1, and NF1 (Khamidullina et al., 2024). HGSOC exhibits genomic complexity, and analysis of copy number alterations has identified seven signatures. Signature 1 is associated with breakage–fusion–bridge (BFB) cycles and active RAS signaling; Signature 4 correlates with whole-genome doubling and amplification of CCNE1 and MYC; and Signature 6 is characterized by aberrant G1/S cell cycle checkpoint control (Macintyre et al., 2018). In response to the replication stress induced by DNA damage, the ATR-CHK1-WEE1 axis is crucial for the survival and proliferation of cancer cells (Gaillard et al., 2015). However, in tumor cells, cell cycle regulation is abnormal, and the cell may proceed to mitosis with unrepaired DNA damage, ultimately leading to mitotic catastrophe, cell death, and micronuclei formation (Dobbelstein et al., 2015; Zhang et al., 2022; Kwon et al., 2020). These DNA fragments are released into the cytoplasm, where the cyclic GMP-AMP synthase (cGAS) sensor recognizes the self-derived DNA in the cytosol, leading to the production of cyclic guanosine monophosphate–adenosine monophosphate (cGAMP), a second messenger, which activates stimulator of interferon genes (STING) and triggers the transcription of type 1 interferon-related genes. This pro-inflammatory signal promotes an anti-tumor immune response and upregulates programmed cell death ligand 1 (PD-L1) expression (Tong et al., 2024; Sato et al., 2017).
3 Targeting replication stress in ovarian cancer
Numerous clinical trials have investigated the potential of inhibiting kinases involved in replication stress, such as ATR, Chk1, and WEE1, in HGSOC. However, efficacy was modest as a single agent in not selected patients, with objective response rates (ORR) in platinum-resistant ovarian cancer (PROC), ranging from 5% to 15% and 20%–25% in selected patients with sensitive alterations, such as ataxia telangiectasia (ATM) mutations and CCNE1 amplification. Response rates tend to improve when combined with chemotherapy or PARPi; however, hematologic toxicity remains a major limitation (Supplementary Table S1) (Yap et al., 2024; Tan et al., 2022; Yap et al., 2023; Shah et al., 2021; Simpkins et al., 2024; Mahdi et al., 2021; Konstantinopoulos et al., 2024; Konstantinopoulos et al., 2022; Giudice et al., 2024a; Kristeleit et al., 2023; Jones et al., 2023; Miller et al., 2022; Fu et al., 2023; Westin et al., 2021; Lheureux et al., 2021; Moore et al., 2022; Leijen et al., 2016; Au-Yeung et al., 2022; Oza et al., 2020; Liu et al., 2023; Gelderblom et al., 2023; Schram et al., 2025).
4 PARP inhibition and immune regulation
The interaction between PARP1/2 inhibition and the cGAS-STING pathway has driven clinical trials investigating the use of PARPi and anti-PD-(L)1 therapies in HGSOC (Ghisoni et al., 2024). PARP inhibition leads to the accumulation of cytosolic DNA, which is recognized by cGAS. This recognition activates the STING pathway in the endoplasmic reticulum. Upon activation, STING recruits TANK-binding kinase 1 (TBK1), which activates transcription factors such as interferon regulatory factor 3 (IRF3) and nuclear factor kappa B (NF-κB). These factors translocate to the nucleus and induce the expression of genes involved in modulating the immune response (Figure 2) (Zhu et al., 2021; Dunphy et al., 2018; Shen et al.).
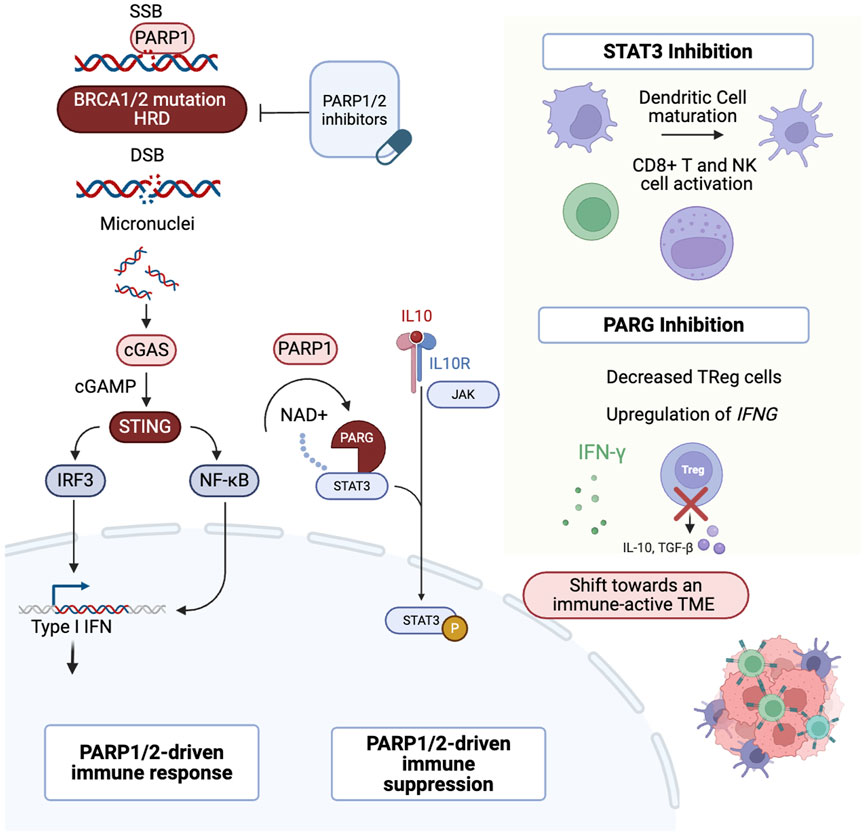
Figure 2. Multifaceted role of PARP1. After DNA damage, PARP1 binds to SSBs. In cells with HRD, PARP1/2 inhibition leads to DSBs. DNA fragments are then released into the cytoplasm and recognized by the cGAS sensor, activating the cGAS–STING pathway. This triggers IRF3 translocation to the nucleus and activates the IFN response. In addition, PARP1 modulates STAT3 through PARylation, promoting an immunosuppressive TME. Inhibition of STAT3 or PARG can shift the TME towards an immune-active state (Zhu et al., 2021; Dunphy et al., 2018; Shen et al.; Yue et al., 2012; Z et al., 2025; Martincuks et al., 2021a; Yu et al., 2007; Ding et al., 2019; Houl et al., 2019; Martincuks et al., 2024). PARP1/2: Poly (ADP-ribose) polymerase 1 and 2, SSBs: Single-Strand Breaks, HRD: Homologous Recombination Deficiency, DSBs: Double-Strand Breaks, cGAS: cyclic GMP-AMP synthase, STING: Stimulator of Interferon Genes, IRF3: Interferon Regulatory Factor 3, IFN: Interferon, STAT3: Signal Transducer and Activator of Transcription 3, TME: Tumor Microenvironment, PARG: Poly (ADP-ribose) glycohydrolase. Created in BioRender. venegas, l. (2025) https://BioRender.com/p3z3594.
Additional mechanisms of interaction of PARP inhibition with the TME have been studied in preclinical models. However, further clinical validation is needed. PARP1/2 inhibition activates signal transducer and activator of transcription 3 (STAT3), a key factor implicated in immune evasion and treatment resistance (Yue et al., 2012; Z et al., 2025), by inhibiting TH1-type immune responses and promoting the overexpression of IL-6, IL-10, and VEGF, which contributes to an immunosuppressive TME (Martincuks et al., 2021a). Poly (ADP-ribose) glycohydrolase (PARG) is an enzyme that counteracts PARP by reversing the PARylation process (Houl et al., 2019). PARG inhibition decreased pSTAT3 levels in vitro and promoted antitumor immunity in vivo by increasing interferon-gamma expression, activating CD8+ T cells, and reducing the population of regulatory T cells (Martincuks et al., 2024). Despite the preclinical rationale, this has not yet been translated into the clinic. One limitation is the lack of models that accurately replicate the dynamic interactions between the tumor and the immune microenvironment, reflecting the evolving genomic and immune landscape (Stur et al., 2025).
Clinical trial results combining anti-PD(L)1 and PARP1/2i are inconsistent, and to date, none of these combinations have been approved for clinical practice in HGSOC (Ghisoni et al., 2024). While the triple combination of durvalumab, olaparib, and cediranib did not improve progression-free survival (PFS) (Lee et al., 2025), the combination of olaparib, durvalumab, and bevacizumab demonstrated encouraging results in patients without BRCA mutations (Drew et al., 2024). Therefore, the prolonged response observed in some patients warrants further investigation to better understand the interactions between the TME and the DNA repair pathways (Fu et al., 2023).
Resistance to PARP1/2 inhibitors is frequent (Soberanis P et al., 2023), and preclinical studies have shown that these inhibitors can interact differently with replication stress (Shih et al., 2024). Replication stress kinase inhibition have been explored in clinical trials, either as monotherapy or in combination with PARP1/2i, as a strategy to overcome resistance; however, modest response rates emphasize the need for novel therapeutic combinations (Stur et al., 2025). The contribution of tumor-extrinsic factors, particularly the role of TME, to acquired therapeutic resistance represents an important area of investigation.
5 Modulating immune response through replication stress kinase inhibition
Preclinical evidence has shown that inhibiting kinases involved in replication stress can modulate the immune response (Figure 3) (Taniguchi et al., 2024).
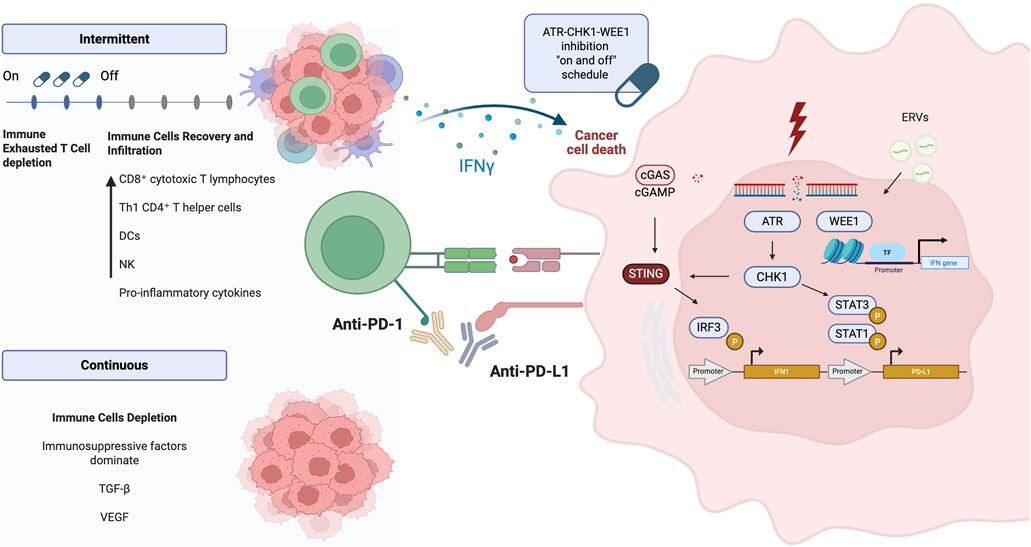
Figure 3. ATR-CHK1-WEE1 pathway and immune interaction. DSBs activate the ATR–Chk1 pathway, which subsequently phosphorylates STAT1 and STAT3, leading to the overexpression of PD-L1. In parallel, the cGAS–STING pathway promotes the transcription of type I interferon genes in response to cytosolic DNA. WEE1 kinase modulates interferon gene expression through recognition of ERVs and regulation of chromatin. An intermittent dosing schedule of ATR, Chk1, and WEE1 inhibitors allows immune cell recovery, enhances immune cell infiltration, and promotes activation of anti-tumor immune responses (Sato et al., 2017; Carlsen and El-Deiry, 2022; Iwai et al., 2017). DNA: Deoxyribonucleic Acid, DSBs: Double-Strand Breaks, ATR: Ataxia Telangiectasia and Rad3-related protein, Chk1: Checkpoint kinase 1, STAT1: Signal Transducer and Activator of Transcription 1, STAT3: Signal Transducer and Activator of Transcription 3, PD-L1: Programmed Death-Ligand 1, cGAS: Cyclic GMP-AMP Synthase, STING: Stimulator of Interferon Genes, WEE1: WEE1 G2 Checkpoint Kinase, ERVs: Endogenous Retroviral Elements. Created in BioRender. venegas, l. (2025) https://BioRender.com/kedtvdt.
In an in vivo colorectal cancer mice model, the ATR inhibitor M6620 (VX-970), when combined with cisplatin, carboplatin, or irinotecan and the anti–PD-L1 antibody avelumab, demonstrated significant anti-tumor activity; similarly, in the MB49 urothelial tumor model, the combination of carboplatin and avelumab also exhibited therapeutic efficacy (Alimzhanov et al., 2020).
An intermittent schedule, in a colorectal cancer mice model, ceralasertib 7 days on, 7 days off, combined with the anti-PD-L1 antibody durvalumab, significantly improved survival through a CD8+ T-cell-dependent mechanism. The intermittent schedule led to superior tumor control compared to continuous treatment. CyTOF and scRNAseq analysis of the TME revealed that ceralasertib reshapes the TME by decreasing the exhausted CD8+ T-cell phenotype and reducing monocytic myeloid derived suppressor cells (M-MDSCs) and tumor-associated macrophages (TAMs). Additionally, ceralasertib increased the presence of CD11c+ MHC II + dendritic cells (DCs). While low-dose ceralasertib showed minimal or no anti-tumor effect in vitro or in vivo when used alone, its combination with PD-L1 blockade resulted in significant anti-tumor activity (Hardaker et al., 2024b).
The Chk1 inhibitor prexasertib (12 mg/kg, BID, 2/7 days) elicited a immune-mediated anti-tumor response in both in vitro and in vivo in Small Cell Lung Cancer (SCLC) models. Treatment with prexasertib induced dynamic remodeling of the TME, characterized by increased infiltration of CD3+ total T cells and CD8+ cytotoxic T cells, and reduction in exhausted T cells by day 7. When combined with anti–PD-L1 therapy (300 μg, administered once weekly on day 3), prexasertib significantly enhanced therapeutic efficacy. Mechanistically, this immune activation was associated with activation of the cGAS–STING–TBK1–IRF3 signaling axis, leading to induction of type I interferon responses, upregulation of PD-L1 expression, and CXCL10 and CCL5 cytokines (Sen et al., 2019a).
The combination of the Chk1 inhibitor SRA737, anti-PD-L1, and low-dose gemcitabine (LDG) was assessed in a SCLC model. While no significant anti-tumor activity was observed with any of the single-agent treatments, the combination led to substantial tumor regressions. Flow cytometry analysis demonstrated a significant increase in CD3+ and CD8+ T-cell infiltration compared to vehicle or single-agent treatments, and a reduction in CD4+ helper T-cells, regulatory T-cells, and exhausted CD8+ T-cells. The combination therapy increased M1 macrophage populations and DCs, while decreasing M2 macrophages and MDSCs (Sen et al., 2019b).
In ovarian cancer cell lines, the WEE1 inhibitor AZD1775 modulates the immune response by inducing expression of endogenous retroviral elements (ERVs), which produces double-stranded RNA (dsRNA), activating IFN-mediated anti-tumor signaling and upregulating PD-L1. This effect was driven by downregulation of the histone H3K9me3. In vivo, STING-deficient ID8 ovarian cancer mice model, AZD1775 (5 days on, 2 days off) combined with anti–PD-L1 antibody significantly enhanced anti-tumor efficacy (Guo et al., 2022).
In a phase I clinical trial involving patients with advanced solid tumors, Prexasertib, in combination with the PD-L1 inhibitor LY3300054, exploratory analysis of immune cell samples collected before and after treatment revealed significant increases in activated CD8+ T cells and natural killer T cells following treatment. Of the 17 patients enrolled, 10 had high-grade serous cancer. The majority of patients exhibited notable signs of T-cell activation (Do et al., 2021).
In a Phase II clinical trial of prexasertib monotherapy in BRCAwt, platinum-resistant HGSOC, exploratory analysis of immune cell subsets revealed that patients with non-clinical benefit exhibited an increase in M-MDSCs, while patients with clinical benefit showed decreased expression of immune suppressive marker TIM-3 on CD8+ Tregs (Giudice et al., 2024b).
These findings are primarily based on non-ovarian models across various solid tumors, where the TME differs from that of HGSOC. Clinical evidence is limited, as the interaction between CHK1 inhibition and TME modulation is derived from a single phase 1 clinical trial. These results require further validation through dedicated models in ovarian cancer.
6 Inhibition of replication stress kinases and anti-PD(L)1
Most clinical trials investigating PD-(L)1 inhibitors combined with ATR, WEE1, or Chk1 inhibitors have been performed in non-ovarian cancers, which have different TME (Kim et al., 2022; Besse et al., 2024; Kwon et al., 2022; Brond et al., 2021). These studies have focused on tumor types with established sensitivity to immune checkpoint blockade, such as melanoma and small cell lung cancer. Supplementary Table S2 summarizes ongoing clinical trials evaluating the combination of ATR inhibitors and immune checkpoint inhibitors across multiple tumor types, many of which are still actively enrolling participants, highlighting sustained interest in this therapeutic strategy.
To date, results have been reported from four trials (Table 1). Two of these specifically investigated the potential of ATR inhibitors to overcome resistance to ICB, and one study included patients with HGSOC (Kim et al., 2022; Besse et al., 2024; Kwon et al., 2022; Brond et al., 2021).
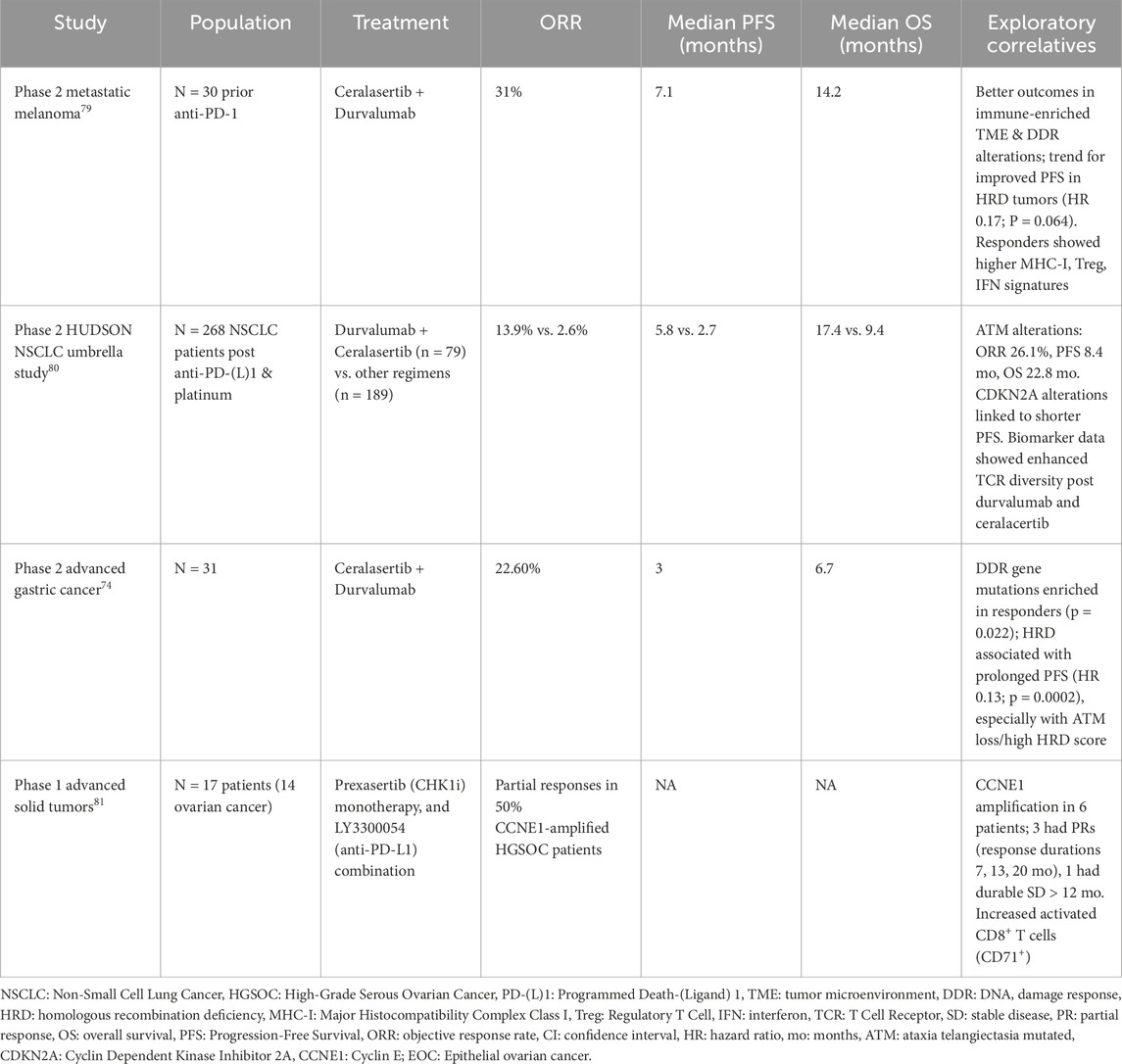
Table 1. Reported clinical trials investigating ATR or Chk1 inhibition and anti-PDL1 therapy in solid tumors.
In a Phase II study of 30 patients with metastatic melanoma who had progressed on prior anti–PD-1 therapy, the combination of ceralasertib and durvalumab demonstrated an ORR of 31% (95% CI, 13.6%–46.4%), a median PFS of 7.1 months (range, 3.8–11.7) and a median overall survival (OS) of 14.2 months (95% CI, 9.3–19.1); 44.4% patients with primary resistance achieved a response. Exploratory biomarker analyses demonstrated that patients with an immune-enriched TME or alterations in the DDR pathway derived the greatest benefit. Responders exhibited a higher expression of major histocompatibility complex class I (MHC-I) and interferon-related gene signatures (Kim et al., 2022). Similarly, in the phase 2 HUDSON umbrella study in patients with NSCLC post anti-PD-L1 and platinum-doublet therapy, durvalumab–ceralasertib combination demonstrated superior efficacy compared to other regimens; responses were particularly pronounced in patients with ATM alterations, correlative biomarker analyses revealed downregulation of monocyte, CD8+ T cell, and exhaustion-associated gene signatures along with upregulation of TNF-α, interferon-γ, and interferon-α pathways (Besse et al., 2024). Based on these findings, LATIFY (NCT05450692) is an ongoing phase III, open-label, randomized, multicenter trial evaluating the efficacy and safety of ceralasertib plus durvalumab versus docetaxel in patients with locally advanced or metastatic NSCLC who have progressed after anti–PD-(L)1 therapy and a platinum-based doublet.
In a phase II study, patients with advanced gastric cancer treated with ceralasertib and durvalumab demonstrated an ORR of 22.6%, mPFS of 3.0 months, and mOS of 6.7 months; only 6.5% have received prior anti-PD1 therapy. Whole-exome sequencing of pretreatment tumor biopsies revealed enrichment of mutations in DDR pathway genes among patients who achieved partial responses, and HRD was associated with prolonged PFS. Correlative analyses showed that responders exhibited an increase in intratumoral lymphocyte infiltration and expansion of circulating tumor-reactive CD8+ T-cell clones. In contrast, treatment resistance was associated with enriched tumor vasculature signatures and decreased T-cell receptor (TCR) clonality (Kwon et al., 2022).
Prexasertib, a Chk1 inhibitor, and anti-PD-L1 LY3300054 were evaluated in a Phase I study, anti-PD-L1 monotherapy, or combination. The study included 14 patients with recurrent ovarian cancer. The most common histology was HGSOC. CCNE1 amplification was present in six patients, 50% achieved PR (Do et al., 2021).
7 Discussion
We summarize how the ATR-CHK1-WEE1 signaling axis is critical for maintaining genomic stability and how cancer cells often rely on this pathway for survival. Therefore, the development of drugs targeting these cell cycle checkpoint kinases is of interest and has shown some encouraging results in cancer treatment. While the molecular mechanisms of this pathway are well understood, its connection to the TME remains poorly characterized. Emerging evidence suggests that modulation of the immune response through inhibition of these kinases, particularly via the cGAS-STING pathway and STAT1/STAT3 transcription factors, which activate a type I interferon response and upregulate PD-L1, contributes to anti-tumor immunity (Sato et al., 2017; Taniguchi et al., 2024). However, other players, such as PARG and epigenetic regulators (Martincuks et al., 2024), may also be involved.
Chromosomal instability in HGSOC arises from cumulative alterations in cell cycle regulators, rather than from a single genetic alteration or mutation, which accumulates over time (Brond et al., 2021). Supporting this, retrospective genomic analysis of tumor samples from patients with stage I–II versus stage III–IV HGSOC revealed a higher frequency of whole-genome duplication in late-stage tumors compared to early-stage tumors (Cheng et al., 2022). Interestingly, copy number signatures appeared largely stable over time, from initial diagnosis through relapse or progression. These findings raise important questions about whether the TME differs in these patients. For example, patients with primary platinum resistance exhibited higher rates of CCNE1 and KRAS amplification at diagnosis, along with increased exposure to copy number signature 1 that is linked to a type of DNA instability known as breakage-fusion-bridge, which was negatively correlated with CD3 and CD8 expression (Smith et al., 2023).
This review highlights that targeting the replication stress response may induce a favorable shift in the TME. Serial tumor biopsies and paired peripheral blood mononuclear cell (PBMC) sampling can capture temporal tumor heterogeneity. To address this gap, patient-derived organoid cultures may serve as functional assays and facilitate the study of tumor–TME interactions. In preclinical models, fiber assays in organoids have been used to assess replication fork instability and predict sensitivity to prexasertib (a CHEK1 inhibitor) and VE-822 (an ATR inhibitor) (Hill et al., 2018); however, their reproducibility in clinical settings remains limited. Future studies should aim to develop tools capable of simultaneously evaluating replication stress and immune modulation in FFPE tissue or plasma.
The TME in HGSOC is particularly complex and unique; the peritoneal cavity provides a permissive niche for tumor dissemination through intricate interactions between metastatic tumor cells and TME components (Tan et al., 2006). Key cellular contributors include TAMs, cancer-associated adipocytes (CAAs), cancer-associated fibroblasts (CAFs), and cancer-associated mesothelial cells (CAMs), all of which play roles in promoting immune evasion (Tan et al., 2006). Additionally, a recently identified HGSOC subtype—C2 IGF2+ tumors—has been shown to engage fibroblasts via paracrine signaling, facilitating their transition into CAFs. This subtype is associated with stromal remodeling, genomic instability, stem-like features, and more advanced disease (Zhao et al., 2025).
A key area of investigation is how the TME may change in response to PARP or replication stress kinase inhibition, and the development of secondary resistance, and whether those changes promote immunosuppression through mechanisms such as senescence and activation of the STAT3 pathway, which increases expression of VEGF (Kamii et al., 2025; Zh et al., 2025; Martincuks et al., 2021b; Sumimoto et al., 2006). In addition to the immune microenvironment, angiogenesis is critical for tumor survival in hypoxic conditions, as high levels of VEGF promote the formation of abnormal vasculature that delivers oxygen and nutrients to cancer cells (Zhou et al., 2024). This pro-angiogenic TME has been associated with resistance to combinations of ATR inhibitors and anti–PD-L1 therapies (Kwon et al., 2022). Notably, triple therapy combining PARP inhibition, anti–PD-(L)1, and antiangiogenic agents has demonstrated clinical benefit in some clinical trials (Lee et al., 2025; Drew et al., 2024). However, whether this strategy can be extended to combinations involving ATR, CHK1, or WEE1 inhibitors remains unexplored.
The conventional on-and-off administration of replication stress kinase inhibitors may represent an interesting strategy to modulate the TME. Intermittent dosing enables active T cells to exert an anti-tumor response during the ‘off’ days, while selectively depleting exhausted T cells during the ‘on’ days; this approach could sensitize the cell to immunotherapies (Hardaker et al., 2024b).
Predictive biomarkers of response to replication stress kinase inhibitors and anti-PD-(L)1 therapies remain limited, in part due to the heterogeneity in HGSOC (Stur et al., 2025; Parvathareddy et al., 2021). However, studies have suggested that tumor immune infiltration and the expansion of CD8+ T cells may be associated with response to the combination of ceralasertib and durvalumab (Hardaker et al., 2024b). Interestingly, WEE1 inhibition has been shown to induce the recognition of endogenous retroviral RNA, leading to activation of interferon-stimulated genes (Brond et al., 2021). The presence of endogenous retrotransposable elements has been identified as a predictive biomarker of response to ICB in melanoma and non-small cell lung cancer (Herrera et al., 2025).
Our review aims to generate hypotheses and stimulate future research in HGSOC before immediate clinical application, given the initial disappointment of PD-1/PDL-1 in this disease. The dual-targeting approach focusing on replication stress response inhibition and anti–PD-(L)1 therapy—is based on mechanistic rationale and supported by emerging early-phase clinical trials in other tumor types. We acknowledge the limited availability of preclinical and clinical data specific to HGSOC and emphasize the need for the development of more representative preclinical models and clinical trial designs capable of capturing the dynamic changes in the tumor microenvironment, which could lead to the development of more effective treatment strategies.
Author contributions
LV: Writing – original draft, Writing – review and editing. SL: Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2025.1638964/full#supplementary-material
References
Alimzhanov, M., George, A., Shah, P., Zimmerman, A., Schroeder, A., Falk, M., et al. (2020). Abstract 964: induction of immunogenic cell death and interferon signaling by carboplatin and the ATR inhibitor M6620 may contribute to anti-tumor activity of M6620-carboplatin-avelumab triplet combination in MC38 tumor model. Cancer Res. 80 (16 Suppl. l), 964. doi:10.1158/1538-7445.am2020-964
Au-Yeung, G., Bressel, M., Prall, O., Surace, D., Andrews, J., Mongta, S., et al. (2022). IGNITE: a phase II signal-seeking trial of adavosertib targeting recurrent high-grade, serous ovarian cancer with cyclin E1 overexpression with and without gene amplification. J. Clin. Oncol. 40 (16_Suppl. l), 5515. doi:10.1200/JCO.2022.40.16_suppl.5515
Aziz, K., Limzerwala, J. F., Sturmlechner, I., Hurley, E., Zhang, C., Jeganathan, K. B., et al. (2019). Ccne1 overexpression causes chromosome instability in liver cells and liver tumor development in mice. Gastroenterology 157, 210–226. doi:10.1053/j.gastro.2019.03.016
Baslan, T., Morris, J. P., Zhao, Z., Reyes, J., Ho, Y. J., Tsanov, K. M., et al. (2022). Ordered and deterministic cancer genome evolution after p53 loss. Nature 608 (7924), 795–802. doi:10.1038/s41586-022-05082-5
Besse, B., Pons-Tostivint, E., Park, K., Hartl, S., Forde, P. M., Hochmair, M. J., et al. (2024). Biomarker-directed targeted therapy plus durvalumab in advanced non-small-cell lung cancer: a phase 2 umbrella trial. Nat. Med. 30 (3), 716–729. doi:10.1038/s41591-024-02808-y
Bronder, D., Tighe, A., Wangsa, D., Zong, D., Meyer, T. J., Wardenaar, R., et al. (2021). TP53 loss initiates chromosomal instability in fallopian tube epithelial cells. Dis. Model Mech. 14 (11), dmm049001. doi:10.1242/dmm.04901
Bronder, D., Tighe, A., Wangsa, D., Zong, D., Meyer, T. J., Wardenaar, R., et al. (2021). TP53 loss initiates chromosomal instability in fallopian tube epithelial cells. Dis. Model Mech. 14 (11), dmm049001. doi:10.1242/dmm.049001
Cancer Genome Atlas Research Network (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474 (7353), 609–615. doi:10.1038/nature10166
Carlsen, L., and El-Deiry, W. S. (2022). Anti-cancer immune responses to DNA damage response inhibitors: molecular mechanisms and progress toward clinical translation. Front. Oncol. 12, 998388. doi:10.3389/fonc.2022.998388
Cheng, Z., Mirza, H., Ennis, D. P., Smith, P., Morrill Gavarró, L., Sokota, C., et al. (2022). The genomic landscape of early-stage ovarian high-grade serous carcinoma. Clin. Cancer Res. 28 (13), 2911–2922. doi:10.1158/1078-0432.CCR-21-1643
Ding, L., Chen, X., Xu, X., Qian, Y., Liang, G., Yao, F., et al. (2019). PARP1 suppresses the transcription of PD-L1 by poly(ADP-ribosyl)ating STAT3. Cancer Immunol. Res. 7 (1), 136–149. doi:10.1158/2326-6066.CIR-18-0071
Do, K. T., Manuszak, C., Thrash, E., Giobbie-Hurder, A., Hu, J., Kelland, S., et al. (2021). Immune modulating activity of the CHK1 inhibitor prexasertib and anti-PD-L1 antibody LY3300054 in patients with high-grade serous ovarian cancer and other solid tumors. Cancer Immunol. Immunother. 70 (10), 2991–3000. doi:10.1007/s00262-021-02910-x
Dobbelstein, M., and Sørensen, C. S. (2015). Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 14 (6), 405–423. doi:10.1038/nrd4553
Domínguez-Kelly, R., Martín, Y., Koundrioukoff, S., Tanenbaum, M. E., Smits, V. A. J., Medema, R. H., et al. (2011). Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J. Cell Biol. 194 (4), 567–579. doi:10.1083/jcb.201101047
Drew, Y., Kim, J. W., Penson, R. T., O'Malley, D. M., Parkinson, C., Roxburgh, P., et al. (2024). Olaparib plus durvalumab, with or without bevacizumab, as treatment in PARP inhibitor-naïve platinum-sensitive relapsed ovarian cancer: a phase II multi-cohort study. Clin. Cancer Res. 30 (1), 50–62. doi:10.1158/1078-0432.CCR-23-2249
Dunphy, G., Flannery, S. M., Almine, J. F., Connolly, D. J., Paulus, C., Jønsson, K. L., et al. (2018). Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol. Cell 71 (6), 745–760. doi:10.1016/j.molcel.2018.07.034
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N. J., Johnson, D. A., Richardson, T. B., et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. doi:10.1038/nature03445
Fu, S., Yao, S., Yuan, Y., Previs, R. A., Elias, A. D., Carvajal, R. D., et al. (2023). Multicenter phase II trial of the WEE1 inhibitor adavosertib in refractory solid tumors harboring CCNE1 amplification. J. Clin. Oncol. 41 (9), 1725–1734. doi:10.1200/JCO.22.00830
Gaillard, H., García-Muse, T., and Aguilera, A. (2015). Replication stress and cancer. Nat. Rev. Cancer 15 (5), 276–289. doi:10.1038/nrc3916
Gelderblom, H., Jalving, M., Desar, I., Saavedra, O., Gietema, J. A., van Ravensteijn, S., et al. (2023). Debio 0123-101: a phase 1 trial of Debio 0123 in combination with carboplatin in advanced solid tumors—safety, pharmacokinetic, and preliminary antitumor activity data. J. Clin. Oncol. 41 (16_Suppl. l), 3012. doi:10.1200/JCO.2023.41.16_suppl.3012
Ghisoni, E., Morotti, M., Sarivalasis, A., Grimm, A. J., Kandalaft, L., Laniti, D. D., et al. (2024). Immunotherapy for ovarian cancer: towards a tailored immunophenotype-based approach. Nat. Rev. Clin. Oncol. 21 (11), 801–817. doi:10.1038/s41571-024-00937-4
Giudice, E., Huang, T. T., Nair, J. R., Zurcher, G., McCoy, A., Nousome, D., et al. (2024a). The CHK1 inhibitor prexasertib in BRCA wild-type platinum-resistant recurrent high-grade serous ovarian carcinoma: a phase 2 trial. Nat. Commun. 15 (1), 2805. doi:10.1038/s41467-04-47215-6
Giudice, E., Huang, T. T., Nair, J. R., Zurcher, G., McCoy, A., Nousome, D., et al. (2024b). The CHK1 inhibitor prexasertib in BRCA wild-type platinum-resistant recurrent high-grade serous ovarian carcinoma: a phase 2 trial. Nat. Commun. 15 (1), 2805. doi:10.1038/s41467-024-47215-6
Guo, E., Xiao, R., Wu, Y., Lu, F., Liu, C., Yang, B., et al. (2022). WEE1 inhibition induces anti-tumor immunity by activating ERV and the dsRNA pathway. J. Exp. Med. 219 (1), e20210789. doi:10.1084/jem.20210789
Hardaker, E. L., Sanseviero, E., Karmokar, A., Taylor, D., Milo, M., Michaloglou, C., et al. (2024a). The ATR inhibitor ceralasertib potentiates cancer checkpoint immunotherapy by regulating the tumor microenvironment. Nat. Commun. 15 (1), 1700.
Hardaker, E. L., Sanseviero, E., Karmokar, A., Taylor, D., Milo, M., Michaloglou, C., et al. (2024b). The ATR inhibitor ceralasertib potentiates cancer checkpoint immunotherapy by regulating the tumor microenvironment. Nat. Commun. 15 (1), 1700. doi:10.1038/s41467-024-45996-4
Herrera, M., Marhon, S. A., Abbas-Aghababazadeh, F., Chen, D., Liu, A., LooYau, H., et al. (2025). Abstract 2060: independent validation of endogenous retrotransposable elements as predictive biomarkers of immune checkpoint blockade response. Cancer Res. 85 (8_Suppl. ment_1), 2060. doi:10.1158/1538-7445.AM2025-2060
Hill, S. J., Decker, B., Roberts, E. A., Horowitz, N. S., Muto, M. G., Worley, M. J., et al. (2018). Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discov. 8 (11), 1404–1421. doi:10.1158/2159-8290.CD-18-0474
Hillmann, J., Maass, N., Bauerschlag, D. O., and Flörkemeier, I. (2025). Promising new drugs and therapeutic approaches for treatment of ovarian cancer—targeting the hallmarks of cancer. BMC Med. 23 (1), 10. doi:10.1186/s12916-024-03826-w
Houl, J. H., Ye, Z., Brosey, C. A., Balapiti-Modarage, L. P. F., Namjoshi, S., Bacolla, A., et al. (2019). Selective small molecule PARG inhibitor causes replication fork stalling and cancer cell death. Nat. Commun. 10, 5654. doi:10.1038/s41467-019-13508-4
Huang, J., Chan, W. C., Ngai, C. H., Lok, V., Zhang, L., Lucero-Prisno, D. E., et al. (2022). Worldwide burden, risk factors, and temporal trends of ovarian cancer: a global study. Cancers (Basel) 14 (9), 2230. doi:10.3390/cancers14092230
Ishak, C. A., Marhon, S. A., Tchrakian, N., Hodgson, A., Loo Yau, H., Gonzaga, I. M., et al. (2025). Chronic viral mimicry induction following p53 loss promotes immune evasion. Cancer Discov. 15 (4), 793–817. doi:10.1158/2159-8290.CD-24-0094
Iwai, Y., Hamanishi, J., Chamoto, K., and Honjo, T. (2017). Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 24 (1), 26. doi:10.1186/s12929-017-0329-9
Jones, R., Plummer, R., Moreno, V., Carter, L., Roda, D., Garralda, E., et al. (2023). A phase I/II trial of oral SRA737 (a Chk1 Inhibitor) given in combination with low-dose gemcitabine in patients with advanced cancer. Clin. Cancer Res. 29, 331–340. doi:10.1158/1078-0432.CCR-22-2074
Kader, T., Lin, J. R., Hug, C. B., Coy, S., Chen, Y. A., de Bruijn, I., et al. (2024). Multimodal spatial profiling reveals immune suppression and microenvironment remodeling in fallopian tube precursors to high-grade serous ovarian carcinoma. Cancer Discov. 20, 1180–1202. doi:10.1158/2159-8290.CD-24-1366
Kamii, M., Kamata, R., Saito, H., Yamamoto, G., Mashima, C., Yamauchi, T., et al. (2025). PARP inhibitors elicit a cellular senescence mediated inflammatory response in homologous recombination proficient cancer cells. Sci. Rep. 15 (1), 15458. doi:10.1038/s41598-025-00336-4
Khamidullina, A. I., Abramenko, Y. E., Bruter, A. V., and Tatarskiy, V. V. (2024). Key proteins of replication stress response and cell cycle control as cancer therapy targets. Int. J. Mol. Sci. 25 (2), 1263. doi:10.3390/ijms25021263
Kim, R., Kwon, M., An, M., Kim, S. T., Smith, S. A., Loembé, A. B., et al. (2022). Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced/metastatic melanoma who have failed prior anti-PD-1 therapy. Ann. Oncol. 33 (2), 193–203. doi:10.1016/j.annonc.2021.10.009
Konstantinopoulos, P. A., Ceccaldi, R., Shapiro, G. I., and D’Andrea, A. D. (2015). Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 5 (11), 1137–1154. doi:10.1158/2159-8290.CD-15-0714
Konstantinopoulos, P. A., Lee, J. M., Gao, B., Miller, R., Lee, J. Y., Colombo, N., et al. (2022). A Phase 2 study of prexasertib (LY2606368) in platinum resistant or refractory recurrent ovarian cancer. Gynecol. Oncol. 167 (2), 213–225. doi:10.1016/j.ygyno.2022.09.019
Konstantinopoulos, P. A., Cheng, S. C., Lee, E. K., da Costa, AABA, Gulhan, D., Wahner Hendrickson, A. E., et al. (2024). Randomized Phase II Study of Gemcitabine with or without ATR inhibitor Berzosertib in platinum-resistant ovarian cancer: final overall survival and biomarker analyses. JCO Precis. Oncol. 8, e2300635. doi:10.1200/PO.23.00635
Kristeleit, R., Plummer, R., Jones, R., Carter, L., Blagden, S., Sarker, D., et al. (2023). A Phase 1/2 trial of SRA737 (a Chk1 inhibitor) administered orally in patients with advanced cancer. Br. J. Cancer 129 (1), 38–45. doi:10.1038/s41416-023-02279-x
Kwon, M., Leibowitz, M. L., and Lee, J. H. (2020). Small but mighty: the causes and consequences of micronucleus rupture. Exp. Mol. Med. 52 (11), 1777–1786. doi:10.1038/s12276-020-00529-z
Kwon, M., Kim, G., Kim, R., Kim, K. T., Kim, S. T., Smith, S., et al. (2022). Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced gastric cancer. J. Immunother. Cancer 10 (7), e005041. doi:10.1136/jitc-2022-005041
Lee, J. M., Miller, A., Rose, P. G., AlHilli, M., Washington, C., John, V. S., et al. (2025). Comparing durvalumab, olaparib, and cediranib monotherapy, combination therapy, or chemotherapy in patients with platinum-resistant ovarian cancer with prior bevacizumab: the phase II NRG-GY023 trial. Clin. Cancer Res. 7, 2370–2378. doi:10.1158/1078-0432.CCR-24-3877
Leijen, S., van Geel, R. M., Sonke, G. S., de Jong, D., Rosenberg, E. H., Marchetti, S., et al. (2016). Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patients with TP53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J. Clin. Oncol. 34 (39), 4354–4361. doi:10.1200/JCO.2016.67.5942
Lheureux, S., Cristea, M. C., Bruce, J. P., Garg, S., Cabanero, M., Mantia-Smaldone, G., et al. (2021). Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 397 (10271), 281–292. doi:10.1016/S0140-6736(20)32554-X
Li, T., and Chen, Z. J. (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 215 (5), 1287–1299. doi:10.1084/jem.20180139
Liu, J. F., Fu, S., Richardson, G. E., Vranjes, Z., Meniawy, T., Shannon, C. M., et al. (2023). Correlation of cyclin E1 expression and clinical outcomes in a phase 1b dose-escalation study of azenosertib (ZN-c3), a WEE1 inhibitor, in combination with chemotherapy (CT) in patients (pts) with platinum-resistant or refractory (R/R) epithelial ovarian, peritoneal, or fallopian tube cancer (EOC). J. Clin. Oncol. 41 (16_Suppl. l), 5513. doi:10.1200/JCO.2023.41.16_suppl.5513
Macintyre, G., Goranova, T. E., De Silva, D., Ennis, D., Piskorz, A. M., Eldridge, M., et al. (2018). Copy number signatures and mutational processes in ovarian carcinoma. Nat. Genet. 50 (9), 1262–1270. doi:10.1038/s41588-018-0179-8
Mahdi, H., Hafez, N., Doroshow, D., Sohal, D., Keedy, V., Do, K. T., et al. (2021). Ceralasertib-mediated ATR inhibition combined with olaparib in advanced cancers harboring DNA damage response and repair alterations (Olaparib combinations). JCO Precis. Oncol. 5, 1432–1442. doi:10.1200/PO.20.00439
Martincuks, A., Song, J., Kohut, A., Zhang, C., Li, Y. J., Zhao, Q., et al. (2021a). PARP inhibition activates STAT3 in both tumor and immune cells underlying therapy resistance and immunosuppression in ovarian cancer. Front. Oncol. 11, 724104. doi:10.3389/fonc.2021.74104
Martincuks, A., Song, J., Kohut, A., Zhang, C., Li, Y. J., Zhao, Q., et al. (2021b). PARP inhibition activates STAT3 in both tumor and immune cells underlying therapy resistance and immunosuppression in ovarian cancer. Front. Oncol. 11, 724104. doi:10.3389/fonc.2021.724104
Martincuks, A., Zhang, C., Austria, T., Li, Y. J., Huang, R., Lugo Santiago, N., et al. (2024). Targeting PARG induces tumor cell growth inhibition and antitumor immune response by reducing phosphorylated STAT3 in ovarian cancer. J. Immunother. Cancer 12 (4), e007716. doi:10.1136/jitc-2023-007716
Miller, W. H., Shields, A., Provencher, D., Gilbert, L., Shapiro, G., Oza, A., et al. (2022). 537P A phase I/II study of oral chk1 inhibitor LY2880070 in combination with low-dose gemcitabine in patients with advanced or metastatic ovarian cancer. Ann. Oncol. 33, S793–S794. doi:10.1016/j.annonc.2022.07.665
Moore, K. N., Chambers, S. K., Hamilton, E. P., Chen, L. M., Oza, A. M., Ghamande, S. A., et al. (2022). Adavosertib with chemotherapy in patients with primary platinum-resistant ovarian, fallopian tube, or peritoneal cancer: an open-label, four-arm, phase II study. Clin. Cancer Res. 28 (1), 36–44. doi:10.1158/1078-0432.CCR-21-0158
Oh, J., and Symington, L. S. (2018). Role of the Mre11 complex in preserving genome integrity. Genes (Basel) 9 (12), 589. doi:10.3390/genes9120589
Oza, A. M., Estevez-Diz, M., Grischke, E. M., Hall, M., Marmé, F., Provencher, D., et al. (2020). A biomarker-enriched, randomized phase II trial of adavosertib (AZD1775) plus paclitaxel and carboplatin for women with platinum-sensitive TP53-mutant ovarian cancer. Clin. Cancer Res. 26 (18), 4767–4776. doi:10.1158/1078-0432.CCR-20-0219
Parvathareddy, S. K., Siraj, A. K., Al-Badawi, I. A., Tulbah, A., Al-Dayel, F., and Al-Kuraya, K. S. (2021). Differential expression of PD-L1 between primary and metastatic epithelial ovarian cancer and its clinico-pathological correlation. Sci. Rep. 11 (1), 3750. doi:10.1038/s41598-021-83276-z
Saldivar, J. C., Cortez, D., and Cimprich, K. A. (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 18, 622–636. doi:10.1038/nrm.2017.67
Sato, H., Niimi, A., Yasuhara, T., Permata, T. B. M., Hagiwara, Y., Isono, M., et al. (2017). DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 8 (1), 1751. doi:10.1038/s41467-017-01883-9
Saxena, S., and Zou, L. (2022). Hallmarks of DNA replication stress. Mol. Cell. 82 (12), 2298–2314. doi:10.1016/j.molcel.2022.05.004
Schoonen, P. M., Guerrero Llobet, S., and van Vugt, MATM (2019). Replication stress: driver and therapeutic target in genomically instable cancers. Adv. Protein Chem. Struct. Biol. 115, 157–201. doi:10.1016/bs.apcsb.2018.10.006
Schram, A. M., Lee, E. K., Højgaard, M., Simpkins, F., LoRusso, P., Duska, L. R., et al. (2025). Abstract CT262: efficacy and safety of the combination PKMYT1-inhibitor lunresertib and ATR-inhibitor camonsertib in patients with ovarian and endometrial cancers: phase I MYTHIC study (NCT04855656). Cancer Res. 85 (8_Suppl. ment_2), CT262. doi:10.1158/1538-7445.AM2025-CT262
Sen, T., Rodriguez, B. L., Chen, L., Corte, C. M. D., Morikawa, N., Fujimoto, J., et al. (2019a). Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 9 (5), 646–661. doi:10.1158/2159-8290.CD-18-1020
Sen, T., Della Corte, C. M., Milutinovic, S., Cardnell, R. J., Diao, L., Ramkumar, K., et al. (2019b). Combination treatment of the oral CHK1 inhibitor, SRA737, and low-dose gemcitabine enhances the effect of programmed death ligand 1 blockade by modulating the immune microenvironment in SCLC. J. Thorac. Oncol. 14 (12), 2152–2163. doi:10.1016/j.jtho.2019.08.009
Shah, P. D., Wethington, S. L., Pagan, C., Latif, N., Tanyi, J., Martin, L. P., et al. (2021). Combination ATR and PARP Inhibitor (CAPRI): a phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol. Oncol. 163 (2), 246–253. doi:10.1016/j.ygyno.2021.08.024
Shen, J., Zhao, W., Ju, Z., Wang, L., Peng, Y., Labrie, M., et al. (2019). PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 79 (2), 311–319. doi:10.1158/0008-5472.CAN-18-1003
Shih, C. T., Huang, T. T., Nair, J. R., Ibanez, K. R., and Lee, J. M. (2024). Poly (ADP-Ribose) polymerase inhibitor olaparib-resistant BRCA1-mutant ovarian cancer cells demonstrate differential sensitivity to PARP inhibitor Rechallenge. Cells 13 (22), 1847. doi:10.3390/cells13221847
Simpkins, F., Nasioudis, D., Wethington, S. L., Martin, L. P., Tanyi, J. L., Latif, N. A., et al. (2024). Combination ATR and PARP Inhibitor (CAPRI): a phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-sensitive epithelial ovarian cancer (cohort A). J. Clin. Oncol. 42 (16_Suppl. l), 5510. doi:10.1200/JCO.2024.42.16_suppl.5510
Smith, P., Bradley, T., Gavarró, L. M., Goranova, T., Ennis, D. P., Mirza, H. B., et al. (2023). The copy number and mutational landscape of recurrent ovarian high-grade serous carcinoma. Nat. Commun. 14 (1), 4387. doi:10.1038/s41467-023-39867-7
Soberanis Pina, P., and Lheureux, S. (2023). Overcoming PARP inhibitor resistance in ovarian cancer. Int. J. Gynecol. Cancer 33 (3), 364–376. doi:10.1136/ijgc-2022-003698
Sørensen, C. S., and Syljuåsen, R. G. (2012). Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 40 (2), 477–486. doi:10.1093/nar/gkr697
Stur, E., Peng, F., Teng, P. N., Bayraktar, E., Hu, M., Corvigno, S., et al. (2025). The dynamic immune behavior of primary and metastatic ovarian carcinoma. npj Precis. Oncol. 9, 120. doi:10.1038/s41698-025-00818-8
Sumimoto, H., Imabayashi, F., Iwata, T., and Kawakami, Y. (2006). The BRAF–MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 203 (7), 1651–1656. doi:10.1084/jem.20051848
Tan, D. S., Agarwal, R., and Kaye, S. B. (2006). Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 7 (11), 925–934. doi:10.1016/S1470-2045(06)70939-1
Tan, D. S., Castonguay, V., Cote, G., De Bono, J. S., El-Rayes, B., Gabrail, N., et al. (2022). EP294/#883 Elimusertib, an oral ataxia telangiectasia and RAD3-related inhibitor, in advanced gynecologic cancers with DNA damage response defects. Int. J. Gynecol. Cancer 32, A172.
Taniguchi, H., Chakraborty, S., Takahashi, N., Banerjee, A., Caeser, R., Zhan, Y. A., et al. (2024). ATR inhibition activates cancer cell cGAS/STING-interferon signaling and promotes antitumor immunity in small-cell lung cancer. Sci. Adv. 10 (39), eado4618. doi:10.1126/sciadv.ado4618
Tong, J., Song, J., Zhang, W., Zhai, J., Guan, Q., Wang, H., et al. (2024). When DNA-damage responses meet innate and adaptive immunity. Cell Mol. Life Sci. 81, 185. doi:10.1007/s00018-024-05214-2
Wang, Y. W., Allen, I., Funingana, G., Tischkowitz, M., and Joko-Fru, Y. W. (2025a). Predictive biomarkers for the efficacy of PARP inhibitors in ovarian cancer: an updated systematic review. BJC Rep. 3 (1), 14. doi:10.1038/s44276-025-00122-9
Wang, Y., Zhu, N., Liu, J., Chen, F., Song, Y., Ma, Y., et al. (2025b). Role of tumor microenvironment in ovarian cancer metastasis and clinical advancements. J. Transl. Med. 23 (1), 539. doi:10.1186/s12967-025-06508-0
Westin, S. N., Coleman, R. L., Fellman, B. M., Yuan, Y., Sood, A. K., Soliman, P. T., et al. (2021). EFFORT: EFFicacy of adavosertib in parp ResisTance: a randomized two-arm non-comparative phase II study of adavosertib with or without olaparib in women with PARP-resistant ovarian cancer. J. Clin. Oncol. 39 (15_Suppl. l), 5505. doi:10.1200/JCO.2021.39.15_suppl.5505
Xu, H., George, E., Gallo, D., Medvedev, S., Wang, X., Kryczka, R., et al. (2024). Targeting CCNE1 amplified ovarian and endometrial cancers by combined inhibition of PKMYT1 and ATR. Res. Sq. doi:10.21203/rs.3.rs-3854682/v1
Yap, T. A., Fontana, E., Lee, E. K., Spigel, D. R., Højgaard, M., Lheureux, S., et al. (2023). Camonsertib in DNA damage response-deficient advanced solid tumors: phase 1 trial results. Nat. Med. 29 (6), 1400–1411. doi:10.1038/s41591-023-02399-0
Yap, T. A., Tolcher, A. W., Plummer, R., Mukker, J. K., Enderlin, M., Hicking, C., et al. (2024). First-in-human study of the ataxia telangiectasia and Rad3-related (ATR) inhibitor tuvusertib (M1774) as monotherapy in patients with solid tumors. Clin. Cancer Res. 30 (10), 2057–2067. doi:10.1158/1078-0432.CCR-23-2409
Yu, H., Kortylewski, M., and Pardoll, D. (2007). Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 7 (1), 41–51. doi:10.1038/nri1995
Yue, P., Zhang, X., Paladino, D., Sengupta, B., Ahmad, S., Holloway, R. W., et al. (2012). Hyperactive EGF receptor, JAKs and STAT3 signaling promote enhanced colony-forming ability, motility and migration of cisplatin-resistant ovarian cancer cells. Oncogene 31 (18), 2309–2322. doi:10.1038/onc.2011.409
Zhang, T., and Xiaohan, C. (2025). Unveiling the role of JAK2/STAT3 signaling in chemoresistance of gynecological cancers: from mechanisms to therapeutic implications. Crit. Rev. Oncol. Hematol. 211, 104712. doi:10.1016/j.critrevonc.2025.10712
Zeman, M. K., and Cimprich, K. A. (2014). Causes and consequences of replication stress. Nat. Cell Biol. 16 (1), 2–9. doi:10.1038/ncb2897
Zhang, T., and Xiaohan, C. (2025). Unveiling the role of JAK2/STAT3 signaling in chemoresistance of gynecological cancers: from mechanisms to therapeutic implications. Crit. Rev. Oncol. Hematol. 211, 104712. doi:10.1016/j.critrevonc.2025.104712
Zhang, Y., Wu, L., Wang, Z., Wang, J., Roychoudhury, S., Tomasik, B., et al. (2022). Replication stress: a review of novel targets to enhance radiosensitivity—from bench to clinic. Front. Oncol. 12, 838637. doi:10.3389/fonc.2022.838637
Zhao, F., Jiang, X., Li, Y., Huang, T., Xiahou, Z., Nie, W., et al. (2025). Characterizing tumor biology and immune microenvironment in high-grade serous ovarian cancer via single-cell RNA sequencing: insights for targeted and personalized immunotherapy strategies. Front. Immunol. 15, 1500153. doi:10.3389/fimmu.2024.1500153
Zhou, W., Zeng, T., Chen, J., Tang, X., Yuan, Y., Hu, D., et al. (2024). Aberrant angiogenic signaling pathways: Accomplices in ovarian cancer progression and treatment. Cell Signal 120, 111240. doi:10.1016/j.cellsig.2024.111240
Zhu, H., Tang, Y. D., Zhan, G., Su, C., and Zheng, C. (2021). Corrigendum: the critical role of PARPs in regulating innate immune responses. Front. Immunol. 14, 1253094. doi:10.3389/fimmu.2023.1253094
Glossary
ATM Ataxia telangiectasia mutated
ATR Ataxia telangiectasia and Rad3-related protein
ATRIP ATR Interacting Protein
BID Twice a day (bis in die)
BFB Breakage–fusion–bridge
BRCA Breast Cancer gene (1 or 2)
CAAs Cancer-associated adipocytes
CAFs Cancer-associated fibroblasts
CAMs Cancer-associated mesothelial cells
CB Clinical benefit
CD8+T cells Cytotoxic T lymphocytes expressing CD8
CDK1/CDC2 Cyclin-dependent kinase 1
CDK2 Cyclin-dependent kinase 2
CCL5 C-C motif chemokine ligand 5
CCNE1 Cyclin E1
CIN Chromosomal instability
cGAMP Cyclic guanosine monophosphate–adenosine monophosphate
cGAS Cyclic GMP-AMP synthase
CyTOF Cytometry by Time Of Flight
DCs Dendritic cells
DDR DNA Damage Response
DNA Deoxyribonucleic acid
ERVs Endogenous retroviral elements
FFPE Formalin-Fixed, Paraffin-Embedded
G1/S Gap 1/Synthesis phase transition of the cell cycle
H3K9me3 Histone 3 lysine 9 trimethylation
HGSOC High-grade serous ovarian cancer
HRD Homologous recombination deficiency
HRR Homologous recombination repair
IFN-I Type I interferon
IL-6 Interleukin 6
IL-10 Interleukin 10
ID8 Mouse ovarian cancer cell line/model
ICB Immune checkpoint blockade
IRF3 Interferon regulatory factor 3
LDG Low-dose gemcitabine
MB49 Murine bladder carcinoma cell line/model
MHC I Major histocompatibility complex class I
MHC II Major histocompatibility complex class II
M-MDSCs Monocytic myeloid-derived suppressor cells
MOS Median overall survival
mPFS Median progression-free survival
MYC MYC proto-oncogene
NHEJ Non-homologous end joining
NF1 Neurofibromin 1
NF-κB Nuclear factor kappa B
ORR Objective response rate
PARP Poly (ADP-ribose) polymerase
PARPi Poly (ADP-ribose) polymerase 1 and 2 inhibitors
PARG Poly (ADP-ribose) glycohydrolase
PBMC Peripheral blood mononuclear cell
PD-(L)1 Programmed death-(ligand) 1
PROC Platinum-resistant ovarian cancer
RAS Rat sarcoma (family of related GTPases; commonly mutated in cancer)
RB1 Retinoblastoma 1
RPA Replication protein A
ROS Reactive oxygen species
RSR Replication stress response
S phase Synthesis phase
scRNAseq Single-cell RNA sequencing
siRNA Small interfering RNA
SCLC Small cell lung cancer
STAT3 Signal transducer and activator of transcription 3
STIC Serous tubal intraepithelial carcinoma
STING Stimulator of interferon genes
TBK1 TANK-binding kinase 1
TH1 T-helper 1 (immune response type)
TIM3 T-cell immunoglobulin and mucin-domain containing-3
TCR T-cell receptor
TME Tumor microenvironment
TP53 Tumor protein p53 (commonly abbreviated as p53)
Tregs Regulatory T cells
TopBP1 Topoisomerase II Binding Protein 1
VEGF Vascular endothelial growth factor
WEE1 WEE1 G2 checkpoint kinase
Keywords: TME (tumor microenvironment), replication stress, HGSOC, Wee1, Chk1, ATR, immunotherapy, PDL1 inhibitors
Citation: Venegas L and Lheureux S (2025) Interplay of replication stress response and immune microenvironment in high-grade serous ovarian cancer. Front. Cell Dev. Biol. 13:1638964. doi: 10.3389/fcell.2025.1638964
Received: 31 May 2025; Accepted: 18 August 2025;
Published: 05 September 2025.
Edited by:
Tanay Thakar, Broad Institute, United StatesReviewed by:
Nataša Lisica-Šikić, Sveuciliste u Zadru Odjel za zdravstvene studije, CroatiaNikolaos Gavalas, National and Kapodistrian University of Athens, Greece
Copyright © 2025 Venegas and Lheureux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephanie Lheureux, c3RlcGhhbmllLmxoZXVyZXV4QHVobi5jYQ==
†ORCID: Stephanie Lheureux, orcid.org/0000-0002-6228-1800; Laura Venegas, orcid.org/0009-0004-1263-1069