 Yuanpei He1†
Yuanpei He1† Xinge Wang2,3†
Xinge Wang2,3† Boya Xu4†
Boya Xu4† Shichao Chen1
Shichao Chen1 Hongcai Li5
Hongcai Li5 Bei Chang6
Bei Chang6 Chao Hu7
Chao Hu7 Xiaorong Lan1*
Xiaorong Lan1* Shiting Li1*
Shiting Li1* Guangwen Li1*
Guangwen Li1*- 1The Affiliated Stomatological Hospital, Southwest Medical University, Luzhou Key Laboratory of Oral and Maxillofacial Reconstruction and Regeneration, Luzhou, China
- 2Department of Stomatology, Norman Bethune International Peace Hospital, Shijiazhuang, China
- 3School of Stomatology, The Fourth Military Medical University, Xian, China
- 4Department of Stomatology, The 941 Hospital of the Joint Service Support Force of the People’s Liberation Army of China, Xining, China
- 5Department of Stomatology, Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 6The PLA Rocket Force Characteristic Medical Center, Beijing, China
- 7Department of General Dentistry, The Integrated Traditional Chinese and Western Medicine Hospital, Wenzhou Medical University, WenZhou, China
Mitochondrial transfer is defined the process through which specific cell types release their mitochondria and subsequently transfer them to unrelated cell types in response to various physiological or pathological stimuli. This process enhances cellular function and alters disease states. Recent research has begun to explore the potential of intercellular mitochondrial transfer as a therapeutic strategy for human diseases. Mitochondrial dysfunction represents a significant pathological alteration in osteoarthritis, and studies indicate that mitochondrial transfer may serve as an effective modulatory treatment approach for osteoarthritis. Mitochondrial transfer, as an innovative subcellular therapeutic technique, presents the advantages of diverse acquisition methods and multiple transmission pathways. This paper aims to summarize the current understanding of the mechanisms of mitochondrial transfer in relation to osteoarthritis, emphasizing the existing research on mitochondrial transfer in osteoarthritis and its potential as a disease-modifying therapy.
1 Introduction
Within eukaryotic cells, mitochondria are considered some of the earliest internal membrane systems. They create adenosine triphosphate (ATP) through the mechanism of oxidative phosphorylation (OXPHOS), thus supplying the essential energy for cell functions and serving as a key driving force for numerous cellular processes. This is why they are commonly referred to as the “powerhouses of the cell” (Lane and Martin, 2010). Mitochondria were initially recognized for their critical role in OXPHOS, but as a diverse family of organelles, they exhibit a multitude of additional functions—up to a dozen—beyond OXPHOS, all of which are essential for cellular viability. To systematically delineate these various functions, researchers have categorized them into five dimensions: molecular level, biological characteristics, bioactivity, functions, and biological behaviors, Furthermore, they have established three levels of histological organization, which include cell types and subtypes, and mitochondrial characterization (Monzel et al., 2023). Mitochondrial functions encompass a range of molecular activities, including amino acid metabolism (Ryu et al., 2024)., ion uptake (Naón et al., 2023), and molecular damage (Verkerke et al., 2024); biological characteristics related to protein input (Tracy et al., 2022), lipid synthesis (Wedan et al., 2024), molecular modifications (Zhang F. et al., 2023; Miriam et al., 2021), and the maintenance and expression of mitochondrial DNA (mtDNA) sequences (Saurer et al., 2023; Rahul et al., 2023); bioactivity categories involving transport (Zhu et al., 2024) and protein complexes/supercomplexes (Milenkovic et al., 2023); functional aspects including OXPHOS (Manford et al., 2021), permeability transition (Zhou et al., 2019)., and the biosynthesis of iron-sulfur (Fe/S) clusters and hormones (Antoine et al., 2017; Olga et al., 2025); and biological behaviors such as fusion/fission dynamics (Di Bona et al., 2024), mitotic nuclear signaling (Zierhut et al., 2019), and intercellular transfer (Baldwin et al., 2024). These categories illustrate a hierarchical progression of mitochondrial functional behaviors, wherein higher-level biological functions exert regulatory feedback on lower-level biological functions.
Mitochondrial transfer is defined as the targeted release of functional mitochondria from donor cells and their subsequent uptake by recipient cells, modulating critical cellular activities (Borcherding and Brestoff, 2023; Frisbie et al., 2024). Research has demonstrated that intercellular mitochondrial transfer can occur through multiple mechanisms in different tissues, serving several critical functions, including supporting the metabolism of recipient cells (McCully et al., 2009; Caicedo et al., 2015; Kim et al., 2018), regulating cell quality (Hutto et al., 2023; Huang et al., 2023), promoting wound healing (Boudreau et al., 2014), modulating the immune system (Tanmoy et al., 2021), and maintaining metabolic homeostasis (Brestoff et al., 2021; Crewe et al., 2021). Mitochondrial transfer provides novel insights for disease treatment: if extracellular mitochondrial transfer into cells is indeed feasible, it raises the question of whether we can artificially introduce free mitochondria via specific pathways to modulate physiological processes or to influence the occurrence and progression of diseases. Consequently, research pertaining to mitochondrial transplantation has gradually begun to emerge (Lin et al., 2024; Wu et al., 2024). Mitochondrial transplantation involves the replacement or compensation of damaged mitochondria within cells with healthy mitochondria, thereby impacting cellular metabolism, signal transduction, and promoting cell survival. Numerous studies have investigated mitochondrial transplantation utilizing various carriers (Cai W. et al., 2024; Long et al., 2024; Sun et al., 2022), establishing a robust research foundation for the treatment of diverse diseases, such as wound healing (Yao et al., 2024), pulmonary diseases (Ding et al., 2024), and cancer (Marabitti et al., 2024), while providing promising therapeutic directions.
Mitochondrial transfer is regulated by specialized intercellular mechanisms that exhibit precise tissue and cell type selectivity, directly modulating essential cellular functions (Table1) (Baldwin et al., 2024; Levoux et al., 2021; Gao et al., 2019; Feng et al., 2023; Wang et al., 2020; Fahey et al., 2022; Rosina et al., 2022; Brestoff et al., 2021; Kawano et al., 2023; Jacoby et al., 2022; Mistry et al., 2019; Ritsuko et al., 2024; Yuan et al., 2021; Morrison et al., 2017; Nicolás-Ávila et al., 2020; Ikeda et al., 2021; Zhou et al., 2024; van der Vlist et al., 2022; Eo et al., 2024; Peruzzotti-Jametti et al., 2021; Jia et al., 2023; Norat et al., 2023; Weinhäuser et al., 2023; Kidwell et al., 2023; Zhang H. et al., 2023; Lazar and Goldfinger, 2021; Xu et al., 2021a). At the cellular level, mitochondrial transfer can enhance the energy metabolism of recipient cells, maintain homeostasis, and activate cellular functions (Cai J. et al., 2024). Notably, cancer cells are known to actively acquire mitochondria from immune cells such as T-cells and macrophages. The acquisition of mitochondria by cancer cells can weaken anti-tumor immunity, support their metabolic demands, and promote proliferation (Zhang H. et al., 2023). At the tissue level, mitochondrial transfer contributes to the stability of various organ systems and initiates coordinated defense and protective mechanisms (Nicolás-Ávila et al., 2020). This protective role is exemplified in neurodegenerative pathologies and brain injury (Zhou et al., 2024; Hannah et al., 2024). Specifically, microglia transfer healthy mitochondria to compromised neurons via tunneling nanotubes (TNTs), which reduces oxidative stress and normalizes pathogenic gene expression. During acute brain injury, mitochondrial transfer further replenishes energy reserves while scavenging toxic reactive oxygen species (ROS), thereby establishing neuroprotection (Fu et al., 2025). Beyond endogenous processes, exogenous induction of mitochondrial transfer represents a promising therapeutic strategy for tissue repair. Osteoarthritis (OA) is a condition associated with this process, wherein mesenchymal stem cells (MSCs) transfer mitochondria to chondrocytes, thereby promoting their regeneration and repair (Fahey et al., 2022; Altuntaş et al., 2025). Interestingly, under different circumstances, the identities of the receptor and donor in mitochondrial transfer can be interchangeable (Nicolás-Ávila et al., 2020; Ikeda et al., 2021). In other words, certain cells may act as mitochondrial donors under specific conditions, and under other conditions, they may switch to become receptors. This suggests that many cells have the ability to both transfer and receive mitochondria, and this ability may be regulated. Furthermore, mitochondrial intercellular transfer involves not only the relationship between receptors and donors, but also potentially complex multicellular interactions, which warrant further investigation in future research.
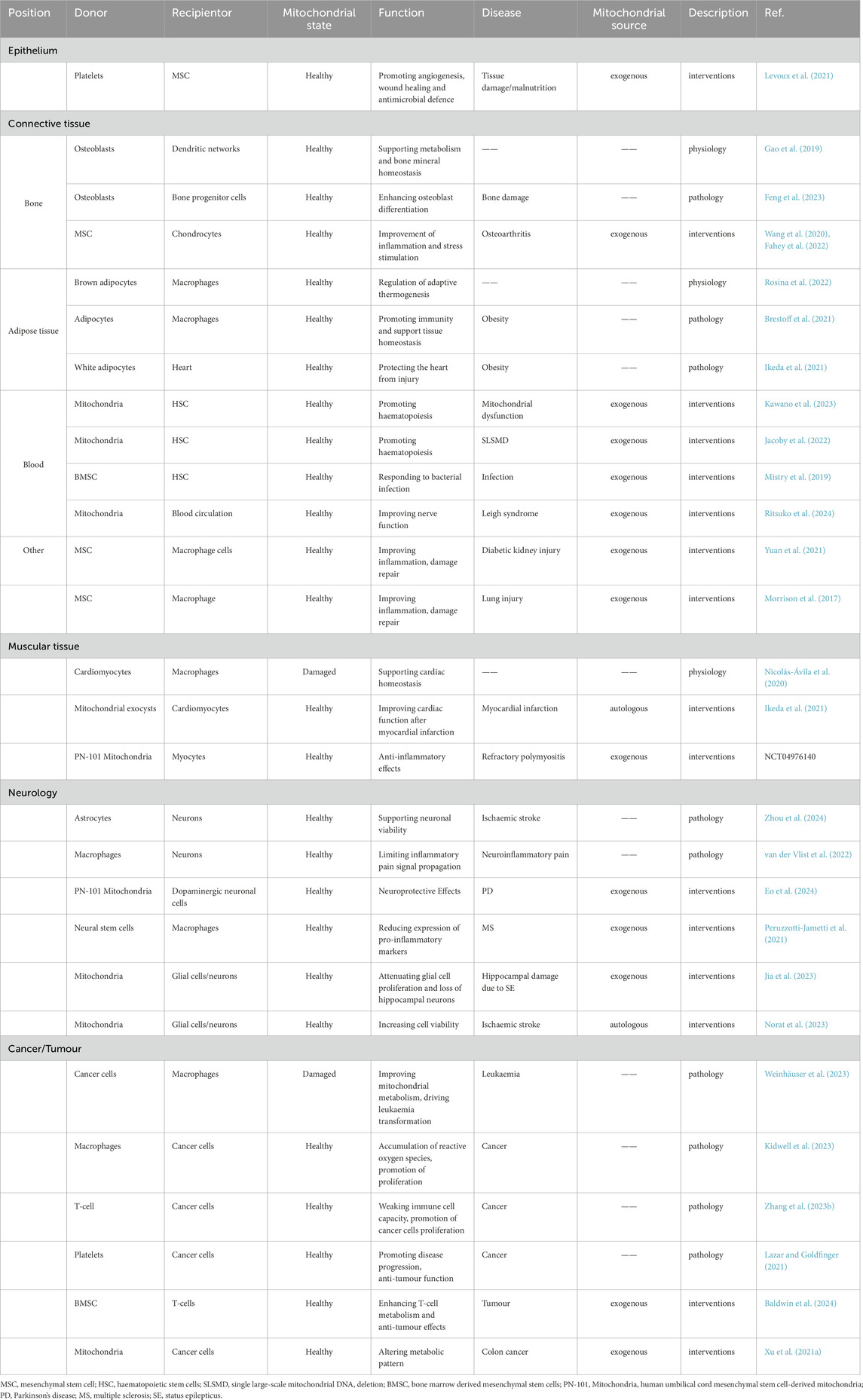
Table 1. The role of mitochondria transfer in physiological functions and disease participation.
In the senior population, OA is a prevalent joint disorder and plays a major role in causing disability. The mechanisms underlying OA are closely associated with mitochondrial function. A substantial body of research indicates that mitochondrial dysfunction results in energy depletion (Singh et al., 2010), the accumulation of ROS (Shi et al., 2025), variation in mtDNA (Mercedes et al.), and calcium metabolism (Wenyu et al., 2024). The initiation and advancement of OA are influenced by these combined factors (Sun et al., 2021; Durán-Sotuela et al., 2023; Blanco et al., 2018). For these reasons, targeted interventions aimed at mitochondrial function have emerged as a potential modifying therapy for OA. Such interventions primarily encompass drug-targeted regulation (Zhang Y. et al., 2023; Chen et al., 2022) and biological interventions using mesenchymal stem cells (Kim et al., 2022; Zhai et al., 2022). Artificial mitochondrial transfer has been shown to effectively treat experimental OA phenotypes in recent studies, establishing novel therapeutic avenues for OA (Kim et al., 2023; Lee et al., 2022). This review discusses the definitions and attributes of mitochondrial transfer and OA, as well as the established mitochondrial-related molecular mechanisms implicated OA pathogenesis. Furthermore, we will explore artificial mitochondrial transfer as a potential a therapeutic option for OA and propose pertinent scientific questions and research methodologies that warrant further investigation in the domain of mitochondrial therapy for OA.
2 The biological basis of mitochondrial transfer
In the early stages of mitochondrial research, it was widely believed that mitochondria within cells were solely inherited through vertical transmission during cell division or through mitochondrial biogenesis. However, in 2006, researchers observed the phenomenon of mitochondrial transfer between co-cultured cells, a process that could rescue ρ0 cells lacking mtDNA (88). Since then, mitochondrial transfer has been recognized as an important higher-order biological function of mitochondria, leading to increased attention and extensive investigation in the field. The biological mechanism of mitochondrial transfer is a highly complex and multi-layered process, involving various molecules, the cytoskeletal system, and intracellular dynamic processes. The biological structural foundation of mitochondrial transfer includes mitochondrial membrane proteins (Ahmad et al., 2014), motor proteins (Joshi et al., 2019), ATP generation and utilization (Spees et al., 2006), regulatory mechanisms, as well as the cytoskeleton and extracellular vesicles. The unique biological structure of mitochondria allows them to exhibit diversity in physiological processes and functions, enabling them to dynamically and reversibly adapt to energy demands, environmental changes, and other. These structures and mechanisms work in coordination to ensure that mitochondria can undergo dynamic transfer within cells as needed, in order to meet energy demands, respond to environmental changes, and support the normal functioning of the cell.
2.1 Mitochondrial membrane proteins and motor proteins
The outer and inner membranes of mitochondria are the core of their structure. They not only participate in maintaining mitochondrial functions (such as ATP synthesis, metabolite transport, etc.) but also play crucial roles during the transfer process (Hu et al., 2024; MacVicar et al., 2019; López-Doménech et al., 2018). Receptor proteins on the mitochondrial outer membrane interact with motor proteins in the cytoskeleton, helping mitochondria attach and move along the cytoskeleton on microtubules or microfilaments. Some proteins are located on the mitochondrial inner membrane and are involved in transport and positioning functions; they may regulate the stability and transfer of mitochondria by interacting with microtubules or microfilaments. Mitochondrial Rho GTPase 1 (Miro 1) is a mitochondrial-associated Rho-GTPase, a type of GTPase found on the mitochondrial outer membrane. The primary function of Miro one is to regulate the positioning and movement of mitochondria. During mitochondrial transfer, mitochondria use Miro one to shuttle along the actin-microtubule highways into the cytoplasm of the recipient cell (Ahmad et al., 2014).
Mitochondrial transfer relies on intracellular motor proteins, which drive the movement of mitochondria. Motor proteins typically move along the negative end of microtubules (toward the centrosome), pulling mitochondria from the cell membrane toward the cell center or other regions. On the other hand, dynein proteins move along the positive end of microtubules (toward the distal end), pushing mitochondria toward the cell membrane or protrusions. These motor proteins form stable complexes with mitochondrial membrane proteins, enabling mitochondria to move in a specific direction (López-Doménech et al., 2018). Studies have reported that mitochondrial quality control processes mediated by migration bodies involve the regulation of motor proteins. When mitochondria are exposed to mild stress, damaged mitochondria are transported to migration bodies, where they are subsequently expelled from the migrating cells (Jiao et al., 2021). Research has also shown that microglial cells rely on dynamin-related protein 1 (DRP1) and mitochondrial fission protein 1 (FIS1) to release free mitochondria. This indicates that both the movement of mitochondria within the cell and their quality control during stress responses are tightly regulated by motor proteins, ensuring proper mitochondrial distribution and function in the cell (Joshi et al., 2019).
2.2 ATP generation and utilization
All mitochondria possess a core OXPHOS system, which consists of five multi-protein complexes, with four being part of the electron transport chain (ETC.). On the inner membrane of the mitochondria, the free energy (ΔG) generated by these complexes establishes an electrochemical gradient, which in turn creates a membrane potential, serving as a major source of energy (Edman et al., 2024). The formation of membrane potential is not only essential for maintaining mitochondrial permeability, channel integrity, and regulating the morphology of mitochondrial cristae, as well as mitochondrial fusion/fission dynamics, but also drives several other mitochondrial functions. These include ion uptake (such as Ca2+, Na+, and Mn2+), synthesis of nuclear-encoded proteins and antioxidant defense regenerated, precursor Fe/S clusters, antiviral signaling, and ATP synthesis by the Fo ATP synthase F1 subunit, among others. ATP also powers motor proteins (such as dynein and kinesin), driving the transfer of mitochondria, and ultimately serves as the energy source for the recipient cell to uptake mitochondria (Figure 1) (Spees et al., 2006).
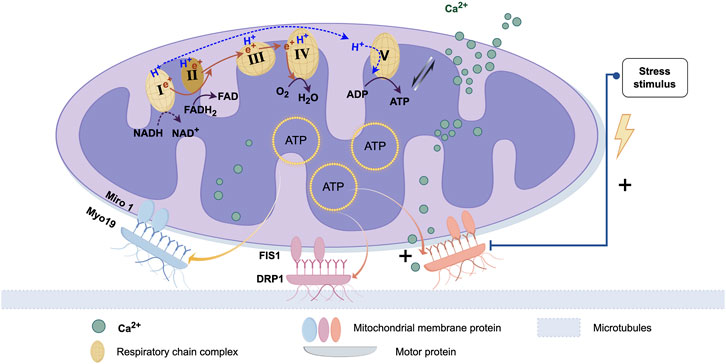
Figure 1. Proteins associated with mitochondrial transfer and regulatory processes. The biological basis of mitochondrial transfer lies in the pairing and binding of membrane proteins and dynamin proteins. After protein binding, movement is typically facilitated by microtubules. The energy for this movement primarily comes from ATP produced by the mitochondria. This process is also regulated by calcium ion uptake and bio-mechanical stress stimuli. (Graphic is created by FigDraw. Copyright Code:ORURW7e3c7).
2.3 Regulation of mitochondrial transfer
Mitochondrial transfer is not only dependent on the action of motor proteins but is also regulated by various signaling pathways. The regulation of mitochondrial transfer involves calcium ion (Ca2+) uptake (Wang and Schwarz, 2009), cellular stress responses (Wang et al., 2018), and the stability of microtubules. Changes in the Ca2+ concentration can affect the position and movement of mitochondria (Emily et al., 2018), especially in neurons, where calcium signaling is crucial for the transfer of mitochondria to and from the synaptic sites (Hayakawa et al., 2016). When cells encounter stress (such as hypoxia (Hu et al., 2023), oxidative stress (Wang et al., 2018), etc.), mitochondria may alter their position or accumulate in specific regions to adapt to environmental changes. The stability and arrangement of the microtubules inside the cell directly influence the movement of mitochondria. Mitochondrial morphology changes, such as fission and fusion, also play a major role in transfer process (Valm et al., 2017). Mitochondrial fission typically occurs when the cell needs to regulate mitochondrial number (for example, during cell division) (Jiao et al., 2021), while fusion helps mitochondria merge into a more powerful functional unit (Nasoni et al., 2021). These morphological changes are closely related to the cell’s energy needs and also affect the distribution and transfer of mitochondria within the cell (Fu et al., 2025).
2.4 Cytoskeleton and extracellular vesicles
Mitochondrial transfer between cells occurs in various tissues in vivo and can be classified into mediated and unmediated mechanisms. Mediated transfer occurs through transient cell connections, based on the cytoskeletal system, which provides “tracks” and “power” for mitochondrial movement (Boldogh and Pon, 2007). Cells establish connection channels, which are elongated, membrane-wrapped structures that allow organelles to transfer between cells. Mitochondria are transferred from 1 cell to another through these connections (Figure 2). These cell connections are typically formed by TNTs and/or gap junction channels (GJC) mediated by connexin 43 (Cx43). TNTs are dynamic structures that permit the exchange of proteins, soluble molecules, and organelles. The transfer of organelles within TNTs is crucial for regulating cell growth, signaling, and disease progression (Nasoni et al., 2021; Saha et al., 2022). Cx43 exists in the mitochondrial inner membrane in a hemichannel form, is related to gap junction channels, and may also regulate the formation of TNTs (Tishchenko et al., 2020).
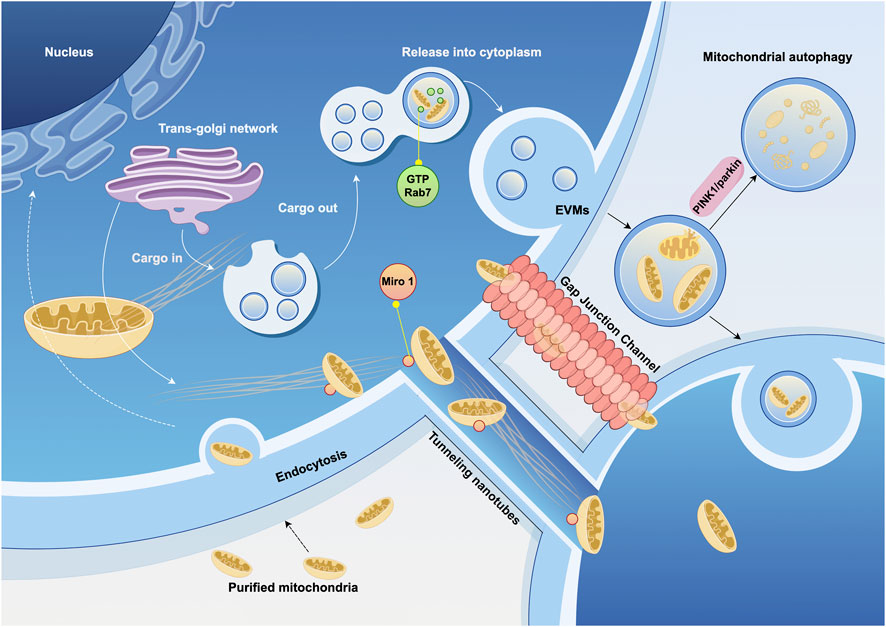
Figure 2. Cytoskeleton and extracellular vesicles associated with mitochondrial transfer. Mitochondrial transfer utilizes microtubules as “tracks” to move mitochondria to specific compartments through the pairing of specialized mitochondrial membrane proteins and dynamin proteins. Mitochondria can also be transported loaded into vesicles and released extracellularly under the regulation of GTPases. The ultimate fate of extracellular vesicles derived from mitochondria—whether they are internalized, released, or trigger autophagy—is also regulated by signaling pathways. EVMs, extracellular vesicles contain mature mitochondria. (Graphic is created by FigDraw. Copyright Code: SSWIW00a0a).
Extracellular vesicle (EV) can also serve as medium for mitochondrial transfer, with mature mitochondria present in the vesicles secreted by cells, which are then delivered to recipient cells. Mitochondrial-derived vesicles (MDVs) have been found in a variety of cell types, including osteoblasts (Suh et al., 2023), bone marrow adipocytes (Crewe et al., 2021), and cardiomyocytes (Ikeda et al., 2021), as well as in migrating cells (Duan et al., 2020). In the extracellular vesicles of these cells, damaged mitochondria are more commonly observed. These structures may be regulated by the PINK1/parkin pathway. Parkin is an E3 ubiquitin ligase that binds PINK1, marking damaged mitochondria for degradation by mitophagy. In the absence of parkin, this elimination process is impaired (Ikeda et al., 2021). Rab7, a small GTPase, plays a critical role in regulating the maturation of endosomes. When extracellular vesicles contain mature mitochondria (EVMs), GTP-bound Rab7 is the determinant of EVM secretion out of the cell (Liang et al., 2023).
Mitochondrial transfer can also occur in a free form. One way is through direct cell fusion, which results in the mixing of cytoplasm and organelles. Another way is through direct contact, where mitochondria are transferred via direct contact between cell membranes, and the mitochondria are released into the extracellular space in a free state, with recipient cells actively capturing these mitochondria (Brestoff et al., 2021). This free transfer is more commonly observed in blood components, where free mitochondria, lacking extracellular vesicles, are approximately 0.5–1 μm in diameter and comprise the full-length mtDNA genome. However, their origin is heterogeneous, suggesting that free mitochondria in blood may originate from different cell types (Boudreau et al., 2014; Borcherding et al., 2022).
3 Overview of the characteristics of OA
3.1 Clinical features and pathological process of OA
OA is a leading cause of musculoskeletal pain and disability and is a common type of arthritis. As a chronic inflammatory condition associated with the aging process, OA primarily affects individuals aged 65 and older, with its prevalence significantly increasing with advancing age (Motta et al., 2023). Clinically, OA is characterized by symptoms such as joint stiffness, chronic pain, instability, deformity, radiographic evidence of joint space narrowing (Heidari, 2011). Given the complexity of its pathogenesis, current treatment primarily focus on alleviating joint stiffness and pain, aiming to provide palliative symptom relief and improve the quality of life for affected individuals.
The onset of this disease is attributed to an active dynamic imbalance between repairing and destroying joint tissue, rather than the passive degenerative process or mere wear-and-tear typically described (Prieto-Alhambra et al., 2014; Fu et al., 2018). And then, alterations occur in the components of cartilage, resulting in the loss of its integrity (Loeser et al., 2016). These compositional changes alter the properties of the cartilage and increase its susceptibility to physical damage. Initial erosion usually occurs at the surface, followed by deeper fissures in the cartilage and subsequent expansion of calcified cartilage areas. During the repair process, hypertrophied chondrocytes demonstrate enhanced synthetic activity, leading to the production of matrix degradation byproducts and pro-inflammatory mediators. These factors disrupt the regulation of chondrocyte function and influence the adjacent synovium, stimulating both proliferation and inflammatory responses. The proliferating synovial cells also release pro-inflammatory products, a process that is accompanied by tissue hypertrophy and increased vascular distribution. Additionally, in the subchondral bone, there is an elevation in bone turnover and vascular invasion, which facilitates the extension from subchondral bone into the cartilage. Furthermore, the formation of bone spurs at the joint margins due to endochondral ossification are significantly influenced by inflammatory biological factors, as well as by mechanical overload and abnormal joint kinematics (Hunter and Bierma-Zeinstra, 2019). Collectively, these factors disrupt the dynamic balance of cartilage, leading to an increase in stiffness and changes in the biological properties, ultimately resulting in the development of OA.
3.2 Etiological analysis of OA
Among the etiologies of OA, limited cartilage regeneration is a key factor in disease progression. Articular cartilage is a thin layer of tissue that covers the surfaces of articulating bones, typically measuring between 2 and 4 mm in thickness, and is referred to as “hyaline cartilage” due to its clear and transparent appearance (Fujii et al., 2022). This type of cartilage exhibits considerable elasticity and lubricity, enabling it to effectively support mechanical loads and facilitate smooth movement between bones, thereby significantly reducing friction during joint activities (Fujii et al., 2022; Iwamoto et al., 2013). However, the regenerative capacity of cartilage is relatively limited and is influenced by various factors. Consequently, self-repair following injury often proves inadequate, which can lead to the development of degenerative diseases, particularly OA (123).The academic community has proposed several hypotheses to explain the failure of cartilage regeneration. These hypotheses include the scarcity of regenerative potential cells, pathological mechanical alterations, persistent inflammation, and metabolic stress, among others.
It is important to note that the physiological characteristics of chondrocytes significantly influence their regenerative capacity. The low density of chondrocytes, coupled with the relatively limited proliferative ability of mature chondrocytes, is considered the main reason for the insufficient self-regeneration capacity of cartilage (Kai-di et al., 2021; Jerry and Kyriacos, 2004). Additionally, aging is associated with pronounced senescence in chondrocytes, resulting in a reduction in cell density and in vitro proliferative capacity, a phenomenon that is particularly evident in patients with post-traumatic OA (Barbero et al., 2004; David et al., 2017). Pathological mechanical changes within the joint can lead to cartilage loss. Trauma or chronic degenerative changes may modify the load distribution across the joint, thereby affecting the contact area of the load. This mechanical alteration may stimulate the activation of osteoclasts, fibroblasts, and macrophages, which release pro-inflammatory mediators that gradually degrade the ECM, ultimately resulting in damage to the collagen network and inflammation of the synovium (Caravaggi et al., 2021; Assirelli et al., 2022). The widely studied concept of “mechanical inflammation” refers to the pathological process of inflammatory signaling caused by mechanical stress. Meanwhile, non-resolving inflammation may result in histological changes in cartilage. Inflammatory stress impairs the activity and matrix synthesis of chondrocytes and promotes matrix degradation by stimulating the production of various matrix metalloproteinases and interleukin-2 (Andriacchi et al., 2004).
Moreover, chondrocytes depend on molecular mechanisms that are specifically adapted to low-oxygen and low-nutrient environments under physiological conditions for normal operation. Following an injury, it becomes difficult to maintain the local partial oxygen pressure at an ideal level, leading to increased energy demands that can affect the functionality of chondrocytes and the requisite microenvironment. At this point, cells experience metabolic changes, particularly the dysregulation of the glycolytic pathway, resulting in the accumulation of lactic acid, which further alters the local microenvironment (Dolzani et al., 2019). This metabolic change inhibit matrix synthesis and accelerates the degradation process of cartilage.
4 Mitochondrial dysfunction in OA
OA is a heterogeneous disease with various potential pathogenic pathways. Each common risk factor for OA may trigger different mechanisms of disease, particularly those involving oxidative stress, impaired biosynthesis and growth responses of chondrocytes and osteoblasts, synovial inflammation, and increased degradation and calcification of the cartilage matrix (Mathiessen and Conaghan, 2017; Lepetsos and Papavassiliou, 2016; Blanco et al., 2011). OA is also associated with phenotypic differences, such as age and obesity, and the pathogenesis of OA varies markedly from one population to the next, with possible interactions between the mechanisms (Muthu et al., 2023). In this context, OA can be viewed as a syndrome rather than a singular disease, with potential interactions among its mechanisms that remain to be fully elucidated. Currently, there are no clear classification standards for mitochondrial dysfunction in OA. To describe the changes occurring in mitochondria within OA more clearly, this article categorizes them into three distinct categories: structural abnormalities, alterations in biological function, and biological interactions.
4.1 Structural abnormalities
When mitochondrial homeostasis or mitosis is impaired, dysfunctional mitochondria cannot be removed in a timely manner, leading to a disturbance in the dynamic balance of mitochondria and further affecting chondrocyte health. The regulatory mechanisms of mitochondrial quality control are impaired in individuals diagnosed with OA. This damage involves mitochondrial biogenesis, mitophagy, and mitochondrial dynamics, which include processes of fission and fusion (Guo et al., 2025). Observations have shown that mtDNA in patients with OA exhibits damage and mutations, resulting in a decrease in the size, quantity, and overall content of mitochondria (Alejandro et al., 2024; Morena et al., 2022). Genomic instability can cause damage to mtDNA, further affecting mitochondrial function.
Damaged mtDNA can cause mitochondrial respiratory chain to malfunction, subsequently resulting in the production of ROS. Excessive production of ROS is a prominent feature of mitochondrial dysfunction. It can cause diverse forms of mtDNA damage. The phenomenon encompasses a range of structural alterations, including oxidative base damage, strand breaks, and telomere shortening. It is hypothesised that such damage may ultimately result in cell cycle arrest and cellular senescence (Macip et al., 2002). In addition, research findings have indicated that particular mtDNA variants, encompassing specific haplotypes, exhibit a notable correlation with the initiation and progression of OA. (Vermulst et al., 2007). The relationship between mtDNA haplotypes and OA may exhibit heterogeneity across different geographic populations, indicating that these haplotypes could serve as potential biomarkers for the diagnosis and prognosis of OA (Fang et al., 2014).
Mitochondrial dysfunction can induce alterations in autophagy, which may lead to the accumulation of defective mitochondria, thereby potentially increasing mitochondrial levels in chondrocytes of patients with OA. Furthermore, chronic oxidative damage may result in a reduction of FIS1, which promotes mitochondrial elongation and the formation of giant mitochondria (Lee et al., 2007). This elongation may contribute to the excessive expansion of the mitochondrial network, which plays a significant role in the progression of age-related diseases such as OA.
The mitochondrial permeability transition pore (mPTP) comprises subunits that are situated within both the inner and outer mitochondrial membranes (Bonora et al., 2022). In the chondrocytes of OA patients, factors such as oxidative stress, calcium overload, and depolarization of the inner membrane may lead to the pathological opening of mPTP. The transient activation of mPTP, triggered by a variety of stimuli, can induce protective pathways, a phenomenon referred to as “mitochondrial excitability.” Conversely, a persistent opening of mPTP can culminate in the release of apoptotic factors, which, if prolonged and unregulated, may ultimately lead to the demise of the cartilage cells (Patel et al., 2021).
4.2 Alterations in biological function
Oxidative stress-mediated mitochondrial dysfunction in chondrocytes constitutes a key driver of OA pathogenesis (Figure 3). ROS are widely recognized as contributors to chondrocyte damage and the progression of OA. The excessive production of ROS disrupts joint homeostasis, triggering cell apoptosis and accelerating metabolic degradation, thereby impairing various structures of the joint (Loeser et al., 2016). Interleukin-1 beta (IL-1β) and tumor necrosis factor-alpha (TNF-α) are major inflammatory mediators in the pathogenesis of OA, and their actions are closely related to mitochondrial dysfunction (van Vulpen et al., 2015). Oxidative stress is primarily attributed to dysfunctional mitochondria, which generate ROS such as superoxide anions and hydroxyl radicals. Excessive ROS has been observed to induce an inflammatory response, which has been linked to the expression of matrix metalloproteinases (MMPs) and the activation of associated signalling pathways, including the MAPK/ERK pathway. The aforementioned alterations have been shown to impede the process of glycosaminoglycan and type II collagen synthesis, whilst concurrently inducing an escalation in the concentrations of both type I collagen and MMPs. Furthermore, there is a concomitant increase in pro-inflammatory cytokine production (Rieder et al., 2018). Furthermore, excessive ROS can also damage mtDNA and reduce its repair capacity, highlighting the complex relationship between mitochondrial dysfunction and OA, making it a potential therapeutic target. In a rat model of OA, a drug delivery system (DDS) containing melatonin was administered via intra-articular injection. Melatonin was found to mitigate cartilage degeneration through its antioxidant properties, which indirectly enhanced mitochondrial function and facilitated the repair of the cartilage matrix (Zhang Y. et al., 2022).
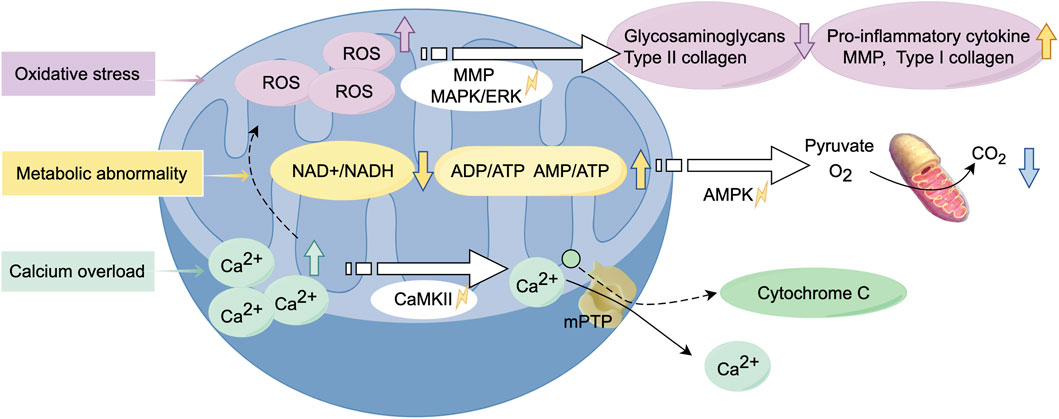
Figure 3. Alterations in biological function of mitochondria during the development of osteoarthritis. Mitochondrial dysfunction leads to oxidative stress, resulting in the excessive production of ROS. This process activates relevant signaling pathways, triggering inflammatory responses and metabolic abnormalities. Calcium overload exacerbates this process, causing alterations in mitochondrial membrane permeability, ultimately leading to chondrocyte cell death. (Graphic is created by FigDraw. Copyright Code: SSTIY92b49).
Damaged mitochondria lead to insufficient energy production, producing impaired energy metabolism and an inability to maintain normal cellular physiological functions, thereby exacerbating chondrocyte damage and apoptosis. In response to environmental stress, chondrocytes exhibit a tendency to switch between different metabolic pathways to adapt to these changes (Lee et al., 2012). The alterations in metabolic pathways are closely related to mitochondrial dysfunction, which may result in a marked decrease in the NAD+/NADH equilibrium, an increased rate of anaerobic glycolysis, and alterations in lipid and amino acid metabolism (Stein and Imai, 2012). The decline in the NAD+/NADH equilibrium, in combination with an increase in the ADP/ATP and AMP/ATP ratios, leads to the activation of AMP-activated protein kinase (AMPK). The balance between OXPHOS and glycolysis is predominantly regulated by the AMPK and mTOR signaling pathways (Herzig and Shaw, 2018). Changes in the NAD + pathway can severely impact mitochondrial function, potentially leading to cell growth arrest or impairing cellular metabolism through the depletion of sirtuins (SIRT) 3 and SIRT 5 (Miwa et al., 2022). It has been reported to improve respiratory chain function through the induction of SIRT3, with particular emphasis on its potential application in the treatment of OA (Zhang Y. et al., 2023).
Abnormal calcium regulation leads to overproduction of reactive oxygen species, mitochondrial depolarisation and reduced mitochondrial membrane potential, which further contributes to chondrocyte apoptosis. In the context of OA, the elevation of cytosolic Ca2+ concentration prompts mitochondria to rapidly absorb Ca2+, thereby preventing calcium overload within the cells (Bravo-Sagua et al., 2017). However, excessive calcium accumulation within mitochondria can result in heightened production of ROS, leading to mitochondrial dysfunction and senescence. This phenomenon has the capacity to exert a deleterious effect on various facets of mitochondrial metabolism, including nitric oxide synthesis, the process of releasing cytochrome c, the process of altering mitochondrial membrane permeability and the process of activation of calcium/calmodulin-dependent protein kinase II (CaMKII) signalling pathways (Brookes et al., 2004; Peng and Jou, 2010). In response to this pathological alteration, the utilization of nanoparticles carrying siRNA alleviates mitochondrial calcium overload in MSCs, thereby regulating dysfunctional mitochondrial autophagy. This approach has demonstrated efficacy in the treatment of OA (Zhai et al., 2022).
In an environment characterized by oxidative stress, chondrocytes may undergo regenerative mitochondrial transfer. Research has observed mitochondrial transfer to chondrocytes through fluorescence imaging, finding that this process is associated with enhanced mitochondrial function, likely correlated with the upregulation of gap junction protein Cx43 expression (Wang et al., 2021; Mayan et al., 2015). In the context of an inflammatory environment, the transfer of mitochondria from MSCs to chondrocytes is significantly increased, with Cx43 playing a critical mediating role in this process (Irwin et al., 2024). Upon contact with damaged cartilage, MSCs localize to the injury site and deliver mitochondria to chondrocytes through cellular protrusions (Fahey et al., 2022). Experimental results indicate that combined inflammatory and mechanical stress enhances mitochondrial transfer between chondrocytes, suggesting that mitochondrial transfer may occur spontaneously in the progression of OA. However, further experimental validation is necessary to substantiate these observations.
4.3 Biological interactions
Chondrocytes have been observed to exhibit a response to mechanical stimuli, which is mediated by the extracellular matrix (ECM). The ECM plays a crucial role in the regulation of the epigenetics in diseases. Mitochondria may serve as a potential communication medium between mechanical signals and epigenetics modifications (Figure 4). The relationship between inhibition of matrix synthesis and activation of matrix degradation is also affected in OA. Redox abnormalities in the mitochondria have been shown to inhibite matrix synthesis whilst concomitantly activating matrix metalloproteinases, thus promoting the degradation of matrix components and, by extension, degenerative cartilage lesions. Within the context of OA, an elevated level of cartilage matrix rigidity in regions that are subjected to substantial stress has been observed to result in considerable impairment to the mitochondria of the cartilage cells. This, in turn, initiates the demethylation of histone H3 lysine 27 trimethylation (H3K27me3). The opening of the mPTP promotes the translocation of plant homeodomain finger protein 8 (Phf8) to the nucleus, catalyzing the demethylation of H3K27me3 (Kan et al., 2024).
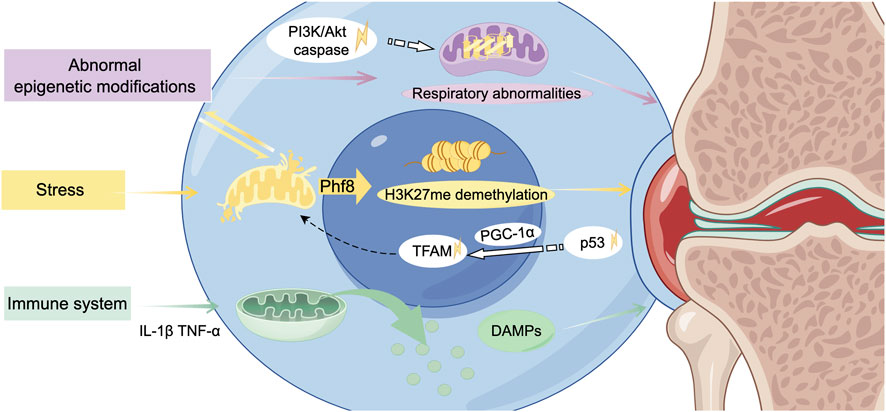
Figure 4. Mitochondria-related biological interactions in the development of osteoarthritis. Under biomechanical stress, mitochondrial dysfunction interacts with aberrant epigenetic modifications, contributing to alterations and damage in the chondrocyte nucleus. The subsequent release of release danger-associated molecular patterns (DAMPs) exacerbates the inflammatory response, ultimately driving the progression of osteoarthritis. TFAM, nuclear-encoded mitochondrial transcription factor A. (Graphic is created by FigDraw. Copyright Code: SSTIY92b49).
Abnormal epigenetic modifications are considered preliminary factors in the occurrence and development of OA, with a strong correlation to mitochondrial dysfunction (Jiang et al., 2024). Genetic and epigenetic abnormalities have been demonstrated to result in increased mitochondrial respiration and glycolysis, elevated levels of free radicals and pro-inflammatory cytokines, and augmented rates of apoptosis. Research has demonstrated that exposure to a hyperglycemic environment in utero increases the vulnerability to OA in later adulthood, attributable to persistently diminished expression levels of Sirt3. The downregulation of Sirt3 results in impaired mitotic function of the cells responsible for cartilage production (chondrocytes), abnormal mitochondrial respiration, and the incapacity to efficiently eliminate aged and damaged mitochondria in a timely manner. The consequence of this is an imbalance in mitochondrial homeostasis, which in turn triggers disease through epigenetic regulation (Li et al., 2024a).
During the occurrence of OA, dysfunctional mitochondria induce the production of cytokines. These cytokines further promote chondrocyte apoptosis and accelerate the progression of OA. In the initial stages, it has been observed that both the Phosphatidylinositol 3 kinase (PI3K)/Protein kinase B (Akt) signalling pathway (relevant to cartilage cell death) and the caspase pathway (pertinentto the initiation of cell death) are activated. This results in oxidative stress-induced cartilage cell death, a process involving the mitochondria (Li et al., 2019). These signaling cascades are crucial for various biological processes, and any imbalances within these pathways can disrupt normal biological processes and lead to diseases. Additionally, this dysregulation may activate the tumor protein p53 signaling pathway, inhibiting peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) and peroxisome proliferator-activated receptor gamma co-activator 1β (PGC-1β). This process may contribute to the exacerbation of mitochondrial dysfunction (Sahin et al., 2011).
During the process known as mitochondrial biogenesis, the nuclear-encoded mitochondrial transcription factor A (TFAM) is an essential component of mtDNA transcription and the regulation of mitochondrial biogenesis (Picca and Lezza, 2015). PGC-1α is activated by physiological stressors, which in turn activate transcription factors and regulate the transcription of TFAM (Wu et al., 1999). Furthermore, PGC-1α can also modulate the activity of other transcription factors such as peroxisome proliferator-activated receptor γ (PPARγ), Yin Yang 1(YY-1), and GA (Guanine-Adenine)binding protein transcription factor (GABPA), thereby impacting mitochondrial function (Scarpulla, 2011). The intravenous administration of meta-Defensomes polymer nanoparticles in collagenase-induced OA mice resulted in the transformation of pro-inflammatory classically activated macrophages (M1 macrophages) into anti-inflammatory alternatively activated macrophages (M2 phenotypes). This intervention led to an increase in the expression of mitochondrial transcription factor A, restoration of aerobic respiration, and a significant reduction in synovitis, thereby effectively inhibiting the early progression of OA (Zhang L. et al., 2022).
The relationship between mitochondrial dysfunction and the activation of the immune system is complex and intertwined, serving as a significant factor in the pathogenesis of OA. Dysfunctional mitochondria release danger-associated molecular patterns (DAMPs), which can trigger innate immune responses and induce inflammation (Miwa et al., 2022; Wan et al., 2014). As the human organism ages, there is an observed increase in the concentration of free mtDNA within cells. This increase has been shown to correlate with the presence of markers associated with sterile inflammation. This increase activates immune response pathways, including the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, which belongs to the family of nucleotide-binding oligomerization domain-like receptors. The activation of the NLRP3 inflammasome consequently results in the maturation of the second messenger GMP-AMP, which in turn activates the immune response, thereby promoting inflammatory responses (White et al., 2014).
5 Practice and prospects of mitochondrial transfer involvement in OA therapy
5.1 Mitochondrial transfer in OA therapy
In the management of OA, in addition to conventional symptomatic treatments and surgical interventions, disease-modifying therapy (DMT) is increasingly receiving scholarly attention (Li Z. et al., 2024). As a precision intervention, DMT corrects core OA etiologies through biomarker-directed pathway modulation, not only arresting disease trajectory but also activating endogenous chondrocyte regenerative mechanisms (Yulong et al., 2021; Ronghui et al., 2024; Giuliana et al., 2023; Kai et al., 2024).
Presently, numerous clinical trials in 2–4 Phase are in progress (https://www.clinicaltrials.gov/), with mesenchymal stem cell therapy and blood-derived therapies emerging as significant areas of research (Naomasa et al., 2017; Yong Sang et al., 2015). The sources of mesenchymal stem utilized in these therapies primarily include adipose-derived mesenchymal stem cells (Chen et al., 2021), bone marrow-derived mesenchymal stem cells (Vives et al., 2015; Orozco et al., 2014), umbilical cord-derived mesenchymal stem (Lim et al., 2021) and other types of MSCs(NCT03866330,NCT02037204). In these therapeutic modalities, MSCs are believed to ameliorate mitochondrial dysfunction in chondrocytes through mitochondrial transfer, thereby providing substantial foundational research support for their clinical application (Fahey et al., 2022; Angela et al., 2024). Recent investigations have identified a significant relationship between Cx43 and its truncated isoform gap junction protein alpha 1–20 kDa (GJA1-20k) in the transfer of mitochondria between highly oxidative cells. The overexpression of GJA1-20k in MSCs has been shown to enhance the transfer of mitochondria to chondrocytes, thereby facilitating the repair of cartilage tissue affected by OA (156). Consequently, the direct transfer of mitochondria to diseased joints emerges as a key facilitator.
Blood-derived treatment strategies predominantly encompass stromal vascular fraction (SVF) (Krześniak et al., 2021)and platelet-rich plasma (PRP) (Mayoly et al., 2019). PRP stimulates cell proliferation and reduces inflammation, oxidative stress and chondrocyte senescence. A related clinical trial in Phase 4 (NCT05660824) is a multicenter, parallel-group, triple-blind trial enrolling 130 patients. The aim of the study is to evaluate the clinical efficacy of SVF as an adjunctive therapy to PRP for functional and tissue regeneration in OA. PRP contain various organelles, including mitochondria, lysosomes, dense granules, and alpha granules (Zhai et al., 2022). After intra-articular injection, mitochondria in platelets are transferred to damaged chondrocytes, promoting chondrocyte repair and regeneration, which is also an effective treatment option for OA.
5.2 Current status of research on mitochondrial artificial transfer in OA therapy
Based on the complexity of mitochondrial dysfunction in OA, research conducted on 450 participants in the Osteoarthritis Initiative (OAI) study has demonstrated that indicators associated with mitochondrial dysfunction are linked to mtDNA variants and the risk of rapid progression of knee OA (80). This evidence supports the potential for mitochondrial-targeted therapies in the management of OA. Mitochondrial artificial transfer, which entails the supplementation of healthy and functionally normal mitochondria, represents a novel and effective therapeutic strategy for degenerative diseases related to bones and cartilage. Currently, many studies are exploring the artificial mitochondria transfer from other environments or cell types to treat various diseases (Ritsuko et al., 2024; Vijith et al., 2024). It is also known as mitochondrial transplantation (Caicedo et al., 2024; Christoph et al., 2022).
In the study of artificial mitochondrial transfer, various methods have been implemented, which can be primarily categorized into prenatal and postnatal artificial mitochondrial transfer. Early prenatal artificial mitochondrial transfer treatments have utilized ooplasm as a mitochondrial carrier, where healthy donor oocyte mitochondria and their mtDNA are directly injected into a recipient oocyte with pathogenic mtDNA mutations through micromanipulation (Barritt et al., 2001). The resultant cells contain both donor and recipient mtDNA and are subsequently utilized for in vitro fertilization. Recently, there has been a growing inclination towards utilizing stem cell-derived mitochondria in mitochondrial-assisted in vitro fertilization treatments. Mitochondria are extracted and purified from autologous ovarian stem cells (Labarta et al., 2019), autologous bone marrow stem cells (NCT03639506), or autologous urine-derived stem cells (NCT06020742), and are then co-injected with sperm into oocytes to improve embryo quality. Presently, several of these therapeutic strategies have progressed into the early clinical research.
Postnatal artificial mitochondrial transfer represents another area of exploration (Mitch, 2022). This therapeutic approach is based on the ability of exogenous mitochondria to enter cells. Mitochondria may be directly administered to patients following purification (Ying-Ting et al., 2019), or alternatively, the purified mitochondria can be incorporated into autologous cells prior to injection into the patient (Jacoby et al., 2022). Furthermore, purified mitochondria can be directly given to patients (Ikeda et al., 2021). In the treatment of OA, several experiments have been reported in animal experiments using autologous mitochondria and exogenous mitochondria, using intra-articular injections, to improve osteoarthritic symptoms and slow down the course of OA in various ways (Table 2). The results confirm the favourable biosafety of mitochondrial transfer following intra-articular injection in mammals. Mitochondrial injections are more biocompatible than cellular level modification treatments. Therefore, it may be more efficient to introduce purified, highly concentrated mitochondria into the lesion site artificially without the aid of exogenous cellular mediators (cells, plasma, etc.) to improve the therapeutic effect.
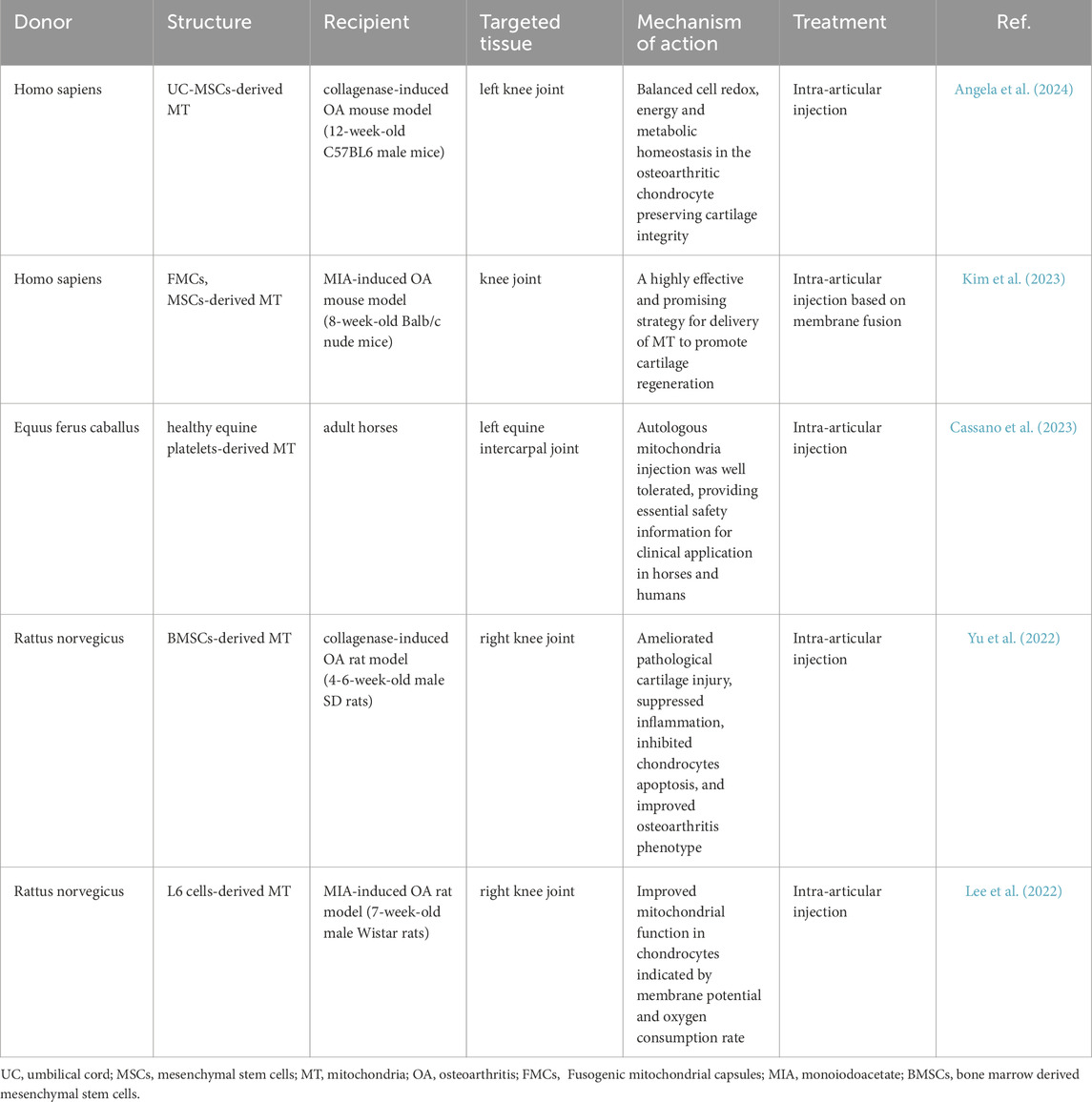
Table 2. Current studies applying purified mitochondrial transfer for osteoarthritis treatment.
5.3 Prospects for mitochondrial transfer in the treatment of OA
The application of the artificial transfer method of mitochondria is a very promising therapeutic strategy for the treatment of OA because of the obvious advantages of this method. However, mitochondria is large size about 0.5–1 μm and the efficiency of delivery by conventional endocytosis is very low, thereby constraining their clinical applicability (Sun et al., 2019). This limitation has prompted recent research to concentrate on optimizing the efficiency of mitochondrial transfer, primarily through two avenues: optimizing carriers and augmenting transfer efficiency (Figure 5). In addition to conventional stem cells, it has been identified that mitochondria can adapt to form structures known as mitochondrial-derived vesicles (MDVs), which exhibit repair functions (Min et al., 2025; Reut et al., 2023). Exosomes from adipose-derived mesenchymal stem cells (AdMSC-Exos) has been found to effectively donate mitochondrial components and improve macrophage mitochondrial integrity and OXPHOS levels. This restores metabolic and immune homeostasis of airway macrophages and attenuates inflammatory lung pathology (Miriam et al., 2021). Therefore, utilizing exosomes as carriers for mitochondrial transfer is supported by a physiological basis for therapeutic interventions.
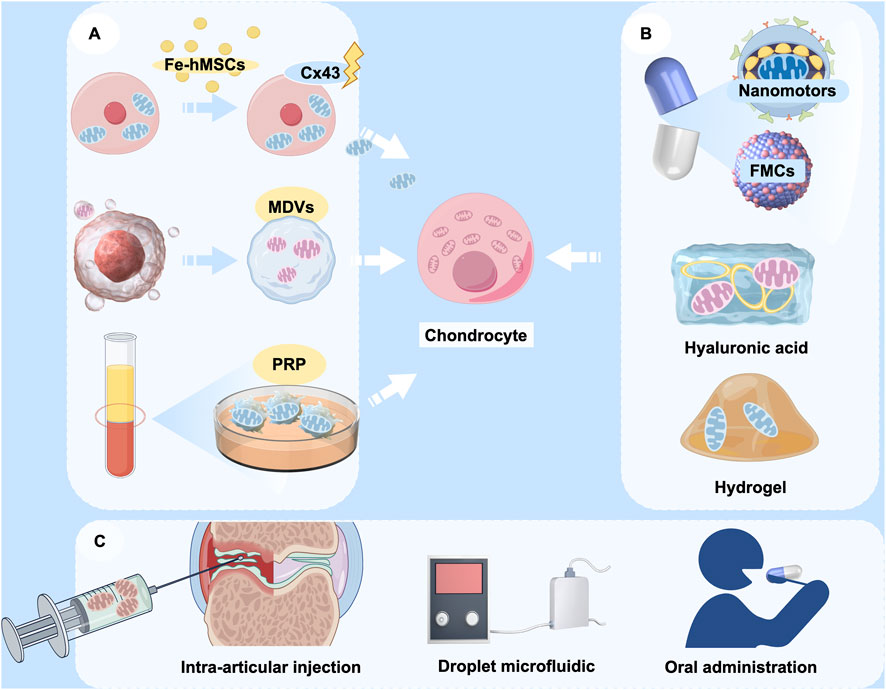
Figure 5. The potential application of mitochondrial transfer in OA therapy. (A) Various sources of donor mitochondria, including mesenchymal stem cells, mitochondrial-derived vesicles (MDVs), and platelet-rich plasma (PRP); (B) Encapsulation of therapeutic mitochondria with various delivery vehicles, including nanomotors, fusogenic mitochondrial capsules (FMCs), hyaluronic acid, and hydrogels; (C) Modes of cargo delivery to target cells, including intra-articular injection, droplet microfluidics, and oral nanoparticle capsules. (Graphic is created by FigDraw. Copyright Code: UYUWA7822c).
Advancements in bioengineering have led to increased diversity and efficiency in the design of biological carriers. Fusogenic mitochondrial capsules (FMCs), which are composed of various lipids and liposomes, facilitate the rapid and efficient transport of mitochondria to chondrocytes, thereby promoting cartilage regeneration (Kim et al., 2023). Besides liposomes, hyaluronic acid has been employed in conventional carrier systems. Liposomes that are coated with hyaluronic acid have the capability to co-deliver the natural cyclic peptide RA-XII and mitochondrial-targeting photosensitizers, exhibiting high selectivity and potential for precise combination therapy in colorectal cancer (Xu et al., 2021b). Targeted modification of mitochondria is also an effective approach. Nanomotorized mitochondria has recently been developed with chemotactic targeting ability for damaged heart tissue (Wu et al., 2024). This mitochondrion is packaged in an enteric gel capsule. The orally administered mitochondria are rapidly absorbed by intestinal cells and subsequently released into the bloodstream, from where they are transported to the damaged heart tissue. Modulation of the disease microenvironment by nanomitochondria not only facilitates the rapid uptake and prolonged retention of mitochondria by damaged cells, but also preserves the high functional activity of exogenous mitochondria. It would be an intriguing attempt if the chemotactic targeting ability of mitochondrial transfer to chondrocytes could be enhanced through a similar approach. Given that hyaluronic acid is frequently utilized in palliative treatment for OA through intra-articular injection, exploring its potential to deliver mitochondria presents novel approach to for OA treatment. Hydrogel is also one of the candidate carriers, and an engineered layered hydrogel with immunoreactive properties that can adapt to the bone regeneration environment and mediate targeted mitochondrial transfer between cells was recently published (Cai W. et al., 2024). This finding provides a new therapeutic strategy to promote bone regeneration and repair, and also has research value and practical applications in the treatment of OA.
Moreover, the indirect enhancement of mitochondrial transfer efficiency represents a viable strategy. The mitochondrial transfer technology based on droplet microfluidics has demonstrated efficient, high-throughput quantitative mitochondrial transfer at the single-cell level. This advancement is expected to significantly promote mitochondrial transfer in clinical applications and optimize cellular function, thereby creating new avenues for the treatment of OA (36). Currently, most of the clinical applications alter the efficiency of mitochondrial transfer by modulating key signalling pathways. For example, magnetic iron oxide nanoparticle (IONP)-activated hMSCs (Fe-hMSCs) can induce the overexpression of Cx43, thereby promoting mitochondrial transfer and enabling human placenta-derived MSCs to efficiently and safely transfer mitochondria to target cells (Huang et al., 2021). This could also be a therapeutic reference for mitochondrial transfer in OA.
5.4 Challenges of artificial mitochondrial transfer in the treatment of OA
Artificial mitochondrial transfer is emerging as a significant therapeutic approach in the treatment of OA, and researchers are actively investigating its potential. However, this procedure encounters several challenges. In the pre-mitochondrial transfer phase, implementers need to refine the treatment plan. OA as a syndrome has different etiologies and significant differences in the pathological stages of presentation (Glyn-Jones et al., 2015; Li et al., 2024c; Papatriantafyllou, 2024; Coryell et al., 2021). Consequently, the efficacy of artificial mitochondrial transfer in addressing specific etiologies, as well as the precise criteria for patient selection for personalized treatment, remains an unresolved issue. Moreover, the optimal therapeutic window for artificial mitochondrial transfer during disease progression to maximize efficacy remains undefined.
Moreover, although mitochondria exhibit a certain affinity for specific tissues, their targeting of chondrocytes within the human body requires enhancement (Tracy et al., 2022; Justin et al., 2024; Qinglian et al., 2003). Ensuring that mitochondria can accurately localize to damaged joint tissues or cells represents a critical issue in current research. It is reported that cells can capture purified mitochondria for aerobic respiration, which raises the possibility that non-targeted cells can capture mitochondria and affect the therapeutic effect (Borcherding et al., 2022). Additionally, artificial mitochondrial transfer may elicit an immune response because mitochondria possess mtDNA, which can be recognized by the immune system as foreign material, potentially leading to an immune attack (Tobias et al., 2019). Such a response may result in transfer failure, cause other complications, and even exacerbate the condition of OA. At present, the clinical application of mitochondrial transfer remains in its infancy, with a limited number of cases reported, thereby complicating a comprehensive assessment of its efficacy and safety (Craven et al., 2018).
Following mitochondrial transfer, it is imperative for the mitochondria to sustain stable functionality within the recipient cells. However, variations in the cellular environment of aging of the transplanted mitochondria, may lead to a gradual decline in their functionality (Yi et al., 2024). Additionally, the processes of mitochondrial fusion and degradation present significant challenges: transplanted mitochondria must successfully fuse with the recipient cells' mitochondria to fulfill their intended functions, a process that may be influenced by a variety of regulatory factors (National Academies of Sciences E and Medicine, 2016; Gammage et al., 2018). It was found that a portion of naked mitochondria can escape from recipient cells after capture, and that some donor-derived mitochondria may also be degraded by recipient cells and fail to fulfil their intended role (Cowan et al., 2017). While some studies have demonstrated positive outcomes of mitochondrial transfer in animal models, the long-term efficacy and safety of this intervention in human subjects necessitate further investigation.
6 Outlook
Mitochondrial dysfunction in osteoarthritis presents as a multifaceted disease mechanism, featuring diverse manifestations with interconnected regulatory networks. Future therapies targeting mitochondrial restoration represent a promising frontier for OA intervention. Given the operability and favorable biocompatibility of artificial mitochondrial transfer, this strategy appears promising as an effective intervention for future OA therapies. Artificial mitochondrial transfer can be conducted through various carriers, including stem cells, subcellular organelles, bioengineering materials, and even the direct transfer of isolated mitochondria. Additionally, targeted interventions, physical stimulation, bio-protein mediation, and modulation of signaling pathway can also be employed to enhance the efficiency of mitochondrial transfer.
Currently, the research directions pertaining to artificial mitochondrial transfer are varied, offering numerous opportunities for achieving the objectives of mitochondrial transfer. Nevertheless, investigations in this domain remain in the nascent stages, necessitating a clarification of the differences in responses observed between animal models and humans subjects. It is imperative to validate these findings through more experimental data prior to advancing to clinical applications. Advancing phase-targeted osteoarthritis therapies while resolving key technical challenges, including mitochondrial delivery optimization and therapeutic durability, represents critical research priorities. With continued investigation, artificial mitochondrial transfer is anticipated to provide new therapeutic strategies for OA, accommodating its diverse stages and underlying causes.
Author contributions
YH: Conceptualization, Project administration, Writing – review and editing, Funding acquisition, Writing – original draft. XW: Software, Visualization, Writing – review and editing. BX: Writing – review and editing, Visualization, Software. SC: Writing – review and editing, Software, Visualization. HL: Writing – review and editing, Visualization, Software. BC: Visualization, Writing – review and editing, Software. CH: Writing – review and editing, Software, Visualization. XL: Supervision, Writing – review and editing, Validation. SL: Writing – review and editing, Supervision, Validation. GL: Writing – original draft, Funding acquisition, Visualization, Software, Supervision, Conceptualization, Project administration, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by the Sichuan Science and Technology Program (No. 2024NSFSC0680), Health Commission of Sichuan Province Medical Science and Technology Program (No. 24QNMP018), Program of Southwest Medical University (No. 2019ZQN031).
Acknowledgments
The authors would like to acknowledge the support from Southwest Medical University and The Affiliated Stomatological Hospital for providing all the facilities required for the completion of this review.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmad, T., Mukherjee, S., Pattnaik, B., Kumar, M., Singh, S., Kumar, M., et al. (2014). Miro1 regulates intercellular mitochondrial transport and enhances mesenchymal stem cell rescue efficacy. Embo J. 33 (9), 994–1010. doi:10.1002/embj.201386030
Alejandro, D.-S., Natividad, O., Mercedes, F.-M., Jorge, V.-G., Sara, R.-F., Vanesa, B.-B., et al. (2024). Mitonuclear epistasis involving TP63 and haplogroup Uk: risk of rapid progression of knee OA in patients from the OAI. Osteoarthr. Cartil. 32 (5), 526–534. doi:10.1016/j.joca.2023.12.008
Altuntaş, C., Duruksu, G., Hunç, F., and Yazir, Y. (2025). Mitochondria transfer from mesenchymal stem cells into osteoarthritic chondrocytes ameliorate cellular functions and alleviate both inflammation and oxidative stress. Tissue Cell 96, 102990. doi:10.1016/j.tice.2025.102990
Andriacchi, T. P., Mündermann, A., Smith, R. L., Alexander, E. J., Dyrby, C. O., and Koo, S. (2004). A framework for the in vivo pathomechanics of osteoarthritis at the knee. Ann. Biomed. Eng. 32, 447–457. doi:10.1023/b:abme.0000017541.82498.37
Angela, C. C., Ana María, V.-L., Eliseo, P.-C., Francesca, V., Cynthia, G., Alexander, O., et al. (2024). Mitochondrial transfer balances cell redox, energy and metabolic homeostasis in the osteoarthritic chondrocyte preserving cartilage integrity. Theranostics 14 (17), 6471–6486. doi:10.7150/thno.96723
Antoine, P., Anthony, D., Floriane, P., Delphine Dupin, D., Christelle, V., Myriam, O., et al. (2017). FDXR mutations cause sensorial neuropathies and expand the spectrum of mitochondrial Fe-S-Synthesis diseases. Am. J. Hum. Genet. 101 (4), 630–637. doi:10.1016/j.ajhg.2017.09.007
Assirelli, E., Caravaggi, P., Mazzotti, A., Ursini, F., Leardini, A., Belvedere, C., et al. (2022). Location-dependent human osteoarthritis cartilage response to realistic cyclic loading: ex-vivo analysis on different knee compartments. Front. Bioeng. Biotechnol. 10, 862254. doi:10.3389/fbioe.2022.862254
Baldwin, J. G., Heuser-Loy, C., Saha, T., Schelker, R. C., Slavkovic-Lukic, D., Strieder, N., et al. (2024). Intercellular nanotube-mediated mitochondrial transfer enhances T cell metabolic fitness and antitumor efficacy. Cell 187 (23), 6614–30.e21. doi:10.1016/j.cell.2024.08.029
Barbero, A., Grogan, S., Schäfer, D., Heberer, M., Mainil-Varlet, P., and Martin, I. (2004). Age related changes in human articular chondrocyte yield, proliferation and post-expansion chondrogenic capacity. Osteoarthr. Cartil. 12 (6), 476–484. doi:10.1016/j.joca.2004.02.010
Barritt, J. A., Brenner, C. A., Malter, H. E., and Cohen, J. (2001). Mitochondria in human offspring derived from ooplasmic transplantation: brief communication. Hum. Reprod. 16 (3), 513–516. doi:10.1093/humrep/16.3.513
Blanco, F. J., Rego, I., and Ruiz-Romero, C. (2011). The role of mitochondria in osteoarthritis. Nat. Rev. Rheumatol. 7 (3), 161–169. doi:10.1038/nrrheum.2010.213
Blanco, F. J., Valdes, A. M., and Rego-Pérez, I. (2018). Mitochondrial DNA variation and the pathogenesis of osteoarthritis phenotypes. Nat. Rev. Rheumatol. 14 (6), 327–340. doi:10.1038/s41584-018-0001-0
Boldogh, I. R., and Pon, L. A. (2007). Mitochondria on the move. Trends Cell Biol. 17 (10), 502–510. doi:10.1016/j.tcb.2007.07.008
Bonora, M., Giorgi, C., and Pinton, P. (2022). Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. cell Biol. 23 (4), 266–285. doi:10.1038/s41580-021-00433-y
Borcherding, N., and Brestoff, J. R. (2023). The power and potential of mitochondria transfer. Nature 623 (7986), 283–291. doi:10.1038/s41586-023-06537-z
Borcherding, N., Jia, W., Giwa, R., Field, R., Moley, J., Kopecky, B., et al. (2022). Dietary lipids inhibit mitochondria transfer to macrophages to divert adipocyte-derived mitochondria into the blood. Cell metab. 34 (10), 1499–513.e8. doi:10.1016/j.cmet.2022.08.010
Boudreau, L. H., Duchez, A.-C., Cloutier, N., Soulet, D., Martin, N., Bollinger, J., et al. (2014). Platelets release mitochondria serving as substrate for bactericidal group IIA-Secreted phospholipase A2 to promote inflammation. Blood, J. Am. Soc. Hematol. 124 (14), 2173–2183. doi:10.1182/blood-2014-05-573543
Bravo-Sagua, R., Parra, V., López-Crisosto, C., Díaz, P., Quest, A., and Lavandero, S. (2017). Calcium transport and signaling in mitochondria. Compr. Physiol. 7 (2), 623–634. doi:10.1002/cphy.c160013
Brestoff, J. R., Wilen, C. B., Moley, J. R., Li, Y., Zou, W., Malvin, N. P., et al. (2021). Intercellular mitochondria transfer to macrophages regulates white adipose tissue homeostasis and is impaired in obesity. Cell metab. 33 (2), 270–82. e8. doi:10.1016/j.cmet.2020.11.008
Brookes, P. S., Yoon, Y., Robotham, J. L., Anders, M., and Sheu, S.-S. (2004). Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiology-Cell Physiology 287, C817–C833. doi:10.1152/ajpcell.00139.2004
Cai, W., Mao, S., Wang, Y., Gao, B., Zhao, J., Li, Y., et al. (2024a). An engineered hierarchical hydrogel with immune responsiveness and targeted mitochondrial transfer to augmented bone regeneration. Adv. Sci. 11, 2406287. doi:10.1002/advs.202406287
Cai, J., Chen, Y., She, Y., He, X., Feng, H., Sun, H., et al. (2024b). Aerobic exercise improves astrocyte mitochondrial quality and transfer to neurons in a mouse model of alzheimer's disease. Brain Pathol. 35, e13316. doi:10.1111/bpa.13316
Caicedo, A., Fritz, V., Brondello, J.-M., Ayala, M., Dennemont, I., Abdellaoui, N., et al. (2015). MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci. Rep. 5 (1), 9073. doi:10.1038/srep09073
Caicedo, A., Morales, E., Moyano, A., Peñaherrera, S., Peña-Cisneros, J., Benavides-Almeida, A., et al. (2024). Powering prescription: Mitochondria as “Living Drugs” - definition, clinical applications, and industry advancements. Pharmacol. Res. 199, 107018. doi:10.1016/j.phrs.2023.107018
Caravaggi, P., Assirelli, E., Ensini, A., Ortolani, M., Mariani, E., Leardini, A., et al. (2021). Biomechanical-based protocol for in vitro study of cartilage response to cyclic loading: a proof-of-concept in knee osteoarthritis. Front. Bioeng. Biotechnol. 9, 634327. doi:10.3389/fbioe.2021.634327
Cassano, J. M., Marycz, K., Horna, M., Nogues, M. P., Morgan, J. M., Herrmann, D. B., et al. (2023). Evaluating the safety of intra-articular mitotherapy in the equine model: a potential novel treatment for osteoarthritis. J. Equine Vet. Sci. 120, 104164. doi:10.1016/j.jevs.2022.104164
Chen, C. F., Hu, C. C., Wu, C. T., Wu, H. H., Chang, C. S., Hung, Y. P., et al. (2021). Treatment of knee osteoarthritis with intra-articular injection of allogeneic adipose-derived stem cells (ADSCs) ELIXCYTE®: a phase I/II, randomized, active-control, single-blind, multiple-center clinical trial. Stem Cell Res. Ther. 12 (1), 562. doi:10.1186/s13287-021-02631-z
Chen, X., Li, C., Cao, X., Jia, X., Chen, X., Wang, Z., et al. (2022). Mitochondria-targeted supramolecular coordination container encapsulated with exogenous itaconate for synergistic therapy of joint inflammation. Theranostics 12 (7), 3251–3272. doi:10.7150/thno.70623
Christoph, G. G., Qian, F., Edin, S., Tomaso, Z., Orane, G.-G., Benoît, K., et al. (2022). Mitochondria transplantation between living cells. PLoS Biol. 20 (3), e3001576. doi:10.1371/journal.pbio.3001576
Coryell, P., Diekman, B., and Loeser, R. (2021). Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat. Rev. Rheumatol. 17 (1), 47–57. doi:10.1038/s41584-020-00533-7
Cowan, D. B., Yao, R., Thedsanamoorthy, J. K., Zurakowski, D., Del Nido, P. J., and McCully, J. D. (2017). Transit and integration of extracellular mitochondria in human heart cells. Sci. Rep. 7 (1), 17450. doi:10.1038/s41598-017-17813-0
Craven, L., Murphy, J., Turnbull, D. M., Taylor, R. W., Gorman, G. S., and McFarland, R. (2018). Scientific and ethical issues in mitochondrial donation. new Bioeth. 24 (1), 57–73. doi:10.1080/20502877.2018.1440725
Crewe, C., Funcke, J.-B., Li, S., Joffin, N., Gliniak, C. M., Ghaben, A. L., et al. (2021). Extracellular vesicle-based interorgan transport of mitochondria from energetically stressed adipocytes. Cell metab. 33 (9), 1853–68. e11. doi:10.1016/j.cmet.2021.08.002
David, M. A., Smith, M. K., Pilachowski, R. N., White, A. T., Locke, R. C., and Price, C. (2017). Early, focal changes in cartilage cellularity and structure following surgically induced meniscal destabilization in the mouse. J. Orthop. Res. 35 (3), 537–547. doi:10.1002/jor.23443
Di Bona, M., Chen, Y., Agustinus, A. S., Mazzagatti, A., Duran, M. A., Deyell, M., et al. (2024). Micronuclear collapse from oxidative damage. Science 385 (6712), eadj8691. doi:10.1126/science.adj8691
Ding, F., Zhou, M., Ren, Y., Li, Y., Xiang, J., Li, Y., et al. (2024). Mitochondrial extracellular vesicles: a promising avenue for diagnosing and treating lung diseases. ACS nano 18 (37), 25372–25404. doi:10.1021/acsnano.4c02940
Dolzani, P., Assirelli, E., Pulsatelli, L., Meliconi, R., Mariani, E., and Neri, S. (2019). Ex vivo physiological compression of human osteoarthritis cartilage modulates cellular and matrix components. PLoS One 14 (9), e0222947. doi:10.1371/journal.pone.0222947
Duan, X., Li, Y., Yi, K., Guo, F., Wang, H., Wu, P., et al. (2020). Dynamic organelle distribution initiates actin-based spindle migration in mouse oocytes. Nat. Commun. 11 (1), 277. doi:10.1038/s41467-019-14068-3
Durán-Sotuela, A., Fernandez-Moreno, M., Suárez-Ulloa, V., Vázquez-García, J., Relaño, S., Hermida-Gómez, T., et al. (2023). A meta-analysis and a functional study support the influence of mtDNA variant M. 16519C on the risk of rapid progression of knee osteoarthritis. Ann. Rheumatic Dis. 82 (7), 974–984. doi:10.1136/ard-2022-223570
Edman, S., Flockhart, M., Larsen, F. J., and Apró, W. (2024). Need for speed: human fast-twitch mitochondria favor power over efficiency. Mol. Metab. 79, 101854. doi:10.1016/j.molmet.2023.101854
Emily, K. N., Olha, M. K., Paige, N., Kim, B., Chantal, A., Meng, W., et al. (2018). CaMKII (Ca(2+)/Calmodulin-Dependent kinase II) in Mitochondria of smooth muscle cells controls mitochondrial mobility, migration, and Neointima Formation. Arterioscler. Thromb. Vasc. Biol. 38 (6), 1333–1345. doi:10.1161/ATVBAHA.118.310951
Eo, H., Yu, S. H., Choi, Y., Kim, Y., Kang, Y. C., Lee, H., et al. (2024). Mitochondrial transplantation exhibits neuroprotective effects and improves behavioral deficits in an animal model of Parkinson's disease. Neurotherapeutics 21 (4), e00355. doi:10.1016/j.neurot.2024.e00355
Fahey, M., Bennett, M., Thomas, M., Montney, K., Vivancos-Koopman, I., Pugliese, B., et al. (2022). Mesenchymal stromal cells donate mitochondria to articular chondrocytes exposed to mitochondrial, environmental, and mechanical stress. Sci. Rep. 12 (1), 21525. doi:10.1038/s41598-022-25844-5
Fang, H., Liu, X., Shen, L., Li, F., Liu, Y., Chi, H., et al. (2014). Role of mtDNA haplogroups in the prevalence of knee osteoarthritis in a southern Chinese population. Int. J. Mol. Sci. 15 (2), 2646–2659. doi:10.3390/ijms15022646
Feng, Z., Jin, M., Liang, J., Kang, J., Yang, H., Guo, S., et al. (2023). Insight into the effect of biomaterials on osteogenic differentiation of mesenchymal stem cells: a review from a mitochondrial perspective. Acta Biomater. 164, 1–14. doi:10.1016/j.actbio.2023.03.032
Frisbie, L., Pressimone, C., Dyer, E., Baruwal, R., Garcia, G., Croix, C. S., et al. (2024). Carcinoma-associated mesenchymal stem cells promote ovarian cancer heterogeneity and metastasis through mitochondrial transfer. Cell Rep. 43 (8), 114551. doi:10.1016/j.celrep.2024.114551
Fu, K., Robbins, S. R., and McDougall, J. J. (2018). Osteoarthritis: the genesis of pain. Rheumatology 57 (Suppl. l_4), iv43–iv50. doi:10.1093/rheumatology/kex419
Fu, H., Cheng, J., Hu, L., Heng, B. C., Zhang, X., Deng, X., et al. (2025). Mitochondria-targeting materials and therapies for regenerative engineering. Biomaterials 316, 123023. doi:10.1016/j.biomaterials.2024.123023
Fujii, Y., Liu, L., Yagasaki, L., Inotsume, M., Chiba, T., and Asahara, H. (2022). Cartilage homeostasis and osteoarthritis. Int. J. Mol. Sci. 23 (11), 6316. doi:10.3390/ijms23116316
Gammage, P. A., Moraes, C. T., and Minczuk, M. (2018). Mitochondrial genome engineering: the revolution may not be CRISPR-Ized. Trends Genet. 34 (2), 101–110. doi:10.1016/j.tig.2017.11.001
Gao, J., Qin, A., Liu, D., Ruan, R., Wang, Q., Yuan, J., et al. (2019). Endoplasmic reticulum mediates mitochondrial transfer within the osteocyte dendritic network. Sci. Adv. 5 (11), eaaw7215. doi:10.1126/sciadv.aaw7215
Giuliana, M. B., Marie, M., Christian, J., and Danièle, N. (2023). Therapeutic potential in rheumatic diseases of extracellular vesicles derived from mesenchymal stromal cells. Nat. Rev. Rheumatol. 19 (11), 682–694. doi:10.1038/s41584-023-01010-7
Glyn-Jones, S., Palmer, A., Agricola, R., Price, A. J., Vincent, T. L., Weinans, H., et al. (2015). Lancet 386 (9991), 376–387. doi:10.1016/S0140-6736(14)60802-3
Guo, P., Alhaskawi, A., Adel Abdo Moqbel, S., and Pan, Z. (2025). Recent development of mitochondrial metabolism and dysfunction in osteoarthritis. Front. Pharmacol. 16, 1538662. doi:10.3389/fphar.2025.1538662
Hannah, S., Frederik, E., Lena, W., Sabine, O., Kay, J., Csaba, C., et al. (2024). Microglia rescue neurons from aggregate-induced neuronal dysfunction and death through tunneling nanotubes. Neuron 112 (18), 3106–3125.e8. doi:10.1016/j.neuron.2024.06.029
Hayakawa, K., Esposito, E., Wang, X., Terasaki, Y., Liu, Y., Xing, C., et al. (2016). Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535 (7613), 551–555. doi:10.1038/nature18928
Heidari, B. (2011). Knee osteoarthritis prevalence, risk factors, pathogenesis and features: part I. Casp. J. Intern. Med. 2 (2), 205–212.
Herzig, S., and Shaw, R. J. (2018). AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. cell Biol. 19 (2), 121–135. doi:10.1038/nrm.2017.95
Hu, Z., Wang, D., Gong, J., Li, Y., Ma, Z., Luo, T., et al. (2023). MSCs deliver hypoxia-treated Mitochondria reprogramming Acinar metabolism to alleviate severe Acute Pancreatitis injury. Adv. Sci. (Weinh) 10 (25), e2207691. doi:10.1002/advs.202207691
Hu, L., Tang, D., Qi, B., Guo, D., Wang, Y., Geng, J., et al. (2024). Mfn2/Hsc70 complex mediates the Formation of Mitochondria-Lipid droplets membrane contact and regulates myocardial lipid metabolism. Adv. Sci. (Weinh) 11 (14), e2307749. doi:10.1002/advs.202307749
Huang, T., Zhang, T., Jiang, X., Li, A., Su, Y., Bian, Q., et al. (2021). Iron oxide nanoparticles augment the intercellular mitochondrial transfer–mediated therapy. Sci. Adv. 7 (40), eabj0534. doi:10.1126/sciadv.abj0534
Huang, N., Chen, Z., Yang, X., Gao, Y., Zhong, J., Li, Y., et al. (2023). Upstream open reading frame-encoded MP31 disrupts the mitochondrial quality control process and inhibits tumorigenesis in glioblastoma. Neuro-oncology 25 (11), 1947–1962. doi:10.1093/neuonc/noad099
Hunter, D., and Bierma-Zeinstra, S. (2019). Osteoarthr. Lancet 393 (10182), 1745–1759. doi:10.1016/S0140-6736(19)30417-9
Hutto, R. A., Rutter, K. M., Giarmarco, M. M., Parker, E. D., Chambers, Z. S., and Brockerhoff, S. E. (2023). Cone photoreceptors transfer damaged mitochondria to Müller glia. Cell Rep. 42 (2), 112115. doi:10.1016/j.celrep.2023.112115
Ikeda, G., Santoso, M. R., Tada, Y., Li, A. M., Vaskova, E., Jung, J. H., et al. (2021). Mitochondria-Rich extracellular vesicles from autologous stem cell-derived cardiomyocytes restore energetics of ischemic myocardium. J. Am. Coll. Cardiol. 77 (8), 1073–1088. doi:10.1016/j.jacc.2020.12.060
Irwin, R. M., Thomas, M. A., Fahey, M. J., Mayán, M. D., Smyth, J. W., and Delco, M. L. (2024). Connexin 43 regulates intercellular mitochondrial transfer from human mesenchymal stromal cells to chondrocytes. Stem Cell Res. and Ther. 15 (1), 359. doi:10.1186/s13287-024-03932-9
Iwamoto, M., Ohta, Y., Larmour, C., and Enomoto-Iwamoto, M. (2013). Toward regeneration of articular cartilage. Birth Defects Res. Part C Embryo Today Rev. 99 (3), 192–202. doi:10.1002/bdrc.21042
Jacoby, E., Bar-Yosef, O., Gruber, N., Lahav, E., Varda-Bloom, N., Bolkier, Y., et al. (2022). Mitochondrial augmentation of hematopoietic stem cells in children with single large-scale mitochondrial DNA deletion syndromes. Sci. Transl. Med. 14 (676), eabo3724. doi:10.1126/scitranslmed.abo3724
Jerry, C. H., and Kyriacos, A. A. (2004). Low-density cultures of bovine chondrocytes: effects of scaffold material and culture system. Biomaterials 26 (14). doi:10.1016/j.biomaterials.2004.06.038
Jia, X., Wang, Q., Ji, J., Lu, W., Liu, Z., Tian, H., et al. (2023). Mitochondrial transplantation ameliorates hippocampal damage following status epilepticus. Anim. Model Exp. Med. 6 (1), 41–50. doi:10.1002/ame2.12310
Jiang, H., Zhang, Y., Hu, G., Ji, P., Ming, J., Li, Y., et al. (2024). RNA-binding protein HNRNPD promotes chondrocyte senescence and osteoarthritis progression through upregulating FOXM1. Commun. Biol. 7 (1), 1695. doi:10.1038/s42003-024-07407-8
Jiao, H., Jiang, D., Hu, X., Du, W., Ji, L., Yang, Y., et al. (2021). Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell 184 (11), 2896–910.e13. doi:10.1016/j.cell.2021.04.027
Joshi, A. U., Minhas, P. S., Liddelow, S. A., Haileselassie, B., Andreasson, K. I., Dorn, G. W., et al. (2019). Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 22 (10), 1635–1648. doi:10.1038/s41593-019-0486-0
Justin, F., Lydia-Marie, J., Neda, F., Michael, F. S., Andrey, V. M., Wah, C., et al. (2024). Dramatic changes in mitochondrial subcellular location and morphology accompany activation of the CO(2) concentrating mechanism. Proc. Natl. Acad. Sci. U. S. A. 121 (43), e2407548121. doi:10.1073/pnas.2407548121
Kai, F., Teng, Y., Xuetao, X., Jiashuo, L., Liangzhi, G., Zhengsheng, C., et al. (2024). ESC-sEVs alleviate non-early-stage osteoarthritis progression by rejuvenating senescent chondrocytes via FOXO1A-autophagy axis but not inducing apoptosis. Pharmacol. Res. 209 (0), 107474. doi:10.1016/j.phrs.2024.107474
Kai-di, W., Xiang, D., Nan, J., Chao, Z., Jing, W., Xian-Yi, C., et al. (2021). Digoxin targets low density lipoprotein receptor-related protein 4 and protects against osteoarthritis. Ann. Rheum. Dis. 81 (4). doi:10.1136/annrheumdis-2021-221380
Kan, T., Li, H., Hou, L., Cui, J., Wang, Y., Sun, L., et al. (2024). Matrix stiffness aggravates osteoarthritis progression through H3K27me3 demethylation induced by mitochondrial damage. Iscience 27 (8), 110507. doi:10.1016/j.isci.2024.110507
Kawano, H., Kawano, Y., Yu, C., LaMere, M. W., McArthur, M. J., Becker, M. W., et al. (2023). Mitochondrial transfer to host cells from Ex Vivo expanded donor hematopoietic stem cells. Cells 12 (11), 1473. doi:10.3390/cells12111473
Kidwell, C. U., Casalini, J. R., Pradeep, S., Scherer, S. D., Greiner, D., Bayik, D., et al. (2023). Transferred mitochondria accumulate reactive oxygen species, promoting proliferation. eLife 12, e85494. doi:10.7554/eLife.85494
Kim, M. J., Hwang, J. W., Yun, C.-K., Lee, Y., and Choi, Y.-S. (2018). Delivery of exogenous mitochondria via centrifugation enhances cellular metabolic function. Sci. Rep. 8 (1), 3330. doi:10.1038/s41598-018-21539-y
Kim, K.-I., Lee, W.-S., Kim, J.-H., Bae, J.-K., and Jin, W. (2022). Safety and efficacy of the intra-articular injection of mesenchymal stem cells for the treatment of osteoarthritic knee: a 5-year follow-up study. Stem cells Transl. Med. 11 (6), 586–596. doi:10.1093/stcltm/szac024
Kim, H.-R., Cho, H. B., Lee, S., Park, J.-I., Kim, H. J., and Park, K.-H. (2023). Fusogenic liposomes encapsulating mitochondria as a promising delivery system for osteoarthritis therapy. Biomaterials 302, 122350. doi:10.1016/j.biomaterials.2023.122350
Krześniak, A. M., Radzimowski, K., and Stolarczyk, A. (2021). Comparison of the treatment results of knee osteoarthritis using adipose tissue mesenchymal stromal cells derived through enzymatic digestion and mechanically fragmented adipose tissue. Med. Baltim. 100 (9), e24777. doi:10.1097/MD.0000000000024777
Labarta, E., de Los Santos, M. J., Herraiz, S., Escribá, M. J., Marzal, A., Buigues, A., et al. (2019). Autologous mitochondrial transfer as a complementary technique to intracytoplasmic sperm injection to improve embryo quality in patients undergoing in vitro fertilization—a randomized pilot study. Fertil. Steril. 111 (1), 86–96. doi:10.1016/j.fertnstert.2018.09.023
Lane, N., and Martin, W. (2010). The energetics of genome complexity. Nature 467 (7318), 929–934. doi:10.1038/nature09486
Lazar, S., and Goldfinger, L. E. (2021). Platelets and extracellular vesicles and their cross talk with cancer. Blood 137 (23), 3192–3200. doi:10.1182/blood.2019004119
Lee, S., Jeong, S.-Y., Lim, W.-C., Kim, S., Park, Y.-Y., Sun, X., et al. (2007). Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J. Biol. Chem. 282 (31), 22977–22983. doi:10.1074/jbc.M700679200
Lee, S.-M., Dho, S. H., Ju, S.-K., Maeng, J.-S., Kim, J.-Y., and Kwon, K.-S. (2012). Cytosolic malate dehydrogenase regulates senescence in human fibroblasts. Biogerontology 13, 525–536. doi:10.1007/s10522-012-9397-0
Lee, A. R., Woo, J. S., Lee, S.-Y., Na, H. S., Cho, K.-H., Lee, Y. S., et al. (2022). Mitochondrial transplantation ameliorates the development and progression of osteoarthritis. Immune Netw. 22 (2), e14. doi:10.4110/in.2022.22.e14
Lepetsos, P., and Papavassiliou, A. G. (2016). ROS/oxidative stress signaling in osteoarthritis. Biochimica Biophysica Acta (BBA)-Molecular Basis Dis. 1862 (4), 576–591. doi:10.1016/j.bbadis.2016.01.003
Levoux, J., Prola, A., Lafuste, P., Gervais, M., Chevallier, N., Koumaiha, Z., et al. (2021). Platelets facilitate the wound-healing capability of mesenchymal stem cells by mitochondrial transfer and metabolic reprogramming. Cell Metab. 33 (2), 688–690. doi:10.1016/j.cmet.2021.02.003
Li, D., Ni, S., Miao, K.-S., and Zhuang, C. (2019). PI3K/Akt and caspase pathways mediate oxidative stress-induced chondrocyte apoptosis. Cell Stress Chaperones 24, 195–202. doi:10.1007/s12192-018-0956-4
Li, X., Zhu, W., Cheng, Y., Ren, Z., Liu, X., Yang, H., et al. (2024a). Intrauterine hyperglycemia induces SIRT3-mediated mitochondrial dysfunction: the fetal origin pathogenesis of precocious osteoarthritis. Osteoarthr. Cartil. 32, 950–962. doi:10.1016/j.joca.2024.05.003
Li, Z., Zhenhui, L., Guojie, X., Xing, N., and Jinmin, Z. (2024b). Dual-targeted disease-modifying therapies for osteoarthritis. Lancet 403 (10444), 2591. doi:10.1016/s0140-6736(24)00475-6
Li, X., Zhang, Z., Jiang, W., Ju, Y., Guo, W., and Huang, Z. (2024c). Dipeptidyl peptidase 4 (DPP4) exacerbates osteoarthritis progression in an enzyme-independent manner. Adv. Sci. (Weinh) 12, e2410525. doi:10.1002/advs.202410525
Liang, W., Sagar, S., Ravindran, R., Najor, R. H., Quiles, J. M., Chi, L., et al. (2023). Mitochondria are secreted in extracellular vesicles when lysosomal function is impaired. Nat. Commun. 14 (1), 5031. doi:10.1038/s41467-023-40680-5
Lim, H. C., Park, Y. B., Ha, C. W., Cole, B. J., Lee, B. K., Jeong, H. J., et al. (2021). Allogeneic umbilical cord blood-derived mesenchymal stem cell implantation versus microfracture for large, full-thickness cartilage defects in older patients: a multicenter randomized clinical trial and extended 5-Year clinical Follow-up. Orthop. J. Sports Med. 9 (1), 2325967120973052. doi:10.1177/2325967120973052
Lin, R. Z., Im, G. B., Luo, A. C., Zhu, Y., Hong, X., Neumeyer, J., et al. (2024). Mitochondrial transfer mediates endothelial cell engraftment through mitophagy. Nature 629 (8012), 660–668. doi:10.1038/s41586-024-07340-0
Loeser, R. F., Collins, J. A., and Diekman, B. O. (2016). Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 12 (7), 412–420. doi:10.1038/nrrheum.2016.65
Long, X., Liu, M., Nan, Y., Chen, Q., Xiao, Z., Xiang, Y., et al. (2024). Revitalizing Ancient mitochondria with Nano-Strategies: mitochondria-remedying nanodrugs concentrate on disease control. Adv. Mater. 36 (18), 2308239. doi:10.1002/adma.202308239
López-Doménech, G., Covill-Cooke, C., Ivankovic, D., Halff, E. F., Sheehan, D. F., Norkett, R., et al. (2018). Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. Embo J. 37 (3), 321–336. doi:10.15252/embj.201696380
Macip, S., Igarashi, M., Fang, L., Chen, A., Pan, Z. Q., Lee, S. W., et al. (2002). Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 21, 2180–2188. doi:10.1093/emboj/21.9.2180
MacVicar, T., Ohba, Y., Nolte, H., Mayer, F. C., Tatsuta, T., Sprenger, H. G., et al. (2019). Lipid signalling drives proteolytic rewiring of mitochondria by YME1L. Nature 575 (7782), 361–365. doi:10.1038/s41586-019-1738-6
Manford, A. G., Mena, E. L., Shih, K. Y., Gee, C. L., McMinimy, R., Martínez-González, B., et al. (2021). Structural basis and regulation of the reductive stress response. Cell 184 (21), 5375–90.e16. doi:10.1016/j.cell.2021.09.002
Marabitti, V., Vulpis, E., Nazio, F., and Campello, S. (2024). Mitochondrial transfer as a strategy for enhancing cancer cell fitness: current insights and future directions. Pharmacol. Res. 208, 107382. doi:10.1016/j.phrs.2024.107382
Mathiessen, A., and Conaghan, P. G. (2017). Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res. and Ther. 19, 18–19. doi:10.1186/s13075-017-1229-9
Mayan, M. D., Gago-Fuentes, R., Carpintero-Fernandez, P., Fernandez-Puente, P., Filgueira-Fernandez, P., Goyanes, N., et al. (2015). Articular chondrocyte network mediated by gap junctions: role in metabolic cartilage homeostasis. Ann. rheumatic Dis. 74 (1), 275–284. doi:10.1136/annrheumdis-2013-204244
Mayoly, A., Iniesta, A., Curvale, C., Kachouh, N., Jaloux, C., Eraud, J., et al. (2019). Development of autologous platelet-rich plasma mixed-microfat as an advanced therapy medicinal product for intra-articular injection of radio-carpal osteoarthritis: from validation data to preliminary clinical results. Int. J. Mol. Sci. 20 (5), 1111. doi:10.3390/ijms20051111
McCully, J. D., Cowan, D. B., Pacak, C. A., Toumpoulis, I. K., Dayalan, H., and Levitsky, S. (2009). Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiology-Heart Circulatory Physiology 296 (1), H94-H105–H105. doi:10.1152/ajpheart.00567.2008
Mercedes, F.-M., Angel, S.-H., María, E. V.-M., Estefanía, C.-P., Sara, R., Tamara, H.-G., et al. (2025). Mitochondrial DNA haplogroups influence the risk of incident knee osteoarthritis in OAI and CHECK cohorts. A meta-analysis and functional study. Ann. Rheum. Dis. 76 (6). doi:10.1136/annrheumdis-2016-210131
Milenkovic, D., Misic, J., Hevler, J. F., Molinié, T., Chung, I., Atanassov, I., et al. (2023). Preserved respiratory chain capacity and physiology in mice with profoundly reduced levels of mitochondrial respirasomes. Cell Metab. 35 (10), 1799–813.e7. doi:10.1016/j.cmet.2023.07.015
Min, T., Yingfeng, T., Yanqiu, G., Qin, Y., Jinrui, W., Zhenzhen, Z., et al. (2025). β-hydroxybutyrate facilitates mitochondrial-derived vesicle biogenesis and improves mitochondrial functions. Mol. Cell 85 (7), 1395–1410.e5. doi:10.1016/j.molcel.2025.02.022
Miriam, L., Philippa, R. B., Lyra, O. R., Claire, Y. M., Julia, M. M., Doreen, A. C., et al. (2021). Mitochondrial translation is required for sustained killing by cytotoxic T cells. Science. 374 (6565). doi:10.1126/science.abe9977
Mistry, J. J., Marlein, C. R., Moore, J. A., Hellmich, C., Wojtowicz, E. E., Smith, J. G. W., et al. (2019). ROS-mediated PI3K activation drives mitochondrial transfer from stromal cells to hematopoietic stem cells in response to infection. Proc. Natl. Acad. Sci. U. S. A. 116 (49), 24610–24619. doi:10.1073/pnas.1913278116
Mitch, L. (2022). Moms' mitochondria may refresh cells in sick kids. Science. 378 (6626), 1267. doi:10.1126/science.adg3936
Miwa, S., Kashyap, S., Chini, E., and von Zglinicki, T. (2022). Mitochondrial dysfunction in cell senescence and aging. J. Clin. investigation 132 (13), e158447. doi:10.1172/JCI158447
Monzel, A. S., Enríquez, J. A., and Picard, M. (2023). Multifaceted mitochondria: moving mitochondrial science beyond function and dysfunction. Nat. Metab. 5 (4), 546–562. doi:10.1038/s42255-023-00783-1
Morena, S., Carlos, V.-G., Ana Victoria, L.-V., Alberto Centeno, C., María Concepción Jiménez, G., Purificación, F.-F., et al. (2022). mtDNA variability determines spontaneous joint aging damage in a conplastic mouse model. Aging (Albany NY) 14 (15), 5966–5983. doi:10.18632/aging.204153
Morrison, T. J., Jackson, M. V., Cunningham, E. K., Kissenpfennig, A., McAuley, D. F., O'Kane, C. M., et al. (2017). Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. Am. J. Respir. Crit. Care Med. 196 (10), 1275–1286. doi:10.1164/rccm.201701-0170OC
Motta, F., Barone, E., Sica, A., and Selmi, C. (2023). Inflammaging and osteoarthritis. Clin. Rev. Allergy and Immunol. 64 (2), 222–238. doi:10.1007/s12016-022-08941-1
Muthu, S., Korpershoek, J. V., Novais, E. J., Tawy, G. F., Hollander, A. P., and Martin, I. (2023). Failure of cartilage regeneration: emerging hypotheses and related therapeutic strategies. Nat. Rev. Rheumatol. 19 (7), 403–416. doi:10.1038/s41584-023-00979-5
Naomasa, Y., Masayuki, Y., Tomohiko, S., Tetsuya, K., and Hideto, K. (2017). Clinical results following intra-articular injection of adipose-derived stromal vascular fraction cells in patients with osteoarthritis of the knee. Regen. Ther. 6 (0), 108–112. doi:10.1016/j.reth.2017.04.002
Naón, D., Hernández-Alvarez, M. I., Shinjo, S., Wieczor, M., Ivanova, S., Martins de Brito, O., et al. (2023). Splice variants of mitofusin 2 shape the endoplasmic reticulum and tether it to mitochondria. Science. 380 (6651), eadh9351. doi:10.1126/science.adh9351
Nasoni, M. G., Carloni, S., Canonico, B., Burattini, S., Cesarini, E., Papa, S., et al. (2021). Melatonin reshapes the mitochondrial network and promotes intercellular mitochondrial transfer via tunneling nanotubes after ischemic-like injury in hippocampal HT22 cells. J. Pineal Res. 71 (1), e12747. doi:10.1111/jpi.12747
National Academies of Sciences E, Medicine (2016). “Committee on the ethical and social Policy considerations of novel techniques for prevention of maternal transmission of mitochondrial DNA diseases,” in Mitochondrial replacement techniques: ethical, social, and Policy considerations. Washington (DC): Institute of Medicine.
Nicolás-Ávila, J. A., Lechuga-Vieco, A. V., Esteban-Martínez, L., Sánchez-Díaz, M., Díaz-García, E., Santiago, D. J., et al. (2020). A network of macrophages supports mitochondrial homeostasis in the heart. Cell 183 (1), 94–109.e23. doi:10.1016/j.cell.2020.08.031
Norat, P., Sokolowski, J. D., Gorick, C. M., Soldozy, S., Kumar, J. S., Chae, Y., et al. (2023). Intraarterial transplantation of Mitochondria after ischemic stroke reduces cerebral infarction. Stroke Vasc. Interv. Neurol. 3 (3). doi:10.1161/svin.122.000644
Olga, A. E., Douglas, M. W., Syam Sundar, N., John, N. A., and Squire, J. B. (2025). Structural basis for catalysis by human lipoyl synthase. Nat. Commun. 16 (1), 6355. doi:10.1038/s41467-025-61393-x
Orozco, L., Munar, A., Soler, R., Alberca, M., Soler, F., Huguet, M., et al. (2014). Treatment of knee osteoarthritis with autologous mesenchymal stem cells: two-year follow-up results. Transplantation 97 (11), e66–e68. doi:10.1097/TP.0000000000000167
Papatriantafyllou, M. (2024). PDZK1 downregulation linked to mitochondrial dysfunction in OA. Nat. Rev. Rheumatol. 20 (10), 597. doi:10.1038/s41584-024-01157-x
Patel, P., Mendoza, A., Robichaux, D. J., Wang, M. C., Wehrens, X. H., and Karch, J. (2021). Inhibition of the anti-apoptotic Bcl-2 family by BH3 mimetics sensitize the mitochondrial permeability transition pore through Bax and Bak. Front. Cell Dev. Biol. 9, 765973. doi:10.3389/fcell.2021.765973
Peng, T. I., and Jou, M. J. (2010). Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 1201 (1), 183–188. doi:10.1111/j.1749-6632.2010.05634.x
Peruzzotti-Jametti, L., Bernstock, J. D., Willis, C. M., Manferrari, G., Rogall, R., Fernandez-Vizarra, E., et al. (2021). Neural stem cells traffic functional mitochondria via extracellular vesicles. PLoS Biol. 19 (4), e3001166. doi:10.1371/journal.pbio.3001166
Picca, A., and Lezza, A. M. S. (2015). Regulation of mitochondrial biogenesis through TFAM–mitochondrial DNA interactions: useful insights from aging and calorie restriction studies. Mitochondrion 25, 67–75. doi:10.1016/j.mito.2015.10.001
Prieto-Alhambra, D., Arden, N., and Hunter, D. J. (2014). Osteoarthritis: the facts. New York, NY: OUP Oxford.
Qinglian, L., Patrick, D. S., William, W., Jaroslaw, M., and Elizabeth, A. C. (2003). Regulated cycling of mitochondrial Hsp70 at the protein import channel. Science. 300 (5616), 139–141. doi:10.1126/science.1083379
Rahul, G., Masahiro, K., Timothy, J. D., Kristin, T., Jason, G. M., Anna, V. K., et al. (2023). Nuclear genetic control of mtDNA copy number and heteroplasmy in humans. Nature 620 (7975), 839–848. doi:10.1038/s41586-023-06426-5
Reut, H. B.-M., Dvora, L., Tamar, Z., Koyeli, D., Irit, R.-G., Ziv, P., et al. (2023). Mitochondrial-derived vesicles retain membrane potential and contain a functional ATP synthase. EMBO Rep. 24 (5), e56114. doi:10.15252/embr.202256114
Rieder, B., Weihs, A. M., Weidinger, A., Szwarc, D., Nürnberger, S., Redl, H., et al. (2018). Hydrostatic pressure-generated reactive oxygen species induce osteoarthritic conditions in cartilage pellet cultures. Sci. Rep. 8 (1), 17010. doi:10.1038/s41598-018-34718-8
Ritsuko, N., Stella, V., Rachael, L. F., Henyun, S., Rocky, G., Wentong, J., et al. (2024). Mitochondria transfer-based therapies reduce the morbidity and mortality of Leigh syndrome. Nat. Metab. 6 (10), 1886–1896. doi:10.1038/s42255-024-01125-5
Ronghui, D., Ruifang, Z., Zining, Z., Yang, C., Meng, Y., Yixuan, L., et al. (2024). Chondrocyte membrane-coated nanoparticles promote drug retention and halt cartilage damage in rat and canine osteoarthritis. Sci. Transl. Med. 16 (735), eadh9751. doi:10.1126/scitranslmed.adh9751
Rosina, M., Ceci, V., Turchi, R., Chuan, L., Borcherding, N., Sciarretta, F., et al. (2022). Ejection of damaged mitochondria and their removal by macrophages ensure efficient thermogenesis in brown adipose tissue. Cell Metab. 34 (4), 533–48.e12. doi:10.1016/j.cmet.2022.02.016
Ryu, K. W., Fung, T. S., Baker, D. C., Saoi, M., Park, J., Febres-Aldana, C. A., et al. (2024). Cellular ATP demand creates metabolically distinct subpopulations of mitochondria. Nature 635 (8039), 746–754. doi:10.1038/s41586-024-08146-w
Saha, T., Dash, C., Jayabalan, R., Khiste, S., Kulkarni, A., Kurmi, K., et al. (2022). Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat. Nanotechnol. 17 (1), 98–106. doi:10.1038/s41565-021-01000-4
Sahin, E., Colla, S., Liesa, M., Moslehi, J., Müller, F. L., Guo, M., et al. (2011). Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470 (7334), 359–365. doi:10.1038/nature09787
Saurer, M., Leibundgut, M., Nadimpalli, H. P., Scaiola, A., Schönhut, T., Lee, R. G., et al. (2023). Molecular basis of translation termination at noncanonical stop codons in human mitochondria. Science. 380 (6644), 531–536. doi:10.1126/science.adf9890
Scarpulla, R. C. (2011). Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochimica biophysica acta (BBA)-molecular cell Res. 1813 (7), 1269–1278. doi:10.1016/j.bbamcr.2010.09.019
Shi, G., Lan, S., Zhang, Q., Wang, J., Shu, F., Hao, Z., et al. (2025). Molybdenum nanodots act as antioxidants for photothermal therapy osteoarthritis. Biomaterials 315, 122909. doi:10.1016/j.biomaterials.2024.122909
Singh, B., Tripathi, M., Pandey, P., and Kakkar, P. (2010). Nimesulide aggravates redox imbalance and calcium dependent mitochondrial permeability transition leading to dysfunction in vitro. Toxicology 275, 1–9. doi:10.1016/j.tox.2010.05.001
Spees, J. L., Olson, S. D., Whitney, M. J., and Prockop, D. J. (2006). Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. U. S. A. 103 (5), 1283–1288. doi:10.1073/pnas.0510511103
Stein, L. R., and Imai, S.-i. (2012). The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. and Metabolism 23 (9), 420–428. doi:10.1016/j.tem.2012.06.005
Suh, J., Kim, N., Shim, W., Lee, S., Kim, H., Moon, E., et al. (2023). Mitochondrial fragmentation and donut formation enhance mitochondrial secretion to promote osteogenesis. Cell metab. 35 (2), 345–60.e7. doi:10.1016/j.cmet.2023.01.003
Sun, C., Liu, X., Wang, B., Wang, Z., Liu, Y., Di, C., et al. (2019). Endocytosis-mediated mitochondrial transplantation: transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 9 (12), 3595–3607. doi:10.7150/thno.33100
Sun, K., Jing, X., Guo, J., Yao, X., and Guo, F. (2021). Mitophagy in degenerative joint diseases. Autophagy 17 (9), 2082–2092. doi:10.1080/15548627.2020.1822097
Sun, J., Lo, H. T. J., Fan, L., Yiu, T. L., Shakoor, A., Li, G., et al. (2022). High-efficiency quantitative control of mitochondrial transfer based on droplet microfluidics and its application on muscle regeneration. Sci. Adv. 8 (33), eabp9245. doi:10.1126/sciadv.abp9245
Tanmoy, S., Chinmayee, D., Ruparoshni, J., Sachin, K., Arpita, K., Kiran, K., et al. (2021). Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat. Nanotechnol. 17 (1), 98–106. doi:10.1038/s41565-021-01000
Tishchenko, A., Azorín, D. D., Vidal-Brime, L., Muñoz, M. J., Arenas, P. J., Pearce, C., et al. (2020). Cx43 and associated cell signaling pathways regulate tunneling nanotubes in breast cancer cells. Cancers (Basel) 12 (10), 2798. doi:10.3390/cancers12102798
Tobias, D., Xiaomeng, H., Sean, A.-E., Martina, K., Matthew, H. S., Alessia, G., et al. (2019). De novo mutations in mitochondrial DNA of iPSCs produce immunogenic neoepitopes in mice and humans. Nat. Biotechnol. 37 (10), 1137–1144. doi:10.1038/s41587-019-0227-7
Tracy, T. E., Madero-Pérez, J., Swaney, D. L., Chang, T. S., Moritz, M., Konrad, C., et al. (2022). Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell 185 (4), 712–28.e14. doi:10.1016/j.cell.2021.12.041
Valm, A. M., Cohen, S., Legant, W. R., Melunis, J., Hershberg, U., Wait, E., et al. (2017). Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546 (7656), 162–167. doi:10.1038/nature22369
van der Vlist, M., Raoof, R., Willemen, H., Prado, J., Versteeg, S., Martin Gil, C., et al. (2022). Macrophages transfer mitochondria to sensory neurons to resolve inflammatory pain. Neuron 110 (4), 613–26.e9. doi:10.1016/j.neuron.2021.11.020
van Vulpen, L. F., Schutgens, R. E., Coeleveld, K., Alsema, E. C., Roosendaal, G., Mastbergen, S. C., et al. (2015). IL-1β, in contrast to TNFα, is pivotal in blood-induced cartilage damage and is a potential target for therapy. Blood, J. Am. Soc. Hematol. 126 (19), 2239–2246. doi:10.1182/blood-2015-03-635524
Verkerke, A. R. P., Wang, D., Yoshida, N., Taxin, Z. H., Shi, X., Zheng, S., et al. (2024). BCAA-nitrogen flux in brown fat controls metabolic health independent of thermogenesis. Cell 187 (10), 2359–74.e18. doi:10.1016/j.cell.2024.03.030
Vermulst, M., Bielas, J. H., Kujoth, G. C., Ladiges, W. C., Rabinovitch, P. S., Prolla, T. A., et al. (2007). Mitochondrial point mutations do not limit the natural lifespan of mice. Nat. Genet. 39 (4), 540–543. doi:10.1038/ng1988
Vijith, V., Hao, Y., Juliane, K. L., Kaylin, A. P., Rachna, V. P., Giulia, B., et al. (2024). Extracellular release of damaged mitochondria induced by prehematopoietic stem cell transplant conditioning exacerbates GVHD. Blood Adv. 8 (14), 3691–3704. doi:10.1182/bloodadvances.2023012328
Vives, J., Oliver-Vila, I., and Pla, A. (2015). Quality compliance in the shift from cell transplantation to cell therapy in non-pharma environments. Cytotherapy 17 (8), 1009–1014. doi:10.1016/j.jcyt.2015.02.002
Wan, M., Hua, X., Su, J., Thiagarajan, D., Frostegård, A. G., Haeggström, J. Z., et al. (2014). Oxidized but not native cardiolipin has pro-inflammatory effects, which are inhibited by Annexin A5. Atherosclerosis 235 (2), 592–598. doi:10.1016/j.atherosclerosis.2014.05.913
Wang, X., and Schwarz, T. L. (2009). The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136 (1), 163–174. doi:10.1016/j.cell.2008.11.046
Wang, J., Liu, X., Qiu, Y., Shi, Y., Cai, J., Wang, B., et al. (2018). Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J. Hematol. Oncol. 11 (1), 11. doi:10.1186/s13045-018-0554-z
Wang, R., Maimaitijuma, T., Ma, Y. Y., Jiao, Y., and Cao, Y. P. (2020). Mitochondrial transfer from bone-marrow-derived mesenchymal stromal cells to chondrocytes protects against cartilage degenerative mitochondrial dysfunction in rats chondrocytes. Chin. Med. J. Engl. 134 (2), 212–218.
Wang, R., Maimaitijuma, T., Ma, Y.-Y., Jiao, Y., and Cao, Y.-P. (2021). Mitochondrial transfer from bone-marrow-derived mesenchymal stromal cells to chondrocytes protects against cartilage degenerative mitochondrial dysfunction in rats chondrocytes. Chin. Med. J. 134 (2), 212–218. doi:10.1097/CM9.0000000000001057
Wedan, R. J., Longenecker, J. Z., and Nowinski, S. M. (2024). Mitochondrial fatty acid synthesis is an emergent central regulator of mammalian oxidative metabolism. Cell Metab. 36 (1), 36–47. doi:10.1016/j.cmet.2023.11.017
Weinhäuser, I., Pereira-Martins, D. A., Almeida, L. Y., Hilberink, J. R., Silveira, D. R. A., Quek, L., et al. (2023). M2 macrophages drive leukemic transformation by imposing resistance to phagocytosis and improving mitochondrial metabolism. Sci. Adv. 9 (15), eadf8522. doi:10.1126/sciadv.adf8522
Wenyu, F., Dmytro, V., Yufei, B., Mingshuang, Z., Guodong, S., Asya, K., et al. (2024). Na(v)1.7 as a chondrocyte regulator and therapeutic target for osteoarthritis. Nature 625 (7995), 557–565. doi:10.1038/s41586-023-06888-7
White, M. J., McArthur, K., Metcalf, D., Lane, R. M., Cambier, J. C., Herold, M. J., et al. (2014). Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159 (7), 1549–1562. doi:10.1016/j.cell.2014.11.036
Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98 (1), 115–124. doi:10.1016/S0092-8674(00)80611-X
Wu, Z., Chen, L., Guo, W., Wang, J., Ni, H., Liu, J., et al. (2024). Oral mitochondrial transplantation using nanomotors to treat ischaemic heart disease. Nat. Nanotechnol. 19 (9), 1375–1385. doi:10.1038/s41565-024-01681-7
Xu, Y., Yao, Y., Wang, L., Chen, H., and Tan, N. (2021a). Hyaluronic acid coated liposomes Co-Delivery of natural cyclic peptide RA-XII and mitochondrial targeted photosensitizer for highly selective precise combined treatment of Colon cancer. Int. J. Nanomedicine 16, 4929–4942.
Xu, Y., Yao, Y., Wang, L., Chen, H., and Tan, N. (2021b). Hyaluronic acid coated liposomes Co-delivery of natural cyclic peptide RA-XII and mitochondrial targeted photosensitizer for highly selective precise combined treatment of colon cancer. Int. J. Nanomedicine 16, 4929–4942. doi:10.2147/IJN.S311577
Yao, W.-D., Zhou, J.-N., Tang, C., Zhang, J.-L., Chen, Z.-Y., Li, Y., et al. (2024). Hydrogel microneedle patches loaded with stem cell Mitochondria-Enriched microvesicles boost the chronic wound healing. ACS nano 18 (39), 26733–26750. doi:10.1021/acsnano.4c06921
Yi, W., Li-Fan, H., Na-Hui, L., Jing-Song, Y., Lei, X., Jee-Heon, J., et al. (2024). Mitophagy-Enhanced nanoparticle-engineered mitochondria restore homeostasis of mitochondrial pool for alleviating pulmonary fibrosis. ACS Nano 18 (47), 32705–32722. doi:10.1021/acsnano.4c10328
Ying-Ting, L., Sung-Tzu, C., Jui-Chih, C., Ren-Jie, T., Chin-San, L., and Gou-Jen, W. (2019). Green extraction of healthy and additive free mitochondria with a conventional centrifuge. Lab. Chip 19 (22), 3862–3869. doi:10.1039/c9lc00633h
Yong Sang, K., Oh Ryong, K., Yun, J. C., Dong Suk, S., Dong Beom, H., and Yong Gon, K. (2015). Comparative matched-pair analysis of the injection versus implantation of mesenchymal stem cells for knee osteoarthritis. Am. J. Sports Med. 43 (11), 2738–2746. doi:10.1177/0363546515599632
Yu, M., Wang, D., Chen, X., Zhong, D., and Luo, J. (2022). BMSCs-derived mitochondria improve osteoarthritis by ameliorating mitochondrial dysfunction and promoting mitochondrial biogenesis in chondrocytes. Stem Cell Rev. Rep. 18 (8), 3092–3111. doi:10.1007/s12015-022-10436-7
Yuan, Y., Yuan, L., Li, L., Liu, F., Liu, J., Chen, Y., et al. (2021). Mitochondrial transfer from mesenchymal stem cells to macrophages restricts inflammation and alleviates kidney injury in diabetic nephropathy mice via PGC-1α activation. Stem cells Dayt. Ohio 39 (7), 913–928. doi:10.1002/stem.3375
Yulong, W., Lijun, L., Tao, G., Feifan, Y., Lesan, Y., Lutian, Y., et al. (2021). Targeting cartilage EGFR pathway for osteoarthritis treatment. Sci. Transl. Med. 13 (576), eabb3946. doi:10.1126/scitranslmed.abb3946
Zhai, Q., Chen, X., Fei, D., Guo, X., He, X., Zhao, W., et al. (2022). Nanorepairers rescue inflammation-induced mitochondrial dysfunction in mesenchymal stem cells. Adv. Sci. 9 (4), 2103839. doi:10.1002/advs.202103839
Zhang, Y., Hou, M., Liu, Y., Liu, T., Chen, X., Shi, Q., et al. (2022a). Recharge of chondrocyte mitochondria by sustained release of melatonin protects cartilage matrix homeostasis in osteoarthritis. J. Pineal Res. 73 (2), e12815. doi:10.1111/jpi.12815
Zhang, L., Chen, X., Cai, P., Sun, H., Shen, S., Guo, B., et al. (2022b). Reprogramming mitochondrial metabolism in synovial macrophages of early osteoarthritis by a camouflaged meta-defensome. Adv. Mater. 34 (30), 2202715. doi:10.1002/adma.202202715
Zhang, F., Yoon, K., Zhang, D. Y., Kim, N. S., Ming, G. L., and Song, H. (2023a). Epitranscriptomic regulation of cortical neurogenesis via Mettl8-dependent mitochondrial tRNA m(3)C modification. Cell Stem Cell 30 (3), 300–11.e11. doi:10.1016/j.stem.2023.01.007
Zhang, H., Yu, X., Ye, J., Li, H., Hu, J., Tan, Y., et al. (2023b). Systematic investigation of mitochondrial transfer between cancer cells and T cells at single-cell resolution. Cancer Cell 41 (10), 1788–802.e10. doi:10.1016/j.ccell.2023.09.003
Zhang, Y., Liu, Y., Hou, M., Xia, X., Liu, J., Xu, Y., et al. (2023c). Reprogramming of mitochondrial respiratory chain complex by targeting SIRT3-COX4I2 axis attenuates osteoarthritis progression. Adv. Sci. 10 (10), 2206144. doi:10.1002/advs.202206144
Zhou, B., Kreuzer, J., Kumsta, C., Wu, L., Kamer, K. J., Cedillo, L., et al. (2019). Mitochondrial permeability uncouples elevated autophagy and lifespan extension. Cell 177 (2), 299–314. doi:10.1016/j.cell.2019.02.013
Zhou, J., Zhang, L., Peng, J., Zhang, X., Zhang, F., Wu, Y., et al. (2024). Astrocytic LRP1 enables mitochondria transfer to neurons and mitigates brain ischemic stroke by suppressing ARF1 lactylation. Cell Metab. 36 (9), 2054–68.e14. doi:10.1016/j.cmet.2024.05.016
Zhu, Y., Fujimaki, M., Snape, L., Lopez, A., Fleming, A., and Rubinsztein, D. C. (2024). Loss of WIPI4 in neurodegeneration causes autophagy-independent ferroptosis. Nat. Cell Biol. 26 (4), 542–551. doi:10.1038/s41556-024-01373-3
Zierhut, C., Yamaguchi, N., Paredes, M., Luo, J. D., Carroll, T., and Funabiki, H. (2019). The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell 178 (2), 302–315. doi:10.1016/j.cell.2019.05.035
Glossary
AdMSC-Exos Exosomes from adipose-derived mesenchymal stem cells
ADP Adenosine diphosphate
AKT Protein kinase B
AMP Adenosine monophosphate
AMPK Adenosine 5′-monophosphate (AMP)-activated protein kinase
ATP Adenosine triphosphate
BMSCs Bone marrow derived mesenchymal stem cells
CaMKII Calcium/calmodulin-dependent protein kinase II
Cx43 Connexin 43
DAMP Danger-associated molecular pattern
DDS Drug delivery system
DMT Disease-modifying therapy
DRP1 Dynamin-related protein one
ECM Extracellular matrix
ERK Extracellular regulated protein kinases
ETC Electron transport chain
EV Extracellular vesicle
Fe-hMSCs Magnetic iron oxide nanoparticle (IONP)-activated human MSCs
Fe/S clusters Iron-Sulfur clusters
FIS1 Fission protein 1
FMCs Fusogenic mitochondrial capsules
GABPA Guanine-adenine(GA)binding protein transcription factor
GJA1-20k Gap junction protein alpha 1–20 kDa
GJC Gap junction channels
GMP-AMP Guanosine monophosphate-adenosine monophosphate
GTPase Guanosine triphosphate
H3K27me3 Histone H3 lysine 27 trimethylation
HSCs Haematopoietic stem cells
IL-1β Interleukin-1 beta
M1 Macrophages Classically activated macrophages
M2 Macrophages Alternatively activated macrophages
MAPK Mitogen-activated protein kinase
MDVs Mitochondrial-derived vesicles
MIA Monoiodoacetate
Miro 1 Mitochondrial Ras homology guanosine triphosphate one
MMPs Matrix metalloproteinases
mPTP Mitochondrial permeability transition pore
MS Multiple sclerosis
MSCs Mesenchymal stem cells
MT Mitochondria
mtDNA Mitochondrial deoxyribonucleic acid
mTOR Mammalian target of rapamycin
NAD+ Nicotinamide adenine dinucleotide (oxidized form)
NADH Nicotinamide adenine dinucleotide (reduced form)
NLRP3 Nucleotide-binding domain(NOD)-like receptor family pyrin domain containing 3
OA Osteoarthritis
OAI Osteoarthritis initiative
OXPHOS Oxidative phosphorylation
PD Parkinson’s disease
PGC-1α Peroxisome proliferator-activated receptor-γ coactivator-1α
PGC-1β Peroxisome proliferator-activated receptor gamma co-activator 1β
Phf8 Plant homeodomain finger protein 8
PI3K Phosphatidylinositol 3 kinase
PINK1 Phosphatase and tensin homolog induced putative kinase one
PN-101 Mitochondria Human umbilical cord mesenchymal stem cell-derived mitochondria
PPARγ Peroxisome proliferator-activated receptor γ
PRP Platelet-rich plasma
ROS Reactive oxygen species
SE Status epilepticus
siRNA Small interfering ribonucleic acid
SIRT Sirtuins
SLSMD Single large-scale mitochondrial deoxyribonucleic acid deletion
SVF Stromal vascular fraction
T-cells Thymus dependent lymphocyte
TFAM Nuclear-encoded mitochondrial transcription factor A
TNF-α Tumor necrosis factor-alpha
TNTs Tunneling nanotubes
UC Umbilical cord
YY-1 Yin yang one
ρ0 cells Cells depleted of mitochondrial deoxyribonucleic acid
Keywords: mitochondrial transfer, osteoarthritis, disease-modifying, therapymitochondrial dysfunction, artificial mitochondrial transfer
Citation: He Y, Wang X, Xu B, Chen S, Li H, Chang B, Hu C, Lan X, Li S and Li G (2025) The potential of mitochondrial transfer as the modifying therapy for osteoarthritis. Front. Cell Dev. Biol. 13:1643141. doi: 10.3389/fcell.2025.1643141
Received: 08 June 2025; Accepted: 12 August 2025;
Published: 26 August 2025.
Edited by:
Martina Di Rienzo, National Institute for Infectious Diseases Lazzaro Spallanzani (IRCCS), ItalyReviewed by:
Sapan Borah, National Institute of Pharmaceutical Education and Research, IndiaPeriyasamy Govindaraj, National Institute of Mental Health and Neurosciences, India
Copyright © 2025 He, Wang, Xu, Chen, Li, Chang, Hu, Lan, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guangwen Li, bGlndWFuZ3dlbkBzd211LmVkdS5jbg==; Shiting Li, dGluZ3RpbmdoaWdoQHNpbmEuY29t; Xiaorong Lan, eGlhb3JvbmdsYW5jZG1AMTYzLmNvbQ==
†These authors have contributed equally to this work