 Yong Qiu1,2
Yong Qiu1,2 Xiao Xiao Tang3,4*
Xiao Xiao Tang3,4* Shigeyuki Shichino5
Shigeyuki Shichino5 Prem Prakash Kushwaha6Yan Y. Sanders7
Prem Prakash Kushwaha6Yan Y. Sanders7 Remo Castro Russo8
Remo Castro Russo8 Chunheng Mo1*
Chunheng Mo1*- 1Key Laboratory of Birth Defects and Related Diseases of Women and Children of MOE, State Key Laboratory of Biotherapy, West China Second University Hospital, Sichuan University, Chengdu, China
- 2Department of Anesthesiology, Zigong First People's Hospital, Zigong Academy of Medical Sciences, Zigong, Sichuan, China
- 3State Key Laboratory of Respiratory Disease, National Clinical Research Center for Respiratory Disease, National Center for Respiratory Medicine, Guangzhou Institute of Respiratory Health, The First Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
- 4Guangzhou Laboratory, Guangzhou, China
- 5Division of Molecular Regulation of Inflammatory and Immune Diseases, Research Institute of Biomedical Sciences, Tokyo University of Science, Chiba, Japan
- 6School of Medicine, Case Western Reserve University, Cleveland, United States
- 7Department of Biomedical and Translational Sciences, Eastern Virginia Medical School, Old Dominion University Norfolk, Norfolk, VA, United States
- 8Laboratory of Pulmonary Immunology and Mechanics, Department of Physiology and Biophysics, Institute of Biological Sciences, Federal University of Minas Gerais (UFMG), Belo Horizonte, Brazil
Editorial on the Research Topic
Cellular and molecular mechanisms of lung regeneration, repair, and fibrosis, volume II
Organ fibrosis represents a substantial a major contributor to global disease burden, implicated in over 30% of cases leading to disability-associated and fatal conditions. Among these conditions, idiopathic pulmonary fibrosis (IPF) stands as a progressive and frequently fatal interstitial lung disease characterized by aberrant extracellular matrix deposition and parenchymal scarring. The prognosis for IPF patients is poor, with a median survival time of just 3–5 years (Bhattacharya and Ramachandran, 2023; Lederer and Martinez, 2018, Ma et al., 2024, Wei et al., 2024). Current FDA-approved therapeutics, Pirfenidone and Nintedanib, demonstrate capacity to attenuate functional decline in pulmonary fibrosis but remain disease-modifying rather than curative. Consequently, elucidating the underlying cellular and molecular mechanisms while developing more effective treatments constitutes an imperative research priority (Bonella et al., 2023, Podolanczuk et al., 2023, Zhang et al., 2023, Zhou et al., 2024).
Impaired pulmonary regeneration and repair mechanisms represent critical determinants in the pathogenesis of lung injury-induced fibrosis. As the primary interface for gas exchange, lungs process >10,000 L of air daily to facilitate oxygen delivery and carbon dioxide clearance. This direct environmental exposure predisposes pulmonary tissue to damage from diverse insults including airborne pollutants, tobacco smoke, chemical agents, and viral or bacterial pathogens (Mo et al., 2024, Moss et al., 2022, Neupane et al., 2020). Post-injury, the lungs activate intrinsic regenerative programs by recruiting multiple stem cell populations—such as type 2 alveolar epithelial (AT2) cells, basal cells, club cells, lineage-negative epithelial progenitors (LNEPs), bronchioalveolar stem cells (BASCs), and respiratory airway secretory cells (RASCs). Efficient execution of these repair cascades is essential for restoring pulmonary homeostasis. However, persistent injury or dysregulated healing responses can compromise regenerative capacity, promoting pathological extracellular matrix deposition that manifests as fibrotic scarring and progressive functional impairment (Basil et al., 2020, Chen et al., 2024, Jones et al., 2024, Liu et al., 2024, Niethamer et al., 2025, Wei et al., 2025) (Figure 1). Elucidating the cellular and molecular basis of lung regeneration and repair is therefore fundamental for deciphering fibrotic pathogenesis and designing targeted therapies.
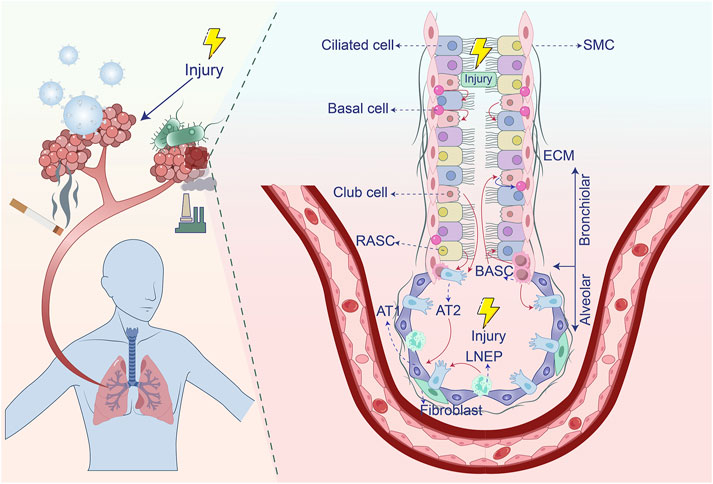
Figure 1. Stem cell populations involved in lung regeneration and repair after injury. After airway damage, basal cells have the capacity to differentiate into both club cells and ciliated cells. Club cells, in turn, can give rise to ciliated cells and are capable of dedifferentiating into basal cells when basal cells are lost. BASCs contribute to the regeneration of both club cells and ciliated cells. In response to alveolar injury, Club cells and BASCs play a key role in replenishing AT2 cells. LNEPs are able to differentiate into AT2 cells, while RASCs primarily give rise to AT2 cells. Additionally, AT2 cells have the ability to differentiate into AT1 cells. Extracellular matrix, ECM; Type 1 alveolar epithelial cells, AT1; Type 2 alveolar epithelial cells, AT2; Alveolar macrophages, AM; Smooth muscle cell, SMC; Lineage-negative epithelial progenitors, LNEPs; Bronchioalveolar stem cells, BASCs; Respiratory airway secretory cells, RASCs.
Current understanding of the cellular and molecular mechanisms governing lung regeneration, repair, and fibrosis remains incomplete. This Research Topic addresses this pressing knowledge gap through two original research article and two comprehensive reviews, collectively advancing mechanistic insights into these interconnected processes. The presented findings elucidate novel aspects of pulmonary regenerative biology and fibrotic pathogenesis while proposing translational diagnostic frameworks and targeted therapeutic strategies for pulmonary fibrosis and related interstitial lung diseases.
Early intervention is critical for effective IPF management. Improving outcomes necessitates identifying precise biomarkers and targets, particularly in early disease stages, to enable timely therapeutic strategies (Karampitsakos et al., 2023, Oldham et al., 2024). Huang et al. synthesized molecular and clinical evidence highlighting SIRT3, SIRT6, and SIRT7 as pivotal regulators in pulmonary fibrosis pathogenesis. These sirtuins exhibit significantly reduced expression in IPF patient-derived fibroblasts, with SIRT7 demonstrating the most pronounced deficiency. SIRT3/6/7 deficiencies disrupt mitochondrial homeostasis, promote oxidative stress, and drive fibroblast-to-myofibroblast transition (FMT) and epithelial-mesenchymal transition (EMT) via dysregulation of TGF-β/Smad, NF-κB, and cGAS-STING pathways. A “sirtuin activity score,” reflecting expression and functional status of SIRT3/6/7 and their downstream targets (e.g., SOD2, OGG1, Smad2/3), correlates significantly with clinical fibrosis severity and disease progression. Pharmacological activation of SIRT3 (e.g., by honokiol, resveratrol) or SIRT6 (e.g., by IMU-856) attenuates mitochondrial damage, inflammation, and fibrogenesis in preclinical models, demonstrating therapeutic potential. In addition, Clinical trials (e.g., NCT05417061 for SIRT6 activators) are evaluating sirtuin-targeted interventions, supported by biomarker analyses showing SIRT3 cytoplasmic levels predict disease outcomes in related conditions. These findings establish SIRT3/6/7 as multifunctional biomarkers and therapeutic targets, offering new diagnostic avenues and mechanistic insights into IPF pathogenesis, while paving the way for sirtuin-modulating therapies to alter disease trajectories.
Bronchopulmonary dysplasia (BPD) represents a developmental disorder of prematurity characterized by impaired alveolarization and pulmonary microvascular dysgenesis, stemming from disrupted lung development and aberrant repair mechanisms. Successful lung regeneration and structural maturation require coordinated signaling within the alveolar niche, involving precise epithelial-endothelial-mesenchymal crosstalk (Gilfillan et al., 2021, Sucre et al., 2018). However, dysregulation of hepatocyte growth factor (HGF)-mediated signaling in the epithelial-endothelial-mesenchymal network disrupts these critical interactions, exacerbating alveolar simplification and fibrosis. Sang and Qiao investigated the role of HGF/c-Met signaling in BPD pathogenesis. They analyzed hyperoxia-induced BPD models and observed that HGF deficiency diminished pulmonary vascular density and promoted epithelial-mesenchymal transition (EMT). Conversely, recombinant HGF (rhHGF) administration activated downstream PI3K/Akt and ERK pathways, stimulating angiogenesis and suppressing TGFβ-driven EMT. Furthermore, HGF synergized with VEGF to stabilize nascent vasculature and countered ECM stiffness by upregulating MMP-9. These findings demonstrate that HGF insufficiency disrupts vascular-epithelial crosstalk, impeding lung repair in BPD. Critically, HGF augmentation promotes alveolar maturation through dual mechanisms: pro-angiogenic activation and anti-fibrotic pathway modulation.
Circular RNAs (circRNAs) represent emerging candidates as diagnostic biomarkers and therapeutic targets in asthma pathogenesis. Non-coding RNAs, including miRNAs and circRNAs, play crucial roles in regulating airway inflammation and remodeling. Alongside miRNA-mediated gene silencing, the competing endogenous RNA (ceRNA) mechanism has emerged as a key regulatory pathway in asthma pathogenesis (Jia et al., 2024, Liu et al., 2023). Liu et al. investigated the role of circ-0001454 in asthma. Circ-0001454 was characterized by its cytoplasmic localization, ability to sponge miRNAs, and involvement in modulating oxidative stress and mitochondrial function. They identified a negative correlation between circ-0001454 and miR-770-5p, demonstrating that circ-0001454 acts as a molecular sponge to sequester miR-770-5p, thereby alleviating its suppression of the target gene cbl-b. They also established that the circ-0001454/miR-770-5p/cbl-b axis regulates downstream signaling proteins (EGFR, AKT1, MAPK1), reducing inflammation, oxidative stress, apoptosis, and mitochondrial dysfunction in bronchial epithelial cells and murine models. In vivo, circ-0001454 overexpression significantly attenuated airway hyperresponsiveness, inflammatory cytokine levels (IL-4, IL-5, IL-13), mucus hypersecretion, collagen deposition, and α-SMA expression. These findings establish circ-0001454 as a pleiotropic regulator of asthmatic pathophysiology while highlighting its translational potential as both a biomarker and RNA-based therapeutic target for mitigating airway inflammation and remodeling.
In addition to asthma and lung fibrosis, chronic obstructive pulmonary disease (COPD), characterized by persistent airflow limitation and chronic airway inflammation often linked to smoking, represents a major cause of global lung function impairment. Transcriptomic analysis of differentiating airway epithelial cells provides a powerful tool for identifying key regulators of COPD-associated epithelial remodeling (Lahmar et al., 2022, Petit et al., 2023). Laure et al. employed this approach combined with Hedgehog pathway inhibition to investigate altered epithelial repair mechanisms in COPD. They highlighted POU5F1 (OCT3/4) as a critical factor bridging Hedgehog signaling and epithelial plasticity. Critically, the alteration of POU5F1 was validated in primary COPD airway epithelial cells and lung tissues. Although deeper exploration is needed, this discovery uncovers essential clues linking Hedgehog signaling dysregulation to the defective epithelial remodeling characteristic of COPD pathogenesis. Thus, POU5F1-mediated signaling contributes to impaired regenerative capacity. Therefore, these findings establish Hedgehog-POU5F1 axis dysregulation as a fundamental mechanism driving pathological remodeling in COPD, revealing actionable targets for therapeutic intervention.
Collectively, the research presented in this Research Topic elucidates fundamental cellular and molecular mechanisms governing lung regeneration, aberrant repair, and fibrotic pathogenesis. Furthermore, these contributions significantly advance therapeutic development for pulmonary fibrosis and enhance diagnostic approaches for affected patients. These integrated discoveries establish new conceptual frameworks for intercepting fibrotic progression and restoring pulmonary homeostasis.
Author contributions
YQ: Visualization, Writing – original draft, Writing – review and editing. XT: Resources, Funding acquisition, Writing – review and editing. SS: Writing – review and editing. PK: Writing – review and editing. YS: Writing – review and editing. RR: Funding acquisition, Writing – review and editing. CM: Project administration, Supervision, Conceptualization, Software, Funding acquisition, Visualization, Resources, Writing – review and editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82200084 to CM and 82270077 XT), and the Natural Science Foundation of Sichuan Province (2023NSFSC1456 to CM). RR is supported by the Fundação de Amparo à Pesquisa de Minas Gerais (FAPEMIG) (Process: APQ-02571-21) and the Rede Mineira de Pesquisa Translacional em Imunobiológicos e Biofármacos no Câncer–REMITRIBIC (Process: RED-00031-21); and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) Brazil for PQ fellowship support (Process: 313839/2023-9).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Basil, M. C., Katzen, J., Engler, A. E., Guo, M., Herriges, M. J., Kathiriya, J. J., et al. (2020). The cellular and physiological basis for lung repair and regeneration: past, present, and future. Cell. Stem Cell. 26, 482–502. doi:10.1016/j.stem.2020.03.009
Bhattacharya, M., and Ramachandran, P. (2023). Immunology of human fibrosis. Nat. Immunol. 24, 1423–1433. doi:10.1038/s41590-023-01551-9
Bonella, F., Spagnolo, P., and Ryerson, C. (2023). Current and future treatment landscape for idiopathic pulmonary fibrosis. Drugs 83, 1581–1593. doi:10.1007/s40265-023-01950-0
Chen, Y., Li, Z., Ji, G., Wang, S., Mo, C., and Ding, B. S. (2024). Lung regeneration: diverse cell types and the therapeutic potential. MedComm 5, e494. doi:10.1002/mco2.494
Gilfillan, M., Bhandari, A., and Bhandari, V. (2021). Diagnosis and management of bronchopulmonary dysplasia. BMJ Clin. Res. Ed. 375, n1974. doi:10.1136/bmj.n1974
Jia, Y., Wang, H., Ma, B., Zhang, Z., Wang, J., Wang, J., et al. (2024). Lipid metabolism-related genes are involved in the occurrence of asthma and regulate the immune microenvironment. BMC genomics 25, 129. doi:10.1186/s12864-023-09795-3
Jones, D. L., Morley, M. P., Li, X., Ying, Y., Zhao, G., Schaefer, S. E., et al. (2024). An injury-induced mesenchymal-epithelial cell niche coordinates regenerative responses in the lung. Science 386, eado5561. doi:10.1126/science.ado5561
Karampitsakos, T., Juan-Guardela, B. M., Tzouvelekis, A., and Herazo-Maya, J. D. (2023). Precision medicine advances in idiopathic pulmonary fibrosis. EBioMedicine 95, 104766. doi:10.1016/j.ebiom.2023.104766
Lahmar, Z., Ahmed, E., Fort, A., Vachier, I., Bourdin, A., and Bergougnoux, A. (2022). Hedgehog pathway and its inhibitors in chronic obstructive pulmonary disease (COPD). Pharmacol. Ther. 240, 108295. doi:10.1016/j.pharmthera.2022.108295
Lederer, D. J., and Martinez, F. J. (2018). Idiopathic pulmonary fibrosis. N. Engl. J. Med. 378, 1811–1823. doi:10.1056/NEJMra1705751
Liu, X., Ali, M. K., Dua, K., Mao, Y., and Liu, J. (2023). Circular RNAs: emerging players in asthma and COPD. Front. Cell. Dev. Biol. 11, 1267792. doi:10.3389/fcell.2023.1267792
Liu, K., Meng, X., Liu, Z., Tang, M., Lv, Z., Huang, X., et al. (2024). Tracing the origin of alveolar stem cells in lung repair and regeneration. Cell. 187, 2428–45.e20. doi:10.1016/j.cell.2024.03.010
Ma, J., Li, G., Wang, H., and Mo, C. (2024). Comprehensive review of potential drugs with anti-pulmonary fibrosis properties. Biomed. Pharmacother. 173, 116282. doi:10.1016/j.biopha.2024.116282
Mo, C., Li, H., Yan, M., Xu, S., Wu, J., Li, J., et al. (2024). Dopaminylation of endothelial TPI1 suppresses ferroptotic angiocrine signals to promote lung regeneration over fibrosis. Cell. Metab. 36, 1839–57.e12. doi:10.1016/j.cmet.2024.07.008
Moss, B. J., Ryter, S. W., and Rosas, I. O. (2022). Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 17, 515–546. doi:10.1146/annurev-pathol-042320-030240
Neupane, A. S., Willson, M., Chojnacki, A. K., Vargas, ESCF, Morehouse, C., Carestia, A., et al. (2020). Patrolling alveolar macrophages conceal bacteria from the immune System to maintain homeostasis. Cell. 183, 110–25.e11. doi:10.1016/j.cell.2020.08.020
Niethamer, T. K., Planer, J. D., Morley, M. P., Babu, A., Zhao, G., Basil, M. C., et al. (2025). Longitudinal single-cell profiles of lung regeneration after viral infection reveal persistent injury-associated cell states. Cell. Stem Cell. 32, 302–21.e6. doi:10.1016/j.stem.2024.12.002
Oldham, J. M., Huang, Y., Bose, S., Ma, S. F., Kim, J. S., Schwab, A., et al. (2024). Proteomic biomarkers of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 209, 1111–1120. doi:10.1164/rccm.202301-0117OC
Petit, L. M. G., Belgacemi, R., Ancel, J., Saber Cherif, L., Polette, M., Perotin, J. M., et al. (2023). Airway ciliated cells in adult lung homeostasis and COPD. Eur. Respir. Rev. Official J. Eur. Respir. Soc. 32, 230106. doi:10.1183/16000617.0106-2023
Podolanczuk, A. J., Thomson, C. C., Remy-Jardin, M., Richeldi, L., Martinez, F. J., Kolb, M., et al. (2023). Idiopathic pulmonary fibrosis: state of the art for 2023. Eur. Respir. J. 61, 2200957. doi:10.1183/13993003.00957-2022
Sucre, J. M. S., Deutsch, G. H., Jetter, C. S., Ambalavanan, N., Benjamin, J. T., Gleaves, L. A., et al. (2018). A shared pattern of β-Catenin activation in bronchopulmonary Dysplasia and idiopathic pulmonary fibrosis. Am. J. Pathol. 188, 853–862. doi:10.1016/j.ajpath.2017.12.004
Wei, Q., Gan, C., Sun, M., Xie, Y., Liu, H., Xue, T., et al. (2024). BRD4: an effective target for organ fibrosis. Biomark. Res. 12, 92. doi:10.1186/s40364-024-00641-6
Wei, X., Qian, W., Narasimhan, H., Chan, T., Liu, X., Arish, M., et al. (2025). Macrophage peroxisomes guide alveolar regeneration and limit SARS-CoV-2 tissue sequelae. Science 387, eadq2509. doi:10.1126/science.adq2509
Zhang, J., Zhang, L., Chen, Y., Fang, X., Li, B., and Mo, C. (2023). The role of cGAS-STING signaling in pulmonary fibrosis and its therapeutic potential. Front. Immunol. 14, 1273248. doi:10.3389/fimmu.2023.1273248
Keywords: lung fibrosis, lung regeneration, bronchopulmonary dysplasia, asthma, chronic obstructive pulmonary disease
Citation: Qiu Y, Tang XX, Shichino S, Kushwaha PP, Sanders YY, Russo RC and Mo C (2025) Editorial: Cellular and molecular mechanisms of lung regeneration, repair, and fibrosis, volume II. Front. Cell Dev. Biol. 13:1653034. doi: 10.3389/fcell.2025.1653034
Received: 24 June 2025; Accepted: 31 July 2025;
Published: 12 August 2025.
Edited and reviewed by:
Patrizia Pignatti, Scientific Clinical Institute Maugeri (ICS Maugeri), ItalyCopyright © 2025 Qiu, Tang, Shichino, Kushwaha, Sanders, Russo and Mo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao Xiao Tang, dGFuZ3hpYW94aWFvQGdpcmQuY24=; Chunheng Mo, Y2h1bmhlbmdtb0BnbWFpbC5jb20=, Y2h1bmhlbmdtb0BzY3UuZWR1LmNu