 Anika Nusrat
Anika Nusrat Mingfu Wu
Mingfu Wu- Pharmacological and Pharmaceutical Sciences, College of Pharmacy, University of Houston, Houston, TX, United States
The epicardium is critical in heart development, functioning as a paracrine signaling hub and a source of progenitor cells. Bidirectional communication between the epicardium and myocardium, mediated by tightly regulated signaling networks, is essential for proper cardiac morphogenesis. This review presents a comprehensive overview of epicardium-myocardium crosstalk across species, emphasizing how this crosstalk influences epicardial epithelial-to-mesenchymal transition (EMT), fate specification of epicardium-derived cells (EPDCs), myocardial proliferation and growth, and coronary vasculature development. We critically assess decades of research elucidating key pathways—retinoic acid, fibroblast growth factor (Fgf), insulin-like growth factor (Igf), platelet-derived growth factor (Pdgf), transforming growth factor-β (Tgfβ), various transcriptional and epigenetic regulators, as well as calcium signaling mediated epicardial function—that coordinate these developmental processes. Additionally, we include detailed tables summarizing key experimental models and mechanistic insights that have shaped the field. This integrative analysis advances our current understanding of epicardial-myocardial crosstalk and highlights unresolved questions to guide future investigations into cardiac development and disease.
1 Introduction
The epicardium, the outermost layer of the heart, has emerged as a key orchestrator of cardiac morphogenesis and repair (Kurkiewicz, 1909; Pennisi et al., 2003; Hiruma and Hirakow, 1989; Quijada et al., 2020). During embryogenesis, mesothelial cells from the proepicardial organ (PEO) migrate to the heart surface, forming a continuous epithelial sheet that constitutes the early epicardium (Cao et al., 2020). As development progresses, a subset of epicardial cells undergoes epithelial-to-mesenchymal transition (EMT), delaminating from the surface and differentiating into epicardium-derived cells (EPDCs) (Wu et al., 2010; Wessels and Pérez-Pomares, 2004). These EPDCs contribute to various cardiac lineages within the heart, including fibroblasts, vascular smooth muscle cells, and pericytes (Gittenberger-de Groot et al., 1998; Dettman et al., 1998). Thus, the epicardium not only provides cellular input to the developing heart but also serves as a signaling nexus, coordinating myocardial growth and coronary vessel formation through paracrine factors.
Communication between the epicardium and myocardium occurs via finely tuned paracrine signaling pathways, including those involving retinoic acid (RA) (Merki et al., 2005; Brade et al., 2011), fibroblast growth factors (Fgfs) (Pennisi and Mikawa, 2009; Lavine et al., 2005; Torlopp et al., 2010; Vega-Hernández et al., 2011), insulin-like growth factors (Igfs) (Li et al., 2011; Shen et al., 2015), platelet-derived growth factors (Pdgfs) (Kang et al., 2008; Mellgren et al., 2008), and transforming growth factor-β (Tgfβ) (Austin et al., 2008; Sánchez and Barnett, 2012; Clark et al., 2016). These pathways govern various aspects of myocardial development, while epicardium-intrinsic signals (Wu et al., 2013) and extracellular matrix remodeling (Craig et al., 2010a; Allison et al., 2015) further influence this dynamic interplay. Importantly, epicardial cells also respond to myocardial cues, emphasizing the bi-directional nature of this communication, critical for heart development.
Recent advances have deepened our understanding of epicardial biology, particularly in lineage tracing, cellular heterogeneity, transcriptional regulation, noncoding RNAs, and chromatin remodeling (Lupu et al., 2020; Villa del Campo et al., 2016; Tyser et al., 2021; Harvey et al., 2024; Jang et al., 2022). Investigations using avian, zebrafish, and mammalian models, along with human pluripotent stem cell-derived epicardial cells, have provided valuable platforms for elucidating the roles of the epicardium in heart development (Meier et al., 2023; Männer et al., 2005; Kim et al., 2010).
This review comprehensively synthesizes past and current knowledge of epicardial-myocardial interplay that has shaped our understanding of how the epicardium communicates with myocardium to regulate heart morphogenesis. By systematically organizing and critically evaluating the chronological progression of discoveries, we highlight key regulatory mechanisms and delineate outstanding questions to further drive research in this evolving field.
2 Development of proepicardium, epicardium, and myocardium
At the very beginning of heart development (around embryonic day E7.5 in mouse (Li et al., 2011), 4 weeks of gestation in human (Cao and Cao, 2018)), the heart is composed of inner endothelium and outer myocardium. Around E9.5–E10.5 in mice, a third layer, the epicardium, starts to cover the naked surface of the myocardium. Precursor cells for the epicardium originate from a structure called PEO, which comprises a sheet of mesothelial cells and is located at the venous pole of the heart (Cao et al., 2020). Mesothelial cells from PEO can migrate to the heart surface by four mechanisms. First, a cluster of mesothelial cells form cyst-like structures in mice and travel through the pericardial cavity towards the myocardium (Sengbusch et al., 2002; Li J. et al., 2017; Komiyama et al., 1987). Second, the mesothelial bridge is formed between PEO and myocardium to enable mesothelial cells to travel and attach to the myocardial surface, as reported in chick (Nahirney et al., 2003), xenopus (Jahr et al., 2008), and zebrafish (Plavicki et al., 2014). A connecting bridge made of extracellular matrix components was reported to carry mesothelial cells to the chick myocardium (Nahirney et al., 2003). Third, free-floating mesothelial cells come in direct contact and attach to the myocardium in mice (Li J. et al., 2017; Rodgers et al., 2008). Heartbeat-mediated fluid forces from blood flow and the pericardial fluid can drive this migration through the pericardial cavity (Peralta et al., 2013). Fourth, villi-like structures protrude from the PEO and carry the mesothelial cells near to the myocardium (Li J. et al., 2017; Rodgers et al., 2008). Notably, these migratory modes are not necessarily exclusive, as multiple mechanisms can be adopted simultaneously (Li J. et al., 2017). After establishment of the epicardial layer, a unique set of transcriptional and surface markers defines its identity and regulates its function (Moore et al., 1999; Kraus et al., 2001; Mahtab et al., 2008; Pryce et al., 2007). At around E10.5 in mice, paralleling the epicardium development, thin-layered myocardium starts to expand via cell proliferation and migration (Li et al., 2016) and achieves two distinct zones: the trabecular zone facing inward, and the compact zone close to the epicardium. The development of myocardium has an intrinsic relationship with epicardium (Li et al., 2011), which is the major focus of this review.
3 Epicardial fate mapping and fate switch
Epicardial cells contribute to several lineages of the heart by generating EPDCs via EMT. EMT is a process conserved across species, which starts at chick at HH (Hamburger-Hamilton) stage 16, E12.5 in mice, and fetal stage 3 in humans (Quijada et al., 2020; Risebro et al., 2015). During EMT, epicardial cells undergo perpendicular division or directional migration (Wu et al., 2010) to enable cells to enter the myocardium. At this early stage, epicardial cells are not typical epithelial cells, and disrupting their polarity and cell-cell adhesion (marked by downregulation of epithelial and adhesion markers: cytokeratin (Kim et al., 2010), E-cadherin (von Gise and Pu, 2012), ZO-1 (Compton et al., 2006), β-catenin (Merki et al., 2005)) promotes their mesenchymal migratory properties (marked by upregulation of N-cadherin (Sridurongrit et al., 2008), Vimentin, Snai1, Twist1). Following EMT, EPDCs invade the subepicardial space and myocardium to give rise to multiple cardiac lineages.
However, no single marker can identify exclusively all epicardial cells and EPDCs because of their complexity and heterogeneity. Wt1 (Moore et al., 1999), Tbx18 (Kraus et al., 2001), Raldh2 (Du et al., 2023), Podoplanin (Mahtab et al., 2008), Scx (Pryce et al., 2007), Sema3D (Katz et al., 2012), Tcf21 (Lupu et al., 2020) have been widely used as classical markers to define epicardial cells and derivatives. Additionally, several other markers have been delineated from time to time by several research groups to label epicardial cells and EPDCs (Lupu et al., 2020; Mahtab et al., 2008; Du et al., 2023; Moss et al., 1998; Perez-Pomares et al., 2002; Smith et al., 2011). All the markers have specific context and time-dependent expression patterns. A summary of the epicardial markers (including gold standards as well as experimentally utilized markers) is discussed in Table 1.
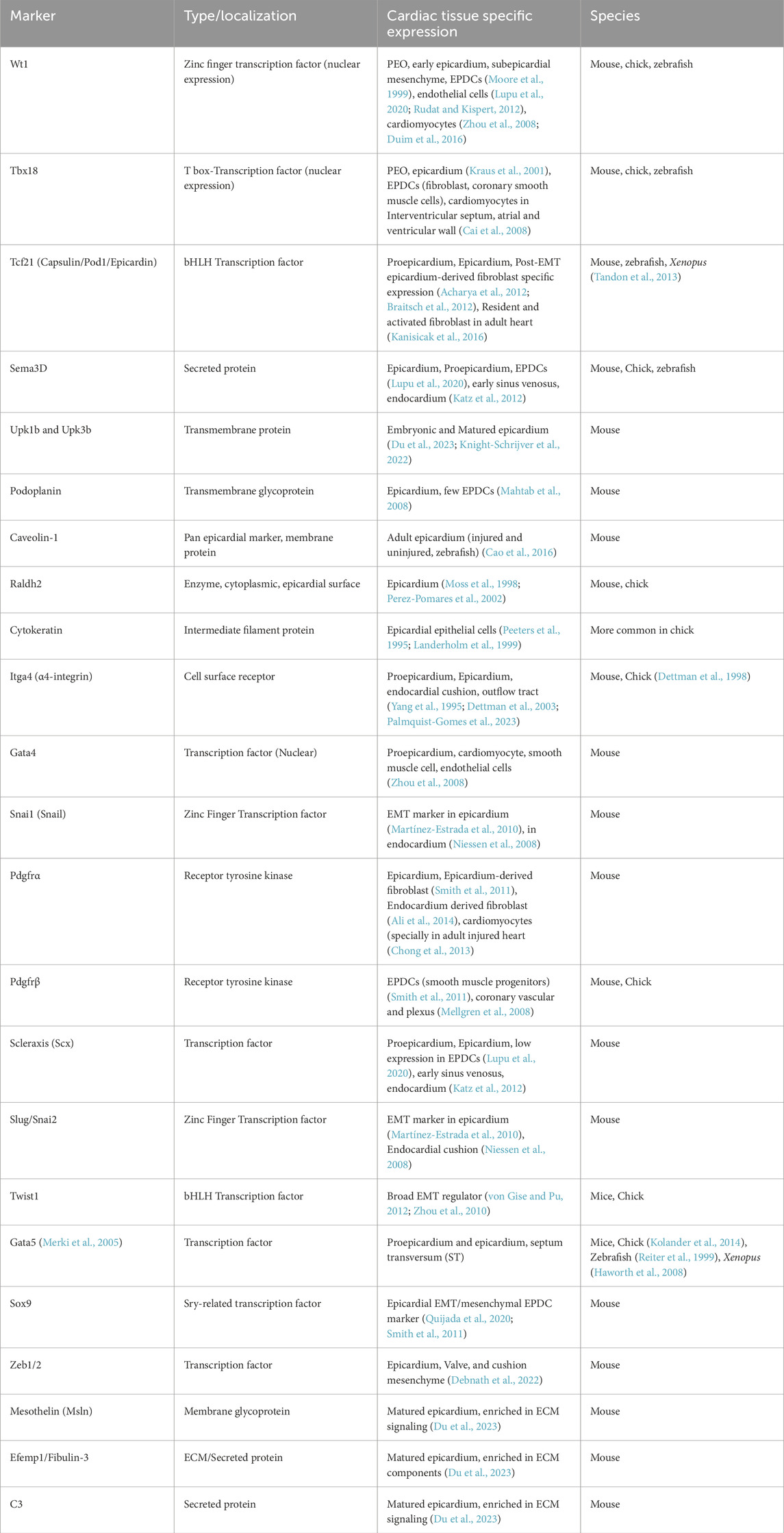
Table 1. List of epicardial cell markers during heart morphogenesis.
Tracing of EPDCs has been intensively investigated for years and is still ongoing across chick, mice, zebrafish, xenopus, and humans. At the beginning, dye or retroviral vectors were employed to label epicardial cells and EPDCs in chick embryos (Peeters et al., 1995; Gittenberger-de Groot et al., 2000; Morabito et al., 2001). Eventually, genetic fate mapping tools were employed, such as epicardial promoter-driven Cre recombinase-mediated recombination to express β-galactosidase or fluorescent reporters, mostly in mouse models. The standard epicardial constitutive Cre lines, as well as the inducible lineages (CreERT2), are discussed in Table 2, detailing their spatiotemporal expression.
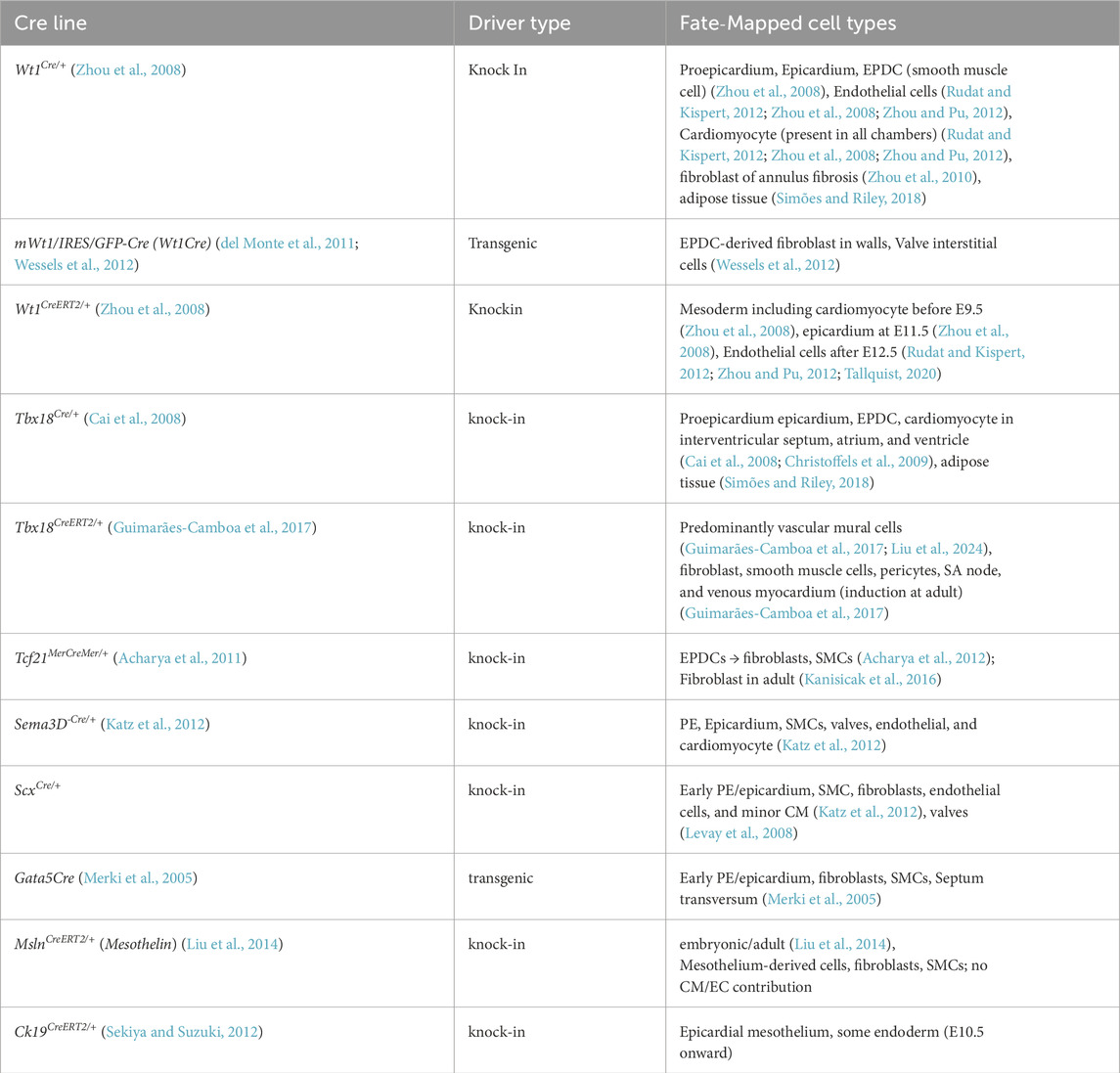
Table 2. List of standard epicardial lineages that label epicardial cells/EPDCs during development.
Decades of epicardial research provide a strong consensus that EPDCs contribute extensively to cardiac fibroblasts, coronary vascular smooth muscle cells (cVSMCs), and pericytes during heart development (Zhou et al., 2008; Acharya et al., 2012; Braitsch et al., 2012; Fang et al., 2016). Lineage tracing via Cre recombinase driven by Wt1, Tbx18, and Tcf21 locus consistently labels fibroblast and smooth muscle cells (SMCs) lineages during normal heart development, confirming that these are the major fates for EPDCs (Zhou et al., 2008; Acharya et al., 2012; Braitsch et al., 2012; Fang et al., 2016). Also, Wt1cre/+ and Tbx18cre/+ lineage-mediated EDPCs’ contribution to adipose tissue has been consistently reported (Yamaguchi et al., 2015; Chau et al., 2014).
However, a long-standing controversy sustains whether EPDCs can also contribute to cardiomyocytes and endothelial cell population of the heart. Several early studies detected cardiomyocyte labeling using constitutive Wt1cre/+ and Tbx18cre/+ lineages, implying that epicardial cells may contribute to myocardial lineage (Zhou et al., 2008; Cai et al., 2008; Zhou and Pu, 2012). If so, it raises another question: whether epicardial marker expression detected in cardiomyocytes conveys myogenic potential of epicardial differentiation, or instead, indicates early co-expression in common progenitor pools.
Villa del Campo et al. (2016) addressed these questions and showed that constitutive Wt1-Cre recombination can label a minor cardiomyocyte population—likely from early Wt1+ progenitors prior to epicardial adhesion to the ventricle (extra cardiac). Moreover, inducible Wt1creERT2/+ failed to mark cardiomyocytes, suggesting that true epicardium-derived myogenesis is minimal or absent once the epicardium is established. Similarly, Tyser et al. (2021) identified a juxta-cardiac field (JCF) — the earliest known mesoderm progenitor population contributing to both cardiomyocytes and proepicardial cells in mice. This might explain a partial overlap of Wt1/Tbx18 expression with cardiomyocytes in constitutively expressed lineages. A previous finding by van Wijk et al. (2009) also supported the concept that epicardium and myocardium share a common progenitor pool. Additionally, using Tcf21CreERT2/+ labeled lineage, Kikuchi et al. (2011) and Acharya et al. (2012) failed to detect epicardial Tcf21+ cells’ contribution to cardiomyocytes and endothelial cells in zebrafish and mouse models, respectively.
In the quest for epicardial differentiation to coronary endothelial cells, early avian studies observed PEO-derived endothelium (Perez-Pomares et al., 2002; Guadix et al., 2006), while murine studies indicated minimal (Rudat and Kispert, 2012) or no such contribution (Zhou et al., 2008; Cai et al., 2008). Katz et al. (2012) resolved this conflict by revealing that PEO is a molecularly compartmentalized structure in mice, where specific subpopulations contribute to different cardiac lineages. For example, Scx+ and Sema3D + PEO subpopulations contribute to the sinus venosus and endocardium of the coronary endothelium (Smits et al., 2018).
Whether epicardial fate commitment occurs before or after EMT has also been a central question over the years. Acharya et al. (2012), using Tcf21iCre/+ lineage mapping and Tcf21 null embryos, proved that Tcf21+ epicardial cells specification precedes the onset of EMT, denying the previous assumption that EPDCs fate is not specified until EPDCs enter the myocardium (Dettman et al., 1998). Consistently, Miao et al. (2021) revealed that the fate of smooth muscle cells is pre-specified in PEO. However, a recent report by Lupu et al. (2020) showed broad co-expression of Wt1, Tbx18, Tcf21, Sema3d, and Scx in the PEO up to E13.5 stage, suggesting that exclusive fate specification may not occur until after EMT. Altogether, these findings imply heterogeneity in the timing of epicardial specification commitment across sub-lineages.
3.1 Epicardial fate switch/lineage transition
Epicardium has multipotent plasticity and can be sensitive to extrinsic signaling cues during its lineage specification. Recent studies have refined our understanding of how transcription factors and environmental cues coordinate this fate switch. A proper balance of epicardial signaling is indispensable to ensure correct lineage specification. For example, Tcf21 null embryos manifested the absence of fibroblast differentiation, but SMC differentiation was not affected (Acharya et al., 2012). Interestingly, temporal loss of Tcf21 expression in epicardial cells switched the fate specification from fibroblast towards cVSMC (Braitsch et al., 2012), implying that Tcf21 must repress transition to SMC and maintain EPDCs in a fibroblast state. Recently, Xiao et al. (2018) demonstrated that deletion of Hippo kinases Lats1/2 in the mouse epicardium caused EPDCs to remain in a transitional state, leading to marked disruption in fibroblast differentiation.
Extrinsic signaling can induce epicardial plasticity toward myocardial fate. mYc (regulator of cell growth and proliferation) overexpression led to expansion of Wt1cre/+ expression into cardiomyocyte (Villa del Campo et al., 2016). According to Kruithof et al. (2006), chick PE/ST ex vivo culture causes spontaneous differentiation into beating cardiomyocytes. This cardiomyogenic bias was enhanced by Bone Morphogenetic Protein (Bmp) and inhibited by Fgf signaling. Recently, Dueñas et al. (2020) revealed several non-coding RNAs, such as miR-195 in association with miR-125, miR-146, and miR-223, to regulate Fgf/Bmp-mediated cardiogenic potential of PEO in chicken. Consistently, a previous study by van Wijk et al. (2009) revealed that Bmp-Smad signaling shifts the lineage into the myocardium, while Fgf-Mek1 signaling causes a fate shift to epicardium. Thus, a misbalance between Fgf and Bmp might cause the developmental shift from epicardium into myocardium. However, these findings may not generalize across species, as Garcia-Padilla et al. (2022) could not replicate this cardiomyogenic shift in mouse PE explants, indicating interspecies differences.
Interestingly, the fate switch can be bidirectional. According to Marques et al. (2022), ectopic expression of Wt1a/b in zebrafish cardiomyocytes induces their trans-differentiation into epicardial-like cells. This suggests that in certain contexts, Wt1 reactivation can destabilize cardiomyocyte fate and trigger epicardial gene programs.
All together, these findings indicate that epicardial fate decisions remain highly plastic and context dependent. Balanced transcriptional, epigenetic, and extracellular signaling cues are essential for forming fibroblasts and coronary vasculature. Also, epicardial lineage specification largely excludes myocardial or endothelial fates under normal physiological conditions during development.
4 Epicardium-myocardium crosstalk
Epicardium plays versatile roles in heart development by exerting both non-cell-autonomous and cell-autonomous influences on myocardium. By regulating EMT and multifaceted paracrine signaling, providing EPDCs, ECM regulation, and guiding coronary vasculature formation, the epicardium tightly controls critical aspects of heart development, such as cardiomyocyte proliferation and maturation, ventricular wall compaction, and vascular system formation (Merki et al., 2005; Li et al., 2011; Zamora et al., 2007). Diverse experimental platforms, including mechanical interference, quail–chick chimeras, epicardial explant, organ culture, and more recently, conditional genetic mouse models have guided us to decipher these critical roles over decades. This section extensively reviews the evolution of the findings and our understanding of indispensable epicardial–myocardial communication during cardiac morphogenesis.
4.1 Non-autonomous epicardial regulation of myocardium
4.1.1 1993–2000 | Foundational models defining epicardial necessity in heart development
The pioneer understanding of epicardial significance in heart development emerged with a novel ‘quail-chick chimera’ technique, where quail PEO was grafted into the host chick embryonic heart (Poelmann et al., 1993). This innovation led to myriads of epicardium-focused research in the following years. Utilizing this technique, Gittenberger-de Groot et al. (1998) first demonstrated that EPDCs populate the myocardium, sub-endocardium, and atrioventricular cushion during heart development. In a parallel study, Dettman et al. (1998) established a primary epicardial explant culture system and specified EPDCs as cVSMCs, perivascular and intermyocardial fibroblasts (Zhou et al., 2008; Vrancken Peeters et al., 1999). First evidence of direct epicardial regulation of heart development was revealed when a complete or partial PEO inhibition in chick embryos (Männer, 1999) led to several myocardial defects, i.e., impaired coronary formation, interventricular septation, and myocardium expansion (Gittenberger-de Groot et al., 2000). In those days, implantation of eggshell membrane (Pennisi et al., 2003; Gittenberger-de Groot et al., 2000; Männer, 1993; Poelmann et al., 2002) and mechanical excision of PEO villi (Perez-Pomares et al., 2002) were the microsurgical techniques used to generate PEO/epicardial block. However, these approaches either caused incomplete inhibition or just a delay in the migration of PEO-derived cells. Later, Männer et al. (2005) introduced a photoablation strategy that enabled permanent epicardium blockade.
4.1.1.1 Adhesion molecules and other mediators
Meanwhile, epicardial necessity in heart development was testified with the advent of genetic models, such as deletion of Vcam-1 (vascular cell adhesion molecule 1 (Kwee et al., 1995)) and α4-integrin (cell surface receptor protein mediating ECM and cell-cell adhesion (Yang et al., 1995)) in mice (Pérez-Pomares and de la Pompa, 2011). Vcam-1 null mice exhibited a complete absence of epicardial formation, a thinned myocardium, and septal defects. Similar defects were observed in α4-integrin deficient embryos as well. Phenotypic manifestation of Vcam-1 and α4-integrin knockout (KO) closely mirrored that of Wt1 null mutants (absence of a definitive epicardial layer, thinning of myocardial wall) (Kreidberg et al., 1993). Wt1 (Wilms Tumor 1) is a zinc finger transcription factor, which by this time, was already detected in the proepicardium, epicardium and subepicardial mesenchyme of mice and chick heart (Moore et al., 1999). Hence, these phenotypic findings implied epicardium-mediated defects in all three models.
In a follow-up study, Sengbusch et al. (2002) outlined that the migration of epicardial progenitors to the heart surface and maintenance of the already formed epicardium both require α4-integrin. Further, Dettman et al. (2003) applied the adenoviral system to delete α4-integrin and revealed that α4-integrin downregulation causes accelerated EMT, leading to premature invasion into the myocardium. Thus, α4-integrin was the first reported regulator to restrain EMT rather than promote it. Wu et al. (1999) discovered another regulator, Erythropoietin (Epo), to mediate myocardial growth. Mice lacking Epo and Epo receptor (EpoR) showed ventricular hypoplasia with myocyte proliferation and vasculature defects. Notably, significant Epo expression was observed in epicardium and endocardium, but not in cardiomyocytes, implying non-autonomous Epo mediated regulation of myocardium.
4.1.2 2001–2015 | dissecting epicardial regulation: genetic models, paracrine signaling, and transcription factors
4.1.2.1 Retinoic acid (RA) signaling
Based on the implication by earlier studies that the epicardium influences myocardium development, researchers started to dissect the underlying mechanisms. Chen et al. (2002) delineated early evidence of epicardial paracrine regulation of myocardium. They showed that epicardial cells secrete retinoic acid (RA) inducible mitogens to regulate cardiomyocyte cell cycle and proliferation. RA receptor X (RXRα)-deficient epicardial cells do not secrete these mitogens. Next year, Stuckmann et al. (2003), using the chick heart slice culture system, specified that epicardial RA (Chen et al., 2002) and Epo (Wu et al., 1999) signaling regulate myocardium growth via these mitogens, rather than directly stimulating cardiomyocytes. Consistently, Perez-Perez-Pomares et al. (2002) pointed out that ablation of chick PEO impaired Wt1 and Raldh2 (enzyme for RA synthesis) expression in EPDCs, leading to a thinned myocardium and defective vasculature.
Earlier, the global RXRα KO mouse model revealed myocardial thinning and midgestation lethality (Gruber et al., 1996), while myocardium-specific deletion produced no cardiac phenotype (Chen et al., 1998), implying that the myocardial defect is not cell-autonomous. Later, Merki et al. (2005) used the Cre recombinase system and generated an epicardial-specific transgenic mouse line Gata5Cre to delete RXRα, and these mutants mimicked the global RXRα KO phenotype. Mechanistically, reduced expression of Wnt9b, Fgf2, and β-catenin was revealed, indicating an epicardial RA–Wnt–Fgf signaling axis mediated non-autonomous regulation of myocardium. Additionally, Guadix et al. (2011) observed a significant reduction in Raldh2 expression in the epicardium of Wt1 null mice, thus revealing Wt1 as another regulator of RA synthesis.
In a parallel study, Brade et al. (2011) uncovered a novel extracardiac-to-epicardial-to-myocardial signaling array. Authors identified that hepatic RA signaling induces hepatic expression of Epo, essential to induce Insulin growth factor (Igf2) mitogen in the epicardium. In the absence of RA or its receptor (Raldh2−/− (Lin et al., 2010) and Rxrα−/− mutants), hepatic Epo fails to induce epicardial Igf2, causing insufficient stimulation of myocardial proliferation and myocardial thinning. Notably, they revealed intact epicardial RA signaling in Rxrα−/− mutants. This suggests that the compact zone defect reported in Rxrα−/− hearts (Merki et al., 2005; Chen et al., 2002) might arise not from intrinsic epicardial RA loss, but possibly from a non-cardiac RA-dependent mechanism. However, previous reports of myocardial thinning in epicardium-specific Rxrα−/− mutants (Merki et al., 2005) indicate that epicardial Rxrα at least partially, if not all, contributes to the defects of global Rxrα−/− mutants.
4.1.2.2 Igf (insulin growth factor) signaling
Paralleling the identification of Igf2 as epicardial mitogen (Brade et al., 2011), Li et al. (2011) uncovered that epicardial Igf2 is indispensable for myocardial proliferation as it cannot be compensated by endocardial sources. Shen et al. (2015) re-established the epicardial origin as well as sufficiency of epicardial Igf2 for compact zone growth using multiple genetic models. Conditional deletion of Igf2 with Tbx18Cre/+ (epicardial-specific) or Nkx2.5 Cre/+ (cardiac mesoderm-wide) resulted in a significant reduction of ventricular wall thickness. In contrast, deletion with Tie2Cre (endothelial/endocardial) or Myh6Cre (myocardial) had no such effect. They further focused on epicardial-myocardial Igf communication by pinpointing that global Igf2 KO and myocardium-specific deletion of Igf receptors, Igf1r, and Insr, both models manifested ventricular hypoplasia associated with proliferation defects.
Later, Yan et al. (2018) demonstrated that, in addition to myocardial expression (Shen et al., 2015), Igfr1 is also expressed in epicardium and contributes to myocardial growth via the Focal adhesion kinase (FAK) pathway. A recent study by Wang et al. (2019) specified that, between Igfr1 and Insr, Igfr1 predominantly binds with Igf2 while Insr is unable to compensate for the function of Igfr1. Findings in zebrafish model by Huang et al. (2013) further complement these studies, indicating similarity across species. Most recently, Meier et al. (2023) confirmed epicardial Igf2 and myocardial Igfr1 expression in a human “epicardioids” (a 3D human stem cell-derived organoids) and mirrored the necessity of mitogenic role of Igf2 in human fetal heart development. Igfr1 inhibition impaired myocardial proliferation and compaction as previously reported in mice (Yan et al., 2018), and recombinant Igf2 rescued these defects in organoids even in the absence of epicardium (in RA-deficient spheroid that lacks epicardium).
In addition to the extracardiac regulation of epicardial Igf2 revealed in earlier studies (Brade et al., 2011), Shen et al. (2015) further elucidated a biphasic regulation. While hepatic Epo regulates epicardial Igf2 at the earlier E10–11.5 stage, a placenta-dependent regulation takes over this process at E11.5 onward, both controlled by RA signaling. Placental regulation is further driven by optimal glucose uptake and normoxic conditions. Recently, Jang et al. (2022) added another dimension by introducing epigenetic regulation of epicardium to influence myocardium growth. Deletion of Histone deacetylase (Hdac3) in epicardium caused microRNA-mediated suppression of mitogens Fgf9 and Igf2, resulting in myocardial proliferation and compaction defects.
4.1.2.3 Fgf (fibroblast growth factor) signaling
Mikawa (1995) and Mima et al. (1995) were among the first groups to introduce Fibroblast Growth Factor (Fgf) signaling in heart development. They revealed that myocardium-targeted Fgf receptor inhibition by antisense RNA led to a significant but transient defect in cell proliferation. Pennisi et al. (2003) first discovered non-autonomous regulation of Fgf by observing that thinner myocardium in epicardium-blocked chick hearts was caused by disrupted Fgf signaling components. They characterized that the epicardium is essential to maintain the correct level of Fgf mitogens required for myocyte proliferation, but the epicardium is not required for establishing the transmural pattern of mitogen expression or myocyte proliferation.
In mammals, Lavine et al. (2005) specified Fgf ligands Fgf9, Fgf16, and Fgf20 as epicardial- and endocardial-derived mitogens, while receptors Fgfr1 and Fgfr2c were found in the myocardium. Fgf9−/− mice showed reduced cardiomyocyte proliferation and ventricular hypoplasia. Notably, myocardium-specific Fgfr1/2 KO showed more severe hypoplasia than Fgf9−/− mice, indicating possible contributions from additional Fgf ligands (Li et al., 2024). Indeed, both Fgf16 (Hotta et al., 2008) and Fgf20 promoted proliferation in a Fgfr1/2-dependent manner (Lavine et al., 2005).
Similarly, ligands such as Fgf1, Fgf2, Fgf7, Vegf, and Egf were reported to enhance EMT in epicardial explant (Dettman et al., 1998) and avian heart culture system (Morabito et al., 2001). Further work by Torlopp et al. (2010) identified expressions of Fgf2, Fgf10, and Fgf12, and their receptors Fgfr1, Fgfr2, and Fgfr4 in chick PEO, supporting findings in quail (Pennisi and Mikawa, 2009). In their study, exogenous Fgf2 enhanced PE growth and EMT via Mapk and PI3K/Akt pathways, supporting findings by Morabito et al. (2001). Similar to Fgf ligands (Morabito et al., 2001), Fgfr1 overexpression also enhanced epicardial EMT and myocardial invasion, and its knockdown significantly impaired the invasion of PE progenies (Pennisi and Mikawa, 2009). Notably, Fgf inhibition did not affect expression of proepicardial markers Tbx18, Wt1, and Tbx5, indicating that Fgf signaling may not regulate lineage identity (Pennisi and Mikawa, 2009).
Vega-Hernández et al. (2011) introduced bidirectional Fgf-mediated epicardial-myocardial communication in heart development. Authors revealed that myocardial Fgf10 binds with epicardial Fgfr1 and Fgfr2b to promote EPDC invasion into the myocardium. Interestingly, the deficiency in epicardium-derived cardiac fibroblasts was correlated with reduced cardiomyocyte proliferation and thin myocardium observed in Fgf10−/−, Fgfr2b−/−, and Wt1Cre/+; Fgfr1/2 mutants. It indicates that not only epicardial mitogen-driven signaling, but also the presence and function of epicardium-derived fibroblasts, are crucial for myocardial growth. However, conflicting findings were revealed by Rudat et al. (2013), who observed no defect in heart development in Tbx18 Cre/+ mediated loss of function of Fgfr1/Fgfr2 murine embryos, implying compensation pathways or possible Cre-driver or timing-dependent differences.
4.1.2.4 Pdgf (platelet-derived growth factor) signaling
Early evidence of Platelet-Derived Growth Factor (Pdgf) signaling in cardiac development emerged from the embryonic/neonatal lethality observed in germline deletion of Pdgfa or Pdgfrα in mice (Boström et al., 1996; Tallquist and Soriano, 2003). Lu et al. (2001) first discovered that Pdgf-BB induces EMT and SMC marker expression in quail, while Pdgf receptor stimulation similarly enhanced EMT in mice (Mellgren et al., 2008). Likewise, inhibition of Pdgf signaling impaired epicardial cell proliferation and coronary vasculature formation in zebrafish (Kim et al., 2010). In similar periods, Pdgfrβ expression in EPDCs was reported in quail-chick chimera (Guadix et al., 2006), as well as Pdgf-A expression in rat epicardial cell line and mouse epicardium and myocardium was confirmed (Kang et al., 2008). In the human fetal heart, Chong et al. (2013) demonstrated robust Pdgfrα expression in interstitial cells of the epicardium, myocardium, endocardium, and cVSMCs, with rare observation in endothelial cells and cardiomyocytes. However, significant Pdgfrα+ cardiomyocytes were observed in the adult diseased heart (Chong et al., 2013).
To assess tissue-specific roles of Pdgf signaling, Kang et al. (2008) deleted both Pdgf receptors (Pdgfrα and Pdgfrβ) in myocardium and mesoderm via Mesp1Cre/+, but did not observe any myocardial or coronary vascular defects in mutants. However, Smith et al. (2011) deleted Pdgfrα, Pdgfrβ, or both in the mouse epicardium and observed significantly defective EMT. Interestingly, the loss of Pdgfrα resulted in a specific deficiency of cardiac fibroblasts, whereas Pdgfrβ deletion impaired the development of cVSMCs, highlighting distinct roles of epicardial Pdgfrα and Pdgfrβ in fate specification. Supporting the role of epicardial Pdgfrα in fibroblast formation, Rudat et al. (2013) further revealed that Tbx18 Cre/+ mediated loss of Pdgfrα prevents differentiation of EPDCs into mature fibroblasts. Interestingly, Ali et al. (2014) revealed strong Pdgfrα expression in endocardium-derived fibroblasts as well, expanding the understanding of Pdgfrα as a marker beyond epicardium-derived fibroblasts (pan fibroblast marker). This was further validated using the collagen1a1-GFP mouse model that labeled both epicardium- and endocardium-derived Pdgfrα+ fibroblasts (Moore-Morris et al., 2014).
Focusing on Pdgfrβ, Mellgren et al. (2008) revealed the absence of coronary vasculature and thinner myocardium in Pdgfrβ−/− mutant hearts. Interestingly, epicardium-specific deletion of Pdgfrβ showed region-specific defects in the coronary vasculature. This indicates that Pdgfrβ signaling from other sources, in addition to the epicardium, might contribute to the development of coronary vasculature. Further contribution of Pdgf signaling in coronary vasculature formation will be discussed in Section 4.4 later in this review.
Multiple downstream effectors have been reported to modulate Pdgf signaling. According to Mellgren et al. (2008), Pdgf receptor stimulation enhanced EMT via Pdgfrβ-PI3K pathway. A recent finding in zebrafish models (Shrestha et al., 2023) further revealed PI3K-Pdgfrα signaling to regulate latero-medial migration of cardiomyocytes in early heart tube formation. Additionally, Smith et al. (2011) demonstrated the Pdgfr-Sox9 axis regulating migration and cytoskeletal organization of epicardial cells. Furthermore, Guadix et al. (2011) introduced the epicardial Pdgf/Pdgfrα-RA axis in heart development. They observed low expression of Pdgfrα in Wt1 null-embryos and immortalized epicardial cells, which were rescued by exogenous RA.
4.1.2.5 Tgfβ signaling
Involvement of transforming growth factor β (Tgfβ) signaling in non-autonomous epicardial regulation of myocardium was also prevalent. Takahashi et al. (2014) delineated that epicardial block-driven chick myocardial thinning results from immature sarcomere formation (reduced Z-line spacing) and smaller cardiomyocyte size (increased cell density). These defects were linked to reduced p-Smad2, p-Erk, and lower expression of Tgfβ2 and Fgf2. These suggest that Tgfβ and Fgf signaling play an essential role in epicardium-regulated cardiomyocyte growth and sarcomere maturation. To note, Tgfβ signaling mostly correlates with epicardial EMT-based regulation of heart development and will be discussed in detail under 4.2.1 section later in this review.
4.1.2.6 Transcription factors
4.1.2.6.1 Wt1
Multiple transcription factors have been reported over time as influencers of non-autonomous epicardial roles. Following the striking phenotype of Wt1 null embryos (Moore et al., 1999; Kreidberg et al., 1993) and confirmed myocardial invasion of Wt1+ EPDCs via genetic labeling (Zhou et al., 2008; Zhou et al., 2010), Martínez-Estrada et al. (2010) generated epicardium-specific Wt1 KO, inducible Wt1 KO epicardial cell line, and Wt1 KO embryoid bodies (differentiated from Wt1 null embryonic stem cells). All three systems showed reduced EMT markers and ectopic expression of epithelial markers in epicardial cells. They also implied that Wt1-regulated EMT might influence the formation of cardiomyocytes. Later, a study by von Gise et al. (2011) proved that Wt1 KO hearts showed defective EPDCs migration and invasion in the myocardium, resulting in diminished proliferation of compact myocardium. Mechanistically, impaired expressions of canonical Wnt/β-catenin signaling components (Lef1 and β-catenin) and downstream targets (Axin2, Cyclin D1, and Cyclin D2), and activity of Wnt/β-catenin reporter transgene were observed in Wt1 KO epicardial cells. Additionally, Wnt5a, a non-canonical Wnt, and Raldh2 were markedly downregulated, supporting findings by Guadix et al. (2011).
Furthermore, Wt1 represses chemokines Ccl5 and Cxcl10 (inhibitors of EPDC migration and cardiomyocyte proliferation) in epicardial cells by upregulating Irf7 (interferon 7) (Velecela et al., 2013; Sanchez-Fernandez et al., 2022), revealing interferon-dependent regulation of myocardial growth.
4.1.2.6.2 NF-κB
A growing body of evidence highlights nuclear factor kappa B (NF-κB) as a central modulator of epicardial-myocardial communication. Early insight came from Craig et al. (2010a), who demonstrated that high molecular weight hyaluronan (HMW-HA) stimulates epicardial cell invasion and differentiation in vitro, via NF-κB signaling. According to Clark et al. (2016) and DeLaughter et al. (2016), NF-κB also regulates Tgfβ receptor 3 (Tgfβr3) mediated epicardial cell invasion. Tgfbr3+/+ epicardial cells treated with NF-κB inhibitor failed to invade in response to a variety of pro-migratory ligands. Li Y. et al. (2017) performed 2D LC-MS/MS–based secretome profiling of chick epicardial–myocardial co-cultures and identified NF-κB as a top-predicted transcriptional regulator. Although NF-κB inhibition blocks EMT in both chick and mouse epicardial cells, the authors noted a species-specific difference in NF-κB localization: it was predominantly nuclear in chick epicardial cells but largely perinuclear in mice. Thus, a potential difference in NF-κB activation dynamics can be implied.
4.1.2.6.3 Ets, Hif-1α, Tcf21, TFEB, Snai1b, Sox9
Expression of transcription factor Ets1 was observed in EMT regions during heart development (Macías et al., 1998). Lie-Venema et al. (2003) introduced an antisense Ets sequence via bloodstream in chick embryos. Despite this broad distribution, strong phenotypes such as reduced EMT, thinner mesenchyme, disrupted coronary vascular system with myocardial-to-subepicardial fistulae, and impaired myocardial expansion (thinner wall, broader and fewer trabeculae)—all implicated epicardial dysfunction as the primary source of the defects. However, in mammals, detailed cardiac analysis for Ets functioning is not available yet, possibly because of mid-gestational lethality in Ets1 and Ets2 null mice (Barton et al., 1998; Yamamoto et al., 1998; Lie-Venema et al., 2007).
Tao et al. (2013) revealed Hypoxia inducible factor-1α (Hif-1α) as another critical regulator. In their study, adenoviral delivery of constitutively active Hif-1α (ca Hif1α) in avian embryos caused enhanced EMT but marked defects in myocardial invasion of EPDCs. They revealed that Hif-1α–driven upregulation of Flt-1 impaired Flk-1 signaling required for proper myocardial invasion.
Boezio et al. (2023) revealed a temporal regulation of epicardial-myocardial crosstalk by using a temporally controlled tcf21:NTR/MTZ ablation system in zebrafish. While early loss of epicardial cells led to reduced CM proliferation, restoration of epicardial coverage at a late stage via MTZ washout successfully rescued myocardial growth. This suggests that epicardial influence might be dispensable once complete coverage is achieved in zebrafish. In addition to reduced Fgf24 and Vegfaa expression, mitochondrial and ribosomal genes were significantly downregulated in mutant cardiomyocytes. This suggests that the epicardial defect may cause mitochondrial insufficiency and defects in CM maturation.
Astanina et al. (2022) uncovered transcription factor EB (TFEB) as an EMT suppressor. TFEB overexpression in the mouse epicardium caused severely impaired EMT and EPDC differentiation along with defective myocardial and interventricular septal thickening. Functionally, TFEB acts through TGIF1 to suppress Tgfβ/Smad-mediated EMT. Gentile et al. (2021) identified an additional role of transcription factor Snai1b beyond its traditional EMT function in zebrafish. Snai1b-deficient embryos showed cardiomyocyte extrusion into the pericardial cavity, especially in the regions under mechanical stress. These CMs showed increased contractility with weakened adhesion. Thus, Snai1b suppresses cardiac contraction to reduce extrusion and preserve CM integrity during heart development.
Most recently, epicardial deletion of transcription factor Sox9 revealed impaired EPDCs invasion into the walls and AV valves, resulting in a thinner myocardial wall. Postnatally, mutants’ phenotype resembles human myxomatous mitral valve disease (MVD), characterized by extracellular matrix disorganization. Authors further identified a novel role of Cd109s association behind the observed valve pathology (Harvey et al., 2024).
4.1.3 2016–2025: emergence of noncoding RNAs and epigenetic regulators
Epigenetic regulation of the epicardium-myocardium crosstalk has become the focus of recent explorations. Deletion of micro-RNA processing enzyme Dicer in proepicardium led to impaired EMT and reduced epicardial proliferation and differentiation into cVSMCs (Singh et al., 2011). Pontemezzo et al. (2021) revealed a novel microRNA mediated mechanism behind Tgfβ1-induced EMT in murine epicardial-mesothelial cells (EMC). Specifically, Tgfβ1 stimulation upregulated epicardium-derived cardiogenic factor Follistatin-like 1 (Fstl1) by suppressing its repressor miR-200c-3p. These findings establish a novel Fstl1-miR-200c-3p regulatory axis for epicardial behavior.
A novel post-transcriptional splicing mechanism emerged from a study by Jackson-Weaver et al. (2020), who identified Prmt1 as a splicing regulator of epicardial EMT and differentiation. Deletion of Prmt1 in the mouse epicardium resulted in blocked EMT, impaired invasion, and reduced formation of EPDCs. Functionally, loss of Prmt1 causes accumulation of p53, which suppresses EMT via enhanced Slug degradation. Prmt1 affects p53 expression by regulating alternative splicing of p53 regulator Mdm4, hence fine-tuning the Prmt1-Mdm4-p53-Slug axis. A deeper look into epigenetic repression was provided by Jang et al. (2022), who demonstrated that Hdac3 regulates epicardial mitogen expression via microRNA suppression. Conditional deletion of Hdac3 in the epicardium led to significant thinning of the compact myocardial layer with unaffected trabeculae, reduced EPDCs, and EMT markers. Mechanistically, they identified that microRNA (miR)-322 and miR-503 were upregulated, repressing Fgf9 and Igf2 expression in Hdac3 KO cells. This study uncovered a double-negative axis where Hdac3 represses key mitogen suppressing miRNA, thus placing the epicardium as a non-cell-autonomous epigenetic regulator of compact myocardium expansion.
4.1.4 Others
Beyond epigenetic regulators, several additional mediators have been identified that regulate epicardial behavior and, in turn, influence myocardial development. Mahtab et al. (2008) revealed that podoplanin KO mice show hypoplastic and perforated compact and septal myocardium and reduced EMT. Weeke-Klimp et al. (2010) explored how EPDCs influence cardiomyocyte development by co-culturing quail EPDCs with neonatal mouse cardiomyocytes. They revealed that only direct co-culture enhanced CM proliferation, maturation, and alignment. In contrast, indirect (using transwell inserts to separate cells while sharing medium) culture with EPDCs conditioned medium failed to induce these effects. However, their findings contrast with Takahashi et al. (2014), who rescued myocardial defects in chick using EPDCs conditioned media.
Another study by Iyer et al. (2016) observed that Loss of Crim1 (transmembrane protein expressed in epicardium and EPDCs) function leads to epicardial defects and hypoplasia. Epicardium-restricted deletion of Crim1 further resulted in reduced proliferation of EPDCs and differentiation into cardiac fibroblasts in mutants, which is mediated by phosphorylation of Smad2 and Erk1/2. At a similar time point, Arora et al. (2016) reported the role of angiogenic hormone, prokineticin-2 (Pk2) and its receptor Pkr1 as epicardial regulators (Olivey and Svensson, 2010). Pk2 induced EMT in epicardial cells, and Pkr1 deletion in epicardium caused markedly diminished EPDC formation, reduced ventricular wall thickness with impaired proliferation, and disrupted coronary vessel development via PI3K/Akt pathway.
Ridge et al. (2017) investigated the role of non-muscle myosin heavy chain IIB (NMHC IIB), encoded by Myh10, in heart development using a mouse model possessing a splice-donor site mutation in Myh10. Mutants showed NF-κB-mediated disorganized, hyperproliferative epicardium, impaired EMT, thin ventricles, and septal defects. Junghof et al. (2022) recently identified type II classical cadherin Cdh18 as a novel biomarker for epicardial cells in a human induced pluripotent stem cell (hiPSC)-derived epicardial differentiation model (Junghof et al., 2022). They further identified that siRNA-mediated knockdown of Cdh18 led to a shift in differentiation towards SMCs, rendering Cdh18 essential for epicardial fate specification.
4.1.4.1 Calcium signaling mediated epicardial regulation of heart development
Proper Calcium (Ca+2) homeostasis is essential for cardiac contraction, and epicardium/EPDCs can influence heart morphogenesis by interfering with Ca+2 signaling-regulated contractility. Weeke-Klimp et al. (2010) revealed that EPDCs can affect cardiomyocytes’ Ca+2 handling to regulate their alignment, maturation, and contraction. A key player in Ca+2 homeostasis in sarcoplasmic reticulum (SR) Ca+2 ATP ase (SERCA2a), which regulates Ca+2 mediated cardiac muscle contraction and relaxation by sequestration of cytosolic Ca+2 and uptake into SR. Co-culturing quail EPDCs with neonatal mice cardiomyocytes revealed increased expression of electrical and mechanical junction proteins as well as upregulated SERCA2 in cardiomyocytes, indicating enhanced Ca2+ handling and more mature sarcomeric organization. Aligned sarcomere patterns observed further correlated with improved cardiomyocyte organization. Similarly, Iyer et al. (2015) demonstrated that human pluripotent stem cell-derived epicardial cells can recapitulate epicardial morphology and marker expression. Moreover, these cells differentiate into SMCs upon ligand stimulation. Interestingly, these SMCs displayed contractility, evidenced by increased intracellular Ca2+ signaling when exposed to cholinergic receptor agonists. Kelder et al. (2015) further supported the epicardium’s role in regulating cardiac physiology, showing that the chick epicardium expresses β-adrenergic receptors and modulates heart rate, contraction, and conduction by responding to epinephrine. In their study, epicardium-blocked hearts exhibited significantly reduced epinephrine responses, leading to slower heart rates. Given the central role of Ca2+ in cardiac contraction, these results also imply dysregulated Ca2+ signaling under epicardial dysfunction, which could be a new area to be explored with this model.
Together, these studies highlight the critical roles of the epicardium/EPDCs in regulating cardiomyocyte contractility and alignment via Ca2+ signaling, thereby influencing heart development and function.
A comprehensive summary of key experimental models and associated signaling that advances our understanding of epicardial-myocardial crosstalk has been provided in Table 3.
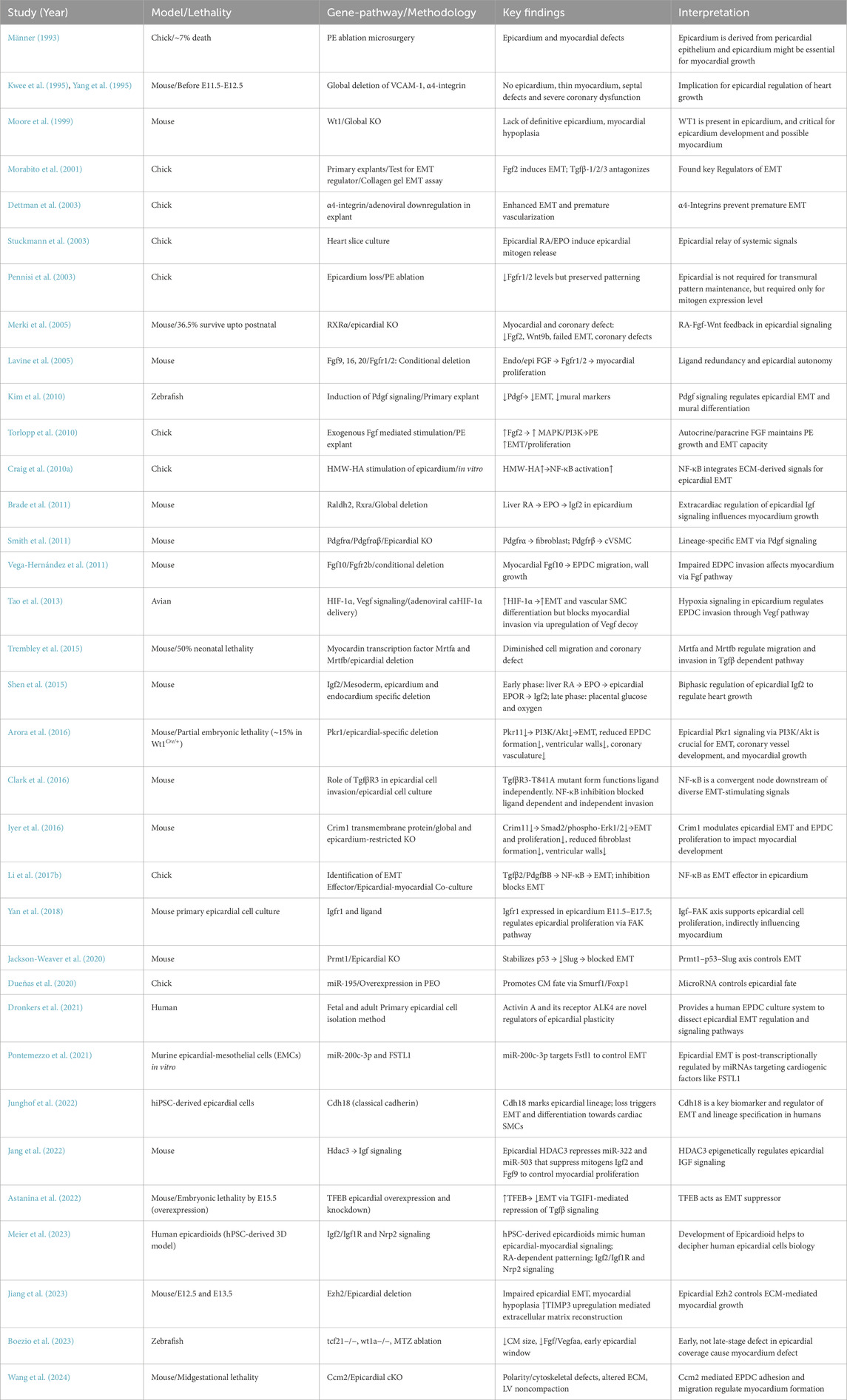
Table 3. A comprehensive summary of key experimental models and associated signaling involved in epicardium-myocardium crosstalk during development.
4.2 Cell-autonomous regulation of epicardium
Several aspects of epicardial regulation, such as polarity, cell division, proliferation, survival, and EMT, shape cardiac development by ensuring proper epicardial coverage. Wu et al. (2010) revealed the requirement of a specific orientation of cell division: perpendicular division to undergo EMT. β-catenin/Numb-N-cadherin complex regulates EMT by maintaining spindle orientation, cell polarity, and directed cell division. Wu et al. (2013) revealed defects in epicardial cell’s proliferation and coronary vessel formation in Tbx18 null mice models, associated with alterations in several components of Hedgehog and Vegf signaling. However, Greulich et al. (2012) did not observe obvious defects in epicardial EMT and differentiation by either loss of Tbx18 function or prolonged functioning of Tbx18. Rather, epicardial mis-expression of a transcriptionally active form Tbx18VP16 resulted in premature differentiation to SMCs. Supporting this, Wu et al. (2013) revealed Tbx18 as a SRF/CArG box dependent repressor of SMCs differentiation, evident by reduced SMCs marker in Tbx18 overexpressed cells in vitro. Interestingly, Takeichi et al. (2013) demonstrated that Tbx18 and Wt1 bi-directionally regulate the epicardial EMT by affecting Slug expression in murine primary epicardial cells. Knockdown of Wt1 induced the epicardial EMT, while knockdown of Tbx18 inhibited the Wt1 and Tgfβ1-induced mesenchymal transition. This dynamic regulation suggests that fine-tuning of Tbx18-Wt1 activity is essential to ensure proper EMT.
Beyond transcriptional regulation, Baek and Tallquist (2012) identified another EMT regulator, Neurofibromin (Nf-1), a Ras-GTPase activating protein (GAP), mutation of which is traditionally connected to tumorigenesis. Specific deletion of Nf-1 in epicardium enhanced EMT and increased the cVSMCs and fibroblasts in myocardium in the Pdgf-dependent pathway. Similarly, Li J. et al. (2017) revealed another small GTPase, Cdc42, as a crucial regulator of PEO translocation. CDC42 conditional deletion disrupted the formation of villous projections and cysts that carry PEO cells to the myocardium, leading to incomplete epicardial coverage.
4.2.1 Tgfβ signaling mediated epicardial EMT
Many studies have dissected the complex, often context-dependent role of Tgfβ signaling in epicardial EMT and differentiation. Initial evidence by Morabito et al. (2001) revealed that Tgfβ1 weakly stimulates EMT in chick epicardial explants, while Tgfβ2/3 showed no EMT induction, instead, inhibited Fgf2-induced EMT. In contrast, Compton et al. (2006) and Austin et al. (2008) identified Tgfβ1/2/3 as a strong promoter of EMT and smooth muscle differentiation in both chick and mouse epicardial models, in which Tgfβ receptor 1 (Alk5) and downstream RhoA/p38 MAPK play as key effectors. Sánchez and Barnett (2012) further revealed that Tgfβ and Bmp2-induced EMT mediates through Alk5 and identified a different Par6–Smurf1–RhoA regulatory axis. Genetic studies in mice by Sridurongrit et al. (2008) demonstrated that epicardial—but not myocardial or endocardial—deletion of Alk5 impaired EMT, smooth muscle formation, and myocardial growth, indicating a cell-autonomous requirement of Alk5 in the epicardium (Dronkers et al., 2020). Meanwhile, Olivey et al. (2006) listed another type 1 Tgfβ receptor isoform (Alk2) for early-stage epicardial EMT regulation, functioning through the Smad pathway.
Interestingly, Sánchez et al. (2011) discovered that GIPC1-binding sites on the Tgfbr3 cytoplasmic tail are indispensable for ligand interaction, followed by EMT and invasion. Supportively, Hill et al. (2012) highlighted distinction in ligand-receptor interaction: Bmp2 binds with Alk3 to induce epithelial loss but not SMC differentiation, while Tgfβ requires Alk5 and supports both EMT and SMC fate. Both require Tgfβr3–GIPC1 interaction, again underscoring the importance of cytoplasmic domain signaling.
A pivotal discovery by Clark et al. (2016) and DeLaughter et al. (2016) established NF-κB as a shared, essential effector downstream of Tgfβr3. A specific T841A-TGFβR3 mutant variant, possibly resistant to internalization, enabled ligand-independent but still NF-κB-dependent EMT induction, suggesting parallel and converging Alk–NF-κB axes. Additionally, Dronkers et al. (2021) uncovered Activin A–Alk4 signaling as an independent epicardial EMT pathway. Adding to this, Lupu et al. (2020) revealed that Alk4 was highly expressed in fetal epicardial cells (Moerkamp et al., 2016), and overexpressing Alk4 in adult epicardial cells mimicked fetal epiMT, highlighting a role of Alk4 in the developmental switch.
Interestingly, Tgfβ-neutralizing antibodies could not block spontaneous EMT in fetal EPDCs, but the combination with Activin A inhibitor blocked, implying compensatory interplay between Activin A and Tgfβ. Their work also addressed a methodological issue: SB431542, which is widely used in myriads of previous studies as a Tgfβ/Alk5 inhibitor, also blocks ALK4/7, thus potentially could overlook activin’s contributions in Tgfβ signaling in previous studies.
Moerkamp et al. (2016) introduced a novel human fetal and adult EPDCs isolation and culture method. Using this method, they characterized that Alk5 inhibition is essential to retain the epithelial phenotype and inhibit spontaneous EMT of fetal EPDCs, making it a tool to study epicardial state transitions. Interestingly, adult EPDCs showed Tgfβ stimulated invasion and formation of tube-like structures, while fetal EPDCs reduced their migration upon Tgfβ stimulation and failed to show tube-like structures, pinpointing potentially different mechanisms of Tgfβ responsiveness.
4.3 Epicardium-derived cardiac fibroblasts influences myocardial growth
A subset of EPDCs differentiates into interstitial fibroblasts and plays critical roles in myocardial growth and organization. Vega-Hernández et al. (2011) discovered that interference of Fgf signaling in murine hearts resulted in a deficiency in epicardium-derived cardiac fibroblasts in the myocardium, leading to reduced cardiomyocyte proliferation. This highlights that the presence of EPDCs underlying the myocardium is crucial for myocardial growth.
Expanding on this, Barnes et al. (2011) revealed that deletion of transcription factors Hand2 in Hand1-or Wt1-expressing cells caused epicardial disorganization, non-compaction and outflow tract defects. Mutants had a deficiency in fibroblasts because of lineage shifting from Pdgfrα+ (fibroblast-biased) to Pdgfrβ+ (vascular-biased) EPDCs. They further identified that a disrupted balance between fibronectin and its receptor disrupts proper ECM assembly, which is critical for fibroblast to anchor in the myocardium, thereby causing myocardial compaction defects, as reviewed in George and Firulli (2019). Also, ablation of cardiac fibroblast mediated by Pdgfrα-CreER controlled DTA system caused myocardial and vasculature defects in developing heart (Deng et al., 2025).
Recently, Xiao et al. (2018) demonstrated that deletion of Hippo signaling kinases Lats1/2 in mice epicardium manifests a significant deficiency in EPDCs differentiated cardiac fibroblasts. Instead of achieving a fibroblast fate, EPDCs remained in a transitional state, co-expressing both epicardial and fibroblast markers. Mechanistically, the increased nuclear localization of Yap interfered with fibroblast differentiation by disrupting RA synthesis and extracellular matrix remodeling. Supporting this finding, cells grown on stiffer substrates exhibited more nuclear Yap as well as reduced fibroblast marker expression. These findings suggest that mechanical environments can modulate Hippo signaling to affect epicardial cell fate and myocardial growth. Further involvement of Hippo pathway has been discussed in Section 4.4.
4.4 Epicardium regulated coronary vasculature formation influences heart development
Epicardial signaling also exerts essential control over coronary vasculature formation of the heart during development. For example, epicardial Gata4/6 deletion affected endothelial cell recruitment and plexus development, indicating epicardial necessity in vascular formation (Kolander et al., 2014). Multiple downstream pathways direct EPDCs differentiation into cVSMCs. Landerholm et al. (1999) showed that Serum Response Factor (SRF) is indispensable for cVSMCs differentiation but not for EMT. Similarly, RhoA signaling promotes cVSMCs development by supporting EPDC survival and migration (Lu et al., 2001). Epicardial Wnt/β-catenin signaling, as illustrated by Zamora et al. (2007), is dispensable for epicardial formation, but essential specifically for EPDC invasion and SMCs differentiation. Furthermore, as studied by Smart et al. (2007), Thymosin β4 (Tβ4) was shown to be both necessary for EPDC migration and differentiation into coronary vascular lineages. Loss of Tβ4 in the heart leads to the entrapment of Tie2+ endothelial and α-actin+ smooth muscle cells in the epicardium, leading to defective coronary vasculature.
Environmental conditions also regulate epicardial signaling. Embryonic hypoxia impaired coronary development as well as compact myocardium formation (Nanka et al., 2008). Hypoxia promotes EMT and causes premature differentiation into cVSMCs through HIF-1α–Snail signaling, as delineated by Jing et al. (2016). Tao et al. (2018) further linked hypoxia to non-canonical Tgfβ-RhoA signaling, showing its necessity for EMT and SMC marker expression.
Additionally, Pdgfrβ signaling plays a critical role in cVSMCs formation. Mellgren et al. (2008) revealed that both epicardial and non-epicardial Pdgfrβ signaling are crucial for cVSMCs formation. Another major signaling pathway, Notch, functions in multiple axes to regulate epicardium. Grieskamp et al. (2011) showed that epicardial deletion of Rbpj (transcriptional regulator of Notch) disrupted EPDC differentiation into SMCs while differentiation into fibroblasts remained unaffected. del Monte et al. (2011) further dissected Notch-specific roles: epicardial Notch1 deletion impaired coronary arteries and myocardial compaction via Raldh2 downregulation, while Notch2/3 were enriched in perivascular regions and drove SMCs maturation. These findings show spatially distinct roles for Notch in coordinating myocardial growth and coronary differentiation.
Myocardin-related transcription factors (MRTF) Mrtfa/b were shown by Trembley et al. (2015) to orchestrate EPDC motility and pericyte formation. Deletion of Mrtfa/b impaired EPDCs migration, causing subepicardial hemorrhage and pericyte loss (Quijada et al., 2020). Hippo signaling also intersects with vascular formation. Singh et al. (2016) found that Sema3D Cre/+ mediated epicardial deletion of Yap/Taz disrupted EMT and coronary formation.
Cilia-related signaling, specifically Wdpcp, also modulates epicardial responses. Liu et al. (2018) showed Wdpcp mutant hearts had premature subepicardial plexus formation and reduced SMC coverage. They found cilia-mediated Shh responsiveness as a regulator of coronary remodeling. Recently, Palmquist-Gomes et al. (2023) demonstrated that Itga4 deletion causes disrupted epicardial formation, leading to myocardial discontinuities and endocardial extrusion, which resembles congenital coronary artery fistula (CAF) morphology. Assessment of CAF structure with lineage tracing revealed epicardial origin of the outer smooth muscle walls and endocardial origin of the inner lining. This highlights the consequences of premature epicardial-myocardial contact and reinforces the structural role of epicardial adhesion in cardiac integrity.
4.5 Epicardium regulates myocardium via extracellular matrix (ECM) modulation
Multiple reports underscored the role of ECM components mediating epicardial regulation of myocardium in recent years (Bowers et al., 2022). Craig et al. (2010a) revealed that hyaluronan HMW-HA stimulates EPDCs differentiation and invasion into the myocardium, which is regulated by MEKK1 phosphorylation. Allison et al. (2015) identified that HA-mediated EPDCs invasion is regulated through Tgfbr3, further dependent on the downstream Src-RhoA/Rac1pathway. Craig et al. (2010b) further demonstrated that Tgfβ2 stimulates epicardial motility in mouse and human epicardial explants by indirectly enhancing hyaluronan synthase 2 (Has2) expression and HA. Likewise, Sun et al. (2022) revealed that targeted ablation of hapln1a+/Tcf21+ EPDCs impairs HA deposition, reduces cardiomyocyte proliferation, and leads to a significantly thinner and disorganized compact layer in zebrafish (Foglio et al., 2024).
Matrix degradation has also been correlated with epicardial invasion. Combs et al. (2011) revealed that Nfatc1 regulates matrix degradation via Ctsk. Loss of Nfatc1 impaired ECM breakdown, reducing EPDC invasion into the myocardium. Similarly, a recent study by Jiang et al. (2023) showed that epicardial Ezh2 is essential for matrix degradation during invasion. Ezh2 was found in human and mouse epicardium, and it acts as a repressor of TIMP3 (tissue inhibitor of metalloproteinase 3) to promote matrix degradation and epicardial cell migration. Wt1 Cre/+ mediated epicardial deletion of Ezh2 showed impaired EMT associated with defective basement membrane degradation and myocardial hypoplasia.
Additional ECM regulators have been reported. According to Sun et al. (2021), basement membrane-associated proteoglycan agrin plays as a crucial regulator of epicardial EMT. Deletion of agrin caused impaired EMT, disruption in basement membrane integrity, and a reduction in Wt1+ EPDCs in myocardium. Functionally, agrin decreases β-catenin and promotes pFAK localization at focal adhesions to enhance EMT, according to findings from human embryonic stem cell-derived epicardial-like cells. Similarly, Bonet et al. (2022) reported that Ccbe1 (ECM protein) KO mouse manifested thinner and hyper-trabeculated ventricular myocardium with reduced cardiomyocyte and epicardial cells proliferation.
At an earlier developmental stage, Nahirney et al. (2003) identified an extracellular matrix bridge (ECMB) in the pericardial cavity, which enables migration of PEO cells to the myocardium. One of the components of ECMB is heparan sulfate, degradation of which resulted in aberrant development of the chick primordial epicardium. This underscores the requirement of ECM components for proper transfer of PEO cells to the myocardium (Nahirney et al., 2003).
Most recently, Wang et al. (2024) revealed Ccm2, a novel signaling molecule to regulate epicardium mediated heart development. Ccm2 is traditionally known for its association with cerebral cavernous malformations, and its epicardial-specific deletion resulted in myocardial thinning and early lethality. Functionally, Ccm2-deficient epicardial cells lose polarity and cell shape, exhibit Golgi mis-localization and disorganized actin stress with altered matrix and cytoskeletal genes. Thus, it highlights the requirement of cytoskeletal integrity and adhesion of EPDCs to myocardium for proper heart development. A comprehensive schematic diagram highlighting major pathways involved in epicardium myocardium crosstalk during development have been provided in Figure 1.
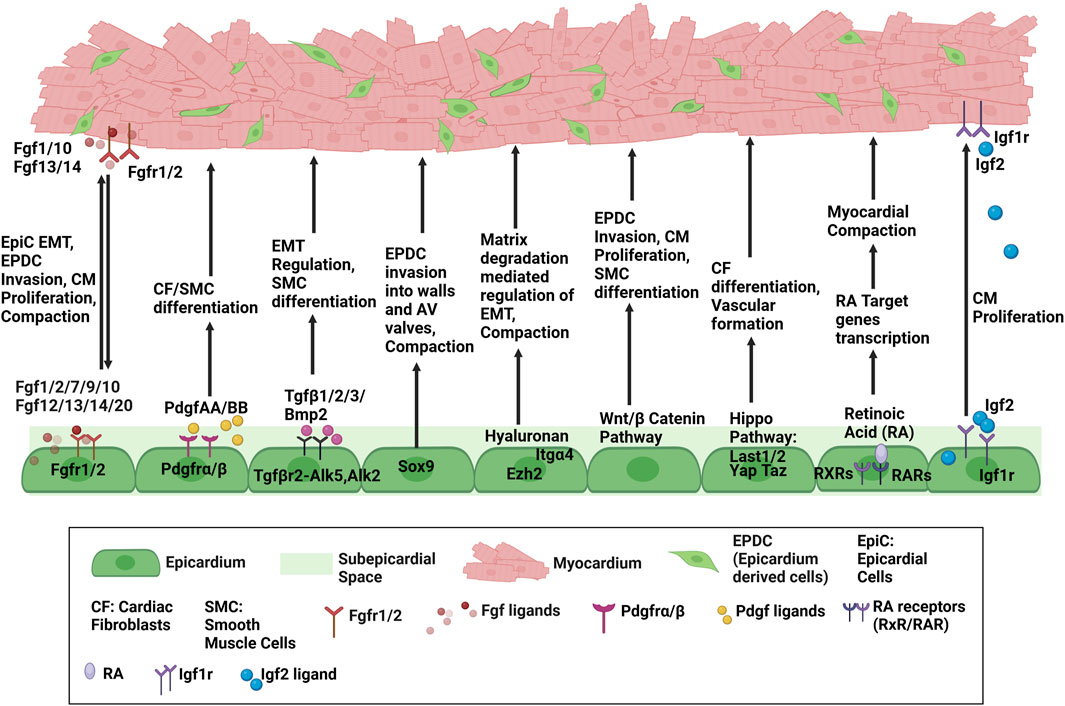
Figure 1. Schematic diagram highlighting major pathways regulating Epicardium-Myocardium Crosstalk.
5 Conclusion and future prospects
In summary, the epicardium undergoes complex and dynamic interactions with the myocardium to regulate myocardial proliferation, growth and wall formation, EPDCs invasion, fibroblast differentiation, coronary vasculature formation, and extracellular matrix remodeling. Recent advances have heavily focused on unraveling the molecular complexity underlying these processes, yet myriads of fundamental aspects of epicardium-myocardium crosstalk remain unresolved and need attention to elucidate. For example, the lineage potential, molecular heterogeneity, and spatiotemporal dynamics of the epicardium remain an active area of controversy.
To fully understand the sophistication of epicardial regulation of heart development, a clear understanding of epicardial lineage contribution to the heart is necessary. As discussed throughout this review, several studies have reported epicardial contributions to cardiomyocytes and endothelial cells under certain experimental conditions; however, many of these conclusions came from in vitro or ex vivo systems. Also, recent studies reveal that even a trivial shift in intrinsic signaling or external cues can shift epicardial lineage fate. Therefore, precise control of experimental variables is required to avoid a misleading interpretation from epicardial lineage tracing.
Proepicardial and epicardial cell’s molecular heterogeneity is another complex field which is sparsely investigated. Weinberger et al. (2020) in zebrafish identified three distinct epicardial subpopulations, each with unique gene expression profile and role in cardiac morphogenesis. Dong et al. (2018) uncovered epithelial-mesenchymal hybrid state in many organs, including the heart, during mouse organogenesis via single-cell RNA sequencing (scRNA-seq). Recently, Farah et al. (2024) discovered several sub populations of EPDCs/cardiac fibroblasts in the ventricle with different expression profiles. Therefore, extensive high-resolution molecular profiling, such as scRNA-seq and ATAC-seq of mammalian models, is required to advance the understanding of the molecular and epigenetic heterogeneity within epicardial/EPDCs populations.
Spatiotemporal analysis of EPDCs populating the heart chambers is essential as epicardial contributions may vary between right and left ventricles during development, as suggested by Vicente-Steijn et al. (2015). They observed spatiotemporally distinct distribution of Wt1+ and Tcf21+ EPDCs between the two ventricles. Understanding these chamber-specific differences will provide insight into ventricle-specific differences in cardiomyopathies.
Another pressing knowledge gap is how epicardial dysfunction contributes to ventricular non-compaction cardiomyopathy (LVNC). A recent study Farah et al. (2024) has highlighted the necessity of Sema-Plexin signaling to mediate communication between compact zone fibroblasts with trabecular cardiomyocytes, during ventricular compaction. This highlights the breadth of unexplored areas of EPDCs and cardiomyocyte signaling to mediate the compaction process. Furthermore, epicardial-myocardial crosstalk likely involves intricate cellular communication that is still poorly explored. While long-distance endocardial cell-cardiomyocytes communication during development was intensively studied Miao et al. (2025), parallel studies exploring the bidirectional communication between epicardial cells/EPDCs and cardiomyocytes are urgently needed.
Translational research in epicardial biology is very promising yet remains at a very primary stage. Human stem cell-derived epicardial-like cells and epicardioids have emerged as valuable tools for studying epicardial behavior (Meier et al., 2023; Moerkamp et al., 2016; Sun et al., 2021). However, the conclusions from these models still need to be validated because of their imperfect recapitulation of the in vivo settings. Improving 3D organoid models, co-culture systems with cardiomyocytes, and testing with biomechanical factors are essential to refine these systems and advance human epicardial development and disease.
Finally, epicardial biology remains an underexplored therapeutic target in congenital heart disease and cardiomyopathies. A deeper understanding of epicardial heterogeneity, cellular and molecular level dynamics of EPDC-cardiomyocyte signaling may open new avenues for developmental studies and congenital heart disease research.
Author contributions
AN: Conceptualization, Investigation, Validation, Data curation, Formal Analysis, Methodology, Writing – original draft. MW: Conceptualization, Validation, Funding acquisition, Project administration, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the following: American Heart Association Pre-Doctoral Fellowship grant 23PRE1025768 to AN. National Heart, Lung, and Blood Institute grant 2R01HL121700-06A1 and 1R01HL172834-01 to MW. American Heart Association 20TPA35490051, 24TPA1303770 and 25EIA1422015 awards to MW.
Acknowledgments
We thank Wu lab members for their scientific discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Generative AI was used to help Grammar check and organize the information in tables.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Acharya, A., Baek, S. T., Banfi, S., Eskiocak, B., and Tallquist, M. D. (2011). Efficient inducible cre-mediated recombination in Tcf21cell lineages in the heart and kidney. Genesis 49, 870–877. doi:10.1002/dvg.20750
Acharya, A., Baek, S. T., Huang, G., Eskiocak, B., Goetsch, S., Sung, C. Y., et al. (2012). The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 139, 2139–2149. doi:10.1242/dev.079970
Ali, S. R., Ranjbarvaziri, S., Talkhabi, M., Zhao, P., Subat, A., Hojjat, A., et al. (2014). Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circulation Res. 115, 625–635. doi:10.1161/CIRCRESAHA.115.303794
Allison, P., Espiritu, D., Barnett, J. V., and Camenisch, T. D. (2015). Type III TGFβ receptor and Src direct hyaluronan-mediated invasive cell motility. Cell. Signal. 27, 453–459. doi:10.1016/j.cellsig.2014.11.037
Arora, H., Boulberdaa, M., Qureshi, R., Bitirim, V., Gasser, A., Messaddeq, N., et al. (2016). Prokineticin receptor-1 signaling promotes epicardial to mesenchymal transition during heart development. Sci. Rep. 6, 25541. doi:10.1038/srep25541
Astanina, E., Doronzo, G., Corà, D., Neri, F., Oliviero, S., Genova, T., et al. (2022). The TFEB-TGIF1 axis regulates EMT in mouse epicardial cells. Nat. Commun. 13, 5191. doi:10.1038/s41467-022-32855-3
Austin, A. F., Compton, L. A., Love, J. D., Brown, C. B., and Barnett, J. V. (2008). Primary and immortalized mouse epicardial cells undergo differentiation in response to TGFbeta. Dev. Dyn. 237, 366–376. doi:10.1002/dvdy.21421
Baek, S. T., and Tallquist, M. D. (2012). Nf1 limits epicardial derivative expansion by regulating epithelial to mesenchymal transition and proliferation. Development 139, 2040–2049. doi:10.1242/dev.074054
Barnes, R. M., Firulli, B. A., VanDusen, N. J., Morikawa, Y., Conway, S. J., Cserjesi, P., et al. (2011). Hand2 loss-of-function in Hand1-expressing cells reveals distinct roles in epicardial and coronary vessel development. Circulation Res. 108, 940–949. doi:10.1161/CIRCRESAHA.110.233171
Barton, K., Muthusamy, N., Fischer, C., Ting, C.-N., Walunas, T. L., Lanier, L. L., et al. (1998). The Ets-1 transcription factor is required for the development of natural killer cells in mice. Immunity 9, 555–563. doi:10.1016/s1074-7613(00)80638-x
Boezio, G. L., Zhao, S., Gollin, J., Priya, R., Mansingh, S., Guenther, S., et al. (2023). The developing epicardium regulates cardiac chamber morphogenesis by promoting cardiomyocyte growth. Dis. Models Mech. 16, dmm049571. doi:10.1242/dmm.049571
Bonet, F., Añez, S. B., Inácio, J. M., Futschik, M. E., and Belo, J. A. (2022). CCBE1 is essential for epicardial function during myocardium development. Int. J. Mol. Sci. 23, 12642. doi:10.3390/ijms232012642
Boström, H., Willetts, K., Pekny, M., Levéen, P., Lindahl, P., Hedstrand, H., et al. (1996). PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 85, 863–873. doi:10.1016/s0092-8674(00)81270-2
Bowers, S. L., Meng, Q., and Molkentin, J. D. (2022). Fibroblasts orchestrate cellular crosstalk in the heart through the ECM. Nat. Cardiovasc. Res. 1, 312–321. doi:10.1038/s44161-022-00043-7
Brade, T., Kumar, S., Cunningham, T. J., Chatzi, C., Zhao, X., Cavallero, S., et al. (2011). Retinoic acid stimulates myocardial expansion by induction of hepatic erythropoietin which activates epicardial Igf2. Development 138, 139–148. doi:10.1242/dev.054239
Braitsch, C. M., Combs, M. D., Quaggin, S. E., and Yutzey, K. E. (2012). Pod1/Tcf21 is regulated by retinoic acid signaling and inhibits differentiation of epicardium-derived cells into smooth muscle in the developing heart. Dev. Biol. 368, 345–357. doi:10.1016/j.ydbio.2012.06.002
Cai, C.-L., Martin, J. C., Sun, Y., Cui, L., Wang, L., Ouyang, K., et al. (2008). A myocardial lineage derives from Tbx18 epicardial cells. Nature 454, 104–108. doi:10.1038/nature06969
Cao, Y., and Cao, J. (2018). Covering and re-covering the heart: development and regeneration of the epicardium. J. Cardiovasc. Dev. Dis. 6, 3. doi:10.3390/jcdd6010003
Cao, J., Navis, A., Cox, B. D., Dickson, A. L., Gemberling, M., Karra, R., et al. (2016). Single epicardial cell transcriptome sequencing identifies caveolin 1 as an essential factor in zebrafish heart regeneration. Development 143, 232–243. doi:10.1242/dev.130534
Cao, Y., Duca, S., and Cao, J. (2020). Epicardium in heart development. Cold Spring Harb. Perspect. Biol. 12, a037192. doi:10.1101/cshperspect.a037192
Chau, Y.-Y., Bandiera, R., Serrels, A., Martínez-Estrada, O. M., Qing, W., Lee, M., et al. (2014). Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nat. cell Biol. 16, 367–375. doi:10.1038/ncb2922
Chen, J., Kubalak, S. W., and Chien, K. R. (1998). Ventricular muscle-restricted targeting of the RXRalpha gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development 125, 1943–1949. doi:10.1242/dev.125.10.1943
Chen, T. H.-P., Chang, T.-C., Kang, J.-O., Choudhary, B., Makita, T., Tran, C. M., et al. (2002). Epicardial induction of fetal cardiomyocyte proliferation via a retinoic acid-inducible trophic factor. Dev. Biol. 250, 198–207. doi:10.1006/dbio.2002.0796
Chong, J. J., Reinecke, H., Iwata, M., Torok-Storb, B., Stempien-Otero, A., and Murry, C. E. (2013). Progenitor cells identified by PDGFR-alpha expression in the developing and diseased human heart. Stem cells Dev. 22, 1932–1943. doi:10.1089/scd.2012.0542
Christoffels, V. M., Grieskamp, T., Norden, J., Mommersteeg, M. T., Rudat, C., and Kispert, A. (2009). Tbx18 and the fate of epicardial progenitors. Nature 458, E8–E9. doi:10.1038/nature07916
Clark, C. R., Robinson, J. Y., Sanchez, N. S., Townsend, T. A., Arrieta, J. A., Merryman, W. D., et al. (2016). Common pathways regulate type III TGFβ receptor-dependent cell invasion in epicardial and endocardial cells. Cell. Signal. 28, 688–698. doi:10.1016/j.cellsig.2016.03.004
Combs, M. D., Braitsch, C. M., Lange, A. W., James, J. F., and Yutzey, K. E. (2011). NFATC1 promotes epicardium-derived cell invasion into myocardium. Development 138, 1747–1757. doi:10.1242/dev.060996
Compton, L. A., Potash, D. A., Mundell, N. A., and Barnett, J. V. (2006). Transforming growth factor-β induces loss of epithelial character and smooth muscle cell differentiation in epicardial cells. Dev. Dyn. 235, 82–93. doi:10.1002/dvdy.20629
Craig, E. A., Parker, P., Austin, A. F., Barnett, J. V., and Camenisch, T. D. (2010a). Involvement of the MEKK1 signaling pathway in the regulation of epicardial cell behavior by hyaluronan. Cell. Signal. 22, 968–976. doi:10.1016/j.cellsig.2010.02.004
Craig, E. A., Austin, A. F., Vaillancourt, R. R., Barnett, J. V., and Camenisch, T. D. (2010b). TGFβ2-mediated production of hyaluronan is important for the induction of epicardial cell differentiation and invasion. Exp. Cell Res. 316, 3397–3405. doi:10.1016/j.yexcr.2010.07.006
Debnath, P., Huirem, R. S., Dutta, P., and Palchaudhuri, S. (2022). Epithelial–mesenchymal transition and its transcription factors. Biosci. Rep. 42, BSR20211754. doi:10.1042/BSR20211754
del Monte, G., Casanova, J. C., Guadix, J. A., MacGrogan, D., Burch, J. B., Pérez-Pomares, J. M., et al. (2011). Differential notch signaling in the epicardium is required for cardiac inflow development and coronary vessel morphogenesis. Circulation Res. 108, 824–836. doi:10.1161/CIRCRESAHA.110.229062
DeLaughter, D. M., Clark, C. R., Christodoulou, D. C., Seidman, C. E., Baldwin, H. S., Seidman, J., et al. (2016). Transcriptional profiling of cultured, embryonic epicardial cells identifies novel genes and signaling pathways regulated by TGFβR3 in vitro. PLoS One 11, e0159710. doi:10.1371/journal.pone.0159710
Deng, Y., He, Y., Xu, J., He, H., Zhang, M., and Li, G. (2025). Cardiac fibroblasts regulate myocardium and coronary vasculature development in the murine heart via the collagen signaling pathway. eLife 13, RP102305. doi:10.7554/eLife.102305
Dettman, R. W., Denetclaw, Jr W., Ordahl, C. P., and Bristow, J. (1998). Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev. Biol. 193, 169–181. doi:10.1006/dbio.1997.8801
Dettman, R. W., Pae, S. H., Morabito, C., and Bristow, J. (2003). Inhibition of alpha4-integrin stimulates epicardial-mesenchymal transformation and alters migration and cell fate of epicardially derived mesenchyme. Dev. Biol. 257, 315–328. doi:10.1016/s0012-1606(03)00064-2
Dong, J., Hu, Y., Fan, X., Wu, X., Mao, Y., Hu, B., et al. (2018). Single-cell RNA-seq analysis unveils a prevalent epithelial/mesenchymal hybrid state during mouse organogenesis. Genome Biol. 19, 31–20. doi:10.1186/s13059-018-1416-2
Dronkers, E., Wauters, M. M., Goumans, M. J., and Smits, A. M. (2020). Epicardial TGFβ and BMP signaling in cardiac regeneration: what lesson can we learn from the developing heart? Biomolecules 10, 404. doi:10.3390/biom10030404
Dronkers, E., van Herwaarden, T., van Brakel, T. J., Sanchez-Duffhues, G., Goumans, M.-J., and Smits, A. M. (2021). Activin A and ALK4 identified as novel regulators of epithelial to mesenchymal transition (EMT) in human epicardial cells. Front. Cell Dev. Biol. 9, 765007. doi:10.3389/fcell.2021.765007
Du, J., Yuan, X., Deng, H., Huang, R., Liu, B., Xiong, T., et al. (2023). Single-cell and spatial heterogeneity landscapes of mature epicardial cells. J. Pharm. Analysis 13, 894–907. doi:10.1016/j.jpha.2023.07.011
Dueñas, A., Expósito, A., Muñoz, M. M., De Manuel, M. J., Cámara-Morales, A., Serrano-Osorio, F., et al. (2020). MiR-195 enhances cardiomyogenic differentiation of the proepicardium/septum transversum by Smurf1 and Foxp1 modulation. Sci. Rep. 10, 9334. doi:10.1038/s41598-020-66325-x
Duim, S. N., Goumans, M.-J., and Kruithof, B. P. (2016). WT1 in cardiac development and disease. Brisbane, Australia: Exon Publications, 211–233.
Fang, M., Xiang, F.-L., Braitsch, C. M., and Yutzey, K. E. (2016). Epicardium-derived fibroblasts in heart development and disease. J. Mol. Cell. Cardiol. 91, 23–27. doi:10.1016/j.yjmcc.2015.12.019
Farah, E. N., Hu, R. K., Kern, C., Zhang, Q., Lu, T.-Y., Ma, Q., et al. (2024). Spatially organized cellular communities form the developing human heart. Nature 627, 854–864. doi:10.1038/s41586-024-07171-z
Foglio, E., D’Avorio, E., Nieri, R., Russo, M. A., and Limana, F. (2024). Epicardial EMT and cardiac repair: an update. Stem Cell Res. Ther. 15, 219. doi:10.1186/s13287-024-03823-z
Garcia-Padilla, C., Hernandez-Torres, F., Lozano-Velasco, E., Dueñas, A., Muñoz-Gallardo, M. M., Garcia-Valencia, I. S., et al. (2022). The role of bmp-and fgf signaling modulating mouse proepicardium cell fate. Front. Cell Dev. Biol. 9, 757781. doi:10.3389/fcell.2021.757781
Gentile, A., Bensimon-Brito, A., Priya, R., Maischein, H.-M., Piesker, J., Guenther, S., et al. (2021). The EMT transcription factor Snai1 maintains myocardial wall integrity by repressing intermediate filament gene expression. Elife 10, e66143. doi:10.7554/eLife.66143
George, R. M., and Firulli, A. B. (2019). Hand factors in cardiac development. Anatomical Rec. 302, 101–107. doi:10.1002/ar.23910
Gittenberger-de Groot, A. C., Vrancken Peeters, M.-P. F., Mentink, M. M., Gourdie, R. G., and Poelmann, R. E. (1998). Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circulation Res. 82, 1043–1052. doi:10.1161/01.res.82.10.1043
Gittenberger-de Groot, A. C., Vrancken Peeters, M.-P. F., Bergwerff, M., Mentink, M. M., and Poelmann, R. E. (2000). Epicardial outgrowth inhibition leads to compensatory mesothelial outflow tract collar and abnormal cardiac septation and coronary formation. Circulation Res. 87, 969–971. doi:10.1161/01.res.87.11.969
Greulich, F., Farin, H. F., Schuster-Gossler, K., and Kispert, A. (2012). Tbx18 function in epicardial development. Cardiovasc. Res. 96, 476–483. doi:10.1093/cvr/cvs277
Grieskamp, T., Rudat, C., Lüdtke, T. H.-W., Norden, J., and Kispert, A. (2011). Notch signaling regulates smooth muscle differentiation of epicardium-derived cells. Circulation Res. 108, 813–823. doi:10.1161/CIRCRESAHA.110.228809
Gruber, P. J., Kubalak, S. W., Pexieder, T., Sucov, H. M., Evans, R. M., and Chien, K. R. (1996). RXR alpha deficiency confers genetic susceptibility for aortic sac, conotruncal, atrioventricular cushion, and ventricular muscle defects in mice. J. Clin. Investigation 98, 1332–1343. doi:10.1172/JCI118920
Guadix, J. A., Carmona, R., Muñoz-Chápuli, R., and Pérez-Pomares, J. M. (2006). In vivo and in vitro analysis of the vasculogenic potential of avian proepicardial and epicardial cells. Dev. Dyn. 235, 1014–1026. doi:10.1002/dvdy.20685
Guadix, J. A., Ruiz-Villalba, A., Lettice, L., Velecela, V., Muñoz-Chápuli, R., Hastie, N. D., et al. (2011). Wt1 controls retinoic acid signalling in embryonic epicardium through transcriptional activation of Raldh2. Development 138, 1093–1097. doi:10.1242/dev.044594
Guimarães-Camboa, N., Cattaneo, P., Sun, Y., Moore-Morris, T., Gu, Y., Dalton, N. D., et al. (2017). Pericytes of multiple organs do not behave as mesenchymal stem cells in vivo. Cell Stem Cell 20, 345–359. e5. doi:10.1016/j.stem.2016.12.006
Harvey, A. B., Wolters, R. A., Deepe, R. N., Tarolli, H. G., Drummond, J. R., Trouten, A., et al. (2024). Epicardial deletion of Sox9 leads to myxomatous valve degeneration and identifies Cd109 as a novel gene associated with valve development. J. Mol. Cell. Cardiol. 186, 16–30. doi:10.1016/j.yjmcc.2023.11.002
Haworth, K. E., Kotecha, S., Mohun, T. J., and Latinkic, B. V. (2008). GATA4 and GATA5 are essential for heart and liver development in xenopus embryos. BMC Dev. Biol. 8, 74–16. doi:10.1186/1471-213X-8-74
Hill, C. R., Sanchez, N. S., Love, J. D., Arrieta, J. A., Hong, C. C., Brown, C. B., et al. (2012). BMP2 signals loss of epithelial character in epicardial cells but requires the type III TGFβ receptor to promote invasion. Cell. Signal. 24, 1012–1022. doi:10.1016/j.cellsig.2011.12.022
Hiruma, T., and Hirakow, R. (1989). Epicardial formation in embryonic chick heart: computer-aided reconstruction, scanning, and transmission electron microscopic studies. Am. J. Anat. 184, 129–138. doi:10.1002/aja.1001840204
Hotta, Y., Sasaki, S., Konishi, M., Kinoshita, H., Kuwahara, K., Nakao, K., et al. (2008). Fgf16 is required for cardiomyocyte proliferation in the mouse embryonic heart. Dev. Dyn. 237, 2947–2954. doi:10.1002/dvdy.21726
Huang, Y., Harrison, M. R., Osorio, A., Kim, J., Baugh, A., Duan, C., et al. (2013). Igf signaling is required for cardiomyocyte proliferation during zebrafish heart development and regeneration. PloS one 8, e67266. doi:10.1371/journal.pone.0067266
Iyer, D., Gambardella, L., Bernard, W. G., Serrano, F., Mascetti, V. L., Pedersen, R. A., et al. (2015). Robust derivation of epicardium and its differentiated smooth muscle cell progeny from human pluripotent stem cells. Development 142, 1528–1541. doi:10.1242/dev.119271
Iyer, S., Chou, F. Y., Wang, R., Chiu, H. S., Raju, V. K. S., Little, M. H., et al. (2016). Crim1 has cell-autonomous and paracrine roles during embryonic heart development. Sci. Rep. 6, 19832. doi:10.1038/srep19832
Jackson-Weaver, O., Ungvijanpunya, N., Yuan, Y., Qian, J., Gou, Y., Wu, J., et al. (2020). PRMT1-p53 pathway controls epicardial EMT and invasion. Cell Rep. 31, 107739. doi:10.1016/j.celrep.2020.107739
Jahr, M., Schlueter, J., Brand, T., and Männer, J. (2008). Development of the proepicardium in Xenopus laevis. Dev. Dyn. 237, 3088–3096. doi:10.1002/dvdy.21713
Jang, J., Song, G., Pettit, S. M., Li, Q., Song, X., Cai, C.-l., et al. (2022). Epicardial HDAC3 promotes myocardial growth through a novel microRNA pathway. Circulation Res. 131, 151–164. doi:10.1161/CIRCRESAHA.122.320785
Jiang, H., Bai, L., Song, S., Yin, Q., Shi, A., Zhou, B., et al. (2023). EZH2 controls epicardial cell migration during heart development. Life Sci. Alliance 6, e202201765. doi:10.26508/lsa.202201765
Jing, X., Gao, Y., Xiao, S., Qin, Q., Wei, X., Yan, Y., et al. (2016). Hypoxia induced the differentiation of Tbx18-positive epicardial cells to CoSMCs. Sci. Rep. 6, 30468. doi:10.1038/srep30468
Junghof, J., Kogure, Y., Yu, T., Verdugo-Sivianes, E. M., Narita, M., Lucena-Cacace, A., et al. (2022). CDH18 is a fetal epicardial biomarker regulating differentiation towards vascular smooth muscle cells. NPJ Regen. Med. 7, 14. doi:10.1038/s41536-022-00207-w
Kang, J., Gu, Y., Li, P., Johnson, B. L., Sucov, H. M., and Thomas, P. S. (2008). PDGF-A as an epicardial mitogen during heart development. Dev. Dyn. 237, 692–701. doi:10.1002/dvdy.21469
Kanisicak, O., Khalil, H., Ivey, M. J., Karch, J., Maliken, B. D., Correll, R. N., et al. (2016). Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 7, 12260. doi:10.1038/ncomms12260
Katz, T. C., Singh, M. K., Degenhardt, K., Rivera-Feliciano, J., Johnson, R. L., Epstein, J. A., et al. (2012). Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev. cell 22, 639–650. doi:10.1016/j.devcel.2012.01.012
Kelder, T. P., Duim, S. N., Vicente-Steijn, R., Végh, A. M., Kruithof, B. P., Smits, A. M., et al. (2015). The epicardium as modulator of the cardiac autonomic response during early development. J. Mol. Cell. Cardiol. 89, 251–259. doi:10.1016/j.yjmcc.2015.10.025
Kikuchi, K., Gupta, V., Wang, J., Holdway, J. E., Wills, A. A., Fang, Y., et al. (2011). tcf21+ epicardial cells adopt non-myocardial fates during zebrafish heart development and regeneration. Development 138, 2895–2902. doi:10.1242/dev.067041
Kim, J., Wu, Q., Zhang, Y., Wiens, K. M., Huang, Y., Rubin, N., et al. (2010). PDGF signaling is required for epicardial function and blood vessel formation in regenerating zebrafish hearts. Proc. Natl. Acad. Sci. 107, 17206–17210. doi:10.1073/pnas.0915016107
Knight-Schrijver, V. R., Davaapil, H., Bayraktar, S., Ross, A. D., Kanemaru, K., Cranley, J., et al. (2022). A single-cell comparison of adult and fetal human epicardium defines the age-associated changes in epicardial activity. Nat. Cardiovasc. Res. 1, 1215–1229. doi:10.1038/s44161-022-00183-w
Kolander, K. D., Holtz, M. L., Cossette, S. M., Duncan, S. A., and Misra, R. P. (2014). Epicardial GATA factors regulate early coronary vascular plexus formation. Dev. Biol. 386, 204–215. doi:10.1016/j.ydbio.2013.12.033
Komiyama, M., Ito, K., and Shimada, Y. (1987). Origin and development of the epicardium in the mouse embryo. Anat. embryology 176, 183–189. doi:10.1007/BF00310051
Kraus, F., Haenig, B., and Kispert, A. (2001). Cloning and expression analysis of the mouse T-box gene tbx20. Mech. Dev. 100, 87–91. doi:10.1016/s0925-4773(00)00499-8
Kreidberg, J. A., Sariola, H., Loring, J. M., Maeda, M., Pelletier, J., Housman, D., et al. (1993). WT-1 is required for early kidney development. Cell 74, 679–691. doi:10.1016/0092-8674(93)90515-r
Kruithof, B. P., van Wijk, B., Somi, S., Kruithof-de Julio, M., Pomares, J. M. P., Weesie, F., et al. (2006). BMP and FGF regulate the differentiation of multipotential pericardial mesoderm into the myocardial or epicardial lineage. Dev. Biol. 295, 507–522. doi:10.1016/j.ydbio.2006.03.033
Kurkiewicz, T. (1909). O histogenezie miesna sercowego zwierzat kregowych-Zur Histogenese des Herzmuskels der Wirbeltiere. Poland: Bull. Int. Acad. Sci. Cracovie.
Kwee, L., Baldwin, H. S., Shen, H. M., Stewart, C. L., Buck, C., Buck, C. A., et al. (1995). Defective development of the embryonic and extraembryonic circulatory systems in vascular cell adhesion molecule (VCAM-1) deficient mice. Development 121, 489–503. doi:10.1242/dev.121.2.489
Landerholm, T. E., Dong, X.-R., Lu, J., Belaguli, N. S., Schwartz, R. J., and Majesky, M. W. (1999). A role for serum response factor in coronary smooth muscle differentiation from proepicardial cells. Development 126, 2053–2062. doi:10.1242/dev.126.10.2053
Lavine, K. J., Yu, K., White, A. C., Zhang, X., Smith, C., Partanen, J., et al. (2005). Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev. cell 8, 85–95. doi:10.1016/j.devcel.2004.12.002
Levay, A. K., Peacock, J. D., Lu, Y., Koch, M., Hinton, Jr R. B., Kadler, K. E., et al. (2008). Scleraxis is required for cell lineage differentiation and extracellular matrix remodeling during murine heart valve formation in vivo. Circulation Res. 103, 948–956. doi:10.1161/CIRCRESAHA.108.177238
Li, P., Cavallero, S., Gu, Y., Chen, T. H., Hughes, J., Hassan, A. B., et al. (2011). IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development 138, 1795–1805. doi:10.1242/dev.054338
Li, J., Miao, L., Shieh, D., Spiotto, E., Li, J., Zhou, B., et al. (2016). Single-cell lineage tracing reveals that oriented cell division contributes to trabecular morphogenesis and regional specification. Cell Rep. 15, 158–170. doi:10.1016/j.celrep.2016.03.012
Li, J., Miao, L., Zhao, C., Shaikh Qureshi, W. M., Shieh, D., Guo, H., et al. (2017a). CDC42 is required for epicardial and pro-epicardial development by mediating FGF receptor trafficking to the plasma membrane. Development 144, 1635–1647. doi:10.1242/dev.147173
Li, Y., Urban, A., Midura, D., Simon, H.-G., and Wang, Q. T. (2017b). Proteomic characterization of epicardial-myocardial signaling reveals novel regulatory networks including a role for NF-κB in epicardial EMT. PloS one 12, e0174563. doi:10.1371/journal.pone.0174563
Li, Y., Du, J., Deng, S., Liu, B., Jing, X., Yan, Y., et al. (2024). The molecular mechanisms of cardiac development and related diseases. Signal Transduct. Target. Ther. 9, 368. doi:10.1038/s41392-024-02069-8
Lie-Venema, H., Gittenberger-de Groot, A. C., van Empel, L. J., Boot, M. J., Kerkdijk, H., de Kant, E., et al. (2003). Ets-1 and Ets-2 transcription factors are essential for normal coronary and myocardial development in chicken embryos. Circulation Res. 92, 749–756. doi:10.1161/01.RES.0000066662.70010.DB
Lie-Venema, H., Van Den Akker, N. M., Bax, N. A., Winter, E. M., Maas, S., Kekarainen, T., et al. (2007). Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. Sci. World J. 7, 1777–1798. doi:10.1100/tsw.2007.294
Lin, S.-C., Dollé, P., Ryckebüsch, L., Noseda, M., Zaffran, S., Schneider, M. D., et al. (2010). Endogenous retinoic acid regulates cardiac progenitor differentiation. Proc. Natl. Acad. Sci. 107, 9234–9239. doi:10.1073/pnas.0910430107
Liu, Q., Huang, X., Oh, J.-H., Lin, R.-Z., Duan, S., Yu, Y., et al. (2014). Epicardium-to-fat transition in injured heart. Cell Res. 24, 1367–1369. doi:10.1038/cr.2014.125
Liu, X., Wang, Y., Liu, F., Zhang, M., Song, H., Zhou, B., et al. (2018). Wdpcp promotes epicardial EMT and epicardium-derived cell migration to facilitate coronary artery remodeling. Sci. Signal. 11, eaah5770. doi:10.1126/scisignal.aah5770
Liu, X., Li, B., Wang, S., Zhang, E., Schultz, M., Touma, M., et al. (2024). Stromal Cell-SLIT3/Cardiomyocyte-ROBO1 axis regulates pressure overload-induced cardiac hypertrophy. Circulation Res. 134, 913–930. doi:10.1161/CIRCRESAHA.122.321292
Lu, J., Landerholm, T. E., Wei, J. S., Dong, X.-R., Wu, S.-P., Liu, X., et al. (2001). Coronary smooth muscle differentiation from proepicardial cells requires rhoA-mediated actin reorganization and p160 rho-kinase activity. Dev. Biol. 240, 404–418. doi:10.1006/dbio.2001.0403
Lupu, I.-E., Redpath, A. N., and Smart, N. (2020). Spatiotemporal analysis reveals overlap of key proepicardial markers in the developing murine heart. Stem Cell Rep. 14, 770–787. doi:10.1016/j.stemcr.2020.04.002
Macías, D., Pérez-Pomares, J. M., García-Garrido, L., Carmona, R., and Muñoz-Chápuli, R. (1998). Immunoreactivity of the ets-1 transcription factor correlates with areas of epithelial-mesenchymal transition in the developing avian heart. Anat. Embryology 198, 307–315. doi:10.1007/s004290050186
Mahtab, E. A., Wijffels, M. C., Van Den Akker, N. M., Hahurij, N. D., Lie-Venema, H., Wisse, L. J., et al. (2008). Cardiac malformations and myocardial abnormalities in podoplanin knockout mouse embryos: correlation with abnormal epicardial development. Dev. Dyn. 237, 847–857. doi:10.1002/dvdy.21463
Männer, J. (1993). Experimental study on the formation of the epicardium in chick embryos. Anat. Embryology 187, 281–289. doi:10.1007/BF00195766
Männer, J. (1999). Does the subepicardial mesenchyme contribute myocardioblasts to the myocardium of the chick embryo heart? A quail-chick chimera study tracing the fate of the epicardial primordium. Anatomical Rec. 255, 212–226. doi:10.1002/(sici)1097-0185(19990601)255:2<212::aid-ar11>3.3.co;2-o
Männer, J., Schlueter, J., and Brand, T. (2005). Experimental analyses of the function of the proepicardium using a new microsurgical procedure to induce loss-of-proepicardial-function in chick embryos. Dev. Dyn. 233, 1454–1463. doi:10.1002/dvdy.20487
Marques, I. J., Ernst, A., Arora, P., Vianin, A., Hetke, T., Sanz-Morejón, A., et al. (2022). Wt1 transcription factor impairs cardiomyocyte specification and drives a phenotypic switch from myocardium to epicardium. Development 149, dev200375. doi:10.1242/dev.200375
Martínez-Estrada, O. M., Lettice, L. A., Essafi, A., Guadix, J. A., Slight, J., Velecela, V., et al. (2010). Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of snail and E-cadherin. Nat. Genet. 42, 89–93. doi:10.1038/ng.494
Meier, A. B., Zawada, D., De Angelis, M. T., Martens, L. D., Santamaria, G., Zengerle, S., et al. (2023). Epicardioid single-cell genomics uncovers principles of human epicardium biology in heart development and disease. Nat. Biotechnol. 41, 1787–1800. doi:10.1038/s41587-023-01718-7
Mellgren, A. M., Smith, C. L., Olsen, G. S., Eskiocak, B., Zhou, B., Kazi, M. N., et al. (2008). Platelet-derived growth factor receptor β signaling is required for efficient epicardial cell migration and development of two distinct coronary vascular smooth muscle cell populations. Circulation Res. 103, 1393–1401. doi:10.1161/CIRCRESAHA.108.176768
Merki, E., Zamora, M., Raya, A., Kawakami, Y., Wang, J., Zhang, X., et al. (2005). Epicardial retinoid X receptor α is required for myocardial growth and coronary artery formation. Proc. Natl. Acad. Sci. 102, 18455–18460. doi:10.1073/pnas.0504343102
Miao, L., Li, J., Lu, Y., Yin, C., Sun, D., Lo, E., et al. (2021). Pro-epicardial cells are heterogeneous with specified smooth muscle-like and pacemaker progenitor cells. SSRN Electron. J. doi:10.2139/ssrn.3832987
Miao, L., Lu, Y., Nusrat, A., Fan, G., Zhang, S., Zhao, L., et al. (2025). Tunneling nanotube–like structures regulate distant cellular interactions during heart formation. Science. 387, eadd3417. doi:10.1126/science.add3417
Mikawa, T. (1995). Retroviral targeting of FGF and FGFR in cardiomyocytes and coronary vascular cells during heart development. Ann. N. Y. Acad. Sci. 752, 506–516. doi:10.1111/j.1749-6632.1995.tb17459.x
Mima, T., Ueno, H., Fischman, D., Williams, L., and Mikawa, T. (1995). Fibroblast growth factor receptor is required for in vivo cardiac myocyte proliferation at early embryonic stages of heart development. Proc. Natl. Acad. Sci. 92, 467–471. doi:10.1073/pnas.92.2.467
Moerkamp, A. T., Lodder, K., Van Herwaarden, T., Dronkers, E., Dingenouts, C. K., Tengström, F. C., et al. (2016). Human fetal and adult epicardial-derived cells: a novel model to study their activation. Stem cell Res. and Ther. 7, 174–12. doi:10.1186/s13287-016-0434-9
Moore, A. W., McInnes, L., Kreidberg, J., Hastie, N. D., and Schedl, A. (1999). YAC complementation shows a requirement for Wt1 in the development of epicardium, adrenal gland and throughout nephrogenesis. Development 126, 1845–1857. doi:10.1242/dev.126.9.1845
Moore-Morris, T., Guimarães-Camboa, N., Banerjee, I., Zambon, A. C., Kisseleva, T., Velayoudon, A., et al. (2014). Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J. Clin. Investigation 124, 2921–2934. doi:10.1172/JCI74783
Morabito, C. J., Dettman, R. W., Kattan, J., Collier, J. M., and Bristow, J. (2001). Positive and negative regulation of epicardial–mesenchymal transformation during avian heart development. Dev. Biol. 234, 204–215. doi:10.1006/dbio.2001.0254
Moss, J. B., Xavier-Neto, J., Shapiro, M. D., Nayeem, S. M., McCaffery, P., Dräger, U. C., et al. (1998). Dynamic patterns of retinoic acid synthesis and response in the developing mammalian heart. Dev. Biol. 199, 55–71. doi:10.1006/dbio.1998.8911
Nahirney, P. C., Mikawa, T., and Fischman, D. A. (2003). Evidence for an extracellular matrix bridge guiding proepicardial cell migration to the myocardium of chick embryos. Dev. Dyn. 227, 511–523. doi:10.1002/dvdy.10335
Nanka, O., Krizova, P., Fikrle, M., Tuma, M., Blaha, M., Grim, M., et al. (2008). Abnormal myocardial and coronary vasculature development in experimental hypoxia. Anatomical Rec. Adv. Integr. Anat. Evol. Biol. 291, 1187–1199. doi:10.1002/ar.20738
Niessen, K., Fu, Y., Chang, L., Hoodless, P. A., McFadden, D., and Karsan, A. (2008). Slug is a direct notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 182, 315–325. doi:10.1083/jcb.200710067
Olivey, H. E., and Svensson, E. C. (2010). Epicardial–myocardial signaling directing coronary vasculogenesis. Circulation Res. 106, 818–832. doi:10.1161/CIRCRESAHA.109.209197
Olivey, H. E., Mundell, N. A., Austin, A. F., and Barnett, J. V. (2006). Transforming growth factor-β stimulates epithelial–mesenchymal transformation in the proepicardium. Dev. Dyn. 235, 50–59. doi:10.1002/dvdy.20593
Palmquist-Gomes, P., Ruiz-Villalba, A., Guadix, J., Romero, J., Bessiéres, B., MacGrogan, D., et al. (2023). Origin of congenital coronary arterio-ventricular fistulae from anomalous epicardial and myocardial development. Exp. Mol. Med. 55, 228–239. doi:10.1038/s12276-022-00913-x
Peeters, M.-P. F. V., Mentink, M., Poelmann, R., and Groot, A. G. (1995). Cytokeratins as a marker for epicardial formation in the quail embryo. Anat. Embryology 191, 503–508. doi:10.1007/BF00186740
Pennisi, D. J., and Mikawa, T. (2009). FGFR-1 is required by epicardium-derived cells for myocardial invasion and correct coronary vascular lineage differentiation. Dev. Biol. 328, 148–159. doi:10.1016/j.ydbio.2009.01.023
Pennisi, D. J., Ballard, V. L., and Mikawa, T. (2003). Epicardium is required for the full rate of myocyte proliferation and levels of expression of myocyte mitogenic factors FGF2 and its receptor, FGFR-1, but not for transmural myocardial patterning in the embryonic chick heart. Dev. Dyn. 228, 161–172. doi:10.1002/dvdy.10360
Peralta, M., Steed, E., Harlepp, S., González-Rosa, J. M., Monduc, F., Ariza-Cosano, A., et al. (2013). Heartbeat-driven pericardiac fluid forces contribute to epicardium morphogenesis. Curr. Biol. 23, 1726–1735. doi:10.1016/j.cub.2013.07.005
Pérez-Pomares, J. M., and de la Pompa, J. L. (2011). Signaling during epicardium and coronary vessel development. Circulation Res. 109, 1429–1442. doi:10.1161/CIRCRESAHA.111.245589
Perez-Pomares, J., Phelps, A., Sedmerova, M., Carmona, R., Gonzalez-Iriarte, M., Munoz-Chapuli, R., et al. (2002). Experimental studies on the spatiotemporal expression of WT1 and RALDH2 in the embryonic avian heart: a model for the regulation of myocardial and valvuloseptal development by epicardially derived cells (EPDCs). Dev. Biol. 247, 307–326. doi:10.1006/dbio.2002.0706
Plavicki, J. S., Hofsteen, P., Yue, M. S., Lanham, K. A., Peterson, R. E., and Heideman, W. (2014). Multiple modes of proepicardial cell migration require heartbeat. BMC Dev. Biol. 14, 18–13. doi:10.1186/1471-213X-14-18
Poelmann, R. E., Gittenberger-de Groot, A. C., Mentink, M., Bökenkamp, R., and Hogers, B. (1993). Development of the cardiac coronary vascular endothelium, studied with antiendothelial antibodies, in chicken-quail chimeras. Circulation Res. 73, 559–568. doi:10.1161/01.res.73.3.559
Poelmann, R. E., Lie-Venema, H., and Gittenberger-de Groot, A. C. (2002). The role of the epicardium and neural crest: as extracardiac contributors to coronary vascular development. Tex. Heart Inst. J. 29, 255–261.
Pontemezzo, E., Foglio, E., Vernucci, E., Magenta, A., D’Agostino, M., Sileno, S., et al. (2021). miR-200c-3p regulates epitelial-to-mesenchymal transition in epicardial mesothelial cells by targeting epicardial follistatin-related protein 1. Int. J. Mol. Sci. 22, 4971. doi:10.3390/ijms22094971
Pryce, B. A., Brent, A. E., Murchison, N. D., Tabin, C. J., and Schweitzer, R. (2007). Generation of transgenic tendon reporters, ScxGFP and ScxAP, using regulatory elements of the scleraxis gene. Dev. Dyn. 236, 1677–1682. doi:10.1002/dvdy.21179
Quijada, P., Trembley, M. A., and Small, E. M. (2020). The role of the epicardium during heart development and repair. Circulation Res. 126, 377–394. doi:10.1161/CIRCRESAHA.119.315857
Reiter, J. F., Alexander, J., Rodaway, A., Yelon, D., Patient, R., Holder, N., et al. (1999). Gata5 is required for the development of the heart and endoderm in zebrafish. Genes Dev. 13, 2983–2995. doi:10.1101/gad.13.22.2983
Ridge, L. A., Mitchell, K., Al-Anbaki, A., Shaikh Qureshi, W. M., Stephen, L. A., Tenin, G., et al. (2017). Non-muscle myosin IIB (Myh10) is required for epicardial function and coronary vessel formation during mammalian development. PLoS Genet. 13, e1007068. doi:10.1371/journal.pgen.1007068
Risebro, C. A., Vieira, J. M., Klotz, L., and Riley, P. R. (2015). Characterisation of the human embryonic and foetal epicardium during heart development. Development 142, 3630–3636. doi:10.1242/dev.127621
Rodgers, L. S., Lalani, S., Runyan, R. B., and Camenisch, T. D. (2008). Differential growth and multicellular villi direct proepicardial translocation to the developing mouse heart. Dev. Dyn. 237, 145–152. doi:10.1002/dvdy.21378
Rudat, C., and Kispert, A. (2012). Wt1 and epicardial fate mapping. Circulation Res. 111, 165–169. doi:10.1161/CIRCRESAHA.112.273946
Rudat, C., Norden, J., Taketo, M. M., and Kispert, A. (2013). Epicardial function of canonical Wnt-Hedgehog-Fgfr1/2-and Pdgfra-signalling. Cardiovasc. Res. 100, 411–421. doi:10.1093/cvr/cvt210
Sánchez, N. S., and Barnett, J. V. (2012). TGFβ and BMP-2 regulate epicardial cell invasion via TGFβR3 activation of the Par6/Smurf1/RhoA pathway. Cell. Signal. 24, 539–548. doi:10.1016/j.cellsig.2011.10.006
Sánchez, N. S., Hill, C. R., Love, J. D., Soslow, J. H., Craig, E., Austin, A. F., et al. (2011). The cytoplasmic domain of TGFβR3 through its interaction with the scaffolding protein, GIPC, directs epicardial cell behavior. Dev. Biol. 358, 331–343. doi:10.1016/j.ydbio.2011.08.008
Sanchez-Fernandez, C., Rodriguez-Outeiriño, L., Matias-Valiente, L., Ramirez de Acuña, F., Hernandez-Torres, F., Lozano-Velasco, E., et al. (2022). Regulation of epicardial cell fate during cardiac development and disease: an overview. Int. J. Mol. Sci. 23, 3220. doi:10.3390/ijms23063220
Sekiya, S., and Suzuki, A. (2012). Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J. Clin. Investigation 122, 3914–3918. doi:10.1172/JCI63065
Sengbusch, J. K., He, W., Pinco, K. A., and Yang, J. T. (2002). Dual functions of [alpha]4[beta]1 integrin in epicardial development: initial migration and long-term attachment. J. Cell Biol. 157, 873–882. doi:10.1083/jcb.200203075
Shen, H., Cavallero, S., Estrada, K. D., Sandovici, I., Kumar, S. R., Makita, T., et al. (2015). Extracardiac control of embryonic cardiomyocyte proliferation and ventricular wall expansion. Cardiovasc. Res. 105, 271–278. doi:10.1093/cvr/cvu269
Shrestha, R., McCann, T., Saravanan, H., Lieberth, J., Koirala, P., and Bloomekatz, J. (2023). The myocardium utilizes a platelet-derived growth factor receptor alpha (Pdgfra)–phosphoinositide 3-kinase (PI3K) signaling cascade to steer toward the midline during zebrafish heart tube formation. Elife 12, e85930. doi:10.7554/eLife.85930
Simões, F. C., and Riley, P. R. (2018). The ontogeny, activation and function of the epicardium during heart development and regeneration. Development 145, dev155994. doi:10.1242/dev.155994
Singh, M. K., Lu, M. M., Massera, D., and Epstein, J. A. (2011). MicroRNA-processing enzyme dicer is required in epicardium for coronary vasculature development. J. Biol. Chem. 286, 41036–41045. doi:10.1074/jbc.M111.268573
Singh, A., Ramesh, S., Cibi, D. M., Yun, L. S., Li, J., Li, L., et al. (2016). Hippo signaling mediators Yap and Taz are required in the epicardium for coronary vasculature development. Cell Rep. 15, 1384–1393. doi:10.1016/j.celrep.2016.04.027
Smart, N., Risebro, C. A., Melville, A. A., Moses, K., Schwartz, R. J., Chien, K. R., et al. (2007). Thymosin beta4 induces adult epicardial progenitor mobilization and neovascularization. Nature 445, 177–182. doi:10.1038/nature05383
Smith, C. L., Baek, S. T., Sung, C. Y., and Tallquist, M. D. (2011). Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circulation Res. 108, e15–e26. doi:10.1161/CIRCRESAHA.110.235531
Smits, A. M., Dronkers, E., and Goumans, M.-J. (2018). The epicardium as a source of multipotent adult cardiac progenitor cells: their origin, role and fate. Pharmacol. Res. 127, 129–140. doi:10.1016/j.phrs.2017.07.020
Sridurongrit, S., Larsson, J., Schwartz, R., Ruiz-Lozano, P., and Kaartinen, V. (2008). Signaling via the Tgf-β type I receptor Alk5 in heart development. Dev. Biol. 322, 208–218. doi:10.1016/j.ydbio.2008.07.038
Stuckmann, I., Evans, S., and Lassar, A. B. (2003). Erythropoietin and retinoic acid, secreted from the epicardium, are required for cardiac myocyte proliferation. Dev. Biol. 255, 334–349. doi:10.1016/s0012-1606(02)00078-7
Sun, X., Malandraki-Miller, S., Kennedy, T., Bassat, E., Klaourakis, K., Zhao, J., et al. (2021). The extracellular matrix protein agrin is essential for epicardial epithelial-to-mesenchymal transition during heart development. Development 148, dev197525. doi:10.1242/dev.197525
Sun, J., Peterson, E. A., Wang, A. Z., Ou, J., Smith, K. E., Poss, K. D., et al. (2022). hapln1 defines an epicardial cell subpopulation required for cardiomyocyte expansion during heart morphogenesis and regeneration. Circulation 146, 48–63. doi:10.1161/CIRCULATIONAHA.121.055468
Takahashi, M., Yamagishi, T., Narematsu, M., Kamimura, T., Kai, M., and Nakajima, Y. (2014). Epicardium is required for sarcomeric maturation and cardiomyocyte growth in the ventricular compact layer mediated by transforming growth factor β and fibroblast growth factor before the onset of coronary circulation. Congenit. Anomalies 54, 162–171. doi:10.1111/cga.12048
Takeichi, M., Nimura, K., Mori, M., Nakagami, H., and Kaneda, Y. (2013). The transcription factors Tbx18 and Wt1 control the epicardial epithelial-mesenchymal transition through bi-directional regulation of slug in murine primary epicardial cells. PloS one 8, e57829. doi:10.1371/journal.pone.0057829
Tallquist, M. D. (2020). Developmental pathways of cardiac fibroblasts. Cold Spring Harb. Perspect. Biol. 12, a037184. doi:10.1101/cshperspect.a037184
Tallquist, M. D., and Soriano, P. (2003). Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development 130, 507–518. doi:10.1242/dev.00241
Tandon, P., Miteva, Y. V., Kuchenbrod, L. M., Cristea, I. M., and Conlon, F. L. (2013). Tcf21 regulates the specification and maturation of proepicardial cells. Development 140, 2409–2421. doi:10.1242/dev.093385
Tao, J., Doughman, Y., Yang, K., Ramirez-Bergeron, D., and Watanabe, M. (2013). Epicardial HIF signaling regulates vascular precursor cell invasion into the myocardium. Dev. Biol. 376, 136–149. doi:10.1016/j.ydbio.2013.01.026
Tao, J., Barnett, J. V., Watanabe, M., and Ramírez-Bergeron, D. (2018). Hypoxia supports epicardial cell differentiation in vascular smooth muscle cells through the activation of the TGFβ pathway. J. Cardiovasc. Dev. Dis. 5, 19. doi:10.3390/jcdd5020019
Torlopp, A., Schlueter, J., and Brand, T. (2010). Role of fibroblast growth factor signaling during proepicardium formation in the chick embryo. Dev. Dyn. 239, 2393–2403. doi:10.1002/dvdy.22384
Trembley, M. A., Velasquez, L. S., de Mesy Bentley, K. L., and Small, E. M. (2015). Myocardin-related transcription factors control the motility of epicardium-derived cells and the maturation of coronary vessels. Development 142, 21–30. doi:10.1242/dev.116418
Tyser, R. C., Ibarra-Soria, X., McDole, K., Arcot Jayaram, S., Godwin, J., van den Brand, T. A., et al. (2021). Characterization of a common progenitor pool of the epicardium and myocardium. Science 371, eabb2986. doi:10.1126/science.abb2986
van Wijk, B., van den Berg, G., Abu-Issa, R., Barnett, P., van der Velden, S., Schmidt, M., et al. (2009). Epicardium and myocardium separate from a common precursor pool by crosstalk between bone morphogenetic protein–and fibroblast growth factor–signaling pathways. Circulation Res. 105, 431–441. doi:10.1161/CIRCRESAHA.109.203083
Vega-Hernández, M., Kovacs, A., De Langhe, S., and Ornitz, D. M. (2011). FGF10/FGFR2b signaling is essential for cardiac fibroblast development and growth of the myocardium. Development 138, 3331–3340. doi:10.1242/dev.064410
Velecela, V., Lettice, L. A., Chau, Y.-Y., Slight, J., Berry, R. L., Thornburn, A., et al. (2013). WT1 regulates the expression of inhibitory chemokines during heart development. Hum. Mol. Genet. 22, 5083–5095. doi:10.1093/hmg/ddt358
Vicente-Steijn, R., Scherptong, R. W., Kruithof, B. P., Duim, S. N., Goumans, M. J. T., Wisse, L. J., et al. (2015). Regional differences in WT-1 and Tcf21 expression during ventricular development: implications for myocardial compaction. PLoS One 10, e0136025. doi:10.1371/journal.pone.0136025
Villa del Campo, C., Lioux, G., Carmona, R., Sierra, R., Muñoz-Chápuli, R., Clavería, C., et al. (2016). Myc overexpression enhances of epicardial contribution to the developing heart and promotes extensive expansion of the cardiomyocyte population. Sci. Rep. 6, 35366. doi:10.1038/srep35366
von Gise, A., and Pu, W. T. (2012). Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circulation Res. 110, 1628–1645. doi:10.1161/CIRCRESAHA.111.259960
von Gise, A., Zhou, B., Honor, L. B., Ma, Q., Petryk, A., and Pu, W. T. (2011). WT1 regulates epicardial epithelial to mesenchymal transition through β-catenin and retinoic acid signaling pathways. Dev. Biol. 356, 421–431. doi:10.1016/j.ydbio.2011.05.668
Vrancken Peeters, M.-P. F., Gittenberger-de Groot, A. C., Mentink, M. M., and Poelmann, R. E. (1999). Smooth muscle cells and fibroblasts of the coronary arteries derive from epithelial-mesenchymal transformation of the epicardium. Anat. Embryology 199, 367–378. doi:10.1007/s004290050235
Wang, K., Shen, H., Gan, P., Cavallero, S., Kumar, S. R., Lien, C.-L., et al. (2019). Differential roles of insulin like growth factor 1 receptor and insulin receptor during embryonic heart development. BMC Dev. Biol. 19, 5–8. doi:10.1186/s12861-019-0186-8
Wang, R., Lu, D., Song, R., Du, L., Yang, X., Wu, S.-t., et al. (2024). Epicardial CCM2 promotes cardiac development and repair via its regulation on cytoskeletal reorganization. Basic Transl. Sci. 9, 203–219. doi:10.1016/j.jacbts.2023.09.004
Weeke-Klimp, A., Bax, N. A., Bellu, A. R., Winter, E. M., Vrolijk, J., Plantinga, J., et al. (2010). Epicardium-derived cells enhance proliferation, cellular maturation and alignment of cardiomyocytes. J. Mol. Cell. Cardiol. 49, 606–616. doi:10.1016/j.yjmcc.2010.07.007
Weinberger, M., Simões, F. C., Patient, R., Sauka-Spengler, T., and Riley, P. R. (2020). Functional heterogeneity within the developing zebrafish epicardium. Dev. cell 52, 574–590. e6. doi:10.1016/j.devcel.2020.01.023
Wessels, A., and Pérez-Pomares, J. (2004). The epicardium and epicardially derived cells (EPDCs) as cardiac stem cells. Anatomical Rec. Part A Discov. Mol. Cell. Evol. Biol. 276, 43–57. doi:10.1002/ar.a.10129
Wessels, A., van den Hoff, M. J., Adamo, R. F., Phelps, A. L., Lockhart, M. M., Sauls, K., et al. (2012). Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 366, 111–124. doi:10.1016/j.ydbio.2012.04.020
Wu, H., Lee, S. H., Gao, J., Liu, X., and Iruela-Arispe, M. L. (1999). Inactivation of erythropoietin leads to defects in cardiac morphogenesis. Development 126, 3597–3605. doi:10.1242/dev.126.16.3597
Wu, M., Smith, C. L., Hall, J. A., Lee, I., Luby-Phelps, K., and Tallquist, M. D. (2010). Epicardial spindle orientation controls cell entry into the myocardium. Dev. Cell 19, 114–125. doi:10.1016/j.devcel.2010.06.011
Wu, S.-P., Dong, X.-R., Regan, J. N., Su, C., and Majesky, M. W. (2013). Tbx18 regulates development of the epicardium and coronary vessels. Dev. Biol. 383, 307–320. doi:10.1016/j.ydbio.2013.08.019
Xiao, Y., Hill, M. C., Zhang, M., Martin, T. J., Morikawa, Y., Wang, S., et al. (2018). Hippo signaling plays an essential role in cell state transitions during cardiac fibroblast development. Dev. cell 45, 153–169. doi:10.1016/j.devcel.2018.03.019
Yamaguchi, Y., Cavallero, S., Patterson, M., Shen, H., Xu, J., Kumar, S. R., et al. (2015). Adipogenesis and epicardial adipose tissue: a novel fate of the epicardium induced by mesenchymal transformation and PPARγ activation. Proc. Natl. Acad. Sci. 112, 2070–2075. doi:10.1073/pnas.1417232112
Yamamoto, H., Flannery, M. L., Kupriyanov, S., Pearce, J., McKercher, S. R., Henkel, G. W., et al. (1998). Defective trophoblast function in mice with a targeted mutation of Ets2. Genes Dev. 12, 1315–1326. doi:10.1101/gad.12.9.1315
Yan, Y., Qin, Q., Wu, L., Jing, X., Deng, S., and She, Q. (2018). Insulin-like growth factor 1 receptor signaling regulates embryonic epicardial cell proliferation through focal adhesion kinase pathway. Acta Biochimica Biophysica Sinica 50, 976–983. doi:10.1093/abbs/gmy103
Yang, J. T., Rayburn, H., and Hynes, R. O. (1995). Cell adhesion events mediated by α4 integrins are essential in placental and cardiac development. Development 121, 549–560. doi:10.1242/dev.121.2.549
Zamora, M., Männer, J., and Ruiz-Lozano, P. (2007). Epicardium-derived progenitor cells require β-catenin for coronary artery formation. Proc. Natl. Acad. Sci. 104, 18109–18114. doi:10.1073/pnas.0702415104
Zhou, B., and Pu, W. T. (2012). Genetic Cre-loxP assessment of epicardial cell fate using Wt1-driven Cre alleles. Circulation Res. 111, e276–e280. doi:10.1161/CIRCRESAHA.112.275784
Zhou, B., Ma, Q., Rajagopal, S., Wu, S. M., Domian, I., Rivera-Feliciano, J., et al. (2008). Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 454, 109–113. doi:10.1038/nature07060
Keywords: epicardium, myocardium, crosstalk, morphogenesis, development
Citation: Nusrat A and Wu M (2025) Epicardium-myocardium crosstalk orchestrates heart development. Front. Cell Dev. Biol. 13:1655878. doi: 10.3389/fcell.2025.1655878
Received: 28 June 2025; Accepted: 10 September 2025;
Published: 24 September 2025.
Edited by:
Elizabeth Vafiadaki, Biomedical Research Foundation of the Academy of Athens (BRFAA), GreeceReviewed by:
Amelia Eva Aranega, University of Jaén, SpainGuang Li, University of Pittsburgh, United States
Copyright © 2025 Nusrat and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingfu Wu, bXd1MjVAY2VudHJhbC51aC5lZHU=