 Mariana Cooke
Mariana Cooke Nahuel Peinetti
Nahuel Peinetti Kerry L. Burnstein
Kerry L. Burnstein- Department of Molecular and Cellular Pharmacology, University of Miami Miller School of Medicine and Sylvester Comprehensive Cancer Center, Miami, FL, United States
Introduction
In a recent study, Baker et al. (2025) identified the Rac guanine nucleotide exchange factor VAV2 as a marker of poor prognosis and a signaling link that contributes to the proliferation and aggressiveness of castration-resistant prostate cancer (CRPC) cells. Prostate cancer (PCa) is the most common noncutaneous malignancy among men worldwide, with 1 in 8 men diagnosed with this disease during their lifetime. While patients with organ-confined, locally advanced, or regionally spread disease display a 5-year survival rate greater than 99%, the survival rate for advanced-stage disease with distant metastatic spread declines to 30%–40% (American Cancer Society, 2025). Androgen deprivation therapy (ADT) remains the cornerstone of treatment for patients with high-risk localized and advanced PCa. Despite initial biochemical or radiological remission after ADT, most patients eventually progress to metastatic castration-resistant prostate cancer (mCRPC), a highly heterogeneous, aggressive, and lethal disease. Deregulation of oncogenic and invasive signaling pathways represents a major hallmark of CRPC cells, enabling their escape from the primary tumor to secondary sites, particularly the axial skeleton (Rebello et al., 2021).
Rac1, a member of the Rho GTPase family, represents a crucially deregulated signaling player leading to tumor progression, particularly in the metastatic spread of cancer cells. Rac1 has been recognized as a major regulator of actin cytoskeleton reorganization, which promotes the formation of cell surface projections (e.g., lamellipodia, membrane ruffles) necessary for cell migration and invasion during metastasis. Additionally, Rac1 regulates a diverse range of cellular functions in cancer cells, including proliferation, gene expression, metabolism, and epithelial-to-mesenchymal transition (EMT), making it an attractive target for cancer therapy (Bustelo, 2018; Kazanietz and Caloca, 2017; De et al., 2020; Casado-Medrano et al., 2019). Like most members of the Rho GTPase family, Rac1 functions as a binary switch, being active in its GTP-bound form and inactive in its GDP-bound form. This “on-off” cycling is tightly regulated by Rac Guanine nucleotide Exchange Factors (Rac-GEFs), which facilitate GTP loading and thus activate Rac1. Inactivation of Rac1 is mediated by GTPase-activating proteins (Rac-GAPs) that accelerate GTP hydrolysis. Active (GTP-bound) Rac1 relays through various effectors, triggering a complex network of signaling events that influence both actin dynamics and diverse cellular processes independent of actin cytoskeleton remodeling. Extracellular cues, such as those involving ligand-mediated stimulation of receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs), represent the most common upstream inputs that confer Rac1 activation (Bustelo, 2018; Kazanietz and Caloca, 2017; Kazanietz et al., 2022). The large size of the Rac-GEF family, which comprises 32 Dbl-like and 11 DOCK Rac-GEFs, along with their distinctive expression based on cell type (Casado-Medrano et al., 2018), suggests multifaceted coupling mechanisms that depend on the nature of the receptor and Rac-GEF, resulting in the activation of discrete intracellular Rac1 pools and exquisite selectivity for downstream responses. Mechanistically, the diversity of Rac-GEF/Rac1 signaling likely relies on strict spatiotemporal regulation of Rac-GEFs by specific receptors and their coupling to effectors (e.g., PI3K), ultimately influencing downstream responses through a complex modulation of the Rac1 interactome (Kazanietz et al., 2022; Banka et al., 2022).
Rac-GEF signaling in prostate cancer: identification of VAV2 as an RTK effector
Rac1 is often deregulated in pathological conditions, including neurological diseases and cancer (Bustelo, 2018; Kazanietz and Caloca, 2017; De et al., 2020; Casado-Medrano et al., 2019; Casado-Medrano et al., 2018; Banka et al., 2022). While cutaneous melanoma can harbor activated Rac1 mutants, this is rare (Krauthammer et al., 2012). Instead, Rac1 deregulation is due to abnormally elevated Rac-GEF expression or hyperactivation of receptors that promote Rac-GEF activation (Bustelo, 2018; Kazanietz and Caloca, 2017; Casado-Medrano et al., 2018). In PCa, constitutively elevated Rac1 activity has been observed in several cellular models of androgen receptor (AR) negative PCa, including DU145, PC3, and PC3-ML cell lines, compared to normal prostate epithelial cells or androgen-dependent PCa cells (Baker et al., 2020). In their recent study, Baker et al. (2025) demonstrated that Rac1 deficiency leads to significant defects in the migratory and proliferative capacities of CRPC cellular models. The migratory defect aligns with the expected role of Rac1 in actin cytoskeleton-dependent motility and invasion signaling. Furthermore, it correlates strongly with bioinformatics analysis in the TCGA-PRAD human prostate carcinoma database, which reveals worse progression-free survival in PCa patients with signatures predicting “high Rac1 cell motility activity.” Rac1 deficiency also leads to significant changes in gene expression, particularly affecting transcriptional networks related to cell adhesion, ECM functions, migration, proliferation, and inflammation. Despite the negative regulation of E-cadherin expression by Rac1, the loss of Rac1 was insufficient to reverse the mesenchymal phenotype typical of AR-null PCa cells.
Identifying the GEF(s) responsible for Rac activation in any given model is daunting due to limited knowledge about the spatial and temporal expression of individual members of the large Rac-GEF family and their activation statuses. Overcoming this challenge is critical to assigning specific functional roles to individual Rac-GEFs in processes associated with oncogenesis and metastasis. Using a pre-designed Q-PCR array, Baker et al. (2025) defined the Rac-GEF mRNA abundance in both castration-resistant and androgen-dependent PCa cell lines. This analysis revealed a relatively common expression pattern among the two groups and a shared subset of Rac-GEFs compared to cell lines derived from other cancer types, namely, adrenocortical and lung cancer (Cooke et al., 2023; Cooke et al., 2021). ECT2, TRIO, FARP1, PLEKHG2, VAV2, PREX1, and FARP2 were identified as the top-expressed Dbl-like Rac-GEFs in PCa cells, while DOCK1, DOCK5, DOCK7, and DOCK9 were the top-expressed DOCK family Rac-GEFs. Through the use of the PARADIGM algorithm, statistically significant positive correlations were identified between the expression of discrete Rac-GEFs and the “Rac1 cell motility pathway,” with the highest correlation found for the Rac-GEF VAV2 (p = 6.7 × 10−10). Functional studies using VAV2-deficient DU145 PCa cells established this Rac-GEF as a key cell migration and proliferation driver. Interestingly, RNAi screening revealed VAV2 to be the only Rac-GEF capable of driving Rac1 activation in response to ligand-mediated stimulation of EGFR (Baker et al., 2025), an RTK with established roles in PCa progression, including metastatic dissemination (Day et al., 2017). VAV2 was also found to mediate the invasiveness of PCa cells (Cooke et al., manuscript in preparation).
Aberrantly elevated VAV expression in human prostate cancer
The mammalian VAV family of Rac-GEFs comprises three members: VAV1, VAV2, and VAV3 (Bustelo, 2014). According to mRNA expression, VAV2 is the most highly expressed VAV isoform in PCa cells, followed by VAV3 (Figure 1A). In contrast, VAV1, which is primarily expressed in hematopoietic cells, is essentially undetectable in PCa cell lines (Baker et al., 2025). Baker et al. conducted an immunohistochemical analysis using a large number of human PCa specimens, establishing prominent upregulation of VAV2 in tumoral areas compared to non-tumoral areas. No significant VAV2 staining could be observed in the prostate stroma, ruling out the possibility of microenvironmental effects of VAV2 in PCa progression. These results were strongly supported by bioinformatic analysis of databases, including TCGA-PRAD, which shows VAV2 as the top upregulated Rac-GEF in PCa compared to normal tissue. Database analysis also revealed the progressive upregulation of VAV2 with increasing Gleason score, as well as in metastasis (Baker et al., 2025), in agreement with Magani et al. (2017). Kaplan-Meier analysis revealed VAV2 to be a negative predictor for disease-specific survival (DSS), disease-free interval (DFI), and progression-free interval (PFI), underscoring the potential prognostic value of this Rac-GEF in human PCa (Baker et al., 2025). Despite VAV2 being the most highly expressed VAV isoform in PCa, studies have also revealed that VAV3 levels are upregulated during the in vivo progression of PCa cell lines to castration resistance (Lyons and Burnstein, 2006; Lin et al., 2012; Lyons et al., 2008). VAV3 expression is elevated in late-stage and metastatic PCa, and its expression in early-stage tumors is associated with a lower overall biochemical failure-free survival rate (Lin et al., 2012). Notably, its expression as a transgene in mouse prostates leads to the development of prostatic intraepithelial neoplasia (PIN) and PCa (Liu et al., 2008). Similar to VAV2, VAV3 has been established as an EGFR effector and can mediate Rac1 activation in response to EphA2 RTK stimulation (Lin et al., 2012). Therefore, it is plausible that both VAV isoforms may participate in PCa progression. Since VAV2 and VAV3 are structurally related, possible functional redundancy may occur in prostate cancer, although co-expression of these VAV isoforms in human prostate tumors has not been thoroughly investigated. Nonetheless, unique non-redundant roles for VAV isoforms have also been described (Pearce et al., 2004; Conde et al., 2021; Fujikawa et al., 2003). The reported upregulation in VAV3 expression and activation observed in VAV1/VAV2-deficient models suggests the existence of compensatory mechanisms controlling VAV isoform expression and is indicative of their complex functional interdependence (Chang et al., 2012). The availability of genetically engineered VAV2/VAV3 mouse models (Pearce et al., 2004; Conde et al., 2021; Fujikawa et al., 2003; Chang et al., 2012; Sauzeau et al., 2007; Quevedo et al., 2010; Menacho-Márquez et al., 2013) would be instrumental in establishing unique and/or distinctive roles in prostate cancer progression in vivo.
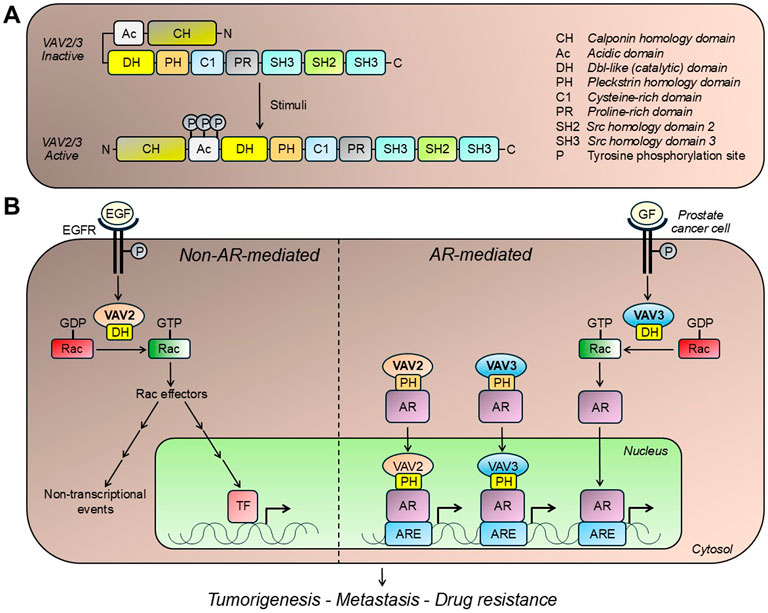
Figure 1. VAV isoforms and prostate cancer. (A) Structure of VAV isoforms expressed in prostate cancer. VAV2 and VAV3 activation occurs upon stimulation of receptor tyrosine kinases, such as EGFR. This process involves a conformational rearrangement that exposes the DH catalytic domain, as well as domains implicated in lipid interactions (e.g., PH and C1 domains) and protein interactions (e.g., SH2 and SH3 domains). (B) VAV isoforms mediate effects in prostate cancer cells both in androgen receptor (AR)-independent and -dependent manners. VAV2 and VAV3 promote GDP/GTP exchange on Rac1, the main Rac small GTPase expressed in prostate cancer cells. This small G-protein has been widely implicated in proliferative and migratory signaling, therefore contributing to prostate tumorigenesis and metastasis. VAV isoforms also enhance ligand-independent AR nuclear translocation and transcriptional activity, contributing to the proliferative and tumorigenic capacities of prostate cancer cells. See text for details. ARE, androgen receptor response element; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; GF, growth factor; P, phosphorylation; TF, transcription factor.
VAV isoforms: roles in androgen receptor function and drug resistance
Research from our group and others demonstrated a complex crosstalk between VAV family members and AR in PCa (Magani et al., 2017; Lyons and Burnstein, 2006; Dong et al., 2006; Peacock et al., 2012; Rao et al., 2012) (Figure 1B). Through its GEF activity, VAV3 can trigger Rac1 signaling, enhancing ligand-independent AR nuclear translocation and transcriptional activity, which in turn increases PCa cell proliferation (Lyons et al., 2008). Alternatively, both VAV2 and VAV3 can act independently of their GEF activity and serve as co-activators of full-length AR and constitutively active AR splice variants (e.g., AR-V7) (Wu et al., 2013; Wang et al., 2025). This AR coactivation is mediated by direct binding to AR through the DH domain as well as by binding to AR co-chaperones such as Cdc31, and possibly SRC-1 and SRC-2 (Magani et al., 2017; Wu et al., 2013). The PH domain has been shown to promote AR N/C interactions, leading to nuclear translocation and the formation of a transcriptional complex that regulates AR target gene expression (Magani et al., 2017; Rao et al., 2012). This role as a coactivator has been linked to CRPC progression (Peacock et al., 2012; Rao et al., 2012). Despite the well-defined differences in the mechanisms of action by which VAVs activate AR in PCa, it remains unclear whether one can prevail over the others during disease progression. While publicly available patient data sets show a correlation of VAV2 and VAV3 with AR-V7 in bone metastatic CRPC (Magani et al., 2017), the interplay between these two VAV isoforms and AR activity is another crucial aspect that requires further elucidation.
With the rise of advanced targeted and hormone-based therapies, overcoming therapeutic resistance has become a primary challenge in PCa management. Increased expression of AR coactivators has been identified as a mechanism by which PCa escapes AR-targeted therapies. Recently, binding of VAV2 to AR and AR splice variants has been shown to stabilize these receptors and mediate enzalutamide resistance (Wang et al., 2025). Additionally, genetically engineered cells with reduced expression levels of VAV3 exhibit an improved response to docetaxel in preclinical models of PCa (Nomura et al., 2013). Disrupting the interaction between the DH domain of VAV3 and the TAU5 region of AR using protein fragments decreased AR-V7 nuclear localization and, as a result, reduced cell proliferation and migration while increasing apoptosis, thus demonstrating the clinical relevance of targeting VAVs in PCa (Magani et al., 2017). Direct targeting of VAVs poses a challenge - as is also the case for most Rho-family GEFs - since these molecules are subject to intricate regulatory mechanisms (i.e., phosphorylation, protein-protein interactions) and lack druggable pockets for selective pharmacological targeting (Neurath and Berg, 2024; Smithers and Overduin, 2016). Drug discovery efforts exploiting unique interfaces involved in GEF/GTPase interactions have led to the development of promising antitumor and antimetastatic small-molecule inhibitors (Bustelo, 2018; Kazanietz and Caloca, 2017). A notable example that highlights the strong feasibility for the design of inhibitors of VAV-Rac/Cdc42 interactions is the development of Ehop-016 and Ehop-097 (Montalvo-Ortiz et al., 2012; Medina et al., 2022). Ehop-016 shows excellent pharmacological activity in mouse models of experimental metastasis with no significant toxicity (Castillo-Pichardo et al., 2014; Humphries-Bickley et al., 2015). Proof-of-principle for the potent anti-migratory activity of Ehop-097 in CRPC cells has been established in Baker et al. (2025). Recently, Nassar and coworkers identified IODVA1 as a first-in-class small-molecule VAV3 inhibitor, likely acting by locking this GEF into an autoinhibitory state that prevents Rac access to the DH catalytic domain (Hegde et al., 2022). With the development of new small-molecule inhibitors for VAVs, preclinical testing efforts will be crucial in determining the translational potential of VAV inhibitors in PCa.
Concluding remarks
Vav family members represent key therapy-resistant nodes in advanced PCa (AR and non-AR expressing) and serve as potential biomarkers of poor clinical outcomes. As GEFs for Rac1, these proteins relay diverse oncogenic signals and control crucial steps in the metastatic dissemination process. VAV2 and VAV3 coactivate AR, a primary driver of PCa, promoting proliferation and therapy resistance. The enhancement of AR activity by VAVs can occur in a GEF-independent manner, posing a unique therapeutic challenge. Developing specific VAV inhibitors that can be utilized in distinct clinical settings alongside patient-risk stratification would be instrumental in advancing PCa therapeutic strategies. Although PCa treatment has made significant strides, new resistance mechanisms have emerged, including tumors with neuroendocrine features as well as “double negative” tumors that lack both AR expression and neuroendocrine markers. Given the fact that VAVs can promote both AR-dependent and AR-independent growth, targeting VAV/Rac signaling pathways offers a novel and promising approach for enhancing PCa management.
Author contributions
MC: Writing – review and editing, Writing – original draft. NP: Writing – original draft, Writing – review and editing. MGK: Writing – original draft, Writing – review and editing. KLB: Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
American Cancer Society (2025). Survival rates for prostate cancer. Available online at: https://www.cancer.org/cancer/types/prostate-cancer/detection-diagnosis-staging/survival-rates.html.
Baker, M. J., Abba, M. C., Garcia-Mata, R., and Kazanietz, M. G. (2020). P-REX1-Independent, calcium-dependent RAC1 hyperactivation in prostate cancer. Cancers 12 (2), 480. doi:10.3390/cancers12020480
Baker, M. J., Zhang, S., Zhang, D., Searle, J., Lal, P., Vlaar, C. P., et al. (2025). “VAV2 drives EGFR-Mediated Rac1 responses in prostate cancer,” in Molecular cancer research: MCR. New York, NY: Advance Online Publication. doi:10.1158/1541-7786.MCR-24-0957
Banka, S., Bennington, A., Baker, M. J., Rijckmans, E., Clemente, G. D., Ansor, N. M., et al. (2022). Activating RAC1 variants in the switch II region cause a developmental syndrome and alter neuronal morphology. Brain a J. neurology 145 (12), 4232–4245. doi:10.1093/brain/awac049
Bustelo, X. R. (2014). Vav family exchange factors: an integrated regulatory and functional view. Small GTPases 5 (2), 9. doi:10.4161/21541248.2014.973757
Bustelo, X. R. (2018). RHO GTPases in cancer: known facts, open questions, and therapeutic challenges. Biochem. Soc. Trans. 46 (3), 741–760. doi:10.1042/BST20170531
Casado-Medrano, V., Baker, M. J., Lopez-Haber, C., Cooke, M., Wang, S., Caloca, M. J., et al. (2018). The role of Rac in tumor susceptibility and disease progression: from biochemistry to the clinic. Biochem. Soc. Trans. 46 (4), 1003–1012. doi:10.1042/BST20170519
Casado-Medrano, V., Barrio-Real, L., Wang, A., Cooke, M., Lopez-Haber, C., and Kazanietz, M. G. (2019). Distinctive requirement of PKCε in the control of Rho GTPases in epithelial and mesenchymally transformed lung cancer cells. Oncogene 38 (27), 5396–5412. doi:10.1038/s41388-019-0796-4
Castillo-Pichardo, L., Humphries-Bickley, T., De La Parra, C., Forestier-Roman, I., Martinez-Ferrer, M., Hernandez, E., et al. (2014). The rac inhibitor EHop-016 Inhibits mammary tumor growth and metastasis in a nude mouse model. Transl. Oncol. 7 (5), 546–555. doi:10.1016/j.tranon.2014.07.004
Chang, K. H., Sanchez-Aguilera, A., Shen, S., Sengupta, A., Madhu, M. N., Ficker, A. M., et al. (2012). Vav3 collaborates with p190-BCR-ABL in lymphoid progenitor leukemogenesis, proliferation, and survival. Blood 120 (4), 800–811. doi:10.1182/blood-2011-06-361709
Conde, J., Fernández-Pisonero, I., Cuadrado, M., Abad, A., Robles-Valero, J., and Bustelo, X. R. (2021). Distinct roles of vav family members in adaptive and innate immune models of arthritis. Biomedicines 9 (6), 695. doi:10.3390/biomedicines9060695
Cooke, M., Kreider-Letterman, G., Baker, M. J., Zhang, S., Sullivan, N. T., Eruslanov, E., et al. (2021). FARP1, ARHGEF39, and TIAM2 are essential receptor tyrosine kinase effectors for Rac1-dependent cell motility in human lung adenocarcinoma. Cell Rep. 37 (5), 109905. doi:10.1016/j.celrep.2021.109905
Cooke, M., Zhang, S., Cornejo Maciel, F., and Kazanietz, M. G. (2023). Gi/o GPCRs drive the formation of actin-rich tunneling nanotubes in cancer cells via a Gβγ/PKCα/FARP1/Cdc42 axis. J. Biol. Chem. 299 (8), 104983. doi:10.1016/j.jbc.2023.104983
Day, K. C., Lorenzatti Hiles, G., Kozminsky, M., Dawsey, S. J., Paul, A., Broses, L. J., et al. (2017). HER2 and EGFR overexpression support metastatic progression of prostate cancer to bone. Cancer Res. 77 (1), 74–85. doi:10.1158/0008-5472.CAN-16-1656
De, P., Rozeboom, B. J., Aske, J. C., and Dey, N. (2020). Active RAC1 promotes tumorigenic phenotypes and therapy resistance in solid tumors. Cancers 12 (6), 1541. doi:10.3390/cancers12061541
Dong, Z., Liu, Y., Lu, S., Wang, A., Lee, K., Wang, L. H., et al. (2006). Vav3 oncogene is overexpressed and regulates cell growth and androgen receptor activity in human prostate cancer. Mol. Endocrinol. Baltim. Md. 20 (10), 2315–2325. doi:10.1210/me.2006-0048
Fujikawa, K., Miletic, A. V., Alt, F. W., Faccio, R., Brown, T., Hoog, J., et al. (2003). Vav1/2/3-null mice define an essential role for Vav family proteins in lymphocyte development and activation, but a differential requirement in MAPK signaling in T and B cells. J. Exp. Med. 198 (10), 1595–1608. doi:10.1084/jem.20030874
Hegde, S., Gasilina, A., Wunderlich, M., Lin, Y., Buchholzer, M., Krumbach, O. H. F., et al. (2022). Inhibition of the RacGEF VAV3 by the small molecule IODVA1 impedes RAC signaling and overcomes resistance to tyrosine kinase inhibition in acute lymphoblastic leukemia. Leukemia 36 (3), 637–647. doi:10.1038/s41375-021-01455-3
Humphries-Bickley, T., Castillo-Pichardo, L., Corujo-Carro, F., Duconge, J., Hernandez-O'Farrill, E., Vlaar, C., et al. (2015). Pharmacokinetics of Rac inhibitor EHop-016 in mice by ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B, Anal. Technol. Biomed. life Sci. 981-982, 19–26. doi:10.1016/j.jchromb.2014.12.021
Kazanietz, M. G., and Caloca, M. J. (2017). The rac GTPase in cancer: from old concepts to new paradigms. Cancer Res. 77 (20), 5445–5451. doi:10.1158/0008-5472.CAN-17-1456
Kazanietz, M. G., Cooke, M., and Garcia-Mata, R. (2022). Nonredundant Rac-GEF control of actin cytoskeleton reorganization. Trends Cell Biol. 32 (10), 815–818. doi:10.1016/j.tcb.2022.06.003
Krauthammer, M., Kong, Y., Ha, B. H., Evans, P., Bacchiocchi, A., McCusker, J. P., et al. (2012). Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 44 (9), 1006–1014. doi:10.1038/ng.2359
Lin, K. T., Gong, J., Li, C. F., Jang, T. H., Chen, W. L., Chen, H. J., et al. (2012). Vav3-rac1 signaling regulates prostate cancer metastasis with elevated Vav3 expression correlating with prostate cancer progression and posttreatment recurrence. Cancer Res. 72 (12), 3000–3009. doi:10.1158/0008-5472.CAN-11-2502
Liu, Y., Mo, J. Q., Hu, Q., Boivin, G., Levin, L., Lu, S., et al. (2008). Targeted overexpression of vav3 oncogene in prostatic epithelium induces nonbacterial prostatitis and prostate cancer. Cancer Res. 68 (15), 6396–6406. doi:10.1158/0008-5472.CAN-08-0645
Lyons, L. S., and Burnstein, K. L. (2006). Vav3, a Rho GTPase guanine nucleotide exchange factor, increases during progression to androgen independence in prostate cancer cells and potentiates androgen receptor transcriptional activity. Mol. Endocrinol. Baltim. Md. 20 (5), 1061–1072. doi:10.1210/me.2005-0346
Lyons, L. S., Rao, S., Balkan, W., Faysal, J., Maiorino, C. A., and Burnstein, K. L. (2008). Ligand-independent activation of androgen receptors by Rho GTPase signaling in prostate cancer. Mol. Endocrinol. Baltim. Md. 22 (3), 597–608. doi:10.1210/me.2007-0158
Magani, F., Peacock, S. O., Rice, M. A., Martinez, M. J., Greene, A. M., Magani, P. S., et al. (2017). Targeting AR variant-coactivator interactions to exploit prostate cancer vulnerabilities. Mol. cancer Res. MCR 15 (11), 1469–1480. doi:10.1158/1541-7786.MCR-17-0280
Medina, J. I., Cruz-Collazo, A., Del Mar Maldonado, M., Gascot, T. M., Borrero-Garcia, L. D., Cooke, M., et al. (2022). Characterization of novel derivatives of MBQ-167, an inhibitor of the GTP-binding proteins Rac/Cdc42. Cancer Res. Commun. 2 (12), 1711–1726. doi:10.1158/2767-9764.crc-22-0303
Menacho-Márquez, M., García-Escudero, R., Ojeda, V., Abad, A., Delgado, P., Costa, C., et al. (2013). The Rho exchange factors Vav2 and Vav3 favor skin tumor initiation and promotion by engaging extracellular signaling loops. PLoS Biol. 11 (7), e1001615. doi:10.1371/journal.pbio.1001615
Montalvo-Ortiz, B. L., Castillo-Pichardo, L., Hernández, E., Humphries-Bickley, T., De la Mota-Peynado, A., Cubano, L. A., et al. (2012). Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J. Biol. Chem. 287 (16), 13228–13238. doi:10.1074/jbc.M111.334524
Neurath, M. F., and Berg, L. J. (2024). VAV1 as a putative therapeutic target in autoimmune and chronic inflammatory diseases. Trends Immunol. 45 (8), 580–596. doi:10.1016/j.it.2024.06.004
Nomura, T., Yamasaki, M., Hirai, K., Inoue, T., Sato, R., Matsuura, K., et al. (2013). Targeting the Vav3 oncogene enhances docetaxel-induced apoptosis through the inhibition of androgen receptor phosphorylation in LNCaP prostate cancer cells under chronic hypoxia. Mol. cancer 12, 27. doi:10.1186/1476-4598-12-27
Peacock, S. O., Fahrenholtz, C. D., and Burnstein, K. L. (2012). Vav3 enhances androgen receptor splice variant activity and is critical for castration-resistant prostate cancer growth and survival. Mol. Endocrinol. Baltim. Md. 26 (12), 1967–1979. doi:10.1210/me.2012-1165
Pearce, A. C., Senis, Y. A., Billadeau, D. D., Turner, M., Watson, S. P., and Vigorito, E. (2004). Vav1 and vav3 have critical but redundant roles in mediating platelet activation by collagen. J. Biol. Chem. 279 (52), 53955–53962. doi:10.1074/jbc.M410355200
Quevedo, C., Sauzeau, V., Menacho-Márquez, M., Castro-Castro, A., and Bustelo, X. R. (2010). Vav3-deficient mice exhibit a transient delay in cerebellar development. Mol. Biol. Cell 21 (6), 1125–1139. doi:10.1091/mbc.e09-04-0292
Rao, S., Lyons, L. S., Fahrenholtz, C. D., Wu, F., Farooq, A., Balkan, W., et al. (2012). A novel nuclear role for the Vav3 nucleotide exchange factor in androgen receptor coactivation in prostate cancer. Oncogene 31 (6), 716–727. doi:10.1038/onc.2011.273
Rebello, R. J., Oing, C., Knudsen, K. E., Loeb, S., Johnson, D. C., Reiter, R. E., et al. (2021). Prostate cancer. Nat. Rev. Dis. Prim. 7 (1), 9. doi:10.1038/s41572-020-00243-0
Sauzeau, V., Jerkic, M., López-Novoa, J. M., and Bustelo, X. R. (2007). Loss of Vav2 proto-oncogene causes tachycardia and cardiovascular disease in mice. Mol. Biol. Cell 18 (3), 943–952. doi:10.1091/mbc.e06-09-0877
Smithers, C. C., and Overduin, M. (2016). Structural mechanisms and drug discovery prospects of rho GTPases. Cells 5 (2), 26. doi:10.3390/cells5020026
Wang, Q., Wang, X., Ye, H., Yao, Y., Li, H., Qin, X., et al. (2025). VAV2 exists in extrachromosomal circular DNA and contributes Enzalutamide resistance of prostate cancer via stabilization of AR/ARv7. Int. J. Biol. Sci. 21 (6), 2843–2863. doi:10.7150/ijbs.109271
Keywords: Vav2, Vav3, Rac1, EGFR, prostate cancer, migration, cancer signaling
Citation: Cooke M, Peinetti N, Kazanietz MG and Burnstein KL (2025) RAC1 signaling in prostate cancer: VAV GEFs take center stage. Front. Cell Dev. Biol. 13:1658639. doi: 10.3389/fcell.2025.1658639
Received: 08 July 2025; Accepted: 31 July 2025;
Published: 15 August 2025.
Edited by:
James T. Murray, Swansea University Medical School, United KingdomReviewed by:
Manisha Tripathi, Texas Tech University Health Sciences Center, United StatesCopyright © 2025 Cooke, Peinetti, Kazanietz and Burnstein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariana Cooke, bXhjMzI0NUBtZWQubWlhbWkuZWR1