 Ying Cheng
Ying Cheng Guangxin Pei
Guangxin Pei Lei Sun
Lei Sun Xiuyun Wu
Xiuyun Wu- School of Life Sciences, Zhengzhou University, Zhengzhou, China
Super enhancers (SEs) are clusters of enhancers with exceptionally high transcriptional activity, crucial for determining cell identity and regulating gene expression. They function as key regulatory hubs, governing gene networks essential for normal hematopoiesis while also driving the pathogenesis of hematological malignancies. This review summarizes the role of SEs in maintaining hematopoietic lineage identity and examines how their dysregulation in acute myeloid leukemia (AML), myelodysplastic neoplasms (MDS), adult T-cell leukemia (ATL), acute lymphoblastic leukemia (ALL), and multiple myeloma (MM) leads to oncogenic activation. By regulating key oncogenes, SEs represent promising therapeutic targets. Emerging strategies-such as BET inhibitors, CDK7/9 inhibitors, and rational drug combinations-effectively disrupt SE-driven transcriptional programs and show potential to overcome treatment resistance in these cancers.
1 Introduction
Eukaryotic transcription is a tightly regulated process, dependent on coordinated interactions between transcription factors, co-regulators, and cis-acting DNA elements, including enhancers, promoters, and silencers. Enhancers were first identified in 1981 when Banerji et al. discovered a class of short DNA sequences capable of activating transcription in a manner independent of their position, distance, or orientation relative to gene promoters (Banerji et al., 1981). Beyond enhancers’ classical role as transcription factor platforms, enhancers are actively transcribed into non-coding RNAs. These enhancer-derived RNAs (eRNAs) cooperate with architectural proteins to stabilize chromatin looping configurations, thereby potentiating enhancer-promoter communication for transcriptional activation (Orom et al., 2010; Wang et al., 2025; Akiki et al., 2024). Super enhancers (SEs) are large clusters of transcription enhancers that play a crucial role in regulating genes necessary for cellular identity and various biological processes (Hnisz et al., 2013). Distinguished from typical enhancers, SEs amplify transcriptional output, including abundant eRNAs, and enrich histone modifications (e.g., H3K27ac), thereby driving high-level gene expression in developmental and disease contexts (Khan et al., 2018; Blayney et al., 2023; Shin, 2018). Given the powerful regulatory role of SEs in gene expression, research on SEs and diseases in recent years has mainly focused on malignant tumors, indicating that they play important regulatory roles in important biological processes such as malignant tumor occurrence, cell differentiation, and immune response (Wang et al., 2023; van Groningen et al., 2017; Kai et al., 2021; Higashijima and Kanki, 2022).
Hematopoiesis is a precisely regulated developmental process whereby multipotent hematopoietic stem cells (HSCs) in the bone marrow microenvironment undergo progressive lineage commitment to generate all mature blood cell types, including erythrocytes, leukocytes, and platelets. Recent evidence increasingly highlights the important role of SEs in hematopoietic cell commitment and differentiation by facilitating dense transcription factor binding and sustaining high-level gene expression of key regulatory genes (Walker et al., 2023; Xie and Dean, 2025; Alvarez-Dominguez et al., 2017). Malignant hematopoiesis frequently arises from the aberrant activation of oncogenic pathways, wherein dysregulated expression or mutation of key oncogenes disrupts normal hematopoietic differentiation and promotes uncontrolled proliferation. Accumulating evidence demonstrates that SEs play pivotal roles in the initiation and progression of malignant hematopoiesis by dysregulating key oncogenic transcriptional programs (Jia et al., 2019; Fang et al., 2022; Wan et al., 2024). Given the critical role of SEs in defining cellular identity and driving oncogene expression in hematopoiesis, elucidating their molecular mechanisms may yield novel therapeutic approaches for hematological malignancies.
In this review, we systematically summarize recent advances in SEs research within the hematopoietic system, focusing on their role in activating gene expression during both normal and malignant hematopoiesis. Moreover, we critically evaluate emerging therapeutic strategies that target SEs regulatory networks in hematological malignancies, providing a conceptual framework for the development of novel targeted therapies against hematological disorders.
2 The characteristics and definition of super enhancers
In 2013, the concept of SEs was first proposed as a cluster of gene motifs containing multiple enhancers. Through ChIP-seq analysis of transcription factors and histone modification markers like H3K27ac and H3K4me1, SEs were characterized by their extensive genomic span and high enrichment of regulatory elements, significantly surpassing traditional enhancers in activity and complexity (Whyte et al., 2013; Lovén et al., 2013), as shown in Figure 1, compared to traditional enhancers, SE regions exhibit dense enrichment of histone modifications such as H3K27ac and H3K4me1, as well as strong accumulation of RNA polymerase II(RNAP II), thereby promoting efficient transcriptional activity and generating super enhancer-derived RNAs (seRNAs). Enhancer RNAs (eRNAs), a class of long non-coding RNAs (lncRNAs), are typically transcribed unidirectionally from enhancer regions, which can recruit transcription factors and promote the enrichment of H3K27ac at these sites, also as a well-established marker of active enhancers (Kim et al., 2010; Hsieh et al., 2014; Mousavi et al., 2013; Schaukowitch et al., 2014). Not surprisingly, SEs accumulate more RNAP II and are more highly transcribed, generating a large amount of long non-coding RNAs, known as seRNAs (Alvarez-Dominguez et al., 2017). Unlike classical eRNAs, seRNAs are transcribed bidirectionally from SEs regions and exist in various lengths with different functions. Among them, non-polyadenylated short seRNAs mainly exert cis-regulatory functions, while polyadenylated seRNAs in the nucleus, which are more stable, can participate in trans-regulatory roles (He et al., 2025; Font-Tello et al., 2020).
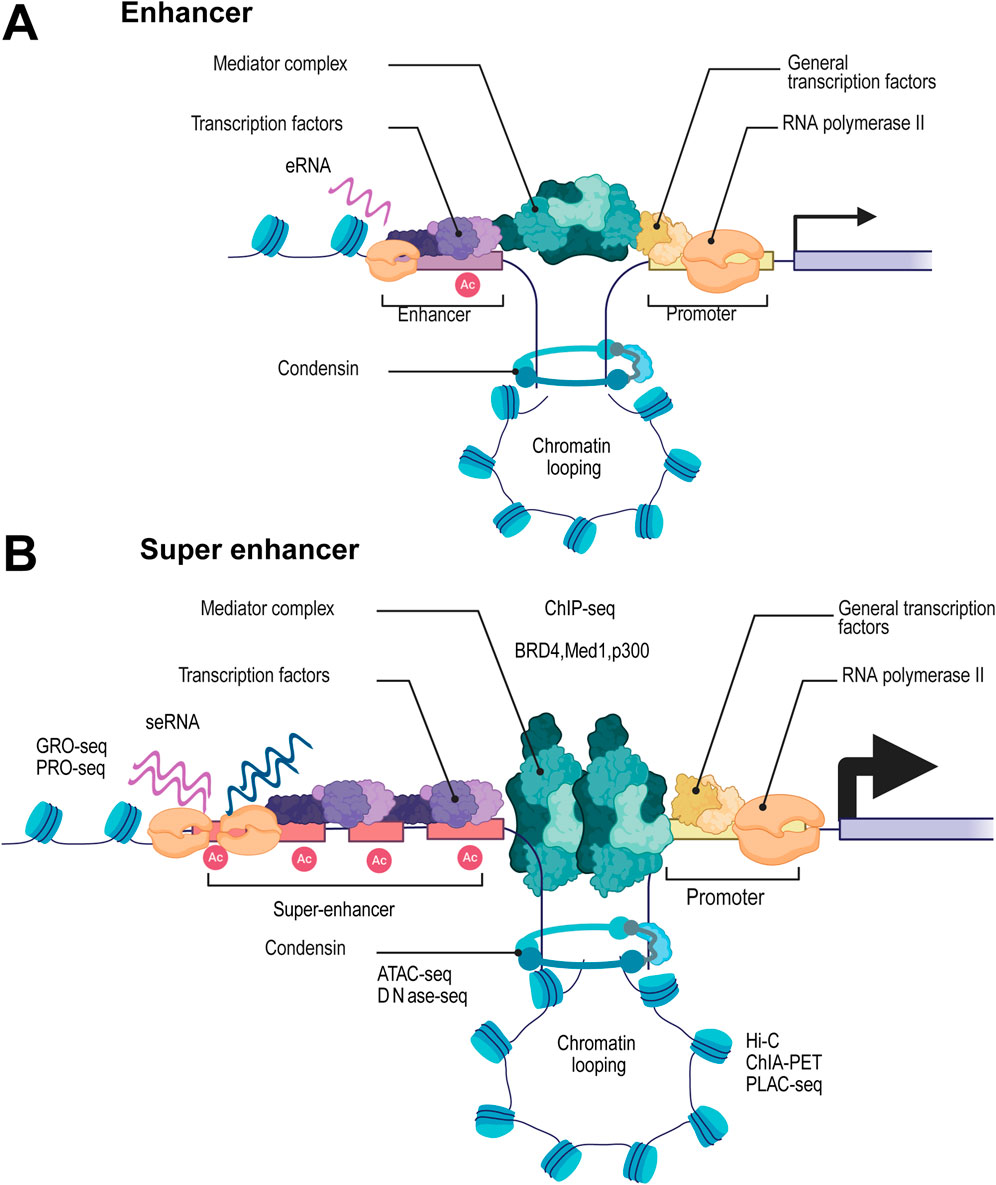
Figure 1. Structural and Functional Characteristics of Super Enhancers and Their Regulatory Role in Transcription Compared to Traditional Enhancers. Schematic diagram showing the structural and functional characteristics of traditional enhancers (A) compared with super enhancers (SEs) (B). SEs are characterized by their extensive genomic span and high enrichment of regulatory elements. Compared with traditional enhancers, SEs exhibit significantly higher activity and complexity, which is reflected in the dense accumulation of histone modifications such as H3K27ac (marked as “Ac”) and H3K4me1. Additionally, SEs show strong enrichment of RNA polymerase II, facilitating robust transcriptional activity and the generation of super enhancer-derived RNAs (seRNAs). These features collectively enable SEs to exert a more potent regulatory role in gene expression compared to traditional enhancers.
Given the unique features of SEs, high-throughput sequencing techniques have become essential tools for SE research and identification. Traditional approaches, particularly those analyzing histone modifications, predominantly use ChIP-seq technology. This method enables precise SE localization by detecting the enrichment of specific histone marks, such as H3K27ac and H3K4me1 (Font-Tello et al., 2020; Kang et al., 2021) Moreover, SE regions exhibit strong enrichment of transcription factors and coactivators, including MED1, BRD4, and p300. Combining ChIP-seq data from multiple factors allows for even more accurate SE prediction (Cai et al., 2024; Zheng et al., 2022; Liu et al., 2022). SEs are usually located in highly open chromatin regions. High-throughput sequencing techniques for checking chromatin accessibility, such as ATAC-seq and DNase-seq, can further verify the presence of SEs (Hnisz et al., 2013; Zhou et al., 2025). SE regions display enhanced bidirectional transcription, typically associated with more active enhancer RNA (eRNA) synthesis. Techniques such as GRO-seq and PRO-seq can directly capture these nascent eRNAs and the pronounced bidirectional transcription signals, enabling precise identification of SE regions (Wang et al., 2025). Furthermore, SE frequently interacts with multiple target gene promoters through chromatin loops. Hi-C technology enables genome-wide analysis of chromatin 3D structure, quantifying interaction frequencies between SEs and their target gene promoters (Huang et al., 2018). When integrated with methods like ChIA-PET and PLAC-seq, these approaches provide deeper insights into SE-mediated regulatory networks, allowing for more accurate functional characterization of SEs (Cao et al., 2017; Nott et al., 2019). These multidimensional approaches enable not only precise mapping of SEs but also uncover their central role in gene regulation.
3 SE-mediated gene activation orchestrates hematopoiesis
Hematopoiesis is the process by which various blood cells develop and mature in hematopoietic organs or sites. HSCs possess the dual capacity for self-renewal and multilineage differentiation, enabling them to generate all lineages of mature blood cells. In the process of differentiation from HSCs to mature blood cells, it goes through the stages of multipotent and directed hematopoietic progenitor cells, ultimately differentiating into all series of mature blood cells, including red blood cells, megakaryocytes (platelets), macrophages, eosinophils, basophils, neutrophils, T cells, B cells, and natural killer cells (Pouzolles et al., 2016; Wei et al., 2020). Studies have demonstrated that genes associated with SEs exhibit more pronounced expression changes during hematopoietic cell differentiation compared to those regulated by typical enhancers (Huang et al., 2016). Here, we review recent studies to elaborate in detail on elucidating the crucial role of SEs in driving gene activation that governs the function of hematopoietic stem and progenitor cells, lineage differentiation, as well as cell fate inheritance.
3.1 SEs maintain HSCs function by activating the expression of lineage-specific genes
SEs critically regulate HSCs fate by fine-tuning gene expression to ensure proper lineage output. For instance, an evolutionarily conserved SE located distally from MYC is essential for its expression in both normal and leukemic HSCs in mice and humans. Deletion of this enhancer leads to loss of c-MYC expression, resulting in differentiation defects and loss of myeloid and B-cell lineages-a phenotype resembling that of conditional MYC knockout (Bahr et al., 2018). Similarly, the RUNX1 intronic enhancer (eR1), embedded within a large HSC-specific SE, serves as a key hub where RUNX1 cooperates with factors such as TAL1, GATA2, and PU.1, making it a pivotal regulatory element in HSCs biology (Liau et al., 2017; Ng et al., 2010). Additionally, NFIX has been shown to co-localize with other transcription factors at SEs to support progenitor differentiation and homeostasis (Walker et al., 2023). Together, these studies underscore that SE-mediated transcriptional activation is fundamental to HSCs maintenance, lineage allocation, and functional integrity.
3.2 SEs dynamically regulate macrophage inflammation and polarization through transcriptional and epigenetic mechanisms
In macrophages, inflammatory signal transduction responds to external stimuli, triggering immediate and drastic changes in gene transcription levels (Ghisletti et al., 2010). SEs play a crucial role in regulating gene expression in macrophages, particularly those involved in immune responses and inflammation. They achieve this goal by recruiting transcription factors, dynamic eRNA transcription, and epigenetic regulation. In macrophages, Toll-like receptor 4 (TLR4) signaling dynamically reorganizes SE activity, inducing enhancer RNA (eRNA) transcription at activated immune genes while suppressing it at repressed loci (Hah et al., 2015). This SE landscape can be reshaped by metabolic stimuli such as palmitic acid, which activates inflammatory SEs and suppresses those associated with phagocytosis (Tanwar et al., 2025). The epigenetic reader ZMYND8 further fine-tunes macrophage inflammation by partnering with NF-κB/p65 to silence specific SEs (Jia et al., 2021). Furthermore, SE integrity is essential for macrophage polarization. BRD4 inhibition disrupts SE-driven expression of polarization master regulators like IRF4, impairing both M1 and M2 programs in alveolar macrophages (Li et al., 2025). Similarly, BRD4/P300-dependent SEs facilitate M2 polarization during L. donovani infection (Das et al., 2021). Targeting SE mechanisms thus offers promising therapeutic avenues for inflammatory and macrophage-related diseases.
3.3 SEs orchestrate erythroid development and globin gene expression
SEs are master regulators of erythroid-specific gene expression, coordinating transcriptional programs essential for red blood cell formation. Although H3K27ac marking and SE activity generally decrease during erythropoiesis, SEs remain critical for lineage commitment (Romano et al., 2020). Functional studies highlight key SE-derived components: the long non-coding RNA lncRNA-EC7/Bloodlinc promotes terminal maturation (Alvarez-Dominguez et al., 2017); The SLC25A37 SE exhibits a tiered architecture ensuring precise gene control (Huang et al., 2016); and CPOX-eRNA, transcribed from a SE spanning the Cpox locus, drives erythroblast proliferation and enucleation (Xie and Dean, 2025). Beyond the essential role of Epo in erythropoiesis, genome-wide mapping has revealed Epo-responsive SEs that regulate key erythroid factors like TAL1, establishing SEs as master regulators of the transcriptional circuitry governing erythroid maturation (Hay et al., 2016).
SEs play an essential role in ensuring balanced expression of globin genes, which is critical for hemoglobin synthesis and erythrocyte function. Studies on the α-globin locus demonstrate that its SEs function autonomously yet cumulatively to control transcriptional output and chromatin architecture (Hay et al., 2016). Disruption of individual enhancers leads to α-globin downregulation and embryonic lethality in mice (Blayney et al., 2023), while inversion of the SE results in α-thalassemia-like phenotypes (Kassouf et al., 2025). Similarly, at the β-globin locus, SE-derived seRNA facilitates RNA polymerase II (Pol II) recruitment to promote β-globin expression (Gurumurthy et al., 2021). Targeting the SE-mediated regulation of globin expression could provide a promising therapeutic approach for hemoglobinopathies such as thalassemia and sickle cell anemia, potentially opening new avenues for clinical intervention.
3.4 SEs serve as transcriptional hubs that orchestrate lymphocyte fate
SEs serve as transcriptional hubs that orchestrate lymphocyte fate by integrating diverse signals to control the development, differentiation, and immune function of B cells, T cells, and NK cells. In B cells, SEs function as critical platforms for NF-κB signaling, which drives chromatin opening and threshold gene expression during B cell receptor (BCR) activation (Basso and Dalla-Favera, 2015; Michida et al., 2020). Beyond NF-κB, SEs recruit factors like PU.1 and are hijacked by Epstein-Barr virus (EBV) to utilize STAT5/NFAT for survival (Wibisana et al., 2022; Zhou et al., 2015). SEs also directly regulate the immunoglobulin heavy chain (IgH) locus, enabling VDJ recombination and antibody production (Saintamand et al., 2017). This multifaceted regulation establishes SEs as master integrators of B cell identity and adaptive immune responses.
SEs in T cells are enriched for single nucleotide polymorphisms linked to immune-mediated diseases, highlighting their significance in genetic predisposition (Witte et al., 2015; Vahedi et al., 2015). Studies have shown that the transcription regulator Aire binds to and activates SEs, and topoisomerase 1 is the main Aire partner co localized on SEs and necessary for Aire to interact with all its other binding partners (Bansal et al., 2017). Recent research has shown that B4galt5 within an SE is essential for NK cell survival and the expansion of cytotoxic CD8+ T cells (Morrison et al., 2025). Together, these findings position SEs as central nodes integrating genetic, epigenetic, and functional axes of T cell biology, with broad implications for understanding immune pathogenesis and therapeutic targeting.
SEs serve as pivotal transcriptional hubs that govern cell identity and function across hematopoietic lineages. As we have shown in Figure 2, SEs are essential for maintaining HSPCs by activating key genes like MYC and RUNX1, and SEs dynamically regulate inflammation and polarization of macrophages by integrating external signals, undergoing epigenetic remodeling, and controlling master regulators. During erythroid development, SEs orchestrate maturation, proliferation, and globin gene expression, Furthermore, SEs integrate diverse signals to direct lymphocyte fate in B cells, T cells, and NK cells, thereby regulating adaptive immunity and being implicated in immune pathologies. Collectively, these findings underscore the fundamental role of SE-mediated transcriptional mechanisms in hematopoiesis and immunity.
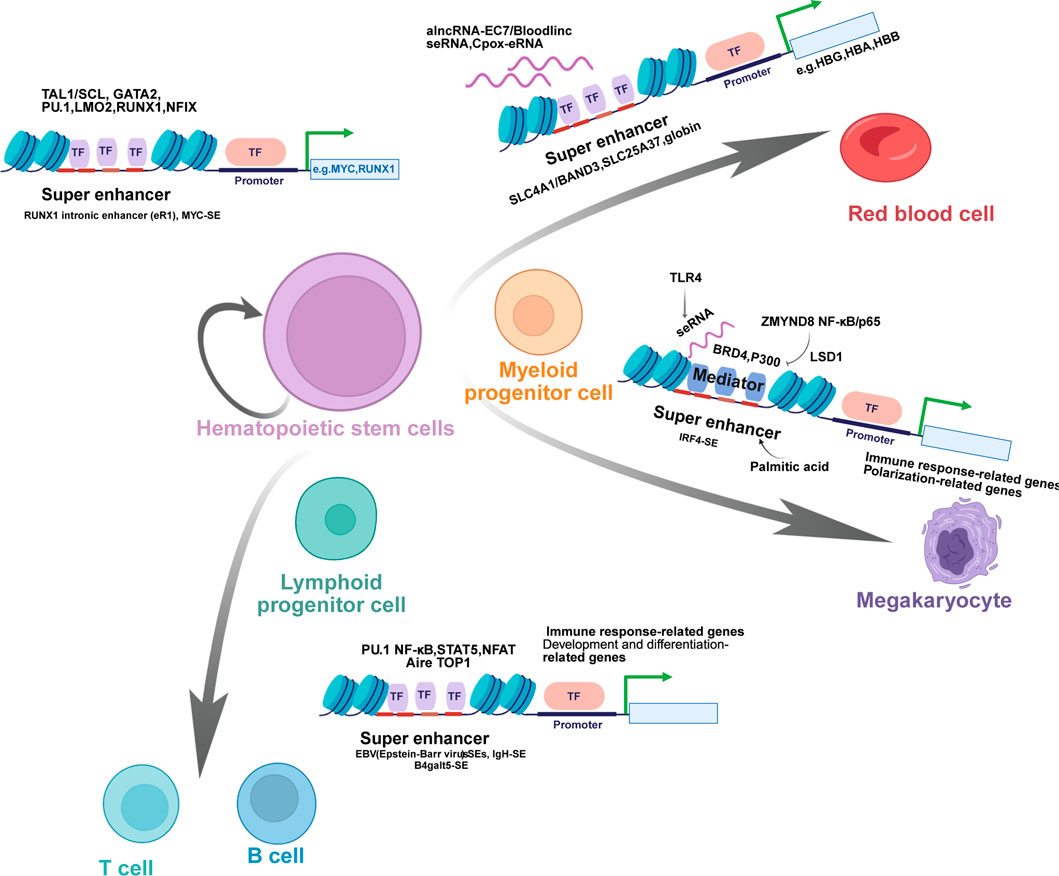
Figure 2. Super Enhancers in the Regulation of Hematopoietic Stem Cell Differentiation and Lineage-Specific Gene Expression. Schematic diagram illustrates the critical role of super enhancers (SEs) in governing the differentiation of hematopoietic stem cells (HSCs) into various lineages of mature blood cells and regulating lineage-specific gene expression. HSCs, with the capacity for self-renewal and multilineage differentiation, give rise to multipotent and lineage-committed progenitor cells, including myeloid progenitor cells, lymphoid progenitor cells, and megakaryocytes, which further differentiate into specialized cell types such as red blood cells, T cells, B cells, and natural killer (NK) cells. SEs mediate this process by targeting key genes and recruiting specific transcription factors (TFs). For instance, SEs such as the RUNX1 intronic enhancer (eR1) and MYC-SE regulate the self-renewal and differentiation of HSCs through interactions with TFs like TAL1/SCL, GATA2, PU.1, LMO2, RUNX1, and NFIX. In red blood cells, SE-derived RNAs (e.g., alncRNA-EC7/Bloodlinc seRNA, Cpox-eRNA) and SEs controlling globin genes (e.g., HBG, HBA, HBB) are involved in terminal differentiation. For lymphoid lineages, SEs such as EBV SEs, IgH-SE, and B4galt5-SE coordinate the development and function of T cells and B cells, with enrichment of TFs including PU.1, NF-κB, STAT5, NFAT, Aire, and TOP1. Collectively, these SE-mediated regulatory networks ensure precise lineage commitment and functional specialization during hematopoiesis, highlighting their pivotal role in maintaining hematopoietic homeostasis.
4 SEs drive oncogene transcription in hematological malignancies
SEs play a pivotal role in the transcriptional regulation of critical oncogenes, including MYC, by orchestrating the formation of high-density transcriptional complexes that drive aberrant gene expression. In various hematological malignancies including leukemia, lymphoma, myelodysplastic neoplasms (MDS) and multiple myeloma (MM), SEs-mediated dysregulation of key oncogenes has been mechanistically established as a critical driver of tumorigenesis through multiple interconnected pathways. Firstly, SEs facilitate long-range genomic interactions by forming chromatin loops that bridge distal regulatory elements with target gene promoters, dramatically amplifying oncogenic transcription; SEs coalesce transcriptional coactivators and RNA polymerase II into high-density nuclear hubs, creating a permissive environment for runaway gene expression; additionally malignancy-defining transcription factors exhibit preferential binding to SEs, where they nucleate the assembly of oncogenic transcriptional complexes that override normal regulatory checkpoints (Debek and Juszczynski, 2022; Belloucif and Lobry, 2022). This SE-driven transcriptional addiction not only sustains hyperproliferative signaling but also underlies acquired resistance to conventional therapies, positioning SEs as high-priority epigenetic targets for next-generation precision oncology interventions.
4.1 SEs driven oncogene activation in AML
Acute myeloid leukemia (AML) represents an aggressive hematological malignancy originating from the malignant transformation of myeloid progenitor cells, comprising 7.6% of all hematopoietic system cancers. This disease is characterized by uncontrolled proliferation of immature myeloblasts in the bone marrow, leading to bone marrow failure and impaired hematopoiesis. Current therapeutic challenges are reflected in the dismal 5-year survival rate of <30%, highlighting the urgent need for novel treatment strategies (Kayser and Levis, 2023; Récher, 2021; Pasquer et al., 2021). Dysregulation of key leukemogenic drivers, such as MYC, HOXA, and MEIS1, triggers a cascade of carcinogenic events in AML (Luo et al., 2005; Xie et al., 2025; Zhou and Lu, 2023), and SEs play a central role in these dysregulation. In 2013, a lineage-specific SE cluster spanning 1.7 Mb downstream of the MYC transcription start site was identified at the MYC locus. These distal regulatory elements harbor focal DNA repeats that are essential for MYC expression. Importantly, these SEs depend on the chromatin remodeler BRG1 (SMARCA4) to sustain the MYC oncogenic transcriptional program, thereby driving AML progression (Shi et al., 2013). Integrative genetic, genomic, and biochemical analyses have demonstrated that the MYC SE region is co-occupied by BRD4, the histone acetyltransferase p300/CBP, and key hematopoietic transcription factors (including PU.1, FLI1, and MYB). This coordinated assembly forms a chromatin-based signaling hub that drives oncogenic transcription and sustains leukemia maintenance in AML (Roe et al., 2015). Studies have demonstrated that chromosomal rearrangements involving inv (3) (q21q26.2) or t (3; 3) (q21; q26.2) in AML induce leukemogenesis through dual dysregulation of GATA2 and EVI1. These structural alterations generate a SE at the EVI1 locus, driving its aberrant overexpression while simultaneously causing haploinsufficient GATA2 expression - a pathogenic combination that promotes leukemic transformation. (Gröschel et al., 2014). Ottema et al. found that a translocation of a MYC super enhancer (MYC SE) to the EVI1 locus, therefore overexpression of EVI1 in AMLs harboring a t (3; 8) (q26; q24) (Ottema et al., 2021). Mechanistic studies have revealed that the t (10; 17) (p15; q21) chromosomal translocation generates an oncogenic ZMYND11-MBTD1 fusion protein. This chimeric protein recruits the NuA4/Tip60 histone acetyltransferase complex to SEs of key leukemogenic drivers (including HOXA, MEIS1, MYB, MYC, and SOX4), thereby establishing a pro-leukemic transcriptional program that promotes malignant transformation (Li et al., 2021). Another researcher highlighted that TRIB1 orchestrates the eviction of C/EBPα specifically from SEs, unleashing a HOXA9-dependent oncogenic cascade comprising ERG, SPNS2, RGL1, and PIK3CD, which drives leukemogenesis in AML (Yoshino et al., 2021). These studies converge to reveal SEs as critical epigenetic nodes in AML, hijacked by diverse genetic lesions to activate oncogenic networks that fuel leukemogenesis.
4.2 SEs silencing as a sore pathogenic mechanism in MDS
MDS formerly known as myelodysplastic syndromes, are clonal hematopoietic malignancies that cause morphologic bone marrow dysplasia along with anemia, neutropenia, or thrombocytopenia. MDS are associated with an increased risk of AML. The yearly incidence of MDS is approximately 4 per 100 000 people in the United States and is higher among patients with advanced age (Sekeres and Taylor, 2022). In AML with MLL gene rearrangements, leukaemia stem cells exhibit unique gene expression patterns and chromatin states, which are thought to function as enhancers. Concurrently, enhancer dysfunction is commonly observed in leukemia stem cells (Pan et al., 2023). Lysine-specific demethylase 1 (LSD1) is a critical epigenetic regulator in myeloid malignancies, including MDS that progress to overt leukemia. The novel LSD1 inhibitor NCD38 targets MDS-related leukemic cells by derepressing abnormally silenced SEs of hematopoietic differentiation genes. Specifically, NCD38 elevates H3K27ac levels on SEs, activating ∼500 new SEs and a core set of 62 LSD1-suppressed genes (including key regulators GFI1 and ERG). SE activation precedes transcriptional upregulation and drives myeloid differentiation programs, while simultaneously disrupting leukemogenic pathways. Depletion of SE-driven GFI1 attenuates differentiation, confirming the functional link. Crucially, SE reactivation by NCD38 eradicates primary MDS-derived leukemia cells with complex karyotypes in vivo, demonstrating that LSD1-mediated SE silencing sustains MDS leukemogenesis, and its reversal offers a potent therapeutic strategy against high-risk MDS (Sugino et al., 2017).
4.3 SEs as central orchestrators of oncogenic transcription in ATL
Adult T-cell leukemia/lymphoma (ATL) is a T-cell lymphoma caused by human T-cell leukemia virus type I (Cook et al., 2019). Controlling gene expression through splice enhancers, or SEs, plays a vital role in the regulation of alternative splicing events, which are essential in the context of ATL. Wong et al. performed enhancer profiling using primary leukemia samples from ATL, defined that the SEs at several known cancer gene loci, including CCR4, PIK3R1, and TP73, which are involved in the T-cell activation pathway in ATL. Notably, the study revealed that THZ1-mediated CDK7 inhibition ablates SE-driven oncogenic transcription in ATL, concomitantly suppressing proliferation and inducing apoptosis, underscoring SE-directed therapy as a viable treatment paradigm for this malignancy (Wong et al., 2017). Wong et al. also illustrated that IRF4 and NF-κB form a feed-forward regulatory loop that coordinately controls oncogenic transcription programs, with their binding sites being significantly enriched in SEs to directly modulate the expression of critical leukemia drivers including MYC, CCR4, and BIRC3, further promoting the ATL development (Wong et al., 2020). The HTLV-1-encoded bZIP gene (HBZ) is the sole viral transcription factor that continues to be expressed in all cases of ATL. Recent studies have demonstrated that HBZ binds to an ATL-cell–specific BATF3 SE, thereby regulating the transcription of the BATF3 factor and its downstream targets, including IRF4. BATF3 and IRF4 act synergistically to drive the expression of ATL-cell–specific genes and are essential for sustaining ATL cell proliferation. This HBZ-mediated SE activation allows the viral protein to directly perturb the host-cell gene-regulatory network, thereby promoting malignant proliferation of ATL cells (Nakagawa et al., 2018). SE-driven overexpression of TP73, a TP53 homolog, promotes ATL maintenance by coordinately regulating cell cycle progression and DNA repair mechanisms (Ong et al., 2022). RUNX1 is a pivotal transcription factor in the development of hematopoiesis, and its dysregulation has been linked to various hematological malignancies. Research has indicated that the disruption of SE-mediated gene regulation can result in the suppression of RUNX1 expression, the induction of apoptosis, and the inhibition of ATL cell proliferation (Kobayashi et al., 2024). Collectively, these findings establish SEs as central epigenetic orchestrators of ATL pathogenesis, where they coordinate oncogenic transcription program, sustain proliferative signaling, highlighting the therapeutic potential of SE-directed interventions in ATL.
4.4 Dysregulated SEs in B-ALL and T-ALL pathogenesis
Acute lymphoblastic leukemia (ALL) is an aggressive hematological malignancy characterized by the uncontrolled proliferation of immature lymphoid precursors (B- or T-cell lineage) (Malard and Mohty, 2020). Recent advances highlight the critical role of SEs-mediated transcriptional dysregulation in ALL pathogenesis. In B-ALL, recurrent alterations targeting B-cell lineage transcription factors involve SE acquisition or disruption. Katerndahl et al. suggested a model in which the balance between STAT5 and a specific transcription factor network at SEs acts as a molecular switch to govern appropriate progenitor B cell proliferation, survival and differentiation (Katerndahl et al., 2017). Altering the balance between these two antagonistic pathways drives B cell transformation, while the degree of imbalance underlies how patients with B-ALL will respond to therapy. In addition, PAX5 deletions or point mutations frequently co-occur with the formation of novel SEs at proto-oncogenes like MYC (Shah et al., 2013). Similarly, IKZF1 (Ikaros) deletions or dominant-negative isoforms compromise its tumor-suppressive enhancer-silencing function, permitting SE-driven expression of cytokine receptors (CRLF2) and kinases (JAK2) (Raca et al., 2021; Mullighan, 2011).
SEs are crucial in regulating the expression of oncogenes in T-ALL. TAL1, one of the most frequently dysregulated genes in T-ALL is overexpressed in ∼50% of T-ALL cases, driven by mutations creating a 5′SE via MYB transcription factor binding (Smith et al., 2023). Noura et al. Have shown that KLF4 can downregulate MYB expression by directly binding to its promoter and inhibits the formation of 5′TAL1 SE, further suppresses SE-driven TAL1 expression in T-ALL cells (Noura et al., 2024). The FYB1 gene driven by SEs is overexpressed in T-ALL and plays an important role in the self-renewal and survival of T-ALL cells. Knocking down FYB1 leads to reduced tumor growth and increased cell apoptosis, highlighting its potential as a therapeutic target (Zhang et al., 2023). In addition, a recent study has shown that another SEs driven gene, IRF2BP2, is activated by specific SEs regions in T-ALL. This gene is crucial for the growth and survival of T-ALL cells, affecting pathways such as MYC and E2F, and IRF2BP2 deficiency can impair the proliferation and survival of T-ALL cells (Yu et al., 2025). Targeting SEs and their related transcription mechanisms provides a promising therapeutic strategy in T-ALL. The BET inhibitor GNE-987 targets SEs related genes such as LCK, effectively inhibiting the progression of T-ALL (Yu et al., 2024). Understanding the mechanism of SEs formation and its related genes provides valuable insights into potential therapeutic targets and offers new avenues for treating T-ALL.
4.5 SEs driven oncogenic networks in MM
MM although a rare disease, is the second most common hematological malignancies. It is found in the spectrum of plasma cell dyscrasias, which begins with monoclonal gammopathy of unknown significance (MGUS) to overt plasma cell leukemia and extramedullary myeloma (Leung and Rajkumar, 2023). MM pathogenesis heavily relies on SE-driven transcription factor networks. MYC locus rearrangements, as a late progression event in MM, reposition MYC mostly with SEs which are associated with a significant increase of MYC expression (Affer et al., 2014). HJURP was identified as an SE-associated gene, which transcription is activated via chromatin interaction and binding of NSD2 and BRD4 to the SE associated with HJURP. SE, further promotes growth and survival of t (4; 14)-positive MM (Jia et al., 2022). As a member of the BET protein family, BRD4 recognises acetylated histones and promotes gene expression driven by SEs. BRD4 is highly enriched on SEs in MM. BET protein inhibitors can specifically displace BET proteins bound to SEs, leading to the potent, selective downregulation of SE target genes and the inhibition of MM cell proliferation (Fulciniti et al., 2015). SEs contribute significantly to drug resistance. Oncogenic overexpression of integrin-β7 (ITGB7) in high-risk MM has been reported to enhance interactions between neoplastic plasma cells and stromal cells, thereby promoting cell-adhesion-mediated drug resistance. Induction of DNA methylation at ITGB7 SE further increases ITGB7 expression promotes overall malignant growth in MM (Chou et al., 2023). MM is often driven by MYC and that is sustained by IRF4, which are upregulated by SEs. IKZF1 and IKZF3 bind to SEs and can be degraded using immunomodulatory imide drugs (IMiDs) (Welsh et al., 2024). The dependence of MM on IKZF1-bound SEs, which can be effectively targeted by a potent therapeutic combination pairing IMiD-mediated degradation of IKZF1 and IKZF3 with EP300 inhibition. Therefore, targeting SE components disrupts these resistance pathways and synergizes with conventional therapies.
In summary, SEs serve as pivotal drivers of oncogenic transcription across hematological malignancies (as shown in Figure 3). In AML, SEs are hijacked through various genetic alterations to activate key oncogenes such as MYC, HOXA, and EVI1, sustaining leukemogenic programs. In MDS, pathological silencing of SEs at hematopoietic differentiation genes contributes to disease pathogenesis, and pharmacological reactivation of these SEs represents a promising therapeutic strategy. Similarly, in ATL, SEs coordinate oncogenic transcription through feed-forward loops involving IRF4 and NF-κB, maintaining malignant phenotypes. In both B-cell and T-cell acute lymphoblastic leukemia (ALL), SEs dysregulation drives transformation by overexpressing oncogenes like MYC and TAL1, often through alterations in lineage-specific transcription factors. MM utilizes SE-driven networks centered on MYC and IRF4 to promote growth and drug resistance, with targeting of SE-associated complexes showing therapeutic potential. Collectively, these findings establish SEs as central epigenetic regulators in hematological malignancies and highlight SE-directed transcriptional inhibition as a viable therapeutic paradigm across cancer types.
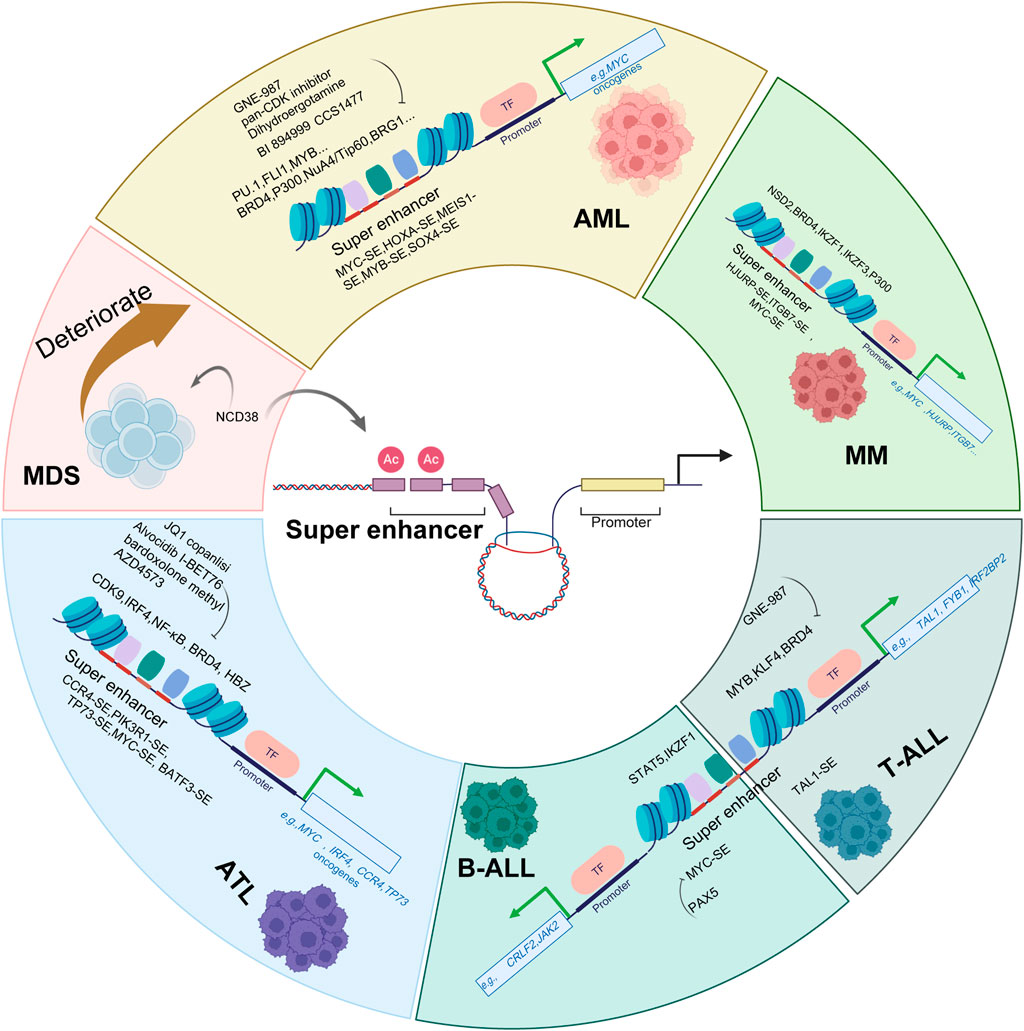
Figure 3. Targeting Super Enhancers: Therapeutic Strategies in Hematological Malignancies. Schematic diagram depicts the role of super enhancers (SEs) in driving hematological malignancies and the corresponding therapeutic strategies targeting SEs and their associated regulatory components. In various hematological malignancies, including acute myeloid leukemia (AML), adult T-cell leukemia/lymphoma (ATL), T-cell acute lymphoblastic leukemia (T-ALL), B-cell acute lymphoblastic leukemia (B-ALL), myelodysplastic neoplasms (MDS), and multiple myeloma (MM), aberrant SEs (e.g., MYC-SE, TAL1-SE, CCR4-SE, TP73-SE) drive oncogenic gene expression by recruiting transcription factors (e.g., PU.1, FLI1, MYB, STAT5, NF-κB) and coactivators (e.g., BRD4, p300, NuA4/Tip60 complex). Therapeutic agents such as GNE-987 (a BRD4 inhibitor), JQ1, pan-CDK inhibitors, alvocidib and AZD4573 (CDK9 inhibitor), bardoxolone-methyl (a NF-κB inhibitor), dihydroergotamine, BI894999 and I-BET76 (as BET inhibitor), CCS1477(a P300 inhibitor), copanlisi (a PI3K inhibitor), and NCD38 (an LSD1 inhibitor) disrupt SE-mediated oncogenic transcription by targeting key components (e.g., BRD4, CDK9, IRF4, NSD2, IKZF1/3, NF-κB) or directly interfering with SE function. These strategies aim to suppress the expression of critical oncogenes (e.g., MYC, TAL1, HOXA, IRF4) and thereby inhibit tumor progression in hematological malignancies.
5 SEs as promising therapeutic targets in hematological malignancies
SEs are potent transcriptional regulatory elements that drive the expression of genes fundamental to cell identity and survival. In hematological malignancies, the dysregulation or hijacking of SEs to activate oncogenes presents a compelling therapeutic vulnerability. Direct pharmacological targeting of the SE transcriptional machinery has emerged as a viable strategy and can be categorized by distinct mechanisms of action.
5.1 Targeting epigenetic readers and co-activators
A primary approach involves inhibiting bromodomain and extra-terminal (BET) proteins, which are critical for recognizing acetylated histones at SEs (Zhang et al., 2021). BET inhibitors, such as BI894999, have been shown to repress SE-associated transcription and control AML in preclinical models, often synergizing with CDK9 inhibition (Gerlach et al., 2018). Similarly, the BRD4 inhibitor GNE-987 exerts anticancer effects in AML by targeting a SE-related oncogen (Fang et al., 2022; Sang et al., 2022). Beyond BET proteins, inhibiting the EP300/CBP bromodomain (e.g., CCS1477 inhibitor) also represents a promising therapeutic avenue across various hematological malignancies (Nicosia et al., 2023). Additionally, JQ1, another BRD4 inhibitor, suppresses RUNX1 expression by disrupting SE-mediated gene regulation, further downregulating c-MYC and inducing apoptosis in ATL cells (Kobayashi et al., 2024).
5.2 Inhibiting transcriptional kinases
Targeting key transcriptional kinases such as CDK7 and CDK9, which facilitate RNA polymerase II-mediated transcription at SEs, provides another therapeutic strategy. Small molecules co-targeting CKIα and CDK7/9 have shown efficacy in AML (Minzel et al., 2018). The CDK9 inhibitor alvocidib, for instance, suppresses ATL proliferation through SE-dependent downregulation of the oncogenic transcription factor IRF4 (Sakamoto et al., 2022).
5.3 Modulating oncogenic expression via SE interference
No-epigenetic pharmacological agents can also disrupt SE-driven oncogenic networks. Dihydroergotamine (DHE), for example, exhibits anti-AML activity by interfering with MYC expression through SE modulation (Call et al., 2020).
5.4 Rational combination therapies
The synergistic potential of SE-directed therapy is illustrated in ATL, where a triple combination of BET (I-BET762), PI3K (copanlisib), and NF-κB (bardoxolone methyl) inhibitors exhibits potent synergistic activity (Daenthanasanmak et al., 2022). Such combinations may enhance efficacy and mitigate resistance.
5.5 Resistance mechanisms and overcoming strategies
Despite promising results, therapeutic resistance remains a challenge. In multiple myeloma, transcriptional heterogeneity can enable escape from SE-targeting drug combinations (Welsh et al., 2024). However, targeting SE-driven dependencies can also overcome resistance, as demonstrated with the CDK9 inhibitor AZD4573, which induces epigenetic reprogramming to circumvent resistance in lymphoma models (Thieme et al., 2023).
In conclusion, the critical role of SEs in maintaining oncogenic programs positions them as attractive therapeutic targets. Targeting SE complexes and their co-factors, particularly through BET and CDK inhibitors, alone or in rational combinations, represents a promising direction for future cancer therapeutics in hematological malignancies.
6 Discussion and outlook
SEs have emerged as pivotal regulators of gene expression in both normal hematopoiesis and hematological malignancies. Their unique ability to amplify transcriptional output through dense clustering of enhancer elements and coactivators underscores their critical role in maintaining cellular identity and driving oncogenic programs. As research progresses, the dual nature of SEs, as guardians of normal development and instigators of malignant transformation, has become increasingly apparent. As we have shown in Figure 4, SEs govern fundamental processes such as stem cell self-renewal and lineage differentiation in HSCs, and specialized functions including inflammation in immune cells and hemoglobin synthesis in erythroid cells, following malignant transformation, dysregulated SEs drive oncogenic programs, leading to uncontrolled proliferation and the maintenance of the malignant state across hematological malignancies.
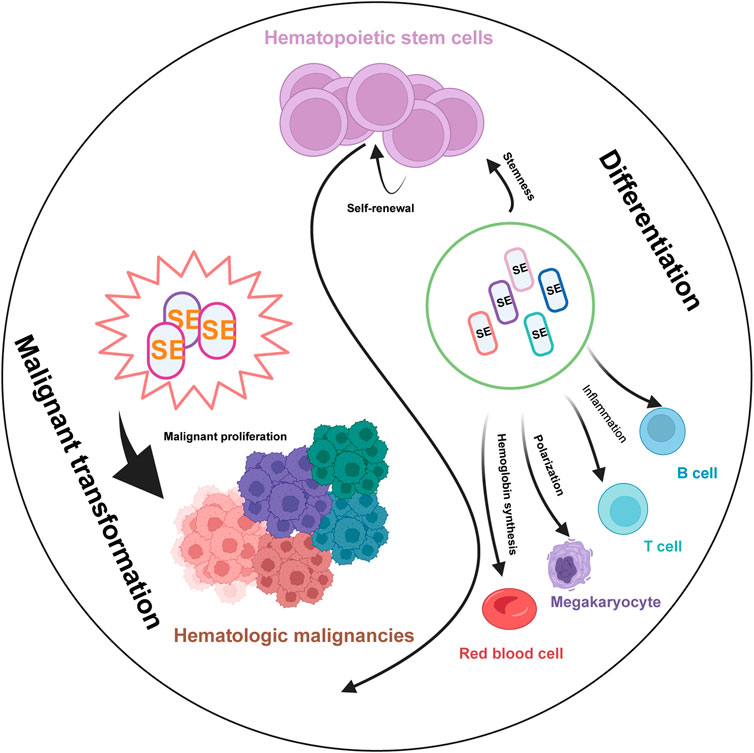
Figure 4. The Pivotal Role of SEs in Normal Hematopoiesis and Hematological Malignancies. Schematic illustrates the critical functions of SEs in regulating key biological processes during normal blood cell development (left) and their contribution to the pathogenesis of various hematologic malignancies (right). In normal hematopoiesis, Lineage-specific SEs (marks in different colors) govern fundamental processes such as stem cell self-renewal, lineage differentiation, and specialized functions including inflammation in immune cells and hemoglobin synthesis in erythroid cells. Following malignant transformation, dysregulated SEs drive oncogenic programs, leading to uncontrolled proliferation and the maintenance of the malignant state across different hematological malignancies.
In normal hematopoiesis, SEs function as master regulators of lineage-specific gene expression, ensuring the precise balance between self-renewal and differentiation of HSCs. For instance, SEs associated with MYC, RUNX1, and BRD4 are indispensable for HSCs maintenance and lineage commitment, as their disruption leads to aberrant differentiation and hematopoietic failure (Bahr et al., 2018; Liau et al., 2017). In macrophages, SEs dynamically regulate immune responses and inflammation related genes, which are essential for immune defense and maintaining inflammation homeostasis (Hah et al., 2015; Tanwar et al., 2025; Jia et al., 2021). Similarly, in erythroid cells, SEs not only regulate the differentiation but also orchestrate the spatiotemporal expression of globin genes (Xie and Dean, 2025; Alvarez-Dominguez et al., 2017; Hay et al., 2016; Kassouf et al., 2025; Gurumurthy et al., 2021), while in lymphoid lineages, they govern immune cell development and function through dynamic interactions with transcription factors like NF-κB and STAT5 (Michida et al., 2020; Wibisana et al., 2022; Zhou et al., 2015). The plasticity of SEs evidenced by their rapid remodeling in response to stimuli such as inflammation or infection, highlights their role as epigenetic hubs integrating environmental cues into transcriptional outputs.
In contrast, the hijacking of SEs in hematological malignancies exemplifies their pathogenic potential. By aberrantly activating oncogenes such as MYC, TAL1, and IRF4, SEs create a permissive environment for uncontrolled proliferation and survival of malignant clones. For example, in AML, chromosomal rearrangements reposition SEs to drive EVI1 or MYC overexpression (Roe et al., 2015; Gröschel et al., 2014; Ottema et al., 2021), while in T-ALL, SE-mediated TAL1 activation sustains leukemogenesis (Smith et al., 2023). Notably, SEs also contribute to therapeutic resistance, as seen in multiple myeloma, where SE-driven ITGB7 overexpression fosters cell-adhesion-mediated drug resistance (Chou et al., 2023). These findings underscore SEs as central nodes in the oncogenic networks of hematological cancers, often rendering tumors dependent on SE activity, a phenomenon termed “transcriptional addiction.”
The molecular characterization of SEs has spurred the development of targeted therapies aimed at disrupting their regulatory hubs. BET inhibitors (e.g., JQ1, GNE-987) and CDK inhibitors (e.g., THZ1, alvocidib) have shown preclinical efficacy by selectively downregulating SE-driven oncogenes (Wong et al., 2017; Kobayashi et al., 2024; Yu et al., 2024; Sakamoto et al., 2022; Fang et al., 2022). In AML and T-ALL, these agents induce apoptosis and differentiation by dismantling SE-associated transcriptional complexes. Similarly, LSD1 inhibitors (e.g., NCD38) reactivate silenced SEs to restore differentiation programs in MDS (Sugino et al., 2017). However, challenges remain, such as the fact that SEs regulate both oncogenic and normal hematopoietic genes, raising concerns about off-target effects; and the tumor cells may bypass SE dependency via compensatory mutations or alternative enhancer activation.
The convergence of multi-omics technologies and artificial intelligence is fundamentally reshaping our understanding of SEs in hematological malignancies. In diseases like AML and T-cell lymphoma, the integration of ChIP-seq with Hi-C has begun to precisely map the dynamic chromatin landscapes and three-dimensional interactions that underlie oncogenic SE hijacking (Hnisz et al., 2016; Koh et al., 2015). Crucially, single-cell multi-omics (e.g., scATAC-seq) is now unraveling the epigenetic heterogeneity of tumors, identifying distinct cellular subpopulations driven by unique SE programs that may confer therapy resistance (Satpathy et al., 2019). Complementing these approaches, deep learning models are being leveraged to predict functional enhancers directly from DNA sequence and to prioritize non-coding mutations that disrupt SE architecture. Together, these synergistic technologies are transitioning the field from descriptive mapping to the predictive modeling of SE networks, enabling the discovery of novel, context-specific therapeutic vulnerabilities directly from patient genomic data (Avsec et al., 2021).
SEs sit at the nexus of gene regulation, bridging normal development and malignant transformation. Their study has not only deepened our understanding of hematopoietic biology but also unveiled actionable targets for precision medicine. While challenges persist, the continued integration of epigenetic, genomic, and pharmacological approaches holds promise for transforming SE research into tangible clinical benefits, ultimately improving outcomes for patients with hematological malignancies.
Author contributions
YC: Writing – original draft, Writing – review and editing. GP: Writing – review and editing. HZ: Supervision, Writing – review and editing. YH: Writing – review and editing. LS: Visualization, Writing – review and editing. HX: Writing – review and editing. YL: Writing – original draft, Writing – review and editing. XW: Project administration, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from the National Natural Science Foundation of China (82400140 and 81900112), China Postdoctoral Science Foundation (2024M752919, GZC20241558) and the China Medicine Education Association (CMEA-2025-008).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Affer, M., Chesi, M., Chen, W. D., Keats, J. J., Demchenko, Y. N., Tamizhmani, K., et al. (2014). Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia 28, 1725–1735. doi:10.1038/leu.2014.70
Akiki, R. M., Cornbrooks, R. G., Magami, K., Greige, A., Snyder, K. K., Wood, D. J., et al. (2024). A long noncoding eRNA forms R-loops to shape emotional experience-induced behavioral adaptation. Science 386, 1282–1289. doi:10.1126/science.adp1562
Alvarez-Dominguez, J. R., Knoll, M., Gromatzky, A. A., and Lodish, H. F. (2017). The super-enhancer-derived alncRNA-EC7/Bloodlinc potentiates red blood cell development in trans. Cell Rep. 19, 2503–2514. doi:10.1016/j.celrep.2017.05.082
Avsec, Z., Agarwal, V., Visentin, D., Ledsam, J. R., Grabska-Barwinska, A., Taylor, K. R., et al. (2021). Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 18, 1196–1203. doi:10.1038/s41592-021-01252-x
Bahr, C., von Paleske, L., Slu, V. V. U., Emeseiro, S. R., Takayama, N., Ng, S. W., et al. (2018). A myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 553, 515–520. doi:10.1038/nature25193
Banerji, J., Rusconi, S., and Schaffner, W. (1981). Expression of a beta-globin gene is enhanced by remote Sv40 DNA-sequences. Cell 27, 299–308. doi:10.1016/0092-8674(81)90413-x
Bansal, K., Yoshida, H., Benoist, C., and Mathis, D. (2017). The transcriptional regulator Aire binds to and activates super-enhancers. Nat. Immunol. 18, 263–273. doi:10.1038/ni.3675
Basso, K., and Dalla-Favera, R. (2015). Germinal centres and B cell lymphomagenesis. Nat. Rev. Immunol. 15, 172–184. doi:10.1038/nri3814
Belloucif, Y., and Lobry, C. (2022). Super-Enhancers dysregulations in hematological malignancies. Cells-Basel 11, 196. doi:10.3390/cells11020196
Blayney, J. W., Francis, H., Rampasekova, A., Camellato, B., Mitchell, L., Stolper, R., et al. (2023). Super-enhancers include classical enhancers and facilitators to fully activate gene expression. Cell 186, 5826–5839.e18. doi:10.1016/j.cell.2023.11.030
Cai, H. S., Liang, J. F., Jiang, Y. Q., Wang, Z. Y., Li, H. Y., Wang, W. J., et al. (2024). KLF7 regulates super-enhancer-driven IGF2BP2 overexpression to promote the progression of head and neck squamous cell carcinoma. J. Exp. Clin. Canc Res. 43, 69. doi:10.1186/s13046-024-02996-y
Call, S. G., Duren, R. P., Panigrahi, A. K., Nguyen, L., Freire, P. R., Grimm, S. L., et al. (2020). Targeting oncogenic super enhancers in MYC-Dependent AML using a small molecule activator of NR4A nuclear receptors. Sci. Rep-Uk 10, 2851. doi:10.1038/s41598-020-59469-3
Cao, F., Fang, Y. W., Tan, H. K., Goh, Y., Choy, J. Y. H., Koh, B. T. H., et al. (2017). Super-Enhancers and broad H3K4me3 domains form complex gene regulatory circuits involving chromatin interactions. Sci. Rep-Uk 7, 2186. doi:10.1038/s41598-017-02257-3
Choudhury, S. R., Byrum, S. D., Alkam, D., Ashby, C., Zhan, F. H., Tackett, A. J., et al. (2023). Expression of integrin β-7 is epigenetically enhanced in multiple myeloma subgroups with high-risk cytogenetics. Clin. Epigenetics 15, 18. doi:10.1186/s13148-023-01433-9
Cook, L. B., Fuji, S., Hermine, O., Bazarbachi, A., Ramos, J. C., Ratner, L., et al. (2019). Revised adult T-Cell leukemia-lymphoma international consensus meeting report. J. Clin. Oncol. 37, 677–687. doi:10.1200/JCO.18.00501
Daenthanasanmak, A., Bamford, R. N., Yoshioka, M., Yang, M., Homan, P., Karim, B., et al. (2022). Triple combination of BET plus PI3K and NF-κB inhibitors exhibit synergistic activity in adult T-cell leukemia/lymphoma. Blood Adv. 6, 2346–2360. doi:10.1182/bloodadvances.2021005948
Das, S., Mukherjee, S., and Ali, N. (2021). Super enhancer-mediated transcription of miR146a-5p drives M2 polarization during infection. PLoS Pathog. 17 (2), e1009343. doi:10.1371/journal.ppat.1009343
Debek, S., and Juszczynski, P. (2022). Super enhancers as master gene regulators in the pathogenesis of hematologic malignancies. Bba-Rev Cancer 1877, 188697. doi:10.1016/j.bbcan.2022.188697
Fang, F., Lu, J., Sang, X., Tao, Y. F., Wang, J. W., Zhang, Z. M., et al. (2022). Super-enhancer profiling identifies novel critical and targetable cancer survival gene LYL1 in pediatric acute myeloid leukemia. J. Exp. Clin. Canc Res. 41, 225. doi:10.1186/s13046-022-02428-9
Font-Tello, A., Kesten, N., Xie, Y. T., Taing, L., Vareslija, D., Young, L. S., et al. (2020). FiTAc-seq: fixed-tissue ChIP-seq for H3K27ac profiling and super-enhancer analysis of FFPE tissues. Nat. Protoc. 15, 2503–2518. doi:10.1038/s41596-020-0340-6
Fulciniti, M., Lin, C. Y., Samur, M. K., Lawlor, M., Anderson, K. C., Bradner, J. E., et al. (2015). Discovery and characterization of promoter and super-enhancer-associated dependencies through E2F and BET bromodomains in multiple myeloma. Blood 126, 838. doi:10.1182/blood.v126.23.838.838
Gerlach, D., Tontsch-Grunt, U., Baum, A., Popow, J., Scharn, D., Hofmann, M. H., et al. (2018). The novel BET bromodomain inhibitor BI 894999 represses super-enhancer-associated transcription and synergizes with CDK9 inhibition in AML. Oncogene 37, 2687–2701. doi:10.1038/s41388-018-0150-2
Ghisletti, S., Barozzi, I., Mietton, F., Polletti, S., De Santa, F., Venturini, E., et al. (2010). Identification and characterization of enhancers controlling the Inflammatory Gene Expression Program in macrophages. Immunity 32, 317–328. doi:10.1016/j.immuni.2010.02.008
Gröschel, S., Sanders, M. A., Hoogenboezem, R., de Wit, E., Bouwman, B. A. M., Erpelinck, C., et al. (2014). A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157, 369–381. doi:10.1016/j.cell.2014.02.019
Gurumurthy, A., Yu, D. T., Stees, J. R., Chamales, P., Gavrilova, E., Wassel, P., et al. (2021). Super-enhancer mediated regulation of adult β-globin gene expression: the role of eRNA and integrator. Nucleic Acids Res. 49, 1383–1396. doi:10.1093/nar/gkab002
Hah, N., Benner, C., Chong, L. W., Yu, R. T., Downes, M., and Evans, R. M. (2015). Inflammation-sensitive super enhancers form domains of coordinately regulated enhancer RNAs. P Natl. Acad. Sci. U. S. A. 112, E297–E302. doi:10.1073/pnas.1424028112
Hay, D., Hughes, J. R., Babbs, C., Davies, J. O. J., Graham, B. J., Hanssen, L. L. P., et al. (2016). Genetic dissection of the α-globin super-enhancer in vivo. Nat. Genet. 48, 895–903. doi:10.1038/ng.3605
He, Y., Cai, Y. W., Cao, Y. Y., Wang, Y., Wang, J., and Ding, H. (2025). Application strategies of super-enhancer RNA in cardiovascular diseases. Biomedicines 13, 117. doi:10.3390/biomedicines13010117
Higashijima, Y., and Kanki, Y. (2022). Potential roles of super enhancers in inflammatory gene transcription. Febs J. 289, 5762–5775. doi:10.1111/febs.16089
Hnisz, D., Abraham, B. J., Lee, T. I., Lau, A., Saint-André, V., Sigova, A. A., et al. (2013). Super-Enhancers in the control of cell identity and disease. Cell 155, 934–947. doi:10.1016/j.cell.2013.09.053
Hnisz, D., Weintraub, A. S., Day, D. S., Valton, A. L., Bak, R. O., Li, C. H., et al. (2016). Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351, 1454–1458. doi:10.1126/science.aad9024
Hsieh, C. L., Fei, T., Chen, Y. W., Li, T. T., Gao, Y. F., Wang, X. D., et al. (2014). Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. P Natl. Acad. Sci. U. S. A. 111, 7319–7324. doi:10.1073/pnas.1324151111
Huang, J. L., Liu, X., Li, D., Shao, Z., Cao, H., Zhang, Y. Y., et al. (2016). Dynamic control of enhancer repertoires drives lineage and stage-specific transcription during hematopoiesis. Dev. Cell 36, 9–23. doi:10.1016/j.devcel.2015.12.014
Huang, J. L., Li, K. L., Cai, W. Q., Liu, X., Zhang, Y. Y., Orkin, S. H., et al. (2018). Dissecting super-enhancer hierarchy based on chromatin interactions. Nat. Commun. 9, 943. doi:10.1038/s41467-018-03279-9
Jia, Y. L., Chng, W. J., and Zhou, J. B. (2019). Super-enhancers: critical roles and therapeutic targets in hematologic malignancies. J. Hematol. Oncol. 12, 77. doi:10.1186/s13045-019-0757-y
Jia, P., Li, X., Wang, X. L., Yao, L. J., Xu, Y. Y., Hu, Y., et al. (2021). ZMYND8 mediated liquid condensates spatiotemporally decommission the latent super-enhancers during macrophage polarization. Nat. Commun. 12, 6535. doi:10.1038/s41467-021-26864-x
Jia, Y. L., Zhou, J. B. A., Tan, T. K., Chung, T. H., Chen, Y. X., Chooi, J. Y., et al. (2022). Super enhancer-mediated upregulation of HJURP promotes growth and survival of t(4;14)-Positive multiple myeloma. Cancer Res. 82, 406–418. doi:10.1158/0008-5472.can-21-0921
Kai, Y., Li, B. E., Zhu, M., Li, G. Y., Chen, F., Han, Y. L., et al. (2021). Mapping the evolving landscape of super-enhancers during cell differentiation. Genome Biol. 22, 269. doi:10.1186/s13059-021-02485-x
Kang, Y., Kang, J., and Kim, A. (2021). Histone H3K4me1 strongly activates the DNase I hypersensitive sites in super-enhancers than those in typical enhancers. Biosci. Rep. 41. doi:10.1042/BSR20210691
Kassouf, M. T., Francis, H. S., Gosden, M., Suciu, M. C., Downes, D. J., Harrold, C., et al. (2025). The α-globin super-enhancer acts in an orientation-dependent manner. Nat. Commun. 16, 1033. doi:10.1038/s41467-025-56380-1
Katerndahl, C. D. S., Heltemes-Harris, L. M., Willette, M. J. L., Henzler, C. M., Frietze, S., Yang, R. D., et al. (2017). Antagonism of B cell enhancer networks by STAT5 drives leukemia and poor patient survival. Nat. Immunol. 18, 694–704. doi:10.1038/ni.3716
Kayser, S., and Levis, M. J. (2023). The clinical impact of the molecular landscape of acute myeloid leukemia. Haematologica 108, 308–320. doi:10.3324/haematol.2022.280801
Khan, A., Mathelier, A., and Zhang, X. G. (2018). Super-enhancers are transcriptionally more active and cell type-specific than stretch enhancers. Epigenetics-Us 13, 910–922. doi:10.1080/15592294.2018.1514231
Kim, T. K., Hemberg, M., Gray, J. M., Costa, A. M., Bear, D. M., Wu, J., et al. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187. doi:10.1038/nature09033
Kobayashi, Y., Ando, K., Imaizumi, Y., Sakamoto, H., Kitanosono, H., Taguchi, M., et al. (2024). RUNX1 expression is regulated by a super-enhancer and is a therapeutic target in adult T-cell leukemia/lymphoma. Leuk. Lymphoma 65, 2116–2128. doi:10.1080/10428194.2024.2393258
Koh, C. M., Bezzi, M., Low, D. H. P., Ang, W. X., Teo, S. X., Gay, F. P. H., et al. (2015). MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 523, 96–100. doi:10.1038/nature14351
Leung, N., and Rajkumar, S. V. (2023). Multiple myeloma with acute light chain cast nephropathy. Blood Cancer J. 13, 46. doi:10.1038/s41408-023-00806-w
Li, J., Galbo, P. M., Gong, W. D., Storey, A. J., Tsai, Y. H., Yu, X. F., et al. (2021). ZMYND11-MBTD1 induces leukemogenesis through hijacking NuA4/TIP60 acetyltransferase complex and a PWWP-mediated chromatin association mechanism. Nat. Commun. 12, 1045. doi:10.1038/s41467-021-21357-3
Li, D. F., Shi, X., Yang, Y. Q., Deng, Y., Chen, D. D., Chen, S. Y., et al. (2025). Targeting BRD4 ameliorates experimental emphysema by disrupting super-enhancer in polarized alveolar macrophage. Resp. Res. 26, 46. doi:10.1186/s12931-025-03120-0
Liau, W. S., Ngoc, P. C. T., and Sanda, T. (2017). Roles of the RUNX1 enhancer in normal hematopoiesis and leukemogenesis. Adv. Exp. Med. Biol. 962, 139–147. doi:10.1007/978-981-10-3233-2_10
Liu, B. B., Liu, X. H., Han, L. L., Chen, X., Wu, X. D., Wu, J. J., et al. (2022). BRD4-directed super-enhancer organization of transcription repression programs links to chemotherapeutic efficacy in breast cancer. P Natl. Acad. Sci. U. S. A. 119, e2109133119. doi:10.1073/pnas.2109133119
Lovén, J., Hoke, H. A., Lin, C. Y., Lau, A., Orlando, D. A., Vakoc, C. R., et al. (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. doi:10.1016/j.cell.2013.03.036
Luo, H., Li, C., O'Neal, J., Kreisel, F., Le Beau, M. M., and Tomasson, M. H. (2005). c-Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood 106, 2452–2461. doi:10.1182/blood-2005-02-0734
Malard, F., and Mohty, M. (2020). Acute lymphoblastic leukaemia. Lancet 395, 1146–1162. doi:10.1016/s0140-6736(19)33018-1
Michida, H., Imoto, H., Shinohara, H., Yumoto, N., Seki, M., Umeda, M., et al. (2020). The number of transcription factors at an enhancer determines switch-like gene expression. Cell Rep. 31, 107724. doi:10.1016/j.celrep.2020.107724
Minzel, W., Venkatachalam, A., Fink, A., Hung, E., Brachya, G., Burstain, I., et al. (2018). Small molecules Co-targeting CKIα and the transcriptional kinases CDK7/9 control AML in preclinical models. Cell 175, 171–185.e25. doi:10.1016/j.cell.2018.07.045
Morrison, T. A., Vigee, J., Tovar, K. A., Talley, T. A., Mujal, A. M., Kono, M., et al. (2025). Selective requirement of glycosphingolipid synthesis for natural killer and cytotoxic T cells. Cell 188, 3497–3512.e16. doi:10.1016/j.cell.2025.04.007
Mousavi, K., Zare, H., Dell'Orso, S., Grontved, L., Gutierrez-Cruz, G., Derfoul, A., et al. (2013). eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol. Cell 51, 606–617. doi:10.1016/j.molcel.2013.07.022
Mullighan, C. G. (2011). New strategies in acute lymphoblastic leukemia: translating advances in genomics into clinical practice. Clin. Cancer Res. 17, 396–400. doi:10.1158/1078-0432.CCR-10-1203
Nakagawa, M., Shaffer, A. L., Ceribelli, M., Zhang, M. L., Wright, G., Huang, D. W., et al. (2018). Targeting the HTLV-I-Regulated BATF3/IRF4 transcriptional network in adult T cell Leukemia/Lymphoma. Cancer Cell 34, 286–297.e10. doi:10.1016/j.ccell.2018.06.014
Ng, C. E. L., Yokomizo, T., Yamashita, N., Cirovic, B., Jin, H., Wen, Z. L., et al. (2010). A Runx1 intronic enhancer marks hemogenic endothelial cells and hematopoietic stem cells. Stem Cells 28, 1869–1881. doi:10.1002/stem.507
Nicosia, L., Spencer, G. J., Brooks, N., Amaral, F. M. R., Basma, N. J., Chadwick, J. A., et al. (2023). Therapeutic targeting of EP300/CBP by bromodomain inhibition in hematologic malignancies. Cancer Cell 41, 2136–2153.e13. doi:10.1016/j.ccell.2023.11.001
Nott, A., Holtman, I. R., Coufal, N. G., Schlachetzki, J. C. M., Yu, M., Hu, R., et al. (2019). Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139. doi:10.1126/science.aay0793
Noura, M., Matsuo, H., Yasuda, T., Tsuzuki, S., Kiyoi, H., and Hayakawa, F. (2024). Suppression of super-enhancer-driven TAL1 expression by KLF4 in T-cell acute lymphoblastic leukemia. Oncogene 43, 447–456. doi:10.1038/s41388-023-02913-1
Ong, J. Z., Yokomori, R., Wong, R. W. J., Tan, T. K., Ueda, R., Ishida, T., et al. (2022). Requirement for TP73 and genetic alterations originating from its intragenic super-enhancer in adult T-cell leukemia. Leukemia 36, 2293–2305. doi:10.1038/s41375-022-01655-5
Orom, U. A., Derrien, T., Beringer, M., Gumireddy, K., Gardini, A., Bussotti, G., et al. (2010). Long noncoding RNAs with enhancer-like function in human cells. Cell 143, 46–58. doi:10.1016/j.cell.2010.09.001
Ottema, S., Mulet-Lazaro, R., Erpelinck-Verschueren, C., van Herk, S., Havermans, M., Varea, A. A., et al. (2021). The leukemic oncogene EVI1 hijacks a MYC super-enhancer by CTCF-facilitated loops. Nat. Commun. 12, 5679. doi:10.1038/s41467-021-25862-3
Pan, F., Iwasaki, M., Wu, W. Q., Jiang, Y. A., Yang, X., Zhu, L., et al. (2023). Enhancer remodeling drives MLL oncogene-dependent transcriptional dysregulation in leukemia stem cells. Blood Adv. 7, 2504–2519. doi:10.1182/bloodadvances.2022008787
Pasquer, H., Tostain, M., Kaci, N., Roux, B., and Benajiba, L. (2021). Descriptive and functional genomics in Acute Myeloid Leukemia (AML): paving the road for a cure. Cancers 13, 748. doi:10.3390/cancers13040748
Pouzolles, M., Oburoglu, L., Taylor, N., and Zimmermann, V. S. (2016). Hematopoietic stem cell lineage specification. Curr. Opin. Hematol. 23, 311–317. doi:10.1097/MOH.0000000000000260
Raca, G., Abdel-Azim, H., Yue, F., Broach, J., Payne, J. L., Reeves, M. E., et al. (2021). Increased incidence of IKZF1 deletions and IGH-CRLF2 translocations in B-ALL of Hispanic/Latino children-a novel health disparity. Leukemia 35, 2399–2402. doi:10.1038/s41375-021-01133-4
Récher, C. (2021). Clinical implications of inflammation in Acute Myeloid leukemia. Front. Oncol. 11, 623952. doi:10.3389/fonc.2021.623952
Roe, J. S., Mercan, F., Rivera, K., Pappin, D. J., and Vakoc, C. R. (2015). BET bromodomain inhibition suppresses the function of hematopoietic transcription factors in acute Myeloid leukemia. Mol. Cell 58, 1028–1039. doi:10.1016/j.molcel.2015.04.011
Romano, O., Petiti, L., Felix, T., Meneghini, V., Portafax, M., Antoniani, C., et al. (2020). GATA factor-mediated gene regulation in human erythropoiesis. Iscience 23, 101018. doi:10.1016/j.isci.2020.101018
Saintamand, A., Vincent-Fabert, C., Marquet, M., Ghazzaui, N., Magnone, V., Pinaud, E., et al. (2017). Eμ and 3'RR IgH enhancers show hierarchic unilateral dependence in mature B-cells. Sci. Rep-Uk 7, 442. doi:10.1038/s41598-017-00575-0
Sakamoto, H., Ando, K., Imaizumi, Y., Mishima, H., Kinoshita, A., Kobayashi, Y., et al. (2022). Alvocidib inhibits IRF4 expression via super-enhancer suppression and adult T-cell leukemia/lymphoma cell growth. Cancer Sci. 113, 4092–4103. doi:10.1111/cas.15550
Sang, X., Zhang, Y. P., Fang, F., Gao, L., Tao, Y. F., Li, X. L., et al. (2022). BRD4 inhibitor GNE-987 exerts anticancer effects by targeting super-enhancer-related gene LYL1 in acute Myeloid leukemia. J. Immunol. Res. 2022, 7912484. doi:10.1155/2022/7912484
Satpathy, A. T., Granja, J. M., Yost, K. E., Qi, Y. Y., Meschi, F., McDermott, G. P., et al. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 37, 925–936. doi:10.1038/s41587-019-0206-z
Schaukowitch, K., Joo, J. Y., Liu, X. H., Watts, J. K., Martinez, C., and Kim, T. K. (2014). Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell 56, 29–42. doi:10.1016/j.molcel.2014.08.023
Sekeres, M. A., and Taylor, J. (2022). Diagnosis and treatment of myelodysplastic syndromes A review. Jama-J Am. Med. Assoc. 328, 872–880. doi:10.1001/jama.2022.14578
Shah, S., Schrader, K. A., Waanders, E., Timms, A. E., Vijai, J., Miething, C., et al. (2013). A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat. Genet. 45, 1226–1231. doi:10.1038/ng.2754
Shi, J., Whyte, W. A., Zepeda-Mendoza, C. J., Milazzo, J. P., Shen, C., Roe, J. S., et al. (2013). Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Gene Dev. 27, 2648–2662. doi:10.1101/gad.232710.113
Shin, H. Y. (2018). Targeting super-enhancers for disease treatment and diagnosis. Mol. Cells 41, 506–514. doi:10.14348/molcells.2018.2297
Smith, C., Touzart, A., Simonin, M., Tran-Quang, C., Hypolite, G., Latiri, M., et al. (2023). Harnessing the MYB-dependent TAL1 5'super-enhancer for targeted therapy in T-ALL. Mol. Cancer 22, 12. doi:10.1186/s12943-022-01701-x
Sugino, N., Kawahara, M., Tatsumi, G., Kanai, A., Matsui, H., Yamamoto, R., et al. (2017). A novel LSD1 inhibitor NCD38 ameliorates MDS-related leukemia with complex karyotype by attenuating leukemia programs via activating super-enhancers. Leukemia 31, 2303–2314. doi:10.1038/leu.2017.59
Tanwar, V. S., Reddy, M. A., Dey, S., Malek, V., Lanting, L., Chen, Z., et al. (2025). Palmitic acid alters enhancers/super-enhancers near inflammatory and efferocytosis-associated genes in human monocytes. J. Lipid Res. 66, 100774. doi:10.1016/j.jlr.2025.100774
Thieme, E., Bruss, N., Sun, D. C., Dominguez, E. C., Coleman, D., Liu, T. T., et al. (2023). CDK9 inhibition induces epigenetic reprogramming revealing strategies to circumvent resistance in lymphoma. Mol. Cancer 22, 64. doi:10.1186/s12943-023-01762-6
Vahedi, G., Kanno, Y., Furumoto, Y., Jiang, K., Parker, S. C. J., Erdos, M. R., et al. (2015). Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature 520, 558–562. doi:10.1038/nature14154
van Groningen, T., Koster, J., Valentijn, L. J., Zwijnenburg, D. A., Akogul, N., Hasselt, N. E., et al. (2017). Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 49, 1261–1266. doi:10.1038/ng.3899
Walker, M., Li, Y. C., Morales-Hernandez, A., Qi, Q., Parupalli, C., Brown, S., et al. (2023). An NFIX-mediated regulatory network governs the balance of hematopoietic stem and progenitor cells during hematopoiesis. Blood Adv. 7, 4677–4689. doi:10.1182/bloodadvances.2022007811
Wan, X. M., Wang, J. W., Fang, F., Hu, Y. X., Zhang, Z. M., Tao, Y. F., et al. (2024). Super enhancer related gene ANP32B promotes the proliferation of acute myeloid leukemia by enhancing MYC through histone acetylation. Cancer Cell Int. 24, 81. doi:10.1186/s12935-024-03271-y
Wang, M., Chen, Q., Wang, S., Xie, H., Liu, J., Huang, R., et al. (2023). Super-enhancers complexes zoom in transcription in cancer. J. Exp. Clin. Cancer Res. 42, 183. doi:10.1186/s13046-023-02763-5
Wang, L. S., Yuan, W., Gamliel, A., Ma, W. B., Lee, S. W., Tan, Y. L., et al. (2025). An eRNA transcription checkpoint for diverse signal-dependent enhancer activation programs. Nat. Genet. 57, 962–972. doi:10.1038/s41588-025-02138-w
Wei, C. J., Yu, P., and Cheng, L. (2020). Hematopoietic reprogramming entangles with hematopoiesis. Trends Cell Biol. 30, 752–763. doi:10.1016/j.tcb.2020.07.006
Welsh, S. J., Barwick, B. G., Meermeier, E. W., Riggs, D. L., Shi, C. X., Zhu, Y. X., et al. (2024). Transcriptional heterogeneity overcomes super-enhancer disrupting drug combinations in multiple myeloma. Blood Cancer Discov. 5, 34–55. doi:10.1158/2643-3230.BCD-23-0062
Whyte, W. A., Orlando, D. A., Hnisz, D., Abraham, B. J., Lin, C. Y., Kagey, M. H., et al. (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. doi:10.1016/j.cell.2013.03.035
Wibisana, J. N., Inaba, T., Shinohara, H., Yumoto, N., Hayashi, T., Umeda, M., et al. (2022). Enhanced transcriptional heterogeneity mediated by NF-κB super-enhancers. Plos Genet. 18, e1010235. doi:10.1371/journal.pgen.1010235
Witte, S., O'Shea, J. J., and Vahedi, G. (2015). Super-enhancers: asset management in immune cell genomes. Trends Immunol. 36, 519–526. doi:10.1016/j.it.2015.07.005
Wong, R. W. J., Ngoc, P. C. T., Leong, W. Z., Yam, A. W. Y., Zhang, T. H., Asamitsu, K., et al. (2017). Enhancer profiling identifies critical cancer genes and characterizes cell identity in adult T-cell leukemia. Blood 130, 2326–2338. doi:10.1182/blood-2017-06-792184
Wong, R. W. J., Tan, T. K., Amanda, S., Ngoc, P. C. T., Leong, W. Z., Tan, S. H., et al. (2020). Feed-forward regulatory loop driven by IRF4 and NF-κB in adult T-cell leukemia/lymphoma. Blood 135, 934–947. doi:10.1182/blood.2019002639
Xie, B. N., and Dean, A. (2025). A Super enhancer-derived enhancer RNA acts together with CTCF/Cohesin in trans to regulate erythropoiesis. Genes-Basel 16, 389. doi:10.3390/genes16040389
Xie, F., Zhang, T. J., Zhang, X. L., Xu, Z. J., Qiao, L., Wang, Y., et al. (2025). Identification of HOXA9 methylation as an epigenetic biomarker predicting prognosis and guiding treatment choice in acute myeloid leukemia. Bmc Cancer 25, 215. doi:10.1186/s12885-025-13633-y
Yoshino, S., Yokoyama, T., Sunami, Y., Takahara, T., Nakamura, A., Yamazaki, Y., et al. (2021). Trib1 promotes acute myeloid leukemia progression by modulating the transcriptional programs of Hoxa9. Blood 137, 75–88. doi:10.1182/blood.2019004586
Yu, J. J., Yang, Y., Zhou, R. F., Tao, Y. F., Zhu, F., Jiao, W. Y., et al. (2024). The BET inhibitor GNE-987 effectively induces anti-cancer effects in T-cell acute lymphoblastic leukemia by targeting enhancer regulated genes. Carcinogenesis 45, 424–435. doi:10.1093/carcin/bgae006
Yu, J. J., Zhang, Z. M., Chen, Y. L., Wang, J. W., Li, G., Tao, Y. F., et al. (2025). Super-Enhancer-Driven IRF2BP2 is activated by master transcription factors and sustains T-ALL cell growth and survival. Adv. Sci. 12, e2407113. doi:10.1002/advs.202407113
Zhang, L., Cai, T. Y., Lin, X. Y., Huang, X. L., Bui, M. H., Plotnik, J. P., et al. (2021). Selective inhibition of the second bromodomain of BET family proteins results in robust antitumor activity in preclinical models of acute Myeloid leukemia. Mol. Cancer Ther. 20, 1809–1819. doi:10.1158/1535-7163.MCT-21-0029
Zhang, K. L., Lu, J., Fang, F., Zhang, Y. P., Yu, J. J., Tao, Y. F., et al. (2023). Super enhancer regulatory gene FYB1 promotes the progression of T cell acute lymphoblastic leukemia by activating IGLL1. J. Immunol. Res. 2023, 3804605. doi:10.1155/2023/3804605
Zheng, Z. Z., Xia, L., Hu, G. S., Liu, J. Y., Hu, Y. H., Chen, Y. J., et al. (2022). Super-enhancer-controlled positive feedback loop BRD4/ERα-RET-ERα promotes ERα-positive breast cancer. Nucleic Acids Res. 50, 10230–10248. doi:10.1093/nar/gkac778
Zhou, X. Y., and Lu, R. (2023). HOXA9/MEIS1 targets in leukemia: reinforced signaling networks and therapeutic opportunities. Haematologica 108, 1205–1207. doi:10.3324/haematol.2022.281779
Zhou, H. F., Schmidt, S. C. S., Jiang, S. Z., Willox, B., Bernhardt, K., Liang, J., et al. (2015). Epstein-Barr virus oncoprotein super-enhancers control B cell growth. Cell Host Microbe 17, 205–216. doi:10.1016/j.chom.2014.12.013
Zhou, X. J., Zhou, X. Q., Li, J., He, Y. Z., Qiu, S. Z., Xu, Y., et al. (2025). Bclaf1 mediates super-enhancer-driven activation of POLR2A to enhance chromatin accessibility in nitrosamine-induced esophageal carcinogenesis. J. Hazard Mater 492, 138218. doi:10.1016/j.jhazmat.2025.138218
Glossary
Aire Autoimmune regulator
ALL Acute lymphoblastic leukemia
AML Acute myeloid leukemia
ATAC-seq Assay for Transposase-Accessible Chromatin using sequencing
ATL Adult T-cell leukemia/lymphoma
B-ALL B-cell acute lymphoblastic leukemia
BET Bromodomain and extra-terminal domain
BIRC3 Baculoviral IAP repeat containing 3
BRD4 Bromodomain-containing protein 4
C/EBPα CCAAT/enhancer binding protein alpha
CBP CREB-binding protein
CCR4 C-C motif chemokine receptor 4
CDK9 Cyclin-dependent kinase 9
CDKs Cyclin-dependent kinases
ChIA-PET Chromatin Interaction Analysis by Paired-End Tag sequencing
ChIP-seq Chromatin Immunoprecipitation sequencing
Cpox-eRNA Coproporphyrinogen oxidase enhancer RNA
CRLF2 Cytokine receptor-like factor 2
DHE Dihydroergotamine
DNase-seq DNase I hypersensitive site sequencing
E2F E2F transcription factor
Epo Erythropoietin
ERG V-ets erythroblastosis virus E26 oncogene homolog
eRNAs Enhancer-derived RNAs
FLI1 Friend leukemia integration 1 transcription factor
FYB1 FYN binding protein 1
GATA2 GATA binding protein 2
GFI1 Growth factor independent 1 transcriptional repressor
GNE-987 A BRD4 inhibitor (no full name specified in manuscript)
GRO-seq Global Run-On Sequencing
H3K4me1 Histone H3 lysine 4 mono-methylation
H3K27ac Histone H3 lysine 27 acetylation
Hi-C High-throughput Chromosome Conformation Capture
HJURP Holliday junction recognition protein
HOXA Homeobox A
HOXA9 Homeobox A9
HSCs Hematopoietic stem cells
HTLV-I Human T-cell leukemia virus type I
IgH Immunoglobulin heavy chain
IKZF1 Ikaros family zinc finger 1
IKZF3 Ikaros family zinc finger 3
IMiD Immunomodulatory imide drugs
IRF2BP2 Interferon regulatory factor 2 binding protein 2
IRF4 Interferon regulatory factor 4
ITGB7 Integrin beta 7
JAK2 Janus kinase 2
LCK Lymphocyte-specific protein tyrosine kinase
LDB1 LIM domain binding 1
LMO2 LIM domain only 2
lncRNAs Long non-coding RNAs
LSD1 Lysine-specific demethylase 1
LYL1 LYL1 basic helix-loop-helix transcription factor
MDS Myelodysplastic neoplasms
MED1 Mediator complex subunit 1
MEIS1 Meis homeobox 1
MGUS Monoclonal gammopathy of unknown significance
MM Multiple myeloma
MYB V-myb myeloblastosis viral oncogene homolog
MYC myelocytomatosis oncogene
NCD38 A novel LSD1 inhibitor (no full name specified in manuscript)
NFIX Nuclear factor I X
NF-κB Nuclear factor kappa B
NK cell Natural killer cell
NSD2 Nuclear receptor binding SET domain protein 2
NuA4/Tip60 Nucleosome acetyltransferase of H4/Tat-interactive protein 60 kDa
p300/CBP E1A-binding protein p300/CREB-binding protein
PAX5 Paired box 5
PIK3CD Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit delta
PIK3R1 Phosphoinositide-3-kinase regulatory subunit 1
PLAC-seq Proximity Ligation-Assisted ChIP-seq
PRO-seq Precision Run-On Sequencing
PU.1 Putative oncogene PU.1
RGL1 Ral guanine nucleotide dissociation stimulator-like 1
RNAP II RNA polymerase II
RUNX1 Runt-related transcription factor 1
seRNAs Super enhancer-derived RNAs
SEs Super enhancers
SLC25A37 Solute carrier family 25 member 37
SLC4A1/BAND3 Solute carrier family 4 member 1/Band 3
SMARCA4 SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 4
SOX4 SRY-box transcription factor 4
SPNS2 Spinster homolog 2
STAT5 Signal transducer and activator of transcription 5
TAL1/SCL T-cell acute lymphocytic leukemia protein 1/stem cell leukemia
T-ALL T-cell acute lymphoblastic leukemia
THZ1 A CDK7 inhibitor (no full name specified in manuscript)
TLR4 Toll-like receptor 4
TP53 Tumor protein p53
TP73 Tumor protein p73
TRIB1 Tribbles pseudokinase 1
ZMYND11-MBTD1 Zinc finger MYND-type containing 11-MBT domain containing 1 fusion protein
ZMYND8 Zinc finger MYND-type containing 8
Keywords: super enhancers, gene expression regulation, hematopoiesis, hematologicalmalignancies, oncogene regulation
Citation: Cheng Y, Pei G, Zhang H, Hou Y, Sun L, Xu H, Lv Y and Wu X (2025) Super enhancers as key drivers of gene regulatory networks in normal and malignant hematopoiesis. Front. Cell Dev. Biol. 13:1674470. doi: 10.3389/fcell.2025.1674470
Received: 28 July 2025; Accepted: 29 October 2025;
Published: 14 November 2025.
Edited by:
Liliana Burlibasa, University of Bucharest, RomaniaReviewed by:
Gratiela Gradisteanu Pircalabioru, University of Bucharest, RomaniaPraveen Kumar, University of Virginia, United States
Copyright © 2025 Cheng, Pei, Zhang, Hou, Sun, Xu, Lv and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiuyun Wu, d3V4eUB6enUuZWR1LmNu