 Xia Huang
Xia Huang Yujie Wang
Yujie Wang Qiushuang Li
Qiushuang Li Xinyi Li
Xinyi Li Congcong Wang
Congcong Wang- College of Life Science and Health, Wuhan University of Science and Technology, Wuhan, Hubei, China
Stem cells are undifferentiated cells characterized by their self-renewal capacity and pluripotency. The multipotent differentiation potential of stem cells grants them significant promise in clinical therapies for tissue injury and organ regeneration. Therefore, the molecular mechanisms underlying the maintenance of stem cell self-renewal and pluripotency have been a major focus of research in the field. In recent years, increasing evidence suggests that cell cycle is not only a central driver of cell division but also participate in controlling stem cell self-renewal and differentiation fate through various pathways. Stem cells, especially embryonic stem cells (ESCs), exhibit unique cell cycle features, with a notably short overall cycle duration, a significantly shortened G1 phase, and a prolonged S phase. This rapid cell cycle not only results in increased cell numbers but is also closely associated with the maintenance of their self-renewal capacity. Pluripotency states (such as naïve, formative, and primed) are tightly linked to specific cell cycle patterns, and this association exhibits species specificity. Elucidating the molecular mechanisms coupling the cell cycle with stemness maintenance is of great significance for the clinical application of stem cells. This review focuses on the cell cycle regulatory network centered around Cyclins and their inhibitors in stem cells, as well as the molecular mechanisms by which core pluripotency factors and cell cycle proteins influence stem cell fate determination. We discuss signaling pathways such as Jak1/Stat3, PI3K/Akt, and Hippo/YAP, and the role of epigenetic regulation, particularly histone modifications, in modulating the expression of differentiation-related and cell cycle-associated genes. Additionally, a brief overview is provided of the unique glycolytic metabolic mode and one-carbon metabolism in stem cells, along with their relationship with epigenetic modifications and rapid proliferative characteristics. Moreover, we analyze the regulatory functions of cell cycle regulators such as Cyclins and checkpoint protein p53 in somatic cell reprogramming and the fate determination of adult stem cells including neural and hematopoietic stem cells (HSCs). Practical strategies based on cell cycle regulation are discussed, along with prospects and challenges for their applications in regenerative medicine.
1 Introduction
Embryonic stem cells (ESCs), primarily derived from the inner cell mass of blastocysts, are a population of undifferentiated cells with the capacity to unlimited proliferation, self-renewal, and maintenance of pluripotency (Evans and Kaufman, 1981; Martin, 1981). Pluripotency refers to the ability to differentiate into the three germ layers: ectoderm, mesoderm, and endoderm (Lu et al., 2001). Previous studies have shown that ESC pluripotency is regulated by core pluripotency factors such as Sox2, Oct4, and Nanog, which maintain the undifferentiated state of ESCs, and suppress the expression of differentiation genes (Boyer et al., 2005; Hall et al., 2009; Mitsui et al., 2003; Surani et al., 2007). Since the establishment of the first mouse ESC (mESC) line, significant breakthroughs have been achieved in ESC-related research (Evans and Kaufman, 1981; Martin, 1981). More recently, the mechanisms by which ESCs couple cell cycle regulation to maintain pluripotency have been progressively elucidated (Chaigne et al., 2020).
Many studies in the ESC field have confirmed that the pluripotent state of ESCs is associated with a specific cell cycle profile (Coronado et al., 2013; Pauklin and Vallier, 2013). The cell cycle is divided into four phases: G1 (pre-DNA synthesis), S (DNA synthesis), G2 (post-DNA synthesis), and M (mitosis). Its progression is primarily regulated by a set of proteins centered on Cyclins, including Cyclin-dependent kinases (CDKs) and Cyclin-dependent kinase inhibitors (CKIs). The cell cycle relies on two peaks of gene expression: during the G1/S transition and the G2/M transition. During G1, growth factors induce the expression of CyclinD (D1, D2, or D3), which binds to CDK4/6 to form active complexes, initiating the sequential phosphorylation of the retinoblastoma protein (RB) (Hydbring et al., 2016). This process partially relieves RB’s inhibition of E2F transcription factors, promoting the expression of key G1/S transition genes such as CyclinE (Liu et al., 2019). After the restriction point, the CyclinE-CDK2 complex further hyperphosphorylates RB via positive feedback, fully activating E2F and phosphorylating replication initiation factors such as CDC6 to facilitate pre-replication complex assembly, preparing for DNA synthesis (Narasimha et al., 2014). Upon entry into S phase, the Cyclin A-CDK2 complex supersedes Cyclin E-CDK2, ensuring the timely initiation and progression of DNA replication (Coverley et al., 2002). In G2 phase, CyclinA levels decline, and the activity of CyclinB-CDK1 is regulated by CDC25 phosphatases, forming a molecular switch that governs the transition from G2 to M phase. During the G2/M transition, CDK1 binds to CyclinA or B, with these complexes being essential for proper mitotic entry (Errico et al., 2010).
Cyclins and CDKs, as the primary executors of cell cycle regulation, are negatively modulated by two major CKI families: INK4 and Cip/Kip. The INK4 family mainly includes p16, p15, p18, and p19, which selectively inhibit the CDK4/CDK6 kinase complexes in the G1 phase by directly binding to CDK4/6 and preventing their association with and activation by D-type Cyclins, thereby maintaining the Rb in a hypo-phosphorylated, active state (Serrano et al., 1993; Sherr and Roberts, 1999). The Cip/Kip family includes p21, p27, and p57, which have a broader inhibitory spectrum, capable of suppressing multiple Cyclin-CDK complexes in G1 and S phases, and exhibit a dual regulatory role on the assembly of Cyclin-CDK complexes at different concentrations, either promoting or inhibiting their formation (Besson et al., 2008; Sherr and Roberts, 1999). In naïve-state ESCs, the expression levels of Cip/Kip family members (especially p21) are typically low, which helps sustain high CDK2 activity and ensure rapid passage through G1, thereby supporting self-renewal and rapid proliferation (Neganova and Lako, 2008). Expression of p57 is essential for genomic imprinting and normal development (Matsumoto et al., 2011). As ESCs initiate differentiation, expression of p21 and p27 is markedly upregulated. At this stage, they inhibit the activity of the Cyclin E-CDK2 complex, forcing cells out of the high-proliferation cycle, lengthening G1, and promoting differentiation (DeVeale et al., 2022). p57 is a core molecule for maintaining the quiescent state of hematopoietic stem cells (HSCs) and neural stem cells (NSCs) (Furutachi et al., 2013; Zou et al., 2011). In quiescent stem cells, p27 protein levels are high; by inhibiting the Cyclin E-CDK2 complex, they effectively lock cells in G0/G1 to prevent excessive proliferation and attrition. Upon receiving activating signals, p27 is degraded via phosphorylation and ubiquitination pathways, relieving inhibition on CDK2 and allowing cells to re-enter the proliferative cycle (Besson et al., 2006; Doetsch et al., 2002). p16 plays a dominant role in reinforcing quiescence and aging-related proliferative blockades. In young adult stem cells (ASCs), p16 expression is relatively low; however, with aging or persistent stress, its expression rises significantly (Molofsky et al., 2006). High levels of p16 inhibit CDK4/6, augment RB-mediated cell-cycle arrest, and lock stem cells into a deeper, more irreversible quiescent state, potentially leading to senescence (D'Arcangelo et al., 2017). In ASCs such as HSCs and NSCs, high levels of CKIs constitute a key mechanism for actively maintaining quiescence, functioning as reversible “molecular brakes” that keep stem cells in a standby state, with INK4 family members expressed at comparatively lower levels. When stem cells are activated and enter division, if their progeny decide to proceed toward terminal differentiation, CKIs will be upregulated again, leading to an irreversible exit from the cell cycle and promotion of differentiation.
Some scholars believe that because stem cells exhibit high expression of lineage-specific genes during G1, the prolongation of G1 phase makes them more susceptible to differentiation signals, thus G1 is considered a “sensitive period” for differentiation (Chaigne et al., 2020; Coronado et al., 2013; Dalton, 2013; Dalton, 2015; Pauklin and Vallier, 2013; Pauklin and Vallier, 2014; Singh et al., 2014). Compared with somatic cells, ESCs demonstrate rapid proliferation, with a markedly shortened cell cycle, predominantly characterized by a significantly reduced G1 phase. In typical ESC populations, S phase cells can account for 60%–70%, while G1 phase cells occupy only 15%–20%, a stark contrast to the G1-dominant cycle of somatic cells (Ter Huurne and Stunnenberg, 2021). Research indicates that the abbreviated G1 phase in ESCs is regulated in part by the interplay between the classical oncogenes MEK1/2 and the tumor suppressor gene TP53, which collectively drive the G1/S transition (Jiang et al., 2022). This distinctive cell cycle pattern is considered a fundamental basis for maintaining the undifferentiated state of ESCs. At the same time, this pattern also ensures rapid cell proliferation during early embryonic development (White and Dalton, 2005). During mammalian embryogenesis, pluripotent cells initially undergo rapid division phases from the preimplantation to the early postimplantation stages (Stead et al., 2002). This distinctive mode of cell division effectively promotes the rapid expansion of pluripotent stem cell (PSC) populations before gastrulation. This conserved regulation of the cell cycle has been confirmed across various model organisms, including fruit flies (Edgar and Lehner, 1996), zebrafish (Yarden and Geiger, 1996), and African clawed frogs (Murray and Kirschner, 1989), in the context of unipotent cells transitioning to a differentiated state, with marked changes in proliferation rates. These findings thoroughly demonstrate that the mechanisms governing cell cycle regulation are highly conserved evolutionarily and play a core role in determining cell fate and maintaining cell characteristics (Boward et al., 2016; Liu et al., 2019).
PSCs represent a heterogeneous population comprising a spectrum of functional states. These states are characterized by distinct developmental potentials, metabolic profiles, epigenetic configurations, and signaling pathway dependencies, and are broadly classified into naïve, formative, and primed pluripotency states (Table 1). A deep understanding of the cell cycle characteristics of these states is crucial for elucidating mechanisms of pluripotency maintenance and exit. Cell cycle dynamics are not only a passive readout of these states but also an active regulator of their maintenance and transitions. Notably, the key properties of these states show important differences between mouse and human cells.
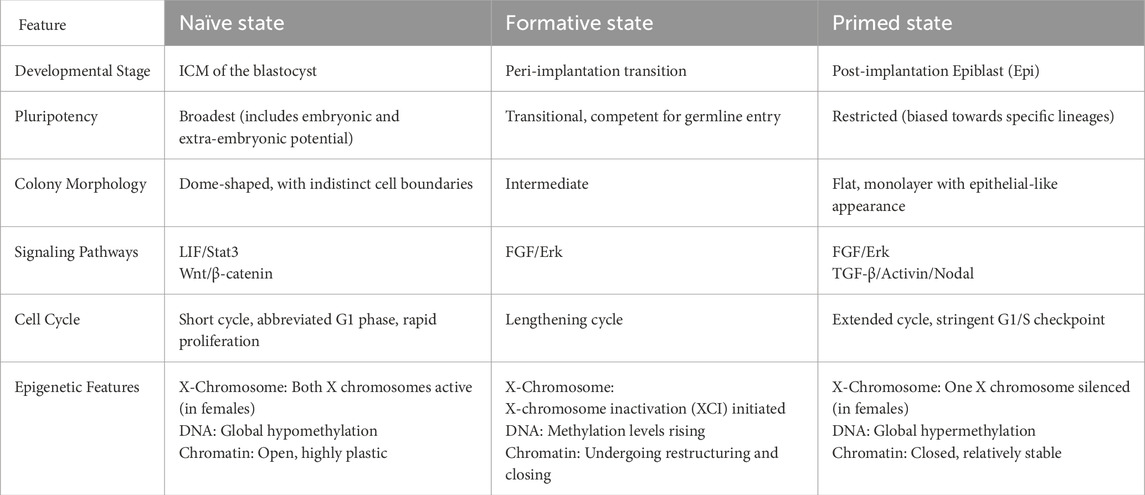
Table 1. Characteristics of ESCs in different states.
Naïve pluripotency corresponds to the pre-implantation ICM or the epiblast of the mouse embryo. This state exhibits the broadest developmental potential, enabling differentiation into all embryonic and extraembryonic lineages and facilitating efficient chimera formation. A hallmark of naïve pluripotent cells is their distinct cell cycle structure, characterized by a shortened G1 phase and an extended S phase, resulting in a total cell cycle length of approximately 12–14 h. This unique cycle architecture is driven by high activity of CDK2, Cyclin A, and Cyclin B, accompanied by silenced expression of G1-phase CDK inhibitors such as p21 and p27, resulting in a hyperphosphorylated, inactive Rb protein. This configuration is considered favorable for rapid proliferation and minimizes the time for external differentiation signals to influence cells in G1, thereby passively maintaining pluripotency. Naïve-state cells rely on glycolytic metabolism to supply biosynthetic precursors while maintaining low reactive oxygen species (ROS) levels, aligning with their short cell cycle. Epigenetically, Naïve cells exhibit global DNA hypomethylation and abundant H3K27me3 marks to suppress differentiation programs, with X chromosome reactivation observed in female cells. Maintenance of this state strictly depends on dual inhibition of the LIF/Stat3 pathway and the GSK3β/MEK/Erk pathways (“2i” conditions) (Coronado et al., 2013; Weinberger et al., 2016).
Primed pluripotency is characteristic of the post-implantation mouse epiblast. This state exhibits restricted developmental potential, lacking the ability to differentiate into the trophoblast lineage and displaying increased responsiveness to differentiation signals. Unlike naïve pluripotency, primed pluripotent cells undergo a substantially prolonged cell cycle, often lasting more than 24 h, primarily attributable to an elongated G1 phase. The upregulation of G1-phase CDK inhibitors, diminished CDK2 activity, and reduced Rb phosphorylation collectively reinforce the G1 checkpoint and decelerate cell cycle progression. This extended G1 phase offers a crucial temporal window for the integration of extracellular cues and the initiation of lineage-specific transcriptional programs. Metabolically, primed cells shift from glycolysis toward oxidative phosphorylation (OXPHOS) to generate more ATP to support cellular activities. Epigenetically, DNA methylation is increased, and chromatin adopts a more closed and compact state, with X chromosome inactivation in female cells. Maintenance of the primed state largely depends on activation of the FGF2 and Activin A/Nodal signaling pathways (Nichols and Smith, 2009; Singh et al., 2015).
Formative pluripotency represents a recently defined intermediate state situated between naïve and primed pluripotency, corresponding to the epiblast at implantation onset. Cells in this state have lost key naïve pluripotency features but have not yet fully acquired primed characteristics. Regarded as a transitional phase, formative pluripotency enables cells to respond efficiently to inductive signals. Evidence suggests that these cells display an intermediate cell cycle structure, marked by a significantly lengthened G1 phase, yet the total cycle duration remains shorter than that of primed cells—reflecting their dynamic and metastable nature. Concurrently, their signaling dependencies and epigenetic landscape undergo rapid remodeling in preparation for lineage specification (Smith, 2017).
Notably, interspecies differences are key to understanding pluripotent states. Human PSCs (hPSC) sunder standard culture conditions (using FGF2 and Activin A) are typically in a primed state rather than the naïve state observed in mouse. Even under optimized “naïve” culture conditions, the G1 phase of hPSCs is generally longer than that of mouse naïve ESCs, and their cell cycle architecture more closely resembles mouse primed or formative states. Additionally, in human cells, the p53 and p16/Rb pathways exert a stronger barrier to reprogramming and maintenance of the naïve state than in the mouse, directly affecting cell cycle dynamics and proliferative potential (Table 2). Consequently, findings from mESC research do not fully translate to human systems (Becker et al., 2010; Theunissen et al., 2014).

Table 2. Comparison of characteristics between mouse and human ESCs.
Pluripotent states of stem cells are not static endpoints. Rather, they are a “quasi-steady state” that is dynamically shaped by the cell cycle, and supported by energy metabolism and the epigenetic landscape in a finely balanced state. Their unique rapid proliferation cycle, especially the shortened G1 phase, serves both as the engine that maintains pluripotency and as a source of genomic instability risk. Therefore, one goal of the pluripotency network is to build a coupled regulatory system that integrates the cell cycle, metabolism, epigenetics, and signaling pathways to achieve self-renewal, fate determination, and genomic safeguarding amid rapid proliferation. This article systematically articulates the coupled mechanism between the cell cycle and stemness maintenance in stem cells, analyzes how the cell cycle regulatory network—centered on Cyclins and CDKs—regulates stem cell pluripotency maintenance, and provides a comprehensive discussion of signaling pathways such as LIF/Stat3, epigenetic regulation like histone modifications, metabolism, and somatic cell reprogramming as regulators of the cell cycle.
2 Specific mode of cell cycle control by Cyclins in ESCs
2.1 Crosstalk of cell cycle regulators and pluripotent factors in ESCs
The distinctive cell cycle architecture observed in ESCs results from the synergistic effects of multiple regulatory layers, including transcription factors, epigenetic modifications, signaling pathways, metabolism, and cell cycle regulators (Nardiello et al., 2011). Studies indicate that, unlike highly differentiated cells, ESCs maintain expression levels of cell cycle genes such as CyclinE-CDK2 and CyclinA-CDK2 throughout the cell cycle, with CyclinE-CDK2 playing a crucial role in the G1/S transition, whereas CyclinA-CDK2 and CyclinB-CDK1 drive rapid progression through S phase and G2/M phase, respectively (Figure 1) (Liu et al., 2019). Furthermore, the expression levels of CDK inhibitors are relatively low in ESCs, further relieving the inhibition of CDK activity (Calder et al., 2013; Egozi et al., 2007; Zhu H. et al., 2014). Concurrently, the highly phosphorylated state of RB, leading to a continuous release of its suppression of E2F transcription factors, enabling rapid passage through the G1/S checkpoint (Koledova et al., 2010; Krishnan et al., 2022).
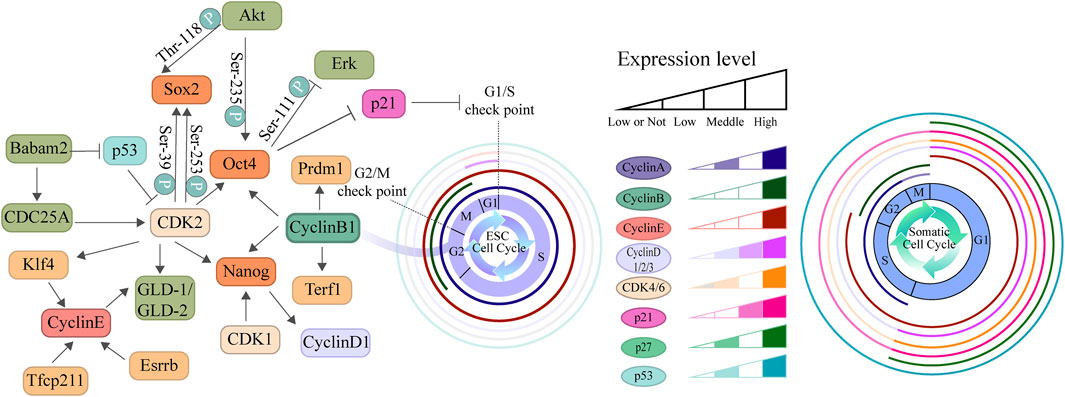
Figure 1. Comparative diagram of cell cycle regulation in ESCs and somatic cells. This diagram illustrates the differences in cell cycle structure and the expression patterns of regulatory factors between ESCs and somatic cell. The left panel depicts the core cell cycle regulatory network centered around cyclins, CKIs, and pluripotency factors. The two cell cycle diagrams on the right represent the typical cell cycle structures of ESCs and somatic cells, respectively, highlighting the significant differences in the duration of the G1, S, G2, and M phases between the two cell types. The relative expression levels of key cell cycle regulatory factors across these phases are indicated by color gradients within concentric rings. The results demonstrate that the enhanced expression of Cyclin-CDK complexes in ESCs, coupled with their abbreviated G1 phase and prolonged S phase, collectively contribute to the maintenance of their pluripotent state.
In recent years, numerous studies have reported the associations between cell cycle regulators and pluripotency control. Knockdown of individual CyclinD isoforms (D1, D2, D3) in human ESCs (hESCs) allows for self-renewal but results in the upregulation of lineage markers, including mesodermal and endodermal genes. Double knockdown of CyclinD promotes spontaneous differentiation toward the endodermal lineage but reduces the capacity for ectodermal differentiation. When all CyclinD isoforms are suppressed, hESCs lose their self-renewal ability and differentiate predominantly into the endoderm (Pauklin and Vallier, 2014). Gonzales et al. found that prolonging the G2 phase specifically upregulates CyclinB1 expression, suggesting that CyclinB1 may be a key factor in main-taining pluripotency during G2 phase. Knockdown of CyclinB1 in hESCs results in a sharp decrease in the expression of pluripotency markers such as Oct4, Nanog, Terf1, and Prdm1, directly confirming its close association with pluripotency. Conversely, overexpression of CyclinB1 can enhance the expression of these markers. These findings collectively indicate that CyclinB1 serves as a central hub connecting G2 phase regulation to the maintenance of pluripotency (Gonzales et al., 2015). Downregulation of CDK2 also reduces the expression of pluripotent factors (Oct4, Sox2, Nanog, Klf4), induces the expression of three germ layer differentiation markers (Cdx2, Pax6, Nestin, Fgr5, Brachyur, Afp), and triggers lineage commitment (Neganova et al., 2009; Spelat et al., 2012). Additional research suggests that Cyclin-CDK complexes prevent ubiquitin-mediated degradation of Sox2, Oct4, and Nanog by promoting their interaction with membrane proteins (Savatier et al., 1994). CDK2-mediated phosphorylation of Sox2 at Ser39 and Ser253 is critical for maintaining its activity (Bar-On et al., 2010). These studies demonstrate that cell cycle mechanisms play a central role in regulating stem cell pluripotency and self-renewal. Similarly, core pluripotency factors are directly involved in cell cycle regulation. To illustrate, Akt-mediated phosphorylation of Sox2 at Thr-118 contributes to the maintenance of the pluripotent state (Jeong et al., 2010). Erk-mediated phosphorylation of Oct4 at Ser-111 promotes its ubiquitination and subsequent degradation (Spelat et al., 2012). Conversely, Akt-mediated phosphorylation of Oct4 at Ser-235 stabilizes the protein and facilitates its interaction with Sox2, thereby supporting stem cell self-renewal (Lin Y. J. et al., 2012). Oct4 can promote the G1/S transition by downregulating p21 expression (Lu Y. et al., 2019), while Nanog is capable of upregulating CyclinD1 expression (Han et al., 2012). The initial pluripotency network transcription factors, including Klf4 and Esrrb, participate in the transcriptional regulation of CyclinE (Gonnot et al., 2019). This bidirectional regulation ensures that ESCs can proliferate rapidly while maintaining their undifferentiated state.
Recent studies have revealed that Cyclins and CDKs actively participate in regulating the pluripotency network through various pathways. CDK1 modulates H3K79me2 methylation by phosphorylating Dot1L, thereby influencing the differentiation tendency of ESCs toward the endoderm (Michowski et al., 2020). Simultaneously, the inhibition of CDK1 activity can activate the p53-Noxa-Mcl1 axis, leading to selective apoptosis of ESCs and further affecting the expression of Oct4 and Nanog, ultimately resulting in stem cell differentiation (Huskey et al., 2015). Additionally, in germline stem cells, CyclinB3 sustains self-renewal through bam-dependent mechanisms, while CyclinE/CDK2 promotes germ cell proliferation via the Gld-1/Gld-2 pathway (Chen D. S. et al., 2018; Fox et al., 2011). These findings suggest that cell cycle proteins exert multi-layered regulatory roles in cell fate determination.
During embryonic germ layer differentiation, the functions of cell cycle proteins are equally crucial. The loss of G1 phase Cyclins weakens the stability of pluripotency factors in ESCs, making them more prone to differentiate into the trophectoderm (TE) lineage (Liu L. J. et al., 2017). Meanwhile, CyclinD promotes neural ectoderm differentiation by binding to regulatory regions of developmental genes and inhibits endodermal gene expression (Pauklin et al., 2016; Pauklin and Vallier, 2013). Furthermore, decreased expression of CDK4 reduces phosphorylation of the Smad-Stat3 axis, favoring the differentiation of mesenchymal stem cells (MSCs) into neuroprogenitors (Kim et al., 2016; Pauklin and Vallier, 2013). Notably, in hESCs, differentiation tendencies vary across different phases of the cell cycle. For instance, the early G1 phase favors endodermal differentiation, whereas the late G1 phase promotes neuroectodermal fate. Additionally, the CyclinD-CDK4/6 complex regulates Activin/Nodal signaling through phosphorylation of Smad2/3, further influencing endodermal differentiation (Pauklin and Vallier, 2013).
Certain cell cycle regulatory complexes are closely associated with the maintenance of stem cell pluripotency. DREAM complex proteins were first discovered in C. elegans, where they primarily maintain normal cellular function by regulating the expression of genes involved in cell proliferation (Mages et al., 2017). DREAM is widely present across multiple species and spans all stages of organismal growth and development; aberrant expression is often closely linked to tumorigenesis. In mammals, the DREAM complex is mainly composed of p107/p130, E2F4/5, DP, and MuvB (Sadasivam and DeCaprio, 2013). This complex is highly conserved evolutionarily. DREM and dREAM complexes in C. elegans and Drosophila are homologs of the DREAM complex in mammals (Schade et al., 2019). The MuvB complex, a key regulator during mitosis, comprises Lin9, Lin37, Lin52, Lin54, and Rbbp4, with Lin54 and Lin52 maintaining the characteristic extended S-phase of ESCs by regulating CyclinB1/CDK1 (Litovchick et al., 2007; Wang C. C. et al., 2022). Notably, the MuvB complex undergoes dynamic remodeling during the cell cycle: at the G1/S transition, it dissociates from the repressive DREAM (Drosophila, RB, E2F, and Myb) complex and associates with B-Myb to form the Myb-MuvB complex, which activates G2/M phase gene expression and suppresses mesoderm/endoderm differentiation markers, thus coupling the cell cycle process with pluripotency maintenance (Guiley et al., 2015; Litovchick et al., 2007). Rbbp4 is not only a major component of the MuvB complex but also serves as an important histone chaperone, playing a key role in maintaining pluripotency in mESCs. Rbbp4 specifically binds to endogenous retrotransposon (TE) elements; on the one hand, it recruits G9a to deposit H3K9me2 modifications at ERVL-class transposons, and on the other hand, it recruits Kap1 to establish H3K9me3 marks at ERVK-class elements. Meanwhile, it cooperates with the chromatin remodeling factor CHD4 to maintain nucleosome occupancy density in heterochromatic regions, collectively repressing the transcriptional activity of totipotency-related genes and transposons. When Rbbp4 function is lost, this heterochromatin barrier is dismantled, leading to aberrant activation of TEs, which drives mESCs to reprogram into totipotent 2C-like cells (2CLCs), accompanied by typical 2CLC phenotypes such as delayed proliferation, increased apoptosis, and G1/S phase arrest. Meanwhile, the repression of key genes in the trophoblast lineage is alleviated, significantly enhancing the potential for differentiation into the outer trophoblast lineage (Ping et al., 2023). The dynamic activation and repression of DREAM subunits directly regulate the activity of cell-cycle proteins. In the G0 quiescent state, p107/p130, DP, E2F, and MuvB interact to suppress the expression of cell-cycle genes (Uxa et al., 2019). After cells receive extracellular growth factor signals and pass the restriction (R) point—the classical checkpoint marking the transition from G1 to S—the expression of genes required for DNA synthesis is activated, the RB-mediated repression of E2F transcription factors is relieved, and Cyclin E and Cyclin D, together with CDKs, can mitigate RB-mediated E2F inhibition; activator E2Fs (E2F1, E2F2, and E2F3) contribute to the expression of early cell-cycle genes during the G1/S transition (Litovchick et al., 2007). Subsequently, MuvB, through interactions with B-myb and FoxM1, induces the expression of late cell-cycle genes. It is the periodic activation and repression of these complex members that ensures the orderly expression of downstream cell-cycle proteins (Fischer and Müller, 2017; Kelleher and O'Sullivan, 2016; Liu et al., 2019; Sadasivam et al., 2012).
Some researchers propose that E2F2 directly binds the B-Myb promoter to promote its transcription. B-Myb, by binding the E2F2 promoter to enhance its expression, together with E2F2, co-activates FoxM1, forming the E2F2/B-Myb/FoxM1 core regulatory network that drives cell-cycle progression (Werwein et al., 2020). E2F has been demonstrated to be a principal target of the tumor suppressor RB (Roufayel et al., 2021). E2F proteins bind RB, and their transcriptional activity is repressed in G0 and early G1 (Inoshita et al., 1999). The RB–E2F complex represses transcription of these genes, an repression relieved by RB phosphorylation and the concomitant release of E2F transcription factors, thereby enabling the expression of factors regulating the cell cycle (Engeland, 2022; Kato et al., 1993). In pluripotent cells, complexes of CDK2 with Cyclin A/E continuously phosphorylate RB family proteins, resulting in sustained release of E2F transcription factors and activation of downstream target genes. On one hand, the persistently activated E2F targets drive pluripotent cells past the G1 checkpoint, directly contributing to a shortened G1 phase; on the other hand, E2F–driven activation of the Cyclin A/E–CDK2 complex creates a positive feedback loop that further phosphorylates RB, reinforces E2F activity, and ensures the irreversibility of the G1/S transition (Stead et al., 2002). Precise regulation of E2F activity is crucial for maintaining cellular pluripotency: when RB is activated, its E2F-binding domain inhibits E2F transcription factor activity, leading to an H3K27me3-enriched repressive chromatin environment at the promoters of pluripotency genes, thereby hindering establishment and maintenance of pluripotency; conversely, excessive E2F activity can trigger genomic stress responses, increase apoptosis, and disrupt chromatin modifications, thereby hindering the generation of high-quality PSCs. Experiments show that pluripotency can be effectively promoted only when E2F activity is moderately activated, by relieving transcriptional repression of pluripotency genes while avoiding cellular homeostasis imbalance (Kareta et al., 2015).
Although extensive evidence suggests that a shortened G1 phase is a typical feature of pluripotency, the causal relationship between them remains controversial. Specifically, whether the shortened G1 phase drives the maintenance of pluripotency or whether the pluripotent state actively shapes a brief G1 phase is debated. On one hand, some scholars argue that a shortened G1 phase itself constitutes the reason for maintaining pluripotency. Cells are more receptive to differentiation signals during G1, and their differentiation propensity changes as G1 progresses (Pauklin and Vallier, 2013). Therefore, shortening G1 can limit the time window during which cells respond to external differentiation cues, thereby passively “locking” cells into the pluripotent state. Studies have shown that artificially lengthening the G1 phase of mESCs through chemical or genetic means is sufficient to disrupt the pluripotency network and induce spontaneous differentiation, directly demonstrating that a short G1 phase is an intrinsic determinant of the naïve pluripotent state (Coronado et al., 2013). Conversely, enforced overexpression of Cyclin E or CDK2 to actively shorten G1 can suppress differentiation programs and maintain the expression of pluripotency markers. In contrast, pharmacological inhibition of CDK2/Cyclin E activity to lengthen G1 promotes differentiation (Pauklin and Vallier, 2013).
Another important hypothesis holds that a short G1 is the consequence of core pluripotency factors regulating the cell cycle, rather than a driving force. Studies indicate that pluripotency transcription factors such as Sox2, Oct4, Klf4, and c-Myc actively promote G1 shortening by regulating the activity of Cyclin–CDK complexes and repressing p21 expression, thereby promoting rapid proliferation, constraining the differentiation time window, and sustaining self-renewal (Fleifel and Cook, 2023). Moreover, the core pluripotency factor Nanog can directly bind and activate CDK6 and CDC25A, thereby promoting the G1/S transition (Boward et al., 2016). Consequently, G1 extension is a consequence of the differentiation program rather than a trigger for differentiation (Coronado et al., 2013).
Based on the aforementioned research, we propose that the short G1 phase and the pluripotent state are not in a simple one-way causal relationship, but are more likely to form a tightly coupled relationship (Dalton, 2015). This coupling manifests as a self-reinforcing, dynamically maintained feedback loop: the core pluripotency network actively configures the cell cycle toward a short G1 by repressing cell-cycle inhibitors and dismantling the DREAM complex; conversely, the short G1 indirectly limits exposure to extracellular differentiation signals while also enabling activated E2F transcription factors to co-occupy promoters with pluripotency factors, thereby creating an epigenetic and transcriptional environment that reinforces the pluripotent state. This bidirectional, interlocked coupling not only ensures self-renewal and rapid proliferation of PSCs but also substantially enhances their stability at differentiation thresholds. Consequently, the system operates as a robust whole: disrupting any link (whether by forcibly extending G1 or downregulating core pluripotency factors) would unravel the coupling and drive cells toward differentiation. Future studies that can dynamically track the interactions between pluripotency factors and cell-cycle regulatory elements in real time hold promise for more precisely delineating the temporal logic and hierarchical control of this coupling network in cell fate determination.
The universality of CDK1 as a core driver of the cell cycle is also debated. On one side, CDK1 is viewed as an indispensable “master switch” that can compensate for the loss of other CDKs. In nearly all somatic cells and most stem cells, complete inhibition of CDK1 leads to cell cycle arrest at G2/M. CRISPR-Cas9 screens also show that once CDK1 is activated, it is sufficient to drive cells into mitosis. Conversely, CDK1 inactivation directly causes G2/M arrest. This underscores the non-redundant nature of CDK1 for proliferation and survival (Santamaría et al., 2007). From an evolutionary perspective, CDK1 homologs in yeast act as the sole cell cycle CDKs controlling the entire cycle, underscoring its central role (Lee M. G. and Nurse, 1987). Another viewpoint does not deny the core role of CDK1 but suggests that its “absolute necessity” may be context-dependent and potentially possesses cell-cycle–independent functions. In certain cell types or states, other CDKs (such as CDK2) may assume more prominent roles. The necessity of CDK1 may vary with cellular metabolic state, DNA damage stress, or differentiation stage (Kozar et al., 2004). CDK1 loss leads to cell death, which may reflect not only cell-cycle arrest but also CDK1 involvement in transcriptional regulation, RNA splicing, DNA damage repair, and metabolism. Therefore, its “essentiality” likely reflects a combination of crisis due to cell-cycle arrest and disruption of key cellular processes. Additionally, technical limitations contribute to the controversy. Traditional gene knockout is long-term and complete; cells may activate compensatory or adaptive mechanisms during chronic CDK1 loss, confounding phenotypes. Acute, rapid knockout systems better reveal the most direct, primary functions.
In future studies, it will be valuable to precisely compare the speed and extent of cell-cycle arrest across different cell types after CDK1 deletion. Constructing a CDK1 mutant that retains only non-Cyclin functions and assessing whether it can rescue all phenotypes caused by complete CDK1 loss would be of great significance for resolving the relationship between CDK1’s non-cyclin functions and stemness.
The functional redundancy and specificity of multi-subunit proteins have long been a focal point across disciplines. Cyclin D comprises Cyclin D1, Cyclin D2, and Cyclin D3. On one hand, Cyclin D1, D2, and D3 can all bind CDK4/6 and phosphorylate the same site on the Rb protein, thereby releasing its inhibition of E2F and promoting G1/S transition. This core biochemical function is highly conserved across the three subtypes. Studies have shown that placing the coding region of Cyclin D1 under the promoter control of Cyclin D2 can fully rescue the lymphoid developmental defects in Cyclin D2–deficient mice. This indicates that as long as Cyclin D1 protein is expressed, normal function can be performed, strongly supporting functional redundancy (Kalaszczynska et al., 2009). Conversely, other views argue that these Cyclin D subtypes have acquired unique functions during evolution. Expression profiles of Cyclin D subtypes differ: Cyclin D1 is highly expressed in most proliferative cells, Cyclin D2 in the hematopoietic system, and Cyclin D3 in early embryos and muscle (Sicinski et al., 1995). The phenotypes following individual deletions of Cyclin D1, Cyclin D2, or Cyclin D3 are not identical, suggesting specific interactions with distinct transcription factors and kinases (Sicinska et al., 2003). Future studies on rescue experiments may provide a more comprehensive understanding of the functions of Cyclin D subtypes. For example, in mouse models with Cyclin D2 and Cyclin D3 double knockout, overexpression of Cyclin D1 could be tested to determine whether it can fully rescue the defect phenotype. Proteomic analyses could identify the specific binding partners of each Cyclin D subtype. These subtype-specific interactors are likely the molecular basis for functional specificity.
The maintenance of pluripotency in ESCs and their unique cell cycle are not independent processes; they are interwoven through a robust bidirectional regulatory network. In this section, we synthesize extensive evidence showing that core cell cycle factors participate in stabilizing the pluripotency transcriptional network by directly phosphorylating and modifying proteins such as Sox2 and Oct4, affecting their activity, stability, or interactions. Conversely, core pluripotency factors Oct4, Sox2, and Nanog are not passive recipients; they actively shape rapid cell cycling. They function by transcriptionally repressing key CDK inhibitors and activating promoters of proliferation-promoting Cyclins. This mutual regulation forms a self-reinforcing feedback loop: the pluripotency network shapes a short G1 phase, and the short G1 phase, in turn, minimizes the time window for integrating differentiation signals and favors the establishment of an epigenetic landscape that supports self-renewal. This complex dialog ensures seamless coupling between rapid proliferation and the undifferentiated state.
2.2 The mechanisms of genomic stability maintenance in stem cells
Although the rapid proliferation of ESCs facilitates development, it also poses risks for genomic instability. To counteract this challenge, ESCs have evolved distinctive DNA damage response mechanisms, including Babam2’s role in stabilizing CDC25A and promoting the ubiquitination of p53, which helps to maintain CDK2 activity and preserve pluripotency factors (Biswas et al., 2018). The loss of CDK1 results in increased DNA double-strand breaks (DSBs), mitotic abnormalities, and chromosomal aberrations, indicating genomic instability (Neganova et al., 2014; Wang X. Q. et al., 2017). APE1 exerts its effects through a dual mechanism, specifically. On the one hand, it enhances the activity of the Gdnf/Gfrα1 axis, thereby activating the Src/Erk pathway to promote proliferation. On the other hand, it regulates the activity of Cyclin-CDK complexes to facilitate G1/S and G2/M phase progression, thereby preventing cell cycle arrest caused by frequent DNA damage (Liu L. et al., 2023; Malfatti et al., 2021). Simultaneously, ESC proliferation is accompanied by the accumulation of ROS(Wang K. et al., 2013). Dnm1L maintains ROS at physiological levels by regulating mitochondrial fission dynamics and the Keap1-Nrf2 antioxidant pathway, thus protecting genomic stability (Seo et al., 2023). Additionally, Cops5 regulates glycolytic metabolism by stabilizing mitochondrial transport protein Mtch2 and enhances DNA repair capacity, providing support for rapid cell cycle progression (Li P. et al., 2020).
ESCs with a shortened G1 phase are capable of rapid self-renewal, yet this may also result in insufficient assembly of the pre-replication complex (pre-RC) and inadequate nucleotide preparation. Such deficiencies can induce replication stress during accelerated DNA synthesis, leading to replication fork deceleration, stalling, or even collapse. These events ultimately cause DNA damage and genomic instability (Matson et al., 2017). To counteract this intrinsic challenge, ESCs have developed a sophisticated multilayered protective mechanism that is functionally linked to their pluripotent status. Low levels of basal replication stress, such as the formation of RPA-coated ssDNA, activate the ATR/CHK1 signaling pathway (Zhu X. F. et al., 2022). Once activated, ATR-CHK1 phosphorylates proteins including CLASPIN and FANCM to stabilize stalled replication forks and prevent their collapse. It also phosphorylates and stabilizes p53, which in turn induces p21 expression. As a broad-spectrum CDK inhibitor, p21 can initiate G1/S checkpoint arrest. Sustained activation of p53 not only enforces cell cycle arrest but also promotes the expression of differentiation-related genes while suppressing core pluripotency factors such as Nanog and Oct4. This facilitates the exit of ESCs from the pluripotent state (Lin T. et al., 2005). Under severe or irreparable stress conditions, p53 activation shifts toward inducing pro-apoptotic genes including Puma and Bax, thereby eliminating damaged cells. Furthermore, ESCs display heightened sensitivity to ATR-CHK1 inhibition, underscoring the critical role of this pathway in managing intrinsic replication stress. Inhibition of ATR or CHK1 results in markedly increased replication fork collapse and DNA damage, rapidly leading to apoptotic cell death. Additionally, studies have shown that various cellular stresses (including replication stress, oxidative stress, and osmotic stress) can induce a subfraction of ESCs to enter a 2-cell-like (2C-like) state. Cells in this state express 2C-stage–specific endogenous retroelements (such as Mervl) and display a more “naïve” epigenetic and transcriptional state, with a markedly slowed cell cycle (Oleksiewicz et al., 2017). It is hypothesized that this transition may be an evolutionarily conserved stress response pattern, wherein transcriptional programs are reprogrammed and cell cycle is slowed to cope with adverse conditions. When ESCs face severe stress, some cells survive by “rebooting” into a more primitive, protected state. A slowed cell cycle affords more time for DNA repair and metabolic adjustment.
In summary, ESCs employ a finely tuned, multilayered defense system to balance rapid proliferation with genomic integrity. The shortened G1 phase leads to inadequate replication preparation and increased replication conflicts, resulting in persistent basal replication stress. Consequently, ESCs depend critically on the ATR-CHK1 pathway for continuous monitoring and stabilization of replication forks. Depending on the severity of replication stress, cells activate distinct fate-determining mechanisms. Under mild stress, buffering networks—involving factors such as Babam2 and APE1—help maintain pluripotency by suppressing p53 and sustaining CDK activity. When stress escalates, strong activation of the p53–p21 axis induces cell cycle arrest, promotes differentiation, or initiates apoptosis to eliminate damaged cells and protect the stem cell pool. Under severe stress, certain cells may enter a 2C-like state through reprogramming, which slows the cell cycle and remodels the epigenome to facilitate survival. This multi-tiered response strategy collectively constitutes a core mechanism through which stem cells maintain genomic integrity and functional competence despite rapid proliferation.
Beyond their classical roles in driving cell cycle phase transitions, Cyclins and CDKs exhibit notable functional diversity and specificity in regulating stem cell fate. This section delves into these nonclassical functions of core regulatory factors, highlighting their direct involvement in epigenetic regulation, signal transduction integration, and lineage specification. We discuss how specific Cyclin-CDK complexes (e.g., Cyclin B1–CDK1) act as critical nodes linking G2/M to pluripotency maintenance. Additionally, this part examines functional redundancy and specificity among Cyclin subtypes and CDKs (e.g., the necessity of CDK1), emphasizing that their roles in the ESC context are highly context-dependent. The discussion also covers the key role of multi-protein complexes such as the DREAM complex, which acts as a master coordinator. It suppresses cell-cycle genes during quiescence and, during proliferation, ensures orderly, temporally specific expression of Cyclins and other factors, thereby correctly integrating developmental signals with core cell cycle operation to determine stem cell fate.
3 Signaling pathways connect the cell cycle and pluripotent genes in stem cells
The fate determination of stem cells is precisely regulated through multi-layered interactions among key signaling pathways and phase-specific cell cycle machinery (Guglielmi et al., 2021; Wulansari et al., 2021; Xu et al., 2023). Leukemia inhibitory factor (LIF) is a key factor in maintaining the pluripotency of mESCs. LIF binds to the cell surface receptor complex composed of LIF receptor (LIFR) and gp130, activating the Jak1 kinase, which then phosphorylates the transcription factor Stat3 (Darnell et al., 1994; Gearing et al., 1991; Stahl et al., 1994). Once phosphorylated, Stat3 dimerizes and translocates into the nucleus, where it directly binds to and activates the promoters of core pluripotency genes such as Oct4, Sox2, Nanog, and Klf4 (Deng et al., 2021; Do et al., 2013; Li J. et al., 2024; Onishi and Zandstra, 2015; Stirparo et al., 2021). Recent studies have identified Gpr160 as an important regulator of the Jak1/Stat3 pathway; it enhances Stat3 phosphorylation and nuclear translocation, promoting the expression of CyclinD1-CDK4/6 and accelerating G1/S phase transition (Fan et al., 2024; Zhou C. H. et al., 2016). Notably, LIF not only activates the canonical Jak/Stat3 pathway but also bypasses other pathways to activate Erk1/2 and PI3K/Akt signaling networks, forming a complex regulatory system (Boulton et al., 1994; Oh et al., 1998).
MAGEA2 (Melanoma-Associated Antigen A2) contributes to sustaining stem cell pluripotency and promoting proliferation, in conjunction with other core pluripotency factors such as Oct4, Sox2, and Nanog (Barker and Salehi, 2002; Lifantseva et al., 2011; Yang B. et al., 2007). Studies have found that knockdown of MAGEA2 leads to increased phosphorylation of Erk1/2, and decreased expression of cell cycle-related genes such as CDK1, CDK2, CyclinA1, CyclinD1, and CDC25a. These molecular changes result in cell cycle arrest, accompanied by a reduction in the expression of pluripotency markers in mESCs. These findings suggest that the MAGEA2/Erk1/2 signaling pathway plays a crucial role in both maintaining the expression of core pluripotency genes and cell cycle genes, as well as inhibiting embryonic lineage differentiation (Park et al., 2020).
Under energy stress conditions, the AMPK signaling pathway exhibits a bidirectional role in the regulation of ESC pluripotency, depending on the cellular state. In pluripotent cells, AMPK inhibits Nanog transcription and promotes its proteasomal degradation via the p53/p21 pathway, driving differentiation. Conversely, in the absence of LIF or under low pluripotency conditions, AMPK stabilizes β-catenin through PI3K/Akt-mediated inhibition of GSK3β, thereby activating the Wnt pathway. The activated β-catenin forms a transcriptional complex with Oct4 and cooperatively upregulates naive pluripotency factors such as Klf4 and Esrrb, significantly increasing Nanog expression. Simultaneously, AMPK induces G1 phase arrest and G2/M phase suppression; through p21-dependent cell cycle reprogramming, it slows proliferation, providing a temporal window for the re-establishment of pluripotency transcription factors (Alba et al., 2020; Chae et al., 2012; Liu Y. J. and Yamashita, 2019). This metabolic-cycle-pluripotency tri-regulatory network dynamically coordinates ESC fate decisions under energy stress, enabling adaptation to the environment.
The Hippo/YAP-TAZ pathway modulates the balance between proliferation and differentiation in a cell cycle phase-dependent manner. In trophoblast stem cells, nuclear localized YAP interacts via its WW2 domain with the stemness factor Cdx2, represses the expression of the G1-phase regulator Cyclin D1, and reduces CDK4/6 activity, thereby restraining the G1 progression rate. This “YAP–CDX2–Cyclin D1” axis specifically maps to G1-phase regulation (Basak and Ain, 2022). In the G2/M phase or endoreplication stage, downregulation of the core kinase LATS1 attenuates the formation of the LATS1–LIMK2 complex, relieving inhibition of LIMK2 and leading to elevated pLIMK2Thr505 and subsequent pCOFILINSer3 levels. This stabilizes F-actin and promotes endoreplication (polyploidization) in trophoblast giant cells, corresponding to the G2/M-to-endoreplication transition (Basak and Ain, 2022).
The Notch pathway antagonizes Hippo/YAP-TAZ and is linked to G1 exit. In epidermal stem cells, YAP/TAZ transcriptionally activate Delta-like ligands (DLL1, DLL3), which inhibit Notch signaling in a cis-inhibitory manner to maintain stemness. Upon YAP/TAZ inactivation—triggered by soft substrates or high cell density—Notch signaling is activated, inducing G1 exit and initiating differentiation. This switch depends on the transcriptional control of Notch ligands by YAP/TAZ and is explicitly associated with the transition between stemness maintenance and differentiation in G1 (Totaro et al., 2017).
Phase-specific CDK activities act as downstream effectors of these pathways: G1 CDK4/6 activity is directly suppressed by YAP, whereas dysregulated CDK activity during endoreplication is modulated indirectly via the LATS1–LIMK2 axis. Notch activation likely promotes cell cycle exit and differentiation by downregulating G1 Cyclin–CDK complexes (Basak and Ain, 2022; Meyer et al., 2023; Totaro et al., 2017). Furthermore, oscillatory YAP activation optimizes G1 CDK-mediated proliferation, while sustained low YAP activity synergistically promotes differentiation through Notch activation and CDK downregulation, reinforcing the phase-specific mapping of pathway activities (Meyer et al., 2023).
The TGF-β/Activin/Nodal–Smad pathway cooperates with cell cycle regulators to control pluripotency maintenance and lineage specification. Early in G1, when Cyclin D–CDK4/6 activity is low, Smad2/3 translocate into the nucleus to bind endodermal gene promoters and initiate differentiation programs. Concurrently, they upregulate the DNA methyltransferase Dnmt3b to establish methylation patterns, facilitating the binding to epiblast markers such as Fgf5 and Dnmt3a. This Smad4-independent process is critical for the transition from naive to primed pluripotency (Zhao et al., 2024). By late G1, elevated Cyclin D–CDK4/6 activity phosphorylates the linker regions of Smad2/3, inhibiting their nuclear translocation and thereby diverting cells toward neuroectodermal differentiation instead of Activin/Nodal-induced endodermal commitment (Pauklin and Vallier, 2013; Yang J. and Jiang, 2020). Additionally, Nodal/Activin signaling regulates the dosage of pSmad2, which binds the Oct4 promoter in a concentration-dependent manner to sustain the pluripotency network (Lee K. L. et al., 2011). Smad4 functions primarily during primed-to-mesendodermal differentiation, forming complexes with Smad2/3 to activate lineage-specific genes such as Wnt3 and Eomes (Zhao et al., 2024). The PI3K/Akt pathway further fine-tunes this balance via mTORC2-mediated degradation of Smad2/3, antagonizing their differentiation-inducing effects (Yang J. and Jiang, 2020).
The Wnt/β-catenin pathway contributes to stem cell regulation by maintaining epigenetic stability. Its decline accelerates the cell cycle, impairs proper pluripotency exit, and compromises differentiation potential. Mechanistically, β-catenin cooperates with the KAP1/DNMT1 complex to maintain DNA methylation and heterochromatic states at imprinting control regions (ICRs), suppressing retrotransposon activity and ensuring genomic stability and cellular homeostasis (Theka et al., 2019).
The Hedgehog (HH) pathway orchestrates NSC dynamics through its downstream GLI transcription factors (particularly GLI1 and GLI2). It shortens the cell cycle of activated NSCs (aNSCs) by accelerating both G1 and S/G2/M phases, thereby enhancing proliferation and self-renewal capacity, as reflected by increased neurosphere formation (Daynac et al., 2016). Moreover, GLI2 directly binds to Sox2 enhancers to drive its expression in NSCs. Sox2 in turn activates Hes5, contributing to the maintenance of an undifferentiated state (Takanaga et al., 2009). However, prolonged HH activation induces accumulation of quiescent NSCs (qNSCs) but ultimately exhausts the aNSC pool, leading to loss of pluripotency, premature cell cycle exit, and aberrant differentiation, underscoring its dual role in NSC homeostasis (Agathocleous et al., 2007; Daynac et al., 2016).
Signal transduction pathways are the key bridge connecting extracellular signals to intracellular responses. They seamlessly integrate environmental cues with core cell cycle control and pluripotency. This section explains how major signaling pathways—including LIF/Jak/Stat3, PI3K/Akt, TGF-β/Smad, Wnt/β-catenin, and Hippo/YAP—orchestrate stem cell fate by directly regulating the activity of Cyclin-CDK complexes, CKIs, and pluripotency transcription factors. These pathways are not independent. They are interwoven and exhibit temporal specificity. The Hippo/YAP pathway can produce opposite effects depending on the relative expression of Cyclins and CDKs at different cell cycle phases, while TGF-β/Smad signaling is gated by G1-phase CDK activity. In addition, AMPK signaling can bidirectionally influence pluripotency by affecting the cell cycle and key transcriptional networks such as Wnt/β-catenin. This complex interplay ensures that decisions of self-renewal and differentiation are precisely calibrated according to cellular metabolic state and the external microenvironment.
4 Linking of cell cycle and epigenetic modification in ESCs
Epigenetic regulation also plays a crucial role in maintaining the cell cycle characteristics of ESCs(Roy et al., 2022). The enrichment patterns of histone modifications, such as H3K27ac and H3K4me3, at the promoters of cell cycle genes vary significantly between ESCs and somatic cells. In particular, the promoters of pluripotency-related genes maintain an “open” chromatin conformation in ESCs, enabling rapid responsiveness to environmental signals and facilitating adjustments to the cell cycle.
Studies have shown that deletion of Jmjd2 family proteins, which are histone demethylases, either individually or collectively, leads to impaired ESC self-renewal and early embryonic lethality. Their primary role involves removing H3K9 methylation to ensure pluripotency (Pedersen et al., 2016). The Polycomb Group (PcG), comprising PRC1 and PRC2, is a highly conserved epigenetic repressive complex throughout evolution. PRC1 catalyzes monoubiquitination of histone H2A at lysine 119 (H2AK119ub1), while PRC2 mediates trimethylation of histone H3 at lysine 27 (H3K27me3), jointly establishing a repressive chromatin state that silences differentiation-related genes (Qin et al., 2021; Zhu Y. R. et al., 2022). Research indicates that PRC1 and PRC2 also promote the maintenance of the short G1 phase characteristic of ESCs by repressing the expression of G1 extension-related genes, such as p21, and by enhancing the activity of the CyclinE/CDK2 complex (Huang et al., 2021; Zhu Y. R. et al., 2022). DNA demethylase Tet1 collaborates with PRC2 to remove DNA methylation marks and enhance H3K27me3 modification, forming a multilayered epigenetic regulatory network. This synergistic effect is crucial for maintaining the activity of pluripotency genes and coordinating the balance between proliferation and differentiation (Chrysanthou et al., 2022a; Chrysanthou et al., 2022b; Ficz et al., 2011).
The unique cell cycle of ESCs is both a cause and a consequence of their distinctive epigenetic features. We discusses the deep bidirectional relationship between the epigenome and the cell cycle in this section. Key epigenetic modifiers, such as Polycomb repressive complexes (PRC1/2) and histone demethylases, are essential for maintaining a shortened G1 phase by repressing the expression of cell cycle inhibitors and pro-differentiation genes. Conversely, core cell cycle regulatory proteins participate in maintaining these epigenetic characteristics. CDKs can directly phosphorylate epigenetic enzymes, altering their activity and affecting histone modification patterns that are critical for fate decisions. This coordinated regulatory loop ensures ESCs proliferate rapidly while maintaining an epigenetically reinforced pluripotent state and suppressing premature differentiation, indicating that epigenetic regulation is a dynamic process tightly synchronized with the cell cycle.
5 Role of metabolic mode in stem cells
ESCs exhibit distinctive metabolic characteristics that provide energy and biosynthetic precursors necessary for rapid proliferation. ESCs primarily depend on glycolysis for energy production. This metabolic pattern not only meets the energetic demands of rapid cell division but also regulates histone acetylation and gene expression by modulating levels of metabolic intermediates such as acetyl-CoA. Recent studies have identified the mTOR signaling pathway as a key hub integrating nutrient signals with cell cycle regulation. Reducing the activity of the mTOR pathway can induce hPSCs and blastocysts to enter a quiescent state, in which cell proliferation, developmental processes, and attachment to the uterine epithelium are all limited (Iyer et al., 2024). Sustained activation of mTORC1 promotes protein synthesis and cell growth, providing the material foundation for frequent cell division (Joshi et al., 2024). Meanwhile, mTORC2 influences mitosis by regulating cytoskeletal reorganization (Palma et al., 2023). This metabolism-cycle coupling mechanism ensures that ESCs can rapidly expand during early embryonic development.
Even under high oxygen conditions, ESCs mainly rely on glycolysis rather than (OXPHOS) for ATP production. Studies have shown that the long non-coding RNA (lncRNA) Lx8-SINE B2 enhances glycolytic flux by binding to the glycolytic enzyme Enolase 1 (ENO1), stabilizing and activating ENO1, thereby significantly increasing glucose utilization. When glycolysis is inhibited, ATP supply becomes insufficient, leading to decreased activity of Cyclin-CDK complexes, delayed G1/S phase transition, and ultimately the loss of pluripotency (Chen F. Q. et al., 2022b; Chen F. Q. et al., 2021a).
Stem cell metabolism constitutes a sophisticated regulatory network that governs energy supply, epigenetic modifications, and the elimination of toxic metabolites to maintain pluripotency and cell cycle homeostasis. This network exhibits state-specific and species-dependent characteristics. Ground-state PSCs, such as mESCs cultured in 2i/LIF conditions (mESCs-2iL) and human induced pluripotent stem cells (iPSCs), predominantly rely on a high glycolytic flux. Even in the presence of homologous or heterologous mitochondrial DNA (mtDNA) mutations, human iPSCs sustain elevated expression of key glycolytic genes, including the glucose transporter Glut3 and hexokinase HK3. Moreover, they suppress pyruvate dehydrogenase (PDH) activity via upregulation of pyruvate dehydrogenase kinase 1 (PDK1), thereby preventing the entry of pyruvate into the tricarboxylic acid (TCA) cycle and promoting the glycolytic pathway. This metabolic preference not only facilitates rapid ATP generation to meet the proliferative demands of the short G1 phase but also minimizes ROS accumulation from OXPHOS, thereby avoiding DNA damage and senescence pathway activation. Consequently, it helps stabilize the expression of core pluripotency factors (Prigione et al., 2014). The PTEN-induced kinase 1 (PINK1)-mediated mitophagy is a critical mechanism enabling this glycolytic switch. Loss of PINK1 leads to accumulation of damaged mitochondria, increased ROS levels, and activation of the p53-dependent cell cycle checkpoint, thereby causing an 80% reduction in reprogramming efficiency and a 3–4 days delay in the emergence of SSEA-1-positive colonies in mESCs. In contrast, functional mitophagy eliminates mature tubular mitochondria and promotes the formation of immature spherical mitochondria, providing the structural basis for a glycolytic metabolic profile (Vazquez-Martin et al., 2016).
Acetyl-CoA serves as a central metabolic effector of pluripotency. Its cellular level and subcellular distribution profoundly influence stem cell fate by modulating histone acetylation and pluripotency gene expression. Both mouse and human PSCs require high acetyl-CoA levels to support active histone marks such as H3K9ac and H3K27ac, as well as an open chromatin configuration. mESCs primarily generate acetyl-CoA through threonine metabolism, while hPSCs rely on glucose-derived pyruvate, which is converted to citrate and transported to the cytoplasm, where ATP-citrate lyase (ACLY) catalyzes its conversion to acetyl-CoA. Exogenous acetate supplementation increases acetyl-CoA levels and delays differentiation of PSCs(Chakrabarty and Chandel, 2021). NAD+ promotes the deacetylation and activation of acetyl-CoA synthetase 1 (AceCS1) via Sirt1, facilitating acetate conversion to acetyl-CoA, thereby maintaining acetyl-CoA and H3K27ac levels in mESCs and supporting pluripotency (Wu et al., 2022). Additionally, nuclear-localized TCA cycle enzymes, such as Pdha1, can directly enhance the nuclear acetyl-CoA pool, augment histone H3 acetylation, activate pluripotency genes including Oct4 and Nanog, and promote somatic reprogramming as well as the naïve-to-primed transition (Li et al., 2022a). In NSCs, TIGAR enhances OXPHOS, elevates acetyl-CoA levels, increases H3K9ac modification, and activates differentiation-related genes such as Ngn1 and Neurod1, thereby regulating neurogenic progression (Zhou W. et al., 2019).
One-carbon metabolism represents a core pathway linking metabolic flux to epigenetic regulation of pluripotency, with notable species-specific variations. mESCs depend on threonine (Thr) metabolism to obtain one-carbon units: Thr is catabolized by threonine dehydrogenase (TDH) to generate glycine and acetyl-CoA. Glycine is further metabolized to formate, which enters the S-adenosylmethionine (SAM) cycle to provide methyl donors for histone H3K4me2 and H3K4me3 modifications. These marks are enriched at the promoters of pluripotency genes such as Esrrb and Klf4, maintaining an open chromatin state. Threonine deprivation leads to a significant reduction in H3K4me2/3 levels, causing proliferation arrest and initiation of differentiation in mESCs, without affecting DNA methylation or other histone modifications such as H3K9me3 (Shyh-Chang et al., 2013; Van Winkle and Ryznar, 2019). In contrast, hESCs, which lack functional TDH, depend on methionine (Met) metabolism. Met cycles through the Met-SAM pathway to generate SAM, which serves as the methyl donor for H3K4me3 and DNA methylation. Methionine deficiency results in near-complete loss of H3K4me3, proliferation arrest, and apoptosis in hESCs, phenotypes that can be partially rescued by exogenous SAM supplementation (Shiraki et al., 2014; Van Winkle and Ryznar, 2019). Furthermore, the glycine cleavage system (GCS) is highly active in PSCs. Its rate-limiting enzyme, glycine decarboxylase (Gldc), is co-regulated by Sox2 and Lin28A. GCS generates one-carbon units via glycine cleavage to support SAM synthesis and maintain H3K4me3 levels, while also clearing methylglyoxal (MG), a toxic byproduct of aberrant glycine metabolism. This prevents accumulation of advanced glycation end products (AGEs) and activation of senescence markers (P15, P16, P21), thereby avoiding irreversible G1 arrest and ensuring normal cell cycle progression (Tian et al., 2019).
Alpha-ketoglutarate (α-KG), a key intermediate of the TCA cycle, is an important regulator of pluripotency. In mESCs, α-KG is primarily generated by mitochondrial isocitrate dehydrogenase 2 (IDH2). Exogenous supplementation with cell-permeable dimethyl-α-ketoglutarate (dm-αKG) can substitute for 2i inhibitors (GSK3 and MEK inhibitors) in maintaining ground-state pluripotency. dm-αKG promotes DNA demethylation by activating TET dioxygenases and reduces H3K9me2 levels via Kdm3a/b histone demethylases, thereby sustaining the proportion of Rex1-GFP-positive cells and dome-shaped colony morphology. During differentiation, dm-αKG extends the developmental competence window of epiblast-like cells (EpiLCs) for differentiation into primordial germ cell-like cells (PGCLCs), enabling EpiLCs cultured for 72 h to differentiate into PGCLCs with efficiency comparable to that of 48-h controls, while maintaining H3K27me3 levels to ensure precise activation of key PGC genes such as Prdm1 and Prdm14 (Carey et al., 2015; Tischler et al., 2019). The transcription factor Spic further reinforces the regulation of ground-state pluripotency by one-carbon metabolism. In 2iL culture, upon inhibition of MEK/Erk signaling, Spic is transcriptionally activated and stabilizes Nanog binding at chromatin regions of betaine-dependent one-carbon metabolism genes, enhancing betaine-to-methionine conversion and maintaining a low SAM/SAH ratio. This process upregulates activating histone marks such as H3R17me2a while suppressing H3K4me3 (to avoid differentiation gene activation), thereby delaying exit from ground-state pluripotency. Concurrently, it provides precursors for dTMP synthesis and other DNA biosynthesis requirements, enhancing cell cycle adaptability under metabolic stress (Azad et al., 2023).
The maintenance of pluripotency is closely associated with mitochondrial remodeling and metabolic reprogramming. Despite the potential presence of homologous or heteroplasmic mtDNA mutations, hiPSCs maintain a highly glycolytic phenotype similar to hESCs by upregulating glycolytic enzymes, increasing glucose-6-phosphate (G6P) levels, and enhancing PDK1 expression (Prigione et al., 2014). Naïve-state ESCs and iPSCs possess immature, spherical mitochondria with underdeveloped cristae and rely predominantly on glycolysis. Upon differentiation into NSCs, mitochondria become elongated with well-defined cristae, and cells shift toward OXPHOS(Choi et al., 2015). Additionally, PINK1-mediated mitophagy promotes mitochondrial rejuvenation and is essential for efficient reprogramming. Its loss significantly impairs reprogramming efficiency, diminishes glycolytic capacity, and reduces α-KG production, thereby compromising pluripotency establishment (Vazquez-Martin et al., 2016).
ESCs rely heavily on glycolytic metabolism, which is not merely a passive adaptation to rapid proliferation but an active regulator of pluripotency and cell cycle progression. This section clarifies how metabolism, the cell cycle, and pluripotency are precisely coupled. A high glycolytic flux provides ample biosynthetic precursors and ATP while maintaining low ROS, supporting rapid biomass accumulation and minimizing DNA damage. Importantly, key metabolic pathways and intermediates directly influence epigenetic modifiers and signaling. Metabolites such as acetyl-CoA, α-ketoglutarate (α-KG), and S-adenosylmethionine (SAM) are essential cofactors for histone acetylation and methylation, directly linking cellular metabolism to the epigenetic regulation of pluripotency genes. Moreover, species-specific metabolic dependencies (e.g., threonine metabolism in mESCs and methionine cycling in hESCs) underscore the evolutionary adaptability of this coupling. Sensors such as AMPK and mTOR integrate energy status with cell cycle decisions, regulating CDK activity and the stability of pluripotency factors. Thus, metabolism acts as a central hub, coordinating energy production, biosynthetic demands, and epigenetic signaling to sustain self-renewal and rapid proliferation.
6 Cell cycle control and somatic cell reprogramming
The breakthroughs in somatic cell reprogramming have effectively enabled the regulation of cell fate, with significant implications for clinical applications, drug screening, in vitro modeling of human diseases, and understanding the early stages of pre-implantation embryonic development. Somatic cell reprogramming refers to the process of reversing mature somatic cells to a pluripotent, embryo-like state or iPSCs by introducing specific transcription factors. This process involves not only reorganization of transcriptional networks but also profound reconfiguration of cell cycle proteins and pluripotent factors. Gurdon and colleagues were the first to discover the method of reprogramming through somatic cell nuclear transfer, which resets the somatic cell nucleus during fertilization by oocyte factors (Campbell et al., 1996; Gurdon, 1962; Washburn et al., 2022).
Yamanaka and colleagues identified the so-called “Yamanaka factors”—Oct3/4, Sox2, c-Myc, and Klf4—from a pool of 24 candidates. The successful reprogramming of fibroblasts into iPSCs with these four factors culminated in the 2012 Nobel Prize in Physiology or Medicine. This landmark discovery opened the door to reprogramming somatic cells into PSCs using defined factors (Takahashi and Yamanaka, 2006). One of the main objectives of cell fate manipulation in reprogramming is to expand the pool of scarce cell sources, and as reprogramming techniques and methods become increasingly refined, these functional cells are expected to be used in the treatment of various major diseases. Moreover, somatic cell reprogramming provides an unlimited source of PSCs for cell replacement therapies, circumventing the ethical issues associated with embryo destruction, and enables the construction of disease models using patient-specific iPSCs to investigate pathogenic mechanisms (Wattanapanitch, 2019).
Increasing evidence indicates that somatic cell reprogramming is not simply the overexpression of four transcription factors (OSKM), but a dramatic identity conversion process impeded by multiple barriers (Figure 2). The cell cycle regulatory network is one of the most critical gatekeeping mechanisms. It not only limits the speed of reprogramming but also acts as a quality controller, eliminating cells that fail to meet criteria. Successful reprogramming is accompanied by profound remodeling of the cell cycle, enabling escape from these intrinsic inhibitory barriers. In normal somatic cells, the cell cycle is composed of tightly regulated phases, including G1, S, G2, and M phases, with Cyclins and CDKs collaboratively controlling the process. During their maintenance of division and metabolic homeostasis, somatic cells typically exhibit a prolonged G1 phase and a relatively slow cell cycle to ensure accurate DNA replication and cellular stability. Studies have shown that cells in the quiescent G0 phase usually display low levels of Cyclin D and E expression, with reduced CDK activity, making entry into the S phase difficult (Liu et al., 2019).
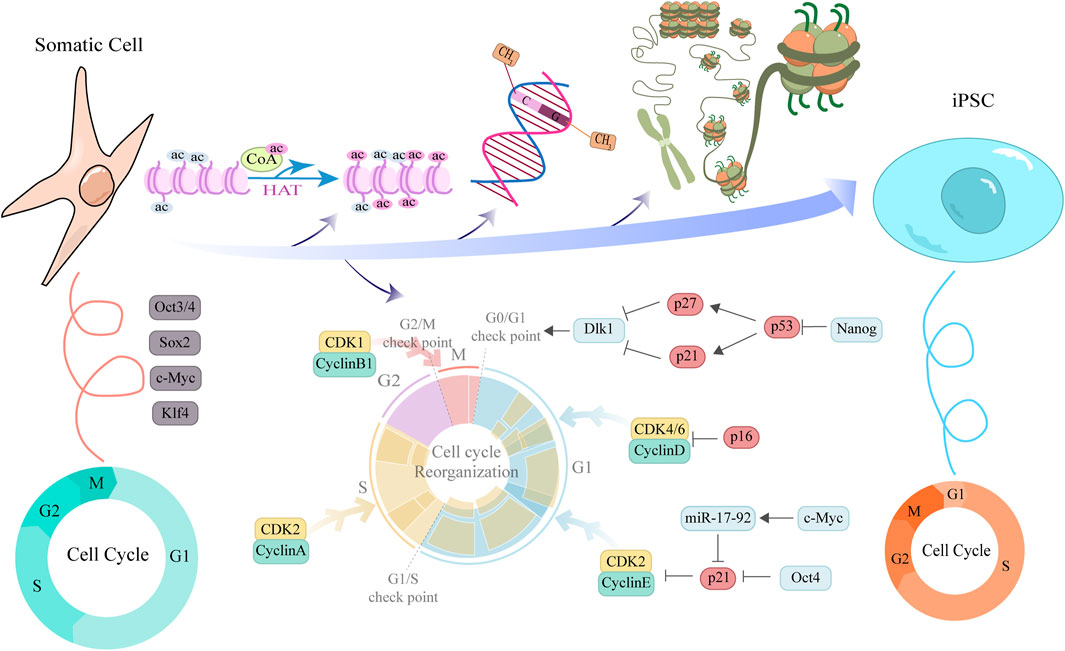
Figure 2. Schematic illustration of cell cycle remodeling and associated factors function during somatic cell reprogramming to iPSCs. This diagram elucidates the molecular mechanisms by which cyclins and pluripotency factors interact to promote the reprogramming of somatic cells into iPSCs. The upper section depicts key epigenetic events underlying the transition from a tightly compacted to an open chromatin structure during reprogramming, including the regulation of histone acetylation, DNA methylation, and chromatin remodeling. The central portion illustrates the ongoing cell cycle remodeling, highlighting the dynamic expression of cyclins across distinct cell cycle phases. Cyclin expression facilitates cell cycle progression and contributes to pluripotency maintenance, whereas CKIs act at cell cycle checkpoints to modulate reprogramming efficiency. Concentric ring diagrams flanking the central illustration provide a comparative analysis of cell cycle structural features between somatic cells and PSCs, underscoring the pivotal role of cell cycle remodeling in the somatic cell reprogramming process.
In contrast, during reprogramming—especially in the later stages—dedifferentiated cells acquire the distinctive cell cycle features of PSCs, including a shortened G1 phase and an accelerated proliferation rate. These features support the restoration of a high proliferative state, which facilitates—but is not solely sufficient for—the establishment of pluripotency (Ruiz et al., 2011). The restructuring of the cell cycle is particularly pronounced in somatic cell reprogramming (Cacchiarelli et al., 2015). A team has proposed a reprogramming strategy in which the inhibition of cell cycle inhibitors (such as p27 or p18) in somatic cells significantly enhances reprogramming efficiency (Zhan et al., 2019). This is primarily because these inhibitors play a key role in maintaining cell quiescence and the early G1 phase, thereby hindering entry into the S phase. Blocking these inhibitory signals can promote cells to advance, shortening the G1 phase, accelerating cell proliferation, and rapidly accumulating the expression of pluripotency transcription factors. Studies have shown that, among the four reprogramming factors proposed by Yamanaka, Myc is most closely associated with cell cycle regulation (Daksis et al., 1994; Washburn et al., 2022). Consistently, overexpression of CyclinB1-CDK1 (Wang X. Q. et al., 2017; Yang G. Q. et al., 2023), CyclinD-CDK4/6, or CyclinE/A-CDK2, as well as downregulation of pRb, can increase reprogramming efficiency by promoting rapid cell proliferation (Ruiz et al., 2011; Zhan et al., 2019). Conversely, ectopic expression of cell cycle inhibitors such as p15, p16, or p21 (Ruiz et al., 2011), or use of small-molecule inhibitors targeting CDK2 or CDK4 (Zhan et al., 2019), impedes cell cycle progression and reduces reprogramming efficiency. CDK2-mediated phosphorylation of Sox2 at Ser-39/Ser-253 is essential for reprogramming mouse embryo fibroblasts (MEFs) to a pluripotent state but is not required for maintaining mESCs(Ouyang et al., 2015). The p53 pathway is the central guardian of genomic integrity in somatic cells and is likewise activated to cope with the drastic cellular state remodeling and consequent replication stress during reprogramming. Ectopic expression of OSKM directly induces DNA damage and replication stress, thereby activating p53. p53 upregulates p21 transcriptionally, and p21, as a broad-spectrum CKI, induces G1 arrest. This provides the cells with a window to repair DNA damage or to initiate apoptosis, thereby effectively preventing the emergence of potentially mutated cells, but at the cost of substantially reduced reprogramming efficiency (Marión R. M. et al., 2009a). p16 is another important senescence-associated CDK inhibitor that maintains the tumor-suppressive function of the Rb protein by specifically inhibiting CDK4/6, thus triggering G1 arrest. In senescent or aged donor somatic cells, p16 expression is higher, constituting an age-related barrier to reprogramming efficiency (Li H. et al., 2009). The intact functions of p53 and p16 are crucial to ensuring the genomic quality of iPSCs. Transient inhibition of the p53–p21 axis or p16 can markedly improve reprogramming efficiency by allowing more cells to pass the G1/S checkpoint and enter cell-cycle states that are more conducive to reprogramming. Complete ablation of their function, while it can dramatically increase efficiency, leads to iPSC clones with genomic instability and a significantly heightened risk of tumorigenicity. Therefore, during reprogramming, the ideal strategy is to transiently dampen rather than completely ablate the functions of p53–p21 and p16. Most somatic cells have short telomeres and low telomerase (TERT) activity.
The aggressive proliferation induced by OSKM accelerates telomere shortening and telomere dysfunction, which is recognized by the cell as DNA damage and similarly activates the p53–p21 pathway, eliciting senescence or apoptosis (Marion et al., 2009b). The Shelterin complex serves as the protective cap of telomeres. During reprogramming, alterations occur in the expression and function of Shelterin components. As a result, exposed telomeres are recognized as double-strand breaks. This recognition activates the ATM/ATR–mediated DNA damage response pathway. In connection with reprogramming, co-expression of TERT can mitigate telomere erosion, lessen the DNA damage response, and thereby improve reprogramming efficiency, particularly in late-passage somatic cells. Studies show that OSKM itself upregulates TERT and certain Shelterin proteins, and successful reprogramming is accompanied by telomere elongation and restoration of function, marking a key step toward achieving true pluripotency.
DNA replication occurs in the S phase of the cell cycle. However, replication is not uniform but follows a strict spatiotemporal order. The replication timing (RT) is highly correlated with the three-dimensional (3D) chromatin structure and transcriptional activity. Somatic cells exhibit RT patterns that are unique to their cell type. PSCs exhibit another distinct, tightly coordinated RT pattern, characterized by more early-replication domains and clearer temporal switches (Marion et al., 2009b). In the reprogramming process, remodeling of RT is a late-stage event, yet it is crucial (Matson et al., 2017). Resetting the RT pattern of somatic cells to a pluripotent-state RT pattern embodies and preconditions comprehensive epigenomic reorganization. Incomplete RT remodeling leads to fork progression at the wrong time and place, exacerbating transcription–replication conflicts (TRCs), thereby triggering replication stress, DNA double-strand breaks, and genomic instability. This renders the S phase a potential bottleneck for reprogramming failure. Efficient reprogramming requires the coordination of RT remodeling with epigenetic modifications and changes in 3D genome architecture. CDK activity directly influences chromatin structure and the selection of replication origins by phosphorylating histones, chromatin regulators, and replication initiation proteins, thereby indirectly regulating the remodeling of RT. Moreover, CDK kinase activity participates in a positive feedback loop with pluripotency factors, directly catalyzing the reprogramming process. CDK1/2-mediated phosphorylation of Oct4 enhances its transcriptional activity and protein stability. CDK1/2-mediated phosphorylation of Sox2 affects its DNA-binding capacity and transcriptional activity. OSKM also exerts feedback regulation on CDKs: the OSKM factors upregulate genes that promote cell cycle progression while suppressing CDK inhibitors such as p16 and p21, thereby actively creating a proliferation-promoting cell-cycle environment. The use of small-molecule CDK inhibitors or CKIs can modulate the cell cycle to indirectly influence reprogramming efficiency. This synergistic interaction forms a positive feedback loop: OSKM promotes cell cycle progression, enabling more cells to enter a permissive state, and CDK-mediated phosphorylation enhances OSKM activity, leading to more efficient reprogramming.
Alterations in the expression of pluripotency factors also play a central role in cell cycle restructuring during reprogramming. During the initial phase of reprogramming, the expression levels of exogenous transcription factors such as Oct4, Sox2, Klf4, and Nanog gradually increase. Studies indicate that core pluripotency factors not only activate pluripotency-related genes but also influence cell cycle status by regulating the expression of cell cycle proteins. For example, Oct4 has been shown to suppress p21 expression, thereby promoting entry into the S phase and accelerating cell division to improve proliferative efficiency (Lu Y. et al., 2019). c-Myc-driven upregulation of miR-17-92 enhances reprogramming efficiency by targeting p21 for inhibition (Ishida et al., 2023). Knockdown of Nanog significantly inhibits cell proliferation by upregulating p27 and p21 via the p53 pathway, leading to G0/G1 phase cell cycle arrest and regulation of Dlk1. Furthermore, Nanog suppression reduces Dnmt1 expression, which also be involved in cell cycle regulation (Pitrone et al., 2019). Additional studies have demonstrated that upon radiation-induced DNA damage, Klf4 can inhibit the expression of CyclinD1 and CyclinB1, thereby blocking the transition of the cell cycle at both the G1/S and G2/M checkpoints. This highlights the critical role of Klf4 in maintaining the integrity of the G2/M checkpoint following DNA damage (Yoon and Yang, 2004). These pluripotency factors modulate cell cycle protein expression and are reciprocally regulated by cell cycle states, forming a bidirectional control system that ensures sufficient proliferation capacity while activating pluripotency genes to complete reprogramming.
Furthermore, the restructuring of the cell cycle during reprogramming involves epigenetic regulation. Activation of epigenetic modifications—such as histone deacetylation, DNA demethylation, and the establishment of an open chromatin state—contributes to the activation and expression of pluripotency genes (Guo et al., 2025). In this process, the cooperation between cell cycle proteins and pluripotency factors facilitates a stably reprogrammed state by modulating the cell cycle, thereby maintaining chromatin in a transcriptionally permissive configuration conducive to the establishment of pluripotency.
It is important to note that while cell cycle reprogramming enhances reprogramming efficiency, it also poses potential risks of genomic instability. Rapidly proliferating cell states are prone to chromosomal breaks, aneuploidy, and mutation accumulation, thereby increasing the risk of genomic damage. Consequently, balancing rapid cell proliferation with the stability of genomic integrity has become a key focus of current research. During reprogramming, by regulating the expression of cell cycle proteins and simultaneously enhancing pathways involved in DNA repair and chromosome stability, reprogramming efficiency can be improved while reducing the risk of genomic instability.
In summary, cell cycle reprogramming during somatic cell reprogramming is jointly driven by reciprocal regulation between cell cycle proteins and pluripotency factors. The upregulation of cell cycle proteins such as CyclinD, CyclinE, and CyclinA, combined with the activation of pluripotency factors, promotes a rapid entry into the S phase, facilitating the transition of cells from a quiescent to a highly proliferative state. This process relies not only on the regulation of cell cycle protein expression but also involves the activation of pluripotency gene networks, which form positive feedback loops to reinforce the pluripotent state. Furthermore, epigenetic modifications further promote chromatin accessibility and the expression of pluripotency genes, significantly enhancing reprogramming efficiency. However, the rapid proliferation associated with this process also increases the risk of genomic instability. Researchers are continuously exploring strategies to maintain genomic integrity while achieving high reprogramming efficiency, aiming to enable the safer and more effective transformation of somatic cells into PSCs. These studies not only reveal the complex relationship between cell cycle proteins and pluripotency factors but also provide a theoretical foundation and practical guidance for optimizing reprogramming technologies and developing safer cell-based therapies. The p53 pathway is a major barrier to cellular reprogramming, and researchers have explored transient inhibition of the p53 pathway with small molecules during reprogramming and withdrawal of these compounds in the late stage to improve reprogramming efficiency without overly compromising genomic safeguarding functions (Marión et al., 2009a). In addition, Vitamin C, a commonly used antioxidant, can serve as a cofactor for epigenetic modifiers (e.g., promoting histone demethylation) to remodel the epigenome, alleviating oxidative stress and associated DNA damage, thereby generating higher-quality iPSCs more efficiently (Esteban et al., 2010). Non-integrating gene delivery systems (such as episomal plasmids or mRNA transfection) are developed to avoid insertional mutations and ensure genomic stability (Yu J. et al., 2009). These strategies aim to balance reprogramming efficiency with safety, producing high-quality iPSCs for clinical applications.
Somatic cell reprogramming is accompanied by remodeling of the somatic cell cycle, endowing cells with cycle characteristics similar to those of PSCs. Synthesizing prior research, we propose that cell cycle remodeling is not a passive outcome but an active driver and a key barrier of reprogramming. The longer G1 phase in somatic cells, maintained by high levels of CKIs such as p21 and p16, constitutes a major obstacle because it provides a window for differentiation signals and activates tumor suppressor pathways under reprogramming pressure. Successful reprogramming requires overcoming these barriers by inhibiting the p53/p21 and p16/Rb pathways and by exogenous expression of core pluripotency factors to upregulate proliferative Cyclins and CDKs. This drives metabolic and epigenetic shifts toward a glycolytic state and open chromatin, further promoting cell cycle acceleration. The resulting shortened G1 phase limits exposure to differentiation signals and creates favorable conditions for establishing pluripotency. However, this forced rapid proliferation comes at the cost of increased genomic instability due to replication stress. Therefore, transiently modulating cell cycle checkpoints or strategies targeting specific cell cycle phases are crucial for improving the efficiency and quality of iPSC generation.
7 A practical toolkit for manipulating the cell cycle in stem cell biology and reprogramming
The intricate coupling between the cell cycle and cell fate presents both a challenge and an opportunity for experimental manipulation. This toolbox summarizes practical strategies for harnessing cell cycle regulators to improve the efficiency and quality of stem cell maintenance, differentiation, and reprogramming (Table 3). Key reagents, applications, and crucial considerations are outlined below.
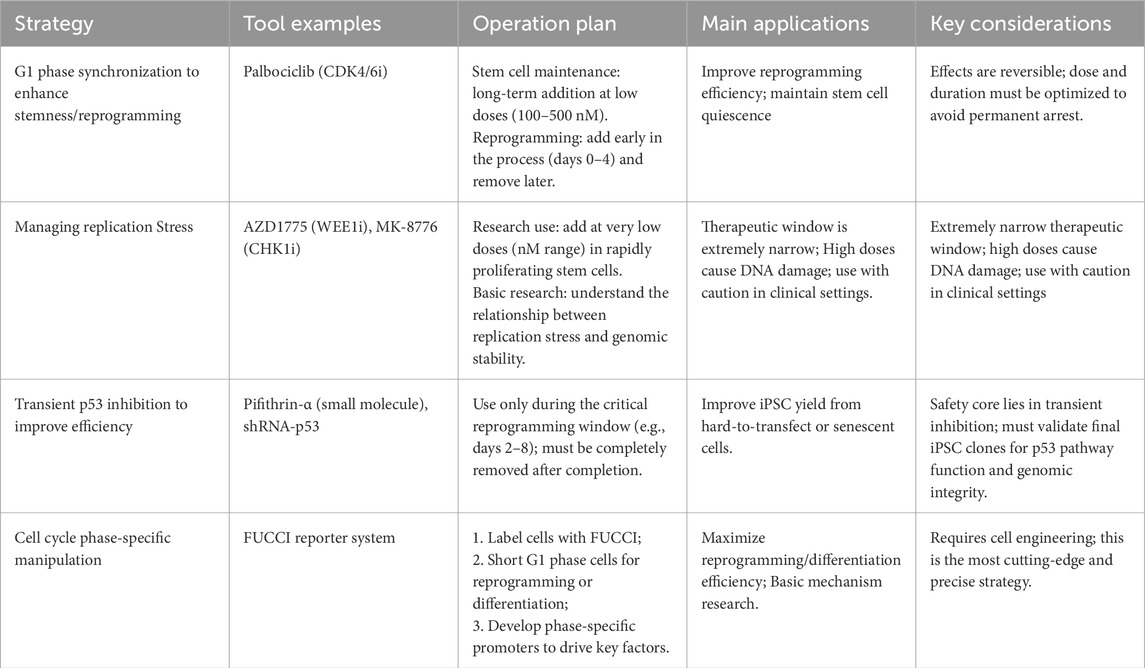
Table 3. Summary of the experimental toolkit for cell cycle manipulation.
7.1 Modulating CDK4/6 and CDK2 activity: driving proliferation and fate decisions
1. Tools:
• CDK4/6 inhibition: Palbociclib (PD-0332991), Abemaciclib, Ribociclib (highly selective ATP-competitive inhibitors).
• CDK2 inhibition: CVT-313, SU9516.
2. Strategy:
• Stem Cell Maintenance: Long-term, low-dose treatment (100–500 nM Palbociclib) can reversibly arrest cells in G1, reduce spontaneous differentiation, and promote the preservation of a quiescent stem cell pool. The inhibitor must be withdrawn for subsequent expansion or differentiation.
• Enhancing Reprogramming: Adding a CDK4/6 or CDK2 inhibitor during the early phase (days 0–4) of somatic cell reprogramming synchronizes the population in G1, a state perceived to be more permissive for reprogramming initiation. Subsequent withdrawal in the middle/late phase, potentially combined with pro-proliferative signals (e.g., Vitamin C), can enhance clonal expansion of successfully reprogrammed cells, thereby significantly increasing iPSC yield (Rais et al., 2013).
• Prospects and Notes: This approach is valuable for optimizing organoid cultures, iPSC generation, and the expansion of stem cells for therapeutic products. The well-established safety profiles of CDK4/6 inhibitors in oncology provide a valuable reference for translational applications.
7.2 Fine-tuning replication stress management via WEE1/CHK1
1. Tools:
• WEE1 inhibition: AZD1775 (Adavosertib).
• CHK1 inhibition: MK-8776 (SCH900776), LY2606368 (Prexasertib).
2. Strategy:
• Alleviating Replication Stress: Low-dose (nM range, requires meticulous titration) inhibition of WEE1 or CHK1 in rapidly proliferating PSCs can promote replication fork progression and alleviate endogenous replication stress, thereby reducing DNA damage accumulation (Rogers et al., 2020; Saini et al., 2015).
• Purging Low-Quality Cells: Transient, low-dose CHK1 inhibition can be applied after high-throughput screening to selectively eliminate subpopulations with high replication stress and DNA damage, thereby enriching for healthier cells.
• Prospects and Notes: Primarily a research tool for investigating the role of replication stress in cell fate decisions. Clinical translation requires extreme caution due to the risk of genomic instability.
7.3 Transient p53 inhibition: safely enhancing reprogramming efficiency
1. Tools:
• siRNA/shRNA: targeting TP53 mRNA.
• Small molecule inhibitors: Pifithrin-α (PFTα, soluble), Pifithrin-μ (PFTμ, mitochondrial p53 inhibitor).
2. Strategy and Safety Protocol:
• Core Principle: Inhibition must be strictly transient. Apply inhibitors or induce shRNA expression only during the critical window of reprogramming (e.g., days 2–8).
• Safety Imperative: The inhibitor must be completely withdrawn or shRNA expression shut off after reprogramming is complete (i.e., upon iPSC colony formation) to allow full restoration of the p53 pathway in the resulting iPSCs. This is non-negotiable for genomic fidelity.
• Validation: iPSC clones must be rigorously validated for genomic integrity (karyotyping, genome sequencing) and functional p53 pathway response.
• Prospects and Notes: This is a gold-standard method for reprogramming recalcitrant or senescent cells. Safety is entirely contingent on the transience of inhibition (Marión R. M. et al., 2009a).
7.4 FUCCI/CDK biosensors and timed factor delivery: Precision control
1. Tools:
• FUCCI system: Lentivectors expressing mVenus-hGeminin (107-130)/mCherry-hCdt1 (30-120). Red (G1), Yellow (G1/S), Green (S/G2/M) (Sakaue-Sawano et al., 2008).
• CDK activity sensors: CDK2 activity reporter (based on Cyclin E-AKAR FRET sensor).
• Timed delivery: Inducible expression systems (e.g., tetracycline-inducible promoters), optogenetic tools for protein degradation (e.g., B-LID system).
2. Strategy:
• Infect the target cell population with the FUCCI system to enable real-time, non-invasive monitoring and sorting of cells based on cell cycle phase via flow cytometry or live-cell imaging.
• Experimental discovery: FACS-sorted G1 cells exhibit higher reprogramming competence.
• Precision intervention: Engineer inducible expression systems activated in specific cell cycle phases (e.g., using a CDT1 promoter active in late G1) to drive the expression of critical reprogramming factors (e.g., GLIS1).
• Prospects and Notes: It enables the dissection of cell cycle-specific events in fate decisions and paves the way for synchronized, high-quality, large-scale stem cell production protocols.
7.5 How to use this toolkit
1. Define the Goal: Determine if the aim is expansion, reprogramming, differentiation, or mechanistic study.
2. Select the Strategy: Choose the most appropriate approach from the table below.
3. Optimize via Pilot Studies: Perform dose- and time-response experiments to define optimal conditions for your specific cell type.
4. Rigorously Validate: Any intervention must be followed by functional assays to confirm the desired outcome (e.g., improved reprogramming efficiency, maintained pluripotency, successful differentiation) and, crucially, genomic stability.
Translating the theory of coupling between the cell cycle and pluripotency into practical applications requires a precise toolkit. This section provides a procedural guide for experimentally manipulating the cell cycle to achieve desirable outcomes in stem cell culture, reprogramming, and differentiation. We offer a toolbox that includes pharmacological inhibitors (such as CDK4/6 inhibitor Palbociclib, WEE1/CHK1 inhibitors, transient p53 inhibitors), genetic tools (siRNA/shRNA, inducible systems), and advanced biosensors (for example, FUCCI systems for real-time cell cycle phase tracking). The strategic use of these tools depends on the context: inhibiting CDK4/6 can be employed to synchronize cells in G1 to boost the initiation of reprogramming or to maintain stem cell quiescence. Transient p53 inhibition can bypass aging barriers in reprogramming but must be carefully controlled to avoid genomic instability. Additionally, real-time monitoring with FUCCI enables isolation of cells in specific cell cycle phases with diverse fate potentials. The overarching principle is the transient and precise nature of these interventions, aiming to mimic physiological transitions and to ensure that resultant stem cell populations are of high quality and safety for research and therapeutic applications.
8 Cell cycle machinery in ASCs
8.1 Cell fate decisions and cell cycle regulation in NSC
NSCs constitute a population capable of self-renewal and multipotent differentiation, capable of giving rise to neurons, astrocytes, and oligodendrocytes. They play a critical role in neural development, homeostasis, and repair following injury (Li X. Y. et al., 2022). The biological behavior of NSCs is tightly controlled by the cell cycle, and the precision of this regulation is essential for maintaining NSC stability and ensuring timely differentiation. In recent years, increasing evidence has shown that disruptions in the neural cell cycle regulatory network not only impairs normal neural development but are also closely associated with the occurrence of various neurodegenerative diseases and brain tumors.
In NSCs, the progression of the G1 phase is particularly critical, as it determines whether the cell continues proliferation or exits the cell cycle to undergo differentiation or remain quiescent. Similar to the process of somatic cell reprogramming, the complex of CyclinD and CDK4/6 phosphorylates Rb, relieving its inhibition of E2F transcription factors. This promotes the expression of CyclinE and activates CDK2, thereby facilitating the cell’s transition through the G1/S checkpoint into the S phase (Grison and Atanasoski, 2020). Rb family proteins inhibit the expression of cell cycle genes such as CyclinA2/E2 and CDK1/2 by suppressing the activation of E2F1/3, thus maintaining NSCs in a quiescent state. When Rb is absent, E2F is released from repression, leading to the expression of CyclinA2/E2-CDK1/2 complexes, promoting G1/S transition and triggering NSC activation and cell cycle progression (Fong et al., 2022). In NSCs, E2F3a and E2F3b function as cell cycle effectors that precisely regulate Sox2 expression level, which counteract each other to modulate the transcription of the pluripotency factors. An overexpression of Sox2 promotes progenitor self-renewal and S retention, whereas reduced Sox2 levels drive cell cycle exit and differentiation (Julian et al., 2013). Recent studies have shown that overexpression of CDK4 shortens the G1 phase, while the loss of CDK6 results in decreased proliferative capacity of NSCs, suggesting that CDK6 may have a unique role in the cell cycle regulation of NSCs(Grison and Atanasoski, 2020; Shih et al., 2022). Additionally, research indicates that under hypoxic conditions, fetal NSC cell cycle progression is delayed, mainly characterized by G2/M phase arrest during the interphase nuclear migration process; short-term exposure to hyperoxia significantly shortens the cell cycle duration, whereas prolonged hyperoxia prolongs it. Both types of oxygen treatment promote the transition of cells from G0/G1 into the S or G2/M phases (Chou et al., 2022; Lanto et al., 2025). Notably, cell cycle regulation in NSCs exhibits clear spatiotemporal specificity, presenting distinct regulatory patterns across different brain regions and developmental stages. This heterogeneity may form a fundamental basis for the diversity within NSC populations.
In the adult mammalian brain, most NSCs remain in a quiescent state (Morales and Mira, 2019; Morshead et al., 1994), but specific external signals can trigger their exit from quiescence into a proliferative state—a dynamic transition termed reactivation. Among the regulatory factors, RingoA directly binds and activates CDK1/2, increasing the activation threshold of CDKs and promoting the re-entry of shallow quiescent NSCs into the cell cycle (Gonzalez et al., 2023). Moreover, Lrig1 facilitates cell cycle re-entry and EGFR response readiness by allowing an increase in EGFR levels while simultaneously limiting its signal activation (Marqués-Torrejón et al., 2021).
At the metabolic level, the mitochondrial pyruvate carrier (MPC), composed of heterodimers of MPC1 and MPC2, is an essential structural complex required for pyruvate transport (Herzig et al., 2012; Vanderperre et al., 2016). Studies have shown that Mpc1 expression is highest in quiescent NSCs and gradually downregulated during differentiation. MPC-mediated mitochondrial import of pyruvate is a key fundamental for maintaining the metabolic homeostasis of the quiescent state (Petrelli et al., 2023). The core of NSC fate decisions lies in a dual regulatory mechanism of the cell cycle, with recent research revealing key pathways involved in its dynamic balance. In physiological quiescence, the Notch-Hey1 pathway continuously suppresses neuronal differentiation genes, collaborating with G1 phase arrest to sustain stemness. Meanwhile, Cend1 promotes cell cycle exit by blocking Notch signaling and remodeling the G1/S transition checkpoint (Harada et al., 2021; Wang R. et al., 2022; Zhang et al., 2020). Notably, both mechanisms ultimately converge at the G1/S transition hub. Furthermore, SUMOylation also plays a crucial role by modifying the Wts kinase to inhibit Hippo pathway activity, thereby relieving its repression of Yki and promoting NSC exit from quiescence into the cell cycle. This process is driven by amino acid-activated Akt signaling and provides a key molecular switch for neurogenesis and brain development in Drosophila (Gao et al., 2024).
In addition to positive regulators, cell cycle inhibitory proteins are equally important in NSC cycle regulation. CDK inhibitors such as p21 and p27 can bind to and inhibit CDK-Cyclin complexes, inducing NSCs to exit the cell cycle (Coqueret, 2003). Research indicates that p21 is expressed in adult NSCs and early neural progenitors, maintaining stem cell quiescence by inhibiting cell cycle progression; its absence leads to excessive proliferation and accelerated differentiation of NSCs(Chiani et al., 2024; Maeda et al., 2023; Pechnick et al., 2008). Foxg1 promotes neural progenitor cell proliferation by suppressing p21 and facilitates cell cycle exit and differentiation through the extension of the G1 phase, thereby coordinating the amplification and differentiation of neural progenitor cells (Wang J. et al., 2022).
Recent studies have revealed that non-coding RNAs, particularly microRNAs, play finely tuned roles in NSC cell cycle regulation. For instance, miR-199a-5p, miR-9, and miR-103-3p target key regulators of the Wnt/β-catenin pathway, such as GSK-3β, Hes1, and Ndel1, mediating G1/S transition via CyclinD1 and bidirectionally regulating the balance of NSC proliferation and differentiation (Li et al., 2022b; Yang Y. et al., 2023; Yuan et al., 2021). MiR-9-5p modulates self-renewal and differentiation by targeting the transcription factor Onecut2 (OC2) (Sharma et al., 2024). lncRNAs, such as Pnky, have been found to regulate NSC cell cycle exit and neuronal differentiation through interaction with PTBP1(Ramos et al., 2015). In vitro hypoxia/reoxygenation (OGD/R) induced NSCs, the lncRNA H19 alleviate p53-mediated G1/S checkpoint arrest by inhibiting p53 transcriptional activity and expression levels, thereby promoting cell proliferation and differentiation after OGD/R (Gan et al., 2022).
Aberrant regulation of NSC cell cycle progression is closely associated with the occurrence and progression of various neurological diseases. In neurodegenerative disorders such as Alzheimer’s disease, endoplasmic reticulum (ER) stress activates the Ire1/Xbp1 pathway, which suppresses the transcription of E2F1, leading to G1/S cell cycle arrest in NSCs. This impedes their normal proliferation and induces apoptosis, thereby damaging neural homeostasis (Aceves et al., 2024). Abnormal activation of the p16-Rb pathway is considered a significant factor contributing to decreased neurogenesis in aged individuals. Gene manipulation that inhibits p16 expression can partially restore neurogenic capacity in aged mice (Si et al., 2021). Studies have also demonstrated that CDK4/6 inhibitors such as Palbociclib and N1J effectively suppress the proliferation of glioblastoma stem cells (GSCs), showing promising clinical prospects for glioblastoma treatment (Li M. et al., 2017; Sun et al., 2025). However, the presence of NSC-like tumor cells renders CDK4/6 inhibitors ineffective in significantly improving the survival of glioblastoma patients. Recent studies have revealed that tGLI1 is a key driver of multi-drug resistance in glioblastoma. The FDA-approved drug KCZ targeting tGLI1, in combination with CDK4/6 inhibitors, has demonstrated synergistic antitumor effects in preclinical models, offering a mechanism-dependent combination therapeutic strategy for glioblastoma (Yu et al., 2025).
In NSCs, the cell cycle does not merely govern cell proliferation. It is a central hub that integrates internal and external signals to determine the balance among quiescence, activation, and differentiation. This section discusses how cell cycle progression, particularly the G1/S transition, governs NSC fate within neurogenic niches. Quiescent NSCs are maintained in a reversible G0 state by high levels of CKIs such as p21 and p27. Activation to enter the cell cycle is a tightly regulated process involving downregulation of CKIs, upregulation of D-type Cyclins, and participation of specific signals such as RingoA and LRIG1. Metabolic status, especially mitochondrial pyruvate input mediated by MPC, is closely linked to maintenance of quiescence. Additionally, this section summarizes the role of noncoding RNAs in fine-tuning the expression of cell cycle regulators and key signaling pathways (Notch, Wnt/β-catenin). Disruptions in this precise control, such as persistent endoplasmic reticulum stress or abnormal expression of p16, can lead to age-related neurogenesis decline and neuropathologies (including gliomas), for which cell cycle targets offer therapeutic prospects. Therefore, in NSCs, the cell cycle is a key determinant of their regenerative potential and homeostatic function.
8.2 Commonalities and specificities of cell cycle regulation in ASCs
HSCs are a typical cell type in a deeply quiescent state. Studies have shown that p57 is a key molecule maintaining HSC quiescence, and its loss leads to accelerated cell cycle progression and exhaustion of HSCs(Matsumoto et al., 2011). p21 and p27 also fulfill essential regulatory roles. Their expression is upregulated following external stressors, including DNA damage, thereby contributing to the maintenance of HSC quiescence and homeostasis (Cheng et al., 2000). This “triple safety net” ensures that HSCs are activated only in response to strong regenerative signals. Intestinal stem cells (ISCs) are classic quiescent reserve stem cells. They also express high levels of p57 and p21 to sustain quiescence. Upon injury, these quiescent ISCs can be rapidly activated to replenish the rapidly cycling Lgr5+ ISCs(Buczacki et al., 2013). This indicates that even in a rapidly renewing tissue, a quiescent stem cell protected by CKIs is required as a backup. Muscle satellite cells (MuSCs) at steady state are almost entirely quiescent. p27 and p21 are central to maintaining their quiescence. Studies show that MuSCs lacking p27 spontaneously enter the cell cycle and eventually exhaust due to excessive proliferation, losing regenerative capacity (Chakkalakal et al., 2012). Thus, CDKIs are a common key group of proteins maintaining quiescence across multiple ASCs. Their function is to establish a high cell-cycle threshold to prevent unnecessary proliferation in the absence of proper cues, thereby preventing premature cellular senescence.
Despite the universal regulation by CDKIs, signals from distinct niches are also organized in a tissue-specific manner to tailor these cell-cycle components to their unique physiological functions. In HSCs, TGF-β strongly inhibits cell-cycle progression by upregulating CKIs such as p57 and p21 (Yamazaki et al., 2009). Angpt1 promotes adhesion of HSCs to the bone marrow niche via its receptor Tie2 and likewise maintains quiescence by upregulating p27 (Arai et al., 2004). Therefore, HSCs primarily maintain quiescence by restricting entry into the cell cycle. In contrast, Lgr5+ ISCs at the crypt base reside in a microenvironment with high Wnt signaling. The core downstream effector of Wnt signaling, β-catenin, directly transcriptionally activates proto-oncogenes such as Cyclin D1 and c-Myc, driving G1/S transition (van der Flier and Clevers, 2009). Consequently, ISCs primarily promote cell-cycle progression to sustain rapid renewal of the intestinal epithelium. Quiescent ISCs reside in a region with a lower Wnt signaling gradient at the upper part of the crypt. MuSC quiescence is largely maintained by physical adhesion to muscle fibers and by its specialized basement membrane. Notch signaling plays a major role in maintaining MuSC quiescence. Upon injury or after exercise, inhibition of TGF-β family members is relieved, while mitogens such as FGF and HGF are released. These signals act synergistically to downregulate CKIs such as p27 and upregulate Cyclins/CDKs, driving MuSC activation and entry into the cell cycle for repair (Yin et al., 2013). Distinct types of ASCs precisely modulate the activity of core cell cycle proteins through unique signaling configurations to adapt to the physiological demands of their respective niches.
8.3 HSC quiescence control: mechanisms and transplantation applications
HSCs are a well-established model for research on cell cycle quiescence and the maintenance of stemness, both of which have important clinical implications. The functional homeostasis of HSCs depends on the preservation of their quiescent state, the precise regulation of signaling pathways, and the dynamic balance of the cell cycle. Elucidating the core molecular mechanisms not only deepens our understanding of hematopoiesis but also provides critical targets for optimizing hematopoietic stem cell transplantation (HSCT) strategies.
The quiescent state of HSCs (G0 phase) is a key safeguard of their long-term self-renewal capacity. This state is finely regulated by CKIs. p21 directly inhibits the association of cyclin–kinase complexes by forming regulatory networks with molecules such as Dkk1, NF-Y, and Skp2, thereby hindering the G0-to-G1 transition. p57 augments the quiescence of HSCs by associating with cell-cycle regulators such as p27 and Cyclin D2. p18 similarly participates in maintaining quiescence through a comparable mechanism. Collectively, these CKIs constitute a regulatory network that prevents HSC exhaustion due to excessive proliferation. In addition, the quiescent state of HSCs is regulated through multiple signals from the bone marrow niche. For example, the TGF-β pathway inhibits proliferation by downregulating c-Kit and IL6R expression while also enhancing the activity of p21 and p57. Furthermore, Angiopoietin-1 (Angpt1) binds to its receptor Tie2, which strengthens adhesion between HSCs and osteolineage cells in the niche. This interaction helps anchor HSCs within a microenvironment that supports low proliferation. Notably, Tie2+ HSCs have been confirmed to be an Long-Term Self-Renewing population at the apex of the hematopoietic hierarchy (Mann et al., 2022).
Activation of HSCs from quiescence and entry into the cell cycle is a multi-signal-coordinated process in which the c-Kit signaling axis plays a pivotal “on/off” role. c-Kit, a type III receptor tyrosine kinase, dimerizes upon binding its ligand stem cell factor (SCF) and undergoes autophosphorylation of intracellular tyrosine residues. In particular, the juxtamembrane region (JM) contains core phosphorylation sites Y568 and Y570: Y568 recruits signaling molecules such as Src family kinases (SFK) and SHP-2, while Y570 mediates binding of SHP-1 and Shc, thereby initiating downstream signaling. However, c-Kit–mediated proliferative signaling is constrained by multiple negative regulators, including SHP-1 (binding Y570) and SHP-2 (binding Y568), which, as protein tyrosine phosphatases (PTPs), can dephosphorylate key tyrosine residues on c-Kit to inhibit activation of pro-proliferative pathways such as PI3K/MAPK, creating a “signal brake” mechanism (Raghav and Gangenahalli, 2018). The small-molecule compound NSC87877 can specifically inhibit the enzymatic activities of SHP-1 (IC50 = 0.355 μM) and SHP-2 (IC50 = 0.318 μM), thereby relieving their inhibition of c-Kit signaling and cooperatively promoting proliferation with SCF(Raghav et al., 2018a). In K562 cells sorted in the G0/G1 phases, pre-treatment with NSC87877 followed by SCF addition (Pre-treatment) increased the proliferation rate to 40.28%, markedly higher than the control (29.25%) and SCF-alone treatment (33.27%) groups (Raghav et al., 2018b). In the human megakaryoblastic cell line MO7e, this pre-treatment raised the S-phase cell fraction from 17.89% (control) to 53.88%, and the proportion of Ki-67+ cells also rose significantly, indicating that the synergistic action of NSC87877 and SCF effectively promotes the transition of HSCs from quiescence to the proliferative cycle (Raghav et al., 2018a).
Activation of the c-Kit signaling pathway triggers deeper metabolic and signaling rewiring. Resting HSCs rely on FoxO3a-mediated transcriptional control to maintain high expression of antioxidant enzymes, ensuring a low ROS environment that sustains self-renewal and quiescence (Miyamoto et al., 2007). Following SCF binding to c-Kit, PI3K/Akt signaling is activated, inhibiting FoxO3 function and steering the metabolic program toward mitochondrial OXPHOS to supply energy for proliferation (Liang and Ghaffari, 2017; Mann et al., 2022). Concurrently, the MAPK pathway is co-activated, downregulating cell cycle inhibitors such as p21, thereby relieving cell cycle arrest and collectively driving the transition of HSCs from quiescence to proliferation (Mann et al., 2022; Raghav and Gangenahalli, 2018).
Notably, although SCF treatment reduces surface c-Kit expression, inhibition of SHP-1/SHP-2 with NSC87877 markedly increases phosphorylation at key c-Kit tyrosine residues (e.g., pY568/pY570), indicating that the activation state of c-Kit, rather than its expression level, is the critical regulator of HSC proliferation. This finding explains why low-dose SCF (20 ng/mL) in combination with NSC87877 (5 μM) robustly promotes proliferation, reducing reliance on high-dose SCF in clinical applications (Raghav et al., 2018a; Raghav et al., 2018b).
The above molecular mechanisms provide multiple potential targets to optimize HSCT strategies. For example, improved HSC quiescence can be achieved by inhibiting Dkk1, whereas activating Angpt1/Tie2 signaling can enhance engraftment efficiency post-transplantation. The synergistic activation strategy of the c-Kit pathway, particularly the NSC87877 and SCF combination, has become a core approach for ex vivo HSC expansion. Studies show that pre-treating bone marrow–derived CD34+ HSCs with NSC87877 for 1 h before adding SCF increases proliferation by approximately 50%, and significantly enhances clonogenic capacity while preserving multipotency (Raghav and Gangenahalli, 2018; Raghav et al., 2018a). This strategy can also reduce SCF usage from 80 ng/mL to 20 ng/mL, lowering clinical costs and minimizing potential side effects (Raghav et al., 2018a; Raghav et al., 2018b).
In summary, quiescence maintenance and activation-driven proliferation of HSCs are governed by multi-layer regulation, including the CKI network, multiple signaling pathways (TGF-β, Angpt1–Tie2, c-Kit), and metabolic reprogramming. Among these, the cooperative activation of c-Kit signaling by SCF and NSC87877, by counteracting the negative regulation of SHP-1/SHP-2, provides an effective strategy for ex vivo expansion and maintenance of HSC stemness in HSCT. Further optimization of pretreatment regimens is expected to significantly enhance both the quantity and quality of HSCs required for transplantation, ultimately improving the clinical outcomes of HSCT (Raghav and Gangenahalli, 2018; Raghav et al., 2018a; Raghav et al., 2018b).
The regulation of quiescence and proliferation in ASCs is a highly tailored process that is essential for long-term tissue maintenance and regenerative capacity. This section compares the cell cycle control mechanisms across different types of ASCs, highlighting their general dependence on CKIs such as p21, p27, and p57 to enforce a deep quiescent state (G0) and prevent exhaustion. However, responses to niche signals that trigger activation show significant tissue specificity. HSCs predominantly respond to signals that reinforce quiescence, with activation involving precise coordination through receptors such as c-Kit and their negative regulators (SHP-1/SHP-2). In contrast, ISCs exist in a high-Wnt environment, which, via expression of Cyclin D1, actively promotes proliferation while maintaining quiescent reserve stem cells as a backup. MuSCs represent another pattern, in which the quiescent phase is maintained by inhibitory signals and is disrupted by pro-activation signals after injury. This comparative analysis indicates that core cell cycle machinery is broadly conserved but is finely tuned by niche-specific signaling networks to meet each tissue’s unique homeostasis and regenerative needs.
9 Conclusion
In recent years, with the continuous advancement of stem cell research, the coupling mechanisms between stemness maintenance and cell cycle regulation have become a focal point in life sciences. Multiple studies confirm that the unique cell cycle characteristics of stem cells, particularly their rapid proliferation ability, are fundamental to maintaining pluripotency and self-renewal. This process is regulated not only by classical cell cycle regulators such as Cyclins and CDKs, but also involves complex regulatory networks with pluripotency-associated transcription factors. The interaction between cell cycle proteins and pluripotency factors in stem cells not only modulates cell cycle progression but also directly influences the expression levels of pluripotency genes, establishing a robust link between proliferation and the maintenance of stemness. Meanwhile, important cell cycle inhibitory proteins such as p21 and p27 play significant roles in regulating self-renewal, ensuring that stem cells proliferate rapidly while avoiding genomic instability by controlling cell cycle entry and arrest.
Regarding regulatory mechanisms, the unique cell cycle structure of ESCs results from the coordinated action of multiple layers of regulation, including epigenetic modifications, signaling pathways, metabolism, and cell cycle regulators (Nardiello et al., 2011). Epigenetic modifications play an indispensable role in ESC cell cycle regulation; chromatin remodeling and changes in its modification status regulate the expression of cell cycle genes. For example, the cooperation between DNA demethylase Tet1 and PRC2 enhances H3K27me3 marks, stabilizing the expression of pluripotency genes (Chrysanthou et al., 2022a; Ficz et al., 2011). Moreover, signaling pathways provide external regulatory cues that support the cell cycle architecture of ESCs. Pathways such as Jak/Stat3, MAPK/Erk, and PI3K/Akt play dual roles in maintaining pluripotency and regulating the cell cycle (Singh et al., 2014).
On the one hand, these mechanisms promote rapid cell proliferation; on the other hand, they activate mitotic regulators, integrating environmental signals with intracellular cell cycle regulation. Furthermore, alterations in glycolytic metabolism are intimately linked to cell cycle control. Elevated glycolytic activity not only supplies rapid energy for stem cells but also regulates the expression of cell cycle proteins, thereby facilitating stem cell proliferation and the maintenance of pluripotency (Dong et al., 2024). This network—the interplay among metabolism, the cell cycle, and stemness—offers a novel perspective for understanding stem cell self-renewal and presents potential targets for metabolic regulation of stem cell states.
Looking ahead, there remains significant potential for research into the coupling mechanisms between stem cells and the cell cycle. Our preliminary studies utilizing CRISPR/Cas9 systems have explored precise modulation of specific regulatory factors to achieve controllable stem cell states (Wang C. C. et al., 2022). As high-throughput sequencing, single-cell analysis, and gene editing technologies continue to advance, it is anticipated that more interactions between cell cycle regulators and pluripotency factors will be uncovered, including their differential regulatory mechanisms across various stem cell types and developmental stages. Future research directions also include the synchronized regulation of cell cycle dynamics and chromatin structural changes. Specifically, how to ensure rapid stem cell proliferation while maximizing genomic stability and preserving genetic integrity. From a clinical perspective, understanding the intricacies of cell cycle regulation in stem cells may help optimize in vitro culture systems, thereby enhancing the safety and efficacy of regenerative medicine therapies.
In addition, strengthening research on the mechanisms of stem cell tumorigenesis should be regarded as a key future direction. Because rapidly proliferating stem cells share characteristics with tumor cells, investigating the relationship between cell cycle dysregulation and tumor development holds potential significance for cancer prevention and treatment. Recently, a research team in Canada examined how cell cycle duration influences tumor susceptibility, discovering that in various tumor types, the overall cell cycle length can predict susceptibility to malignant transformation (Chen D. N. et al., 2025). Future studies should also focus on developing novel strategies that ensure efficient cell proliferation while effectively preventing genomic instability and tumorigenesis.
Cell cycle regulators play a dual role in stem cell fate determination and safety. Traditional stem cell expansion strategies often emphasize proliferative signals while neglecting the genomic instability and tumorigenic risk arising from cell cycle abnormalities. In recent years, cell-cycle–targeted drugs, exemplified by CDK4/6 inhibitors, have provided new approaches to maintaining stemness while preserving genomic integrity and safety. Using CDK4/6 inhibitors (such as Palbociclib, Abemaciclib) can artificially arrest stem cells in the G0/G1 phase, forcing them into a quiescent state. Theoretically, this can prevent exhaustion due to excessive proliferation and sustain long-term self-renewal. For example, in HSCs, high expression of CIP/KIP family CDK inhibitors such as p57 and p27 is central to maintaining quiescence. Studies show that CDK6 loss or pharmacological inhibition can promote HSC quiescence, reduce differentiation, and thereby protect stem cells from chemotherapy-induced damage (Laurenti et al., 2015). This inhibitory effect is typically reversible. Upon drug withdrawal, the cell cycle arrest is lifted and stem cells can re-enter the cycle to proliferate and differentiate. This reversibility is one of the greatest advantages for stem cell culture applications. Researchers can switch flexibly between conditions that require expansion (no inhibitor) and those that require maintenance of quiescence or protection from damage (with inhibitors). For instance, in hPSC culture, brief, low-dose Palbociclib application effectively suppresses spontaneous differentiation and maintains the expression of pluripotency markers, and after withdrawal, normal proliferation and differentiation capabilities can be restored without observed permanent cell-cycle arrest (Pauklin and Vallier, 2013). It is important to note that this forced quiescence acts as a double-edged sword. While it can preserve stem cell function, excessive or irreversible inhibition may prevent stem cells from responding to normal regenerative signals. This could result in functional exhaustion and impaired tissue repair.
In clinical-stage stem cell applications, manipulating the cell cycle can optimize proliferation and differentiation strategies to reduce tumorigenic risk. One approach is to rapidly expand cells using pro-proliferation signals, followed by a short introduction of CDK4/6 inhibitors or other cell cycle blockers such as mTOR inhibitors to induce a “pause” and allow DNA repair, thereby increasing genomic integrity during expansion. This dynamic regulation more closely mimics the cyclic control of in vivo stem cells and is advantageous for maintaining a healthy stem cell compartments compared with continuous stimulation or inhibition. Additionally, residual undifferentiated hPSCs pose a major tumorigenic risk due to their limitless proliferative potential. Targeting cell cycle checkpoints to eliminate these cells is a highly promising strategy. Antiproliferative agents such as 5-fluorouracil (5-FU) can selectively kill rapidly proliferating undifferentiated hPSCs while having relatively little impact on differentiated cells that have slowed or exited the cell cycle.
Our review synthesizes prior work in the field and proposes that the cell cycle is not merely the central driver of cell division but also a key component of stemness maintenance and fate determination. The coupling between a shortened cell cycle (especially a short G1 phase) and pluripotent states is mediated by a multi-faceted network, including reciprocal regulation between core pluripotency factors and Cyclin-CDK complexes, integration of key signaling pathways, dynamic epigenetic remodeling, and metabolic reprogramming. This interconnected system ensures tight coupling of rapid proliferation with differentiation suppression. Looking forward, to dissect causality in depth, advanced single-cell technologies and real-time biosensors should be employed to resolve the temporal sequence of these interactions during fate transitions. A major translational challenge lies in how to harness this knowledge to safely and effectively manipulate the cell cycle. For example, by using transient CDK inhibition to enhance reprogramming efficiency or expand clinically relevant stem cells without compromising genomic integrity. Moreover, studying how cell cycle dysregulation contributes to stem cell–derived pathologies (such as teratoma formation) is crucial for developing safer regenerative therapies. Cracking the precise regulation of the cell cycle will accelerate the translation of stem cell research into medical and biological applications.
Author contributions
XH: Conceptualization, Data curation, Investigation, Validation, Visualization, Writing – original draft, Writing – review and editing. YW: Writing – review and editing, Investigation, Validation. QL: Validation, Writing – review and editing. XL: Writing – review and editing. CW: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the National Natural Science Foundation of China (32300615).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aceves, M., Granados, J., Leandro, A. C., Peralta, J., Glahn, D. C., Williams-Blangero, S., et al. (2024). Role of neurocellular endoplasmic reticulum stress response in alzheimer's disease and related dementias risk. Genes. 15 (5), 569. doi:10.3390/genes15050569
Agathocleous, M., Locker, M., Harris, W. A., and Perron, M. (2007). A general role of hedgehog in the regulation of proliferation. Cell Cycle 6 (2), 156–159. doi:10.4161/cc.6.2.3745
Alba, G., Martínez, R., Postigo-Corrales, F., López, S., Santa-María, C., Jiménez, J., et al. (2020). AICAR stimulates the pluripotency transcriptional complex in embryonic stem cells mediated by PI3K, GSK3β, and β-Catenin. ACS Omega 5 (32), 20270–20282. doi:10.1021/acsomega.0c02137
Arai, F., Hirao, A., Ohmura, M., Sato, H., Matsuoka, S., Takubo, K., et al. (2004). Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 118 (2), 149–161. doi:10.1016/j.cell.2004.07.004
Azad, F. M., Struys, E. A., Wingert, V., Hannibal, L., Mills, K., Jansen, J. H., et al. (2023). Spic regulates one-carbon metabolism and histone methylation in ground-state pluripotency. Sci. Adv. 9 (33), 14. doi:10.1126/sciadv.adg7997
Bar-On, O., Shapira, M., Skorecki, K., Hershko, A., and Hershko, D. D. (2010). Regulation of APC/C Cdh1 ubiquitin ligase in differentiation of human embryonic stem cells. Cell Cycle 9 (10), 1986–1989. doi:10.4161/cc.9.10.11727
Barker, P. A., and Salehi, A. (2002). The MAGE proteins: emerging roles in cell cycle progression, apoptosis, and neurogenetic disease. J. Neurosci. Res. 67 (6), 705–712. doi:10.1002/jnr.10160
Basak, T., and Ain, R. (2022). Molecular regulation of trophoblast stem cell self-renewal and giant cell differentiation by the Hippo components YAP and LATS1. Stem Cell Res. Ther. 13 (1), 189. doi:10.1186/s13287-022-02844-w
Becker, K. A., Ghule, P. N., Lian, J. B., Stein, J. L., Van Wijnen, A. J., and Stein, G. S. (2010). Cyclin D2 and the CDK substrate p220(NPAT) are required for self-renewal of human embryonic stem cells. J. Cell Physiol. 222 (2), 456–464. doi:10.1002/jcp.21967
Besson, A., Gurian-West, M., Chen, X. Y., Kelly-Spratt, K. S., Kemp, C. J., and Roberts, J. M. (2006). A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes. Dev. 20 (1), 47–64. doi:10.1101/gad.1384406
Besson, A., Dowdy, S. F., and Roberts, J. M. (2008). CDK inhibitors: cell cycle regulators and beyond. Dev. Cell 14 (2), 159–169. doi:10.1016/j.devcel.2008.01.013
Biswas, K., Philip, S., Yadav, A., Martin, B. K., Burkett, S., Singh, V., et al. (2018). BRE/BRCC45 regulates CDC25A stability by recruiting USP7 in response to DNA damage. Nat. Commun. 9, 537. doi:10.1038/s41467-018-03020-6
Boulton, T. G., Stahl, N., and Yancopoulos, G. D. (1994). Ciliary neurotrophic factor/leukemia inhibitory factor/interleukin 6/oncostatin M family of cytokines induces tyrosine phosphorylation of a common set of proteins overlapping those induced by other cytokines and growth factors. J. Biol. Chem. 269 (15), 11648–11655. doi:10.1016/s0021-9258(19)78174-5
Boward, B., Wu, T., and Dalton, S. (2016). Concise review: control of cell fate through cell cycle and pluripotency networks. Stem Cells 34 (6), 1427–1436. doi:10.1002/stem.2345
Boyer, L. A., Lee, T. I., Cole, M. F., Johnstone, S. E., Levine, S. S., Zucker, J. P., et al. (2005). Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122 (6), 947–956. doi:10.1016/j.cell.2005.08.020
Buczacki, S. J. A., Zecchini, H. I., Nicholson, A. M., Russell, R., Vermeulen, L., Kemp, R., et al. (2013). Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature 495 (7439), 65–69. doi:10.1038/nature11965
Cacchiarelli, D., Trapnell, C., Ziller, M. J., Soumillon, M., Cesana, M., Karnik, R., et al. (2015). Integrative analyses of human reprogramming reveal dynamic nature of induced pluripotency. Cell 162 (2), 412–424. doi:10.1016/j.cell.2015.06.016
Calder, A., Roth-Albin, I., Bhatia, S., Pilquil, C., Lee, J. H., Bhatia, M., et al. (2013). Lengthened G1 phase indicates differentiation status in human embryonic stem cells. Stem Cells Dev. 22 (2), 279–295. doi:10.1089/scd.2012.0168
Campbell, K. H., McWhir, J., Ritchie, W. A., and Wilmut, I. (1996). Sheep cloned by nuclear transfer from a cultured cell line. Nature 380 (6569), 64–66. doi:10.1038/380064a0
Carey, B. W., Finley, L. W. S., Cross, J. R., Allis, C. D., and Thompson, C. B. (2015). Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518 (7539), 413–416. doi:10.1038/nature13981
Chae, H. D., Lee, M. R., and Broxmeyer, H. E. (2012). 5-Aminoimidazole-4-carboxyamide ribonucleoside induces G1/S arrest and nanog downregulation via p53 and enhances erythroid differentiation. Stem Cells 30 (2), 140–149. doi:10.1002/stem.778
Chaigne, A., Labouesse, C., Agnew, M., Hannezo, E., Chalut, K. J., Paluch, E. K., et al. (2020). Abscission couples cell division to embryonic stem cell fate. Dev. Cell 55 (2), 195–208. doi:10.1016/j.devcel.2020.09.001
Chakkalakal, J. V., Jones, K. M., Basson, M. A., and Brack, A. S. (2012). The aged niche disrupts muscle stem cell quiescence. Nature 490 (7420), 355–360. doi:10.1038/nature11438
Chakrabarty, R. P., and Chandel, N. S. (2021). Mitochondria as signaling organelles control mammalian stem cell fate. Cell Stem Cell 28 (3), 394–408. doi:10.1016/j.stem.2021.02.011
Chen, D. S., Zhou, L. J., Sun, F. L., Sun, M. Z., and Tao, X. Q. (2018). Cyclin B3 deficiency impairs germline stem cell maintenance and its overexpression delays cystoblast differentiation in Drosophila ovary. Int. J. Mol. Sci. 19 (1), 298. doi:10.3390/ijms19010298
Chen, F. Q., Zhang, M., Feng, X., Li, X. M., Sun, H. T., and Lu, X. Y. (2021a). Discovery of a novel long noncoding RNA Lx8-SINE B2 as a marker of pluripotency. Stem Cells Int. 2021, 6657597. doi:10.1155/2021/6657597
Chen, F. Q., Li, X. M., Feng, X., Gao, T. T., Zhang, W. Y., Cheng, Z., et al. (2022b). Long noncoding RNA Lx8-SINE B2 interacts with Eno1 to regulate self-renewal and metabolism of embryonic stem cells. Stem Cells 40 (12), 1094–1106. doi:10.1093/stmcls/sxac067
Chen, D. N., Lu, S. Y., Huang, K., Pearson, J. D., Pacal, M., Peidis, P., et al. (2025). Cell cycle duration determines oncogenic transformation capacity. Nature 641 (8065), 1309–1318. doi:10.1038/s41586-025-08935-x
Cheng, T., Rodrigues, N., Shen, H., Yang, Y., Dombkowski, D., Sykes, M., et al. (2000). Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 287 (5459), 1804–1808. doi:10.1126/science.287.5459.1804
Chiani, F., Mastrorilli, V., Marchetti, N., Macioce, A., Nappi, C., Strimpakos, G., et al. (2024). Essential role of p21Waf1/Cip1 in the modulation of post-traumatic hippocampal neural stem cells response. Stem Cell Res. Ther. 15 (1), 197. doi:10.1186/s13287-024-03787-0
Choi, H. W., Kim, J. H., Chung, M. K., Hong, Y. J., Jang, H. S., Seo, B. J., et al. (2015). Mitochondrial and metabolic remodeling during reprogramming and differentiation of the reprogrammed cells. Stem Cells Dev. 24 (11), 1366–1373. doi:10.1089/scd.2014.0561
Chou, F. S., Chen, C. Y., Lee, A. C., and Wang, P. S. (2022). Impaired cell cycle progression and self-renewal of fetal neural stem and progenitor cells in a murine model of intrauterine growth restriction. Front. Cell Dev. Biol. 10, 821848. doi:10.3389/fcell.2022.821848
Chrysanthou, S., Flores, J. C., and Dawlaty, M. M. (2022a). Tet1 suppresses p21 to ensure proper cell cycle progression in embryonic stem cells. Cells 11 (8), 1366. doi:10.3390/cells11081366
Chrysanthou, S., Tang, Q., Lee, J., Taylor, S. J., Zhao, Y. L., Steidl, U., et al. (2022b). The DNA dioxygenase Tet1 regulates H3K27 modification and embryonic stem cell biology independent of its catalytic activity. Nucleic Acids Res. 50 (6), 3169–3189. doi:10.1093/nar/gkac089
Coqueret, O. (2003). New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 13 (2), 65–70. doi:10.1016/s0962-8924(02)00043-0
Coronado, D., Godet, M., Bourillot, P. Y., Tapponnier, Y., Bernat, A., Petit, M., et al. (2013). A short G1 phase is an intrinsic determinant of naive embryonic stem cell pluripotency. Stem Cell Res. 10 (1), 118–131. doi:10.1016/j.scr.2012.10.004
Coverley, D., Laman, H., and Laskey, R. A. (2002). Distinct roles for cyclins E and A during DNA replication complex assembly and activation. Nat. Cell Biol. 4 (7), 523–528. doi:10.1038/ncb813
D'Arcangelo, D., Tinaburri, L., and Dellambra, E. (2017). The role of p16INK4a pathway in human epidermal stem cell self-renewal, aging and cancer. Int. J. Mol. Sci. 18 (7), 1591. doi:10.3390/ijms18071591
Daksis, J. I., Lu, R. Y., Facchini, L. M., Marhin, W. W., and Penn, L. J. (1994). Myc induces cyclin D1 expression in the absence of de novo protein synthesis and links mitogen-stimulated signal transduction to the cell cycle. Oncogene 9 (12), 3635–3645.
Dalton, S. (2013). G1 compartmentalization and cell fate coordination. Cell 155 (1), 13–14. doi:10.1016/j.cell.2013.09.015
Dalton, S. (2015). Linking the cell cycle to cell fate decisions. Trends Cell Biol. 25 (10), 592–600. doi:10.1016/j.tcb.2015.07.007
Darnell, J. E., Kerr, I. M., and Stark, G. R. (1994). Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264 (5164), 1415–1421. doi:10.1126/science.8197455
Daynac, M., Tirou, L., Faure, H., Mouthon, M. A., Gauthier, L. R., Hahn, H., et al. (2016). Hedgehog controls quiescence and activation of neural stem cells in the adult ventricular-subventricular zone. Stem Cell Rep. 7 (4), 735–748. doi:10.1016/j.stemcr.2016.08.016
Deng, L., Zhang, X. P., Xiang, X. C., Xiong, R., Xiao, D. Q., Chen, Z., et al. (2021). NANOG promotes cell proliferation, invasion, and stemness via IL-6/STAT3 signaling in esophageal squamous carcinoma. Technol. Cancer Res. Treat. 20, 15330338211038492. doi:10.1177/15330338211038492
DeVeale, B., Liu, L. Q., Boileau, R., Swindlehurst-Chan, J., Marsh, B., Freimer, J. W., et al. (2022). G1/S restriction point coordinates phasic gene expression and cell differentiation. Nat. Commun. 13 (1), 3696. doi:10.1038/s41467-022-31101-0
Do, D. V., Ueda, J., Messerschmidt, D. M., Lorthongpanich, C., Zhou, Y., Feng, B., et al. (2013). A genetic and developmental pathway from STAT3 to the OCT4-NANOG circuit is essential for maintenance of ICM lineages in vivo. Genes. Dev. 27 (12), 1378–1390. doi:10.1101/gad.221176.113
Doetsch, F., Verdugo, J.M.-G., Caille, I., Alvarez-Buylla, A., Chao, M. V., and Casaccia-Bonnefil, P. (2002). Lack of the cell-cycle inhibitor p27Kip1 results in selective increase of transit-amplifying cells for adult neurogenesis. J. Neurosci. 22 (6), 2255–2264. doi:10.1523/jneurosci.22-06-02255.2002
Dong, Q. M., Zhang, Q. Y., Yang, X. Q., Nai, S. S., Du, X. L., and Chen, L. Y. (2024). Glycolysis-stimulated esrrb lactylation promotes the self-renewal and extraembryonic endoderm stem cell differentiation of embryonic stem cells. Int. J. Mol. Sci. 25 (5), 2692. doi:10.3390/ijms25052692
Edgar, B. A., and Lehner, C. F. (1996). Developmental control of cell cycle regulators: a fly's perspective. Science 274 (5293), 1646–1652. doi:10.1126/science.274.5293.1646
Egozi, D., Shapira, M., Paor, G., Ben-Izhak, O., Skorecki, K., and Hershko, D. D. (2007). Regulation of the cell cycle inhibitor p27 and its ubiquitin ligase Skp2 in differentiation of human embryonic stem cells. FASEB J. 21 (11), 2807–2817. doi:10.1096/fj.06-7758com
Engeland, K. (2022). Cell cycle regulation: P53-p21-RB signaling. Cell Death Differ. 29 (5), 946–960. doi:10.1038/s41418-022-00988-z
Errico, A., Deshmukh, K., Tanaka, Y., Pozniakovsky, A., and Hunt, T. (2010). Identification of substrates for cyclin dependent kinases. Adv. Enzyme Regul. 50 (1), 375–399. doi:10.1016/j.advenzreg.2009.12.001
Esteban, M. A., Wang, T., Qin, B., Yang, J., Qin, D., Cai, J., et al. (2010). Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell 6 (1), 71–79. doi:10.1016/j.stem.2009.12.001
Evans, M. J., and Kaufman, M. H. (1981). Establishment in culture of pluripotential cells from mouse embryos. Nature 292 (5819), 154–156. doi:10.1038/292154a0
Fan, S. S., Guo, C. L., Yang, G. H., Hong, L., Li, H. Y., Ma, J., et al. (2024). GPR160 regulates the self-renewal and pluripotency of mouse embryonic stem cells via JAK1/STAT3 signaling pathway. J. Genet. Genomics 51 (10), 1055–1065. doi:10.1016/j.jgg.2024.05.003
Ficz, G., Branco, M. R., Seisenberger, S., Santos, F., Krueger, F., Hore, T. A., et al. (2011). Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 473 (7347), 398–402. doi:10.1038/nature10008
Fischer, M., and Müller, G. A. (2017). Cell cycle transcription control: dream/muvb and RB-E2F complexes. Crit. Rev. Biochem. Mol. Biol. 52 (6), 638–662. doi:10.1080/10409238.2017.1360836
Fleifel, D., and Cook, J. G. (2023). G1 dynamics at the crossroads of pluripotency and cancer. Cancers 15 (18), 4559. doi:10.3390/cancers15184559
Fong, B. C., Chakroun, I., Iqbal, M. A., Paul, S., Bastasic, J., O'Neil, D., et al. (2022). The Rb/E2F axis is a key regulator of the molecular signatures instructing the quiescent and activated adult neural stem cell state. Cell Rep. 41 (5), 111578. doi:10.1016/j.celrep.2022.111578
Fox, P. M., Vought, V. E., Hanazawa, M., Lee, M. H., Maine, E. M., and Schedl, T. (2011). Cyclin E and CDK-2 regulate proliferative cell fate and cell cycle progression in the C. elegans germline. Development 138 (11), 2223–2234. doi:10.1242/dev.059535
Furutachi, S., Matsumoto, A., Nakayama, K. I., and Gotoh, Y. (2013). p57 controls adult neural stem cell quiescence and modulates the pace of lifelong neurogenesis. EMBO J. 32 (7), 970–981. doi:10.1038/emboj.2013.50
Gan, L., Liao, S. T., Tong, Y. Q., Li, W. H., Peng, W. L., and Deng, S. X. (2022). Long noncoding RNA H19 mediates neural stem/progenitor cells proliferation, differentiation and apoptosis through the p53 signaling pathway after ischemic stroke. Biochem. Biophys. Res. Commun. 597, 8–15. doi:10.1016/j.bbrc.2022.01.095
Gao, Y., Tan, Y. S., Lin, J. E., Chew, L. Y., Aung, H. Y., Palliyana, B., et al. (2024). SUMOylation of Warts kinase promotes neural stem cell reactivation. Nat. Commun. 15 (1), 8557. doi:10.1038/s41467-024-52569-y
Gearing, D. P., Thut, C. J., VandeBos, T., Gimpel, S. D., Delaney, P. B., King, J., et al. (1991). Leukemia inhibitory factor receptor is structurally related to the IL-6 signal transducer, gp130. EMBO J. 10 (10), 2839–2848. doi:10.1002/j.1460-2075.1991.tb07833.x
Gonnot, F., Langer, D., Bourillot, P. Y., Doerflinger, N., and Savatier, P. (2019). Regulation of Cyclin E by transcription factors of the naive pluripotency network in mouse embryonic stem cells. Cell Cycle 18 (20), 2697–2712. doi:10.1080/15384101.2019.1656475
Gonzales, K. A. U., Liang, H., Lim, Y. S., Chan, Y. S., Yeo, J. C., Tan, C. P., et al. (2015). Deterministic restriction on pluripotent state dissolution by cell-cycle pathways. Cell 162 (3), 564–579. doi:10.1016/j.cell.2015.07.001
Gonzalez, L., Domingo-Muelas, A., Duart-Abadia, P., Nuñez, M., Mikolcevic, P., Llonch, E., et al. (2023). The atypical CDK activator RingoA/Spy1 regulates exit from quiescence in neural stem cells. iScience 26 (3), 106202. doi:10.1016/j.isci.2023.106202
Grison, A., and Atanasoski, S. (2020). Cyclins, cyclin-dependent kinases, and cyclin-dependent kinase inhibitors in the mouse nervous system. Mol. Neurobiol. 57 (7), 3206–3218. doi:10.1007/s12035-020-01958-7
Guglielmi, L., Heliot, C., Kumar, S., Alexandrov, Y., Gori, I., Papaleonidopoulou, F., et al. (2021). Smad4 controls signaling robustness and morphogenesis by differentially contributing to the Nodal and BMP pathways. Nat. Commun. 12 (1), 6374. doi:10.1038/s41467-021-26486-3
Guiley, K. Z., Liban, T. J., Felthousen, J. G., Ramanan, P., Litovchick, L., and Rubin, S. M. (2015). Structural mechanisms of DREAM complex assembly and regulation. Genes. Dev. 29 (9), 961–974. doi:10.1101/gad.257568.114
Guo, L., Lin, J. C., Ren, Q. W., Sun, H., Wu, Y. H., Ge, H. F., et al. (2025). Enhanced activities of OCT4 and SOX2 promote epigenetic reprogramming by shortening G1 phase. Adv. Sci. (Weinh). 12, e15528. doi:10.1002/advs.202415528
Gurdon, J. B. (1962). The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J. Embryol. Exp. Morphol. 10, 622–640. doi:10.1242/dev.10.4.622
Hall, J., Guo, G., Wray, J., Eyres, I., Nichols, J., Grotewold, L., et al. (2009). Oct4 and LIF/Stat3 additively induce kruppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell 5 (6), 597–609. doi:10.1016/j.stem.2009.11.003
Han, J. H., Zhang, F., Yu, M., Zhao, P. Q., Ji, W., Zhang, H. C., et al. (2012). RNA interference-mediated silencing of NANOG reduces cell proliferation and induces G0/G1 cell cycle arrest in breast cancer cells. Cancer Lett. 321 (1), 80–88. doi:10.1016/j.canlet.2012.02.021
Harada, Y., Yamada, M., Imayoshi, I., Kageyama, R., Suzuki, Y., Kuniya, T., et al. (2021). Cell cycle arrest determines adult neural stem cell ontogeny by an embryonic Notch-nonoscillatory Hey1 module. Nat. Commun. 12 (1), 6562. doi:10.1038/s41467-021-26605-0
Herzig, S., Raemy, E., Montessuit, S., Veuthey, J. L., Zamboni, N., Westermann, B., et al. (2012). Identification and functional expression of the mitochondrial pyruvate carrier. Science 337 (6090), 93–96. doi:10.1126/science.1218530
Huang, Y. K., Su, T., Wang, C. C., Dong, L. X., Liu, S., Zhu, Y. R., et al. (2021). Rbbp4 suppresses premature differentiation of embryonic stem cells. Stem Cell Rep. 16 (3), 566–581. doi:10.1016/j.stemcr.2021.01.009
Huskey, N. E., Guo, T. X., Evason, K. J., Momcilovic, O., Pardo, D., Creasman, K. J., et al. (2015). CDK1 inhibition targets the p53-NOXA-MCL1 axis, selectively kills embryonic stem cells, and prevents teratoma formation. Stem Cell Rep. 4 (3), 374–389. doi:10.1016/j.stemcr.2015.01.019
Hydbring, P., Malumbres, M., and Sicinski, P. (2016). Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat. Rev. Mol. Cell Biol. 17 (5), 280–292. doi:10.1038/nrm.2016.27
Inoshita, S., Terada, Y., Nakashima, O., Kuwahara, M., Sasaki, S., and Marumo, F. (1999). Regulation of the G1/S transition phase in mesangial cells by E2F1. Kidney Int. 56 (4), 1238–1241. doi:10.1046/j.1523-1755.1999.00705.x
Ishida, T., Ueyama, T., Ihara, D., Harada, Y., Nakagawa, S., Saito, K., et al. (2023). c-Myc/microRNA-17-92 axis phase-dependently regulates PTEN and p21 expression via ceRNA during reprogramming to mouse pluripotent stem cells. Biomedicines 11 (6), 1737. doi:10.3390/biomedicines11061737
Iyer, D. P., Khoei, H. H., van der Weijden, V. A., Kagawa, H., Pradhan, S. J., Novatchkova, M., et al. (2024). mTOR activity paces human blastocyst stage developmental progression. Cell 187 (23), 6566–6583.e22. doi:10.1016/j.cell.2024.08.048
Jeong, C. H., Cho, Y. Y., Kim, M. O., Kim, S. H., Cho, E. J., Lee, S. Y., et al. (2010). Phosphorylation of Sox2 cooperates in reprogramming to pluripotent stem cells. Stem Cells 28 (12), 2141–2150. doi:10.1002/stem.540
Jiang, J. F., Qiu, T., Yang, C., Yuan, Y., Qin, L., and Zhang, P. X. (2022). Atypical cell cycle profile of mouse embryonic stem cell is regulated by classic oncogenic and tumor suppressive genes in vitro. Heliyon 8 (12), e11979. doi:10.1016/j.heliyon.2022.e11979
Joshi, J. N., Lerner, A. D., Scallo, F., Grumet, A. N., Matteson, P., Millonig, J. H., et al. (2024). mTORC1 activity oscillates throughout the cell cycle, promoting mitotic entry and differentially influencing autophagy induction. Cell Rep. 43 (8), 114543. doi:10.1016/j.celrep.2024.114543
Julian, L. M., Vandenbosch, R., Pakenham, C. A., Andrusiak, M. G., Nguyen, A. P., McClellan, K. A., et al. (2013). Opposing regulation of Sox2 by cell-cycle effectors E2f3a and E2f3b in neural stem cells. Cell Stem Cell 12 (4), 440–452. doi:10.1016/j.stem.2013.02.001
Kalaszczynska, I., Geng, Y., Iino, T., Mizuno, S. I., Choi, Y., Kondratiuk, I., et al. (2009). Cyclin A is redundant in fibroblasts but essential in hematopoietic and embryonic stem cells. Cell 138 (2), 352–365. doi:10.1016/j.cell.2009.04.062
Kareta, M. S., Gorges, L. L., Hafeez, S., Benayoun, B. A., Marro, S., Zmoos, A. F., et al. (2015). Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 16 (1), 39–50. doi:10.1016/j.stem.2014.10.019
Kato, J., Matsushime, H., Hiebert, S. W., Ewen, M. E., and Sherr, C. J. (1993). Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes. Dev. 7 (3), 331–342. doi:10.1101/gad.7.3.331
Kelleher, F. C., and O'Sullivan, H. (2016). FOXM1 in sarcoma: role in cell cycle, pluripotency genes and stem cell pathways. Oncotarget 7 (27), 42792–42804. doi:10.18632/oncotarget.8669
Kim, D. Y., Lee, J., Kang, D., Lee, D. H., Kim, Y. J., Hwang, S. G., et al. (2016). Multipotent neurogenic fate of mesenchymal stem cell is determined by Cdk4-mediated hypophosphorylation of Smad-STAT3. Cell Cycle 15 (13), 1787–1795. doi:10.1080/15384101.2016.1188230
Koledova, Z., Krämer, A., Kafkova, L. R., and Divoky, V. (2010). Cell-cycle regulation in embryonic stem cells: centrosomal decisions on self-renewal. Stem Cells Dev. 19 (11), 1663–1678. doi:10.1089/scd.2010.0136
Kozar, K., Ciemerych, M. A., Rebel, V. I., Shigematsu, H., Zagozdzon, A., Sicinska, E., et al. (2004). Mouse development and cell proliferation in the absence of D-cyclins. Cell 118 (4), 477–491. doi:10.1016/j.cell.2004.07.025
Krishnan, B., Yasuhara, T., Rumde, P., Stanzione, M., Lu, C. Y., Lee, H., et al. (2022). Active RB causes visible changes in nuclear organization. J. Cell Biol. 221 (3), e202102144. doi:10.1083/jcb.202102144
Lanto, J., Vehlken, M. M. N., Abramenko, V., Storch, A., and Markert, F. (2025). Hyperoxia shows duration-dependent effects on the lengths of cell cycle phases in fetal cortical neural stem cells. Front. Cell Dev. Biol. 13, 1546131. doi:10.3389/fcell.2025.1546131
Laurenti, E., Frelin, C., Xie, S., Ferrari, R., Dunant, C. F., Zandi, S., et al. (2015). CDK6 levels regulate quiescence exit in human hematopoietic stem cells. Cell Stem Cell 16 (3), 302–313. doi:10.1016/j.stem.2015.01.017
Lee, M. G., and Nurse, P. (1987). Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature 327 (6117), 31–35. doi:10.1038/327031a0
Lee, K. L., Lim, S. K., Orlov, Y. L., Yit, L. Y., Yang, H., Ang, L. T., et al. (2011). Graded Nodal/activin signaling titrates conversion of quantitative Phospho-Smad2 levels into qualitative embryonic stem cell fate decisions. PLoS Genet. 7 (6), e1002130. doi:10.1371/journal.pgen.1002130
Li, H., Collado, M., Villasante, A., Strati, K., Ortega, S., Cañamero, M., et al. (2009). The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460 (7259), 1136–1139. doi:10.1038/nature08290
Li, M., Xiao, A. Z., Floyd, D., Olmez, I., Lee, J., Godlewski, J., et al. (2017). CDK4/6 inhibition is more active against the glioblastoma proneural subtype. Oncotarget 8 (33), 55319–55331. doi:10.18632/oncotarget.19429
Li, P., Gao, L. L., Cui, T. X., Zhang, W. Y., Zhao, Z. X., and Chen, L. Y. (2020). Cops5 safeguards genomic stability of embryonic stem cells through regulating cellular metabolism and DNA repair. Proc. Natl. Acad. Sci. U. S. A. 117 (5), 2519–2525. doi:10.1073/pnas.1915079117
Li, W., Long, Q., Wu, H., Zhou, Y. S., Duan, L. F., Yuan, H., et al. (2022a). Nuclear localization of mitochondrial TCA cycle enzymes modulates pluripotency via histone acetylation. Nat. Commun. 13 (1), 7414. doi:10.1038/s41467-022-35199-0
Li, W., Wang, S. S., Shan, B. Q., Qin, J. B., Zhao, H. Y., Tian, M. L., et al. (2022b). miR-103-3p targets Ndel1 to regulate neural stem cell proliferation and differentiation. Neural Regen. Res. 17 (2), 401–408. doi:10.4103/1673-5374.317987
Li, X. Y., Jiang, X., Gao, Q. S., and Zhao, P. (2022c). FOXO3 regulates sevoflurane-induced neural stem cell differentiation in fetal rats. Cell Mol. Neurobiol. 42 (6), 1777–1786. doi:10.1007/s10571-021-01055-w
Li, J., Wang, Y. H., Wang, Z. Y., Wei, Y. X., Diao, P. F., Wu, Y. P., et al. (2024). Super-enhancer driven LIF/LIFR-STAT3-SOX2 regulatory feedback loop promotes cancer stemness in head and neck squamous cell carcinoma. Adv. Sci. (Weinh). 11 (40), e2404476. doi:10.1002/advs.202404476
Liang, R., and Ghaffari, S. (2017). Mitochondria and FOXO3 in stem cell homeostasis, a window into hematopoietic stem cell fate determination. J. Bioenerg. Biomembr. 49 (4), 343–346. doi:10.1007/s10863-017-9719-7
Lifantseva, N., Koltsova, A., Krylova, T., Yakovleva, T., Poljanskaya, G., and Gordeeva, O. (2011). Expression patterns of cancer-testis antigens in human embryonic stem cells and their cell derivatives indicate lineage tracks. Stem Cells Int. 2011, 795239. doi:10.4061/2011/795239
Lin, T., Chao, C., Saito, S., Mazur, S. J., Murphy, M. E., Appella, E., et al. (2005). p53 induces differentiation of mouse embryonic stem cells by suppressing nanog expression. Nat. Cell Biol. 7 (2), 165–171. doi:10.1038/ncb1211
Lin, Y. J., Yang, Y., Li, W. H., Chen, Q., Li, J., Pan, X., et al. (2012). Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol. Cell 48 (4), 627–640. doi:10.1016/j.molcel.2012.08.030
Litovchick, L., Sadasivam, S., Florens, L., Zhu, X. P., Swanson, S. K., Velmurugan, S., et al. (2007). Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol. Cell 26 (4), 539–551. doi:10.1016/j.molcel.2007.04.015
Liu, Y. J., and Yamashita, J. K. (2019). AMPK activators contribute to maintain naive pluripotency in mouse embryonic stem cells. Biochem. Biophys. Res. Commun. 509 (1), 24–31. doi:10.1016/j.bbrc.2018.11.164
Liu, L. J., Michowski, W., Inuzuka, H., Shimizu, K., Nihira, N. T., Li, N., et al. (2017). G1 cyclins link proliferation, pluripotency and differentiation of embryonic stem cells. Nat. Cell Biol. 19 (3), 177–188. doi:10.1038/ncb3474
Liu, L., Wu, Q., Wang, Z., Niu, B. B., Jiao, Y. G., and An, H. B. (2023). APE1 promotes embryonic stem cell proliferation and teratoma formation by regulating GDNF/GFRα1 axis. Reprod. Biol. 23 (3), 100792. doi:10.1016/j.repbio.2023.100792
Liu, L., Michowski, W., Kolodziejczyk, A., and Sicinski, P. (2019). The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell Biol. 21 (9), 1060–1067. doi:10.1038/s41556-019-0384-4
Lu, C. C., Brennan, J., and Robertson, E. J. (2001). From fertilization to gastrulation: axis formation in the mouse embryo. Curr. Opin. Genet. Dev. 11 (4), 384–392. doi:10.1016/s0959-437x(00)00208-2
Lu, Y., Qu, H. N., Qi, D., Xu, W. H., Liu, S. T., Jin, X. A., et al. (2019). OCT4 maintains self-renewal and reverses senescence in human hair follicle mesenchymal stem cells through the downregulation of p21 by DNA methyltransferases. Stem Cell Res. Ther. 10, 28. doi:10.1186/s13287-018-1120-x
Maeda, Y., Isomura, A., Masaki, T., and Kageyama, R. (2023). Differential cell-cycle control by oscillatory versus sustained Hes1 expression via p21. Cell Rep. 42 (5), 112520. doi:10.1016/j.celrep.2023.112520
Mages, C. F. S., Wintsche, A., Bernhart, S. H., and Müller, G. A. (2017). The DREAM complex through its subunit Lin37 cooperates with Rb to initiate quiescence. eLife 6, e26876. doi:10.7554/eLife.26876
Malfatti, M. C., Antoniali, G., Codrich, M., and Tell, G. (2021). Coping with RNA damage with a focus on APE1, a BER enzyme at the crossroad between DNA damage repair and RNA processing/decay. DNA Repair 104, 103133. doi:10.1016/j.dnarep.2021.103133
Mann, Z., Sengar, M., Verma, Y. K., Rajalingam, R., and Raghav, P. K. (2022). Hematopoietic stem cell factors: their functional role in self-renewal and clinical aspects. Front. Cell. Dev. Biol. 10, 664261. doi:10.3389/fcell.2022.664261
Marión, R. M., Strati, K., Li, H., Murga, M., Blanco, R., Ortega, S., et al. (2009a). A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 460 (7259), 1149–1153. doi:10.1038/nature08287
Marion, R. M., Strati, K., Li, H., Tejera, A., Schoeftner, S., Ortega, S., et al. (2009b). Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 4 (2), 141–154. doi:10.1016/j.stem.2008.12.010
Marqués-Torrejón, M. A., Williams, C. A. C., Southgate, B., Alfazema, N., Clements, M. P., Garcia-Diaz, C., et al. (2021). LRIG1 is a gatekeeper to exit from quiescence in adult neural stem cells. Nat. Commun. 12 (1), 2594. doi:10.1038/s41467-021-22813-w
Martin, G. R. (1981). Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. U. S. A. 78 (12), 7634–7638. doi:10.1073/pnas.78.12.7634
Matson, J. P., Dumitru, R., Coryell, P., Baxley, R. M., Chen, W. L., Twaroski, K., et al. (2017). Rapid DNA replication origin licensing protects stem cell pluripotency. eLife 6, e30473. doi:10.7554/eLife.30473
Matsumoto, A., Takeishi, S., Kanie, T., Susaki, E., Onoyama, I., Tateishi, Y., et al. (2011). p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell 9 (3), 262–271. doi:10.1016/j.stem.2011.06.014
Meyer, K., Lammers, N. C., Bugaj, L. J., Garcia, H. G., and Weiner, O. D. (2023). Optogenetic control of YAP reveals a dynamic communication code for stem cell fate and proliferation. Nat. Commun. 14 (1), 6929. doi:10.1038/s41467-023-42643-2
Michowski, W., Chick, J. M., Chu, C., Kolodziejczyk, A., Wang, Y. C., Suski, J. M., et al. (2020). Cdk1 controls global epigenetic landscape in embryonic stem cells. Mol. Cell 78 (3), 459–476. doi:10.1016/j.molcel.2020.03.010
Mitsui, K., Tokuzawa, Y., Itoh, H., Segawa, K., Murakami, M., Takahashi, K., et al. (2003). The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 113 (5), 631–642. doi:10.1016/s0092-8674(03)00393-3
Miyamoto, K., Araki, K. Y., Naka, K., Arai, F., Takubo, K., Yamazaki, S., et al. (2007). Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 1 (1), 101–112. doi:10.1016/j.stem.2007.02.001
Molofsky, A. V., Slutsky, S. G., Joseph, N. M., He, S. H., Pardal, R., Krishnamurthy, J., et al. (2006). Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443 (7110), 448–452. doi:10.1038/nature05091
Morales, A. V., and Mira, H. (2019). Adult neural stem cells: born to last. Front. Cell Dev. Biol. 7, 96. doi:10.3389/fcell.2019.00096
Morshead, C. M., Reynolds, B. A., Craig, C. G., McBurney, M. W., Staines, W. A., Morassutti, D., et al. (1994). Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron 13 (5), 1071–1082. doi:10.1016/0896-6273(94)90046-9
Murray, A. W., and Kirschner, M. W. (1989). Cyclin synthesis drives the early embryonic cell cycle. Nature 339 (6222), 275–280. doi:10.1038/339275a0
Narasimha, A. M., Kaulich, M., Shapiro, G. S., Choi, Y. J., Sicinski, P., and Dowdy, S. F. (2014). Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 3, e02872. doi:10.7554/eLife.02872
Nardiello, T., Jungbluth, A. A., Mei, A., DiLiberto, M., Huang, X. G., Dabrowski, A., et al. (2011). MAGE-A inhibits apoptosis in proliferating myeloma cells through repression of Bax and maintenance of survivin. Clin. Cancer Res. 17 (13), 4309–4319. doi:10.1158/1078-0432.Ccr-10-1820
Neganova, I., and Lako, M. (2008). G1 to S phase cell cycle transition in somatic and embryonic stem cells. J. Anat. 213 (1), 30–44. doi:10.1111/j.1469-7580.2008.00931.x
Neganova, I., Zhang, X., Atkinson, S., and Lako, M. (2009). Expression and functional analysis of G1 to S regulatory components reveals an important role for CDK2 in cell cycle regulation in human embryonic stem cells. Oncogene 28 (1), 20–30. doi:10.1038/onc.2008.358
Neganova, I., Tilgner, K., Buskin, A., Paraskevopoulou, I., Atkinson, S. P., Peberdy, D., et al. (2014). CDK1 plays an important role in the maintenance of pluripotency and genomic stability in human pluripotent stem cells. Cell Death Dis. 5, e1508. doi:10.1038/cddis.2014.464
Nichols, J., and Smith, A. (2009). Naive and primed pluripotent states. Cell Stem Cell 4 (6), 487–492. doi:10.1016/j.stem.2009.05.015
Oh, H., Fujio, Y., Kunisada, K., Hirota, H., Matsui, H., Kishimoto, T., et al. (1998). Activation of phosphatidylinositol 3-kinase through glycoprotein 130 induces protein kinase B and p70 S6 kinase phosphorylation in cardiac myocytes. J. Biol. Chem. 273 (16), 9703–9710. doi:10.1074/jbc.273.16.9703
Oleksiewicz, U., Gładych, M., Raman, A. T., Heyn, H., Mereu, E., Chlebanowska, P., et al. (2017). TRIM28 and interacting KRAB-ZNFs control self-renewal of human pluripotent stem cells through epigenetic repression of pro-differentiation genes. Stem Cell Rep. 9 (6), 2065–2080. doi:10.1016/j.stemcr.2017.10.031
Onishi, K., and Zandstra, P. W. (2015). LIF signaling in stem cells and development. Development 142 (13), 2230–2236. doi:10.1242/dev.117598
Ouyang, J., Yu, W., Liu, J., Zhang, N., Florens, L., Chen, J. K., et al. (2015). Cyclin-dependent kinase-mediated Sox2 phosphorylation enhances the ability of Sox2 to establish the pluripotent state. J. Biol. Chem. 290 (37), 22782–22794. doi:10.1074/jbc.M115.658195
Palma, M., Riffo, E., Farias, A., Coliboro-Dannich, V., Espinoza-Francine, L., Escalona, E., et al. (2023). NUAK1 coordinates growth factor-dependent activation of mTORC2 and Akt signaling. Cell Biosci. 13 (1), 232. doi:10.1186/s13578-023-01185-2
Park, S., Han, J. E., Kim, H. G., Kim, H. Y., Kim, M. G., Park, J. K., et al. (2020). Inhibition of MAGEA2 regulates pluripotency, proliferation, apoptosis, and differentiation in mouse embryonic stem cells. J. Cell Biochem. 121 (11), 4667–4679. doi:10.1002/jcb.29692
Pauklin, S., and Vallier, L. (2013). The cell-cycle State of stem cells determines cell fate propensity. Cell 155 (1), 135–147. doi:10.1016/j.cell.2013.08.031
Pauklin, S., and Vallier, L. (2014). The cell-cycle state of stem cells determines cell fate propensity. Cell 156 (6), 1338. doi:10.1016/j.cell.2014.02.044
Pauklin, S., Madrigal, P., Bertero, A., and Vallier, L. (2016). Initiation of stem cell differentiation involves cell cycle-dependent regulation of developmental genes by Cyclin D. Genes. Dev. 30 (4), 421–433. doi:10.1101/gad.271452.115
Pechnick, R. N., Zonis, S., Wawrowsky, K., Pourmorady, J., and Chesnokova, V. (2008). p21Cip1 restricts neuronal proliferation in the subgranular zone of the dentate gyrus of the hippocampus. Proc. Natl. Acad. Sci. U. S. A. 105 (4), 1358–1363. doi:10.1073/pnas.0711030105
Pedersen, M. T., Kooistra, S. M., Radzisheuskaya, A., Laugesen, A., Johansen, J. V., Hayward, D. G., et al. (2016). Continual removal of H3K9 promoter methylation by Jmjd2 demethylases is vital for ESC self-renewal and early development. EMBO J. 35 (14), 1550–1564. doi:10.15252/embj.201593317
Petrelli, F., Scandella, V., Montessuit, S., Zamboni, N., Martinou, J. C., and Knobloch, M. (2023). Mitochondrial pyruvate metabolism regulates the activation of quiescent adult neural stem cells. Sci. Adv. 9 (9), eadd5220. doi:10.1126/sciadv.add5220
Ping, W. F., Sheng, Y. L., Hu, G. C., Zhong, H. X., Li, Y. Y., Liu, Y. J., et al. (2023). RBBP4 is an epigenetic barrier for the induced transition of pluripotent stem cells into totipotent 2C-like cells. Nucleic Acids Res. 51 (11), 5414–5431. doi:10.1093/nar/gkad219
Pitrone, M., Pizzolanti, G., Coppola, A., Tomasello, L., Martorana, S., Pantuso, G., et al. (2019). Knockdown of NANOG reduces cell proliferation and induces G0/G1 cell cycle arrest in human adipose stem cells. Int. J. Mol. Sci. 20 (10), 2580. doi:10.3390/ijms20102580
Prigione, A., Rohwer, N., Hoffmann, S., Mlody, B., Drews, K., Bukowiecki, R., et al. (2014). HIF1α modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1–3 and PKM2. Stem Cells 32 (2), 364–376. doi:10.1002/stem.1552
Qin, J. Z., Wang, C. C., Zhu, Y. R., Su, T., Dong, L. X., Huang, Y. K., et al. (2021). Mga safeguards embryonic stem cells from acquiring extraembryonic endoderm fates. Sci. Adv. 7 (4), eabe5689. doi:10.1126/sciadv.abe5689
Raghav, P. K., and Gangenahalli, G. (2018). Hematopoietic stem cell molecular targets and factors essential for hematopoiesis. J. Stem Cell Res. Ther. 8 (11). doi:10.4172/2157-7633.1000441
Raghav, P. K., Singh, A. K., and Gangenahalli, G. (2018a). Stem cell factor and NSC87877 combine to enhance c-Kit mediated proliferation of human megakaryoblastic cells. PLoS One 13 (11), e0206364. doi:10.1371/journal.pone.0206364
Raghav, P. K., Singh, A. K., and Gangenahalli, G. (2018b). Stem cell factor and NSC87877 synergism enhances c-Kit mediated proliferation of human erythroid cells. Life Sci. 214, 84–97. doi:10.1016/j.lfs.2018.09.055
Rais, Y., Zviran, A., Geula, S., Gafni, O., Chomsky, E., Viukov, S., et al. (2013). Deterministic direct reprogramming of somatic cells to pluripotency. Nature 502 (7469), 65–70. doi:10.1038/nature12587
Ramos, A. D., Andersen, R. E., Liu, S. J., Nowakowski, T. J., Hong, S. J., Gertz, C. C., et al. (2015). The long noncoding RNA pnky regulates neuronal differentiation of embryonic and postnatal neural stem cells. Cell Stem Cell 16 (4), 439–447. doi:10.1016/j.stem.2015.02.007
Rogers, R. F., Walton, M. I., Cherry, D. L., Collins, I., Clarke, P. A., Garrett, M. D., et al. (2020). CHK1 inhibition is synthetically lethal with loss of B-Family DNA polymerase function in human lung and colorectal cancer cells. Cancer Res. 80 (8), 1735–1747. doi:10.1158/0008-5472.Can-19-1372
Roufayel, R., Mezher, R., and Storey, K. B. (2021). The role of retinoblastoma protein in cell cycle regulation: an updated review. Curr. Mol. Med. 21 (8), 620–629. doi:10.2174/1566524020666210104113003
Roy, A., Padhi, S. S., Khyriem, I., Nikose, S., Sankar, S. H. H., and Bharathavikru, R. S. (2022). Resetting the epigenome: methylation dynamics in cancer stem cells. Front. Cell Dev. Biol. 10, 909424. doi:10.3389/fcell.2022.909424
Ruiz, S., Panopoulos, A. D., Herrerías, A., Bissig, K. D., Lutz, M., Berggren, W. T., et al. (2011). A high proliferation rate is required for cell reprogramming and maintenance of human embryonic stem cell identity. Curr. Biol. 21 (1), 45–52. doi:10.1016/j.cub.2010.11.049
Sadasivam, S., and DeCaprio, J. A. (2013). The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat. Rev. Cancer 13 (8), 585–595. doi:10.1038/nrc3556
Sadasivam, S., Duan, S. H., and DeCaprio, J. A. (2012). The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes. Dev. 26 (5), 474–489. doi:10.1101/gad.181933.111
Saini, P., Li, Y., and Dobbelstein, M. (2015). Wee1 is required to sustain ATR/Chk1 signaling upon replicative stress. Oncotarget 6 (15), 13072–13087. doi:10.18632/oncotarget.3865
Sakaue-Sawano, A., Kurokawa, H., Morimura, T., Hanyu, A., Hama, H., Osawa, H., et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132 (3), 487–498. doi:10.1016/j.cell.2007.12.033
Santamaría, D., Barrière, C., Cerqueira, A., Hunt, S., Tardy, C., Newton, K., et al. (2007). Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448 (7155), 811–815. doi:10.1038/nature06046
Savatier, P., Huang, S., Szekely, L., Wiman, K. G., and Samarut, J. (1994). Contrasting patterns of retinoblastoma protein expression in mouse embryonic stem cells and embryonic fibroblasts. Oncogene 9 (3), 809–818.
Schade, A. E., Oser, M. G., Nicholson, H. E., and DeCaprio, J. A. (2019). Cyclin D-CDK4 relieves cooperative repression of proliferation and cell cycle gene expression by DREAM and RB. Oncogene 38 (25), 4962–4976. doi:10.1038/s41388-019-0767-9
Seo, B. J., Na, S. B., Choi, J., Ahn, B., Habib, O., Park, C., et al. (2023). Metabolic and cell cycle shift induced by the deletion of Dnm1l attenuates the dissolution of pluripotency in mouse embryonic stem cells. Cell Mol. Life Sci. 80 (10), 302. doi:10.1007/s00018-023-04962-x
Serrano, M., Hannon, G. J., and Beach, D. (1993). A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 366 (6456), 704–707. doi:10.1038/366704a0
Sharma, S., Majumdar, A., and Basu, A. (2024). Regulation of Onecut2 by miR-9-5p in Japanese encephalitis virus infected neural stem/progenitor cells. Microbiol. Spectr. 12 (3), e0323823. doi:10.1128/spectrum.03238-23
Sherr, C. J., and Roberts, J. M. (1999). CDK inhibitors: positive and negative regulators of G1-phase progression. Genes. Dev. 13 (12), 1501–1512. doi:10.1101/gad.13.12.1501
Shih, L. K., Mukherjee, S., and Brat, D. J. (2022). Off the clock: the non-canonical roles of cyclin-dependent kinases in neural and glioma stem cell self-renewal. Mol. Neurobiol. 59 (11), 6805–6816. doi:10.1007/s12035-022-03009-9
Shiraki, N., Shiraki, Y., Tsuyama, T., Obata, F., Miura, M., Nagae, G., et al. (2014). Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 19 (5), 780–794. doi:10.1016/j.cmet.2014.03.017
Shyh-Chang, N., Locasale, J. W., Lyssiotis, C. A., Zheng, Y. X., Teo, R. Y., Ratanasirintrawoot, S., et al. (2013). Influence of threonine metabolism on S-Adenosylmethionine and histone methylation. Science 339 (6116), 222–226. doi:10.1126/science.1226603
Si, Z. Z., Sun, L. L., and Wang, X. D. (2021). Evidence and perspectives of cell senescence in neurodegenerative diseases. Biomed. Pharmacother. 137, 111327. doi:10.1016/j.biopha.2021.111327
Sicinska, E., Aifantis, I., Le Cam, L., Swat, W., Borowski, C., Yu, Q., et al. (2003). Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell 4 (6), 451–461. doi:10.1016/s1535-6108(03)00301-5
Sicinski, P., Donaher, J. L., Parker, S. B., Li, T., Fazeli, A., Gardner, H., et al. (1995). Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 82 (4), 621–630. doi:10.1016/0092-8674(95)90034-9
Singh, A. M., Chappell, J., Trost, R., Lin, L., Wang, T., Tang, J., et al. (2014). Cell-cycle control of developmentally regulated transcription factors accounts for heterogeneity in human pluripotent cells. Stem Cell Rep. 2 (3), 398. doi:10.1016/j.stemcr.2014.02.009
Singh, A. M., Sun, Y. H., Li, L., Zhang, W. J., Wu, T. M., Zhao, S. Y., et al. (2015). Cell-cycle control of bivalent epigenetic domains regulates the exit from pluripotency. Stem Cell Rep. 5 (3), 323–336. doi:10.1016/j.stemcr.2015.07.005
Smith, A. (2017). Formative pluripotency: the executive phase in a developmental continuum. Development 144 (3), 365–373. doi:10.1242/dev.142679
Spelat, R., Ferro, F., and Curcio, F. (2012). Serine 111 phosphorylation regulates OCT4A protein subcellular distribution and degradation. J. Biol. Chem. 287 (45), 38279–38288. doi:10.1074/jbc.M112.386755
Stahl, N., Boulton, T. G., Farruggella, T., Ip, N. Y., Davis, S., Witthuhn, B. A., et al. (1994). Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science 263 (5143), 92–95. doi:10.1126/science.8272873
Stead, E., White, J., Faast, R., Conn, S., Goldstone, S., Rathjen, J., et al. (2002). Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene 21 (54), 8320–8333. doi:10.1038/sj.onc.1206015
Stirparo, G. G., Kurowski, A., Yanagida, A., Bates, L. E., Strawbridge, S. E., Hladkou, S., et al. (2021). OCT4 induces embryonic pluripotency via STAT3 signaling and metabolic mechanisms. Proc. Natl. Acad. Sci. U. S. A. 118 (3), e2008890118. doi:10.1073/pnas.2008890118
Sun, J. H., Liang, S. S., Liu, X. G., Zhang, S. L., Li, M., Zhang, Q. G., et al. (2025). Insights into the selectivity of a brain-penetrant CDK4/6 vs CDK1/2 inhibitor for glioblastoma used in multiple replica molecular dynamics simulations. J. Biomol. Struct. Dyn. 43 (5), 2223–2242. doi:10.1080/07391102.2023.2294175
Surani, M. A., Hayashi, K., and Hajkova, P. (2007). Genetic and epigenetic regulators of pluripotency. Cell 128 (4), 747–762. doi:10.1016/j.cell.2007.02.010
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126 (4), 663–676. doi:10.1016/j.cell.2006.07.024
Takanaga, H., Tsuchida-Straeten, N., Nishide, K., Watanabe, A., Aburatani, H., and Kondo, T. (2009). Gli2 is a novel regulator of Sox2 expression in telencephalic neuroepithelial cells. Stem Cells 27 (1), 165–174. doi:10.1634/stemcells.2008-0580
Ter Huurne, M., and Stunnenberg, H. G. (2021). G1-phase progression in pluripotent stem cells. Cell Mol. Life Sci. 78 (10), 4507–4519. doi:10.1007/s00018-021-03797-8
Theka, I., Sottile, F., Cammisa, M., Bonnin, S., Sanchez-Delgado, M., Di Vicino, U., et al. (2019). Wnt/β-catenin signaling pathway safeguards epigenetic stability and homeostasis of mouse embryonic stem cells. Sci. Rep. 9, 948. doi:10.1038/s41598-018-37442-5
Theunissen, T. W., Powell, B. E., Wang, H. Y., Mitalipova, M., Faddah, D. A., Reddy, J., et al. (2014). Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell 15 (4), 524–526. doi:10.1016/j.stem.2014.09.003
Tian, S., Feng, J., Cao, Y., Shen, S., Cai, Y., Yang, D., et al. (2019). Glycine cleavage system determines the fate of pluripotent stem cells via the regulation of senescence and epigenetic modifications. Life Sci. Alliance 2 (5), e201900413. doi:10.26508/lsa.201900413
Tischler, J., Gruhn, W. H., Reid, J., Allgeyer, E., Buettner, F., Marr, C., et al. (2019). Metabolic regulation of pluripotency and germ cell fate through α-ketoglutarate. EMBO J. 38 (1), e99518. doi:10.15252/embj.201899518
Totaro, A., Castellan, M., Battilana, G., Zanconato, F., Azzolin, L., Giulitti, S., et al. (2017). YAP/TAZ link cell mechanics to notch signalling to control epidermal stem cell fate. Nat. Commun. 8, 15206. doi:10.1038/ncomms15206
Uxa, S., Bernhart, S. H., Mages, C. F. S., Fischer, M., Kohler, R., Hoffmann, S., et al. (2019). DREAM and RB cooperate to induce gene repression and cell-cycle arrest in response to p53 activation. Nucleic Acids Res. 47 (17), 9087–9103. doi:10.1093/nar/gkz635
van der Flier, L. G., and Clevers, H. (2009). Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 71, 241–260. doi:10.1146/annurev.physiol.010908.163145
Van Winkle, L. J., and Ryznar, R. (2019). One-carbon metabolism regulates embryonic stem cell fate through epigenetic DNA and histone modifications: implications for transgenerational metabolic disorders in adults. Front. Cell Dev. Biol. 7, 300. doi:10.3389/fcell.2019.00300
Vanderperre, B., Cermakova, K., Escoffier, J., Kaba, M., Bender, T., Nef, S., et al. (2016). MPC1-like is a placental mammal-specific mitochondrial pyruvate carrier subunit expressed in postmeiotic Male germ cells. J. Biol. Chem. 291 (32), 16448–16461. doi:10.1074/jbc.M116.733840
Vazquez-Martin, A., Van den Haute, C., Cufí, S., Corominas-Faja, B., Cuyàs, E., Lopez-Bonet, E., et al. (2016). Mitophagy-driven mitochondrial rejuvenation regulates stem cell fate. Aging-US 8 (7), 1330–1352. doi:10.18632/aging.100976
Wang, C. C., Hao, K. Y., Dong, L. X., Wang, J. N., Zhao, L. C., Xu, L. J., et al. (2022). The MuvB complex safeguards embryonic stem cell identity through regulation of the cell cycle machinery. J. Biol. Chem. 298 (3), 101701. doi:10.1016/j.jbc.2022.101701
Wang, K., Zhang, T., Dong, Q., Nice, E. C., Huang, C. H., and Wei, Y. Q. (2013). Redox homeostasis: the linchpin in stem cell self-renewal and differentiation. Cell Death Dis. 4, e537. doi:10.1038/cddis.2013.50
Wang, X. Q., Lo, C. M., Chen, L., Ngan, E. S. W., Xu, A. M., and Poon, R. Y. C. (2017). CDK1-PDK1-PI3K/Akt signaling pathway regulates embryonic and induced pluripotency. Cell Death Differ. 24 (1), 38–48. doi:10.1038/cdd.2016.84
Wang, J., Zhai, H. R., Ma, S. F., Shi, H. Z., Zhang, W. J., Yun, Q., et al. (2022). FOXG1 contributes adult hippocampal neurogenesis in mice. Int. J. Mol. Sci. 23 (23), 14979. doi:10.3390/ijms232314979
Wang, R., Yang, D. X., Liu, Y. L., Ding, J., Guo, Y., Ding, W. H., et al. (2022). Cell cycle exit and neuronal differentiation 1-engineered embryonic neural stem cells enhance neuronal differentiation and neurobehavioral recovery after experimental traumatic brain injury. Neural Regen. Res. 17 (1), 130–136. doi:10.4103/1673-5374.314316
Washburn, R. L., Hibler, T., Thompson, L. A., Kaur, G., and Dufour, J. M. (2022). Therapeutic application of sertoli cells for treatment of various diseases. Semin. Cell Dev. Biol. 121, 10–23. doi:10.1016/j.semcdb.2021.04.007
Wattanapanitch, M. (2019). Recent updates on induced pluripotent stem cells in hematological disorders. Stem Cells Int. 2019, 5171032. doi:10.1155/2019/5171032
Weinberger, L., Ayyash, M., Novershtern, N., and Hanna, J. H. (2016). Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 17 (3), 155–169. doi:10.1038/nrm.2015.28
Werwein, E., Biyanee, A., and Klempnauer, K. H. (2020). Intramolecular interaction of B-MYB is regulated through Ser-577 phosphorylation. FEBS Lett. 594 (24), 4266–4279. doi:10.1002/1873-3468.13940
White, J., and Dalton, S. (2005). Cell cycle control of embryonic stem cells. Stem Cell Rev. 1 (2), 131–138. doi:10.1385/scr:1:2:131
Wu, Y., Liu, Y., Hao, Z., and Liu, X. (2022). NAD+ is critical for maintaining acetyl-CoA and H3K27ac in embryonic stem cells by Sirt1-dependent deacetylation of AceCS1. Life Med. 1 (3), 401–405. doi:10.1093/lifemedi/lnac046
Wulansari, N., Sulistio, Y. A., Darsono, W. H. W., Kim, C. H., and Lee, S. H. (2021). LIF maintains mouse embryonic stem cells pluripotency by modulating TET1 and JMJD2 activity in a JAK2-dependent manner. Stem Cells 39 (6), 750–760. doi:10.1002/stem.3345
Xu, T., Su, P., Wu, L. H., Li, D. L., Qin, W., Li, Q., et al. (2023). OCT4 regulates WNT/β-catenin signaling and prevents mesoendoderm differentiation by repressing EOMES in porcine pluripotent stem cells. J. Cell Physiol. 238 (12), 2855–2866. doi:10.1002/jcp.31135
Yamazaki, S., Iwama, A., Takayanagi, S., Eto, K., Ema, H., and Nakauchi, H. (2009). TGF-β as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood 113 (6), 1250–1256. doi:10.1182/blood-2008-04-146480
Yang, J., and Jiang, W. (2020). The role of SMAD2/3 in Human embryonic stem cells. Front. Cell Dev. Biol. 8, 653. doi:10.3389/fcell.2020.00653
Yang, B., O'Herrin, S. M., Wu, J., Reagan-Shaw, S., Ma, Y., Bhat, K. M. R., et al. (2007). MAGE-A, mMage-b, and MAGE-C proteins form complexes with KAP1 and suppress p53-dependent apoptosis in MAGE-positive cell lines. Cancer Res. 67 (20), 9954–9962. doi:10.1158/0008-5472.Can-07-1478
Yang, G. Q., Xie, H., Wang, C. H., Zhang, C., Yu, L. P., Zhang, L. Y., et al. (2023). Design, synthesis, and discovery of Eudistomin Y derivatives as lysosome-targeted antiproliferation agents. Eur. J. Med. Chem. 250, 115193. doi:10.1016/j.ejmech.2023.115193
Yang, Y., Li, Y. Y., Zhang, S. Q., Cao, L. Y., Zhang, Y. S., and Fang, B. (2023). miR-199a-5p from bone marrow mesenchymal stem cell exosomes promotes the proliferation of neural stem cells by targeting GSK-3β. Acta Biochim. Biophys. Sin. 55 (5), 783–794. doi:10.3724/abbs.2023024
Yarden, A., and Geiger, B. (1996). Zebrafish cyclin E regulation during early embryogenesis. Dev. Dyn. 206 (1), 1–11. doi:10.1002/(SICI)1097-0177(199605)206:1<1::AID-AJA1>3.0.CO;2-M
Yin, H., Price, F., and Rudnicki, M. A. (2013). Satellite cells and the muscle stem cell niche. Physiol. Rev. 93 (1), 23–67. doi:10.1152/physrev.00043.2011
Yoon, H. S., and Yang, V. W. (2004). Requirement of Kruppel-like factor 4 in preventing entry into mitosis following DNA damage. J. Biol. Chem. 279 (6), 5035–5041. doi:10.1074/jbc.M307631200
Yu, J., Hu, K., Smuga-Otto, K., Tian, S., Stewart, R., Slukvin, I. I., et al. (2009). Human induced pluripotent stem cells free of vector and transgene sequences. Science 324 (5928), 797–801. doi:10.1126/science.1172482
Yu, Y., Arrigo, A., Chandra, A., Zhuang, C., Najjar, M. K., Khan, M. S., et al. (2025). Targeting tGLI1, a novel mediator of tumor therapeutic resistance, using ketoconazole sensitizes glioblastoma to CDK4/6 therapy and chemoradiation. bioRxiv, 2025.02.20.639359. doi:10.1101/2025.02.20.639359
Yuan, P., Ding, L., Chen, H. L., Wang, Y., Li, C. H., Zhao, S., et al. (2021). Neural stem cell-derived exosomes regulate neural stem cell differentiation through miR-9-Hes1 axis. Front. Cell Dev. Biol. 09, 601600. doi:10.3389/fcell.2021.601600
Zhan, Z., Song, L., Zhang, W., Gu, H., Cheng, H., Zhang, Y., et al. (2019). Absence of cyclin-dependent kinase inhibitor p27 or p18 increases efficiency of iPSC generation without induction of iPSC genomic instability. Cell Death Dis. 10 (4), 271. doi:10.1038/s41419-019-1502-8
Zhang, L. L., Ye, M. S., Zhu, L., Cha, J. M., Li, C. C., Yao, Y. G., et al. (2020). Loss of ZC4H2 and RNF220 inhibits neural stem cell proliferation and promotes neuronal differentiation. Cells 9 (7), 1600. doi:10.3390/cells9071600
Zhao, B. N., Yu, X. W., Shi, J. T., Ma, S. Y., Li, S. Z., Shi, H. T., et al. (2024). A stepwise mode of TGFβ-SMAD signaling and DNA methylation regulates naïve-to-primed pluripotency and differentiation. Nat. Commun. 15 (1), 10123. doi:10.1038/s41467-024-54433-5
Zhou, C. H., Dai, X. C., Chen, Y., Shen, Y. Y., Lei, S. F., Xiao, T., et al. (2016). G protein-coupled receptor GPR160 is associated with apoptosis and cell cycle arrest of prostate cancer cells. Oncotarget 7 (11), 12823–12839. doi:10.18632/oncotarget.7313
Zhou, W., Zhao, T., Du, J., Ji, G., Li, X., Ji, S., et al. (2019). TIGAR promotes neural stem cell differentiation through acetyl-coa-mediated histone acetylation. Cell Death Dis. 10 (3), 198. doi:10.1038/s41419-019-1434-3
Zhu, H., Hu, S. J., and Baker, J. (2014). JMJD5 regulates cell cycle and pluripotency in human embryonic stem cells. Stem Cells 32 (8), 2098–2110. doi:10.1002/stem.1724
Zhu, X. F., Zheng, X. Y., Gong, P., and Xu, X. Z. (2022). Regulation of ATR-CHK1 signaling by ubiquitination of CLASPIN. Biochem. Soc. Trans. 50 (5), 1471–1480. doi:10.1042/bst20220729
Zhu, Y. R., Dong, L. X., Wang, C. C., Hao, K. Y., Wang, J. N., Zhao, L. C., et al. (2022). Functional redundancy among polycomb complexes in maintaining the pluripotent state of embryonic stem cells. Stem Cell Rep. 17 (5), 1198–1214. doi:10.1016/j.stemcr.2022.02.020
Keywords: pluripotent stem cells, Cyclin, cyclin-dependent kinases, Jak1/Stat3 pathway, epigenetic modification, Metabolism, somatic cell reprogramming, adult stem cells
Citation: Huang X, Wang Y, Li Q, Li X and Wang C (2025) Coupling of stemness maintenance with cell cycle control in stem cells. Front. Cell Dev. Biol. 13:1693489. doi: 10.3389/fcell.2025.1693489
Received: 27 August 2025; Accepted: 23 September 2025;
Published: 08 October 2025.
Edited by:
Mustapha Najimi, UCLouvain, BelgiumReviewed by:
Hassan Azari, Barry University, United StatesPawan Kumar Raghav, University of California, San Francisco, United States
Copyright © 2025 Huang, Wang, Li, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Congcong Wang, d2FuZ2Nvbmdjb25nQHd1c3QuZWR1LmNu