 Xinxin Li
Xinxin Li Chen Huang
Chen Huang Kul Raj Rai
Kul Raj Rai Quanming Xu
Quanming Xu- 1Meizhouwan Vocational Technology College, Putian, China
- 2Scientific Research and Experiment Center, Fujian Police College, Fuzhou, China
- 3Key Laboratory of Fujian-Taiwan Animal Pathogen Biology, College of Animal Sciences, Fujian Agriculture and Forestry University, Fuzhou, China
- 4Hanjiang District Agriculture and Rural Affairs Bureau, Putian, China
Interleukin-6 (IL-6), a pleiotropic cytokine, is induced by infection of influenza A virus (IAV), where it plays a pivotal role in immune defense and the regulation of inflammation. IAV induces IL-6 transcription upon recognition by pattern recognition receptors, which activate downstream signaling cascades, leading to the activation of transcription factors. Activated transcription factors subsequently regulate the expression of IL-6 in innate immune cells, such as macrophages, dendritic cells, and epithelial cells. IL-6 contributes to antiviral immunity by promoting the recruitment of immune cells to sites of infection and amplifying the inflammatory response. While optimal IL-6 production is essential for effective anti-IAV immunity, excessive IL-6 production can contribute to a dysregulated immune response, leading to a cytokine storm. In this context, IL-6 signaling, in coordination with other proinflammatory cytokines such as TNF-α and IL-1β, not only enhances its own production but can also serve as a key mediator of inflammation. This cascade can result in exaggerated immune responses, causing tissue damage and potentially leading to severe outcomes, including organ failure and death. Understanding the molecular mechanisms underlying cytokine storms presents important therapeutic opportunities. However, the precise pathways responsible for excessive IL-6 production and its dysregulation during IAV infection is not fully understood. This review explores the reported mechanisms regulating IL-6 induction in response to IAV and its innate immune role in IAV pathogenesis, highlighting existing research gaps in understanding IAV-induced IL-6 production and its impact on immune modulation. A deeper understanding of IAV-induced IL-6 production and signaling could inform the development of targeted therapies to more effectively manage influenza.
1 Introduction
Influenza A virus (IAV) is a highly contagious respiratory pathogen responsible for seasonal flu outbreaks and periodic pandemics, posing a significant global health burden (Monto and Fukuda, 2020). A member of orthomyxoviridae family, IAV is characterized by its segmented RNA genome, which enables frequent genetic reassortment and rapid mutation, allowing it to evade immune surveillance (Shao et al., 2017). IAV encodes multiple structural and nonstructural proteins, such as hemagglutinin (HA), neuraminidase (NA), nucleoprotein (NP), components of the RNA polymerase complex including polymerase basic 1 (PB1) and PB2, matrix 1 (M1), and nonstructural protein 1 (NS1) (Krammer et al., 2018). The virus infects epithelial cells of the respiratory tract, causing symptoms ranging from mild illness to severe complications such as viral pneumonia, acute respiratory distress syndrome (ARDS), and multi-organ failure (Kalil and Thomas, 2019). The World Health Organization estimates that IAV infects nearly 10% of the world’s population annually, resulting in approximately 3 to 5 million cases of severe illness and between 290,000 to 650,000 respiratory-related deaths worldwide (Uyeki et al., 2022). The most devastating “Spanish flu” pandemic in 1918 took over 50 million lives. The subsequent emergence of flu pandemics, such as “Asian flu” and “Hong Kong flu” in 1957 and 1968, respectively, killed about three million people (Krammer et al., 2018; Rai et al., 2021). The continuous antigenic evolution of IAV presents an ongoing challenge for vaccine development and antiviral strategies, underscoring the need for a deeper understanding of host immune responses to the virus for alternative therapeutics.
The immune response to IAV consists of both innate and adaptive immunity, with the innate immunity playing a crucial role in the initial defense against viral invasion (Rai et al., 2021). Upon infection, pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) and retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) detect viral components, leading to the activation of signaling pathways that stimulate the production of interferons (IFNs) and pro-inflammatory cytokines, including Interleukin-6 (IL-6) (Carty et al., 2021). This early immune response is vital for controlling viral replication and promoting the activation of the adaptive immune system, which provides long-term immunity (Kawai and Akira, 2010). However, an excessive or dysregulated immune response can contribute to immunopathology, exacerbating tissue damage and inflammation. Understanding the balance between protective and detrimental immune responses is critical for developing effective therapeutic strategies against IAV infections.
During IAV infection, cytokines mediate antiviral defenses and immune cell recruitment (Chan and Gack, 2016; Chen et al., 2017). PRRs detect pathogen-associated molecular patterns (PAMPs), such as viral RNA and proteins, as well as damage-associated molecular patterns (DAMPs) from infected host cells (Pillai, 2014; Chen et al., 2018; Ramos et al., 2019). Key PRRs—including TLR, RLR, and NLR (NOD-like receptor) pathways—enable influenza recognition, triggering antiviral and inflammatory responses (Tanaka et al., 2014). TLRs, such as TLR3, TLR7, and TLR8, are membrane-bound PRRs that recognize viral RNA: TLR3 detects double-stranded RNA (dsRNA), a byproduct of influenza replication, TLR7/8 sense single-stranded RNA (ssRNA) from the viral genome, while TLR2 and TLR4 recognize influenza-induced DAMPs (Shinya et al., 2012; Kim et al., 2022). TLRs activate signaling cascades via immune adaptor proteins like MyD88 or TRIF, leading to the production of type I IFN-α/β and pro-inflammatory cytokines (e.g., IL-6, TNF-α) through transcription factors like nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and interferon regulatory factors (IRFs) (Iwasaki and Pillai, 2014; Maarouf and Rai, 2018). This response is critical for early antiviral defense and immune cell recruitment (Pillai, 2014).
In the cytoplasm, RLRs, including RIG-I and MDA5, act as PRRs to detect viral RNA (Pillai, 2014). RIG-I recognizes short dsRNA and 5’-triphosphate RNA, while MDA5 senses long dsRNA (Chan and Gack, 2015). In influenza virus induced RLRs signaling, the mitochondrial antiviral-signaling protein (MAVS) plays a key role as immune adaptor protein that not only activates the downstream protein kinase TANK-binding kinase 1 (TBK1), but also license TBK1 to phosphorylate transcriptional factor IRF3, triggering the production of interferons (Gaur et al., 2011; Jensen and Thomsen, 2012; Pillai, 2014; Liu et al., 2015; Hagenaars et al., 2016). The NLR pathway, particularly NLRP3, is another cytosolic PRR that detects cellular stress or damage caused by influenza infection, often in response to PAMPs like viral proteins or DAMPs such as ATP or reactive oxygen species (ROS) (Niu and Meng, 2023). NLRP3 forms an inflammasome complex, activating caspase-1, which processes pro-IL-1β and pro-IL-18 into their active forms, driving inflammation and immune cell recruitment (Negash et al., 2013; Guo et al., 2015; Niu and Meng, 2023).
Among the various cytokines involved in the innate immune response to IAV, IL-6 stands out as a pivotal player in the innate immune response to IAV infection (Gentile et al., 1998; Thomas et al., 2009; Chiaretti et al., 2013; Richetta and Faure, 2013; Killip et al., 2015; Yang et al., 2017; Kalil and Thomas, 2019; Choy et al., 2020; Carty et al., 2021). IL-6, a pleiotropic cytokine with diverse functions, was identified in 1973 as a soluble factor secreted by T cells (Choy et al., 2020). Over the past 50 years, it has become recognized as a crucial immune regulator, modulating acute phase responses, promoting B and T cell differentiation, and regulating inflammatory processes (Grebenciucova and VanHaerents, 2023). It is primarily produced by macrophages, dendritic cells, and epithelial cells in response to viral infections and other inflammatory stimuli (Grebenciucova and VanHaerents, 2023). IL-6 exerts its effects through binding to the IL-6 receptor (IL-6R) and subsequent activation of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, leading to the expression of genes involved in immune regulation (Grebenciucova and VanHaerents, 2023).
Despite its the role in antiviral defense, IL-6 exhibits a dual nature in the immune response to IAV, with both protective and detrimental effects (Gentile et al., 1998; Bradley-Stewart et al., 2013; Chiaretti et al., 2013, Chi et al, 2013; Radigan et al., 2019; Zuo et al., 2021; Zhang et al., 2022; Grebenciucova and VanHaerents, 2023). On one hand, IL-6 contributes to viral clearance by promoting the activation and proliferation of immune cells, enhancing antibody production, and supporting tissue repair (Tanaka et al., 2014; Zuo et al., 2021). Studies have shown that IL-6-deficient mice exhibit impaired immune responses to IAV, suggesting a protective role in host defense (Dienz et al., 2012; Lauder et al., 2013; Yang et al., 2017). On the other hand, excessive IL-6 production has been associated with severe inflammatory responses and immunopathology, particularly in cases of hypercytokinemia or ‘cytokine storm’ (Fajgenbaum and June, 2020; Gu et al., 2021). In severe influenza infections, elevated IL-6 levels correlate with increased disease severity, lung inflammation, and poor clinical outcomes (Paquette et al., 2012; Radigan et al., 2019; Gu et al., 2021). Thus, dysregulated IL-6 can contribute to respiratory failure and an increased risk of mortality.
Given the complex and context-dependent role of IL-6 in IAV infection, a comprehensive understanding of its functions is essential for developing targeted therapeutic approaches. This review discusses the mechanisms of IAV-induced IL-6 production and IL-6 signaling pathways, emphasizing its potential as an inflammation amplifier that may contribute to a dysregulated immune response during IAV infection. We also examine the dual role of IL-6 in IAV pathogenesis. A deeper understanding of IL-6 regulation within the innate immune response could offer critical insights for improving treatment strategies for influenza infections.
2 Cytokine storm in influenza
Cytokines play a dual role in influenza infection (Jiang and Zhang, 2023). Initially, they are crucial for viral clearance by promoting immune cell recruitment and activation (Zuo et al., 2021). IL-6, TNF-α, and IL-1β enhance the infiltration of neutrophils and monocytes into infected tissues, aiding in viral elimination (Dienz et al., 2012; Lauder et al., 2013). Chemokines such as CXCL10 and CCL2 further mediate immune cell trafficking to the lungs (Alon et al., 2021). However, excessive cytokine production can be detrimental, leading to immune dysregulation and severe tissue damage. A cytokine storm refers to the excessive and uncontrolled release of pro-inflammatory cytokines, which can cause significant immunopathology (Fajgenbaum and June, 2020; Gu et al., 2021; Jiang and Zhang, 2023). In severe influenza cases, including those caused by highly virulent strains such as H5N1 and H1N1, an overwhelming cytokine response leads to widespread inflammation, lung injury, and ARDS (Short et al., 2017; Kalil and Thomas, 2019). The cytokine storm results in increased vascular permeability, leading to pulmonary edema and impaired gas exchange (Chen et al., 2018; Jiang and Zhang, 2023). Overactivation of immune cells, causing collateral damage to lung tissue. Dysfunctional coregulation pathways, increasing the risk of thrombosis and multi-organ failure (Vincent et al, 2019). The severity of influenza is often correlated with the magnitude of the cytokine storm, and individuals with heightened immune responses may suffer from more severe disease outcomes (Bradley-Stewart et al., 2013; Vincent and Tang BM, Cootes T, 2019; Fajgenbaum and June, 2020). Excessively elevated levels of cytokines such as IFN-γ, IL-6, IL-1α, IL-1β, TNF-α, IL-15, IL-12p70, IL-17, IL-10, MCP-1, MIP-1β, IL-8, MIG, IP-10, MIP-1α, GM-CSF, and RANTES were observed in cytokine storm caused by H1N1 (“Pandemic strain”) influenza virus (reviewed in (Morris et al., 2021)). Among the inflammatory cytokines, IL-6 is a critical cytokine contributing to cytokine storm in the pathogenesis of IAV virus. For a more detailed understanding of the mechanisms underlying the cytokine storm induced by influenza virus, readers can refer to review articles (Gu et al., 2019, Gu et al, 2021).
3 Roles of IL-6 in innate immune response to IAV
IL-6 plays a crucial role in innate immunity, particularly in response to IAV. It contributes to antiviral defense, inflammation regulation, and immune cell activation. While essential for host defense, excessive IL-6 production can lead to pathological inflammation, highlighting its dual and complex role in immunity.
3.1 IL-6 Induction in response to IAV
The upregulation of IL-6 during IAV infection has been reported since the late 1980s, with further characterization in the 1990s (Sprenger et al., 1994; Pahl and Baeuerle, 1995; Matsukura et al., 1996; Gentile et al., 1998; Visseren et al., 1999). IL-6 is induced in response to various subtypes of IAV infection by both immune and non-immune cells, including macrophages, dendritic cells, endothelial cells, and epithelial cells (Matsukura et al., 1996; Hn et al., 2013; Short et al., 2017; Yang et al., 2017; Gou et al., 2020; Gu et al., 2021; Xie et al., 2021). The production of IL-6 is primarily triggered by the activation of PRRs, such as TLRs and RLRs, which recognize viral components and DAMPs (Shinya et al., 2012; Prantner et al., 2017; Chen et al., 2018; Liu et al., 2019; Gu et al., 2021; Jiang and Zhang, 2023). Among TLRs, the role of TLR9 in IAV-induced DAMP recognition remains less clear, studied only in the co-infection model (Tuvim et al., 2012; Maarouf et al., 2018; Sun and Metzger, 2019). RLRs (RIG-I and MDA5), on the other hand, sense viral RNA within the cytoplasm and initiate antiviral responses. Upon activation, these PRRs trigger intracellular signaling cascades, including the NF-κB, AP-1, and MAPK (ERK, JNK, p38) pathways (Eastman et al., 1996; Brydon et al., 2005; Klemm et al., 2017). NF-κB and AP-1 are the primary transcription factors driving IAV-induced IL-6 gene expression, while MAPK signaling further enhances cytokine production by promoting transcription factor phosphorylation (Visseren et al., 1999; Ludwig et al., 2001; Pinto et al., 2011; Liu et al., 2019; Gu et al., 2021).
The magnitude of cytokine induction, including IL-6, in IAV infection is determined by a multifactorial interplay involving influenza strain specificity, viral protein polymorphisms, replication kinetics, and host-specific immune responses (Li et al., 2006, Li et al, 2018; Newby et al., 2007). Cumulative evidence suggests that certain influenza virus strains induce higher levels cytokines including IL-6 and are more prone to triggering cytokine storms, with pathogenicity linked to strain-specific viral protein functions (De Jong et al., 2006; Perrone et al., 2008; Hn et al., 2013; Li et al., 2018). For example, highly pathogenic strains such as H5N1 and the 1918 H1N1 pandemic virus elicit elevated IL-6 production and severe immunopathology compared to seasonal H1N1 strains (De Jong et al., 2006; Boon et al., 2011; Kang et al., 2011; Kalaiyarasu et al., 2016; Li et al., 2018). Among IAV proteins, the NS1 protein is a potent inhibitor of cytokine production and innate immune signaling (García-Sastre et al., 1998; Marc, 2014; Rai et al., 2020), which exhibits strain-specific sequence differences (Hale et al., 2008). NS1 mutation affects IAV replication efficiency by modulating cytokine expression (Newby et al., 2007; Chen et al., 2021a). Of note, strains with higher replication efficiency generate increased viral loads (Boon et al., 2011; Li et al., 2018). Elevated viral loads can hyperactivate PRRs, boosting IL-6 overproduction and increasing the risk of cytokine storm (Li et al., 2018). Certain strain-specific IAV proteins, including HA (Pahl and Baeuerle, 1995), NA (Kim et al., 2022), M1 (Kim et al., 2023), PB1-F2 (Zeng et al., 2021), contribute to IL-6 production by activating the NF-κB signaling pathway, while dampening IFN-mediated antiviral responses (Kochs et al., 2007; Iwai et al., 2010; Zeng et al., 2021; Zhang et al., 2023; Ouyang et al., 2024), indicating polymorphisms, or variations in these viral proteins, may lead to differential cytokine responses (Newby et al., 2007; Williams et al., 2024). Moreover, other factors such as host factors and receptor tropism also influence immune activation resulting into differential IL-6 induction in the response to IAV (reviewed in (Long et al., 2019)).
Table 1 summarizes representative evidence regarding IL-6 induction by IAV and viral components, such as HA and NP, across various experimental models, including human cell cultures, animal models, and human populations. The data demonstrate that IL-6 is significantly upregulated during infections with different IAV strains, including H5N1 and H7N9, contributing to proinflammatory responses, viral clearance, and tissue protection. Elevated IL-6 levels are often correlated with the severity of infection. Furthermore, IAV-induced IL-6 exerts an immunomodulatory role in both respiratory and extra-respiratory tissues, including the central nervous system.
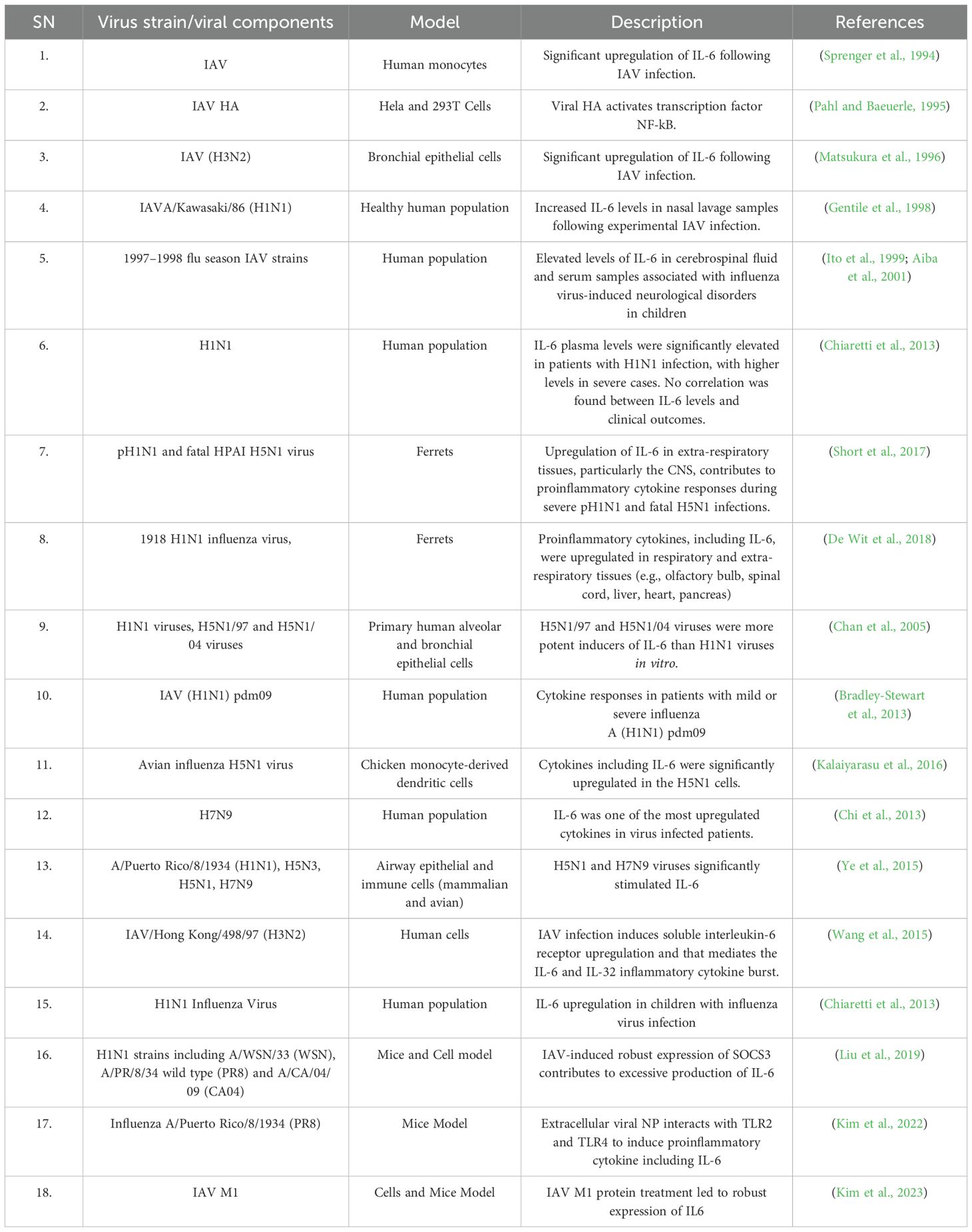
Table 1. Early and some recent representative evidences for IL-6 upregulation following IAV infection.
3.2 IL-6 signaling pathways: classical and trans-signaling
IAV-induced IL-6 orchestrates innate immune responses through two distinct IL-6 signaling pathways: (i) classical signaling, which involves membrane-bound IL-6 receptor (mIL-6R), and (ii) trans-signaling, which relies on the soluble IL-6 receptor (sIL-6R). Binding of the ligand to the receptor activates a cascade of events that regulate downstream gene expression and trigger various cellular responses, including inflammation (Rose-John, 2012; Tanaka et al., 2014; Wang et al., 2015; Pullamsetti et al., 2018; Radigan et al., 2019; Velazquez-Salinas et al., 2019) (Please also refer the Figure 1 below).
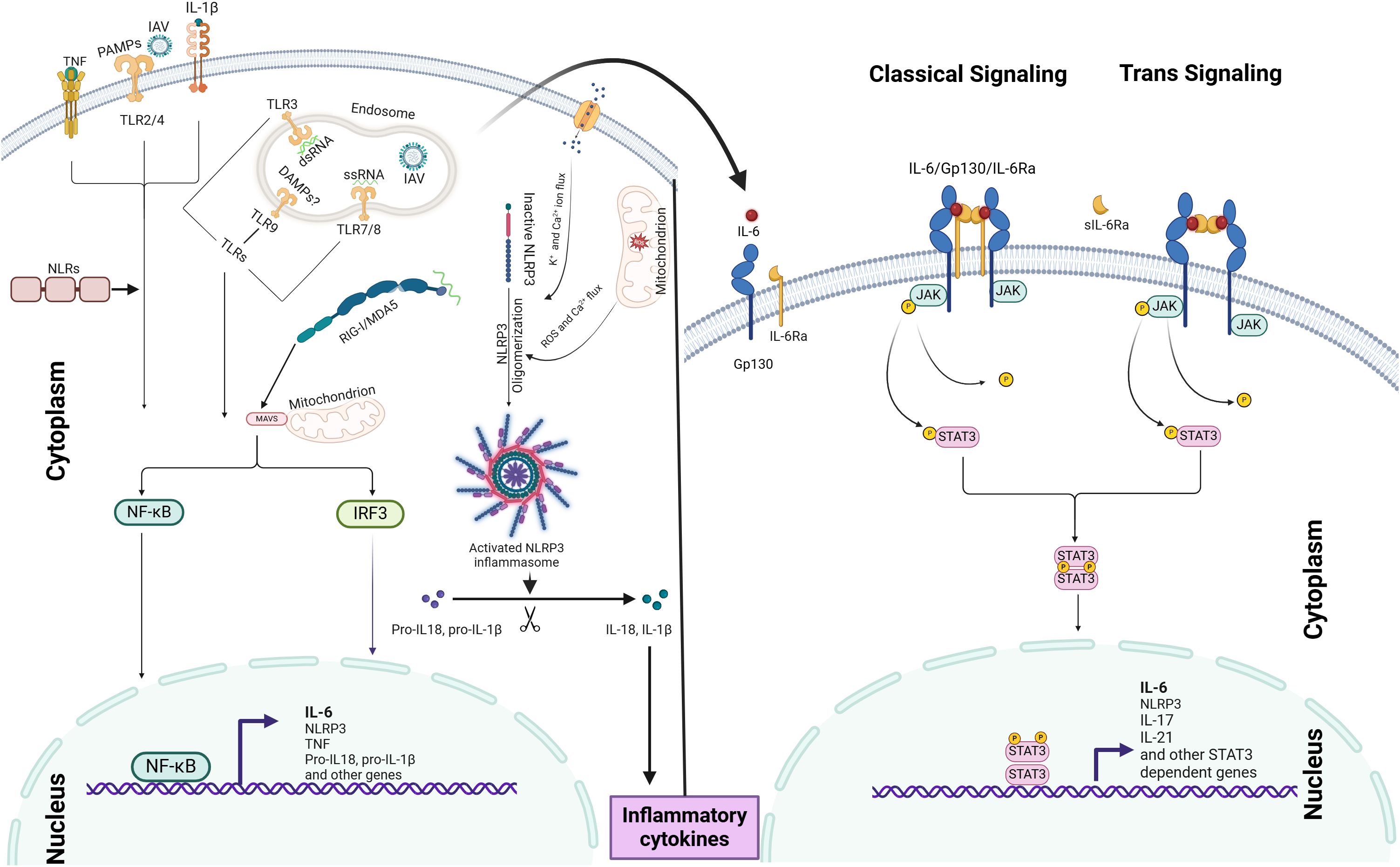
Figure 1. IAV-induced IL-6 induction mechanism and IL-6 signaling pathways: IL-6 production is initiated by PRRs, such as TLRs and RLRs, which recognize viral PAMPs and DAMPs, leading to the activation of NF-κB signaling and IL-6 expression. The NLRP3 inflammasome further amplifies IL-6 production through IL-1β and IL-18 release. IAV-induced IL-6 modulates immune responses via two distinct signaling pathways: (i) classical signaling, involving membrane-bound IL-6R, and (ii) trans-signaling, mediated by soluble IL-6R. Ligand binding to these receptors activates the JAK-STAT3 pathway, driving downstream gene expression. The coordinated activation of both NF-κB and JAK-STAT3 pathways acts as an inflammation amplifier.
IL-6 classical pathway: The activation of the IL-6 receptor begins when IL-6 binds to IL-6Rα, which causes a conformational change in the receptor complex. This change allows for the recruitment and dimerization of gp130, forming a hexameric signaling complex (Pullamsetti et al., 2018). The hexamerization of gp130 triggers intracellular signaling by inducing phosphorylation events and activating downstream signaling pathways, such as JAK/STAT, MAPK/ERK, and PI3K/AKT (Rose-John, 2012; Pullamsetti et al., 2018). These pathways play a crucial role in mediating IL-6-induced gene expression, cellular proliferation, differentiation, survival, and immune responses.
IL-6 Trans signaling pathway: This pathway involves the soluble form of the IL-6 receptor (sIL-6R) and gp130 (Wang et al., 2015). It enables IL-6 to influence cells that do not express the membrane-bound IL-6 receptor (mIL-6R). The trans signaling pathway is initiated when IL-6 binds to sIL-6R in the extracellular space. The IL-6/sIL-6R complex can then diffuse and reach cells lacking the mIL-6R. When it encounters such cells, the complex binds to the ubiquitously expressed gp130 on their surfaces (Rose-John, 2012; Liu et al., 2024). This binding induces conformational changes in gp130 that activate JAKs associated with the receptor’s cytoplasmic domain. Activated JAKs phosphorylate specific tyrosine residues on gp130, creating docking sites for downstream signaling molecules.
In the STAT pathway, phosphorylated gp130 serves as a docking site for STAT proteins, particularly STAT3 (Zhang et al., 2000). Upon recruitment to the phosphorylated gp130, JAK1 and JAK2 carry out phosphorylation. Once phosphorylated, STAT3 forms either homodimers or heterodimers with other STAT isoforms and translocates from the cytoplasm to the nucleus (Liu et al., 2024). Within the nucleus, STAT3 dimers bind to specific DNA sequences called STAT response elements located in the promoter regions of target genes (Arshad et al., 2020), driving expression of various STAT3 dependent genes.
3.3 IL-6 amplifier and IL-6-mediated crosstalk in IAV infection
An IL-6 amplifier, also referred to as an inflammation amplifier, comprises molecules, receptors, or signaling pathways that enhance IL-6 activity (Lee et al., 2012). This system establishes a self-perpetuating inflammatory cycle through the coordinated activation of IL-6, NF-κB, and STAT3 signaling (Lee et al., 2012). As illustrated in the Figure 1, upon IAV infection, viral components are detected by PRRs, initiating the first wave of IL-6 production via NF-κB activation. The secreted IL-6 then binds to its receptor (IL-6R), activating JAK-STAT3 signaling, which promotes the expression of additional inflammatory mediators, including TNF and IL-1β. These secondary cytokines reinforce feed-forward loops that further stimulate IL-6 production through autocrine and paracrine mechanisms, potentially resulting in excessive cytokine release and tissue injury (Gu et al., 2021; Niu and Meng, 2023).
The IL-6-mediated crosstalk during IAV infection is intricate (Thomas et al., 2009; Malik and Zhou, 2020). Ligand-receptor complex formation via either classic or trans-signaling pathways trigger the activation of multiple intracellular signaling cascades, including the JAK-STAT pathway, Ras-MAPK pathway, p38 and JNK MAPK pathways, PI3K-Akt pathway, and the MEK-ERK5 pathway (Klemm et al., 2017). Notably, both classic and trans-signaling complexes activate similar signaling pathways (Liu et al., 2024). NF-κB and STAT3 cooperatively regulate the expression of several inflammatory cytokines, including IL-6 itself, as well as other cytokines like TNF, CCL2, CCL5, IL-8, and IL-17 and so forth. Additionally, they modulate the expression of inflammation amplifiers such as NLRP3, further intensifying the inflammatory response (Pandey et al., 2023).
Although, NLRs (NOD1 and NOD2) are not primary sensors of IAV but can upregulate inflammation via crosstalk with other PRRs and inflammatory signaling pathways like NF-κB and the NLRP3 inflammasome. Importantly, the NLRP3 inflammasome plays a critical role in IL-6 production and amplification during IAV infection. Its activation involves two sequential steps: the priming signal, triggered by viral recognition via PRRs, leading to the upregulation of NLRP3, as well as the transcription of pro-IL-1β and pro-IL-18. Subsequently, viral components or host-derived DAMPs induce the activation of NLRP3, resulting in the oligomerization of the inflammasome mechanisms such as the ion channel activity of the IAV M2 protein, mitochondrial ROS production, and disruption of cellular homeostasis (Zamarin et al., 2006; Thomas et al., 2009; Ichinohe et al., 2010; Pinar et al., 2017; Niu and Meng, 2023) (please refer the Figure 1 below). This leads to caspase-1 activation and subsequent IL-1β and IL-18 release, which can further amplify IL-6 production by binding with their cell membrane bound receptors resulting into the activation of NF-κB (Chanturiya et al., 2004; Thomas et al., 2009; Ichinohe et al., 2010; Kinoshita et al., 2015; Zeng et al., 2021; Kim et al., 2022) (see Figure 1).
In parallel, the PI3K-Akt pathway can also contribute to IL-6 induction, with viral proteins such as hemagglutinin and non-structural protein 1 triggering Akt phosphorylation (Ehrhardt et al., 2007), which sustains NF-κB activation and IL-6 transcription (Arora et al., 2016). Although IRF3 and IRF7 primarily drive type I interferon responses (Chen et al., 2018), IFNs can also secondarily amplify IL-6 production through autocrine and paracrine signaling mechanisms (Murray et al., 2015). The relative contribution of these pathways varies across different cell types; for instance, bronchial epithelial cells predominantly rely on TLR3 and TLR7, whereas macrophages primarily utilize RLRs. For example, bronchial epithelial cells predominantly use TLR3 and TLR7, while macrophages rely more on RLRs (Malik and Zhou, 2020).
3.4 The dual role of IL-6 in influenza infection
IL-6 has a dual role in influenza infections, exerting both protective and detrimental effects (Longhi et al., 2008; Vogel et al., 2014; Hn et al., 2013; Zuo et al., 2021). It facilitates viral clearance by enhancing the innate immune response, aids in lung tissue repair, and regulates immune cell (T- and B-cell) activity to modulate inflammation and promote protective immunity (Dienz et al., 2012; Velazquez-Salinas et al., 2019). Additionally, IL-6 supports neutrophil function and preserves epithelial integrity, thereby limiting viral replication and preventing secondary bacterial infections (Dienz et al., 2012). However, excessive IL-6 production can lead to immune dysregulation and a cytokine storm, which exacerbates disease severity (Yu et al., 2011; Bradley-Stewart et al., 2013; Short et al., 2017). Elevated IL-6 levels are associated with poor outcomes, including ARDS and increased mortality (Bradley-Stewart et al., 2013; Short et al., 2017). In co-infection scenarios, dysregulated IL-6 exacerbates lung damage and impairs bacterial containment (Kalil and Thomas, 2019; Gou et al., 2020). However, its effects are context-dependent. Table 2 presents a brief summary when IL-6 is beneficial versus when it becomes detrimental.
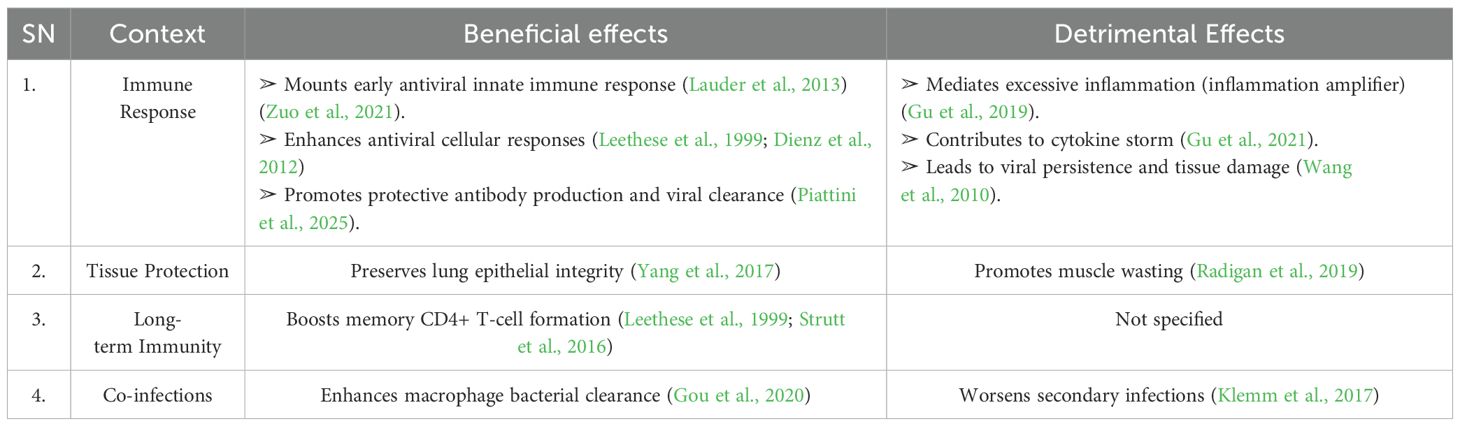
Table 2. Beneficial vs. detrimental effects of IL-6 in influenza infection (please refer Supplementary Table 1 for more detail).
3.4.1 Host protective function of Il-6
Multiple lines of evidence support that IL-6 protects the host against influenza by enhancing antiviral responses. Studies using IL-6 knockout models suggest that IL-6 boosts both innate and adaptive immune responses to influenza. For instance, IL-6 was shown to be crucial for resolving infection by protecting neutrophils from virus-induced death and enhancing their viral clearance activity (Dienz et al., 2012). IL-6 knockout resulted in prolonged viral persistence in the lung, leading to severe damage and eventual death (Dienz et al., 2012). Mice lacking IL-6 exhibited a profound defect in mounting an anti-viral T-cell response, accompanied by increased infiltration of inflammatory monocytes and elevated production of pro-inflammatory cytokines like IFN-α and TNF-α (Lauder et al., 2013). Early IL-6 signaling was also shown to promote IL-27-dependent maturation of regulatory T cells, aiding in the resolution of viral immunopathology (Pyle et al., 2017). Additionally, IL-6 has been suggested to facilitate lung repair during influenza infection (Yang et al., 2017).
A study also demonstrated that IL-6 interacts with IL-27 to form IL-6–IL-27 complexes, which exhibit antiviral activity. These complexes enhance the expression of influenza-induced inflammatory cytokines, IFNs, and ISGs, while promoting the phosphorylation of STAT1 and STAT3 (Zuo et al., 2021). In an IAV-Streptococcus pneumoniae co-infection model, IL-6 was shown to reduce cell death in the lungs and enhance bacterial phagocytosis through upregulation of MARCO expression in macrophages, limiting infection-related inflammation and controlling bacterial invasion (Gou et al., 2020). Overall, it appears that early and optimal IL-6 production is beneficial for host protection. However, IL-6 must also be tightly regulated during the early immune response to balance effective immunity and inflammation. Additional host-protective functions of IL-6 in influenza virus infection are summarized in Supplementary Table 1.
3.4.2 Detrimental role of IL-6 in influenza virus infection
In certain cases, especially during severe influenza infections, IL-6 can contribute to a hyper-inflammatory state known as a “cytokine storm.” This is characterized by an excessive release of pro-inflammatory cytokines that can cause tissue damage, organ failure, and even death. In severe influenza infections, IL-6, along with other cytokines like TNF-α and IL-1β, can exacerbate the disease, leading to complications such as ARDS. For example, fatal cases of human influenza A H5N1 exhibited elevated IL-6 levels, indicating IL-6-mediated hypercytokinemia, which contributed to reactive hemophagocytic syndrome, multiorgan failure, and ultimately, death (To et al., 2001). IL-6 promoted muscle degradation via JAK/STAT, FOXO3a, and atrogin-1 upregulation and the treatment with treatment with a Food and Drug Administration-approved Ab antagonist to the IL-6R (tocilizumab) attenuated the severity of IAV-induced muscle dysfunction (Radigan et al., 2019). Another study has shown that the influenza virus-cytokine-protease cycle as one of the main mechanisms for vascular dysfunction in severe influenza (Wang et al., 2010). IL-6 can act as a bridge between innate immunity and oxidative stress, leading to worsened pathology. For example, inactivated influenza virus induces the production of oxidized phospholipids through the TLR4-IL-6-ROS signaling cascade, contributing to acute lung injury (Imai et al., 2008). Moreover, immunocompromised individuals, particularly the elderly, are prone to elevated risk of severe influenza, largely driven by IL-6-mediated hyperinflammation (Harpur et al., 2021). Notably, an IL-6 amplifier mechanism appears crucial in the pathogenic cascade triggered by dysregulated IL-6 signaling in IAV infection. Other representative pathogenic implications IAV-induced IL-6 is described in the Supplementary Table 1.
4 Clinical and therapeutic implications
4.1 IL-6 as a biomarker
IL-6 has been shown as a potential biomarker for assessing influenza severity, with elevated levels correlating with disease progression and poor clinical outcomes (Ito et al., 1999; Paquette et al., 2012). Studies have shown that patients with severe influenza, particularly those developing ARDS, exhibit significantly higher IL-6 levels compared to those with mild infections (Gentile et al., 1998; Kalil and Thomas, 2019). This positions IL-6 as a valuable prognostic and predictive biomarker, aiding in risk stratification and early identification of high-risk patients Beyond prognosis, IL-6 holds significant utility in guiding therapeutic development, as its levels can inform the efficacy and safety of anti-inflammatory interventions (Denisova et al., 2014; Tanaka et al., 2014; Radigan et al., 2019; Grebenciucova and VanHaerents, 2023). However, the acceptable threshold of IL-6 production remains debated, particularly in preclinical studies where biomarker standardization is critical but not yet fully established. The strong association between IL-6 and disease severity has spurred interest in targeting IL-6 or it signaling pathways, highlighting its dual role as both a biomarker and a therapeutic target in influenza and other inflammatory conditions.
4.2 Targeting IL-6 in influenza treatment
Targeting IL-6 signaling pathway is an important alternative therapeutic strategy for mitigating hyperinflammation in severe influenza cases. Although various therapeutic approaches have been attempted, completely blocking IL-6 would impair the innate immune responses (Kishimoto, 2005; Kang and Kishimoto, 2021). Instead, protection by IL-6 inhibition using short term antibodies to block is good therapeutic pathway to reduce the detrimental effect of IL-6 (Kishimoto, 2005; Narazaki and Kishimoto, 2018). Accordingly, anti-inflammatory drugs that target IL-6 signaling, such as monoclonal antibodies and Janus kinase (JAK) inhibitors, have been explored in various viral infections. Oclacitinib, a JAK inhibitor, and arbidol hydrochloride, known for its anti-inflammatory properties, are among the drugs studied in this context (Liu et al., 2013; Yu et al., 2024). Monoclonal antibodies like tocilizumab, which blocks the IL-6 receptor, have been particularly explored as potential treatments for reducing inflammation in severe cases of influenza (Tanaka et al., 2014; Radigan et al., 2019; Grebenciucova and VanHaerents, 2023). While IL-6 blockade has shown promise in conditions like rheumatoid arthritis (Davies and Choy, 2014) and COVID-19 (The REMAP−CAP Investigators, 2021). However, their application in influenza treatment has not been widely established. The main challenge lies in striking a balance between reducing harmful inflammation while preserving the essential antiviral functions of IL-6. Suppressing IL-6 aggressively may impair immune responses, potentially prolonging viral persistence or increasing susceptibility to secondary infections. Experimental trials evaluating IL-6 inhibitors in viral infections, including influenza and co-infections, have yielded mixed results (Pandey et al., 2023). While some studies indicate reduced inflammatory damage and improved respiratory function, others highlight concerns about increased risk of secondary bacterial infections due to immune suppression. Indeed, therapeutic targeting of IL-6 requires precise timing, as IL-6 levels rise early in infection, before peak viral clearance (Leethese et al., 1999; Chiaretti et al., 2013; Yang et al., 2017). Late inhibition (after viral peak) may improve outcomes by dampening excessive inflammation (Kishimoto, 2005; Choy et al., 2020; Kang and Kishimoto, 2021) but early blockade compromise antiviral immunity (Choy et al., 2020), suggesting most effective during the transition from viral replication to hyperinflammation. Future research should focus on refining therapeutic strategies, potentially using combination approaches that modulate IL-6 activity without entirely abolishing its protective effects (Pandey et al., 2023). Additionally, further clinical trials are needed to determine optimal patient selection, timing, and dosing for IL-6-targeted therapies in managing IAV infections.
5 Perspectives
IL-6, a key mediator of inflammation, is induced by the activation of innate immune responses during IAV infection. While optimal IL-6 production is beneficial for the host by promoting anti-IAV defenses, excessive IL-6 production can lead to hyperinflammation and contribute to a cytokine storm. Various amplifiers of IL-6, including NLRP3, play a critical role in amplifying inflammation through a positive feedback loop. However, the molecular mechanisms underlying the balance between optimal and excessive IL-6 production during IAV infection remain poorly understood. This complexity arises from the intricate and interconnected nature of innate immune signaling pathways, which involve crosstalk that is not yet fully elucidated.
During IAV infection, both PAMPs and DAMPs activate multiple signaling pathways. For instance, in co-infection models involving bacteria and IAV, enhanced activity of MAPKs such as p38 and ERK1/2 leads to excessive IL-6 production (Klemm et al., 2017). Additionally, it has been proposed that robust expression of SOCS3, induced by IAV infection, contributes to excessive IL-6 production, likely through the hyperactivation of IAV-induced NF-κB signaling (Liu et al., 2019). NF-κB, a key transcription factor, regulates IL-6 production during IAV infection, as shown in several studies (Pahl and Baeuerle, 1995; Liu et al., 2019; Zuo et al., 2021). Unlike IRF3, which is mainly activated by IAV nucleic acids via the MAVS-dependent RIG-I/MDA5 pathway and plays a central role in type I and III interferon production (Liu et al., 2015). NF-κB activation may be predominantly triggered by the sensing of IAV proteins and DAMPs (Pahl and Baeuerle, 1995; Flory et al., 2000; Imai et al., 2008; Tuvim et al., 2012; Holm et al., 2016; Fernández-Arjona et al., 2019; Sun and Metzger, 2019; Kim et al., 2022, Kim et al., 2023), also involving the cGAS-independent STING pathway (Holm et al., 2016), potentially leading to expression of inflammatory cytokines, including IL-6 expression (Kim et al., 2022, Kim et al, 2023). Notably, most IAV proteins, such as PB1-F2, NP, and HA, inhibit RIG-I/TRIM25/MAVS-mediated signaling, thereby impairing type I and III interferon production and subsequent IFN-mediated antiviral responses (Kochs et al., 2007; Iwai et al., 2010; Zeng et al., 2021; Zhang et al., 2023; Ouyang et al., 2024). For example, IAV PB1-F2 binds and inhibits RIG-I/TRIM25/MAVS signaling, suppressing type I and III interferon transcription (Zeng et al., 2021), while NP and HA similarly impair MAVS-mediated antiviral immunity (Zhang et al., 2023; Ouyang et al., 2024). However, while these IAV proteins suppress interferon production, they can also simultaneously activate NF-κB, further contributing to IL-6 expression (Pahl and Baeuerle, 1995; Flory et al., 2000). Moreover, PB1-F2 has been shown to form fibrillar aggregates that promote the release of pyrogenic IL-1β through NLRP3-dependent inflammasome activation, which can amplify IL-6 production by activating NF-κB in both autocrine and paracrine manner (Chanturiya et al., 2004; Zamarin et al., 2006; Pinar et al., 2017; Zeng et al., 2021).
An imbalanced cytokine response, marked by excessive IL-6, correlates with severe disease outcomes in influenza (Gentile et al., 1998; Chiaretti et al., 2013; Zuo et al., 2021; Zhang et al., 2022). A similar dysregulated immune response, marked by low levels of type I and III IFNs alongside elevated chemokines and high IL-6 expression, contributes to the pathogenesis of SARS-CoV-2 infection and the development of COVID-19 (Blanco-Melo et al., 2020). This pattern mirrors observations in severe seasonal influenza, where ferrets exhibited reduced interferon induction and heightened IL-6 levels (Svitek et al., 2008). Interestingly, disruption of STAT3 phosphorylation, a key downstream event in IL-6 signaling, has been shown to result in excessive production of IAV-induced type I and III IFNs (Liu et al., 2023). Additionally, IAV can suppress type I IFN signaling through NF-κB-dependent induction of SOCS3 expression (Pauli et al., 2008; Ye et al., 2015). Despite these findings, the precise molecular mechanisms by which virus-induced IL-6 modulates interferon responses remain unclear. On the other hand, it has been reported that there is competitive inhibition between IRF3 and NF-κB activation (Popli et al., 2022; Chakravarty et al., 2024), underscoring the need for further investigation into the molecular interplay between IL-6 signaling and antiviral IFN pathways during IAV infection.
IL-6 knockout animal models, as described above, indicate that IL-6 depletion impairs both innate and adaptive immune responses, leading to increased morbidity and mortality during influenza infection. However, defining the non-redundant function of IL-6 in influenza virus pathogenesis using these models remains challenging. Some studies have shown that IL-6 knockout mice exhibit similar morbidity and mortality to wild-type mice when infected with highly pathogenic H5N1 influenza virus (Salomon et al., 2007; Szretter et al., 2007).These discrepancies may arise from differences in influenza strain, experimental conditions, or other variables, suggesting a need for further investigation with more rigorous and controlled experimental designs. Interestingly, a recent study has proposed that IL-6 primarily contributes to defense against influenza by promoting protective antibody responses, rather than by mediating innate inflammation (Piattini et al., 2025).
Substantial evidence underscores the pivotal role of IL-6 in viral infections, where it mediates diverse immunological processes. These include the activation of the acute-phase response, modulation of inflammatory cytokine production, induction of chemokine secretion, recruitment of immune cells, and activation of NK cells and cytotoxic T lymphocytes. However, dysregulated IL-6 production can drive a hyperinflammatory state (cytokine storm), which leads to severe pathological consequences for the host. This supports the hypothesis that a finely tuned innate immune response is crucial for effective viral clearance while minimizing immunopathology. In contrast, an alternative perspective posits that disease severity is primarily dictated by viral load rather than immune dysregulation (Wang et al., 2010). For instance, a study utilizing quantitative real-time PCR to assess the cytokine-to-viral RNA ratio found no significant differences between susceptible and resistant mouse strains. These findings suggest that host genetic determinants influencing viral replication and viral load play a dominant role in susceptibility to influenza infection (Wang et al., 2010).
Indeed, elucidating the complex molecular mechanisms underlying host-virus interactions is important for distinguishing between protective and pathological immune responses. To this end, emerging evidence has identified several new host factors as key regulators of innate immunity during influenza virus infection. In particular, non-coding RNAs—including long non-coding RNAs (lncRNA), microRNAs, and circular RNAs—as well as micropeptides have been increasingly recognized as crucial modulators of IAV-induced innate immune responses (Ouyang et al., 2014; Ma et al., 2016; Maarouf et al., 2019; Xiao et al., 2021; Rai et al., 2022; Chen et al., 2024; Chi et al., 2024; Sun et al., 2025). For examples, RDUR, a lncRNA, promotes innate antiviral responses and provides feedback control of NF-κB activation to suppress excessive inflammatory response to IAV infection (Chen et al., 2021b). HOTAIR regulates the activation of NF-κB and its target gene IL-6 expression by facilitating the degradation of IκBα (Obaid et al., 2018). LncRNA MALAT1 regulates inflammatory cytokine production in lipopolysaccharide-stimulated human gingival fibroblasts through sponging miR-20a and activating TLR4 pathways (Li et al., 2020). Targeting of such host factors involved in IAV-induced IL-6 regulation may hold promising therapeutic strategy in the treatment of influenza (Zhang and Chu, 2019). Investigating the roles of these new regulatory elements in innate immune signaling presents exciting avenues for research and may provide critical insights into the molecular mechanisms governing IL-6-mediated cytokine storms.
Given the dual role of IL-6, therapeutic strategies targeting this cytokine should aim to enhance its beneficial antiviral effects while preventing excessive IL-6 production to mitigate tissue damage and multiple organ failure. Especially, the cytokine storm is not solely driven by IL-6 but results from the combined effects of various cytokines and chemokines. Consequently, no single pharmacological intervention has proven entirely effective in preventing tissue damage and inflammation associated with severe influenza. Therefore, it is crucial to explore other cellular pathways, such as apoptosis, pyroptosis, necroptosis, and ferroptosis, which influence innate immune signaling and contribute to cytokine storm pathogenesis (Tripathi et al., 2013; Arora et al., 2016; Shrivastava et al., 2016; Fang and Peng, 2022; Resnik et al., 2023; Ouyang et al., 2024). This is because IAV induces apoptotic, necrotic, and necroptotic death of lung epithelial and pulmonary immune cells (Kim et al., 2023; Boyd et al., 2025). Supportively, a recent study indicated that that blocking cell death (necroptosis blockage) can reduce lung injury in severe influenza by limiting inflammation and tissue damage (Gautam et al., 2024). Z-DNA binding protein 1 (ZBP1) is another innate immune sensor (recently gained a wide research attention) that recognizes Z-RNA generated during IAV-infection leading to RHIM-mediated recruitment and activation of receptor interacting kinase 3 (RIPK3) followed by apoptosis and necroptosis (Oltean et al., 2024). It would be interesting to study the role of IL-6 in ZBP1-initiated necroinflammatory cell death.
6 Summary
IL-6 is a key cytokine mediating the innate immune response to IAV, linking innate and adaptive immunity. While optimal IL-6 production appears to exert antiviral defense, excessive IL-6 production can contribute to hyperinflammatory responses and severe disease outcomes, such as ARDS. The molecular mechanisms behind IL-6 excessive production and its contributing to cytokine storm are complex, but evidence suggests that the hyperactivation of inflammatory pathways by DAMPs, PAMPs and IL-6 amplifier contribute to this dysregulation. The magnitude of IL-6 induction depends on a multifactorial interplay of viral factors (e.g., strain specificity, protein polymorphisms, replication kinetics) and host immune responses. Although IL-6 knockout models indicate its importance in host immunity, discrepancies in findings highlight the need for more controlled studies. Emerging research into new host factors like non-coding RNAs and micropeptides that regulate IL-6 is essential to elaborate the precise role of IL-6 in antiviral innate immunity. Given the contribution of IL-6 in cytokine storms, therapeutic strategies require balancing its antiviral effects while preventing excessive production. Future studies are warranted to explore other cellular pathways and processes, such as mitophagy, autophagy, apoptosis, and necroptosis, to better understand how innate immune signaling pathways governing IL-6 induction interact with these processes and mitigate the pathological effects of immune dysregulation and tissue damage during influenza infection.
Author contributions
XL: Formal analysis, Funding acquisition, Writing – original draft. CH: Writing – original draft. KR: Writing – review & editing. QX: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Educational and Scientific Research Project for Young and Middle-aged Teachers of Fujian Province (JAT241367).
Acknowledgments
We are grateful to all our colleagues for their valuable discussions and contributions during the drafting of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1605446/full#supplementary-material
Abbreviations
MAVS, Mitochondrial Antiviral-signaling Protein; NLRP3, NOD-, LRR- and Pyrin Domain-containing Protein 3; MyD88, Myeloid Differentiation Primary Response 88; TRIF, TIR-domain-containing Adapter-inducing Interferon-β; TNF-α, Tumor Necrosis Factor-alpha; ISGs, Interferon-stimulated Genes; IL-1β, Interleukin-1 Beta; CXCL10, C-X-C motif chemokine Ligand 10; CCL2, C-C motif chemokine Ligand 2; HMGB1, High-mobility Group Box 1; MAPK, Mitogen-activated Protein Kinase; AP-1, Activator protein 1; ERK, Extracellular signal-regulated Kinase; JNK, c-Jun N-terminal Kinase; PI3K, Phosphoinositide 3-kinase; Akt, Protein Kinase B; IL-18, Interleukin-18; Caspase-1, Cysteine Aspartate Protease-1; IL-1β, Interleukin-1 Beta; FOXO3a, Forkhead Box O3a; COVID-19, Coronavirus Disease 2019; BALF, Bronchoalveolar Lavage Fluid; SOCS3, Suppressor of cytokine signaling 3; HOTAIR, HOX Transcript Antisense RNA; MALAT1, Metastasis-associated Lung Adenocarcinoma Transcript 1; RDUR, RIG-I-dependent IAV-upregulated Noncoding RNA; TRIM25, Tripartite motif-containing protein 25; MARCO, Macrophage Receptor with Collagenous Structure.
References
Aiba, H., Mochizuki, M., Kimura, M., and Hojo, H. (2001). Predictive value of serum interleukin-6 level in influenza virus-associated encephalopathy. Neurology 57, 295–299. doi: 10.1212/WNL.57.2.295
Alon, R., Sportiello, M., Kozlovski, S., Kumar, A., Reilly, E. C., Zarbock, A., et al. (2021). Leukocyte trafficking to the lungs and beyond: lessons from influenza for COVID-19. Nat. Rev. Immunol. 21, 49–64. doi: 10.1038/s41577-020-00470-2
Arora, S., Lim, W., Bist, P., Perumalsamy, R., Lukman, H. M., Li, F., et al. (2016). Influenza A virus enhances its propagation through the modulation of Annexin-A1 dependent endosomal trafficking and apoptosis. Cell Death Differentiation 23, 1243–1256. doi: 10.1038/cdd.2016.19
Arshad, S., Naveed, M., Ullia, M., Javed, K., Butt, A., Khawar, M., et al. (2020). Targeting STAT-3 signaling pathway in cancer for development of novel drugs: Advancements and challenges. Genet. Mol. Biol. 43, 1–20. doi: 10.1590/1678-4685-GMB-2018-0160
Blanco-Melo, D., Nilsson-Payant, B. E., Liu, W. C., Uhl, S., Hoagland, D., Møller, R., et al. (2020). Imbalanced host response to SARS-coV-2 drives development of COVID-19. Cell 181, 1036–1045.e9. doi: 10.1016/j.cell.2020.04.026
Boon, A. C. M., Finkelstein, D., Zheng, M., Liao, G., Allard, J., Klumpp, K., et al. (2011). H5n1 influenza virus pathogenesis in genetically diverse mice is mediated at the level of viral load. mBio 2, p10–1128. doi: 10.1128/mBio.00171-11
Boyd, D. F., Jordan, S. V., and Balachandran, S. (2025). ZBP1-driven cell death in severe influenza. Trends Microbiol. 33, 521–532. doi: 10.1016/j.tim.2024.12.008
Bradley-Stewart, A., Jolly, L., Adamson, W., Gunson, R., Frew-Gillespie, C., Templeton, K., et al. (2013). Cytokine responses in patients with mild or severe influenza A(H1N1)pdm09. J. Clin. Virol. 58, 100–107. doi: 10.1016/j.jcv.2013.05.011
Brydon, E. W. A., Morris, S. J., and Sweet, C. (2005). Role of apoptosis and cytokines in influenza virus morbidity. FEMS Microbiol. Rev. 29, 837–850. doi: 10.1016/j.femsre.2004.12.003
Carty, M., Guy, C., and Bowie, A. G. (2021). Detection of viral infections by innate immunity. Biochem. Pharmacol. 183, 114316. doi: 10.1016/j.bcp.2020.114316
Chakravarty, S., Varghese, M., Fan, S., Taylor, R. T., Chakravarti, R., and Chattopadhyay, S. (2024). IRF3 inhibits inflammatory signaling pathways in macrophages to prevent viral pathogenesis. Sci. Adv. 10, 1–13. doi: 10.1126/sciadv.adn2858
Chan, M. C. W., Cheung, C. Y., Chui, W. H., Tsao, G. S. W., Nicholls, J. M., Chan, Y. O., et al. (2005). Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir. Res. 6, 1–13. doi: 10.1186/1465-9921-6-135
Chan, Y. K. and Gack, M. U. (2015). RIG-I-like receptor regulation in virus infection and immunity. Curr. Opin. Virol. 12, 7–14. doi: 10.1016/j.coviro.2015.01.004
Chan, Y. K. and Gack, M. U. (2016). Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 14, 360–373. doi: 10.1038/nrmicro.2016.45
Chanturiya, A. N., Basan, G., Schubert, U., Henklein, P., Yewdell, J. W., and Zimmerberg, J. (2004). PB1F2 creates pores in lipid membranes.pdf. J. Virol. 78, 6304–6312. doi: 10.1128/JVI.78.12.6304
Chen, B., Guo, G., Wang, G., Zhu, Q., Wang, L., Shi, W., et al. (2024). ATG7/GAPLINC/IRF3 axis plays a critical role in regulating pathogenesis of influenza A virus. PloS Pathog. 20, e, 1011958. doi: 10.1371/journal.ppat.1011958
Chen, Y., Hu, J., Liu, S., Chen, B., Xiao, M., Li, Y., et al. (2021b). RDUR, a lncRNA, promotes innate antiviral responses and provides feedback control of NF-κB activation. Front. Immunol. 12. doi: 10.3389/fimmu.2021.672165
Chen, X., Liu, S., Goraya, M. U., Maarouf, M., Huang, S., and Chen, J. L. (2018). Host immune response to influenza A virus infection. Front. Immunol. 9. doi: 10.3389/fimmu.2018.00320
Chen, S., Miao, X., Huangfu, D., Zhao, X., Zhang, M., Qin, T., et al. (2021a). H5N1 avian influenza virus without 80–84 amino acid deletion at the NS1 protein hijacks the innate immune system of dendritic cells for an enhanced mammalian pathogenicity. Transboundary Emerging Dis. 68, 2401–2413. doi: 10.1111/tbed.13904
Chen, N., Xia, P., Li, S., Zhang, T., Wang, T. T., and Zhu, J. (2017). RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life 69, 297–304. doi: 10.1002/iub.1625
Chi, X., Huang, G., Wang, L., Zhang, X., Liu, J., Yin, Z., et al. (2024). A small protein encoded by PCBP1-AS1 is identified as a key regulator of influenza virus replication via enhancing autophagy. PloS Pathog. 20, 1–29. doi: 10.1371/journal.ppat.1012461
Chi, Y., Zhu, Y., Wen, T., Cui, L., Ge, Y., Jiao, Y., et al. (2013). Cytokine and chemokine levels in patients infected with the novel avian influenza A (H7N9) virus in China. J. Infect. Dis. 208, 1962–1967. doi: 10.1093/infdis/jit440
Chiaretti, A., Pulitanò, S., Barone, G., Ferrara, P., Romano, V., Capozzi, D., et al. (2013). IL-1 β and IL-6 upregulation in children with H1N1 influenza virus infection. Mediators Inflammation 2013, 495848. doi: 10.1155/2013/495848
Choy, E. H., De Benedetti, F., Takeuchi, T., Hashizume, M., John, M. R., and Kishimoto, T. (2020). Translating IL-6 biology into effective treatments. Nat. Rev. Rheumatol. 16, 335–345. doi: 10.1038/s41584-020-0419-z
Davies, R. and Choy, E. (2014). Clinical experience of IL-6 blockade in rheumatic diseases—Implications on IL-6 biology and disease pathogenesis. Semin. Immunol. 26, 97–104. doi: 10.1016/j.smim.2013.12.002
De Jong, M. D., Simmons, C. P., Thanh, T. T., Hien, V. M., Smith, G. J. D., Chau, T. N. B., et al. (2006). Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 12, 1203–1207. doi: 10.1038/nm1477
Denisova, O. V., Söderholm, S., Virtanen, S., Von Schantz, C., Bychkov, D., Vashchinkina, E., et al. (2014). Akt inhibitor MK2206 prevents influenza pH1N1 virus infection in vitro. Antimicrobial Agents Chemotherapy 58, 3689–3696. doi: 10.1128/AAC.02798-13
De Wit, E., Siegers, J. Y., Cronin, J. M., Weatherman, S., Van Den Brand, J. M., Leijten, L. M., et al. (2018). 1918 H1N1 influenza virus replicates and induces proinflammatory cytokine responses in extrarespiratory tissues of ferrets. J. Infect. Dis. 217, 1237–1246. doi: 10.1093/infdis/jiy003
Dienz, O., Rud, J. G., Eaton, S. M., Lanthier, P. A., Burg, E., Drew, A., et al. (2012). Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol. 5, 258–266. doi: 10.1038/mi.2012.2
Eastman, A., Jacoby, D. B., Eastman, A., and Jacoby, D. B. (1996). Oxidant stress responses in influenza virus pneumonia: gene expression and transcription factor activation. Am. J. Pathophysiology. 271, L383–L391. doi: 10.1152/ajplung.1996.271.3.L383
Ehrhardt, C., Wolff, T., Pleschka, S., Planz, O., Beermann, W., Bode, J. G., et al. (2007). Influenza A virus NS1 protein activates the PI3K/akt pathway to mediate antiapoptotic signaling responses. J. Virol. 81, 3058–3067. doi: 10.1128/JVI.02082-06
Fajgenbaum, D. C. and June, C. H. (2020). Cytokine storm. New Engl. J. Med. 383, 2255–2273. doi: 10.1056/nejmra2026131
Fang, Y. and Peng, K. (2022). Regulation of innate immune responses by cell death-associated caspases during virus infection. FEBS J. 289, 4098–4111. doi: 10.1111/febs.16051
Fernández-Arjona, M. D. M., Grondona, J. M., Fernández-Llebrez, P., and López-Ávalos, M. D. (2019). Microglial activation by microbial neuraminidase through TLR2 and TLR4 receptors. J. Neuroinflamm. 16, 1–14. doi: 10.1186/s12974-019-1643-9
Flory, E., Kunz, M., Scheller, C., Jassoy, C., Stauber, R., Rapp, U. R., et al. (2000). Influenza virus-induced NF-κB-dependent gene expression is mediated by overexpression of viral proteins and involves oxidative radicals and activation of IκB kinase. J. Biol. Chem. 275, 8307–8314. doi: 10.1074/jbc.275.12.8307
García-Sastre, A., Egorov, A., Matassov, D., Brandt, S., Levy, D. E., Durbin, J. E., et al. (1998). Influenza A virus lacking the NS1 gene replicates in interferon- deficient systems. Virology 252, 324–330. doi: 10.1006/viro.1998.9508
Gaur, P., Munjhal, A., and Lal, S. K. (2011). Influenza virus and cell signaling pathways. Med. Sci. monitor : Int. Med. J. Exp. Clin. Res. 17, RA148–RA154. doi: 10.12659/MSM.881801
Gautam, A., Boyd, D. F., Nikhar, S., Zhang, T., Siokas, I., Van de Velde, L.-A., et al. (2024). Necroptosis blockade prevents lung injury in severe influenza. Nature 628, 835–843. doi: 10.1038/s41586-024-07265-8
Gentile, D., Doyle, W., Whiteside, T., Fireman, P., Hayden, F. G., and Skoner, D. (1998). Increased interleukin-6 levels in nasal lavage samples following experimental influenza a virus infection. Clin. Diagn. Lab. Immunol. 5, 604–608. doi: 10.1128/cdli.5.5.604-608.1998
Gou, X., Yuan, J., Wang, H., Wang, X., Xiao, J., Chen, J., et al. (2020). IL-6 during influenza-streptococcus pneumoniae co-infected pneumonia—A protector. Front. Immunol. 10. doi: 10.3389/fimmu.2019.03102
Grebenciucova, E. and VanHaerents, S. (2023). Interleukin 6: at the interface of human health and disease. Front. Immunol. 14. doi: 10.3389/fimmu.2023.1255533
Gu, Y., Hsu, A. C. Y., Pang, Z., Pan, H., Zuo, X., Wang, G., et al. (2019). Role of the innate cytokine storm induced by the influenza A virus. Viral Immunol. 32, 244–251. doi: 10.1089/vim.2019.0032
Gu, Y., Zuo, X., Zhang, S., Ouyang, Z., Jiang, S., Wang, F., et al. (2021). The mechanism behind influenza virus cytokine storm. Viruses 13, 1362. doi: 10.3390/v13071362
Guo, H., Jin, Y., Zhi, X., Yan, D., and Sun, S. (2015). NLRP3 inflammasome activation by viroporins of animal viruses. Viruses. 3380–3391. doi: 10.3390/v7062777
Hagenaars, T. J., Fischer, E. A. J., Jansen, C. A., Rebel, J. M. J., Spekreijse, D., Vervelde, L., et al. (2016). Modelling the innate immune response against avian influenza virus in chicken. PloS One. 1–17. doi: 10.1371/journal.pone.0157816
Hale, B. G., Randall, R. E., Ortin, J., and Jackson, D. (2008). The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 89, 2359–2376. doi: 10.1099/vir.0.2008/004606-0
Harpur, C. M., Le Page, M. A., and Tate, M. D. (2021). Too young to die? How aging affects cellular innate immune responses to influenza virus and disease severity. Virulence 12, 1629–1646. doi: 10.1080/21505594.2021.1939608
Hn, P.İ.A., Tnf-, H., Düzeyleri, I.-, and Etkisi, M. (2013). TNF- a, IL-1 b and IL-6 Levels in Pandemic Influenza A (H1N1) 2009 Patients and Effect on Mortality. MJIMA 2–7, 2.
Holm, C. K., Rahbek, S. H., Gad, H. H., Bak, R. O., Jakobsen, M. R., Jiang, Z., et al. (2016). Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat. Commun. 7, 10680. doi: 10.1038/ncomms10680
Ichinohe, T., Pang, I. K., and Iwasaki, A. (2010). Influenza virus activates inflammasomes through intracellular M2 channel. Nat. Immunol. 11, 404–410. doi: 10.1038/ni.1861.Influenza
Imai, Y., Kuba, K., Neely, G. G., Yaghubian-Malhami, R., Perkmann, T., van Loo, G., et al. (2008). Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133, 235–249. doi: 10.1016/j.cell.2008.02.043
Ito, Y., Ichiyama, T., Kimura, H., Shibata, M., Ishiwada, N., Kuroki, H., et al. (1999). Detection of influenza virus RNA by reverse transcription-PCR and proinflammatory cytokines in influenza-virus-associated encephalopathy. J. Med. Virol. 58, 420–425. doi: 10.1002/(SICI)1096-9071(199908)58:4<420::AID-JMV16>3.0.CO;2-T
Iwai, A., Shiozaki, T., Kawai, T., Akira, S., Kawaoka, Y., Takada, A., et al. (2010). Influenza A virus polymerase inhibits type I interferon induction by binding to interferon β promoter stimulator 1. J. Biol. Chem. 285, 32064–32074. doi: 10.1074/jbc.M110.112458
Iwasaki, A. and Pillai, P. S. (2014). Innate immunity to influenza virus infection. Nat. Rev. Immunol. 14, 315–328. doi: 10.1038/nri3665
Jensen, S. and Thomsen, A. R. (2012). Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 86, 2900–2910. doi: 10.1128/JVI.05738-11
Jiang, H. and Zhang, Z. (2023). Immune response in influenza virus infection and modulation of immune injury by viral neuraminidase. Virol. J. 20, 1–10. doi: 10.1186/s12985-023-02164-2
Kalaiyarasu, S., Kumar, M., Senthil Kumar, D., Bhatia, S., Dash, S. K., Bhat, S., et al. (2016). Highly pathogenic avian influenza H5N1 virus induces cytokine dysregulation with suppressed maturation of chicken monocyte-derived dendritic cells. Microbiol. Immunol. 60, 687–693. doi: 10.1111/1348-0421.12443
Kalil, A. C. and Thomas, P. G. (2019). Influenza virus-related critical illness: pathophysiology and epidemiology. Critical Care 23 (1), 258. doi: 10.1186/s13054-019-2539-x
Kang, S. and Kishimoto, T. (2021). Interplay between interleukin-6 signaling and the vascular endothelium in cytokine storms. Exp. Mol. Med. 53, 1116–1123. doi: 10.1038/s12276-021-00649-0
Kang, Y. M., Song, B. M., Lee, J. S., Kim, H. S., and Seo, S. H. (2011). Pandemic H1N1 influenza virus causes a stronger inflammatory response than seasonal H1N1 influenza virus in ferrets. Arch. Virol. 156, 759–767. doi: 10.1007/s00705-010-0914-7
Killip, M. J., Fodor, E., and Randall, R. E. (2015). Influenza virus activation of the interferon system. Virus Res. 209, 11–22. doi: 10.1016/j.virusres.2015.02.003
Kim, C. U., Jeong, Y. J., Lee, P., Lee, M. S., Park, J. H., Kim, Y. S., et al. (2022). Extracellular nucleoprotein exacerbates influenza virus pathogenesis by activating Toll-like receptor 4 and the NLRP3 inflammasome. Cell. Mol. Immunol. 19, 715–725. doi: 10.1038/s41423-022-00862-5
Kim, C., Lim, D., Kim, Y. S., Ku, B., and Kim, D. (2023). Influenza viral matrix 1 protein aggravates viral pathogenicity by inducing TLR4-mediated reactive oxygen species production and apoptotic cell death. Cell Death Dis. 1–12, 228. doi: 10.1038/s41419-023-05749-5
Kinoshita, T., Imamura, R., Kushiyama, H., and Suda, T. (2015). NLRP3 Mediates NF-κB activation and cytokine induction in microbially induced and sterile inflammation. PloS One 10, 1–15. doi: 10.1371/journal.pone.0119179
Kishimoto, T. (2005). Interleukin-6: From basic science to medicine - 40 Years in immunology. Annu. Rev. Immunol. 23, 1–21. doi: 10.1146/annurev.immunol.23.021704.115806
Klemm, C., Bruchhagen, C., Van Krüchten, A., Niemann, S., Löffler, B., Peters, G., et al. (2017). Mitogen-activated protein kinases (MAPKs) regulate IL-6 over-production during concomitant influenza virus and Staphylococcus aureus infection. Sci. Rep. 7, 1–15. doi: 10.1038/srep42473
Kochs, G., Garcia-Sastre, A., and Martinez-Sobrido, L. (2007). Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 81, 7011–7021. doi: 10.1128/jvi.02581-06
Krammer, F., Smith, G. J. D., Fouchier, R. A. M., Peiris, M., Kedzierska, K., Doherty, P. C., et al. (2018). Influenza. Nat. Rev. Dis. Primers 4, 3. doi: 10.1038/s41572-018-0002-y
Lauder, S. N., Jones, E., Smart, K., Bloom, A., Williams, A. S., Hindley, J. P., et al. (2013). Interleukin-6 limits influenza-induced inflammation and protects against fatal lung pathology. Eur. J. Immunol. 43, 2613–2625. doi: 10.1002/eji.201243018
Lee, J., Nakagiri, T., Oto, T., Harada, M., Morii, E., Shintani, Y., et al. (2012). IL-6 amplifier, NF-κB–triggered positive feedback for IL-6 signaling, in grafts is involved in allogeneic rejection responses. J. Immunol. 189, 1928–1936. doi: 10.4049/jimmunol.1103613
Leethese, S. W., Youn, J. W., Seong, B. L., and Sung, Y. C. (1999). IL-6 induces long-term protective immunity against a lethal challenge of influenza virus. Vaccine 17, 490–496. doi: 10.1016/S0264-410X(98)00223-0
Li, H., Bradley, K. C., Long, J. S., Frise, R., Ashcroft, J. W., Hartgroves, L. C., et al. (2018). Internal genes of a highly pathogenic H5N1 influenza virus determine high viral replication in myeloid cells and severe outcome of infection in mice. PloS Pathog. 14, e1006821. doi: 10.1371/journal.ppat.1006821
Li, Z., Jiang, Y., Jiao, P., Wang, A., Zhao, F., Tian, G., et al. (2006). The NS1 gene contributes to the virulence of H5N1 avian influenza viruses. J. Virol. 80, 11115–11123. doi: 10.1128/jvi.00993-06
Li, J., Wang, M., Song, L., Wang, X., Lai, W., and Jiang, S. (2020). LncRNA MALAT1 regulates inflammatory cytokine production in lipopolysaccharide-stimulated human gingival fibroblasts through sponging miR-20a and activating TLR4 pathway. J. Periodontal Res. 55, 182–190. doi: 10.1111/jre.12700
Liu, S., Cai, X., Wu, J., Cong, Q., Chen, X., Li, T., et al. (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Sci. (New York N.Y.) 347, aaa2630. doi: 10.1126/science.aaa2630
Liu, S., Liu, S., Yu, Z., Zhou, W., Zheng, M., Gu, R., et al. (2023). STAT3 regulates antiviral immunity by suppressing excessive interferon signaling. Cell Rep. 42, 112806. doi: 10.1016/j.celrep.2023.112806
Liu, S., Qiu, F., Gu, R., and Xu, E. (2024). Functional involvement of signal transducers and activators of transcription in the pathogenesis of influenza A virus. Int. J. Mol. Sci. 25, 1–14. doi: 10.3390/ijms252413589
Liu, Q., Xiong, H. R., Lu, L., Liu, Y. Y., Luo, F., Hou, W., et al. (2013). Antiviral and anti-inflammatory activity of arbidol hydrochloride in influenza A (H1N1) virus infection. Acta Pharmacologica Sin. 34, 1075–1083. doi: 10.1038/aps.2013.54
Liu, S., Yan, R., Chen, B., Pan, Q., Chen, Y., Hong, J., et al. (2019). Influenza virus-induced robust expression of SOCS3 contributes to excessive production of IL-6. Front. Immunol. 10. doi: 10.3389/fimmu.2019.01843
Long, J. S., Mistry, B., Haslam, S. M., and Barclay, W. S. (2019). Host and viral determinants of influenza A virus species specificity. Nat. Rev. Microbiol. 17, 67–81. doi: 10.1038/s41579-018-0115-z
Longhi, M. P., Wright, K., Lauder, S. N., Nowell, M. A., Jones, G. W., Godkin, A. J., et al. (2008). Interleukin-6 is crucial for recall of influenza-specific memory CD4 + T cells. PloS Pathog. 4, 2–9. doi: 10.1371/journal.ppat.1000006
Ludwig, S., Ehrhardt, C., Neumeier, E. R., Kracht, M., Rapp, U. R., and Pleschka, S. (2001). Influenza virus-induced AP-1-dependent gene expression requires activation of the JNK signaling pathway. J. Biol. Chem. 276, 10990–10998. doi: 10.1074/jbc.M009902200
Ma, Y., Ouyang, J., Wei, J., Maarouf, M., and Chen, J. L. (2016). Involvement of host non-coding RNAs in the pathogenesis of the influenza virus. Int. J. Mol. Sci. 18, 39. doi: 10.3390/ijms18010039
Maarouf, M., Chen, B., Chen, Y., Wang, X., Rai, K. R., Zhao, Z., et al. (2019). Identification of lncRNA-155 encoded by MIR155HG as a novel regulator of innate immunity against influenza A virus infection. Cell. Microbiol. 21, e13036. doi: 10.1111/cmi.13036
Maarouf, M. and Rai, K. R. (2018). Immune ecosystem of virus-infected host tissues. Int. J. Mol. Sci. 13, 1–18. doi: 10.3390/ijms19051379
Malik, G. and Zhou, Y. (2020). Innate immune sensing of influenza a virus. Viruses 12, 755. doi: 10.3390/v12070755
Marc, D. (2014). Influenza virus non-structural protein NS1: Interferon antagonism and beyond. J. Gen. Virol. 95, 2594–2611. doi: 10.1099/vir.0.069542-0
Matsukura, S., Kokubu, F., Noda, H., Tokunaga, H., and Adachi, M. (1996). Expression of IL-6, IL-8, and RANTES on human bronchial epithelial cells, NCI-H292, induced by influenza virus A. J. Allergy Clin. Immunol. 98, 1080–1087. doi: 10.1016/S0091-6749(96)80195-3
Kawai, T. and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nature Immunology 11, 373–384. doi: 10.1038/ni.1863
Maarouf, M., Rai, K. R., Goraya, M. U., and Chen, J. L. (2018). Immune Ecosystem of Virus-Infected Host Tissues. International journal of molecular sciences 13, 1–18. doi: 10.3390/ijms19051379
Monto, A. S. and Fukuda, K. (2020). Lessons from influenza pandemics of the last 100 years. Clin. Infect. Dis. 70, 951–957. doi: 10.1093/cid/ciz803
Morris, G., Bortolasci, C. C., Puri, B. K., Marx, W., O’Neil, A., Athan, E., et al. (2021). The cytokine storms of COVID-19, H1N1 influenza, CRS and MAS compared. Can one sized treatment fit all? Cytokine 144, 155593. doi: 10.1016/j.cyto.2021.155593
Murray, C., Griffin, É.W., O’Loughlin, E., Lyons, A., Sherwin, E., Ahmed, S., et al. (2015). Interdependent and independent roles of type I interferons and IL-6 in innate immune, neuroinflammatory and sickness behaviour responses to systemic poly I: C. Brain Behavior Immun. 48, 274–286. doi: 10.1016/j.bbi.2015.04.009
Narazaki, M. and Kishimoto, T. (2018). The two-faced cytokine IL-6 in host defense and diseases. Int. J. Mol. Sci. 19, 3528. doi: 10.3390/ijms19113528
Negash, A. A., Ramos, H. J., Crochet, N., Lau, D. T. Y., Doehle, B., Papic, N., et al. (2013). IL-1β Production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PloS Pathog. 9, 1–13. doi: 10.1371/journal.ppat.1003330
Newby, C. M., Sabin, L., and Pekosz, A. (2007). The RNA binding domain of influenza A virus NS1 protein affects secretion of tumor necrosis factor alpha, interleukin-6, and interferon in primary murine tracheal epithelial cells. J. Virol. 81, 9469–9480. doi: 10.1128/jvi.00989-07
Niu, J. and Meng, G. (2023). Roles and mechanisms of NLRP3 in influenza viral infection. Viruses 15, 1339. doi: 10.3390/v15061339
Obaid, M., Udden, S. M. N., Deb, P., Shihabeddin, N., Zaki, M. H., and Mandal, S. S. (2018). LncRNA HOTAIR regulates lipopolysaccharide-induced cytokine expression and inflammatory response in macrophages. Sci. Rep. 8, 1–18. doi: 10.1038/s41598-018-33722-2
Oltean, T., Maelfait, J., Saelens, X., and Vandenabeele, P. (2024). Need for standardization of Influenza A virus-induced cell death in vivo to improve consistency of inter-laboratory research findings. Cell Death Discov. 10, 1–10. doi: 10.1038/s41420-024-01981-w
Ouyang, A., Chen, T., Feng, Y., Zou, J., Tu, S., Jiang, M., et al. (2024). The hemagglutinin of influenza A virus induces ferroptosis to facilitate viral replication. Advanced Sci. 2404365, 1–16. doi: 10.1002/advs.202404365
Ouyang, J., Zhu, X., Chen, Y., Wei, H., Chen, Q., Chi, X., et al. (2014). NRAV, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe 16, 616–626. doi: 10.1016/j.chom.2014.10.001
Pahl, H. L. and Baeuerle, P. A. (1995). Expression of influenza virus hemagglutinin activates transcription factor NF-kappa B. J. Virol. 69, 1480–1484. doi: 10.1128/jvi.69.3.1480-1484.1995
Pandey, P., Al Rumaih, Z., Kels, M. J. T., Ng, E., Kc, R., Malley, R., et al. (2023). Therapeutic targeting of inflammation and virus simultaneously ameliorates influenza pneumonia and protects from morbidity and mortality. Viruses 15, 318. doi: 10.3390/v15020318
Paquette, S. G., Banner, D., Zhao, Z., Fang, Y., Huang, S. S. H., León, A. J., et al. (2012). Interleukin-6 is a potential biomarker for severe pandemic H1N1 influenza a infection. PloS One 7, e38214. doi: 10.1371/journal.pone.0038214
Pauli, E., Schmolke, M., Wolff, T., Viemann, D., Roth, J., Johannes, G., et al. (2008). ). Influenza A Virus Inhibits Type I IFN Signaling via NF- k B- Dependent Induction of SOCS-3 Expression. PLoS pathogens 4, 1–15. doi: 10.1371/journal.ppat.1000196
Perrone, L. A., Plowden, J. K., García-Sastre, A., Katz, J. M., and Tumpey, T. M. (2008). H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PloS Pathog. 4, e1000115. doi: 10.1371/journal.ppat.1000115
Piattini, F., Sidiropoulos, N. D., Berest, I., and Kopf, M. (2025). IL-6 mediates defense against influenza virus by promoting protective antibody responses but not innate inflammation. Mucosal Immunol. 18, 596–606. doi: 10.1016/j.mucimm.2025.02.001
Pillai, A. I. (2014). Innate immunity to influenza virus infection. Nat. Rev. Immunol. 14, 315–328. doi: 10.1038/nri3665.Innate
Pinar, A., Dowling, J. K., Bitto, N. J., Robertson, A. A. B., Latz, E., Stewart, C. R., et al. (2017). PB1-F2 peptide derived from avian influenza A virus H7N9 induces inflammation via activation of the NLRP3 inflammasome. J. Biol. Chem. 292, 826–836. doi: 10.1074/jbc.M116.756379
Pinto, R., Herold, S., Cakarova, L., Hoegner, K., Lohmeyer, J., Planz, O., et al. (2011). Inhibition of influenza virus-induced NF-kappaB and Raf/MEK/ERK activation can reduce both virus titers and cytokine expression simultaneously in vitro and in vivo. Antiviral Res. 92, 45–56. doi: 10.1016/j.antiviral.2011.05.009
Popli, S., Chakravarty, S., Fan, S., Glanz, A., Aras, S., Nagy, L. E., et al. (2022). IRF3 inhibits nuclear translocation of NF-κB to prevent viral inflammation. Proc. Natl. Acad. Sci. United States America 119, 1–11. doi: 10.1073/pnas.2121385119
Prantner, D., Shirey, K. A., Lai, W., Lu, W., Cole, A. M., Vogel, S. N., et al. (2017). The θ-defensin retrocyclin 101 inhibits TLR4- and TLR2-dependent signaling and protects mice against influenza infection. J. leukocyte Biol. 102, 1103–1113. doi: 10.1189/jlb.2A1215-567RR
Pullamsetti, S. S., Seeger, W., and Savai, R. (2018). Classical IL-6 signaling: A promising therapeutic target for pulmonary arterial hypertension. J. Clin. Invest. 128, 1720–1723. doi: 10.1172/JCI120415
Pyle, C. J., Uwadiae, F. I., Swieboda, D. P., and Harker, J. A. (2017). Early IL-6 signalling promotes IL-27 dependent maturation of regulatory T cells in the lungs and resolution of viral immunopathology. PloS Pathog. 13, 1–27. doi: 10.1371/journal.ppat.1006640
Radigan, K. A., Nicholson, T. T., Welch, L. C., Chi, M., Amarelle, L., Angulo, M., et al. (2019). Influenza A virus infection induces muscle wasting via IL-6 regulation of the E3 ubiquitin ligase atrogin-1. J. Immunol. 202, 484–493. doi: 10.4049/jimmunol.1701433
Rai, K. R., Chen, B., Zhao, Z., Chen, Y., Hu, J., Liu, S., et al. (2020). Robust expression of p27Kip1 induced by viral infection is critical for antiviral innate immunity. Cell. Microbiol. 22, e13242. doi: 10.1111/cmi.13242
Rai, K. R., Liao, Y., Cai, M., Qiu, H., Wen, F., and Min Peng, S. W. (2022). MIR155HG plays a bivalent role in regulating innate antiviral. mBio 13, e02510–e02522. doi: 10.1128/mbio.02510-22
Rai, K. R., Shrestha, P., Yang, B., Chen, Y., Liu, S., Maarouf, M., et al. (2021). Acute infection of viral pathogens and their innate immune escape. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.672026
Ramos, I., Smith, G., Ruf-Zamojski, F., Martínez-Romero, C., Fribourg, M., Carbajal, E. A., et al. (2019). Innate immune response to influenza virus at single-cell resolution in human epithelial cells revealed paracrine induction of interferon lambda 1. J. Virol. 93, 1–22. doi: 10.1128/jvi.00559-19
Resnik, R., Lopez Mingorance, F., Rivera, F., Mitchell, F., Gonzalez, C. D., and Vaccaro, M. I. (2023). Autophagy in inflammatory response against SARS-coV-2. Int. J. Mol. Sci. 24, 4928. doi: 10.3390/ijms24054928
Richetta, C. and Faure, M. (2013). Autophagy in antiviral innate immunity. Cell. Microbiol. 15, 368–376. doi: 10.1111/cmi.12043
Rose-John, S. (2012). Il-6 trans-signaling via the soluble IL-6 receptor: Importance for the proinflammatory activities of IL-6. Int. J. Biol. Sci. 8, 1237–1247. doi: 10.7150/ijbs.4989
Salomon, R., Hoffmann, E., and Webster, R. G. (2007). Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc. Natl. Acad. Sci. United States America 104, 12479–12481. doi: 10.1073/pnas.0705289104
Shao, W., Li, X., Goraya, M. U., Wang, S., and Chen, J. L. (2017). Evolution of influenza a virus by mutation and re-assortment. Int. J. Mol. Sci. 18, 1650. doi: 10.3390/ijms18081650
Shinya, K., Ito, M., Makino, A., Tanaka, M., Miyake, K., Eisfeld, A. J., et al. (2012). The TLR4-TRIF pathway protects against H5N1 influenza virus infection. J. Virol. 86, 19–24. doi: 10.1128/jvi.06168-11
Short, K. R., Veeris, R., Leijten, L. M., Van Den Brand, J. M., Jong, V. L., Stittelaar, K., et al. (2017). Proinflammatory cytokine responses in extra-respiratory tissues during severe influenza. J. Infect. Dis. 216, 829–833. doi: 10.1093/infdis/jix281
Shrivastava, G., León-Juárez, M., García-Cordero, J., Meza-Sánchez, D. E., and Cedillo-Barrón, L. (2016). Inflammasomes and its importance in viral infections. Immunologic Res. 64, 1101–1117. doi: 10.1007/s12026-016-8873-z
Sprenger, H., Bacher, M., Rischkowsky, E., Bender, A., Nain, M., and Gemsa, D. (1994). Characterization of a high molecular weight tumor necrosis factor-alpha mRNA in influenza A virus-infected macrophages. J. Immunol. (Baltimore Md. : 1950) 152, 280–289. doi: 10.4049/jimmunol.152.1.280
Strutt, T. M., McKinstry, K. K., Kuang, Y., Finn, C. M., Hwang, J. H., Dhume, K., et al. (2016). Direct IL-6 signals maximize protective secondary CD4 T cell responses against influenza. J. Immunol. 197, 3260–3270. doi: 10.4049/jimmunol.1600033
Sun, H., Gu, R., Tang, T., and Rai, K. R. (2025). Current perspectives on functional involvement of micropeptides in virus – host interactions. Int. J. Mol. Sci. 26, 1–14. doi: 10.3390/ijms26083651
Sun, K. and Metzger, D. W. (2019). Influenza and Staphylococcus aureus Coinfection: TLR9 at Play. Trends Microbiol. 27, 383–384. doi: 10.1016/j.tim.2019.02.006
Svitek, N., Rudd, P. A., Obojes, K., Pillet, S., and von Messling, V. (2008). Severe seasonal influenza in ferrets correlates with reduced interferon and increased IL-6 induction. Virology 376, 53–59. doi: 10.1016/j.virol.2008.02.035
Szretter, K. J., Gangappa, S., Lu, X., Smith, C., Shieh, W.-J., Zaki, S. R., et al. (2007). Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J. Virol. 81, 2736–2744. doi: 10.1128/jvi.02336-06
Tanaka, T., Narazaki, M., and Kishimoto, T. (2014). IL-6 in inflammation, immunity, and disease. Cold Spring Harbor Perspect. Biol. 6 (10), a016295. doi: 10.1101/cshperspect.a016295
The REMAP−CAP Investigators (2021). Interleukin-6 receptor antagonists in critically ill patients with covid-19. New Engl. J. Med. 384, 1491–1502. doi: 10.1056/nejmoa2100433
Thomas, P. G., Dash, P., Aldridge, J. R., Ellebedy, A. H., Reynolds, C., Funk, A. J., et al. (2009). The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30, 566–575. doi: 10.1016/j.immuni.2009.02.006
To, K. F., Chan, P. K. S., Chan, K. F., Lee, W. K., Lam, W. Y., Wong, K. F., et al. (2001). Pathology of fatal human infection associated with avian influenza A H5N1 virus. J. Med. Virol. 63, 242–246. doi: 10.1002/1096-9071(200103)63:3<242::AID-JMV1007>3.0.CO;2-N
Tripathi, S., Batra, J., Cao, W., Sharma, K., Patel, J. R., Ranjan, P., et al. (2013). Influenza A virus nucleoprotein induces apoptosis in human airway epithelial cells: Implications of a novel interaction between nucleoprotein and host protein clusterin. Cell Death Dis. 4, e562–e512. doi: 10.1038/cddis.2013.89
Tuvim, M. J., Gilbert, B. E., Dickey, B. F., and Evans, S. E. (2012). Synergistic TLR2/6 and TLR9 activation protects mice against lethal influenza pneumonia. PloS One 7, 1–9. doi: 10.1371/journal.pone.0030596
Uyeki, T. M., Hui, D. S., Zambon, M., Wentworth, D. E., and Monto, A. S. (2022). Influenza. Lancet 400, 693–706. doi: 10.1016/S0140-6736(22)00982-5
Velazquez-Salinas, L., Verdugo-Rodriguez, A., Rodriguez, L. L., and Borca, M. V. (2019). The role of interleukin 6 during viral infections. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.01057
Vincent, J.-L., Tang, B. M., and Cootes T, M. A. (2019). Annual update in intensive care 2019. Annu. Update Intensive Care Emergency Med. 653. doi: 10.1007/978-3-030-06067-1
Visseren, F. L. J., Verkerk, M. S. A., Bouter, K. P., Diepersloot, R. J. A., and Erkelens, D. W. (1999). Interleukin-6 production by endothelial cells after infection with influenza virus and cytomegalovirus. J. Lab. Clin. Med. 134, 623–630. doi: 10.1016/S0022-2143(99)90103-8
Vogel, A. J., Harris, S., Marsteller, N., Condon, S. A., and Brown, D. M. (2014). Early cytokine dysregulation and viral replication are associated with mortality during lethal influenza infection. Viral Immunol. 27, 214–224. doi: 10.1089/vim.2013.0095
Wang, S., Le, T. Q., Kurihara, N., Chida, J., Cisse, Y., Yano, M., et al. (2010). Influenza virus-cytokine-protease cycle in the pathogenesis of vascular hyperpermeability in severe influenza. J. Infect. Dis. 202, 991–1001. doi: 10.1086/656044
Wang, J., Wang, Q., Han, T., Li, Y. K., Zhu, S. L., Ao, F., et al. (2015). Soluble interleukin-6 receptor is elevated during influenza A virus infection and mediates the IL-6 and IL-32 inflammatory cytokine burst. Cell. Mol. Immunol. 12, 633–644. doi: 10.1038/cmi.2014.80
Williams, S. L., Qi, L., Sheng, Z. M., Xiao, Y., Freeman, A., Matthews, L., et al. (2024). Effect of pandemic influenza A virus PB1 genes of avian origin on viral RNA polymerase activity and pathogenicity. Sci. Adv. 10, eads573. doi: 10.1126/sciadv.ads5735
Xiao, M., Chen, Y., Wang, S., Liu, S., Rai, K. R., Chen, B., et al. (2021). LncRNA IFITM4P regulates host antiviral responses by acting as a ceRNA. J. Virol. 95, 10–128. doi: 10.1128/jvi.00277-21
Xie, Y., Yu, Y., Zhao, L., Ning, P., Luo, Q., Zhang, Y., et al. (2021). Specific cytokine profiles predict the severity of influenza A pneumonia: A prospectively multicenter pilot study. BioMed. Res. Int. 2021, 9533044. doi: 10.1155/2021/9533044
Yang, M. L., Wang, C. T., Yang, S. J., Leu, C. H., Chen, S. H., Wu, C. L., et al. (2017). IL-6 ameliorates acute lung injury in influenza virus infection. Sci. Rep. 7, 1–11. doi: 10.1038/srep43829
Ye, S., Lowther, S., and Stambas, J. (2015). Inhibition of reactive oxygen species production ameliorates inflammation induced by influenza A viruses via upregulation of SOCS1 and SOCS3. J. Virol. 89, 2672–2683. doi: 10.1128/jvi.03529-14
Yu, Y., Chen, S., Zhang, H., Duan, Y., Li, Z., Jiang, L., et al. (2024). Inflammatory effects protect mice from lethal influenza virus infection. Antimicrob Agents Chemother. 68.
Yu, X., Zhang, X., Zhao, B., Wang, J., Zhu, Z., Teng, Z., et al. (2011). Intensive cytokine induction in pandemic H1N1 influenza virus infection accompanied by robust production of IL-10 and IL-6. PloS One 6, 1–9. doi: 10.1371/journal.pone.0028680
Zamarin, D., Ortigoza, M. B., and Palese, P. (2006). Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J. Virol. 80, 7976–7983. doi: 10.1128/jvi.00415-06
Zeng, Y., Xu, S., Wei, Y., Zhang, X., Wang, Q., Jia, Y., et al. (2021). The PB1 protein of influenza A virus inhibits the innate immune response by targeting MAVS for NBR1-mediated selective autophagic degradation. PloS Pathog. 17, 1–27. doi: 10.1371/JOURNAL.PPAT.1009300
Zhang, J. and Chu, M. (2019). Targeting of IL-6-relevant long noncoding RNA profiles in inflammatory and tumorous disease. Inflammation 42, 1139–1146. doi: 10.1007/s10753-019-00995-2
Zhang, T., Kee, W. H., Seow, K. T., Fung, W., and Cao, X. (2000). The coiled-coil domain of stat3 is essential for its SH2 domain-mediated receptor binding and subsequent activation induced by epidermal growth factor and interleukin-6. Mol. Cell. Biol. 20, 7132–7139. doi: 10.1128/mcb.20.19.7132-7139.2000
Zhang, J., Wang, J., Gong, Y., Gu, Y., Xiang, Q., and Tang, L. L. (2022). Interleukin-6 and granulocyte colony-stimulating factor as predictors of the prognosis of influenza-associated pneumonia. BMC Infect. Dis. 22, 1–12. doi: 10.1186/s12879-022-07321-6
Zhang, B., Xu, S., Liu, M., Wei, Y., Wang, Q., Shen, W., et al. (2023). The nucleoprotein of influenza A virus inhibits the innate immune response by inducing mitophagy. Autophagy 19, 1916–1933. doi: 10.1080/15548627.2022.2162798
Keywords: IL-6, IAV, innate immunity, inflammation, cytokine storm
Citation: Li X, Huang C, Rai KR and Xu Q (2025) Innate immune role of IL-6 in influenza a virus pathogenesis. Front. Cell. Infect. Microbiol. 15:1605446. doi: 10.3389/fcimb.2025.1605446
Received: 03 April 2025; Accepted: 12 June 2025;
Published: 07 July 2025.
Edited by:
Pratibha Gaur, International Centre for Genetic Engineering and Biotechnology (India), IndiaReviewed by:
Saikat De, The Scripps Research Institute, United StatesPhaneendra Chennampally, Novavax (United States), United States
Copyright © 2025 Li, Huang, Rai and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kul Raj Rai, a3VscmFqcmFpNzAxQGdtYWlsLmNvbQ==; Quanming Xu, eHF1YW5taW5nQGZqcHNjLmVkdS5jbg==