 Fatma F. Mohamed1
Fatma F. Mohamed1 Michael B. Chavez1
Michael B. Chavez1 Flavia Amadeu de Oliveira2
Flavia Amadeu de Oliveira2 Sonoko Narisawa2
Sonoko Narisawa2 Colin Farquharson3
Colin Farquharson3 José Luis Millán2
José Luis Millán2 Brian L. Foster1*
Brian L. Foster1*- 1Division of Biosciences, College of Dentistry, The Ohio State University, Columbus, OH, United States
- 2Sanford Children's Health Research Center, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA, United States
- 3The Royal (Dick) School of Veterinary Studies (RDSVS), The Roslin Institute, University of Edinburgh, Edinburgh, United Kingdom
Mineralization of the skeleton occurs by several physicochemical and biochemical processes and mechanisms that facilitate the deposition of hydroxyapatite (HA) in specific areas of the extracellular matrix (ECM). Two key phosphatases, phosphatase, orphan 1 (PHOSPHO1) and tissue-non-specific alkaline phosphatase (TNAP), play complementary roles in the mineralization process. The actions of PHOSPHO1 on phosphocholine and phosphoethanolamine in matrix vesicles (MVs) produce inorganic phosphate (Pi) for the initiation of HA mineral formation within MVs. TNAP hydrolyzes adenosine triphosphate (ATP) and the mineralization inhibitor, inorganic pyrophosphate (PPi), to generate Pi that is incorporated into MVs. Genetic mutations in the ALPL gene-encoding TNAP lead to hypophosphatasia (HPP), characterized by low circulating TNAP levels (ALP), rickets in children and/or osteomalacia in adults, and a spectrum of dentoalveolar defects, the most prevalent being lack of acellular cementum leading to premature tooth loss. Given that the skeletal manifestations of genetic ablation of the Phospho1 gene in mice resemble many of the manifestations of HPP, we propose that Phospho1 gene mutations may underlie some cases of “pseudo-HPP” where ALP may be normal to subnormal, but ALPL mutation(s) have not been identified. The goal of this perspective article is to compare and contrast the loss-of-function effects of TNAP and PHOSPHO1 on the dentoalveolar complex to predict the likely dental phenotype in humans that may result from PHOSPHO1 mutations. Potential cases of pseudo-HPP associated with PHOSPHO1 mutations may resist diagnosis, and the dental manifestations could be a key criterion for consideration.
Introduction
Mineralization of skeleton occurs by several physicochemical and biochemical processes and mechanisms that facilitate the deposition of hydroxyapatite (HA) in specific areas of the extracellular matrix (ECM). Experimental evidence has pointed to the presence of HA crystals inside, and outside, collagen fibrils in the ECM (1, 2), and also within the lumen of matrix vesicles (MVs) (3–5). Production of inorganic phosphate (Pi) for the initiation of HA mineral formation within the lumen of MVs takes place by the action of phosphatase, orphan 1 (PHOSPHO1) on phosphocholine and phosphoethanolamine in the inner leaflet of the MV membrane (6, 7). On the outer surface of the MV membrane, the glycosylphosphatidylinositol (GPI)-anchored tissue-non-specific alkaline phosphatase (TNAP) isozyme hydrolyzes both adenosine triphosphate (ATP) and mineralization inhibitor inorganic pyrophosphate (PPi) to generate Pi that is incorporated into MVs by the action of phosphate transporter 1 (PiT-1) (6, 8). These backup mechanisms explain why MVs isolated from chondro-osteogenic cells deficient in either TNAP or PHOSPHO1 retain the ability to initiate mineralization, although at reduced levels (9–11).
Genetic mutations in the ALPL gene-encoding TNAP lead to hypophosphatasia (HPP; OMIM# 241500, 241510, and 146300) characterized by low circulating alkaline phosphatase (ALP), rickets in children and/or osteomalacia in adults, and with a very broad range of severity with several clinical types (from most to least severe: perinatal lethal, infantile, severe or mild childhood, adult, and odontohypophosphatasia) (12). Those with the gravest forms of HPP (perinatal and infantile types) often die in utero or soon after birth because of severe skeletal hypomineralization, respiratory failure due to thoracic cage dysplasia and hypoplastic lungs, and elevated intracranial pressure due to craniosynostosis (13–16). Dentoalveolar phenotypes (described in detail below), particularly premature exfoliation of primary teeth, are commonly observed in patients with mild and severe subtypes of HPP (17–19). Alpl knockout (Alpl−/−) mice phenocopy infantile HPP extremely well, including their failure to thrive, high-pitched cries, rickets and extensive hypomineralization of their skeleton, and severe seizures that precede death within a few weeks after birth (though a pyridoxal-enforced diet can reduce seizures and slightly extend lifespan) (20–22). The term pseudo-HPP has been used to describe apparent cases of HPP where ALP levels are within normal range and may also apply when no genetic variant in ALPL can be identified (23–28).
Mice with a Phospho1 gene ablation (Phospho1−/−) live to adulthood but develop scoliosis starting from birth, osteomalacia, greenstick fractures, accompanied by the elevation in plasma PPi and a decrease in plasma TNAP activity (11, 29). Less pronounced and distinct dentoalveolar abnormalities (described in detail below) accompany PHOSPHO1 loss-of-function (29). Importantly, the simultaneous ablation of both the Alpl and Phospho1 genes leads to embryonic lethality with a complete absence of skeletal and dental mineralization (11).
To our knowledge, no pathogenic, loss-of-function mutations in PHOSPHO1 have been reported in humans. Given that the symptomatology of Phospho1−/− mice (osteomalacia, spontaneous fractures, scoliosis) resembles many of the manifestations of HPP, we propose that a subset of individuals with HPP-like manifestations that resist diagnosis (i.e., some in the pseudo-HPP group) could potentially carry unreported pathogenic PHOSPHO1 variants. The goal of this perspective article is to compare and contrast the loss-of-function effects of key mineralization-associated phosphatases, TNAP and PHOSPHO1, on the dentoalveolar complex to predict the dental phenotype in humans that may result from PHOSPHO1 mutations. Potential cases of pseudo-HPP associated with PHOSPHO1 mutations may resist diagnosis, and dental manifestations could be a key criterion for consideration.
Dental Mineralization Defects Associated With Hypophosphatasia
An array of dental manifestations can accompany HPP, including enamel defects, thin and/or hypomineralized dentin, wide pulp chambers, root size and shape abnormalities, acellular cementum hypoplasia, alveolar bone loss, periodontal disease, tooth mobility, and malocclusion (17, 18, 30–32). Dental defects are variable across clinical HPP subtypes, though the premature loss of primary teeth is a hallmark of all types of HPP and can be a key diagnostic criterion, as teeth are exfoliated “fully rooted” (i.e., lacking substantial root resorption that normally marks primary tooth loss) spontaneously or resulting from minor traumas. Tooth loss is the result of acellular cementum hypoplasia and loss of periodontal attachment. As with other clinical manifestations of HPP, genotype –phenotype correlations are not well-understood for dental aspects of the disease. The mildest clinical form of HPP referred to as odontohypophosphatasia (odonto-HPP), features primarily tooth loss and other dental disorders in combination with low ALP but in the absence of other clinically evident signs.
Alpl−/− mice phenocopy severe infantile HPP with <1% of circulating ALP activity compared to control mice, reflecting the range of severe dental defects observed in individuals with HPP, and serving as an important model to study disease mechanisms (20, 21, 31, 33). Compared to wild-type (WT) controls, Alpl−/− mice show severe enamel and dentin hypoplasia and reduced alveolar bone volume and mineral density (Figures 1A–E) (30, 35–39). Some local defects in Alpl−/− dentin and alveolar bone are so severe that whole regions of these tissues fall below the threshold for the detection by micro-CT, appearing invisible in 3D and 2D renderings (yellow arrows and stars in Figure 1D), and contributing to reduced volumes in affected tissues (35). Alveolar bone levels are already decreased in Alpl−/− mice by 24 days postnatal (dpn), shortly after first molar eruption (yellow dotted lines showing alveolar crest height in Figures 1A vs. 1C). All mineralizing dentoalveolar cells express TNAP, including ameloblasts, odontoblasts, cementoblasts, and osteoblasts (30, 37, 40). Studies analyzing mechanisms for enamel defects indicate disruptions in ameloblast organization and enamel ultrastructure (37), while dentin defects arise from inhibition of mineralization outside of MVs in the dentin matrix (30). These dentin defects initiate in the outer mantle dentin layer where MVs are critical for the initiation of mineralization, and in more severe cases, dentin hypomineralization also spreads to circumpulpal dentin, as indicated in mouse, sheep, and human studies of HPP (30, 35, 41).
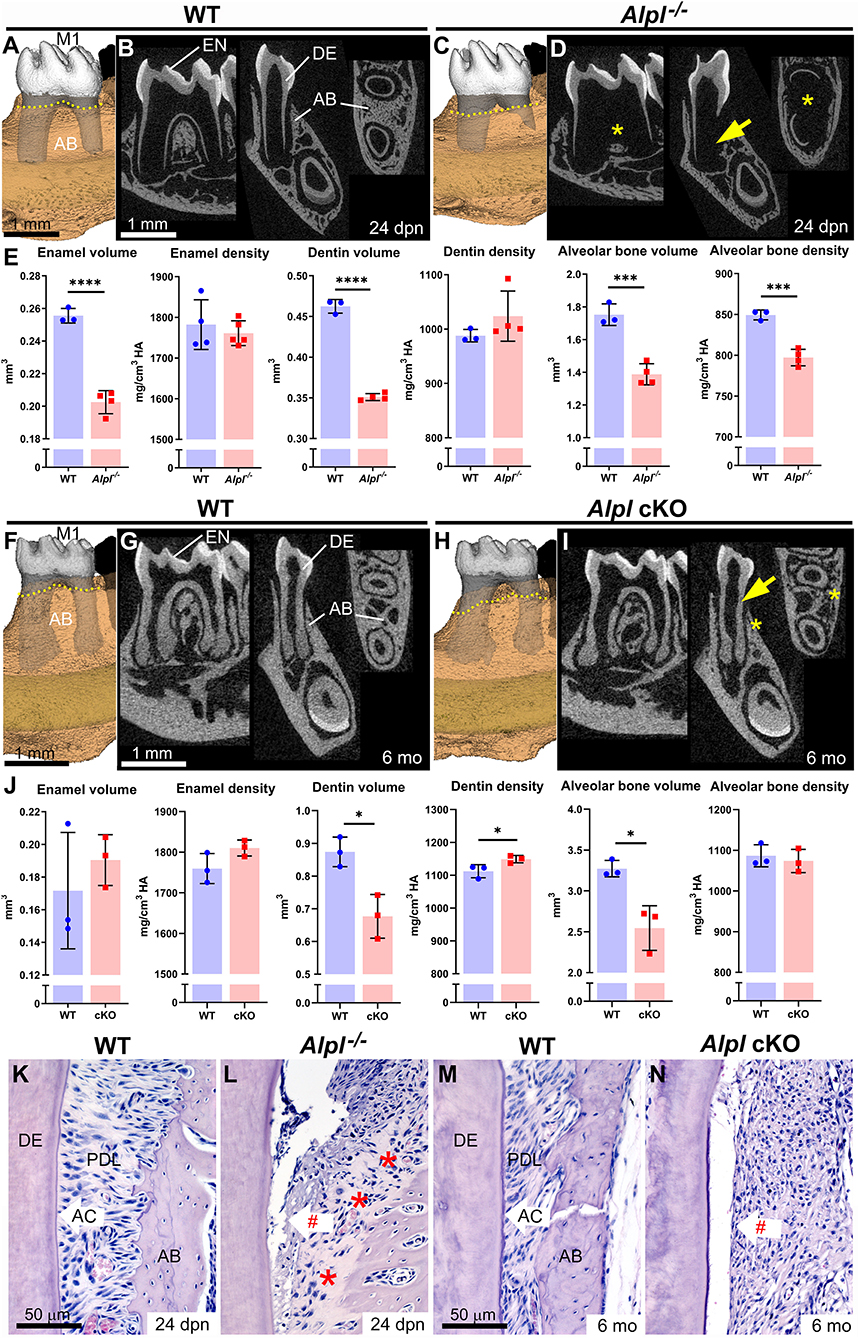
Figure 1. Dentoalveolar defects in mouse models of hypophosphatasia. (A–D) 3D and 2D micro-CT renderings of first molars (M1) reveal severe dentin (DE) defects (yellow arrows) and alveolar bone (AB) defects (yellow *) in Alpl−/− vs. wild-type (WT) mice at 24 days postnatal (dpn). AB levels (yellow dotted lines in A and C) are already reduced in Alpl−/− vs. WT mice. (E) Compared to WT, Alpl−/− have reduced enamel (EN) volume, DE volume, and AB volume and density. (F–I) 3D and 2D micro-CT renderings of M1 reveal thin DE (yellow arrow) and AB loss (yellow *) in Alpl cKO vs. WT mice. AB levels (yellow dotted lines in F and H) have declined significantly in Alpl cKO vs. WT mice by 6 months (mo). (J) Compared to WT, Alpl cKO has reduced DE and AB volumes. (K–N) Histology confirms that both Alpl−/− and Alpl cKO mouse models lack acellular cementum (AC), resulting in periodontal ligament (PDL) detachment (red #). Alpl−/− mice feature large areas of alveolar bone osteoid (red stars in panel L). Detailed analytical methods are included in the original publications, and as described in Chavez et al., JBMR Plus 5(3): e10474, 2021. Briefly, micro-CT generated DICOM images were analyzed with AnalyzePro 1.0 (AnalyzeDirect, Overland Park, KS), calibrated to five known densities of HA (mg/cm3 HA), and density thresholds were defined as >1,600 mg/cm3 HA for enamel and 450–1,600 mg/cm3 HA for dentin and bone. Statistical analyses included independent samples t-tests where graphs show means ± SD and results are indicated by: *p < 0.05; **p < 0.01; ***p < 0.001; ****P < 0.0001. (A–E) are republished with permission from Kramer et al., Bone 143:115732, 2021. The remaining images are from expanded, previously unpublished analyses from data presented in Foster et al. (34) and presented with permission.
Alpl−/− mice have lethality within the first few weeks after birth, precluding their use in longer-term studies on pathology and treatment effects. A mouse that harbored a floxed Alpl allele was designed to produce tissue-specific conditional deletion of Alpl when crossed with Cre recombinase-expressing mouse lines, allowing more refined deletion and less severe and/or later onset of disease. Col1a1-Cre-mediated deletion of Alpl in conditional knockout (cKO) mice affected reduced circulating ALP levels by about 75% and mice lived to the termination of the study at 6 months (34). These cKO mice resembled childhood-onset HPP, with axial and appendicular skeletal defects, long bone fractures, and defects in dentin and alveolar bone that became apparent prior to adulthood (Figures 1F–J).
While enamel and dentin defects in mouse models of HPP are well-represented in reports on developmental phenotypes, the longer lifespan of cKO vs. Alpl−/− mice allowed assessment of tissues later in life, leading to the first observation of severe periodontal bone loss in a mouse HPP model (yellow dotted lines showing alveolar crest height in Figures 1F vs. 1H). Histology reveals that both Alpl−/− and cKO mouse models of HPP have prominent acellular cementum hypoplasia, resulting in the poor periodontal attachment (Figures 1K–N). This defect is considered the primary driver of primary and secondary tooth loss in individuals with HPP and contributes to periodontal disease, mobility, and malocclusion.
In summary, while effects of HPP are highly variable, they can potentially impact all dentoalveolar mineralized tissues, in part because TNAP is highly expressed by all mineralizing cells during dental development and tissue mineralization. A mouse model that harbored a knock-in Alpl A116T substitution mutation associated with odonto-HPP (42) showed about 50% decreased circulating ALP levels and mild effects on acellular cementum and alveolar bone, possibly the two most sensitive tissues (33).
Dental Effects of Phospho1 Loss-of-Function In Mice
Dental development in Phospho1−/− mice has been previously described (29, 43); however, in this study, we reanalyzed samples using high-resolution micro-CT in combination with previous histological observations to provide additional insights into the consequences of Phospho1 deletion on dental development. Analyses were performed to parallel those reported for HPP mouse models described above (34, 35), to allow more direct comparisons. Overall, deletion of Phospho1 leads to milder mineralization defects than global or conditional deletion of Alpl (Figures 2A–D). Compared to WT controls, Phospho1−/− mice show enamel hypoplasia (Figure 2E), and a previous report indicated disturbed enamel ultrastructure and reduced mineralization in continuously erupting mouse incisors (44). Dentin volume increases in Phospho1−/− vs. WT mice by 3 months, although a small but significant reduction in dentin mineral density is evident. Alveolar bone density is also negatively affected by the loss of PHOSPHO1.
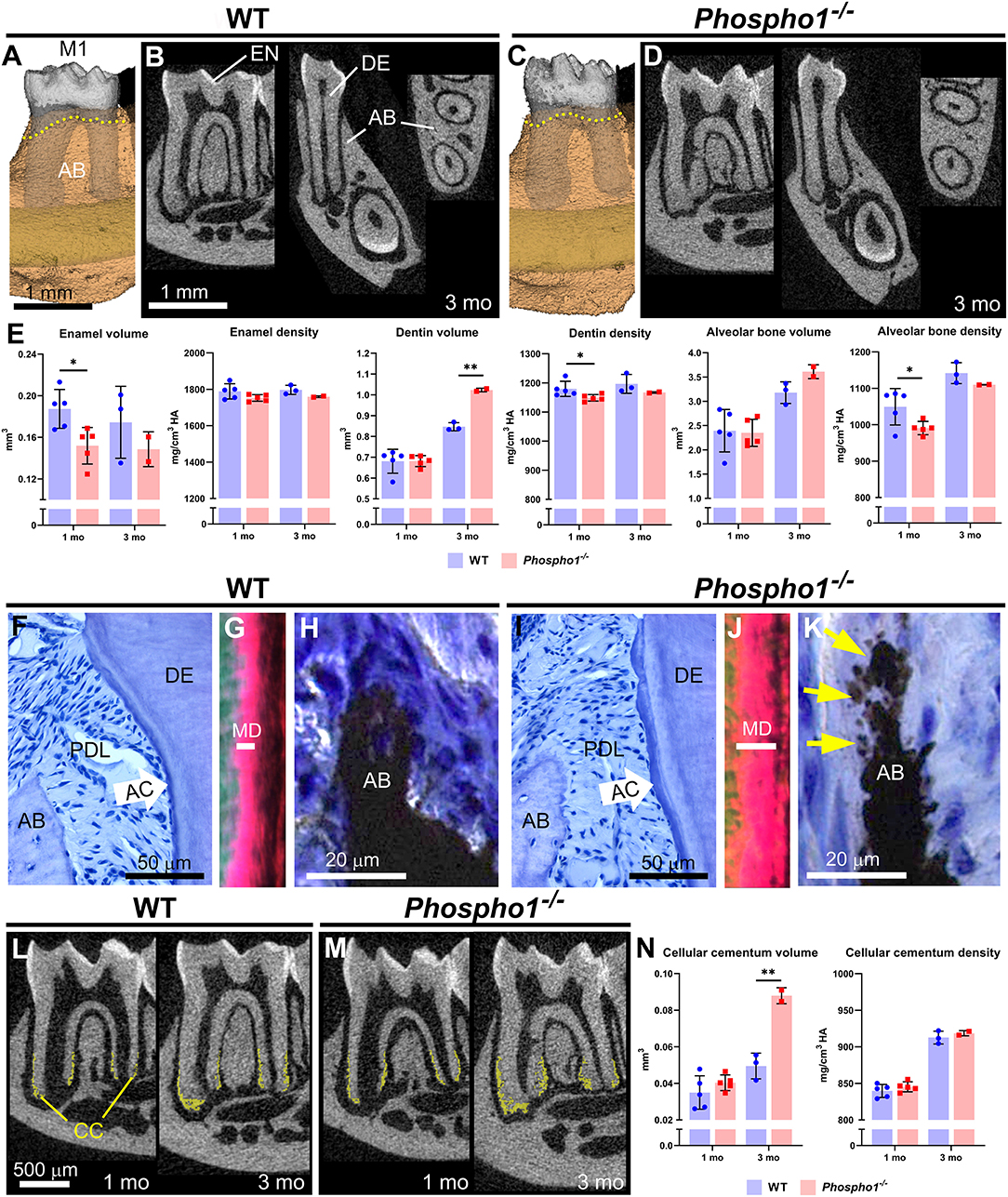
Figure 2. Dentoalveolar defects in Phospho1−/− mice. (A–D) 3D and 2D renderings of first molars (M1) from WT and Phospho1−/− mice at 3 months (mo), including enamel (EN), dentin (DE), and alveolar bone (AB). AB levels (yellow dotted lines in A and C) are similar in Phospho1−/− vs. WT mice at 3 mo. (E) Compared to wild-type (WT), Phospho1−/− mice have mild enamel hypoplasia and reduced DE and AB mineral densities at 3 mo. (F, I) H&E stain of WT and Phospho1−/− dentoalveolar tissues at 3 mo showing intact acellular cementum (AC) layer and periodontal ligament (PDL) attachment. (G, J) Picrosirius red stain viewed under polarized light shows abnormally expanded mantle dentin (MD) layer (note length of the white bar) in Phospho1−/− vs. WT mice, where DE mineralization defects are focused. (H, K) von Kossa stain shows patchy osteomalacia (yellow arrows) characteristic of Phospho1−/− mouse AB defects. (L, M) 2D micro-CT renderings with cellular cementum (CC) segmented (shown in yellow) reveal increased CC in Phospho1−/− vs. WT mice by 3 mo. (N) Quantitative analysis confirms increased CC volume and similar mineral density in Phospho1−/− vs. WT mice. Detailed analytical methods are included in the original publication. Micro-CT analysis was performed as described in Figure 1, and as described in Chavez et al., JBMR Plus 5(3): e10474, 2021, with the segmentation of cellular cementum from dentin accomplished using a median filter. Statistical analyses included independent samples t-tests where graphs show means ± SD and results are indicated by: *p < 0.05; **p < 0.01. (F, H, I, and K) are republished with permission from Zweifler et al. (29). The remaining images are expanded, previously unpublished analyses of samples presented in the above publication.
Histology provides further insights into how the absence of PHOSPHO1 affects dentoalveolar tissues, and we focus on periodontal tissues to contrast important differences arising from loss-of-function of TNAP vs. PHOSPHO1. Unlike in HPP models, an apparently normal acellular cementum layer is present in Phospho1−/− mice and PDL attachment remains intact (Figures 2F–I), corresponding to normal alveolar crest height (yellow dotted lines in Figures 2A vs. 2C). The outer dentin layer of Phospho1−/− vs. WT mice shows altered collagen organization, indicative of the mineralization defect that is limited to mantle dentin (Figures 2G–J). Alveolar bone in Phospho1−/− shows “patchy” osteomalacia (Figures 2H,K) that has been described for bone in other skeletal locations (45). Intriguingly, cellular cementum volume nearly doubles in Phospho1−/− vs. WT mice by 3 months (Figures 2L–N).
The pattern of defects with loss-of-function of PHOSPHO1 closely match the developmental expression, as explored by mRNA in situ hybridization, and protein immunohistochemistry, immunogold labeling, and western blot (29, 43, 44). Expression was noted in ameloblasts, odontoblasts, alveolar bone osteoblasts, and cementoblasts associated with cellular cementum. MVs, wherein PHOSPHO1 functions, have been associated with mineralization by all these cells (46, 47). Strikingly, PHOSPHO1 expression was not identified in cementoblasts during acellular cementum formation, no acellular cementum defect was noted in mice lacking PHOSPHO1, and MVs have not been associated with acellular cementum (29, 46).
Predicting Dental Manifestations in Patients With Pseudo-HPP From Phospho1 Mutations
The nomenclature of pseudo-HPP has sometimes been used to describe apparent cases of HPP wherein clinical presentation overlaps with manifestations of HPP, but where ALP levels are within the normal range (23, 24, 26, 27, 48–50). Many studies also report cases where clinical signs of HPP are present and ALP is low or normal, and no genetic variant in ALPL can be identified (51–55). By Sanger sequencing, up to 5% of suspected HPP cases resist the identification of a genetic factor (54–57). These cases are sometimes attributed to ALPL variants that are difficult to locate by sequencing (e.g., in regulatory regions of the gene). It should also be noted that a comprehensive differential diagnosis for low ALP will include other genetic conditions (e.g., osteogenesis imperfecta, forms of hypophosphatemic rickets, cleidocranial dysplasia, and Wilson's disease), altered nutritional states (e.g., vitamin C, magnesium, or zinc deficiency, Celiac disease, malnutrition, or starvation), endocrine and mineral metabolism disorders (e.g., hypothyroidism, Cushing's syndrome, and milk-alkali syndrome), and potential effects of medications (e.g., bisphosphonates, other antiresorptive drugs, and chronic corticosteroid treatment) (31, 58). Additionally, we propose that a subset of these individuals with low ALP that resist diagnosis could potentially carry unreported PHOSPHO1 variants.
In a patient with suspected HPP that had no detectable ALPL variants, Taillandier et al. mapped an inherited heterozygous c. 95_97CCT deletion in PHOSPHO1, in addition to a heterozygous COL1A2 mutation in a newborn with severe mineralization defects (57). In vitro functional tests using COS-1 fibroblast-like cells that transfected with a pCMV expression vector carrying the PHOSPHO1 deletion did not indicate enzymatic loss-of-function using a modified PEA substrate. However, these tests may not recapitulate in vivo function (particularly with this complex genetic background), and long-term clinical manifestations in this patient carrying PHOSPHO1 and COL1A2 variants were not reported. Little work has been done defining the effects of mutation on PHOSPHO1 using functional assays, though mutation of key active site residues resulted in the loss of enzymatic activity (59). It is also possible that PHOSPHO1 loss-of-function could be compensated by additional regulators, including TNAP. Compensation and competition between mineralization regulators are complex, sometimes with age-dependent changes in compensatory mechanisms (11, 43, 60–63), and we propose that this is an aspect for further study in HPP and pseudo-HPP.
Skeletal manifestations resulting from loss-of-function of TNAP vs. PHOSPHO1 share significant overlap based on the mouse models (6, 11, 43). Consideration of the dentoalveolar effects from loss-of-function of TNAP vs. PHOSPHO1 provides a valuable opportunity to not only contrast the physiological functions of these phosphatases but may also provide clinical utility. While enamel, (mantle) dentin, and alveolar bone are affected similarly by loss-of-function of these phosphatases, there is one key difference that stands out from the accumulated preclinical data. PHOSPHO1 deficiency does not appear to negatively affect acellular cementum or periodontal function. This observation can guide a prediction of the dentoalveolar phenotype to be expected in individuals who may carry pathogenic PHOSPHO1 variants. First, relatively mild enamel and dentin defects may exist in such an individual and could manifest as enamel hypoplasia and dentin abnormalities focused in the mantle dentin (e.g., altered appearance by histology, such as interglobular patterns). Both types of alterations might be subclinical in that they may be so mild as to not cause detectable pathology. Second, and most importantly, periodontal attachment would likely not be impaired by PHOSPHO1 mutations. Acellular cementum appeared undiminished (even increased in thickness) in mice lacking Phospho1, PDL attachment was intact, and alveolar bone levels were unaltered (29). This paints a picture in contrast to HPP, where premature tooth loss from cementum and periodontal attachment defects represents the most consistent and common dental effect.
Such a hypothetical individual presenting mild enamel and/or dentin defects and normal periodontal function could potentially be diagnosed from the dental presentation with amelogenesis imperfecta (AI; OMIM# 301200, 204650, 104500, 612529, 204700, and many more), dentinogenesis imperfecta (DI) type II or III (OMIM# 125490, 125500), dentin dysplasia type II (DD; OMIM# 125420), while potential skeletal effects of a PHOSPHO1 mutation (e.g., scoliosis, fractures, and osteomalacia) may suggest one of the many forms of osteogenesis imperfecta (OMIM# 166200, 166210, and several others). HPP would also be in the differential diagnosis, and normal or low ALP in combination with lack of detectable ALPL mutation(s) could be labeled as an unusual manifestation of HPP or pseudo-HPP. Based on the accumulated evidence presented in this perspective article, we encourage clinicians to consider PHOSPHO1 as a genetic sequencing target in cases of suspected HPP where mutations in ALPL cannot be identified and include it in genetic testing panels for endocrine and mineral metabolism disorders, an approach increasingly gaining in popularity (57, 64, 65).
A dissenting opinion may be that PHOSPHO1 loss-of-function variants have not been identified in humans because they are not compatible with life. For example, the types of mineralization defects that are tolerable in Phospho1−/− mice may be more severe and embryonic lethal in humans, though harmless when heterozygous (an unusual scenario). PHOSPHO1 expression in testes may affect fertility and thus the heritability of mutations. This is an area deserving further study.
Conclusion
In this perspective article, we compared and contrasted the loss-of-function effects of mineralization-associated phosphatases, TNAP and PHOSPHO1, on the dentoalveolar complex, based on preclinical and case report studies. To date and to our knowledge, PHOSPHO1 loss-of-function variants have not been reported in humans. Based on the accumulated evidence, we propose that there may be a subset of cases of pseudo-HPP associated with PHOSPHO1 mutations. These would be expected to have skeletal effects such as osteomalacia, and fractures, consistent with HPP, but featuring dental phenotypes with mild enamel and dentin effects and intact cementum, periodontal attachment, and no premature tooth loss. Further research is clearly needed to identify such individuals if they remain undiagnosed or misdiagnosed under current medical paradigms.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA, United States.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study was funded by the National Institute of Dental and Craniofacial Research (NIDCR) grants R01DE12889 to JM, R03DE028411 to BF, and T32DE014320 in support of MC, and research grants from Soft Bones, Inc. to BF and FA, and Biotechnology and Biological Sciences Research Council (BBSRC) for Institute Strategic Programme Grant Funding BB/J004316/1 to CF.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Glimcher M. Bone: nature of the calcium phosphate crystals and cellular, structural, and physical chemical mechanisms in their formation. Rev Mineral Geochem. (2006) 64:223–82. doi: 10.1515/9781501509421-009
2. McNally EA, Schwarcz HP, Botton GA, Arsenault AL. A model for the ultrastructure of bone based on electron microscopy of ion-milled sections. PLoS ONE. (2012)7:e29258. doi: 10.1371/journal.pone.0029258
3. Anderson HC. Vesicles associated with calcification in the matrix of epiphyseal cartilage. J Cell Biol. (1969) 41:59–72. doi: 10.1083/jcb.41.1.59
4. Ali SY, Sajdera SW, Anderson HC. Isolation and characterization of calcifying matrix vesicles from epiphyseal cartilage. Proc Natl Acad Sci U S A. (1970) 67:1513–20. doi: 10.1073/pnas.67.3.1513
5. Bonucci E. Fine structure and histochemistry of “calcifying globules” in epiphyseal cartilage. Z Zellforsch Mikrosk Anat. (1970) 103:192–217. doi: 10.1007/BF00337312
6. Millan JL. The role of phosphatases in the initiation of skeletal mineralization. Calcif Tissue Int. (2013) 93:299–306. doi: 10.1007/s00223-012-9672-8
7. Roberts SJ, Stewart AJ, Sadler PJ, Farquharson C. Human PHOSPHO1 exhibits high specific phosphoethanolamine and phosphocholine phosphatase activities. Biochem J. (2004) 382:59–65. doi: 10.1042/BJ20040511
8. Ciancaglini P, Yadav MC, Simao AM, Narisawa S, Pizauro JM, Farquharson C, et al. Kinetic analysis of substrate utilization by native and TNAP-, NPP1-, or PHOSPHO1-deficient matrix vesicles. J Bone Miner Res. (2010) 25:716–23. doi: 10.1359/jbmr.091023
9. Anderson HC, Hsu HH, Morris DC, Fedde KN, Whyte MP. Matrix vesicles in osteomalacic hypophosphatasia bone contain apatite-like mineral crystals. Am J Pathol. (1997) 151:1555–61.
10. Anderson HC, Sipe JB, Hessle L, Dhanyamraju R, Atti E, Camacho NP, et al. Impaired calcification around matrix vesicles of growth plate and bone in alkaline phosphatase-deficient mice. Am J Pathol. (2004) 164:841–7. doi: 10.1016/S0002-9440(10)63172-0
11. Yadav MC, Simao AM, Narisawa S, Huesa C, McKee MD, Farquharson C, et al. Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: a unified model of the mechanisms of initiation of skeletal calcification. J Bone Miner Res. (2011)26:286–97. doi: 10.1002/jbmr.195
12. Millan JL, Whyte MP. Alkaline phosphatase and hypophosphatasia. Calcif Tissue Int. (2016) 98:398–416. doi: 10.1007/s00223-015-0079-1
13. Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. (2012) 366:904–13. doi: 10.1056/NEJMoa1106173
14. Silver MM, Vilos GA, Milne KJ. Pulmonary hypoplasia in neonatal hypophosphatasia. Pediatr Pathol. (1988) 8:483–93. doi: 10.3109/15513818809022304
15. Shohat M, Rimoin DL, Gruber HE, Lachman RS. Perinatal lethal hypophosphatasia; clinical, radiologic and morphologic findings. Pediatr Radiol. (1991) 21:421–7. doi: 10.1007/BF02026677
16. Whyte MP, Leung E, Wilcox WR, Liese J, Argente J, Martos-Moreno G, et al. Natural history of perinatal and infantile hypophosphatasia: a retrospective study. J Pediatr. (2019) 209:116–24.e114. doi: 10.1016/j.jpeds.2019.01.049
17. Foster BL, Nociti FH Jr, Somerman MJ. The rachitic tooth. Endocr Rev. (2014) 35:1–34 doi: 10.1210/er.2013-1009
18. Reibel A, Maniere MC, Clauss F, Droz D, Alembik Y, Mornet E, et al. Orodental phenotype and genotype findings in all subtypes of hypophosphatasia. Orphanet J Rare Dis. (2009) 4:6. doi: 10.1186/1750-1172-4-6
19. Foster BL, Ramnitz MS, Gafni RI, Burke AB, Boyce AM, Lee JS, et al. Rare bone diseases and their dental, oral, craniofacial manifestations. J Dent Res. (2014) 93:7S–19. doi: 10.1177/0022034514529150
20. Narisawa S, Frohlander N, Millan JL. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev Dyn. (1997) 208:432–46.
21. Fedde KN, Blair L, Silverstein J, Coburn SP, Ryan LM, Weinstein RS, et al. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res. (1999) 14:201–26. doi: 10.1359/jbmr.1999.14.12.2015
22. Waymire KG, Mahuren JD, Jaje JM, Guilarte TR, Coburn SP, MacGregor GR. Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat Genet. (1995) 11:45–51. doi: 10.1038/ng0995-45
23. Wenkert D, McAlister WH, Mumm S, Whyte MP. c.1250A>G, p.N417S is a common American TNSALP mutation involved in all clinical forms of hypophosphatasia (HPP), including pseudo-HPP. J Bone Miner Res. (2008) 23:S242.
24. Sarkar AK, Ghosh SK, Mitra P, Mandal S, Mukhopadhyay S, Mathew J. Pseudohypophosphatasia. Indian J Pediatr. (1997) 64:256–61. doi: 10.1007/BF02752460
25. Fedde KN, Cole DE, Whyte MP. Pseudohypophosphatasia: aberrant localization and substrate specificity of alkaline phosphatase in cultured skin fibroblasts. Am J Hum Genet. (1990) 47:776–83.
26. Heaton BW, McClendon JL. Childhood pseudohypophosphatasia. Clinical and laboratory study of two cases. Tex Dent J. (1986) 103:4–8.
27. Scriver CR, Cameron D. Pseudohypophosphatasia. N Engl J Med. (1969) 281:604–6. doi: 10.1056/NEJM196909112811107
28. Whyte MP. Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. (2016) 12:233–46. doi: 10.1038/nrendo.2016.14
29. Zweifler LE, Ao M, Yadav M, Kuss P, Narisawa S, Kolli TN, et al. Role of PHOSPHO1 in periodontal development and function. J Dent Res. (2016) 95:742–51. doi: 10.1177/0022034516640246
30. Foster BL, Nagatomo KJ, Tso HW, Tran AB, Nociti FH Jr, et al. Tooth root dentin mineralization defects in a mouse model of hypophosphatasia. J Bone Miner Res. (2013) 28:271–82. doi: 10.1002/jbmr.1767
31. Bowden SA, Foster BL. Alkaline phosphatase replacement therapy for hypophosphatasia in development and practice. Adv Exp Med Biol. (2019) 1148:279–322. doi: 10.1007/978-981-13-7709-9_13
32. Bowden SA, Foster BL. Profile of asfotase alfa in the treatment of hypophosphatasia: design, development, and place in therapy. Drug Des Devel Ther. (2018) 12:3147–61. doi: 10.2147/DDDT.S154922
33. Foster BL, Sheen CR, Hatch NE, Liu J, Cory E, Narisawa S, et al. Periodontal defects in the A116T knock-in murine model of odontohypophosphatasia. J Dent Res. (2015) 94:706–14. doi: 10.1177/0022034515573273
34. Foster BL, Kuss P, Yadav MC, Kolli TN, Narisawa S, Lukashova L, et al. Conditional alpl ablation phenocopies dental defects of hypophosphatasia. J Dent Res. (2017) 96:81–91. doi: 10.1177/0022034516663633
35. Kramer K, Chavez MB, Tran AT, Farah F, Tan MH, Kolli TN, et al. Dental defects in the primary dentition associated with hypophosphatasia from biallelic ALPL mutations. Bone. (2021) 143:115732. doi: 10.1016/j.bone.2020.115732
36. Gasque KC, Foster BL, Kuss P, Yadav MC, Liu J, Kiffer-Moreira T, et al. Improvement of the skeletal and dental hypophosphatasia phenotype in Alpl-/- mice by administration of soluble (non-targeted) chimeric alkaline phosphatase. Bone. (2015) 72:137–47. doi: 10.1016/j.bone.2014.11.017
37. Yadav MC, de Oliveira RC, Foster BL, Fong H, Cory E, Narisawa S, et al. Enzyme replacement prevents enamel defects in hypophosphatasia mice. J Bone Miner Res. (2012) 27:1722–34. doi: 10.1002/jbmr.1619
38. McKee MD, Nakano Y, Masica DL, Gray JJ, Lemire I, Heft R, et al. Enzyme replacement therapy prevents dental defects in a model of hypophosphatasia. J Dent Res. (2011) 90:470–6. doi: 10.1177/0022034510393517
39. Millan JL, Narisawa S, Lemire I, Loisel TP, Boileau G, Leonard P, et al. Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res. (2008) 23:777–87. doi: 10.1359/jbmr.071213
40. Zweifler LE, Patel MK, Nociti FH Jr, Wimer HF, Millan JL, et al. Counter-regulatory phosphatases TNAP and NPP1 temporally regulate tooth root cementogenesis. Int J Oral Sci. (2015) 7:27–41. doi: 10.1038/ijos.2014.62
41. Williams DK, Pinzon C, Huggins S, Pryor JH, Falck A, Herman F, et al. Genetic engineering a large animal model of human hypophosphatasia in sheep. Sci Rep. (2018) 8:16945. doi: 10.1038/s41598-018-35079-y
42. Hu JC, Plaetke R, Mornet E, Zhang C, Sun X, Thomas HF, et al. Characterization of a family with dominant hypophosphatasia. Eur J Oral Sci. (2000) 108:189–94. doi: 10.1034/j.1600-0722.2000.108003189.x
43. McKee MD, Yadav MC, Foster BL, Somerman MJ, Farquharson C, Millan JL. Compounded PHOSPHO1/ALPL deficiencies reduce dentin mineralization. J Dent Res. (2013) 92:721–7. doi: 10.1177/0022034513490958
44. Pandya M, Rosene L, Farquharson C, Millan JL, Diekwisch TGH. Intravesicular Phosphatase PHOSPHO1 function in enamel mineralization and prism formation. Front Physiol. (2017) 8:805. doi: 10.3389/fphys.2017.00805
45. Boyde A, Staines KA, Javaheri B, Millan JL, Pitsillides AA, Farquharson C. A distinctive patchy osteomalacia characterises Phospho1-deficient mice. J Anat. (2017) 231:298–308. doi: 10.1111/joa.12628
46. Takano Y, Sakai H, Baba O, Terashima T. Differential involvement of matrix vesicles during the initial and appositional mineralization processes in bone, dentin, and cementum. Bone. (2000) 26:333–9. doi: 10.1016/S8756-3282(00)00243-X
47. Takano Y, Sakai H, Baba O, Sakamoto Y, Terashima T, Ohya K, et al. Demonstration of putative Ca-binding domains in dentin matrix of rat incisors after daily injections of 1-hydroxyethylidene-1,1-bisphosphonate (HEBP). Eur J Oral Sci. (1998) 106(Suppl. 1):274–81. doi: 10.1111/j.1600-0722.1998.tb02187.x
48. Whyte MP, Walkenhorst DA, Fedde KN, Henthorn PS, Hill CS. Hypophosphatasia: levels of bone alkaline phosphatase immunoreactivity in serum reflect disease severity. J Clin Endocrinol Metab. (1996) 81:2142–8. doi: 10.1210/jcem.81.6.8964842
49. Whyte MP. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev. (1994) 15:439–61. doi: 10.1210/er.15.4.439
50. Cole DE, Salisbury SR, Stinson RA, Coburn SP, Ryan LM, Whyte MP. Increased serum pyridoxal-5'-phosphate in pseudohypophosphatasia. N Engl J Med. (1986) 314:992–3. doi: 10.1056/NEJM198604103141515
51. Mornet E, Taillandier A, Domingues C, Dufour A, Benaloun E, Lavaud N, et al. Hypophosphatasia: a genetic-based nosology and new insights in genotype-phenotype correlation. Eur J Hum Genet. (2021) 29:289–99. doi: 10.1038/s41431-020-00732-6
52. Garcia-Fontana C, Villa-Suarez JM, Andujar-Vera F, Gonzalez-Salvatierra S, Martinez-Navajas G, Real PJ, et al. Epidemiological clinical and genetic study of hypophosphatasia in a spanish population: identification of two novel mutations in the alpl gene. Sci Rep. (2019) 9:9569. doi: 10.1038/s41598-019-46004-2
53. Fauvert D, Brun-Heath I, Lia-Baldini AS, Bellazi L, Taillandier A, Serre JL, et al. Mild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate alleles. BMC Med Genet. (2009) 10:51. doi: 10.1186/1471-2350-10-51
54. Hepp N, Frederiksen AL, Duno M, Praest Holm J, Rye Jorgensen N, Beck Jensen JE. Biochemical clinical and genetic characteristics in adults with persistent hypophosphatasaemia; data from an endocrinological outpatient clinic in Denmark. Bone Rep. (2021) 15:101101. doi: 10.1016/j.bonr.2021.101101
55. Jandl NM, Schmidt T, Rolvien T, Sturznickel J, Chrysostomou K, von Vopelius E, et al. Genotype-phenotype associations in 72 adults with suspected ALPL-associated hypophosphatasia. Calcif Tissue Int. (2021) 108:288–301. doi: 10.1007/s00223-020-00771-7
56. Mornet E. Hypophosphatasia: the mutations in the tissue-nonspecific alkaline phosphatase gene. Hum Mutat. (2000) 15:309–15. doi: 10.1002/(SICI)1098-1004(200004)15:4<309::AID-HUMU2>3.0.CO;2-C
57. Taillandier A, Domingues C, De Cazanove C, Porquet-Bordes V, Monnot S, Kiffer-Moreira T, et al. Molecular diagnosis of hypophosphatasia and differential diagnosis by targeted next generation sequencing. Mol Genet Metab. (2015) 116:215–20. doi: 10.1016/j.ymgme.2015.09.010
58. Schmidt T, Schmidt C, Amling M, Kramer J, Barvencik F. Prevalence of low alkaline phosphatase activity in laboratory assessment: Is hypophosphatasia an underdiagnosed disease? Orphanet J Rare Dis. (2021) 16:452. doi: 10.1186/s13023-021-02084-w
59. Roberts SJ, Stewart AJ, Schmid R, Blindauer CA, Bond SR, Sadler PJ, et al. Probing the substrate specificities of human PHOSPHO1 and PHOSPHO2. Biochim Biophys Acta. (2005) 1752:73–82. doi: 10.1016/j.bbapap.2005.06.009
60. Harmey D, Hessle L, Narisawa S, Johnson KA, Terkeltaub R, Millan JL. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: an integrated model of the pathogenesis of mineralization disorders. Am J Pathol. (2004) 164:1199–209. doi: 10.1016/S0002-9440(10)63208-7
61. Harmey D, Johnson KA, Zelken J, Camacho NP, Hoylaerts MF, Noda M, et al. Elevated skeletal osteopontin levels contribute to the hypophosphatasia phenotype in Akp2(-/-) mice. J Bone Miner Res. (2006) 21:1377–86. doi: 10.1359/jbmr.060619
62. Murshed M, Harmey D, Millan JL, McKee MD, Karsenty G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. (2005) 19:1093–104. doi: 10.1101/gad.1276205
63. Ao M, Chavez MB, Chu EY, Hemstreet KC, Yin Y, Yadav MC, et al. Overlapping functions of bone sialoprotein and pyrophosphate regulators in directing cementogenesis. Bone. (2017) 105:134–47. doi: 10.1016/j.bone.2017.08.027
64. Rush ET, Johnson B, Aradhya S, Beltran D, Bristow SL, Eisenbeis S, et al. Molecular diagnoses of X-linked and other genetic hypophosphatemias: results from a sponsored genetic testing program. J Bone Miner Res. (2021). doi: 10.1002/jbmr.4454. [Epub ahead of print].
Keywords: hypophosphatasia, mineralization, dental, Periodontal, genetic models
Citation: Mohamed FF, Chavez MB, Amadeu de Oliveira F, Narisawa S, Farquharson C, Millán JL and Foster BL (2022) Perspective on Dentoalveolar Manifestations Resulting From PHOSPHO1 Loss-of-Function: A Form of Pseudohypophosphatasia? Front. Dent. Med. 3:826387. doi: 10.3389/fdmed.2022.826387
Received: 30 November 2021; Accepted: 03 January 2022;
Published: 03 February 2022.
Edited by:
Yong-Hee Patricia Chun, The University of Texas Health Science Center at San Antonio, United StatesReviewed by:
Yan Zhang, University of California, San Francisco, United StatesCatherine Chaussain, Université de Paris, France
Copyright © 2022 Mohamed, Chavez, Amadeu de Oliveira, Narisawa, Farquharson, Millán and Foster. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian L. Foster, Zm9zdGVyLjEwMDRAb3N1LmVkdQ==