 Kirk M. Habegger1
Kirk M. Habegger1 Erin Grant1
Erin Grant1 Paul Thomas Pfluger1,2
Paul Thomas Pfluger1,2 Diego Perez-Tilve1 Alan Daugherty3 Dennis Bruemmer3 Matthias H. Tschöp1,2* Susanna M. Hofmann1,4
Diego Perez-Tilve1 Alan Daugherty3 Dennis Bruemmer3 Matthias H. Tschöp1,2* Susanna M. Hofmann1,4
- 1 Division of Endocrinology, Department of Internal Medicine, Metabolic Diseases Institute, University of Cincinnati, Cincinnati, OH, USA
- 2 Institute for Diabetes and Obesity, German Research Center for Environmental Health, München/Neuherberg, Germany
- 3 Saha Cardiovascular Research Center, University of Kentucky College of Medicine, Lexington, KY, USA
- 4 Institute of Experimental Genetics, Helmholtz Zentrum München, German Research Center for Environmental Health, München/Neuherberg, Germany
Objective: Ghrelin, a stomach-derived, secreted peptide, and its receptor (growth hormone secretagogue receptor, GHSR) are known to modulate food intake and energy homeostasis. The ghrelin system is also expressed broadly in cardiovascular tissues. Since ghrelin has been associated with anti-inflammatory and anti-atherogenic properties, but is also well known to promote obesity and impair glucose metabolism, we investigated whether ghrelin has any impact on the development of atherosclerosis. The hypothesis that endogenous ghrelin signaling may be involved in atherosclerosis has not been tested previously. Methods and Results: We crossed ghrelin receptor knockout mice (GHSr−/−) into a low-density lipoprotein receptor-null (Ldlr−/−) mouse line. In this model, atherosclerotic lesions were promoted by feeding a high-fat, high-cholesterol Western-type diet for 13 months, following a standard protocol. Body composition and glucose homeostasis were similar between Ldlr−/− and Ldlr/GHSR−/−ko mice throughout the study. Absence or presence of GHSr did not alter the apolipoprotein profile changes in response to diet exposure on an LDLRko background. Atherosclerotic plaque volume in the aortic arch and thoracic aorta were also not affected differentially in mice without ghrelin signaling due to GHSR gene disruption as compared to control LDLRko littermates. In light of the associations reported for ghrelin with cardiovascular disease in humans, the lack of a phenotype in these loss-of-function studies in mice suggests no direct role for endogenous ghrelin in either the inhibition or the promotion of diet-induced atherosclerosis. Conclusion: These data indicate that, surprisingly, the complex and multifaceted actions of endogenous ghrelin receptor mediated signaling on the cardiovascular system have minimal direct impact on atherosclerotic plaque progression as based on a loss-of-function mouse model of the disease.
Introduction
As obesity rates continue to rise in the United States, the risk for mortality from its comorbidities is also accelerating. Among the most concerning of these associated disease states are cardiovascular events, which besides being correlated with obesity (Jousilahti et al., 1996; McGill et al., 2002), are also associated with many other obesity-derived comorbidities such as hypertension, dyslipidemia, hyperinsulinemia, and diabetes mellitus (McGill et al., 1997, 1998; Calle et al., 1999; Fontaine et al., 2003; Banerjee et al., 2011). This increase in obesity-related atherosclerosis has spurred intense research into shared pathways that may be essential to mediating both pathologies. Among the mutual regulators, recent associations have implicated ghrelin as a protective agent against atherosclerosis (Zhang et al., 2010).
The 28-amino acid peptide ghrelin is produced in the stomach and has been identified as the endogenous ligand for the growth hormone secretagogue receptor (GHSr; (Kojima et al., 1999). It circulates in two distinct forms, acylated ghrelin and unacylated ghrelin; its acylated form is essential for binding to the GHSr and for its primary endocrine functions, including stimulation of GH secretion from the hypophysis, induction of food intake and regulation of energy homeostasis (Gutierrez et al., 2008). Recently, ghrelin and its receptors have been detected in cardiovascular tissues (Matsubara et al., 1995; Christodoulou et al., 2006) indicating that the peptide may play a role in cardiovascular disease. Indeed, ghrelin has been shown to inhibit cardiomyocyte and endothelial cell inflammation and apoptosis (Iglesias et al., 2004; Li et al., 2004). Furthermore, ghrelin enhances left ventricular function during ischemia–reperfusion injury in rodents (Chang et al., 2004), improves left ventricular dysfunction and attenuates the development of cardiac cachexia in rats (Xu et al., 2005). Ghrelin has also been suggested to reduce mortality and correct the hemodynamic and metabolic abnormalities associated with endotoxic shock in rats (Chang et al., 2003). Similarly, in humans, ghrelin exerts vasodilatory effects, improves cardiac function and decreases systemic vascular resistance in chronic heart failure (for review see Granata et al., 2011).
Thus, ghrelin has been repeatedly inferred to modulate the atherogenetic process, although the reports in this area are not all aligned: Ghrelin may play an anti-atherosclerotic role by inhibiting proinflammatory cytokine production, mononuclear cell binding, and nuclear factor-kappaB activation in human endothelial cells in vitro and endotoxin-induced cytokine production in rats (Li et al., 2004). In addition, ghrelin may improve endothelial function and decrease apoptosis by enhancing NO production (Suematsu et al., 2005; Zhao et al., 2007). Circulating ghrelin concentrations are reportedly increased in men with atherosclerosis (Poykko et al., 2006) and analyses of atherosclerotic lesions in rats and humans have detected expression of elevated levels of the ghrelin receptor, underscoring a potential role of the novel vasodilator ghrelin in the pathogenesis of cardiovascular disease. To further complicate the picture, circulating ghrelin concentrations are reduced in obese patients raising the possibility that a reduction in endogenous ghrelin could potentially contribute to inflammatory states and increased incidence of atherosclerosis in obese patients. However, none of these studies allowed for conclusions on the functions of endogenous ghrelin as no in vivo loss-of-function models were used and applied to an appropriate and well-established mouse model of atherosclerosis.
In the present investigation, we therefore generated such a unique model and analyzed ghrelin receptor-dependent effects on the progression of atherosclerosis in mice. We examined lesion size in ghrelin receptor (GSHr−/−) and low-density lipoprotein receptor (LDLr−/−) –deficient mouse models. In our experimental paradigm, ablation of the ghrelin receptor did not reveal beneficial effects on the course of cardiovascular disease.
Materials and Methods
Animals and Diet
Growth hormone secretagogue receptor-(Wortley et al., 2004) and LDLr-deficient mice were obtained from Regeneron Pharmaceuticals (Abizaid et al., 2006; Tarrytown, NY, USA) and the Jackson Laboratory (Bar Harbor, ME, USA), respectively. Mice from both lines were bred in our facilities and maintained on a C57/B6 background. LDLr knockout (LDLr−/−) mice of both sexes were intercrossed with GHSr−/− mice to generate a double-heterozygous generation. Double-heterozygous mice were inbred to produce mice deficient for both LDLr and GHSr (LDLr/GSHr−/−), or for LDLr only (LDLr−/−). DNA was extracted from tail snips and PCR used to establish genotypes. All animals were housed on a 12:12-h light–dark cycle at 22°C. At 12 weeks old, mice were placed on a Western-style diet obtained from Teklad Custom Research Diets (TD.88137, Harlan Laboratories, Inc, Indianapolis, IN, USA) with free access to food and water. All studies were conducted in male, age-matched mice provided with western diet for 13 months. All studies were approved by, and performed according to, the guidelines of the Institutional Animal Care and Use Committee of the University of Cincinnati.
Body Composition Analysis and Glucose Tolerance Test
Whole body composition (fat and lean mass) was measured using NMR technology (EchoMRI, Houston, TX, USA). For measurements of glucose tolerance, mice were fasted for 6 h and injected intraperitoneally with 50% D-glucose (Sigma) in 0.9% saline, such that the final dose was 1.5 g glucose/kg body wt in all animals. Glucose levels (mg/dl) were measured from tail blood using a handheld glucometer (Freestyle lite) before (0 min) and at 15, 30, 60, and 120 min after injection.
Blood Parameters, Tissue Preparation, and Morphometric Analysis of Vascular Lesion
Total cholesterol concentrations were measured with standard enzymatic assays (Infinity, Cholesterol and Triglyceride kit; Thermo Fisher Scientific, Waltham, MA, USA). Samples were analyzed individually except for lipoprotein separation in which pooled samples (0.25 ml) from 10 animals per group were subjected to fast-performance liquid chromatography (FPLC) gel filtration on two Superose six columns connected in series. Atherosclerosis was quantified using en-face analysis of the aortic intima of the arch and thorax, as described previously (Daugherty and Whitman, 2003).
Statistical Analysis
All statistical analysis was performed with Prism 5.0 (GraphPad Software, La Jolla, CA, USA). Statistical differences between groups were determined by Student’s t-test. A P value less than 0.05 was considered significant.
Results
Endogenous Ghrelin Signaling had No Effect on Hypercholesterolemia in Mice
Studies described in this report were conducted in LDLr-deficient mice, one of the most utilized genetic mouse models for the study of atherosclerosis. Lack of LDLr in mice (LDLr−/−) induces a profound increase in circulating cholesterol concentrations when mice are fed a saturated fat diet. LDLr−/− mice were crossed with GHSr−/− mice, creating double knockout mice (GHSr/LDLr−/−) that allowed us to evaluate the role of ghrelin signaling in lipoprotein metabolism and the progression of atherosclerosis. Male, age-matched mice lacking LDLr displayed profound hypercholesterolemia (1505 ± 82.6 mg/dl). Deletion of both LDLr and GHSr resulted in similar levels of plasma cholesterol (1478 ± 175.3 mg/dl) as those observed in LDLr−/−mice (Figure 1A). FPLC analyses of plasma from these mice elucidated similar characteristics in terms of cholesterol particle distribution. Specifically, mice of both genotypes displayed considerable peaks in very low-density lipoprotein (VLDL) and LDL fractions (Figure 1B). Furthermore, deletion of the ghrelin receptor was unable to rescue the depressed concentrations of HDL-cholesterol observed with deletion of the LDL receptor (Figure 1B).
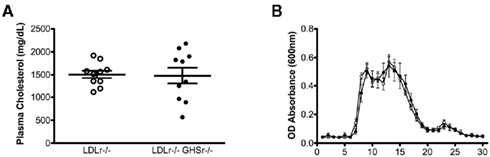
Figure 1. Total plasma cholesterol concentrations (A) and analysis of plasma lipoproteins (B) in male mice fasted overnight. Pooled plasma (0.25 ml) from 10 GSHr/LDLr−/− (open circles) and 10 LDLr−/− (closed circles) mice maintained on atherogenic diet was applied to two FPLC Superose columns, and 0.5-ml fractions were collected for cholesterol determinations. According to calibration with standard lipoproteins, the peak fractions 5–9 represent VLDL, fractions 10–20 are mainly IDL/LDL particles, and fractions 20–26 contain mainly HDL.
Endogenous Ghrelin Signaling Did not Affect Atherosclerosis
We next evaluated the protective role of ghrelin signaling via GHSr in the context of atherosclerotic progression. LDLr−/− mice maintained on a saturated fat diet displayed considerable atherosclerosis in the aortic arch, with lesion areas of 12.01 ± 0.99 mm2 (Figure 2A) that covered 55.77 ± 3.45% of the assayed area (Figure 2B). As with LDLr−/− mice, GHSr/LDLr−/− mice also showed considerable atherosclerosis of the aortic arch. Mice with the double knockout mutation displayed lesion areas of 13.46 ± 1.31 mm2 (Figure 2A) and covered 54.86 ± 3.75% of the intimal area (Figure 2B). In addition to the aortic arch, we also analyzed atherosclerotic development along the thoracic aorta. Like lesions present in the arch, those of the thoracic aorta were of similar size (Figures 3A,B) in both LDLr−/− and GHSr/LDLr−/− mice (11.29 ± 2.59 vs. 6.74 ± 1.46 mm2) and covered similar portions of the assayed area (30.62 ± 5.75 vs. 20.15 ± 4.13%).
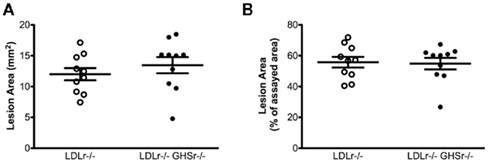
Figure 2. Atherosclerosis was measured on aortic arches from male GHSr+/+LDLr−/− (open circles, n = 10) and GHSr−/−LDLr−/− (closed circles, n = 10) mice. The extent of intimal surface covered by grossly discernible lesions was determined from en-face preparations of the aortic intima in the arch region. Data were expressed as lesion area of the arch (A) and as percent area covered by atherosclerosis (B).
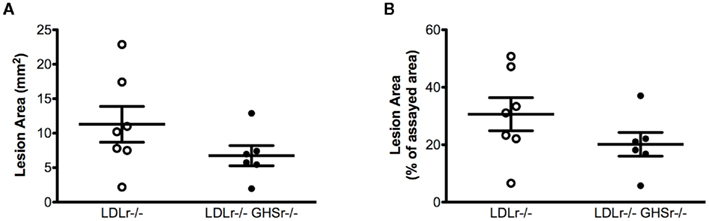
Figure 3. Atherosclerosis was measured on the thoracic aorta from male GHSr+/+LDLr−/− (open circles, n = 7) and GHSr−/−LDLr−/− (closed circles, n = 7) mice. The extent of intimal surface covered by grossly discernible lesions was determined from en-face preparations of the aortic intima in the thoracic region. Data were expressed as lesion area of the thoracic aorta (A) and as percent area covered by atherosclerosis (B).
Ghrelin Regulation of Body Composition and Glucose Homeostasis in LDLr-Deficient Mice
Low-density lipoprotein receptor-null and GHSr/LDLr−/− mice had similar body weight (Figure 4A), lean, and fat mass (Figures 4B,C), suggesting that lack of ghrelin signaling through GHSr does not alter body composition in LDLr−/− mice. Similar to our earlier findings (Pfluger et al., 2008), and consistent with our body weight and fat mass results, lack of GHSr-mediated ghrelin signaling had no effect on glucose tolerance, resulting in similar glucose excursions in both LDLr−/− and GHSr/LDLr−/− mice (AUC = 73371 ± 1098 and 74706 ± 1443, respectively; Figures 4D,E).
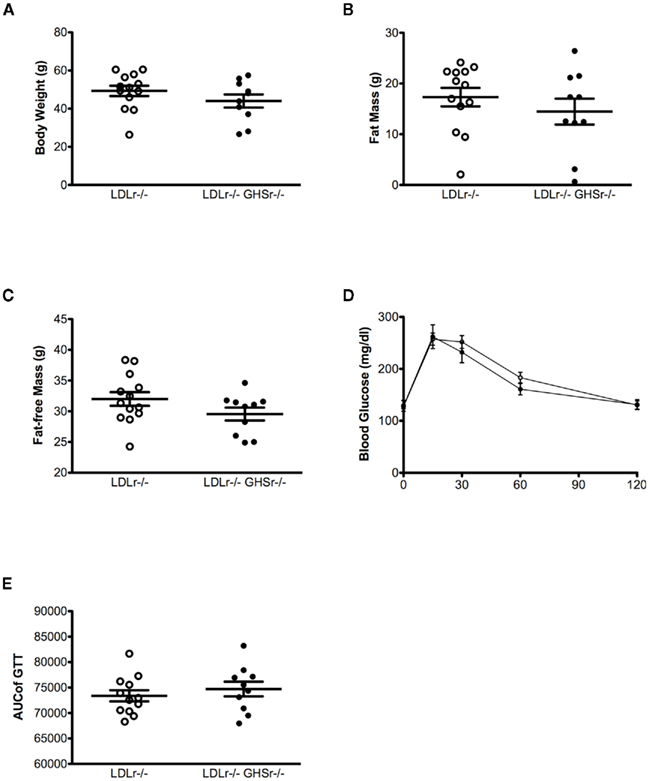
Figure 4. Body weight (A), fat mass (B), and glucose tolerance (C) were assessed after 13 months of western-style diet in male mice. A glucose tolerance test (GTT) was conducted using an i.p. dose of 1.5 g of glucose per kg of body weight in GHSr+/+LDLr−/− (open circles, n = 12) and GHSr−/−LDLr−/− (closed circles, n = 10) mice fasted for 6 h. (D,E) are data from the ipGTT. (D) displays ipGTT data as collected at each time point, and (E) displays this data as the integrated, area under the curve.
Discussion
The connection between obesity and cardiovascular disease has been the focus of intense research. Recent findings have brought forth several mutual pathways that may be dysregulated in both disease states. Among these regulators, the role of ghrelin in atherosclerosis has yet to be fully elucidated. Specifically, it has been argued that ghrelin is protective against the development of atherosclerosis.
We therefore carefully evaluated the role of GHSr-mediated ghrelin signaling in a common mouse model of atherosclerosis. As such, this study is the first to our knowledge that assesses the mechanistic role of ghrelin in atherosclerosis in an in vivo setting. While we were able to elicit a profound atherosclerotic state in LDLr−/− mice in the presence of an atherogenic diet, we failed to detect an effect on the degree of atherosclerosis in the ghrelin receptor loss-of-function mice.
A potential caveat of this study is the choice of the atherosclerotic mouse model, the LDLr−/− mouse. These mice develop extreme hypercholesterolemia that may not be amendable by a single genetic manipulation. Accordingly, this is not the first example of an observation made in humans that could not be directly translated to the LDLr−/− model of atherosclerosis (Nawrocki et al., 2010). In addition, our finding that fasting glucose levels and glucose tolerance are similar between LDLr−/− mice with or without GHSr also shows that the previously reported enhancement of insulin sensitivity in GHSr−/− mice (Longo et al., 2008) did not translate into our mouse model of atherosclerosis and thus may have interfered with an otherwise potential beneficial indirect effect of GHSr on the atherogenetic process. Furthermore, at the time of lesion analysis, these mice had been maintained on the high-fat, high-cholesterol “Western” diet for 13 months. This extended exposure to the atherogenic diet may have masked any temporary effects exerted through the ghrelin receptor.
On the surface, these findings seem difficult to reconcile with the considerable number of descriptive, pharmacological, or in vitro reports suggesting a role for ghrelin signaling in the pathogenesis of atherosclerosis. However, the ghrelin system plays numerous and pleiotropic roles in the control of cardiovascular risk factors associated with energy, glucose and lipid metabolism. It is important to note that in this experimental model ghrelin signaling, or lack thereof, is restricted to GHSr-mediated events. As such this model focuses on the physiological role of acylated ghrelin and its direct signaling through GHSr in the progression of atherosclerosis. However, this paradigm does not exclude a potential effect that is regulated by desacyl-ghrelin, obestatin, or other ghrelin prepropetide variants that are mediated by receptors other than GHSr (see review by Muccioli et al., 2007). In addition, ghrelin has been reported to directly modulate several targets and processes with direct relevance for atherosclerosis (see review by Zhang et al., 2010). It therefore appears possible that direct and indirect impacts of ghrelin action may have mutually opposite effects on the development of atherosclerosis, at least in this double-loss-of-function mouse model.
Our findings may indicate potential limitations of the LDL receptor mouse model in reproducing associations that have been established in humans. Unlike humans, mice are resistant to plaque rupture and myocardial infarction. In our studies, genetic manipulation of ghrelin signaling through GHSr, in a traditional rodent model of atherosclerosis did not correlate with atherosclerosis. These data suggest that ghrelin, or at least ghrelin signaling mediated via the ghrelin receptor, is not involved in advanced plaque progression. Alternatively, ghrelin-induced effects on the pathogenesis of atherosclerosis in one or the other direction may be mediated by an alternative ghrelin receptor, which has repeatedly been postulated to exist (Cassoni et al., 2004; Granata et al., 2007), but remains to be identified. Until then, the present data suggest that ghrelin-based therapeutics may not provide the previously assumed considerable potential for the prevention or treatment of atherosclerosis.
Conflict of Interest Statement
Susanna M. Hofmann has no conflicts to declare, Matthias H. Tschöp is a consultant for Roche Pharmaceuticals and receives Roche research funds.
Acknowledgments
Susanna M. Hofmann has received basic science grant 1-10-BS-72 from the American Diabetes Association and National Institutes of Health Grant 2K12HD051953-06. Kirk M. Habegger is supported by the University of Cincinnati Training Program in Neuroendocrinology of Homeostasis grant T32 DK059803.
References
Abizaid, A., Liu, Z. W., Andrews, Z. B., Shanabrough, M., Borok, E., Elsworth, J. D., Roth, R. H., Sleeman, M. W., Picciotto, M. R., Tschop, M. H., Gao, X. B., and Horvath, T. L. (2006). Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J. Clin. Invest. 116, 3229–3239.
Banerjee, D., Recio-Mayoral, A., Chitalia, N., and Kaski, J. C. (2011). Insulin resistance, inflammation, and vascular disease in nondiabetic predialysis chronic kidney disease patients. Clin. Cardiol. 34, 360–365.
Calle, E. E., Thun, M. J., Petrelli, J. M., Rodriguez, C., and Heath, C. W. Jr. (1999). Body-mass index and mortality in a prospective cohort of U.S. adults. N. Engl. J. Med. 341, 1097–1105.
Cassoni, P., Ghe, C., Marrocco, T., Tarabra, E., Allia, E., Catapano, F., Deghenghi, R., Ghigo, E., Papotti, M., and Muccioli, G. (2004). Expression of ghrelin and biological activity of specific receptors for ghrelin and des-acyl ghrelin in human prostate neoplasms and related cell lines. Eur. J. Endocrinol. 150, 173–184.
Chang, L., Ren, Y., Liu, X., Li, W. G., Yang, J., Geng, B., Weintraub, N. L., and Tang, C. (2004). Protective effects of ghrelin on ischemia/reperfusion injury in the isolated rat heart. J. Cardiovasc. Pharmacol. 43, 165–170.
Chang, L., Zhao, J., Yang, J., Zhang, Z., Du, J., and Tang, C. (2003). Therapeutic effects of ghrelin on endotoxic shock in rats. Eur. J. Pharmacol. 473, 171–176.
Christodoulou, C., Schally, A. V., Chatzistamou, I., Kondi-Pafiti, A., Lamnissou, K., Kouloheri, S., Kalofoutis, A., and Kiaris, H. (2006). Expression of growth hormone-releasing hormone (GHRH) and splice variant of GHRH receptors in normal mouse tissues. Regul. Pept. 136, 105–108.
Daugherty, A., and Whitman, S. C. (2003). Quantification of atherosclerosis in mice. Methods Mol. Biol. 209, 293–309.
Fontaine, K. R., Redden, D. T., Wang, C., Westfall, A. O., and Allison, D. B. (2003). Years of life lost due to obesity. JAMA 289, 187–193.
Granata, R., Isgaard, J., Alloatti, G., and Ghigo, E. (2011). Cardiovascular actions of the ghrelin gene-derived peptides and growth hormone-releasing hormone. Exp. Biol. Med. 236, 505–514.
Granata, R., Settanni, F., Biancone, L., Trovato, L., Nano, R., Bertuzzi, F., Destefanis, S., Annunziata, M., Martinetti, M., Catapano, F., Ghe, C., Isgaard, J., Papotti, M., Ghigo, E., and Muccioli, G. (2007). Acylated and unacylated ghrelin promote proliferation and inhibit apoptosis of pancreatic beta-cells and human islets: involvement of 3’,5’-cyclic adenosine monophosphate/protein kinase A, extracellular signal-regulated kinase 1/2, and phosphatidyl inositol 3-kinase/Akt signaling. Endocrinology 148, 512–529.
Gutierrez, J. A., Solenberg, P. J., Perkins, D. R., Willency, J. A., Knierman, M. D., Jin, Z., Witcher, D. R., Luo, S., Onyia, J. E., and Hale, J. E. (2008). Ghrelin octanoylation mediated by an orphan lipid transferase. Proc. Natl. Acad. Sci. U.S.A. 105, 6320–6325.
Iglesias, M. J., Pineiro, R., Blanco, M., Gallego, R., Dieguez, C., Gualillo, O., Gonzalez-Juanatey, J. R., and Lago, F. (2004). Growth hormone releasing peptide (ghrelin) is synthesized and secreted by cardiomyocytes. Cardiovasc. Res. 62, 481–488.
Jousilahti, P., Tuomilehto, J., Vartiainen, E., Pekkanen, J., and Puska, P. (1996). Body weight, cardiovascular risk factors, and coronary mortality. 15-year follow-up of middle-aged men and women in eastern Finland. Circulation 93, 1372–1379.
Kojima, M., Hosoda, H., Date, Y., Nakazato, M., Matsuo, H., and Kangawa, K. (1999). Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402, 656–660.
Li, W. G., Gavrila, D., Liu, X., Wang, L., Gunnlaugsson, S., Stoll, L. L., Mccormick, M. L., Sigmund, C. D., Tang, C., and Weintraub, N. L. (2004). Ghrelin inhibits proinflammatory responses and nuclear factor-kappaB activation in human endothelial cells. Circulation 109, 2221–2226.
Longo, K. A., Charoenthongtrakul, S., Giuliana, D. J., Govek, E. K., Mcdonagh, T., Qi, Y., Distefano, P. S., and Geddes, B. J. (2008). Improved insulin sensitivity and metabolic flexibility in ghrelin receptor knockout mice. Regul. Pept. 150, 55–61.
Matsubara, S., Sato, M., Mizobuchi, M., Niimi, M., and Takahara, J. (1995). Differential gene expression of growth hormone (GH)-releasing hormone (GRH) and GRH receptor in various rat tissues. Endocrinology 136, 4147–4150.
McGill, H. C. Jr., Mcmahan, C. A., Herderick, E. E., Zieske, A. W., Malcom, G. T., Tracy, R. E., and Strong, J. P. (2002). Obesity accelerates the progression of coronary atherosclerosis in young men. Circulation 105, 2712–2718.
McGill, H. C. Jr., Mcmahan, C. A., Malcom, G. T., Oalmann, M. C., and Strong, J. P. (1997). Effects of serum lipoproteins and smoking on atherosclerosis in young men and women. The PDAY Research Group. Pathobiological Determinants of Atherosclerosis in Youth. Arterioscler. Thromb. Vasc. Biol. 17, 95–106.
McGill, H. C. Jr., Mcmahan, C. A., Tracy, R. E., Oalmann, M. C., Cornhill, J. F., Herderick, E. E., and Strong, J. P. (1998). Relation of a postmortem renal index of hypertension to atherosclerosis and coronary artery size in young men and women. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Arterioscler. Thromb. Vasc. Biol. 18, 1108–1118.
Muccioli, G., Baragli, A., Granata, R., Papotti, M., and Ghigo, E. (2007). Heterogeneity of ghrelin/growth hormone secretagogue receptors. Toward the understanding of the molecular identity of novel ghrelin/GHS receptors. Neuroendocrinology 86, 147–164.
Nawrocki, A. R., Hofmann, S. M., Teupser, D., Basford, J. E., Durand, J. L., Jelicks, L. A., Woo, C. W., Kuriakose, G., Factor, S. M., Tanowitz, H. B., Hui, D. Y., Tabas, I., and Scherer, P. E. (2010). Lack of association between adiponectin levels and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 30, 1159–1165.
Pfluger, P. T., Kirchner, H., Gunnel, S., Schrott, B., Perez-Tilve, D., Fu, S., Benoit, S. C., Horvath, T., Joost, H. G., Wortley, K. E., Sleeman, M. W., and Tschop, M. H. (2008). Simultaneous deletion of ghrelin and its receptor increases motor activity and energy expenditure. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G610–G618.
Poykko, S. M., Kellokoski, E., Ukkola, O., Kauma, H., Paivansalo, M., Kesaniemi, Y. A., and Horkko, S. (2006). Plasma ghrelin concentrations are positively associated with carotid artery atherosclerosis in males. J. Intern. Med. 260, 43–52.
Suematsu, M., Katsuki, A., Sumida, Y., Gabazza, E. C., Murashima, S., Matsumoto, K., Kitagawa, N., Akatsuka, H., Hori, Y., Nakatani, K., Togashi, K., Yano, Y., and Adachi, Y. (2005). Decreased circulating levels of active ghrelin are associated with increased oxidative stress in obese subjects. Eur. J. Endocrinol. 153, 403–407.
Wortley, K. E., Anderson, K. D., Garcia, K., Murray, J. D., Malinova, L., Liu, R., Moncrieffe, M., Thabet, K., Cox, H. J., Yancopoulos, G. D., Wiegand, S. J., and Sleeman, M. W. (2004). Genetic deletion of ghrelin does not decrease food intake but influences metabolic fuel preference. Proc. Natl. Acad. Sci. U.S.A. 101, 8227–8232.
Xu, X. B., Pang, J. J., Cao, J. M., Ni, C., Xu, R. K., Peng, X. Z., Yu, X. X., Guo, S., Chen, M. C., and Chen, C. (2005). GH-releasing peptides improve cardiac dysfunction and cachexia and suppress stress-related hormones and cardiomyocyte apoptosis in rats with heart failure. Am. J. Physiol. Heart Circ. Physiol. 289, H1643–H1651.
Zhang, G., Yin, X., Qi, Y., Pendyala, L., Chen, J., Hou, D., and Tang, C. (2010). Ghrelin and cardiovascular diseases. Curr. Cardiol. Rev. 6, 62–70.
Keywords: ghrelin, LDL receptor, atherosclerosis
Citation: Habegger KM, Grant E, Pfluger PT, Perez-Tilve D, Daugherty A, Bruemmer D, Tschöp MH and Hofmann SM (2011) Ghrelin receptor deficiency does not affect diet-induced atherosclerosis in low-density lipoprotein receptor-null mice. Front. Endocrin. 2:67. doi: 10.3389/fendo.2011.00067
Received: 13 September 2011; Paper pending published: 24 September 2011;
Accepted: 15 October 2011; Published online: 02 November 2011.
Edited by:
Francisco Gracia-Navarro, University of Cordoba, SpainReviewed by:
Rhonda D. Kineman, University of Illinois at Chicago, USARaul Miguel Luque Huertas, University of Cordoba, Spain
Sabrina Diano, Yale University School of Medicine, USA
Copyright: © 2011 Habegger, Grant, Pfluger, Perez-Tilve, Daugherty, Bruemmer, Tschöp and Hofmann. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Matthias H. Tschöp, Division of Endocrinology, Department of Medicine, Metabolic Disease Institute, University of Cincinnati College of Medicine, 2170 E Galbraith Road, Cincinnati, OH 45237, USA. e-mail:dHNjaG9lbWhAdWNtYWlsLnVjLmVkdQ==