 Robert Frank Spurney
Robert Frank Spurney- Division of Nephrology, Department of Medicine, Duke University and Durham VA Medical Centers, Durham, NC, USA
Diseases affecting the glomerulus are the most common cause of end-stage kidney disease in developed countries (1). These disorders are characterized by significant proteinuria, and the level of proteinuria is an independent risk factor for disease progression (2). Podocytes are thought to play a key role in the pathogenesis of glomerular diseases (3, 4). The importance of podocytes in glomerular diseases is highlighted by genetic studies, which have identified mutant podocyte proteins that cause familial forms of nephrosis (5). Because podocytes are terminally differentiated cells with little capacity for replication, their ability to compensate for podocyte loss is limited (3). A component of current therapy is, therefore, focused on reducing podocyte injury by decreasing systemic blood pressure (BP) and inhibition of the renin–angiotensin system (2, 6).
Historically, the immune system was thought to play a significant role in non-genetic forms of nephrosis including acquired diseases such as minimal-change disease (MCD) and focal segmental glomerulosclerosis (FSGS) (7). As a result, corticosteroids and CNIs are often used to treat these disorders (6). Indeed, the response to steroid therapy is an important prognostic indicator for both MCD and FSGS (8, 9). Recent studies, however, suggest that these agents may have actions that are independent of their immunosuppressive properties. For example, while not a universal finding (10, 11), steroids and/or CNIs are reported to induce partial or complete remissions of proteinuria in a subset of patients with genetic forms of nephrosis (7, 11, 12). Although we acknowledge that these reports have significant limitations, the data support the concept that that steroids and/or CNIs may have beneficial effects unrelated to their immunosuppressive actions. Similarly, CNIs inhibit death of cultured podocytes after apoptotic stimuli despite the absence of immune effector mechanisms in the tissue culture model (13, 14). Moreover, genetic activation of the CN effector NFAT (nuclear factor of activated T cells) in podocytes promotes proteinuria, glomerulosclerosis, and a decrease in podocyte numbers in mice despite restricting the experimental manipulation to glomerular podocytes (15).
As shown in Figure 1, non-immunological actions of CNIs can be broadly divided into effects on the podocyte cytoskeleton and effects on podocyte survival. A seminal observation was that the actin-associated protein synaptopodin (SYN) was phosphorylated by either protein kinase A (PKA) or calcium/calmodulin-dependent protein kinase II (CaMKII). Phosphorylation of SYN provided a docking site for 14–3–3 proteins and prevented degradation of SYN by the cysteine proteinase cathepsin L (16). Dephosphorylation of the 14–3–3 docking site by calcium sensitive phosphatase CN promoted SYN degradation. This group further demonstrated that SYN competitively antagonized ubiquitination of Rho A by Smurf1 (SMAD specific E3 ubiquitin protein ligase 1), and promoted Rho A activation and stress fiber formation (17). While Rho A activity is also stimulated by calcium-dependent mechanisms (18, 19), the SYN dependence of these effects appeared relevant to glomerular diseases because expression of a degradation resistant SYN in podocytes protected mice from proteinuric stimuli (16). In this scenario, CNIs promote a podocyte phenotype that is resistant to the development of proteinuria by stabilizing the actin cytoskeleton.
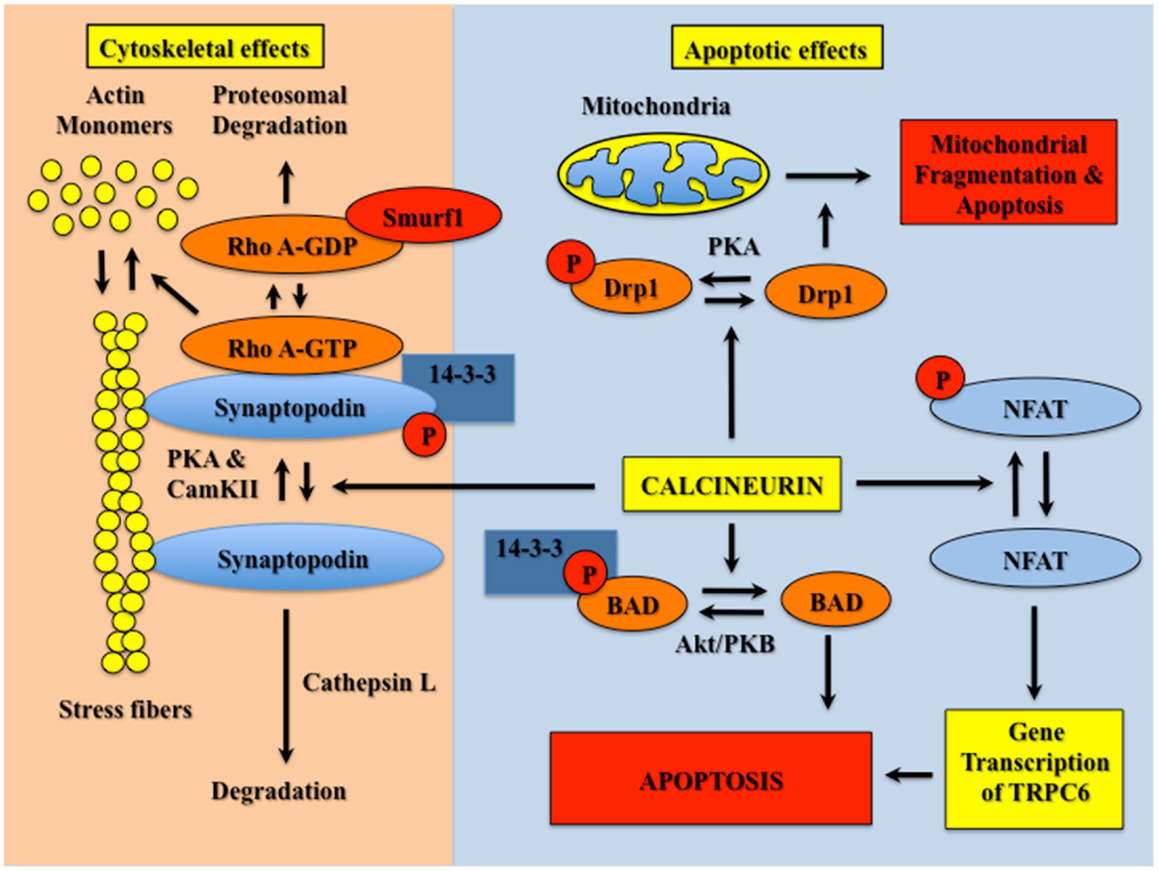
Figure 1. CN activation destabilizes the actin cytoskeleton and causes podocyte apoptosis. Phosphorylation of SYN is mediated by PKA and CamKII. Phosphorylated SYN promotes 14–3–3 binding, which protects SYN from degradation by cathepsin L. SYN also binds Rho A, and competitively inhibits binding of Rho A to the ubiquitin ligase Smurf1, which prevents targeting of Rho A for proteasomal degradation. Binding of Rho A to SYN activates Rho A (GTP bound Rho A) and, in turn, induces stress fiber formation and stabilizes the podocyte cytoskeleton. CN dephosphorylates the 14–3–3 docking site in SYN and promotes SYN degradation by cathepsin L. In the absence of SYN, Rho A is targeted for proteosomal degradation, which reduces stress fiber formation and destabilizes the actin cytoskeleton. CN also promotes podocyte apoptosis by dephosphorylation of either NFAT isoforms, Drp1 or BAD. This apoptotic effect is mediated both directly by Drp1- or BAD-dependent activation of mitochondrial apoptotic pathways, as well as indirectly by stimulation of NFAT-dependent gene transcription.
CNIs also protect podocytes from apoptotic stimuli (13, 14). At least one mechanism is dependent on gene transcription induced by NFAT (13, 14). NFAT transcription factors were originally discovered in cells of the lymphoid lineage, but abundant evidence indicates that NFAT isoforms are expressed in non-immune cells with some family members expressed ubiquitously (20). In quiescent cells, NFAT isoforms are phosphorylated and located in the cytoplasm (20). CN dephosphorylates NFAT, which permits translocation to the nucleus and stimulation of gene transcription. In cultured podocytes, expression of a constitutively active CN construct causes apoptosis, and this apoptotic effect is blocked by the pharmacologic CNI FK506 as well as by a peptide inhibitor of CN termed VIVIT (13). Similarly, hyperglycemia induces nuclear localization of NFAT isoforms as well as promotes apoptosis of cultured podocytes, and this apoptotic effect is also attenuated by VIVIT (14). Moreover, CN activity is enhanced in kidneys of diabetic rodents (13, 21), and treatment with FK506 attenuates hyperglycemia-induced podocyte apoptosis in diabetic mice (13). Because VIVIT specifically inhibits CN-dependent NFAT activation (22), these data suggest that CN causes podocyte apoptosis by mechanisms that require NFAT mediated gene transcription. In this regard, TRPC6 (transient receptor potential channel C6) is an important gene target of NFAT transcription factors (23). Indeed, gain-of-function mutations in TRPC6 cause FSGS (24, 25). TRPC6 is also up-regulated in primary glomerular diseases (26) and over-expression of TRPC6 in podocytes causes proteinuric kidney disease (27). Thus, TRPC6 may be an important downstream gene target of CN signaling in glomerular disorders. In contrast, one study reported that CNIs induced podocyte apoptosis (28). This report, however, is controversial, and we and others (7) have not been able to reproduce this observation.
As shown in Figure 1, other mechanisms of CN-mediated podocyte apoptosis include induction of mitochondrial fragmentation by Drp1 (dynamin related protein 1) as well as activation of the apoptosis inducing Bcl-2 family member BAD (Bcl-2 associated death promoter). Drp1 is phosphorylated and inhibited by PKA (29); BAD is phosphorylated by Akt, which causes sequestration of BAD by 14–3–3 proteins and inhibits apoptosis (30). CN dephosphorylates both proteins and induces apoptotic cell death through the mitochondrial pathway (29, 30). Both Drp1 and BAD have been implicated in the pathogenesis of glomerular diseases by promoting podocyte apoptosis (31–33), with the extent of apoptosis presumably dependent on the relative activities of CN and the relevant kinases. Based on these observations as well as the NFAT-dependent apoptotic effects described above, we speculate that CNIs might be useful therapies for attenuating podocyte apoptosis in diseases with either enhanced CN activity or in diseases associated with reduced activity of the relevant kinase.
In summary, CNIs may have important beneficial effects for both the podocyte cytoskeleton and podocyte viability. These agents attenuate podocyte apoptosis as well as promote a podocyte phenotype that is resistant to the development of proteinuria. The beneficial effects of CNIs may be mediated by mechanisms that are independent of the immune system. Given the potential role of CN in diverse glomerular diseases, the use of CNIs might be useful for a broader range of kidney disorders. We acknowledge that CNI nephrotoxicity is a concern (34), but the development of more specific agents with fewer off-target effects (35, 36) may be an effective strategy for expanding the use of CN inhibition to a broader range of glomerular disease processes.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
These studies were supported by grants RO1-DK087707 from the National Institutes of Health and BX000791 from the Veterans Administration Merit Review Program.
References
1. Collins AJ, Foley RN, Chavers B, Gilbertson D, Herzog C, Johansen K, et al. United States renal data system 2011 annual data report: atlas of chronic kidney disease & end-stage renal disease in the United States. Am J Kidney Dis (2012) 59(1 Suppl 1):e1–420. doi: 10.1053/j.ajkd.2011.11.015
2. Jafar TH, Stark PC, Schmid CH, Landa M, Maschio G, Marcantoni C, et al. Proteinuria as a modifiable risk factor for the progression of non-diabetic renal disease. Kidney Int (2001) 60(3):1131–40. doi:10.1046/j.1523-1755.2001.0600031131.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int (1998) 54(3):687–97. doi:10.1046/j.1523-1755.1998.00044.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Shankland SJ. The podocyte’s response to injury: role in proteinuria and glomerulosclerosis. Kidney Int (2006) 69(12):2131–47. doi:10.1038/sj.ki.5000410
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Winn MP, Daskalakis N, Spurney RF, Middleton JP. Unexpected role of TRPC6 channel in familial nephrotic syndrome: does it have clinical implications? J Am Soc Nephrol (2006) 17(2):378–87. doi:10.1681/ASN.2005090962
6. Hogan J, Mohan P, Appel GB. Diagnostic tests and treatment options in glomerular disease: 2014 update. Am J Kidney Dis (2014) 63(4):656–66. doi:10.1053/j.ajkd.2013.09.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Schonenberger E, Ehrich JH, Haller H, Schiffer M. The podocyte as a direct target of immunosuppressive agents. Nephrol Dial Transplant (2011) 26(1):18–24. doi:10.1093/ndt/gfq617
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Waldman M, Crew RJ, Valeri A, Busch J, Stokes B, Markowitz G, et al. Adult minimal-change disease: clinical characteristics, treatment, and outcomes. Clin J Am Soc Nephrol (2007) 2(3):445–53. doi:10.2215/CJN.03531006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Schwartz MM, Evans J, Bain R, Korbet SM. Focal segmental glomerulosclerosis: prognostic implications of the cellular lesion. J Am Soc Nephrol (1999) 10(9):1900–7.
10. Ruf RG, Lichtenberger A, Karle SM, Haas JP, Anacleto FE, Schultheiss M, et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol (2004) 15(3):722–32. doi:10.1097/01.ASN.0000113552.59155.72
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Buscher AK, Kranz B, Buscher R, Hildebrandt F, Dworniczak B, Pennekamp P, et al. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol (2010) 5(11):2075–84. doi:10.2215/CJN.01190210
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Bensman A, Niaudet P. Non-immunologic mechanisms of calcineurin inhibitors explain its antiproteinuric effects in genetic glomerulopathies. Pediatr Nephrol (2010) 25(7):1197–9. doi:10.1007/s00467-010-1469-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Wang L, Chang JH, Paik SY, Tang Y, Eisner W, Spurney RF. Calcineurin (CN) activation promotes apoptosis of glomerular podocytes both in vitro and in vivo. Mol Endocrinol (2011) 25(8):1376–86. doi:10.1210/me.2011-0029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Li R, Zhang L, Shi W, Zhang B, Liang X, Liu S, et al. NFAT2 mediates high glucose-induced glomerular podocyte apoptosis through increased Bax expression. Exp Cell Res (2013) 319(7):992–1000. doi:10.1016/j.yexcr.2013.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Wang Y, Jarad G, Tripathi P, Pan M, Cunningham J, Martin DR, et al. Activation of NFAT signaling in podocytes causes glomerulosclerosis. J Am Soc Nephrol (2010) 21(10):1657–66. doi:10.1681/ASN.2009121253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med (2008) 14(9):931–8. doi:10.1038/nm.1857
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol (2006) 8(5):485–91. doi:10.1038/ncb1400
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Vassiliadis J, Bracken C, Matthews D, O’Brien S, Schiavi S, Wawersik S. Calcium mediates glomerular filtration through calcineurin and mTORC2/Akt signaling. J Am Soc Nephrol (2011) 22(8):1453–61. doi:10.1681/ASN.2010080878
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol (2004) 6(11):1122–8. doi:10.1038/ncb1183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Horsley V, Pavlath GK. NFAT: ubiquitous regulator of cell differentiation and adaptation. J Cell Biol (2002) 156(5):771–4. doi:10.1083/jcb.200111073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Gooch JL, Barnes JL, Garcia S, Abboud HE. Calcineurin is activated in diabetes and is required for glomerular hypertrophy and ECM accumulation. Am J Physiol Renal Physiol (2003) 284(1):F144–54. doi:10.1152/ajprenal.00158.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Aramburu J, Yaffe MB, Lopez-Rodriguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science (1999) 285(5436):2129–33. doi:10.1126/science.285.5436.2129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest (2006) 116(12):3114–26. doi:10.1172/JCI27702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science (2005) 308(5729):1801–4. doi:10.1126/science.1106215
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet (2005) 37(7):739–44. doi:10.1038/ng1592
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Moller CC, Wei C, Altintas MM, Li J, Greka A, Ohse T, et al. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol (2007) 18(1):29–36. doi:10.1681/ASN.2006091010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Krall P, Canales CP, Kairath P, Carmona-Mora P, Molina J, Carpio JD, et al. Podocyte-specific overexpression of wild type or mutant trpc6 in mice is sufficient to cause glomerular disease. PLoS One (2010) 5(9):e12859. doi:10.1371/journal.pone.0012859
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Fornoni A, Li H, Foschi A, Striker GE, Striker LJ. Hepatocyte growth factor, but not insulin-like growth factor I, protects podocytes against cyclosporin A-induced apoptosis. Am J Pathol (2001) 158(1):275–80. doi:10.1016/S0002-9440(10)63966-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep (2007) 8(10):939–44. doi:10.1038/sj.embor.7401062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Shibasaki F, Hallin U, Uchino H. Calcineurin as a multifunctional regulator. J Biochem (2002) 131(1):1–15. doi:10.1093/oxfordjournals.jbchem.a003063
31. Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab (2012) 15(2):186–200. doi:10.1016/j.cmet.2012.01.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Huber TB, Hartleben B, Kim J, Schmidts M, Schermer B, Keil A, et al. Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol Cell Biol (2003) 23(14):4917–28. doi:10.1128/MCB.23.14.4917-4928.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Yu SY, Qi R. Role of bad in podocyte apoptosis induced by puromycin aminonucleoside. Transplant Proc (2013) 45(2):569–73. doi:10.1016/j.transproceed.2012.07.160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol (2009) 4(2):481–508. doi:10.2215/CJN.04800908
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Yu H, van Berkel TJ, Biessen EA. Therapeutic potential of VIVIT, a selective peptide inhibitor of nuclear factor of activated T cells, in cardiovascular disorders. Cardiovasc Drug Rev (2007) 25(2):175–87. doi:10.1111/j.1527-3466.2007.00011.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Noguchi H, Matsushita M, Okitsu T, Moriwaki A, Tomizawa K, Kang S, et al. A new cell-permeable peptide allows successful allogeneic islet transplantation in mice. Nat Med (2004) 10(3):305–9. doi:10.1038/nm994
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: glomerular podocyte, cell signaling pathways, calcineurin, calcineurin inhibitors, synaptopodin, Rho GTPases
Citation: Spurney RF (2014) Non-immunologic actions of calcineurin inhibitors in proteinuric kidney diseases. Front. Endocrinol. 5:181. doi: 10.3389/fendo.2014.00181
Received: 12 July 2014; Paper pending published: 10 September 2014;
Accepted: 07 October 2014; Published online: 12 November 2014.
Edited by:
Barbara Lewko, Medical University of Gdansk, PolandReviewed by:
Herman Pavenstädt, Universitätsklinikum Münster, GermanyCopyright: © 2014 Spurney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:cm9iZXJ0LnNwdXJuZXlAZG0uZHVrZS5lZHU=