 Enzo Lalli1,2,3*
Enzo Lalli1,2,3* Bonald C. Figueiredo3,4,5*
Bonald C. Figueiredo3,4,5*- 1Institut de Pharmacologie Moléculaire et Cellulaire CNRS, Valbonne, France
- 2University of Nice-Sophia-Antipolis, Valbonne, France
- 3Associated International Laboratory (LIA) NEOGENEX, CNRS, Valbonne, France
- 4Federal University of Paraná, Curitiba, Brazil
- 5Instituto de Pesquisa Pelé Pequeno Principe, Curitiba, Brazil
Adrenocortical tumors (ACT) in children are very rare and are most frequently diagnosed in the context of the Li-Fraumeni syndrome, a multiple cancer syndrome linked to germline mutations of the tumor suppressor gene TP53 with loss of heterozygosity in the tumors. A peak of children ACT incidence is present in the states of southern Brazil, where they are linked to the high prevalence in the population of a specific TP53 mutation (R337H). Children ACT have specific features distinguishing them from adult tumors in their pathogenetic mechanisms, genomic profiles, and prognosis. Epidemiological and molecular evidence suggests that in most cases they are derived from the fetal adrenal.
Dynamics of Human Adrenocortical Morphology and Hormone Secretion during Development and Postnatal Life
The adrenal gland is a continuously evolving endocrine organ from the developmental to the elder age. In humans, adrenal gland development begins at 3–4 weeks of gestation by a condensation of the coelomic epithelium lining the abdominal cavity, followed at 4–6 weeks of gestation by proliferation and migration of coelomic epithelial cells, and subsequent differentiation of fetal adrenal cortical cells into two distinct zones (inner fetal zone and outer definitive zone) at 8–10 weeks of gestation, while neural crest-derived cells start to infiltrate the gland at 7–8 weeks of gestation to give origin to adrenomedullary cells (1). Starting from around the ninth week of gestation, the embryonal adrenal is surrounded by the adrenal capsule formed by mesenchymal cells. Fetal adrenal cells, which are large and rich in lipids, express the steroidogenic enzyme CYP17, which enables them to produce high levels of DHEA and its sulfoconjugate DHEAS, which play a key role for the maintenance of pregnancy, being metabolized into estrogens by the placenta (1, 2). By the end of the second trimester of gestation, a distinct zone (transitional zone) differentiates between the definitive and fetal zones, which express HSD3B2, this way starting cortisol synthesis in the fetus. Close to birth, HSD3B2 is expressed in the definitive zone, which acquires the capacity to synthesize the mineralocorticoid hormone aldosterone. Cell proliferation in the fetal adrenal is mainly localized in the outer definitive zone, followed by centripetal migration and differentiation into fetal zone cells, which subsequently die from apoptosis in the center of the gland. This streaming process of adrenocortical cell differentiation continues during the whole life, as shown by studies in the mouse (3–5).
Starting shortly after birth, a rapid, dramatic remodeling of adrenal cortex structure takes place, with massive shrinkage of the gland due to apoptosis of the fetal zone and progressive differentiation of the glomerulosa, fasciculata, and reticularis zona, which are the hallmark of the adult adrenal (1). Defects in this process may cause the cytomegalic form of adrenal hypoplasia congenita, a syndrome of adrenal insufficiency due to altered postnatal adrenocortical differentiation due to mutations in the NR0B1 (DAX-1) gene [reviewed in Ref. (6)]. Studies in pre-term neonates have shown that parturition itself is the cause for fetal adrenal involution (7), suggesting a crosstalk between placenta and fetal adrenal in reciprocal maintenance. Remarkably, postnatal adrenal remodeling also takes place in the mouse adrenal cortex, where an inner zone adjacent to the medulla termed zone X, that lineage tracing experiments have shown to be derived from the fetal adrenal (8), regresses after puberty in males and after the first pregnancy in females.
After being suppressed following the regression of the fetal zone, adrenal production of DHEA/DHEAS starts to progressively increase again by around 8 years of age. This phenomenon is termed adrenarche and is concomitant with full differentiation of the reticularis zone, which expresses CYP17 but not HSD3B2. Moreover, in the zona reticularis, CYP17 has an increased ratio of 17,20-lyase to 17α hydroxylase activity (which favors DHEA production) compared to the zona fasciculata, probably due to increased serine phosphorylation and increased abundance of cytochrome b5 (CYB5), which allosterically stimulates 17,20-lyase activity of CYP17 (9). DHEA/DHEAS levels continue to increase until adulthood and then progressively decline (adrenopause) reaching pre-adrenarche levels by the ninth decade, correlating with progressive atrophy of the zona reticularis (10).
Adrenocortical Tumors in Children and Adults: Similarities and Differences
Adrenocortical tumors (ACT) are among the most common neoplasms in humans and are frequently detected by hazard during diagnostic procedures for other medical issues (incidentalomas), in the great majority of cases remaining clinically silent and having a completely benign prognosis. In contrast, adrenocortical malignancies (adrenocortical carcinomas or ACC) are very rare, with a general incidence of 0.7–2 cases/million/year, with a maximum between 40 and 50 years of age and a higher frequency in women than in men (11). They become clinically evident with signs and symptoms due to hormone excess (Cushing’s syndrome, androgen excess) and/or local symptoms (pain, abdominal discomfort). The prognosis of ACC is still poor, with an average 5-year overall survival around 40%, which is influenced to a great extent by tumor stage at diagnosis. Some histopathological parameters (Weiss score ≥3, Ki-67 index >10%) also have negative prognostic value (11).
Adrenocortical tumors in children under 15 years of age are even rarer. Their worldwide incidence has been estimated at 0.3/million/year with a bimodal peak under the age of 5 and after 10 years and they also affect girls more frequently than boys. The main reason why ACT in children become clinically evident is virilization, which may be associated to Cushing’s syndrome. Overall survival at 5 years after diagnosis in children with ACT is better than in adult patients, approximating 50%. Favorable prognostic factors are younger age (<4 years), stage I at diagnosis, tumor weight ≤200 g, volume <200 cm3, and presence of virilization alone (12). It is noteworthy that in children ACT, the Weiss score is not a reliable system to assess malignancy (13–15) (Table 1).
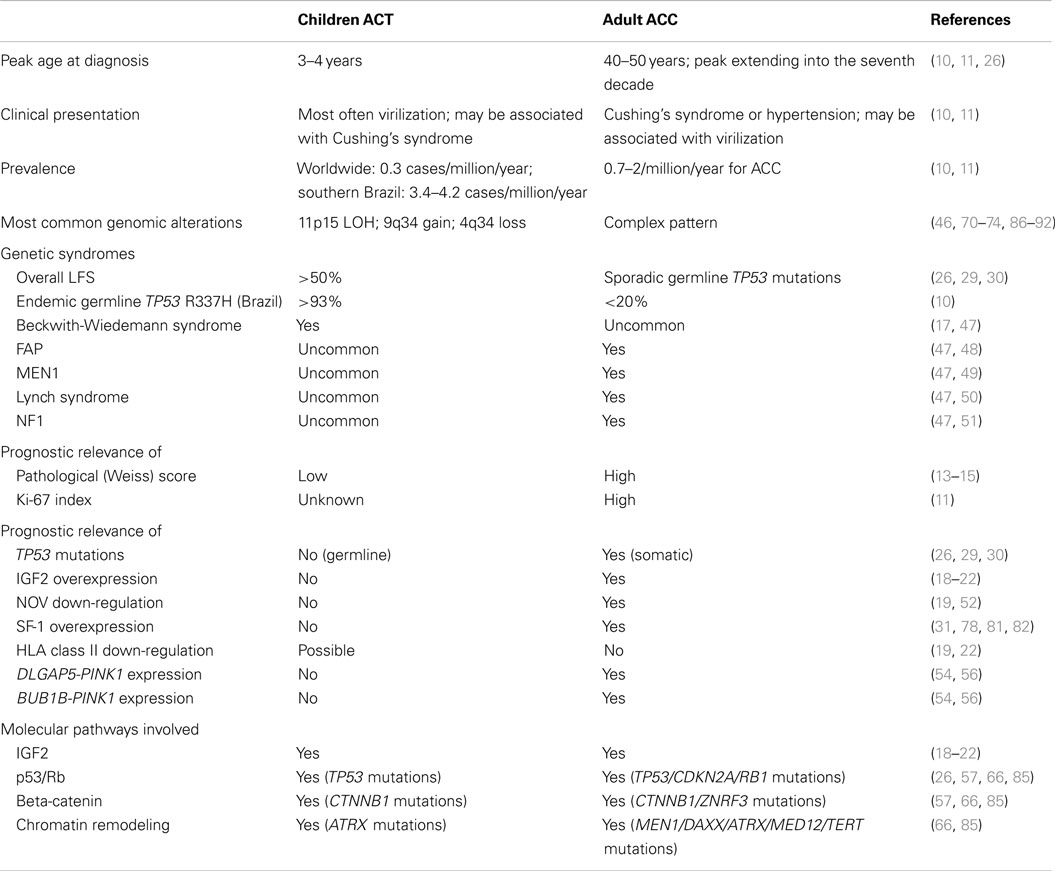
Table 1. Distinctive features and common characteristics of ACT in children and adults.
Childhood malignancies have long been associated to congenital defects (16), which suggest that they may be considered as a degeneration of normal developmental processes. Children ACT are a typical example since they can be found in the context of two genetically determined syndromes, Beckwith-Wiedemann and Li-Fraumeni.
(1) Adrenocortical hyperplasia and neoplasms of variable malignancy are common in Beckwith-Wiedemann syndrome, a systemic overgrowth syndrome caused by genetic defects as uniparental disomy in the 11p15 chromosomal region (17), which cause overexpression of the IGF2 growth factor in the great majority of cases. Loss of heterozygosity (LOH) of the 11p15 region is a systematic finding, not related to prognosis (18–20), in children ACT, leading to IGF2 overexpression from the paternal allele. Similarly, IGF2 is expressed at high levels in the fetal adrenal where it has an important role to regulate proliferation and steroid production (1). Conversely, IGF2 overexpression and abnormalities in the 11p15 region are a marker of malignancy in ACT of adults (21, 22). In mouse models, Igf2 overexpression in the adrenal induces tissue hyperplasia but is not able to induce malignant tumorigenesis per se (23, 24).
(2) Adrenocortical tumors are a distinctive feature of Li-Fraumeni syndrome (LFS), a multiple cancer syndrome due to germline mutations in the TP53 tumor suppressor gene [(25); reviewed in Ref. (26)] encoding p53, a transcription factor that has a pivotal role in preserving genome integrity and activating apoptosis of cells bearing irreparable DNA damage (27). It has been shown that in LFS, excessive DNA copy number variation exists in the patients’ germline, which may predispose to cancer (28). Due to its rarity and its characteristic association with LFS, discovery of an ACT in a child is an absolute indication for researching TP53 mutations in the proband and in his/her parents as well indicative for genetic counseling. Conversely, germline TP53 mutations are much less common in adults with ACC (29, 30) (Table 1). The high incidence of children ACT in LFS suggests that normal p53 function is required for the physiological process of postnatal fetal adrenal regression (Figure 1). In the absence of p53, genetic alterations may accumulate in the adrenal driving proliferation [such as NR5A1 overexpression, (31, 32); see below section on Whole Genome Studies in Children and Adult ACT Reveal Important Drivers for Tumorigenesis and LOH of 11p15 leading to IGF2 overexpression (18–20)] of specific cellular clones. This increased proliferative capacity may favor the emergence of further genetic alterations ultimately leading to clonal expansion and tumorigenesis [reviewed in Ref. (33)].
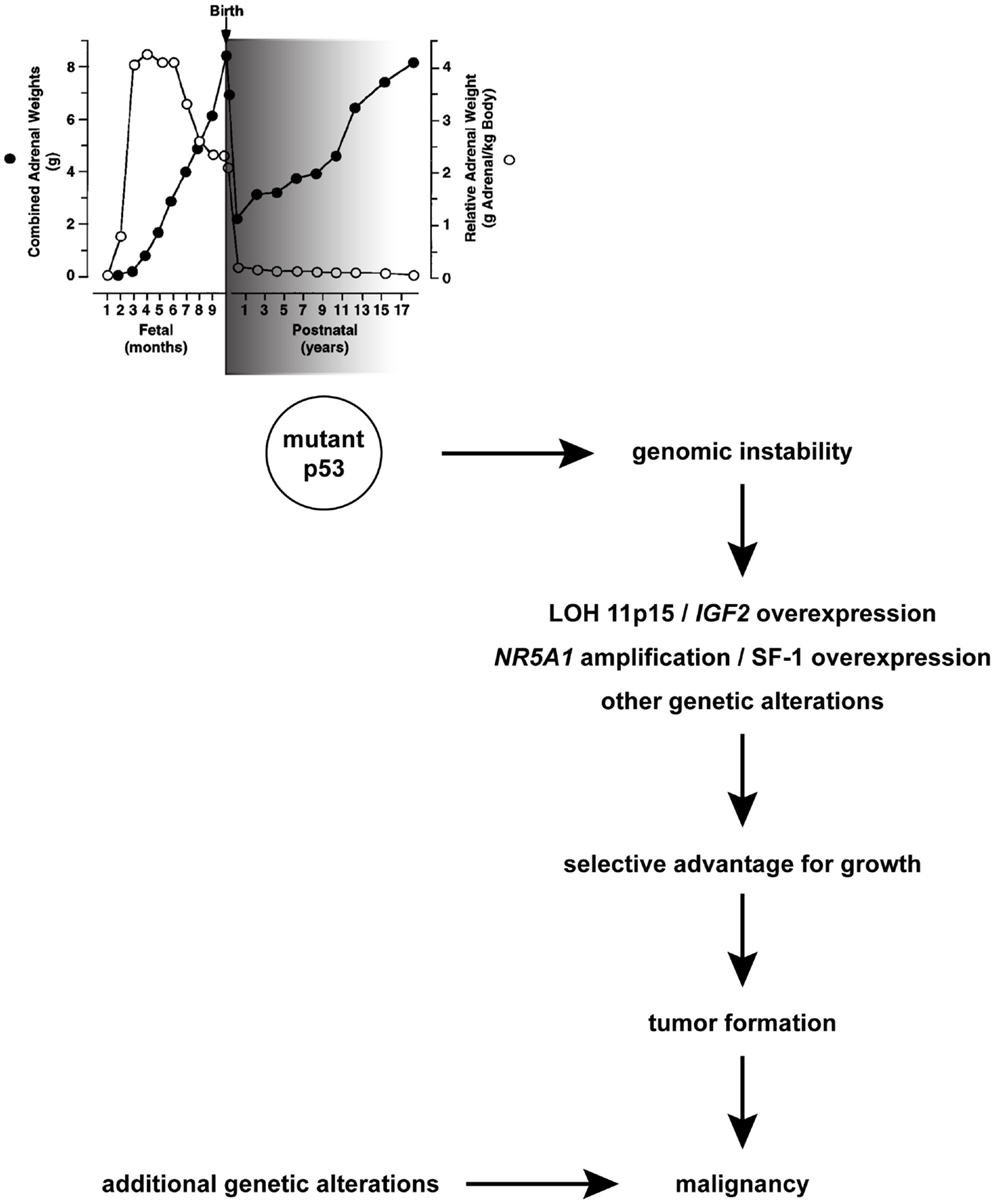
Figure 1. A hypothesis for TP53 role during physiological adrenocortical remodeling in early postnatal life and pathogenesis of children ACT. A window of sensitivity (with an early peak; shaded in grey) of human adrenal to defective p53 function exists during the first years after birth when its physiological involution takes place. Mutant p53 may favor genomic instability, which in some cells may cause LOH of 11p15 and gain/amplification of NR5A1, which confer those cells a selective advantage for growth and lead to tumor formation. Additional genetic alterations arising in this mutation-prone background may cause malignancy. Combined adrenal weight is indicated with black circles. Relative adrenal weight in relationship to body weight is indicated with white circles. Adapted in part from Ref. (1) with permission from Endocrine Society Press.
In classical LFS, due to TP53 mutations that completely abolish protein function, the lifetime incidence of cancer in carriers is close to 100%. However, low-penetrance mutated TP53 alleles exist that can increase the risk of developing cancer only in a fraction of carriers (34). A remarkable example of that situation exists in southern Brazil. In that geographical region, children ACT prevalence is at least 15-fold higher than in the rest of the world (10). This is related to a specific germline TP53 mutation (R337H) (35, 36), whose prevalence is very high (0.3%) in the population but whose penetrance to produce ACT in children has been estimated at only about 2% (37). However, the TP53 R337H mutation has also been reported to be associated to other cancers in the Li-Fraumeni spectrum (38–41) and so its overall penetrance is still unknown. R337 is a conserved arginine residue in the C-terminal tetramerization domain of p53 whose mutation to histidine destabilizes p53 tetramer formation in conditions of elevated temperature and pH (42). It has been shown that a founder effect is responsible for the spreading of the TP53 R337H mutation in the population of southern Brazil (43, 44). An about 0.5 Mb identical by descent haplotype in 17p13 encompassing the TP53 gene carrying the R337H mutation is conserved in all carriers of the mutation (45, 46). A newborn screening and surveillance program of the TP53 R337H mutation carriers in the state of Paraná has proven to be successful to detect ACT in children at an early stage and to treat it with better therapeutic results compared to children who did not undergo surveillance (37).
Apart from rare cases of germline TP53 mutations, as mentioned before (29, 30), ACC in adults may also be associated to other hereditary conditions in some uncommon cases [reviewed in Ref. (47)]: familial adenomatous polyposis (FAP) (48), multiple endocrine neoplasia type1 (MEN1) (49), Lynch syndrome (50), and neurofibromatosis type 1 (NF1) (51) (Table 1).
Genome-Wide Studies in Children and Adult ACT
Distinct Patterns of Coding Genes Expression in Children vs. Adult ACT
Children ACT can be readily differentiated from age-matched normal adrenals by unsupervised clustering based on their gene expression profiles (19). As reported before, IGF2 is the single gene that is most highly up-regulated in children ACT, while genes in the 11p15 region expressed from the maternal allele (KCNQ1, CDKN1C) are among the most strongly down-regulated transcripts. These data are consistent with the systematic LOH of 11p15 in those tumors, with conservation of the paternal allele and loss of the maternal allele (18, 20). Genes belonging to growth factor receptor and mitogen-activated kinase pathways are also dysregulated in children ACT. This suggests that those signaling pathways may be targets for therapeutic intervention. Furthermore, HSD3B2, a steroidogenic enzyme involved in the synthesis of aldosterone and cortisol and expressed in the glomerulosa and reticularis zones of the adult adrenal cortex and its transcriptional regulators NR4A1 and NR4A2 are strongly down-regulated in children ACT, lending further support to the hypothesis of their derivation from the fetal adrenal. This is also suggested by the finding that global gene expression profiles of children ACT are significantly correlated with those present in the fetal adrenal. Another strongly down-regulated gene in children ACT is NOV, encoding a secreted multimodular protein that has a pro-apoptotic function on adrenocortical cancer cells (52). In the study by West et al. (19), a set of 52 differentially expressed genes between adrenocortical adenomas and carcinomas (as distinguished by histological parameters) was identified. It is noteworthy that those included some transcripts encoding HLA class II molecules. Down-regulation of class II expression may represent a mechanism to escape immune surveillance, which could contribute to malignancy. Since malignancy markers are dramatically lacking for children ACT, it will be very important to confirm those data in larger series of patients. However, a recent immunohistochemical study failed to detect consistent HLA class II immunoreactivity in children ACT, both benign and malignant (53).
Unsupervised clustering of gene expression profiles of adult ACT allowed to distinguish two groups, termed C1 and C2 in the study by de Reyniès et al. (54). The C1 group could be further subdivided into C1A and C1B, which correspond to unfavorable and favorable outcome, respectively. Those results were confirmed by another independent study (55), which also confirmed that IGF2 overexpression is associated to malignancy in adult ACC. From gene expression data, de Reyniès and collaborators identified both a signature for malignancy based on the analysis of the expression of two genes (DLGAP5/PINK1) and a two-gene (BUB1B/PINK1) molecular predictor of overall survival for patients with ACC (54). Remarkably, those molecular markers were confirmed to be valid prognostic indicators in adult but not in children ACT in a study on patients from southern Brazil (56) (Table 1). Further studies showed that tumors classified in the C1A group could be further divided into two subgroups each one bearing either TP53 or CTNNB1 (beta-catenin) mutations and in a third subgroup with no other known mutation (57). The importance of the activation of the beta-catenin pathway for adrenocortical tumorigenesis is also shown by studies in mouse models [(23, 24, 58); reviewed in Ref. (59)].
microRNA Sets Differentially Expressed in Children and Adult ACT
In the only study published to date investigating miRNA expression profiles in children ACT, a distinct subgroup of miRNA was found to be differentially expressed in tumor samples compared to age-matched normal adrenal cortex (60). This subgroup included miR-99a and miR-100, which are down-regulated in children ACT and are able to down-regulate expression of IGF-1R (the receptor for IGF2), mTOR, and its associated protein raptor in adrenocortical cell lines. These proteins are up-regulated in children ACT and their pharmacological blockade is able to significantly decrease adrenocortical cancer cell proliferation (60–63). These results show that miR-99a and miR-100 have an important role in children ACT by the modulation of growth factor signaling through the IGF-1R–mTOR pathway.
On the other hand, several studies reported data on miRNA expression profiles in adult ACT. Those studies show in general only limited overlap [reviewed in Ref. (64)]. Nevertheless, most studies detected overexpression in ACC of miR-483-5p and/or -3p, whose gene is situated in an intron of IGF2 and may have an independent oncogenic function (65). miR-483-3p was also found up-regulated in children ACT in the study by Doghman et al. (60). Other miRNAs that display similar differential regulation in children and adult ACT are miR-503 (up-regulated), miR-195, miR-214, and miR-375 (down-regulated). A recently published integrative analysis of genomic alterations in adult ACC (66) found up-regulation of miRNAs belonging to the miR-506-514 cluster on chromosome Xq27 and down-regulation of the expression of the DLK1-MEG3 miRNA cluster on chromosome 14q in one subgroup of samples with favorable prognosis (C1B; see below section on Whole Genome Studies in Children and Adult ACT Reveal Important Drivers for Tumorigenesis). There is of great interest for the potential use of circulating miRNAs as biomarkers of malignancy in ACC (67–69).
Whole Genome Studies in Children and Adult ACT Reveal Important Drivers for Tumorigenesis
The first studies analyzing children ACT genome copy number alterations by comparative genomic hybridization (CGH) reported patterns of recurrent gains and losses (70–72). In particular, one of the most common alterations found in almost all cases of children ACT investigated was the gain/amplification of 9q34. Gains in this region were also reported in some studies of chromosomal alterations in adult ACT (73, 74). In close proximity to this chromosomal region (9q33) is situated the gene (NR5A1) encoding the transcription factor SF-1, a master regulator of adrenocortical and gonadal development [reviewed in Ref. (75, 76)]. Further studies showed that the NR5A1 gene is amplified and the SF-1 protein is overexpressed in the large majority of Brazilian children ACT (31, 77, 78). Interestingly, the SF-1 protein was overexpressed even in cases lacking gene amplification (31, 78), suggesting that mechanisms in addition to copy number gain may also account for SF-1 overexpression. The dosage-dependent effect of SF-1 in boosting adrenocortical cell proliferation was shown by studies in human cell lines and in transgenic mice (32) by regulation of a fairly large set of dosage-dependent target genes far exceeding its classical steroidogenic targets [(79); reviewed in Ref. (80)]. In children ACT, SF-1 overexpression appears to be a widespread finding, with no relationship with malignancy [(31, 78); see Figure 1]. Conversely, SF-1 overexpression in adult ACT is less common than in children (78) and is an unfavorable prognostic marker (81, 82) (Table 1). Remarkably, SF-1 transcriptional regulatory activity can be pharmacologically targeted leading to a decrease of adrenocortical cancer cell proliferation (83), suggesting that this transcription factor may represent a novel therapeutic target in ACT.
A subsequent SNP array study on both Brazilian and non-Brazilian ACT cases precisely defined recurrent genomic alterations in children ACT (46), the most frequent being loss of 4q34, gain of 9q33-q34 and 19p, and LOH of the whole chromosome 17 (harboring TP53) and 11p15 (harboring IGF2). Remarkably, a number of focal deletions were detected at 4q34, defining a common deleted region surrounding the non-coding RNA LINC00290 gene. It is also noteworthy that the extent of the peak region of gain in 9q33-q34 suggests that other genes lying in a telomeric position with respect to NR5A1 may also be important for ACT pathogenesis. In addition, focal amplifications and homozygous deletions comprising well-known oncogenes (MYC, MDM2, PDGFRA, KIT, MCL1, BCL2L1) and tumor suppressors (TP53, RB1, RPH3AL) were identified. Although genomic profiles in non-Brazilian tumors with a mutated TP53 (other than R337H) were similar to Brazilian tumors, those with a wild-type TP53 displayed distinct genomic alterations, harboring significantly fewer rearrangements. Remarkably, 50% of TP53 wild-type tumors investigated in this study displayed as sole rearrangement a copy-neutral LOH of the imprinted region at 11p15, providing further evidence for a major role of this region in ACT development.
The landscape of genomic alterations in a worldwide series of children ACT enrolled at IPACTR (84) has been more precisely defined by a very recent study integrating whole exome, whole genome, and RNA-sequencing data (85). This work confirmed LOH in the 11p15 region in the large majority of cases and systematic overexpression of IGF2, together with frequent TP53 mutations, widespread 9q copy number gain, and 4q34 loss. By comparing the mutant allele fraction of SNV in copy-neutral LOH regions to allelic imbalance values, it was possible to establish that in most cases copy-neutral LOH of chromosomes 11p and 17 occurred early during tumorigenesis, suggesting that those events drive tumor formation. Additional recurrent genetic alterations in children ACT were somatic mutations in the ATRX (a DNA helicase) and CTNNB1 genes. Intriguingly, some tumors bore integration of human herpesvirus-6 (HHV6) in the telomeric region of chromosome 11p. A poor outcome was predicted by concomitant TP53/ATRX mutations and associated genomic abnormalities, including massive structural variations and a high background mutation rate (Table 1).
In adult ACT, earlier CGH studies showed a significantly increased prevalence of genomic imbalances in carcinomas compared to adenomas and sometimes contrasting patterns of gain and losses (73, 74, 86–88). CGH array studies evidenced a set of chromosomal aberrations in ACC associated with survival in a fashion dependent on their accumulation (89). Carcinomas were confirmed to harbor a higher number of chromosomal alterations than adenomas (90–92). Recently, activating mutations of the PKA catalytic subunit were shown to be associated with cortisol-secreting adrenocortical adenomas in adults (93–96). In general, gains had a higher impact than losses on gene expression profiles (91). A comparison between genome alterations in children ACT and adult ACC is shown in Figure 2. Methylome studies were also performed in adult ACC (97–99). According to their DNA methylation levels, malignant tumors could be divided into two groups, one displaying low and the other one elevated levels of methylation in CpG islands (CpG island methylator phenotype, CIMP). This hypermethylated tumors group could in turn be subdivided into two subgroups (CIMP-high and CIMP-low), which had prognostic relevance, with the CIMP-high phenotype clearly being associated to worse prognosis (99).
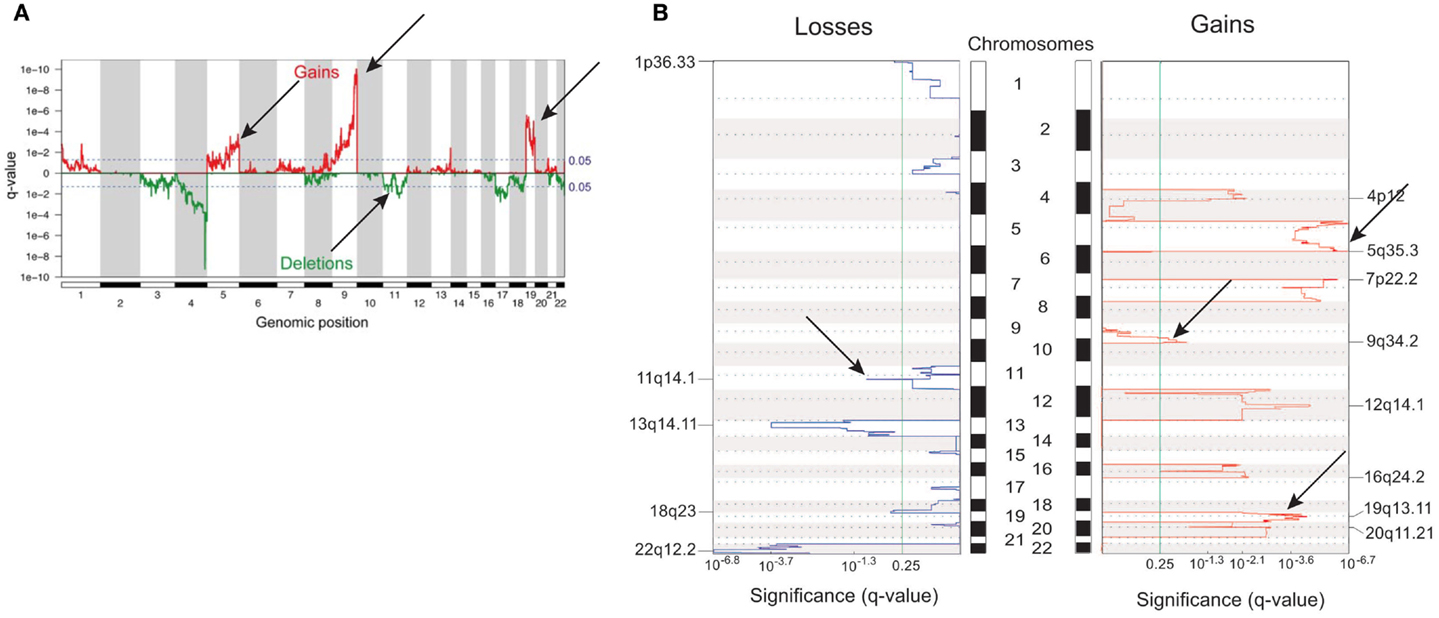
Figure 2. Differences and similarities in genomic profiles between children and adult ACT. (A) Genomic alterations in children ACT. (B) Genomic alterations in adult ACC. Common regions of gains (chr. 5, 9q, 19) and losses (chr. 11) are indicated with arrows. Adapted from Ref. (46, 91) with permission from Endocrine Society Press.
A study integrating transcriptome, miRNome, copy number alterations, methylome, and whole exome sequencing data in adult ACC was recently published (66), showing that major pathways involved by mutation or homozygous deletion include beta-catenin (CTNNB1 and ZNRF3), p53/Rb signaling (TP53, CDKN2A, and RB1), and chromatin remodeling (MEN1, DAXX, ATRX, MED12, and TERT) (Table 1). In addition, recurrent homozygous deletions were found in 4q34, similarly to children ACT. This study also showed that a substantial overlap exists among the different omics classifications of ACC: the previously identified gene expression profile clusters (C1A, C1B, and C2; see section on Distinct Patterns of Coding Genes Expression in Children vs. Adult ACT) (54) are strongly correlated with subgroups based on DNA methylation and miRNA expression, mutation rate, and alteration of key molecular pathways.
Perspectives
Children ACT represent a distinct pathological entity compared to tumors in adults concerning their origin, clinical manifestations, molecular alterations, and prognostic evolution. Important fields of investigation in the future will be the search for genetic and environmental factors that modulate penetrance of ACT in carriers of germline TP53 mutations in order to orient screening procedures to detect disease at an early stage, the identification of robust biomarkers of malignancy, which are still lacking, and the clinical testing of targeted therapies against the major molecular pathways that are altered in this disease (100).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank E. Pinto, G. Zambetti, and R. Ribeiro for communicating results before publication. Work in our laboratories is supported by CNRS and Ciência sem Fronteiras program from the Brazilian government.
References
1. Mesiano S, Jaffe RB. Developmental and functional biology of the primate fetal adrenal cortex. Endocr Rev (1997) 18:378–401. doi: 10.1210/edrv.18.3.0304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Chamoux E, Otis M, Gallo-Payet N. A connection between extracellular matrix and hormonal signals during the development of the human fetal adrenal gland. Braz J Med Biol Res (2005) 38:1495–503. doi:10.1590/S0100-879X2005001000006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. King P, Paul A, Laufer E. Shh signaling regulates adrenocortical development and identifies progenitors of steroidogenic lineages. Proc Natl Acad Sci USA (2009) 106:21185–90. doi:10.1073/pnas.0909471106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Freedman BD, Kempna PB, Carlone DL, Shah MS, Guagliardo NA, Barrett PQ, et al. Adrenocortical zonation results from lineage conversion of differentiated zona glomerulosa cells. Dev Cell (2013) 26:666–73. doi:10.1016/j.devcel.2013.07.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Bandiera R, Vidal VPI, Motamedi FJ, Clarkson M, Sahut-Barnola I, von Gise A, et al. WT1 maintains adrenal-gonadal primordium identity and marks a population of AGP-like progenitors within the adrenal gland. Dev Cell (2013) 27:5–18. doi:10.1016/j.devcel.2013.09.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Lalli E. Role of orphan nuclear receptor DAX-1/NR0B1 in development, physiology and disease. Adv Biol (2014) 2014:582749. doi:10.1155/2014/582749
7. Ben-David S, Zuckerman-Levin N, Epelman M, Shen-Orr Z, Levin M, Sujov P, et al. Parturition itself is the basis for fetal adrenal involution. J Clin Endocrinol Metab (2007) 92:93–7. doi:10.1210/jc.2005-2720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Zubair M, Ishihara S, Oka S, Okumura K, Morohashi KI. Two-step regulation of Ad4BP/SF-1 gene transcription during fetal adrenal development: initiation by a Hox-Pbx1-Prep1 complex and maintenance via autoregulation by Ad4BP/SF-1. Mol Cell Biol (1996) 26:1411–21.
9. Grumbach MM, Styne DM. Puberty: ontogeny, neuroendocrinology, physiology and disorders. In: Larsen PR, Kronenberg HM, Melmed S, Polonsky KS, editors. Williams Textbook of Endocrinology. Philadelphia, PA: Saunders (2003). p. 1115–286.
10. Custódio G, Komechen H, Figueiredo FRO, Fachin ND, Pianovski MAD, Figueiredo BC. Molecular epidemiology of adrenocortical tumors in southern Brazil. Mol Cell Endocrinol (2012) 351:44–51. doi:10.1016/j.mce.2011.10.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Fassnacht M, Libé R, Kroiss M, Allolio B. Adrenocortical carcinoma: a clinician’s update. Nat Rev Endocrinol (2011) 7:323–35. doi:10.1038/nrendo.2010.235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Michalkiewicz E, Sandrini R, Figueiredo B, Miranda EC, Caran E, Oliveira-Filho AG, et al. Clinical and outcome characteristics of children with adrenocortical tumors. An analysis of 254 cases from the international pediatric adrenocortical tumor registry. J Clin Oncol (2004) 22:838–45. doi:10.1200/JCO.2004.08.085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Wieneke JA, Thompson LD, Heffess CS. Adrenal cortical neoplasms in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol (2003) 27:867–81. doi:10.1097/00000478-200307000-00001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Lau SK, Weiss LM. The Weiss system for evaluating adrenocortical neoplasms: 25 years later. Hum Pathol (2009) 40:757–68. doi:10.1016/j.humpath.2009.03.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Dehner LP, Hill DA. Adrenal cortical neoplasms in children: why so many carcinomas and yet so many survivors? Pediatr Dev Pathol (2009) 12:284–91. doi:10.2350/08-06-0489.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Miller RW. Relation between cancer and congenital defects: an epidemiologic evaluation. J Natl Cancer Inst (1968) 40:1079–85.
17. Weksberg R, Smith AC, Squire J, Sadowski P. Beckwith-Wiedemann syndrome demonstrates a role for epigenetic control of normal development. Hum Mol Genet (2003) 12:R61–8. doi:10.1093/hmg/ddg067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Wilkin F, Gagné N, Paquette J, Oligny LL, Deal C. Pediatric adrenocortical tumors: molecular events leading to insulin-like growth factor II gene overexpression. J Clin Endocrinol Metab (2000) 85:2048–56. doi:10.1210/jcem.85.5.6589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. West AN, Neale GA, Pounds S, Figueredo BC, Rodriguez-Galindo C, Pianovski MA, et al. Gene expression profiling of childhood adrenocortical tumors. Cancer Res (2007) 67:600–8. doi:10.1158/0008-5472.CAN-06-3767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Rosati R, Cerrato F, Doghman M, Pianovski MAD, Parise GA, Custódio G, et al. High frequency of loss of heterozygosity at 11p15 and IGF2 overexpression is not associated with clinical outcome in childhood adrenocortical tumors positive for the R337H TP53 mutation. Cancer Genet Cytogenet (2008) 186:19–24. doi:10.1016/j.cancergencyto.2008.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Gicquel C, Raffin-Sanson ML, Gaston V, Bertagna X, Plouin PF, Schlumberger M, et al. Structural and functional abnormalities at 11p15 are associated with the malignant phenotype in sporadic adrenocortical tumors: study on a series of 82 tumors. J Clin Endocrinol Metab (1997) 82:2559–65. doi:10.1210/jc.82.8.2559
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Giordano TJ, Thomas DG, Kuick R, Lizyness M, Misek DE, Smith AL, et al. Distinct transcriptional profiles of adrenocortical tumors uncovered by DNA microarray analysis. Am J Pathol (2003) 162:521–31. doi:10.1016/S0002-9440(10)63846-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Drelon C, Berthon A, Ragazzon B, Tissier F, Bandiera R, Sahut-Barnola I, et al. Analysis of the role of Igf2 in adrenal tumour development in transgenic mouse models. PLoS One (2012) 7:e44171. doi:10.1371/journal.pone.0044171
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Heaton JH, Wood MA, Kim AC, Lima LO, Barlaskar FM, Almeida MQ, et al. Progression to adrenocortical tumorigenesis in mice and humans through insulin-like growth factor 2 and β-catenin. Am J Pathol (2012) 181:1017–33. doi:10.1016/j.ajpath.2012.05.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, Kim DH, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science (1990) 250:1233–8. doi:10.1126/science.1978757
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Wasserman JD, Zambetti GP, Malkin D. Towards an understanding of the role of p53 in adrenocortical carcinogenesis. Mol Cell Endocrinol (2012) 351:101–10. doi:10.1016/j.mce.2011.09.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Shlien A, Tabori U, Marshall CR, Pienkowska M, Feul L, Novokmet A, et al. Excessive genomic DNA copy number variation in the Li-Fraumeni cancer predisposition syndrome. Proc Natl Acad Sci USA (2008) 105:11264–9. doi:10.1073/pnas.0802970105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Herrmann LJM, Heinze B, Fassnacht M, Willenberg HS, Quinkler M, Reisch N, et al. TP53 germline mutations in adult patients with adrenocortical carcinoma. J Clin Endocrinol Metab (2012) 97:E476–85. doi:10.1210/jc.2011-1982
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Raymond VM, Else T, Everett JN, Long JM, Gruber SB, Hammer GD. Prevalence of germline TP53 mutations in a prospective series of unselected patients with adrenocortical carcinoma. J Clin Endocrinol Metab (2013) 98:E119–25. doi:10.1210/jc.2012-2198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Pianovski MA, Cavalli LR, Figueiredo BC, Santos SC, Doghman M, Ribeiro RC, et al. SF-1 overexpression in childhood adrenocortical tumours. Eur J Cancer (2006) 42:1040–3. doi:10.1016/j.ejca.2006.01.022
32. Doghman M, Karpova T, Rodrigues GA, Arhatte M, De Moura J, Cavalli LR, et al. Increased steroidogenic factor-1 dosage triggers adrenocortical cell proliferation and cancer. Mol Endocrinol (2007) 21:2968–87. doi:10.1210/me.2007-0120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. El Wakil A, Doghman M, Latre de Late P, Zambetti GP, Figueiredo BC, Lalli E. Genetics and genomics of childhood adrenocortical tumors. Mol Cell Endocrinol (2011) 336:169–73. doi:10.1016/j.mce.2010.11.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Varley JM, McGown G, Thorncroft M, James LA, Margison GP, Forster G, et al. Are there low-penetrance TP53 alleles? Evidence from childhood adrenocortical tumors. Am J Hum Genet (1999) 65:995–1006. doi:10.1086/302575
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Ribeiro RC, Sandrini F, Figueiredo B, Zambetti GP, Michalkiewicz E, Lafferty AR, et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A (2001) 98:9330–5. doi:10.1073/pnas.161479898
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Latronico AC, Pinto EM, Domenice S, Fragoso MC, Martin RM, Zerbini MC, et al. An inherited mutation outside the highly conserved DNA-binding domain of the p53 tumor suppressor protein in childhood and adults with sporadic adrenocortical tumors. J Clin Endocrinol Metab (2001) 86:4970–3. doi:10.1210/jcem.86.10.7957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Custódio G, Parise GA, Kiesel FN, Komechen H, Sabbaga CC, Rosati R, et al. Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. J Clin Oncol (2013) 31:2619–26. doi:10.1200/JCO.2012.46.3711
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Achatz MI, Olivier M, Le Calvez F, Martel-Planche G, Lopes A, Rossi BM, et al. The TP53 mutation, R337H, is associated with Li-Fraumeni and Li-Fraumeni-like syndromes in Brazilian families. Cancer Lett (2007) 245:96–102. doi:10.1016/j.canlet.2005.12.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Assumpção JG, Seidinger AL, Mastellaro MJ, Ribeiro RC, Zambetti GP, Ganti R, et al. Association of the germline TP53 R337H mutation with breast cancer in southern Brazil. BMC Cancer (2008) 8:357. doi:10.1186/1471-2407-8-357
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Seidinger AL, Mastellaro MJ, Paschoal Fortes F, Godoy Assumpção J, Aparecida Cardinalli I, Aparecida Ganazza M, et al. Association of the highly prevalent TP53 R337H mutation with pediatric choroid plexus carcinoma and osteosarcoma in southeast Brazil. Cancer (2011) 117:2228–35. doi:10.1002/cncr.25826
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Custódio G, Taques GR, Figueiredo BC, Gugelmin ES, Oliveira Figueiredo MM, Watanabe F, et al. Increased incidence of choroid plexus carcinoma due to the germline TP53 R337H mutation in southern Brazil. PLoS One (2011) 6:e18015. doi:10.1371/journal.pone.0018015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. DiGiammarino EL, Lee AS, Cadwell C, Zhang W, Bothner B, Ribeiro RC, et al. A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nat Struct Biol (2002) 9:12–6. doi:10.1038/nsb730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Pinto EM, Billerbeck AE, Villares MC, Domenice S, Mendonça BB, Latronico AC. Founder effect for the highly prevalent R337H mutation of tumor suppressor p53 in Brazilian patients with adrenocortical tumors. Arq Bras Endocrinol Metabol (2004) 48:647–50. doi:10.1590/S0004-27302004000500009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Garritano S, Gemignani F, Palmero EI, Olivier M, Martel-Planche G, Le Calvez-Kelm F, et al. Detailed haplotype analysis at the TP53 locus in p.R337H mutation carriers in the population of southern Brazil: evidence for a founder effect. Hum Mutat (2010) 31:143–50. doi:10.1002/humu.21151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Letouzé E, Sow A, Petel F, Rosati R, Figueiredo BC, Burnichon N, et al. Identity by descent mapping of founder mutations in cancer using high-resolution tumor SNP data. PLoS One (2012) 7:e35897. doi:10.1371/journal.pone.0035897
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Letouzé E, Rosati R, Komechen H, Doghman M, Marisa L, Flück C, et al. SNP array profiling of childhood adrenocortical tumors reveals distinct pathways of tumorigenesis and highlights candidate driver genes. J Clin Endocrinol Metab (2012) 97:E1284–93. doi:10.1210/jc.2012-1184
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Else T. Association of adrenocortical carcinoma with familial cancer susceptibility syndromes. Mol Cell Endocrinol (2012) 351:66–70. doi:10.1016/j.mce.2011.12.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Gaujoux S, Pinson S, Gimenez-Roqueplo AP, Amar L, Ragazzon B, Launay P, et al. Inactivation of the APC gene is constant in adrenocortical tumors from patients with familial adenomatous polyposis but not frequent in sporadic adrenocortical cancers. Clin Cancer Res (2010) 16:5133–41. doi:10.1158/1078-0432.CCR-10-1497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Gatta-Cherifi B, Chabre O, Murat A, Niccoli P, Cardot-Bauters C, Rohmer V, et al. Adrenal involvement in MEN1. Analysis of 715 cases from the Groupe d’etude des tumeurs endocrines database. Eur J Endocrinol (2012) 166:269–79. doi:10.1530/EJE-11-0679
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Raymond VM, Everett JN, Furtado LV, Gustafson SL, Jungbluth CR, Gruber SB, et al. Adrenocortical carcinoma is a Lynch syndrome-associated cancer. J Clin Oncol (2013) 31:3012–8. doi:10.1200/JCO.2012.48.0988
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Menon RK, Ferrau F, Kurzawinski TR, Rumsby G, Freeman A, Amin Z, et al. Adrenal cancer in neurofibromatosis type 1: case report and DNA analysis. Endocrinol Diabetes Metab Case Rep (2014) 2014:140074. doi:10.1530/EDM-14-0074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Doghman M, Arhatte M, Thibout H, Rodrigues G, De Moura J, Grosso S, et al. Nephroblastoma overexpressed/cysteine-rich protein 61/connective tissue growth factor/nephroblastoma overexpressed gene-3 (NOV/CCN3), a selective adrenocortical cell proapoptotic factor, is down-regulated in childhood adrenocortical tumors. J Clin Endocrinol Metab (2007) 92:3253–60. doi:10.1210/jc.2007-0342
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Magro G, Esposito G, Cecchetto G, Dall’Igna P, Marcato R, Gambini C, et al. Pediatric adrenocortical tumors: morphological diagnostic criteria and immunohistochemical expression of matrix metalloproteinase type 2 and human leucocyte-associated antigen (HLA) class II antigens. Results from the Italian Pediatric Rare Tumor (TREP) Study project. Hum Pathol (2012) 43:31–9. doi:10.1016/j.humpath.2011.04.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. de Reyniès A, Assié G, Rickman DS, Tissier F, Groussin L, René-Corail F, et al. Gene expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J Clin Oncol (2009) 27:1108–15. doi:10.1200/JCO.2008.18.5678
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Giordano TJ, Kuick R, Else T, Gauger PG, Vinco M, Bauersfeld J, et al. Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin Cancer Res (2009) 15:668–76. doi:10.1158/1078-0432.CCR-08-1067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Fragoso MC, Almeida MQ, Mazzuco TL, Mariani BM, Brito LP, Gonçalves TC, et al. Combined expression of BUB1B, DLGAP5, and PINK1 as predictors of poor outcome in adrenocortical tumors: validation in a Brazilian cohort of adult and pediatric patients. Eur J Endocrinol (2012) 166:61–7. doi:10.1530/EJE-11-0806
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Ragazzon B, Libé R, Gaujoux S, Assié G, Fratticci A, Launay P, et al. Transcriptome analysis reveals that p53 and beta-catenin alterations occur in a group of aggressive adrenocortical cancers. Cancer Res (2010) 70:8276–81. doi:10.1158/0008-5472.CAN-10-2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Berthon A, Sahut-Barnola I, Lambert-Langlais S, de Joussineau C, Damon-Soubeyrand C, Louiset E, et al. Constitutive beta-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum Mol Genet (2010) 19:1561–76. doi:10.1093/hmg/ddq029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. El Wakil A, Lalli E. The Wnt/beta-catenin pathway in adrenocortical development and cancer. Mol Cell Endocrinol (2011) 332:32–7. doi:10.1016/j.mce.2010.11.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Doghman M, El Wakil A, Cardinaud B, Thomas E, Wang J, Zhao W, et al. Regulation of insulin-like growth factor – mammalian target of rapamycin signalling by microRNA in childhood adrenocortical tumors. Cancer Res (2010) 70:4666–75. doi:10.1158/0008-5472.CAN-09-3970
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Almeida MQ, Fragoso MC, Lotfi CF, Santos MG, Nishi MY, Costa MH, et al. Expression of insulin-like growth factor-II and its receptor in pediatric and adult adrenocortical tumors. J Clin Endocrinol Metab (2008) 93:3524–31. doi:10.1210/jc.2008-0065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Barlaskar FM, Spalding AC, Heaton JH, Kuick R, Kim AC, Thomas DG, et al. Preclinical targeting of the type I insulin-like growth factor receptor in adrenocortical carcinoma. J Clin Endocrinol Metab (2009) 94:204–12. doi:10.1210/jc.2008-1456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Doghman M, Axelson M, Lalli E. Potent inhibitory effect of the cyclolignan picropodophyllin (PPP) on human adrenocortical carcinoma cells proliferation. Am J Cancer Res (2011) 1:356–61.
64. Singh P, Soon PSH, Feige J-J, Chabre O, Zhao JT, Cherradi N, et al. Dysregulation of microRNAs in adrenocortical tumors. Mol Cell Endocrinol (2012) 351:118–28. doi:10.1016/j.mce.2011.09.041
65. Veronese A, Lupini L, Consiglio J, Visone R, Ferracin M, Fornari F, et al. Oncogenic role of miR-483-3p at the IGF2/483 locus. Cancer Res (2010) 70:3140–9. doi:10.1158/0008-5472.CAN-09-4456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Assié G, Letouzé E, Fassnacht M, Jouinot A, Luscap W, Barreau O, et al. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet (2014) 46:607–12. doi:10.1038/ng.2953
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Chabre O, Libé R, Assié G, Barreau O, Bertherat J, Bertagna X, et al. Serum miR-483-5p and miR-195 are predictive of recurrence risk in adrenocortical cancer patients. Endocr Relat Cancer (2013) 20:579–94. doi:10.1530/ERC-13-0051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Patel D, Boufraqech M, Jain M, Zhang L, He M, Gesuwan K, et al. MiR-34a and miR-483-5p are candidate serum biomarkers for adrenocortical tumors. Surgery (2013) 154:1224–8. doi:10.1016/j.surg.2013.06.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Szabó DR, Luconi M, Szabó PM, Tóth M, Szücs N, Horányi J, et al. Analysis of circulating microRNAs in adrenocortical tumors. Lab Invest (2014) 94:331–9. doi:10.1038/labinvest.2013.148
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Figueiredo BC, Stratakis CA, Sandrini R, DeLacerda L, Pianovski MAD, Giatzakis C, et al. Comparative genomic hybridization analysis of adrenocortical tumors of childhood. J Clin Endocrinol Metab (1999) 84:1116–21. doi:10.1210/jcem.84.3.5526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. James LA, Kelsey AM, Birch JM, Varley JM. Highly consistent genetic alterations in childhood adrenocortical tumours detected by comparative genomic hybridization. Br J Cancer (1999) 81:300–4. doi:10.1038/sj.bjc.6990691
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Loncarevic IF, Hering A, Posorski N, Linden T, Hoyer H, Bucsky P. Number of genomic imbalances correlates with the overall survival for adrenocortical cancer in childhood. Pediatr Blood Cancer (2008) 51:356–62. doi:10.1002/pbc.21603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Zhao J, Speel EJM, Muletta-Feurer S, Rütimann K, Saremaslani P, Roth J, et al. Analysis of genomic alterations in sporadic adrenocortical lesions. Am J Pathol (1999) 155:1039–45. doi:10.1016/S0002-9440(10)65205-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Dohna M, Reincke M, Mincheva A, Allolio B, Solinas-Toldo S, Lichter P. Adrenocortical carcinoma is characterized by a high frequency of chromosomal gains and high-level amplifications. Genes Chromosomes Cancer (2000) 28:145–52. doi:10.1002/(SICI)1098-2264(200006)28:2<145::AID-GCC3>3.0.CO;2-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Schimmer BP, White PC. Steroidogenic factor 1: its roles in differentiation, development, and disease. Mol Endocrinol (2010) 24:1322–37. doi:10.1210/me.2009-0519
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Lalli E. Adrenocortical development and cancer: focus on SF-1. J Mol Endocrinol (2010) 44:301–7. doi:10.1677/JME-09-0143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Figueiredo BC, Cavalli LR, Pianovski MA, Lalli E, Sandrini R, Ribeiro RC, et al. Amplification of the steroidogenic factor 1 gene in childhood adrenocortical tumors. J Clin Endocrinol Metab (2005) 90:615–9. doi:10.1210/jc.2004-0942
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Almeida MQ, Soares IC, Ribeiro TC, Fragoso MC, Marins LV, Wakamatsu A, et al. Steroidogenic factor 1 overexpression and gene amplification are more frequent in adrenocortical tumors from children than from adults. J Clin Endocrinol Metab (2010) 95:1458–62. doi:10.1210/jc.2009-2040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Doghman M, Figueiredo BC, Volante M, Papotti M, Lalli E. Integrative analysis of SF-1 transcription factor dosage impact on genome-wide binding and gene expression regulation. Nucleic Acids Res (2013) 41:8896–907. doi:10.1093/nar/gkt658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Lalli E, Doghman M, Latre de Late P, El Wakil A, Mus-Veteau I. Beyond steroidogenesis: novel target genes for SF-1 discovered by genomics. Mol Cell Endocrinol (2013) 371:154–9. doi:10.1016/j.mce.2012.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Sbiera S, Schmull S, Assié G, Voelker HU, Kraus L, Beyer M, et al. High diagnostic and prognostic value of steroidogenic factor-1 expression in adrenal tumors. J Clin Endocrinol Metab (2010) 95:E161–71. doi:10.1210/jc.2010-0653
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Duregon E, Volante M, Giorcelli J, Terzolo M, Lalli E, Papotti M. Diagnostic and prognostic role of steroidogenic factor-1 in adrenocortical carcinoma: a validation study focusing on clinical and pathological correlates. Hum Pathol (2013) 44:822–8. doi:10.1016/j.humpath.2012.07.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Doghman M, Cazareth J, Douguet D, Madoux F, Hodder P, Lalli E. Inhibition of adrenocortical carcinoma cell proliferation by SF-1 inverse agonists. J Clin Endocrinol Metab (2009) 94:2178–83. doi:10.1210/jc.2008-2163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Ribeiro RC, Pinto EM, Zambetti GP, Rodriguez-Galindo C. The international pediatric adrenocortical tumor registry initiative: contributions to clinical, biological, and treatment advances in pediatric adrenocortical tumors. Mol Cell Endocrinol (2012) 351:37–43. doi:10.1016/j.mce.2011.10.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Pinto EM, Chen X, Easton J, Finkelstein D, Liu Z, Pounds S, et al. Genomic landscape of pediatric adrenocortical tumors. Nat Commun (Forthcoming).
86. Kjellman M, Kallioniemi OP, Karhu R, Hög A, Farnebo LO, Auer G, et al. Genetic aberrations in adrenocortical tumors detected using comparative genomic hybridization correlate with tumor size and malignancy. Cancer Res (1996) 56:4219–23.
87. Sidhu S, Marsh DJ, Theodosopoulos G, Philips J, Bambach CP, Campbell P, et al. Comparative genomic hybridization analysis of adrenocortical tumors. J Clin Endocrinol Metab (2002) 87:3467–74. doi:10.1210/jcem.87.7.8697
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Zhao J, Roth J, Bode-Lesniewska B, Pfaltz M, Heitz PU, Komminoth P. Combined comparative genomic hybridization and genomic microarray for detection of gene amplifications in pulmonary artery intimal sarcomas and adrenocortical tumors. Genes Chromosomes Cancer (2002) 34:48–57. doi:10.1002/gcc.10035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Stephan EA, Chung TH, Grant CS, Kim S, Von Hoff DD, Trent JM, et al. Adrenocortical carcinoma survival rates correlated to copy number variants. Mol Cancer Ther (2008) 7:425–31. doi:10.1158/1535-7163.MCT-07-0267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Ronchi CL, Leich E, Sbiera S, Weismann D, Rosenwald A, Allolio B, et al. Single nucleotide polymorphism microarray analysis in cortisol-secreting adrenocortical adenomas identifies new candidate genes and pathways. Neoplasia (2012) 14:206–18.
91. Barreau O, de Reynies A, Wilmot-Roussel H, Guillaud-Bataille M, Auzan C, René-Corail F, et al. Clinical and pathophysiological implications of chromosomal alterations in adrenocortical tumors: an integrated genomic approach. J Clin Endocrinol Metab (2012) 97:E301–11. doi:10.1210/jc.2011-1588
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Ronchi CL, Sbiera S, Leich E, Henzel K, Rosenwald A, Allolio B, et al. Single nucleotide polymorphism array profiling of adrenocortical tumors – evidence for an adenoma carcinoma sequence? PLoS One (2013) 8:e73959. doi:10.1371/journal.pone.0073959
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Beuschlein F, Fassnacht M, Assié G, Calebiro D, Stratakis CA, Osswald A, et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med (2014) 370:1019–28. doi:10.1056/NEJMoa1310359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet (2014) 46:613–7. doi:10.1038/ng.2956
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Cao Y, He M, Gao Z, Peng Y, Li Y, Li L, et al. Activating hotspot L205R mutation in PRKACA and adrenal Cushing’s syndrome. Science (2014) 344:913–7. doi:10.1126/science.1249480
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, Shiraishi Y, et al. Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science (2014) 344:917–20. doi:10.1126/science.1252328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Fonseca AL, Kugelberg J, Starker LF, Scholl U, Choi M, Hellman P, et al. Comprehensive DNA methylation analysis of benign and malignant adrenocortical tumors. Genes Chromosomes Cancer (2012) 51:949–60. doi:10.1002/gcc.21978
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Rechache NS, Wang Y, Stevenson HS, Killian JK, Edelman DC, Merino M, et al. DNA methylation profiling identifies global methylation differences and markers of adrenocortical tumors. J Clin Endocrinol Metab (2012) 97:E1004–13. doi:10.1210/jc.2011-3298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Barreau O, Assié G, Wilmot-Roussel H, Ragazzon B, Baudry C, Perlemoine K, et al. Identification of a CpG island methylator phenotype in adrenocortical carcinomas. J Clin Endocrinol Metab (2013) 98:E174–84. doi:10.1210/jc.2012-2993
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Assié G, Jouinot A, Bertherat J. The ‘omics’ of adrenocortical tumours for personalized medicine. Nat Rev Endocrinol (2014) 10:215–28. doi:10.1038/nrendo.2013.272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: cancer, adrenal glands, adrenal gland neoplasms, adrenal cortex, genetic
Citation: Lalli E and Figueiredo BC (2015) Pediatric adrenocortical tumors: what they can tell us on adrenal development and comparison with adult adrenal tumors. Front. Endocrinol. 6:23. doi: 10.3389/fendo.2015.00023
Received: 26 December 2014; Accepted: 08 February 2015;
Published online: 18 February 2015.
Edited by:
Antoine Martinez, Centre National de la Recherche Scientifique, FranceReviewed by:
Norifumi Iijima, Yale University School of Medicine, USAYewei Xing, University of Michigan, USA
Copyright: © 2015 Lalli and Figueiredo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Enzo Lalli, Institut de Pharmacologie Moléculaire et Cellulaire CNRS, 660 route des Lucioles – Sophia Antipolis, Valbonne 06560, France e-mail:bmluaW5vQGlwbWMuY25ycy5mcg==;
Bonald C. Figueiredo, Instituto de Pesquisa Pelé Pequeno Principe, Av. Silva Jardim, 1632, Curitiba, Paraná CEP 80250-060, Brazil e-mail:Ym9uYWxkZkB5YWhvby5jb20uYnI=