 Laura W. Bowers
Laura W. Bowers Emily L. Rossi
Emily L. Rossi Ciara H. O’Flanagan
Ciara H. O’Flanagan Linda A. deGraffenried2
Linda A. deGraffenried2 Stephen D. Hursting
Stephen D. Hursting- 1Department of Nutrition, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
- 2Department of Nutritional Sciences, University of Texas at Austin, Austin, TX, USA
Numerous epidemiological and pre-clinical studies have demonstrated that the insulin/insulin-like growth factor (IGF) system plays a key role in the development and progression of several types of cancer. Insulin/IGF signaling, in cooperation with chronic low-grade inflammation, is also an important contributor to the cancer-promoting effects of obesity. However, clinical trials for drugs targeting different components of this system have produced largely disappointing results, possibly due to the lack of predictive biomarker use and problems with the design of combination therapy regimens. With careful attention to the identification of likely patient responders and optimal drug combinations, the outcome of future trials may be improved. Given that insulin/IGF signaling is known to contribute to obesity-associated cancer, further investigation regarding the efficacy of drugs targeting this system and its downstream effectors in the obese patient population is warranted.
Introduction
The insulin and insulin-like growth factor (IGF) signaling system, a key regulator of growth and energy metabolism, is involved in the pathogenesis and progression of numerous malignancies. Population studies have clearly established a link between insulin and IGF-I levels and the risk and prognosis of certain cancers, with much of this research driven by the rapidly growing incidence of obesity and metabolic syndrome across the globe (1–5). While a number of factors likely contribute to the increased cancer risk and cancer-related mortality that accompanies obesity, dysregulated insulin and IGF signaling is thought to play a significant role. Epidemiological evidence linking the insulin/IGF system with cancer has been reinforced by a large body of pre-clinical work in cell culture and animal models that has established many of the mechanisms underlying these associations.
Consequently, enthusiasm regarding the development and testing of pharmaceutical agents targeting the type 1 IGF receptor (IGF-IR) was very high in the 1990s and early 2000s. However, while the early phase clinical trials for these agents showed some promise, large randomized phase III trials failed to demonstrate that the addition of these drugs to a conventional treatment regimen results in a significant clinical benefit (6, 7), and many drug development programs targeting IGF-IR were shut down. Several researchers have argued that these programs were abandoned too quickly, though. They suggest that the poor phase III trial results were due to a number of factors, including the failure to use predictive biomarkers for the identification of probable responders and non-optimal drug combinations and timing (8, 9). With greater attention to these considerations and a better understanding of the complex insulin/IGF system, future trials may have more success. Drugs targeting other components of this system (alone or in combination), including the insulin receptor (IR) as well as various ligands and downstream signaling molecules, may also prove to be more efficacious.
This review will examine how dysregulation of the insulin/IGF system, which occurs with obesity and metabolic syndrome, promotes cancer risk and progression. Several cancer prevention and treatment interventions targeting insulin/IGF signaling or downstream factors will also be described, as well as recent results from trials testing these agents. Finally, following an analysis of the lessons learned from previous clinical trials, we will conclude with a discussion of potential new strategies, including both pharmaceutical and lifestyle interventions. However, in order to understand the links between dysregulated insulin/IGF signaling and cancer as well as the variety of ideas regarding how to most effectively block this signaling, one must first appreciate the system’s complexity. We will thus begin with an overview of the structure and functions of the components of the insulin/IGF system.
The Insulin/IGF Signaling System
Receptors
The IR and IGF-IR are transmembrane tyrosine kinase receptors with a high degree of homology (10). Their functions also partially overlap and can vary depending on cell type. However, the IR is traditionally considered a regulator of metabolism, specifically the storage and release of glucose, protein, and lipids, while the IGF-IR controls whole body and organ growth. Both are heterotetramers composed of two half receptors formed when the receptor gene products are processed to glycosylated alpha and beta subunits that associate together. The extracellular alpha subunit contains the ligand-binding domain while the transmembrane beta subunit possesses the tyrosine kinase domain (11). The exception to this structural homology among the receptors is the type 2 IGF receptor (IGF-IIR), which lacks tyrosine kinase function. This receptor instead serves to clear IGF-II from circulation by binding and internalizing its ligand, which is then subject to lysosomal degradation (12). There are also two forms of the IR that are generated by alternate splicing of exon 11 in the receptor’s gene. IR-A results from the exclusion of exon 11 and has greater mitogenic function than IR-B, which is formed when exon 11 is included and has greater metabolic function. Both IR-A and IGF-IR are ubiquitously expressed in normal adult tissues, but IR-B expression is typically limited to metabolic tissues like the liver, muscle, and adipose (13, 14). Hybrid receptors consisting of a half IR and a half IGF-IR can also form. Given that most cancer cells express both IR and IGF-IR, multiple homo- and heterodimer variations of these receptors may be found in tumors (11). However, gene amplification or mutations resulting in overexpression or ligand-independent activation of the insulin/IGF system receptors are rare (8).
Ligands and Binding Proteins
In addition to the four receptors described above, the insulin/IGF system involves three ligands: insulin, IGF-I, and IGF-II. Insulin is produced by pancreatic beta cells and primarily in response to elevated blood glucose levels. IGF expression is more widespread, but the liver is the predominant site of production. IGF-I expression in the liver is stimulated by growth hormone (GH), but tissue-specific factors also play a role in its regulation elsewhere (8). Both genetic and lifestyle factors impact an individual’s circulating IGF-I levels to an approximately equal degree (15). IGF-II expression is also regulated by hormones and affected by lifestyle factors like obesity (16). Cancer cells can produce the IGFs, so while insulin must travel through circulation to reach a tumor, IGFs have the potential to interact with a cancer cell via autocrine, paracrine, and endocrine mechanisms (17). The insulin/IGF system receptors have varying affinities for these different ligands. While the metabolic IR-B primarily binds insulin only, IR-A binds insulin and IGF-II with equal affinity (18). IGF-IR preferentially binds the IGFs over insulin, but can bind all three. In addition, the IGFs can bind the IGF-IR/IR heterodimers (Figure 1). Ligand binding stimulates the kinase activity of these receptors via transphosphorylation of their beta subunits, resulting in phosphorylation of adaptor proteins, including the IR substrates (IRS 1–6) and Shc. These activate the phosphatidylinositol 3′-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways, which regulate cell proliferation, survival, migration, metabolism, and angiogenesis (11) (Figure 2). Aberrant expression of the IGFs, particularly IGF-II (19), is common in many malignancies and may represent one mechanism by which the tumor stimulates its own growth. In fact, the primary purpose of the IGF-IIR, which lacks a tyrosine kinase domain, may be to counteract excessive IR-A and IGF-IR bioactivity by sequestering IGF-II.
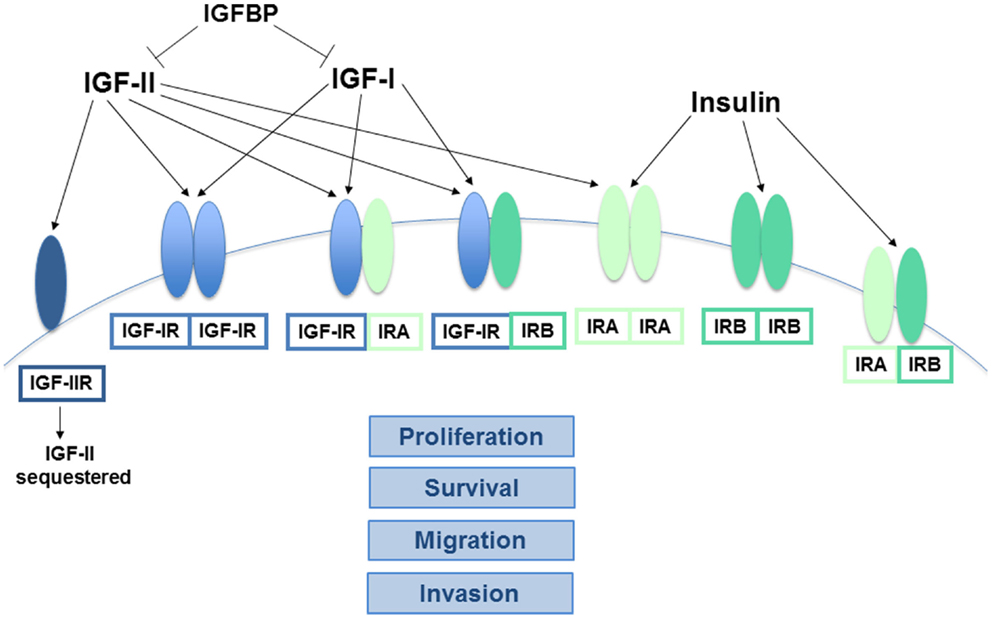
Figure 1. Interactions between the key components of the insulin/IGF system. The receptors of the insulin/IGF system are tetramers comprised of two half receptors, each with an extracellular ligand-binding domain and an intracellular tyrosine kinase domain. IGF-IIR is the exception, as it lacks a kinase domain. Both IGF-IR and IR homodimers and IGF-IR/IR heterodimers can form, depending on the relative abundance of the half receptors. Alternative splicing also results in two different forms of the IR half receptor, IR-A and IR-B. The ligands of this system (insulin, IGF-I, and IGF-II) vary in their affinity for the different receptors. IGF-I primarily binds to homo or heterodimers containing an IGF-IR half receptor, while insulin has the greatest affinity for IR-A and IR-B. In contrast, IGF-II binds IR-A with high affinity and can also bind IGF-IR homo or heterodimers, but binding to IGF-IIR limits its bioavailability. Similarly, the IGFBPs sequester both the IGFs, preventing their ability to bind and activate their cognate receptors. The signaling pathways downstream of the activated IR-A and IGF-IR homodimers are known to stimulate cancer cell proliferation, survival, migration, and invasion, while IR-B is more closely linked to metabolic regulation. The exact functions of the various heteroreceptor combinations have not been clearly defined, but it is likely that receptors containing an IR-A or IGF-IR holoreceptor will modulate cancer growth and metastasis to some degree.
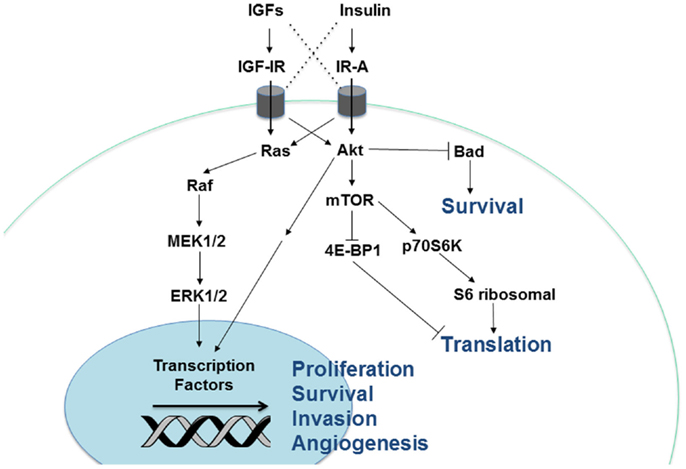
Figure 2. Cellular signaling pathways downstream of the insulin/IGF receptors. Insulin and IGF activate two major signaling pathways, Akt and Ras-MAPK. Stimulation of Akt activates the mTOR signaling complex, leading to greater protein translation. In addition, Akt and Ras-MAPK enhance cellular proliferation, survival, angiogenesis, and invasion via regulation of gene transcription.
Insulin-like growth factor bioactivity also has an additional level of regulation. Six IGF binding proteins (IGFBP) bind both IGF-I and IGF-II with high affinity and protect them from proteases while in circulation, which also effectively prevents their ability to interact with receptors until released (Figure 1). The IGFBPs must deliver the IGFs to a target tissue and dissociate from them via an enzymatic process in order to free them for activity. This suggests that the IGFBPs may act as tumor suppressors by limiting IGF activity, but conflicting study results and the fact that the IGFBPs are also thought to have IGF-independent effects on cancer (20) indicates that their role may be more complicated than originally hypothesized.
Insulin/IGF Connection to Cancer
Epidemiological Evidence
Numerous studies have linked the insulin/IGF system with an increased risk of several cancers as well as a worse cancer prognosis, driving researchers’ interest in the enhancement of our understanding of these signaling pathways and the development of agents targeting them. For example, two prospective studies of postmenopausal women have found that hyperinsulinemia is associated with an elevated risk of breast cancer, though the link was limited to women not receiving hormone replacement therapy in one of the studies (21, 22). A meta-analysis of earlier population studies examining insulin levels and cancer incidence confirmed that hyperinsulinemia and high C-peptide levels (another measure of insulin production) are correlated with an increased risk of breast cancer as well as colorectal and pancreatic cancer (4). The link between serum IGF-I levels and cancer risk has also been investigated extensively. Intriguingly, a nested case-control study conducted within the Nurse’s Health Study found that an elevation in circulating IGF-I is associated with greater breast cancer risk in premenopausal women (23). Subsequent meta-analyses of several studies have substantiated these results (5, 24). Prostate cancer incidence has also been positively correlated with IGF-I in multiple prospective studies, with these findings confirmed by meta-analysis (5, 25, 26). These links appear to be independent of obesity-associated elevations in IGF-I, as obesity does not enhance prostate cancer or premenopausal breast cancer risk. Finally, researchers have demonstrated that both higher IGF-I and lower IGFBP-3 levels are associated with increased colorectal cancer risk in men and women (27, 28). Reports regarding IGFBP-3 levels and breast or prostate cancer risk have been contradictory, with some finding no association and others a positive or negative correlation (5, 23–26). These inconsistencies may be due to methodological issues and the complicated role that the IGFBPs seem to play in tumor growth, as argued by Renehan et al. (29). Regardless of the explanation, the highly variable results indicate that IGFBP levels are likely not an ideal indicator of cancer risk.
Researchers have also examined whether insulin/IGF signaling impacts cancer prognosis and have generally found that circulating insulin levels appear to be more predictive than the IGFs. This may be due to the relative importance of local IGF production and autocrine/paracrine signaling within the tumor. That is, systemic levels of the IGFs may not be as relevant to the growth of an established tumor that is capable of producing its own IGF-I and 2. In contrast, insulin is produced solely by the pancreatic beta cells, except in very rare cases, so serum insulin is a relatively good measure of the level of insulin signaling occurring at the tumor site. In fact, several prospective studies have demonstrated strong positive associations between different measures of insulin signaling and a poor breast cancer prognosis, including an increased risk of distant recurrence and mortality (1, 30, 31). Other investigators have shown similar correlations between high C-peptide levels and increased prostate and colorectal cancer mortality (2, 3). Breast tumor total IR levels have also been analyzed and found to be positively associated with poor survival (32, 33). In contrast, reports regarding the prognostic significance of IGF-IR levels in breast tumors are mixed, with some suggesting that its link to a better or worse outcome may depend on the breast cancer subtype (34–36). The reason(s) for these differences remain unclear. Greater tumor IGF-IR expression has been positively associated with a worse disease outcome for several other forms of cancer, including prostate cancer, gastric cancer, and renal cell carcinoma (37–39).
Pre-Clinical Evidence
This epidemiological research has been accompanied by extensive mechanistic investigations that have established the pro-tumorigenic effects of insulin/IGF signaling. The mitogenic effect of insulin on mammary tumors was first demonstrated more than 40 years ago, when researchers found that insulin-deficiency reduced chemically induced mammary tumor growth in rats (40). Administration of exogenous insulin was also shown to reverse these effects (41). Researchers later determined that insulin has a similar effect on the growth of other forms of cancer (42, 43). These early findings may not have been pursued further because it was assumed that insulin signaling could not be targeted without the stimulation of unacceptable metabolic side effects. However, there has been renewed interest recently in the role of insulin in carcinogenesis, possibly due to the realization that resistance to IGF-IR inhibitors may be driven by continued IR signaling. In addition, small-molecule IR/IGF-IR inhibitors have been better tolerated metabolically than anticipated, suggesting that it is possible to target IR signaling without triggering hyperglycemia (8). In a recent series of papers, LeRoith and colleagues utilized a mouse model of genetically induced insulin resistance that develops hyperinsulinemia without obesity to examine how insulin signaling impacts mammary tumor growth in vivo. Using multiple models of mammary carcinoma, these investigators demonstrated that hyperinsulinemia promotes mammary tumor growth and pulmonary metastasis and that treatment with an insulin sensitizer or a small-molecule IR/IGF-IR inhibitor can attenuate these effects (44–47). Zhang et al. (48) have also shown that silencing the IR using shRNA blocks breast cancer cells’ ability to form pulmonary metastases in vivo. These results clearly illustrate the important role that insulin signaling can play in cancer growth and invasion.
Early experiments also validated the hypothesis that IGF-I signaling promotes tumorigenesis as well as cancer growth and invasion. In fact, IGF-IR expression was found to be necessary for the transforming action of the simian virus 40 large tumor antigen as well as numerous other oncogenes (49, 50). With these findings and indications that IGF-I stimulates cancer cell proliferation and metastasis (51–54), interest in this pathway as a potential pharmaceutical target grew. Animal models employing mutations that reduce circulating IGF-I have also confirmed that this growth factor does play a significant role in both mammary and prostate tumor growth in vivo (55, 56). IGF-IR monoclonal antibodies were the first IGF-I signaling drugs to be developed, and the pre-clinical studies testing these produced promising results that further reinforced earlier conclusions regarding this pathway’s important impact on tumorigenesis. An early study from Arteaga et al. (57) demonstrated that an IGF-IR monoclonal antibody inhibits the growth of human triple negative breast cancer cell xenografts in athymic mice. Other monoclonal antibodies targeting this receptor were later shown to inhibit the growth of multiple forms of cancer in vivo, including breast, pancreatic, renal, lung, and colon (58, 59). IGF-IR and dual IR/IGF-IR tyrosine kinase inhibitors (TKIs) have also been found to significantly attenuate the growth of several cancer cell lines (60–62). Overall, the pre-clinical evidence strongly supports the hypothesis that insulin/IGF signaling promotes the progression of numerous cancer types. The continued rise in global rates of obesity, which is typically accompanied by elevations in systemic insulin and IGF-I levels, suggests that efforts to develop pharmaceutical agents that reduce this signaling for cancer prevention and treatment are particularly warranted. The role that elevated or dysregulated insulin/IGF signaling, like that seen with obesity and other conditions, may play in the development of cancer will be discussed further below.
Clinical Trial Evidence
Numerous pharmaceutical agents designed to specifically target the insulin/IGF system have been developed and tested for their tumor-inhibiting effects (Table 1). These can be divided into two general categories: receptor-targeting agents and drugs that reduce ligand bioactivity. The first includes both anti-receptor antibodies and small-molecule TKIs (Figure 3). Many of the agents that performed well in pre-clinical studies and early clinical trials have been further tested in phase II and III trials, with largely disappointing results that led to the discontinuation of many insulin/IGF-targeting programs. However, several experts in the field of insulin/IGF signaling have suggested that these findings do not indicate that this system is a poor cancer treatment target. They argue that these studies suffered from flaws in design, including a failure to use predictive biomarkers and to identify optimal drug combinations based on a clear understanding of insulin/IGF signaling (8, 9). Here, we will review the most current clinical trial results and recent developments based on the lessons learned from these.
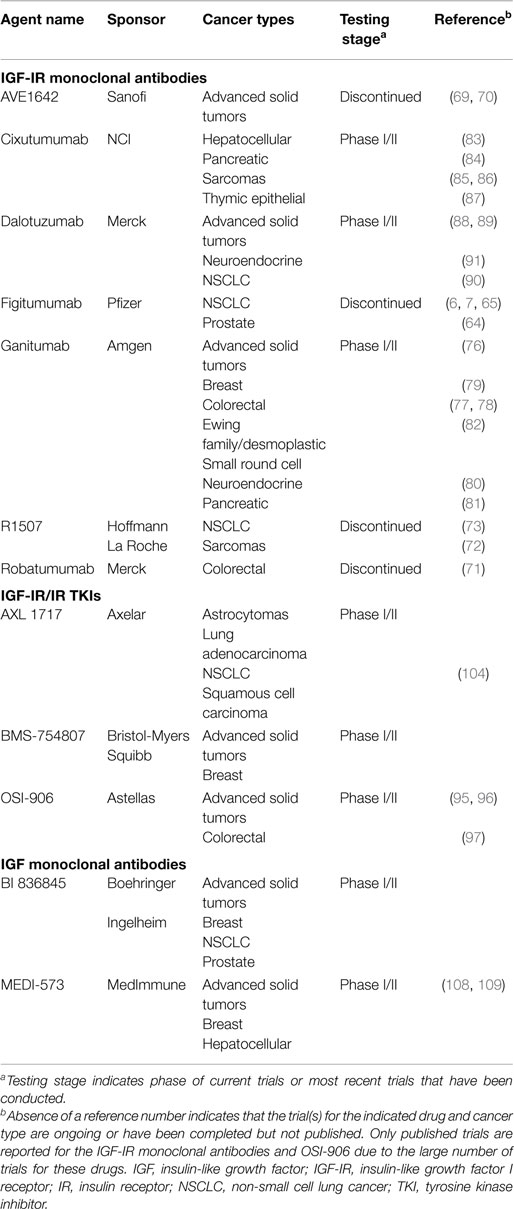
Table 1. Insulin/IGF-targeting drug trials.
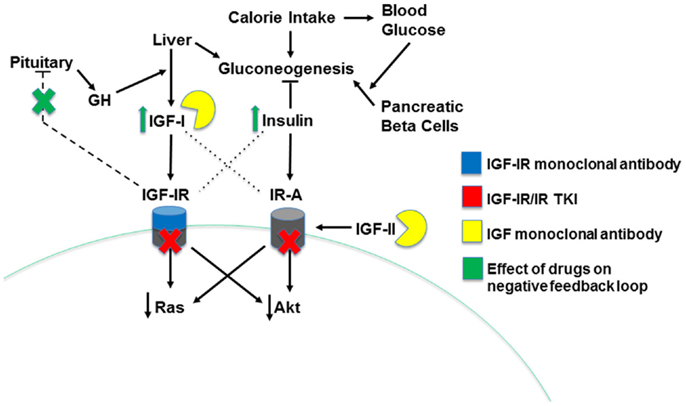
Figure 3. Mechanisms of insulin/IGF system-targeting drugs. The three types of insulin/IGF system-targeting drugs are illustrated. IGF-IR monoclonal antibodies bind IGF-IR, leading to its internalization and degradation. IGF-IR/IR TKI drugs decrease receptor activity by competing for the ATP binding site on the receptor’s kinase domain, blocking transduction of a signal to downstream effectors. Finally, IGF monoclonal antibodies directly bind both IGF-I and IGF-II, preventing them from binding and activating the system’s receptors. Drug-induced decreases in IGF-IR signaling disrupt the pituitary-mediated negative feedback loop regulating IGF-I production, leading to higher IGF-I levels and, indirectly, greater insulin levels.
Receptor-Targeting Agents: IGF-IR Monoclonal Antibodies
These agents prevent ligand activation of the IGF-IR by binding the receptor, which leads to its internalization and degradation. While they do not cross-react with the IR, they can bind and inhibit hybrid receptor activity (63). Figitumumab, an IGF-IR monoclonal antibody produced by Pfizer, is perhaps the most well-known member of this drug class. Despite promising phase II trial data (64, 65), phase III trials examining the combination of figitumumab with cytotoxic chemotherapy or the epidermal growth factor receptor (EGFR) inhibitor erlotinib for non-small cell lung cancer (NSCLC) were terminated early due to the drug’s failure to improve overall survival (OS) and problems with toxicity (6, 7). In addition, some phase II results were later retracted (66), and Pfizer discontinued its development of figitumumab. Unfortunately, several other IGF-IR antibodies have met a similar fate. AVE1642 (Sanofi) inhibited mammary tumor growth and metastases in pre-clinical studies (67, 68) and exhibited anti-tumor activity in phase I trials for patients with advanced solid tumors (69, 70). However, it failed in phase II trials, and its development was discontinued. R1507 (Hoffmann-La Roche) and robatumumab (Merck) also performed poorly in phase II trials (71–73), and several studies were ended early along with the research programs for these drugs.
Some IGF-IR monoclonal antibodies remain in active clinical trials, though. Ganitumab (Amgen), like other drugs of this class, demonstrated promising pre-clinical anti-tumor activity (74, 75), and has moved into phase I and II trials with mixed results. Most of the published phase II trial data have been negative, with no improvement shown with ganitumab treatment for multiple cancer types (76–80). A couple of the phase II trials did produce positive results, though (81, 82), and while no new trials are planned, studies examining this drug’s use in patients with newly diagnosed metastatic Ewing sarcoma, NSCLC, and advanced solid tumors are still actively recruiting. The results from phase II trials for cixutumumab (NCI) have also been mixed, with this agent showing some benefit in patients with thymic epithelial tumors and adipocytic sarcoma, but none in several other cancer types (83–87). Dalotuzumab (Merck) demonstrated anti-tumor activity in pre-clinical and phase I clinical trials of advanced solid tumors (88, 89), but provided no improvement in patient outcome in two phase II studies (90, 91). Both cixutumumab and dalotuzumab remain the subject of a few active phase I and II clinical trials, though, and have been utilized in several completed trials whose results remain unpublished.
Receptor-Targeting Agents: Tyrosine Kinase Inhibitors
One hypothesis regarding the IGF-IR antibodies’ poor performance is that IGF-IR inhibition alone allows IR-A signaling to continue and even significantly increase. This may then promote cancer progression, as tumor IR-A expression and activity have been correlated with worse survival (32, 33). Consequently, excess IR-A signaling may be driving resistance to the IGF-IR antibodies. Researchers have posited that one solution to this problem may be dual inhibition of IGF-IR and IR using TKIs. These agents prevent receptor activity by competing for the ATP-binding site in the catalytic domain of the IR and IGF-IR, which is found on the receptors’ beta subunits. Due to the high degree of beta subunit homology between the two receptors, most of these drugs inhibit both IR and IGF-IR activity. Pre-clinical studies demonstrated that one TKI, OSI-906 (Astellas), exhibits anti-proliferative activity on a variety of cancer cell types and enhances the effects of doxorubicin (92, 93). It also performed better than an IGF-IR inhibitor against tumors with both IR and IGF-IR activation (94). The phase I trial results for this agent have been partially positive, with two recent papers reporting observations of anti-tumor activity and good tolerance in patients with advanced solid tumors (95, 96). However, the combination of OSI-906 and the mTOR inhibitor everolimus did not produce an objective response in metastatic colorectal cancer patients (97). In addition, some trials have been terminated early due to safety concerns and/or patient progression, though it appears that some patients are benefiting from treatment with this TKI, as a rollover study is available for subjects from completed trials. There are also active and completed trials that have not yet published their results. BMS-754807 (Bristol-Myers Squibb) and AXL 1717 (Axelar), are two additional TKIs that produced positive pre-clinical data (98–102). Hou et al. (103) demonstrated synergistic inhibition of tumor growth in a mouse model of postmenopausal estrogen receptor positive (ER+) breast cancer following treatment with BMS-754807 plus letrozole or tamoxifen. A phase II trial examining a BMS-754807 plus letrozole combination in aromatase inhibitor (AI)-resistant breast cancer patients has been completed, along with other phase I and II trials, but results have not yet been published. The absence of any new trials with this agent suggests that little to no benefit was produced, though. AXL 1717, which only targets IGF-IR, was tolerated well in a phase I trial for NSCLC. Patients also had indications of a possible tumor response to the drug (104), but published data from other completed trials involving this drug are also lacking. A phase I/II trial for AXL 1717 in patients with recurrent astrocytomas is still recruiting. Overall, as with the IGF-IR antibodies, the TKIs have largely produced disappointing trial results. However, some of the more recent ongoing or completed trials may have benefited from the lessons learned from the early trial failures, including the need for more rational therapy combination. These lessons and future directions based on what we have learned will be discussed in more detail below, following a review of the ligand-targeting agents.
Prevention of Ligand Activity
Another approach to the inhibition of IR/IGF-IR signaling is utilization of agents that target the ligands of this system. Insulin secretion and action cannot be directly reduced because this would result in unacceptable metabolic side effects. However, IGF-targeting monoclonal antibodies have been tested pre-clinically and in early clinical trials with some success. Because these drugs cross-react with IGF-I and IGF-II, they result in the inhibition of both IGF-IR and IR-A activity (105–107). Two completed phase I trials for MEDI-573 (MedImmune) demonstrated that this agent is well-tolerated and has some anti-tumor activity in patients with advanced solid tumors (108, 109). Additional phase Ib/II trials for MEDI-573 remain active or are completed but unpublished. Several phase I and II trials testing the safety and efficacy of another IGF antibody, BI 836845 (Boehringer Ingelheim), are currently recruiting patients. While it is too early to declare these drugs more successful than the receptor-targeting agents, their combined inhibition of both IR-A and IGF-IR (but not IR-B) activity suggests that they may lead to an improved clinical response with less metabolic toxicity.
Lessons Learned from Clinical Trials
The Need for Biomarkers
Perhaps the most prominent criticism of past phase II and III trials for insulin/IGF-targeting agents is the researchers’ failure to utilize any predictive biomarkers for the selection of likely responders. To be fair, it should be acknowledged that there were no validated biomarkers for sensitivity to these agents at the start of these trials. In fact, there is still no consensus regarding which biomarker(s) to use for patient selection. However, several have now been proposed and tested in pre-clinical studies. In addition, retrospective analysis of patient biospecimens for the identification of biomarkers linked to drug response has occurred following some trials. These candidate biomarkers can be divided into two categories: insulin/IGF system members and other markers connected to the insulin/IGF axis.
Insulin/IGF system biomarkers. One of the first biomarkers proposed was serum IGF-I. Despite the retraction of one study suggesting that elevated pre-treatment free IGF-I levels were associated with NSCLC patient response to figitumumab (110), additional evidence supporting these findings has been published. Figitumumab response in another trial for NSCLC patients was correlated with higher baseline total serum IGF-I concentrations (65). Other IGF-IR monoclonal antibody trials have reported a similar link between circulating IGF levels and drug response (73, 111, 112). In addition, studies utilizing pre-clinical models of multiple forms of cancer have found that response to insulin/IGF-targeting agents is associated with greater IGF-I and IGF-II levels (113–115). While these positive results are encouraging, it is potentially problematic to use systemic IGF levels as biomarkers when tumors are capable of autocrine IGF signaling. Measurement of serum IGFs does not account for local IGF production and could fail to detect tumors that are addicted to autocrine IGF signaling and possibly responsive to insulin/IGF signaling inhibition. While more invasive than a simple blood test, this could be remedied by measuring tumor IGF expression as well as serum levels. Others have also shown that a high IGFBP-5/4 ratio predicted sensitivity to an IR/IGF-IR TKI in a pre-clinical breast cancer model and was correlated with a worse breast cancer outcome, suggesting that this ratio may be indicative of patient response (116). In addition, some studies have reported that tumor IGF-IR expression can predict response (114, 115, 117), but the results have been conflicting (118, 119). Two factors may explain these mixed findings. First, these studies do not account for the possible expression of IR/IGF-IR heteroreceptors, and varying levels of these hybrids versus holoreceptors may also be affecting drug response. Second, there is no standardized protocol for the measurement of tumor IGF-IR expression, so differences in detection methods could contribute to variations in results.
Other biomarkers. The remaining candidate biomarkers include gene signatures indicative of elevated tumor insulin/IGF system activity, signaling molecules connected to the insulin/IGF axis that may mediate drug resistance, and other markers signifying drug response (120). In the last category is a pre-clinical study demonstrating that a reduced 2-deoxy-2-[18F]fluoro-d-glucose positron emission tomography (18FDG-PET) reading following a single dose of the TKI OSI-906 was a marker for cellular sensitivity to this drug (121). This represents a relatively simple method for determining drug response, but may not be practical for trial enrollment since the patient must be started on the drug to measure sensitivity. Others have noted that constitutive Akt and MAPK pathway activation is associated with resistance to the insulin/IGF-targeting drugs (122–124), leading to the idea that the presence of activating mutations in these pathways may be a negative marker for drug sensitivity. However, this idea is confounded by the fact that KRAS mutant NSCLC cells do respond to IGF-IR inhibition (125). This speaks to the complexity of the insulin/IGF signaling system, whose activity can differ by cancer subtype and molecular environment. Finally, an IGF-I gene signature has been identified that was associated with a poor breast cancer prognosis and could be reversed by different anti-IGF-IR agents in the pre-clinical setting. In addition, the gene signature, when combined with measurement of IGF-IR expression, was predictive of IR/IGF-IR TKI sensitivity in breast cancer cells (113, 126). The one clear conclusion that can be drawn from these studies is that more research is needed to test and validate these biomarkers before a consensus can be formed around one candidate.
Optimal Drug Combination
The second major criticism of past trials for the insulin/IGF-targeting drugs is that inadequate consideration was given to determining the optimal combination of agents and timing of their administration. Many of the initial trials used the pragmatic approach of simply adding an insulin/IGF-targeting drug to the current standard of care for randomized patient populations where that standard was not performing adequately. This approach, however does not take into consideration the effect previous treatments can have on the biology of the cancer cell, in particular the emergence of resistant cells. Exposure to chemotherapy will kill the majority of cells, but those which can adapt and persist may do so by derailing signaling pathways and reducing their dependence on systems such as insulin/IGF. There is now abundant pre-clinical and clinical evidence that can be used in the design of future trials to achieve improved efficacy. For example, many trials have examined the impact of combining insulin/IGF-targeting drugs to standard chemotherapy regimens, and several phase I trials demonstrated that IGF-IR inhibition is tolerable with standard chemotherapeutic regimens (127, 128). Traditional chemotherapy, in particular platinum-based compounds, targets rapidly dividing cells primarily by inducing DNA damage. Pre-clinical studies suggest IR/IGF-IR signaling protects cells from DNA damage and induces DNA damage repair via non-homologous end joining (NHEJ) and homologous repair (129, 130). Consistent with this, inhibition of IGF-IR causes sensitization to cisplatin (98, 131, 132), doxorubicin, and trabectedin (133), as well as ionizing radiation (129, 134–136) in ovarian, prostate, colon, and breast cancer cells and in mouse xenograft models. Consequently, it may be most beneficial to combine insulin/IGF-targeting drugs with DNA-damaging chemotherapy agents versus those with an alternate cytotoxic mechanism, like the taxanes.
In addition, timing of drug administration may be a key factor in the efficacy of anti-insulin/IGF therapies in combination with chemotherapeutic regimens. Pre-clinical data indicate that the most effective inhibition of tumorigenesis occurs when the drug is administered following chemotherapeutic treatment (68, 92). This is likely due to the fact that IGF-IR inhibition blocks cell cycle progression, thereby reducing the pool of cells being targeted by the chemotherapy. The timing of drug administration was not incorporated into the design of the failed clinical trials. Furthermore, the half-life of the inhibitor should be taken into consideration, as monoclonal antibodies with extended activity may still affect the chemotherapy given in the next treatment cycle. While the clinical trials focused on insulin/IGF system inhibition as a monotherapy or in combination with standard chemotherapy, the effect of inhibition of the system in conjunction with radiotherapy was not addressed. From the abundant pre-clinical data on IGF-IR and DNA damage and repair, such a combination would be appropriate in future clinical trials. As with chemotherapy combinations, certain considerations should be taken in the design of such clinical trials, such as resistance to radiation, timing of therapies, and predictive biomarkers of response. IGF-IR expression may be a useful predictive biomarker for radiation response, as cervical cancer patients with high levels of IGF-IR have a 28.6-fold greater risk of treatment failure (137).
Additional drug combinations have generally been chosen based on IGF-IR’s ability to mediate resistance to inhibitors of other receptor tyrosine kinase (RTK) and hormone signaling pathways. These other pathways can also be upregulated in response to IGF-IR inhibition. The most obvious example of this reciprocal interaction is the increase in IR activity that can occur following IGF-IR inhibition. Reduced IGF-IR signaling results in a pituitary feedback loop that stimulates increased GH secretion, resulting in elevated IGF-I production and hyperinsulinemia (Figure 3). These side effects have been evident in many clinical trials for IGF-IR-targeting agents (89, 138, 139). Insulin, IGF-II, and elevated IGF-I levels can all activate IR-A, driving resistance to IGF-IR antibodies (140–142). Dual IR/IGF-IR TKIs and ligand-targeting antibodies may be able to block this compensatory signaling, leading to improved efficacy over the IGF-IR antibodies (94).
Crosstalk between the insulin/IGF system and ER signaling pathway has been well-established (143–146), and IGF-IR is a known mediator of endocrine therapy resistance in breast cancer (147). The combined inhibition of both pathways has also been shown to be effective pre-clinically (148). Consequently, there was substantial evidence supporting trials examining treatment with IGF-IR antibodies plus endocrine therapy for hormone-resistant breast cancer. The phase II trial results have been disappointing, though, showing no clinical benefit (79). This may be because a complete blockade of insulin/IGF signaling using a TKI or ligand-targeting antibody is needed. Trials investigating the efficacy of an AI plus the TKI BMS-754807 or the IGF antibody MEDI-573 in ER+ breast cancer patients are completed and ongoing, respectively, but no results have been published. Pre-clinical evidence indicates that tamoxifen-resistance is associated with decreased breast cancer cell IGF-IR expression (149, 150), and this would likely reduce the effectiveness of a tamoxifen plus IGF-IR antibody combination treatment. The addition of an insulin/IGF-targeting agent to androgen deprivation therapy for prostate cancer is also supported by pre-clinical evidence, as insulin has been shown to promote androgen production by prostate cancer cells, possibly hastening the development of castration-resistant prostate cancer (151). There are ongoing trials examining the combination of anti-hormone medications with the IGF-IR antibody cixutumumab or the IGF antibody BI 836845.
Finally, several trials have combined insulin/IGF-targeting drugs with EGFR inhibitors in a number of different cancers based on pre-clinical studies showing that EGFR signaling mediates resistance to IGF-IR inhibition and vice versa (152–154). But like so many others, the results from these trials have been largely disappointing, with no clinical benefit shown (7, 73, 77, 90). However, these studies may also have suffered from the failure to utilize biomarkers for patient selection. Some recently completed or ongoing trials for an EGFR antibody plus TKI OSI-906 or IGF antibody BI 836845 combination treatment were open only to NSCLC patients with activating EGFR mutations, which may have improved efficacy. No results have been published yet from the completed studies. Promising data have emerged from in vitro and in vivo pre-clinical investigations regarding anti-EGFR/IGF-IR inhibitor “nanobullets,” EGFR nanobody liposomes loaded with the IGF-IR inhibitor AG538. This therapy may aid specificity, but would also benefit from stratification of patients through predictive biomarkers (155).
A number of researchers are trying a new direction in their quest to the overcome resistance to the insulin/IGF system inhibitors, looking to the common pathways found downstream of the RTKs for different targets. Specifically, the PI3K/Akt/mTOR pathway has become the focus of intense interest, as a feedback loop allows Akt signaling to continue despite IGF-IR inhibition (156). Conversely, mTOR inhibition also promotes Akt activation through a feedback loop, but this compensatory mechanism can be blocked via IGF-IR inhibition (157). Phase I and II clinical trials combining insulin/IGF-targeting agents with mTOR inhibitors have produced some promising results (118, 158–160) as well as some negative data indicating no clinical benefit (85). Additional phase II trials are ongoing or have been completed, but remain unpublished. One ongoing trial for advanced solid tumors is comparing a ganitumab and everolimus combination with this dual treatment plus the EGFR antibody panitumumab. It appears that many of the current trials are utilizing more carefully considered treatment regimens and screening participants for biomarkers. It remains to be seen whether this translates into better response rates. Interesting pre-clinical data also exist for the combination of insulin/IGF system inhibitors with other drugs, including the lipid modulator simvastatin in prostate cancer (161), the VEGF antibody bevacizumab in ovarian cancer (162), and methyl jasmonate in endometrial cancer (163).
Energy Balance, Insulin/IGF-I, and Cancer
Role of Insulin and IGF-I in the Obesity-Cancer Link
The prevalence of obesity, defined as having a body mass index [BMI, body weight (in kilograms) divided by height (in meters) squared] ≥30 kg/m2, has tripled in the past 50 years in the United States (US). Today, nearly 40% of adults and 20% of American children are considered obese (164). Worldwide, an estimated 750 million people are currently obese (165). Among obese adults, approximately 60% meet the criteria for the metabolic syndrome, a state of metabolic dysregulation characterized by insulin resistance, hyperglycemia, hypertension, and dyslipidemia (166). The hyperinsulinemia induced by insulin resistance is a hallmark of obesity and/or metabolic syndrome (167), and bioavailable IGF-I also increases in the obese state, possibly via hyperglycemia-induced suppression of IGFBP synthesis and/or hyperinsulinemia-induced promotion of hepatic GH receptor expression and IGF-I synthesis (168). Through these mediators, obesity and metabolic syndrome are linked to various chronic diseases, including cardiovascular disease, type II diabetes, and the focus of this review, cancer.
The American Society of Clinical Oncology’s recent position statement on obesity and cancer (169) calls obesity the leading preventable cause of cancer in the US and a central challenge to cancer prevention and care. It estimates that by 2030, 500,000 Americans will be diagnosed with obesity-caused cancers each year unless corrective action is taken. Overall, an estimated 20–25% of all cancer deaths in in the US are attributable to overweight and obesity (170).
Insulin/IGF-I, Inflammation, and Cancer
The link between chronic inflammation and cancer development was first noticed more than 100 years ago by Rudolph Virchow, who observed an abundance of leukocytes in neoplastic tissue (171). Now, several tissue-specific inflammatory lesions are established neoplastic precursors for invasive cancer, including gastritis for gastric cancer, inflammatory bowel disease for colon cancer, and pancreatitis for pancreatic cancer (172, 173). In addition to elevated levels of circulating insulin and IGF-I, obesity and metabolic syndrome are associated with a low-grade, chronic (smoldering) state of inflammation characterized by increased circulating free fatty acids and the chemoattraction of immune cells, like macrophages, into the local adipose tissue milieu (174–176). These effects are further amplified by the immune cells’ release of inflammatory cytokines, including interleukin (IL)-1β, IL-6, TNF-α, and monocyte chemoattractant protein (MCP)-1. Adipocytes can enlarge past the point of effective oxygen diffusion, which results in hypoxia and eventually necrosis. Dannenberg and colleagues have established that crown-like structures, rings of activated macrophages surrounding these dead or dying adipocytes, are common in the adipose tissue of obese subjects and are important contributors to the proinflammatory and pro-cancer effects of obesity (177, 178). In addition, free fatty acids escape the engorged/necrotic adipocytes and deposit in other tissues, which in turn promotes insulin resistance and diabetes (through downregulation of IRs and glucose transporters), hypertension, and fatty liver disease. Fatty acid deposition also activates signaling molecules involved in epithelial carcinogenesis, including NF-κB (174–176). This transcription factor is activated in response to bacterial and viral stimuli, growth factors, and inflammatory molecules (e.g., TNF-α, IL-6, and IL-1β) and is responsible for inducing the expression of genes associated with cell proliferation, survival, angiogenesis, metastasis, and further inflammation. Activation of NF-κB is a common characteristic of many tumors and has been associated with insulin resistance and elevated circulating levels of insulin and/or IGF-I (176, 179–181). In summary, there is close reciprocal relationship between obesity-associated elevations in insulin/IGF and inflammatory signaling, such that both factors should be considered in the development of interventions to improve cancer prevention and treatment in the obese patient population (Figure 4).
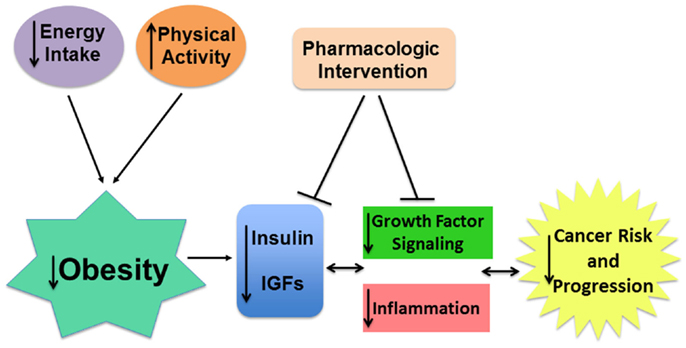
Figure 4. Targeting insulin/IGF signaling may attenuate the pro-tumor effects of obesity. Obesity is associated with higher insulin and bioavailable IGF-I levels which, in cooperation with chronic inflammation, contributes to greater risk and progression of many cancers. The pro-tumor effects of obesity can be mitigated directly by obesity reversal using calorie restriction and increased physical activity. Pharmaceutical interventions targeting obesity-associated insulin/IGF and inflammatory signaling or their downstream effectors may also improve patient risk and outcome.
Alternative Pharmaceutical Targets
In addition to pharmacological agents targeting insulin/IGFRs or ligands, including emerging work on microRNA-based approaches (182), a wide variety of natural agents with demonstrated cancer chemopreventive or chemotherapeutic activity have recently been reported to target components of the insulin/IGF pathway (183). These agents, which likely exert only modest inhibitory effects on insulin/IGFR activity, may provide a promising and safe approach, especially if effective combinations can be identified, for breaking the obesity-cancer link.
Pharmacological mTOR inhibitors have emerged as lead candidates for so-called calorie restriction (CR) mimetics, agents that mimic the anti-cancer or anti-aging effects of CR without the restriction of dietary energy intake. Rapamycin treatment extends lifespan and delays cancer in mice, providing additional support for mTOR as a target for mimicking the effects of CR (184). We have shown that rapamycin or its analog, Afinitor® (everolimus), can offset the obesity-associated increased growth of mammary or pancreatic tumors (185–187). Rapamycin and the so-called rapalogs are potent inhibitors of mTOR complex 1, but chronic rapamycin exposure has been linked in some studies to disruption of mTOR complex 2 signaling, resulting in impaired glucose tolerance and insulin action (188). Thus, while inhibiting mTOR complex 1 appears to be a good strategy for mimicking many of the anti-cancer effects of CR, the search for agents that can do so without disrupting mTOR complex 2 signaling is ongoing.
Metformin, a biguanide commonly used to treat type 2 diabetes, is an mTOR-inhibiting drug with great promise as a CR mimetic that overcomes the concerns about glucose intolerance associated with rapamycin/rapalogs. It inhibits gluconeogenesis through indirect activation of AMPK in the liver and may also exert direct effects on AMPK in cancer cells. Administration of metformin suppresses tumor development and/or growth in multiple experimental models, including colon, mammary, and hematopoietic cancer models (189). Epidemiological studies have suggested that type 2 diabetic patients treated with metformin have lower risk of developing or dying from cancer relative to diabetic patients receiving sulfonylureas, insulin, or other therapies (190–192). A randomized trial is now underway to evaluate the effect of metformin on breast cancer recurrence (193). Phenformin, another biguanide that has been abandoned for diabetes therapy due to its toxicity from lactic acidosis, is a more potent AMPK inhibitor than metformin and may also have some potential as a CR mimetic at lower, non-toxic doses (189).
In addition to these pharmaceutical strategies, dietary modulation may also be useful in controlling the high serum IGF-I and insulin levels present in obese cancer patients. While CR is restrictive and difficult to employ, low carbohydrate/high fat diets such as the ketogenic diet can mimic CR in many ways (194) and may be a more amenable dietary alteration for obese individuals at risk for, or diagnosed with, cancer. Low carbohydrate/high fat diets rewire energy metabolism to utilize ketones derived from fatty acids, in particular medium chain triglycerides (MCTs), as an energy source rather than glucose. The ketogenic diet has long been used successfully as a means to reduce epileptic seizures (195) and more recently to regulate blood glucose in type 2 diabetes (196), and studies have shown that the diet has no adverse effects in cancer patients either as an adjuvant or monotherapy (197). Pre-clinical studies using the ketogenic diet have shown promising results in reducing tumor growth in mammary (198), prostate (199, 200), brain (201), and gastric cancer models (202), and it has been shown to promote response to adjuvant therapy (197, 203). In addition to restricting glucose as fuel for tumors, the tumor suppressive effect of the ketogenic diet appears to be mediated through reduction of serum IGF-I and insulin levels via GH resistance (204). Intriguingly, a switch to a low carbohydrate diet can also prevent cachexia in patients undergoing chemotherapy, assisting in the retention of lean body mass (205, 206). The application of low carbohydrate/high fat diets is a promising strategy not only for obesity reversal, but also as a potential treatment in conjunction with normal anti-cancer therapies.
Conclusion
Insulin and IGF signaling play an important role in the development and progression of many cancers, as they can promote tumor cell proliferation, survival, migration, and invasion as well as angiogenesis. However, despite strong pre-clinical support for their efficacy, most clinical trials testing inhibitors of the insulin/IGF system have produced disappointing results. By increasing the use of validated predictive biomarkers and optimized drug combination treatments, trial outcomes may be improved in the future. Researchers should consider specifically focusing on whether drugs targeting the insulin/IGF system and its downstream signaling molecules may particularly benefit obese cancer patients, who generally have higher circulating insulin and IGF-I levels as well as a greater risk of treatment failure and cancer mortality. There is also a need for more research regarding cancer prevention interventions that counteract the effects of obesity-related elevations in insulin/IGF signaling. Together, these initiatives may lead to a significant reduction in the burden of obesity on cancer risk and mortality.
Author Contributions
LB, ER, COF, LD, and SH contributed to the writing and revision of this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Irwin ML, Duggan C, Wang CY, Smith AW, McTiernan A, Baumgartner RN, et al. Fasting C-peptide levels and death resulting from all causes and breast cancer: the health, eating, activity, and lifestyle study. J Clin Oncol (2011) 29:47–53. doi: 10.1200/JCO.2010.28.4752
2. Wolpin BM, Meyerhardt JA, Chan AT, Ng K, Chan JA, Wu K, et al. Insulin, the insulin-like growth factor axis, and mortality in patients with nonmetastatic colorectal cancer. J Clin Oncol (2009) 27:176–85. doi:10.1200/JCO.2008.17.9945
3. Ma J, Li H, Giovannucci E, Mucci L, Qiu W, Nguyen PL, et al. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: a long-term survival analysis. Lancet Oncol (2008) 9:1039–47. doi:10.1016/S1470-2045(08)70235-3
4. Pisani P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch Physiol Biochem (2008) 114:63–70. doi:10.1080/13813450801954451
5. Renehan AG, Zwahlen M, Minder C, O’Dwyer ST, Shalet SM, Egger M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: systematic review and meta-regression analysis. Lancet (2004) 363:1346–53. doi:10.1016/S0140-6736(04)16044-3
6. Langer CJ, Novello S, Park K, Krzakowski M, Karp DD, Mok T, et al. Randomized, phase III trial of first-line figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients with advanced non-small-cell lung cancer. J Clin Oncol (2014) 32:2059–66. doi:10.1200/JCO.2013.54.4932
7. Scagliotti GV, Bondarenko I, Blackhall F, Barlesi F, Hsia TC, Jassem J, et al. Randomized, phase III trial of figitumumab in combination with erlotinib versus erlotinib alone in patients with nonadenocarcinoma nonsmall-cell lung cancer. Ann Oncol (2015) 26:497–504. doi:10.1093/annonc/mdu517
8. Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer (2012) 12:159–69. doi:10.1038/nrc3215
9. Yee D. Insulin-like growth factor receptor inhibitors: baby or the bathwater? J Natl Cancer Inst (2012) 104:975–81. doi:10.1093/jnci/djs258
10. Massague J, Czech MP. The subunit structures of two distinct receptors for insulin-like growth factors I and II and their relationship to the insulin receptor. J Biol Chem (1982) 257:5038–45.
11. Belfiore A. The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr Pharm Des (2007) 13:671–86. doi:10.2174/138161207780249173
12. Kornfeld S. Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu Rev Biochem (1992) 61:307–30. doi:10.1146/annurev.bi.61.070192.001515
13. Mosthaf L, Grako K, Dull TJ, Coussens L, Ullrich A, McClain DA. Functionally distinct insulin receptors generated by tissue-specific alternative splicing. EMBO J (1990) 9:2409–13.
14. Moller DE, Yokota A, Caro JF, Flier JS. Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol Endocrinol (1989) 3:1263–9. doi:10.1210/mend-3-8-1263
15. Harrela M, Koistinen H, Kaprio J, Lehtovirta M, Tuomilehto J, Eriksson J, et al. Genetic and environmental components of interindividual variation in circulating levels of IGF-I, IGF-II, IGFBP-1, and IGFBP-3. J Clin Invest (1996) 98:2612–5. doi:10.1172/JCI119081
16. Livingstone C, Borai A. Insulin-like growth factor-II: its role in metabolic and endocrine disease. Clin Endocrinol (Oxf) (2014) 80:773–81. doi:10.1111/cen.12446
17. Clemmons DR. Modifying IGF1 activity: an approach to treat endocrine disorders, atherosclerosis and cancer. Nat Rev Drug Discov (2007) 6:821–33. doi:10.1038/nrd2359
18. Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol (1999) 19:3278–88.
19. Zhang L, Zhou W, Velculescu VE, Kern SE, Hruban RH, Hamilton SR, et al. Gene expression profiles in normal and cancer cells. Science (1997) 276:1268–72. doi:10.1126/science.276.5316.1268
20. Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev (2002) 23:824–54. doi:10.1210/er.2001-0033
21. Gunter MJ, Hoover DR, Yu H, Wassertheil-Smoller S, Rohan TE, Manson JE, et al. Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J Natl Cancer Inst (2009) 101:48–60. doi:10.1093/jnci/djp159
22. Kabat GC, Kim M, Caan BJ, Chlebowski RT, Gunter MJ, Ho GY, et al. Repeated measures of serum glucose and insulin in relation to postmenopausal breast cancer. Int J Cancer (2009) 125:2704–10. doi:10.1002/ijc.24609
23. Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet (1998) 351:1393–6. doi:10.1016/S0140-6736(97)10384-1
24. Shi R, Yu H, McLarty J, Glass J. IGF-I and breast cancer: a meta-analysis. Int J Cancer (2004) 111:418–23. doi:10.1002/ijc.20233
25. Harman SM, Metter EJ, Blackman MR, Landis PK, Carter HB. Baltimore longitudinal study on A. Serum levels of insulin-like growth factor I (IGF-I), IGF-II, IGF-binding protein-3, and prostate-specific antigen as predictors of clinical prostate cancer. J Clin Endocrinol Metab (2000) 85:4258–65. doi:10.1210/jcem.85.11.6990
26. Chan JM, Stampfer MJ, Giovannucci E, Gann PH, Ma J, Wilkinson P, et al. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science (1998) 279:563–6. doi:10.1126/science.279.5350.563
27. Giovannucci E, Pollak MN, Platz EA, Willett WC, Stampfer MJ, Majeed N, et al. A prospective study of plasma insulin-like growth factor-1 and binding protein-3 and risk of colorectal neoplasia in women. Cancer Epidemiol Biomarkers Prev (2000) 9:345–9.
28. Ma J, Pollak MN, Giovannucci E, Chan JM, Tao Y, Hennekens CH, et al. Prospective study of colorectal cancer risk in men and plasma levels of insulin-like growth factor (IGF)-I and IGF-binding protein-3. J Natl Cancer Inst (1999) 91:620–5. doi:10.1093/jnci/91.7.620
29. Renehan AG, Harvie M, Howell A. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and breast cancer risk: eight years on. Endocr Relat Cancer (2006) 13:273–8. doi:10.1677/erc.1.01219
30. Duggan C, Irwin ML, Xiao L, Henderson KD, Smith AW, Baumgartner RN, et al. Associations of insulin resistance and adiponectin with mortality in women with breast cancer. J Clin Oncol (2011) 29:32–9. doi:10.1200/JCO.2009.26.4473
31. Goodwin PJ, Ennis M, Pritchard KI, Trudeau ME, Koo J, Madarnas Y, et al. Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J Clin Oncol (2002) 20:42–51. doi:10.1200/JCO.20.1.42
32. Law JH, Habibi G, Hu K, Masoudi H, Wang MY, Stratford AL, et al. Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res (2008) 68:10238–46. doi:10.1158/0008-5472.CAN-08-2755
33. Mathieu MC, Clark GM, Allred DC, Goldfine ID, Vigneri R. Insulin receptor expression and clinical outcome in node-negative breast cancer. Proc Assoc Am Physicians (1997) 109:565–71.
34. Yerushalmi R, Gelmon KA, Leung S, Gao D, Cheang M, Pollak M, et al. Insulin-like growth factor receptor (IGF-1R) in breast cancer subtypes. Breast Cancer Res Treat (2012) 132:131–42. doi:10.1007/s10549-011-1529-8
35. Fu P, Ibusuki M, Yamamoto Y, Hayashi M, Murakami K, Zheng S, et al. Insulin-like growth factor-1 receptor gene expression is associated with survival in breast cancer: a comprehensive analysis of gene copy number, mRNA and protein expression. Breast Cancer Res Treat (2011) 130:307–17. doi:10.1007/s10549-011-1605-0
36. Hartog H, Horlings HM, van der Vegt B, Kreike B, Ajouaou A, van de Vijver MJ, et al. Divergent effects of insulin-like growth factor-1 receptor expression on prognosis of estrogen receptor positive versus triple negative invasive ductal breast carcinoma. Breast Cancer Res Treat (2011) 129:725–36. doi:10.1007/s10549-010-1256-6
37. Zu K, Martin NE, Fiorentino M, Flavin R, Lis RT, Sinnott JA, et al. Protein expression of PTEN, insulin-like growth factor I receptor (IGF-IR), and lethal prostate cancer: a prospective study. Cancer Epidemiol Biomarkers Prev (2013) 22:1984–93. doi:10.1158/1055-9965.EPI-13-0349
38. Sichani MM, Yazdi FS, Moghaddam NA, Chehrei A, Kabiri M, Naeimi A, et al. Prognostic value of insulin-like growth factor-I receptor expression in renal cell carcinoma. Saudi J Kidney Dis Transpl (2010) 21:69–74.
39. Matsubara J, Yamada Y, Hirashima Y, Takahari D, Okita NT, Kato K, et al. Impact of insulin-like growth factor type 1 receptor, epidermal growth factor receptor, and HER2 expressions on outcomes of patients with gastric cancer. Clin Cancer Res (2008) 14:3022–9. doi:10.1158/1078-0432.CCR-07-1898
40. Heuson JC, Legros N. Influence of insulin deprivation on growth of the 7,12-dimethylbenz(a)anthracene-induced mammary carcinoma in rats subjected to alloxan diabetes and food restriction. Cancer Res (1972) 32:226–32.
41. Heuson JC, Legros N, Heimann R. Influence of insulin administration on growth of the 7,12-dimethylbenz(a)anthracene-induced mammary carcinoma in intact, oophorectomized, and hypophysectomized rats. Cancer Res (1972) 32:233–8.
42. Tran TT, Medline A, Bruce WR. Insulin promotion of colon tumors in rats. Cancer Epidemiol Biomarkers Prev (1996) 5:1013–5.
43. Sharon R, Pillemer G, Ish-Shalom D, Kalman R, Ziv E, Berry EM, et al. Insulin dependence of murine T-cell lymphoma. II. Insulin-deficient diabetic mice and mice fed low-energy diet develop resistance to lymphoma growth. Int J Cancer (1993) 53:843–9. doi:10.1002/ijc.2910530523
44. Ferguson RD, Gallagher EJ, Cohen D, Tobin-Hess A, Alikhani N, Novosyadlyy R, et al. Hyperinsulinemia promotes metastasis to the lung in a mouse model of Her2-mediated breast cancer. Endocr Relat Cancer (2013) 20:391–401. doi:10.1530/ERC-12-0333
45. Ferguson RD, Novosyadlyy R, Fierz Y, Alikhani N, Sun H, Yakar S, et al. Hyperinsulinemia enhances c-Myc-mediated mammary tumor development and advances metastatic progression to the lung in a mouse model of type 2 diabetes. Breast Cancer Res (2012) 14:R8. doi:10.1186/bcr3089
46. Fierz Y, Novosyadlyy R, Vijayakumar A, Yakar S, LeRoith D. Insulin-sensitizing therapy attenuates type 2 diabetes-mediated mammary tumor progression. Diabetes (2010) 59:686–93. doi:10.2337/db09-1291
47. Novosyadlyy R, Lann DE, Vijayakumar A, Rowzee A, Lazzarino DA, Fierz Y, et al. Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Res (2010) 70:741–51. doi:10.1158/0008-5472.CAN-09-2141
48. Zhang H, Fagan DH, Zeng X, Freeman KT, Sachdev D, Yee D. Inhibition of cancer cell proliferation and metastasis by insulin receptor downregulation. Oncogene (2010) 29:2517–27. doi:10.1038/onc.2010.17
49. Baserga R. The IGF-I receptor in cancer research. Exp Cell Res (1999) 253:1–6. doi:10.1006/excr.1999.4667
50. Sell C, Rubini M, Rubin R, Liu JP, Efstratiadis A, Baserga R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc Natl Acad Sci U S A (1993) 90:11217–21. doi:10.1073/pnas.90.23.11217
51. Sachdev D, Hartell JS, Lee AV, Zhang X, Yee D. A dominant negative type I insulin-like growth factor receptor inhibits metastasis of human cancer cells. J Biol Chem (2004) 279:5017–24. doi:10.1074/jbc.M305403200
52. Dunn SE, Ehrlich M, Sharp NJ, Reiss K, Solomon G, Hawkins R, et al. A dominant negative mutant of the insulin-like growth factor-I receptor inhibits the adhesion, invasion, and metastasis of breast cancer. Cancer Res (1998) 58:3353–61.
53. Long L, Rubin R, Baserga R, Brodt P. Loss of the metastatic phenotype in murine carcinoma cells expressing an antisense RNA to the insulin-like growth factor receptor. Cancer Res (1995) 55:1006–9.
54. Myal Y, Shiu RP, Bhaumick B, Bala M. Receptor binding and growth-promoting activity of insulin-like growth factors in human breast cancer cells (T-47D) in culture. Cancer Res (1984) 44:5486–90.
55. Majeed N, Blouin MJ, Kaplan-Lefko PJ, Barry-Shaw J, Greenberg NM, Gaudreau P, et al. A germ line mutation that delays prostate cancer progression and prolongs survival in a murine prostate cancer model. Oncogene (2005) 24:4736–40. doi:10.1038/sj.onc.1208572
56. Wu Y, Cui K, Miyoshi K, Hennighausen L, Green JE, Setser J, et al. Reduced circulating insulin-like growth factor I levels delay the onset of chemically and genetically induced mammary tumors. Cancer Res (2003) 63:4384–8.
57. Arteaga CL, Kitten LJ, Coronado EB, Jacobs S, Kull FC Jr, Allred DC, et al. Blockade of the type I somatomedin receptor inhibits growth of human breast cancer cells in athymic mice. J Clin Invest (1989) 84:1418–23. doi:10.1172/JCI114315
58. Rowinsky EK, Youssoufian H, Tonra JR, Solomon P, Burtrum D, Ludwig DL. IMC-A12, a human IgG1 monoclonal antibody to the insulin-like growth factor I receptor. Clin Cancer Res (2007) 13:5549s–55s. doi:10.1158/1078-0432.CCR-07-1109
59. Burtrum D, Zhu Z, Lu D, Anderson DM, Prewett M, Pereira DS, et al. A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res (2003) 63:8912–21.
60. Ji QS, Mulvihill MJ, Rosenfeld-Franklin M, Cooke A, Feng L, Mak G, et al. A novel, potent, and selective insulin-like growth factor-I receptor kinase inhibitor blocks insulin-like growth factor-I receptor signaling in vitro and inhibits insulin-like growth factor-I receptor dependent tumor growth in vivo. Mol Cancer Ther (2007) 6:2158–67. doi:10.1158/1535-7163.MCT-07-0070
61. Haluska P, Carboni JM, Loegering DA, Lee FY, Wittman M, Saulnier MG, et al. In vitro and in vivo antitumor effects of the dual insulin-like growth factor-I/insulin receptor inhibitor, BMS-554417. Cancer Res (2006) 66:362–71. doi:10.1158/0008-5472.CAN-05-1107
62. Garcia-Echeverria C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J, et al. In vivo antitumor activity of NVP-AEW541-A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell (2004) 5:231–9. doi:10.1016/S1535-6108(04)00051-0
63. Pandini G, Wurch T, Akla B, Corvaia N, Belfiore A, Goetsch L. Functional responses and in vivo anti-tumour activity of h7C10: a humanised monoclonal antibody with neutralising activity against the insulin-like growth factor-1 (IGF-1) receptor and insulin/IGF-1 hybrid receptors. Eur J Cancer (2007) 43:1318–27. doi:10.1016/j.ejca.2007.03.009
64. Chi KN, Gleave ME, Fazli L, Goldenberg SL, So A, Kollmannsberger C, et al. A phase II pharmacodynamic study of preoperative figitumumab in patients with localized prostate cancer. Clin Cancer Res (2012) 18:3407–13. doi:10.1158/1078-0432.CCR-12-0482
65. Goto Y, Sekine I, Tanioka M, Shibata T, Tanai C, Asahina H, et al. Figitumumab combined with carboplatin and paclitaxel in treatment-naive Japanese patients with advanced non-small cell lung cancer. Invest New Drugs (2012) 30:1548–56. doi:10.1007/s10637-011-9715-4
66. Karp DD, Paz-Ares LG, Novello S, Haluska P, Garland L, Cardenal F, et al. Phase II study of the anti-insulin-like growth factor type 1 receptor antibody CP-751,871 in combination with paclitaxel and carboplatin in previously untreated, locally advanced, or metastatic non-small-cell lung cancer. J Clin Oncol (2009) 27:2516–22. doi:10.1200/JCO.2008.19.9331
67. Sachdev D, Zhang X, Matise I, Gaillard-Kelly M, Yee D. The type I insulin-like growth factor receptor regulates cancer metastasis independently of primary tumor growth by promoting invasion and survival. Oncogene (2010) 29:251–62. doi:10.1038/onc.2009.316
68. Zeng X, Sachdev D, Zhang H, Gaillard-Kelly M, Yee D. Sequencing of type I insulin-like growth factor receptor inhibition affects chemotherapy response in vitro and in vivo. Clin Cancer Res (2009) 15:2840–9. doi:10.1158/1078-0432.CCR-08-1401
69. Macaulay VM, Middleton MR, Protheroe AS, Tolcher A, Dieras V, Sessa C, et al. Phase I study of humanized monoclonal antibody AVE1642 directed against the type 1 insulin-like growth factor receptor (IGF-1R), administered in combination with anticancer therapies to patients with advanced solid tumors. Ann Oncol (2013) 24:784–91. doi:10.1093/annonc/mds511
70. Soria JC, Massard C, Lazar V, Ozoux ML, Mery-Mignard D, Deslandes A, et al. A dose finding, safety and pharmacokinetic study of AVE1642, an anti-insulin-like growth factor-1 receptor (IGF-1R/CD221) monoclonal antibody, administered as a single agent and in combination with docetaxel in patients with advanced solid tumours. Eur J Cancer (2013) 49:1799–807. doi:10.1016/j.ejca.2013.01.003
71. Lin EH, Lenz HJ, Saleh MN, Mackenzie MJ, Knost JA, Pathiraja K, et al. A randomized, phase II study of the anti-insulin-like growth factor receptor type 1 (IGF-1R) monoclonal antibody robatumumab (SCH 717454) in patients with advanced colorectal cancer. Cancer Med (2014) 3:988–97. doi:10.1002/cam4.263
72. Pappo AS, Vassal G, Crowley JJ, Bolejack V, Hogendoorn PC, Chugh R, et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a Sarcoma Alliance for Research Through Collaboration study. Cancer (2014) 120:2448–56. doi:10.1002/cncr.28728
73. Ramalingam SS, Spigel DR, Chen D, Steins MB, Engelman JA, Schneider CP, et al. Randomized phase II study of erlotinib in combination with placebo or R1507, a monoclonal antibody to insulin-like growth factor-1 receptor, for advanced-stage non-small-cell lung cancer. J Clin Oncol (2011) 29:4574–80. doi:10.1200/JCO.2011.36.6799
74. Beltran PJ, Chung YA, Moody G, Mitchell P, Cajulis E, Vonderfecht S, et al. Efficacy of ganitumab (AMG 479), alone and in combination with rapamycin, in Ewing’s and osteogenic sarcoma models. J Pharmacol Exp Ther (2011) 337:644–54. doi:10.1124/jpet.110.178400
75. Beltran PJ, Mitchell P, Chung YA, Cajulis E, Lu J, Belmontes B, et al. AMG 479, a fully human anti-insulin-like growth factor receptor type I monoclonal antibody, inhibits the growth and survival of pancreatic carcinoma cells. Mol Cancer Ther (2009) 8:1095–105. doi:10.1158/1535-7163.MCT-08-1171
76. Tabernero J, Chawla SP, Kindler H, Reckamp K, Chiorean EG, Azad NS, et al. Anticancer activity of the type I insulin-like growth factor receptor antagonist, ganitumab, in combination with the death receptor 5 agonist, conatumumab. Target Oncol (2015) 10:65–76. doi:10.1007/s11523-014-0315-z
77. Van Cutsem E, Eng C, Nowara E, Swieboda-Sadlej A, Tebbutt NC, Mitchell E, et al. Randomized phase Ib/II trial of rilotumumab or ganitumab with panitumumab versus panitumumab alone in patients with wild-type KRAS metastatic colorectal cancer. Clin Cancer Res (2014) 20:4240–50. doi:10.1158/1078-0432.CCR-13-2752
78. Cohn AL, Tabernero J, Maurel J, Nowara E, Sastre J, Chuah BY, et al. A randomized, placebo-controlled phase 2 study of ganitumab or conatumumab in combination with FOLFIRI for second-line treatment of mutant KRAS metastatic colorectal cancer. Ann Oncol (2013) 24:1777–85. doi:10.1093/annonc/mdt057
79. Robertson JF, Ferrero JM, Bourgeois H, Kennecke H, de Boer RH, Jacot W, et al. Ganitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: a randomised, controlled, double-blind, phase 2 trial. Lancet Oncol (2013) 14:228–35. doi:10.1016/S1470-2045(13)70026-3
80. Strosberg JR, Chan JA, Ryan DP, Meyerhardt JA, Fuchs CS, Abrams T, et al. A multi-institutional, phase II open-label study of ganitumab (AMG 479) in advanced carcinoid and pancreatic neuroendocrine tumors. Endocr Relat Cancer (2013) 20:383–90. doi:10.1530/ERC-12-0390
81. Kindler HL, Richards DA, Garbo LE, Garon EB, Stephenson JJ Jr, Rocha-Lima CM, et al. A randomized, placebo-controlled phase 2 study of ganitumab (AMG 479) or conatumumab (AMG 655) in combination with gemcitabine in patients with metastatic pancreatic cancer. Ann Oncol (2012) 23:2834–42. doi:10.1093/annonc/mds142
82. Tap WD, Demetri G, Barnette P, Desai J, Kavan P, Tozer R, et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol (2012) 30:1849–56. doi:10.1200/JCO.2011.37.2359
83. Abou-Alfa GK, Capanu M, O’Reilly EM, Ma J, Chou JF, Gansukh B, et al. A phase II study of cixutumumab (IMC-A12, NSC742460) in advanced hepatocellular carcinoma. J Hepatol (2014) 60:319–24. doi:10.1016/j.jhep.2013.09.008
84. Philip PA, Goldman B, Ramanathan RK, Lenz HJ, Lowy AM, Whitehead RP, et al. Dual blockade of epidermal growth factor receptor and insulin-like growth factor receptor-1 signaling in metastatic pancreatic cancer: phase Ib and randomized phase II trial of gemcitabine, erlotinib, and cixutumumab versus gemcitabine plus erlotinib (SWOG S0727). Cancer (2014) 120:2980–5. doi:10.1002/cncr.28744
85. Wagner LM, Fouladi M, Ahmed A, Krailo MD, Weigel B, DuBois SG, et al. Phase II study of cixutumumab in combination with temsirolimus in pediatric patients and young adults with recurrent or refractory sarcoma: a report from the children’s oncology group. Pediatr Blood Cancer (2014) 62:440–4. doi:10.1002/pbc.25334
86. Schoffski P, Adkins D, Blay JY, Gil T, Elias AD, Rutkowski P, et al. An open-label, phase 2 study evaluating the efficacy and safety of the anti-IGF-1R antibody cixutumumab in patients with previously treated advanced or metastatic soft-tissue sarcoma or Ewing family of tumours. Eur J Cancer (2013) 49:3219–28. doi:10.1016/j.ejca.2013.06.010
87. Rajan A, Carter CA, Berman A, Cao L, Kelly RJ, Thomas A, et al. Cixutumumab for patients with recurrent or refractory advanced thymic epithelial tumours: a multicentre, open-label, phase 2 trial. Lancet Oncol (2014) 15:191–200. doi:10.1016/S1470-2045(13)70596-5
88. Di Cosimo S, Sathyanarayanan S, Bendell JC, Cervantes A, Stein MN, Brana I, et al. Combination of the mTOR inhibitor ridaforolimus and the anti-IGF1R monoclonal antibody dalotuzumab: preclinical characterization and phase I clinical trial. Clin Cancer Res (2015) 21:49–59. doi:10.1158/1078-0432.CCR-14-0940
89. Atzori F, Tabernero J, Cervantes A, Prudkin L, Andreu J, Rodriguez-Braun E, et al. A phase I pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res (2011) 17:6304–12. doi:10.1158/1078-0432.CCR-10-3336
90. Moran T, Felip E, Keedy V, Borghaei H, Shepherd FA, Insa A, et al. Activity of dalotuzumab, a selective anti-IGF1R antibody, in combination with erlotinib in unselected patients with Non-small-cell lung cancer: a phase I/II randomized trial. Exp Hematol Oncol (2014) 3:26. doi:10.1186/2162-3619-3-26
91. Reidy-Lagunes DL, Vakiani E, Segal MF, Hollywood EM, Tang LH, Solit DB, et al. A phase 2 study of the insulin-like growth factor-1 receptor inhibitor MK-0646 in patients with metastatic, well-differentiated neuroendocrine tumors. Cancer (2012) 118:4795–800. doi:10.1002/cncr.27459
92. Zeng X, Zhang H, Oh A, Zhang Y, Yee D. Enhancement of doxorubicin cytotoxicity of human cancer cells by tyrosine kinase inhibition of insulin receptor and type I IGF receptor. Breast Cancer Res Treat (2012) 133:117–26. doi:10.1007/s10549-011-1713-x
93. Mulvihill MJ, Cooke A, Rosenfeld-Franklin M, Buck E, Foreman K, Landfair D, et al. Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med Chem (2009) 1:1153–71. doi:10.4155/fmc.09.89
94. Buck E, Gokhale PC, Koujak S, Brown E, Eyzaguirre A, Tao N, et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol Cancer Ther (2010) 9:2652–64. doi:10.1158/1535-7163.MCT-10-0318
95. Jones RL, Kim ES, Nava-Parada P, Alam S, Johnson FM, Stephens AW, et al. Phase I study of intermittent oral dosing of the insulin-like growth factor-1 and insulin receptors inhibitor OSI-906 in patients with advanced solid tumors. Clin Cancer Res (2015) 21:693–700. doi:10.1158/1078-0432.CCR-14-0265
96. Puzanov I, Lindsay CR, Goff LW, Sosman JA, Gilbert J, Berlin J, et al. A phase I study of continuous oral dosing of OSI-906, a dual inhibitor of insulin-like growth factor-1 and insulin receptors in patients with advanced solid tumors. Clin Cancer Res (2014) 21:701–11. doi:10.1158/1078-0432.CCR-14-0303
97. Bendell JC, Jones SF, Hart L, Spigel DR, Lane CM, Earwood C, et al. A phase Ib study of linsitinib (OSI-906), a dual inhibitor of IGF-1R and IR tyrosine kinase, in combination with everolimus as treatment for patients with refractory metastatic colorectal cancer. Invest New Drugs (2015) 33:187–93. doi:10.1007/s10637-014-0177-3
98. Singh RK, Gaikwad SM, Jinager A, Chaudhury S, Maheshwari A, Ray P. IGF-1R inhibition potentiates cytotoxic effects of chemotherapeutic agents in early stages of chemoresistant ovarian cancer cells. Cancer Lett (2014) 354:254–62. doi:10.1016/j.canlet.2014.08.023
99. Awasthi N, Zhang C, Ruan W, Schwarz MA, Schwarz RE. BMS-754807, a small-molecule inhibitor of insulin-like growth factor-1 receptor/insulin receptor, enhances gemcitabine response in pancreatic cancer. Mol Cancer Ther (2012) 11:2644–53. doi:10.1158/1535-7163.MCT-12-0447
100. Feng X, Aleem E, Lin Y, Axelson M, Larsson O, Stromberg T. Multiple antitumor effects of picropodophyllin in colon carcinoma cell lines: clinical implications. Int J Oncol (2012) 40:1251–8. doi:10.3892/ijo.2011.1281
101. Lemaire M, Fristedt C, Agarwal P, Menu E, Van Valckenborgh E, De Bruyne E, et al. The HDAC inhibitor LBH589 enhances the antimyeloma effects of the IGF-1RTK inhibitor picropodophyllin. Clin Cancer Res (2012) 18:2230–9. doi:10.1158/1078-0432.CCR-11-1764
102. Carboni JM, Wittman M, Yang Z, Lee F, Greer A, Hurlburt W, et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol Cancer Ther (2009) 8:3341–9. doi:10.1158/1535-7163.MCT-09-0499
103. Hou X, Huang F, Macedo LF, Harrington SC, Reeves KA, Greer A, et al. Dual IGF-1R/InsR inhibitor BMS-754807 synergizes with hormonal agents in treatment of estrogen-dependent breast cancer. Cancer Res (2011) 71:7597–607. doi:10.1158/0008-5472.CAN-11-1080
104. Ekman S, Frodin JE, Harmenberg J, Bergman A, Hedlund A, Dahg P, et al. Clinical phase I study with an insulin-like growth factor-1 receptor inhibitor: experiences in patients with squamous non-small cell lung carcinoma. Acta Oncol (2011) 50:441–7. doi:10.3109/0284186X.2010.499370
105. Zhong H, Fazenbaker C, Breen S, Chen C, Huang J, Morehouse C, et al. MEDI-573, alone or in combination with mammalian target of rapamycin inhibitors, targets the insulin-like growth factor pathway in sarcomas. Mol Cancer Ther (2014) 13:2662–73. doi:10.1158/1535-7163.MCT-14-0144
106. Friedbichler K, Hofmann MH, Kroez M, Ostermann E, Lamche HR, Koessl C, et al. Pharmacodynamic and antineoplastic activity of BI 836845, a fully human IGF ligand-neutralizing antibody, and mechanistic rationale for combination with rapamycin. Mol Cancer Ther (2014) 13:399–409. doi:10.1158/1535-7163.MCT-13-0598
107. Gao J, Chesebrough JW, Cartlidge SA, Ricketts SA, Incognito L, Veldman-Jones M, et al. Dual IGF-I/II-neutralizing antibody MEDI-573 potently inhibits IGF signaling and tumor growth. Cancer Res (2011) 71:1029–40. doi:10.1158/0008-5472.CAN-10-2274
108. Iguchi H, Nishina T, Nogami N, Kozuki T, Yamagiwa Y, Yagawa K. Phase I dose-escalation study evaluating safety, tolerability and pharmacokinetics of MEDI-573, a dual IGF-I/II neutralizing antibody, in Japanese patients with advanced solid tumours. Invest New Drugs (2015) 33:194–200. doi:10.1007/s10637-014-0170-x
109. Haluska P, Menefee M, Plimack ER, Rosenberg J, Northfelt D, LaVallee T, et al. Phase I dose-escalation study of MEDI-573, a bispecific, antiligand monoclonal antibody against IGFI and IGFII, in patients with advanced solid tumors. Clin Cancer Res (2014) 20:4747–57. doi:10.1158/1078-0432.CCR-14-0114
110. Gualberto A, Hixon ML, Karp DD, Li D, Green S, Dolled-Filhart M, et al. Pre-treatment levels of circulating free IGF-1 identify NSCLC patients who derive clinical benefit from figitumumab. Br J Cancer (2011) 104:68–74. doi:10.1038/sj.bjc.6605972
111. McCaffery I, Tudor Y, Deng H, Tang R, Suzuki S, Badola S, et al. Putative predictive biomarkers of survival in patients with metastatic pancreatic adenocarcinoma treated with gemcitabine and ganitumab, an IGF1R inhibitor. Clin Cancer Res (2013) 19:4282–9. doi:10.1158/1078-0432.CCR-12-1840
112. Weickhardt A, Doebele R, Oton A, Lettieri J, Maxson D, Reynolds M, et al. A phase I/II study of erlotinib in combination with the anti-insulin-like growth factor-1 receptor monoclonal antibody IMC-A12 (cixutumumab) in patients with advanced non-small cell lung cancer. J Thorac Oncol (2012) 7:419–26. doi:10.1097/JTO.0b013e31823c5b11
113. Litzenburger BC, Creighton CJ, Tsimelzon A, Chan BT, Hilsenbeck SG, Wang T, et al. High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clin Cancer Res (2011) 17:2314–27. doi:10.1158/1078-0432.CCR-10-1903
114. Huang F, Greer A, Hurlburt W, Han X, Hafezi R, Wittenberg GM, et al. The mechanisms of differential sensitivity to an insulin-like growth factor-1 receptor inhibitor (BMS-536924) and rationale for combining with EGFR/HER2 inhibitors. Cancer Res (2009) 69:161–70. doi:10.1158/0008-5472.CAN-08-0835
115. Zha J, O’Brien C, Savage H, Huw LY, Zhong F, Berry L, et al. Molecular predictors of response to a humanized anti-insulin-like growth factor-I receptor monoclonal antibody in breast and colorectal cancer. Mol Cancer Ther (2009) 8:2110–21. doi:10.1158/1535-7163.MCT-09-0381
116. Becker MA, Hou X, Harrington SC, Weroha SJ, Gonzalez SE, Jacob KA, et al. IGFBP ratio confers resistance to IGF targeting and correlates with increased invasion and poor outcome in breast tumors. Clin Cancer Res (2012) 18:1808–17. doi:10.1158/1078-0432.CCR-11-1806
117. Gong Y, Yao E, Shen R, Goel A, Arcila M, Teruya-Feldstein J, et al. High expression levels of total IGF-1R and sensitivity of NSCLC cells in vitro to an anti-IGF-1R antibody (R1507). PLoS One (2009) 4:e7273. doi:10.1371/journal.pone.0007273
118. Schwartz GK, Tap WD, Qin LX, Livingston MB, Undevia SD, Chmielowski B, et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol (2013) 14:371–82. doi:10.1016/S1470-2045(13)70049-4
119. Browne BC, Eustace AJ, Kennedy S, O’Brien NA, Pedersen K, McDermott MS, et al. Evaluation of IGF1R and phosphorylated IGF1R as targets in HER2-positive breast cancer cell lines and tumours. Breast Cancer Res Treat (2012) 136:717–27. doi:10.1007/s10549-012-2260-9
120. Boone DN, Lee AV. Targeting the insulin-like growth factor receptor: developing biomarkers from gene expression profiling. Crit Rev Oncog (2012) 17:161–73. doi:10.1615/CritRevOncog.v17.i2.30
121. McKinley ET, Bugaj JE, Zhao P, Guleryuz S, Mantis C, Gokhale PC, et al. 18FDG-PET predicts pharmacodynamic response to OSI-906, a dual IGF-1R/IR inhibitor, in preclinical mouse models of lung cancer. Clin Cancer Res (2011) 17:3332–40. doi:10.1158/1078-0432.CCR-10-2274
122. Flanigan SA, Pitts TM, Newton TP, Kulikowski GN, Tan AC, McManus MC, et al. Overcoming IGF1R/IR resistance through inhibition of MEK signaling in colorectal cancer models. Clin Cancer Res (2013) 19:6219–29. doi:10.1158/1078-0432.CCR-13-0145
123. Bao XH, Takaoka M, Hao HF, Wang ZG, Fukazawa T, Yamatsuji T, et al. Esophageal cancer exhibits resistance to a novel IGF-1R inhibitor NVP-AEW541 with maintained RAS-MAPK activity. Anticancer Res (2012) 32:2827–34.
124. Yeh J, Litz J, Hauck P, Ludwig DL, Krystal GW. Selective inhibition of SCLC growth by the A12 anti-IGF-1R monoclonal antibody correlates with inhibition of Akt. Lung Cancer (2008) 60:166–74. doi:10.1016/j.lungcan.2007.09.023
125. Molina-Arcas M, Hancock DC, Sheridan C, Kumar MS, Downward J. Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov (2013) 3:548–63. doi:10.1158/2159-8290.CD-12-0446
126. Creighton CJ, Casa A, Lazard Z, Huang S, Tsimelzon A, Hilsenbeck SG, et al. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol (2008) 26:4078–85. doi:10.1200/JCO.2007.13.4429
127. Mahadevan D, Sutton GR, Arteta-Bulos R, Bowden CJ, Miller PJ, Swart RE, et al. Phase 1b study of safety, tolerability and efficacy of R1507, a monoclonal antibody to IGF-1R in combination with multiple standard oncology regimens in patients with advanced solid malignancies. Cancer Chemother Pharmacol (2014) 73:467–73. doi:10.1007/s00280-013-2372-x
128. Okusaka T, Ikeda M, Fukutomi A, Kobayashi Y, Shibayama K, Takubo T, et al. Safety, tolerability, pharmacokinetics and antitumor activity of ganitumab, an investigational fully human monoclonal antibody to insulin-like growth factor type 1 receptor, combined with gemcitabine as first-line therapy in patients with metastatic pancreatic cancer: a phase 1b study. Jpn J Clin Oncol (2014) 44:442–7. doi:10.1093/jjco/hyu034
129. Chitnis MM, Lodhia KA, Aleksic T, Gao S, Protheroe AS, Macaulay VM. IGF-1R inhibition enhances radiosensitivity and delays double-strand break repair by both non-homologous end-joining and homologous recombination. Oncogene (2014) 33:5262–73. doi:10.1038/onc.2013.460
130. Lodhia KA, Gao S, Aleksic T, Esashi F, Macaulay VM. Suppression of homologous recombination sensitizes human tumor cells to IGF-1R inhibition. Int J Cancer (2015) 136:2961–6. doi:10.1002/ijc.29327
131. Beltran PJ, Calzone FJ, Mitchell P, Chung YA, Cajulis E, Moody G, et al. Ganitumab (AMG 479) inhibits IGF-II-dependent ovarian cancer growth and potentiates platinum-based chemotherapy. Clin Cancer Res (2014) 20:2947–58. doi:10.1158/1078-0432.CCR-13-3448
132. Ferte C, Loriot Y, Clemenson C, Commo F, Gombos A, Bibault JE, et al. IGF-1R targeting increases the antitumor effects of DNA-damaging agents in SCLC model: an opportunity to increase the efficacy of standard therapy. Mol Cancer Ther (2013) 12:1213–22. doi:10.1158/1535-7163.MCT-12-1067
133. Amaral AT, Garofalo C, Frapolli R, Manara MC, Mancarella C, Uboldi S, et al. Trabectedin efficacy in Ewing sarcoma is greatly increased by combination with anti-IGF signaling agents. Clin Cancer Res (2015) 21(6):1373–82. doi:10.1158/1078-0432.CCR-14-1688
134. Zhao H, Gu X. Silencing of insulin-like growth factor-1 receptor enhances the radiation sensitivity of human esophageal squamous cell carcinoma in vitro and in vivo. World J Surg Oncol (2014) 12:325. doi:10.1186/1477-7819-12-325
135. Isebaert SF, Swinnen JV, McBride WH, Haustermans KM. Insulin-like growth factor-type 1 receptor inhibitor NVP-AEW541 enhances radiosensitivity of PTEN wild-type but not PTEN-deficient human prostate cancer cells. Int J Radiat Oncol Biol Phys (2011) 81:239–47. doi:10.1016/j.ijrobp.2011.03.030
136. Rochester MA, Riedemann J, Hellawell GO, Brewster SF, Macaulay VM. Silencing of the IGF1R gene enhances sensitivity to DNA-damaging agents in both PTEN wild-type and mutant human prostate cancer. Cancer Gene Ther (2005) 12:90–100. doi:10.1038/sj.cgt.7700775
137. Moreno-Acosta P, Gamboa O, Sanchez de Gomez M, Cendales R, Diaz GD, Romero A, et al. IGF1R gene expression as a predictive marker of response to ionizing radiation for patients with locally advanced HPV16-positive cervical cancer. Anticancer Res (2012) 32:4319–25.
138. Tolcher AW, Sarantopoulos J, Patnaik A, Papadopoulos K, Lin CC, Rodon J, et al. Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1. J Clin Oncol (2009) 27:5800–7. doi:10.1200/JCO.2009.23.6745
139. Haluska P, Shaw HM, Batzel GN, Yin D, Molina JR, Molife LR, et al. Phase I dose escalation study of the anti insulin-like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors. Clin Cancer Res (2007) 13:5834–40. doi:10.1158/1078-0432.CCR-07-1118
140. Bid HK, Zhan J, Phelps DA, Kurmasheva RT, Houghton PJ. Potent inhibition of angiogenesis by the IGF-1 receptor-targeting antibody SCH717454 is reversed by IGF-2. Mol Cancer Ther (2012) 11:649–59. doi:10.1158/1535-7163.MCT-11-0575
141. Ulanet DB, Ludwig DL, Kahn CR, Hanahan D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc Natl Acad Sci U S A (2010) 107:10791–8. doi:10.1073/pnas.0914076107
142. Avnet S, Sciacca L, Salerno M, Gancitano G, Cassarino MF, Longhi A, et al. Insulin receptor isoform A and insulin-like growth factor II as additional treatment targets in human osteosarcoma. Cancer Res (2009) 69:2443–52. doi:10.1158/0008-5472.CAN-08-2645
143. Bowers LW, Cavazos DA, Maximo IX, Brenner AJ, Hursting SD, deGraffenried LA. Obesity enhances nongenomic estrogen receptor crosstalk with the PI3K/Akt and MAPK pathways to promote in vitro measures of breast cancer progression. Breast Cancer Res (2013) 15:R59. doi:10.1186/bcr3453
144. Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med (2011) 62:233–47. doi:10.1146/annurev-med-070909-182917
145. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer (2009) 9:631–43. doi:10.1038/nrc2713
146. Fagan DH, Yee D. Crosstalk between IGF1R and estrogen receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia (2008) 13:423–9. doi:10.1007/s10911-008-9098-0
147. Fox EM, Miller TW, Balko JM, Kuba MG, Sanchez V, Smith RA, et al. A kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer Res (2011) 71:6773–84. doi:10.1158/0008-5472.CAN-11-1295
148. Chakraborty AK, Welsh A, Digiovanna MP. Co-targeting the insulin-like growth factor I receptor enhances growth-inhibitory and pro-apoptotic effects of anti-estrogens in human breast cancer cell lines. Breast Cancer Res Treat (2010) 120:327–35. doi:10.1007/s10549-009-0382-5
149. Fagan DH, Uselman RR, Sachdev D, Yee D. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor: implications for breast cancer treatment. Cancer Res (2012) 72:3372–80. doi:10.1158/0008-5472.CAN-12-0684
150. Drury SC, Detre S, Leary A, Salter J, Reis-Filho J, Barbashina V, et al. Changes in breast cancer biomarkers in the IGF1R/PI3K pathway in recurrent breast cancer after tamoxifen treatment. Endocr Relat Cancer (2011) 18:565–77. doi:10.1530/ERC-10-0046
151. Lubik AA, Gunter JH, Hendy SC, Locke JA, Adomat HH, Thompson V, et al. Insulin increases de novo steroidogenesis in prostate cancer cells. Cancer Res (2011) 71:5754–64. doi:10.1158/0008-5472.CAN-10-2470
152. Schmitz S, Kaminsky-Forrett MC, Henry S, Zanetta S, Geoffrois L, Bompas E, et al. Phase II study of figitumumab in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck: clinical activity and molecular response (GORTEC 2008-02). Ann Oncol (2012) 23:2153–61. doi:10.1093/annonc/mdr574
153. Buck E, Eyzaguirre A, Rosenfeld-Franklin M, Thomson S, Mulvihill M, Barr S, et al. Feedback mechanisms promote cooperativity for small molecule inhibitors of epidermal and insulin-like growth factor receptors. Cancer Res (2008) 68:8322–32. doi:10.1158/0008-5472.CAN-07-6720
154. Chakravarti A, Loeffler JS, Dyson NJ. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res (2002) 62:200–7.
155. van der Meel R, Oliveira S, Altintas I, Heukers R, Pieters EH, van Bergen en Henegouwen PM, et al. Inhibition of tumor growth by targeted anti-EGFR/IGF-1R nanobullets depends on efficient blocking of cell survival pathways. Mol Pharm (2013) 10:3717–27. doi:10.1021/mp400212v
156. Cao L, Yu Y, Darko I, Currier D, Mayeenuddin LH, Wan X, et al. Addiction to elevated insulin-like growth factor I receptor and initial modulation of the AKT pathway define the responsiveness of rhabdomyosarcoma to the targeting antibody. Cancer Res (2008) 68:8039–48. doi:10.1158/0008-5472.CAN-08-1712
157. O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res (2006) 66:1500–8. doi:10.1158/0008-5472.CAN-05-2925
158. Naing A, Lorusso P, Fu S, Hong D, Chen HX, Doyle LA, et al. Insulin growth factor receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with metastatic adrenocortical carcinoma. Br J Cancer (2013) 108:826–30. doi:10.1038/bjc.2013.46
159. Naing A, LoRusso P, Fu S, Hong DS, Anderson P, Benjamin RS, et al. Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing’s sarcoma family tumors. Clin Cancer Res (2012) 18:2625–31. doi:10.1158/1078-0432.CCR-12-0061
160. Quek R, Wang Q, Morgan JA, Shapiro GI, Butrynski JE, Ramaiya N, et al. Combination mTOR and IGF-1R inhibition: phase I trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors. Clin Cancer Res (2011) 17:871–9. doi:10.1158/1078-0432.CCR-10-2621
161. Nordstrand A, Lundholm M, Larsson A, Lerner UH, Widmark A, Wikstrom P. Inhibition of the insulin-like growth factor-1 receptor enhances effects of simvastatin on prostate cancer cells in co-culture with bone. Cancer Microenviron (2013) 6:231–40. doi:10.1007/s12307-013-0129-z
162. Shao M, Hollar S, Chambliss D, Schmitt J, Emerson R, Chelladurai B, et al. Targeting the insulin growth factor and the vascular endothelial growth factor pathways in ovarian cancer. Mol Cancer Ther (2012) 11:1576–86. doi:10.1158/1535-7163.MCT-11-0961
163. Bruchim I, Sarfstein R, Reiss A, Flescher E, Werner H. IGF1R tyrosine kinase inhibitor enhances the cytotoxic effect of methyl jasmonate in endometrial cancer. Cancer Lett (2014) 352:214–9. doi:10.1016/j.canlet.2014.06.013
164. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA (2014) 311:806–14. doi:10.1001/jama.2014.732
165. Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the global burden of disease study 2013. Lancet (2014) 384:766–81. doi:10.1016/S0140-6736(14)60460-8
166. Bjorntorp P. Abdominal obesity and the metabolic syndrome. Ann Med (1992) 24:465–8. doi:10.3109/07853899209166997
167. Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest (2013) 123:2764–72. doi:10.1172/JCI67227
168. Ruan W, Lai M. Insulin-like growth factor binding protein: a possible marker for the metabolic syndrome? Acta Diabetol (2010) 47:5–14. doi:10.1007/s00592-009-0142-3
169. Ligibel JA, Alfano CM, Courneya KS, Demark-Wahnefried W, Burger RA, Chlebowski RT, et al. American society of clinical oncology position statement on obesity and cancer. J Clin Oncol (2014) 32:3568–74. doi:10.1200/JCO.2014.58.4680
170. Colditz GA, Wolin KY, Gehlert S. Applying what we know to accelerate cancer prevention. Sci Transl Med (2012) 4:127rv4. doi:10.1126/scitranslmed.3003218
171. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet (2001) 357:539–45. doi:10.1016/S0140-6736(00)04046-0
173. Foltz CJ, Fox JG, Cahill R, Murphy JC, Yan L, Shames B, et al. Spontaneous inflammatory bowel disease in multiple mutant mouse lines: association with colonization by Helicobacter hepaticus. Helicobacter (1998) 3:69–78. doi:10.1046/j.1523-5378.1998.08006.x
174. Harvey AE, Lashinger LM, Hursting SD. The growing challenge of obesity and cancer: an inflammatory issue. Ann N Y Acad Sci (2011) 1229:45–52. doi:10.1111/j.1749-6632.2011.06096.x
175. Subbaramaiah K, Howe LR, Bhardwaj P, Du B, Gravaghi C, Yantiss RK, et al. Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland. Cancer Prev Res (Phila) (2011) 4:329–46. doi:10.1158/1940-6207.CAPR-10-0381
176. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi:10.1146/annurev-physiol-021909-135846
177. Subbaramaiah K, Morris PG, Zhou XK, Morrow M, Du B, Giri D, et al. Increased levels of COX-2 and prostaglandin E2 contribute to elevated aromatase expression in inflamed breast tissue of obese women. Cancer Discov (2012) 2:356–65. doi:10.1158/2159-8290.CD-11-0241
178. Morris PG, Hudis CA, Giri D, Morrow M, Falcone DJ, Zhou XK, et al. Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prev Res (Phila) (2011) 4:1021–9. doi:10.1158/1940-6207.CAPR-11-0110
179. O’Rourke RW. Inflammation in obesity-related diseases. Surgery (2009) 145:255–9. doi:10.1016/j.surg.2008.08.038
180. Renehan AG, Roberts DL, Dive C. Obesity and cancer: pathophysiological and biological mechanisms. Arch Physiol Biochem (2008) 114:71–83. doi:10.1080/13813450801954303
181. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature (2006) 441:431–6. doi:10.1038/nature04870
182. McKinsey EL, Parrish JK, Irwin AE, Niemeyer BF, Kern HB, Birks DK, et al. A novel oncogenic mechanism in Ewing sarcoma involving IGF pathway targeting by EWS/Fli1-regulated microRNAs. Oncogene (2011) 30:4910–20. doi:10.1038/onc.2011.197
183. Lee KW, Bode AM, Dong Z. Molecular targets of phytochemicals for cancer prevention. Nat Rev Cancer (2011) 11:211–8. doi:10.1038/nrc3017
184. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature (2009) 460:392–5. doi:10.1038/nature08221
185. De Angel RE, Conti CJ, Wheatley KE, Brenner AJ, Otto G, Degraffenried LA, et al. The enhancing effects of obesity on mammary tumor growth and Akt/mTOR pathway activation persist after weight loss and are reversed by RAD001. Mol Carcinog (2013) 52:446–58. doi:10.1002/mc.21878
186. Nogueira LM, Dunlap SM, Ford NA, Hursting SD. Calorie restriction and rapamycin inhibit MMTV-Wnt-1 mammary tumor growth in a mouse model of postmenopausal obesity. Endocr Relat Cancer (2012) 19:57–68. doi:10.1530/ERC-11-0213
187. Lashinger LM, Malone LM, Brown GW, Daniels EA, Goldberg JA, Otto G, et al. Rapamycin partially mimics the anticancer effects of calorie restriction in a murine model of pancreatic cancer. Cancer Prev Res (Phila) (2011) 4:1041–51. doi:10.1158/1940-6207.CAPR-11-0023
188. Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science (2012) 335:1638–43. doi:10.1126/science.1215135
189. Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov (2012) 2:778–90. doi:10.1158/2159-8290.CD-12-0263
190. Dowling RJ, Niraula S, Stambolic V, Goodwin PJ. Metformin in cancer: translational challenges. J Mol Endocrinol (2012) 48:R31–43. doi:10.1530/JME-12-0007
191. Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila) (2010) 3:1451–61. doi:10.1158/1940-6207.CAPR-10-0157
192. Currie CJ, Poole CD, Gale EA. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia (2009) 52:1766–77. doi:10.1007/s00125-009-1440-6
193. Goodwin PJ, Stambolic V, Lemieux J, Chen BE, Parulekar WR, Gelmon KA, et al. Evaluation of metformin in early breast cancer: a modification of the traditional paradigm for clinical testing of anti-cancer agents. Breast Cancer Res Treat (2011) 126:215–20. doi:10.1007/s10549-010-1224-1
194. Brehm BJ, Seeley RJ, Daniels SR, D’Alessio DA. A randomized trial comparing a very low carbohydrate diet and a calorie-restricted low fat diet on body weight and cardiovascular risk factors in healthy women. J Clin Endocrinol Metab (2003) 88:1617–23. doi:10.1210/jc.2002-021480
195. Levy RG, Cooper PN, Giri P. Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst Rev (2012) 3:CD001903. doi:10.1002/14651858.CD001903.pub2
196. Yancy WS, Foy M, Chalecki AM, Vernon MC, Westman EC. A low-carbohydrate, ketogenic diet to treat type 2 diabetes. Nutr Metab (2005) 2:34. doi:10.1186/1743-7075-2-34
197. Schwartz K, Chang HT, Nikolai M, Pernicone J, Rhee S, Olson K, et al. Treatment of glioma patients with ketogenic diets: report of two cases treated with an IRB-approved energy-restricted ketogenic diet protocol and review of the literature. Cancer Metab (2015) 3:3. doi:10.1186/s40170-015-0129-1
198. Ho VW, Leung K, Hsu A, Luk B, Lai J, Shen SY, et al. A low carbohydrate, high protein diet slows tumor growth and prevents cancer initiation. Cancer Res (2011) 71:4484–93. doi:10.1158/0008-5472.CAN-10-3973
199. Caso J, Masko EM, Ii JA, Poulton SH, Dewhirst M, Pizzo SV, et al. The effect of carbohydrate restriction on prostate cancer tumor growth in a castrate mouse xenograft model. Prostate (2013) 73:449–54. doi:10.1002/pros.22586
200. Mavropoulos JC, Buschemeyer WC, Tewari AK, Rokhfeld D, Pollak M, Zhao Y, et al. The effects of varying dietary carbohydrate and fat content on survival in a murine LNCaP prostate cancer xenograft model. Cancer Prev Res (2009) 2:557–65. doi:10.1158/1940-6207.CAPR-08-0188
201. Stafford P, Abdelwahab MG, Kim Y, Preul MC, Rho JM, Scheck AC. The ketogenic diet reverses gene expression patterns and reduces reactive oxygen species levels when used as an adjuvant therapy for glioma. Nutr Metab (2010) 7:74. doi:10.1186/1743-7075-7-74
202. Otto C, Kaemmerer U, Illert B, Muehling B, Pfetzer N, Wittig R, et al. Growth of human gastric cancer cells in nude mice is delayed by a ketogenic diet supplemented with omega-3 fatty acids and medium-chain triglycerides. BMC Cancer (2008) 8:122. doi:10.1186/1471-2407-8-122
203. Abdelwahab MG, Fenton KE, Preul MC, Rho JM, Lynch A, Stafford P, et al. The ketogenic diet is an effective adjuvant to radiation therapy for the treatment of malignant glioma. PLoS One (2012) 7:e36197. doi:10.1371/journal.pone.0036197
204. Bielohuby M, Sawitzky M, Stoehr BJ, Stock P, Menhofer D, Ebensing S, et al. Lack of dietary carbohydrates induces hepatic growth hormone (GH) resistance in rats. Endocrinology (2011) 152:1948–60. doi:10.1210/en.2010-1423
205. Tisdale MJ, Brennan RA. A comparison of long-chain triglycerides and medium-chain triglycerides on weight loss and tumour size in a cachexia model. Br J Cancer (1988) 58:580–3. doi:10.1038/bjc.1988.263
Keywords: insulin-like growth factor, insulin, biomarkers, obesity, energy balance
Citation: Bowers LW, Rossi EL, O’Flanagan CH, deGraffenried LA and Hursting SD (2015) The role of the insulin/IGF system in cancer: lessons learned from clinical trials and the energy balance-cancer link. Front. Endocrinol. 6:77. doi: 10.3389/fendo.2015.00077
Received: 23 March 2015; Accepted: 29 April 2015;
Published: 15 May 2015
Edited by:
Antonino Belfiore, University Magna Graecia of Catanzaro, ItalyReviewed by:
Maximilian Bielohuby, Sanofi, GermanyNaoyuki Kataoka, Kyoto University School of Medicine, Japan
Copyright: © 2015 Bowers, Rossi, O’Flanagan, deGraffenried and Hursting. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen D. Hursting, Department of Nutrition, University of North Carolina at Chapel Hill, 135 Dauer Drive, McGavran-Greenberg Hall, Chapel Hill, NC 27599, USA,aHVyc3RpbmdAZW1haWwudW5jLmVkdQ==