 Parveena Firdous1Kamran Nissar2
Parveena Firdous1Kamran Nissar2 Sajad Ali1
Sajad Ali1 Bashir Ahmad Ganai1*Uzma Shabir1Toyeeba Hassan1Shariq Rashid Masoodi3
Bashir Ahmad Ganai1*Uzma Shabir1Toyeeba Hassan1Shariq Rashid Masoodi3
- 1Centre of Research for Development (CORD), University of Kashmir, Srinagar, India
- 2Department of Biochemistry, University of Kashmir, Srinagar, India
- 3Department of Endocrinology, Sher-I-Kashmir Institute of Medical Sciences, Srinagar, India
Diabetes is a global epidemic problem growing exponentially in Asian countries posing a serious threat. Among diabetes, maturity-onset diabetes of the young (MODY) is a heterogeneous group of monogenic disorders that occurs due to β cell dysfunction. Genetic defects in the pancreatic β-cells result in the decrease of insulin production required for glucose utilization thereby lead to early-onset diabetes (often <25 years). It is generally considered as non-insulin dependent form of diabetes and comprises of 1–5% of total diabetes. Till date, 14 genes have been identified and mutation in them may lead to MODY. Different genetic testing methodologies like linkage analysis, restriction fragment length polymorphism, and DNA sequencing are used for the accurate and correct investigation of gene mutations associated with MODY. The next-generation sequencing has emerged as one of the most promising and effective tools to identify novel mutated genes related to MODY. Diagnosis of MODY is mainly relying on the sequential screening of the three marker genes like hepatocyte nuclear factor 1 alpha (HNF1α), hepatocyte nuclear factor 4 alpha (HNF4α), and glucokinase (GCK). Interestingly, MODY patients can be managed by diet alone for many years and may also require minimal doses of sulfonylureas. The primary objective of this article is to provide a review on current status of MODY, its prevalence, genetic testing/diagnosis, possible treatment, and future perspective.
Introduction
Maturity-onset diabetes of the young (MODY) is a monogenic, clinically and genetically heterogeneous form of diabetes showing autosomal dominant mode of inheritance spanning up to three generations. It accounts for 1–5% of all the diabetic forms of young and is specified by anomalous pancreatic β-cell activity (1–3). Neonatal diabetes mellitus (NDM) and MODY are the two monogenic forms of diabetes, with MODY showing higher occurrence than NDM (Figure 1). Based on previous reports, MODY is included in a group of genetic defects in the pancreatic β-cells limiting the ability of pancreas to produce insulin required for glucose utilization (4, 5). These patients exhibit mild or no diabetic symptoms, and their elevated glucose level can be detected only during routine blood tests. MODY has numerous subtypes depending upon the gene involved and the clinical phenotypes. Previously, 13 MODY subtypes were identified and now 14th subtype has been recently added to the list (Table 1) (6, 7).
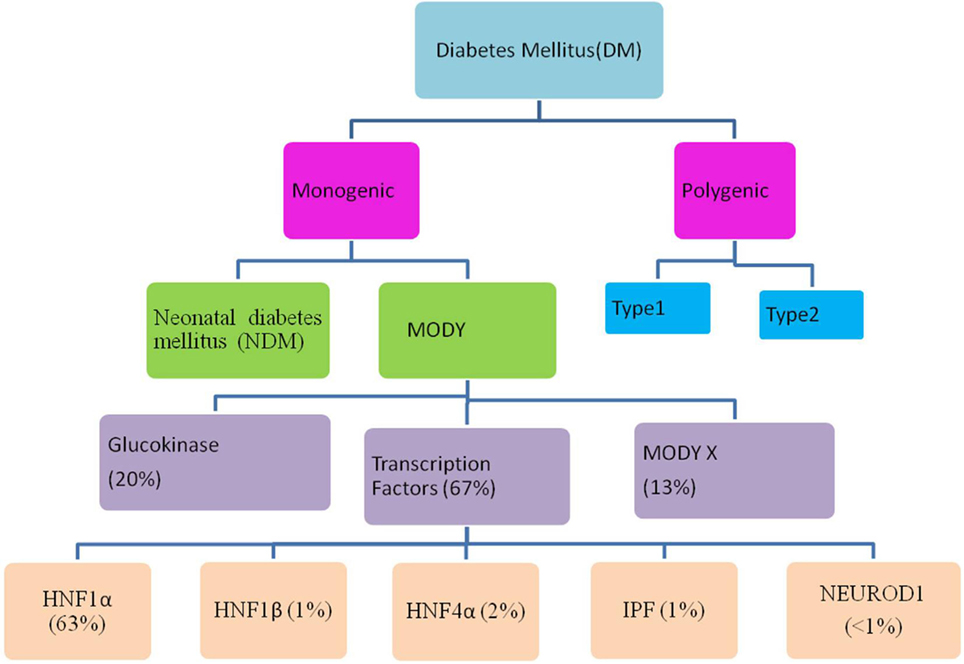
Figure 1. Flow through diagram showing types of diabetes mellitus including the relative prevalence of various maturity-onset diabetes of the young (MODY) causing genes.
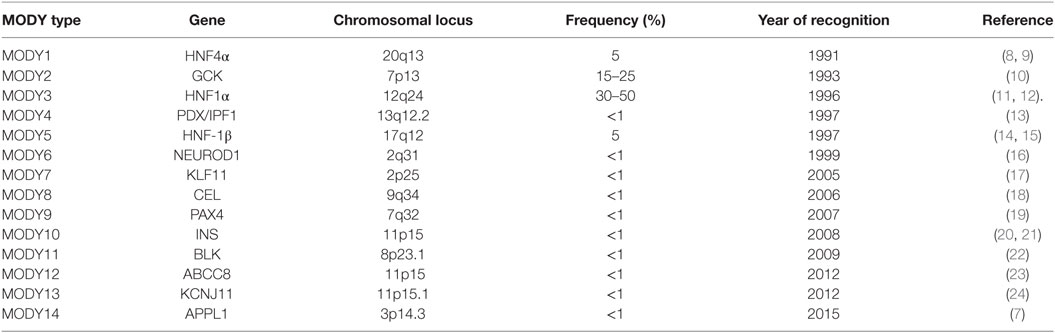
Table 1. Genes associated with various maturity-onset diabetes of the young (MODY) types including frequencies and year of recognition of various MODY types.
History
The symptomless diabetes was reported in the non-obese children and teenagers in 1958 whose glucose level was maintained by sulfonyl-urea treatment (25). The term “maturity-onset type diabetes of Young or of the children” was given by Fajan in 1964 at the Fifth Congress of the International Diabetes Federation in Toronto to highlight its firm hereditary basis. During this period, MODY was named as type 2 diabetes, which could be managed by diet, insulin, and oral drugs (26). In 1973, it was reported that the carbohydrate intolerance in 45 patients below the age group of 25 years never reached extreme conditions, although they were treated with sulfonylureas for 16 years; however, 43 of these patients were having a strong diabetic history. A “mild” form of diabetes was reported in three families having inherited autosomal dominant and early-onset diabetes treated with sulfonylureas (27). The acronym “Maturity-onset Diabetes of the Young” (MODY) was jointly used by Tattersall and Fajans and defined MODY as “Fasting hyperglycemia” (28). The genetic studies on MODY was started on Michigan family viz R-W pedigree in which 74 members suffered from non-insulin dependent diabetes mellitus (29). The co-segregation of DNA polymorphism in adenosine deaminase (ADA) gene was observed after analyzing 75 DNA markers (30). Bowden et al. (31) confirmed that MODY is linked to D20S16. The monogenetic nature of MODY diabetes was confirmed after the identification of mutation in glucokinase (GCK) gene (32). The non-sense gene mutation (P.Q268X) in hepatocyte nuclear factor 4 alpha (HNF4α) gene was reported as the cause of MODY5 (11). At present, 14 MODY types specified by mutation in respective 14 genes with their etiologies are known.
Prevalence of MODY
Maturity-onset diabetes of the young is not the prevalent diabetic form in the world population and is usually misclassified with type 1 and type 2 diabetes (33, 34). In UK, 80% of MODY patients are misdiagnosed as type 1 or type 2 diabetes (35). In developed countries, the reported frequency of MODY is 1–2% (36). Amed et al. (37) reported the minimum incidence rate of MODY in Canadian population that is 0.4 cases per 100,000 children and youth of <18 years age. However, it has been reported to be highly prevalent in certain communities including Pima Indians (38), Nauru population (39), and southern India (40). Mohan et al. (41) reported a high prevalence of MODY in Asian Indian patients. According, to Pihoker et al. (42), the minimum prevalence of MODY increases to 2.1/100,000 individuals of <20 years of age.
Till now, 14 subtypes of MODY with distinct genetic etiologies have been identified. The relative prevalence of MODY subtypes vary greatly with MODY2 and MODY3 being the most prevalent (Table 2). The relative frequency of each MODY type varies due to different recruitment data and ethnic variability.
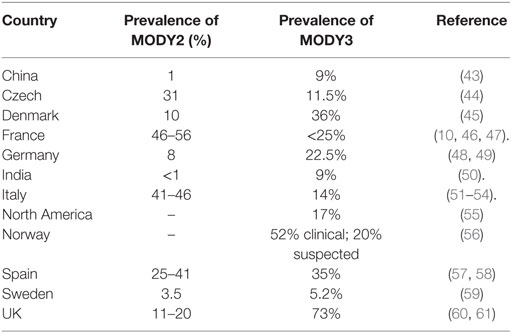
Table 2. Prevalence of MODY2 and MODY3 in different countries.
MODY Subtypes
As per Vaxillaire and Froguel (62), MODY represents a combination of genetic, metabolic, and clinical heterogenicity. MODY has several subtypes (Table 1) depending upon the involvement of genes and their mutations (deletion, splice-site, non-sense, etc.) with MODY2 being associated with enzyme GCK, while rest being associated with transcription factors.
HNF4α-MODY (MODY1)
Hepatic nuclear factor 4 alpha (HNF4α) is an important gene located on chromosome 20 and plays a significant role in regulating the expression of liver and pancreatic β-cells (63, 64). Mutation in exon 4 (Thr130 → Ile) and exon 7 (Gly268 → Thr) of HNF4α gene is responsible for causing MODY1 (8, 9). HNF4α regulates the proteins essential for glucose transport metabolism as well as lipoprotein biosynthesis in liver cells (65–67). HNF4α mutation causes decline in insulin production except during neonatal period, where it results in hyperinsulinemic hypoglycemia thereby exhibit phenotype similar to MODY3 (8, 9, 68). MODY1 has the penetrance of less than 5% where most carriers exhibit diabetic signs before 25 years of age and about 15% patients show diazoxide-responsive hyperinsulinemic hypoglycemia history (69, 70). HNF4α mutation also leads number of pancreatic disorders like decline in high-density lipoprotein (HDL), triglyceride levels and increment in low density lipoprotein (LDL), respectively.
GCK-MODY (MODY2)
Glucokinase enzyme also named as hexokinase D belongs to hexokinase gene family which acts as glucose sensor in pancreatic β-cells (71, 72) thus, playing critical role in glucose homeostasis. This enzyme is constitutively expressed and catalyzes the initial rate limiting step in the glycolytic pathway by ATP dependent phosphorylation of glucose to glucose-6-phosphate (71) in presence of Mg ions. GCK gene mutations upregulating the insulin secretion and resetting the glucose threshold results in enhanced fasting glucose level (73). Ellard et al. (74) reported GCK mutations in 5–6% of women who were suffering from gestational diabetes. The frequent heterozygous inactivating GCK mutations are responsible for the second most common type of MODY termed as MODY2 (75). MODY2 is characterized by mild persistent fasting hyperglycemia and low glucose-stimulated insulin secretion (75). So for as the clinical complications of GCK mutations are concerned, activating and heterozygous mutations results in persistent hyperinsulinemic hypoglycemia of infants (PHIH), while as homozygous mutations in both the alleles can result in permanent neonatal diabetes mellitus (PNDM) and mutations in single allele can result in GCK-MODY (76–78). The corresponding amino acids are very much important for GCK activity as per the reports of Galán et al. (79) who demonstrated the occurrence of two new GCK mutations Leu165 → Phe, Glu265 → lys and missense Thr206 → Met in Spanish families. However, some mutations do not have any visible effect on the GCK activity like Val62 → Met (80). Previously more than 600 GCK mutations have been reported in 10 exons of GCK gene (81).
HNF1α-MODY (MODY3)
The hepatocyte nuclear factor 1 alpha (HNF1α) gene plays an important role in regulating expression of insulin gene in the mature β-cell and glucose transporter GLUT2 gene (82, 83). Fajans (12) revealed the association of HNF1α gene mutation with MODY 3 diabetes by conducting linkage analysis of R-W pedigree. Hyperglycemia due to HNF1α mutations can be controlled with sulfonylureas for a number of years (84). Anuradha et al. (85) studied the gene polymorphism of HNF1α and showed the high prevalence of amino acid polymorphism at codon (Ala98 → Val) which is associated with MODY3 diabetes. Earlier, it was demonstrated that patients with HNF1α mutations develop diabetes after the first decade, preceded by abnormal glucose-induced insulin secretion (86). MODY3 is characterized by a progressive reduction in insulin secretion. It has been shown that carriers of HNF1α mutation show increased β-cell apoptosis (87).
PDX1/IPF1-MODY (MODY4)
Pancreatic and duodenal homeobox 1 (PDX1)/insulin promoter factor 1 (IPF1) plays significant role in regulating transcription of genes encoding: glucagon, Insulin, GLUT2, and GCK enzymes (88). IPF1 acts as key switch in maintaining the hormonal and enzymatic functions of pancreas on binding with the TAAT element of the target gene (IPF1) promoter region (89). However, mutation of IPF1 gene located on chromosome 13 results in MODY4 (13, 90). Pancreatic anomaly termed agenesis can occur due to disrupted IPF gene activity and heterozygous IPF mutations can cause defective insulin secretion thus MODY 4 diabetes, while as, homozygous mutation resulting in permanent neonatal diabetes (PND) and pancreatic exocrine insufficiency (13, 91, 92).
HNF-1β-MODY (MODY5)
Hepatocyte nuclear factor-1β (HNF-1β) gene functions in the various developmental processes in humans like nephron and embryonic pancreas development. Mutation in HNF-1β gene results in MODY type 5 and the patients commonly exhibit notable histological anomalies showing meganephrons, and renal cysts referred as renal cysts and diabetes syndrome (RCAD) (48, 93). Additional complications associated with MODY5 are vaginal aplasia, rudimentary uterus (48), hyperuricemia, gout (94), and reduced birth weight to 900 g (15). Heterozygous Pro159 → Leu mutation in HNF1β gene results in impaired glucose metabolism (95).
NEUROD1-MODY (MODY6)
Neurogenic differentiation-1 gene (NEUROD1) transcription factor is significant role in pancreatic and neuronal development and is functionally expressed in both pancreatic and neurons cells. However, its impairment results in autosomal dominant inheritance mutations similar to type 2 (16, 96, 97). Modified activity of NEUROD1 gene in carriers of Arg111 → Leu and insertion of cytosine residue in a polyC tract in codon 206 (206 + C) of exon2 were largely responsible for development of diabetes (16). Mutation in basic-loop-helix transcription factor NEUROD1 is responsible for MODY6 (75).
KLF11-MODY (MODY7)
Krueppel-like factor 11 (KLF11) gene is located on chromosome 2 and plays important role in pancreatic β-cell functioning (98, 99). KLF11 mutation affects the pancreatic β-cell function by modulating the expression of certain free radical scavengers such as catalase and superoxide dismutase (SOD). The expression level of these antioxidant enzymes is reported to be low in pancreatic islets; however, the over expression of these enzymes protects β-cells against glucolipotoxicity (100). The occurrence of several uncommon variants Ala347 → ser, Thr220 → Met, and Gln62 → Arg of KLF11 gene associated with familial diabetes which severely disrupts transcriptional machinery was analyzed by genetic analysis and mutation in KLF11 results in MODY termed MODY7 (17).
CEL-MODY (MODY8)
The main ingredient of pancreatic juice is carboxyl-ester lipase (CEL) encoded by carboxyl-ester lipase gene (9q) and plays an important role in newborns by digesting milk and hydrolyzing dietary esters in duodenum (101, 102). A single base deletion resulted in the altered C-terminal sequencing, thus causing the monogenetic disease, i.e., MODY8. On the other hand, a single base insertion (1686delT; C563fsx673) causes polygenic diabetes resulted by truncation of the CEL (103), thus suggesting a frame shift deletion mutation in variable number tandom repeats (VNTR) in the 11th exon of CEL gene. In addition, common as well as rare CEL mutations also affect exocrine and endocrine functioning of pancreas leading to MODY diabetic syndrome (MODY8).
PAX4-MODY (MODY9)
Paired box gene 4 (PAX4) is a transcription factor which is a member of PAX family located on chromosome 7 and regulates fetal development, cancer growth, commitment of progenitor cells to various islet cells and also represses the promoter activity of insulin and glucagon (104, 105). PAX4 is required for the regeneration of β-cells in adults and its mutation blocks or inhibits β-cell proliferation (106). Two PAX4 gene mutations viz Arg164 → trp and IVS7-IG>A were reported as the cause of monogenetic form of diabetes in Thai population termed MODY 9 (19).
INS-MODY (MODY10)
The insulin gene (INS) which is located on chromosome 11p15.5 encodes the proinsulin precursor of insulin, its mutation cause defect in NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) and thus leads to MODY10 (107, 108). The mutations in cysteine residues (example Cys96 → tyr) leads to disruption of disulfide bonds, other mutations like Ala24 → Asp, phe48 → Cys, Arg89 → Cys, His29 → Asp, Leu35 → Pro, Gly84 → Arg, Cys96 → Ser, S101 → Cys, and Tyr103 → Cys have been observed in young diabetic patients (21).
BLK-MODY (MODY11)
Lymphocyte kinase tyrosine kinase (BLK-B) encoding the receptor protein tyrosine kinase, plays a significant role in thymopoiesis in immature T cells (109). Drebin et al. (110) cloned human homolog of murine BLK and concluded that like murine BLK gene human BLK is expressed only in β-cells. The obese feature of diabetes was reported to be linked with chromosome 8p23 gene locus (111). BLK expression is altered in response to this Ala71 → Thr substitution (22). The BLK gene mutation essentially affects MIN6 B-cells (a highly differentiated B-cell line) and is responsible for MODY11 (22).
ABCC8-MODY (MODY12)
Before 40 years of age, the ABCC8 mutations were reported in the cohort of adult type 2 diabetic patients (112). The insulin secretion which regulates blood sugar levels is mediated by the ATP-binding cassette transporter subfamily C member 8 (ABCC8) encoding sulfonyl-urea receptor 1 ATP-binding cassette transporter subfamily C member 8 (ABCC8), and encodes sulfonyl-urea receptor 1 (SUR1) which is the subunit of K-ATP channel (ATP-sensitive potassium channel) found across β-cell membrane. ABCC8 gene mutation results in sulfonylureas responsive MODY termed MODY12 and clinical anomalies can occur both due to activating and inactivating mutations of ABCC8 gene. Congenital hyperinsulinism occurs as a result of dominantly inherited inactivating mutations, while the permanent or transient neonatal diabetes (TNDM OR PNDM) is caused due to activating mutations or recessive loss-of-function mutations (113). Four unique mutations (Glu100 → Lys, Gly214 → Arg, Gln985 → Arg, and Asn125 → Asp) in ABBC8 gene was found in susceptible MODY patients (23), the heterozygous ABCC8 mutations were reported in 7/85 (8%) non-obese sulfonylureas sensitive patients with BMI < 30 kg/m2 with clinical features similar to HNF1α/4α MODY (23).
KCNJ11-MODY (MODY13)
The potassium channel, rectifying subfamily J, having member 11 (KCNJ11) which encodes human BIR (beta cell inward rectifier or Kir6.20) that is intron less coding gene was determined (114). Earlier, it was predicted that KCNJ11 gene mutations might result in neonatal diabetes. The sequence analysis revealed the occurrence of six new heterozygous missense mutations in 10 out of 29 patients and among these four patients exhibit Arg201 → His mutation concluding that neonatal diabetes is caused by heterozygous mutations of the KCNJ11 gene (115). The onset and severity of diabetes occurring due to the missense KCNJ11 gene mutation is variable and spans up to three generations (116). Massa et al. (117) revealed the occurrence of five different heterozygous mutations including two novel mutations by screening the KCNJ11 gene in eight Italian patients and concluded that KCNJ11 gene mutations are the common cause of PNDM. The KCNJ11 gene mutation due to the disrupted subunit interaction results in severe conditions like channel inactivation, which was found to be associated with Arg301 mutation that determinately cause hyperinsulinism (118).
APPL1-MODY (MODY14)
The knockdown mutation of APPL1 gene results in apoptosis and the overexpression resulted in dysmorphic phenotypes and delay in development, as was revealed from the previous reports of APPL1 gene expression in Zebra fish (119). In adapter protein, phosphotyrosine interaction domain and leucine zipper containing protein-1 (APPL1), both missense and non-sense heterozygous loss of function results in asp-to-asn (Asp94 → Asn) substitution, while non-sense mutation results in Leu552 → X (Leu552 → X) substitution (7). The mutation in APPL1 gene was reported by Prundate et al. (7) as a cause of MODY14.
Diagnosis/Clinical Features and Management
Some of the major diagnostic characteristics of MODY given by Vaxillaire and Froguel (62) are as follows:
• Elevated glucose level (hyperglycemia) is diagnosed in early age (before 25 years) in one or two suspects of the diabetic family.
• Autosomal dominant inheritance is showing vertical transmission through at least three generations. Similar phenotype shared by diabetic family members.
• Insulin is not required by patients up to 5 years after initial diagnosis. As far as C-peptide level is concerned it tends to remain low even in patients on insulin therapy.
• Functional impairment in the pancreatic β cells.
• Does not show the normal diabetic features like obesity, etc.
Besides the clinical features, probability of MODY occurrence can be calculated by using standardized MODY probability calculator (120) (Figure 2), and the clinical features may vary with the MODY type in consideration. However, clinical diagnostic characteristics of the most common MODY types are as follows.
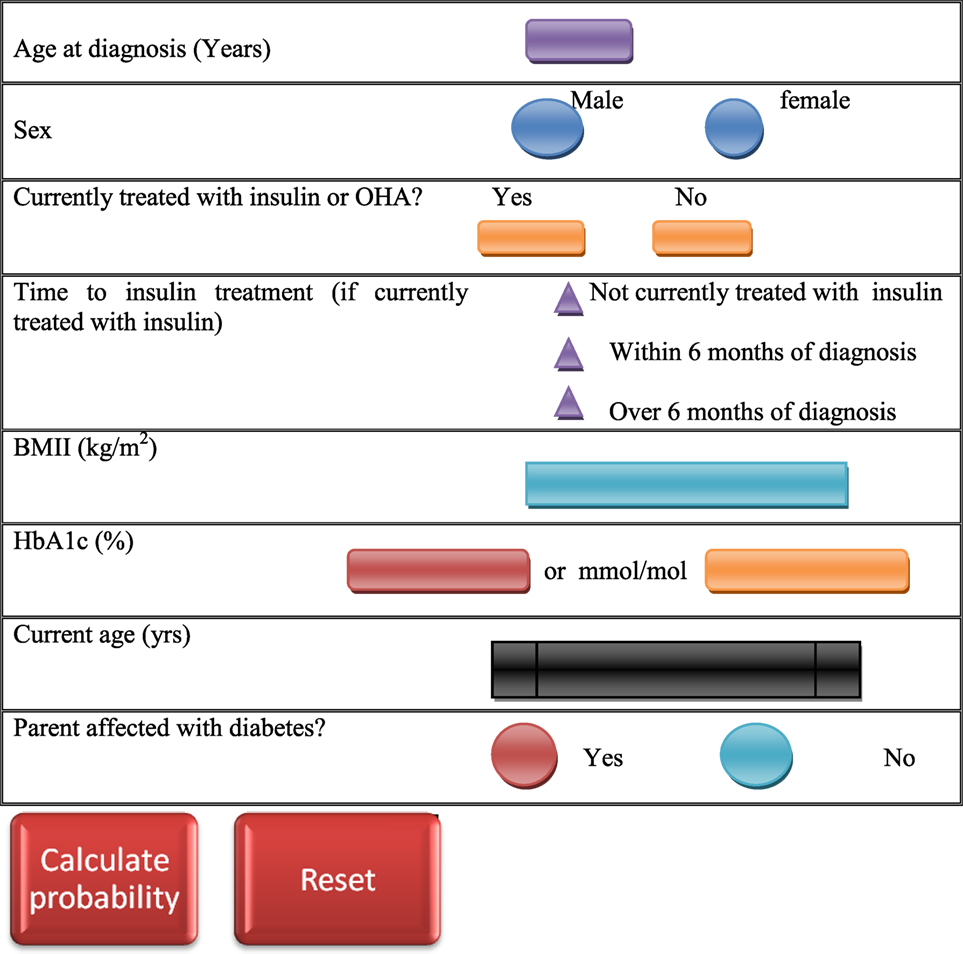
Figure 2. Maturity-onset diabetes of the young (MODY) testing calculator (120). The MODY probability is calculated by entering clinical features of the patient in the probability calculator.
GCK-MODY2
1. The fasting elevated glucose level observed is ≥5.5 mmol/l (98%) which is stable (from months up to years) and persistent (for at least three independent occurrences).
2. It has been observed that the Hb1Ac is typically just above the normal level and in some cases rarely exceeds 7.5% (3).
3. As per the reports of Stride and Hattersley (3), there had been a little (in European population 71% of the patients had <3 mmol/l) increment in an Oral Glucose Tolerance Test (OGTT) [(2 h glucose) − (fasting glucose)].
4. Parents of the affected individuals may have mild type 2 diabetes with lesser complications or may not be diabetic at all. However, further testing revealed that one parent will commonly have a moderately raised fasting hyperglycemia (5.5–8 mmol/l) except in the subjects where the mutation has originated de novo (3).
HNF1α-MODY3
1. Lower insulinogenic index and lower early insulin response is observed in HNF1α mutants compared with the familial non-mutants even when blood glucose level is normal (86).
2. Patients are typically lean and without characteristic insulin resistance (121).
3. Positive β-auto-antibodies (IA-2) were reported in 25% (7/28) of HNF1α patients, and these patients develop diabetes much later than those with negative autoantibody. It was further reported that in such patient’s glycemia was difficult to control (122).
4. C-reactive protein level tends to be lower in MODY3 patients (123).
5. Glycosuria often observed in patients with low renal threshold due to diminished expression of high affinity low-capacity co-transporter 2 (SGLT2) which decreases the glucose reabsorption from kidney renal tubules (124).
For the genetic diagnosis of MODY, only those subjects are selected which fit into the standard clinical criteria of disease (125). For testing the monogenetic diabetes, DNA is isolated from the patient’s blood sample which is then analyzed for mutation(s). Next-generation sequencing (NGS) strategies according to Ellard et al., (126), Johanson et al. (127) can be employed to identify MODY gene mutation. By direct sequencing, MODY can be diagnosed approximately with 100% sensitivity (128). According to Nyunt et al. (129) suspected cases when not diagnosed by routine screening technique can be detected by multiple ligase dependent probe amplification (MLPA) technique. There are some biomarkers that effectively differentiate MODY from other forms of diabetes and are thus effectively used for accurate testing. MicroRNAs possibly can be used as biomarkers of HNF1α-MODY, as it has been observed that the inducible expression of Pro291 → fsinsC-HNF1A in INS-1 cells resulted in a significant upregulation of three miRNAs (miR-292-3p, miR-103, and miR-224) (130). Furthermore, the elevated expression levels of miR-103 and miR-224 could be detected in the serum of HNF1α-MODY carriers compared with MODY-negative family controls. The high HDL cholesterol level in MODY3 patients acts as the biomarker for differentiating MODY from type 2 diabetes (131). Sanger sequencing is the most reliable method for MODY diagnosis (70). A correct molecular diagnosis helps in the optimal treatment of disease. As β-cell function deteriorates over time, pharmacologic therapy becomes necessary to prevent diabetes-related complications. Once the gene mutation is accurately detected, only then the first degree relatives with MODY symptoms can be accurately diagnosed with the disease (132).
Maturity-onset diabetes of the young patients can be managed by diet alone for many years and sulfonylureas is also recommended to be very effective for managing glucose level for more than 30 years (12). Patients diagnosed with GCK mutations usually need no treatment, but require medication in case of pregnancy and gestational diabetes mellitus (GDM). HNF1α and HNF4α MODY patients are particularly sensitive to sulfonylureas, but in the later stages should be supplemented with the addition of insulin (133). In HNF1α MODY patients (134) reported higher mean A1C levels in those taking insulin (7.5%) vs. oral agents (6.7%). Patients with HNF1α and HNF4α mutations are typically sensitive to sulfonylureas. Low-dose sulfonylureas (20–40 mg gliclazide regularly) can aid in sustaining the MODY patients for decades (135). Drugs like nateglinide and liraglutide lower the postprandial glucose levels in HNF1α MODY subjects (136, 137). The pathophysiology associated with MODY gene mutations and various possible treatments for different MODY types are shown in (Table 3).
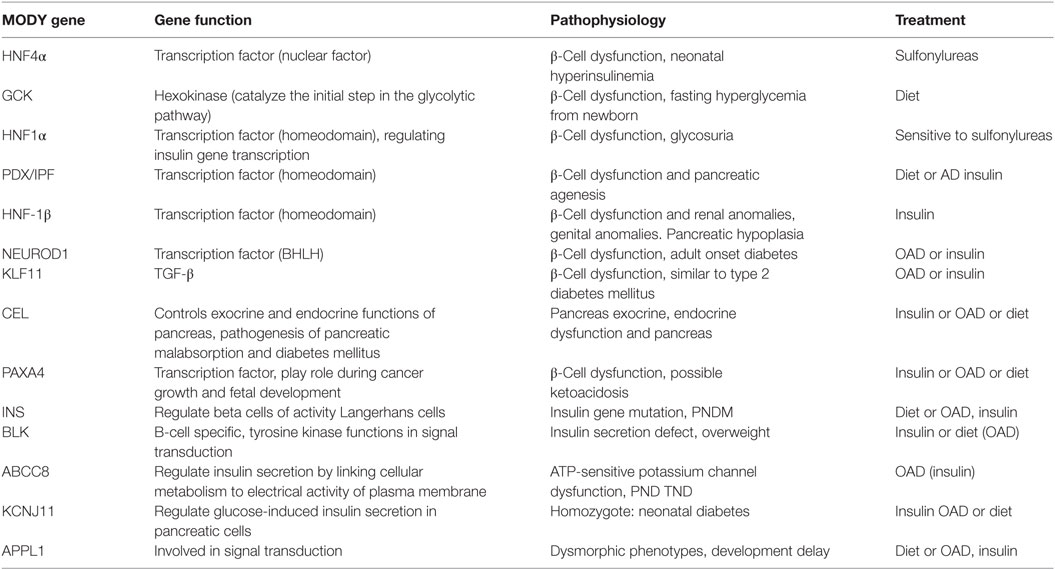
Table 3. Maturity-onset diabetes of the young (MODY) gene functions and pathophysiology and possible treatment.
Genetic Testing Methodologies
Different genetic testing methodologies like linkage analysis and direct sequencing were used by different workers for investigating gene mutations associated with MODY diabetes, which are summarized below (Table 4). Fajans (12) by linkage analysis investigated 360 members of the diabetic family (R-W Pedigree) by taking into account MODY patients. Linkage analysis confirmed that the HNF1α gene which is located on chromosome 12q24.2 and is responsible for causing MODY3. Association of the GCK gene mutations with the early-onset diabetes has been reported with the help of linkage analysis (32). Froguel et al. (32) reported hyperglycemia in 125 out of 304 examined subjects, with 33% exhibiting clinical characteristics of maturity early-onset diabetes termed MODY2. In addition to this, they also analyzed that there was statistically significant evidence of genetic heterogeneity with an estimated 45–95% of the 16 families showing linkage to GCK. Yamagata et al. (8, 9) isolated and partially sequenced the human HNF4α gene in R-W pedigree; following genetic screening identified thr130ile mutation in exon 4 and Gly268 → Thr mutation in exon 7 of the HNF1α gene. Finally, confirmed the association of early-onset diabetes with HNF4α gene mutation.
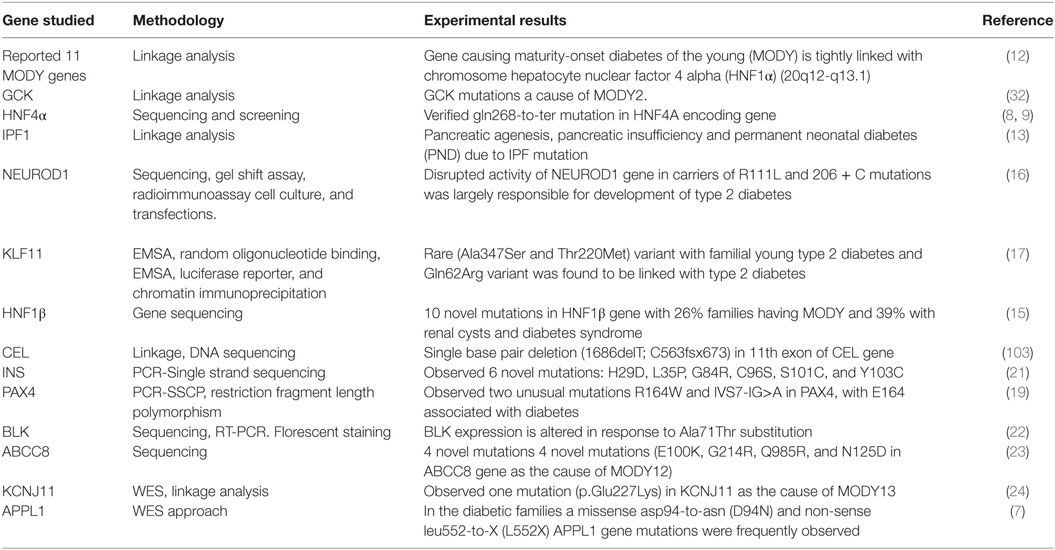
Table 4. Genetic testing methodologies (genetic screening and linkage analysis).
Stoffers et al. (13) on the basis of linkage analysis, mode of transmission and phenotypic variations suggest the occurrence of fourth type of early-onset diabetes termed MODY 4 caused due to mutation of IPF1 gene locus. Stoffers et al. (13) reported that pancreatic agenesis is caused by homozygous mutation in IPF, which results in exocrine pancreatic insufficiency and permanent neonatal diabetes (PND). Malecki et al. (16) by direct sequencing of DNA samples from 94 subjects with type 2 diabetes, verified that the disrupted activity of NEUROD1 gene in carriers of Arg111 → Leu (arginine to leucine substitution at codon 111) and insertion of cytosine residue in a polyC tract in codon 206 (206 + C) in exon 2 of NEUROD1 were largely responsible for development of type 2 diabetes. The NEUROD1 results in autosomal dominant inheritances similar to type 2 diabetes (16). Neve et al. (17) on investigating KLF11 gene by direct sequencing and various other testing methodologies like-random nucleotide binding; chromatin immune-precipitation demonstrated that KLF11 gene by binding to the insulin promoter maintain pancreatic β-cell activity. Genetic analysis revealed the occurrence of two uncommon variants Ala347 → Ser and Thr220 → Met which are associated with familial young type 2 diabetes and severely disrupts transcriptional machinery. Moreover, in North European population while analyzing 1,696 type 2 diabetic patients and 1,776 normal individuals. Neve et al. (17) reported that polymorphic Gln62 → Arg variant was found to be linked with type 2 diabetes. This Gln162 → Arg variant was found to deviate the KLF11 co-repressors mSin3A-binding property which results in altered insulin promoter activation and hence results in reduced insulin gene expression in pancreatic β-cells along with other complications like reduced plasma insulin. Thus, it was concluded that KLF11 variants assist in developing diabetes.
Edghill et al. (15) sequenced HNF-1β gene in 160 non-associated patients suffering from renal diseases and observed HNF1β mutations in 14% (23/160) including 10 unique mutations (Val61 → Gly, Val110 → Gly, Ser148 → Leu, Lys1565 → Glu, Gln176 → X, Arg276 → Gln, Ser281 → fsinsC, R295 → P, His324 → fsdelCA, and Gln470 → X). Furthermore, out of these 23 novel mutations; diagnosed renal cysts in 19 (83%) and diabetes in 11 (48%). Only 6% (26%) of families fulfill the diagnostic characteristics for MODY, and 39% families had RCAD. Thus, by sequence analysis observed a definite association between HNF1β gene mutation and renal abnormalities. Raeder et al. (103) examined a large family with autosomal dominant diabetes typically detected before 40 years of age with primary β-cell dysfunction. Linkage analysis and DNA sequencing of CEL gene reveals that the single base deletion (1686delThy; C563fsx673) at 11th exon was found in all risk family members having both diabetes and fecal elastase deficiency. Plengvidhya et al. (19) tested 46 Thia type 2 diabetic patients of <35 years of age by PCR-single-stranded conformation polymorphism (PCR-SSCP), this was succeeded by direct screening of PAX4 mutations; final observation of variants was performed by restriction fragment length polymorphism. Two unusual mutations Arg164 → Trp and IVS7-IG > A of PAX4 gene were identified. R164W mutation was shown to be associated with diabetes, by affecting the function of PAX4 gene (suppressing the insulin and glucagon promoter action). Edghill et al. (21) in European population observed INS mutation in 33 of 141 probands (diagnosed at < 6 months age), 2 of 86 (diagnosed between 6 and 12 months age) and none of 58 (diagnosed between 12 and 24 years of age) by PCR-single strand sequencing. Edghill et al. (21) observed that 46% of the complications were due to the three already described mutations viz Arg24 → Asp, Phe48 → Cys, and Arg89 → Cys and also identified six unusual mutations: His29 → Asp, Leu35 → Pro, Gly84 → Arg, Cys96 → Ser, Ser101 → Cys, and Tyr103 → Cys. Borowiec et al. (22) by using the techniques of immune florescence staining, western blotting, sequencing, RT-PCR, examined 6 MODY families and identified 410 sequence variations in 732 kb of genomic sequence at 8p23. Based on the results observed—five mutations with the frequency of <1% comprises four non-coding mutations and Ala71 → Thr substitution, they summarized that BLK expression is altered in response to this Ala71 → Thr substitution.
Bowman et al. (23) sequenced ABCC8 gene in 85 patients (BMI < 30 kg/m2) with no familial diabetic history and observed ABBC8 mutations in 7 (8%) probands. In addition, to four unique mutations (Glu100 → Lys, G2ly14 → Arg, Gln985 → Arg, and Asn125 → Asp), some previously reported heterozygous mutations were also reported in four patients. Final results showed that only four probands fulfill the clinical criteria of early-onset diabetes termed MODY 12 which was having clinical pathologies identical to that of HNF1α MODY and can be diagnosed before 25 years of age. Bonnefond et al. (24) performed WES (Agilent-sureselect capture/Illumina Golden Gate assay) and linkage analysis in three affected and one unaffected subjects in MODY-X family, and observed 69 mutations with only one mutation (p.Glu227 → Lys) in KCNJ11 gene associated with diabetes (LOD-score of 3.68). Thus, confirming KCNJ11 gene mutation as the cause of early-onset diabetes termed MODY13. Prundate et al. (7) studied loss-of-function mutations in APPL1 gene in 60 families (52 from US and 8 from Italy) by following WES approach. However, in Italian families, normal subjects of >55 age were also included in the study. Both missense and non-sense heterozygous loss-of-function mutations have been reported to occur in APPL1 gene, and missense mutation results in asp-to-asn (Asp94 → Asn) substitution while non-sense mutation can result in Leu552-to-X (Leu552 → X) substitution.
Conclusion and Future Perspective
Researchers have been effectively working to investigate the genetic determinants and pathophysiology of MODY by means of various powerful approaches (based on genome and cell biology). These studies have been very promising and insightful which has greatly extended our understanding of both normal and pathologic β-cell biology as well as regulation of insulin secretion in humans. At clinical level, MODY requires correct and accurate methods of diagnosis as well as to distinguish it from diabetes type 1 or type 2 to avoid the unnecessary insulin or sulfonylureas treatment, which may severely affect the patient’s health.
In future, translational biology and integrative genomic research and studies on both monogenic and polygenic forms of diabetes are required, which will not only provide novel insights but, also broaden our understanding in terms of pathophysiology, treatment and cure of diabetes. Currently, application of NGS has emerged a key factor to decipher the large amount of known genetic defects underlying pancreatic islet β-cell dysfunction in diabetic patients. As there is increased rate of MODY across the globe, NGS can serve as one of the potential and rapid molecular based diagnostic tool as well as to identify novel genetic etiologies of familial or atypical early-onset diabetes. Transcriptomic and secretomic analysis related to MODY may provide a significant molecular insight into the disease and its procurement. Besides genomic approaches, stem cell technology can also be another novel and effective disease model to study and therapy for the diabetes. Use of pluripotent stem cells (PSCs) can help us to better understand the molecular mechanisms underlying different forms of diabetes (MODY). In addition, metabolomic profiling can also be one of the important approaches to study and differentiate MODY with other diabetes.
Author Contributions
PF and BG conceived and designed this review. PF, KN, US, and TH collected the literature for this review. PF, KN, and SA wrote the manuscript draft. BG, SA, and SM edited this manuscript. All the authors gave final shape to this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer LB and handling Editor declared their shared affiliation.
Acknowledgments
The authors are highly thankful to the Centre of Research for development (CORD), University of Kashmir for providing the necessary facilities. They are thankful to Director CORD, other faculty Members, and Non-teaching staff for always helping them whenever needed.
References
1. Gloyn AL, Ellard S, Shepherd M, Howell RT, Parry EM, Jefferson A, et al. Maturity-onset diabetes of the young caused by a balanced translocation where the 20q12 break point results in disruption upstream of the coding region of hepatocyte nuclear factor-4A gene. Diabetes (2001) 51(7):2329–33. doi:10.2337/diabetes.51.7.2329
2. Owen K, Hattersley A. Maturity-onset diabetes of the young: from clinical description to molecular genetic characterization. Best Pract Res Clin Endocrinol Metab (2001) 15:309–23. doi:10.1053/beem.2001.0148
3. Stride A, Hattersley AT. Different genes, different diabetes: lessons from maturity onset diabetes of the young. Ann Med (2002) 34:207–16.
4. World Health Organisation. Definition and Diagnosis of Diabetes Mellitus and Intermediate Hyperglycaemia. Geneva, Switzerland: World Health Organisation (2006).
5. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care (2012) 35(Suppl 1):S64–71. doi:10.2337/dc12-s064
6. Kim EH. Maturity-onset diabetes of the young: what do clinicians need to know? Diabetes Metab J (2015) 39:468–77. doi:10.4093/dmj.2015.39.6.468
7. Prundate S, Jungtrakoon P, Marucci A, Ludovico O, Buranasupkajorn P, Mazza T, et al. Loss-of-function mutations in APPL1 in familial diabetes mellitus. Am J Hum Genet (2015) 97:177–85. doi:10.1016/j.ajhg.2015.05.011
8. Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ, et al. Mutations in the hepatocyte nuclear factor-4a gene in maturity-onset diabetes of the young (MODY1). Nature (1996) 384:458–60. doi:10.1038/384458a0
9. Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature (1996) 384:455–8. doi:10.1038/384455a0
10. Froguel P, Zouali H, Vionnet N, Velho G, Vaxillaire M, Sun F, et al. Familial hyperglycemia due to mutations in glucokinase—definition of a subtype of diabetes mellitus. N Engl J Med (1993) 328:697–702. doi:10.1056/NEJM199303113281005
11. Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al. Mutation in hepatocyte nuclear factor-1beta gene (TCF2) associated with MODY. Nat Genet (1997) 17:384-5. doi:10.1038/ng1297-384
12. Fajans SS. Maturity onset diabetes of the young (MODY). Diabet Metab Rev (1989) 5:579–606. doi:10.1002/dmr.5610050705
13. Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet (1997) 15:106–10. doi:10.1038/ng0197-106
14. McCarthy MI, Hattersley AT. Learning from molecular genetics: novel insights arising from the definition of genes for monogenic and type 2 diabetes. Diabetes (2008) 57(11):2889–989. doi:10.2337/db08-0343
15. Edghill EL, Bingham C, Slingerland AS, Minton JA, Noordam C, Ellard S, et al. Hepatocyte nuclear factor-1 beta mutations cause neonatal diabetes and intrauterine growth retardation: support for a critical role of HNF1B in human pancreatic development. Diabet Med (2006) 23:1301–6. doi:10.1111/j.1464-5491.2006.01999.x
16. Malecki MT, Jhala U, Antonellis A, Fields L, Doria A, Orban T, et al. Mutations in NEUROD1 are associated with the development of type two diabetes. Nat Genet (1999) 23:323–8. doi:10.1038/15500
17. Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V, Dina C, Hamid YH, Joly E, et al. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci U S A (2005) 102:4807–12. doi:10.1073/pnas.0409177102
18. Torsvick J, Johannson S, Johansen A, EK J, Minton J, Raeder H, et al. Mutations in the VNTR of the carboxyl-ester lipase gene (CEL) are a rare cause of monogenetic diabetes. Hum Genet (2010) 127:55–64. doi:10.1007/s00439-009-0740-8
19. Plengvidhya N, Kooptiwut S, Songtawee N, Doi A, Furuta H, Nishi M, et al. PAX4 mutations in Thais with maturity onset diabetes of the young. J Clin Endocrinol Metab (2007) 92:2821–6. doi:10.1210/jc.2006-1927
20. Molven A, Ringdal M, Nordbø AM, Raeder H, Støy J, Lipkind GM, et al. Mutations in the insulin gene can cause MODY and autoantibody-negative type 1 diabetes. Diabetes (2008) 57(4):1131–5. doi:10.2337/db07-1467
21. Edghill EL, Flanagan SE, Patch AM, Boustred C, Parrish A, Shields B, et al. Insulin mutation screening in 1.044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes (2008) 57:11034–42. doi:10.2337/db07-1405
22. Borowiec M, Liew CW, Thompson R, Boonyasrisawat W, Hu J, Mlynarski WM, et al. Mutations at the BLK locus linked to maturity onset diabetes of the young and beta-cell dysfunction. Proc Natl Acad Sci U S A (2009) 106:14460–5. doi:10.1073/pnas.0906474106
23. Bowman P, Flanagan SE, Edghill EL, Damhuis A, Shepherd MH, Paisey R, et al. Heterozygous ABCC8 mutations are a cause of MODY. Diabetologia (2012) 55:123-7. doi:10.1007/s00125-011-2319-x
24. Bonnefond A, Philippe J, Durand E, Dechaume A, Huyvaert M, Montagne L, et al. Whole exome sequencing and high throughput genotyping identified KCNJ11 as the thirteenth MODY gene. PLoS One (2012) 7:37423. doi:10.1371/journal.pone.0037423
25. Fajans SS, Conn JW. The use of tolbutamide in the treatment of young people with mild diabetes mellitus—a progress report. Diabetes (1962) 11:123–6.
26. Fajans SS, Conn JW. The early recognition of diabetes mellitus. Ann N Y Acad Sci (1959) 82:208–18. doi:10.1111/j.1749-6632.1959.tb44901.x
28. Tattersall R, Fajans S. A difference between the inheritance of classical juvenile onset and maturity-onset type diabetes of young people. Diabetes (1975) 24:44–53. doi:10.2337/diabetes.24.1.44
29. Andreone T, Fajans SS, Rotwein P, Skolnick M, Permutt MA. Insulin gene analysis in a family with maturity-onset diabetes of the young. Diabetes (1985) 34:108–14. doi:10.2337/diab.34.2.108
30. Bell GI, Xiang KS, Newman MV, Wu SH, Wright LG, Fajans SS, et al. Gene for non-insulin-dependent diabetes mellitus (maturity-onset diabetes of the young subtype) is linked to DNA polymorphism on human chromosome 20q. Proc Natl Acad Sci U S A (1991) 1991(88):1484–8. doi:10.1073/pnas.88.4.1484
31. Bowden DW, Gravius TC, Akots G, Fajans SS. Identification of genetic markers flanking the locus for maturity-onset diabetes of the young on human chromosome 20. Diabetes (1992) 41:88–92. doi:10.2337/diab.41.1.88
32. Froguel P, Vaxillaire M, Sun F, Velho G, Zouali H, Butal MO, et al. Close linkage of glucokinase locus on chromosome 7p to early onset non-insulin dependent diabetes mellitus. Nature (1992) 356:162–4. doi:10.1038/356162a0
33. Panzram G, Savage PJ, Nagulesparan M, Howar BV, Pettitt DN, Lisse JR, et al. Obesity, insulin resistance and diabetes mellitus in the Pima Indians. In: Kobberling J, Tattersall RB, editors. The Genetics of Diabetes Mellitus. London: Academic Press (1982). p. 243–50.
34. Gardner DS, Tai ES. Clinical features and treatment of maturity onset diabetes of the young (MODY). Diabetes Metab Syndr Obes (2012) 5:101–8. doi:10.2147/DMSO.S23353
35. Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S. Maturity onset diabetes of the young (MODY): How many cases are we missing? Diabetologia (2010) 53:2504–8. doi:10.1007/s00125-010-1799-4
36. Guja C, Guja L. Landscape of monogenetic diabetes in the third millennium. Proc Rom Acad Ser B (2013) 15(3):217–32.
37. Amed S, Dean HJ, Panagiotopoulos C, Sellers EAC, Hadjiyannakis S, Laubscher TA, et al. Type 2 diabetes, medication-induced diabetes, and monogenic diabetes in Canadian children: a prospective national surveillance study. Diabetes Care (2010) 33:786–91. doi:10.2337/dc09-1013
38. Serjeantson S, Zimmet P. Diabetes in the Pacific: evidence for a major gene. In: Baba S, Gould MK, Zimmet P, editors. Diabetes Mellitus: Recent Knowledge on a Etiology, Complications and Treatment. Sydney: Academic Press. (1984). 23–30.
39. Asmal AC, Dayal B, Jialal I, Leary WP, Omar MA, Pillay NL, et al. Non-insulin-dependent-diabetes with early onset in blacks and Indians. S Afr Med J (1981) 60:93–6.
40. Fajans SS. Heterogeneity between various families with non-insulin dependent diabetes of the MODY type. In: Kobberling J, Tattersall RB, editors. The Genetics of Diabetes Mellitus. New York: Academic Press (1982). p. 251–60.
41. Mohan V, Ramchandran A, Snehalatha C, Mohan R, Bharani G, Viswanathan M. High prevalence of maturity onset diabetes of the young (MODY) among Indians. Diabetes Care (1985) 8:371–4. doi:10.2337/diacare.8.4.371
42. Pihoker C, Gilliam LK, Ellard S, Dabelea D, Davis C, Dolan LM, et al. Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for diabetes in youth. J Clin Endocrinol Metab (2013) 98:4055–62. doi:10.1210/jc.2013-1279
43. Xu JY, Dan QH, Chan V, Wat NM, Tam S, Tiu SC, et al. Genetic and clinical characteristics of maturity-onset diabetes of the young in Chinese patients. Eur J Hum Genet (2004) 13:422–7. doi:10.1038/sj.ejhg.5201347
44. Pruhova S, Ek J, Lebl J, Sumnik Z, Saudek F, Andel M, et al. Genetic epidemiology of MODY in the Czech Republic: new mutations in the MODY genes HNF-4alpha, GCK and HNF-1alpha. Diabetologia (2003) 46:291–5. doi:10.1007/s00125-002-1010-7
45. Johansen A, Ek J, Mortensen HB, Pedersen O, Hansen T. Half of clinically defined maturity onset diabetes of the young patients in Denmark do not have mutations in HNF4A, GCK and TCF1. J Clin Endocrinol Metab (2005) 90:4607–14. doi:10.1210/jc.2005-0196
46. Velho G, Blanche H, Vaxillaire M, Bellanne-Chantelot C, Pardini VC, Timsit J, et al. Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY2 families. Diabetologia (1997) 40:217–24. doi:10.1007/s001250050666
47. Vaxillaire M, Rouard M, Yamagata K, Oda N, Kaisaki PJ, Boriraj VV, et al. Identification of nine novel mutations in the hepatocyte nuclear factor1alpha gene associated with maturity-onset diabetes of the young (MODY3). Hum Mol Genet (1997) 6:583±58. doi:10.1093/hmg/6.4.583
48. Lindner TH, Cockburn BN, Bell GI. Molecular genetics of MODY in Germany. Diabetologia (1999) 42:121–3. doi:10.1007/s001250051128
49. Toaima D, Nake A, Wendenburg J, Praedicow K, Rohayem J, Engel K, et al. Identification of novel GCK and HNF1A/TCF1 mutations and polymorphisms in German families with maturity-onset diabetes of the young (MODY). Hum Mutat (2005) 25:503–4. doi:10.1002/humu.9334
50. Anuradha S, Radha V, Mohan V. Association of novel variants in the hepatocyte nuclear factor-4A gene with maturity onset diabetes of the young and early onset type 2 diabetes. Clin Genet (2011) 80:541–9. doi:10.1111/j.1399-0004.2010.01577.x
51. Massa O, Meschi F, Cuesta-Munoz A, Caumo A, Cerutti F, Toni S, et al. High prevalence of glucokinase mutations in Italian children with MODY. Influence on glucose tolerance, first-phase insulin response, insulin sensitivity and BMI. Diabetologia (2001) 44(7):898–905. doi:10.1007/s001250100530
52. Mantovani V, SalardI S, Cerreta V, Bastia D, Cenci M, Ragni L, et al. Identification of eight novel glucokinase mutations in Italian children with maturity-onset diabetes of the young. Hum Mutat (2003) 22:338. doi:10.1002/humu.9179
53. Gragnoli C, Cockburn BN, Chiaramonte F, Gorini A, Marietti G, Marozzi G, et al. Early onset type II diabetes mellitus in Italian families due to mutations in the genes encoding hepatic nuclear factor-1alpha and glucokinase. Diabetologia (2001) 44:1326–9. doi:10.1007/s001250100644
54. Gragnoli C, Lindner T, Cockburn BN, Kaisaki PJ, Gragnoli F, Marozzi G, et al. Maturity-onset diabetes of the young due to a mutation in the hepatocyte nuclear factor-4 alpha binding site in the promoter of the hepatocyte nuclear factor-1 alpha gene. Diabetes (1997) 46:1648–51. doi:10.2337/diacare.46.10.1648
55. Glucksmann MA, Lehto M, Tayber O, Scotti S, Berkemeier L, Pulido C, et al. Novel mutations and a mutational hotspot in the MODY3 gene. Diabetes (1997) 46:1081–6. doi:10.2337/diab.46.6.1081
56. Bjorkhaug L, Sagen JV, Thorsby P, Sovik O, Molven A, Njolstad PR. Hepatocyte nuclear factor-1 alpha gene mutations and diabetes in Norway. J Clin Endocrinol Metab (2003) 88:920–31. doi:10.1210/jc.2002-020945
57. Costa A, Bescos M, Velho G, Chevre J, Vidal J, Sesmilo G, et al. Genetic and clinical characterisation of maturity-onset diabetes of the young in Spanish families. Eur J Endocrinol (2000) 142:380–6. doi:10.1530/eje.0.1420380
58. Barrio R, Bellanne-Chantelot C, Moreno JC, Morel V, Calle H, Alonso M, et al. Nine novel mutations in maturity-onset diabetes of the young (MODY) candidate genes in 22 Spanish families. J Clin Endocrinol Metab (2002) 87:2532–9. doi:10.1210/jcem.87.6.8530
59. Lehto M, Wipemo C, Ivarsson SA, Lindgren C, Lipsanen-Nyman M, Weng J, et al. High frequency of mutations in MODY and mitochondrial genes in Scandinavian patients with familial early-onset diabetes. Diabetologia (1999) 42:1131–7. doi:10.1007/s001250051281
60. Thomson KL, Gloyn AL, Colclough K, Batten M, Allen LI, Beards F, et al. European Caucasians with maturity-onset diabetes of the young (MODY). Hum Mutat (2003) 22:417. doi:10.1002/humu.9186
61. Frayling T, Bulman MP, Ellard S, Appleton M, Dronsfield M, Mackie A, et al. Mutations in the hepatocyte nuclear factor 1 alpha gene are a common cause of maturity-onset diabetes of the young in the United Kingdom. Diabetes (1997) 46:720–5. doi:10.2337/diab.46.4.720
62. Vaxillaire M, Froguel P. Monogenetic diabetes of young, pharmacogenetics and relevance to multifactorial forms of type 2 diabetes. Endocr Rev (2008) 29:254–64.
63. Sladek FM, Zhong WM, Lai E, Darnell JE Jr. Liver enriched transcription factor HNF-4 Is a novel member of the steroid hormone receptor superfamily. Genes Dev (1990) 4:2353–65. doi:10.1101/gad.4.12b.2353
64. Kuo CJ, Conley PB, Chen L, Sladek FM, Darnell JE Jr., Crabtree GR. A transcriptional hierarchy involved in mammalian cell-type specification. Nature (1992) 355:457–61. doi:10.1038/355457a0
65. Ladias JA, Hadzopoulou-Cladaras M, Kardassis D, Cardot P, Cheng J, Zannis V, et al. Transcriptional regulation of human apolipoprotein genes ApoB, ApoCIII, and ApoAII by members of the steroid hormone receptor superfamily HNF-4, ARP-1, EAR-2, and EAR-3. J Biol Chem (1992) 267:15849–60.
66. Stoffel M, Duncan SA. The maturity-onset diabetes of the young (MODY1) transcription factor HNF4a regulates expression of genes required for glucose transport and metabolism. Proc Natl Acad U S A (1997) 94:13209–14. doi:10.1073/pnas.94.24.13209
67. Wang H, Maechler P, Antinozzi PA, Hagenfeldt KA, Wollheim CB. Hepatocyte nuclear factor 4a regulates the expression of pancreatic b-cell genes implicated in glucose metabolism and nutrient-induced insulin secretion. J Biol Chem (2000) 275:35953–9. doi:10.1074/jbc.M006612200
68. Pingul MM, Hughes N, Wu A, Stanley CA, Gruppuso PA. Hepatocyte nuclear factor-4α gene mutation associated with familial neonatal hyperinsulinism and maturity onset diabetes of the young. J Pediatr (2011) 158:852–4. doi:10.1016/j.jpeds.2011.01.003
69. Flanagan SE, Kapoor RR, Mali G. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF1A gene mutation. Eur J Endocrinol (2010) 162:987–92. doi:10.1530/EJE-09-0861
70. McDonald TJ, Ellard S. Maturity onset diabetes if the young; identification and diagnosis. Ann Clin Biochem (2013) 50(Pt):403–15. doi:10.1177/0004563213483458
71. Matschinsky FM, Liang Y, Kesavan P, Wang L, Froguel P, Velho G, et al. Glucokinase as pancreatic beta cell glucose sensor and diabetes gene. J Clin Invest (1993) 92:2092–8. doi:10.1172/JCI116809
72. Matschinsky FM. Glucokinase, glucose homeostasis, and diabetes mellitus. Curr Diab Rep (2005) 5:171–6. doi:10.1007/s11892-005-0005-4
73. Byrne MM, Sturis J, Clément K, Vionnet N, Pueyo ME, Stoffel M, et al. Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J Clin Invest (1994) 93(3):1120–30. doi:10.1172/JCI117064
74. Ellard S, Beards F, Allen LI, Shepherd M, Ballantyne E, Harvey R, et al. A high prevalence of glucokinase mutation in gestational diabetic subjects selected by clinical criteria. Diabetologia (2000) 43:250–3. doi:10.1007/s001250050038
75. Fajan SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med (2001) 345:971–80. doi:10.1056/NEJMra002168
76. Vionnet N, Stoffel M, Takeda J, Yasuda K, Bell GI, Zouali H, et al. Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes mellitus. Nature (1992) 356:721–2. doi:10.1038/356721a0
77. Njolstad PR, Sovik O, Cuesta-Munoz A, Bjorkhaug L, Massa O, Barbetti F, et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med (2001) 344:1588–92. doi:10.1056/NEJM200105243442104
78. Gloyn AL. Glucokinase (GCK) mutations in hyper- and hypoglycemia: maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemia of infancy. Hum Mutat (2003) 22:353–62. doi:10.1002/humu.10277
79. Galán M, Vincent O, Roncero I, Azriel S, Boix-Pallares P, Delgado-Alvarez E, et al. Effects of novel maturity-onset diabetes of the young (MODY)-associated mutations on glucokinase activity and protein stability. Biochem J (2006) 393:386–96. doi:10.1042/BJ20051137
80. Gloyn AL, Odili S, Zelent D, Buettger C, Castleden HAJ, Steele AM, et al. Insights into the structure and regulation of glucokinase from a novel mutation (V62M), which causes maturity-onset diabetes of the young. J Biol Chem (2005) 280:14105–13. doi:10.1074/jbc.M413146200
81. Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanné-Chantelot C, Ellard S, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat (2009) 30(11):1512–26. doi:10.1002/humu.21110
82. Cerf ME. Transcription factors regulating beta cell function. Eur J Endocrinol (2006) 155:671-9. doi:10.1530/eje.1.02277
83. Galan M, Garcia-Herrero CM, Azriel S, Gargallo M, Durán M, Gorgojo JJ, et al. Differential effects of HNF-1α mutations associated with familial young onset diabetes on target gene regulation. Mol Med (2011) 17:256-65. doi:10.2119/molmed.2010.00097
84. Fajans SS, Brown MB. Administration of sulfonylureas can increase glucose-induced insulin secretion for decades in patients with maturity-onset diabetes of the young. Diabetes Care (1993) 16:1254–61. doi:10.2337/diacare.16.9.1254
85. Anuradha S, Radha V, Deepa R, Hansen T, Carstensen B, Pedersen O, et al. A prevalent amino acid polymorphism at codon 98 (Ala98Val) of the hepatocyte nuclear factor-1alpha is associated with maturity-onset diabetes of the young and younger age at onset of type 2 diabetes in Asian Indians. Diabetes Care (2005) 28(10):2430–5. doi:10.2337/diacare.28.10.2430
86. Stride A, Ellard S, Clark P, Shakespeare L, Salzmann M, Shepherd M, et al. Cell dysfunction, insulin sensitivity, and glycosuria precede diabetes in hepatocyte nuclear factor-1alpha mutation carriers. Diabetes Care (2005) 28:1751–6. doi:10.2337/diacare.28.7.1751
87. Bacon S, Kyithar MP, Schmid J, Rizvi SR, Bonner C, Graf R, et al. Serum levels of pancreatic stone protein (PSP)/reg1A as an indicator of beta cell apoptosis suggest an increased apoptosis rate in hepatocyte nuclear factor-1 alpha (HNF1A-MODY) carriers from the third decade of life onward. BMC Endocr Disord (2012) 12:13. doi:10.1186/1472-6823-12-13
88. Kim SK, Selleri L, Lee JS, Zhang AY, Gu X, Jacobs Y, et al. L.Pbx1 inactivation disrupts pancreas development and in Ipf1-deficient mice promotes diabetes mellitus. Nature Genet (2002) 30:430–5. doi:10.1038/ng860
89. Schwitzgebel VM, Mamin A, Brun T, Ritz-Laser B, Zaiko M, Maret A, et al. Agenesis of human pancreas due to decreased half-life of insulin promoter factor 1. J Clin Endocr Metab (2003) 88:4398–406. doi:10.1210/jc.2003-030046
90. Hansen L, Urioste S, Petersen HV, Jensen JN, Eiberg H, Barbetti F, et al. Mis-sense mutation in the human insulin promoter factor-1 gene and their relation to maturity-onset diabetes of the young and late-onset type 2 diabetes mellitus in Caucasians. J Clin Endocrinol Metab (2000) 85:1323–6. doi:10.1210/jc.85.3.1323
91. Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature (1994) 371:606–9. doi:10.1038/371606a0
92. Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. Beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev (1998) 12:1763–8. doi:10.1101/gad.12.12.1763
93. Bellanne-Chantelot C, Chauveau D, Gautier JF, Dubois-Laforgue D, Clauin S, Wilhelm JM, et al. Clinical spectrum associated with hepatocyte nuclear factor-1 β mutations. Ann Intern Med (2004) 140:510–7. doi:10.7326/0003-4819-140-7-200404060-00009
94. Bingham C, Ellard S, Van’t Hoff WG, Simmonds HA, Marinaki AM, Badman MK, et al. Atypical familial juvenile hyperuricemic nephropathy associated with a hepatocyte nuclear factor-1-beta gene mutation. Kidnet Int (2003) 63:1645–51. doi:10.1046/j.1523-1755.2003.00903.x
95. Kim EK, Lee JS, Cheong HI, Chung SS, Kwak SH, Park KS. Idetification and functional characterization of P159L mutation in HNF1B in a family with maturity-onset diabetes of the young5 (MODY5). Genomics Inform (2014) 12:240–6. doi:10.5808/GI.2014.12.4.240
96. Naya FJ, Huang H-P, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev (1997) 11:2323–34. doi:10.1101/gad.11.18.2323
97. Kristinsson SY, Thorolfsdottir ET, Talseth B, Steingrimsson E, Thorsson AV, Helgason T, et al. MODY in Iceland is associated with mutations in HNF-1alpha and a novel mutation in Neuro-D1. Diabetologia (2001) 44:2098-103. doi:10.1007/s001250100016
98. Cook T, Gebelein B, Mesa K, Mladek A, Urrutia R. Molecular cloning and characterization of TIEG2 reveals a new subfamily of transforming growth factor-beta-inducible Sp1-like zinc finger-encoding genes involved in the regulation of cell growth. J Biol Chem (1998) 273(40):25929–36. doi:10.1074/jbc.273.40.25929
99. Scohy S, Gabant P, Van Reeth T, Hertveldt V, Dreze PL, Van Vooren P, et al. Identification of KLF13 and KLF14 (SP6), novel members of the SP/XKLF transcription factor family. Genomics (2001) 70(1):93–101. doi:10.1006/geno.2000.6362
100. Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes (2004) 53:S119–24. doi:10.2337/diabetes.53.2007.S119
101. Lombardo DY, Howles PN. Carboxyl ester lipase: its pathophysiological implications. Biochem Biophy Acta (2001) 1533:1–28.
102. Hui DY, Howles PN. Carboxyl ester lipase: structure-function relationship and physiological role in lipoprotein metabolism and arterosclerosis. J Lipid Res (2002) 43:2017–30. doi:10.1194/jlr.R200013-JLR200
103. Raeder H, Johansson S, Holm PI, Haldorsen IS, Mas E, Sbarra V, et al. Mutations in the CEL-VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat Genet (2006) 38:54–62. doi:10.1038/ng1708
104. Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing cells in the mammalian pancreas. Nature (1997) 386:399–402. doi:10.1038/386399a0
105. Smith SB, Ee HC, Conners JR, German MS. Paired-homeodomain transcription factor PAX4 acts as a transcriptional repressor in early pancreatic development. Mol Cell Biol (1999) 19:8272–80. doi:10.1128/MCB.19.12.8272
106. Biason-Lauber A, Boehm B, Lang-Muritano M, Gauthier BR, Brun T, Wollheim CB, et al. Association of childhood type 1 diabetes mellitus with a variant of PAX4: possible link to cell regenerative capacity. Diabetologia (2005) 48:900–5. doi:10.1007/s00125-005-1723-5
107. Bell GI, Pictet RL, Rutter WJ, Cordell B, Tischer E, Goodman HM. Sequence of the human insulin gene. Nature (1980) 284(5751):26–32. doi:10.1038/284026a0
108. Dandona P, Aljada A, Mohanty P, Ghanim H, Hamouda W, Assian E, et al. Insulin inhibits intranuclear nuclear factor kappa-B and stimulates I-kappa-B in mononuclear cells in obese subjects: evidence for an anti-inflammatory effect? J Clin Endocr Metab (2001) 86:3257–65. doi:10.1210/jcem.86.7.7623
109. Islam KB, Rabbani H, Larsson C, Sanders R, Smith CI. Molecular cloning, characterization, and chromosomal localization of a human lymphoid tyrosine kinase relayed to murine BLK. J Immunol (1995) 154:1265–72.
110. Drebin JA, Hartzell SW, Griffin C, Campbell MJ, Niederhuber JE. Molecular cloning and chromosomal localization of the human homologue of a B-lymphocyte specific protein tyrosine kinase (Blk). Oncogene (1995) 10(3):477–86.
111. Kim SH, Ma X, Weremowicz S, Ercolino T, Powers C, Mlynarski W, et al. Identification of a locus for maturity-onset diabetes of the young on chromosome 8p23. Diabetes (2004) 53:1375–84. doi:10.2337/diabetes.53.5.1375
112. Tarasov AI, Nicolson TJ, Riveline JP, Taneja TK, Baldwin SA, Baldwin JM, et al. A rare mutation in ABCC8/SUR1 leading to altered ATP-sensitive K+ channel activity and beta-cell glucose sensing is associated with type 2 diabetes in adults. Diabetes (2008) 57:1595–604. doi:10.2337/db07-1547
113. Kapoor RR, Flanagan SE, James C, Shield J, Ellard S, Hussain K. Hyperinsulinaemic hypoglycemia. Arch Dis Child (2009) 94:450–7. doi:10.1136/adc.2008.148171
114. Inagaki N, Gonoi T, Clement JP, Namba N, Inazawa J, Gonzalez G, et al. Reconstruction of I (KATP): an inward rectifier subunit plus the sulfonylurea receptor. Science (1995) 270:1166–70. doi:10.1126/science.270.5239.1166
115. Gloyn AL, Cummings EA, Edghill EL, Harries LW, Scott R, Costa T, et al. Permanent neonatal diabetes due to paternal germline mosaicism for an activating mutation of the KCNJ11 gene encoding the kir6.2 subunit of the beta-cell potassium adenosine triphosphate channel. J Clin Endocrinol Metab (2004) 89:3932–5. doi:10.1210/jc.2004-0568
116. Yorifuji T, Nagashima K, Kurokawa K, Kawai M, Oishi M, Akazawa Y, et al. The C42R mutation in the Kir6.2(KCNJ11) gene as a cause of transient neonatal diabetes, or later-onset, apparently type 2 diabetes mellitus. J Clin Endocrinol Metab (2005) 90:3174–8. doi:10.1210/jc.2005-0096
117. Massa O, Iafusco D, D’Amato E, Gloyn AL, Hattersly AT, Pasquino B, et al. KCNJ11 activating mutations in Italian patients with permanent neonatal diabetes. Hum Mutat (2005) 25:22–7. doi:10.1002/humu.20124
118. Lin Y-W, Bushman JD, Yan F-F, Haidar S, MacMullen C, Ganguly A, et al. Destabilization of ATP-sensitive potassium channel activity by novel KCNJ11 mutations identified in congenital hyperinsulinism. J Biol Chem (2008) 283:9146–56. doi:10.1074/jbc.M708798200
119. Schenck A, Goto-Silva L, Collinet C, Rhinn M, Giner A, Habermann B, et al. The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell (2008) 133:486–97. doi:10.1016/j.cell.2008.02.044
120. Shields BM, McDonald TJ, Ellard S, Campbell MJ, Hyde C, Hattersley AT. The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia (2012) 55(5):1265–72. doi:10.1007/s00125-011-2418-8
121. Owen KR, Shepherd M, Stride A, Ellard S, Hattersley AT. Heterogeneity in young adult onset aetiology alters clinical characteristics. Diabet Med (2002) 19(9):758–61. doi:10.1046/j.1464-5491.2002.00766.x
122. Urbanová J, Rypáčková B, Procházková Z, Kučera P, Cerná M, Anděl M, et al. Positivity for islet cell autoantibodies in patients with monogenic diabetes is associated with later diabetes onset and higher HbA1c level. Diabet Med (2014) 31:466–71. doi:10.1111/dme.12314
123. Thanabalasingham G, Shah N, Vaxillaire M, Hansen T, Tuomi T, Gašperíková D, et al. A large multi-centre European study validates high-sensitivity C-reactive protein (hsCRP) as a clinical biomarker for the diagnosis of diabetes subtypes. Diabetologia (2011) 54(11):2801–10. doi:10.1007/s00125-011-2261-y
124. Pontoglio M, Prié D, Cheret C, Doyen A, Leroy C, Froguel P, et al. HNF1 alpha controls renal glucose reabsorption in mouse and man. EMBO Rep (2000) 1(4):359–65. doi:10.1093/embo-reports/kvd071
125. Ellard S, Bellanné-Chantelot C, Hattersley AT. Best practice guidelines for the molecular genetics diagnosis of maturity onset diabetes of the young. Diabetologia (2008) 51:546–53. doi:10.1007/s00125-008-0942-y
126. Ellard S, Allen HL, Franco ED, Flanagan SE, Hysenaj G, Colclough K, et al. Improved genetic testing for monogenic diabetes using targeted next-generation sequencing. Diabetologia (2013) 56:1958–63. doi:10.1007/s00125-013-2962-5
127. Johansson S, Irgens H, Chudasama KK, Molnes J, Aerts J, Roque FS, et al. Exome Sequencing and Genetic Testing for MODY. PLoS ONE (2012) 7(5):e38050. doi:10.1371/journal.pone.0038050
128. Kavvoura FK, Owen KR. Maturity onset diabetes of the young: clinical characteristics, diagnosis and management. Pediatr Endocrinol Rev (2012) 10:234–42.
129. Nyunt O, Wu J, McGown I, Harris M, Huynh T, Leong GM, et al. Investigating maturity onset diabetes of the young. Clin Biochem Rev (2009) 30:67–70.
130. Bonner C, Nyhan KC, Bacon S, Kyithar MP, Schmid J, Concannon CG, et al. Identification of circulating microRNAs in HNF1A-MODY carriers. Diabetologia (2013) 56:1743–51. doi:10.1007/s00125-013-2939-4
131. McDonald TJ, MC Eneny J, Pearson ER, Thanabalasingham G, Szopa M, Shields BM, et al. Lipoprotein composition in HNF1A-MODY: differentiation between HNF1A and type 2 diabetes. Clin Chem Acta (2012) 413:927–32. doi:10.1016/j.cca.2012.02.005
132. Owen KR. Monogenetic diabetes: old and new approaches of diagnosis. Clin Med (2013) 13(13):278–81. doi:10.7861/clinmedicine.13-3-278
133. Shepherd M, Shields B, Ellard S, Rubio-Cabezas O, Hattersley AT. A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin-treated patients. Diabet Med (2009) 26:437–41. doi:10.1111/j.1464-5491.2009.02690.x
134. Raile K, Schober E, Konrad K, Thon A, Grulich-Henn J, Meissner T, et al. Treatment of young patients with HNF1A mutations (HNF1A-MODY). Diabet Med (2015) 32:526–30. doi:10.1111/dme.12662
135. Rubio-Cabezas O, Hattersley AT, Njolstad PR, Mlynarski W, Ellard S, White N, et al. The diagnosis and management of monogenetic diabetes in child and adolescence. Pediatr Diabetes (2014) 15:47–64. doi:10.1111/pedi.12192
136. Østoft SH, Bagger JI, Hansen T, Pedersen O, Faber J, Holst JJ, et al. Glucose-lowering effects and low risk of hypoglycemia in patients with maturity-onset diabetes of the young when treated with a GLP-1 receptor agonist: a double-blind, randomized, crossover trial. Diabetes Care (2014) 37(7):1797–805. doi:10.2337/dc13-3007
137. Tuomi T, Honkanen EH, Isomaa B, Sarelin L, Groop LC. Improved prandial glucose control with lower risk of hypoglycemia with nateglinide than with glibenclamide in patients with maturity-onset diabetes of the young type 3. Diabetes Care (2006) 23(2):189–94. doi:10.2337/diacare.29.02.06.dc05-1314
Keywords: maturity-onset diabetes of the young, gene mutation, diabetes, hyperglycemia, sulfonylureas, insulin
Citation: Firdous P, Nissar K, Ali S, Ganai BA, Shabir U, Hassan T and Masoodi SR (2018) Genetic Testing of Maturity-Onset Diabetes of the Young Current Status and Future Perspectives. Front. Endocrinol. 9:253. doi: 10.3389/fendo.2018.00253
Received: 16 October 2017; Accepted: 02 May 2018;
Published: 17 May 2018
Edited by:
Jan Polák, Charles University, CzechiaReviewed by:
Ludmila Brunerova, Charles University, CzechiaHelena Barbosa Sampaio, Universidade Estadual de Campinas, Brazil
Copyright: © 2018 Firdous, Nissar, Ali, Ganai, Shabir, Hassan and Masoodi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bashir Ahmad Ganai, YmJjZ2FuYWlAZ21haWwuY29t