 Carola Rotermund
Carola Rotermund Gerrit Machetanz
Gerrit Machetanz Julia C. Fitzgerald
Julia C. Fitzgerald- 1German Centre for Neurodegenerative Diseases, Tübingen, Germany
- 2Department of Neurodegenerative Diseases, Centre of Neurology and Hertie Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany
The search for treatments for neurodegenerative diseases is a major concern in light of today's aging population and an increasing burden on individuals, families, and society. Although great advances have been made in the last decades to understand the underlying genetic and biological cause of these diseases, only some symptomatic treatments are available. Metformin has long since been used to treat Type 2 Diabetes and has been shown to be beneficial in several other conditions. Metformin is well-tested in vitro and in vivo and an approved compound that targets diverse pathways including mitochondrial energy production and insulin signaling. There is growing evidence for the benefits of metformin to counteract age-related diseases such as cancer, cardiovascular disease, and neurodegenerative diseases. We will discuss evidence showing that certain neurodegenerative diseases and diabetes are explicitly linked and that metformin along with other diabetes drugs can reduce neurological symptoms in some patients and reduce disease phenotypes in animal and cell models. An interesting therapeutic factor might be how metformin is able to balance survival and death signaling in cells through pathways that are commonly associated with neurodegenerative diseases. In healthy neurons, these overarching signals keep energy metabolism, oxidative stress, and proteostasis in check, avoiding the dysfunction and neuronal death that defines neurodegenerative disease. We will discuss the biological mechanisms involved and the relevance of neuronal vulnerability and potential difficulties for future trials and development of therapies.
Introduction
The evolution of genomics has greatly advanced our understanding of the genetic contribution to neurodegenerative diseases and provided an entry point for studying the biological cascades leading to neuronal degeneration. The growing research areas of bioinformatics and systems medicine have also opened up opportunities for better targeted treatments and individualized therapies. However, even for diseases such as Alzheimer's and Parkinson's disease, in which much progress has been made, a clear link between genetics, underlying pathological processes and the resulting clinical phenotype seldom exists. Neurodegenerative diseases are currently incurable, debilitating conditions caused by the progressive degeneration and death of nerve cells and their prevalence is rising in today's society (1).
Therefore, despite substantial advances in the development of symptomatic treatments for Alzheimer's disease (AD) and Parkinson's disease (PD) (Figure 1), there is still a major need for novel therapeutic strategies and disease-modifying treatments.
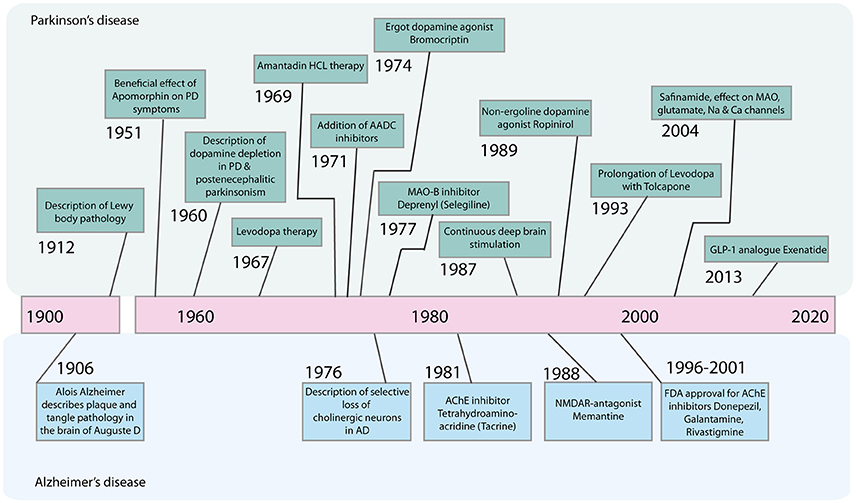
Figure 1. Timeline of major advances in the treatment of Parkinson's disease and Alzheimer's disease in the last century. AADC, Aromatic L-amino acid decarboxylase; AChE, Acetylcholinesterase; GLP-1, Glucagone-like Peptide 1; HCL, Hydrochloride; MAO-B, Monoamine oxidase B; NMDAR, N-Methyl-D-Aspartate Receptor.
Given the complex and heterogeneous molecular basis of neurodegenerative disease the task can appear overwhelming and the previous decades have seen mostly disappointing clinical trial outcomes and subsequent lack of financial investment.
Gallega officinalis (French lilac) contains glucose-lowering guanidines and has been used for treatment of diabetes for centuries. The derivate metformin is a biguanide which was introduced in Europe in the 1950s and in the United States in the 1990s (2, 3). Metformin has recently been reported to decrease cardiovascular risk, restore ovarian function in polycystic ovary syndrome, reduce hepatic lipogenesis, fatty liver disease, and reduce oxidative stress (3, 4). The mechanisms by which metformin exerts its effects are still not fully defined but it is known that metformin inhibits glucose production in the liver and increases glucose uptake in peripheral tissues thereby lowering blood glucose levels (3, 5, 6). It is also accepted that metformin slows mitochondrial respiration via its direct action on complex I of the respiratory chain of mitochondria.
The Therapeutic Potential of Metformin: Rationale
Metformin has the potential to interfere with neuronal longevity mechanisms and is therefore an interesting drug since it has already been approved for human use. However, human aging research in general has been slowed down by the lack of good aging models that can be used in the laboratory. Retaining aging signatures in reprogrammed neurons has been made possible by the use of direct reprogramming protocols (7, 8) but this may not be feasible for some research groups and time is needed for the technologies to be established in non-expert laboratories. New, simple and affordable methods to investigate the role of aging in human cells are still greatly needed.
Nonetheless, data from human and animal studies regardless of cell type have shown that dysregulation of insulin function contributes to aging and the development of neurodegenerative diseases (9). Insulin resistance and diabetes are increasingly recognized as a contributor to disease development especially in the field of dementias (10–12). Therefore, the rationale for using metformin is its potential to slow aging processes by acting on mitochondrial metabolism and insulin signaling. Slowing the aging process will be beneficial because quality of life could be improved in old age by delaying disease.
A link between diabetes and neurodegenerative diseases is for the most part accepted, although data is not unequivocal, and the exact mechanisms are unclear. A large body of data on metformin use in humans and animals with neurodegenerative diseases exists but metformin's therapeutic use is not yet accepted since the results are often conflicting. These different outcomes are dependent on disease, model system, species and the underlying biological pathways involved, which are now briefly reviewed.
Dementia
Dementia is a common neurological disease of heterogeneous origin and the most important risk factor is aging. Dementia affects memory and other cognitive functions, interfering with a person's ability to carry out routine daily activities. According to the UN world population prospects, the number of persons aged 60 or over on the globe is estimated to grow approximately four times over the next 30 years (13) bringing the prediction that diagnoses of dementia will also rise. The most common form of dementia is AD but there are other types of dementia including vascular dementia, mixed dementia, frontotemporal dementia, dementia with Lewy bodies, and Parkinson's disease dementia.
Alzheimer's Disease
Alzheimer's disease (AD) is the most common neurodegenerative disease, with 45 million people worldwide affected (14). AD is characterized by progressive memory loss and decline of cognitive function.
Neurofibrillary tangles (NFTs, composed of abnormal tau protein) and amyloid plaques [composed of extracellular aggregates of amyloid-β (Aβ)] are pathological hallmarks of the AD brain (15–17). The NFT protein tau is associated with microtubules and is responsible for their stabilization (18). Tau pathology and synaptic loss correlates with cognitive impairments in AD patients (19). The amyloid plaque component Aβ derives from the sequential cleavage of the membrane protein APP (Amyloid precursor protein) by β-secretase BACE1 (β-site amyloid precursor protein cleaving enzyme 1) and the γ-secretase complex (20). Dysregulation, abnormal modification, and build-up of these protein structures in the brain are thought to be the major pathologies underlying AD.
From a genetic standpoint, most forms of AD are sporadic and of late onset but familial forms of early onset AD exist and are commonly caused by mutations in APP or presenilin (21–23). The underlying biological mechanisms leading to sporadic forms of AD have still not been defined. Inflammatory response, hormone regulation, mitochondrial dysfunction, and lysosomal dysfunction have been implicated, to name only a few processes. There is also growing genetic evidence for microglial involvement (24–26). Still, the main risk factors for developing AD are aging, genetic risk factors including being an APOE-ε4 allele carrier, variants in TREM2, and several GWAS loci, traumatic brain injury, cardiovascular risk factors, and several environmental risk factors (27–31).
Diabetes and Dementia: Animal Models
Most of the rodent models used to investigate the role of insulin and glucose metabolism in dementia have focused on AD. Insulin signaling and glucose tolerances are altered in APP/PS1 mice fed a high fat diet (32, 33), in partially leptin deficient (db/+)-APP/PS1 mice (34) and APP23-(ob/ob) mice (35). APP load may therefore boost susceptibility to disturbances of energy metabolism.
A high fat diet induces insulin resistance and promotes amyloidosis and memory impairment in both the Tg2576 mouse model of AD as well as in APP transgenic mice (36, 37). High fat diets or obesity could contribute to memory deficits even in wild type animals. However, some studies reviewed by Agusti and colleagues had no effect on cognition at all (38) leaving the topic still debated because of conflicting data. Diabetic rats show increased levels of APP, Aβ, and phosphorylated tau (39). These data suggest that alteration of energy metabolism via insulin signaling may contribute to Aβ generation and altered tau phosphorylation, two well know biochemical events associated with dementias. The modulation of insulin has been proven to be an effective strategy to protect neurons and synapses against toxic Aβ oligomers and to improve cognition in other AD animal models (40, 41). For example, glucagon-like peptide-1 (GLP-1), insulin-like growth factor 1 (IGF-1) as well as caloric restriction have all been shown to exert neuroprotective effects (42–44).
Metformin and Dementia: Animal Models
Until now only few animal studies have assessed the effect of metformin on cognitive decline and the results are not in line (Table 1). There are many different ways in which researchers can modulate energy metabolism in rodents to try to induce cognitive impairments and perhaps this has contributed to the variable data on metformin in this context. Some animals are fed high fat diets, others such as the (db/db) mice have a spontaneous mutation that cause them to be insulin resistant and obese. In three such high fat diet studies, metformin treatment reduces cognitive deficits (57, 58, 60), but one study found no improvement (59). In (db/db) mice, one study found metformin improved memory (53) whereas another study found no effect (55). It should be noted that in one study looking at normal aging in wild type mice, metformin had a detrimental effect on memory impairment (61). In this study activation of AMPK by phosphorylation was not measured and therefore it is not clear whether the metformin diet in these animals was optimal. More studies with proper controls are clearly needed to understand the effect of metformin in normal aging.
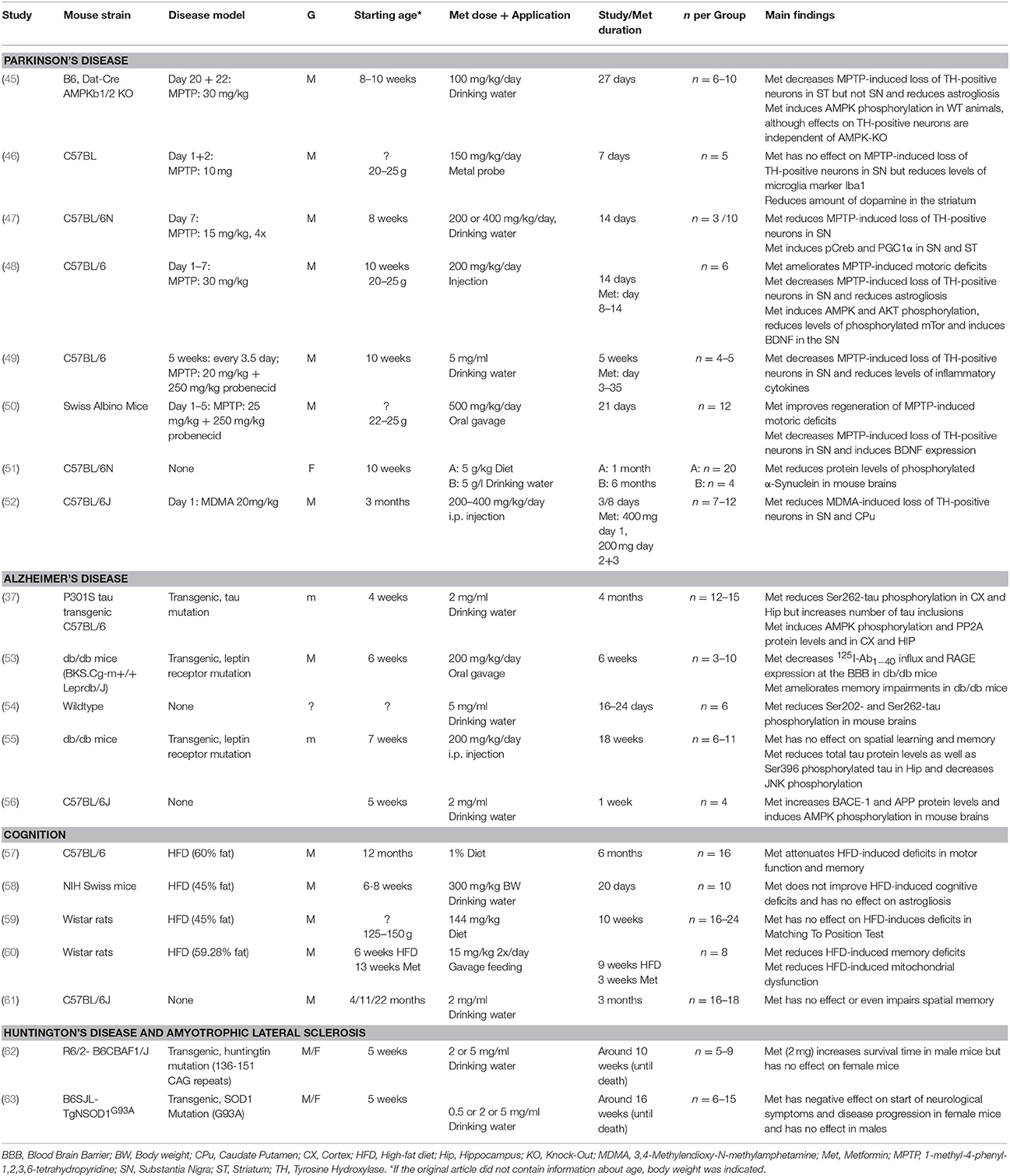
Table 1. Studies investigating the effect of metformin on neurodegeneration in rodent models.
It also seems that metformin is capable of simultaneously having both a negative and positive impact on specific biochemical events within the same disease model. For example, in a P301S tauopathy mouse model, metformin treatment reduced tau phosphorylation but promoted tau aggregation (37). The authors suggest that metformin could be beneficial as a dephosphorylating agent but could promote protein aggregation, the latter being unquestionably the more widely accepted neurodegenerative disease pathology. Similarly, short term metformin treatment again reduced tau phosphorylation but had negative effects since it activates APP and BACE-1 (54, 56). Metformin again seems to have positive effects on reducing total tau and tau phosphorylation at serine 236, whereas the sulfonylurea type diabetes drug glibenclamide performed much better in similar tests (53, 55).
Sex may also influence metformin action, which could complicate the interpretation of animal data. Male rodents are often favored and sometimes the sex of the animals used is either overlooked or omitted entirely. In one metformin study already mentioned, male mice showed impaired cognitive function while female mice were improved after treatment (36).
Diabetes and Dementia: Human Studies
Changes in cognition have been reported in type 2 diabetes mellitus (T2DM) patients who have not received a diagnosis of dementia and meta-analyses have found moderate but significant deficits across cognitive domains (64–66). T2DM also seems to increase the risk of conversion from mild cognitive impairment to dementia and the conversion from amnestic mild cognitive impairment to AD (67).
Brain imaging studies in T2DM patients have shown a reduction of whole and regional gray matter volume including hippocampal volume when compared to non-diabetics (68, 69). Taken together, the clinical data mostly shows that T2DM patients have an increased likelihood of developing dementia (10, 70–73). The relationship between diabetes and dementia is further strengthened by reports that reversely, AD patients have an increased risk of developing T2DM or impaired glucose tolerance (74–76). Furthermore, post mortem brain pathology in AD shows decreased insulin receptors and IGF protein levels, and insulin levels and markers of insulin signaling are altered in the brain (77–80). Hyperglycemia and hyperinsulinemia have also been positively correlated with AD pathology (75, 81, 82). However, it must be stated that the vast majority of neuropathological studies did not find any association between T2DM or indeed glucose levels and extent of AD pathology (83–87) and two studies even suggest a negative association (88, 89).
One explanation for the discrepancy between clinical and neuropathological studies in AD is the influence of vascular pathology. It is now established that concomitant pathologies in the aging brain are rather the rule than the exception (90). The fact that most studies show no association between T2DM and Aβ deposition therefore seems to hint that there is no major effect of T2DM on Alzheimer's pathology. An additional effect of small vessel disease on cognition in patients with T2DM and Alzheimer's pathology could explain the higher likelihood to develop dementia in this group. This implies that even if T2DM does not have a large impact on Alzheimer's pathology the proper management of diabetes in AD is relevant (67, 91). An interplay between T2DM and Alzheimer's beyond vascular pathology should not be disregarded though especially considering evidence on shared pathophysiological features.
Metformin and Dementia: Human Studies
Results from clinical studies assessing the effect of metformin use on cognitive decline and AD mostly show a positive influence (Table 2). Metformin use is associated with significantly lower risk of cognitive impairment in T2DM (102, 107). The incidence of dementia in general is lower in T2DM patients receiving metformin, sulfonylurea or a combination of both drugs compared to those not receiving oral anti-hyperglycemic agents (96). The risk of developing AD was lower in diabetics receiving metformin than in patients receiving sulfonylurea or thiazolidinediones in two studies (97, 101). However, in a single study, long-term use of metformin for T2DM (though not sulfonylureas or thiazolidinediones) was associated with higher risk of developing AD (103). One informative study used latent class analysis to identify groups of men with T2DM receiving metformin who develop different profiles of comorbidities including dementia. They concluded that the effect of metformin may in fact differ depending on the risk-profile of patient receiving the drug (100).
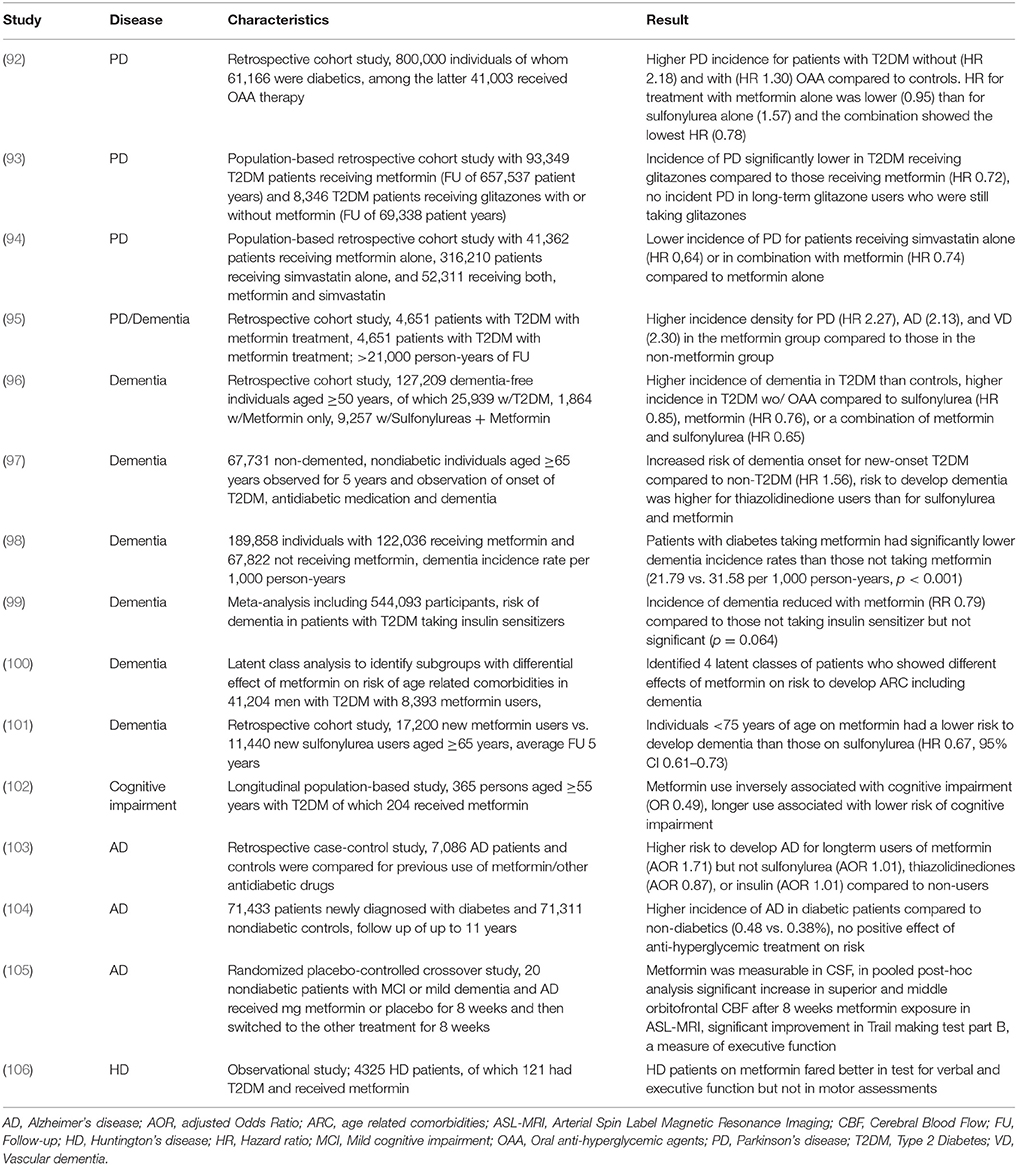
Table 2. Studies evaluating the effect of metformin on incidence and progression of neurodegenerative diseases.
In an interventional study, Luchsinger and colleagues investigated the effect of metformin given daily for 12 months compared to placebo in overweight patients with amnestic mild cognitive impairment. There was improvement in the selective reminding test in the group receiving metformin but not in other cognitive or biomarker outcomes (108). The results were only marginally significant and there was no correction for multiple measurements which at least suggests that the observed improvement must be confirmed in an independent trial. In another interventional, short-term metformin study, nondiabetic patients with mild cognitive impairment or mild dementia due to AD took metformin or a placebo for 8 weeks. Those taking metformin significantly improved in a measure of executive function but not in other cognitive tests or biomarkers. Again, a multitude of test was performed without correction for multiple testing (105).
Although the majority of data on metformin use in dementia with or without T2DM is generally positive, it should be considered that the effect of metformin likely depends on complex underlying pathological processes and may to some extent be related to an effect on vascular rather than neurodegenerative processes. In some instances, metformin could even exert detrimental effects. Prospective interventional studies have not been able to show convincing evidence of a positive effect of metformin in mild cognitive impairment or mild Alzheimer's dementia but were likely underpowered or of too short duration. More, long-term, controlled metformin studies in large, well-defined dementia cohorts are needed.
Parkinson's Disease
Background
Parkinson's disease (PD) is a common neurodegenerative disease, affecting over 1% of the population above the age of 60 and around 4% older than 85 (109). PD is characterized by bradykinesia and a combination of rigidity, resting tremor, postural instability, and a large range of non-motor symptoms (110). Like other neurodegenerative diseases, PD is clinically and pathologically heterogeneous, with a large variation in disease onset and progression. Progressive loss of dopamine-containing neurons in the substantia nigra pars compacta, located in the mid brain, results in a deficit of dopamine in the striatum (111, 112). Insoluble protein inclusions in neurons, termed Lewy bodies, mainly consisting of aggregated α-synuclein (aSyn) are the main neuropathological hallmark of PD (113). Lewy bodies and protein aggregates are found in multiple brain regions and spread with disease progression (114, 115). The exact biological mechanism leading to aSyn aggregation and neuronal loss remains unknown and currently only the symptoms of PD are treated with dopamine-replacement therapy and in some cases deep brain stimulation.
Approximately 5–15% of PD cases can be attributed to disease-causing genetic variants and around 15% of patients have a first degree relative who is also affected (116). The genetic architecture of PD has been well studied but it is complex. 23 loci and 19 genes have so far been associated with familial forms of PD (117). Like in most neurodegenerative diseases, the majority of cases probably result from a complex interplay of risk modifying genetic variation, environmental factors and chance. Knowledge about the genes involved in PD have allowed insight into the underlying biological pathways. Together with multiple environmental factors and epidemiological data, the genetic data has highlighted several cellular functions and pathways including mitochondrial dysfunction, lysosomal function, inflammation, build-up of aggregation-prone proteins and oxidative stress (118, 119).
Despite large investments in research for neuroprotective compounds for PD, none have so far shown any convincing effects in clinical trials (120).
Diabetes and PD: Animal Studies
Rodent studies have shown that there is a link between insulin resistance and development of PD. A high fat diet enhanced vulnerability to the neurotoxins 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6-hydroxydopamine (6-OHDA) as measured by increased nigrostriatal neurodegeneration and motor deficits (121, 122). Likewise, in an aSyn mouse model of PD a high fat diet led to an accelerated development of locomotor phenotype and earlier onset of neurodegeneration (123).
Insulin resistance can directly interfere with dopamine signaling. Rats fed a high fat diet exhibit impaired nigrostriatal dopamine function (124) and overweight and diabetic mice show degeneration of dopaminergic neurons (125).
Metformin and PD: Animal Studies
Only a handful of rodent studies have so far assessed the effects of metformin as a neuroprotective agent in PD. These studies have focused mainly on metformin treatment in combination with acute MPTP induced parkinsonism. Although experimental designs in these studies are quite similar, the results are variable, arguing against differences in modeling as the major cause of metformin's variable effects. However, differences in the dose and duration of MPTP and metformin treatments may be important (Table 1).
Most studies in rodents find that metformin reduces the damaging effect of MPTP on dopaminergic neurons, shown by tyrosine hydroxylase staining (a marker for dopaminergic neurons) in the substantia nigra pars compacta (49, 50), striatum (45), or both (48). Two studies suggest that metformin's protective effect may not be specific. A study by Ismaiel and colleagues however, reported that metformin had no protective effect against MPTP-induced neuronal loss in the SN (46) and Bayliss reported no protective effect on dopaminergic neurons in the SN, only in striatum (45).
Metformin's supposed ability to protect against dopaminergic neuronal death induced by the neurotoxin MPTP correlates in three studies to improvements in the motor function of rodents (48–50). Given that both, MPTP and metformin act on complex 1 of the respiratory chain, a mutual influence of the drugs on mitochondrial survival cannot be excluded. It is possible that in these studies metformin primarily reduced the damaging effects of MPTP itself rather than restoring damaged neurons. Therefore, examination of metformin's action in transgenic mouse models rather than acute toxin models of PD might give better insight about its potential. An interesting first hint comes from a study using healthy non-transgenic mice that showed that metformin could reduce aSyn phosphorylation in the brain (51).
Diabetes and PD: Human Studies
Studies assessing the risk of developing PD in patients with diabetes have very mixed outcomes (126–132). In one meta-analysis comprising 14 case-control studies, PD risk was decreased in T2DM patients (133). Conversely, Cereda and colleagues describe an increased risk for developing PD in diabetics in four prospective cohort studies but not a higher prevalence of diabetes in in patients with PD in five case-control trials (134). It has to be noted that the case-control trials with the largest populations did consistently show a similar of even higher prevalence of diabetes in PD compared to controls. More recently a meta-analysis including seven population-based cohort studies which also found an increased PD risk in patients with diabetes (135). Taken together the meta-analyses seem to hint toward an increased incidence of PD in T2DM. A potential pitfall is the inclusion of vascular PD in some of the studies. T2DM does contribute to cerebral small vessel disease and therefore non-exclusion of patients with vascular lesions may skew the results toward more patients with T2DM exhibiting signs of parkinsonism. This particular problem was addressed in some studies showing an increased incidence of PD in T2DM and therefore cannot sufficiently explain the discrepancies. From a neuropathological view one study describes an association between increased blood glucose levels with increased risk of Lewy body formation in the substantia nigra pars compacta and locus coeruleus further supporting a role of T2DM in the pathogenesis of PD (136).
Dementia with Lewy bodies (DLB) and Parkinson's disease dementia (PDD) are common causes of dementia in the elderly (137). PD patients with T2DM are reported to have a greater rate of cognitive decline and lower gray matter and white matter volume, although the group was small (138). PDD patients are more likely to show insulin resistance in an oral glucose-tolerance-test than PD patients without dementia (139). DLB and PDD were less common in patients with diabetes in one study using data from the Swedish Dementia Registry (140), yet T2DM was not significantly associated with PDD in many others (141–144).
Metformin in PD: Human Studies
Clinical studies have not looked solely at metformin, but rather metformin compared to, or in combination with other oral anti-hyperglycemic agents (see Table 2). Taken together all the studies look at different medications and are hardly comparable. There is lack of clinical data that suggests a positive effect of metformin on PD risk. Wahlqvist and colleagues tried to determine the effect of sulfonylurea, metformin or a combination of both drugs on the incidence of PD in patients with T2DM. Patients with T2DM receiving sulfonylurea had an increased PD risk compared to those not receiving oral anti-hyperglycemic agents. Metformin alone or in combination with sulfonylurea had no impact, suggesting that metformin might rescue the harmful effect of sulfonylurea (92).
Brakedal and colleagues compared the incidence of PD in patients with T2DM from the Norwegian Prescription Database (NorPD) receiving glitazones with or without metformin or metformin alone. Patients taking glitazones had a significantly lower incidence of PD compared to patients on metformin alone. There was no risk reduction in past users of glitazones, indicating the necessity of long-term or even permanent exposure to glitazones for risk reduction (93). Looking at patients from the NorPD receiving statins, metformin or both showed a lower hazard ratio to develop PD for patients using statins in combination with metformin when compared to metformin alone and the risk was lowest in patients only taking statins (93). The positive effect of statins may come through their anti-inflammatory effect and a reduction of microglial inflammatory response, which has been shown to have a positive effect on striatal dopamine activity (145). The question why metformin seems to have a positive effect when added to sulfonylurea while it has a negative effect when taken together with statins must be addressed. The combination of T2DM and hypercholesterolemia might increase the risk of developing PD more than hypercholesterinemia alone and this risk may not be lowered sufficiently by a combination of statins and metformin. The addition of metformin to sulfonylurea may result in a better control of T2DM than the therapy with sulfonylurea alone thereby reducing effects that promote PD risk. Also, the complex interplay between the different drugs has to be taken into account.
To our knowledge there is no data available on metformin use and disease progression. It is also unclear whether metformin use in individuals without insulin resistance may have a beneficial effect on PD development.
Other Neurodegenerative Diseases
There are to our knowledge very few or no reports of metformin studies in other, rarer neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), Huntington disease (HD), motor neuron disease, or atypical parkinsonian disorders. Here we briefly note relevant studies concerning association with diabetes or use of drugs targeting energy metabolism.
Amyotrophic Lateral Sclerosis
ALS is a progressive neurodegenerative disease that is characterized by degeneration of the first and second motor neuron resulting in spasticity and muscle atrophy. Eventually this results in difficulty speaking, swallowing, and breathing and often leads to death within a few years after diagnosis. Neurochemical imbalance and genetic mutations are known to cause ALS, but most cases are sporadic and old-age is an important risk factor. Most drugs available for ALS relieve symptoms only, although the drug riluzole and more recently edaravone have been shown to slow progression of the disease (146, 147).
A protective effect of diabetes in older patients and an increased risk of developing ALS in younger patients with diabetes has been described which is thought to reflect differences in association of ALS with T1DM and T2DM (148, 149). Most studies have shown a decreased risk for developing ALS in patients with T2DM (150, 151). However, other studies reported no significant effect on ALS risk or progression and even a higher risk of developing ALS in T2DM in patients below 65 years of age (152–154). Nutritional status is negatively associated with ALS severity (155) and hypercaloric nutrition has even been suggested as a potential treatment option for ALS. Two trials with the PPAR-γ agonist Pioglitazone (which reduces insulin resistance) (156, 157) have not shown any benefit in disease progression (158).
Huntington Disease
HD is a progressive neurodegenerative disease that causes choreatic movements, psychiatric symptoms, and cognitive decline. The most common form of the disease is of early onset, usually diagnosed around 30–40 years of age. HD is caused by defects in the gene HTT, which encodes the protein huntingtin and the mode of inheritance is autosomal dominant. Expansion of CAG repeats in the HTT gene leads to the production of an abnormally long version of the huntingtin protein. This results in the protein being broken down by the cell into small, toxic fragments and these protein fragments aggregate and accumulate in neurons causing the disease.
Altered glucose metabolism and increased rates of T2DM have been reported in patients with HD (159, 160) and a high prevalence of T2DM has been reported in a Chinese family with HD (161). However, other studies were not able to identify differences in oral glucose tolerance test or pancreatic tissue between HD patients and controls (162, 163). HD patients with T2DM receiving metformin had better cognitive test results than HD patients without diabetes not taking metformin. This was in stark contrast to the non-HD control group where people with T2DM taking metformin fared worse in the cognitive test compared to non-diabetic controls (106).
Metformin: Mechanism of Action in Neurodegenerative Diseases
The in vivo studies conducted so far, regarding the effect of metformin have generated conflicting results. Besides the large differences in study design, these outcomes are probably also due to the many biological pathways influenced by metformin. Here we will discuss some of the biological signaling pathways and biological mechanisms that are the most relevant for metformin's potential as a therapy in neurodegenerative disease (Figure 2).
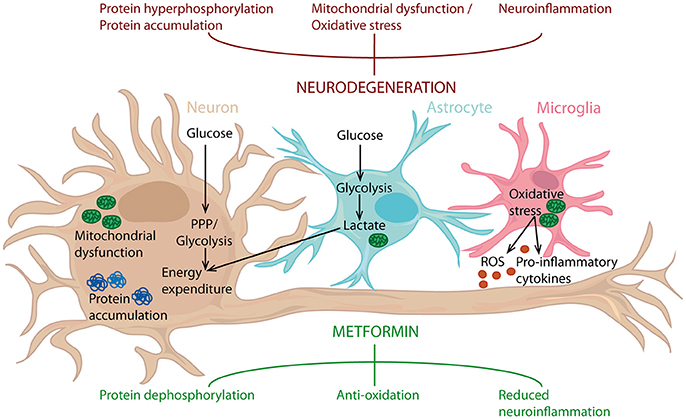
Figure 2. Metformin's potential as a neuroprotective agent. Metformin can counteract protein hyperphosphorylation, oxidative stress and neuroinflammation, processes known to drive neurodegeneration. Metformin can act on neurons, but also targets astrocytes and microglia. Consequently, metformin can influence inflammatory status, along with glucose metabolism in the entire brain and thereby reduce neuroinflammation and act as an antioxidant, leading to protein dephosphorylation. PPP, Pentose phosphate pathway.
Central Metabolism and Signaling
Central metabolism is tied to the overarching cell signaling pathways involved in proliferation, stress and survival, which are heavily implicated in human diseases including cancer and neurodegeneration. Metformin acts on central metabolism and several major signaling pathways including energy sensing (glucose metabolism and AMPK signaling), mTOR signaling, and inflammatory signaling. Mitochondrial signaling will be addressed separately.
Energy Sensing and Metabolism
The brain constitutes only 2% of the total body mass, but it is one of the main energy-demanding organs in the human body utilizing around 20% of total energy expenditure. Brain cells incorporate (i) the neurons (70–80% of brain energy expenditure) and (ii) glial cells, comprising oligodendrocytes, astrocytes and microglia (accounting for the remaining 20–30% of energy expenditure). The high energy demand of neurons is one of several factors partially explaining the selective vulnerability of certain neuronal subtypes in neurodegenerative diseases. Energy metabolism has long since been thought to play a role in the etiology of neurodegenerative diseases and here we will briefly mention some of the related signaling pathways and biological mechanisms that are relevant for metformin's therapeutic potential in neurodegeneration.
AMPK signaling
AMPK is an evolutionarily conserved sensor of cellular energy status. AMPK is activated by increasing AMP levels in conditions of energy deprivation and the enzyme consequently inhibits energy consumption and stimulates catabolic pathways. Activation of AMPK has a wide range of effects, including inhibition of mTor and PI3K-Akt signaling (two important pathways discussed later).
Dysregulation of AMPK is associated with insulin resistance and T2DM (164, 165) and neuroinflammation (166–168). AMPK signaling plays a major role in AD disease progression since AMPK has been shown to regulate both Aβ generation and tau phosphorylation. Inhibition of Aβ production and tau phosphorylation in neuronal cultures is dependent on AMPK activation (169) and activation of AMPK lowers extracellular Aβ accumulation (170). Conversely, in neurons, AMPK activation has been linked to tau phosphorylation as a response to Aβ toxicity (171, 172).
Metformin inhibits complex I of the electron transport chain needed for mitochondrial respiration, thereby leading to an energy deficit and indirectly activating the AMPK pathway (173–175). Thus, stimulation of AMPK can be seen as a key consequence of metformin administration, explaining many of the known effects of the drug (Figure 3).
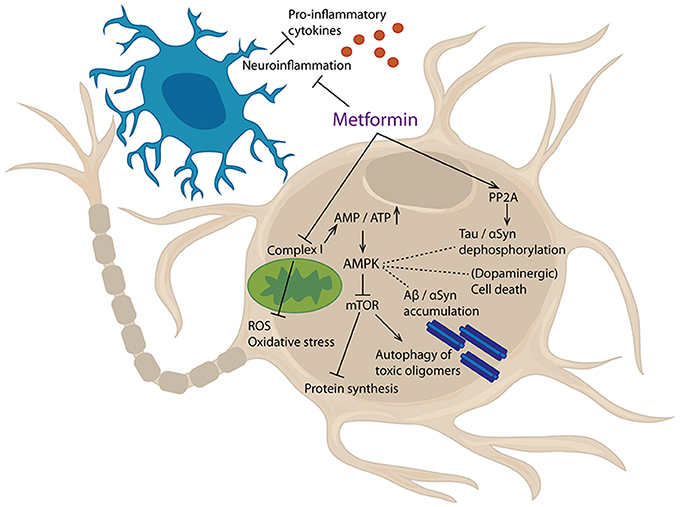
Figure 3. Cellular targets of metformin. Metformin inhibits mitochondrial complex I, thereby increasing AMP/ATP ratio. This lack of energy leads to an activation of AMPK, which, amongst others, inhibits mTor signaling. Furthermore, metformin can activate PP2A and inhibit neuroinflammatory processes. Results of these events are reduced production of pro-inflammatory cytokines and reactive oxygen species (ROS), decreased oxidative stress, inhibition of protein synthesis and augmented autophagy of toxic oligomers. Additionally, protein dephosphorylation, protein aggregation, and cell death are affected.
However, in the context of AD especially, more studies are needed to understand the complex role of AMPK signaling and the action of metformin. A study conducted in human neuronal stem cells proposed that activation of AMPK via metformin is neuroprotective against Aβ (176) and other in vitro studies showed that metformin is able to reduce tau phosphorylation via mTOR/PP2A (Protein phosphates 2A) signaling (54) and that it can reduce molecular pathologies associated with AD (177). An additional level of modulation via AMPK by metformin could come from metformin's ability to reduce BACE1 protein levels in neurons (178). Conversely, metformin was also reported to upregulate BACE1 in neurons and increase the generation of Aβ (179), suggesting detrimental effects of activating AMPK in diseased neurons.
In PD mouse models the AMPK involvement is similarly multifarious. Administration of the neurotoxin MPTP activates AMPK signaling (180). Interestingly, both AMPK overexpression and AMPK inhibition have promoted survival in neurotoxin treated PD models (180) but another study provided evidence for a protective function of AMPK activation in in vivo PD models (181).
Overexpression of aSyn in cell culture reduced AMPK activity, while inhibition of AMPK lowered resistance to aSyn toxicity (182). AMPK's subunits α1 and α2 have neuroprotective effects against aSyn toxicity with low but continuous AMPK activity almost completely preventing loss of dopaminergic neurons (183). Accordingly, in rodent PD models dietary metformin influenced neuronal function via AMPK modulated aSyn phosphorylation status (49, 51). However, several other studies point in a different direction. Over-active AMPK promotes aSyn accumulation (184) and hyperactivation of AMPK leads to aSyn binding to the GTPase PIKE-L and dopaminergic cell death (47). These studies show that lower AMPK activity may in fact be beneficial at least in aSyn models of PD.
As is the case in many neurodegenerative diseases, the underlying genetic and biological causes are heterogeneous, often causing multiple pathologies that can overlap across the disease spectrum. The action of metformin primarily via the mitochondria could have numerous and potentially opposite effects on AMPK depending on the amount of involvement and type of mitochondrial signaling in each patient or disease model at any given moment. One important aspect to consider here is that biological pathways are not necessarily fixed in a single state throughout the disease course. Neurons especially have evolved to carefully adapt to energetic needs in order to survive since they are seldom replaced. Sophisticated compensatory mechanisms are initiated for the purpose of mitochondrial rejuvenation and adaption. Such complexity has made modeling neurodegenerative diseases in human neurons challenging and has contributed to the current situation where no causative or “cure all” therapies are available.
Glucose metabolism
Glucose is an essential energy substrate needed to sustain neuronal activity and is taken up via glucose transporters expressed in the brain endothelium, astrocytes, and neurons (185). Neurons mostly rely on glucose for energy but utilize ketone bodies during fasting. In contrast to other cell types, in neurons the rate limiting glycolytic enzyme Phosphofructokinase B3 is highly turned over by the proteasome, resulting in the preferential metabolism of glucose via the pentose phosphate pathway (PPP) as opposed to glycolysis (119, 186).
A product of the PPP is the electron donor NADPH, which provides reducing power for anabolic reactions and is crucial for maintaining antioxidant potential. The PPP helps neurons to meet high energy demands, but since neurons are predominantly oxidative, maintaining a fine balance between glycolysis and PPP is essential for counteracting oxidative damage and conserving energy.
Glial cells on the other hand, predominantly metabolize glucose via glycolysis producing lactate and have only very low rates of mitochondrial oxidation. Glia metabolically support axons and lactate can be shuttled across a gradient from glia to neurons (Figure 2) (187, 188). Interestingly, in cell culture, neurons favor lactate over glucose (189) preferring a fast supply of energy over metabolic efficiency. In the human brain, energy demand must be tightly regulated to offset oxidative damage and therefore cell culture and cell culture media effects should be taken into consideration when considering the conflicting data on metformin performed in situ.
Inhibiting the PPP and glutathione pathways causes increased levels of oxidative stress and cell death similar to that seen during neurodegeneration (119). Glucose hypometabolism has been shown in PD brains (190) and deregulation of glucose metabolism has been proposed as an early event in the pathogenesis of PD (119). Dunn et al. proposed that dysregulation of glucose metabolism occurs via dysregulation of the PPP, which causes oxidative stress because of less efficient glutathione recycling, and it is this event that underlies the increased levels of oxidative stress observed in PD (119).
Metformin can act in these pathways by slowing oxidative phosphorylation via inhibition of complex I in mitochondria and by inhibiting gluconeogenesis, having the effect of further aiding neurons to reduce their oxidative burden by minimalizing NADH utilization.
Insulin signaling
Insulin plays an important role in the brain. It is used as a hormonal signal to control body weight, food uptake, and metabolic homeostasis (191–193). Insulin has also been shown to influence expression of dopamine receptors and concentration of dopamine (194–196). Disturbances in insulin signaling have been implicated in several neurodegenerative diseases including AD, PD, and HD (197–200). Insulin is secreted in response to high blood sugar and acts in different organs including the brain. Activation of the Phosphoinositide-3-kinase (PI3K)—Akt pathway via insulin receptor activation and insulin receptor substrates plays a central role in the metabolic actions of insulin (201). Akt activation regulates proteins such as mTOR, FOXO, and BAD. Overall, Akt has over 100 known substrates and has diverse effects on cellular growth, cell proliferation, glucose uptake, protein synthesis, glycogen synthesis, and apoptosis (202). Akt is inhibited by PP2A (203), PHLPP1/2 (204), and indirectly by PTEN (205). Insulin resistance has been associated with disturbances in signaling up and downstream of Akt (206–208).
Insulin has been administered to patients to try to improve symptoms of neurodegeneration (209, 210) and has been shown to protect cells from Aβ induced death (211–213). The Insulin Degrading Enzyme (IDE), originally found to play an important role in insulin turnover (214) is involved in Aβ degradation. IDE can degrade secreted Aβ from neurons and microglia and mediate its clearance (215). Furthermore, IDE hypofunction can contribute to in vivo Aβ accumulation (216). In hippocampi of ApoE4 carriers reduced expression levels of IDE have been measured (217) and genetic differences in IDE expression and activity have been suggested to be involved in AD development (218–221). Reduced levels of IDE in liver and brain have been correlated with aging (222) and IDE can counteract damage from oxidative stress, suggesting a neuroprotective role (223–226).
Metformin lowers blood glucose levels through inhibition gluconeogenesis in the liver via AMPK (227, 228). AMPK inhibits PI3K/Akt signaling, the crucial pathway downstream of the insulin and IGF1 receptors (229). Metformin has also been shown to act on insulin signaling independently of AMPK. Metformin is reported to downregulate expression of insulin and IGF-1 receptors (230, 231) and reduces phosphorylation of insulin receptors (232) including IRS-1 (230, 233).
Both acute and chronic metformin administration has been found to increase levels of GLP-1, an incretin known to induce insulin secretion, in humans and mice (234–236). Very recently a randomized, double-blind, placebo-controlled trial for PD showed that a GLP-1 agonist had positive effects on motor symptoms in PD (237), generating a new potential mechanism for metformin action in neurodegeneration.
mTOR Signaling
mTOR signaling is a highly conserved and central signaling pathway integrating upstream signals such as nutrient and redox status and then controlling downstream processes such as cellular growth, motility, survival, and death (238). The mTor pathway is crucial for regulating mitochondrial biogenesis and autophagy, two processes that are defective in many neurodegenerative diseases.
mTOR is a serine/threonine protein kinase, composed of the protein complexes mTORC1 and mTORC2. mTOR signaling is targeted by the PI3K/Akt pathway, the key insulin signaling pathway (239, 240). Both PTEN (241, 242) and AMPK (243, 244) suppress mTor signaling and rapamycin is a well-studied inhibitor of mTORC1 (245–247). Although mTor signaling influences many downstream events, the most important mechanism of action is through the phosphorylation and activation of S6K1 and 4E-BP1 and subsequent control of RNA translation (238) (Figure 4). Interestingly, deficiency in mTor signaling has been implicated with insulin resistance and diabetes. Nutrient dependent stimulation of S6K1 can induce insulin resistance (248, 249) and S6K1 deficiency protects against high fat diet-induced insulin resistance (250).
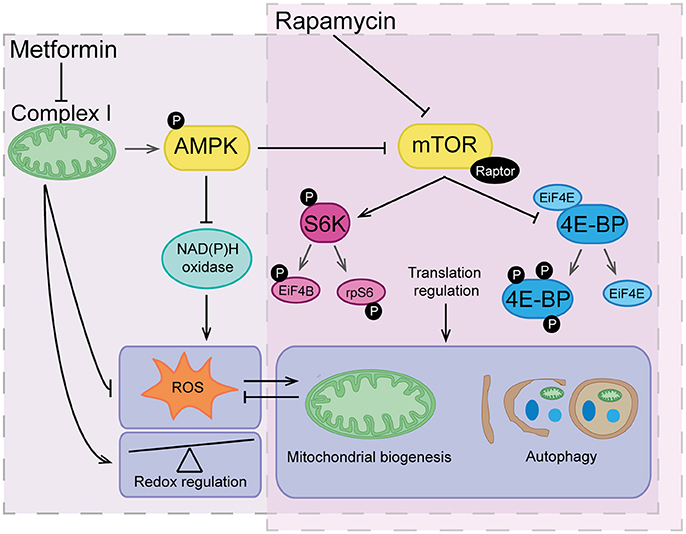
Figure 4. The overlapping actions of metformin and rapamycin. Rapamycin acts by directly inhibiting mTOR and therefore translation regulation, which has a major influence of highly regulated processes such as mitochondrial biogenesis and autophagy. Metformin acts indirectly on the mTOR pathway through inhibition of complex I and activation of AMPK signaling. Metformin also reduces reactive oxygen species (ROS) via inhibitory action on complex I and NAD(P)H oxidase having an overall effect as a redox regulator. Downstream of metformin action, low level ROS can indirectly trigger signals for mitochondrial biogenesis and turnover of organelles and proteins via autophagy. Vice versa, maintenance of healthy mitochondrial networks involving autophagy and mitochondrial biogenesis further reduces build-up of damaging levels of ROS.
The mTOR inhibitor rapamycin suppresses neurodegeneration phenotypes in mice (251) and protects against MPTP-induced loss of dopaminergic neurons (252). Rapamycin also prevents the development of dyskinesia without affecting the therapeutic efficacy of L-DOPA and thus, the mTORC1 signaling cascade represents a promising target for the design of anti-Parkinsonian therapies (253).
Elevated mTOR signaling has been found in AD patients and is linked to diabetes and aging (254, 255). Rapamycin abolishes cognitive deficits and reduces Aβ levels in a mouse model of AD (256). It also ameliorates AD-related phenotypes by restoring hippocampal gene expression signatures (257). Importantly, mTor regulates tau phosphorylation and degradation (258), making this pathway an interesting target for the treatment of tauopathies.
If we compare the therapeutic potential of metformin, a well-known inhibitor of mTOR signaling through activation of AMPK (259) to that of rapamycin, which is more widely accepted in the field, the obvious difference is that metformin action on mTOR is relatively indirect. Rapamycin forms a complex with the FKBP12 binding protein which binds and specifically alters mTORC1. Metformin acts on the mTOR pathway indirectly via multiple routes. The AMPK independent routes include inhibition of transcription factors (260), the PI3K/AKT pathway (261), and induction of REDD (262). In direct contrast to this, one study has shown that metformin can directly inhibit mTORC1 and is dependent on Rag GTPases not AMPK (263). These data support the view that metformin has more than one direct target and is likely to have many more indirect targets, thus explaining why the use of metformin and a research tool or treatment is less accepted than rapamycin.
Nevertheless, the mTOR pathway links several biological pathways underlying neurodegenerative diseases and therefore the ability of metformin to inhibit this signaling cascade endorses the argument that more mechanistic work using metformin and its inclusion in clinical trials should be positively considered.
Inflammation
Neuroinflammation is considered a major driving force in the progression of neurodegenerative diseases and the triggering of innate immune mechanisms is emerging as a crucial component in disease pathogenesis. Microglia and other cell types in the brain can be activated in response to misfolded proteins or aberrantly localized nucleic acids. This diverts microglia from their physiological and beneficial functions, and leads to their sustained release of pro-inflammatory mediators (264).
Intake of non-steroidal anti-inflammatory drugs (NSAIDs) has been reported to decrease incidence of AD later in life (265, 266) and activated microglia are found in brains of AD patients (267, 268).
In AD, an integrated network-based approach identified gene perturbations associated with innate immune pathways and microglia cells in late onset forms of the disease (269). AD patients show increased expression of inducible nitric oxide synthase (iNOS, a product of neuroinflammation) in neurons and glia, leading to augmented nitric oxide production (270, 271). Activated microglia can further induce tau phosphorylation in primary mouse neurons, activating IL1β receptor and p38 MAPK stress signaling (272).
In PD, patients show increased numbers of activated microglia and astrocytes (273, 274) and microglia activation has been associated with disease progression (273, 275, 276). aSyn has been found to activate microglia, enhancing neurotoxicity (277). Activation of microglia increases nitration of aSyn, resulting in neuronal cell death (278).
Immune signaling triggers transcriptional events, but also changes in metabolic flux, redox balance, and metabolite balance via mitochondria (279). Mitochondrial dysfunction is associated with neuroinflammation (280) and even moderate mitochondrial DNA stress can trigger antiviral signaling (281).
Metformin reduces general inflammation parameters and inhibits NF-κB signaling as well as proinflammatory cytokines in different cell types (282–285), suggesting that metformin could protect against neuroinflammation. Interestingly, in two MPTP-induced PD mouse models, metformin reduced levels of the microglia marker Iba1 as well as the pro-inflammatory cytokines TNFα, Il-1β, IL-6, and iNOS in the substantia nigra pars compacta (46, 49). Here, more studies are needed but metformin seems to have a wholly positive effect against general inflammation. Neuroinflammation is a recognized event associated with neurodegenerative diseases and therefore metformin could be both a useful tool and therapy.
Mitochondria
Mitochondria are crucial organelles that produce energy and perform a plethora of other functions needed for central metabolism and cell signaling. Mitochondrial dysfunction is a phenomenon that traverses all neurodegenerative diseases and forms the basis of β-cell dysfunction in T2DM (286). One important aspect of mitochondrial dysfunction in neurological disease is that the need for tightly controlled energy metabolism in neurons can partially explain some of the vulnerabilities involved in their demise.
Parkinson's Disease
In PD, the link between mitochondrial dysfunction and disease has been proven by the identification of environmental factors and disease genes which critically affect mitochondria. The outcome has been a large body of work depicting the role of mitochondrial dysfunction in PD, yet the exact mechanisms underlying sporadic forms of PD are less defined.
Loss of function mutations in PINK1 or parkin cause PD (287–290) as a result of mitochondrial dysfunction and this has been elucidated in vitro (291, 292) and in vivo (293–296). PINK1 and parkin act in a pathway that is important for mitophagy (removal of damaged mitochondria via the lysosome) induced by mitochondrial depolarization. Here, PINK1 functions upstream of parkin (295, 297). Upon mitochondrial damage, PINK1 accumulates on the mitochondrial surface and selectively recruits parkin to mitochondria (298, 299). Mitochondrial substrates are ubiquitinylated, leading to the removal of damaged mitochondria. PINK1 is now known to be a ubiquitin kinase (300) but may have other functions yet unknown. For example, PINK1 is not required for basal mitophagy in vivo (301, 302) and has been proposed to regulate complex I (303), mitochondrial dynamics (304), mitochondrial proteostasis (305), and mitochondrial metabolism via TRAP1 (306, 307).
PINK1 and parkin are upregulated under metabolic stress in the vessel walls of obese and diabetic mice and have a protective action by limiting reactive oxygen species (ROS) production and mitochondrial dysfunction (308). In a diabetic mouse model, PINK1 expression in the hippocampus was in this case reduced following hydrogen peroxide treatment (309), further suggesting that PINK1 plays a role as a stress sensor and functions accordingly in diverse ways. PINK1 is generally associated with neuroprotection since loss of function causes PD, but because PINK1 is normally highly turned over at the mitochondrial outer membrane and therefore overexpression and/or altered expression might also induce unwanted downstream events. In one study, PINK1 overexpression restrained MAPK and ROS signaling and mitigated insulin resistance in cell models (310). Conversely, PINK1 loss corrupts function of islet and β-cells causing impaired glucose uptake and increased levels of plasma insulin (311). Further evidence that PD proteins play important roles in energy metabolism is a study showing that TP53INP1 deficient cells (TP53INP1 is a susceptibility locus in T2DM) causes an increase in ROS that impairs mitophagy via the PINK1-parkin pathway (312).
Parkinson's disease mutations in aSyn are associated with several cellular defects, including reduced mitochondrial integrity and function. Recent work has identified a highly neurotoxic aSyn species which induces mitochondrial damage and mitophagy in the human and animal brain (313). However, the consequences of these mitochondrial changes for bioenergetic functions remains somewhat undefined. Interestingly, aSyn toxicity is mitigated by TRAP1 (314), a mitochondrial ATPase that has been linked to metformin.
In this pathway, TRAP1 and the mitochondrial serine protease HtrA2 are both targets of the PD protein PINK1 (305, 306). HtrA2 and TRAP1 genetic variants have been found in PD patients (307, 315) but the mutations are rare and a controversial topic (316–318). Regardless of the genetic contribution to disease, TRAP1 at least appears to play an important regulatory role in mitochondria that is relevant for the fine tuning of energy metabolism. TRAP1 is well studied in cancer since TRAP1 expression is tightly regulated in tumor cells (319), TRAP1 acts as a metabolic switch (320) by targeting and inhibiting succinate dehydrogenase (321), which is important for metabolic re-purposing and inflammatory responses (322).
In ovarian cancer where TRAP1 expression was altered, metformin was effective in rendering the tumor sensitive to chemotherapy (323), suggesting that metformin might be relevant to TRAP1 mediated signaling. On this basis, metformin was then used to successfully rescue mitochondrial dysfunction in a TRAP1 cell model of PD (307). In a healthy person, fine tuning of mitochondrial energy usage via the PINK1-HtrA2-TRAP1 pathway and other regulatory mechanisms may allow cells to conserve energy and reduce oxidative burden. Metformin's ability to mimic this fine tuning role in vitro was beneficial in one model of sporadic PD (307). However, there are still a lot of questions that remain unanswered such as whether metformin is beneficial in non-diseased neurons, aging neurons and other forms of familial and sporadic PD. One question is whether metformin could specifically target energetic deficits in the dopaminergic neurons of the substantia nigra pars compacta. The question is not yet answered because selective vulnerability is still not yet fully understood. We can speculate that oxidative or metabolic burden over time could contribute to making these cells especially vulnerable. Many redox reactions happen in mitochondria as a result of mitochondrial activity. Neurons in comparison to many other cell types have a high energy demand and because of the autonomous pacemaking in dopaminergic neurons of the substantia nigra (324), these cells are thought to have a higher oxidative burden. The metabolism of dopamine itself is highly oxidative and can form several toxic species. Therefore, if metformin can mildly reduce the oxidative burden at the mitochondria without interfering with normal redox signaling and stimulate autophagy and other processes which can become less effective over time, it could be seen as a very useful drug to counteract neurodegenerative diseases. Neurons have a sophisticated and unique line of quality control defenses which allow them to compensate for stress and survive against all odds because once they die, inflammation often ensues and they are seldom replaced. It just depends whether metformin treatment could be used to intervene at the right time to not interfere with necessary compensatory responses, rather enhance them.
Alzheimer's Disease
The exact mitochondrial events leading to AD are less defined than in PD, yet aging is still the greatest known risk factor. Energy metabolism and mitochondrial dysfunction have been proposed as a primary event in mechanisms underlying AD such as synaptic degeneration, Aβ deposition and formation of neurofibrillary tangles (325). There is a vast amount of evidence that mitochondrial dysfunction occurs after the early cellular events in AD and can contribute to the advancement of further degeneration, but it is often unclear whether mitochondrial dysfunction is indeed just a secondary event or whether it might be involved in primary pathogenesis. For example, in the case of tau, abnormal tau triggers oxidative stress and mitochondrial defects such as mitochondrial depolarization, impaired mitochondrial complex activities and reduced energy output (326, 327). Tau also localizes to the microtubules, the tracks on which mitochondria move along with the help of adapter proteins and defective mitochondrial movement has been shown in several models of AD (328, 329).
There is also evidence that mitochondrial metabolism is altered in AD brains (reviewed in (330)). The tricarboxylic acid (TCA) cycle enzymes pyruvate dehydrogenase, isocitrate dehydrogenase, and alpha-ketoglutarate dehydrogenase are affected in AD brain tissue and in patient-derived fibroblasts (331). Changes in these checkpoint TCA cycle enzymes are associated with metabolic re-wiring often in response to stress and redox changes. In addition to matrix enzymes, deficiencies in oxidative phosphorylation (OXPHOS) have been reported [reviewed in (332)].
In AD research, there are few mechanistic models for mitochondrial dysfunction, mainly due to the fact that there are no mitochondrial causative genes for AD. The mitochondrial mechanism of metformin action in dementia and AD is likely similar in PD, in that metformin can act on mitochondrial quality control via mitochondrial biogenesis and energy conservation.
The Complex I Paradox
Many of metformin's actions are thought to be an indirect result of complex I inhibition. The exact inhibitory mechanism of metformin on complex I is not fully understood. The inhibitory mechanisms of other complex I inhibitors such as MPTP and rotenone are better known in terms of binding site and mechanism of toxicity, especially in disease.
Complex I deficiency has long since been associated with mitochondrial dysfunction and Parkinson's disease risk [for a review see (333)]. Complex I deficiencies have also been reported in AD, HD and ALS (332). The neurotoxins MPTP and rotenone inhibit complex I and generate toxic levels of ROS, which leads to neuronal cell death. It is possible that sub-lethal concentrations of mitochondrial inhibitors that do not generate ROS (or generate less ROS) could be beneficial but little is known.
It is generally accepted that metformin does not generate dangerous levels of ROS. Pharmacologically reducing oxidative phosphorylation and thus the oxidative burden (at the right moment) without generating too much ROS is certainly a challenge. We found that sub lethal concentrations of the specific mitochondrial complex V inhibitor oligomycin, could rescue mitochondrial dysfunction in a TRAP1 deficient PD model to a similar extent as metformin (307) but since metformin is an approved compound for human consumption, we followed up the protective effects of metformin only. It might be interesting to assess the potential neuroprotective action and toxicity with a titration of several respiratory chain inhibitors that act at different sites. For example, the mitochondrial complex III inhibitor, antimycin A is known to generate large amounts of ROS (334), but oligomycin and other disrupters of the respiratory chain have been shown to generate little or no ROS (335).
Aging
The main hallmarks of aging set out by Lopez-Otin are genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication (336). All of these hallmarks in one way or another are associated with the pathogenesis of neurodegenerative diseases. Here we will focus attention on some specific aspects relating to these hallmarks that could be the most relevant to metformin's mechanism of action at the mitochondria.
Mitonuclear protein imbalance
Human mitochondrial DNA (mtDNA) is bound inside nucleoid bundles, has a high copy number, is inherited maternally and has a high mutation rate (337). Mitochondrial damage and/or depletion induces stress-signaling and adaptive metabolic responses. MtDNA instability is a physiologically relevant stress observed in many human diseases and aging (281). Mitonuclear protein imbalance, is a stoichiometric imbalance between nuclear and mitochondrially encoded proteins and is activated as a key longevity response across many species (338). Alterations to mtDNA are directly linked to respiratory chain dysfunction in sporadic PD patients and it has been shown that complex I is initially affected followed next by complex IV (339). It is thought that imbalance in the stoichiometry between mitochondrially translated proteins and nuclear encoded ones is both a cellular signal and marker of mitochondrial adaption. The mTOR inhibitor rapamycin is used as a tool to initiate mitonuclear protein imbalance (338) and metformin is capable of modulating mitonuclear protein imbalance in human cells (307).
Oxidant stress and senescence
The production of reactive species is usually balanced by the cell's antioxidant defenses. An imbalance in the amount of ROS to antioxidant defense results in oxidative stress and can cause damage to proteins, lipids and nucleotides.
Mitochondria are a major source of ROS due to oxygen use in energy production through the electron transport chain. Electrons leak while they are being transferred along the complexes of the electron transport chain. Leaked electrons can react with molecular oxygen to form superoxide radicals. Superoxide can react with Mn-SOD to form hydrogen peroxide, a ROS and a signaling molecule. Hydrogen peroxide is either broken down to form water or it can react with metals to form the highly reactive hydroxyl radical. In mitochondria the main leakage sites are at the transfer of four electrons to oxygen at Complex IV, but also complex I, complex III and via certain dehydrogenases of the TCA cycle in the mitochondrial matrix. Consequences of oxidative stress include proliferation, adaption, damage, senescence, or death depending on the cell type and severity [for review see (340, 341)]. Neurons need to counteract a great deal of ROS because of high energy bursts and catecholamine neurotransmitter metabolism.
Oxidative damage is a major contributor to neurodegenerative diseases [for a review see (342)]. Both oxidative stress and oxidative damage can lead to stress adaption. One such adaptive mechanism in mitochondria might be finely-tuned inhibition of respiratory complexes or mitochondrial uncoupling via uncoupling proteins. There is mounting evidence that mitochondrial uncoupling proteins are neuroprotective [for a review see (343)]. Cellular senescence can occur when adaptive responses are unable to properly protect key molecules from damage to the extent that a cell can no longer divide.
The PD protein DJ-1 provides a link between neurodegeneration and energy metabolism. DJ-1 acts as a chaperone and protease to stabilize mitochondria and protect cells from oxidative stress (344). Several other cellular functions have been attributed to DJ-1, including; binding of Ras as a transcriptional co-activator (345), negative regulation of the phosphoinositide-3-kinase (PI3K)/AKT signaling cascade through inhibition of PTEN (309, 346, 347), chaperone function (348, 349), and RNA binding (350). Although controversial, DJ-1 has also been claimed to have glyoxalase (351) and deglycase (352) enzyme activities (353). DJ-1 also influences insulin secretion as well as β cell viability in the pancreas and DJ-1 knockout mice show increased ROS levels in islet cells, impaired glucose tolerance and decreased insulin secretion (354).
Gaps in the Research
Trials using metformin to treat or protect against neurodegenerative diseases in humans and animals have produced mostly conflicting results. The data shows either positive, no or even detrimental effects of metformin on neurodegenerative processes in cell cultures, animals and humans.
The outcome may depend on the species, cell type or underlying metabolic state. Two promising research areas however are neuroinflammation and aging, yet more work is needed. Very few studies have looked directly at the role of metformin in neuroinflammation, but since this is a growing research focus in the field, more metformin studies may arise. The exact role of metformin in aging is a question that needs to be at least partly understood before we can progress further in understanding its potential to treat neurodegenerative diseases. A major hurdle to this is the lack of good human aging models mainly in vitro but also in vivo.
Another gap in the knowledge is whether there are potential adverse effects of metformin use in non-diabetics. For example, it has been well documented that long term metformin use leads to vitamin B12 deficiency (355). Vitamin B12 and folate are needed for transmethylation and hydroxylation reactions from amino acids that are crucial for neurotransmitter biosynthesis. How much influence could this have in a patient with disturbed neurotransmitter metabolism and/or those receiving other medications.
The Therapeutic Potential of Metformin: Feasibility
There are several reasons why the use of metformin to treat neurodegeneration could bring about doubt from clinicians and scientists when considering its potential as a therapy or as a research tool. The main point being that metformin seems to be acting on a plethora of biological pathways, and therefore it is very difficult to pin down mechanisms. The second point is the controversial subject of “anti-aging” drugs in general. Since we know very little about the biological underpinnings of aging and know even less about how to efficiently model it in the laboratory, the promotion of an “anti-aging” drug often conjures up more questions than it answers. Then there are several other sticking points among researchers, one being the fact that metformin acts by inhibiting mitochondrial respiration, the exact effect that has been shown by years of research in the Parkinson's disease field to in fact contribute development of disease.
In direct contrast, there are several arguments for metformin being a feasible and useful drug. Firstly, glucose metabolism is of central importance to neuronal redox status, therefore to the long-term survival of neurons. Secondly, as a population we are increasingly insulin resistant and therefore metformin is particularly apt. Metformin is a cheap and safe drug with few side effects and therefore more work in vitro, in vivo and in trials will be welcomed.
Nir Barzilai, the director of the Institute for Aging Research at the Albert Einstein College of Medicine suggests that metformin and other related drugs can extend our years of healthy, disease-free living by decades (356). Other scientists have not specifically mentioned metformin but in his 2005 book on mitochondria, Nick Lane suggests that if we live longer to rid ourselves of diseases of old age we need more mitochondria and perhaps a more refined free-radical detection system (357). Whether metformin is capable of modifying the detection system at the right physiological moment without deleterious effects is at least an exciting possibility.
Future Developments in the Field
There is potential that metformin could be beneficial in the task of counteracting aging and clinical studies imply that metformin may have positive effects on cognition in T2DM patients. A better understanding of how metformin works will help researchers in the neurodegeneration field to successfully design future research and trials. Upcoming studies such as TAME (358) will help in this respect.
The anti-aging effects of metformin could be summarized by its ability to interfere with the multistage process of energy production without producing damaging amounts of ROS. This action alone could be seen as neuroprotective and metformin may further protect by activating other biological pathways. For example, slowing mitochondrial energy production can also trigger a cascade of signaling events in the liver that result in reduced glucose and insulin. The key role of insulin in nutrient sensing which balances growth and proliferation with life-extending conservation, makes metformin an interesting drug. The field of aging research is growing and in vivo and in vitro aging models are advancing.
Probably due to the complexity of metformin action, this drug will not likely serve as a potential treatment for neurodegenerative diseases on the current stage because much more work is needed to understand the role of aging in different neurodegenerative disease forms. The greatest value of metformin today might lie in its potential to help decipher those mechanisms underlying neurodegeneration.
Author Contributions
CR, GM, and JF contributed equally to the writing and the editing of the manuscript. All authors approved the final version and submission of this article.
Funding
CR is funded by the German Center for Neurodegenerative Diseases (DZNE). GM is supported by an Edmond J. Safra Fellowship in Movement Disorders from the Michael J. Fox Foundation. JF acknowledges funding from the German Research Council (DFG), German Federal Ministry of Education and Research (BMBF) support code 031 A 430 A, DZNE, The University of Tubingen and the EU Joint Programme—Neurodegenerative Disease Research (JPND) project.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Manu Sharma, Jonas Neher, Anna Schaedler, and Laura Dunn for critically reviewing the manuscript.
References
1. Feigin VL, Abajobir AA, Abate KH, Abd-Allah F, Abdulle AM, Abera SF, et al. Global, regional, and national burden of neurological disorders during 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. (2017) 16, 877–97. doi: 10.1016/S1474-4422(17)30299-5
2. Goodarzi MO, Bryer-Ash M. Metformin revisited: re-evaluation of its properties and role in the pharmacopoeia of modern antidiabetic agents. Diabetes Obes Metab. (2005) 7:654–65. doi: 10.1111/j.1463-1326.2004.00448.x
3. Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci. (2012) 122:253–70. doi: 10.1042/CS20110386
4. Lamanna C, Monami M, Marchionni N, Mannucci E. Effect of metformin on cardiovascular events and mortality: a meta-analysis of randomized clinical trials. Diabetes Obes Metab (2011) 13:221–8. doi: 10.1111/j.1463-1326.2010.01349.x
5. Rojas LB, Gomes MB. Metformin: an old but still the best treatment for type 2 diabetes. Diabetol Metab Syndr. (2013) 5:6. doi: 10.1186/1758-5996-5-6
6. Patrone C, Eriksson O, Lindholm D. Diabetes drugs and neurological disorders: new views and therapeutic possibilities. Lancet Diabetes Endocrinol. (2014) 2:256–62. doi: 10.1016/S2213-8587(13)70125-6
7. Mertens J, Paquola ACM, Ku M, Hatch E, Bohnke L, Ladjevardi S, et al. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell (2015) 17:705–18. doi: 10.1016/j.stem.2015.09.001
8. Kim Y, Zheng X, Ansari Z, Bunnell MC, Herdy JR, Traxler L, et al. Mitochondrial aging defects emerge in directly reprogrammed human neurons due to their metabolic profile. Cell Rep. (2018) 23:2550–8. doi: 10.1016/j.celrep.2018.04.105
9. Craft S, Stennis Watson G. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. (2004) 3:169–78. doi: 10.1016/S1474-4422(04)00681-7
10. Ninomiya T. Diabetes mellitus and dementia. Curr Diab Rep. (2014) 14:487. doi: 10.1007/s11892-014-0487-z
11. Neumann KF, Rojo L, Navarrete LP, Farias G, Reyes P, Maccioni RB. Insulin resistance and Alzheimer's disease: molecular links & clinical implications. Curr Alzheimer Res. (2008) 5:438–47. doi: 10.2174/156720508785908919
12. Verdile G, Fuller SJ, Martins RN. The role of type 2 diabetes in neurodegeneration. Neurobiol Dis. (2015) 84:22–38. doi: 10.1016/j.nbd.2015.04.008
13. United Nations DoEaSA Population Division. World population prospects: The 2015 revision, key findings and advance tables. In: Working Paper No ESA/P/WP241 (2015).
14. Prince M, Comas Herrera A, Knapp M, Karagiannidou M, Guerchet M. World Alzheimer Report 2016: Improving Healthcare for People with Dementia. Coverage, Quality and Costs Now and in the Future. London: Alzheimer's Disease International (ADI) (2016).
15. Brion JP, Couck AM, Passareiro E, Flament-Durand J. Neurofibrillary tangles of Alzheimer's disease: an immunohistochemical study. J Submicrosc Cytol. (1985) 17:89–96.
16. Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. (1986) 83:4913–7. doi: 10.1073/pnas.83.13.4913
17. Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. (2012) 425:534–9. doi: 10.1016/j.bbrc.2012.08.020
18. Johnson GV, Stoothoff WH. Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci. (2004) 117:5721–9. doi: 10.1242/jcs.01558
19. Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology (1992) 42:631–9. doi: 10.1212/WNL.42.3.631
20. Sun X, Bromley-Brits K, Song W. Regulation of beta-site APP-cleaving enzyme 1 gene expression and its role in Alzheimer's disease. J Neurochem. (2012) 120(Suppl). 1:62–70. doi: 10.1111/j.1471-4159.2011.07515.x
21. Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature (1991) 349:704–6. doi: 10.1038/349704a0
22. Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science (1995) 269:973–7. doi: 10.1126/science.7638622
23. Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature (1995) 375:754–60. doi: 10.1038/375754a0
24. Hardy J, Bogdanovic N, Winblad B, Portelius E, Andreasen N, Cedazo-Minguez A, et al. Pathways to Alzheimer's disease. J Intern Med. (2014) 275:296–303. doi: 10.1111/joim.12192
25. Efthymiou AG, Goate AM. Late onset Alzheimer's disease genetics implicates microglial pathways in disease risk. Mol Neurodegener. (2017) 12:43. doi: 10.1186/s13024-017-0184-x
26. Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer's disease. Nat Neurosci. (2017) 20:1052–1061. doi: 10.1038/nn.4587
27. Kivipelto M, Ngandu T, Fratiglioni L, Viitanen M, Kareholt I, Winblad B, et al. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch Neurol. (2005) 62:1556–60. doi: 10.1001/archneur.62.10.1556
28. Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Lince DH, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer's disease. J Clin Psychiatry (2007) 68:613–8. doi: 10.4088/JCP.v68n0419
29. Baumgart M, Snyder HM, Carrillo MC, Fazio S, Kim H, Johns H. Summary of the evidence on modifiable risk factors for cognitive decline and dementia: a population-based perspective. Alzheimer's Dement. (2015) 11:718–26. doi: 10.1016/j.jalz.2015.05.016
30. Killin LO, Starr JM, Shiue IJ, Russ TC. Environmental risk factors for dementia: a systematic review. BMC Geriatr. (2016) 16:175. doi: 10.1186/s12877-016-0342-y
31. Li W, Risacher SL, McAllister TW, Saykin AJ. Traumatic brain injury and age at onset of cognitive impairment in older adults. J Neurol. (2016) 263:1280–5. doi: 10.1007/s00415-016-8093-4
32. Mody N, Agouni A, McIlroy GD, Platt B, Delibegovic M. Susceptibility to diet-induced obesity and glucose intolerance in the APP (SWE)/PSEN1 (A246E) mouse model of Alzheimer's disease is associated with increased brain levels of protein tyrosine phosphatase 1B (PTP1B) and retinol-binding protein 4 (RBP4), and basal phosphorylation of S6 ribosomal protein. Diabetologia (2011) 54:2143–51. doi: 10.1007/s00125-011-2160-2
33. Ruiz HH, Chi T, Shin AC, Lindtner C, Hsieh W, Ehrlich M, et al. Increased susceptibility to metabolic dysregulation in a mouse model of Alzheimer's disease is associated with impaired hypothalamic insulin signaling and elevated BCAA levels. Alzheimer's Dement. (2016) 12:851–61. doi: 10.1016/j.jalz.2016.01.008
34. Jimenez-Palomares M, Ramos-Rodriguez JJ, Lopez-Acosta JF, Pacheco-Herrero M, Lechuga-Sancho AM, Perdomo G, et al. Increased Abeta production prompts the onset of glucose intolerance and insulin resistance. Am J Physiol Endocrinol Metab. (2012) 302:E1373–80. doi: 10.1152/ajpendo.00500.2011
35. Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci USA. (2010) 107:7036–41. doi: 10.1073/pnas.1000645107
36. DiTacchio KA, Heinemann SF, Dziewczapolski G. Metformin treatment alters memory function in a mouse model of Alzheimer's disease. J Alzheimer's Dis. (2015) 44:43–8. doi: 10.3233/JAD-141332
37. Barini E, Antico O, Zhao Y, Asta F, Tucci V, Catelani T, et al. Metformin promotes tau aggregation and exacerbates abnormal behavior in a mouse model of tauopathy. Mol Neurodegener. (2016) 11:16. doi: 10.1186/s13024-016-0082-7
38. Agusti A, Garcia-Pardo MP, Lopez-Almela I, Campillo I, Maes M, Romani-Perez M, et al. Interplay between the gut-brain axis, obesity and cognitive function. Front Neurosci. (2018) 12:155. doi: 10.3389/fnins.2018.00155
39. Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes (2007) 56:1817–24. doi: 10.2337/db07-0171
40. Pedersen WA, McMillan PJ, Kulstad JJ, Leverenz JB, Craft S, Haynatzki GR. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol. (2006) 199:265–73. doi: 10.1016/j.expneurol.2006.01.018
41. De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, et al. Protection of synapses against Alzheimer's-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci USA. (2009) 106:1971–6. doi: 10.1073/pnas.0809158106
42. Piriz J, Muller A, Trejo JL, Torres-Aleman I. IGF-I and the aging mammalian brain. Exp Gerontol. (2011) 46:96–9. doi: 10.1016/j.exger.2010.08.022
43. Salcedo I, Tweedie D, Li Y, Greig NH. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: an emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br J Pharmacol. (2012) 166:1586–99. doi: 10.1111/j.1476-5381.2012.01971.x
44. Mattson MP, Longo VD, Harvie M. Impact of intermittent fasting on health and disease processes. Ageing Res Rev. (2017) 39:46–58. doi: 10.1016/j.arr.2016.10.005
45. Bayliss JA, Lemus MB, Santos VV, Deo M, Davies JS, Kemp BE, et al. Metformin prevents nigrostriatal dopamine degeneration independent of AMPK activation in dopamine neurons. PLoS ONE (2016) 11:e0159381. doi: 10.1371/journal.pone.0159381
46. Ismaiel AA, Espinosa-Oliva AM, Santiago M, Garcia-Quintanilla A, Oliva-Martin MJ, Herrera AJ, et al. Metformin, besides exhibiting strong in vivo anti-inflammatory properties, increases mptp-induced damage to the nigrostriatal dopaminergic system. Toxicol Appl Pharmacol. (2016) 298:19–30. doi: 10.1016/j.taap.2016.03.004
47. Kang H, Khang R, Ham S, Jeong GR, Kim H, Jo M, et al. Activation of the ATF2/CREB-PGC-1alpha pathway by metformin leads to dopaminergic neuroprotection. Oncotarget (2017) 8:48603–18. doi: 10.18632/oncotarget.18122
48. Katila N, Bhurtel S, Shadfar S, Srivastav S, Neupane S, Ojha U, et al. Metformin lowers alpha-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson's disease. Neuropharmacology (2017) 125:396–407. doi: 10.1016/j.neuropharm.2017.08.015
49. Lu M, Su C, Qiao C, Bian Y, Ding J, Hu G. Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of parkinson's disease via autophagy and mitochondrial ROS clearance. Int J Neuropsychopharmacol. (2016) 19:pyw047. doi: 10.1093/ijnp/pyw047
50. Patil SP, Jain PD, Ghumatkar PJ, Tambe R, Sathaye S. Neuroprotective effect of metformin in MPTP-induced Parkinson's disease in mice. Neuroscience (2014) 277:747–54. doi: 10.1016/j.neuroscience.2014.07.046
51. Perez-Revuelta BI, Hettich MM, Ciociaro A, Rotermund C, Kahle PJ, Krauss S, et al. Metformin lowers Ser-129 phosphorylated alpha-synuclein levels via mTOR-dependent protein phosphatase 2A activation. Cell Death Dis. (2014) 5:e1209. doi: 10.1038/cddis.2014.175
52. Porceddu PF, Ishola IO, Contu L, Morelli M. Metformin prevented dopaminergic neurotoxicity induced by 3,4-Methylenedioxymethamphetamine administration. Neurotox Res. (2016) 30:101–9. doi: 10.1007/s12640-016-9633-5
53. Chen F, Dong RR, Zhong KL, Ghosh A, Tang SS, Long Y, et al. Antidiabetic drugs restore abnormal transport of amyloid-beta across the blood-brain barrier and memory impairment in db/db mice. Neuropharmacology (2016) 101:123–36. doi: 10.1016/j.neuropharm.2015.07.023
54. Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci USA. (2010) 107:21830–5. doi: 10.1073/pnas.0912793107
55. Li J, Deng J, Sheng W, Zuo Z. Metformin attenuates Alzheimer's disease-like neuropathology in obese, leptin-resistant mice. Pharmacol Biochem Behav. (2012) 101:564–74. doi: 10.1016/j.pbb.2012.03.002
56. Picone P, Nuzzo D, Caruana L, Messina E, Barera A, Vasto S, et al. Metformin increases APP expression and processing via oxidative stress, mitochondrial dysfunction and NF-kappaB activation: use of insulin to attenuate metformin's effect. Biochim Biophys Acta (2015) 1853:1046–59. doi: 10.1016/j.bbamcr.2015.01.017
57. Allard JS, Perez EJ, Fukui K, Carpenter P, Ingram DK, de Cabo R. Prolonged metformin treatment leads to reduced transcription of Nrf2 and neurotrophic factors without cognitive impairment in older C57BL/6J mice. Behav Brain Res. (2016) 301:1–9. doi: 10.1016/j.bbr.2015.12.012
58. Lennox R, Porter DW, Flatt PR, Holscher C, Irwin N, Gault VA. Comparison of the independent and combined effects of sub-chronic therapy with metformin and a stable GLP-1 receptor agonist on cognitive function, hippocampal synaptic plasticity and metabolic control in high-fat fed mice. Neuropharmacology (2014) 86:22–30. doi: 10.1016/j.neuropharm.2014.06.026
59. McNeilly AD, Williamson R, Balfour DJ, Stewart CA, Sutherland C. A high-fat-diet-induced cognitive deficit in rats that is not prevented by improving insulin sensitivity with metformin. Diabetologia (2012) 55:3061–70. doi: 10.1007/s00125-012-2686-y
60. Pintana H, Apaijai N, Pratchayasakul W, Chattipakorn N, Chattipakorn SC. Effects of metformin on learning and memory behaviors and brain mitochondrial functions in high fat diet induced insulin resistant rats. Life Sci. (2012) 91:409–14. doi: 10.1016/j.lfs.2012.08.017
61. Thangthaeng N, Rutledge M, Wong JM, Vann PH, Forster MJ, Sumien N. Metformin impairs spatial memory and visual acuity in old male mice. Aging Dis. (2017) 8:17–30. doi: 10.14336/AD.2016.1010
62. Ma TC, Buescher JL, Oatis B, Funk JA, Nash AJ, Carrier RL, et al. Metformin therapy in a transgenic mouse model of Huntington's disease. Neurosci Lett. (2007) 411:98–103. doi: 10.1016/j.neulet.2006.10.039
63. Kaneb HM, Sharp PS, Rahmani-Kondori N, Wells DJ. Metformin treatment has no beneficial effect in a dose-response survival study in the SOD1(G93A) mouse model of ALS and is harmful in female mice. PLoS ONE (2011) 6:e24189. doi: 10.1371/journal.pone.0024189
64. Monette MC, Baird A, Jackson DL. A meta-analysis of cognitive functioning in nondemented adults with type 2 diabetes mellitus. Can J Diab. (2014) 38:401–8. doi: 10.1016/j.jcjd.2014.01.014
65. Palta P, Schneider AL, Biessels GJ, Touradji P, Hill-Briggs F. Magnitude of cognitive dysfunction in adults with type 2 diabetes: a meta-analysis of six cognitive domains and the most frequently reported neuropsychological tests within domains. J Int Neuropsychol Soc. (2014) 20:278–91. doi: 10.1017/S1355617713001483
66. Vincent C, Hall PA. Executive function in adults with type 2 diabetes: a meta-analytic review. Psychosom Med. (2015) 77:631–42. doi: 10.1097/PSY.0000000000000103
67. Cooper C, Sommerlad A, Lyketsos CG, Livingston G. Modifiable predictors of dementia in mild cognitive impairment: a systematic review and meta-analysis. Am J Psychiatry (2015) 172:323–34. doi: 10.1176/appi.ajp.2014.14070878
68. Moulton CD, Costafreda SG, Horton P, Ismail K, Fu CH. Meta-analyses of structural regional cerebral effects in type 1 and type 2 diabetes. Brain Imaging Behav. (2015) 9:651–62. doi: 10.1007/s11682-014-9348-2
69. Wu G, Lin L, Zhang Q, Wu J. Brain gray matter changes in type 2 diabetes mellitus: a meta-analysis of whole-brain voxel-based morphometry study. J Diabetes Complicat. (2017). 31:1698–703. doi: 10.1016/j.jdiacomp.2017.09.001
70. Lu FP, Lin KP, Kuo HK. Diabetes and the risk of multi-system aging phenotypes: a systematic review and meta-analysis. PLoS ONE (2009) 4:e4144. doi: 10.1371/journal.pone.0004144
71. Profenno LA, Porsteinsson AP, Faraone SV. Meta-analysis of Alzheimer's disease risk with obesity, diabetes, and related disorders. Biol Psychiatry (2010) 67:505–12. doi: 10.1016/j.biopsych.2009.02.013
72. Vagelatos NT, Eslick GD. Type 2 diabetes as a risk factor for Alzheimer's disease: the confounders, interactions, and neuropathology associated with this relationship. Epidemiol Rev. (2013) 35:152–60. doi: 10.1093/epirev/mxs012
73. Cheng G, Huang C, Deng H, Wang H. Diabetes as a risk factor for dementia and mild cognitive impairment: a meta-analysis of longitudinal studies. Intern Med J. (2012) 42:484–91. doi: 10.1111/j.1445-5994.2012.02758.x
74. Janson J, Laedtke T, Parisi JE, O'Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes (2004) 53:474–81. doi: 10.2337/diabetes.53.2.474
75. Matsuzaki T, Sasaki K, Tanizaki Y, Hata J, Fujimi K, Matsui Y, et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology (2010) 75:764–70. doi: 10.1212/WNL.0b013e3181eee25f
76. Turner R, Craft S, Aisen P. Individuals with Alzheimer's disease exhibit a high prevalence of undiagnosed impaired glucose tolerance and type 2 diabetes mellitus. Alzheimer's Dement. (2013) 9:P284–5. doi: 10.1016/j.jalz.2013.05.573
77. Frolich L, Blum-Degen D, Riederer P, Hoyer S. A disturbance in the neuronal insulin receptor signal transduction in sporadic Alzheimer's disease. Ann NY Acad Sci. (1999) 893:290–3. doi: 10.1111/j.1749-6632.1999.tb07839.x
78. Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer's disease: link to brain reductions in acetylcholine. J Alzheimer's Dis. (2005) 8:247–68. doi: 10.3233/JAD-2005-8304
79. Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer's disease- associated Abeta oligomers. J Clin Invest. (2012) 122:1339–53. doi: 10.1172/JCI57256
80. Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. (2012) 122:1316–38. doi: 10.1172/JCI59903
81. Luchsinger JA, Tang MX, Shea S, Mayeux R. Hyperinsulinemia and risk of Alzheimer disease. Neurology (2004) 63:1187–92. doi: 10.1212/01.WNL.0000140292.04932.87
82. Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch Neurol. (2011) 68:51–7. doi: 10.1001/archneurol.2010.225
83. Arvanitakis Z, Schneider JA, Wilson RS, Li Y, Arnold SE, Wang Z, et al. Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology (2006) 67:1960–5. doi: 10.1212/01.wnl.0000247053.45483.4e
84. Abner EL, Nelson PT, Kryscio RJ, Schmitt FA, Fardo DW, Woltjer RL, et al. Diabetes is associated with cerebrovascular but not Alzheimer's disease neuropathology. Alzheimer's Dement. (2016) 12:882–9. doi: 10.1016/j.jalz.2015.12.006
85. Crane PK, Walker RL, Sonnen J, Gibbons LE, Melrose R, Hassenstab J, et al. Glucose levels during life and neuropathologic findings at autopsy among people never treated for diabetes. Neurobiol Aging (2016) 48:72–82. doi: 10.1016/j.neurobiolaging.2016.07.021
86. Dos Santos Matioli MNP, Suemoto CK, Rodriguez RD, Farias DS, da Silva MM, Leite REP, et al. Diabetes is not associated with Alzheimer's disease neuropathology. J Alzheimer's Dis. (2017) 60:1035–43. doi: 10.3233/JAD-170179
87. Pruzin JJ, Schneider JA, Capuano AW, Leurgans SE, Barnes LL, Ahima RS, et al. Diabetes, hemoglobin A1C, and regional Alzheimer disease and infarct pathology. Alzheimer Dis Assoc Disord. (2017) 31:41–7. doi: 10.1097/WAD.0000000000000172
88. Beeri MS, Silverman JM, Davis KL, Marin D, Grossman HZ, Schmeidler J, et al. Type 2 diabetes is negatively associated with Alzheimer's disease neuropathology. J Gerontol Ser A Biol Sci Med Sci. (2005) 60:471–5. doi: 10.1093/gerona/60.4.471
89. Ahtiluoto S, Polvikoski T, Peltonen M, Solomon A, Tuomilehto J, Winblad B, et al. Diabetes, Alzheimer disease, and vascular dementia: a population-based neuropathologic study. Neurology (2010) 75:1195–202. doi: 10.1212/WNL.0b013e3181f4d7f8
90. Rahimi J, Kovacs GG. Prevalence of mixed pathologies in the aging brain. Alzheimer's Res Ther. (2014) 6:82. doi: 10.1186/s13195-014-0082-1
91. Norton S, Matthews FE, Barnes DE, Yaffe K, Brayne C. Potential for primary prevention of Alzheimer's disease: an analysis of population-based data. Lancet Neurol. (2014) 13:788–94. doi: 10.1016/S1474-4422(14)70136-X
92. Wahlqvist ML, Lee MS, Hsu CC, Chuang SY, Lee JT, Tsai HN. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson's disease occurring with Type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat Disord. (2012) 18:753–8. doi: 10.1016/j.parkreldis.2012.03.010
93. Brakedal B, Flønes I, Reiter SF, Torkildsen Ø, Dölle C, Assmus J, et al. Glitazone use associated with reduced risk of Parkinson's disease. Mov Dis (2017) 32:1594–9. doi: 10.1002/mds.27128
94. Brakedal B, Haugarvoll K, Tzoulis C. Simvastatin is associated with decreased risk of Parkinson disease. Ann Neurol. (2017) 81:329–30. doi: 10.1002/ana.24857
95. Kuan YC, Huang KW, Lin CL, Hu CJ, Kao CH. Effects of metformin exposure on neurodegenerative diseases in elderly patients with type 2 diabetes mellitus. Prog Neuropsychopharmacol Biol Psychiatry (2017) 79:77–83. doi: 10.1016/j.pnpbp.2017.06.002
96. Hsu CC, Wahlqvist ML, Lee MS, Tsai HN. Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. J Alzheimer's Dis. (2011) 24:485–93. doi: 10.3233/JAD-2011-101524
97. Cheng C, Lin CH, Tsai YW, Tsai CJ, Chou PH, Lan TH. Type 2 diabetes and antidiabetic medications in relation to dementia diagnosis. J Gerontol Ser A Biol Sci Med Sci (2014) 69:1299–305. doi: 10.1093/gerona/glu073
98. Heneka MT, Fink A, Doblhammer G. Effect of pioglitazone medication on the incidence of dementia. Ann Neurol. (2015) 78:284–94. doi: 10.1002/ana.24439
99. Ye F, Luo YJ, Xiao J, Yu NW, Yi G. Impact of insulin sensitizers on the incidence of dementia: a meta-analysis. Dement Geriatr Cogn Disord. (2016) 41:251–60. doi: 10.1159/000445941
100. Wang CP, Lorenzo C, Habib SL, Jo B, Espinoza SE. Differential effects of metformin on age related comorbidities in older men with type 2 diabetes. J Diabetes Complicat. (2017) 31:679–86. doi: 10.1016/j.jdiacomp.2017.01.013
101. Orkaby AR, Cho K, Cormack J, Gagnon DR, Driver JA. Metformin vs sulfonylurea use and risk of dementia in US veterans aged >/ = 65 years with diabetes. Neurology (2017) 89:1877–85. doi: 10.1212/WNL.0000000000004586
102. Ng TP, Feng L, Yap KB, Lee TS, Tan CH, Winblad B. Long-term metformin usage and cognitive function among older adults with diabetes. J Alzheimer's Dis. (2014) 41:61–8. doi: 10.3233/JAD-131901
103. Imfeld P, Bodmer M, Jick SS, Meier CR. Metformin, other antidiabetic drugs, and risk of Alzheimer's disease: a population-based case-control study. J Am Geriatr Soc. (2012) 60:916–21. doi: 10.1111/j.1532-5415.2012.03916.x
104. Huang CC, Chung CM, Leu HB, Lin LY, Chiu CC, Hsu CY, et al. Diabetes mellitus and the risk of Alzheimer's disease: a nationwide population-based study. PLoS ONE (2014) 9:e87095. doi: 10.1371/journal.pone.0087095
105. Koenig AM, Mechanic-Hamilton D, Xie SX, Combs MF, Cappola AR, Xie L, et al. Effects of the insulin sensitizer metformin in Alzheimer Disease: pilot data from a randomized placebo-controlled crossover study. Alzheimer Dis Assoc Disord. (2017) 31:107–13. doi: 10.1097/WAD.0000000000000202
106. Hervas D, Fornes-Ferrer V, Gomez-Escribano AP, Sequedo MD, Peiro C, Millan JM, et al. Metformin intake associates with better cognitive function in patients with Huntington's disease. PLoS ONE (2017) 12:e0179283. doi: 10.1371/journal.pone.0179283
107. Yokoyama H, Ogawa M, Honjo J, Okizaki S, Yamada D, Shudo R, et al. Risk factors associated with abnormal cognition in Japanese outpatients with diabetes, hypertension or dyslipidemia. Diabetol Int. (2015) 6:268–74. doi: 10.1007/s13340-014-0194-7
108. Luchsinger JA, Perez T, Chang H, Mehta P, Steffener J, Pradabhan G, et al. Metformin in amnestic mild cognitive impairment: results of a pilot randomized placebo controlled clinical trial. J Alzheimer's Dis. (2016) 51:501–14. doi: 10.3233/JAD-150493
109. de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurol. (2006) 5:525–35. doi: 10.1016/S1474-4422(06)70471-9
110. Jankovic J. Parkinson's disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatr. (2008) 79:368–76. doi: 10.1136/jnnp.2007.131045
111. Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain (1999) 122(Pt 8), 1437–48.
112. Schneider SA, Obeso JA. Clinical and pathological features of Parkinson's disease. Curr Top Behav Neurosci. (2014) 22:205–20. doi: 10.1007/7854_2014_317
113. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature (1997) 388:839–40. doi: 10.1038/42166
114. Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rub U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson's disease (preclinical and clinical stages). J Neurol. (2002) 249(Suppl. 3):iii1–5. doi: 10.1007/s00415-002-1301-4
115. Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, et al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. (2009) 117:613–34. doi: 10.1007/s00401-009-0538-8
116. Gasser T. Identifying PD-causing genes and genetic susceptibility factors: current approaches and future prospects. Prog Brain Res. (2010) 183:3–20. doi: 10.1016/S0079-6123(10)83001-8
117. Deng H, Wang P, Jankovic J. The genetics of Parkinson disease. Ageing Res Rev. (2017) 42:72–85. doi: 10.1016/j.arr.2017.12.007
118. Klein C, Westenberger A. Genetics of Parkinson's disease. Cold Spring Harb Perspect Med. (2012) 2:a008888. doi: 10.1101/cshperspect.a008888
119. Dunn L, Allen GF, Mamais A, Ling H, Li A, Duberley KE, et al. Dysregulation of glucose metabolism is an early event in sporadic Parkinson's disease. Neurobiol Aging (2014) 35:1111–5. doi: 10.1016/j.neurobiolaging.2013.11.001
120. Athauda D, Foltynie T. The ongoing pursuit of neuroprotective therapies in Parkinson disease. Nat Rev Neurol. (2015) 11:25–40. doi: 10.1038/nrneurol.2014.226
121. Choi JY, Jang EH, Park CS, Kang JH. Enhanced susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity in high-fat diet-induced obesity. Free Radic Biol Med. (2005) 38:806–16. doi: 10.1016/j.freeradbiomed.2004.12.008
122. Morris JK, Bomhoff GL, Stanford JA, Geiger PC. Neurodegeneration in an animal model of Parkinson's disease is exacerbated by a high-fat diet. Am J Physiol Regul Integr Compar Physiol. (2010) 299:R1082–90. doi: 10.1152/ajpregu.00449.2010
123. Rotermund C, Truckenmuller FM, Schell H, Kahle PJ. Diet-induced obesity accelerates the onset of terminal phenotypes in alpha-synuclein transgenic mice. J Neurochem. (2014) 131:848–58. doi: 10.1111/jnc.12813
124. Morris JK, Bomhoff GL, Gorres BK, Davis VA, Kim J, Lee PP, et al. Insulin resistance impairs nigrostriatal dopamine function. Exp Neurol. (2011) 231:171–80. doi: 10.1016/j.expneurol.2011.06.005
125. Wang L, Zhai YQ, Xu LL, Qiao C, Sun XL, Ding JH, et al. Metabolic inflammation exacerbates dopaminergic neuronal degeneration in response to acute MPTP challenge in type 2 diabetes mice. Exp Neurol. (2014) 251:22–9. doi: 10.1016/j.expneurol.2013.11.001
126. Hu G, Jousilahti P, Bidel S, Antikainen R, Tuomilehto J. Type 2 diabetes and the risk of Parkinson's disease. Diabetes Care (2007) 30:842–7. doi: 10.2337/dc06-2011
127. Simon KC, Chen H, Schwarzschild M, Ascherio A. Hypertension, hypercholesterolemia, diabetes, and risk of Parkinson disease. Neurology (2007) 69:1688–95. doi: 10.1212/01.wnl.0000271883.45010.8a
128. Driver JA, Smith A, Buring JE, Gaziano JM, Kurth T, Logroscino G. Prospective cohort study of type 2 diabetes and the risk of Parkinson's disease. Diabetes Care (2008) 31:2003–5. doi: 10.2337/dc08-0688
129. D'Amelio M, Ragonese P, Callari G, Di Benedetto N, Palmeri B, Terruso V, et al. Diabetes preceding Parkinson's disease onset. A case-control study. Parkinsonism Relat Disord. (2009) 15:660–4. doi: 10.1016/j.parkreldis.2009.02.013
130. Miyake Y, Tanaka K, Fukushima W, Sasaki S, Kiyohara C, Tsuboi Y, et al. Case-control study of risk of Parkinson's disease in relation to hypertension, hypercholesterolemia, and diabetes in Japan. J Neurol Sci. (2010) 293:82–6. doi: 10.1016/j.jns.2010.03.002
131. Schernhammer E, Hansen J, Rugbjerg K, Wermuth L, Ritz B. Diabetes and the risk of developing Parkinson's disease in Denmark. Diabetes Care (2011) 34:1102–8. doi: 10.2337/dc10-1333
132. Xu Q, Park Y, Huang X, Hollenbeck A, Blair A, Schatzkin A, et al. Diabetes and risk of Parkinson's disease. Diabetes Care (2011) 34:910–5. doi: 10.2337/dc10-1922
133. Lu L, Fu DL, Li HQ, Liu AJ, Li JH, Zheng GQ. Diabetes and risk of Parkinson's disease: an updated meta-analysis of case-control studies. PLoS ONE (2014) 9:e85781. doi: 10.1371/journal.pone.0085781
134. Cereda E, Barichella M, Pedrolli C, Klersy C, Cassani E, Caccialanza R, et al. Diabetes and risk of Parkinson's disease: a systematic review and meta-analysis. Diabetes Care (2011) 34:2614–23. doi: 10.2337/dc11-1584
135. Yue X, Li H, Yan H, Zhang P, Chang L, Li T. Risk of Parkinson disease in diabetes mellitus: an updated meta-analysis of population-based cohort studies. Medicine (2016) 95:e3549. doi: 10.1097/MD.0000000000003549
136. Crane PK, Walker R, Hubbard RA, Li G, Nathan DM, Zheng H, et al. Glucose levels and risk of dementia. N Engl J Med. (2013) 369:540–8. doi: 10.1056/NEJMoa1215740
137. Walker Z, Possin KL, Boeve BF, Aarsland D. Lewy body dementias. Lancet (2015) 386:1683–97. doi: 10.1016/S0140-6736(15)00462-6
138. Ong M, Foo H, Chander RJ, Wen MC, Au WL, Sitoh YY, et al. Influence of diabetes mellitus on longitudinal atrophy and cognition in Parkinson's disease. J Neurol Sci. (2017) 377:122–6. doi: 10.1016/j.jns.2017.04.010
139. Bosco D, Plastino M, Cristiano D, Colica C, Ermio C, De Bartolo M, et al. Dementia is associated with insulin resistance in patients with Parkinson's disease. J Neurol Sci. (2012) 315:39–43. doi: 10.1016/j.jns.2011.12.008
140. Secnik J, Cermakova P, Fereshtehnejad SM, Dannberg P, Johnell K, Fastbom J, et al. Diabetes in a large dementia cohort: clinical characteristics and treatment from the Swedish dementia registry. Diabetes Care (2017) 40:1159–66. doi: 10.2337/dc16-2516
141. Levy G, Tang MX, Cote LJ, Louis ED, Alfaro B, Mejia H, et al. Do risk factors for Alzheimer's disease predict dementia in Parkinson's disease? An exploratory study. Mov Disord. (2002) 17:250–7. doi: 10.1002/mds.10086
142. Malek N, Lawton MA, Swallow DM, Grosset KA, Marrinan SL, Bajaj N, et al. Vascular disease and vascular risk factors in relation to motor features and cognition in early Parkinson's disease. Mov Disord. (2016) 31:1518–26. doi: 10.1002/mds.26698
143. Pilotto A, Turrone R, Liepelt-Scarfone I, Bianchi M, Poli L, Borroni B, et al. Vascular risk factors and cognition in Parkinson's disease. J Alzheimer's Dis. (2016) 51:563–70. doi: 10.3233/JAD-150610
144. Xu Y, Yang J, Shang H. Meta-analysis of risk factors for Parkinson's disease dementia. Transl Neurodegener. (2016) 5:11. doi: 10.1186/s40035-016-0058-0
145. Malfitano AM, Marasco G, Proto MC, Laezza C, Gazzerro P, Bifulco M. Statins in neurological disorders: an overview and update. Pharmacol Res. (2014) 88:74–83. doi: 10.1016/j.phrs.2014.06.007
146. Abe K, Aoki M, Tsuji S, Itoyama Y, Sobue G, Togo M, et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. (2017) 16:505–12. doi: 10.1016/S1474-4422(17)30115-1
148. Kioumourtzoglou MA, Rotem RS, Seals RM, Gredal O, Hansen J, Weisskopf MG. Diabetes mellitus, obesity, and diagnosis of amyotrophic lateral sclerosis: a population-based study. JAMA Neurol. (2015) 72:905–11. doi: 10.1001/jamaneurol.2015.0910
149. Mariosa D, Kamel F, Bellocco R, Ye W, Fang F. Association between diabetes and amyotrophic lateral sclerosis in Sweden. Eur J Neurol. (2015) 22:1436–42. doi: 10.1111/ene.12632
150. Jawaid A, Salamone AR, Strutt AM, Murthy SB, Wheaton M, McDowell EJ, et al. ALS disease onset may occur later in patients with pre-morbid diabetes mellitus. Eur J Neurol. (2010) 17:733–9. doi: 10.1111/j.1468-1331.2009.02923.x
151. D'Ovidio F, d'Errico A, Carna P, Calvo A, Costa G, Chio A. The role of pre-morbid diabetes on developing amyotrophic lateral sclerosis. Eur J Neurol. (2018) 25:164–70. doi: 10.1111/ene.13465
152. Paganoni S, Hyman T, Shui A, Allred P, Harms M, Liu J, et al. Pre-morbid type 2 diabetes mellitus is not a prognostic factor in amyotrophic lateral sclerosis. Muscle Nerve (2015) 52:339–43. doi: 10.1002/mus.24688
153. Sun Y, Lu CJ, Chen RC, Hou WH, Li CY. Risk of amyotrophic lateral sclerosis in patients with diabetes: a nationwide population-based cohort study. J Epidemiol. (2015) 25:445–51. doi: 10.2188/jea.JE20140176
154. Hollinger SK, Okosun IS, Mitchell CS. Antecedent disease and amyotrophic lateral sclerosis: what is protecting whom? Front Neurol. (2016) 7:47. doi: 10.3389/fneur.2016.00047
155. Park Y, Park J, Kim Y, Baek H, Kim SH. Association between nutritional status and disease severity using the amyotrophic lateral sclerosis (ALS) functional rating scale in ALS patients. Nutrition (2015) 31:1362–7. doi: 10.1016/j.nut.2015.05.025
156. Dupuis L, Dengler R, Heneka MT, Meyer T, Zierz S, Kassubek J, et al. A randomized, double blind, placebo-controlled trial of pioglitazone in combination with riluzole in amyotrophic lateral sclerosis. PLoS ONE (2012) 7:e37885. doi: 10.1371/journal.pone.0037885
157. Wills AM, Hubbard J, Macklin EA, Glass J, Tandan R, Simpson EP, et al. Hypercaloric enteral nutrition in patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet (2014) 383:2065–72. doi: 10.1016/S0140-6736(14)60222-1
158. Levine TD, Bowser R, Hank NC, Gately S, Stephan D, Saperstein DS, et al. A pilot trial of pioglitazone HCl and tretinoin in ALS: cerebrospinal fluid biomarkers to monitor drug efficacy and predict rate of disease progression. Neurol Res Int. (2012) 2012:582075. doi: 10.1155/2012/582075
159. Lalic NM, Maric J, Svetel M, Jotic A, Stefanova E, Lalic K, et al. Glucose homeostasis in Huntington disease: abnormalities in insulin sensitivity and early-phase insulin secretion. Arch Neurol. (2008) 65:476–80. doi: 10.1001/archneur.65.4.476
160. Montojo MT, Aganzo M, Gonzalez N. Huntington's disease and diabetes: chronological sequence of its association. J Huntington's Dis. (2017) 6:179–88. doi: 10.3233/JHD-170253
161. Hu Y, Liang J, Yu S. High prevalence of diabetes mellitus in a five-generation Chinese family with Huntington's disease. J Alzheimer's Dis. (2014) 40:863–8. doi: 10.3233/JAD-131847
162. Boesgaard TW, Nielsen TT, Josefsen K, Hansen T, Jorgensen T, Pedersen O, et al. Huntington's disease does not appear to increase the risk of diabetes mellitus. J Neuroendocrinol. (2009) 21:770–6. doi: 10.1111/j.1365-2826.2009.01898.x
163. Russo CV, Salvatore E, Sacca F, Tucci T, Rinaldi C, Sorrentino P, et al. Insulin sensitivity and early-phase insulin secretion in normoglycemic Huntington's disease patients. J Huntington's Dis. (2013) 2:501–7. doi: 10.3233/JHD-130078
164. Sriwijitkamol A, Coletta DK, Wajcberg E, Balbontin GB, Reyna SM, Barrientes J, et al. Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes (2007) 56:836–48. doi: 10.2337/db06-1119
165. Xu XJ, Gauthier MS, Hess DT, Apovian CM, Cacicedo JM, Gokce N, et al. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J Lipid Res. (2012) 53:792–801. doi: 10.1194/jlr.P022905
166. Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. (2001) 17:45–58. doi: 10.1385/JMN:17:1:45
167. Garcia-Gil M, Pesi R, Perna S, Allegrini S, Giannecchini M, Camici M, et al. 5'-aminoimidazole-4-carboxamide riboside induces apoptosis in human neuroblastoma cells. Neuroscience (2003) 117:811–20. doi: 10.1016/S0306-4522(02)00836-9
168. Meares GP, Qin H, Liu Y, Holdbrooks AT, Benveniste EN. AMP-activated protein kinase restricts IFN-gamma signaling. J Immunol. (2013) 190:372–80. doi: 10.4049/jimmunol.1202390
169. Greco SJ, Sarkar S, Johnston JM, Tezapsidis N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem Biophys Res Commun. (2009) 380:98–104. doi: 10.1016/j.bbrc.2009.01.041
170. Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem. (2010) 285:9100–13. doi: 10.1074/jbc.M109.060061
171. Thornton C, Bright NJ, Sastre M, Muckett PJ, Carling D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid beta-peptide exposure. Biochem J. (2011) 434:503–12. doi: 10.1042/BJ20101485
172. Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Abeta oligomers through Tau phosphorylation. Neuron (2013) 78:94–108. doi: 10.1016/j.neuron.2013.02.003
173. Maziere JC, Maziere C, Mora L, Gardette J, Salmon S, Auclair M, et al. The antidiabetic drug metformin decreases cholesterol metabolism in cultured human fibroblasts. Atherosclerosis (1988) 71:27–33. doi: 10.1016/0021-9150(88)90299-7
174. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, DePinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science (2005) 310:1642–6. doi: 10.1126/science.1120781
175. Stephenne X, Foretz M, Taleux N, van der Zon GC, Sokal E, Hue L, et al. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia (2011) 54:3101–10. doi: 10.1007/s00125-011-2311-5
176. Chiang MC, Cheng YC, Chen SJ, Yen CH, Huang RN. Metformin activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against Amyloid-beta-induced mitochondrial dysfunction. Exp Cell Res. (2016) 347:322–31. doi: 10.1016/j.yexcr.2016.08.013
177. Gupta A, Bisht B, Dey CS. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer's-like changes. Neuropharmacology (2011) 60:910–20. doi: 10.1016/j.neuropharm.2011.01.033
178. Hettich MM, Matthes F, Ryan DP, Griesche N, Schroder S, Dorn S, et al. The anti-diabetic drug metformin reduces BACE1 protein level by interfering with the MID1 complex. PLoS ONE (2014) 9:e102420. doi: 10.1371/journal.pone.0102420
179. Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, Ma T, et al. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer's amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci USA. (2009) 106:3907–12. doi: 10.1073/pnas.0807991106
180. Choi JS, Park C, Jeong JW. AMP-activated protein kinase is activated in Parkinson's disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem Biophys Res Commun. (2010) 391:147–51. doi: 10.1016/j.bbrc.2009.11.022
181. Ng CH, Guan MS, Koh C, Ouyang X, Yu F, Tan EK, et al. AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson's disease. J Neurosci. (2012) 32:14311–7. doi: 10.1523/JNEUROSCI.0499-12.2012
182. Dulovic M, Jovanovic M, Xilouri M, Stefanis L, Harhaji-Trajkovic L, Kravic-Stevovic T, et al. The protective role of AMP-activated protein kinase in alpha-synuclein neurotoxicity in vitro. Neurobiol Dis. (2014) 63:1–11. doi: 10.1016/j.nbd.2013.11.002
183. Bobela W, Nazeeruddin S, Knott G, Aebischer P, Schneider BL. Modulating the catalytic activity of AMPK has neuroprotective effects against alpha-synuclein toxicity. Mol Neurodegener. (2017) 12:80. doi: 10.1186/s13024-017-0220-x
184. Jiang P, Gan M, Ebrahim AS, Castanedes-Casey M, Dickson DW, Yen SH. Adenosine monophosphate-activated protein kinase overactivation leads to accumulation of alpha-synuclein oligomers and decrease of neurites. Neurobiol Aging (2013) 34, 1504–15. doi: 10.1016/j.neurobiolaging.2012.11.001
185. Duran-Aniotz C, Hetz C. Glucose metabolism: a sweet relief of Alzheimer's disease. Curr Biol. (2016) 26:R806–9. doi: 10.1016/j.cub.2016.07.060
186. Herrero-Mendez A, Almeida A, Fernandez E, Maestre C, Moncada S, Bolanos JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol. (2009) 11:747–52. doi: 10.1038/ncb1881
187. Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature (2012) 487:443–8. doi: 10.1038/nature11314
188. Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature (2012) 485:517–21. doi: 10.1038/nature11007
189. Bouzier-Sore AK, Voisin P, Canioni P, Magistretti PJ, Pellerin L. Lactate is a preferential oxidative energy substrate over glucose for neurons in culture. J Cereb Blood Flow Metab. (2003) 23:1298–306. doi: 10.1097/01.WCB.0000091761.61714.25
190. Borghammer P. Perfusion and metabolism imaging studies in Parkinson's disease. Dan Med J. (2012) 59:B4466.
191. Woods SC, Lotter EC, McKay LD, Porte D Jr. Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature (1979) 282:503–5. doi: 10.1038/282503a0
192. Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med. (2002) 8:1376–82. doi: 10.1038/nm1202-798
193. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature (2006) 443:289–95. doi: 10.1038/nature05026
194. Lozovsky D, Saller CF, Kopin IJ. Dopamine receptor binding is increased in diabetic rats. Science (1981) 214:1031–3. doi: 10.1126/science.6458088
195. Lozovsky DB, Kopin IJ, Saller CF. Modulation of dopamine receptor supersensitivity by chronic insulin: implication in schizophrenia. Brain Res. (1985) 343:190–3. doi: 10.1016/0006-8993(85)91178-3
196. Danguir J, Elghozi JL, Laude D. Increased dopamine and serotonin metabolites in CSF during severe insulin-induced hypoglycemia in freely moving rats. Neurochem Int (1984) 6:71–5. doi: 10.1016/0197-0186(84)90028-7
197. Colin E, Regulier E, Perrin V, Durr A, Brice A, Aebischer P, et al. Akt is altered in an animal model of Huntington's disease and in patients. Eur J Neurosci. (2005) 21:1478–88. doi: 10.1111/j.1460-9568.2005.03985.x
198. Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, et al. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J Neurochem. (2005) 93:105–17. doi: 10.1111/j.1471-4159.2004.02949.x
199. Malagelada C, Jin ZH, Greene LA. RTP801 is induced in Parkinson's disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J Neurosci. (2008) 28:14363–71. doi: 10.1523/JNEUROSCI.3928-08.2008
200. Timmons S, Coakley MF, Moloney AM, O' Neill C. Akt signal transduction dysfunction in Parkinson's disease. Neurosci Lett. (2009) 467:30–5. doi: 10.1016/j.neulet.2009.09.055
201. Shepherd PR, Withers DJ, Siddle K. Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochem J. (1998) 333(Pt 3):471–90.
202. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell (2007) 129:1261–74. doi: 10.1016/j.cell.2007.06.009
203. Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, et al. Role of translocation in the activation and function of protein kinase B. J Biol Chem. (1997) 272:31515–24. doi: 10.1074/jbc.272.50.31515
204. Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell (2007) 25:917–31. doi: 10.1016/j.molcel.2007.02.017
205. Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell (1998) 95:29–39. doi: 10.1016/S0092-8674(00)81780-8
206. Yamauchi T, Tobe K, Tamemoto H, Ueki K, Kaburagi Y, Yamamoto-Honda R, et al. Insulin signalling and insulin actions in the muscles and livers of insulin-resistant, insulin receptor substrate 1-deficient mice. Mol Cell Biol. (1996) 16:3074–84. doi: 10.1128/MCB.16.6.3074
207. Bruning JC, Michael MD, Winnay JN, Hayashi T, Horsch D, Accili D, et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell (1998) 2:559–69. doi: 10.1016/S1097-2765(00)80155-0
208. Previs SF, Withers DJ, Ren JM, White MF, Shulman GI. Contrasting effects of IRS-1 versus IRS-2 gene disruption on carbohydrate and lipid metabolism in vivo. J Biol Chem. (2000) 275:38990–4. doi: 10.1074/jbc.M006490200
209. Reger MA, Watson GS, Frey WH II, Baker LD, Cholerton B, Keeling ML, et al. Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol Aging (2006) 27:451–8. doi: 10.1016/j.neurobiolaging.2005.03.016
210. McNay EC, Ong CT, McCrimmon RJ, Cresswell J, Bogan JS, Sherwin RS. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol Learn Mem. (2010) 93:546–53. doi: 10.1016/j.nlm.2010.02.002
211. Takadera T, Sakura N, Mohri T, Hashimoto T. Toxic effect of a beta-amyloid peptide (beta 22-35) on the hippocampal neuron and its prevention. Neurosci Lett. (1993) 161:41–4. doi: 10.1016/0304-3940(93)90135-8
212. Rensink AA, Otte-Holler I, de Boer R, Bosch RR, ten Donkelaar HJ, de Waal RM, et al. Insulin inhibits amyloid beta-induced cell death in cultured human brain pericytes. Neurobiol Aging (2004) 25:93–103. doi: 10.1016/S0197-4580(03)00039-3
213. Di Carlo M, Picone P, Carrotta R, Giacomazza D, San Biagio PL. Insulin promotes survival of amyloid-beta oligomers neuroblastoma damaged cells via caspase 9 inhibition and Hsp70 upregulation. J Biomed Biotechnol. (2010) 2010:147835. doi: 10.1155/2010/147835
214. Mirsky IA, Broh-Kahn RH. The inactivation of insulin by tissue extracts; the distribution and properties of insulin inactivating extracts. Arch Biochem. (1949) 20:1–9.
215. Vekrellis K, Ye Z, Qiu WQ, Walsh D, Hartley D, Chesneau V, et al. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J Neurosci. (2000) 20:1657–65. doi: 10.1523/JNEUROSCI.20-05-01657.2000
216. Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA. (2003) 100:4162–7. doi: 10.1073/pnas.0230450100
217. Cook DG, Leverenz JB, McMillan PJ, Kulstad JJ, Ericksen S, Roth RA, et al. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer's disease is associated with the apolipoprotein E-epsilon4 allele. Am J Pathol. (2003) 162:313–9. doi: 10.1016/S0002-9440(10)63822-9
218. Ertekin-Taner N, Allen M, Fadale D, Scanlin L, Younkin L, Petersen RC, et al. Genetic variants in a haplotype block spanning IDE are significantly associated with plasma Abeta42 levels and risk for Alzheimer disease. Hum Mutat. (2004) 23:334–42. doi: 10.1002/humu.20016
219. Zuo X, Jia J. Promoter polymorphisms which modulate insulin degrading enzyme expression may increase susceptibility to Alzheimer's disease. Brain Res. (2009) 1249:1–8. doi: 10.1016/j.brainres.2008.10.034
220. Reitz C, Cheng R, Schupf N, Lee JH, Mehta PD, Rogaeva E, et al. Association between variants in IDE-KIF11-HHEX and plasma amyloid beta levels. Neurobiol Aging (2012) 33:199.e113–197. doi: 10.1016/j.neurobiolaging.2010.07.005
221. Wang S, He F, Wang Y. Association between polymorphisms of the insulin-degrading enzyme gene and late-onset Alzheimer disease. J Geriatr Psychiatry Neurol. (2015) 28:94–8. doi: 10.1177/0891988714554707
222. Kochkina EG, Plesneva SA, Vasilev DS, Zhuravin IA, Turner AJ, Nalivaeva NN. Effects of ageing and experimental diabetes on insulin-degrading enzyme expression in male rat tissues. Biogerontology (2015) 16:473–84. doi: 10.1007/s10522-015-9569-9
223. Barber AJ, Nakamura M, Wolpert EB, Reiter CE, Seigel GM, Antonetti DA, et al. Insulin rescues retinal neurons from apoptosis by a phosphatidylinositol 3-kinase/Akt-mediated mechanism that reduces the activation of caspase-3. J Biol Chem. (2001) 276:32814–21. doi: 10.1074/jbc.M104738200
224. Duarte AI, Santos MS, Seica R, de Oliveira CR. Insulin affects synaptosomal GABA and glutamate transport under oxidative stress conditions. Brain Res. (2003) 977:23–30. doi: 10.1016/S0006-8993(03)02679-9
225. Yu XR, Jia GR, Gao GD, Wang SH, Han Y, Cao W. Neuroprotection of insulin against oxidative stress-induced apoptosis in cultured retinal neurons: involvement of phosphoinositide 3-kinase/Akt signal pathway. Acta Biochim Biophys Sin. (2006) 38:241–8. doi: 10.1111/j.1745-7270.2006.00152.x
226. Moosavi M, Maghsoudi N, Zahedi-Asl S, Naghdi N, Yousefpour M, Trounce IA. The role of PI3/Akt pathway in the protective effect of insulin against corticosterone cell death induction in hippocampal cell culture. Neuroendocrinology (2008) 88:293–8. doi: 10.1159/000150441
227. Cusi K, Consoli A, DeFronzo RA. Metabolic effects of metformin on glucose and lactate metabolism in noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. (1996) 81:4059–67.
228. Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes (2002) 51:2420–5. doi: 10.2337/diabetes.51.8.2420
229. Adak T, Samadi A, Unal AZ, Sabuncuoglu S. A reappraisal on metformin. Regul Toxicol Pharmacol. (2017) 92:324–32. doi: 10.1016/j.yrtph.2017.12.023
230. Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat. (2010) 123:271–9. doi: 10.1007/s10549-010-0763-9
231. Sarfstein R, Friedman Y, Attias-Geva Z, Fishman A, Bruchim I, Werner H. Metformin downregulates the insulin/IGF-I signaling pathway and inhibits different uterine serous carcinoma (USC) cells proliferation and migration in p53-dependent or -independent manners. PLoS ONE (2013) 8:e61537. doi: 10.1371/journal.pone.0061537
232. Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen–induced lung tumorigenesis. Cancer Prevent Res. (2010) 3:1066–76. doi: 10.1158/1940-6207.CAPR-10-0055
233. Karnevi E, Said K, Andersson R, Rosendahl AH. Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer (2013) 13:235. doi: 10.1186/1471-2407-13-235
234. Mannucci E, Ognibene A, Cremasco F, Bardini G, Mencucci A, Pierazzuoli E, et al. Effect of metformin on glucagon-like peptide 1 (GLP-1) and leptin levels in obese nondiabetic subjects. Diabetes Care (2001) 24:489–94. doi: 10.2337/diacare.24.3.489
235. Mannucci E, Tesi F, Bardini G, Ognibene A, Petracca MG, Ciani S, et al. Effects of metformin on glucagon-like peptide-1 levels in obese patients with and without Type 2 diabetes. Diabetes Nutr Metab. (2004) 17:336–42.
236. Maida A, Lamont BJ, Cao X, Drucker DJ. Metformin regulates the incretin receptor axis via a pathway dependent on peroxisome proliferator-activated receptor-alpha in mice. Diabetologia (2011) 54:339–49. doi: 10.1007/s00125-010-1937-z
237. Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, et al. Exenatide once weekly versus placebo in Parkinson's disease: a randomised, double-blind, placebo-controlled trial. Lancet (2017) 390:1664–75. doi: 10.1016/S0140-6736(17)31585-4
238. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149:274–93. doi: 10.1016/j.cell.2012.03.017
239. Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell (2003) 11:1457–66. doi: 10.1016/S1097-2765(03)00220-X
240. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (2005) 307:1098–101. doi: 10.1126/science.1106148
241. Walsh PT, Buckler JL, Zhang J, Gelman AE, Dalton NM, Taylor DK, et al. PTEN inhibits IL-2 receptor-mediated expansion of CD4+ CD25+ Tregs. J Clin Invest. (2006) 116:2521–31. doi: 10.1172/JCI28057
242. Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VC, et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell (2012) 149:49–62. doi: 10.1016/j.cell.2012.02.030
243. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell (2003) 115:577–90. doi: 10.1016/S0092-8674(03)00929-2
244. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell (2008) 30:214–26. doi: 10.1016/j.molcel.2008.03.003
245. Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, Schreiber SL. Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature (1995) 377:441–6. doi: 10.1038/377441a0
246. Brunn GJ, Hudson CC, Sekulic A, Williams JM, Hosoi H, Houghton PJ, et al. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science (1997) 277:99–101. doi: 10.1126/science.277.5322.99
247. Yip CK, Murata K, Walz T, Sabatini DM, Kang SA. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol Cell (2010) 38:768–74. doi: 10.1016/j.molcel.2010.05.017
248. Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology (2005) 146:1473–81. doi: 10.1210/en.2004-0921
249. Tremblay F, Krebs M, Dombrowski L, Brehm A, Bernroider E, Roth E, et al. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes (2005) 54:2674–84. doi: 10.2337/diabetes.54.9.2674
250. Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature (2004) 431:200–5. doi: 10.1038/nature02866
251. Siddiqui A, Bhaumik D, Chinta SJ, Rane A, Rajagopalan S, Lieu CA, et al. Mitochondrial quality control via the PGC1α-TFEB signaling pathway is compromised by parkin Q311X mutation but independently restored by rapamycin. J Neurosci. (2015) 35:12833–44. doi: 10.1523/JNEUROSCI.0109-15.2015
252. Zhang Y, He X, Wu X, Lei M, Wei Z, Zhang X, et al. Rapamycin upregulates glutamate transporter and IL-6 expression in astrocytes in a mouse model of Parkinson's disease. Cell Death Dis. (2017) 8:e2611. doi: 10.1038/cddis.2016.491
253. Santini E, Heiman M, Greengard P, Valjent E, Fisone G. Inhibition of mTOR signaling in Parkinson's disease prevents L-DOPA–induced dyskinesia. Sci Signal. (2009) 2:ra36. doi: 10.1126/scisignal.2000308
254. Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. (2011) 23:744–55. doi: 10.1016/j.ceb.2011.09.003
255. Wang C, Yu JT, Miao D, Wu ZC, Tan MS, Tan L. (2014) Targeting the mTOR signaling network for Alzheimer's disease therapy. Mol Neurobiol. 49:120–35. doi: 10.1007/s12035-013-8505-8
256. Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS ONE (2010) 5:e9979. doi: 10.1371/journal.pone.0009979
257. Caccamo A, De Pinto V, Messina A, Branca C, Oddo S. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer's disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J Neurosc. (2014) 34:7988–98. doi: 10.1523/JNEUROSCI.0777-14.2014
258. Caccamo A, Magri A, Medina DX, Wisely EV, Lopez-Aranda MF, Silva AJ, et al. mTOR regulates tau phosphorylation and degradation: implications for Alzheimer's disease and other tauopathies. Aging Cell (2013) 12:370–80. doi: 10.1111/acel.12057
259. Shi WY, Xiao D, Wang L, Dong LH, Yan ZX, Shen ZX, et al. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. (2012) 3:e275. doi: 10.1038/cddis.2012.13
260. Nair V, Pathi S, Jutooru I, Sreevalsan S, Basha R, Abdelrahim M, et al. Metformin inhibits pancreatic cancer cell and tumor growth and downregulates Sp transcription factors. Carcinogenesis (2013) 34:2870–9. doi: 10.1093/carcin/bgt231
261. Slomovitz BM, Coleman RL. The PI3K/AKT/mTOR pathway as a therapeutic target in endometrial cancer. Clin Cancer Res. (2012) 18:5856–64. doi: 10.1158/1078-0432.CCR-12-0662
262. Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. (2011) 71:4366–72. doi: 10.1158/0008-5472.CAN-10-1769
263. Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. (2010) 11:390–401. doi: 10.1016/j.cmet.2010.03.014
264. Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. (2014) 14:463–77. doi: 10.1038/nri3705
265. Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature (2001) 414:212–6. doi: 10.1038/35102591
266. Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, et al. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci. (2003) 23:9796–804. doi: 10.1523/JNEUROSCI.23-30-09796.2003
267. Perlmutter LS, Barron E, Chui HC. Morphologic association between microglia and senile plaque amyloid in Alzheimer's disease. Neurosci Lett. (1990) 119:32–6. doi: 10.1016/0304-3940(90)90748-X
268. Wisniewski HM, Wegiel J, Wang KC, Lach B. Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer's disease. Acta Neuropathol. (1992) 84:117–27. doi: 10.1007/BF00311383
269. Zhang Y, Zhou B, Deng B, Zhang F, Wu J, Wang Y, et al. Amyloid-beta induces hepatic insulin resistance in vivo via JAK2. Diabetes (2013) 62:1159–66. doi: 10.2337/db12-0670
270. Vodovotz Y, Lucia MS, Flanders KC, Chesler L, Xie QW, Smith TW, et al. Inducible nitric oxide synthase in tangle-bearing neurons of patients with Alzheimer's disease. J Exp Med. (1996) 184:1425–33. doi: 10.1084/jem.184.4.1425
271. Heneka MT, Wiesinger H, Dumitrescu-Ozimek L, Riederer P, Feinstein DL, Klockgether T. Neuronal and glial coexpression of argininosuccinate synthetase and inducible nitric oxide synthase in Alzheimer disease. J Neuropathol Exp Neurol. (2001) 60:906–16. doi: 10.1093/jnen/60.9.906
272. Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron (2010) 68:19–31. doi: 10.1016/j.neuron.2010.08.023
273. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology (1988) 38:1285–91. doi: 10.1212/WNL.38.8.1285
274. Damier P, Hirsch EC, Zhang P, Agid Y, Javoy-Agid F. Glutathione peroxidase, glial cells and Parkinson's disease. Neuroscience (1993) 52:1–6. doi: 10.1016/0306-4522(93)90175-F
275. Czlonkowska A, Kohutnicka M, Kurkowska-Jastrzebska I, Czlonkowski A. Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson's disease mice model. Neurodegeneration (1996) 5:137–43.
276. Sugama S, Yang L, Cho BP, DeGiorgio LA, Lorenzl S, Albers DS, et al. Age-related microglial activation in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurodegeneration in C57BL/6 mice. Brain Res. (2003) 964:288–94. doi: 10.1016/S0006-8993(02)04085-4
277. Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J. (2005) 19:533–42. doi: 10.1096/fj.04-2751com
278. Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, et al. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science (2000) 290:985–9. doi: 10.1126/science.290.5493.985
279. Sancho D, Enamorado M, Garaude J. Innate immune function of mitochondrial metabolism. Front Immunol. (2017) 8:527. doi: 10.3389/fimmu.2017.00527
280. Torres-Odio S, Key J, Hoepken H-H, Canet-Pons J, Valek L, Roller B, et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J Neuroinflammation (2017) 14:154. doi: 10.1186/s12974-017-0928-0
281. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature (2015) 520:553–7. doi: 10.1038/nature14156
282. Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N, et al. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol. (2006) 26:611–7. doi: 10.1161/01.ATV.0000201938.78044.75
283. Gu J, Ye S, Wang S, Sun W, Hu Y. Metformin inhibits nuclear factor-κB activation and inflammatory cytokines expression induced by high glucose via adenosine monophosphate-activated protein kinase activation in rat glomerular mesangial cells in vitro. Chin Med J (Engl) (2014) 127:1755–60. doi: 10.3760/cma.j.issn.0366-6999.20132781
284. Woo SL, Xu H, Li H, Zhao Y, Hu X, Zhao J, et al. Metformin ameliorates hepatic steatosis and inflammation without altering adipose phenotype in diet-induced obesity. PLoS ONE (2014) 9:e91111. doi: 10.1371/journal.pone.0091111
285. Cameron AR, Morrison VL, Levin D, Mohan M, Forteath C, Beall C, et al. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ Res. (2016) 119:652–65. doi: 10.1161/CIRCRESAHA.116.308445
286. Kaufman BA, Li C, Soleimanpour SA. Mitochondrial regulation of beta-cell function: maintaining the momentum for insulin release. Mol Aspects Med. (2015) 42:91–104. doi: 10.1016/j.mam.2015.01.004
287. Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, et al. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med. (2000) 342:1560–7. doi: 10.1056/NEJM200005253422103
288. Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science (2003) 299:256–9. doi: 10.1126/science.1077209
289. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science (2004) 304:1158–60. doi: 10.1126/science.1096284
290. Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson's disease. Biochim Biophys Acta (2010) 1802:29–44. doi: 10.1016/j.bbadis.2009.08.013
291. Exner N, Treske B, Paquet D, Holmstrom K, Schiesling C, Gispert S, et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. (2007) 27:12413–8. doi: 10.1523/JNEUROSCI.0719-07.2007
292. Mortiboys H, Aasly J, Bandmann O. Ursocholanic acid rescues mitochondrial function in common forms of familial Parkinson's disease. Brain (2013) 136:3038–50. doi: 10.1093/brain/awt224
293. Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. (2003) 100:4078–83. doi: 10.1073/pnas.0737556100
294. Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. (2004) 279:18614–22. doi: 10.1074/jbc.M401135200
295. Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature (2006) 441:1157–61. doi: 10.1038/nature04788
296. Gispert S, Ricciardi F, Kurz A, Azizov M, Hoepken HH, Becker D, et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE (2009) 4:e5777. doi: 10.1371/journal.pone.0005777
297. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature (2006) 441:1162–6. doi: 10.1038/nature04779
298. Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. (2010) 12:119–31. doi: 10.1038/ncb2012
299. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. (2010) 8:e1000298. doi: 10.1371/journal.pbio.1000298
300. Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature (2014) 510:162–6. doi: 10.1038/nature13392
301. Lee JJ, Sanchez-Martinez A, Zarate AM, Benincá C, Mayor U, Clague MJ, et al. (2018). Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol. 217:1613–22. doi: 10.1083/jcb.201801044
302. McWilliams TG, Prescott AR, Montava-Garriga L, Ball G, Singh F, Barini E, et al. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. (2018) 27:439–449.e435. doi: 10.1016/j.cmet.2017.12.008
303. Morais VA, Haddad D, Craessaerts K, De Bock PJ, Swerts J, Vilain S, et al. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science (2014) 344:203–7. doi: 10.1126/science.1249161
304. Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, et al. PINK1 and parkin target miro for phosphorylation and degradation to arrest mitochondrial motility. Cell (2011) 147:893–906. doi: 10.1016/j.cell.2011.10.018
305. Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, et al. The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat Cell Biol. (2007) 9:1243–52. doi: 10.1038/ncb1644
306. Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. (2007) 5:e172. doi: 10.1371/journal.pbio.0050172
307. Fitzgerald JC, Zimprich A, Carvajal Berrio DA, Schindler KM, Maurer B, Schulte C, et al. Metformin reverses TRAP1 mutation-associated alterations in mitochondrial function in Parkinson's disease. Brain (2017) 140:2444–59. doi: 10.1093/brain/awx202
308. Wu W, Xu H, Wang Z, Mao Y, Yuan L, Luo W, et al. PINK1-parkin-mediated mitophagy protects mitochondrial integrity and prevents metabolic stress-induced endothelial injury. PLoS ONE (2015) 10:e0132499. doi: 10.1371/journal.pone.0132499
309. Choi J, Ravipati A, Nimmagadda V, Schubert M, Castellani RJ, Russell JW. Potential roles of PINK1 for increased PGC-1α-mediated mitochondrial fatty acid oxidation and their associations with Alzheimer disease and diabetes. Mitochondrion (2014) 18:41–8. doi: 10.1016/j.mito.2014.09.005
310. Cang X, Wang X, Liu P, Wu X, Yan J, Chen J, et al. PINK1 alleviates palmitate induced insulin resistance in HepG2 cells by suppressing ROS mediated MAPK pathways. Biochem Biophys Res Commun. (2016) 478:431–8. doi: 10.1016/j.bbrc.2016.07.004
311. Deas E, Piipari K, Machhada A, Li A, Gutierrez-del-Arroyo A, Withers DJ, et al. (2014). PINK1 deficiency in β-cells increases basal insulin secretion and improves glucose tolerance in mice. Open Biol. 4:140051. doi: 10.1098/rsob.140051
312. Seillier M, Pouyet L, N'Guessan P, Nollet M, Capo F, Guillaumond F, et al. Defects in mitophagy promote redox-driven metabolic syndrome in the absence of TP53INP1. EMBO Mol Med. (2015) 7:802–18. doi: 10.15252/emmm.201404318
313. Grassi D, Howard S, Zhou M, Diaz-Perez N, Urban NT, Guerrero-Given D, et al. Identification of a highly neurotoxic alpha-synuclein species inducing mitochondrial damage and mitophagy in Parkinson's disease. Proc Natl Acad Sci USA. (2018) 115:E2634–43. doi: 10.1073/pnas.1713849115
314. Butler EK, Voigt A, Lutz AK, Toegel JP, Gerhardt E, Karsten P, et al. The mitochondrial chaperone protein TRAP1 mitigates alpha-Synuclein toxicity. PLoS Genet. (2012) 8:e1002488. doi: 10.1371/journal.pgen.1002488
315. Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum Mol Genet. (2005) 14:2099–111. doi: 10.1093/hmg/ddi215
316. Simon-Sanchez J, Singleton AB. Sequencing analysis of OMI/HTRA2 shows previously reported pathogenic mutations in neurologically normal controls. Hum Mol Genet. (2008) 17:1988–93. doi: 10.1093/hmg/ddn096
317. Fitzgerald JC, Zimprich A, Reddy Bobbili D, Sharma M, May P, Kruger R. Reply: No evidence for rare TRAP1 mutations influencing the risk of idiopathic Parkinson's disease. Brain (2018) 141:e17. doi: 10.1093/brain/awx380
318. Gaare JJ, Nido GS, Sztromwasser P, Knappskog PM, Dahl O, Lund-Johansen M, et al. No evidence for rare TRAP1 mutations influencing the risk of idiopathic Parkinson's disease. Brain (2018) 141:e16. doi: 10.1093/brain/awx378
319. Rasola A, Neckers L, Picard D. Mitochondrial oxidative phosphorylation TRAP(1)ped in tumor cells. Trends Cell Biol. (2014) 24:455–63. doi: 10.1016/j.tcb.2014.03.005
320. Yoshida S, Tsutsumi S, Muhlebach G, Sourbier C, Lee MJ, Lee S, et al. Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc Natl Acad Sci USA. (2013) 110:E1604–12. doi: 10.1073/pnas.1220659110
321. Sciacovelli M, Guzzo G, Morello V, Frezza C, Zheng L, Nannini N, et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. (2013) 17:988–99. doi: 10.1016/j.cmet.2013.04.019
322. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell (2016) 167:457–470.e413. doi: 10.1016/j.cell.2016.08.064
323. Matassa DS, Amoroso MR, Lu H, Avolio R, Arzeni D, Procaccini C, et al. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell Death Differ. (2016) 23:1542–54. doi: 10.1038/cdd.2016.39
324. Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci. (2009) 29:11011–9. doi: 10.1523/JNEUROSCI.2519-09.2009
325. Swerdlow RH, Burns JM, Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis. J Alzheimer's Dis. (2010) 20(Suppl. 2):S265–79. doi: 10.3233/JAD-2010-100339
326. David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem. (2005) 280:23802–14. doi: 10.1074/jbc.M500356200
327. Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, et al. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer's disease mice. Proc Natl Acad Sci USA. (2009) 106:20057–62. doi: 10.1073/pnas.0905529106
328. Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, et al. Tau reduction prevents Abeta-induced defects in axonal transport. Science (2010) 330:198. doi: 10.1126/science.1194653
329. Calkins MJ, Reddy PH. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer's disease neurons. Biochim Biophys Acta (2011) 1812:507–13. doi: 10.1016/j.bbadis.2011.01.007
330. Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. (2005) 58:495–505. doi: 10.1002/ana.20624
331. Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. (2005) 57:695–703. doi: 10.1002/ana.20474
332. Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther. (2012) 342:619–30. doi: 10.1124/jpet.112.192138
333. Schapira AH, Jenner P. Etiology and pathogenesis of Parkinson's disease. Mov Disord. (2011) 26:1049–55. doi: 10.1002/mds.23732
334. Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. (2000) 29:222–30. doi: 10.1016/S0891-5849(00)00317-8
335. Li N, Oquendo E, Capaldi RA, Robinson JP, He YD, Hamadeh HK, et al. A systematic assessment of mitochondrial function identified novel signatures for drug-induced mitochondrial disruption in cells. Toxicol Sci (2014) 142:261–73. doi: 10.1093/toxsci/kfu176
336. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
337. Pakendorf B, Stoneking M. MITOCHONDRIAL DNA AND HUMAN EVOLUTION. Annu Rev Genomics Hum Genet. (2005) 6:165–83. doi: 10.1146/annurev.genom.6.080604.162249
338. Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature (2013) 497:451–7. doi: 10.1038/nature12188
339. Dolle C, Flones I, Nido GS, Miletic H, Osuagwu N, Kristoffersen S, et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun. (2016) 7:13548. doi: 10.1038/ncomms13548
340. Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. (2011) 194:7–15. doi: 10.1083/jcb.201102095
341. Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol. (2007) 8:722–8. doi: 10.1038/nrm2240
342. Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. (2004) 10(Suppl.) S18–25. doi: 10.1038/nrn1434
343. Andrews ZB, Diano S, Horvath TL. Mitochondrial uncoupling proteins in the cns: in support of function and survival. Nat Rev Neurosci. (2005) 6:829. doi: 10.1038/nrn1767
344. Kahle PJ, Waak J, Gasser T. DJ-1 and prevention of oxidative stress in Parkinson's disease and other age-related disorders. Free Radic Biol Med. (2009) 47:1354–61. doi: 10.1016/j.freeradbiomed.2009.08.003
345. Xu J, Zhong N, Wang H, Elias JE, Kim CY, Woldman I, et al. The Parkinson's disease-associated DJ-1 protein is a transcriptional co-activator that protects against neuronal apoptosis. Hum Mol Genet. (2005) 14:1231–41. doi: 10.1093/hmg/ddi134
346. Kim RH, Peters M, Jang Y, Shi W, Pintilie M, Fletcher GC, et al. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell (2005) 7:263–73. doi: 10.1016/j.ccr.2005.02.010
347. Wu L, Xu H, Cao L, Li T, Li R, Feng Y, et al. Salidroside protects against MPP+-induced neuronal injury through DJ-1-Nrf2 antioxidant pathway. Evid Based Complement Altern Med. (2017) 2017:5398542. doi: 10.1155/2017/5398542
348. Zhou W, Zhu M, Wilson MA, Petsko GA, Fink AL. The oxidation state of DJ-1 regulates its chaperone activity toward alpha-synuclein. J Mol Biol. (2006) 356:1036–48. doi: 10.1016/j.jmb.2005.12.030
349. Bjorkblom B, Adilbayeva A, Maple-Grodem J, Piston D, Okvist M, Xu XM, et al. Parkinson disease protein DJ-1 binds metals and protects against metal-induced cytotoxicity. J Biol Chem. (2013) 288:22809–20. doi: 10.1074/jbc.M113.482091
350. van der Brug MP, Blackinton J, Chandran J, Hao LY, Lal A, Mazan-Mamczarz K, et al. RNA binding activity of the recessive parkinsonism protein DJ-1 supports involvement in multiple cellular pathways. Proc Natl Acad Sci USA. (2008) 105:10244–9. doi: 10.1073/pnas.0708518105
351. Lee JY, Song J, Kwon K, Jang S, Kim C, Baek K, et al. Human DJ-1 and its homologs are novel glyoxalases. Hum Mol Genet. (2012) 21:3215–25. doi: 10.1093/hmg/dds155
352. Richarme G, Mihoub M, Dairou J, Bui LC, Leger T, Lamouri A. Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J Biol Chem. (2015) 290:1885–97. doi: 10.1074/jbc.M114.597815
353. Richarme G, Dairou J. Parkinsonism-associated protein DJ-1 is a bona fide deglycase. Biochem Biophys Res Commun. (2017) 483:387–91. doi: 10.1016/j.bbrc.2016.12.134
354. Eberhard D, Lammert E. The role of the antioxidant protein DJ-1 in type 2 diabetes mellitus. Adv Exp Med Biol. (2017) 1037:173–86. doi: 10.1007/978-981-10-6583-5_11
355. De Jager J, Kooy A, Lehert P, Wulffelé MG, Van Der Kolk J, Bets D, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. (2010) BMJ 340:c2181. doi: 10.1136/bmj.c2181
356. Warasila W. Forget the Blood of Teens. This Pill Promises to Extend Life for a Nickel a Pop. Wired Magazine (2017).
357. Lane N. Power, Sex, Suicide. Mitochondria and the Meaning of Life. Oxford: Oxford University Press (2005).
Keywords: metformin, neurodegeneration, diabetes, Parkinson's disease, Alzheimer's disease, aging, mitochondria
Citation: Rotermund C, Machetanz G and Fitzgerald JC (2018) The Therapeutic Potential of Metformin in Neurodegenerative Diseases. Front. Endocrinol. 9:400. doi: 10.3389/fendo.2018.00400
Received: 04 April 2018; Accepted: 27 June 2018;
Published: 19 July 2018.
Edited by:
Frederic Bost, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Hudson Sousa Buck, Faculdade de Ciências Médicas, Santa Casa de São Paulo, BrazilGuillermo Romero, University of Pittsburgh, United States
Antonio J. Herrera, Departamento de Bioquímica y Biología Molecular, Facultad de Farmacia, Universidad de Sevilla, Spain
Copyright © 2018 Rotermund, Machetanz and Fitzgerald. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julia C. Fitzgerald, anVsaWEuZml0emdlcmFsZEB1bmktdHVlYmluZ2VuLmRl