 Ivan N. Tyurenkov1
Ivan N. Tyurenkov1 Denis V. Kurkin1
Denis V. Kurkin1 Dmitry A. Bakulin1*Elena V. Volotova1
Dmitry A. Bakulin1*Elena V. Volotova1 Evgeny I. Morkovin1,2Mikhail A. Chafeev3Ruben N. Karapetian3
Evgeny I. Morkovin1,2Mikhail A. Chafeev3Ruben N. Karapetian3- 1Volgograd State Medical University, Volgograd, Russia
- 2Volgograd Medical Research Center, Volgograd, Russia
- 3Chemical Diversity Research Institute, Khimki, Russia
This article is to highlight the chemical properties and primary pharmacology of novel GPR119 agonist ZB-16 and its analogs, which were rejected during the screening. Experiments were performed in vitro (specific activity, metabolism and cell toxicity) and in vivo (hypoglycemic activity and pharmacokinetics). ZB-16 exhibits nanomolar activity (EC50 = 7.3–9.7 nM) on target receptor GPR119 in vitro associated with hypoglycemic activity in vivo. In animals with streptozotocin-nicotinamide induced type 2 diabetes mellitus (STZ-NA T2D) daily oral dose of ZB-16 (1 mg/kg) or sitagliptin (10 mg/kg) for 28 days resulted in the reduction of blood glucose levels. The effects of ZB-16 were comparable to the hypoglycemic action of sitagliptin. ZB-16 demonstrated relatively low plasma exposition, high distribution volume, mild clearance and a prolonged half-life (more than 12 h). The present study demonstrates that the targeted search for selective GPR119 receptor agonists is a well-founded approach for developing novel drugs for the therapy of T2D. Based on the combination of high in vitro activity (compared to competitor standards), a useful ADME profile, distinct hypoglycemic activity which is comparable to the efficacy of sitagliptin in rats with experimental T2D, and the acceptable pharmacokinetic profile, we recommend the ZB-16 compound for further research.
Introduction
Over the past century, there has been a dramatic increase in the prevalence of metabolic disorders such as diabetes mellitus and obesity. In accordance to International Diabetes Federation there are 425 million people with diabetes in the World, and this portion will increase up to 629 million in 2045 (1). About 12% of global health expenditure ($727 billion) is spent on diabetes, thus the development of novel medicines and treatment strategies plays crucial role in the improvement of the disease management. In the last few decades, there has been a surge of interest in the effects of incretins and their analogs on the glucose metabolism and utilization. Evidence suggests that G protein-coupled receptor 119 (GPR119) is implicated in the incretin-depended insulin synthesis and exocytosis (2, 3). Mostly expressed either on intestinal L- and K-cells or pancreatic β-cells, GPR119 had been validated as a potential target for novel pharmacological agents which could become a prominent medicine against type 2 diabetes mellitus (T2D), obesity and metabolic syndrome (4). The activation of GPR119 stimulates the secretion of incretins—glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) (5) are metabolic hormones which augment the secretion of insulin (GLP-1 and GIP) from pancreatic β-cells and inhibit glucagon release (GLP-1) in α-cells. Unlike GIP, the secretion of GLP-1 is impaired in patients with T2D (6, 7).
Both GLP-1 and GIP, being rapidly inactivated by the enzyme dipeptidyl peptidase-4 (DPP-4) expressed on the surface of most cell types, have relatively short life-time. Although several researches resulted in the development of synthetic GLP-1 analogs, resistant to the DPP-4 action, or DPP-4 inhibitors, extending the incretin action in T2D (8), the potency of GPR119 agonists to enhance the incretin action is not well studied (9, 10).
The 10 years of clinical experience shown that GLP-1 analogs and DPP-4 inhibitors exert moderate or potent hypoglycemic activity and are safe in patients with T2D. The secondary effects of GLP-1 analogs include the body weight decline and the decrease in risk of vascular complications of T2D. The main disadvantages are the lack of GLP-1 analogs for oral administration, or insufficient efficacy of DPP-4 inhibitors.
Previously we published our data (11) about the activity of novel GPR119 agonist ZB-16 in vivo, which exerted a potent anti-diabetic action in rat model of T2D. ZB-16 administered orally (1 mg/kg/day) for 28 day decreased the blood glucose levels under fasting conditions and improved the glucose utilization in male Wistar rats with streptozotocin-nicotinamide induced type 2 diabetes mellitus (STZ-NA T2D). These changes were associated with the increase of stimulated secretion of GLP-1 and insulin, accompanied by the growth of insulin-positive cells in pancreas. This article is to highlight the chemical properties and primary pharmacology of ZB-16 both in vitro and in vivo compared with other series of similar compounds rejected during the screening.
Methods
In vitro Studies
Solubility Evaluation
A Millipore filter system was used to measure the kinetic solubility in each of 3 Britton-Robinson universal buffer solutions of pH 2, 4, and 7 (0.04 M H3BO3, 0.04 M H3PO4 and 0.04 M CH3COOH titrated with 0.2 M NaOH) in the presence of 2% DMSO (12). Verapamil and diethylstilbestrol constituted the high and low solubility control solutions, respectively.
Specific Activity in vitro
Specific activity was evaluated using an immortalized Chinese hamster ovary cell line CHO-K1 steadily expressed human GPR119 (hGPR119). The cells (107 ml−1) were cultured in Dulbecco's modified Eagle medium (DMEM) modified with 10% fetal bovine serum (FBS), revolved continuously at 50 rpm at 37°C in 8% CO2 for 3–4 days. Lance Ultra cAMP kit (Perkin Elmer, Waltham, MA, USA) was chosen as the experimental platform (13). The experiments were performed in accordance to the manufacturer's instructions. The agonistic activity of the test compounds was estimated by the increase in intracellular cAMP concentration, assuming the hGPR119 activation is followed by the activation of cellular adenylate cyclase, which increases intracellular cAMP levels (4, 14). Known agonists of hGPR119 receptors were used as positive controls to determine the maximal possible effect (MPE). The activity of test compounds was expressed in terms of median effective concentration (EC50).
Biotransformation in vitro
The metabolism of the compounds was studied in commercially available microsomal fractions derived from human and rat hepatocytes. Test compounds (0.5 μM) were incubated with microsomal fractions in the presence of NADPH during 30 min in a shaker at 37°C. Reactions were stopped at 0, 5, 10, 15, and 30 min by adding acetonitrile. After proteins precipitation, the remaining amount of the test compound in the supernatant was detected with high performance liquid chromatography tandem-mass spectrometry (HPLC-MS/MS) method using Agilent 1260 liquid chromatograph (Agilent Technologies, Santa Clara, CA, USA) and a QTRAP6500 triple quadrupole mass spectrometer with a TurboIonSpray electro-spraying module (ABSciex, Foster City, CA, USA) (15). Incubation was performed twice, with at least 2 measurements for each replicate (n ≥ 3). The half-life (T1/2), the clearance in vitro (CLint) and the remaining amount of substance (% of initial quantity) were calculated.
Inhibition of cytochrome P450 (CYP) isozymes, in particular CYP3A4, CYP1A2, CYP2C9, CYP2C19, and CYP2D6, was studied using Vivid® CYP450 Screening Kits (Invitrogen Corporation, Carlsbad, CA, USA) in accordance to the Invitrogen protocol (Vivid CYP450 screening kit protocol O-13873-r1 US 0405) in a 384-well plate format. Ketoconazole, α-naphtoflavone, sulfaphenazole, miconazole and quinidine (Sigma-Aldrich) were used as the control inhibitors of CYP3A4, CYP1A2, CYP2C9, CYP2C19 and CYP2D6 correspondingly (16). Test compounds were incubated for 15 min in the presence of a NADPH-regenerating system. Briefly, the reaction was initiated by adding NADP+ and a substrate converted to fluorescent derivates by specific CYP isozyme. The reaction was terminated by adding 1M Tris. The fluorescence of the reaction product, which was proportional to CYP isozyme activity, was measured. Test compounds and the control inhibitor were tested twice at concentrations ranging from 0.0046 and 10 μM. The maximal enzyme activity determined in 1% DMSO with no compounds added. The minimal signal was defined as the signal obtained without enzyme incubation. The inhibitory action of test compounds on the CYP isozymes was expressed in terms of median inhibitory concentration (IC50).
Cell Toxicity in vitro
Cell viability was estimated by the quantity of ATP produced by the HepG2 cell culture, derived from human liver hepatocellular carcinoma. Experiments were performed using a Cell Titer Glo Cell Viability (Promega) test system (17). Tubercidin, an inhibitor of various cellular metabolic processes, including RNA processing, nucleic acid synthesis, protein synthesis, and methylation of tRNA through intracellular incorporation into nucleic acids, was used as a positive control (18). Median inhibitory concentration (IC50) was used as a quantitative parameter to evaluate cell toxicity. To evaluate inhibition effectiveness (% Inh), the following formula was used: % Inh = [(Lpos − Lexp)/(Lpos − Lneg)] * 100%, where Lpos is the positive control (luminescence in cells with no compound); Lneg is the negative control (luminescence in cells containing medium but no cells); and Lexp is the luminescence in cells containing the compound at a certain concentration. IC50 values were calculated using Prism 5 and the criteria for minimization of the experimental points squares deviation from the theoretically calculated curve algorithm.
Cardiac toxicity was evaluated by estimating the influence of each substance on hERG potassium channels using the in vitro test system Invitrogen® PredictorTM hERG (Invitrogen Corporation, Carlsbad, CA, USA), containing Predictor hERG membrane preparations, fluorescent tracer stock solution and an assay buffer solution. Experimental procedures were conducted in accordance with the PredictorTM hERG test system's manufacturer instructions. Assays were performed in 384-well microplates. Each well contained 5 μL aliquot of each concentration of test compound, 10 μL of (2X) membrane preparation and 5 μL of working tracer solution. The cumulative reaction volume was 20 μl with 1% of DMSO (19). All procedures were performed at 4 replicates for each concentration. The plate was allowed to incubate at room temperature for at least 1 h prior to fluorescence polarization measurements performed with a microplate reader Infinite M1000 PRO (Tecan).
In vivo Studies
All animal experiments were conducted in accordance with ethical animal research standards defined by Russian law (20), Guidelines for preclinical trials of medicinal products (21), Guide for the care and use of laboratory animals (22) and approved by the local ethical committee (VMRC, Volgograd, Russian Federation; registration number IRB 00005839 IORG 0004900 (OHRP), Protocol Number: 191-2014, February 25, 2014). The present study used female Wistar rats aged 3–4 months weighing 250–280 g. Animals were housed 4–6 per cage in a separate room with controlled environment (20–26°C, relative humidity 50–70%, consistent 12/12 light/dark regimen, 8:00 lights on) with food and water ad libitum (except intentional fasting needed for blood glucose levels determination described below). At the end of the experiment animals were sacrificed by an overdose of chloral hydrate (800 mg/kg).
Evaluation in vivo of the Hypoglycemic Action in Intact Rats
Blood glucose levels were evaluated in the peripheral blood (from the tail vein) of intact fasted (6 h) female Wistar rats using a glucometer Contour TS (Bayer, Germany) at 60, 120, and 180 min after single oral administration of each ZB-09 analogs (in doses of 0.1, 1, and 10 mg/kg). The female animals were used because of their advantages in toxicological studies shown by several authors (23).
Evaluation of the Hypoglycemic Action in a Rat Model of T2D
Procedures were performed in 40 female Wistar rats weighing 250–280 g. Streptozotocin-nicotinamide-induced rat model of T2D was used (24, 25). Briefly, streptozotocin (Sigma, St. Louis, MO, USA) was administered i.p. to overnight fasted rats (65 mg/kg in 100 mmol/l citrate buffer, Ph 4.5). Nicotinamide (Sigma, St. Louis, MO, USA) (230 mg/kg) dissolved in saline was administered i.p. 15 min before streptozotocin. STZ-NA T2D had been confirmed 72 h after in rats with blood glucose levels in a range 9–14 mmol/l measured in fasting (6 h) conditions. Animals failed to reach the criteria of STZ-NA T2D were excluded from the experiment. Following the continuous oral administration of sitagliptin (10 mg/kg), ZB-16 (1 mg/kg) or saline during 28 days STZ-NA T2D confirmation the fasting blood glucose levels were measured, and oral glucose tolerance test (OGTT) was performed. For OGTT rats received an oral dose of 3 mg/kg glucose dissolved in saline; blood glucose levels were monitored every 30 min for 2 h after glucose administration. Sitagliptin dose was chosen in accordance with published data (26).
Evaluation of Pharmacokinetics in vivo
The pharmacokinetics of ZB-16 in a dose of 10 mg/kg after single oral or intravenous administration was studied in 6 male Wistar rats weighing 250–280 g. The blood from the tail vein (200 μL) was collected before and at 0, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 h after ZB-16 administration. The blood concentrations of the test compounds in animals were evaluated using HPLC-MS/MS. Basic pharmacokinetic parameters were calculated using a non-model method and WinNonlin Professional 6.0 (Pharsight Corporation, Mountain View, CA, USA) based on the experimental concentration-time data obtained on each animal. The list of calculated parameters included: AUC0 → t (ng * hour/ml)—area under the “drug concentration—time” curve from the injection until the last measurement; AUC0 → ∞ (ng * hour/ml)—area under the curve from the injection until infinity; Cmax (ng/ml)—maximal blood plasma drug concentration; C0 (ng/ml)—calculated initial blood plasma concentration of the drug after i.v. injection; kel (1/hour)—velocity of elimination constant (the parameter that characterizes the velocity of elimination of the compound from blood plasma); MRT (hour)—average compound lifetime in the organism from the moment of injection; tmax (h)—time required for the compound to achieve its maximum blood plasma concentration; t1/2 (h)—half-life period (the time required for half of the compound to be eliminated from the blood plasma); F (%)—relative bioavailability, calculated using the following formula: F (%) = 100% * [AUC0 → t, p.o. * Di.v.]/[AUC0 → t, i.v. * Dp.o.], where D – p.o. or i.v. dose.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA). The distribution of all data was estimated using the Shapiro-Wilk normality test. Kruskal-Wallis test with Dunn's post hoc test was used for non-parametric data distribution. One-way ANOVA or repeated measures two-way ANOVA with Newman-Keuls post hoc test was performed for data sets obtained from Gaussian distribution (indicated in the figure legends).
Results
Development and Screening
Based on selective high throughput screening of the data library consisting of 1.5 million small organic molecules, 2 prospective compound series were chosen. The structures of the most promising compounds from these series are presented in Figure 1.
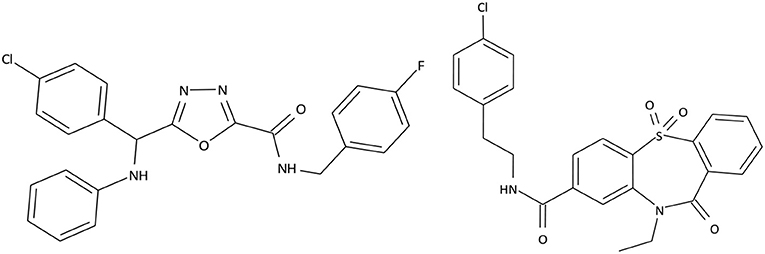
Figure 1. Structures of C301-5947 and C530-0315.
C301-5947, C530-0315, and 40 similar compounds were synthesized. Each compound was screened for primary agonist activity (EC50) to evaluate putative structure-effect patterns and to further target structure modifications. Activity screening results are presented in Supplement 1. Non-active compounds are shown at Supplement 2.
The compounds exhibiting low levels of activity compared with Arena (0119) we rejected from further investigation. After analyzing the open access formulas of compounds synthesized by pharmaceutical companies and introducing new and original elements into their structures, ZB series of test compounds was developed (Figure 2).
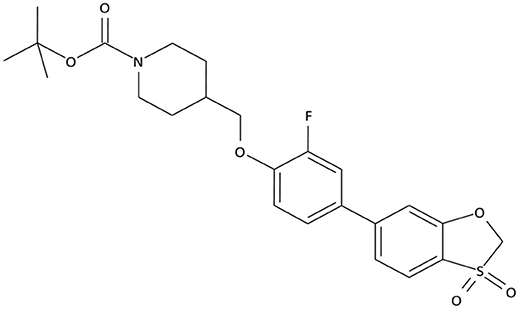
Figure 2. Primary structure of ZB-09.
ZB-09 exhibited an EC50 = 4 nM, comparable to the standard compound Arena (0119). At this stage, absorption, distribution, metabolism and excretion parameters (ADME) were evaluated. The test compound ZB-09 was found to be barely soluble in water at all pH levels investigated, significantly limiting its further use (Table 1).
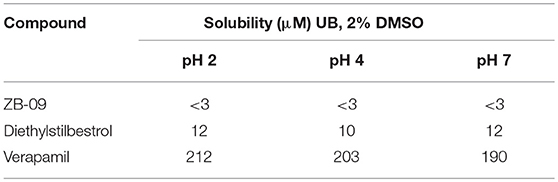
Table 1. Solubility of ZB-09 by pH.
The biotransformation assay revealed that ZB-09 exhibits a high rate of microsomal oxidation (Table 2), which can lead to inferior pharmacokinetics in vivo, including the short life time and low blood concentrations of the compound. Therefore, we focused on improving the properties of the compound by modifying its structure. First, we had to increase the solubility and microsomal stability of the compound.
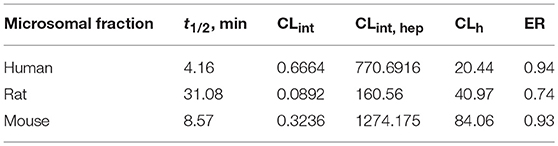
Table 2. Microsomal stability of ZB-09.
The structures and activities of the synthesized analogs of ZB-09 are summarized in Table 3.
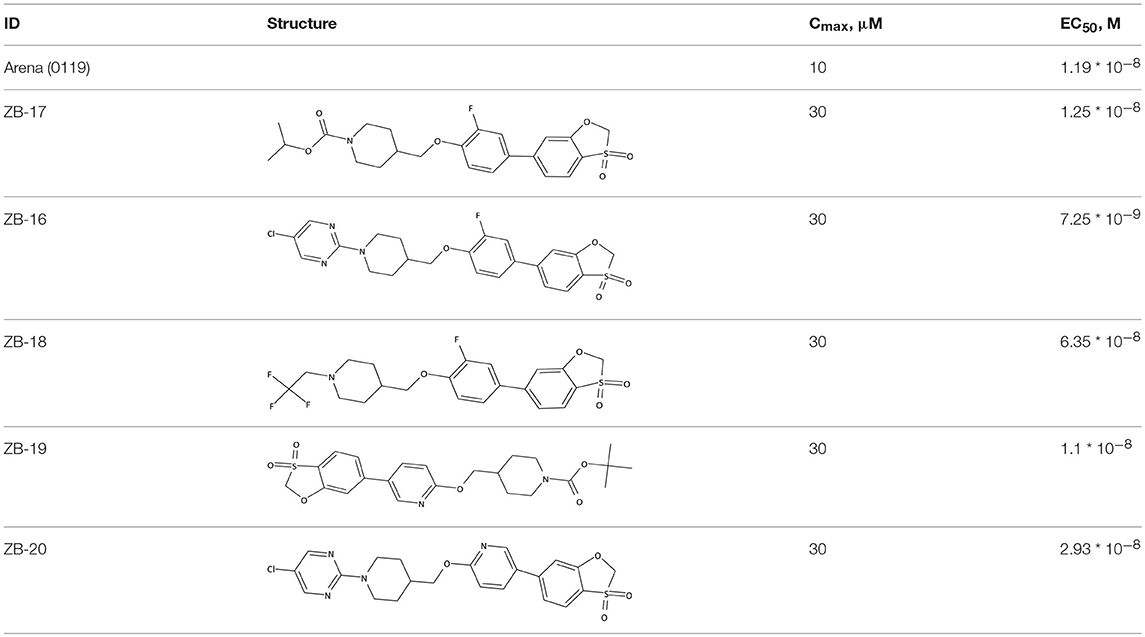
Table 3. Structures and activities of ZB-09 analogs.
ZB-09 analogs showed the activity levels within the Arena (0119) standard range. Table 4 presents pH-depended solubility of these compounds.
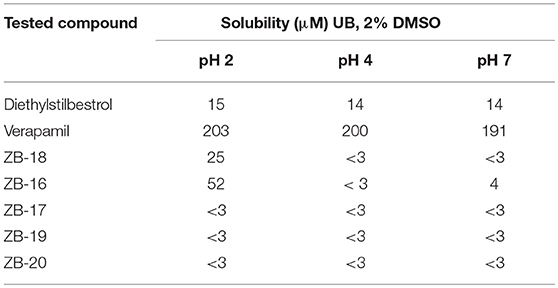
Table 4. Effects of pH on the water solubility of ZB-09 analogs.
ZB-16 was found to be the most effective compound investigated. Nonetheless, we decided to further investigate ZB-19 and ZB-20 due to their high agonistic activity.
Microsomal Stability of ZB-09 Analogs
As seen in Table 5, ZB-16 exhibits significantly higher resistance in human and rat microsomes compared to ZB-09, the primary compound of the series.
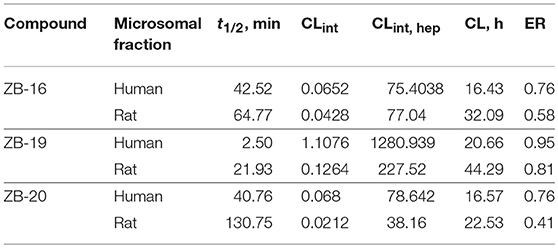
Table 5. Microsomal stability of test compounds.
The ZB-19 compound is the most similar to the standard compound Arena (0119) with regard to in vitro activity, although the presence of the end tert-butyl-oxycarbonyl group makes ZB-19 nonresistant to liver enzymes. The ZB-20 compound, in which, as for ZB-16, the tert-butyl-oxycarbonyl group was replaced with a resistant heterocyclic bioisostere, chlorine-pyrimidine, did not exhibit increased solubility in water (as was the case with ZB-16) but did exhibit increased resistance to liver enzymes. This compound has an in vitro EC50 of 29 nM, which is less than those of the ZB-16 compound and the Arena (0119) but still acceptable.
Cardiac Toxicity of Leader Compounds
The human Ether-à-go-go-Related Gene (hERG) codes the alpha subunit of a potassium ion channel which contributes to the electrical activity of the heart. Inhibition or compromising of activity of this ion channel could result in long QT syndrome creating the concomitant sudden death risk (27). Results obtained with regard to the binding of compounds from the ZB series to hERG are presented in Table 6.

Table 6. Binding of compounds from the ZB series to hERG.
The test compounds bind to less than 50% hERG in both concentrations tested (10 and 1 μM). This results in minimal cardiac toxicity risk in regard to the activity of hERG ion channels.
Cell Toxicity of Leader Compounds
As shown in Figure 3 ZB-16 had no effect on HepG2 cell culture viability.
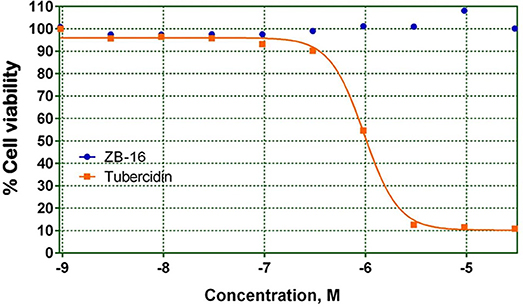
Figure 3. HepG2 cell culture viability on the presence of test compound ZB-16 or the control compound Tubercidin. The data expressed as the means (n ≥ 3); the response was normalized to the values of the cells cultured without test or control compounds.
CYP450 Inhibition by Leader Compounds
Parameters pertaining to CYP450 inhibition by ZB series are presented in Table 7.

Table 7. Inhibition of CYP450 isoforms by ZB-16 and ZB-20 test compounds and standard compounds.
As seen in Table 7, ZB-16 and ZB-20 inhibit neither of the CYP450 isoforms, indicating that the metabolism of other drugs is unlikely to be affected.
GPR119 Agonistic Activity of Leader Compound
The relationship between GPR119 agonistic activity and concentration was analyzed for the ZB-16 compound. The EC50 was calculated from the graph shown in Figure 4.
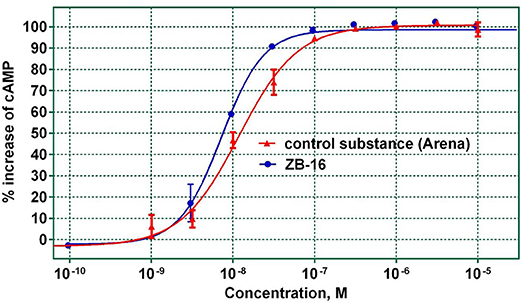
Figure 4. The agonistic activity of ZB-16 and the control substance (Arena) with regard to hGPR119 target receptors. The data expressed as the mean ± SEM (n ≥ 3); the response was normalized to the maximal response value obtained with the control substance.
As seen in Figure 4, ZB-16 exhibits nanomolar activity (EC50 = 7.3–9.7 nM) on target GPR119 receptors, indicating that ZB-16 is a complete agonist. Based on the determined ADME parameter set and high activity in vitro, ZB-16 and ZB-20 were selected for in vivo studies.
Hypoglycemic Activity of Leader Compounds
ZB-09 analogs were tested in vivo for their hypoglycemic action in intact rats. All compounds, except ZB-19, were included in this study. The in vivo results are presented in Figure 5.
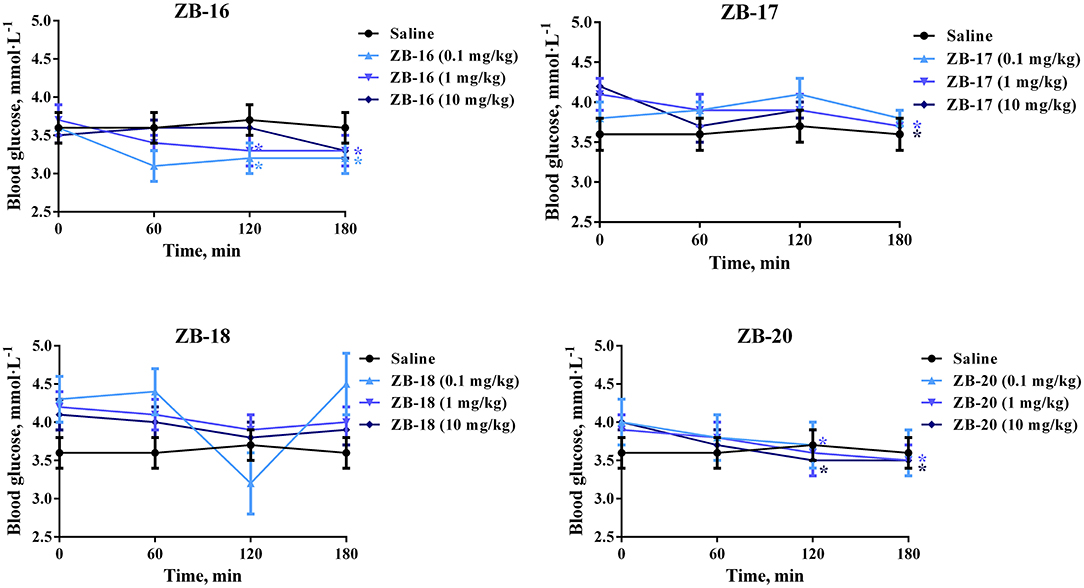
Figure 5. Hypoglycemic action of ZB-09 analogs in intact rats after single oral administration. The data expressed as the mean ± SEM (n = 10); *p < 0.05, significant, compared to initial levels of glycemia (2-way ANOVA with Newman-Keuls post hoc); no significant difference was seen at 0 min (p > 0.05).
ZB-16 caused significant decrease in blood glucose levels after single oral administration of 0.1–1 mg/kg. ZB-17 and ZB-20 both exhibited similar hypoglycemic activity at higher dosages (1–10 mg/kg).
Hypoglycemic Activity of ZB-16 After Single Oral Administration in T2D Rat Model
Single oral administration of ZB-16 (1 mg/kg) in fasted rats 3 days after the confirmation of streptozotocin-nicotinamide (STZ-NA) induced T2D caused a 24% decrease of blood glucose levels over a 3 h period. This hypoglycemic effect was more prominent than that obtained for sitagliptin (10 mg/kg) (p < 0.05). This effect increased over time and continued until the end of the observation period (Figure 6).
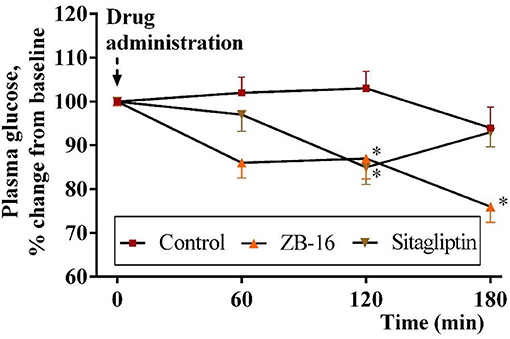
Figure 6. The decrease in plasma glucose level (changes from baseline) in rats with experimental diabetes over a 3 h period after single intragastral administration of ZB-16 or sitagliptin. The data were normalized to initial values and are expressed as the mean ± SEM (n = 10); Intact, the intact group of animals; Control, group of non-treated animals with experimental diabetes; *p < 0.05, significant, compared to the Control group (2-way ANOVA with Newman-Keuls post hoc).
Hypoglycemic Activity of ZB-16 After Continuous Oral Administration in T2D Rat Model
The animals received daily oral dose of ZB-16 (1 mg/kg) or sitagliptin (10 mg/kg) for 28 days after the confirmation of STZ-NA induced T2D. Body weight of rats are shown at Supplement 3. On 28 day of treatment oral glucose tolerance test was conducted.
No significant changes were observed in control animals treated with saline. ZB-16 group showed blood glucose levels by 27% lower than in saline group 30 min after oral glucose administration (3 mg/kg). One hour after glucose administration, glucose blood levels were slightly lower in the treated groups compared to the control group. Two hours after glucose administration the mean blood glucose level in rats received ZB-16 was 9.1 ± 0.37 mmol/l, which was significantly lower than in the control group (Figure 7A).
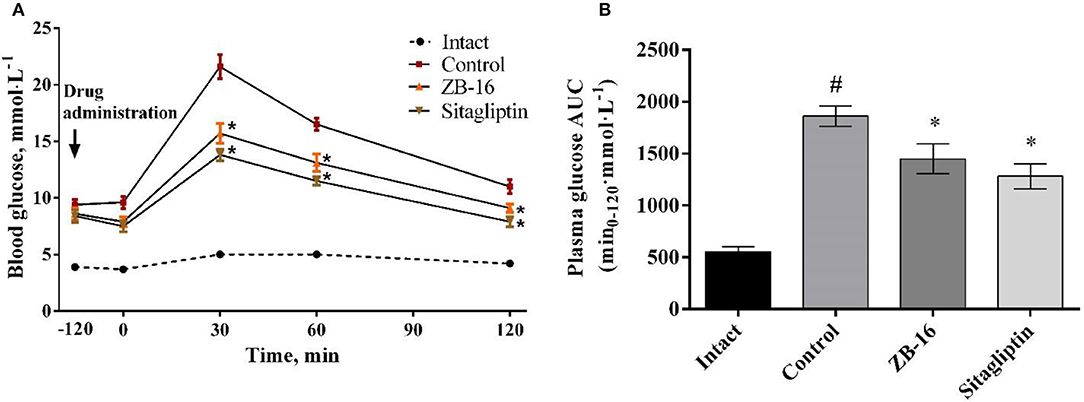
Figure 7. Blood glucose levels during oral glucose tolerance test (A), and the area under the “glucose concentration-time” (B) in rats with T2D, obtained after 28 days of treatment. The data expressed as the mean ± SEM (n = 10); Intact, intact group of animals; Control, group of animals with experimental diabetes without treatment; *p < 0.05, significant, compared to the Control group; #p < 0.05 – compared to the Intact group (2-way ANOVA with Newman-Keuls post-hoc).
Compared to the control group 22% reduction in the area under the “glucose concentration-time” curve was observed in ZB-16 group (Figure 7B). The effects of ZB-16 were comparable to the hypoglycemic action of sitagliptin.
Pharmacokinetic Parameters of Leader Compounds
The pharmacokinetics of ZB-16 was studied in rats after single intravenous or oral administration in a dose of 10 mg/kg (Tables 8, 9).

Table 8. Pharmacokinetic parameters of ZB-16 in rats after i.v. injection (n = 3).

Table 9. Pharmacokinetic parameters of ZB-16 in rats after oral administration (n = 3).
ZB-16 demonstrated relatively low plasma exposition, high distribution volume, mild clearance and a prolonged half-life (more than 12 h). The median absorption time (MAT) of ZB-16 was calculated using average MRT values and the following formula: MAT = MRTp.o. − MRTi.v.. The absolute bioavailability of the ZB-16 was 8%.
Discussion
Diabetes mellitus (DM) is increasingly recognized as a worldwide public health concern. The prevalence of DM has experienced unprecedented growth over the past years. In 1998, King et al. (28) predicted an increase in patients with DM from 135 to 300 million by 2025. Although this prediction seemed to be quite pessimistic, the current population of people affected by DM is foundto reach 425 million (1).
The main challenge faced by many researchers is the lack of effective treatment for T2D due to various etiological, pathogenetic and social causes resulting from the individual characteristics of the course of the disease in the patient and his/her lifestyle (1). However the list of anti-diabetic medications, which are the major way to establish the T2D management, is continuously updating. To date, standards of medical care in diabetes include such pharmacological groups as secretagogues (sulfonylureas), sensitizers (biguanides, thiazolidinediones), glycosurics (inhibitors of the sodium glucose cotransporter type 2), alpha-glucosidase inhibitors, and peptide analogs (GLP-1 analogs and agonists or DPP-4 inhibitors), as well as human or recombinant insulin (29).
In recent years, there has been a growing number of publications focusing on the problem of exogenous influence on the incretin system (30–32). Incretins are gastrointestinal-derived hormones released in response to a meal playing a key role in the regulation of postprandial secretion of insulin and glucagon by the pancreas (33). The interest in the incretin system is based on the fact that the incretin effect is severely reduced or absent in patients with T2D (34). Thus, the restoration of adequate incretin biosynthesis and metabolism could become a promising treatment strategy for T2D (35). In particular, this approach includes the development of drugs able to stimulate the incretin secretion via activation of GPCR, found on the intestinal enteroendocrine cells. This group of receptors acts as the sensors of fatty acids, their derivates and some other digestion products. The activation of such receptors leads to the stimulation of incretin secretion, which, in turn, stimulates the synthesis and secretion of insulin, providing a state of postprandial normoglycemia (36, 37). In particular, the activation of GPR119, expressed in L- and K-cells of intestine as well as in pancreatic β-cells (38), leads to glucose-depended activation of insulin secretion (4, 5). Such mechanism of GPR119 agonists action is proposed to be beneficial because it could provide a pronounced antihyperglycemic effect in T2D without the risk of excess hypoglycemia. Therefore, these substances are considered as promising candidates for the role of drugs for T2D treatment (9).
Previous non-clinical studies and investigations performed in healthy volunteers has established that GPR119 are able to increase the level of circulating incretins including GLP-1, GIP, and tyrosine-tyrosine peptide (PYY), reducing the hyperglycemia after oral glucose load (39). Furthermore, animal studies demonstrated several secondary pharmacodynamic effects including cerebral, cardiac and endothelial protection which are in contrast to the anti-diabetic medications, which have only the hupoglycemic action. These secondary (or “pleiotropic”) effects could make GPR119 agonists essential for the prevention of T2D complications (40, 41).
Following the trends described above this work was to elucidate the particular steps of early non-clinical development of GPR119 agonists as a potential oral hypoglycemic drugs for human use.
The synthesis of test compounds was based on the results of computer modeling. Following this step the most active structure—ZB-09 was developed and consequentially modified to improve its ADME parameters. A highly-lipophilic tert-butoxycarbonyl group was found metabolically unstable. Furthermore, it was chemically unstable, particularly in acid medium, which limits oral bioavailability. Thus, the bioisostere of the tert-butoxycarbonyl group must meet requirements related to size, electronic density, polar distribution and solubility.
The synthesis of ZB-17 was performed by replacement of the tert-butoxycarbonyl group by other urethane fragment—an isopropyl-oxycarbonyl group.
The nonclassical bioisostere of the tert-butoxycarbonyl group is a heterocyclic aromatic structure, where nitrogen atoms replace the oxygen atoms of oxycarbonyl and additional modifications alter the lipophilicity. In the resulting ZB-16 compound, chloropyrimidine replaces urethane near the nitrogen.
We also attempted to change the size of the replacement group without changing its lipophilicity and polarity, which appeared to be possible due to the replacement of the tert-butoxycarbonyl group with a trifluoroethyl group. In this case, the additional fluorine atoms on the aliphatic fragment play a double role: first of all, they modify lipophilicity (more fluorine atoms means higher lipophilicity); secondly, due to their electronegativity, they increase the electrophilic effect of the replacement, bringing the “electronic action” of the alkyl replacement close to that of the oxycarbonyl replacement (i.e., the nitrogen in this group is not basic like the alkyl group). Therefore, the ZB-18 structure was introduced.
The presence of a highly lipophilic fluorine-phenyl structure fragment contributes to the low solubility of the end compound in aqueous solutions. The phenyl bioisostere with slightly higher hydrophilicity is pyridine due to the potential formation of hydrogen bonds near nitrogen atom. Likewise, a ZB-19 compound was designed in which fluorine-phenyl was replaced with pyridine. The ending tert-butyl-oxycarbonyl group, which is primarily responsible for the low microsomal stability, was replaced with chlorine-pyrimidine in the ZB-20 compound.
The data obtained in vitro indicate that all the ZB-09 analogs synthesized have activities in the Arena (0119) standard range. Replacement of the tert-butyl-oxycarbonyl group with its possible bioisosteres did not lead to significant loss of activity (except for a slight decrease in activity in the case of the trifluoroethyl group).
The solubility data obtained for the compounds synthesized indicate that replacing the tert-butyl-oxycarbonyl group to an isopropyl-oxy-carbonyl group did not increase the water solubility in solutions of any pH studied. However, replacing this group on the trifluoroethyl group, and especially on chloropyrimidine, increased solubility in acidic media (pH 2). This may improve the compound's pharmacokinetics and stomach absorption. Therefore, due to its solubility, ZB-16 is the most effective compound investigated. Nonetheless, we decided to further investigate 2 compounds from the series (ZB-19 and ZB-20) due to their high agonistic activity.
ZB-16 exhibits significantly higher resistance in human and rat microsomes compared to ZB-09, the primary compound of the series. Thus, bioisostere replacement of the tert-butyl-oxycarbonyl group with a nonclassical bioisostere such as chloropyrimidine enabled us to increase both problematic parameters of the series: low water solubility (which increases as the pH decreases) and microsomal stability.
ZB-16 exerted nanomolar agonist activity on human GPR119 receptors associated with the absence of cytotoxic effects in standard tests for cellular toxicity. The study of the hypoglycemic activity of ZB-16 in intact animals and animals with experimental diabetes in cases of single and chronic (4 weeks) administration, revealed an extensive hypoglycemic effect and decreased glucose tolerance as assessed by oral glucose tolerance test. This action is associated with a previously reported increase of GLP-1 release. Thus, this study produced results which are consistent with the findings of a great deal of our previous work on the chronic oral administration of ZB-16 to male rats with STZ-NA-induced T2D, demonstrated that ZB-16 exerts a pronounced antidiabetic activity due to the improvement of glucose utilization by stimulating GLP-1 and insulin secretion, as well as protective action on pancreatic β-cells (11).
In conclusion, the present study demonstrates that the targeted search for selective GPR119 receptor agonists is a valid approach for developing novel drugs for the therapy of T2D. Based on the data presented, it can be argued that the GPR119 agonists have a high therapeutic potential. High in vitro activity (compared to competitor standards), a useful ADME profile, distinct hypoglycemic activity comparable to the hypoglycemic activity of sitagliptin in an experimental T2D and the resulting acceptable pharmacokinetic profile, allow us to recommend the ZB-16 compound for further research.
Author Contributions
All authors contributed to the study's conception and design. MC and RK carried out the chemical synthesis and in vitro studies. IT, DK, EV, EM, and DB performed the in vivo analysis. All authors were involved in the analysis and interpretation of data, and each contributed to critical revision of the manuscript. All authors provided final approval of the version to be published.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
These studies were supported by the Ministry of Industry and Trade of the Russian Federation as the part of a governmental contract (Preclinical studies of drugs based on GPR119 receptor agonists for diabetes mellitus treatment № 13411.1008799.154, July 24, 2013).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2018.00543/full#supplementary-material
Abbreviations
ADME, Absorption, Distribution, Metabolism and Excretion; DPP-4, Dipeptidyl peptidase-4; GLP-1, Glucagon-like peptide-1; GPR119, G protein-coupled receptor 119; T2D, type 2 diabetes mellitus; STZ-NA, streptozotocin-nicotinamide.
References
1. International Diabetes Federation. IDF Diabetes Atlas. 8th ed. Brussels: International Diabetes Federation (2017) Available online at: http://www.diabetesatlas.org
2. Ning Y, O'Neill K, Lan H, Pang L, Shan LX, Hawes BE, et al. Endogenous and synthetic agonists of GPR119 differ in signalling pathways and their effects on insulin secretion in MIN6c4 insulinoma cells. Br J Pharmacol. (2008) 155:1056–65. doi: 10.1038/bjp.2008.337
3. Henquin JC. Pathways in beta-cell stimulus-secretion coupling as targets for therapeutic insulin secretagogues. Diabetes (2004) 53:S48–58. doi: 10.2337/diabetes.53.suppl_3.s48
4. Fyfe MC, McCormack JG, Overton HA, Procter MJ, Reynet C. GPR119 agonists as potential new oral agents for the treatment of type 2 diabetes and obesity. Expert Opin Drug Discov. (2008) 3:403–13. doi: 10.1517/17460441.3.4.403
5. Chu ZL, Carroll C, Alfonso J, Gutierrez V, He H, Lucman A, et al. A role for intestinal endocrine cell-expressed g protein-coupled receptor 119 in glycemic control by enhancing glucagon-like Peptide-1 and glucose-dependent insulinotropic Peptide release. Endocrinology (2008) 149:2038–47. doi: 10.1210/en.2007-0966
6. Ahren B. Incretin dysfunction in type 2 diabetes: clinical impact and future perspectives. Diabetes Metab. (2013) 39:195–201. doi: 10.1016/j.diabet.2013.03.001
7. Lund A, Vilsbøll T, Bagger JI, Holst JJ, Knop FK. The separate and combined impact of the intestinal hormones, GIP, GLP-1, and GLP-2, on glucagon secretion in type 2 diabetes. Am J Physiol Endocrinol Metab. (2011) 300:E1038–46. doi: 10.1152/ajpendo.00665.2010
8. Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. (2007) 132:2131–57. doi: 10.1053/j.gastro.2007.03.054
9. Ritter K, Buning C, Halland N, Pöverlein C, Schwink L. G protein-coupled receptor 119 (GPR119) agonists for the treatment of diabetes: recent progress and prevailing challenges. J Med Chem. (2016) 59:3579–92. doi: 10.1021/acs.jmedchem.5b01198
10. Tyurenkov IN, Ozerov AA, Kurkin DV, Logvinova EO, Bakulin DA, Volotova EV, et al. Structure and biological activity of endogenous and synthetic agonists of GPR119. Russ Chem Rev. (2018) 87:151–66. doi: 10.1070/rcr4737
11. Tyurenkov IN, Kurkin DV, Bakulin DA, Volotova EV, Chafeev MA, Smirnov AV, et al. ZB-16, a Novel GPR119 agonist, relieves the severity of Streptozotocin-Nicotinamide-induced diabetes in rats. Front Endocrinol. (2017) 8:152. doi: 10.3389/fendo.2017.00152
12. Baka E, Comer JE, Takács-Novák K. Study of equilibrium solubility measurement by saturation shake-flask method using hydrochlorothiazide as model compound. J Pharm Biomed Anal. (2008) 46(2):335–41. doi: 10.1016/j.jpba.2007.10.030
13. LANCE® Ultra cAMP Kit - PerkinElmer. Available online at: https://www.perkinelmer.com/lab-solutions/resources/docs/MAN_TRF0262_TRF0263_TRF0264_v6.pdf
14. Soga T, Ohishi T, Matsui T, Saito T, Matsumoto M, Takasaki J, et al. Lysophosphatidylcholine enhances glucose-dependent insulin secretion via an orphan G-protein-coupled receptor. Biochem Biophys Res Commun. (2005) 326:744–51. doi: 10.1016/j.bbrc.2004.11.120
15. Chuang CK, Lin HY, Wang TJ, Tsai CC, Liu HL, Lin SP. A modified liquid chromatography/tandem mass spectrometry method for predominant disaccharide units of urinary glycosaminoglycans in patients with mucopolysaccharidoses. Orphanet J Rare Dis. (2014) 9:135. doi: 10.1186/s13023-014-0135-3
16. Vivid CYP450 Screening Kits User Guide - Thermo Fisher Scientific. Available online at: https://assets.thermofisher.com/TFS-Assets/LSG/brochures/VividScreeningKitManual24Apr20121.pdf
17. Pierce CG, Saville SP, Lopez-Ribot JL. High-content phenotypic screenings to identify inhibitors of Candida albicans biofilm formation and filamentation. Pathog Dis. (2014) 70:423–31. doi: 10.1111/2049-632x.12161
18. Drew ME, Morris JC, Wang Z, Wells L, Sanchez M, Landfear SM, et al. The adenosine analog tubercidin inhibits glycolysis in Trypanosoma brucei as revealed by an RNA interference library. J Biol Chem. (2003) 278:46596–600. doi: 10.1074/jbc.m309320200
19. Held P. Invitrogen's Predictor™ hERG Fluorescence Polarization Assay. Using BioTek's Synergy™ 4 Hybrid Microplate Reader. Non-Radioactive High Throughput Screening Assay for hERG Binding. Available online at: https://www.biotek.com/resources/single.html?newsid=8235
20. Russian Federation Federal Law no. 61-FZ “On Circulation of Medicines” dated April 12 (2010). Available online at: https://www.unodc.org/res/cld/document/rus/federal-law-on-circulation-of-medicines_html/Russian_Federation_Federal_Law_On_Circulation_of_Medicines_61-FZ_EN.pdf
21. Mironov AN. Guidelines for Preclinical Trials of Medicinal Products. Part 1. Moscow: Grif i K (2012). 944 p.
22. Guide for the Care and Use of Laboratory Animals. 8th ed. Washington, DC: National Academies Press US, (2011).
23. OECD Guidance Document on Acute Oral Toxicity. Environmental Health and Safety. Monograph Series on Testing and Assessment No 24. (2000). Available online at: https://ntp.niehs.nih.gov/iccvam/suppdocs/feddocs/oecd/oecd-gd24.pdf
24. Masiello P, Broca C, Gross R, Roye M, Manteghetti M, Hillaire-Buys D, et al. Experimental NIDDM: development of a new model in adult rats administered streptozotocin and nicotinamide. Diabetes (1988) 47:224–9. doi: 10.2337/diabetes.47.2.224
25. Szkudelski T. Streptozotocin-nicotinamide-induced diabetes in the rat. Characteristics of the experimental model. Exp Biol Med. (2012) 237:481–90. doi: 10.1258/ebm.2012.011372
26. Ferreira L, Teixeira-de-Lemos E, Pinto F, Parada B, Mega C, Vala H. Effects of sitagliptin treatment on dysmetabolism, inflammation, and oxidative stress in an animal model of type 2 diabetes (ZDF rat). Mediators Inflamm. (2010) 2010:1–11. doi: 10.1155/2010/592760
27. De Bruin ML, Pettersson M, Meyboom RH, Hoes AW, Leufkens HG. Anti-HERG activity and the risk of drug-induced arrhythmias and sudden death. Eur Heart J. (2005) 26:590–7. doi: 10.1016/j.accreview.2005.08.075
28. King H, Aubert RE, Herman WH. Global burden of diabetes, 1995-2025: prevalence, numerical estimates, and projections. Diabetes Care (1998) 21:1414–31. doi: 10.2337/diacare.21.9.1414
29. American Diabetes Association. Standards of Medical Care in Diabetes, with Notable New Recommendations for People with Cardiovascular Disease and Diabetes. (2018) Available online at: http://www.diabetes.org/newsroom/press-releases/2017/american-diabetes-association-2018-release-standards-of-medical-care-in-diabetes.html
30. Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. (2013) 17:819–37. doi: 10.1016/j.cmet.2013.04.008
31. DeFronzo RA, Ferrannini E, Zimmet P, Alberti KGMM (eds.). International Textbook of Diabetes Mellitus. Oxford: Wiley-Blackwell Ltd. (2015).
32. Li Y, Li L, Hölscher C. Incretin-based therapy for type 2 diabetes mellitus is promising for treating neurodegenerative diseases. Rev Neurosci. (2016) 27:689–711. doi: 10.1515/revneuro-2016-0018
33. João AL, Reis F, Fernandes R. The incretin system ABCs in obesity and diabetes - novel therapeutic strategies for weight loss and beyond. Obes Rev. (2016) 17:553–72. doi: 10.1111/obr.12421
34. Holst JJ, Vilsbøll T, Deacon CF. The incretin system and its role in type 2 diabetes mellitus. Mol Cell Endocrinol. (2009) 297:127–36. doi: 10.1016/j.mce.2008.08.012
35. Watterson KR, Hudson BD, Ulven T, Milligan G. Treatment of type 2 diabetes by free Fatty Acid receptor agonists. Front Endocrinol. (2014) 5:137. doi: 10.3389/fendo.2014.00137
36. Poulsen Ll, Siersbæk M, Mandrup S. PPARs: fatty acid sensors controlling metabolism. Semin Cell Dev Biol. (2012) 23:631–9. doi: 10.1016/j.semcdb.2012.01.003
37. Kebede MA, Alquier T, Latour MG, Poitout V. Lipid receptors and islet function: therapeutic implications? Diabetes Obes Metab. (2009) 11:10–20. doi: 10.1111/j.1463-1326.2009.01114.x
38. Hothersall JD, Bussey CE, Brown AJ, Scott JS, Dale I, Rawlins P. Sustained wash-resistant receptor activation responses of GPR119 agonists. Eur J Pharmacol. (2015) 762:430–42. doi: 10.1016/j.ejphar.2015.06.031
39. Nunez DJ, Bush MA, Collins DA, McMullen SL, Gillmor D, Apseloff G, et al. Gut hormone pharmacology of a novel GPR119 agonist (GSK1292263), metformin, and sitagliptin in type 2 diabetes mellitus: results from two randomized studies. PLoS ONE (2014) 9:e92494. doi: 10.1371/journal.pone.0092494
40. Saraiva FK, Sposito AC. Cardiovascular effects of glucagon-like peptide 1 (GLP-1) receptor agonists. Cardiovasc Diabetol. (2014) 13:142. doi: 10.1186/s12933-014-0142-7
Keywords: ZB-16, sitagliptin, T2D, hypoglycemia, GPR119 agonists, rats
Citation: Tyurenkov IN, Kurkin DV, Bakulin DA, Volotova EV, Morkovin EI, Chafeev MA and Karapetian RN (2018) Chemistry and Hypoglycemic Activity of GPR119 Agonist ZB-16. Front. Endocrinol. 9:543. doi: 10.3389/fendo.2018.00543
Received: 03 June 2018; Accepted: 28 August 2018;
Published: 19 September 2018.
Edited by:
Åke Sjöholm, Gävle Hospital, SwedenReviewed by:
Sneha Shah, Harvard Medical School, United StatesAlex Rafacho, Universidade Federal de Santa Catarina, Brazil
Rajakrishnan Veluthakal, City of Hope National Medical Center, United States
Copyright © 2018 Tyurenkov, Kurkin, Bakulin, Volotova, Morkovin, Chafeev and Karapetian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dmitry A. Bakulin, bWJmZG9jQGdtYWlsLmNvbQ==